DOI: https://doi.org/10.1186/s12943-024-02008-9
PMID: https://pubmed.ncbi.nlm.nih.gov/38711083
تاريخ النشر: 2024-05-06
تنظيم التغذية الراجعة الإيجابية بين التحلل السكري والأسيتيل الهستوني يقود الأورام في سرطان القناة البنكرياسية الغدي
الملخص
الخلفية: إعادة برمجة الأيض والتغيرات الوراثية تلعب دورًا في عدوانية سرطان القناة البنكرياسية الغدي (PDAC). يعد تعديل الهيستون المعتمد على اللاكتات نوعًا جديدًا من علامات الهيستون، والذي يربط بين ناتج التحلل السكري وعملية اللاكتيل. ومع ذلك، لا يزال دور اللاكتيل في الهيستون في PDAC غير واضح. الطرق: تم تحديد مستوى اللاكتيل في الهيستون في PDAC من خلال التحليل الغربي والتلوين المناعي، وتم تقييم علاقته بالبقاء العام باستخدام مخطط بقاء كابلان-ماير. تم تأكيد مشاركة اللاكتيل في الهيستون في نمو وتقدم PDAC من خلال تثبيط اللاكتيل في الهيستون بواسطة مثبطات التحلل السكري أو تقليل التعبير عن إنزيم لاكتات ديهيدروجيناز A (LDHA) في كل من المختبر وفي الكائنات الحية. تم تحديد الكتاب والممحيات المحتملة للاكتيل في الهيستون في PDAC من خلال التحليل الغربي والتجارب الوظيفية. تم فحص الجينات المستهدفة المحتملة للاكتيل H3K18 (H3K18la) من خلال تحليلات CUT&Tag وRNA-seq. تم التحقق من الجينات المستهدفة المرشحة TTK كيناز البروتين (TTK) وBUB1 كيناز نقطة التفتيش الانقسامية سيرين/ثريونين B (BUB1B) من خلال تحليلات ChIP-qPCR وRT-qPCR والتحليل الغربي. بعد ذلك، تم تأكيد تأثير هذين الجينين في PDAC من خلال تقليل التعبير أو زيادة التعبير. تم تحديد التفاعل بين TTK وLDHA من خلال اختبار Co-IP. النتائج: كان مستوى اللاكتيل في الهيستون، وخاصة مستوى H3K18la، مرتفعًا في PDAC، وكان المستوى العالي من H3K18la مرتبطًا بتشخيص سيئ. ساهمت قمع النشاط التحللي بواسطة أنواع مختلفة من المثبطات أو تقليل التعبير عن LDHA في التأثيرات المضادة للورم لـ PDAC في المختبر وفي الكائنات الحية. كان بروتين ربط E1A p300 (P300) وديستاز الهيستون 2 هما الكاتب والممحاة المحتملان للاكتيل في الهيستون في خلايا PDAC، على التوالي. تم إثراء H3K18la عند المحفزات وتفعيل نسخ منظمين نقطة التفتيش الانقسامية TTK وBUB1B. ومن المثير للاهتمام، أن TTK وBUB1B يمكن أن يزيدا من تعبير P300 الذي بدوره زاد من التحلل السكري. علاوة على ذلك، TTK
الملخص
LDHA الفسفوري في التيروزين 239 (Y239) وLDHA المنشط، وبالتالي زاد من مستويات اللاكتات وH3K18la. الاستنتاجات حلقة التغذية الراجعة الإيجابية بين الجليكوليز وH3K18la وTTK/BUB1B تفاقم الخلل الوظيفي في PDAC. هذه النتائج قدمت استكشافًا جديدًا وعلاقة مهمة بين إعادة برمجة الأيض للاكتات والتنظيم الوراثي، مما قد يمهد الطريق نحو استراتيجيات علاج جديدة تعتمد على اللاكتيل في علاج PDAC.
مقدمة
ومع ذلك، لم يتم تحقيق استراتيجيات تتضمن علاج اللاكتيل الهستوني للتغلب على وتحويل المشهد الجزيئي لسرطان البنكرياس القنوي. وبالتالي، لا يزال فهم أفضل للاكتيل الهستوني في علم الأمراض لسرطان البنكرياس القنوي يتطلب مزيدًا من البحث.
تهدف دراستنا إلى التحقيق في دور اللاكتيل في الهيستون، وبالتحديد H3K18la، في تقدم سرطان البنكرياس (PDAC) وتوضيح الآلية المحتملة. أظهرت نتائجنا أن تراكم اللاكتات في بيئة الورم في PDAC أدى إلى تحفيز اللاكتيل في الهيستون، سواء في الجسم الحي أو في المختبر، مما ارتبط بقوة مع تكوين الأورام والنتائج السريرية السيئة. من الناحية الآلية، أدى تعزيز التحلل السكري في PDAC إلى زيادة إنتاج اللاكتات وH3K18la، مما عزز نسخ بروتين كيناز TTK.
المواد والأساليب
عينات بشرية
اختبار الميتابولوميات غير المستهدفة
زراعة الخلايا والعلاج
الأيض عن طريق تثبيط الهيكسوكيناز وإيزوميراز الجلوكوز-6-فوسفات [18] أو التحكم في المركبة (محلول فوسفات الملح، PBS). (2) تم نقل الخلايا باستخدام siRNA الخاص بـ LDHA أو siRNA عشوائي كتحكم سلبي، مع إضافة لاكتات الصوديوم (NaLa،
تداخل RNA وزيادة التعبير
عدوى الفيروسات القهقرية
تم تحضير التعبير عن sh-LDHA و oe-TTK بواسطة Hanbio Tech (شنغهاي، الصين). كانت خلايا MIA PaCa-2 عند تداخل
اختبار تكاثر الخلايا
اختبار تشكيل المستعمرات
اختبار شفاء الجروح
تم قياس مناطق الجروح باستخدام برنامج Image J (المعاهد الوطنية للصحة، بيثيسدا، ماريلاند، الولايات المتحدة الأمريكية).
اختبار هجرة ترانسويل
نموذج الورم المزروع في الفئران العارية
المؤسسة الدولية للحقوق
تم حضانة الأقسام طوال الليل في
استخراج RNA و RT-qPCR
استخراج البروتين وويسترن بلوت
قياس محتوى اللاكتات ونشاط إنزيم LDH
تم قياس قيم الامتصاص عند 570 نانومتر باستخدام مطياف الضوء. تم تطبيع محتوى اللاكتات بناءً على عدد الخلايا.
Co-IP
اختبار CUT&Tag
قم بإجراء تحليلات علم الأحياء الجزيئي (GO) و(KEGG).
تسلسل RNA
اختبار ChIP و ChIP-qPCR
التحليل الإحصائي
النتائج
ترتبط مستويات اللاكتيل في الهيستونات المرتفعة بتوقعات غير مواتية لدى المرضى المصابين بسرطان البنكرياس القنوي.
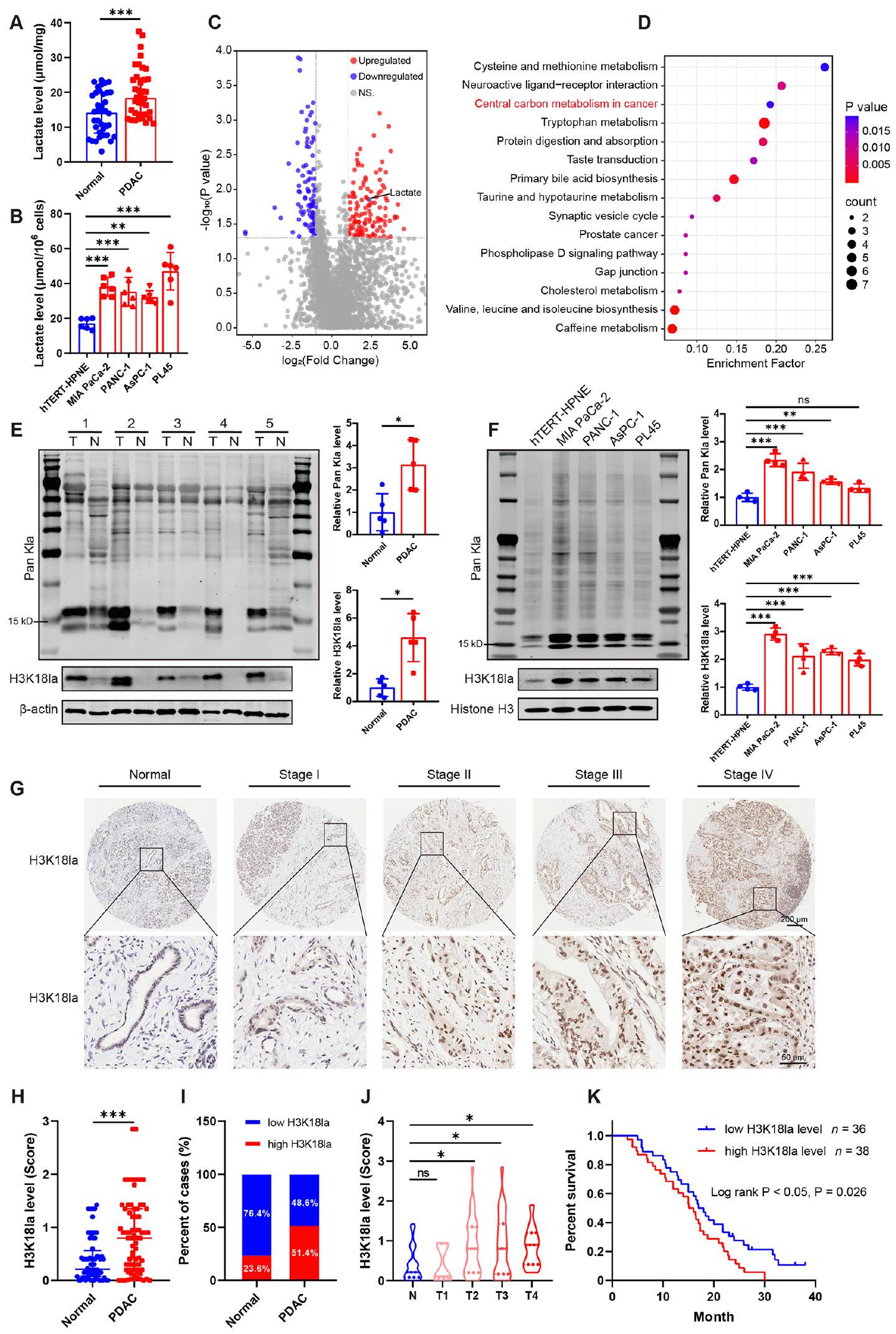
نظرًا لإنتاج كميات كبيرة من اللاكتات المقدمة كركائز لأسيتيل الهيستون، قمنا بفحص مستويات أسيتيل البروتين في 5 أزواج من أنسجة سرطان البنكرياس (PDAC) والأنسجة غير السرطانية المحيطة. لوحظت مستويات أعلى من أسيتيل اللايسين الشامل/العمومي (Pan Kla) في أنسجة PDAC (الشكل 1E). نظرًا لأن تعديلات الهيستون تلعب أدوارًا أساسية في معظم العمليات البيولوجية وقد تم الإبلاغ عن أن H3K18la ينظم عمليات بيولوجية متعددة مثل الأورام [12،19]، ركزنا على
الملخص
الشكل 1 مستوى اللاكتات المرتفع وتعديل الهيستون باللاكتيل مرتبطان بتوقعات غير مواتية لدى المرضى المصابين بسرطان القناة البنكرياسية (PDAC). (A) محتوى اللاكتات في سرطان القناة البنكرياسية (PDAC) والأنسجة المجاورة للسرطان.
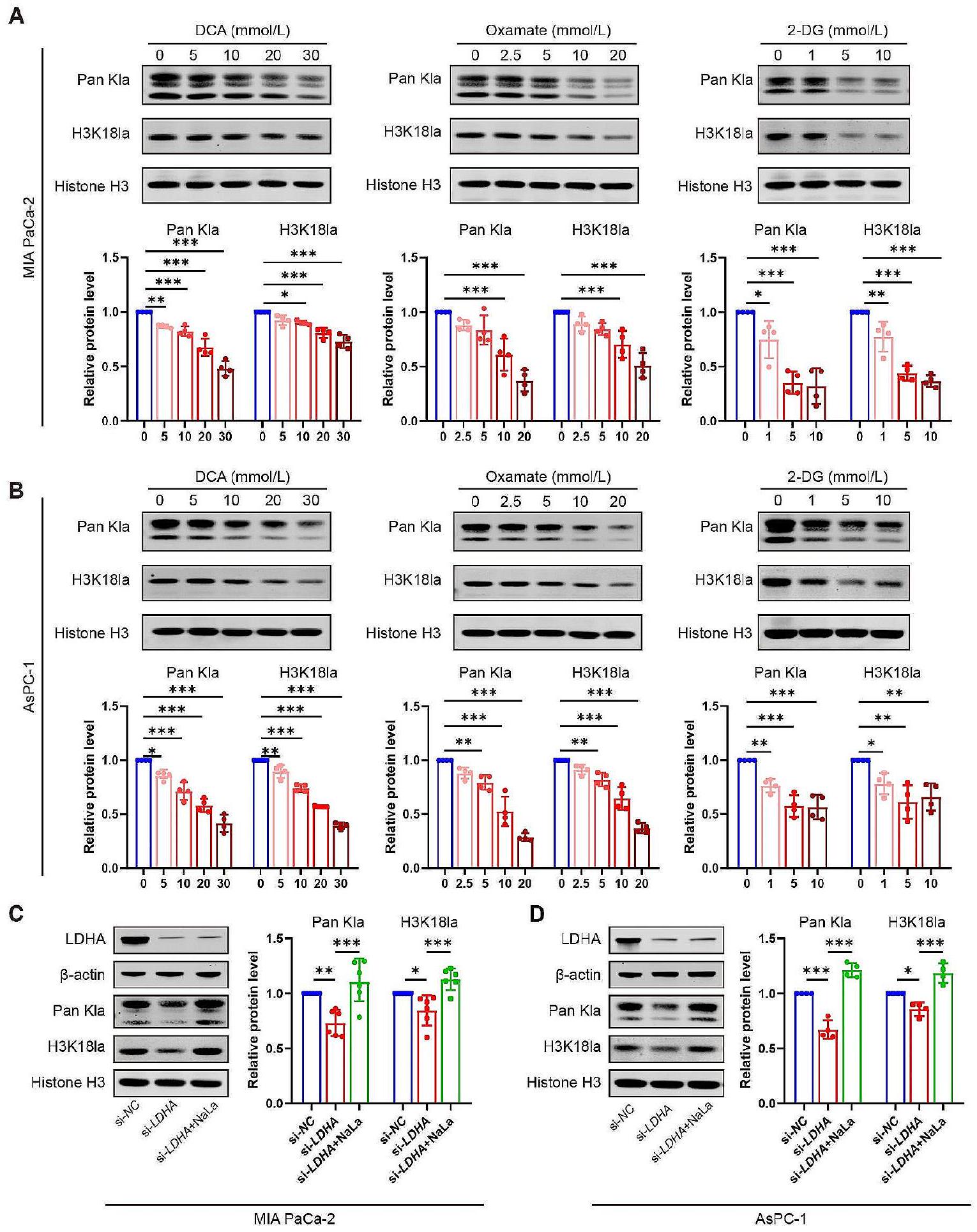
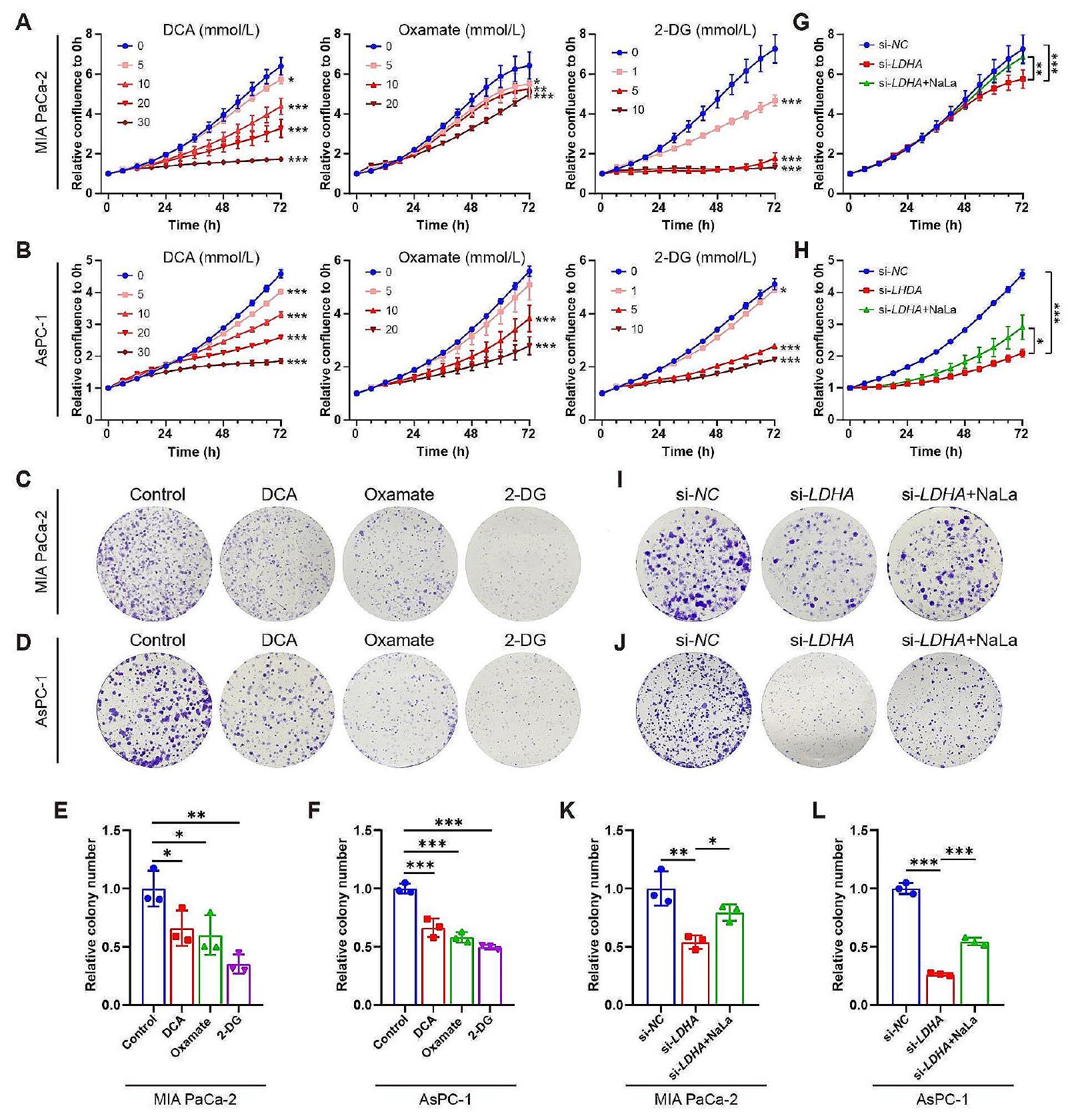
تثبيط التحلل السكري يقلل من لاكتيل الهستون ويعيق تكاثر خلايا سرطان البنكرياس القنوي والهجرة
خفف تأثير تثبيط تكاثر الخلايا لـ
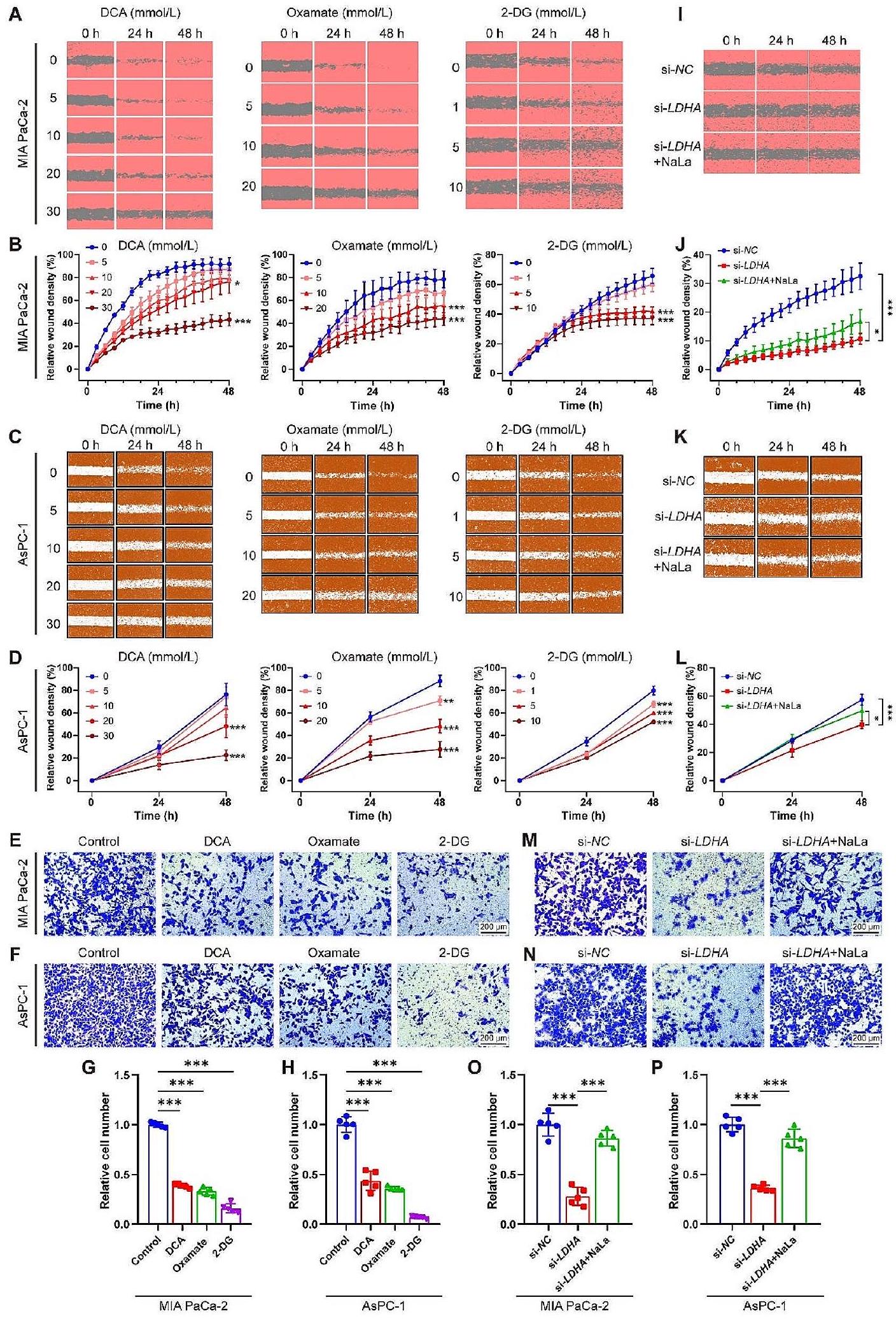
لاستكشاف ما إذا كان يمكن أن تؤثر اللاكتيل على هجرة خلايا PDAC، تم إجراء اختبارات شفاء الجروح واختبارات ترانسويل. أدت العلاجات باستخدام DCA وOxamate و2-DG إلى تقليل هجرة الخلايا بشكل كبير في خلايا MIA PaCa-2 وAsPC-1، وأظهر الجرعة الأعلى تأثيرات تثبيط أكثر وضوحًا على هجرة الخلايا مقارنة بالجرعة الأقل في اختبار شفاء الجروح (الشكل 4A-D). وبالمثل، أدى تقليل LDHA إلى تثبيط قدرات الهجرة مقارنة بمجموعة المتجه، وأدى إضافة Nala إلى تخفيف التأثير المثبط لـ si-LDHA (الشكل 4I-L). أظهرت اختبارات ترانسويل أيضًا أن خلايا MIA PaCa-2 أو AsPC-1 المعالجة بـ DCA وOxamate و2-DG قد قللت بشكل كبير من هجرة الخلايا (الشكل 4E-H). لوحظت نفس النتائج في تقليل LDHA، وعكست Nala تأثير التثبيط الهجري لـ si-LDHA في كلا خطي الخلايا (الشكل 4M-P). بشكل جماعي، تشير هذه النتائج إلى أن اللاكتيل الهيستوني يلعب دورًا حاسمًا في بدء وتقدم PDAC، بينما قد يظهر تثبيط اللاكتيل الهيستوني نشاطًا مضادًا للورم محتمل ضد PDAC.
تثبيط التحلل السكري يقلل من اللاكتيل الهيستوني ويثبط تقدم PDAC في نموذج الفأر الزرعي لـ PDAC
ملخص
الشكل 4 تثبيط التحلل السكري يقلل من اللاكتيل الهيستوني ويثبط هجرة خلايا PDAC. (A-H) تم معالجة خطي خلايا PDAC MIA PaCa-2 وAsPC-1 بمثبطات التحلل السكري DCA (
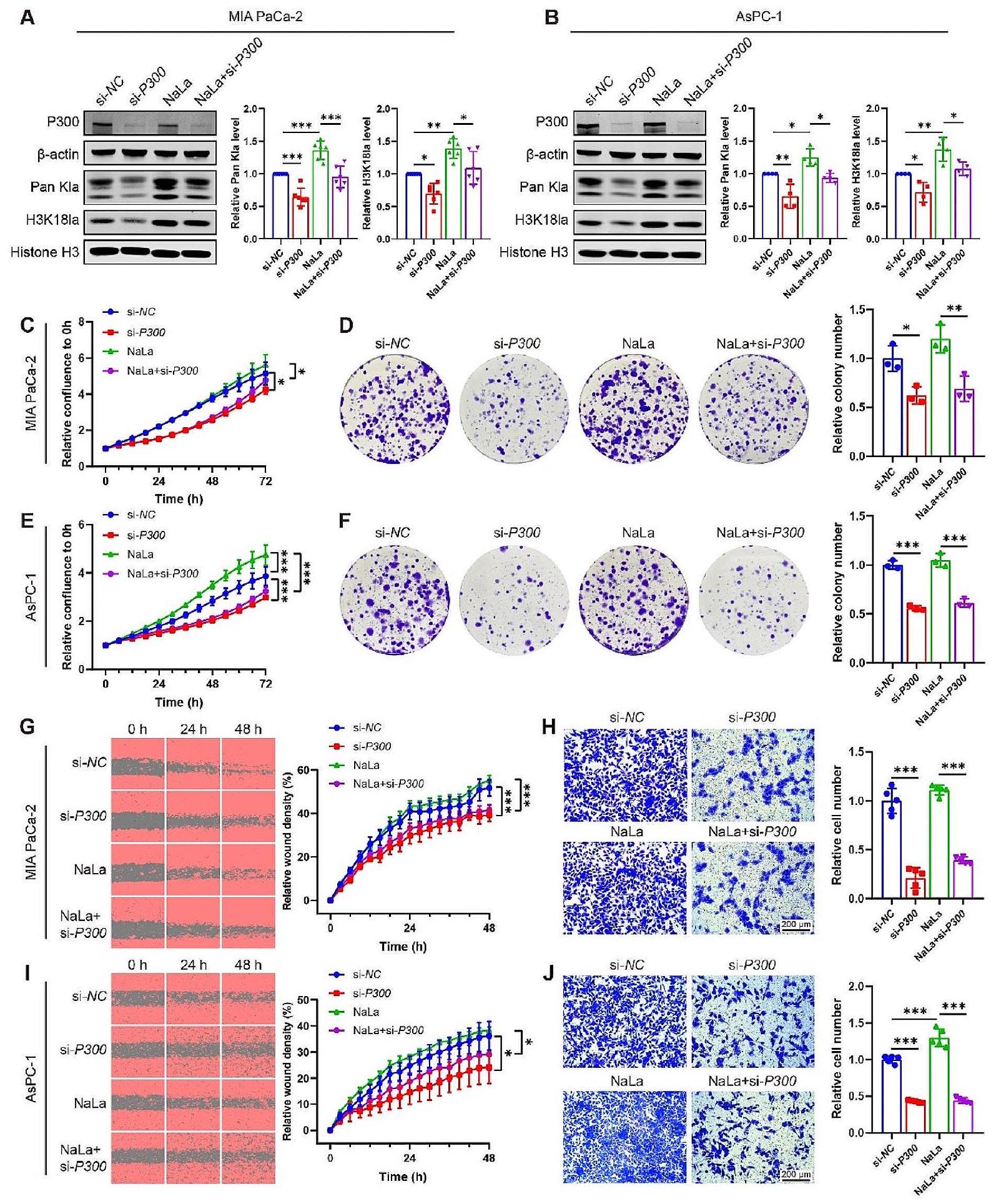
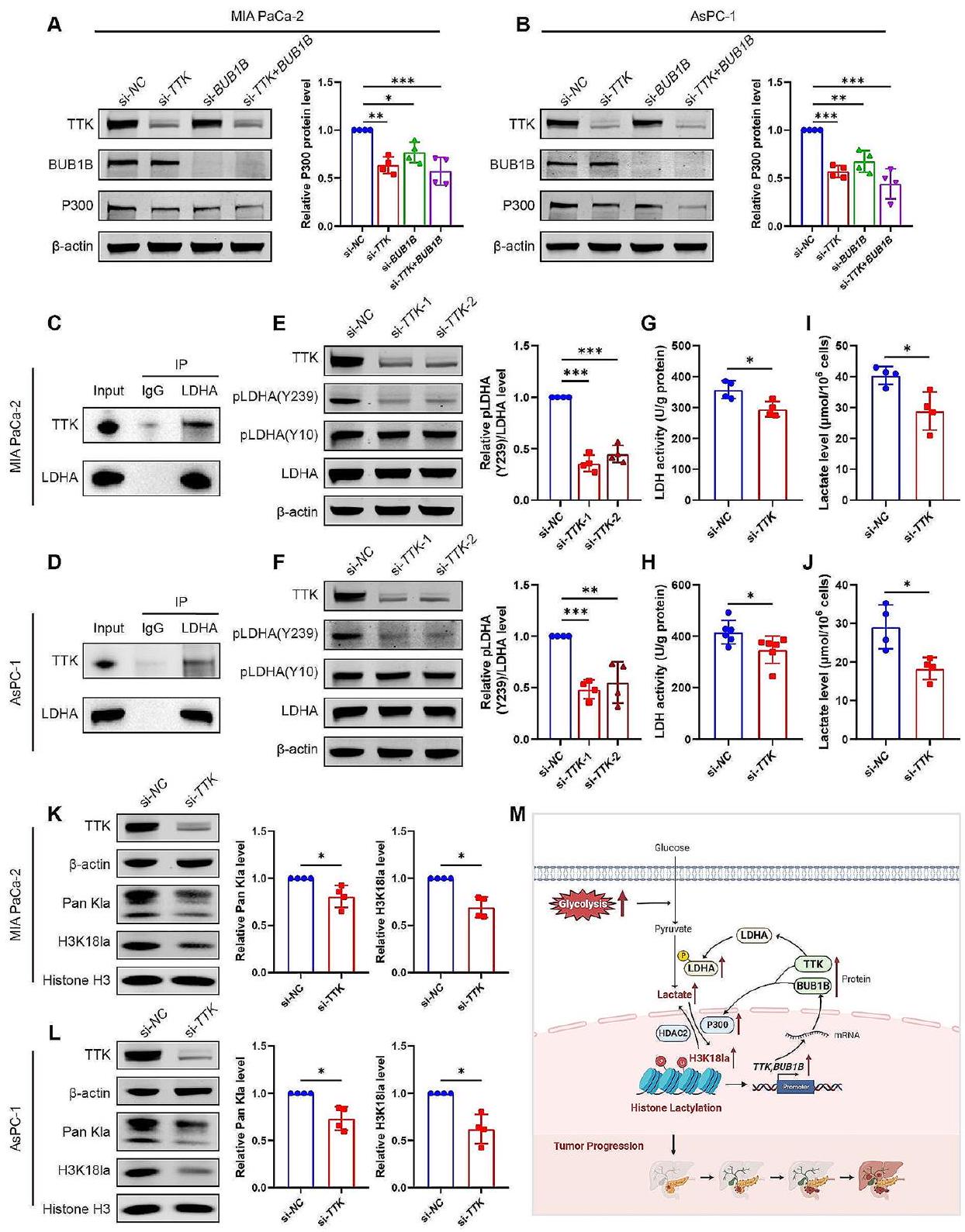
P300 وHDAC2 هما كاتبان ومزيلان محتملان للاكتيل الهيستوني في خلايا PDAC على التوالي
أو معالجة C646 يمكن أن تعكس تأثير NaLa على هجرة PDAC. كما هو متوقع، أظهرت الخلايا التي تم نقلها باستخدام si-P300 أو المعالجة بـ C646 مع NaLa قدرة هجرة مخفضة مقارنة بتلك المعالجة فقط بـ NaLa في خلايا MIA PaCa-2 (الشكل 6G وH، S1G-H) وخلايا AsPC-1 (الشكل 6I وJ، S1I-J). تشير هذه النتائج إلى أن P300، الكاتبة المحتملة للاكتيل الهيستوني، تعزز تقدم PDAC.
لتحديد المحو المحتمل لتعديل اللاكتيل على الهيستون في PDAC، تم استخدام أنواع مختلفة من مثبطات هيستون ديستيلز (HDAC) في خلايا MIA PaCa-2 وAsPC-1. من المثير للاهتمام، أن العلاج بمثبطات HDAC واسعة الطيف TSA، بدلاً من مثبط السرتوين NAM، أدى إلى زيادة المستوى العالمي لتعديل اللاكتيل على الهيستون. بالإضافة إلى ذلك، زاد مثبط HDAC من الفئة I CI994 من تعديل اللاكتيل، بينما لم يكن لمثبط HDAC من الفئة IIa TMP195، ومثبط HDAC من الفئة IIb Bufexamac، ومثبط HDAC من الفئة IV SIS17 أي تأثير مماثل (الشكل S2A-B). للتحقق من دور كل إيزوزيم من HDAC، تم إجراء تجارب التعبير الزائد في خلايا MIA PaCa-2 وAsPC-1. أظهرت النتائج أن التعبير الزائد عن HDAC2 قلل بشكل كبير من مستوى تعديل اللاكتيل، وخاصة H3K18la، بينما لم يؤثر التعبير الزائد عن HDAC1 وHDAC3 على مستويات Pan Kla وH3K18la (الشكل S2C-H). تشير هذه النتائج إلى أن P300 قد تعمل ككاتبة محتملة بينما يعمل HDAC2 كممحاة لتعديل اللاكتيل على الهيستون في PDAC.
H3K18la ينشط نسخ TTK وBUB1B في PDAC
بالإضافة إلى ذلك، أجرينا اختبار RNA-seq لفحص الجينات المستهدفة المحتملة التي تنظمها اللاكتيل (البيانات الأصلية في الملف الإضافي 4). عند مقارنتها بمجموعة التحكم، زادت معالجة Oxamate من
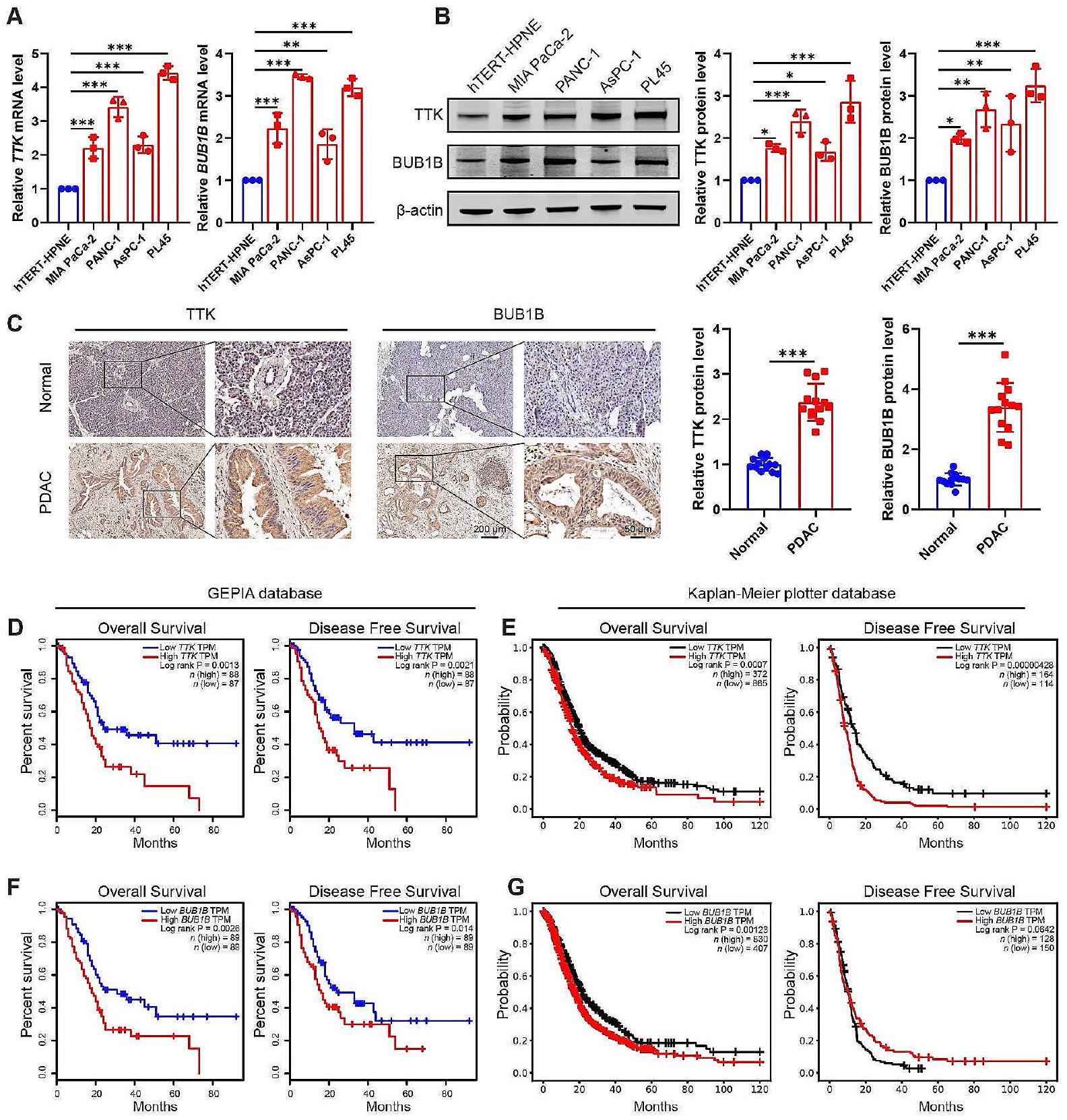
ترتبط المستويات العالية من TTK وBUB1B بخباثة PDAC
ارتفاع تعبير TTK أو BUB1B كان لديهم وقت بقاء إجمالي أقصر أو وقت بقاء خالٍ من المرض مقارنة بأولئك الذين لديهم مستويات تعبير أقل (الشكل 8D-G).
يؤدي حذف TTK وBUB1B إلى قمع خباثة PDAC بينما يمنع التعبير الزائد عن TTK جزئيًا التأثيرات المضادة للسرطان لتثبيط اللاكتيل
للتحقيق في تأثير TTK في تقدم PDAC بواسطة H3K18la، تم التعبير الزائد عن TTK في خلايا MIA PaCa-2 (الشكل 9I) وتم علاجها بمثبطات التحلل السكري. مقارنة بالتحكم السلبي، لم يعزز
حلقة تغذية راجعة إيجابية بين جينات هدف H3K18la والتحلل السكري
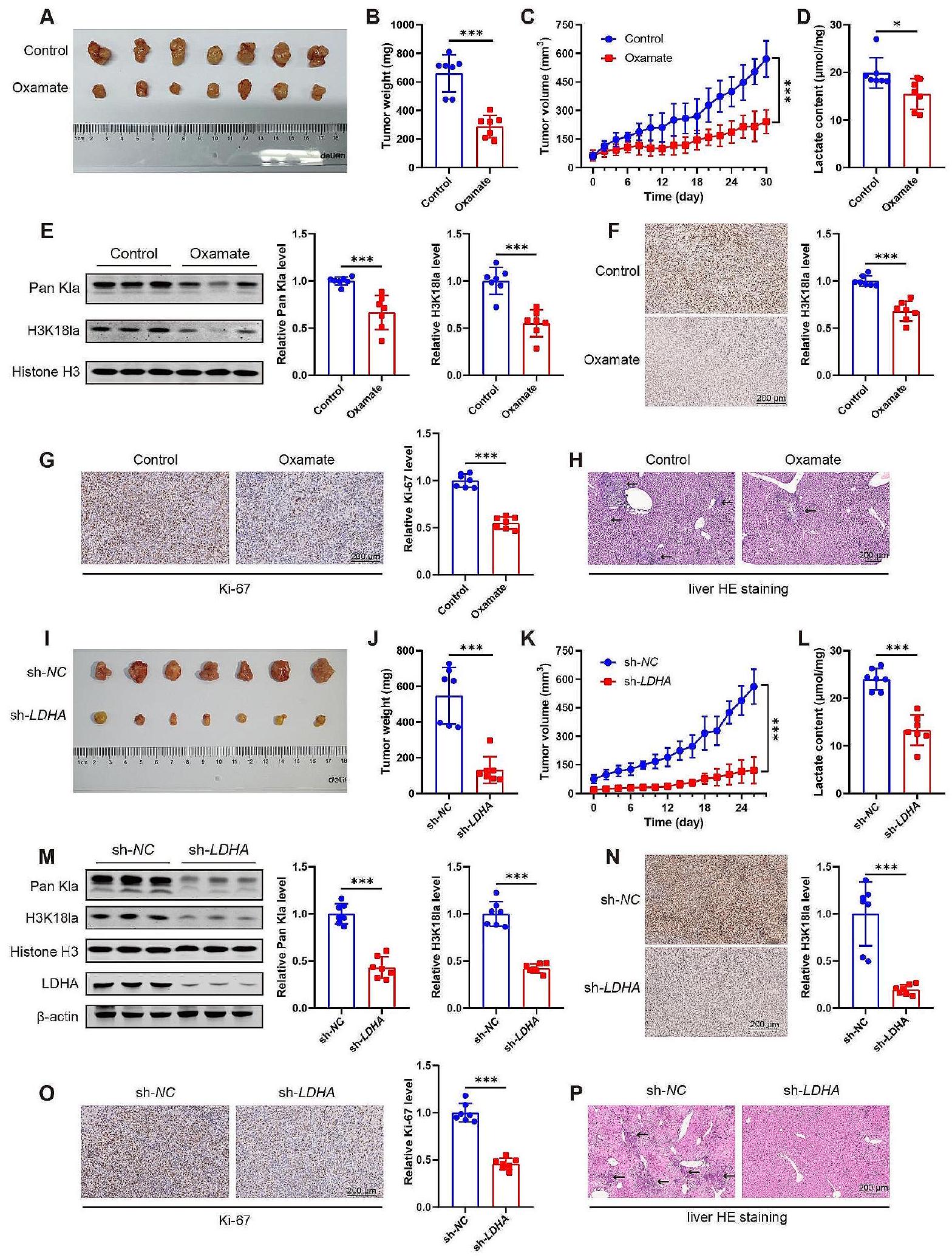
الشكل 5 تثبيط التحلل السكري يقلل من تعديل اللاكتيل على الهيستون ويقمع تقدم السرطان في PDAC المزروع في الفئران العارية. (A-H) تم حقن خلايا MIA PaCa-2 في الفئران العارية تحت الجلد وتم إعطاؤها Oxamate (
كما أن TTK/BUB1B لعبت دورًا إيجابيًا في اللاكتيلation الهيستونية، مما زاد من سوء الخباثة.
نقاش
مزيل لكتلة الهيستون في خلايا PDAC. علاوة على ذلك، فإن H3K18la حفزت
يُعتبر سرطان البنكرياس (PDAC) واحدًا من أكثر أنواع السرطان فتكًا وتحديًا كبيرًا في مجال الطب السرطاني. تُعتبر الاضطرابات الأيضية عوامل خطر لتقدم سرطان البنكرياس. يُعتبر إعادة برمجة الأيض الخلوي لدعم التكاثر والعمليات الخلوية المختلفة علامة رئيسية للسرطان. تلعب عملية التحلل السكري دورًا مركزيًا في إعادة برمجة الأيض لخلايا سرطان البنكرياس وتدعم السلوكيات الخبيثة. يُعرف التحلل السكري الهوائي، المعروف أيضًا بتأثير واربورغ، بأنه يتميز بتوليد الطاقة بشكل تفضيلي من خلال التحلل السكري بدلاً من الفسفرة التأكسدية، حتى في وجود مستويات كافية من الأكسجين، مما يؤدي إلى إنتاج كبير من حمض اللبنيك. تظهر هذه الظاهرة كعلامة مميزة لخلايا الورم، وخاصة في سرطان البنكرياس. لذلك، فإن استهداف تأثير واربورغ يحمل أهمية قصوى في استكشاف استراتيجيات علاجية جديدة ضد سرطان البنكرياس. كان يُعتبر حمض اللبنيك سابقًا مجرد منتج ثانوي لعملية الأيض التحللي حتى اكتشف فوبيرت وزملاؤه أنه يمكن إعادة استخدامه كمصدر الكربون الرئيسي لدورة حمض الكربوكسيليك الميتوكوندري. من جانبهم، قدم زانغ وزملاؤه أدلة على أن حمض اللبنيك لديه القدرة على العمل كمنظم وراثي من خلال تعديل بقايا الليسين في الهيستون عبر عملية اللبنيك. في السنوات الأخيرة، كشفت الدراسات عن مشاركة اللبنيك في عمليات مثل تكوين الأورام، وفشل القلب، والإنتان، وأمراض أخرى. ومع ذلك، لم يتم الإبلاغ عن دراسات حول تعديل اللبنيك في الهيستون في سرطان البنكرياس حتى الآن. في دراستنا الحالية، لاحظنا تراكم حمض اللبنيك في البيئة الدقيقة للورم في سرطان البنكرياس، والذي عمل كركائز لتعديل الهيستون باللبنيك. لقد كشفنا أن
يمكن أن تؤدي اللاكتيلation، وخاصة H3K18la، إلى حدوث الأورام وتقدمها في PDAC في المختبر وفي الجسم الحي للمرة الأولى. علاوة على ذلك، أظهرت المستويات العالية من اللاكتيلation ارتباطًا قويًا بالنتائج السريرية السيئة. تشير جميع هذه النتائج إلى أن اللاكتيلation تشارك في تطور PDAC. ومع ذلك، لم نتمكن من إثبات العلاقة المباشرة بين H3K18la وتطور PDAC، نظرًا لأن الطبيعة المعقدة والديناميكية لتعديلات الهيستون تجعلها أقل عرضة للتأثر بالطفرات النقطية في قاعدة واحدة. هذه هي قيود شائعة في الدراسات التي تهدف إلى إثبات الارتباط المباشر لتعديلات الهيستون (بما في ذلك اللاكتيلation، والأسيتيلation، والفوسفو-تعديل) مع الأمراض. بالإضافة إلى ذلك، فإن آثار اللاكتيلation في بروتينات هيستون أو غير هيستون الأخرى مثيرة للاهتمام أيضًا.
‘كاتب’ و’ممحاة’ يلعبان أدوارًا تنظيمية حاسمة في التعديلات الجينية. الكاتب هو إنزيم مسؤول عن إضافة أو إدخال تعديل محدد لجزيء الركيزة، بينما الممحاة هي إنزيم مسؤول عن إزالة أو عكس تعديل ما بعد الترجمة المحدد من جزيء الركيزة. حاليًا، تم الإشارة إلى أن الأسيتيل ترانسفيراز P300 [20] وGCN5 [33] يمكن أن يعملوا ككتّاب لكتلة الهيستون. بينما تعمل مزيلات الأسيتيل HDAC1-3 [36] جنبًا إلى جنب مع SIRT3 [37] كممحاة للكتلة. هنا، من خلال استخدام المثبطات وتجارب الوظيفة، حددنا P300 وHDAC2 ككاتب وممحاة محتملين على التوالي المعنيين في تنظيم كتلة الهيستون في خلايا PDAC. من خلال إسكاة أو تثبيط P300 لتعطيل كتلة الهيستون، تمكنا من تثبيط تكاثر الخلايا وقدرتها على الهجرة في خلايا PDAC في بيئة غنية باللاكتات. ومن الملاحظ أن تدخل اللاكتات عزز من قدرات التكاثر والهجرة في خلايا AsPC-1، ولكن ليس في خلايا MIA PaCa-2. قد تنشأ هذه الفجوة من الإنتاج الأعلى للاكتات في خلايا MIA PaCa-2 مقارنة بخلايا AsPC-1 [38]، وهو ما يكفي لتعزيز التكاثر والهجرة. سؤال آخر يجب ملاحظته هو أنه عند المقارنة مع خفض P300 وحده، بدا أن إضافة NaLa مع خفض P300 زادت من كتلة الهيستون. وذلك لأننا قمنا فقط بخفض التعبير بدلاً من الإزالة الكاملة. عند المستوى المرتفع للغاية من NaLa العلوي، حتى الكاتب (P300) كان مقيدًا بشكل كبير، قد يتم أيضًا تنظيم الكتلة (كتلة الهيستون) في الأسفل. أيضًا، قد لا يكون P300 هو الكاتب الوحيد لكتلة الهيستون، وقد يكون هناك كتّاب آخرون يشاركون في العملية. بالإضافة إلى ذلك، نظرًا لأن P300 يعمل أيضًا ككتّاب بروتين لتعديلات هيستون متنوعة بما في ذلك الأسيتيل، الكروتونيل، و
تم التحقق من أن زيادة كتلة الهيستون في مناطق المحفز تؤدي إلى تحفيز التعبير عن الجينات المستهدفة، بما في ذلك أرجيناز 1 (Arg1)، وبروتين قارئ N6-methyladenosine YTHDF2 في أنواع خلايا مختلفة [13، 20]. في الدراسة الحالية، كشفنا عن H3K18la الذي ينشط
أكد المزيد والمزيد من الباحثين أن إعادة توصيل الأيض وإعادة تشكيل الجينات مرتبطان ارتباطًا وثيقًا وينظمان بعضهما البعض بشكل متبادل [44]. في سرطان الخلايا الكلوية الواضحة، يؤدي عدم نشاط فون هيبل-لينداو إلى H3K18la، الذي ينشط محفز مستقبل عامل النمو المشتق من الصفائح الدموية
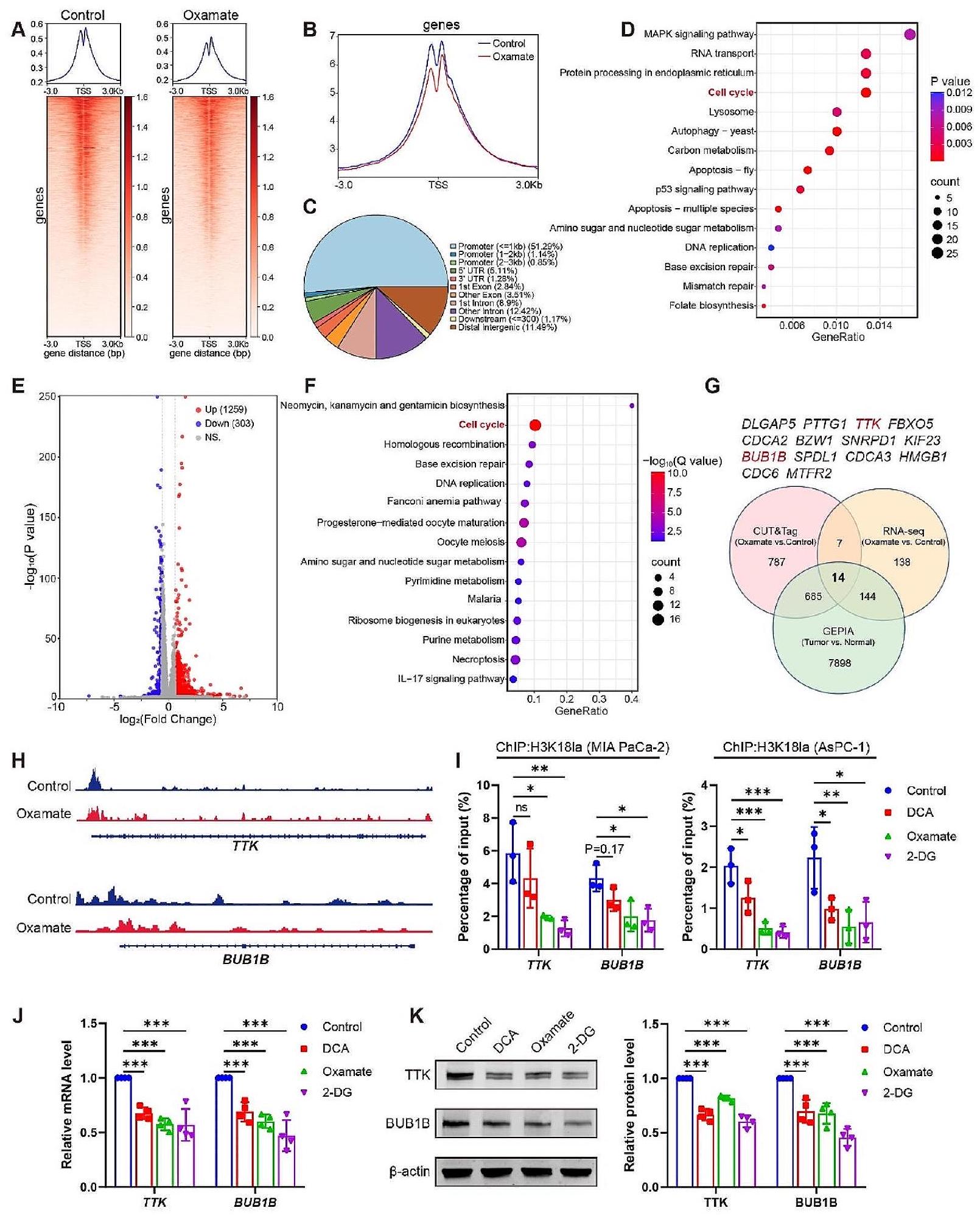
الشكل 7 H3K18la ينشط التعبير عن TTK وBUB1B في PDAC. (A-H) تم معالجة خلايا MIA PaCa-2 بمثبط تحلل سكري Oxamate (
حلقة تغذية راجعة إيجابية مسرطنة [45]. تظهر خلايا الميكروغليا تراكمًا كبيرًا لكتلة الهيستون H4 ليسين 12 في عينات الدماغ من مرضى الزهايمر، والتي تستهدف مناطق المحفز للجينات المرتبطة بالتحلل السكري، مما يؤدي إلى تحفيز التعبير عنها وزيادة إنتاج اللاكتات. هذا يؤسس حلقة تغذية راجعة إيجابية تشمل الأيض-الجينات-الأيض، مما يؤدي إلى تفاقم اختلال الأيض الميكروغلي والضعف الوظيفي [46]. ومع ذلك، لا يزال هناك نقص في الفهم الشامل لإعادة برمجة الأيض وكتلة الهيستون في PDAC. في هذه الدراسة، وجدنا أن خفض التعبير عن
نشاط LDH وإنتاج اللاكتات، مما يقلل من مستويات Pan Kla و H3K18la. وقد تم الإبلاغ عن أن فسفرة LDHA تؤثر على نشاط LDHA، الذي يتم تعديله بشكل رئيسي بواسطة Y10 [48-50]. ومع ذلك، في نظامنا، لاحظنا للمرة الأولى أن
الخاتمة
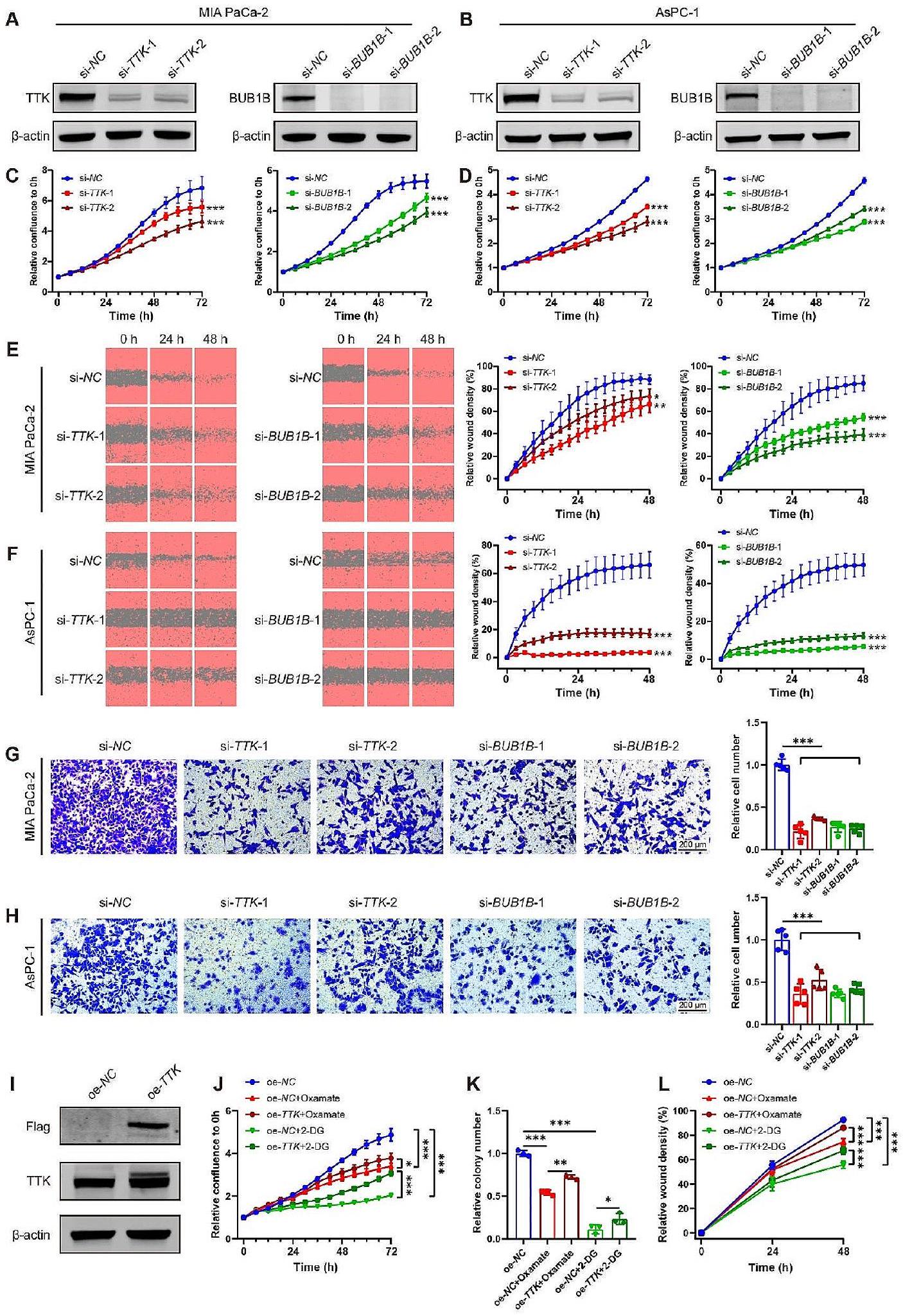
(انظر الشكل في الصفحة السابقة.)
الشكل 10 حلقة تغذية راجعة إيجابية بين جينات الهدف H3K18la (TTK و BUB1B) والجليكوليز. (A-B) تم نقل خلايا MIA PaCa-2 (A) و AsPC-1 (B) باستخدام siRNA للتحكم السلبي (si-NC) و siRNA لـ TTK (si-TTK) و/أو siRNA لـ BUB1B (si-BUB1B). تم الكشف عن مستويات البروتين بواسطة تقنية الويسترن بلوت وتم قياسها.
معلومات إضافية
المادة التكميلية 1
المادة التكميلية 3
المادة التكميلية 4
شكر وتقدير
مساهمات المؤلفين
تمويل
توفر البيانات
الإعلانات
موافقة الأخلاقيات والموافقة على المشاركة
موافقة على النشر
المصالح المتنافسة
تفاصيل المؤلف
تم النشر على الإنترنت: 06 مايو 2024
References
- Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73:17-48.
- Grossberg AJ, Chu LC, Deig CR, Fishman EK, Hwang WL, Maitra A, et al. Multidisciplinary standards of care and recent progress in pancreatic ductal adenocarcinoma. CA Cancer J Clin. 2020;70:375-403.
- Halbrook CJ, Lyssiotis CA, Pasca di Magliano M, Maitra A. Pancreatic cancer: advances and challenges. Cell. 2023;186:1729-54.
- Dreyer SB, Upstill-Goddard R, Legrini A, Biankin AV, Glasgow Precision Oncology L, Jamieson NB, et al. Genomic and molecular analyses identify molecular subtypes of pancreatic cancer recurrence. Gastroenterology. 2022;162:320-4. e4.
- Yang J, Ren B, Yang G, Wang H, Chen G, You L, et al. The enhancement of glycolysis regulates pancreatic cancer metastasis. Cell Mol Life Sci. 2020;77:305-21.
- El Kaoutari A, Fraunhoffer NA, Audebert S, Camoin L, Berthois Y, Gayet O et al. Pancreatic ductal adenocarcinoma ubiquitination profiling reveals specific prognostic and theranostic markers. EBioMedicine.2023;92:104634.
- Qin C, Yang G, Yang J, Ren B, Wang H, Chen G, et al. Metabolism of pancreatic cancer: paving the way to better anticancer strategies. Mol Cancer. 2020;19:50.
- Pan C, Li B, Simon MC. Moonlighting functions of metabolic enzymes and metabolites in cancer. Mol Cell. 2021;81:3760-74.
- Cao L, Wu J, Qu X, Sheng J, Cui M, Liu S, et al. Glycometabolic rearrange-ments-aerobic glycolysis in pancreatic cancer: causes, characteristics and clinical applications. J Exp Clin Cancer Res. 2020;39:267.
- Liu M, Quek LE, Sultani G, Turner N. Epithelial-mesenchymal transition induction is associated with augmented glucose uptake and lactate production in pancreatic ductal adenocarcinoma. Cancer Metab. 2016;4:19.
- Lin J, Liu G, Chen L, Kwok HF, Lin Y. Targeting lactate-related cell cycle activities for cancer therapy. Semin Cancer Biol. 2022;86:1231-43.
- Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381-95.
- Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574:575-80.
- Dai X, Lv X, Thompson EW, Ostrikov KK. Histone lactylation: epigenetic mark of glycolytic switch. Trends Genet. 2022;38:124-7.
- Liberti MV, Locasale JW. Histone lactylation: a new role for glucose metabolism. Trends Biochem Sci. 2020;45:179-82.
- Itoh Y, Esaki T, Shimoji K, Cook M, Law MJ, Kaufman E, et al. Dichloroacetate effects on glucose and lactate oxidation by neurons and astroglia in vitro and on glucose utilization by brain in vivo. Proc Natl Acad Sci U SA. 2003;100:4879-84.
- Zhao Z, Han F, Yang S, Wu J, Zhan W. Oxamate-mediated inhibition of lactate dehydrogenase induces protective autophagy in gastric cancer cells: involvement of the Akt-mTOR signaling pathway. Cancer Lett. 2015;358:17-26.
- Zhang D, Li J, Wang F, Hu J, Wang S, Sun Y. 2-Deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 2014;355:176-83.
- Xie B, Lin J, Chen X, Zhou X, Zhang Y, Fan M, et al. CircXRN2 suppresses tumor progression driven by histone lactylation through activating the Hippo pathway in human bladder cancer. Mol Cancer. 2023;22:151.
- Yu J, Chai P, Xie M, Ge S, Ruan J, Fan X, et al. Histone lactylation drives oncogenesis by facilitating m(6)a reader protein YTHDF2 expression in ocular melanoma. Genome Biol. 2021;22:85.
- Dominguez-Brauer C, Thu KL, Mason JM, Blaser H, Bray MR, Mak TW. Targeting mitosis in cancer: emerging strategies. Mol Cell. 2015;60:524-36.
- Kaistha BP, Honstein T, Müller V, Bielak S, Sauer M, Kreider R, et al. Key role of dual specificity kinase TTK in proliferation and survival of pancreatic cancer cells. Br J Cancer. 2014;111:1780-7.
- Grützmann R, Pilarsky C, Ammerpohl O, Lüttges J, Böhme A, Sipos B, et al. Gene expression profiling of microdissected pancreatic ductal carcinomas using high-density DNA microarrays. Neoplasia. 2004;6:611-22.
- Zhou X, Yuan Y, Kuang H, Tang B, Zhang H, Zhang M. BUB1B (BUB1 mitotic checkpoint serine/threonine kinase
) promotes lung adenocarcinoma by interacting with zinc finger protein ZNF143 and regulating glycolysis. Bioengineered. 2022;13:2471-85. - Comandatore A, Franczak M, Smolenski RT, Morelli L, Peters GJ, Giovannetti E. Lactate dehydrogenase and its clinical significance in pancreatic and thoracic cancers. Semin Cancer Bio. 2022;86:93-100.
- Musacchio A. The molecular biology of spindle assembly checkpoint signaling dynamics. Curr Biol. 2015;25:R1002-18.
- Pavlova NN, Zhu J, Thompson CB. The hallmarks of cancer metabolism: still emerging. Cell Metab. 2022;34:355-77.
- Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325-37.
- Jiang SH, Li J, Dong FY, Yang JY, Liu DJ, Yang XM et al. Increased serotonin signaling contributes to the Warburg effect in pancreatic tumor cells under metabolic stress and promotes growth of pancreatic tumors in mice. Gastroenterology. 2017;153:277-91.e19.
- Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG et al. Lactate metabolism in human lung tumors. Cell. 2017;171:358-71.e9.
- Zhang N, Zhang Y, Xu J, Wang P, Wu B, Lu S, et al. Alpha-myosin heavy chain lactylation maintains sarcomeric structure and function and alleviates the development of heart failure. Cell Res. 2023;33:679-98.
- Yang K, Fan M, Wang X, Xu J, Wang Y, Tu F, et al. Lactate promotes macrophage HMGB1 lactylation, acetylation, and exosomal release in polymicrobial sepsis. Cell Death Differ. 2022;29:133-46.
- Wang N, Wang W, Wang X, Mang G, Chen J, Yan X, et al. Histone lactylation boosts reparative gene activation post-myocardial infarction. Circul Res. 2022;131:893-908.
- Compton SE, Kitchen-Goosen SM, DeCamp LM, Lau KH, Mabvakure B, Vos M et al. LKB1 controls inflammatory potential through CRTC2-dependent histone acetylation. Mol Cell. 2023;83:1872-86.e5.
- Zhao Y, Li S, Chen Y, Wang Y, Wei Y, Zhou T, et al. Histone phosphorylation integrates the hepatic glucagon-PKA-CREB gluconeogenesis program in response to fasting. Mol Cell. 2023;83:1093-108.e8.
- Moreno-Yruela C, Zhang D, Wei W, Baek M, Liu W, Gao J, et al. Class I histone deacetylases (HDAC1-3) are histone lysine delactylases. Sci Adv. 2022;8:eabi6696.
- Mao Y, Zhang J, Zhou Q, He X, Zheng Z, Wei Y, et al. Hypoxia induces mitochondrial protein lactylation to limit oxidative phosphorylation. Cell Res. 2024;34:13-30.
- Daemen A, Peterson D, Sahu N, McCord R, Du XN, Liu BN, et al. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc Natl Acad Sci USA. 2015;112:E4410-7.
- Chen Q, Yang B, Liu X, Zhang XD, Zhang L, Liu T. Histone acetyltransferases CBP/p300 in tumorigenesis and CBP/p300 inhibitors as promising novel anticancer agents. Theranostics. 2022;12:4935-48.
- Huang H, Zhang D, Weng Y, Delaney K, Tang Z, Yan C et al. The regulatory enzymes and protein substrates for the lysine beta-hydroxybutyrylation pathway. Sci Adv. 2021;7:eabe2771.
- Sabari BR, Tang Z, Huang H, Yong-Gonzalez V, Molina H, Kong HE, et al. Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Mol Cell. 2015;58:203-15.
- Yang Z, Yan C, Ma J, Peng P, Ren X, Cai S, et al. Lactylome analysis suggests lactylation-dependent mechanisms of metabolic adaptation in hepatocellular carcinoma. Nat Metab. 2023;5:61-79.
- Dong S, Huang F, Zhang H, Chen Q. Overexpression of BUB1B, CCNA2, CDC20, and CDK1 in tumor tissues predicts poor survival in pancreatic ductal adenocarcinoma. Biosci Rep. 2019;39:BSR20182306.
- Sun L, Zhang H, Gao P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell. 2022;13:877-919.
- Yang J, Luo L, Zhao C, Li X, Wang Z, Zeng Z, et al. A positive feedback loop between inactive VHL-triggered histone lactylation and PDGFRbeta signaling drives clear cell renal cell carcinoma progression. Int J Biol Sci. 2022;18:3470-83.
- Pan RY, He L, Zhang J, Liu X, Liao Y, Gao J, et al. Positive feedback regulation of microglial glucose metabolism by histone H 4 lysine 12 lactylation in Alzheimer’s disease. Cell Metab. 2022;34:634-48.e6.
- Kes MMG, Van den Bossche J, Griffioen AW, Huijbers EJM. Oncometabolites lactate and succinate drive pro-angiogenic macrophage response in tumors. Biochim Biophys Acta Rev Cancer. 2020;1874:188427.
- Xue W, Li X, Li W, Wang Y, Jiang C, Zhou L, et al. Intracellular CYTL1, a novel tumor suppressor, stabilizes NDUFV1 to inhibit metabolic reprogramming in breast cancer. Signal Transduct Target Ther. 2022;7:35.
- Li J, Zhang Q, Guan Y, Liao D, Jiang D, Xiong H, et al. Circular RNA circVAMP3 promotes aerobic glycolysis and proliferation by regulating LDHA in renal cell carcinoma. Cell Death Dis. 2022;13:443.
- Li S, Gao J, Zhuang X, Zhao C, Hou X, Xing X, et al. Cyclin G2 inhibits the Warburg effect and tumour progression by suppressing LDHA phosphorylation in glioma. Int J Biol Sci. 2019;15:544-55.
ملاحظة الناشر
ساهم في العمل كل من في لي، وينزهي سي ولي شيا بالتساوي.
*المراسلة:
تيانبي هونغ
tpho66@bjmu.edu.cn
روي وي
weirui@bjmu.edu.cn
قائمة كاملة بمعلومات المؤلفين متاحة في نهاية المقال
DOI: https://doi.org/10.1186/s12943-024-02008-9
PMID: https://pubmed.ncbi.nlm.nih.gov/38711083
Publication Date: 2024-05-06
Positive feedback regulation between glycolysis and histone lactylation drives oncogenesis in pancreatic ductal adenocarcinoma
Abstract
Background Metabolic reprogramming and epigenetic alterations contribute to the aggressiveness of pancreatic ductal adenocarcinoma (PDAC). Lactate-dependent histone modification is a new type of histone mark, which links glycolysis metabolite to the epigenetic process of lactylation. However, the role of histone lactylation in PDAC remains unclear. Methods The level of histone lactylation in PDAC was identified by western blot and immunohistochemistry, and its relationship with the overall survival was evaluated using a Kaplan-Meier survival plot. The participation of histone lactylation in the growth and progression of PDAC was confirmed through inhibition of histone lactylation by glycolysis inhibitors or lactate dehydrogenase A (LDHA) knockdown both in vitro and in vivo. The potential writers and erasers of histone lactylation in PDAC were identified by western blot and functional experiments. The potential target genes of H3K18 lactylation (H3K18la) were screened by CUT&Tag and RNA-seq analyses. The candidate target genes TTK protein kinase (TTK) and BUB1 mitotic checkpoint serine/threonine kinase B (BUB1B) were validated through ChIP-qPCR, RT-qPCR and western blot analyses. Next, the effects of these two genes in PDAC were confirmed by knockdown or overexpression. The interaction between TTK and LDHA was identified by Co-IP assay. Results Histone lactylation, especially H3K18la level was elevated in PDAC, and the high level of H3K18la was associated with poor prognosis. The suppression of glycolytic activity by different kinds of inhibitors or LDHA knockdown contributed to the anti-tumor effects of PDAC in vitro and in vivo. E1A binding protein p300 (P300) and histone deacetylase 2 were the potential writer and eraser of histone lactylation in PDAC cells, respectively. H3K18la was enriched at the promoters and activated the transcription of mitotic checkpoint regulators TTK and BUB1B. Interestingly, TTK and BUB1B could elevate the expression of P300 which in turn increased glycolysis. Moreover, TTK
Abstract
phosphorylated LDHA at tyrosine 239 (Y239) and activated LDHA, and subsequently upregulated lactate and H3K18la levels. Conclusions The glycolysis-H3K18la-TTK/BUB1B positive feedback loop exacerbates dysfunction in PDAC. These findings delivered a new exploration and significant inter-relationship between lactate metabolic reprogramming and epigenetic regulation, which might pave the way toward novel lactylation treatment strategies in PDAC therapy.
Introduction
metabolism [15]. However, strategies involving histone lactylation treatment to overcome and shift molecular landscape of PDAC have not been achieved. Thus, a better understanding of histone lactylation in the pathology of PDAC still requires further investigation.
Our study aims to investigate the role of histone lactylation, specifically H3K18la, in PDAC progression and to clarify the potential mechanism. Our results demonstrated that lactate accumulation in the PDAC tumor microenvironment drove histone lactylation, both in vivo and in vitro, correlating strongly with tumorigenesis and poor clinical outcomes. Mechanistically, enhanced glycolysis in PDAC led to increased lactate production and H3K18la, which promoted the transcription of TTK protein kinase (
Materials and methods
Human samples
Non-targeted metabolomics assay
Cell culture and treatment
metabolism by inhibiting hexokinase and glucose-6-phosphate isomerase [18] or the vehicle control (phosphate buffer saline, PBS). (2) Cells were transfected with LDHA siRNA or scramble siRNA as negative control, combined with sodium lactate (NaLa,
RNA interference and overexpression
Lentivirus transfection
expressing sh-LDHA and oe-TTK were prepared by Hanbio Tech (Shanghai, China). MIA PaCa-2 cells at a confluence of
Cell proliferation assay
Colony formation assay
Wound healing assay
the wound areas were quantified using Image J software (National Institutes of Health, Bethesda, MD, USA).
Transwell migration assay
Transplanted tumor model in nude mice
IHC
sections were incubated overnight at
RNA extraction and RT-qPCR
Protein extraction and western blot
Lactate content and LDH enzyme activity measurement
assay kit (Solarbio, Beijing, China). The absorbance values at 570 nm were measured using a spectrophotometer. The content of lactate was normalized based on the cell number.
Co-IP
CUT&Tag assay
perform Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses.
RNA-seq
ChIP assay and ChIP-qPCR
Statistical analysis
Results
Elevated histone lactylation levels are associated with unfavorable prognosis in patients with PDAC
Since the production of large amounts of lactate supplied as substrates for histone lactylation, we examined the protein lactylation levels in 5 pairs PDAC tissues and surrounding noncancerous tissues. Higher levels of global/pan-lysine lactylation (Pan Kla) were observed in PDAC tissues (Fig. 1E). As histone modifications play fundamental roles in most biological processes and H3K18la has been reported to regulate multiple biological processes such as oncogenesis [12,19], we focused on
Abstract
Fig. 1 Elevated lactate level and histone lactylation are associated with unfavorable prognosis in patients with PDAC. (A) Lactate content in pancreatic ductal adenocarcinoma (PDAC) and paired para-carcinoma tissues.
Glycolysis inhibition diminishes histone lactylation and inhibits PDAC cell proliferation and migration
attenuated the proliferation inhibition effect of
To explore whether lactylation could affect PDAC cell migration, wound healing and transwell assays were performed. Treatments with DCA, Oxamate and 2-DG significantly decreased cell migration in MIA PaCa-2 and AsPC-1 cells, and the higher dose showed more pronounced inhibition effects on the cell migration than the lower dose in the wound healing assay (Fig. 4A-D). Similarly, LDHA knockdown inhibited migration capacities compared to the vector group, and Nala addition attenuated the suppressive effect of si-LDHA (Fig. 4I-L). Transwell assays also showed that DCA, Oxamate and 2-DG treated MIA PaCa-2 or AsPC-1 cells significantly decreased cell migration (Fig. 4E-H). The same outcomes were observed in LDHA knockdown, and Nala counteracted the migratory suppression effect of si-LDHA in both cell lines (Fig. 4M-P). Collectively, these findings suggest that histone lactylation plays a crucial role in the initiation and progression of PDAC, while inhibiting histone lactylation may exhibit potential antitumor activity against PDAC.
Glycolysis inhibition diminishes histone lactylation and suppresses PDAC progression in the PDAC xenograft mouse model
Abstract
Fig. 4 Glycolysis inhibition diminishes histone lactylation and suppresses PDAC cell migration. (A-H) Two PDAC cell lines MIA PaCa-2 and AsPC-1 cells were treated with glycolysis inhibitors DCA (
P300 and HDAC2 are potential writer and eraser of histone lactylation in PDAC cells respectively
or C646 treatment could reverse the effect of NaLa on PDAC migration. As expected, cells transfected with si-P300 or treatment with C646 in combination with NaLa exhibited reduced migration ability compared to those solely treated with NaLa in MIA PaCa-2 (Fig. 6G and H, S1G-H) and AsPC-1 cells (Fig. 6I and J, S1I-J). These results suggest that P300, the potential writer of histone lactylation, promotes progression of PDAC.
To identify the potential eraser of histone lactylation in PDAC, various types of histone deacetylase (HDAC) inhibitors were employed in MIA PaCa-2 and AsPC-1 cells. Intriguingly, treatment with broad-spectrum HDAC inhibitors TSA, rather than sirtuin inhibitor NAM, resulted in an increased global level of histone lactylation. Additionally, the class I HDAC inhibitor CI994 enhanced lactylation, while class IIa HDAC inhibitor TMP195, class IIb HDAC inhibitor Bufexamac, and class IV HDAC inhibitor SIS17 had no such effect (Fig. S2A-B). To further validate the role of each HDAC isozyme, overexpression experiments were conducted in MIA PaCa-2 and AsPC-1 cells. Results demonstrated that overexpression of HDAC2 significantly decreased the lactylation level, particularly H3K18la, while HDAC1 and HDAC3 overexpression did not affect the levels of Pan Kla and H3K18la (Fig. S2C-H). These findings suggest that P300 may act as a potential writer while HDAC2 functions as an eraser for histone lactylation in PDAC.
H3K18la activates TTK and BUB1B transcription in PDAC
Additionally, we conducted RNA-seq assay to screen the potential target genes regulated by lactylation (the original data is in Additional file 4). When compared with the control group, Oxamate treatment upregulated
The high levels of TTK and BUB1B are associated with the malignancy of PDAC
high TTK or BUB1B expression had shorter overall survival time or disease-free survival time than those with lower expression levels (Fig. 8D-G).
TTK and BUB1B deletion suppresses PDAC malignancy while TTK overexpression partially prevents the anticancer effects of lactylation inhibition
To investigate the effect of TTK in H3K18la-mediated PDAC progression, MIA PaCa-2 cells were overexpressed with TTK (Fig. 9I) and treated with glycolysis inhibitors. Compared with negative control,
A positive feedback loop between H3K18la target genes and glycolysis
Fig. 5 Glycolysis inhibition diminishes histone lactylation and suppresses cancer progression in the transplanted PDAC in nude mice. (A-H) MIA PaCa-2 cells were injected into nude mice subcutaneously and were intraperitoneally administered Oxamate (
also TTK/BUB1B played positive feedback in histone lactylation, which further exacerbate the malignancy.
Discussion
eraser of histone lactylation in PDAC cells. Furthermore, H3K18la stimulated
PDAC is one of the most lethal types of cancer and a significant challenge in the field of cancer medicine. Metabolic disorders are risk factors for PDAC progression. Reprogramming cellular metabolism to support proliferation and various cellular processes is considered a key hallmark of cancer [27]. Glycolysis plays a central role in the metabolic reprogramming of PDAC cells and supports malignant behaviors. Aerobic glycolysis, commonly recognized as the Warburg effect, is characterized by the preferential generation of energy through glycolysis instead of oxidative phosphorylation, even in the presence of adequate oxygen levels, which results in substantial production of lactate [28]. This phenomenon emerges as a hallmark of tumor cells, particularly in PDAC. Therefore, targeting the Warburg effect holds paramount importance in exploring novel therapeutic strategies against PDAC [29]. Lactate was previously regarded solely as a byproduct of glycolytic metabolism until Faubert et al. discovered that it could be reused as the primary carbon source for the mitochondrial tricarboxylic acid cycle [30]. Zhang et al., on their part, provided evidence that lactate has the potential to act as an epigenetic regulator by modifying histone lysine residues through lactylation [13]. In recent years, studies have revealed the participation of lactylation in processes such as tumorigenesis [19, 20], heart failure [31], sepsis [32] and other diseases. However, studies on histone lactylation modification in PDAC have not been reported yet till now. In our present study, we observed lactate accumulation in the tumor microenvironment of PDAC, which served as substrates for histone lactylation. We have revealed that
histone lactylation, particularly H3K18la, could drive tumorigenesis and progression in PDAC in vitro and in vivo for the first time. Furthermore, the high level of lactylation showed a strong correlation with poor clinical outcomes. All these results suggest that lactylation participates in the development of PDAC. However, we could not prove the direct connection between H3K18la and PDAC development, owing that the complex and dynamic nature of histone modifications makes them less likely to be affected by point mutations in a single base. This is a common limitation in studies aimed at demonstrating the direct association of histone modifications (including lactylation, acetylation and phosphorylation) with diseases [19, 20, 33-35]. Besides, the effects of lactylation in other histone or non-histone proteins are also interesting.
‘Writer’ and ‘eraser’ play crucial regulatory roles in epigenetic modifications. A writer is an enzyme responsible for adding or introducing a specific modification to a substrate molecule, while an eraser is an enzyme responsible for removing or reversing a specific post-translational modification from a substrate molecule. Currently, it has been noted that acetyltransferase P300 [20] and GCN5 [33] could act as histone lactylation writers. while deacetylase HDAC1-3 [36] along with SIRT3 [37] serve as lactylation erasers. Here, by using inhibitors and function experiments, we identified P300 and HDAC2 as the potential writer and eraser respectively involved in regulating histone lactylation in PDAC cells. By silencing or inhibiting P300 to disrupt histone lactylation, we were able to inhibit cell proliferation and migration ability in PDAC cells in a high lactate environment. Notably, lactate intervention promoted proliferation and migration abilities in AsPC-1 cells, but not in MIA PaCa-2 cells. This discrepancy may arise from the higher lactate production in MIA PaCa-2 cells compared to AsPC-1 cells [38], which is sufficient to promote proliferation and migration. Another question that needs to be noted is that when compared with P300 knockdown alone, NaLa supplementation together with P300 knockdown seemed to increase histone lactylation. This is because we only did the knockdown rather than the knockout. Upon the extremely high level of the upstream NaLa, even the writer (P300) was largely limited, the downstream (histone lactylation) might also be upregulated. Also, P300 might not be the sole writer of histone lactylation, and there might be other writers participating in the process. Besides, since P300 also acts as a writer protein for diverse histone acylations including acetylation, crotonylation, and
Increased histone lactylation in the promoter regions has been verified to induce the expression of target genes, including Arginase 1 (Arg1), N6-methyladenosinereader protein YTHDF2 in different cell types [13, 20]. In the present study, we revealed H3K18la activated
More and more researchers have confirmed that metabolic rewiring and epigenetic remodeling are closely linked and reciprocally regulate each other [44]. In clear cell renal cell carcinoma, inactive von Hippel-Lindau leads to H3K18la, which activates the promoter of platelet-derived growth factor receptor
Fig. 7 H3K18la activates TTK and BUB1B transcription in PDAC. (A-H) MIA PaCa-2 cells were treated with a glycolysis inhibitor Oxamate (
an oncogenic positive feedback loop [45]. Microglial cells exhibit significant enrichment of histone H4 lysine 12 lactylation in brain specimens from patients with Alzheimer’s disease, which target the promoter regions of genes associated with glycolysis, thereby inducing their transcription and promoting lactate production. This establishes a positive feedback loop involving metabo-lism-epigenetics-metabolism, which exacerbates microglial metabolic dysregulation and functional impairments [46]. However, a comprehensive understanding of metabolic reprogramming and histone lactylation in PDAC is still lacking. In this study, we found the knockdown of
activity of LDH and the lactate production, thus decreasing the levels of Pan Kla and H3K18la. The phosphorylation of LDHA has been reported to influence LDHA activity, which is mainly modulated by Y10 [48-50]. However, in our system, we observed for the first time that
Conclusion
(See figure on previous page.)
Fig. 10 A positive feedback loop between H3K18la target genes (TTK and BUB1B) and glycolysis. (A-B) MIA PaCa-2 (A) and AsPC-1 (B) cells were transfected with negative control siRNA (si-NC), TTK siRNA (si-TTK) and/or BUB1B siRNA (si-BUB1B). The protein levels were detected by western blot and quantified.
Supplementary Information
Supplementary Material 1
Supplementary Material 3
Supplementary Material 4
Acknowledgements
Author contributions
Funding
Data availability
Declarations
Ethics approval and consent to participate
Consent for publication
Competing interests
Author details
Published online: 06 May 2024
References
- Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73:17-48.
- Grossberg AJ, Chu LC, Deig CR, Fishman EK, Hwang WL, Maitra A, et al. Multidisciplinary standards of care and recent progress in pancreatic ductal adenocarcinoma. CA Cancer J Clin. 2020;70:375-403.
- Halbrook CJ, Lyssiotis CA, Pasca di Magliano M, Maitra A. Pancreatic cancer: advances and challenges. Cell. 2023;186:1729-54.
- Dreyer SB, Upstill-Goddard R, Legrini A, Biankin AV, Glasgow Precision Oncology L, Jamieson NB, et al. Genomic and molecular analyses identify molecular subtypes of pancreatic cancer recurrence. Gastroenterology. 2022;162:320-4. e4.
- Yang J, Ren B, Yang G, Wang H, Chen G, You L, et al. The enhancement of glycolysis regulates pancreatic cancer metastasis. Cell Mol Life Sci. 2020;77:305-21.
- El Kaoutari A, Fraunhoffer NA, Audebert S, Camoin L, Berthois Y, Gayet O et al. Pancreatic ductal adenocarcinoma ubiquitination profiling reveals specific prognostic and theranostic markers. EBioMedicine.2023;92:104634.
- Qin C, Yang G, Yang J, Ren B, Wang H, Chen G, et al. Metabolism of pancreatic cancer: paving the way to better anticancer strategies. Mol Cancer. 2020;19:50.
- Pan C, Li B, Simon MC. Moonlighting functions of metabolic enzymes and metabolites in cancer. Mol Cell. 2021;81:3760-74.
- Cao L, Wu J, Qu X, Sheng J, Cui M, Liu S, et al. Glycometabolic rearrange-ments-aerobic glycolysis in pancreatic cancer: causes, characteristics and clinical applications. J Exp Clin Cancer Res. 2020;39:267.
- Liu M, Quek LE, Sultani G, Turner N. Epithelial-mesenchymal transition induction is associated with augmented glucose uptake and lactate production in pancreatic ductal adenocarcinoma. Cancer Metab. 2016;4:19.
- Lin J, Liu G, Chen L, Kwok HF, Lin Y. Targeting lactate-related cell cycle activities for cancer therapy. Semin Cancer Biol. 2022;86:1231-43.
- Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381-95.
- Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574:575-80.
- Dai X, Lv X, Thompson EW, Ostrikov KK. Histone lactylation: epigenetic mark of glycolytic switch. Trends Genet. 2022;38:124-7.
- Liberti MV, Locasale JW. Histone lactylation: a new role for glucose metabolism. Trends Biochem Sci. 2020;45:179-82.
- Itoh Y, Esaki T, Shimoji K, Cook M, Law MJ, Kaufman E, et al. Dichloroacetate effects on glucose and lactate oxidation by neurons and astroglia in vitro and on glucose utilization by brain in vivo. Proc Natl Acad Sci U SA. 2003;100:4879-84.
- Zhao Z, Han F, Yang S, Wu J, Zhan W. Oxamate-mediated inhibition of lactate dehydrogenase induces protective autophagy in gastric cancer cells: involvement of the Akt-mTOR signaling pathway. Cancer Lett. 2015;358:17-26.
- Zhang D, Li J, Wang F, Hu J, Wang S, Sun Y. 2-Deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 2014;355:176-83.
- Xie B, Lin J, Chen X, Zhou X, Zhang Y, Fan M, et al. CircXRN2 suppresses tumor progression driven by histone lactylation through activating the Hippo pathway in human bladder cancer. Mol Cancer. 2023;22:151.
- Yu J, Chai P, Xie M, Ge S, Ruan J, Fan X, et al. Histone lactylation drives oncogenesis by facilitating m(6)a reader protein YTHDF2 expression in ocular melanoma. Genome Biol. 2021;22:85.
- Dominguez-Brauer C, Thu KL, Mason JM, Blaser H, Bray MR, Mak TW. Targeting mitosis in cancer: emerging strategies. Mol Cell. 2015;60:524-36.
- Kaistha BP, Honstein T, Müller V, Bielak S, Sauer M, Kreider R, et al. Key role of dual specificity kinase TTK in proliferation and survival of pancreatic cancer cells. Br J Cancer. 2014;111:1780-7.
- Grützmann R, Pilarsky C, Ammerpohl O, Lüttges J, Böhme A, Sipos B, et al. Gene expression profiling of microdissected pancreatic ductal carcinomas using high-density DNA microarrays. Neoplasia. 2004;6:611-22.
- Zhou X, Yuan Y, Kuang H, Tang B, Zhang H, Zhang M. BUB1B (BUB1 mitotic checkpoint serine/threonine kinase
) promotes lung adenocarcinoma by interacting with zinc finger protein ZNF143 and regulating glycolysis. Bioengineered. 2022;13:2471-85. - Comandatore A, Franczak M, Smolenski RT, Morelli L, Peters GJ, Giovannetti E. Lactate dehydrogenase and its clinical significance in pancreatic and thoracic cancers. Semin Cancer Bio. 2022;86:93-100.
- Musacchio A. The molecular biology of spindle assembly checkpoint signaling dynamics. Curr Biol. 2015;25:R1002-18.
- Pavlova NN, Zhu J, Thompson CB. The hallmarks of cancer metabolism: still emerging. Cell Metab. 2022;34:355-77.
- Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325-37.
- Jiang SH, Li J, Dong FY, Yang JY, Liu DJ, Yang XM et al. Increased serotonin signaling contributes to the Warburg effect in pancreatic tumor cells under metabolic stress and promotes growth of pancreatic tumors in mice. Gastroenterology. 2017;153:277-91.e19.
- Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG et al. Lactate metabolism in human lung tumors. Cell. 2017;171:358-71.e9.
- Zhang N, Zhang Y, Xu J, Wang P, Wu B, Lu S, et al. Alpha-myosin heavy chain lactylation maintains sarcomeric structure and function and alleviates the development of heart failure. Cell Res. 2023;33:679-98.
- Yang K, Fan M, Wang X, Xu J, Wang Y, Tu F, et al. Lactate promotes macrophage HMGB1 lactylation, acetylation, and exosomal release in polymicrobial sepsis. Cell Death Differ. 2022;29:133-46.
- Wang N, Wang W, Wang X, Mang G, Chen J, Yan X, et al. Histone lactylation boosts reparative gene activation post-myocardial infarction. Circul Res. 2022;131:893-908.
- Compton SE, Kitchen-Goosen SM, DeCamp LM, Lau KH, Mabvakure B, Vos M et al. LKB1 controls inflammatory potential through CRTC2-dependent histone acetylation. Mol Cell. 2023;83:1872-86.e5.
- Zhao Y, Li S, Chen Y, Wang Y, Wei Y, Zhou T, et al. Histone phosphorylation integrates the hepatic glucagon-PKA-CREB gluconeogenesis program in response to fasting. Mol Cell. 2023;83:1093-108.e8.
- Moreno-Yruela C, Zhang D, Wei W, Baek M, Liu W, Gao J, et al. Class I histone deacetylases (HDAC1-3) are histone lysine delactylases. Sci Adv. 2022;8:eabi6696.
- Mao Y, Zhang J, Zhou Q, He X, Zheng Z, Wei Y, et al. Hypoxia induces mitochondrial protein lactylation to limit oxidative phosphorylation. Cell Res. 2024;34:13-30.
- Daemen A, Peterson D, Sahu N, McCord R, Du XN, Liu BN, et al. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc Natl Acad Sci USA. 2015;112:E4410-7.
- Chen Q, Yang B, Liu X, Zhang XD, Zhang L, Liu T. Histone acetyltransferases CBP/p300 in tumorigenesis and CBP/p300 inhibitors as promising novel anticancer agents. Theranostics. 2022;12:4935-48.
- Huang H, Zhang D, Weng Y, Delaney K, Tang Z, Yan C et al. The regulatory enzymes and protein substrates for the lysine beta-hydroxybutyrylation pathway. Sci Adv. 2021;7:eabe2771.
- Sabari BR, Tang Z, Huang H, Yong-Gonzalez V, Molina H, Kong HE, et al. Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Mol Cell. 2015;58:203-15.
- Yang Z, Yan C, Ma J, Peng P, Ren X, Cai S, et al. Lactylome analysis suggests lactylation-dependent mechanisms of metabolic adaptation in hepatocellular carcinoma. Nat Metab. 2023;5:61-79.
- Dong S, Huang F, Zhang H, Chen Q. Overexpression of BUB1B, CCNA2, CDC20, and CDK1 in tumor tissues predicts poor survival in pancreatic ductal adenocarcinoma. Biosci Rep. 2019;39:BSR20182306.
- Sun L, Zhang H, Gao P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell. 2022;13:877-919.
- Yang J, Luo L, Zhao C, Li X, Wang Z, Zeng Z, et al. A positive feedback loop between inactive VHL-triggered histone lactylation and PDGFRbeta signaling drives clear cell renal cell carcinoma progression. Int J Biol Sci. 2022;18:3470-83.
- Pan RY, He L, Zhang J, Liu X, Liao Y, Gao J, et al. Positive feedback regulation of microglial glucose metabolism by histone H 4 lysine 12 lactylation in Alzheimer’s disease. Cell Metab. 2022;34:634-48.e6.
- Kes MMG, Van den Bossche J, Griffioen AW, Huijbers EJM. Oncometabolites lactate and succinate drive pro-angiogenic macrophage response in tumors. Biochim Biophys Acta Rev Cancer. 2020;1874:188427.
- Xue W, Li X, Li W, Wang Y, Jiang C, Zhou L, et al. Intracellular CYTL1, a novel tumor suppressor, stabilizes NDUFV1 to inhibit metabolic reprogramming in breast cancer. Signal Transduct Target Ther. 2022;7:35.
- Li J, Zhang Q, Guan Y, Liao D, Jiang D, Xiong H, et al. Circular RNA circVAMP3 promotes aerobic glycolysis and proliferation by regulating LDHA in renal cell carcinoma. Cell Death Dis. 2022;13:443.
- Li S, Gao J, Zhuang X, Zhao C, Hou X, Xing X, et al. Cyclin G2 inhibits the Warburg effect and tumour progression by suppressing LDHA phosphorylation in glioma. Int J Biol Sci. 2019;15:544-55.
Publisher’s Note
Fei Li, Wenzhe Si and Li Xia contributed equally to this work.
*Correspondence:
Tianpei Hong
tpho66@bjmu.edu.cn
Rui Wei
weirui@bjmu.edu.cn
Full list of author information is available at the end of the article