تمديد الشد لمواقع الإريديوم في أكاسيد المنغنيز لمحللات الماء بغشاء تبادل البروتون Tensile straining of iridium sites in manganese oxides for proton-exchange membrane water electrolysers
على الرغم من أن تفاعل تطور الأكسجين الحمضي (OER) يلعب دورًا حاسمًا في أجهزة التحليل الكهربائي للماء بغشاء تبادل البروتون (PEMWE)، إلا أن التحديات لا تزال قائمة بسبب نقص المحفزات الكهربائية الفعالة والمستقرة في الأحماض. هنا، نقدم محفزًا كهربائيًا منخفض الإريديوم حيث يتم توطين ذرات الإريديوم المشدودة في مواقع الكاتيونات على سطح أكسيد المنغنيز (TS-Ir/للنشاط العالي والمستدام في تفاعلات الأكسدة الاختزالية. تكشف التوصيفات في الموقع باستخدام السنكروترون أن TS-Ir/يمكن أن يؤدي إلى تفعيل آلية مستمرة محلية تعتمد على الأكسجين (L-LOM). بشكل خاص، يمكن أن تعزز عملية L-LOM بشكل كبير من امتصاص وتحويلالجزيئات فوق الفراغات الأكسجينية حول مواقع الإروبيوم المتوترة وتمنع المزيد من فقدان ذرات الأكسجين الشبكي في الداخلالكتلة لتحسين السلامة الهيكلية للمحفز. من المهم أن الجهاز الناتج عن PEMWE المصنوع باستخدام TS-Ir/يوفر كثافة تيارويعمل بشكل مستقر لمدة 200 ساعة.
أجهزة التحليل الكهربائي للماء بغشاء تبادل البروتون (PEMWE)، التي تتميز بخسائر مقاومة أقل، وتداخل غاز أقل، وكثافات تيار أعلى مقارنة بأجهزة التحليل الكهربائي للماء القلوي، تمثل طريقًا واعدًا ومستدامًا لإنتاج الهيدروجين النظيف.الوقودحالياً، تقتصر الكفاءة العامة لتحليل الماء كهربائياً بشكل رئيسي على تفاعل تطور الأكسجين الأنودي (OER)، الذي يتضمن عملية نقل إلكترون مرتبطة بأربعة بروتونات بطيئة.علاوة على ذلك، فإن التدهور الشديد لمواقع النشاط في حالة الأكسدة الحمضية العالية، خاصة عند كثافات التيار العالية، يحد بشدة من النشر على نطاق واسع لأجهزة PEMWE.“. على الرغم من أن أكاسيد المعادن القائمة على الإريديوم (لقد تم استخدامها على نطاق واسع في التحليل الكهربائي للماء، لكنها تتميز بنشاط كتلي منخفض، وفوق جهد مرتفع، وتكاليف عالية (الولايات المتحدة)ولا يمكن تحقيق نشاط عالٍ مستمر ودوام عند كثافات تيار عالية. لذلك، على الرغم من تطوير محفزات كهربائية منخفضة الإريديوم مستقرة في الأحماض والتي تظهر أداءً كهربائيًا محسنًا ودوامًا جيدًا على المدى الطويل يُعتبر أمرًا مرغوبًا بشدة لتعزيز أجهزة PEMWE الأكثر تنافسية، إلا أنه لا يزال يمثل تحديًا كبيرًا.
يتم ملاحظة آلية تطور الممتزات المقبولة على نطاق واسع (AEM) لتفاعل الأكسدة الكهربائية للماء (OER) التي يتم تحفيزها بواسطة أكاسيد الإريديوم، وتشمل عدة وسائط، مثل، و “. عادةً ما تكون تشكيل *OOH على مواقع المعادن هي الخطوة المحددة للسرعة، والتي تعتبر عنق الزجاجة في تحسين أداء OER الحمضيلتجاوز العلاقة بين طاقات الامتزاز بين *OH و *OOH، فإن المحفزات (التي تحتوي على روابط تساهمية قويةيمكن أن تؤثر الروابط (حيث M هو ذرة معدن) سلبًا وإيجابًا على مستوى هوبارد المنخفضأشرطة لذرات المعادن، مما يجعل ذرات الأكسجين في الشبكة أكثر احتمالاً لفقدان الإلكترونات من خلال الأكسدةلذلك، يتبع المحفز آلية وسيطة تعتمد على الأكسجين الشبكي (LOM)، والتي يمكن أن تتجاوز إنتاج *OOH الوسيطة الحرجة من أجل تعزيز النشاط.. للأسف، فإن إزالة البروتون ببطء في مواقع ذرات الأكسجين في الشبكة وتدهور
تمنع أنواع المعادن على سطح المحفز الاستقرار التشغيلي على المدى الطويل. بشكل خاص، يمكن أن يؤدي إطلاق الأكسجين من الشبكة بشكل أكسيدي إلى أكسدة مفرطة للمعادن (مثل الروثينيوم) إلى أكاسيد معدنية عالية التكافؤ قابلة للذوبانخلال عمليات الأكسدة الكهربائية الحمضية. علاوة على ذلك، فإن التفاعلات العنيفة التي تشمل ذرات الأكسجين في الشبكة تنتج العديد من فراغات الأكسجين، التي تذوب مباشرة المعادن على سطح المحفز.وما هو أسوأ من ذلك، خلال التفاعلات المطولة، تتغلغل فراغات الأكسجين التي تتشكل بسهولة في عمق المادة، مما يؤدي في النهاية إلى انهيار هيكل المادة وبالتالي تعطيل المحفز.
مؤخراً،هو أكسيد معدن انتقالي وفير ورخيص الثمن يتمتع بنشاط سطحي عالٍ وثبات تحفيزي في الأوساط الحمضية، ويعتبر حاملاً محتملاً لتفاعلات الأكسدة الحمضية.جزيئات نانوية وتحميل منخفض بنسبة 5% من الإريديوم المدعوم علىأظهر المحفز الكهربائي أداءً جيدًا في تفاعل الأكسدة الكهربائي بسبب المساحات السطحية النشطة العالية ووجود إيريديوم بحالة عالية من التكافؤ.قدمت مجموعة ليو استراتيجية مدفوعة بالضوء لتحقيق تجميع منظم لذرات الإيريديوم على مادة مشوبة بالفلور.سطح (IrMnOF) مع إنتروبيا أقل مرتبطة بالدوران تم تحسينها لتقليل الطاقة التنشيطية الجوهرية عند الوسائط المحددة للجهد لعملية OER مستقرةلتفكيك العلاقة الخطية للعديد من الوسائط التفاعلية لزيادة النشاط، تم دعم رقع من مصفوفات ذرات الروثينيوم علىمع المسافات الذرية المناسبة في مواقع المعادن الثنائية المتماثلة، تم تحقيق نشاط محسن في الأكسدة الحمضية (OER) من خلال آلية مسار الأكسيد.. علاوة على ذلك، يحتوي الشبك المتوتر على والترابط مع وجودفي التحميل العاليإير-إنكوربوريتيت – أظهر استقرارًا محسنًا في عملية الأكسدةلذلك، فإن كسر العلاقة الخطية بين عدة وسائط تفاعل في الوقت نفسه وكبح عدد كبير من فراغات الأكسجين يعد أمرًا مرغوبًا فيه بشكل أساسي لتصميم محفزات كهربائية متقدمة، لكنه يمثل تحديًا كبيرًا.
لذلك، في هذه الدراسة، استخدمنا تبادل الكاتيونات واستراتيجية التلدين السريع والتبريد اللاحق لتحضير محفز كهربائي منخفض الإيريديوم حيث يتم توطين ذرات الإيريديوم عند مواقع السطح لمركب أكسيد المنغنيز المتوتر.التمدد الشدّي الذي تم إدخاله بواسطة الشكل المربع المخلوطالجزئيات المحصورة في المقاومة للأحماضيمكن أن يعزز التساهمية لـرابطة لتحسين قدرة إزالة البروتون وزيادة درجة تداخل المدارات للرابطة مع المعدنفرقة خلال التفاعل. بشكل خاص، يتمركز إجهاد الشد على يمكن أن يقوم السطح بتكييف سلوك الامتزاز لمواقع Ir لتسريع إزالة البروتون من *OH عند فراغات الأكسجين على السطح، وبالتالي، منع الأكسدة المحلية لمواقع Ir بشكل فعال لتقليل الذوبان والحفاظ على السلامة الهيكلية للمحفز. وبالتالي، فإن عملية الأكسدة الحمضية التي حدثت على TS-Ir/السطح اتبع آلية أكسجين الشبكة المحلية المستمرة (L-LOM)، مما يجعل المحفز يقدم نشاط كتلة عالي.عند جهد زائد قدره 198 مللي فولت (عادة عند كثافة تيار قدرها )، وهو تقريبًا 19 و 380 مرة أعلى من تلك الخاصة بـ وتجاري ( )، على التوالي. يتم توضيح آلية L-LOM تجريبيًا باستخدام قياسات هيكل الامتصاص الدقيق بالأشعة السينية (XAFS) وقياسات الطيف الكهرومغناطيسي بالأشعة تحت الحمراء من الإشعاع المتزامن (SRIR). والأهم من ذلك، تؤكد قياسات SRIR بتوسيم النظائر في الموقع الامتصاص السريع لـ الجزيئات على فراغات الأكسجين السطحي وإزالة البروتون السريعة لـ *OH فوق ذرات الأكسجين الشبكية لتحفيز تفاعل تحفيزي مستمر L-LOM لتثبيت مواقع النشاط السطحي لإيريديوم. لذلك، فإن TS-Ir/العامل الحفاز يظهر تحلل كهربائي مستقر للماء الحمضي لمدة 100 و200 ساعة عند كثافات تيار تبلغ 200 وفي نظام ثلاثي الأقطاب وجهاز PEMWE الناتج، على التوالي، مع تدهور أداء ضئيل، مما يشير إلى إمكانيات جيدة للتطبيق العملي في PEMWEs.
النتائج والمناقشة
تركيب وتوصيف المحفزات
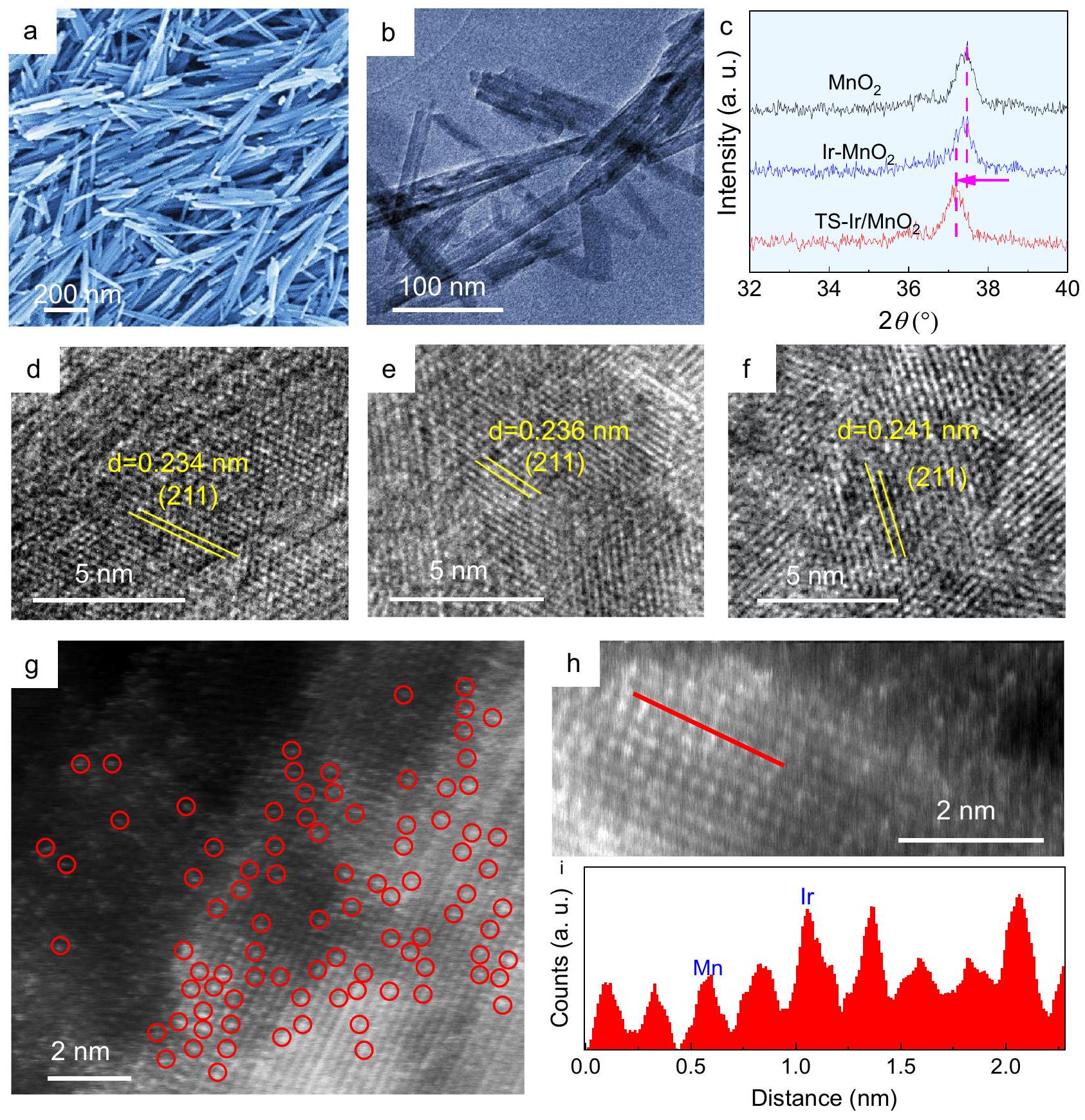
محفز كهربائي منخفض الإريديوم حيث تم حصر مواقع الإريديوم الموزعة ذريًا والمشدودة في أكاسيد المنغنيز (TS-Ir/تمت عملية التخليق الهيدروحراري ثم تم إجراء عملية التلدين الحراري السريع والتبريد بعد ذلك، والتي تُسمى فيما بعد استراتيجية “التلدين الحراري السريع-التبريد” (RTAC). تُظهر صور المجهر الإلكتروني الماسح (SEM) والمجهر الإلكتروني الناقل (TEM) شكل الألياف النانوية، الذي يكشف عن المزيد من المواقع المعدنية المفتوحة (المعلومات التكميلية الشكل 1).تم نقل الألياف النانوية إلى محلول مائيحل لتفاعل تبادل الكاتيونات. تم تسخين العينة الناتجة بسرعة إلىثم تم تبريده بسرعة لإدخال إجهاد الشد في الألياف النانوية. الصور اللاحقة بتقنية SEM (الشكل 1a) وTEM (الشكل 1b) تكشف بوضوح أن شكل الألياف النانوية فائقة الرقة تم الاحتفاظ به بعد تفاعل تبادل Ir والانحلال الحراري السريع لـعينة. كما هو موضح في الشكل 1c والشكل التكميلي 2، فإن أنماط حيود الأشعة السينية (XRD) لـ TS-Ir/عينة تظهر قمم حيادية نموذجية للتشتتدون أي قمم تُنسب إلى مراحل مرتبطة بالإيريديوم. بعد إضافة الإيريديوم والتسخين اللاحق في درجة حرارة الغرفة،قمم انحرفت قليلاً نحو زوايا أقل، مما يشير إلى أن إجهاد الشد تشكل في العينة. علاوة على ذلك، تكشف نتائج حيود الأشعة السينية لعينة الإيريديوم مع تركيزات مختلفة من الدوبينغ وطرق التلدين بوضوح أن معالجة التلدين السريع والتبريد تُدخل إجهاد الشد في المضيف.الشبكة التي تؤدي إلى تحول أكبر في نمط حيود الأشعة السينية، مع استبعاد تأثير ذرات الإيريديوم ذات التحميل المنخفض على شبكة المضيف.
علاوة على ذلك، كما هو موضح في صور TEM عالية الدقة (HRTEM) في الشكل 1d-f، فإن معامل الشبكة للحدود في TS-Ir/تم قياس الألياف النانوية عند، الذي يتوافق مع طائرات. للمقارنة، تم توزيع ذرات الإيريديوم على حالة الشد الخالية من الإجهادسطح الألياف النانوية من خلال التلدين البطيء والتبريد اللاحق، حيث تظهر صور TEM وHRTEM (الأشكال التكميلية 3 و4، على التوالي) أن معامل الشبكة المتقطع هوتمثل المسافة المتزايدة بين الشبكات الإجهاد الشد الناتج عن RTAC على TS-Ir/سطح الألياف النانوية. لتوضيح الهيكل المورفولوجي لمواقع Ir بشكل أكبر، تشير الخرائط العنصرية (الشكل التكميلي 5) إلى التوزيع المتجانس لـ، و O في الألياف النانوية. بالمثل، و O موزعة بشكل موحد في (الشكل التوضيحي التكميلي 6). تظهر صور مجهر المسح الإلكتروني المجهري عالي الزاوية مع تصحيح التشوهات (HAADF-STEM، الشكل 1g، h) بوضوح ذرات الإيريديوم موزعة على المستوى الذري في TS-Ir/الألياف النانوية. ذرات الإيريديوم، المميزة بنقاط ساطعة متناثرة في الشبكة، تقع في نفس مواقع أعمدة ذرات المنغنيز (الشكل 1i)، مما يشير إلى أن الإيريديوم الموزع على المستوى الذري يحل محل مواقع المنغنيز السطحية فيشبكة الألياف النانوية عبر تفاعل تبادل الكاتيون. في هذه العملية RTAC، يقدم علاج التلدين السريع إجهادًا في الشبكة، ثم لا تخضع عملية التبريد السريع لعملية تبريد بطيئة لإطلاق الإجهاد. خلال هذه العملية، تم الاحتفاظ بالتمدد الشدّي في TS-Ir/. وبالمثل، يتم توزيع Ir أيضًا على المستوى الذري على الألياف النانوية في (الشكل التوضيحي الإضافي 7). إن تحميل الإيريديوم الذي تم الحصول عليه باستخدام مطيافية الانبعاث الضوئي الناتج عن البلازما المقترنة بالحث (ICP-OES) هو تقريبًا في TS-Ir/. علاوة على ذلك، تظهر نتيجة ICP-OES أن تحميل الإيريديوم مشابه في، والذي يُعزى إلى نفس تغذية المواد الخام. تؤكد هذه النتائج التشتت المتجانس لذرات الإيريديوم على مواقع المنغنيز السطحية لـالألياف النانوية وأن إجهاد الشد لمواقع Ir تم تقديمه إلى.
الخصائص الإلكترونية للعوامل المساعدة
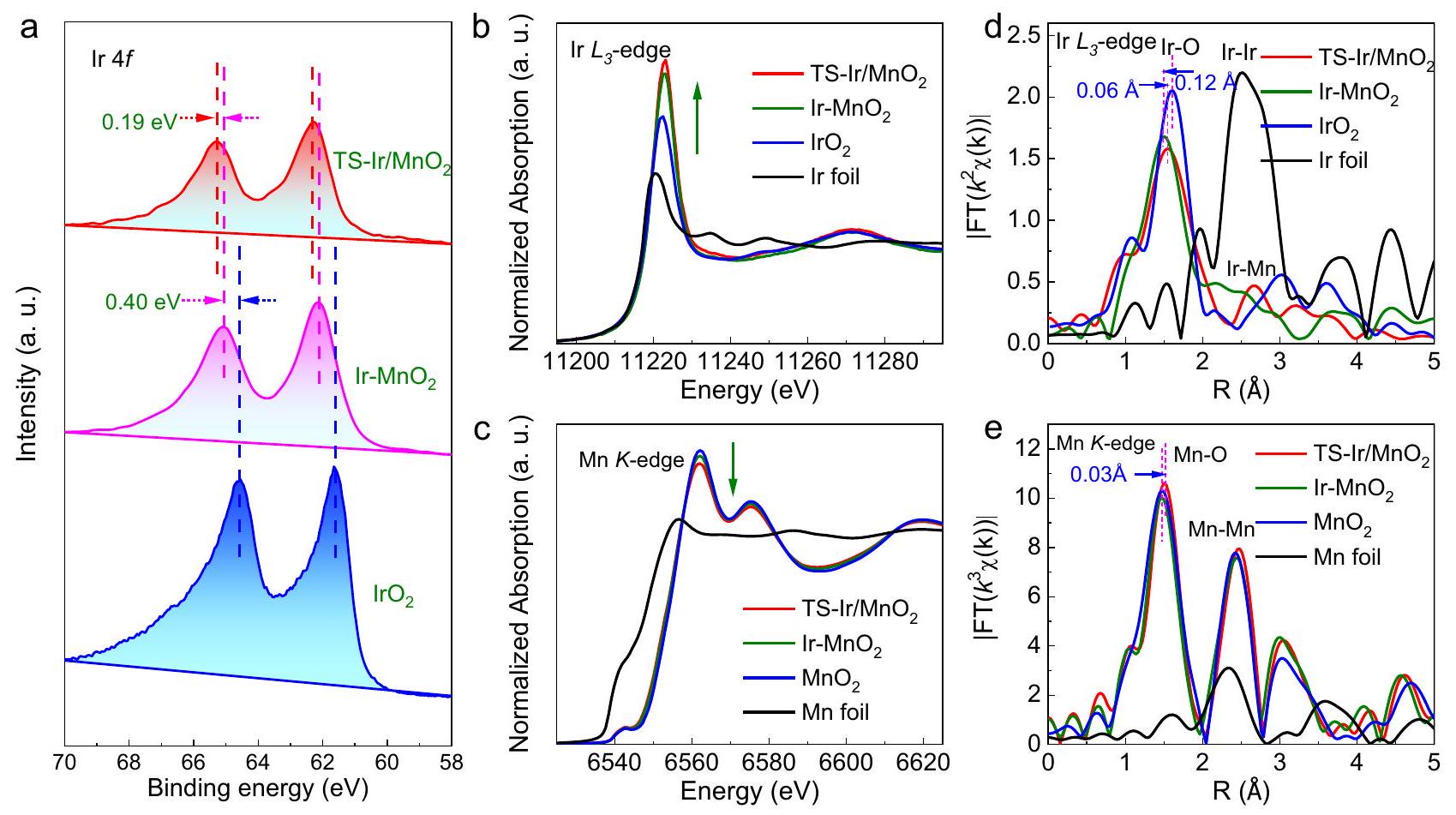
لتوضيح الهيكل الإلكتروني المحلي وبيئة الربط لأنواع الإيريديوم في TS-Ir/تم إجراء دراسة الألياف النانوية، وطيف الإلكترون الضوئي بالأشعة السينية (XPS) وطيف امتصاص الأشعة السينية (XAS). الإريديوميوضح طيف XPS في الشكل 2a قمة واضحة عند 65.36 إلكترون فولت (Ir )، الذي يظهر طاقة ارتباط مرتفعة بمقدار 0.59 إلكترون فولت مقارنة بتلك الخاصة بـ ويقترح أن الحالة المؤكسدة المتزايدة للروثينيوم قد تُعزى إلى التفاعل الإلكتروني بين الروثينيوم و. وبالمثل، الـيوضح طيف XPS في الشكل التكميلية 8 تحولًا في الطاقة السلبية بعد إدخال ذرات Ir، مما يكشف عن انتقال الإلكترونات من Ir إلى Mn. من المهم أن RTAC يمكن أن يزيد من انتقال الإلكترونات إلى
الشكل 1 | التوصيف الهيكلي لـ TS-Ir/المحفز الكهربائي. صور SEM و TEM لـ TS-Ir/محفز كهربائيأنماط وصور HRTEM لـإير-“، و TS-Ir/المحفزات الكهربائية. صورة HAADF-TEM،
حيث يتم نسب النقاط الساطعة المميزة بدوائر حمراء إلى مواقع Ir.صورة HAADF-TEM مكبرةملفات الخطوط لتحليل شدة HAADF المعلمة بـلـ TS-Ir/محفز كهربائي زيادة حالة الأكسدة لأنواع الإيريديوم في TS-Ir/الألياف النانوية. يمكن التحقق من حالة الفالنس المتزايدة للـ Ir من خلال طيف XAS للـ Ir و Mn.طيف امتصاص الأشعة السينية بالقرب من الحافة (XANES) للـ Ir-الحافة في الشكل 2ب تظهر أن ذروة الخط الأبيض لـأكثر كثافة من تلك لكل منو، مما يشير إلى أن حالة التكافؤ قد زادت قليلاً. ومن المثير للاهتمام أن طيف XANES لـ -edge يظهر الاتجاه المعاكس؛ أي أن ذروة الخط الأبيض لمعدن المنغنيز (Mn) تضعف، مما يعني أن حالة الأكسدة لمعدن المنغنيز (Mn) تنخفض بعد إدخال أنواع الإريديوم (Ir) والضغط الشدّي (الشكل 2c). هذه النتائج تتماشى مع طيف XPS للإريديوم (Ir) والمنغنيز (Mn)، مما يشير إلى انتقال الإلكترونات من الإريديوم (Ir) إلى المنغنيز (Mn).
علاوة على ذلك، تكشف طيفيات XAFS الممتدة (EXAFS) عن البيئات التنسيقية المحلية لأنواع الإيريديوم في العينات.. في الـ
منحنيات تحويل فورييه (FT) للـ Ir-طيف EXAFS عند حافة، القمم الرئيسية في نطاقيمكن أن يُعزى إلى الغلاف الأول من تنسيق Ir-O. بالإضافة إلى ذلك، Oتدعم طيفيات XPS وجود الروابط في العينة (الشكل التوضيحي التكميلي 9). علاوة على ذلك، فإن طاقة الربط الأعلى لـفييظهر بشكل أكبر أن حالة الأكسدة المتزايدة للروثينيوم بسبب نقل الإلكترونات المدفوع بالضغط الشدّي. هناك اختلاف طفيف في موضع القمة الرئيسية عند حواليفي الشكل 2d يمثل مظهرًا مختلفًاطول الرابطة في TS-Ir/, ، و “تظهر الشكل 2d، الشكل التكميلي 10 والجدول التكميلي 1 أن الذروة التنسيقية السائدة عندالذي يتم تعيينه للقشرة الأولى منفيأقصر من ذلك في (لـ السندات في )، الذي يُنسب إلى الهيكل المميز للغلاف الثاني بين
الشكل 2 | خصائص الخصائص الإلكترونية للمواد المحفزة الكهربائية. أ Irطيف XPS لـ TS-Ir/، ، و “. إير-طيف XANES الحافة وفويل فورييه وإير.-طيف XANES الحافة و e FTs لـ-تذبذبات EXAFS الحادة لـ TS-Ir/، ، ورقة Mn. تحويلات (FTs) من Ir-تذبذبات EXAFS الحادة لـ TS-Ir/،
إر-و. علاوة على ذلك، فإن طول الرابطة الظاهر بين Ir-O في القشرة الأولى في TS-Ir/أطول قليلاً من قشرة Ir-O الأولى في Ir-، الذي يُنسب إلى إجهاد الشد. في هذه الأثناء، طول الرابطة في TS-Ir/أقصر من ذلك في، مما يشير إلى أن زيادة التساهمية وانتقال الإلكترون لـالرابطة كانت مناسبة في TS-من أجل تقييم الطول الحقيقي للرابطة بالفعلالتنسيق، المنحنيات الملائمة لـ-وزن Ir-edge EXAFS طيف جميع العينات في الشكل التكميلية 11، والتي تم إدراج البيانات المقابلة لها في الجدول التكميلية 2، تظهر أن عدد تنسيق قشرة Ir-O الأولى هووأن طول رابطة الإيريديوم-أوكسجين الحقيقي هوفي TS-Ir/، الذي هو أقصر من ذلك في وأطول من ذلك في إير-“، مما يشير إلى وجود إجهاد شد وزيادة في التساهمية. علاوة على ذلك، بعد إدخال Ir إلىطيف FT-EXAFS لـ-الحافة في الشكل 2e لا تظهر أي تغيير واضح في طول الرابطة لـالقشرة الأولى، التي تعود أساسًا إلى انخفاض محتوى الإيريديوم. ومع ذلك، يمكن العثور على تأثير إجهاد الشد من خلال المقارنةأطوال الروابط في TS-Ir/ ضد (الجدول التكميلي 1)، وزادت أطوال الروابط بمقدارمما يشير إلى وجود إجهاد شد في المضيفلـ TS-Ir/. ومع ذلك، يمكن أيضًا العثور على تأثير إجهاد الشد من خلال المقارنة بينأطوال الروابط في TS-Ir/ ضد ، الذي يكشف عن أصل إجهاد الشد في موقع Ir في الألياف النانوية. علاوة على ذلك، هناك قمة أخرى عندأقصر من عند (لرابطة Ir-Ir عند ) وأكبر من في (لـ رابطة في )، مما يشير إلى أنه يمكن تخصيص الذروة لـ في. هذا يُظهر بشكل أكبر أن الإيريديوم الموزع ذريًا يحل محل مواقع المنغنيز السطحية في شبكة الألياف النانوية من خلال تفاعل تبادل الكاتيونات في TS-Ir/. لذلك، تكشف هذه النتائج أن إطالة السطحالسندات في TS-يعدل بشكل فعال الروابط التساهمية والهياكل الإلكترونية لمواقع الإيريديوم في العينات.
التوصيف الكهروكيميائي في الإلكتروليت الحمضي
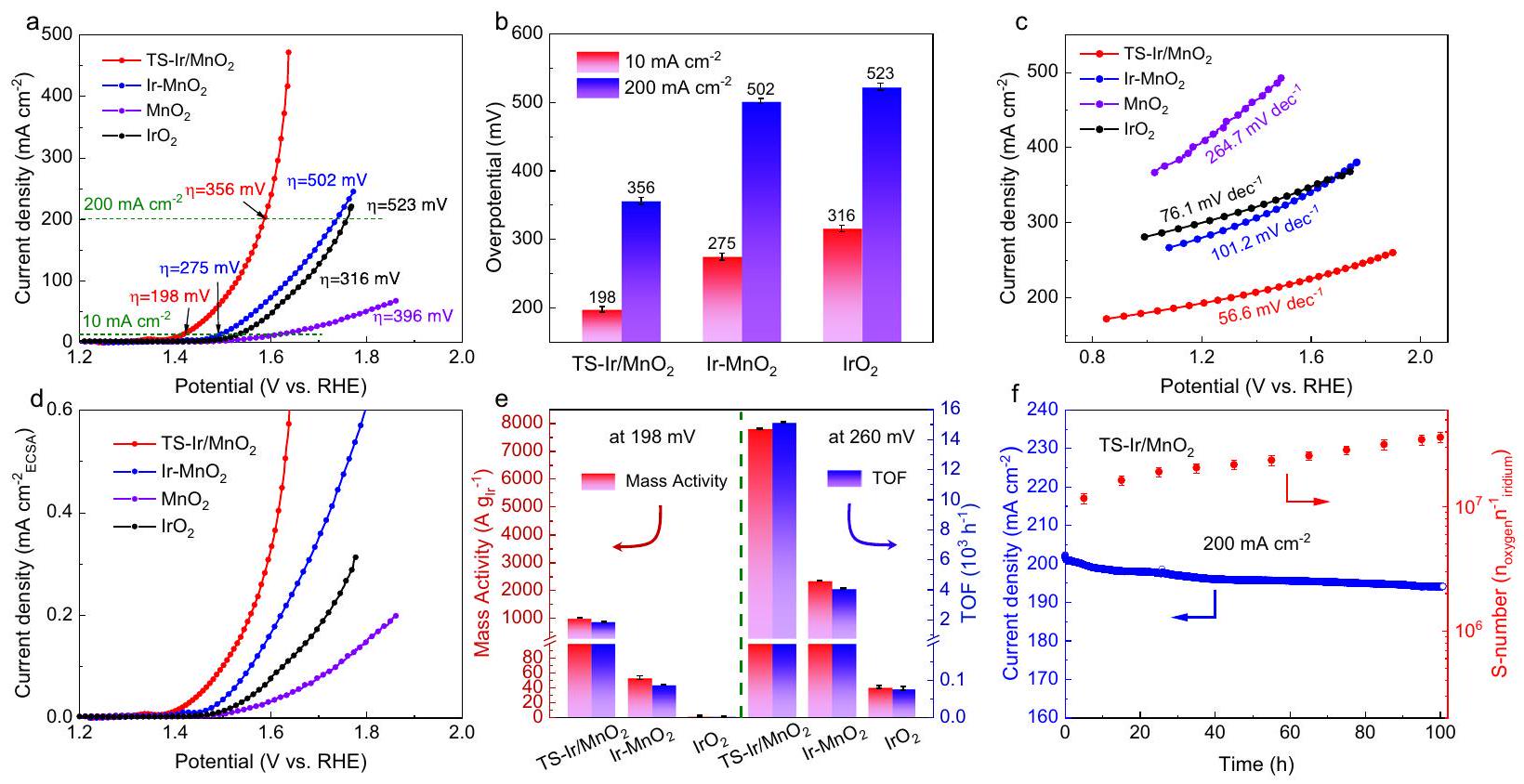
لتقييم نشاط التحفيز الكهربائي لتفاعل الأكسدة (OER) لـ TS-Ir/تم استخدام محطة عمل كهربائية كيميائية ثلاثية الأقطاب لإجراء القياسات في نإلكتروليت، وإجهاد الشد الخالي من الإيريديوم، و “تم استخدام عينات مرجعية. لزيادة عدد مواقع الفتحات والأنواع المتاحة، تم اختيار قماش الكربون لدعم المحفزات للقياسات الكهروكيميائية (الشكل التكميلي 12). منحنيات الفولتمترية المسحية الخطية (LSV) لـ TS-Ir/وعينات المرجع موضحة في الشكل 3أ. لتحقيق كثافة تيار نموذجية من, TS-Ir/ يتطلب فقط جهدًا منخفضًا للغاية ) من 198 مللي فولت، وهو أقل من تلك الخاصة بـ , وتجاري. علاوة على ذلك، تم تحضير المحفزات بتحميلات مختلفة من Ir، من بينها TS-Ir/مع تحميل معتدل من Irأظهرت أفضل نشاط في تفاعل الأكسدة (الشكل التكميلي 13). ومن المثير للاهتمام، TS-Ir/يظهر أفضل نشاط حمضي عند كثافة تيار عالية منويتطلب فقط جهدًا زائدًا منخفضًا قدره 356 مللي فولت (الشكل 3ب). تُظهر بنية الشكل وأداء OER لمحفزات الإلكتروكيميائية من Ir عند درجات حرارة مختلفة للتسخين الحراري في الشكل التكميلي 14، مما يكشف عن إجهاد الشد الأمثل والمواقع النشطة الموزعة ذريًا من Ir في TS-Ir/المحفز الكهربائي. بالنظر إلى تأثير درجة الحرارة على أداء الأكسدة الحمضية، قمنا بإجراء اختبار لتفكيك الماء تحت درجات حرارة تشغيل مختلفة. إن نشاط الأكسدة الحمضية يعتمد بشكل كبير على درجة الحرارة، وتكون الفائض الكهربائي لـ TS-Ir/انخفاض المحفز الكهربائي من 198 إلى 180 مللي فولت عند كثافة تيارمع زيادة درجة الحرارة إلى“، وهو ما يتماشى مع ما تم ملاحظته من أن درجة حرارة أعلى توفر حركيات OER أسرع ونشاط OER أفضل (الشكل التكميلي 15). علاوة على ذلك، فإن حركيات OER الحمضية لـ TS-Ir/تم تحليله، كما هو موضح في الشكل 3c. من الواضح أن TS-Ir/يعرض أقل ميل لطاولة، والتي هي بشكل ملحوظ أكثر لطفًا من تلك الخاصة بـ Ir- وتجاريويكشف عن حركيات OER أسرع وانتقال الإلكترونات علىالمواقع النشطة في TS-Ir/بفضل الروابط التساهمية المناسبة بين إيريديوم والأكسجين.يوفر نشاطًا حركيًا تحفيزيًا فعالًا في الجدول التكميلي 3، وهو يتجاوز ذلك الخاص بمحفزات OER المماثلة. علاوة على ذلك، وفقًا لتحليل طيف الامتياز الكهربائي، TS-Ir/لديه نقل شحنة ضئيل
الشكل 3 | خصائص التحفيز الكهربائي لتفاعل الأكسدة (OER) للمواد المحفزة الكهربائية في ( ). منحنيات الفولتمترية ذات المسح الخطي (LSV)، الجهود الزائدة عند 10 و(أشرطة الخطأ هي الانحرافات المعيارية لثلاث حسابات مكررة). تحميل المحفزات هوومقاومة المحلول هي. ميل الطاولة، ومنحنيات استقطاب OER بناءً على المساحة السطحية الكهروكيميائية (ECSA) لـ TS-Ir/إير-، و “مدعوم على أقمشة الكربون.
المقاومة وبالتالي أسرع حركيات التفاعل (الشكل التكميلي 16).
من المهم أن المساحة السطحية النشطة كيميائيًا (ECSA) لـ TS-Ir/تم تحقيقه من خلال عامل الخشونة، وسعات الطبقة المزدوجةتم قياسها (الأشكال التكميلية 17 و 18 والجدول 4)“. من الجدير بالذكر، TS-Ir/ لديه أعلى من“، مما يشير إلى وجود مواقع نشطة مفتوحة أكثر لتحقيق نشاط OER عالي الكفاءة. كما هو متوقع، تظهر الشكل 3d أن TS-Ir/كما يقدم أفضل نشاط محدد عندما يتم تطبيع كثافة التيار وفقًا لـ ECSA. تكشف هذه النتائج عن النشاط الفائق للأكسدة الحمضية لـ TS-Ir/لتقييم النشاط الجوهري لـ TS-Ir بشكل أكبرتم حساب النشاط الكتلي وتردد الدوران (TOF) بناءً على تحميل مواقع Ir، كما هو موضح في الشكل 3e. TS-Ir/لديه نشاط كتلة عالي منووقت طيران فائق الارتفاع لـعند جهد زائد نموذجي قدره 198 مللي فولت (يصل إلى كثافة تيار قدرها )، وهو أعلى بمقدار 380 مرة من تلك الخاصة بالتجارة وعلى التوالي). مع زيادة إمكانيات العمل، كل من النشاط الكتلي و TOF لـ TS-Ir/زيادة كبيرة مقارنة بتلك الخاصة بـ“، مما يؤكد أن TS-Ir/لديه حركيات OER أسرع من“. والأهم من ذلك، TS-Ir/حقق نشاطًا كتلويًا فائق الارتفاع منعند 1.65 فولت (الجدول التكميلي 5)، وهو أعلى بكثير من الهدف (عند 1.7 فولت) الذي حددته الوكالة الدولية للطاقة المتجددة (IRENA). بعبارة أخرى، هذا يعادل تقليل كمية المعدن الثمين (Ir) المستخدم لتحقيق طاقة كهربائية مماثلة للتطبيقات العملية.
علاوة على ذلك، فإن متانة التشغيل مهمة جدًا للتطبيقات العملية للكهروكاتاليست. لتقييم متانة التشغيل للكاتاليست، تم قياس منحنيات استقطاب OER (الشكل التكميلي 19) والكرونوأمبيرومترية (الشكل التكميلي 21) عند إمكانيات نموذجية. TS-Ir/يظهر استقرارًا تحفيزيًا جيدًا عند كثافة تياربعد 200 ساعة من اختبارات OER المستمرة مع تدهور أداء ضئيل. لتوضيح المتانة الكهروكيميائية عند كثافات تيار أعلى، تم إجراء اختبارات OER المستمرة. تم الأداء لـ TS-Ir/عند كثافة تياركما هو موضح في الشكل 3f. TS-Ir/لا يزال يحتفظ بمستوى مرضٍكثافة التيار الأولية بعد 100 ساعة من التشغيل. في هذه الأثناء، فقطمن Ir المستخرج من TS-Ir/بعد التشغيل المستمر لتفاعل الأكسدة الكهربائي، والذي يُعزى إلى تأثير الشبكة المحصورة القوي للمواد المتينةركيزة لتثبيط ذوبان مواقع النشاط لإيريديوم. كما هو موضح في الشكل التوضيحي 21، TS-Ir/لديه رقم S مرتفع ( ) في ، مما يشير إلى استقرار جيد في الأكسدة الكهربائية الحمضية. شهد تركيز الإريديوم زيادة بطيئة ولم يُلاحظ أي اتجاه تنازلي خلال الساعات القليلة الأولى من عملية التحليل الكهربائي للأكسجين، مما يشير إلى أن الإريديوم المذاب لم يكن له توازن ديناميكي واضح بين الترسيب والذوبان. علاوة على ذلك، تُظهر التوصيفات الشكلية والهيكلية، مثل TEM وXPS وXAFS (الأشكال التكميلية 22-24، على التوالي)، بعد قياسات كهربائية طويلة الأمد بوضوح أن الإريديوم موزع ذريًا فيدون تكتل وأن حالة أكسدة الإيريديوم وهيكل التنسيق المحلي تتغير بشكل ضئيل. ومن الجدير بالذكر أن ذرات الأكسجين في الشبكة تظهر انزياحًا إيجابيًا طفيفًا بعد قياسات OER، مما يكشف أن ذرات الأكسجين المؤكسدة في الشبكة يمكن أن تشارك بشكل أكبر في التفاعل الحفاز (الشكل التكميلي 25). تؤكد هذه النتائج الاستقرار الجيد لـ TS-Ir/العامل المساعد خلال عملية الأكسدة الكهربائية الحمضية المستمرة. باختصار، فإن المتانة العالية لـ TS-Ir/يُنسب أداء المحفز الكهربائي تحت ظروف التشغيل المستمرة إلى الهيكل المستقر وآلية L-LOM المستمرة خلال عملية OER، والتي يمكن أن تثبط الأكسدة الزائدة وذوبان مواقع النشاط لإيريديوم.
استكشاف الآلية من خلال التوصيف في الموقع
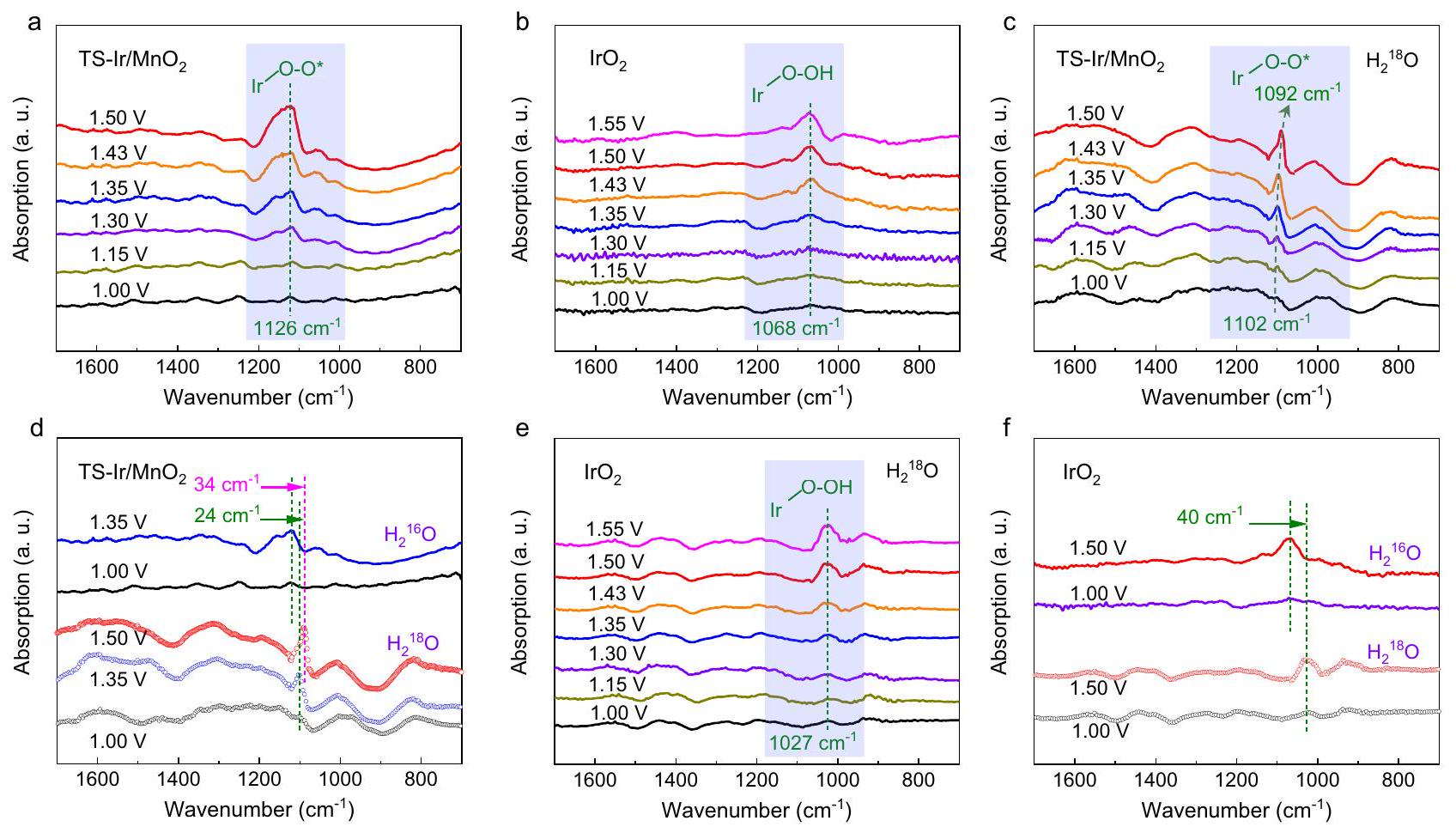
لتوضيح الآلية التحفيزية، تم قياس طيفي SRIR وXAFS في الموقع باستخدام خلايا مصنوعة محليًا تحت ظروف جهد نموذجية.كما هو موضح في الشكل 4a، لا تظهر طيفيات SRIR في الموقع أي نطاقات امتصاص واضحة لـ TS-Ir/في النطاقعند الإمكانياتمقابل قطب هيدروجين عكسي (RHE، الجهود المذكورة أدناه جميعها بالنسبة لـ RHE). مع زيادة
الشكل 4 | قياسات SRIR في الموقعتحميل المحفزات هوومقاومة المحلول. قياسات طيفية SRIR في الموقع في النطاقعند إمكانيات مختلفة لـ TS-Ir/ وإير أوقياسات طيفية SRIR في الموقع باستخدام وسم النظائر نطاق 1700-700عند إمكانيات مختلفة لـتي إس-و. د تم توليد طيف SRIR في الموقع تحت ظروف عدم ووسم النظائر عند جهدين نموذجيين قدرهما 1.00 و 1.35 فولت لـ TS-Ir/. f تم توليد طيف SRIR في الموقع تحت ظروف عدم ووسم النظائر عند إمكانيات نموذجية تبلغ 1.00 و 1.50 فولت لـ. إمكانية الوصول إلى 1.30 فولت مقابل RHE، ظهرت شريحة امتصاص عندويمكن أن يتم تعيينه إلى المفتاحالوسطاء لأن أنواع الأكسجين (عادةً ما تكون اهتزازات الشد في النطاق. من المثير للاهتمام أن شدة هذه النطاق الامتصاصي مرتبطة إيجابياً بالجهد المطبق، مما يكشف عن التراكم السريع للوسطاء الجذريين O-O الرئيسيين على المواقع النشطة. للمقارنة، تم قياس طيف SRIR في الموقع تحت ظروف OER مماثلة لـكما هو موضح في الشكل 4ب. مع زيادة الجهد المطبق تدريجياً، ظهرت شريحة امتصاص الأشعة تحت الحمراء عند. لأن قمم الاهتزاز تحت الحمراء لنوع *OOH عادة ما تظهر في المنطقةنطاق الامتصاص عنديمكن تخصيصه لإنتاج أنواع *OOH على مواقع Ir. نتيجة SRIR لـيظهر أيضًا إنتاج الأنواع الرئيسية * OOH تحت ظروف العمل، مما يشير إلى أن Ir-يحفز تفاعل الأكسدة الذي يتبع مسار AEM البطيء من حيث الديناميكا (الشكل التكميلي 26). ومن الجدير بالذكر أنه خلال تفاعل الأكسدة، تراكمت فقط الجذور الحرة O-O.“، الذي تجاوز تشكيل الأنواع البطيئة *OOH. تشير هذه النتائج إلى أنه على TS-Ir/على السطح، يخضع تفاعل OER لآلية تفاعل مختلفة بدلاً من AEM التقليدي علىسطح.
لتأكيد هذه الفرضية، تم قياس طيف SRIR مع وضع العلامات النظيرية في الموقع لـ TS-Ir/ومع زيادة الجهد المطبق من 1.00 إلى 1.55 فولت مقابل RHE. عادةً، بالنسبة لعينات الأكسجين ( ) ، تبادل النظائر يزاحم نطاق الاهتزاز بواسطة. وبالمثل، بالنسبة لأنواع الأكسجين ( ) ، يمكن لتبادل النظائر أن يحول نطاق الاهتزاز إلى الأحمر بحواليكما هو موضح في الشكل 4c، تحت ظروف العمل، ظهرت نطاق الامتصاص السائد عندلـ TS-Ir/، وتشير زيادة شدة الفرقة إلى احتمال تحمل الاعتماد. والأكثر إثارة للاهتمام، مع زيادة الجهد المطبق إلى 1.50 فولت، فإن السمة السائدة انزياحات نطاق الامتصاص، مما يكشف أن ( تزداد النسبة تدريجياً مع زيادة الجهد المطبق. عند جهد مطبق قدره 1.35 فولت، انزاح نطاق الامتصاص نحو الأحمر. من 1126 إلىعندماتم استبدال الحل بـ، الذي أكد أنه خلال الـ OER، الجذور مشتقة من الممتصالجزيئات وذرات الأكسجين في الشبكة (الشكل 4d). تكشف هذه النتائج عن آلية مشابهة لـ LOM على TS-Ir/سطح المحفز. علاوة على ذلك، مع زيادة الجهد المطبق إلى 1.50 فولت، انزاح نطاق الامتصاص بشكل أكبر نحو الأحمر إلى، مما يشير إلى أن جزيئات الماء تملأ فجوات الأكسجين بسرعة لتحفيز آلية مشابهة لـ LOM بشكل مستمر. في الوقت نفسه، بالنسبة لـ (الشكل 4هـ، و) ،ظهرت شريحة الامتصاص عندوزادت تدريجياً مع زيادة الجهود المطبقة. ومن الجدير بالذكر أن نطاق امتصاص *OOH ينتقل من 1068 إلىعندمايتحول إلى، مما يؤكد أن الناشئة الأنواع الوسيطة مشتقة منالجزيئات الممتصة عند المواقع النشطة. تؤكد هذه النتائج أن تفاعل الأكسدة على TS-Ir/تتبع السطح مسار تفاعل تحفيزي مختلف عن ذلك الموجود على سطح المنتجات التجاريةويتجاوز الـوسيط أثناء التفاعل لتسريع مسار تفاعل الأربعة إلكترونات.
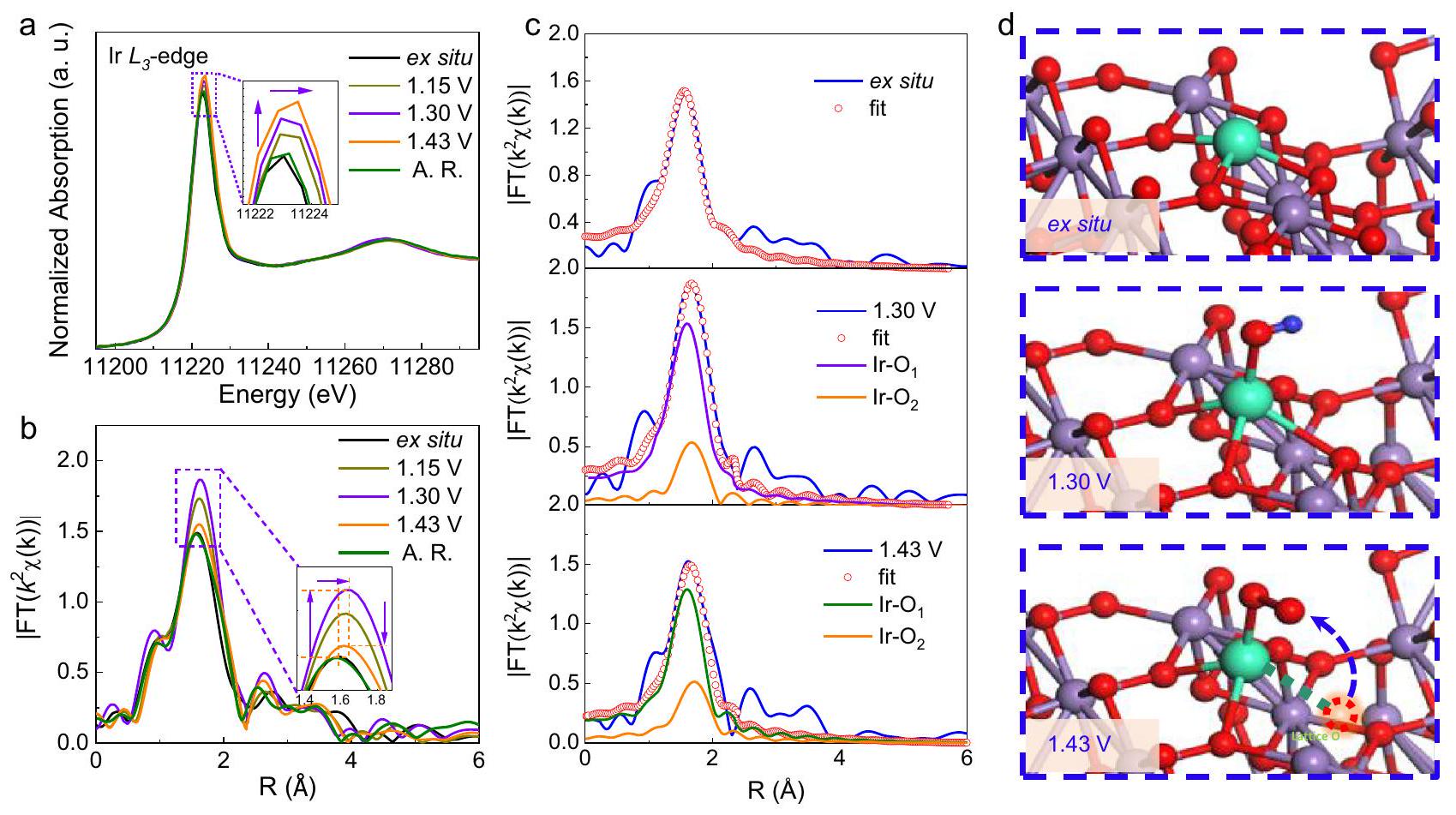
لتوضيح الآلية الأساسية لـ TS-Ir/تم قياس طيف XAFS في الموقع، الذي يتأثر بالتطور الهيكلي المحلي، باستخدام خلية مصنوعة يدويًا تم تصنيعها بناءً على نظام الثلاثة أقطاب.الشكل 5أ يُظهر طيف XANES في الموقع للـ Ir-حافة لـ TS-Ir/سُجلت عند إمكانيات تطبيقية مختلفة. بالمقارنة مع الحالة الخارجية (الغمر في محلول حمضي دون تطبيق أي جهد)، فإن قمة الخط الأبيض تتعزز قليلاً وتتحول إيجابياً مع زيادة الجهد المطبق إلى 1.43 فولت مقابل RHE، مما يشير إلى أن حالة الأكسدة لمواقع Ir قد ارتفعت. ويعزى ذلك إلى حركة المزيد من الإلكترونات من Ir إلى الأنواع الماصة القريبة من الأكسجين، مما يعزز بعد ذلك تفاعل الأكسدة السريع. طيف FTEXAFS لـ Ir-الحافة (الشكل 5ب والشكل التوضيحي 27) تظهر قمة سائدة عند“، الذي يُعزى إلى القشرة الأولى لرابطة Ir-O. بالمقارنة مع الحالة الخارجية، زادت شدة قمة القشرة الأولى عند جهد مُطبق قدره 1.15 فولت وأظهرت انزياحًا طفيفًا نحو القيم الأعلى من 1.54 إلى (الجدول التكميلي 6). مع زيادة
الشكل 5 | قياسات XAFS في الموقعتحميل المحفزات هوومقاومة المحلول. طيف XANES في الموقع للروثينيوم-حافة لـ TS-Ir/ عند الجهود النموذجية خلال عملية تطور الأكسجين.مقابل –
طيف FT-XAFS الموزون.المنحنيات الملائمةمخططات آلية OER لـ TS-تحت ظروف 1.30 و 1.43 فولت. يظهر الشكل المصغروتم الإشارة إلى ذرات الهيدروجين بالأرجواني والأحمر والأخضر والأزرق الملكي، على التوالي. عند تطبيق جهد قدره 1.30 فولت، زادت شدة قمة التنسيق لإيريديوم. تشير هذه النتائج إلى إعادة ترتيب الهيكل التنسيقي المحلي لمواقع الإيريديوم خلال عملية تطوير الأكسجين، وهو ما يرجع إلى امتصاص أنواع الأكسجين على مواقع الإيريديوم. ومن المثير للاهتمام أنه عند تطبيق جهد نموذجي قدره 1.43 فولت على القطب، تضعف قمة التنسيق، مما يشير إلى أن عدد تنسيق الإيريديوم ينخفض تحت ظروف تطوير الأكسجين (توليد فراغات أكسجين). علاوة على ذلك، فإن “تظهر طيفيات XAFS الحافة أن المادة في الطور الكتلي تحافظ على استقرارها الهيكلي (الشكل التوضيحي 28). والأهم من ذلك، بعد قياسات OER طويلة الأمد لـ TS-Ir/، كان عدد تنسيق Ir يقترب من ذلك تحت ظروف خارج الموقع. يكشف طيف الأشعة تحت الحمراء للعينة الموسومة نظائريًا بعد التفاعل الموضح في الشكل التكميلية 29 أن الذروة لـ ( وانزياحات الطيف الأحمر لـ Mn)، مما يؤكد أن TS- يحتوي على بعد التفاعل. تؤكد هذه النتائج أن الفراغات الموضعية للأكسجين على TS-Ir/تم ملء السطح بسرعة بواسطةالجزيئات لتعديل آلية LOM، التي تُسمى هنا آلية “LOM المحلية” (L-LOM).
تظهر المنحنيات الملائمة لـ FT-XAFS في الشكل 5c والأشكال التكميلية 30 و31، والبيانات المقابلة مدرجة في الجدول التكميلية 7 لتكوين الأربعةروابط التنسيق التي تم تحسينها كميًا خارج الموقع. عند 1.30 فولت، تم اعتبار غلاف أول إضافي لرابطة التنسيق الأطول Ir-O. )، مما يعني أن أنواع الأكسجين تم امتصاصها على مواقع الإيريديوم. مع زيادة الجهد المطبق إلى 1.43 فولت، انخفض عدد التنسيق لروابط الإيريديوم-أكسجين الأقصر إلى ثلاثة ( )، وظهرت رابطة تنسيق Ir-O أطول واحدة عند . تشير هذه النتائج إلى أنه بالنسبة لمواقع الإريديوم المتوترة، يحدث تطور هيكلي مدفوع بالجهد خلال عملية تطوير الأكسجين عن طريق إطلاق ذرات الأكسجين الشبكي من TS-Ir/المواقع السطحية المحلية لتحفيز آلية تفاعل L-LOM. التحليل الشامل للمنحنيات الملائمة التي تم الحصول عليها لمواقع Ir والجذور الحرة O-O التي تم استكشافها عبر SRIR في الموقع يشير إلى أن جزيئات الأكسجين التي تمتص على مواقع Ir المتوترة يمكن أن تتفكك بسرعة وتتزاوج مع ذرات الأكسجين الشبكية المجاورة لتكوين جذور O-O. nas *OO-Ir- “، الذي يتجاوز الوسطاء البطيئين *OOH (الشكل 5d). يمكن لآلية L-LOM المقترحة تسريع الوسائط المحتوية على الأكسجين التي تم نزع بروتونها بسرعة ثم ربط ذرات الأكسجين الشبكية لتشكيل ” النشطة الحقيقية الهيكل. بعد يتم إزالة الامتصاص، وتملأ جزيئات الماء بسرعة الفراغات الأكسجينية وتفقد البروتونات بسرعة للحفاظ على السلامة الهيكلية لـ TS-Ir/العامل المساعد. تشير هذه النتائج إلى أنه بالمقارنة مع آلية LOM التقليدية، يمكن لآلية L-LOM تسريع إزالة البروتون من *OH على فراغات الأكسجين السطحية ومنع الأكسدة المحلية لمواقع Ir السطحية بالإضافة إلى فقدان المزيد من ذرات الأكسجين الشبكي في الكتلة الداخلية للعوامل المساعدة. فوق كل شيء، فإن الضغط الشدّي الذي تم تقديمه في TS-Ir/يمكن أن يعزز من تساهمية رابطة Ir-O لتحسين قدرة إزالة البروتون وزيادة حالة أكسدة Ir، مما يؤدي إلى تفعيل آلية L-LOM المحلية المستمرة تحت ظروف العمل. لذلك، يمكن لطريق L-LOM تحقيق حركيات OER الحمضية بشكل أسرع بكثير مع تحسين الاستقرار الهيكلي للمحفز أثناء التفاعل، مما يمنح TS-Ir/محفز ذو إمكانيات جيدة للتطبيقات الصناعية.
أداء جهاز PEMWE
للتحقق من إمكانيات TS-Ir/للتطبيقات الصناعية، قمنا بتصنيع جهاز PEMWE باستخدام نافيونالغشاء و TS-Ir/وتجاريكعوامل حفازة للأنود والكاثود، على التوالي، في إلكتروليت حمضي. الشكلوعرض صور لكل من خلية ومجموعة التحليل الكهربائي PEM. الأنود TS-Ir/والكاثودتشكل القطب الكهربائي، ويتم مطابقة جهاز التسخين ونظام تدوير المحلول لتشكيل جهاز الخلية الكهروكيميائية بالكامل، حيث يتم التحكم في درجة حرارة التشغيل عندوسرعة دوران المحلول هي“. تم تصنيع المحلل الكهربائي الناتج باستخدام كل من TS-Ir/ويوفر التيتانيوم نشاطًا واستقرارًا محسنين في التحليل الكهربائي للماء. على وجه التحديد، عندما تكون كمية المعدن الثمين (الإيريديوم) هيللكهربائي الناتج مع TS-Ir/، يتم تقليل الجرعة بمقدار 54 مرة مقارنة بالهدف ( ) التي وضعتها الوكالة الدولية للطاقة المتجددة (IRENA). وهذا يعني أن تكلفة المحفزات لـ
الشكل 6 | قياسات أداء جهاز PEMWE فيخلية وم系统 (ب) من المحلل الكهربائي PEM (أ).جهد الخلية لنظام التحليل الكهربائي PEM استقر عندتحميل المحفز هوومقاومة المحلول هي.
يمكن تقليل إنتاج الهيدروجين بشكل كبير. في التطبيقات العملية، تعتبر الاستقرار الحفزي أكثر أهمية من النشاط الحفزي. كما هو موضح في الشكل 6c، فإن الخلايا الكهروكيميائية الناتجة توفر كثافة تيار تبلغعند انحياز مطبق لـويمكن أن تعمل بشكل مستمر لمدة 200 ساعة مع تدهور ضئيل تحت ظروف صناعية محاكاة (عندصور TEM (الشكل التكميلي 32) تكشف أن TS-Ir/لم تتغير التركيبة الشكلية بعد 200 ساعة من التشغيل في جهاز التحليل الكهربائي PEM. علاوة على ذلك، أكدت نتائج ICP-OES أن إيريديم (Ir) ذاب بشكل ضئيل (ضمن 8%) في العينات. تشير نتائج اختبار الاستقرار هذه إلى الإمكانيات الجيدة لـ TS-Ir/محفز لتطبيقات PEMWE العملية.
باختصار، فإن المحفز الكهربائي منخفض الإريديوم الذي يتم فيه توطين ذرات الإريديوم في مواقع الكاتيونات على سطح أكسيد المنغنيز المتوتر (TS-Ir/تم تطويره من خلال تبادل الكاتيونات وRTAC اللاحق. كشفت الدراسات الآلية التي استخدمت تقنيات SRIR وXAFS في الموقع أن *OO-Ir- الناتجيمكن أن يتم نزع البروتونات من الوسطيات بسرعة، ثم تملأ جزيئات الماء الفراغات الأكسجينية بسرعة حول مواقع إير المتوترة على السطح للحفاظ على السلامة الهيكلية للمحفز، الذي قام بتحفيز تفاعل الأكسدة الكهربائية للأكسجين بكفاءة من خلال آلية L-LOM المستمرة. خاصة، يمكن أن تسرع هذه الآلية L-LOM بشكل كبير من حركية تفاعل الأكسدة الكهربائية للأكسجين الحمضي مع تحسين السلامة الهيكلية للمادة. لذلك، تم الحصول على TS-Ir/المحفز يوفر نشاطًا عالي الكتلة منعند جهد زائد قدره 198 مللي فولت (عادة عند كثافة تيار من )، وهو ما يزيد بحوالي 19 و 380 مرة عن تلك الخاصة بـ وتجاري“، على التوالي. من المثير للاهتمام، TS-Ir/يظهر التحليل الكهربائي للماء الحمضي المستقر لمدة 100 ساعة عند كثافة تيارمع تدهور أداء ضئيل. والأهم من ذلك، أن جهاز التحليل الكهربائي PEM الناتج قدم كثافة تيارعند انحياز مطبق لـلأكثر من 200 ساعة تحت ظروف صناعية محاكاة (عندتظهر النتائج في هذا العمل ليس فقط وجود محفز قوي وعالي النشاط لتعزيز التطبيقات الصناعية لخلايا الوقود ذات الأغشية البوليمرية، ولكنها تقدم أيضًا استراتيجية لاستخدام الشد التوتري لتحفيز آلية L-LOM من أجل تعزيز النشاط والمتانة لمختلف التفاعلات الحفزية.
طرق
تركيب
تمت عملية التخليق الهيدروحراري باستخدام إجراء نموذجي كما يلي: تم إذابة كبريتات المنغنيز (0.02 مول) وكبريتات الأمونيوم (0.02 مول) وكبريتات الأمونيوم (0.06 مول) في 70 مل من الماء المنزوع الأيونات (DI) وتمت معالجتها بالموجات فوق الصوتية. ثم تم تحريك المحلول بقوة باستخدام مغناطيس لمدة ساعة واحدة، ثم تم نقله إلى وعاء ضغط وتم تسخينه إلىلمدة 12 ساعة. بعد ذلك، تم استخدام لون أسودتم الحصول على المسحوق بعد الغسيل بالطرد المركزي والتجفيف بالفراغ عدة مرات لكل منهما.
تركيب TS-Ir/
عادةً، 120 ملغ منتم إذابته في 30 مل من الماء المنزوع الأيونات وتمت الموجات فوق الصوتية له بشكل مستمر لمدة 30 دقيقة. ثم أضيف 10 مل من محلول كلوريد الإريديوم المائي (5 مللي مول) إلى هذا المحلول وتمت الموجات فوق الصوتية له بشكل مستمر. بعد ذلك، تم تحريك هذا المحلول مغناطيسيًا بشكل مستمر لمدة 12 ساعة ثم تم غسله ثلاث مرات. أخيرًا، تم الحصول على مسحوق جاف وتم نقله مباشرة إلى فرن مافل عندلمدة ساعتين ثم تم نقلها بسرعة إلى الهواء للتبريد السريع. تم إدخال إجهاد الشد الشبكي إلى TS-Ir/عينة من خلال التسخين والتبريد الشديد.
تركيب
على الرغم من أن مسار التخليق هذا مشابه لذلك لـ TS-Ir/لم تكن هناك حاجة للتسخين والتبريد السريع. بدلاً من ذلك، تم نقل المسحوق المجفف إلى الفرن الميكانيكي وتم تسخينه عندإلىتم الاحتفاظ بها هناك لمدة ساعتين، ثم تم تبريدها إلى درجة حرارة الغرفة في الفرن.
الخصائص الهيكلية
تم إجراء التصوير المجهري الإلكتروني النافذ بتصحيح الانحراف الكروي (HAADF-STEM) وتحليل الطيف الطاقي باستخدام مجهر FEI Titan Themis (300 كف). تم إجراء TEM و HRTEM باستخدام مجهر JEM-2100F الذي يعمل بتسارع 200 كف. تم الحصول على صور المجهر الإلكتروني الماسح بالانبعاث الميداني باستخدام مجهر Gemini SEM.
500 مجهر إلكتروني مسح ضوئي. تم قياس أنماط حيود الأشعة السينية باستخدام جهاز حيود الأشعة السينية Philips X’Pert Pro Super المزود بـمصدر الإشعاعتم تسجيل XPS باستخدام جهاز Thermo ESCALAB 250Xi المزود بمصدر أشعة Al K. ( مصدر الإثارة). تم تصحيح طاقات الربط المستخلصة من طيف XPS بالنسبة إلىبلغ الذروة عند 284.5 إلكترون فولت. تم إجراء مطيافية انبعاث الذرة باستخدام بلازما مقترنة بالحث (ICP-OES) باستخدام جهاز أوبتيما 7300 DV (بيركن إلمر).
القياسات الكهروكيميائية
تم إجراء جميع القياسات الكهروكيميائية لأداء الأكسدة الحمضية باستخدام نظام ثلاثي الأقطاب ومحطة العمل CHI 760 E. كان المحلول الكهربائي هو، وقضبان الجرافيت، إلكترود، وتم دعم قماش الكربون (CC) المحفزات كأقطاب مضادة ومرجعية وعاملة، على التوالي. تم تحضير محاليل المحفز عن طريق خلط 5.0 ملغ من المحفز في محلول يحتوي علىالإيثانول،من مياه DI، ومننافيوينحل واهتزاز الخليط لتشكيل أحبار متجانسة. ثم،تم إسقاط كمية من حبر المحفز الموزع بشكل جيد بعناية على CC النظيفة ( ) وتم تجفيفها بشكل طبيعي للتقييم. تم تحويل جميع الإمكانيات النهائية إلى الإمكانيات المرجعية المقابلة لـ RHE باستخدام المعادلة (ضد. RHE) (ضد. تم مسح منحنيات الاستقطاب بالمسح الخطي عندوتم تعويضه باستخدام تصحيح iR. السعة الكهربائية للطبقة الثنائية الكهروكيميائية (تم حساب ( ) استنادًا إلى منحنيات الفولتمترية الدورية التي تم مسحها في النطاقضد RHE في، و “تم حساب ECSA للمحفز بناءً علىوفقًا للمعادلة ECSA (2)، حيث هو استنادًا إلى القيم المبلغ عنها عادةًتم حساب عامل الخشونة (RF) من خلال تقدير المساحة السطحية الكهربائية الفعالة (ECSA) وقسمتها على المساحة الهندسية للقطب وفقًا للمعادلة. (3). تشير النشاط المحدد إلى كثافة التيار المحددة لكل ECSA ) ويمكن حسابها باستخدام المعادلة .
حسابات النشاط الكتلي وتردد الدوران
النشاط الجماهيري (MA) (يمكن حسابه بناءً على تحميل معدن الإيريديوموكثافة التيارعند الفائض الكهربائي النموذجي البالغ 198 و 260 مللي فولت، على التوالي، كما يلي:
يمكن حساب TOF بناءً على عدد المواقع النشطة وكثافة التيار (j) كما يلي:
أينهو ثابت فاراداي، و “يمثل ثابت أفوجادرو. بالنسبة للمحفزات الكهروكيميائية، يُعتبر العدد الإجمالي لمواقع سطح Ir مواقع نشطة على السطح لأن Ir موزع ذريًا ومثبت على رقيقالألياف النانوية.
حسابات رقم S
رقم S هو النسبة بين كميات الأكسجين المنبعث (المُحتسبة منالإجمالي) والإيريديوم المذاب (المستخلص من قياسات ICP-MS).
قياسات SRIR في الموقع
تم قياس طيف SRIR في الموقع باستخدام خلايا مصنوعة محليًا في خط الأشعة تحت الحمراء (BLO1B) من مختبر الإشعاع السنكروتروني الوطني (الصين). تم استخدام مطياف الأشعة تحت الحمراء (Bruker)مزود بمقسم شعاع KBr، وكاشف MCT مبرد بالنيتروجين السائل، وميكروسكوب IR (Bruker Hyperion 3000) الذي كان لديه عدسة الهدف تشكل جهاز اختبار SRIR. يمكن أن توفر طيف SRIR طيفًا واضحًا في النطاقبدقة طيفية عالية منلتقليل تداخل جزيئات الماء مع الإشارة تحت الحمراء أثناء التفاعل الكهروكيميائي، يجب ضبط المسافة بين القطب الكهربائي والنافذة على المقياس الميكروسكوبي. للحصول على إشارة متوسطة تفاعل ذات كثافة كافية، يجب استخدام وضع الانعكاس لجمع الإشارات تحت الحمراء. تم الحصول على طيف الخلفية لقطب الكهروكatalyst عند جهد الدائرة المفتوحة قبل كل قياس لنظام OER، وكانت نطاقات جهد OER المقاسة هيمن الجدير بالذكر أنه تم تنعيم منحنيات SRIR في الموقع باستخدام معالجة البيانات لـ 10 نقاط، مما يعني أنه تم حساب متوسط 10 نقاط متتالية لتحسين نسبة الإشارة إلى الضوضاء في المنحنيات.
قياسات XAFS في الموقع
تم قياس طيف XAFS في الموقع باستخدام خلايا مصنوعة محليًا في كل من محطة 1W1B في منشأة الإشعاع السنكروتروني في بكين (BSRF) وBL14W1 في منشأة الإشعاع السنكروتروني في شنغهاي (SSRF) في الصين. كانت حلقات التخزين في BSRF وSSRF تعمل عند 2.5 و3.5 جيجا فولت، على التوالي، عند أقصى تيار قدره 250 مللي أمبير. تم تلوين الشعاع الناتج من المغناطيس المنحني باستخدام مقياس تلوين مزدوج البلورات Si (111) وتم ضبطه بشكل إضافي بنسبة 15% لإزالة أي توافقيات أعلى. تم جمع أطياف XAFS باستخدام كاشف الحالة الصلبة المكون من 19 عنصرًا والذي يعمل في وضع الفلورية. تم قياس أطياف XAFS في الموقع باستخدام CC المعدل بالكاتاليس كقطب عمل تم لصقه بشريط كابتون.فيلم على الظهر. للحصول على معلومات حول تطور الموقع النشط خلال التفاعل الكهروكيميائي، تم تطبيق سلسلة من الجهود التمثيلية (1.15-1.43 فولت) على القطب. خلال جمع قياسات XAFS، تم قياس حافة الامتصاص (تم معايرة الموضع باستخدام عينة قياسية من Ir.
تحليل بيانات XAFS
تم معالجة بيانات EXAFS المستخرجة باستخدام وحدة ATHENA التي تم تنفيذها في حزم برامج IFEFFIT.. بعد ذلك، لفصل مساهمات EXAFS من قذائف التنسيق المختلفة، تم تحويل بيانات EXAFS إلى الفضاء الحقيقي باستخدام نافذة هانينغ. ) في الـ-نطاق الفضاء. عامل تقليل السعة ) تم الحصول عليه من الملاءمة منحنى EXAFS الحدي لثاني أكسيد الإريديوم. تم ضبط المنحنى باستخدام-دالة EXAFS الموزونةالبيانات في– و-نطاقاتو، على التوالي. يمكن حساب عدد النقاط المستقلة باستخدام المعادلة. لحالة TS-Ir/ خارج الموقع، أظهرت منحنى FT ذروة تنسيق بارزة عندالذي تم تعيينه لـرابطة التنسيق. بالمقارنة مع الحالة الخارجية، كانت قمم التنسيق في الطبقة الأولى عند الجهود المطبقة 1.15 و 1.30 و 1.43 فولت أقوى ولها قيم أعلى.التحولات، التي نُسبت إلى امتصاص أنواع الأكسجين. لذلك، فإن إضافةتم أخذ الروابط التنسيقية في الاعتبار أثناء ملاءمة المنحنى. ومن الجدير بالذكر أن القمة عند 1.43 فولت، التي تت correspond إلى روابط التنسيق Ir-O، قد ضعفت، مما قد يُعزى إلى تطور الهيكل التنسيقي المحلي لـ Ir. أثناء ملاءمة المنحنى، كانت عوامل ديباي-والر (أرقام التنسيق (المسافات بين الذرات ( ) ، وتحولات الطاقة ( ) تم اعتبارها معلمات قابلة للتعديل. لتقليل عدد معلمات التعديل القابلة للتعديل، كان كما هو الحال في العينة خارج الموقع عند جهد مختلف.
القياسات الكهروكيميائية في اختبارات PEMWE
أولاً، Nafion الذي تم الحصول عليهتمت معالجة الغشاء مسبقًا عن طريق تنظيفه بمحلول يحتوي علىوالماء منزوع الأيونات عندلمدة ساعة واحدة وتم الاحتفاظ به في الماء المقطر لتقييمات PEMWE اللاحقة. ثم، TS-Ir/وتم استخدام (ألياف الفelt المغلفة بالتيتانيوم Pt) كعوامل حفازة أنودية وكاثودية لاختبارات PEMWE، على التوالي. لتحضير حبر العامل الحفاز الأنودي، تم استخدام 5 ملغ من تم تعليق المحفز في 1 مل من الإيثانول والماء المختلطين بنسبة حجم 1:3. ثم،منتم إضافة محلول إلى هذا المزيج، وتمت عملية التشتت بالموجات فوق الصوتية لمدة ساعة حتى تم تشكيل حبر محفز متجانس. تم تحضير TS-Ir/حبر المحفزتم رشها على صفائح من ألياف التيتانيوم (مساحة السطح ). ثم، كانت القطب الكهربائي، الذي يتكون من TS-Ir المدعوم بألياف التيتانيوم.نَافْيُون المعالجغشاء، وتم ضغطه ساخنًا عند ضغط معين. أخيرًا، تم تطبيق القطب في PEMWE لتقييم الأداء التحفيزي. تم تشغيل جهاز PEMWE عندباستخدامالكهرباء السائلة تتدفق عنداستقرار الـ PEMWE الذي تم إجراؤه باستخدام TS-Ir/تم تقييم المحفز الأنودي من خلال قياس الكرونو بوتنشيومتر عند كثافة تيار قدرهالمدة 200 ساعة عندوضغط البيئة. تم الإبلاغ عن جميع فولتية الخلايا المقاسة في إلكتروليزرات PEMWE دون تطبيق أي تعويض iR.
توفر البيانات
جميع البيانات المبلغ عنها في هذه الورقة متاحة من المؤلف المراسل عند الطلب.
References
Chu, S. & Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 488, 294-303 (2012).
Seh, Z. W. et al. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 355, eaad4998 (2017).
An, L. et al. Recent development of oxygen evolution electrocatalysts in acidic environment. Adv. Mater. 33, 2006328 (2021).
Su, H. et al. In-situ spectroscopic observation of dynamic-coupling oxygen on atomically dispersed iridium electrocatalyst for acidic water oxidation. Nat. Commun. 12, 6118 (2021).
Wang, N. et al. Doping shortens the metal/metal distance and promotes OH coverage in non-noble acidic oxygen evolution reaction catalysts. J. Am. Chem. Soc. 145, 7829-7836 (2023).
Zhang, X. et al. Fast modulation of d-band holes quantity in the early reaction stages for boosting acidic oxygen evolution. Angew. Chem. 62, e202308082 (2023).
Song, J. et al. A review on fundamentals for designing oxygen evolution electrocatalysts. Chem. Soc. Rev. 49, 2196-2214 (2020).
Zhang, R. et al. A dissolution/precipitation equilibrium on the surface of iridium-based perovskites controls their activity as oxygen evolution reaction catalysts in acidic media. Angew. Chem. 131, 4619-4623 (2019).
Chen, F.-Y., Wu, Z.-Y., Adler, Z. & Wang, H. Stability challenges of electrocatalytic oxygen evolution reaction: From mechanistic understanding to reactor design. Joule 5, 1704-1731 (2021).
Lin, C. et al. In-situ reconstructed Ru atom array on with enhanced performance for acidic water oxidation. Nat. Catal. 4, 1012-1023 (2021).
Reier, T., Nong, H. N., Teschner, D., Schlögl, R. & Strasser, P. Electrocatalytic oxygen evolution reaction in acidic environments-reaction mechanisms and catalysts. Adv. Energy Mater. 7, 1601275 (2017).
Wang, X. et al. Pivotal role of reversible geometric conversion in oxygen evolution. Nature 611, 702-708 (2022).
Zhang, N. & Chai, Y. Lattice oxygen redox chemistry in solid-state electrocatalysts for water oxidation. Energy Environ. Sci. 14, 4647-4671 (2021).
Yao, Y. et al. Engineering the electronic structure of single atom Ru sites via compressive strain boosts acidic water oxidation electrocatalysis. Nat. Catal. 2, 304-313 (2019).
Shi, Z. et al. Confined Ir single sites with triggered lattice oxygen redox: Toward boosted and sustained water oxidation catalysis. Joule 5, 2164-2176 (2021).
Roy, C. et al. Trends in activity and dissolution on under oxygen evolution conditions: particles versus well-defined extended surfaces. ACS Energy Lett. 3, 2045-2051 (2018).
Hodnik, N. et al. New insights into corrosion of ruthenium and ruthenium oxide nanoparticles in acidic media. J. Phys. Chem. C 119, 10140-10147 (2015).
Wu, Z. Y. et al. Non-iridium-based electrocatalyst for durable acidic oxygen evolution reaction in proton exchange membrane water electrolysis. Nat. Mater. 22, 100-108 (2023).
Devaraj, S. & Munichandraiah, N. Effect of crystallographic structure of MnO 2 on its electrochemical capacitance properties. J. Phys. Chem. C 112, 4406-4417 (2008).
Wang, Z. et al. Influence of the phase on oxygen evolution reaction performance for low-loading iridium electrocatalysts. ChemElectroChem 8, 418-424 (2021).
Xu, Z. et al. Light-driven orderly assembly of Ir-atomic chains to integrate a dynamic reaction pathway for acidic oxygen evolution. Angew. Chem. Int. Ed. https://doi.org/10.1002/anie. 202301128 (2023).
Wen, M., Yang, H., Tong, L. & Yuan, L. Study on degradation of goldcyanide wastewater by rare-earth La doped composite photocatalyst. Mol. Catal. 550, 113506 (2023).
Yagi, S. et al. Covalency-reinforced oxygen evolution reaction catalyst. Nat. Commun. 6, 8249 (2015).
Cheng, W. et al. Lattice-strained metal-organic-framework arrays for bifunctional oxygen electrocatalysis. Nat. Energy 4, 115-122 (2019).
Hao, S. et al. Torsion strained iridium oxide for efficient acidic water oxidation in proton exchange membrane electrolyzers. Nat. Nanotechnol. 16, 1371-1377 (2021).
Lübke, M. et al. Transition-metal-doped nanorods as bifunctional catalysts for efficient oxygen reduction and evolution reactions. ChemistrySelect 3, 2613-2622 (2018).
He, C. et al. Molecular evidence for metallic cobalt boosting electroreduction on pyridinic nitrogen. Angew. Chem. 132, 4944-4949 (2020).
Edgington, J., Schweitzer, N., Alayoglu, S. & Seitz, L. C. Constant change: exploring dynamic oxygen evolution reaction catalysis and material transformations in strontium zinc iridate perovskite in acid. J. Am. Chem. Soc. 143, 9961-9971 (2021).
Chen, Y. et al. Exceptionally active iridium evolved from a pseudocubic perovskite for oxygen evolution in acid. Nat. Commun. 10, 572 (2019).
Sun, Z., Liu, Q., Yao, T., Yan, W. & Wei, S. X-ray absorption fine structure spectroscopy in nanomaterials. Sci. China Mater. 58, 313-341 (2015).
Wang, Q. et al. Ultrahigh-loading of Ir single atoms on NiO matrix to dramatically enhance oxygen evolution reaction. J. Am. Chem. Soc. 142, 7425-7433 (2020).
Geiger, S. et al. The stability number as a metric for electrocatalyst stability benchmarking. Nat. Catal. 1, 508-515 (2018).
Su, H. et al. Hetero-N-coordinated Co single sites with high turnover frequency for efficient electrocatalytic oxygen evolution in an acidic medium. ACS Energy Lett. 4, 1816-1822 (2019).
Cheng, W. R., Xu, Y. Z., Yang, C. Y., Su, H. & Liu, Q. H. Monitoring surface dynamics of electrodes during electrocatalysis using in situ synchrotron FTIR spectroscopy. J. Synchron. Rad. 30, 340-346 (2023).
Li, Y. et al. High mass-specific reactivity of a defect-enriched Ru electrocatalyst for hydrogen evolution in harsh alkaline and acidic media. Sci. China Mater. 64, 2467-2476 (2021).
Zhou, W. et al. Regulating the scaling relationship for high catalytic kinetics and selectivity of the oxygen reduction reaction. Nat. Commun. 13, 6414 (2022).
Lang, C. et al. Observation of a potential-dependent switch of water-oxidation mechanism on Co-oxide-based catalysts. Chemistry 7, 2101-2117 (2021).
Rao, R. R. et al. Operando identification of site-dependent water oxidation activity on ruthenium dioxide single-crystal surfaces. Nat. Catal. 3, 516-525 (2020).
Sivasankar, N., Weare, W. W. & Frei, H. Direct observation of a hydroperoxide surface intermediate upon visible light-driven water oxidation at an Ir oxide nanocluster catalyst by rapid-scan FT-IR spectroscopy. J. Am. Chem. Soc. 133, 12976-12979 (2011).
Chen, C. et al. Coupling and in to synthesize urea under ambient conditions. Nat. Chem. 12, 717-724 (2020).
Cheng, W., Su, H. & Liu, Q. Tracking the oxygen dynamics of solid-liquid electrochemical interfaces by correlative in situ synchrotron spectroscopies. Acc. Chem. Res. 55, 1949-1959 (2022).
Su, H. et al. Dynamic evolution of solid-liquid electrochemical interfaces over single-atom active sites. J. Am. Chem. Soc. 142, 12306-12313 (2020).
McCrory, C. C., Jung, S., Peters, J. C. & Jaramillo, T. F. Benchmarking heterogeneous electrocatalysts for the oxygen evolution reaction. J. Am. Chem. Soc. 135, 16977-16987 (2013).
Li, R. et al. IrW nanochannel support enabling ultrastable electrocatalytic oxygen evolution at in acidic media. Nat. Commun. 12, 3540 (2021).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchron. Rad. 12, 537-541 (2005).
الشكر والتقدير
تم دعم هذا العمل من قبل البرنامج الوطني الرئيسي للبحث والتطوير في الصين (2022YFA1502903 (Q.L.))، ومؤسسة العلوم الطبيعية الوطنية في الصين (12205300 (H.S.)، 22241202 (Q.L.)، و12135012 (H.S.))، ومؤسسة العلوم الطبيعية في مقاطعة آنهوي (رقم المنح 2208085JO1 (Q.L.) و2208085QA28 (H.S.)).
مساهمات المؤلفين
ق.ل.، هـ.س.، و س.ل. تصوروا المشروع. صمم هـ.س. و ج.ي. تجارب SRIR و XAFS في الموقع. قام هـ.س. و ج.ي. بإجراء التجارب، بما في ذلك تخليق المحفزات، واختبارات التحفيز، و SRIR في الموقع و
قياسات XAFS. قام M.L. بأداء النشاط التحفيزي. قام X.Z. و K.Z. بإجراء اختبارات TEM. قام W.Z. و Y.Z. و S.L. بتحليل البيانات التجريبية. تم كتابة المخطوطة بواسطة H.S. و C.Y. و Q.L. ناقش جميع المؤلفين النتائج وعلقوا على الورقة.
يجب توجيه المراسلات والطلبات للحصول على المواد إلى هوي سو، شيشون ليان أو تشينغhua ليو.
تُعرب مجلة Nature Communications عن شكرها لليزهي ليو، وشينلونغ تيان، والمراجعين الآخرين المجهولين، على مساهمتهم في مراجعة هذا العمل. يتوفر ملف مراجعة الأقران.
المختبر الرئيسي لمواد تحويل الطاقة الضوئية في كلية هنان، كلية الكيمياء والهندسة الكيميائية، جامعة هنان العادية، تشانغشا 410081 هنان، الصين.مختبر الإشعاع السنكروتروني الوطني، جامعة العلوم والتكنولوجيا في الصين، هيفي 230029 آنهوي، الصين.المختبر الوطني الرئيسي لعلوم المعادن المسحوقة، جامعة جنوب الوسط، تشانغشا 410083 هونان، الصين.مختبر بكين الرئيسي للميكروهيكل وخصائص المواد الصلبة، كلية المواد والتصنيع، جامعة بكين للتكنولوجيا، بكين 100124، الصين.ساهم هؤلاء المؤلفون بالتساوي: هوي سو، تشينيو يانغ. البريد الإلكتروني: سوهوي@هونو.edu.cn؛sxlian@hunnu.edu.cn؛qhliu@ustc.edu.cn
Although the acidic oxygen evolution reaction (OER) plays a crucial role in proton-exchange membrane water electrolysis (PEMWE) devices, challenges remain owing to the lack of efficient and acid-stable electrocatalysts. Herein, we present a low-iridium electrocatalyst in which tensile-strained iridium atoms are localized at manganese-oxide surface cation sites (TS-Ir/ ) for high and sustainable OER activity. In situ synchrotron characterizations reveal that the TS-Ir/ can trigger a continuous localized lattice oxygenmediated (L-LOM) mechanism. In particular, the L-LOM process could substantially boost the adsorption and transformation of molecules over the oxygen vacancies around the tensile-strained Ir sites and prevent further loss of lattice oxygen atoms in the inner bulk to optimize the structural integrity of the catalyst. Importantly, the resultant PEMWE device fabricated using TS-Ir/ delivers a current density of and operates stably for 200 h .
Proton-exchange membrane water electrolysis (PEMWE) devices, which have lower resistance losses, less gas crossover, and higher current densities than alkaline water electrolysis devices, are a promising and sustainable route for producing clean hydrogen ( ) fuels . Presently, the overall efficiency of water electrolysis is mainly limited by the anodic oxygen evolution reaction (OER), which includes a sluggish four proton-coupled electron-transfer process . Moreover, severe degradation of active sites in a highly acidic oxidation state, especially at high current densities, severely limits the large-scale deployment of PEMWE devices . Although iridium-based metal oxides ( ) have been widely used in water electrolysis, they have low mass activities, high overpotentials, and high costs (US ) and cannot achieve continuous high activity and durability at high current densities . Therefore, although the development of intrinsically active acid-stable low-iridium electrocatalysts that exhibit both enhanced electrocatalytic performance and good long-term durability
is highly desired for promoting more competitive PEMWE devices, it remains a formidable challenge.
The widely-accepted adsorbate evolution mechanism (AEM) is observed for the OER that is catalyzed by iridium oxides and involves multiple intermediates, such as , and . Usually, the formation of *OOH over metal sites is the rate-limiting step, which is considered as the bottleneck in improving the acidic-OER performance . To overcome the scaling relation of the adsorption energies between * OH and * OOH , the catalysts ( ) that have strong covalent bonds (where M is a metal atom) can negatively and positively shift the low Hubbard and bands for metal atoms, respectively, rendering the lattice oxygen atoms more likely to lose electrons via oxidation . Therefore, the catalyst follows a lattice oxygen-mediated (LOM) mechanism, which can bypass the production of critical *OOH intermediates for enhanced activity . Unfortunately, slow deprotonation at lattice oxygen-atom sites and degradation of
metal species on the catalyst surface preclude long-term operating stability . In particular, the oxidative release of lattice oxygen atoms can overoxidize metal species (such as Ru ) to dissolvable high-valence metal oxides during acidic-OER processes. Moreover, violent reactions involving lattice oxygen atoms produce numerous oxygen vacancies, which directly dissolve metals on the catalyst surface . Even worse, during prolonged reactions, easily formed oxygen vacancies deeply penetrate the inner bulk of the material, which finally collapses the material structure and thus deactivates the catalyst.
Recently, is an abundant and inexpensive transition metal oxide with high surface activity and catalytic stability in acidic media, which is considered as a potential carrier for acidic-OER reactions. The nanoparticles and a low loading of 5% iridium supported on the electrocatalyst exhibited good electrocatalytic OER performance due to high active specific surface areas and more high-valencestate iridium . Liu’s group presented a light-driven strategy to realize an orderly Ir atomic assembly on a F doped (IrMnOF) surface with a spin-related lower entropy that is optimized to reduce the intrinsic activation energy at potential-determining intermediates for a stable OER process . To break the linear relationship of multiple reaction intermediates for increasing activity, Ru-atom-array patches supported on with appropriate atomic distances in symmetric dual-metal sites delivered enhanced acidic-OER activity following an oxide path mechanism . Furthermore, a strained lattice containing optimized and bonding along with the presence of in the high-loading Ir-incorporated – exhibited enhanced OER stability . Therefore, simultaneously breaking the linear relationship of multiple reaction intermediates and inhibiting a large number of oxygen vacancies is essentially desirable for designing advanced electrocatalysts, but it is a great challenge.
Therefore, in this study, we used cation exchange and a subsequent rapid annealing-cooling strategy to prepare a low-iridium electrocatalyst in which iridium atoms are localized at the surface Mn sites of tensile-strained manganese oxide ( ). The tensile strain introduced by the twisted square planar ( ) moieties confined in the acid-resistant can enhance the covalency of the bond to improve the deprotonation ability and increase the bond’s orbital overlap degree with the metal band during the reaction . In particular, the tensile strain localized on the surface could tailor the adsorption behavior of Ir sites to accelerate the deprotonation of *OH at surface oxygen vacancies and, thus, effectively prevent the local peroxidation of Ir sites to reduce the dissolution and maintain the structural integrity of the catalyst. Consequently, the acidic-OER that occurred on the TS-Ir/ surface followed a continuous localized lattice oxygenmediated (L-LOM) mechanism, which causes the catalyst to deliver a high mass activity of at an overpotential of 198 mV (typically at a current density of ), which is approximately 19 and 380 times higher than those of and commercial ( ), respectively. The L-LOM mechanism is experimentally elucidated using in situ X-ray absorption fine structure (XAFS) and synchrotron radiation infrared (SRIR) spectroscopic measurements. Most importantly, in situ isotope-labeling SRIR measurements confirm the rapid adsorption of molecules on surface oxygen vacancies and the rapid deprotonation of *OH over lattice oxygen atoms to trigger a continuous L-LOM catalytic reaction for stabilizing surface Ir active sites. Therefore, the TS-Ir/ catalyst exhibits stable acidic-water electrolysis of 100 and 200 h at current densities of 200 and in a three-electrode system and the resultant PEMWE device, respectively, with negligible performance degradation, which suggests good potential for practical application in PEMWEs.
Results and discussion
Synthesis and characterization of catalysts
A low-iridium electrocatalyst in which atomically dispersed and tensilestrained iridium sites were confined in manganese oxides
(TS-Ir/ ) was hydrothermally synthesized and then rapidly thermally annealed and subsequently cooled, which is hereafter called the “rapid thermal annealing-cooling” (RTAC) strategy. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images show the nanofiber morphology, which exposes more open metal sites (Supplementary Information Fig. 1). nanofibers were transferred to an aqueous solution for the cation-exchange reaction. The as-obtained sample was rapidly heated to and then rapidly cooled to introduce tensile strain to the nanofibers. Subsequent SEM (Fig. 1a) and TEM (Fig. 1b) images clearly reveal that the ultrathin nanofiber morphology was retained after the Ir exchange reaction and rapid pyrolysis for the sample. As shown in Fig. 1c and Supplementary Fig. 2, the X-ray diffraction (XRD) patterns for the TS-Ir/ sample present typical characteristic diffraction peaks of without any peaks attributed to Ir-related phases. After Ir doping and subsequent RTAC, the peaks slightly shifted toward lower angles, which implies that tensile strain formed in the sample. Furthermore, the XRD results of the Ir samples with different Ir doping concentrations and annealing methods clearly reveal that a rapid annealing-cooling treatment introduces tensile strain in the host lattice resulting in a larger shift in the XRD pattern, excluding the effect of low loading Ir atoms on the lattice of host .
Moreover, as shown in the high-resolution TEM (HRTEM) images in Fig. 1d-f, the fringe lattice parameter of the TS-Ir/ nanofibers was measured at , which corresponds to the planes . For comparison, Ir atoms were dispersed on the tensile strain-free nanofiber surface via slow annealing and subsequent cooling, for which the TEM and HRTEM images (Supplementary Figs. 3 and 4, respectively) show that the fringe lattice parameter is . The increased lattice spacing represents the RTAC-driven tensile strain generated on the TS-Ir/ nanofiber surface. To further clarify the morphological structure of Ir sites, the elemental mappings (Supplementary Fig. 5) indicate the uniform distribution of , and O in the nanofiber. Similarly, , and O are uniformly distributed in (Supplementary Fig. 6). Aberration-corrected high-angle annular darkfield scanning TEM (HAADF-STEM, Fig. 1g, h) images clearly show Ir atomically dispersed in the obtained TS-Ir/ nanofibers. The Ir atoms, highlighted by scattered bright dots in the lattice, are at the same locations as columns of Mn atoms (Fig. 1i), which suggests that atomically dispersed Ir replaces surface Mn sites in the nanofiber lattice via a cation-exchange reaction. In this RTAC process, the rapid annealing treatment introduces stress in the lattice, and then rapid cooling does not undergo a slow cooling process for stress release. During this process, the tensile strain was retained in TS-Ir/ . Similarly, Ir is also atomically dispersed over the nanofibers in (Supplementary Fig. 7). The Ir loading obtained using inductively coupled plasma optical emission spectroscopy (ICP-OES) is approximately in TS-Ir/ . Furthermore, the ICP-OES result shows that the Ir loading is similar in , which is attributed to the same raw material feeding. These results confirm the uniform dispersion of Ir atoms on the surface Mn sites of nanofibers and that the tensile strain of Ir sites was introduced to .
Electronic properties of catalysts
To clarify the local electronic structure and binding environment of Ir species in the TS-Ir/ nanofibers, X-ray photoelectron spectroscopy (XPS) and X-ray absorption spectroscopy (XAS) were performed. The Ir XPS spectrum in Fig. 2a shows an obvious peak at 65.36 eV (Ir ), which exhibits a 0.59 eV positively shifted binding energy compared to that of and suggests that the increased oxidation state of Ir could be attributed to the electronic interaction between Ir and . Correspondingly, the XPS spectrum in Supplementary Fig. 8 shows a negative-energy shift after the introduction of Ir atoms, which reveals the electron transfer from Ir to Mn. Importantly, RTAC can further increase the electron transfer to
Fig. 1 | Structural characterization of the TS-Ir/ electrocatalyst. a SEM and b TEM images of the TS-Ir/ electrocatalyst. patterns and HRTEM images of , Ir- , and TS-Ir/ electrocatalysts. g HAADF-TEM image,
in which bright spots highlighted by red circles are ascribed to Ir sites. Enlarged HAADF-TEM image and line profiles for HAADF intensity analysis labeled in for the TS-Ir/ electrocatalyst.
increase the oxidation state of Ir species in the TS-Ir/ nanofibers. The increased Ir valence state can be further verified by Ir and Mn XAS spectra . The X-ray absorption near-edge spectroscopy (XANES) spectra of the Ir -edge in Fig. 2b show that the white-line peak for is more intense than those for both and , which indicates that the valence state slightly increased. Interestingly, the XANES spectrum of the -edge shows the opposite trend; that is, the white-line peak for Mn weakens, which means that the oxidation state of Mn decreases after the introduction of Ir species and tensile strain (Fig. 2c). These results are consistent with the XPS spectra of Ir and Mn , which indicates the electron transfer from Ir to Mn .
Furthermore, extended XAFS (EXAFS) spectra reveals the local coordination environments of Ir species in the samples . In the
Fourier-transform (FT) curves of the Ir -edge EXAFS spectra, the main peaks in the range of can be assigned to the first shell of the Ir-O coordination. Additionally, the O XPS spectra support the existence of bonds in the sample (Supplementary Fig. 9). Furthermore, the higher binding energy of in further demonstrates that the increased oxidation state of Ir due to electron transfer driven by tensile strain. A slight difference in the position of the main peak at approximately in Fig. 2d represents a different apparent bond length in TS-Ir/ , , and . Figure 2d, Supplementary Fig. 10 and Supplementary Table 1 show that the dominant coordination peak at , which is assigned to the first shell of in , is shorter than that at (for bonds in ), which is attributed to the distinct second shell structure between
Fig. 2 | Electronic property characterizations of electrocatalysts. a Ir XPS spectra of TS-Ir/ , , and . Ir -edge XANES spectra and Fourier and Ir foil. c -edge XANES spectra and e FTs of -edge EXAFS oscillations for TS-Ir/ , , and Mn foil. transforms (FTs) of Ir -edge EXAFS oscillations for TS-Ir/ ,
Ir- and . Moreover, the apparent bond length of the Ir-O first shell in TS-Ir/ is slightly longer than that of the Ir-O first shell in Ir- , which is attributed to the tensile strain. Meanwhile, the bond length in TS-Ir/ is shorter than that in , which suggests that the increased covalency and electron transfer of the bond were suitable in TS- . In order to indeed evaluate the real bond length of the coordination, the fitted curves of the -weighted Ir -edge EXAFS spectra of all samples in Supplementary Fig. 11, for which the corresponding data are listed in Supplementary Table 2, show that the coordination number of the Ir-O first shell is and that the real Ir-O bond length is in TS-Ir/ , which is shorter than that in and longer than that in Ir- , suggesting the existence of tensile strain and increased covalency. Furthermore, after Ir was introduced to , the FT-EXAFS spectra of the -edge in Fig. 2e shows no obvious change in the bond length of the first shell, which is primarily due to the low Ir content. However, the tensile strain effect could be found by comparing the bond lengths in TS-Ir/ versus (Supplementary Table 1), and the bond lengths increased by , suggesting that tensile stress existed in host for TS-Ir/ . However, the tensile strain effect could also be found by comparing the bond lengths in TS-Ir/ vs. , which reveals the origin of the Ir-site tensile strain in the nanofibers. Furthermore, another peak at is shorter than at (for Ir-Ir bond at ) and is larger than at (for bond at ), suggesting that the peak can be assigned to at . This further demonstrates that atomically dispersed Ir replaces surface Mn sites in the nanofiber lattice via a cation-exchange reaction in TS-Ir/ . Therefore, these results reveal that the elongation of surface bonds in TS- effectively tunes the covalency and electronic structures of Ir sites in the samples.
Electrochemical characterization in acidic electrolyte
To evaluate the electrocatalytic OER activity of TS-Ir/ , a threeelectrode electrochemical workstation was used for measurements in
a electrolyte, and tensile strain-free Ir- , and were employed as reference samples. For an increased number of opening sites and accessible species, carbon cloth was selected to support catalysts for electrochemical measurements (Supplementary Fig. 12). The linear sweep voltammetry (LSV) curves of the TS-Ir/ and reference samples are shown in Fig. 3a. To achieve a typical current density of , TS-Ir/ only requires an ultralow overpotential ( ) of 198 mV , which is lower than those of , , and commercial . Moreover, catalysts were prepared with different Ir loadings, among which TS-Ir/ with a moderate Ir loading ( ) exhibited the best OER activity (Supplementary Fig. 13). Interestingly, TS-Ir/ exhibits the best acidic activity at a high current density of and only requires a low overpotential of 356 mV (Fig. 3b). The morphology structure and OER performance of Ir electrocatalysts at different annealing pyrolysis temperatures are shown in Supplementary Fig. 14, revealing the optimal tensile strain and atomically dispersed Ir active sites in the TS-Ir/ electrocatalyst. Considering the effect of temperature on acidic-OER performance, we conducted a water splitting test under different operating temperatures. The OER activity is quite temperature dependent, and the overpotentials of the TS-Ir/ electrocatalyst decrease from 198 to 180 mV at a current density of with the increase of temperature to , which is consistent with what has been observed that a higher temperature provides faster OER kinetics and better OER activity (Supplementary Fig. 15). Moreover, the acidic-OER kinetics of TS-Ir/ was analyzed, as shown in Fig. 3c. Clearly, TS-Ir/ exhibits the gentlest Tafel slope of , which is substantially gentler than those of Ir- , and commercial and reveals faster OER kinetics and electron transfer over active sites in TS-Ir/ owing to suitably covalent Ir-O bonds. TS-Ir/ delivers an efficient catalytic kinetic activity in Supplementary Table 3, which is beyond that of comparable OER catalysts. Furthermore, according to electrochemical impedance spectroscopy analysis, TS-Ir/ has negligible charge-transfer
Fig. 3 | Electrocatalytic OER properties of electrocatalysts in ( ). a Linear sweep voltammetry (LSV) curves, overpotentials at 10 and (error bars are the standard deviations of three replicate calculation). The loading of catalysts is , and the solution resistance is . c Tafel slopes, and OER polarization curves based on electrochemical surface area (ECSA) for TS-Ir/ , Ir- , and supported on carbon cloths.
resistance and therefore the fastest reaction kinetics (Supplementary Fig. 16).
Importantly, the electrochemically active surface area (ECSA) of TS-Ir/ was achieved via the roughness factor, and double-layer capacitances ( ) were measured (Supplementary Figs. 17 and 18 and Table 4) . Notably, TS-Ir/ has the highest of , which suggests more open active sites to achieve high-efficiency OER activity. As expected, Fig. 3d shows that TS-Ir/ also delivers the best specific activity when the current density is normalized per the ECSA. These results reveal the superior acidic-OER activity of TS-Ir/ . To further assess the intrinsic activity of TS-Ir/ , the mass activity and turnover frequency (TOF) were calculated based on the loading of Ir sites, as shown in Fig. 3e. TS-Ir/ has a high mass activity of and an ultrahigh TOF of at a typical overpotential of 198 mV (reaching a current density of ), which is 380 times higher than those of commercial and , respectively). With increasing working potentials, both the mass activity and TOF of TS-Ir/ substantially increase compared to those of , which confirms that TS-Ir/ has faster OER kinetics than . More importantly, TS-Ir/ achieved an ultrahigh mass activity of at 1.65 V (Supplementary Table 5), which is much higher than the target ( at 1.7 V ) set by the International Renewable Energy Agency (IRENA). In other words, this is equivalent to an reduction in the amount of the precious metal (Ir) that is used to achieve a similar current power for practical applications.
Moreover, operation durability is very important for practical applications of electrocatalysts. To evaluate the operational durability of the catalyst, OER polarization curves (Supplementary Fig. 19) and chronoamperometry (Supplementary Fig. 21) were measured at typical potentials. TS-Ir/ presents good catalytic stability at a current density of after 200 h of continuous OER tests with negligible performance degradation. To further clarify the electrochemical durability at higher current densities, continuous OER tests were
performed for TS-Ir/ at a current density of , as shown in Fig. 3f. TS-Ir/ still maintains a satisfactory of the initial current density after 100 h of operation. Meanwhile, only of the Ir leached from TS-Ir/ after continuous OER operation, which is attributed to the strong lattice-confined effect of the robust substrate for inhibiting the dissolution of Ir active sites. As shown in Supplementary Fig. 21, TS-Ir/ has a high S-number ( ) at , which suggests good acidic-OER stability . The Ir concentration underwent a slow increase and no decreasing trend was observed during the first few hours of OER operation, suggesting that the dissolved Ir did not have an obvious dynamic sediment-dissolution equilibrium. Moreover, morphological and structural characterizations, such as TEM, XPS, and XAFS (Supplementary Figs. 22-24, respectively), after long-term electrochemical measurements clearly show that Ir is atomically dispersed in without agglomeration and that the Ir oxidation state and local coordination structure negligibly change. Notably, the lattice oxygen atoms show a slightly positive shift after the OER measurements, which reveals that the oxidized lattice oxygen atoms can further participate in the catalytic reaction (Supplementary Fig. 25). These results confirm the good stability of the TS-Ir/ catalyst during continuous acidic-OER operation. In summary, the high durability of the TS-Ir/ electrocatalyst under continuous operating conditions is attributed to the stable structure and a continuous L-LOM mechanism during the OER process, which can inhibit peroxidation and dissolution of Ir active sites.
Exploration of mechanism via in situ characterization
To elucidate the catalytic mechanism, in situ SRIR and XAFS spectra were measured using homemade cells under typical potential conditions . As shown in Fig. 4a, the in situ SRIR spectra show no obvious absorption bands for TS-Ir/ in the range at potentials vs. a reversible hydrogen electrode (RHE, the potentials mentioned below are all relative to RHE). With increasing
Fig. 4 | In situ SRIR measurements in . The loading of catalysts is , and the solution resistance . In situ SRIR spectroscopy measurements in the range at various potentials for TS-Ir/ and IrO . Isotope-labeling in situ SRIR spectroscopy measurements in the
range 1700-700 at various potentials for TS- and . d In situ SRIR spectra generated under no- and isotope-labeling conditions at typical potentials of 1.00 and 1.35 V for TS-Ir/ . f In situ SRIR spectra generated under no- and isotope-labeling conditions at typical potentials of 1.00 and 1.50 V for .
potential to 1.30 V vs. RHE, an absorption band appeared at and could be assigned to the key intermediates because the oxygen species ( ) stretching vibration is usually in the range . Interestingly, the intensity of this absorption band is positively correlated with the applied potential, which reveals the rapid accumulation of key O-O radical intermediates over the active sites. For comparison, in situ SRIR spectra were measured under similar OER conditions for , as shown in Fig. 4b. With gradually increasing applied potential, an IR absorption band appeared at . Because the infrared vibration peaks of the *OOH species usually appear in the region , the absorption band at can be assigned to the production of *OOH species over the Ir sites . The SRIR result of further shows the production of key * OOH species under working conditions, suggesting that Ir- catalyses the OER that follows a kinetic-slow AEM pathway (Supplementary Fig. 26). Notably, during the OER, only O-O radicals accumulated for , which bypassed the formation of the sluggish *OOH species. These results suggest that on the TS-Ir/ surface, the OER undergoes a different reaction mechanism rather than the conventional AEM on the surface.
To confirm this hypothesis, isotope-labeling in situ SRIR spectra were measured for TS-Ir/ and with increasing applied potential from 1.00 to 1.55 V vs. RHE. Usually, for oxygen species ( ), the isotope exchange redshifts the vibrational band by . Correspondingly, for oxygen species ( ), the isotope exchange can redshift the vibrational band by approximately . As shown in Fig. 4c, under working conditions, the dominant absorption band appeared at for TS-Ir/ , and the band’s intensification indicates potential dependence tolerance. More interestingly, with increasing applied potential to 1.50 V , the dominant absorption band redshifts, which reveals that the ( ) proportion gradually increases with increasing applied potential. At an applied potential of 1.35 V , the absorption band redshifted
from 1126 to when the solution was replaced by , which confirmed that during the OER, radicals are derived from adsorbed molecules and lattice oxygen atoms (Fig. 4d). These results reveal an LOM-like mechanism on the TS-Ir/ catalyst surface. Moreover, with increasing applied potential to 1.50 V , the absorption band further redshifted to , which suggests that water molecules rapidly fill oxygen vacancies to continuously trigger the LOM-like mechanism. Meanwhile, for (Fig. 4e, f), the absorption band appeared at and gradually intensified with increasing applied potentials. Notably, the *OOH absorption band shifts from 1068 to when transforms to , which confirms that emerging intermediate species are derived from molecules adsorbed at active sites. These results confirm that the OER on the TS-Ir/ surface follows a different catalytic reaction path than that on the surface of commercial and bypasses the intermediate during the reaction to accelerate the fourelectron reaction path.
To elucidate the underlying mechanism for the TS-Ir/ catalyst, in situ XAFS spectra, which are sensitive to the local structural evolution, were measured using a homemade cell fabricated based on the three-electrode system . Fig. 5a shows the in situ XANES spectra of the Ir -edge for TS-Ir/ recorded at different applied potentials. Compared with the ex situ state (immersion in an acidic solution without applying any potential), the white-line peak slightly intensifies and positively shifts with increasing applied potential to 1.43 V vs. RHE, which suggests that the oxidation state of Ir sites is elevated. This is attributed to more electrons moving from Ir to nearby adsorbed oxygen species and then promoting a rapid oxidation reaction. The FTEXAFS spectra of the Ir -edge (Fig. 5b and Supplementary Fig. 27) show a dominant peak at , which is assigned to the first shell of the Ir-O bond. Compared with the ex situ state, the first-shell peak intensified at an applied potential of 1.15 V and exhibited a slight high-R shift from 1.54 to (Supplementary Table 6). With increasing
Fig. 5 | In situ XAFS measurements in . The loading of catalysts is , and the solution resistance . a In situ XANES spectra of Ir -edge for TS-Ir/ at typical potentials during OER. Corresponding –
weighted FT-XAFS spectra. Fitted curves and OER mechanism diagrams of TS- under ex situ, 1.30 and 1.43 V conditions. The inset shows , and H atoms indicated by purple, red, green, and royal blue, respectively.
applied potential to 1.30 V , the Ir-O coordination peak further intensified. These results imply the rearrangement of the local coordination structure of Ir sites during the OER, which is due to the adsorption of oxygen species over Ir sites. Interestingly, when a typical potential of 1.43 V is applied to the electrode, the coordination peak weakens, which suggests that the Ir coordination number is reduced under OER conditions (oxygen vacancy generation). Moreover, the in situ edge XAFS spectra further reveal that the bulk phase material maintains its structural stability (Supplementary Fig. 28). Most importantly, after long-term OER measurements for TS-Ir/ , the Ir coordination number approximated that under ex situ conditions. The infrared spectrum of the isotopically labeled sample after the reaction shown in Supplementary Fig. 29 reveals that the peak for the ( and Mn ) band redshifts by , which confirms that TS- contains after the reaction. These results confirm that localized oxygen vacancies on the TS-Ir/ surface were rapidly filled by molecules to modify the LOM mechanism, which is named the “localized LOM” (L-LOM) mechanism hereafter.
The FT-XAFS fitted curves are shown in Fig. 5c and Supplementary Figs. 30 and 31, and the corresponding data are listed in Supplementary Table 7 for the configuration of four coordination bonds that are quantitatively optimized ex situ. At 1.30 V , an additional first shell was considered for the longer Ir-O coordination bond ( ), which implies that oxygen species adsorbed on Ir sites. With increasing applied potential to 1.43 V , the coordination number of the shorter Ir-O bonds reduced to three ( ), and one longer Ir-O coordination bond appeared at . These results imply that for the tensile-strained Ir sites, a potential-driven structural evolution occurs during the OER by releasing lattice oxygen atoms from TS-Ir/ localized surface sites to induce the L-LOM reaction mechanism. The comprehensive analysis of the fitted curves obtained for Ir sites and O-O radicals probed via in situ SRIR suggests that oxygen molecules that adsorb on tensile-strained Ir sites can be quickly deprotonated and coupled with adjacent lattice oxygen atoms to form O-O radicals
as *OO-Ir- , which bypasses sluggish *OOH intermediates (Fig. 5d). The proposed L-LOM mechanism can rapidly accelerate deprotonated oxygen-containing intermediates and then couple lattice oxygen atoms to form the true active structure. After is desorbed, water molecules rapidly fill oxygen vacancies and are rapidly deprotonated to maintain the structural integrity of the TS-Ir/ catalyst. These results suggest that compared with the conventional LOM mechanism, the L-LOM mechanism could accelerate the deprotonation of *OH on the surface oxygen vacancies and prevent the local peroxidation of surface Ir sites as well as the further loss of lattice oxygen atoms in the inner bulk of the catalysts. Above all, tensile strain introduced in TS-Ir/ can enhance the covalency of the Ir-O bond to improve the deprotonation ability and increase the Ir oxidation state, triggering a continuous localized L-LOM mechanism under working conditions. Therefore, the L-LOM route can achieve much faster acidic-OER kinetics while optimizing the structural stability of the catalyst during the reaction, which endows the TS-Ir/ catalyst with good potential for industrial applications.
PEMWE device performance
To verify the potential of TS-Ir/ for industrial applications, we constructed a PEMWE device using a Nafion membrane and TS-Ir/ and commercial as anode and cathode catalysts, respectively, in an acidic electrolyte. Figure and show pictures of both a PEM electrolyzer cell and system. The anode TS-Ir/ and cathode constitute the electrode, and the heating device and solution circulation system are matched to form the entire electrolytic cell device, in which the operating temperature is controlled at and the solution circulation speed is . The resultant electrolyzer fabricated using both TS-Ir/ and Ti delivers improved water electrolysis activity and stability. Specifically, when the preciousmetal (lr) loading is for the resultant electrolyzer with TS-Ir/ , the dosage is reduced by 54 -fold compared to the target ( ) set by IRENA. This means that the cost of catalysts for
Fig. 6 | PEMWE device performance measurements in . The PEM electrolyzer (a) cell and system (b). The cell voltage of the PEM electrolyzer system held at . The loading of catalyst is , and the solution resistance is .
producing hydrogen can be substantially reduced. In practical applications, catalytic stability is even more important than catalytic activity. As shown in Fig. 6c, the resultant electrolyzer delivers a current density of at an applied bias of and can continuously operate for 200 h with negligible attenuation under simulated industrial conditions (at ). TEM images (Supplementary Fig. 32) reveal that the TS-Ir/ morphology did not change after 200 h of operation in the PEM electrolyzer. Moreover, the ICP-OES results further confirmed that Ir negligibly dissolved (within 8%) in the samples. These stability test results indicate the good potential of the TS-Ir/ catalyst for practical PEMWE applications.
In summary, a low-iridium electrocatalyst in which iridium atoms are localized at tensile-strained manganese-oxide surface cation sites (TS-Ir/ ) was developed via cation exchange and subsequent RTAC. Mechanistic studies that employed in situ SRIR and XAFS spectroscopies revealed that the generated *OO-Ir- intermediates could be rapidly deprotonated and then water molecules rapidly filled oxygen vacancies around surface tensile-strained Ir sites to maintain the structural integrity of the catalyst, which efficiently catalyzed the OER via a continuous L-LOM mechanism. Especially, this L-LOM mechanism could considerably accelerate the acidic-OER kinetics while optimizing the material’s structural integrity. Therefore, the asobtained TS-Ir/ catalyst delivers a high mass activity of at an overpotential of 198 mV (typically at a current density of ), which is approximately 19 and 380 times higher than those of and commercial , respectively. Interestingly, TS-Ir/ shows stable acidic-water electrolysis for 100 h at a current density of with negligible performance degradation. More importantly, the resultant PEM electrolyzer delivered a current density of at an applied bias of for over 200 h under simulated industrial conditions (at ). The findings in this work not only demonstrate a robust and highly-active catalyst to promote the industrial applications of PEMWEs, but also provide a strategy of using tensile straining to induce a L-LOM mechanism for enhancing the activity and durability of various catalytic reactions.
Methods
Synthesis of
was hydrothermally synthesized using a typical procedure as follows: manganese sulfate ( 0.02 mol ), ammonium persulfate ( 0.02 mol ), and ammonium sulfate ( 0.06 mol ) were dissolved in 70 mL of deionized (DI) water and ultrasonicated. Then, the solution was vigorously magnetically stirred for 1 h , transferred to an autoclave, and heated to for 12 h . Subsequently, a black powder was obtained after centrifugal washing and subsequent vacuum drying several times each.
Synthesis of TS-Ir/
Typically, 120 mg of was dissolved in 30 mL of deionized water and continuously ultrasonicated for 30 min . Then, 10 mL of aqueous iridium chloride ( 5 mM ) was added to this solution and continuously ultrasonicated. Next, this solution was continuously magnetically stirred for 12 h and then washed three times. Finally, a dried powder was obtained and transferred directly to a muffle furnace at for 2 h and then quickly transferred to the air for rapid cooling. Lattice tensile strain was introduced to the TS-Ir/ sample via extreme heating and cooling.
Synthesis of
Although this synthesis path is similar to that for TS-Ir/ , rapid heating and cooling were not required. Instead, the dried powder was transferred to the muffle furnace and heated at to , held there for 2 h , and cooled to room temperature in the furnace.
Structural characterizations
Spherical aberration-corrected high-angle annular dark-field scanning TEM (HAADF-STEM) and energy-dispersive spectroscopy were conducted using an FEI Titan Themis ( 300 kV ) microscope. TEM and highresolution TEM (HRTEM) were performed using a JEM-2100F microscope operating at 200 kV acceleration. Field-emission scanning electron microscopy images were acquired using a Gemini SEM
500 scanning electron microscope. Powder X-ray diffraction patterns were measured using a Philips X’Pert Pro Super X-ray diffractometer equipped with a radiation source ( Å). XPS was recorded using a Thermo ESCALAB 250Xi equipped with an Al K ( ) excitation source. The binding energies obtained from XPS spectra were corrected with respect to the peak at 284.5 eV . Inductively coupled plasma atomic emission spectroscopy (ICP-OES) was performed using an Optima 7300 DV instrument (Perkin-Elmer).
Electrochemical measurements
All the electrochemical measurements for the acidic-OER performance were performed using a three-electrode system and the CHI 760 E workstation. The electrolyte was , and graphite rods, an electrode, and a carbon cloth (CC) supported the catalysts as the counter, reference, and working electrodes, respectively. Catalyst solutions were prepared by mixing 5.0 mg of the catalyst in a solution containing of ethanol, of DI water, and of a Nafion solution and sonicating the mixture to form homogeneous inks. Then, of well-dispersed catalyst ink was carefully dropped onto the clean CC ( ) and naturally dried for evaluation. All the final potentials were converted to corresponding reference potentials of a RHE with the equation (vs. RHE) (vs. . Linear sweep polarization curves were scanned at and compensated using iR correction. The electrochemical double-layer capacitance ( ) was calculated based on cyclic voltammetry curves that were scanned in the range vs. RHE at , and . The ECSA of the catalyst was calculated based on according to the equation ECSA (2), where is based on typical reported values . The roughness factor (RF) was calculated by estimating the ECSA and dividing it by the geometric area of the electrode according to the equation (3). The specific activity refers to the specific current density per ECSA ( ) and can be calculated using the equation .
Mass activity and turnover frequency calculations
The mass activity (MA) ( ) can be calculated based on the Ir metal loading and current density at typical overpotentials of 198 and 260 mV , respectively, as follows:
The TOF can be calculated based on the number of active sites and current density (j) as follows:
where is the Faraday constant , and represents the Avogadro constant . For the electrocatalysts, the total number of Ir surface sites are regarded as surface-active sites because Ir is atomically dispersed and anchored to thin nanofibers .
S-number calculations
The S -number is the ratio between the amounts of evolved oxygen (calculated from total) and dissolved iridium (obtained from ICP-MS measurements) .
In situ SRIR measurements
In situ SRIR spectra were measured using homemade cells at the infrared beamline (BLO1B) of the National Synchrotron Radiation Laboratory (China). An infrared (IR) spectrometer (Bruker ) equipped with a KBr beam splitter, a liquid nitrogen-cooled MCT detector, and an IR microscope (Bruker Hyperion 3000) that had a
objective lens comprised the SRIR testing device. SRIR spectroscopy can provide a clear spectrum in the range at a high spectral resolution of . To reduce the interference of water molecules on the infrared signal during the electrochemical reaction, the distance between the electrode and window must be adjusted at the microscale. To obtain a sufficiently intense reaction-intermediate signal, the reflection mode must be used to collect infrared signals. The background spectrum was acquired for the electrocatalyst electrode at the open-circuit voltage before each systemic OER measurement, and the measured OER potential ranges were . Notably, in situ SRIR curves were smoothed using data processing for 10 points, which means that 10 consecutive points were averaged to improve the signal-to-noise ratio of the curves.
In situ XAFS measurements
In situ XAFS spectra were measured using homemade cells at both the 1W1B station at the Beijing Synchrotron Radiation Facility (BSRF) and BL14W1 at the Shanghai Synchrotron Radiation Facility (SSRF) in China. The BSRF and SSRF storage rings were operated at 2.5 and 3.5 GeV , respectively, at a maximum current of 250 mA . The beam originating from the bending magnet was monochromatized utilizing a Si (111) double-crystal monochromator and further detuned by 15% to remove any higher harmonics. XAFS spectra were collected using a 19-element solid-state detector operating in fluorescence mode. In situ XAFS spectra were measured using catalyst-modified CC as the working electrode that was taped with Kapton film on the back. To obtain information about the active site evolution during the electrochemical reaction, a series of representative potentials (1.15-1.43 V) were applied to the electrode. During the collection of XAFS measurements, the absorption edge ( ) position was calibrated using a standard Ir sample.
XAFS data analysis
The obtained EXAFS data were processed using the ATHENA module that was implemented in IFEFFIT software packages . Subsequently, to separate EXAFS contributions from different coordination shells, the EXAFS data were Fourier-transformed to real space using a Hanning window ( ) in the -space range . The amplitude reduction factor ( ) was obtained from the fitted edge EXAFS curve for iridium dioxide. The curve was fitted using the -weighted EXAFS function data in – and -ranges and , respectively. The number of independent points can be calculated using the equation . For the ex situ condition of TS-Ir/ , the FT curve showed a prominent coordination peak at , which was assigned to the coordination bond. Compared with the ex situ condition, the first-shell coordination peaks at applied biases of 1.15, 1.30 , and 1.43 V were stronger and had higher shifts, which were ascribed to oxygen species adsorption. Therefore, the addition of coordination bonds was considered during curve fitting. Notably, the peak at 1.43 V , which corresponded to Ir-O coordination bonds, weakened, which may be attributed to the evolution of the Ir local coordination structure. During curve fitting, the Debye-Waller factors ( ), coordination numbers ( ), interatomic distances ( ), and energy shifts ( ) were treated as adjustable parameters. To reduce the number of adjustable fitting parameters, was the same as that of the ex situ sample at different applied potentials.
Electrochemical measurements in PEMWE tests
First, the as-obtained Nafion membrane had be pretreated by cleaning with a solution comprising , and deionized water at for 1 h and was preserved in distilled water for subsequent PEMWE evaluations. Then, TS-Ir/ and (Pt-plated Ti-fiber felt) were used as anodic and cathodic catalysts for the PEMWE tests, respectively. To prepare the anodic catalyst ink, 5 mg of
the catalyst was suspended in 1 mL of ethanol and water mixed in a 1:3 volumetric ratio. Then, of the solution was added to this mixture, and the suspension was ultrasonicated for 1 h until a well-dispersed catalyst ink was formed. The as-prepared TS-Ir/ catalyst ink ( ) was sprayed on sheets of Ti-fiber felt (surface area ). Then, the electrode, which comprised the Ti-fiber felt-supported TS-Ir/ , treated Nafion membrane, and , was hot pressed at a certain pressure. Finally, the electrode was applied in PEMWE for evaluating catalytic performance. The PEMWE device was operated at using a electrolyte flowing at . The stability of the PEMWE conducted using the TS-Ir/ anodic catalyst was evaluated by measuring chronopotentiometry at a current density of for 200 h at and ambient pressure. All the cell voltages measured in PEMWE electrolyzers were reported without applying any iR compensation.
Data availability
All data reported in this paper are available from the corresponding author upon request.
References
Chu, S. & Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 488, 294-303 (2012).
Seh, Z. W. et al. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 355, eaad4998 (2017).
An, L. et al. Recent development of oxygen evolution electrocatalysts in acidic environment. Adv. Mater. 33, 2006328 (2021).
Su, H. et al. In-situ spectroscopic observation of dynamic-coupling oxygen on atomically dispersed iridium electrocatalyst for acidic water oxidation. Nat. Commun. 12, 6118 (2021).
Wang, N. et al. Doping shortens the metal/metal distance and promotes OH coverage in non-noble acidic oxygen evolution reaction catalysts. J. Am. Chem. Soc. 145, 7829-7836 (2023).
Zhang, X. et al. Fast modulation of d-band holes quantity in the early reaction stages for boosting acidic oxygen evolution. Angew. Chem. 62, e202308082 (2023).
Song, J. et al. A review on fundamentals for designing oxygen evolution electrocatalysts. Chem. Soc. Rev. 49, 2196-2214 (2020).
Zhang, R. et al. A dissolution/precipitation equilibrium on the surface of iridium-based perovskites controls their activity as oxygen evolution reaction catalysts in acidic media. Angew. Chem. 131, 4619-4623 (2019).
Chen, F.-Y., Wu, Z.-Y., Adler, Z. & Wang, H. Stability challenges of electrocatalytic oxygen evolution reaction: From mechanistic understanding to reactor design. Joule 5, 1704-1731 (2021).
Lin, C. et al. In-situ reconstructed Ru atom array on with enhanced performance for acidic water oxidation. Nat. Catal. 4, 1012-1023 (2021).
Reier, T., Nong, H. N., Teschner, D., Schlögl, R. & Strasser, P. Electrocatalytic oxygen evolution reaction in acidic environments-reaction mechanisms and catalysts. Adv. Energy Mater. 7, 1601275 (2017).
Wang, X. et al. Pivotal role of reversible geometric conversion in oxygen evolution. Nature 611, 702-708 (2022).
Zhang, N. & Chai, Y. Lattice oxygen redox chemistry in solid-state electrocatalysts for water oxidation. Energy Environ. Sci. 14, 4647-4671 (2021).
Yao, Y. et al. Engineering the electronic structure of single atom Ru sites via compressive strain boosts acidic water oxidation electrocatalysis. Nat. Catal. 2, 304-313 (2019).
Shi, Z. et al. Confined Ir single sites with triggered lattice oxygen redox: Toward boosted and sustained water oxidation catalysis. Joule 5, 2164-2176 (2021).
Roy, C. et al. Trends in activity and dissolution on under oxygen evolution conditions: particles versus well-defined extended surfaces. ACS Energy Lett. 3, 2045-2051 (2018).
Hodnik, N. et al. New insights into corrosion of ruthenium and ruthenium oxide nanoparticles in acidic media. J. Phys. Chem. C 119, 10140-10147 (2015).
Wu, Z. Y. et al. Non-iridium-based electrocatalyst for durable acidic oxygen evolution reaction in proton exchange membrane water electrolysis. Nat. Mater. 22, 100-108 (2023).
Devaraj, S. & Munichandraiah, N. Effect of crystallographic structure of MnO 2 on its electrochemical capacitance properties. J. Phys. Chem. C 112, 4406-4417 (2008).
Wang, Z. et al. Influence of the phase on oxygen evolution reaction performance for low-loading iridium electrocatalysts. ChemElectroChem 8, 418-424 (2021).
Xu, Z. et al. Light-driven orderly assembly of Ir-atomic chains to integrate a dynamic reaction pathway for acidic oxygen evolution. Angew. Chem. Int. Ed. https://doi.org/10.1002/anie. 202301128 (2023).
Wen, M., Yang, H., Tong, L. & Yuan, L. Study on degradation of goldcyanide wastewater by rare-earth La doped composite photocatalyst. Mol. Catal. 550, 113506 (2023).
Yagi, S. et al. Covalency-reinforced oxygen evolution reaction catalyst. Nat. Commun. 6, 8249 (2015).
Cheng, W. et al. Lattice-strained metal-organic-framework arrays for bifunctional oxygen electrocatalysis. Nat. Energy 4, 115-122 (2019).
Hao, S. et al. Torsion strained iridium oxide for efficient acidic water oxidation in proton exchange membrane electrolyzers. Nat. Nanotechnol. 16, 1371-1377 (2021).
Lübke, M. et al. Transition-metal-doped nanorods as bifunctional catalysts for efficient oxygen reduction and evolution reactions. ChemistrySelect 3, 2613-2622 (2018).
He, C. et al. Molecular evidence for metallic cobalt boosting electroreduction on pyridinic nitrogen. Angew. Chem. 132, 4944-4949 (2020).
Edgington, J., Schweitzer, N., Alayoglu, S. & Seitz, L. C. Constant change: exploring dynamic oxygen evolution reaction catalysis and material transformations in strontium zinc iridate perovskite in acid. J. Am. Chem. Soc. 143, 9961-9971 (2021).
Chen, Y. et al. Exceptionally active iridium evolved from a pseudocubic perovskite for oxygen evolution in acid. Nat. Commun. 10, 572 (2019).
Sun, Z., Liu, Q., Yao, T., Yan, W. & Wei, S. X-ray absorption fine structure spectroscopy in nanomaterials. Sci. China Mater. 58, 313-341 (2015).
Wang, Q. et al. Ultrahigh-loading of Ir single atoms on NiO matrix to dramatically enhance oxygen evolution reaction. J. Am. Chem. Soc. 142, 7425-7433 (2020).
Geiger, S. et al. The stability number as a metric for electrocatalyst stability benchmarking. Nat. Catal. 1, 508-515 (2018).
Su, H. et al. Hetero-N-coordinated Co single sites with high turnover frequency for efficient electrocatalytic oxygen evolution in an acidic medium. ACS Energy Lett. 4, 1816-1822 (2019).
Cheng, W. R., Xu, Y. Z., Yang, C. Y., Su, H. & Liu, Q. H. Monitoring surface dynamics of electrodes during electrocatalysis using in situ synchrotron FTIR spectroscopy. J. Synchron. Rad. 30, 340-346 (2023).
Li, Y. et al. High mass-specific reactivity of a defect-enriched Ru electrocatalyst for hydrogen evolution in harsh alkaline and acidic media. Sci. China Mater. 64, 2467-2476 (2021).
Zhou, W. et al. Regulating the scaling relationship for high catalytic kinetics and selectivity of the oxygen reduction reaction. Nat. Commun. 13, 6414 (2022).
Lang, C. et al. Observation of a potential-dependent switch of water-oxidation mechanism on Co-oxide-based catalysts. Chemistry 7, 2101-2117 (2021).
Rao, R. R. et al. Operando identification of site-dependent water oxidation activity on ruthenium dioxide single-crystal surfaces. Nat. Catal. 3, 516-525 (2020).
Sivasankar, N., Weare, W. W. & Frei, H. Direct observation of a hydroperoxide surface intermediate upon visible light-driven water oxidation at an Ir oxide nanocluster catalyst by rapid-scan FT-IR spectroscopy. J. Am. Chem. Soc. 133, 12976-12979 (2011).
Chen, C. et al. Coupling and in to synthesize urea under ambient conditions. Nat. Chem. 12, 717-724 (2020).
Cheng, W., Su, H. & Liu, Q. Tracking the oxygen dynamics of solid-liquid electrochemical interfaces by correlative in situ synchrotron spectroscopies. Acc. Chem. Res. 55, 1949-1959 (2022).
Su, H. et al. Dynamic evolution of solid-liquid electrochemical interfaces over single-atom active sites. J. Am. Chem. Soc. 142, 12306-12313 (2020).
McCrory, C. C., Jung, S., Peters, J. C. & Jaramillo, T. F. Benchmarking heterogeneous electrocatalysts for the oxygen evolution reaction. J. Am. Chem. Soc. 135, 16977-16987 (2013).
Li, R. et al. IrW nanochannel support enabling ultrastable electrocatalytic oxygen evolution at in acidic media. Nat. Commun. 12, 3540 (2021).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchron. Rad. 12, 537-541 (2005).
Acknowledgements
This work was supported by the National Key R&D Program of China (2022YFA1502903 (Q.L.)), the National Natural Science Foundation of China (12205300 (H.S.), 22241202 (Q.L.), and 12135012 (H.S.)), and the Natural Science Foundation of Anhui Province (Grant Nos. 2208085JO1 (Q.L.) and 2208085QA28 (H.S.)).
Author contributions
Q.L., H.S., and S.L. conceived the project. H.S. and C.Y. designed the in situ SRIR and XAFS experiments. H.S. and C.Y. conducted the experiments, including catalyst synthesis, catalytic tests, and in situ SRIR and
XAFS measurements. M.L. performed catalytic activity. X.Z. and K.Z. performed TEM tests. W.Z., Y.Z., and S.L. analyzed the experimental data. The manuscript was written by H.S., C.Y., and Q.L. All authors discussed the results and commented on the paper.
Correspondence and requests for materials should be addressed to Hui Su, Shixun Lian or Qinghua Liu.
Peer review information Nature Communications thanks lizhe liu, Xinlong Tian and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Key Laboratory of Light Energy Conversion Materials of Hunan Province College, College of Chemistry and Chemical Engineering, Hunan Normal University, Changsha 410081 Hunan, China. National Synchrotron Radiation Laboratory, University of Science and Technology of China, Hefei 230029 Anhui, China. State Key Laboratory for Powder Metallurgy, Central South University, Changsha 410083 Hunan, China. Beijing Key Laboratory of Microstructure and Properties of Solids, Faculty of Materials and Manufacturing, Beijing University of Technology, Beijing 100124, China. These authors contributed equally: Hui Su, Chenyu Yang. e-mail: suhui@hunnu.edu.cn; sxlian@hunnu.edu.cn; qhliu@ustc.edu.cn