DOI: https://doi.org/10.1038/s41467-024-45320-0
PMID: https://pubmed.ncbi.nlm.nih.gov/38307871
تاريخ النشر: 2024-02-03
تنشيط الأكسجين الشبكي وتعزيز المجال الكهربائي المحلي من خلال التشارك في التسميم بالحديد والفلور في مصفوفات إبر نانوية من CoO لأكسدة الماء الكهروكيميائية الصناعية
تم القبول: 19 يناير 2024
نُشر على الإنترنت: 03 فبراير 2024
(4) تحقق من التحديثات
الملخص
تفاعل تطور الأكسجين (OER) هو أمر حاسم لتقنيات تحويل الطاقة المتجددة، لكن علاقات الهيكل-النشاط والآليات التحفيزية الأساسية في المحفزات ليست مفهومة تمامًا. نحن هنا نعرض استراتيجية لتعزيز OER مع تحقيق تنشيط الأكسجين الشبكي وزيادة المجال الكهربائي المحلي بشكل متزامن من خلال التسميم المزدوج للكاتيونات والأنيونات. تم بناء مصفوفات خشنة من إبر نانوية CoO المزدوجة بالتسميم من الحديد والفلور، وتم تحقيق جهد زائد منخفض قدره 277 مللي فولت عند
هيكل المحفزات الكهربائية والسلوكيات الكهروكيميائية لها أهمية كبيرة في تطوير محفزات OER عالية التفاعل ودائمة.
النتائج والمناقشة
تركيب وتوصيف المحفزات
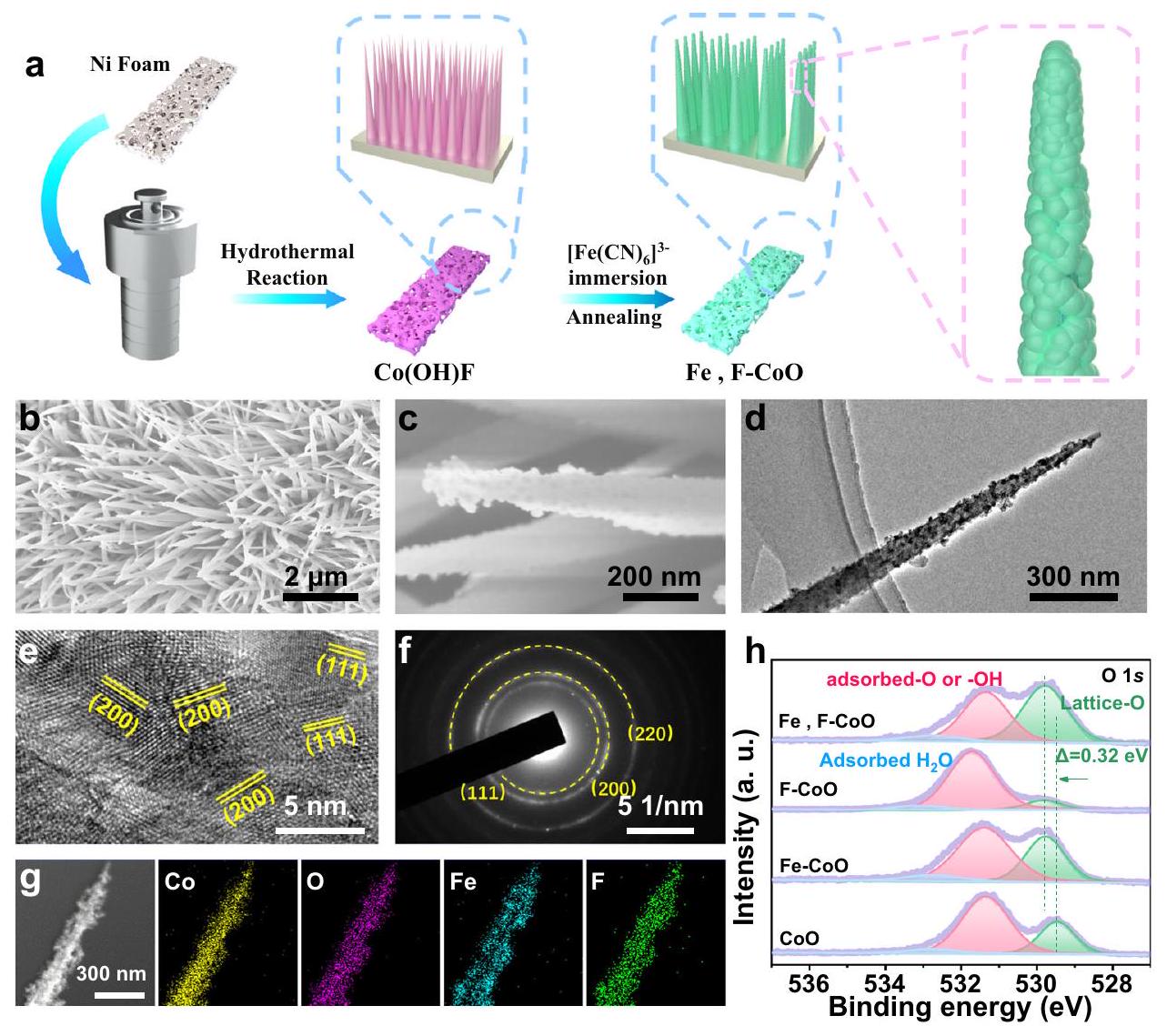
صورة HRTEM. نمط SAED. صورة STEM وصور الخرائط العنصرية المقابلة.
على التوالي، وتغير بشكل مشترك التركيب الإلكتروني لـ CoO. تُظهر طيف O 1s ثلاثة أنواع من الأكسجين بما في ذلك الأكسجين الشبكي، والأكسجين الممتص أو الهيدروكسيل (-OH)، والأكسجين الممتص.
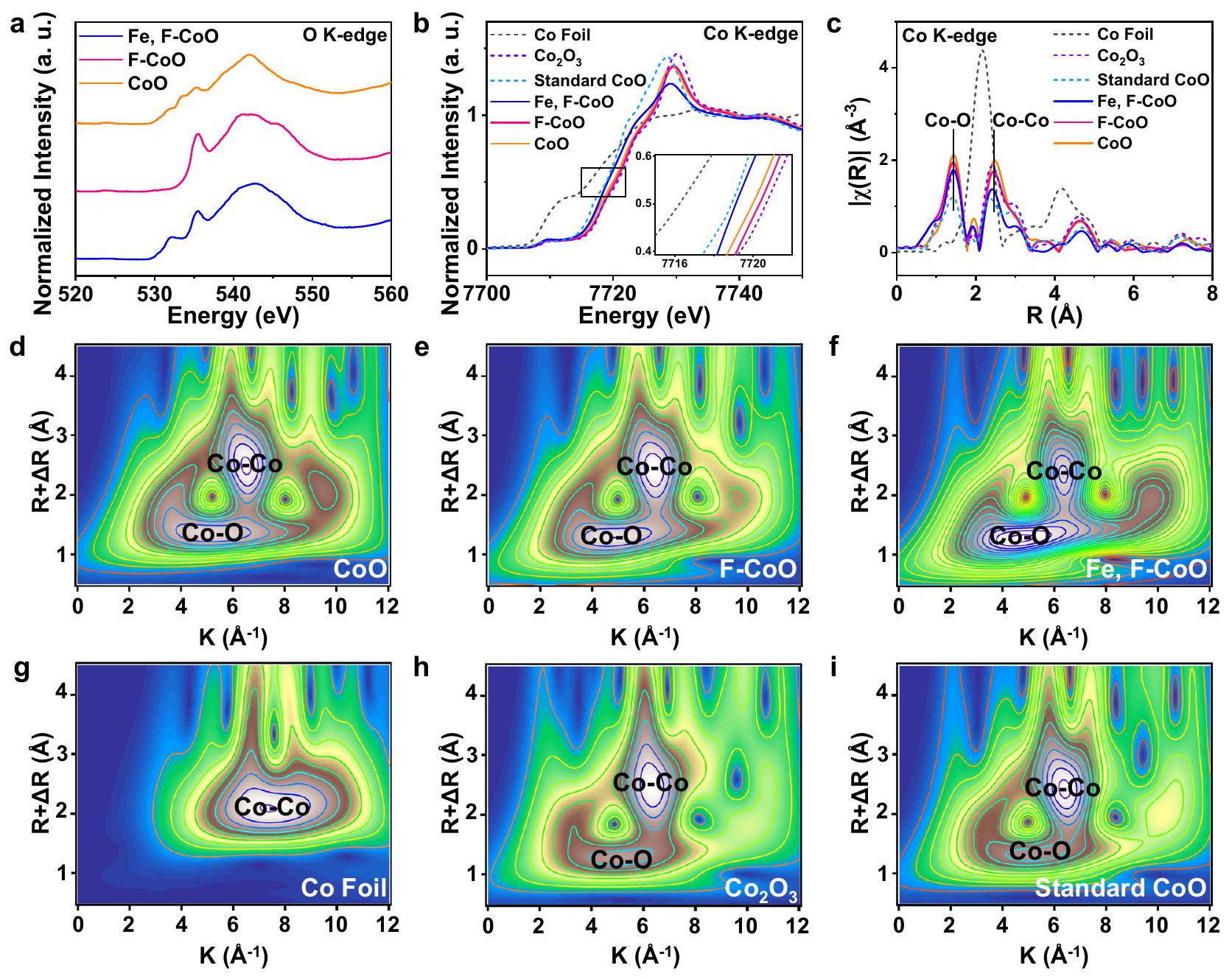
تسبب تغييرات هيكلية كبيرة في CoO (الأشكال التكميلية 10، 11، الجدول التكميلية 2). يتم تأكيد ذلك بشكل أكبر من خلال الرسوم البيانية لتحويل الموجات (WT) المماثلة (الشكل 2d-i)، التي تتوافق جيدًا مع الملاحظات من XRD وRaman.
أداء التحفيز الكهربائي لتفاعل الأكسدة
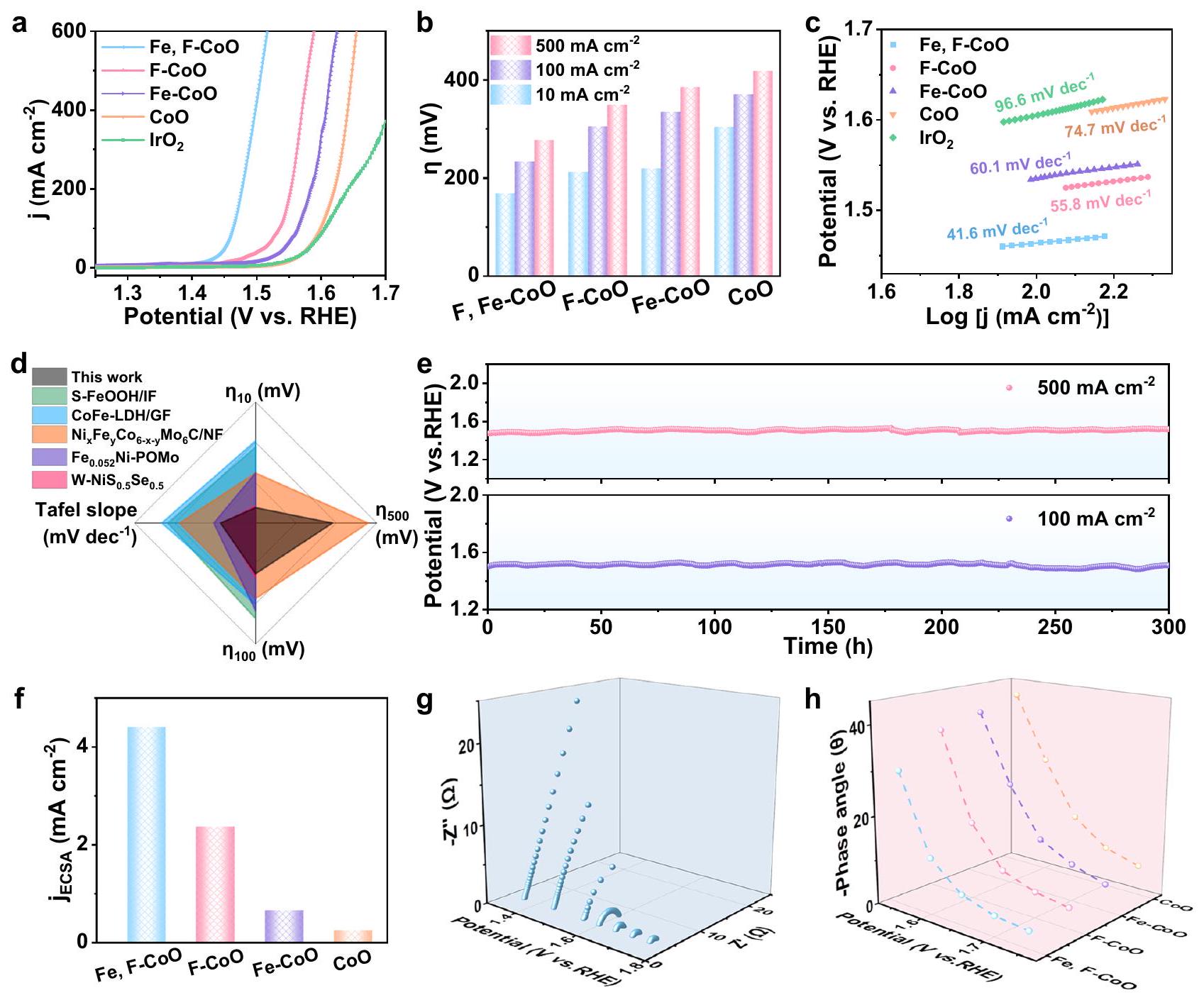
تم التحقق من ذلك مع منحنيات LSV (الشكل التكميلي 13). كما أن FESEM و XRD يبرهنان بشكل أكبر أن الشكل والهيكل البلوري يظهران تغييرات ضئيلة بعد اختبار الدورة، مما يشير إلى استقرارهما القوي وإمكاناتهما الكبيرة في التطبيقات العملية (الشكل التكميلي 14).
دراسة حركية
تمت دراستها بواسطة مطيافية الامتياز الكهروكيميائي (EIS). تظهر مخططات نايكويست أن مقاومة نقل الشحنة
استكشاف آلية التفاعل
آلية التفاعل. كما هو موضح في الشكل التكميلي 21، تزداد كثافات التيار لـ Fe و F-CoO NNAs بشكل حاد مع زيادة الرقم الهيدروجيني من 12.5 إلى 14.0، مما يشير إلى مسار نقل بروتون-إلكترون غير متزامن.
تم إثباتها مباشرةً باستخدام طيف رامان، حيث تظهر القمم النموذجية لـ TMA
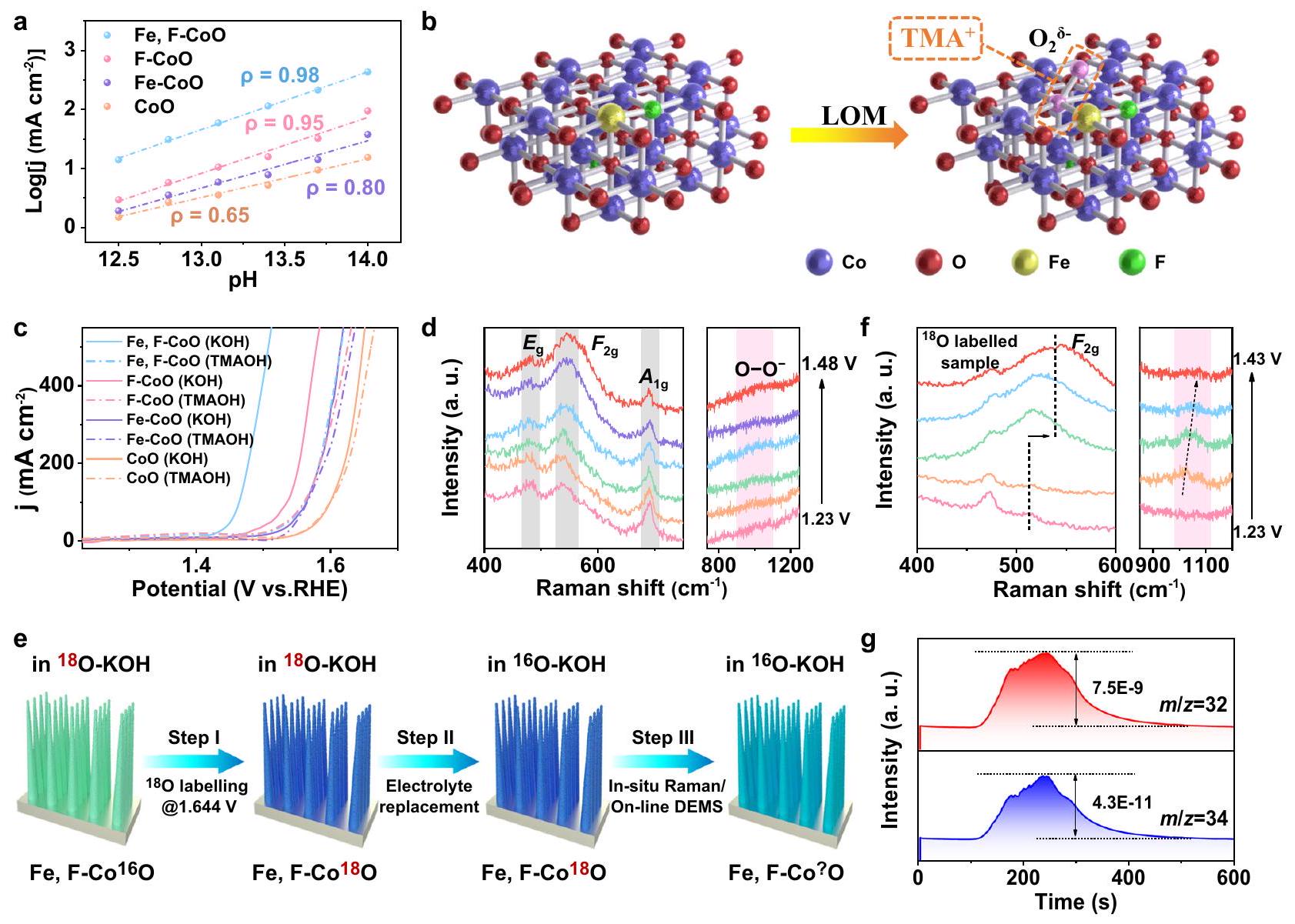
ملفات LSV لمحفزات Fe و F-CoO NNAs و F-CoO NNAs و Fe-CoO NNAs و CoO NNAs في 1 م KOH و 1 م TMAOH مع
طيف عند 1.33 فولت وما فوق، مع ذروة حوالي
في OER. توفر هذه الملاحظات أدلة أكثر إقناعًا على آلية LOM.
رؤى نظرية حول آلية التفاعل
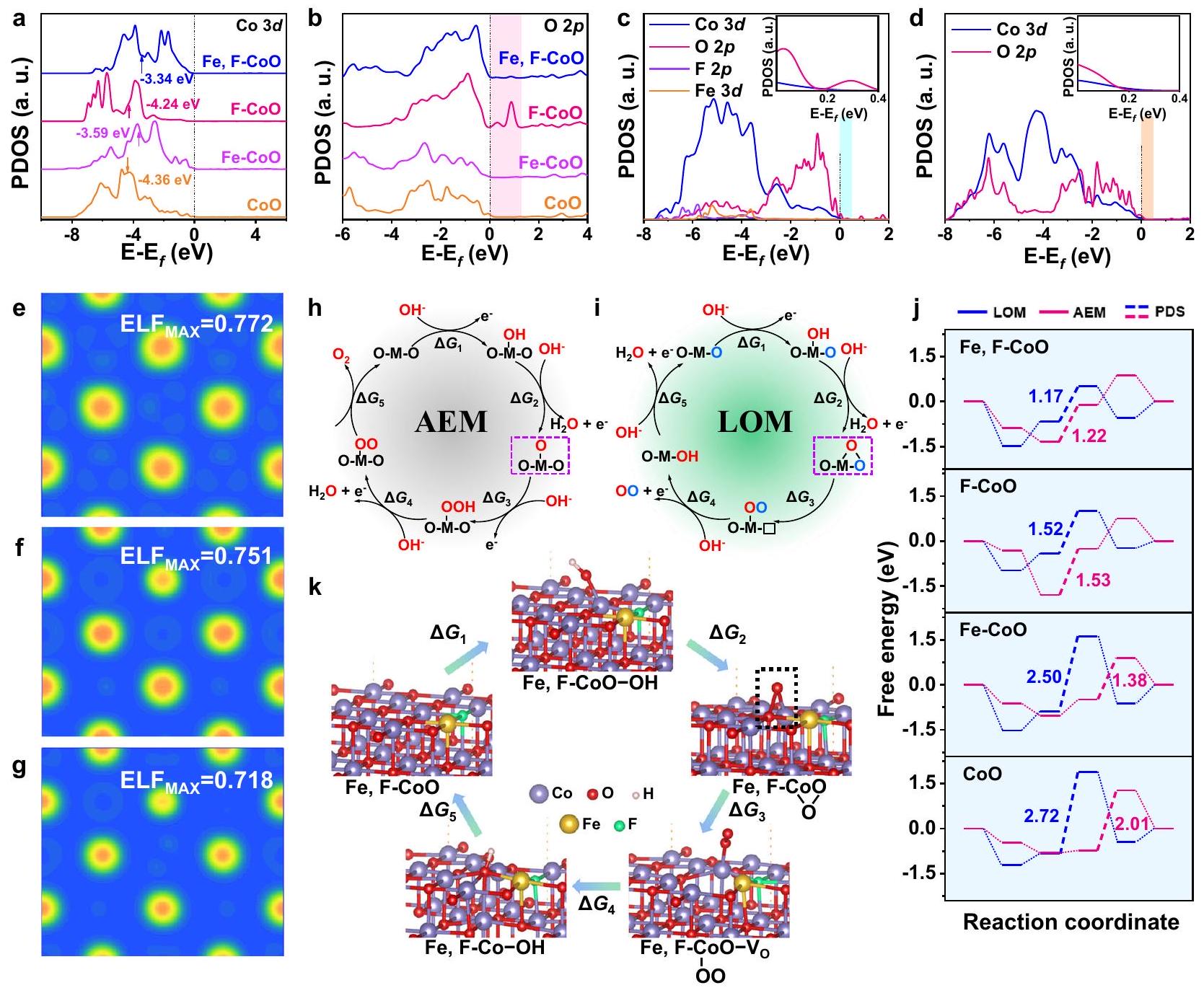
تحليل وظيفة التوطين (ELF)، حيث يتم تقليل المحلية الإلكترونية بعد
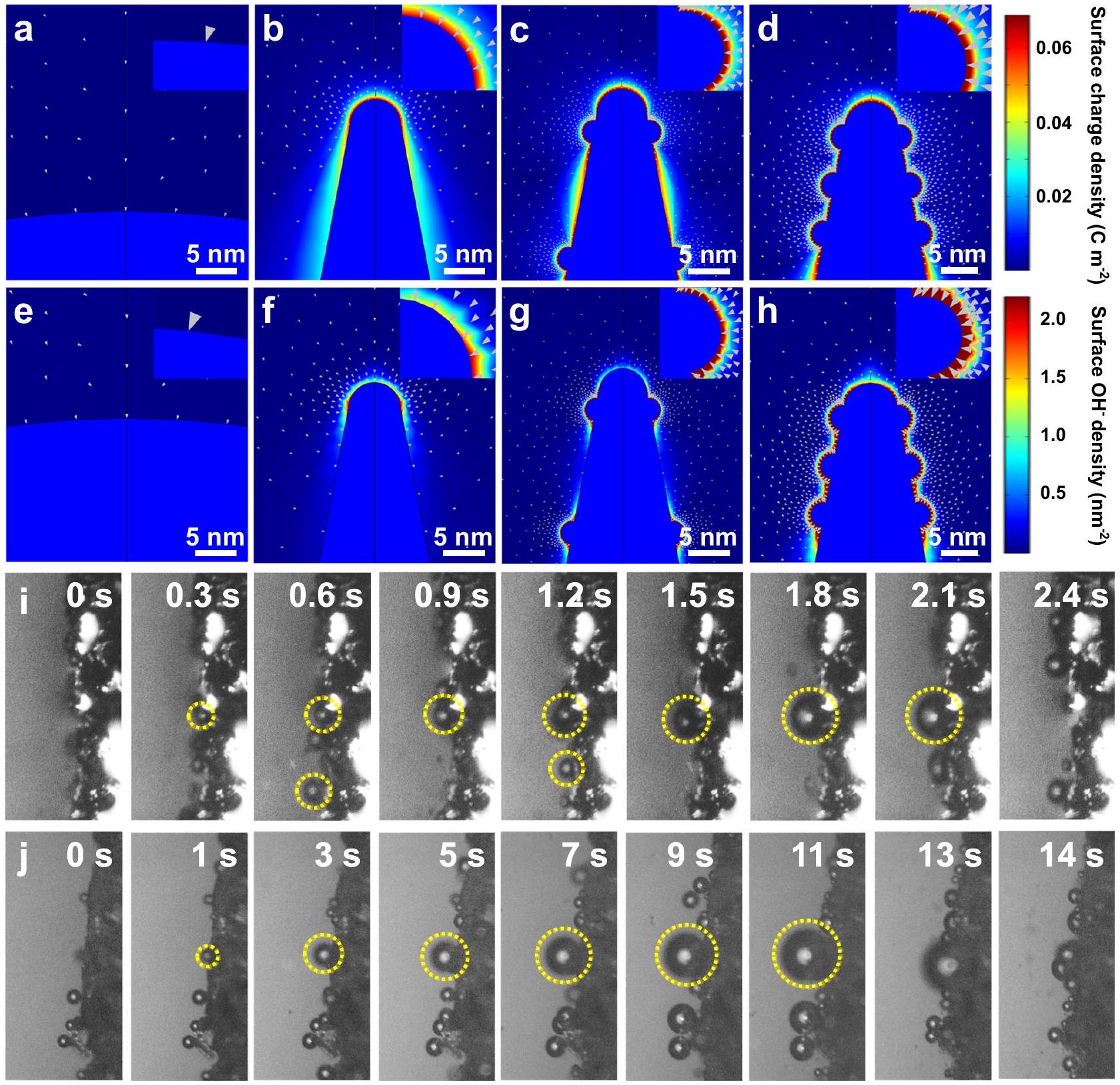
آثار المجال الكهربائي المحلي
0.99 إلكترون فولت، مما يشير إلى تسريع حركيات تنشيط الأكسجين في الشبكة. في الوقت نفسه، فإن معدل الإفراج عن
أداء تقسيم المياه الصناعي الشامل
طرق
المواد الكيميائية
تم الحصول على KOH من شركة سينوفارم للمواد الكيميائية المحدودة. تم استخدام جميع المواد الكيميائية مباشرة دون مزيد من التنقية.
تركيب
الإيثانول اللامائي بشكل متسلسل.
تركيب نانوكومبليكسات الحديد وفوسفات الكوبالت
تركيب NNAs من F-CoO
تركيب
تركيب NNAs من Fe-CoO
تركيب أكسيد الكوبالت NNAs
توصيف
القياسات الكهروكيميائية
معدل المسح. يتم عرض حساب ECSA كما يلي.
توفر البيانات
References
- Vojvodic, A. & Norskov, J. K. Optimizing perovskites for the watersplitting reaction. Science 334, 1355-1356 (2011).
- Hu, E. et al. Construction of hierarchical Ni-Co-P hollow nanobricks with oriented nanosheets for efficient overall water splitting. Energy Environ. Sci. 11, 872-880 (2018).
- Zhao, S. et al. Ultrathin metal-organic framework nanosheets for electrocatalytic oxygen evolution. Nat. Energy 1, 1-10 (2016).
- Ng, J. W. D. et al. Gold-supported cerium-doped
catalysts for water oxidation. Nat. Energy 1, 16053 (2016). - Hu, E. et al. Graphene layers-wrapped
nanoparticles supported on N -doped graphene nanosheets for highly efficient oxygen reduction. Adv. Energy Mater. 8, 1702476 (2018). - Li, J. et al. A general strategy for preparing pyrrolic-
type singleatom catalysts via pre-located isolated atoms. Nat. Commun. 12, 6806 (2021). - Cao, L. et al. Dynamic oxygen adsorption on single-atomic Ruthenium catalyst with high performance for acidic oxygen evolution reaction. Nat. Commun. 10, 4849 (2019).
- Zhai, P. et al. Engineering single-atomic ruthenium catalytic sites on defective nickel-iron layered double hydroxide for overall water splitting. Nat. Commun. 12, 4587 (2021).
- Zhang, R. et al. Tracking the role of defect types in
structural evolution and active motifs during oxygen evolution reaction. J. Am. Chem. Soc. 145, 2271-2281 (2023). - Yan, D. et al. Cation defect engineering of transition metal electrocatalysts for oxygen evolution reaction. Adv. Energy Mater. 12, 2202317 (2022).
- Yan, J. et al. Single atom tungsten doped ultrathin
for enhanced electrocatalytic water oxidation. Nat. Commun. 10, 2149 (2019). - Zhao, Y., Nakamura, R., Kamiya, K., Nakanishi, S. & Hashimoto, K. Nitrogen-doped carbon nanomaterials as non-metal electrocatalysts for water oxidation. Nat. Commun. 4, 2390 (2013).
- Zhang, L. et al. Self-reconstructed metal-organic framework heterojunction for switchable oxygen evolution reaction. Angew. Chem. Int. Ed. 61, e202214794 (2022).
- Qiu, C. et al. Interfacial engineering FeOOH/CoO nanoneedle array for efficient overall water splitting driven by solar energy. Chem. Eur. J. 26, 4120-4127 (2020).
- Yan, L. et al. Engineering self-supported hydrophobic-aerophilic air cathode with
nanoparticles embedded in co-doped carbon plate arrays for long-life rechargeable Zn-air batteries. Adv. Energy Mater. 13, 2204245 (2023). - Yang, X. G. et al. Enhanced activity of enzyme immobilized on hydrophobic ZIF-8 modified by
ions. Angew. Chem. Int. Ed. 62, e202216699 (2023). - Wang, X., Zhong, H., Xi, S., Lee, W. S. V. & Xue, J. Understanding of oxygen redox in the oxygen evolution reaction. Adv. Mater. 34, 2107956 (2022).
- Xiao, K. et al. Activating lattice oxygen in spinel
through filling oxygen vacancies with fluorine for electrocatalytic oxygen evolution. Angew. Chem. Int. Ed. 62, e202301408 (2023). - Song, J. et al. A review on fundamentals for designing oxygen evolution electrocatalysts. Chem. Soc. Rev. 49, 2196-2214 (2020).
- Grimaud, A. et al. Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution. Nat. Chem. 9, 457-465 (2017).
- Wang, C. et al. Engineering lattice oxygen activation of iridium clusters stabilized on amorphous bimetal borides array for oxygen evolution reaction. Angew. Chem. Int. Ed. 60, 27126-27134 (2021).
- Huang, Z. F. et al. Tuning of lattice oxygen reactivity and scaling relation to construct better oxygen evolution electrocatalyst. Nat. Commun. 12, 3992 (2021).
- Zhang, N. et al. Lattice oxygen activation enabled by high-valence metal sites for enhanced water oxidation. Nat. Commun. 11, 4066 (2020).
- Huang, Z. F. et al. Chemical and structural origin of lattice oxygen oxidation in Co-Zn oxyhydroxide oxygen evolution electrocatalysts. Nat. Energy 4, 329-338 (2019).
- Zhang, N. & Chai, Y. Lattice oxygen redox chemistry in solid-state electrocatalysts for water oxidation. Energy Environ. Sci. 14, 4647-4671 (2021).
- Hu, Q. et al. Integrating well-controlled core-shell structures into “superaerophobic” electrodes for water oxidation at large current densities. Appl. Catal. B:Environ. 286, 119920 (2021).
- Liu, M. et al. Enhanced electrocatalytic
reduction via fieldinduced reagent concentration. Nature 537, 382-386 (2016). - Chen, Q. et al. Ordered Ag nanoneedle arrays with enhanced electrocatalytic
reduction via structure-induced inhibition of hydrogen evolution. Nano Lett. 22, 6276-6284 (2022). - Gao, Y. et al. Field-induced reagent concentration and sulfur adsorption enable efficient electrocatalytic semihydrogenation of alkynes. Sci. Adv. 8, eabm9477 (2022).
- Liu, P. et al. Tip-enhanced electric field: A new mechanism promoting mass transfer in oxygen evolution reactions. Adv. Mater. 33, 2007377 (2021).
- Liu, D. et al. Atomically dispersed platinum supported on curved carbon supports for efficient electrocatalytic hydrogen evolution. Nat. Energy 4, 512-518 (2019).
- Nairan, A. et al. Proton selective adsorption on Pt-Ni nano-thorn array electrodes for superior hydrogen evolution activity. Energy Environ. Sci. 14, 1594-1601 (2021).
- Jiang, H., Hou, Z. & Luo, Y. Unraveling the mechanism for the sharptip enhanced electrocatalytic carbon dioxide reduction: The kinetics decide. Angew. Chem. Int. Ed. 56, 15617-15621 (2017).
- Zhang, L. et al. Attenuating a metal-oxygen bond of a double perovskite oxide via anion doping to enhance its catalytic activity for the oxygen reduction reaction. J. Mater. Chem. A 8, 14091-14098 (2020).
- Yu, X. et al. Activation or passivation: Influence of halogen dopant (
) on photothermal activity of in degrading toluene. Appl. Catal. B: Environ. 309, 121236 (2022). - Xie, L., Zhao, D., Dai, J., Wu, Z. & Li, L. Solvothermally doping
nanoparticles on carbon with ferric ions for efficient oxygen evolution catalysis. Catalysts 9, 458 (2019). - Lei, H. et al. Coordination and interface engineering to boost catalytic property of two-dimensional ZIFs for wearable Zn-air batteries. J. Energy Chem. 68, 78-86 (2022).
- Li, M. et al. Scalable dry production process of a duperior 3D netlike carbon-based iron oxide anode material for lithium-ion batteries. Angew. Chem. Int. Ed. 56, 12649-12653 (2017).
- Chen, X., Wei, M. & Zhou, J. Fluorine-doping-assisted vacancy engineering for efficient electrocatalyst toward hydrogen production. J. Mater. Chem. A 9, 22626-22634 (2021).
- Lu, W. et al. Construction of CoO/Co-Cu-S hierarchical tubular heterostructures for hybrid supercapacitors. Angew. Chem. Int. Ed. 58, 15441-15447 (2019).
- Lian, J. et al. Magnetic flower-like Fe-doped CoO nanocomposites with dual enzyme-like activities for facile and sensitive determination of
and dopamine. Inorg. Chem. 60, 1893-1901 (2021). - Mi, J. et al. Activation of partial metal sites in high-entropy oxides for enhancing thermal and electrochemical catalysis. Chin. J. Catal. 48, 235-246 (2023).
- Wu, Y. et al. Triggering lattice oxygen activation of single-atomic Mo sites anchored on Ni-Fe oxyhydroxides nanoarrays for electrochemical water oxidation. Adv. Mater. 34, 2202523 (2022).
- Roychoudhury, S. et al. Deciphering the oxygen absorption preedge: A caveat on its application for probing oxygen redox reactions in batteries. Energy Environ. Mater. 4, 246-254 (2021).
- Wang, X. et al. The role of electron localization in covalency and electrochemical properties of lithium-ion battery cathode materials. Adv. Funct. Mater. 31, 2001633 (2021).
- Huang, J. et al. Modifying redox properties and local bonding of
by enhances oxygen evolution catalysis in acid. Nat. Commun. 12, 3036 (2021). - Su, H. et al. In-situ spectroscopic observation of dynamic-coupling oxygen on atomically dispersed iridium electrocatalyst for acidic water oxidation. Nat. Commun. 12, 6118 (2021).
- Chen, X . et al. S-doping triggers redox reactivities of both iron and lattice oxygen in FeOOH for low-cost and high-performance water oxidation. Adv. Funct. Mater. 32, 2112674 (2022).
- Mefford, J. T. et al. Water electrolysis on
perovskite electrocatalysts. Nat. Commun. 7, 11053 (2016). - Xiao, L. et al. Electronic and nano-structural modulation of
nanosheets by Fe-benzenedicarboxylate for efficient oxygen evolution. Chem. Res. Chin. U. 39, 219-223 (2023). - Guo, W. et al. In situ revealing the reconstruction behavior of monolayer rocksalt CoO nanosheet as water oxidation catalyst. J. Energy Chem. 70, 373-381 (2022).
- Moysiadou, A., Lee, S., Hsu, C. S., Chen, H. M. & Hu, X. Mechanism of oxygen evolution catalyzed by cobalt oxyhydroxide: cobalt superoxide species as a key intermediate and dioxygen release as a ratedetermining step. J. Am. Chem. Soc. 142, 11901-11914 (2020).
- Lee, S., Banjac, K., Lingenfelder, M. & Hu, X. Oxygen isotope labeling experiments reveal different reaction sites for the oxygen evolution reaction on nickel and nickel iron oxides. Angew. Chem. Int. Ed. 58, 10295-10299 (2019).
- Farsi, L. & Deskins, N. A. First principles analysis of surface dependent segregation in bimetallic alloys. Phys. Chem. Chem. Phys. 21, 23626-23637 (2019).
- Grimaud, A., Hong, W. T., Shao-Horn, Y. & Tarascon, J. M. Anionic redox processes for electrochemical devices. Nat. Mater. 15, 121-126 (2016).
- Wang, Z. et al. Elevating the d -band center of six-coordinated octahedrons in
through Fe-incorporated topochemical deintercalation. Adv. Energy Mater. 11, 2003023 (2021). - Han, B. et al. Nanoscale structural oscillations in perovskite oxides induced by oxygen evolution. Nat. Mater. 16, 121-126 (2017).
- Ji, J. et al.
-regulated electron state of for superb aqueous zinc-manganese oxide batteries. Adv. Energy Mater. 11, 2003203 (2021). - Mueller, D. N., Machala, M. L., Bluhm, H. & Chueh, W. C. Redox activity of surface oxygen anions in oxygen-deficient perovskite oxides during electrochemical reactions. Nat. Commun. 6, 6097 (2015).
- Song, Q. et al. General strategy to optimize gas evolution reaction via assembled striped-pattern sduperlattices. J. Am. Chem. Soc. 142, 1857-1863 (2020).
- Lin, Q. et al. Tuning the interface of
by atomic replacement strategy toward high-performance electrocatalytic oxygen evolution. ACS Nano 16, 15460-15470 (2022).
شكر وتقدير
مساهمات المؤلفين
تحليل ومراجعة المخطوطة. ناقش جميع المؤلفين النتائج وعلقوا على المخطوطة.
المصالح المتنافسة
معلومات إضافية
المواد التكميلية متاحة على
https://doi.org/10.1038/s41467-024-45320-0.
يجب توجيه المراسلات والطلبات للحصول على المواد إلى هايان وانغ، هاو هوانغ أو يونغ هو.
http://www.nature.com/reprints
© المؤلف(ون) 2024
المختبر الرئيسي لوزارة التعليم للمواد الحفازة المتقدمة، قسم الكيمياء، جامعة تشجيانغ العادية، جينهوا 321004، الصين.
قسم الأنظمة الدقيقة، جامعة جنوب شرق النرويج، بوري 3184، النرويج. قسم علوم البصريات والهندسة، جامعة فودان، شنغهاي 200438، الصين. كلية الكيمياء وهندسة المواد، جامعة تشجيانغ للزراعة والغابات، هانغتشو 311300، الصين. ساهم هؤلاء المؤلفون بالتساوي: بينغتشنغ يي، كيكينغ فانغ. البريد الإلكتروني: chemwhy@zjnu.edu.cn; huanghao881015@163.com; yonghu@zafu.edu.cn
DOI: https://doi.org/10.1038/s41467-024-45320-0
PMID: https://pubmed.ncbi.nlm.nih.gov/38307871
Publication Date: 2024-02-03
Lattice oxygen activation and local electric field enhancement by co-doping Fe and F in CoO nanoneedle arrays for industrial electrocatalytic water oxidation
Accepted: 19 January 2024
Published online: 03 February 2024
(4) Check for updates
Abstract
Oxygen evolution reaction (OER) is critical to renewable energy conversion technologies, but the structure-activity relationships and underlying catalytic mechanisms in catalysts are not fully understood. We herein demonstrate a strategy to promote OER with simultaneously achieved lattice oxygen activation and enhanced local electric field by dual doping of cations and anions. Rough arrays of Fe and F co-doped CoO nanoneedles are constructed, and a low overpotential of 277 mV at
structure of electrocatalysts and electrochemical behaviors, are of great significance to the development of highly reactive and durable OER catalysts.
Results and discussion
Synthesis and characterization of catalysts
e HRTEM image. f SAED pattern. g STEM image and corresponding elemental mapping images.
respectively, and jointly change the electronic structure of CoO . The O 1s spectra show three oxygen species including lattice-O, adsorbed-O or hydroxyl ( -OH ), and adsorbed
cause significant structural changes in CoO (Supplementary Figs. 10, 11, Supplementary Table 2). This is further confirmed by the similar wavelet transform (WT) plots (Fig. 2d-i), agreeing well with the observations from XRD and Raman.
Electrocatalytic OER performance
verified with the LSV curves (Supplementary Fig. 13). FESEM and XRD further demonstrate that the morphology and crystal structure show negligible changes after the cycling test, suggesting their robust stability and great potential in practical applications (Supplement tary Fig. 14).
Kinetic study
were studied by electrochemical impedance spectroscopy (EIS). Nyquist plots manifest that the charge transfer resistance
Exploration of reaction mechanism
the reaction mechanism. As profiled in Supplementary Fig. 21, the current densities of the Fe, F-CoO NNAs increase steeply with the increase of pH from 12.5 to 14.0, suggesting a nonconcerted protonelectron transfer pathway
were directly demonstrated with Raman spectra, in which the typical peaks of TMA
c LSV profiles of the Fe, F-CoO NNAs, F-CoO NNAs, Fe-CoO NNAs, and CoO NNAs catalysts in 1 M KOH and 1 M TMAOH with
spectra at 1.33 V and above, with a zenith at around
in the OER. These observations provide more compelling evidence for the LOM mechanism.
Theoretical insights into reaction mechanism
localization function (ELF) analysis, in which the electronic locality is reduced after
Effects of local electric field
0.99 eV , indicating the accelerated lattice oxygen activation kinetics. Meanwhile, the release rate of
Industrial overall water splitting performance
Methods
Chemicals
and KOH were obtained from Sinopharm Reagent Co., Ltd. All the reagents were directly used without further purification.
Synthesis of
anhydrous ethanol sequentially.
Synthesis of Fe, F-CoO NNAs
Synthesis of F-CoO NNAs
Synthesis of
Synthesis of Fe-CoO NNAs
Synthesis of CoO NNAs
Characterization
Electrochemical measurements
scanning rate. The calculation for ECSA is shown as follows.
Data availability
References
- Vojvodic, A. & Norskov, J. K. Optimizing perovskites for the watersplitting reaction. Science 334, 1355-1356 (2011).
- Hu, E. et al. Construction of hierarchical Ni-Co-P hollow nanobricks with oriented nanosheets for efficient overall water splitting. Energy Environ. Sci. 11, 872-880 (2018).
- Zhao, S. et al. Ultrathin metal-organic framework nanosheets for electrocatalytic oxygen evolution. Nat. Energy 1, 1-10 (2016).
- Ng, J. W. D. et al. Gold-supported cerium-doped
catalysts for water oxidation. Nat. Energy 1, 16053 (2016). - Hu, E. et al. Graphene layers-wrapped
nanoparticles supported on N -doped graphene nanosheets for highly efficient oxygen reduction. Adv. Energy Mater. 8, 1702476 (2018). - Li, J. et al. A general strategy for preparing pyrrolic-
type singleatom catalysts via pre-located isolated atoms. Nat. Commun. 12, 6806 (2021). - Cao, L. et al. Dynamic oxygen adsorption on single-atomic Ruthenium catalyst with high performance for acidic oxygen evolution reaction. Nat. Commun. 10, 4849 (2019).
- Zhai, P. et al. Engineering single-atomic ruthenium catalytic sites on defective nickel-iron layered double hydroxide for overall water splitting. Nat. Commun. 12, 4587 (2021).
- Zhang, R. et al. Tracking the role of defect types in
structural evolution and active motifs during oxygen evolution reaction. J. Am. Chem. Soc. 145, 2271-2281 (2023). - Yan, D. et al. Cation defect engineering of transition metal electrocatalysts for oxygen evolution reaction. Adv. Energy Mater. 12, 2202317 (2022).
- Yan, J. et al. Single atom tungsten doped ultrathin
for enhanced electrocatalytic water oxidation. Nat. Commun. 10, 2149 (2019). - Zhao, Y., Nakamura, R., Kamiya, K., Nakanishi, S. & Hashimoto, K. Nitrogen-doped carbon nanomaterials as non-metal electrocatalysts for water oxidation. Nat. Commun. 4, 2390 (2013).
- Zhang, L. et al. Self-reconstructed metal-organic framework heterojunction for switchable oxygen evolution reaction. Angew. Chem. Int. Ed. 61, e202214794 (2022).
- Qiu, C. et al. Interfacial engineering FeOOH/CoO nanoneedle array for efficient overall water splitting driven by solar energy. Chem. Eur. J. 26, 4120-4127 (2020).
- Yan, L. et al. Engineering self-supported hydrophobic-aerophilic air cathode with
nanoparticles embedded in co-doped carbon plate arrays for long-life rechargeable Zn-air batteries. Adv. Energy Mater. 13, 2204245 (2023). - Yang, X. G. et al. Enhanced activity of enzyme immobilized on hydrophobic ZIF-8 modified by
ions. Angew. Chem. Int. Ed. 62, e202216699 (2023). - Wang, X., Zhong, H., Xi, S., Lee, W. S. V. & Xue, J. Understanding of oxygen redox in the oxygen evolution reaction. Adv. Mater. 34, 2107956 (2022).
- Xiao, K. et al. Activating lattice oxygen in spinel
through filling oxygen vacancies with fluorine for electrocatalytic oxygen evolution. Angew. Chem. Int. Ed. 62, e202301408 (2023). - Song, J. et al. A review on fundamentals for designing oxygen evolution electrocatalysts. Chem. Soc. Rev. 49, 2196-2214 (2020).
- Grimaud, A. et al. Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution. Nat. Chem. 9, 457-465 (2017).
- Wang, C. et al. Engineering lattice oxygen activation of iridium clusters stabilized on amorphous bimetal borides array for oxygen evolution reaction. Angew. Chem. Int. Ed. 60, 27126-27134 (2021).
- Huang, Z. F. et al. Tuning of lattice oxygen reactivity and scaling relation to construct better oxygen evolution electrocatalyst. Nat. Commun. 12, 3992 (2021).
- Zhang, N. et al. Lattice oxygen activation enabled by high-valence metal sites for enhanced water oxidation. Nat. Commun. 11, 4066 (2020).
- Huang, Z. F. et al. Chemical and structural origin of lattice oxygen oxidation in Co-Zn oxyhydroxide oxygen evolution electrocatalysts. Nat. Energy 4, 329-338 (2019).
- Zhang, N. & Chai, Y. Lattice oxygen redox chemistry in solid-state electrocatalysts for water oxidation. Energy Environ. Sci. 14, 4647-4671 (2021).
- Hu, Q. et al. Integrating well-controlled core-shell structures into “superaerophobic” electrodes for water oxidation at large current densities. Appl. Catal. B:Environ. 286, 119920 (2021).
- Liu, M. et al. Enhanced electrocatalytic
reduction via fieldinduced reagent concentration. Nature 537, 382-386 (2016). - Chen, Q. et al. Ordered Ag nanoneedle arrays with enhanced electrocatalytic
reduction via structure-induced inhibition of hydrogen evolution. Nano Lett. 22, 6276-6284 (2022). - Gao, Y. et al. Field-induced reagent concentration and sulfur adsorption enable efficient electrocatalytic semihydrogenation of alkynes. Sci. Adv. 8, eabm9477 (2022).
- Liu, P. et al. Tip-enhanced electric field: A new mechanism promoting mass transfer in oxygen evolution reactions. Adv. Mater. 33, 2007377 (2021).
- Liu, D. et al. Atomically dispersed platinum supported on curved carbon supports for efficient electrocatalytic hydrogen evolution. Nat. Energy 4, 512-518 (2019).
- Nairan, A. et al. Proton selective adsorption on Pt-Ni nano-thorn array electrodes for superior hydrogen evolution activity. Energy Environ. Sci. 14, 1594-1601 (2021).
- Jiang, H., Hou, Z. & Luo, Y. Unraveling the mechanism for the sharptip enhanced electrocatalytic carbon dioxide reduction: The kinetics decide. Angew. Chem. Int. Ed. 56, 15617-15621 (2017).
- Zhang, L. et al. Attenuating a metal-oxygen bond of a double perovskite oxide via anion doping to enhance its catalytic activity for the oxygen reduction reaction. J. Mater. Chem. A 8, 14091-14098 (2020).
- Yu, X. et al. Activation or passivation: Influence of halogen dopant (
) on photothermal activity of in degrading toluene. Appl. Catal. B: Environ. 309, 121236 (2022). - Xie, L., Zhao, D., Dai, J., Wu, Z. & Li, L. Solvothermally doping
nanoparticles on carbon with ferric ions for efficient oxygen evolution catalysis. Catalysts 9, 458 (2019). - Lei, H. et al. Coordination and interface engineering to boost catalytic property of two-dimensional ZIFs for wearable Zn-air batteries. J. Energy Chem. 68, 78-86 (2022).
- Li, M. et al. Scalable dry production process of a duperior 3D netlike carbon-based iron oxide anode material for lithium-ion batteries. Angew. Chem. Int. Ed. 56, 12649-12653 (2017).
- Chen, X., Wei, M. & Zhou, J. Fluorine-doping-assisted vacancy engineering for efficient electrocatalyst toward hydrogen production. J. Mater. Chem. A 9, 22626-22634 (2021).
- Lu, W. et al. Construction of CoO/Co-Cu-S hierarchical tubular heterostructures for hybrid supercapacitors. Angew. Chem. Int. Ed. 58, 15441-15447 (2019).
- Lian, J. et al. Magnetic flower-like Fe-doped CoO nanocomposites with dual enzyme-like activities for facile and sensitive determination of
and dopamine. Inorg. Chem. 60, 1893-1901 (2021). - Mi, J. et al. Activation of partial metal sites in high-entropy oxides for enhancing thermal and electrochemical catalysis. Chin. J. Catal. 48, 235-246 (2023).
- Wu, Y. et al. Triggering lattice oxygen activation of single-atomic Mo sites anchored on Ni-Fe oxyhydroxides nanoarrays for electrochemical water oxidation. Adv. Mater. 34, 2202523 (2022).
- Roychoudhury, S. et al. Deciphering the oxygen absorption preedge: A caveat on its application for probing oxygen redox reactions in batteries. Energy Environ. Mater. 4, 246-254 (2021).
- Wang, X. et al. The role of electron localization in covalency and electrochemical properties of lithium-ion battery cathode materials. Adv. Funct. Mater. 31, 2001633 (2021).
- Huang, J. et al. Modifying redox properties and local bonding of
by enhances oxygen evolution catalysis in acid. Nat. Commun. 12, 3036 (2021). - Su, H. et al. In-situ spectroscopic observation of dynamic-coupling oxygen on atomically dispersed iridium electrocatalyst for acidic water oxidation. Nat. Commun. 12, 6118 (2021).
- Chen, X . et al. S-doping triggers redox reactivities of both iron and lattice oxygen in FeOOH for low-cost and high-performance water oxidation. Adv. Funct. Mater. 32, 2112674 (2022).
- Mefford, J. T. et al. Water electrolysis on
perovskite electrocatalysts. Nat. Commun. 7, 11053 (2016). - Xiao, L. et al. Electronic and nano-structural modulation of
nanosheets by Fe-benzenedicarboxylate for efficient oxygen evolution. Chem. Res. Chin. U. 39, 219-223 (2023). - Guo, W. et al. In situ revealing the reconstruction behavior of monolayer rocksalt CoO nanosheet as water oxidation catalyst. J. Energy Chem. 70, 373-381 (2022).
- Moysiadou, A., Lee, S., Hsu, C. S., Chen, H. M. & Hu, X. Mechanism of oxygen evolution catalyzed by cobalt oxyhydroxide: cobalt superoxide species as a key intermediate and dioxygen release as a ratedetermining step. J. Am. Chem. Soc. 142, 11901-11914 (2020).
- Lee, S., Banjac, K., Lingenfelder, M. & Hu, X. Oxygen isotope labeling experiments reveal different reaction sites for the oxygen evolution reaction on nickel and nickel iron oxides. Angew. Chem. Int. Ed. 58, 10295-10299 (2019).
- Farsi, L. & Deskins, N. A. First principles analysis of surface dependent segregation in bimetallic alloys. Phys. Chem. Chem. Phys. 21, 23626-23637 (2019).
- Grimaud, A., Hong, W. T., Shao-Horn, Y. & Tarascon, J. M. Anionic redox processes for electrochemical devices. Nat. Mater. 15, 121-126 (2016).
- Wang, Z. et al. Elevating the d -band center of six-coordinated octahedrons in
through Fe-incorporated topochemical deintercalation. Adv. Energy Mater. 11, 2003023 (2021). - Han, B. et al. Nanoscale structural oscillations in perovskite oxides induced by oxygen evolution. Nat. Mater. 16, 121-126 (2017).
- Ji, J. et al.
-regulated electron state of for superb aqueous zinc-manganese oxide batteries. Adv. Energy Mater. 11, 2003203 (2021). - Mueller, D. N., Machala, M. L., Bluhm, H. & Chueh, W. C. Redox activity of surface oxygen anions in oxygen-deficient perovskite oxides during electrochemical reactions. Nat. Commun. 6, 6097 (2015).
- Song, Q. et al. General strategy to optimize gas evolution reaction via assembled striped-pattern sduperlattices. J. Am. Chem. Soc. 142, 1857-1863 (2020).
- Lin, Q. et al. Tuning the interface of
by atomic replacement strategy toward high-performance electrocatalytic oxygen evolution. ACS Nano 16, 15460-15470 (2022).
Acknowledgements
Author contributions
analysis and revision of the manuscript. All authors discussed the results and commented on the manuscript.
Competing interests
Additional information
supplementary material available at
https://doi.org/10.1038/s41467-024-45320-0.
Correspondence and requests for materials should be addressed to Haiyan Wang, Hao Huang or Yong Hu.
http://www.nature.com/reprints
© The Author(s) 2024
Key Laboratory of the Ministry of Education for Advanced Catalysis Materials, Department of Chemistry, Zhejiang Normal University, Jinhua 321004, China.
Department of Microsystems, University of South-Eastern Norway, Borre 3184, Norway. Department of Optical Science and Engineering, Fudan University, Shanghai 200438, China. College of Chemistry and Materials Engineering, Zhejiang A&F University, Hangzhou 311300, China. These authors contributed equally: Pengcheng Ye, Keqing Fang. e-mail: chemwhy@zjnu.edu.cn; huanghao881015@163.com; yonghu@zafu.edu.cn