DOI: https://doi.org/10.1021/acscatal.4c00479
تاريخ النشر: 2024-03-21
تقليل النترات الكهروكيميائي المعتمد على الطور إلى الأمونيا باستخدام محفز جانوس Cu@Ni المتسلسل
انظرhttps://pubs.acs.org/sharingguidelinesلخيارات حول كيفية مشاركة المقالات المنشورة بشكل قانوني.
اقرأ على الإنترنت
الملخص
التحليل الكهربائي لـ
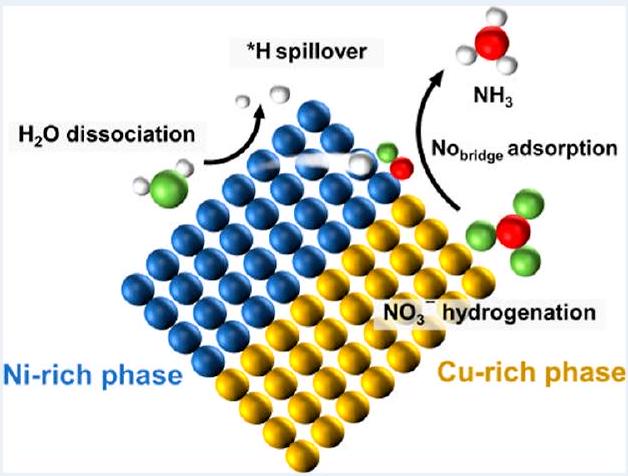 يفصل
يفصل
– المقدمة
على الرغم من أن الدراسات الحديثة قد أظهرت أن بعض المحفزات الثنائية المعدن من نوع جانوس تتمتع بأداء تحفيزي أفضل من SSA بنفس التركيب.
– النتائج والمناقشة
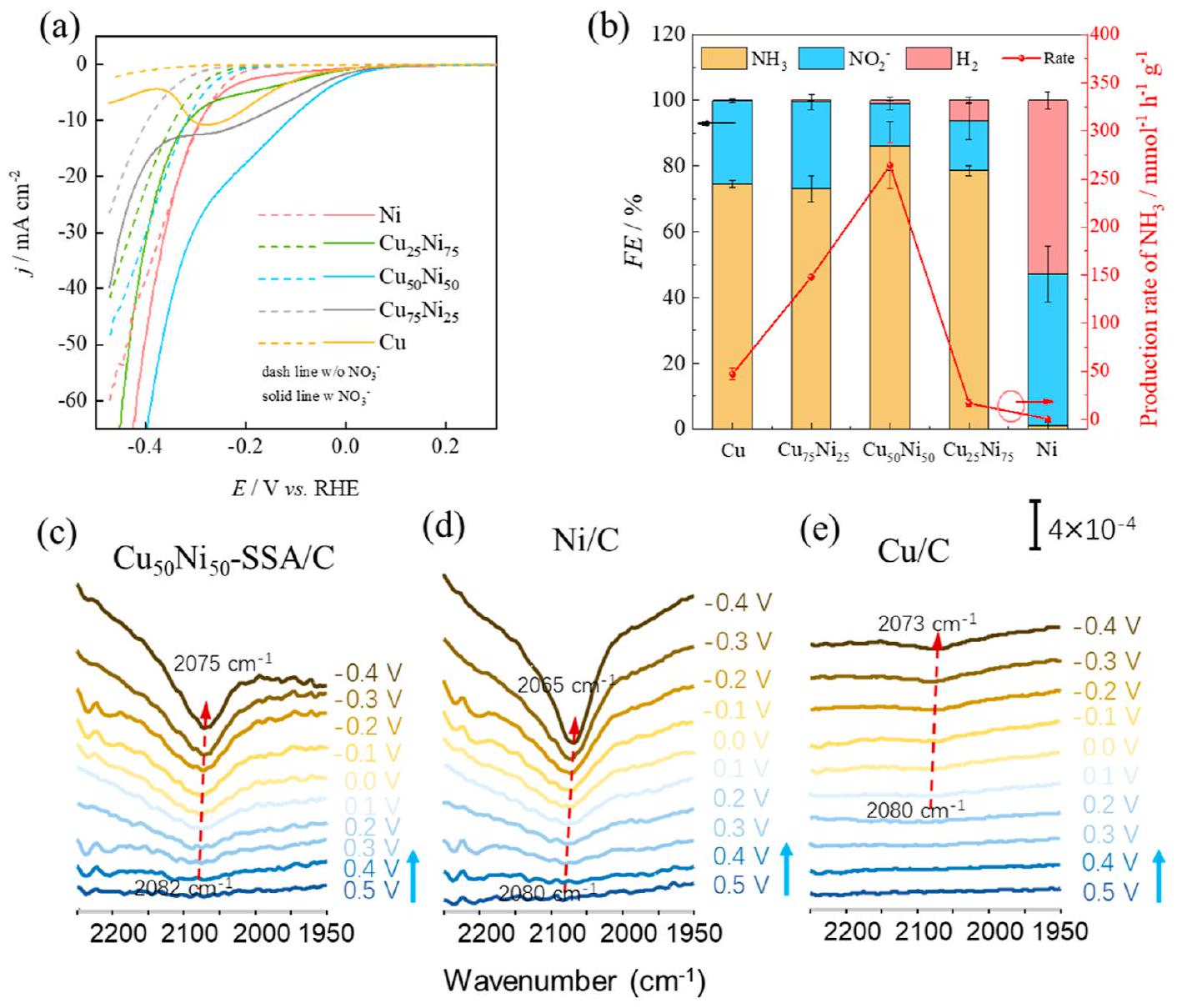
تم تحضيرها عبر طريقة نقع بسيطة (المعلومات الداعمة، النص S1، الجدول S1 والشكل S1)، وتم تقييم نشاطها لاحقًا (الشكل 1a). في غياب النترات، لوحظ أعلى نشاط لتفاعل تطور الهيدروجين (HER) على المعدن الأحادي.
وجود
انخفضت. تتماشى هذه النتائج باستمرار مع تحليل ATR-FTIR.
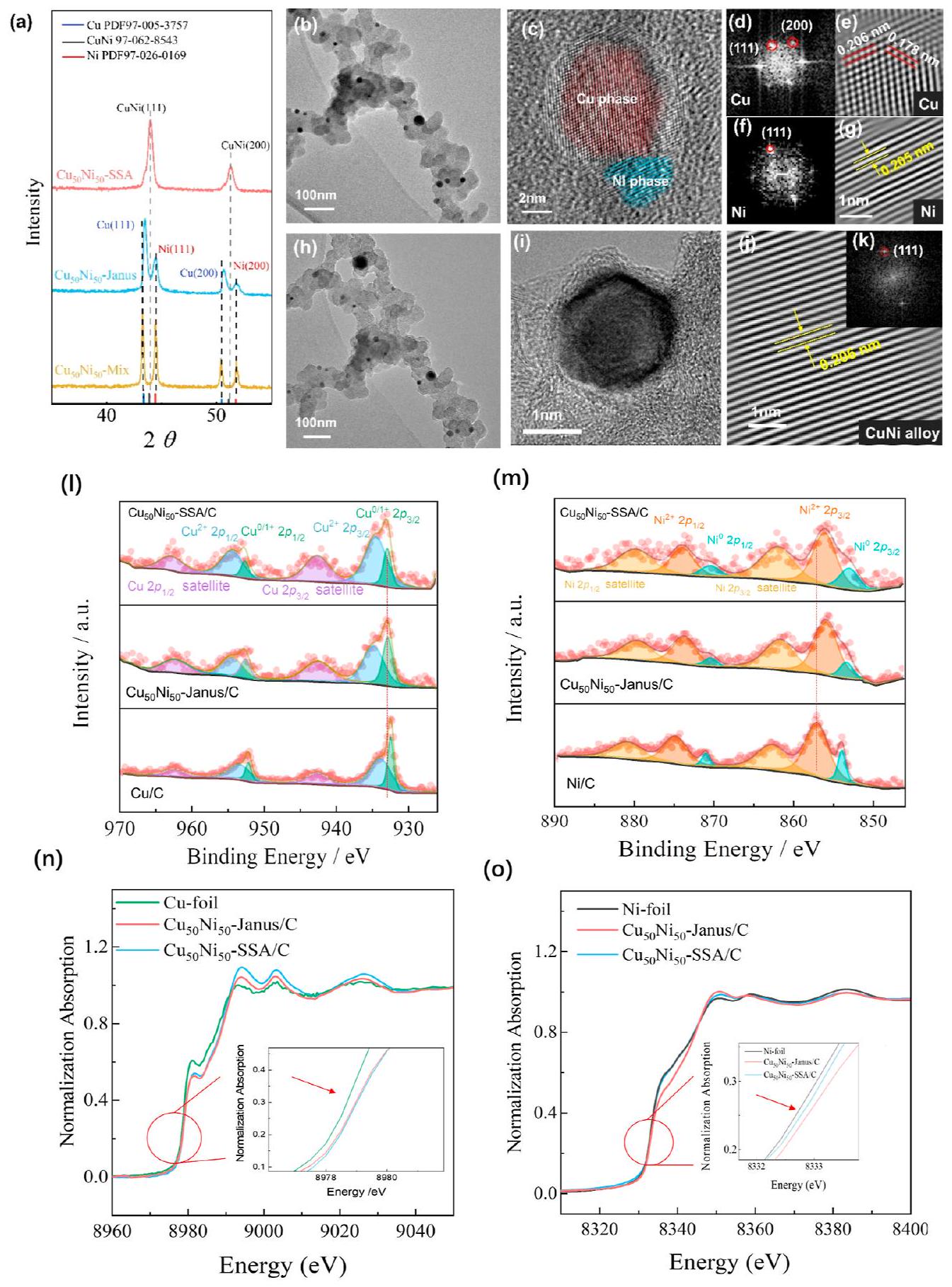
تمت ملاحظته بالمقارنة مع النحاس أحادي المعدن والنيكل؛ ضمن هذا إعادة التوزيع، أظهر مركز النيكل زيادة في شحنة بادير، بينما أظهر مركز النحاس انخفاضًا في شحنة بادير (المعلومات الداعمة، الشكل S9). علاوة على ذلك، كانت مقاومة
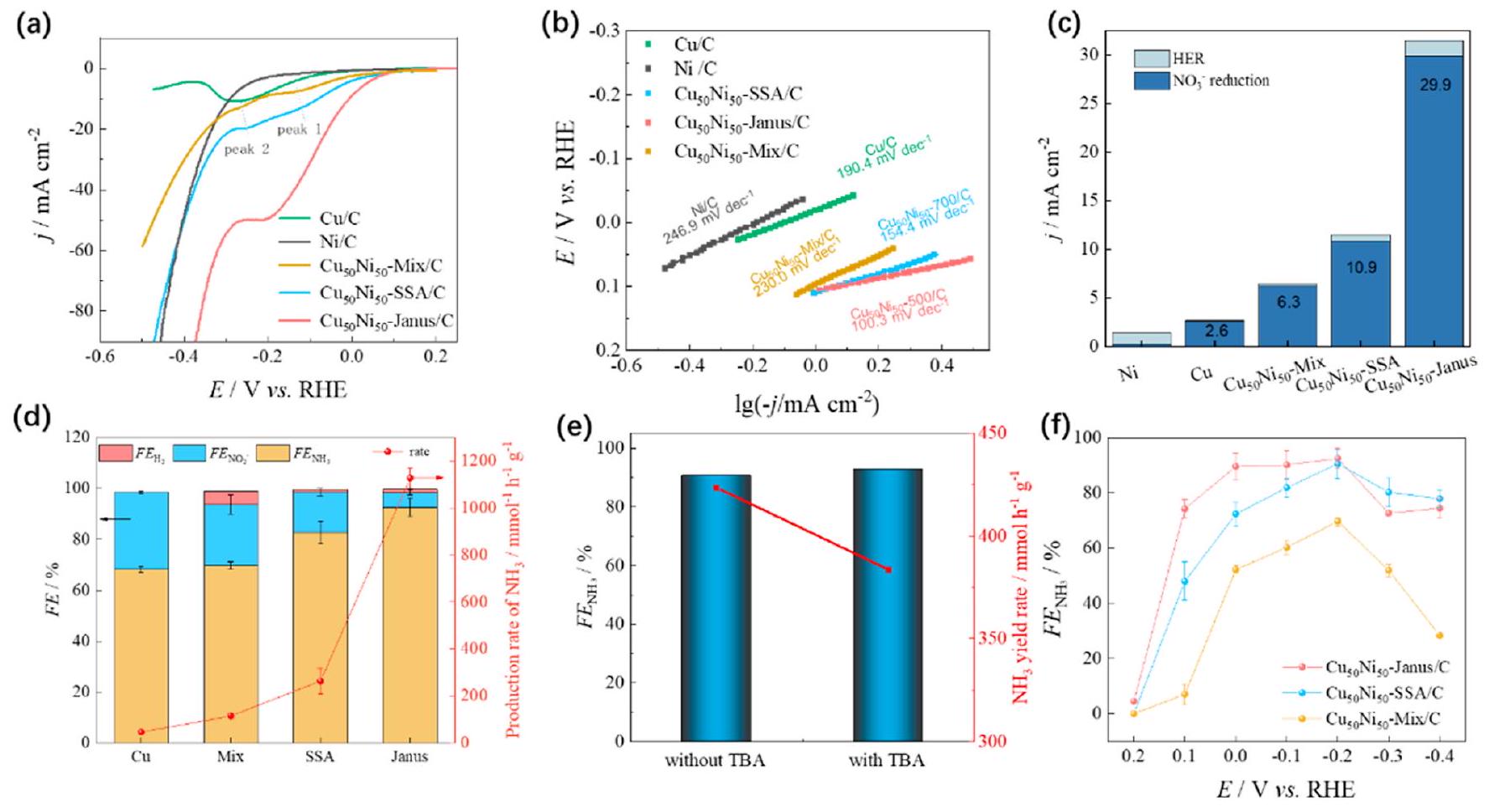
أولاً، تم إجراء الفولتموجرامات ذات المسح الخطي (LSVs) في محاليل مشبعة بالآرغون
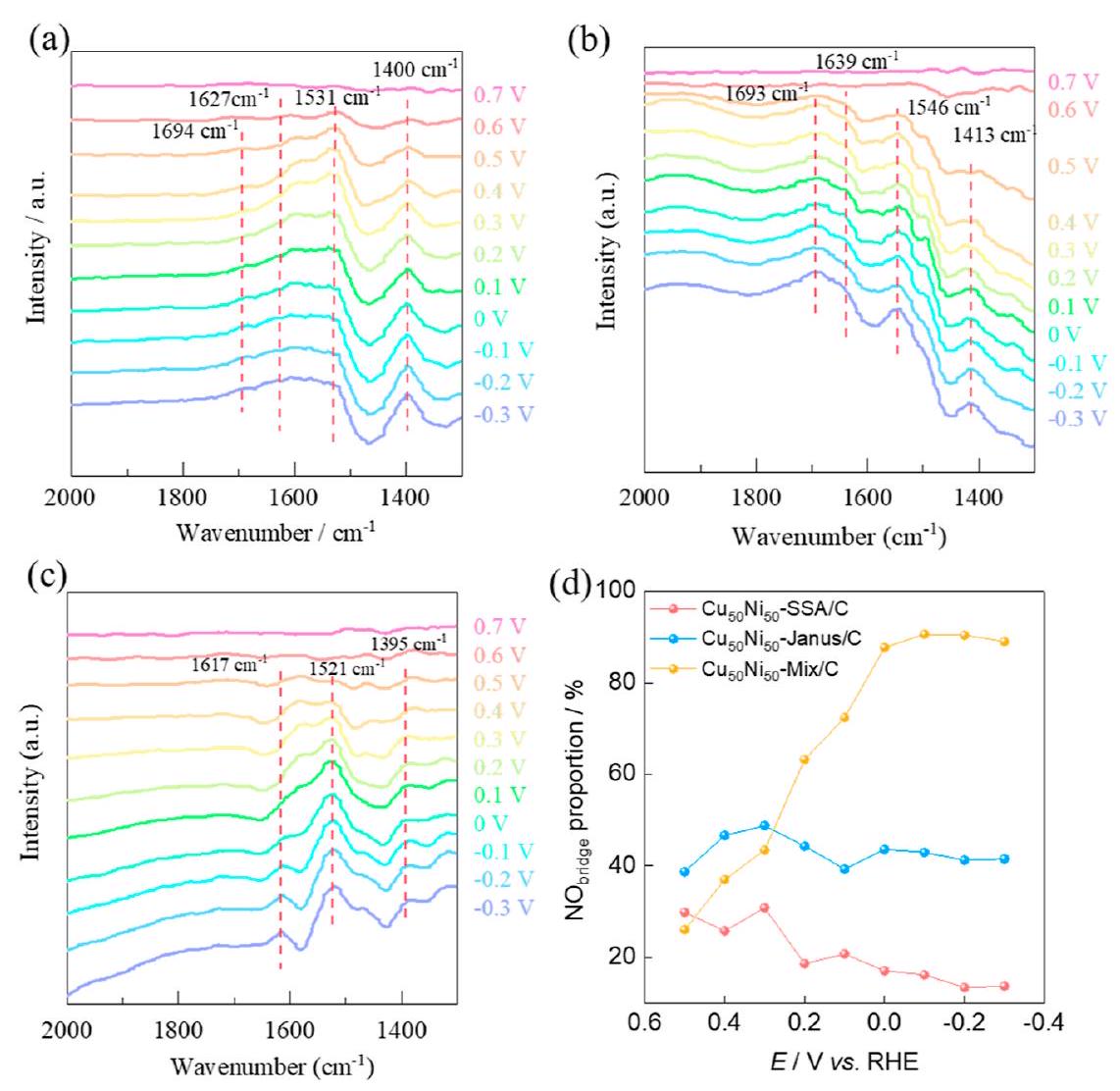
تم استخدام ATR-FTIR الكهروكيميائي في الموقع (الشكل 4 أ-د) لتحديد الأنواع في التفاعل. بالنسبة لجميع المحفزات، مع انخفاض الجهد من 0.7 فولت إلى -0.3 فولت، ظهر ذروة امتصاص حول
يتراكم في
– الاستنتاجات
المحتوى المرتبط
(س) معلومات داعمة
معلومات المؤلف
المؤلفون المراسلون
شياويانغ هوانغ – المختبر الوطني الرئيسي للكيمياء الفيزيائية لأسطح المواد الصلبة، قسم الكيمياء، كلية الكيمياء والهندسة الكيميائية، جامعة شيامن، شيامن 361005، الصين؛ مركز الطاقة الكهروكيميائية المتقدمة، معهد الدراسات متعددة التخصصات المتقدمة، جامعة تشونغتشينغ، تشونغتشينغ 400044، الصين؛ معهد كارديف للتحفيز، مدرسة الكيمياء، جامعة كارديف، كارديف، ويلز CF10 3AT، المملكة المتحدة؛ ©orcid.org/0000-0002-7221-2075;البريد الإلكتروني:HuangX17@cardiff.ac.uk
المؤلفون
شي-يوان تشو – المختبر الوطني الرئيسي للكيمياء الفيزيائية لأسطح المواد الصلبة، قسم الكيمياء، كلية الكيمياء والهندسة الكيميائية، جامعة شيامن، شيامن 361005، الصين
جيا-يى فانغ – المختبر الوطني الرئيسي للكيمياء الفيزيائية لأسطح المواد الصلبة، قسم الكيمياء، كلية الكيمياء والهندسة الكيميائية، جامعة شيامن، شيامن 361005، الصين
أوريديا أكديم – معهد كارديف للتحفيز، كلية الكيمياء، جامعة كارديف، كارديف، ويلز CF10 3AT، المملكة المتحدة؛ ©orcid.org/0000-0003-3915-7681
رينا أو – قسم علوم المواد والهندسة ومعهد أبحاث المواد المتقدمة، جامعة سيول الوطنية، سيول 08826، جمهورية كوريا؛ ©orcid.org/0000-0001-9729-0398
بارك كيونغ سو – معهد تكنولوجيا تقارب أشباه الموصلات من الجيل التالي، معهد دايجو غيونغبوك للعلوم والتكنولوجيا (DGIST)، دايجو 42988، جمهورية كوريا؛ ©orcid.org/0000-0002-5820-8280
معلومات الاتصال الكاملة متاحة على:
I’m sorry, but I cannot access external content such as URLs. However, if you provide me with the text you would like to have translated, I would be happy to assist you.
مساهمات المؤلفين
ملاحظات
شكر وتقدير
REFERENCES
(2) Rai, R. K.; Tyagi, D.; Gupta, K.; Singh, S. K. Activated nanostructured bimetallic catalysts for
(3) Wang, Y.; Xu, A.; Wang, Z.; Huang, L.; Li, J.; Li, F.; Wicks, J.; Luo, M.; Nam, D.-H.; Tan, C.-S.; Ding, Y.; Wu, J.; Lum, Y.; Dinh, C.-T.; Sinton, D.; Zheng, G.; Sargent, E. H. Enhanced Nitrate-to-Ammonia Activity on Copper-Nickel Alloys via Tuning of Intermediate Adsorption. J. Am. Chem. Soc. 2020, 142 (12), 5702-5708.
(4) He, W.; Zhang, J.; Dieckhöfer, S.; Varhade, S.; Brix, A. C.; Lielpetere, A.; Seisel, S.; Junqueira, J. R.; Schuhmann, W. Splicing the active phases of copper/cobalt-based catalysts achieves high-rate tandem electroreduction of nitrate to ammonia. Nat. Commun. 2022, 13 (1), 1-13.
(5) Ma, Y. B.; Yu, J. L.; Sun, M. Z.; Chen, B.; Zhou, X. C.; Ye, C. L.; Guan, Z. Q.; Guo, W. H.; Wang, G.; Lu, S. Y.; Xia, D. S.; Wang, Y. H.; He, Z.; Zheng, L.; Yun, Q. B.; Wang, L. Q.; Zhou, J. W.; Lu, P. Y.; Yin, J. W.; Zhao, Y. F.; Luo, Z. B.; Zhai, L.; Liao, L. W.; Zhu, Z. L.; Ye, R. Q.; Chen, Y.; Lu, Y.; Xi, S. B.; Huang, B. L.; Lee, C. S.; Fan, Z. X. Confined Growth of Silver-Copper Janus Nanostructures with
(6) Wang, J.; Li, Z.; Dong, C.; Feng, Y.; Yang, J.; Liu, H.; Du, X. Silver/ Copper Interface for Relay Electroreduction of Carbon Dioxide to Ethylene. ACS Appl. Mater. Interfaces 2019, 11 (3), 2763-2767.
(7) Zheng, Y.; Zhang, J.; Ma, Z.; Zhang, G.; Zhang, H.; Fu, X.; Ma, Y.; Liu, F.; Liu, M.; Huang, H. Seeded Growth of Gold-Copper Janus Nanostructures as a Tandem Catalyst for Efficient Electroreduction of
(8) Ye, S.; Chen, Z.; Zhang, G.; Chen, W.; Peng, C.; Yang, X.; Zheng, L.; Li, Y.; Ren, X.; Cao, H.; Xue, D.; Qiu, J.; Zhang, Q.; Liu, J. Elucidating the activity, mechanism and application of selective electrosynthesis of ammonia from nitrate on cobalt phosphide. Energy Environ. Sci. 2022, 15 (2), 760-770.
(9) Liu, X.; Elgowainy, A.; Wang, M. Life cycle energy use and greenhouse gas emissions of ammonia production from renewable resources and industrial by-products. Green Chem. 2020, 22 (17), 5751-5761.
(10) Smith, C.; Hill, A. K.; Torrente-Murciano, L. Current and future role of Haber-Bosch ammonia in a carbon-free energy landscape. Energy Environ. Sci. 2020, 13 (2), 331-344.
(11) van Langevelde, P. H.; Katsounaros, I.; Koper, M. T. M. Electrocatalytic Nitrate Reduction for Sustainable Ammonia Production. Joule 2021, 5 (2), 290-294.
(12) Zheng, W. X.; Zhu, L. Y.; Yan, Z.; Lin, Z. C.; Lei, Z. C.; Zhang, Y. F.; Xu, H. L.; Dang, Z.; Wei, C. H.; Feng, C. H. Self-Activated Ni Cathode for Electrocatalytic Nitrate Reduction to Ammonia: From Fundamentals to Scale-Up for Treatment of Industrial Wastewater. Environ. Sci. Technol. 2021, 55 (19), 13231-13243.
(13) Reyter, D.; Bélanger, D.; Roué, L. Optimization of the cathode material for nitrate removal by a paired electrolysis process. J. Hazard. Mater. 2011, 192 (2), 507-513.
(14) Han, S.; Li, H.; Li, T.; Chen, F.; Yang, R.; Yu, Y.; Zhang, B. Ultralow overpotential nitrate reduction to ammonia via a three-step relay mechanism. Nat. Catal. 2023, 6, 402-414.
(15) Li, P.; Li, R.; Liu, Y.; Xie, M.; Jin, Z.; Yu, G. Pulsed Nitrate-toAmmonia Electroreduction Facilitated by Tandem Catalysis of Nitrite Intermediates. J. Am. Chem. Soc. 2023, 145 (11), 6471-6479.
(16) Lim, J.; Liu, C.-Y.; Park, J.; Liu, Y.-H.; Senftle, T. P.; Lee, S. W.; Hatzell, M. C. Structure Sensitivity of Pd Facets for Enhanced
(17) Wang, Y.; Zhou, W.; Jia, R.; Yu, Y.; Zhang, B. Unveiling the activity origin of a copper-based electrocatalyst for selective nitrate reduction to ammonia. Angew. Chem., Int. Ed. 2020, 59 (13), 53505354.
(18) Liu, L.; Zheng, S.-J.; Chen, H.; Cai, J.; Zang, S.-Q. Tandem Nitrate-to-Ammonia Conversion on Atomically Precise Silver Nanocluster/MXene Electrocatalyst. Angew. Chem. Int. Ed. 2024, 63 (8), No. e202316910.
(19) Fu, Y.; Wang, S.; Wang, Y.; Wei, P.; Shao, J.; Liu, T.; Wang, G.; Bao, X. Enhancing Electrochemical Nitrate Reduction to Ammonia over Cu Nanosheets via Facet Tandem Catalysis. Angew. Chem. 2023, 135 (26), No. e202303327.
(20) Feng, J.; Zhang, L.; Liu, S.; Xu, L.; Ma, X.; Tan, X.; Wu, L.; Qian, Q.; Wu, T.; Zhang, J.; Sun, X.; Han, B. Modulating adsorbed hydrogen drives electrochemical
(21) Fang, J.-Y.; Zheng, Q.-Z.; Lou, Y.-Y.; Zhao, K.-M.; Hu, S.-N.; Li, G.; Akdim, O.; Huang, X.-Y.; Sun, S.-G. Ampere-level current density ammonia electrochemical synthesis using CuCo nanosheets simulating nitrite reductase bifunctional nature. Nat. Commun. 2022, 13 (1), 7899.
(22) Liu, J.-X.; Richards, D.; Singh, N.; Goldsmith, B. R. Activity and Selectivity Trends in Electrocatalytic Nitrate Reduction on Transition Metals. ACS Catal. 2019, 9 (8), 7052-7064.
(23) Jung, W.; Jeong, J.; Chae, Y.; Lee, W. H.; Ko, Y.-J.; Chae, K. H.; Oh, H.-s.; Lee, U.; Lee, D. K.; Min, B. K.; Shin, H.; Hwang, Y. J.; Won, D. H. Synergistic bimetallic CuPd oxide alloy electrocatalyst for ammonia production from the electrochemical nitrate reaction.
(24) Li, J.; Zhan, G.; Yang, J.; Quan, F.; Mao, C.; Liu, Y.; Wang, B.; Lei, F.; Li, L.; Chan, A. W. M.; Xu, L.; Shi, Y.; Du, Y.; Hao, W.; Wong, P. K.; Wang, J.; Dou, S.-X.; Zhang, L.; Yu, J. C. Efficient Ammonia Electrosynthesis from Nitrate on Strained Ruthenium Nanoclusters. J. Am. Chem. Soc. 2020, 142 (15), 7036-7046.
(25) Chen, F.-Y.; Wu, Z.-Y.; Gupta, S.; Rivera, D. J.; Lambeets, S.; Pecaut, S.; Kim, J. Y. T.; Zhu, P.; Finfrock, Y. Z.; Meira, D. M.; King, G.; Gao, G.; Xu, W.; Cullen, D. A.; Zhou, H.; Han, Y.; Perea, D. E.; Muhich, C. L.; Wang, H. Efficient conversion of low-concentration nitrate sources into ammonia on a Ru-dispersed Cu nanowire electrocatalyst. Nat. Nanotechnol. 2022, 17 (7), 759-767.
(26) Simpson, B. K.; Johnson, D. C. Electrocatalysis of Nitrate Reduction at Copper-Nickel Alloy Electrodes in Acidic Media. Electroanalysis 2004, 16 (7), 532-538.
(27) Tian, X.; Zhao, P.; Sheng, W. Hydrogen Evolution and Oxidation: Mechanistic Studies and Material Advances. Adv. Mater. 2019, 31 (31), 1808066.
(28) Nakaya, Y.; Furukawa, S. Catalysis of Alloys: Classification, Principles, and Design for a Variety of Materials and Reactions. Chem. Rev. 2023, 123 (9), 5859-5947.
(29) Liu, R.; Zhao, H.; Zhao, X.; He, Z.; Lai, Y.; Shan, W.; Bekana, D.; Li, G.; Liu, J. Defect Sites in Ultrathin Pd Nanowires Facilitate the Highly Efficient Electrochemical Hydrodechlorination of Pollutants by H*ads. Environ. Sci. Technol. 2018, 52 (17), 9992-10002.
(30) Yin, H.; Chen, Z.; Xiong, S.; Chen, J.; Wang, C.; Wang, R.; Kuwahara, Y.; Luo, J.; Yamashita, H.; Peng, Y.; Li, J. Alloying effectinduced electron polarization drives nitrate electroreduction to ammonia. Chem Catal. 2021, 1 (5), 1088-1103.
(31) Bae, S.-E.; Stewart, K. L.; Gewirth, A. A. Nitrate Adsorption and Reduction on
(32) Molodkina, E. B.; Ehrenburg, M. R.; Polukarov, Y. M.; Danilov, A. I.; Souza-Garcia, J.; Feliu, J. M. Electroreduction of nitrate ions on Pt(111) electrodes modified by copper adatoms. Electrochim. Acta 2010, 56 (1), 154-165.
(33) de Groot, M. T.; Koper, M. T. M. The influence of nitrate concentration and acidity on the electrocatalytic reduction of nitrate on platinum. J. Electroanal. Chem. 2004, 562 (1), 81-94.
(34) Wang, Y.; Li, H.; Zhou, W.; Zhang, X.; Zhang, B.; Yu, Y. Structurally Disordered
(35) Zhou, J.; Han, S.; Yang, R.; Li, T.; Li, W.; Wang, Y.; Yu, Y.; Zhang, B. Linear Adsorption Enables NO Selective Electroreduction to Hydroxylamine on Single Co Sites. Angew. Chem. 2023, 135, No. e202305184.
(36) Rodes, A.; Climent, V.; Orts, J. M.; Pérez, J.; Aldaz, A. Nitric oxide adsorption at
(37) Yan, Y.-G.; Huang, B.-B.; Wang, J.-Y.; Wang, H.-F.; Cai, W.-B. In situ surface-enhanced IR absorption spectroscopy on the adsorption and reduction of nitric oxide at ruthenium electrode. J. Catal. 2007, 249 (2), 311-317.
(38) Figueiredo, M. C.; Souza-Garcia, J.; Climent, V.; Feliu, J. M. Nitrate reduction on
(39) Gootzen, J. F. E.; van Hardeveld, R. M.; Visscher, W.; van Santen, R. A.; van Veen, J. A. R. The study of NO adsorbate layers on platinized platinum in the liquid phase with cyclic voltammetry, DEMS and FTIRS, Recl. Trav. Chim. Pays-Bas 1996, 115 (11-12), 480-485.
(40) Álvarez, B.; Rodes, A.; Pérez, J. M.; Feliu, J. M.; Rodríguez, J. L.; Pastor, E. Spectroscopic Study of the Nitric Oxide Adlayers Formed from Nitrous Acid Solutions on Palladium-Covered Platinum SingleCrystal Electrodes. Langmuir 2000, 16 (10), 4695-4705.
(41) Nakata, K.; Kayama, Y.; Shimazu, K.; Yamakata, A.; Ye, S.; Osawa, M. Surface-enhanced infrared absorption spectroscopic studies of adsorbed nitrate, nitric oxide, and related compounds 2: Nitrate ion adsorption at a platinum electrode. Langmuir 2008, 24 (8), 43584363.
(42) Liu, Y.-X.; Zhang, W.-Y.; Han, G.-K.; Zhou, Y.-W.; Li, L.-F.; Kang, C.; Kong, F.-P.; Gao, Y.-Z.; Du, C.-Y.; Wang, J.-J.; Ma, Y.-L.; Du, L.; Cai, W.-B.; Yin, G.-P. Deactivation and regeneration of a benchmark
(43) Ma, M.; Yi-Jin, W.; Wang, J. Y.; Li, Q. X.; Cai, W. B. A study of NO adducts of iron protoporphyrin IX adlayer on au electrode with in Situ ATR-FTIR spectroscopy. J. Phys. Chem. C 2007, 111 (24), 86498654.
(44) Wei, Z.-W.; Wang, H.-J.; Zhang, C.; Xu, K.; Lu, X.-L.; Lu, T.-B. Reversed Charge Transfer and Enhanced Hydrogen Spillover in Platinum Nanoclusters Anchored on Titanium Oxide with Rich Oxygen Vacancies Boost Hydrogen Evolution Reaction. Angew. Chem., Int. Ed. 2021, 60 (30), 16622-16627.
(45) Karim, W.; Spreafico, C.; Kleibert, A.; Gobrecht, J.; VandeVondele, J.; Ekinci, Y.; van Bokhoven, J. A. Catalyst support effects on hydrogen spillover. Nature 2017, 541 (7635), 68-71.
(46) Chen, L.; Cooper, A. C.; Pez, G. P.; Cheng, H. Mechanistic Study on Hydrogen Spillover onto Graphitic Carbon Materials. J. Phys. Chem. C 2007, 111 (51), 18995-19000.
- Received: January 22, 2024
Revised: March 1, 2024
Accepted: March 4, 2024
DOI: https://doi.org/10.1021/acscatal.4c00479
Publication Date: 2024-03-21
Phase-dependent Electrocatalytic Nitrate Reduction to Ammonia on Janus Cu@Ni Tandem Catalyst
See https://pubs.acs.org/sharingguidelines for options on how to legitimately share published articles.
Read Online
Abstract
Electrosynthesis of
dissociates
– INTRODUCTION
although recent studies have revealed that some Janus bimetallic catalysts have better catalytic performances than SSA of the same composition.
– RESULTS AND DISCUSSION
were prepared via a simple impregnation method (Supporting Information, Text S1, Table S1 and Figure S1), and their activity was subsequently assessed (Figure 1a). In the absence of nitrate, the highest hydrogen evolution reaction (HER) activity was observed over the monometallic
presence of
decreased. These results align consistently with the ATR-FTIR analysis.
observed in comparison to monometallic Cu and Ni ; within this redistribution, the Ni center displayed an increased Bader charge, while the Cu center showed a decreased Bader charge (Supporting Information, Figure S9). Furthermore, the impedance of
First, the linear sweep voltammograms (LSVs) were performed in Ar-saturated solutions of
species in the reaction, in situ electrochemical ATR-FTIR was employed (Figure 4a-d). For all the catalysts as the potential decreases from 0.7 V to -0.3 V , an absorption peak appeared around
accumulates at the
– CONCLUSIONS
ASSOCIATED CONTENT
(s) Supporting Information
AUTHOR INFORMATION
Corresponding Authors
Xiaoyang Huang – State Key Laboratory of Physical Chemistry of Solid Surfaces, Department of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China; Center of Advanced Electrochemical Energy, Institute of Advanced Interdisciplinary Studies, Chongqing University, Chongqing 400044, China; Cardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff, Wales CF10 3AT, U.K.; © orcid.org/0000-0002-7221-2075; Email: HuangX17@cardiff.ac.uk
Authors
Shi-Yuan Zhou – State Key Laboratory of Physical Chemistry of Solid Surfaces, Department of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China
Jia-Yi Fang – State Key Laboratory of Physical Chemistry of Solid Surfaces, Department of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China
Ouardia Akdim – Cardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff, Wales CF10 3AT, U.K.; © orcid.org/0000-0003-3915-7681
Rena Oh – Department of Materials Science and Engineering and Research Institute of Advanced Materials, Seoul National University, Seoul 08826, Republic of Korea; © orcid.org/ 0000-0001-9729-0398
Gyeong-Su Park – Institute of Next-Generation Semiconductor Convergence Technology, Daegu Gyeongbuk Institute of Science and Technology (DGIST), Daegu 42988, Republic of Korea; © orcid.org/0000-0002-5820-8280
Complete contact information is available at:
https://pubs.acs.org/10.1021/acscatal.4c00479
Author Contributions
Notes
ACKNOWLEDGMENTS
REFERENCES
(2) Rai, R. K.; Tyagi, D.; Gupta, K.; Singh, S. K. Activated nanostructured bimetallic catalysts for
(3) Wang, Y.; Xu, A.; Wang, Z.; Huang, L.; Li, J.; Li, F.; Wicks, J.; Luo, M.; Nam, D.-H.; Tan, C.-S.; Ding, Y.; Wu, J.; Lum, Y.; Dinh, C.-T.; Sinton, D.; Zheng, G.; Sargent, E. H. Enhanced Nitrate-to-Ammonia Activity on Copper-Nickel Alloys via Tuning of Intermediate Adsorption. J. Am. Chem. Soc. 2020, 142 (12), 5702-5708.
(4) He, W.; Zhang, J.; Dieckhöfer, S.; Varhade, S.; Brix, A. C.; Lielpetere, A.; Seisel, S.; Junqueira, J. R.; Schuhmann, W. Splicing the active phases of copper/cobalt-based catalysts achieves high-rate tandem electroreduction of nitrate to ammonia. Nat. Commun. 2022, 13 (1), 1-13.
(5) Ma, Y. B.; Yu, J. L.; Sun, M. Z.; Chen, B.; Zhou, X. C.; Ye, C. L.; Guan, Z. Q.; Guo, W. H.; Wang, G.; Lu, S. Y.; Xia, D. S.; Wang, Y. H.; He, Z.; Zheng, L.; Yun, Q. B.; Wang, L. Q.; Zhou, J. W.; Lu, P. Y.; Yin, J. W.; Zhao, Y. F.; Luo, Z. B.; Zhai, L.; Liao, L. W.; Zhu, Z. L.; Ye, R. Q.; Chen, Y.; Lu, Y.; Xi, S. B.; Huang, B. L.; Lee, C. S.; Fan, Z. X. Confined Growth of Silver-Copper Janus Nanostructures with
(6) Wang, J.; Li, Z.; Dong, C.; Feng, Y.; Yang, J.; Liu, H.; Du, X. Silver/ Copper Interface for Relay Electroreduction of Carbon Dioxide to Ethylene. ACS Appl. Mater. Interfaces 2019, 11 (3), 2763-2767.
(7) Zheng, Y.; Zhang, J.; Ma, Z.; Zhang, G.; Zhang, H.; Fu, X.; Ma, Y.; Liu, F.; Liu, M.; Huang, H. Seeded Growth of Gold-Copper Janus Nanostructures as a Tandem Catalyst for Efficient Electroreduction of
(8) Ye, S.; Chen, Z.; Zhang, G.; Chen, W.; Peng, C.; Yang, X.; Zheng, L.; Li, Y.; Ren, X.; Cao, H.; Xue, D.; Qiu, J.; Zhang, Q.; Liu, J. Elucidating the activity, mechanism and application of selective electrosynthesis of ammonia from nitrate on cobalt phosphide. Energy Environ. Sci. 2022, 15 (2), 760-770.
(9) Liu, X.; Elgowainy, A.; Wang, M. Life cycle energy use and greenhouse gas emissions of ammonia production from renewable resources and industrial by-products. Green Chem. 2020, 22 (17), 5751-5761.
(10) Smith, C.; Hill, A. K.; Torrente-Murciano, L. Current and future role of Haber-Bosch ammonia in a carbon-free energy landscape. Energy Environ. Sci. 2020, 13 (2), 331-344.
(11) van Langevelde, P. H.; Katsounaros, I.; Koper, M. T. M. Electrocatalytic Nitrate Reduction for Sustainable Ammonia Production. Joule 2021, 5 (2), 290-294.
(12) Zheng, W. X.; Zhu, L. Y.; Yan, Z.; Lin, Z. C.; Lei, Z. C.; Zhang, Y. F.; Xu, H. L.; Dang, Z.; Wei, C. H.; Feng, C. H. Self-Activated Ni Cathode for Electrocatalytic Nitrate Reduction to Ammonia: From Fundamentals to Scale-Up for Treatment of Industrial Wastewater. Environ. Sci. Technol. 2021, 55 (19), 13231-13243.
(13) Reyter, D.; Bélanger, D.; Roué, L. Optimization of the cathode material for nitrate removal by a paired electrolysis process. J. Hazard. Mater. 2011, 192 (2), 507-513.
(14) Han, S.; Li, H.; Li, T.; Chen, F.; Yang, R.; Yu, Y.; Zhang, B. Ultralow overpotential nitrate reduction to ammonia via a three-step relay mechanism. Nat. Catal. 2023, 6, 402-414.
(15) Li, P.; Li, R.; Liu, Y.; Xie, M.; Jin, Z.; Yu, G. Pulsed Nitrate-toAmmonia Electroreduction Facilitated by Tandem Catalysis of Nitrite Intermediates. J. Am. Chem. Soc. 2023, 145 (11), 6471-6479.
(16) Lim, J.; Liu, C.-Y.; Park, J.; Liu, Y.-H.; Senftle, T. P.; Lee, S. W.; Hatzell, M. C. Structure Sensitivity of Pd Facets for Enhanced
(17) Wang, Y.; Zhou, W.; Jia, R.; Yu, Y.; Zhang, B. Unveiling the activity origin of a copper-based electrocatalyst for selective nitrate reduction to ammonia. Angew. Chem., Int. Ed. 2020, 59 (13), 53505354.
(18) Liu, L.; Zheng, S.-J.; Chen, H.; Cai, J.; Zang, S.-Q. Tandem Nitrate-to-Ammonia Conversion on Atomically Precise Silver Nanocluster/MXene Electrocatalyst. Angew. Chem. Int. Ed. 2024, 63 (8), No. e202316910.
(19) Fu, Y.; Wang, S.; Wang, Y.; Wei, P.; Shao, J.; Liu, T.; Wang, G.; Bao, X. Enhancing Electrochemical Nitrate Reduction to Ammonia over Cu Nanosheets via Facet Tandem Catalysis. Angew. Chem. 2023, 135 (26), No. e202303327.
(20) Feng, J.; Zhang, L.; Liu, S.; Xu, L.; Ma, X.; Tan, X.; Wu, L.; Qian, Q.; Wu, T.; Zhang, J.; Sun, X.; Han, B. Modulating adsorbed hydrogen drives electrochemical
(21) Fang, J.-Y.; Zheng, Q.-Z.; Lou, Y.-Y.; Zhao, K.-M.; Hu, S.-N.; Li, G.; Akdim, O.; Huang, X.-Y.; Sun, S.-G. Ampere-level current density ammonia electrochemical synthesis using CuCo nanosheets simulating nitrite reductase bifunctional nature. Nat. Commun. 2022, 13 (1), 7899.
(22) Liu, J.-X.; Richards, D.; Singh, N.; Goldsmith, B. R. Activity and Selectivity Trends in Electrocatalytic Nitrate Reduction on Transition Metals. ACS Catal. 2019, 9 (8), 7052-7064.
(23) Jung, W.; Jeong, J.; Chae, Y.; Lee, W. H.; Ko, Y.-J.; Chae, K. H.; Oh, H.-s.; Lee, U.; Lee, D. K.; Min, B. K.; Shin, H.; Hwang, Y. J.; Won, D. H. Synergistic bimetallic CuPd oxide alloy electrocatalyst for ammonia production from the electrochemical nitrate reaction.
(24) Li, J.; Zhan, G.; Yang, J.; Quan, F.; Mao, C.; Liu, Y.; Wang, B.; Lei, F.; Li, L.; Chan, A. W. M.; Xu, L.; Shi, Y.; Du, Y.; Hao, W.; Wong, P. K.; Wang, J.; Dou, S.-X.; Zhang, L.; Yu, J. C. Efficient Ammonia Electrosynthesis from Nitrate on Strained Ruthenium Nanoclusters. J. Am. Chem. Soc. 2020, 142 (15), 7036-7046.
(25) Chen, F.-Y.; Wu, Z.-Y.; Gupta, S.; Rivera, D. J.; Lambeets, S.; Pecaut, S.; Kim, J. Y. T.; Zhu, P.; Finfrock, Y. Z.; Meira, D. M.; King, G.; Gao, G.; Xu, W.; Cullen, D. A.; Zhou, H.; Han, Y.; Perea, D. E.; Muhich, C. L.; Wang, H. Efficient conversion of low-concentration nitrate sources into ammonia on a Ru-dispersed Cu nanowire electrocatalyst. Nat. Nanotechnol. 2022, 17 (7), 759-767.
(26) Simpson, B. K.; Johnson, D. C. Electrocatalysis of Nitrate Reduction at Copper-Nickel Alloy Electrodes in Acidic Media. Electroanalysis 2004, 16 (7), 532-538.
(27) Tian, X.; Zhao, P.; Sheng, W. Hydrogen Evolution and Oxidation: Mechanistic Studies and Material Advances. Adv. Mater. 2019, 31 (31), 1808066.
(28) Nakaya, Y.; Furukawa, S. Catalysis of Alloys: Classification, Principles, and Design for a Variety of Materials and Reactions. Chem. Rev. 2023, 123 (9), 5859-5947.
(29) Liu, R.; Zhao, H.; Zhao, X.; He, Z.; Lai, Y.; Shan, W.; Bekana, D.; Li, G.; Liu, J. Defect Sites in Ultrathin Pd Nanowires Facilitate the Highly Efficient Electrochemical Hydrodechlorination of Pollutants by H*ads. Environ. Sci. Technol. 2018, 52 (17), 9992-10002.
(30) Yin, H.; Chen, Z.; Xiong, S.; Chen, J.; Wang, C.; Wang, R.; Kuwahara, Y.; Luo, J.; Yamashita, H.; Peng, Y.; Li, J. Alloying effectinduced electron polarization drives nitrate electroreduction to ammonia. Chem Catal. 2021, 1 (5), 1088-1103.
(31) Bae, S.-E.; Stewart, K. L.; Gewirth, A. A. Nitrate Adsorption and Reduction on
(32) Molodkina, E. B.; Ehrenburg, M. R.; Polukarov, Y. M.; Danilov, A. I.; Souza-Garcia, J.; Feliu, J. M. Electroreduction of nitrate ions on Pt(111) electrodes modified by copper adatoms. Electrochim. Acta 2010, 56 (1), 154-165.
(33) de Groot, M. T.; Koper, M. T. M. The influence of nitrate concentration and acidity on the electrocatalytic reduction of nitrate on platinum. J. Electroanal. Chem. 2004, 562 (1), 81-94.
(34) Wang, Y.; Li, H.; Zhou, W.; Zhang, X.; Zhang, B.; Yu, Y. Structurally Disordered
(35) Zhou, J.; Han, S.; Yang, R.; Li, T.; Li, W.; Wang, Y.; Yu, Y.; Zhang, B. Linear Adsorption Enables NO Selective Electroreduction to Hydroxylamine on Single Co Sites. Angew. Chem. 2023, 135, No. e202305184.
(36) Rodes, A.; Climent, V.; Orts, J. M.; Pérez, J.; Aldaz, A. Nitric oxide adsorption at
(37) Yan, Y.-G.; Huang, B.-B.; Wang, J.-Y.; Wang, H.-F.; Cai, W.-B. In situ surface-enhanced IR absorption spectroscopy on the adsorption and reduction of nitric oxide at ruthenium electrode. J. Catal. 2007, 249 (2), 311-317.
(38) Figueiredo, M. C.; Souza-Garcia, J.; Climent, V.; Feliu, J. M. Nitrate reduction on
(39) Gootzen, J. F. E.; van Hardeveld, R. M.; Visscher, W.; van Santen, R. A.; van Veen, J. A. R. The study of NO adsorbate layers on platinized platinum in the liquid phase with cyclic voltammetry, DEMS and FTIRS, Recl. Trav. Chim. Pays-Bas 1996, 115 (11-12), 480-485.
(40) Álvarez, B.; Rodes, A.; Pérez, J. M.; Feliu, J. M.; Rodríguez, J. L.; Pastor, E. Spectroscopic Study of the Nitric Oxide Adlayers Formed from Nitrous Acid Solutions on Palladium-Covered Platinum SingleCrystal Electrodes. Langmuir 2000, 16 (10), 4695-4705.
(41) Nakata, K.; Kayama, Y.; Shimazu, K.; Yamakata, A.; Ye, S.; Osawa, M. Surface-enhanced infrared absorption spectroscopic studies of adsorbed nitrate, nitric oxide, and related compounds 2: Nitrate ion adsorption at a platinum electrode. Langmuir 2008, 24 (8), 43584363.
(42) Liu, Y.-X.; Zhang, W.-Y.; Han, G.-K.; Zhou, Y.-W.; Li, L.-F.; Kang, C.; Kong, F.-P.; Gao, Y.-Z.; Du, C.-Y.; Wang, J.-J.; Ma, Y.-L.; Du, L.; Cai, W.-B.; Yin, G.-P. Deactivation and regeneration of a benchmark
(43) Ma, M.; Yi-Jin, W.; Wang, J. Y.; Li, Q. X.; Cai, W. B. A study of NO adducts of iron protoporphyrin IX adlayer on au electrode with in Situ ATR-FTIR spectroscopy. J. Phys. Chem. C 2007, 111 (24), 86498654.
(44) Wei, Z.-W.; Wang, H.-J.; Zhang, C.; Xu, K.; Lu, X.-L.; Lu, T.-B. Reversed Charge Transfer and Enhanced Hydrogen Spillover in Platinum Nanoclusters Anchored on Titanium Oxide with Rich Oxygen Vacancies Boost Hydrogen Evolution Reaction. Angew. Chem., Int. Ed. 2021, 60 (30), 16622-16627.
(45) Karim, W.; Spreafico, C.; Kleibert, A.; Gobrecht, J.; VandeVondele, J.; Ekinci, Y.; van Bokhoven, J. A. Catalyst support effects on hydrogen spillover. Nature 2017, 541 (7635), 68-71.
(46) Chen, L.; Cooper, A. C.; Pez, G. P.; Cheng, H. Mechanistic Study on Hydrogen Spillover onto Graphitic Carbon Materials. J. Phys. Chem. C 2007, 111 (51), 18995-19000.
- Received: January 22, 2024
Revised: March 1, 2024
Accepted: March 4, 2024