DOI: https://doi.org/10.1038/s41467-024-45959-9
PMID: https://pubmed.ncbi.nlm.nih.gov/38368417
تاريخ النشر: 2024-02-17
تقليل ثاني أكسيد الكربون القابل لتحمل الأكسجين عبر الأطر العضوية التساهمية من خلال استراتيجية التحكم في التمرير الضوئي لتأمين الأكسجين
تم القبول: 8 فبراير 2024
نُشر على الإنترنت: 17 فبراير 2024
(د) التحقق من التحديثات
الملخص
الاستخدام المباشر لغاز العادم في العملية الكهروكيميائية
يجب أن يتم الحصول عليه من مصدر حقيقي
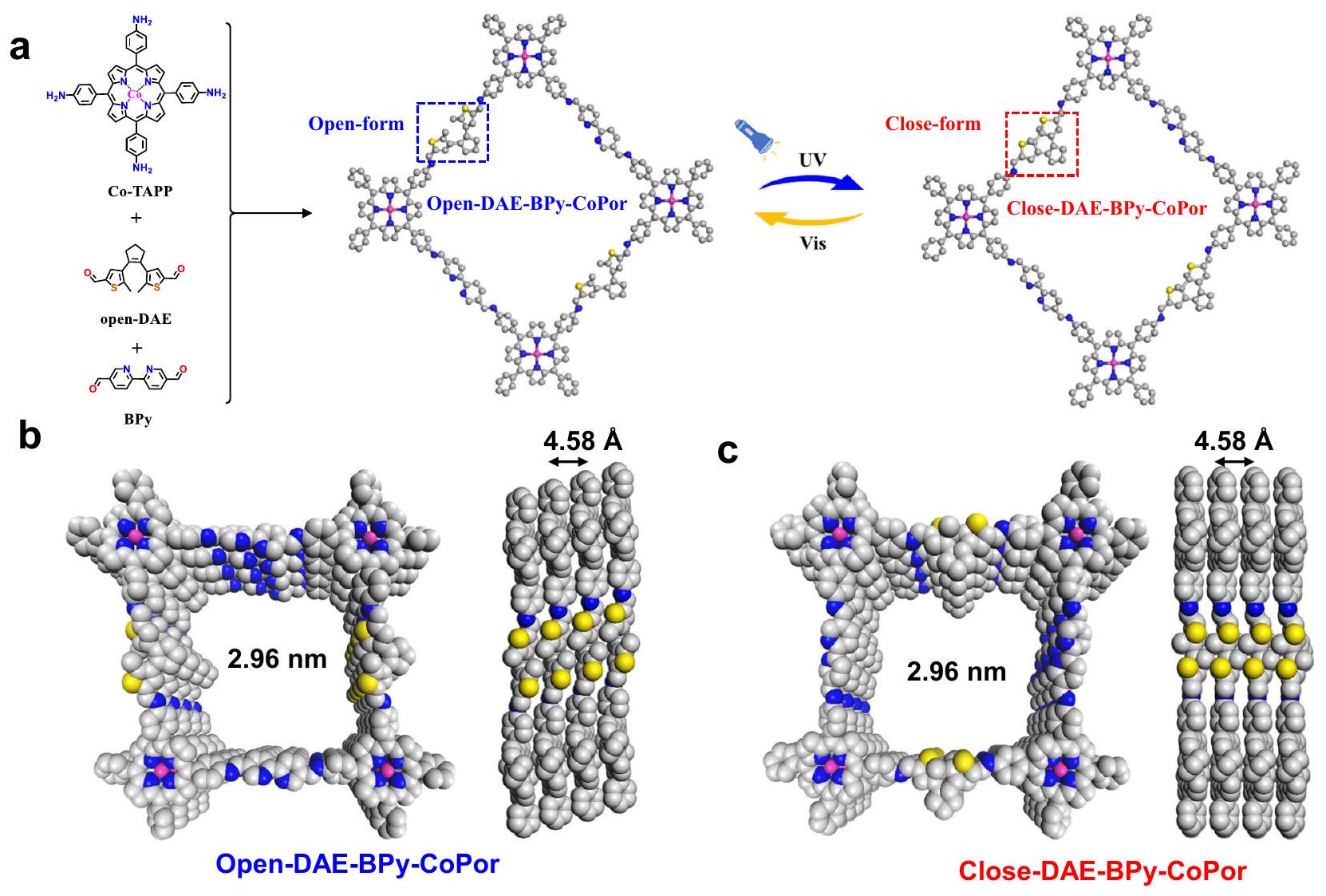
النتائج
تركيب وتوصيف DAE-BPy-CoPor
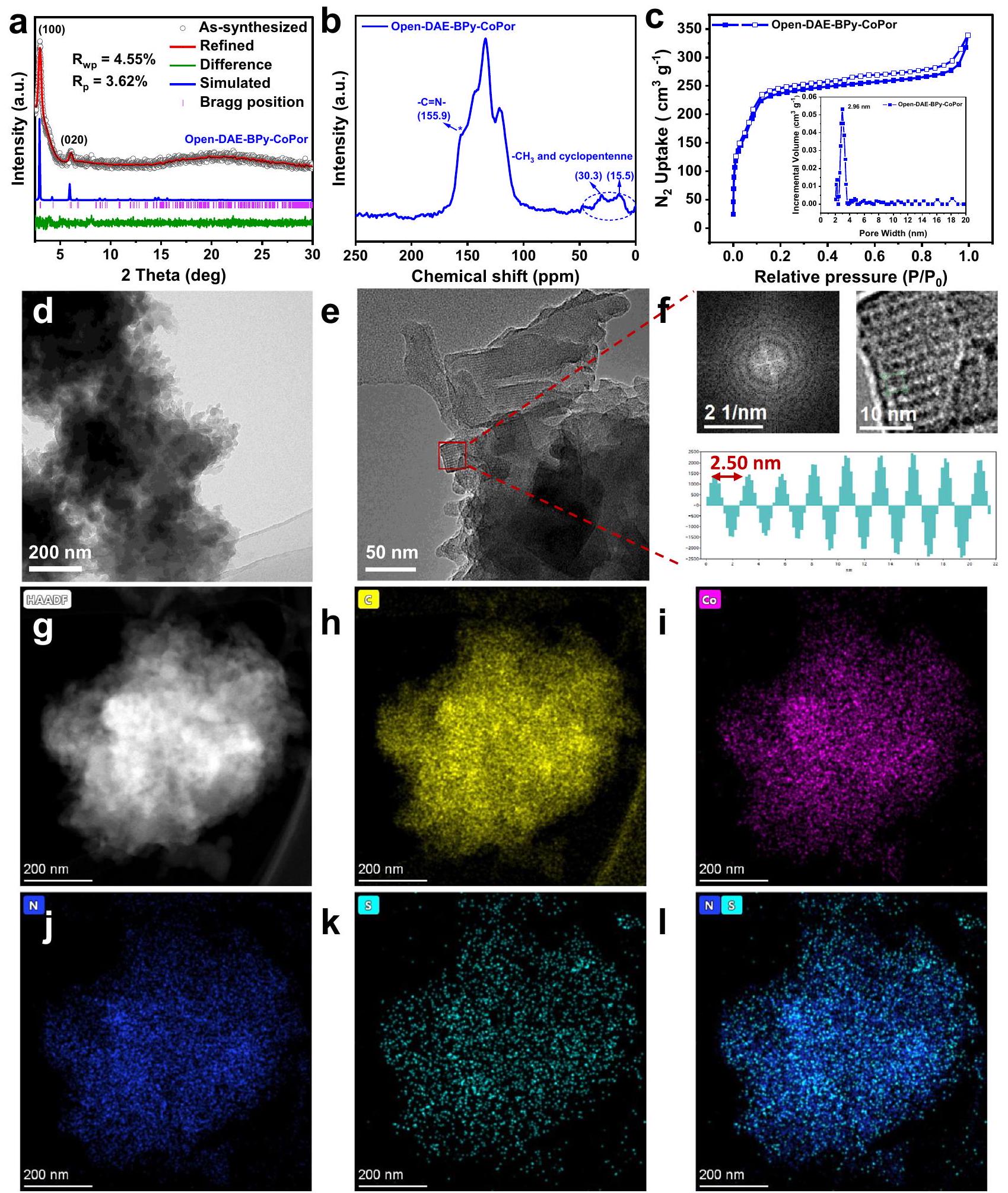
مقارنة بين نمط حيود الأشعة السينية للبودرة التجريبية مع أنماط PXRD المحاكاة. الحالة الصلبة
طاقات الربط المختلفة للإيزومرين
تم نسب الذروة عند 164.7 إلكترون فولت إلى الشكل القريب من DAE في close-DAE-BPy-CoPor. طيف XPS لـ
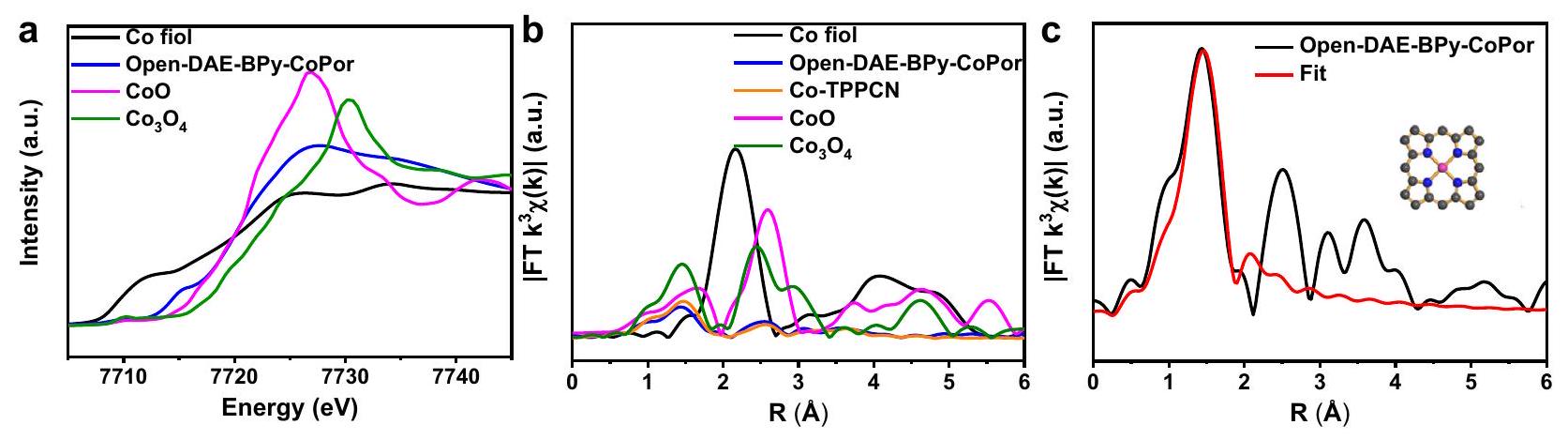
(الشكل التكميلي 15ب)، مما جعل من الصعب الكشف عن تفاعل الفوتوسايكلization لأجزاء DAE في COF. ومع ذلك، يمكن الكشف عن الفوتوسايكلization لمونومر open-DAE في الحالة الصلبة باستخدام UV-Vis. كما هو موضح في الشكل التكميلي 15أ، مقارنةً بطيف مونومر open-DAE، ظهر ذروة جديدة عند 640 نانومتر في طيف close-DAE، مما يدل على تشكيل الحالة المغلقة من مجموعة السيكلوهكسايدين في close-DAE-BPy-CoPor. علاوة على ذلك، أظهر ملف Co K-edge XANES لـ close-DAE-BPy-CoPor ميزة موجية مشابهة لتلك الخاصة بـ open-DAE-BPy-CoPor، مما يشير إلى أن حالة تأكسد مركز Co في close-DAE-BPy-CoPor مشابهة لتلك في open-DAE-BPy-CoPor. ومن الجدير بالذكر أنه مقارنةً بـ open-DAE-BPy-CoPor، انزاحت ذروة الامتصاص لـ close-DAE-BPy-CoPor عند حوالي 7715 eV إلى الجانب ذو الطاقة الأقل. هذا الانزياح يوحي بحالة تأكسد Co أقل قليلاً في close-DAE-BPy-CoPor مقارنةً بـ open-DAE-BPy-CoPor، مما يدل على وجود المزيد من الإلكترونات في مركز Co في close-DAE-BPy-CoPor. علاوة على ذلك، أظهر طيف EXAFS المحول فورييه لـ open-DAE-BPy-CoPor ذروة سائدة عند
التحفيز الكهربائي
مرشحين واعدين لـ
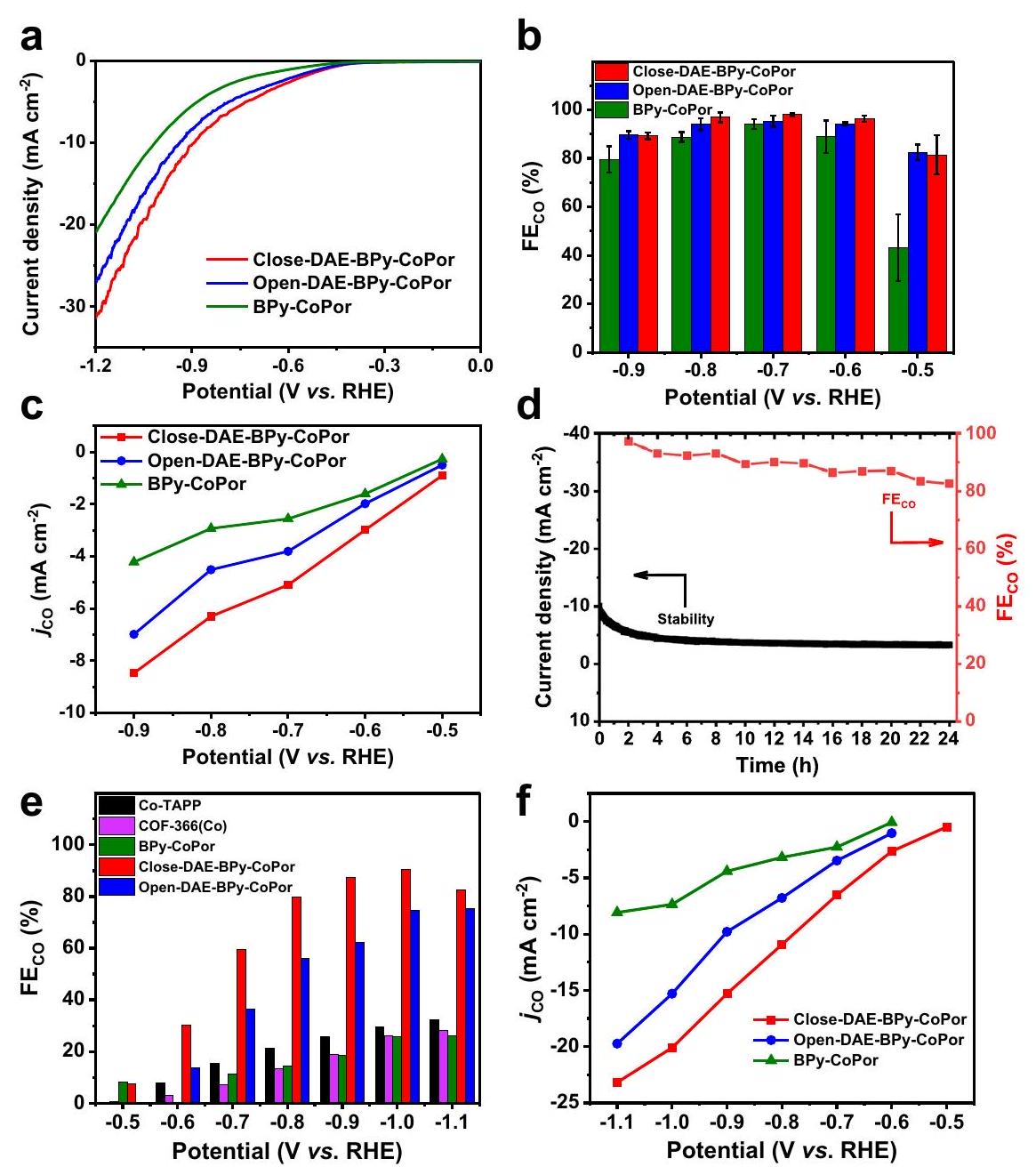
CoPor و close-DAE-BPy-CoPor و open-DAE-BPy-CoPor في التغذية المشتركة لـ CO2 و
الانخفاض الحالي
إلى -1.1 فولت. الأمثل
تمت في
حساب DFT وآلية التفاعل
طرق
تركيب BPy-CoPor
تركيب close-DAE-BPy-CoPor
المواد وإجراءات التصنيع
الخصائص والأدوات
تم قياسها باستخدام جهاز Micromeritics ASAP 2460.
القياسات الكهروكيميائية
أنا: إجمالي تيار الخلية في حالة الاستقرار؛
توفر البيانات
References
- Navarro-Jaén, S. et al. Highlights and challenges in the selective reduction of carbon dioxide to methanol. Nat. Rev. Chem. 5, 564-579 (2021).
- Ross, M. B. et al. Designing materials for electrochemical carbon dioxide recycling. Nat. Catal. 2, 648-658 (2019).
- Wu, Q.-J., Liang, J., Huang, Y.-B. & Cao, R. Thermo-, electro-, and photocatalytic
conversion to value-added products over porous metal/covalent organic frameworks. Acc. Chem. Res. 55, 2978-2997 (2022). - Zhu, H.-J. et al. Efficient electron transmission in covalent organic framework nanosheets for highly active electrocatalytic carbon dioxide reduction. Nat. Commun. 11, 497 (2020).
- Gong, Y.-N. et al. Regulating photocatalysis by spin-state manipulation of cobalt in covalent organic frameworks. J. Am. Chem. Soc. 142, 16723-16731 (2020).
- Zhou, J. et al. Linking oxidative and reductive clusters to prepare crystalline porous catalysts for photocatalytic
reduction with . Nat. Commun. 13, 4681 (2022). - Zhai, S. et al. High-capacity thermochemical
dissociation using iron-poor ferrites. Energy Environ. Sci. 13, 592-600 (2020). - Zhang, X. Y. et al. Operando metalloid
active sites for highly efficient carbon dioxide reduction electrocatalysis. Angew. Chem. Int. Ed. 61, e202202298 (2022). - Wang, Y., Liu, J. & Zheng, G. Designing copper-based catalysts for efficient carbon dioxide electroreduction. Adv. Mater. 33, 2005798 (2021).
- Wang, Y. et al. Local weak hydrogen bonds significantly enhance
electroreduction performances of a metal-organic framework. CCS Chem. 5, 145-151 (2023). - Service, R. F. Cost of carbon capture drops, but does anyone want it? Science 354, 1362-1363 (2016).
- Jouny, M., Luc, W. & Jiao, F. General techno-economic analysis of
electrolysis systems. Ind. Eng. Chem. Res. 57, 2165-2177 (2018). - Wang, J. & Ciucci, F. Boosting bifunctional oxygen electrolysis for N-doped carbon via bimetal addition. Small 13, 1604103 (2017).
- Han, B. et al. Two-dimensional covalent organic frameworks with cobalt (II)-Phthalocyanine sites for efficient electrocatalytic carbon dioxide reduction. J. Am. Chem. Soc. 143, 7104-7113 (2021).
- Lv, H. et al. Promoting exsolution of RuFe alloy nanoparticles on Sr2Fe1.4RuO.1Mo0.5 O6-
via repeated redox manipulations for electrolysis. Nat. Commun. 12, 5665 (2021). - Li, Q.-X. et al. Highly efficient electroreduction of
by defect single-atomic sites anchored on ordered micro-macroporous carbons. Sci. China Chem. 65, 1584-1593 (2022). - Kim, D. et al. Electrocatalytic reduction of low concentrations of
gas in a membrane electrode assembly electrolyzer. ACS Energy Lett. 6, 3488-3495 (2021). -
. et al. A bio-inspired -tolerant catalytic reduction electrode. Sci. Bull. 64, 1890-1895 (2019). - Cheng, Y., Hou, J. & Kang, P. Integrated capture and electroreduction of flue gas
to formate using amine functionalized SnOx nanoparticles. ACS Energy Lett. 6, 3352-3358 (2021). - Majee, D. & Presolski, S. Dithienylethene-based photoswitchable catalysts: state of the art and future perspectives. ACS Catal. 11, 2244-2252 (2021).
- Han, J., Zhang, J., Zhao, T., Liu, M. & Duan, P. Photoswitchable photon upconversion from turn-on mode fluorescent diarylethenes. CCS Chem. 3, 665-674 (2021).
- Endtner, J. M., Effenberger, F., Hartschuh, A. & Port, H. Optical ON/ OFF switching of intramolecular photoinduced charge separation in a donor-bridge-acceptor system containing dithienylethene. J. Am. Chem. Soc. 122, 3037-3046 (2000).
- Park, J., Jiang, Q., Feng, D. & Zhou, H.-C. Controlled generation of singlet oxygen in living cells with tunable ratios of the photochromic switch in metal-organic frameworks. Angew. Chem. Int. Ed. 55, 7188-7193 (2016).
- Cheng, H.-B. et al. Protein-activatable diarylethene monomer as a smart trigger of noninvasive control over reversible generation of
singlet oxygen: a facile, switchable, theranostic strategy for photodynamic-immunotherapy. J. Am. Chem. Soc. 143, 2413-2422 (2021). - Luo, Y.-C. et al. Heterogenization of photochemical molecular devices: embedding a metal-organic cage into a ZIF-8-derived matrix to promote proton and electron transfer. J. Am. Chem. Soc. 141, 13057-13065 (2019).
- Sun, N. et al. Photoresponsive covalent organic frameworks with diarylethene switch for tunable singlet oxygen generation. Chem. Mater. 34, 1956-1964 (2022).
- Park, J., Feng, D., Yuan, S. & Zhou, H. C. Photochromic metal-organic frameworks: reversible control of singlet oxygen generation. Angew. Chem. Int. Ed. 127, 440-445 (2015).
- Park, J., Feng, D., Yuan, S. & Zhou, H. C. Photochromic metal-organic frameworks: reversible control of singlet oxygen generation. Angew. Chem. Int. Ed. 127, 440-445 (2014).
- Hou, L., Zhang, X., Pijper, T. C., Browne, W. R. & Feringa, B. L. Reversible Photochemical Control of Singlet Oxygen Generation Using Diarylethene Photochromic Switches. J. Am. Chem. Soc. 136, 910-913 (2014).
- Yaghi, O. M. et al. Reticular synthesis and the design of new materials. Nature 423, 705-714 (2003).
- Yaghi, O. et al. Reticular Chemistry-Construction, Properties, and Precision Reactions of Frameworks. J. Am. Chem. Soc. 138, 15507-15509 (2016).
- Ruidas, S. et al. Non-fluorinated and robust superhydrophobic modification on covalent organic framework for crude-oil-in-water emulsion separation. Angew. Chem. Int. Ed. 61, e202210507 (2022).
- Furukawa, H. & Yaghi, O. M. Storage of hydrogen, methane, and carbon dioxide in highly porous covalent organic frameworks for clean energy applications. J. Am. Chem. Soc. 131, 8875-8883 (2009).
- Ding, S.-Y. & Wang, W. Covalent organic frameworks (COFs): from design to applications. Chem. Soc. Rev. 42, 548-568 (2013).
- Zhao, X., Pachfule, P. & Thomas, A. Covalent organic frameworks (COFs) for electrochemical applications. Chem. Soc. Rev. 50, 6871-6913 (2021).
- Zhang, T., Zhang, G. & Chen, L. 2D conjugated covalent organic frameworks: defined synthesis and tailor-made functions. Acc. Chem. Res. 55, 795-808 (2022).
- Keller, N. & Bein, T. Optoelectronic processes in covalent organic frameworks. Chem. Soc. Rev. 50, 1813-1845 (2021).
- Ge, L., Qiao, C., Tang, Y., Zhang, X. & Jiang, X. Light-activated hypoxia-sensitive covalent organic framework for tandemresponsive drug delivery. Nano Lett. 21, 3218-3224 (2021).
- Yusran, Y., Fang, Q. & Valtchev, V. Electroactive covalent organic frameworks: design, synthesis, and applications. Adv. Mater. 32, 2002038 (2020).
- Liu, Y. et al. Covalent-Organic-Framework-Based Composite Materials. Chem 6, 3172-3202 (2020).
- Wu, Q. et al. Construction of donor-acceptor heterojunctions in covalent organic framework for enhanced
electroreduction. Small 17, 2004933 (2021). - Huang, N. et al. A stable and conductive metallophthalocyanine framework for electrocatalytic carbon dioxide reduction in water. Angew. Chem. Int. Ed. 132, 16730-16736 (2020).
- Lin, S. et al. Covalent organic frameworks comprising cobalt porphyrins for catalytic
reduction in water. Science 349, 1208-1213 (2015). - Wu, Q.-J. et al. Boosting electroreduction of
over cationic covalent organic frameworks: hydrogen bonding effects of halogen ions. Angew. Chem. Int. Ed. 62, e202215687 (2023). - Diercks, C. S. et al. Reticular electronic tuning of porphyrin active sites in covalent organic frameworks for electrocatalytic carbon dioxide reduction. J. Am. Chem. Soc. 140, 1116-1122 (2018).
- Santoro, C., Bollella, P., Erable, B., Atanassov, P. & Pant, D. Oxygen reduction reaction electrocatalysis in neutral media for bioelectrochemical systems. Nat. Catal. 5, 473-484 (2022).
- Chen, X., Gao, J. & Jiang, D. Designed synthesis of porphyrin-based two-dimensional covalent organic frameworks with highly ordered structures. Chem. Lett. 44, 1257-1259 (2015).
- Yusran, Y. et al. Exfoliated mesoporous 2D covalent organic frameworks for high-rate electrochemical double-layer capacitors. Adv. Mater. 32, 1907289 (2020).
- He, C., Si, D. H., Huang, Y. B. & Cao, R. A CO2-masked carbene functionalized covalent organic framework for highly efficient carbon dioxide conversion. Angew. Chem. Int. Ed. 134, e202207478 (2022).
- Wu, Q.-J. et al. Photocoupled electroreduction of
over photosensitizer-decorated covalent organic frameworks. J. Am. Chem. Soc. 145, 19856-19865 (2023). - Li, N., Lu, W., Pei, K. & Chen, W. Interfacial peroxidase-like catalytic activity of surface-immobilized cobalt phthalocyanine on multiwall carbon nanotubes. RSC Adv. 5, 9374-9380 (2015).
- Yi, J.-D. et al. Cobalt single-atoms anchored on porphyrinic triazinebased frameworks as bifunctional electrocatalysts for oxygen reduction and hydrogen evolution reactions. J. Mater. Chem. A 7, 1252-1259 (2019).
- Dolgopolova, E. A. et al. Connecting wires: Photoinduced electronic structure modulation in metal-organic frameworks. J. Am. Chem. Soc. 141, 5350-5358 (2019).
- Fang, M., Xu, L., Zhang, H., Zhu, Y. & Wong, W.-Y. Metalloporphyrinlinked mercurated graphynes for ultrastable
electroreduction to CO with nearly selectivity at a current density of . J. Am. Chem. Soc. 144, 15143-15154 (2022). - Zhang, X. et al. Molecular engineering of dispersed nickel phthalocyanines on carbon nanotubes for selective
reduction. Nat. Energy 5, 684-692 (2020). - Entradas, T., Waldron, S. & Volk, M. The detection sensitivity of commonly used singlet oxygen probes in aqueous environments. J. Photochem. Photobiol., B 204, 111787 (2020).
- Meng, D. L. et al. Highly selective tandem electroreduction of
to ethylene over atomically isolated nickel-nitrogen site/copper nanoparticle catalysts. Angew. Chem. Int. Ed. 133, 25689-25696 (2021). - Zhu, S., Jiang, B., Cai, W.-B. & Shao, M. Direct observation on reaction intermediates and the role of bicarbonate anions in
electrochemical reduction reaction on Cu surfaces. J. Am. Chem. Soc. 139, 15664-15667 (2017). - Yang, X. et al. Hydrophobic perfluoroalkane modified metalorganic frameworks for the enhanced electrocatalytic reduction of
. SmartMat 3, 163-172 (2022). - Zou, Y. & Wang, S. An investigation of active sites for electrochemical
reduction reactions: from in situ characterization to rational design. Adv. Sci. 8, 2003579 (2021). - Zhou, W. et al. Regulating the scaling relationship for high catalytic kinetics and selectivity of the oxygen reduction reaction. Nat. Commun. 13, 6414 (2022).
- Chan, K. & Nørskov, J. K. Electrochemical barriers made simple. J. Phys. Chem. C. 6, 2663-2668 (2015).
- Chan, K. & Nørskov, J. K. Potential dependence of electrochemical barriers from ab Initio calculations. J. Phys. Chem. C. 7, 1686-1690 (2016).
- Liu, M. et al. Post-synthetic modification of covalent organic frameworks for
electroreduction. Nat. Commun. 14, 3800 (2023). - Wu, Q., Si, D. H., Liang, J., Huang, Y. B. & Cao, R. Highly efficient electrocatalytic
reduction over pyrolysis-free conjugated metallophthalocyanine networks in full pH range. Appl. Catal., B. 333, 122803 (2023).
شكر وتقدير
مساهمات المؤلفين
المصالح المتنافسة
معلومات إضافية
المواد التكميلية متاحة على
https://doi.org/10.1038/s41467-024-45959-9.
© المؤلف(ون) 2024
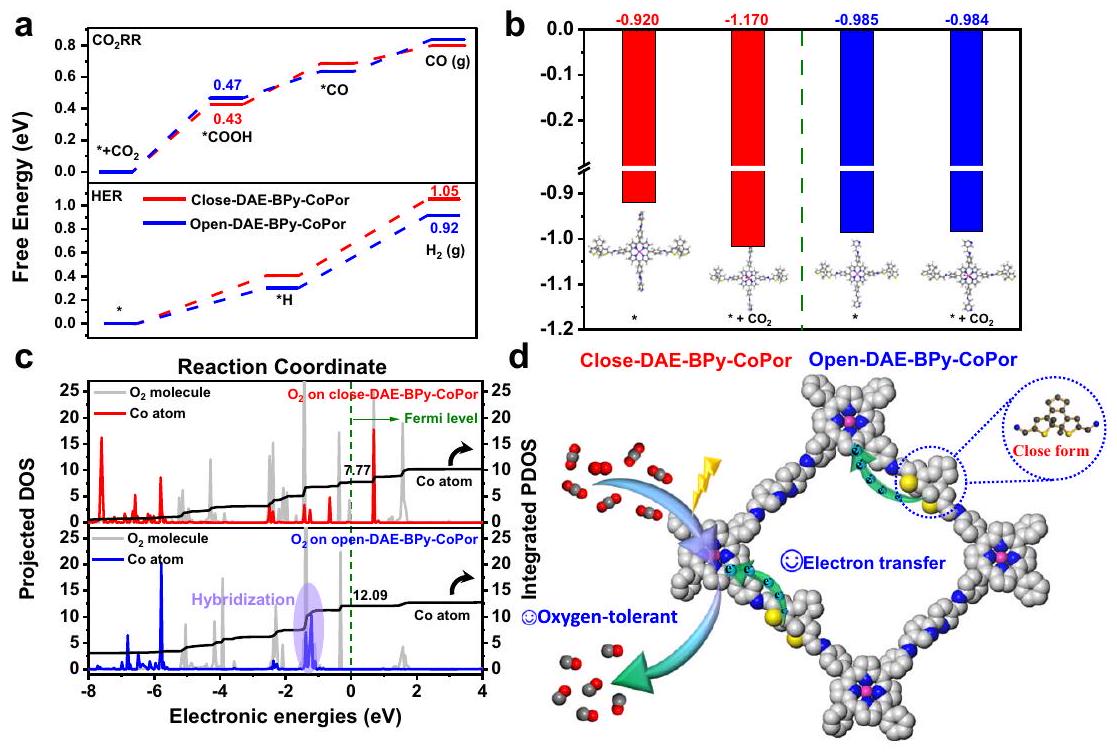
المختبر الوطني الرئيسي للكيمياء الهيكلية، معهد فوجيان للبحث في بنية المادة، الأكاديمية الصينية للعلوم، 350002 فوزهو، جمهورية الصين الشعبية. مختبر الابتكار العلمي والتكنولوجي في فوجيان للمعلومات البصرية والإلكترونية في الصين، 350108 فوزهو، جمهورية الصين الشعبية. جامعة الأكاديمية الصينية للعلوم، 100049 بكين، جمهورية الصين الشعبية. البريد الإلكتروني: ybhuang@fjirsm.ac.cn - c close-DAE-BPy-CoPor، Co أرجواني، N أزرق، C رمادي، S أصفر، من أجل وضوح أكبر تم حذف ذرات الهيدروجين.
- نماذج متوسطة من
). كثافة الحالات المتوقعة وكثافة الحالات المتكاملة لهياكل الامتزاز من على DAE-BPy-CoPor المغلقة وDAE-BPy-CoPor المفتوحة. د الآلية التخطيطية المقترحة لـ على DAE-BPy-CoPor المغلقة تحت ظروف هوائية.
DOI: https://doi.org/10.1038/s41467-024-45959-9
PMID: https://pubmed.ncbi.nlm.nih.gov/38368417
Publication Date: 2024-02-17
Oxygen-tolerant
Accepted: 8 February 2024
Published online: 17 February 2024
(D) Check for updates
Abstract
The direct use of flue gas for the electrochemical
should be sourced from a real
Results
The synthesis and characterization of DAE-BPy-CoPor
a Comparison of the experimental powder X-ray diffraction pattern with simulated PXRD patterns. b Solid-state
different binding energies of the two isomers
peak at 164.7 eV was attributed to the close form of DAE in the close-DAE-BPy-CoPor. The XPS spectrum of
(Supplementary Fig. 15b), which made it difficult to detect the photocyclization reaction of the DAE parts in the COF. Nevertheless, the photocyclization of the open-DAE monomer can be detected in the solid-state UV-Vis. As shown in Supplementary Fig. 15a, compared with the spectrum of the monomer open-DAE, a new peak at 640 nm appeared in that of the close-DAE, which indicated the formation of the closed state of the cyclohexadiene moiety in the close-DAE-BPy-CoPor. Furthermore, the Co K-edge XANES profile of close-DAE-BPy-CoPor exhibited a similar wave feature with that of open-DAE-BPy-CoPor, indicating that the Co center valence of close-DAE-BPy-CoPor similar with open-DAE-BPy-CoPor. Notably, compared with open-DAE-BPyCoPor, the absorption peak of close-DAE-BPy-CoPor at around 7715 eV shifted to the lower-energy side. This shift implied a slightly lower Co oxidation state in close-DAE-BPy-CoPor than open-DAE-BPy-CoPor, indicating the presence of more electrons in the Co center of close-DAE-BPy-CoPor. Moreover, the Fourier-transformed Co K-edge EXAFS spectrum for open-DAE-BPy-CoPor showed a dominant peak at
The electrocatalytic
promising candidates for
CoPor, close-DAE-BPy-CoPor and open-DAE-BPy-CoPor in the co-feeding CO2 and
current drop
to -1.1 V . The optimal
were conducted in the
The DFT calculation and reaction mechanism
Methods
Synthesis of BPy-CoPor
Synthesis of close-DAE-BPy-CoPor
Materials and synthetic procedures
Characterizations and instruments
were measured using Micromeritics ASAP 2460 instrument.
Electrochemical measurements
I: total steady-state cell current;
Data availability
References
- Navarro-Jaén, S. et al. Highlights and challenges in the selective reduction of carbon dioxide to methanol. Nat. Rev. Chem. 5, 564-579 (2021).
- Ross, M. B. et al. Designing materials for electrochemical carbon dioxide recycling. Nat. Catal. 2, 648-658 (2019).
- Wu, Q.-J., Liang, J., Huang, Y.-B. & Cao, R. Thermo-, electro-, and photocatalytic
conversion to value-added products over porous metal/covalent organic frameworks. Acc. Chem. Res. 55, 2978-2997 (2022). - Zhu, H.-J. et al. Efficient electron transmission in covalent organic framework nanosheets for highly active electrocatalytic carbon dioxide reduction. Nat. Commun. 11, 497 (2020).
- Gong, Y.-N. et al. Regulating photocatalysis by spin-state manipulation of cobalt in covalent organic frameworks. J. Am. Chem. Soc. 142, 16723-16731 (2020).
- Zhou, J. et al. Linking oxidative and reductive clusters to prepare crystalline porous catalysts for photocatalytic
reduction with . Nat. Commun. 13, 4681 (2022). - Zhai, S. et al. High-capacity thermochemical
dissociation using iron-poor ferrites. Energy Environ. Sci. 13, 592-600 (2020). - Zhang, X. Y. et al. Operando metalloid
active sites for highly efficient carbon dioxide reduction electrocatalysis. Angew. Chem. Int. Ed. 61, e202202298 (2022). - Wang, Y., Liu, J. & Zheng, G. Designing copper-based catalysts for efficient carbon dioxide electroreduction. Adv. Mater. 33, 2005798 (2021).
- Wang, Y. et al. Local weak hydrogen bonds significantly enhance
electroreduction performances of a metal-organic framework. CCS Chem. 5, 145-151 (2023). - Service, R. F. Cost of carbon capture drops, but does anyone want it? Science 354, 1362-1363 (2016).
- Jouny, M., Luc, W. & Jiao, F. General techno-economic analysis of
electrolysis systems. Ind. Eng. Chem. Res. 57, 2165-2177 (2018). - Wang, J. & Ciucci, F. Boosting bifunctional oxygen electrolysis for N-doped carbon via bimetal addition. Small 13, 1604103 (2017).
- Han, B. et al. Two-dimensional covalent organic frameworks with cobalt (II)-Phthalocyanine sites for efficient electrocatalytic carbon dioxide reduction. J. Am. Chem. Soc. 143, 7104-7113 (2021).
- Lv, H. et al. Promoting exsolution of RuFe alloy nanoparticles on Sr2Fe1.4RuO.1Mo0.5 O6-
via repeated redox manipulations for electrolysis. Nat. Commun. 12, 5665 (2021). - Li, Q.-X. et al. Highly efficient electroreduction of
by defect single-atomic sites anchored on ordered micro-macroporous carbons. Sci. China Chem. 65, 1584-1593 (2022). - Kim, D. et al. Electrocatalytic reduction of low concentrations of
gas in a membrane electrode assembly electrolyzer. ACS Energy Lett. 6, 3488-3495 (2021). -
. et al. A bio-inspired -tolerant catalytic reduction electrode. Sci. Bull. 64, 1890-1895 (2019). - Cheng, Y., Hou, J. & Kang, P. Integrated capture and electroreduction of flue gas
to formate using amine functionalized SnOx nanoparticles. ACS Energy Lett. 6, 3352-3358 (2021). - Majee, D. & Presolski, S. Dithienylethene-based photoswitchable catalysts: state of the art and future perspectives. ACS Catal. 11, 2244-2252 (2021).
- Han, J., Zhang, J., Zhao, T., Liu, M. & Duan, P. Photoswitchable photon upconversion from turn-on mode fluorescent diarylethenes. CCS Chem. 3, 665-674 (2021).
- Endtner, J. M., Effenberger, F., Hartschuh, A. & Port, H. Optical ON/ OFF switching of intramolecular photoinduced charge separation in a donor-bridge-acceptor system containing dithienylethene. J. Am. Chem. Soc. 122, 3037-3046 (2000).
- Park, J., Jiang, Q., Feng, D. & Zhou, H.-C. Controlled generation of singlet oxygen in living cells with tunable ratios of the photochromic switch in metal-organic frameworks. Angew. Chem. Int. Ed. 55, 7188-7193 (2016).
- Cheng, H.-B. et al. Protein-activatable diarylethene monomer as a smart trigger of noninvasive control over reversible generation of
singlet oxygen: a facile, switchable, theranostic strategy for photodynamic-immunotherapy. J. Am. Chem. Soc. 143, 2413-2422 (2021). - Luo, Y.-C. et al. Heterogenization of photochemical molecular devices: embedding a metal-organic cage into a ZIF-8-derived matrix to promote proton and electron transfer. J. Am. Chem. Soc. 141, 13057-13065 (2019).
- Sun, N. et al. Photoresponsive covalent organic frameworks with diarylethene switch for tunable singlet oxygen generation. Chem. Mater. 34, 1956-1964 (2022).
- Park, J., Feng, D., Yuan, S. & Zhou, H. C. Photochromic metal-organic frameworks: reversible control of singlet oxygen generation. Angew. Chem. Int. Ed. 127, 440-445 (2015).
- Park, J., Feng, D., Yuan, S. & Zhou, H. C. Photochromic metal-organic frameworks: reversible control of singlet oxygen generation. Angew. Chem. Int. Ed. 127, 440-445 (2014).
- Hou, L., Zhang, X., Pijper, T. C., Browne, W. R. & Feringa, B. L. Reversible Photochemical Control of Singlet Oxygen Generation Using Diarylethene Photochromic Switches. J. Am. Chem. Soc. 136, 910-913 (2014).
- Yaghi, O. M. et al. Reticular synthesis and the design of new materials. Nature 423, 705-714 (2003).
- Yaghi, O. et al. Reticular Chemistry-Construction, Properties, and Precision Reactions of Frameworks. J. Am. Chem. Soc. 138, 15507-15509 (2016).
- Ruidas, S. et al. Non-fluorinated and robust superhydrophobic modification on covalent organic framework for crude-oil-in-water emulsion separation. Angew. Chem. Int. Ed. 61, e202210507 (2022).
- Furukawa, H. & Yaghi, O. M. Storage of hydrogen, methane, and carbon dioxide in highly porous covalent organic frameworks for clean energy applications. J. Am. Chem. Soc. 131, 8875-8883 (2009).
- Ding, S.-Y. & Wang, W. Covalent organic frameworks (COFs): from design to applications. Chem. Soc. Rev. 42, 548-568 (2013).
- Zhao, X., Pachfule, P. & Thomas, A. Covalent organic frameworks (COFs) for electrochemical applications. Chem. Soc. Rev. 50, 6871-6913 (2021).
- Zhang, T., Zhang, G. & Chen, L. 2D conjugated covalent organic frameworks: defined synthesis and tailor-made functions. Acc. Chem. Res. 55, 795-808 (2022).
- Keller, N. & Bein, T. Optoelectronic processes in covalent organic frameworks. Chem. Soc. Rev. 50, 1813-1845 (2021).
- Ge, L., Qiao, C., Tang, Y., Zhang, X. & Jiang, X. Light-activated hypoxia-sensitive covalent organic framework for tandemresponsive drug delivery. Nano Lett. 21, 3218-3224 (2021).
- Yusran, Y., Fang, Q. & Valtchev, V. Electroactive covalent organic frameworks: design, synthesis, and applications. Adv. Mater. 32, 2002038 (2020).
- Liu, Y. et al. Covalent-Organic-Framework-Based Composite Materials. Chem 6, 3172-3202 (2020).
- Wu, Q. et al. Construction of donor-acceptor heterojunctions in covalent organic framework for enhanced
electroreduction. Small 17, 2004933 (2021). - Huang, N. et al. A stable and conductive metallophthalocyanine framework for electrocatalytic carbon dioxide reduction in water. Angew. Chem. Int. Ed. 132, 16730-16736 (2020).
- Lin, S. et al. Covalent organic frameworks comprising cobalt porphyrins for catalytic
reduction in water. Science 349, 1208-1213 (2015). - Wu, Q.-J. et al. Boosting electroreduction of
over cationic covalent organic frameworks: hydrogen bonding effects of halogen ions. Angew. Chem. Int. Ed. 62, e202215687 (2023). - Diercks, C. S. et al. Reticular electronic tuning of porphyrin active sites in covalent organic frameworks for electrocatalytic carbon dioxide reduction. J. Am. Chem. Soc. 140, 1116-1122 (2018).
- Santoro, C., Bollella, P., Erable, B., Atanassov, P. & Pant, D. Oxygen reduction reaction electrocatalysis in neutral media for bioelectrochemical systems. Nat. Catal. 5, 473-484 (2022).
- Chen, X., Gao, J. & Jiang, D. Designed synthesis of porphyrin-based two-dimensional covalent organic frameworks with highly ordered structures. Chem. Lett. 44, 1257-1259 (2015).
- Yusran, Y. et al. Exfoliated mesoporous 2D covalent organic frameworks for high-rate electrochemical double-layer capacitors. Adv. Mater. 32, 1907289 (2020).
- He, C., Si, D. H., Huang, Y. B. & Cao, R. A CO2-masked carbene functionalized covalent organic framework for highly efficient carbon dioxide conversion. Angew. Chem. Int. Ed. 134, e202207478 (2022).
- Wu, Q.-J. et al. Photocoupled electroreduction of
over photosensitizer-decorated covalent organic frameworks. J. Am. Chem. Soc. 145, 19856-19865 (2023). - Li, N., Lu, W., Pei, K. & Chen, W. Interfacial peroxidase-like catalytic activity of surface-immobilized cobalt phthalocyanine on multiwall carbon nanotubes. RSC Adv. 5, 9374-9380 (2015).
- Yi, J.-D. et al. Cobalt single-atoms anchored on porphyrinic triazinebased frameworks as bifunctional electrocatalysts for oxygen reduction and hydrogen evolution reactions. J. Mater. Chem. A 7, 1252-1259 (2019).
- Dolgopolova, E. A. et al. Connecting wires: Photoinduced electronic structure modulation in metal-organic frameworks. J. Am. Chem. Soc. 141, 5350-5358 (2019).
- Fang, M., Xu, L., Zhang, H., Zhu, Y. & Wong, W.-Y. Metalloporphyrinlinked mercurated graphynes for ultrastable
electroreduction to CO with nearly selectivity at a current density of . J. Am. Chem. Soc. 144, 15143-15154 (2022). - Zhang, X. et al. Molecular engineering of dispersed nickel phthalocyanines on carbon nanotubes for selective
reduction. Nat. Energy 5, 684-692 (2020). - Entradas, T., Waldron, S. & Volk, M. The detection sensitivity of commonly used singlet oxygen probes in aqueous environments. J. Photochem. Photobiol., B 204, 111787 (2020).
- Meng, D. L. et al. Highly selective tandem electroreduction of
to ethylene over atomically isolated nickel-nitrogen site/copper nanoparticle catalysts. Angew. Chem. Int. Ed. 133, 25689-25696 (2021). - Zhu, S., Jiang, B., Cai, W.-B. & Shao, M. Direct observation on reaction intermediates and the role of bicarbonate anions in
electrochemical reduction reaction on Cu surfaces. J. Am. Chem. Soc. 139, 15664-15667 (2017). - Yang, X. et al. Hydrophobic perfluoroalkane modified metalorganic frameworks for the enhanced electrocatalytic reduction of
. SmartMat 3, 163-172 (2022). - Zou, Y. & Wang, S. An investigation of active sites for electrochemical
reduction reactions: from in situ characterization to rational design. Adv. Sci. 8, 2003579 (2021). - Zhou, W. et al. Regulating the scaling relationship for high catalytic kinetics and selectivity of the oxygen reduction reaction. Nat. Commun. 13, 6414 (2022).
- Chan, K. & Nørskov, J. K. Electrochemical barriers made simple. J. Phys. Chem. C. 6, 2663-2668 (2015).
- Chan, K. & Nørskov, J. K. Potential dependence of electrochemical barriers from ab Initio calculations. J. Phys. Chem. C. 7, 1686-1690 (2016).
- Liu, M. et al. Post-synthetic modification of covalent organic frameworks for
electroreduction. Nat. Commun. 14, 3800 (2023). - Wu, Q., Si, D. H., Liang, J., Huang, Y. B. & Cao, R. Highly efficient electrocatalytic
reduction over pyrolysis-free conjugated metallophthalocyanine networks in full pH range. Appl. Catal., B. 333, 122803 (2023).
Acknowledgements
Author contributions
Competing interests
Additional information
supplementary material available at
https://doi.org/10.1038/s41467-024-45959-9.
© The Author(s) 2024
State Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, 350002 Fuzhou, PR China. Fujian Science & Technology Innovation Laboratory for Optoelectronic Information of China, 350108 Fuzhou, PR China. University of Chinese Academy of Science, 100049 Beijing, PR China. e-mail: ybhuang@fjirsm.ac.cn - c close-DAE-BPy-CoPor, Co magenta, N blue, C gray, S yellow, for clear clarity the H atoms were omitted.
- intermediate models of the
). c Projected density of states and integrated density of states of adsorption structures of on close-DAE-BPy-CoPor and open-DAE-BPy-CoPor. d Proposed schematic mechanism for the on close-DAE-BPy-CoPor under aerobic conditions.