تعديل تداخل الروابط بين الروثينيوم والأكسجين بدقة فيلتحقيق استقرار معزز في التحليل الكهربائي للماء باستخدام غشاء تبادل البروتون مطلوب بشدة. ومع ذلك، فإن المعادن الانتقالية مع-إلكترونات التكافؤ، التي تم إضافتها أو سبكها مع، تكون بطبيعتها عرضة لتأثير بيئة التنسيق، مما يجعل من الصعب تعديل التساهمية بين الروثينيوم والأكسجين بطريقة دقيقة ومستدامة. هنا، نستنتج أولاً أن إدخال اللانثانيد بتغيرات إلكترونية تدريجية يمكن أن يعدل التساهمية بين الروثينيوم والأكسجين بشكل مستمر بفضل تأثير التغطية لـالمدارات. تؤكد الحسابات النظرية أن متانةاتباع اتجاه بركاني كدالة لتشارك الروابط بين الروتينيوم والأكسجين. من بين مختلفيتم تحديده كأفضل محفز ويمتلك استقرارًا أعلى بمقدار 35.5 مرة من ذلك لـ. بشكل خاص، الـالجهاز القائم على يتطلب فقط 1.837 فولت للوصول إلىويظهر استقرارًا طويل الأمد عندلمدة 100 ساعة بمعدل تدهور بسيط.
يُعترف على نطاق واسع بأن التحليل الكهربائي للمياه المدفوع بالطاقة المتجددة هو طريق واعد ومستدام للإنتاج القابل للتوسع للهيدروجين الأخضر.تظهر تقنية التحليل الكهربائي للماء بغشاء تبادل البروتون (PEMWE) إمكانيات كبيرة نظرًا لتوافقها العالي مع التغيرات، ومقاومتها الكهربائية المنخفضة، وكثافة التيار العالية، وضغط التشغيل المنخفض، والتفاعلات الجانبية المحدودة.ومع ذلك، فإن التطبيق الواسع النطاق لتقنية PEMWE يعيقه نقص المحفزات الكهروكيميائية الفعالة والميسورة التكلفة لتفاعل تطور الأكسجين الحمضي.يتطلب تحويل الطاقة المتجددة (OER) مع نقل البروتونات المرتبط بالإلكترونات الأربعة جهدًا زائدًا عاليًا بسبب حركته البطيئة، مما يقلل من كفاءة تشغيل خلايا الوقود القابلة للاستخدام في الماء (PEMWE).بالإضافة إلى ذلك، فإن الانحياز الأكسيدي القوي والتآكل الحمضي الشديد يتحدان معًا لتحدي متانة المحفزات الكهروكيميائية المتاحة لتفاعل تطور الأكسجين في عمليات الكثافة العالية للتيار..
حاليًا، المحفزات المعتمدة على الإريديوم (Ir)، مثلتظل الخيار العملي الوحيد لمحفزات الأنود في PEMWE بسبب توازنها الجيد بين المتانة والنشاط.. ومع ذلك، فإن انتشار تقنية PEMWE محدود بشكل كبير بسبب توفر الإيريديوم، مما يؤدي في النهاية إلى نقص حاد في الإيريديوم. في هذا السياق، تم بذل العديد من الجهود لقد تم تكريس الجهود للبحث عن بدائل لـ Ir، حيث تم تحديد أن أكثرها وعدًا هو أكاسيد Ru نظرًا لنشاطها العالي الفطري. ومع ذلك، فإن محفزات OER القائمة على Ru تعاني دائمًا من عدم الاستقرار الكافي عند كثافة التيار الصناعية، مما يعيق استخدامها في PEMWE لصناعة الهيدروجين الأخضر.تنبع عدم استقرار الأنود القائم على الروثينيوم من الأكسدة المفرطة لأنواع الروثينيوم، مما يؤدي إلى توليد مواد قابلة للذوبانتحت إمكانيات التعليم المفتوحعلى الرغم من الجهود الكبيرة المبذولة في استقرار الروثينيوم لعملية التحليل الكهربائي للأكسدة، لا يزال هناك فجوة كبيرة في الأداء لتلبية المتطلبات الصناعية.لسد هذه الفجوة، سيكون من الأكثر كفاءة وضع استراتيجية أساسية وقابلة للإدارة لتثبيت المحفزات الكهربائية القائمة على الروثينيوم، بدلاً من النهج التقليدي القائم على التجربة والخطأ.
استقرار النانوسكرياتالمحفزات القائمة على ترتبط ارتباطًا وثيقًا بالتساهمية لـسنداتإضعافيمكن أن تحدد تساهمية الرابطةوروالمدارات تحت مستوى فيرمي، مما يمنع مشاركة الأكسجين الشبكي في تفاعل أكسدة الأكسجين وتكوين فراغات الأكسجين، وبالتالي يمنع الأكسدة المفرطة لأنواع الروثينيوم إلى شكل قابل للذوبان.خلال. في هذه الأثناء، منخفضة بشكل مفرط
الترابط التساهمي ضار لأنه يجعل الروديوم يتسرب بسهولة، مما يؤدي إلى إزالة المعدن مباشرة من سطح الروديوم والتدهور الهيكلي اللاحق.. الدراسات السابقة ركزت على تعديل الهيكل الإلكتروني لـيستخدم، و ركائز معدنية / مواد مضافة. ومع ذلك، كماتحدد المدارات الإلكترونية المعادن الانتقالية الخارجية، وهي عرضة للتأثيرات الخارجية لحقل البلورة وبيئة التنسيق.. على سبيل المثال، إدخال عناصر متتالية مثل ، و إلىيؤدي إلى تحويل إلكتروناتها التكافؤية إلى, ، و على التواليتجعل هذه التأثيرات الخارجية من الصعب للغاية تعديل التساهمية بين الروثينيوم والأكسجين بطريقة دقيقة ومستدامة.
هنا، نستنتج أن عناصر مجموعة اللانثانيد (Ln) مع مدار مدفون تحتيمكن أن يقلل من التأثيرات الخارجية وبالتالي يمكّن من ضبط دقة ومستمر لروابط Ru-O من أجل تحفيز OER المستدام. تم إجراء حسابات نظرية الكثافة الوظيفية (DFT) علىالأنظمة، مما يظهر أن الناتجالهجين يسبب تباينًا مستمرًا في التساهمية بين الروثينيوم والأكسجين لتحديد أداء تفاعل أكسدة الأكسجين. مستفيدًا من تحسين مثاليالتساهميةتم استبعاده لأنه أظهر أكبر طاقة تشكيل للأكسجين الشبكي وخلل الروديوم. تؤكد التوصيفات التشغيلية الدور الحاسم لمواد الإربيوم في استقرار هيكل Ru-O من أجل تحسين متانة تفاعل تطور الأكسجين. علاوة على ذلك، تم رفع-مستوى الطاقة ( -0.855 eV ) لـذو صلة بالمعيار، مما يؤدي إلى تقليل الشغل الإلكتروني في حالات مضادة للرابطة وزيادة أقوى لامتصاص *OH، كما تم التحقق من ذلك من خلال تجارب بروب الميثانول الجزيئي، مما يعزز بشكل كبير النشاط التحفيزي لـ. هذه العمل يثبت فكرة جديدةنهج تنظيمي لضبط تداخل الروابط التساهمية بين الروثينيوم والأكسجين بدقة واستمرار، مما يساعد على تحقيق تكلفة اقتصادية أكثر ملاءمة في سيناريو اقتصاد الهيدروجين.
النتائج
توصيف تفاعل الأكسدة-الاختزال باستخدام تساهمية Ru-O التي تنظم اللانثانيدات
تقدم عناصر Ln وسيلة مرنة لضبط وتحسين الأداء الكهروكيميائي للمحفزات بسبب تأثير الحماية لـمدارات، مملوءة بالتدرجتكوين الإلكترونات المداري، مستويات الطاقة الإلكترونية الغنية، والقدرة على استيعاب أرقام التنسيق المختلفة. يُظهر الاقتران المداري المتدرج للروثينيوم والأكسجين والعناصر الأرضية النادرة وعدًا في تعزيز أداء تفاعل تطور الأكسجين استنادًا إلى تحليل متماثل موجه بواسطة نظرية المجموعات.. لالترابط المداري للروابط، و مدارات مع-خلط المدارات من ذرات الأكسجين المنسقة ينتج حالات ارتباط معدنية-أكسجين (M-O) تتكون من، مع حالاتهم المضادة للرابطة (MO)*، و مصطلحات غير مرتبطة. لـ [ ]، الاقتران المداري بين و يساهم فيه و مع روابطهم المضادة و الشروط، ومصطلحات غير مرتبطة. شكرًا لـبالاشتراك معيمكن تشكيل اقتران المدارات المتدرج لـ Ru-O-Ln (الشكل 1a)، مما يؤدي إلى تفاعلات إلكترونية أكثر مرونة للتكيف الكهروتحفيزي.
قمنا أولاً بإجراء حسابات DFT مع تكوين Ru-O-Ln الذي تم بناؤه استنادًا إلى الروتيل (الأشكال التكميلية 1-4). تُظهر تحليل شحنة بادير انتقالات الإلكترونات من Ln إلى Ru، مما يقلل شحنة Ru بشكل متسلسل منإلى 1.50، و (الشكل التوضيحي الإضافي 5)، الذي يؤكد الهجين المداري فيمن خلال تحليل ملء المدارات الرابطة وغير الرابطة، تظهر نتائج حسابات كثافة هاملتونية المدارات البلورية (COHP) وكثافة هاملتونية المدارات البلورية المدمجة (ICOHP) أن إدخاليمكن أن يضعف الـشغل حالة الترابط منإلى -1.523 إلكترون فولت، و (الشكل التوضيحي التكميلي 6). إن الضبط الدقيق للتكافؤ بين الروثينيوم والأكسجين ينشأ من تأثير الحماية لـمدارات لعناصر اللانثانيد مع تعبئة متدرجةتكوين الإلكترونات المداري.
لتقييم استقرار الأكسجين الشبكي والروثينيوم، قمنا بحساب طاقة تشكيل الأكسجين الشبكي (شاغر) و شاغر Ru ( )، والتي تُستخدم معًا لتقييم استقرار المحفزات الكهربائية. حيث ستحدث ذوبان Ln أثناء عملية OER تم إنشاء هياكل معيبة تحتوي على فراغات Ln. في وجود فراغات Ln،يعرض أعلىيتجاوز، و بمقدار 0.29 و 0.02 و 0.12 إلكترون فولت، على التوالي (الشكل 1ب). بشكل محدد، يؤدي تنظيم تساهمية Ru-O إلى تعديلفراغ، يزيد من 2.58 إلكترون فولت فيإلى، و 3.44 إلكترون فولت لـ، ، و على التوالي (الشكل 1ج). بالنظر إلىوظيفة شاغرةشغور في التركيبة، استقراريتبع الاتجاه البركاني كدالة لتوافقية Ru-O. وهذا بدوره يؤكد مبدأ التصميم المقترح لدينا بأن الضبط الدقيق لتوافقية Ru-O بواسطة Ln ينظم الاستقرار.
للمزيد من التحقيق في الآلية التي من خلالهانظمنا نشاط OER، حسبنا الطاقة الحرة لجيبس للوسائط الأكسجينية خلال OER، مما أسفر عن الجهد الزائد النظري لـ Ho، و كما هو موضح في الشكل 1d والأشكال التكميلية 7 و 8، فإن الخطوة المحددة للإمكانات (PDS) لـ هو التطور من *O إلى *OOH، مع جهد زائد محسوب قدره 0.53 فولت، بينما ينخفض الجهد الزائد إلى ، و 0.50 فولت لـ، و ، على التوالي. يتم تصوير الجهد الزائد النظري لمختلف المحفزات الكهروكيميائية في الشكل 1e باستخدام مخطط ثلاثي الأبعاد على شكل بركان يوضح الفرق في الطاقة الحرة بين *O و *OH ك intermediates. مستفيدين من التحسيناتالتساهميةيظهر تحسينًاقوة الربط (الشكل التكميلي 9) وتوفر طاقات حرة قريبة من المثالية لكل وسيط، مما يؤدي إلى انخفاض الجهد الزائد النظري لتفاعل الأكسدة. بالنسبة للعينات التي تحتوي على كل من الجهد المنخفض ) وأعلى ( تمت ملاحظة زيادة الطاقة الحرة لـ PDS بسبب تساهمية Ru-O. تعكس تساهمية Ru-O في المحفزات تفاعل الربط بين Ru-*OH، مما يشير إلى أن تساهمية Ru-O المثلى التي ليست ضعيفة جداً ولا قوية جداً هي المفضلة لتفاعل أكسدة الماء (OER).
بشكل عام، يمكن أن يؤدي دمج Ln إلى تحسين تساهمية Ru-O باستمرار ضمن نطاق ضيق، مما يتيح التحكم في حركية الذوبان لـالمحفزات القائمة على – وتأثيرها على حواجز الطاقة للخطوات الرئيسية في التفاعل.
تحضير المواد وخصائصها
كـيمتلك نظريًا نشاطًا عاليًا في OER واستقرارًا، تم اختياره لمزيد من التحقيق. للتحقق تجريبيًا من التوقعات المذكورة أعلاه،تم تحضير المحفز بطريقة واحدة باستخدام طريقة نفخ الجلوكوز. نقيتمت أيضًا عملية التخليق باستخدام نفس الطريقة للمقارنة. تظهر صور المجهر الإلكتروني الماسح (SEM) (الشكل التوضيحي الإضافي 10) أنيمتلك هيكلًا مساميًا على شكل ورقة مع مساحة سطح محددة ( ) من (الشكل التكميلي 11). علاوة على ذلك، تتكون الأوراق المسامية من عدد كبير من الجسيمات النانوية الصغيرة (الشكل التكميلي 12). تكشف أنماط حيود الأشعة السينية (XRD) أن يمتلك هيكلًا من نوع الروتايل دون وجود قمم حيود مميزة تتعلق بـ (الشكل التكميلي 13)، مما يشير إلى الدمج الناجح لذرات Er فيصورة مجهر الإلكترون الناقل عالي الدقة (HRTEM) تظهرتتكون الأوراق النانوية من جزيئات نانوية صغيرة بحجم حواليالتي تتكون من الوجه المهيمن (110) (الشكل التوضيحي 14).
علاوة على ذلك، استخدمنا مجهر الإلكترون الناقل المصحح للانحرافات (AC-TEM) لكشف البنية الدقيقة على المستوى الذري لـالمحفز. تظهر صور المجهر الإلكتروني الناقل الماسح ذو الحقل المظلم الزاوي العالي (HAADF-STEM) (الشكل 2a، d) الحافة الواضحة لـ Er-RuOالحبوب. الشكل 2 ب والشكل التكميلي 15 يعرضان الرسوم البيانية السطحية لـ، مما يسمح بتصوير كثافة الأعمدة الذرية. التكوين الذري لـ Erيمكن رؤيته بوضوح من صور HAADF-STEM الذرية على محاور المنطقة [001] و [110] (الشكل 2c، e)، متوافقًا مع ذلك الخاص بالنقي
الشكل 1 | توقع أداء OER باستخدام حسابات DFT. مخطط المدارات الجزيئية النوعية المستخلص من و . ب و (ج) كنتيجة لـ -ICOHP لـ. الـ تشير قيم upshift لـ ICOHP إلى انخفاض التشارك في الروابط بين الروثينيوم والأكسجين. د مسارات التفاعل على Er و عند 1.23 فولت. مخطط البركان لمختلف المحفزات الكهربائية والهياكل المقابلة. تم تسجيل مسح خط الطيف الطيفي للطاقة المشتتة (EDS) في الشكل 2f من صورة HAADF-STEM لـ (الشكل 2د، الخط الأخضر ” ). الـ، ويتم الكشف عن إشارات Er وتسجيلها في ملف المسح الخطي. خاصة، فإن الإشارات الناتجة عن Er تظهر فقط في مناطق إشارة Ru، مما يشير بشكل أكبر إلى أنه تم إدخال Er بنجاح في تشير شدة الإشارة الضعيفة نسبيًا لعناصر الإربيوم (Er) مقارنة بعنصر الروثينيوم (Ru) إلى انخفاض محتوى الإربيوم، مما يتوافق مع نتيجة طيف EDS (الشكل التكميلي 16). توضح صور رسم العناصر بشكل أكبر التعايش والتوزيع على المستوى الذري لذرات الروثينيوم والإربيوم (الشكل.توزع ذرات الإيريديوم داخل المحفز، مع تفضيل للتواجد على السطح، مما يساهم في الأداء الكهروكيميائي حيث يمكن تنظيم تساهمية الروثينيوم-أكسجين كما تم مناقشته أعلاه.
أداء التحفيز الكهربائي في تكوينات الثلاثة أقطاب وأجهزة PEMWE
أداء الموارد التعليمية المفتوحةوتم قياس عينات التحكم فيحل في نظام ثلاثي الأقطاب. كما هو موضح في الشكل 3a، Er-RuOيمثل نشاط OER المتفوق على الأنشطة التجارية والمصنوعة في المنزلبشكل مثير، الجهد الزائد المطلوب للوصول إلى كثافة التيار منفقطعلى وأقل من تلك الخاصة بالتجارةومصنوع في المنزل، على التوالي. علاوة على ذلك، عند تطبيعها على المساحة النشطة كهربائياً، فإن النشاط التحفيزي لـيبقى أفضل من ذلك التجاري (الشكل التوضيحي 19). الـالمحفز يظهر أيضًا انخفاضًا في ميل تافل بـمقارنة بالتجاري (الشكل 3ب)، مما يشير إلى تسريع حركية التفاعل.
تمت دراسة استقرار المحفزات المُعدة حديثًا بواسطة الفولتمترية الدورية (CV) بين 1 فولت و 1.45 فولت مقابل RHE.يظهر تضعيفًا أقل بكثير من التجاريبعد 30,000 اختبار للدراجات (الشكل 3c). تركيز الروثينيوم المذاب بعددورات لـتم قياسه ليكون 13.7 جزء في البليون، وهو ما كان أقل بكثير من ذلك الخاص بالتجارة.. تشير هذه النتائج إلى أن دمج الإربيوم يقلل من ذوبان . بالإضافة إلى ذلك، تم تقييم متانة التحفيز بواسطة الكرونوپوتنشيومترية (CP) في (الشكل 3د)، مما يوضح ميزة استقرار أكثر وضوحًا لـفوق التجاري. بالتفصيل، الجهد الزائد المطلوب للتجاريزاد بمقدار 674 مللي فولت بعد اختبار الاستقرار لمدة 73 ساعة فيالذي كانأعلى بمرات من، التحقق من الدور المفيد للـ Er في استقرار التحفيز. أداء OER لـ و كان
الشكل 2 | توصيف الهيكل على المستوى الذري لـ Er-RuO. صورة HAADF-STEM و b رسم سطحي لـعلى محاور منطقة [001]، شريط القياس: 2 نانومتر. صورة STEM ذرية على طول محاور منطقة [001] ونموذج هيكلي (داخل الصورة) لـ Er-RuOشريط القياس:. صور HAADF-STEM (مقياس: 2 نانومتر) و (e) صور STEM الذرية (مقياس: ) من
إر-روعلى محاور المنطقة [110]. f مسح خط EDS لـوإشارة Er المسجلة من الخط الأخضرفي (د). صورة HAADF-STEM وصور رسم الخرائط العنصرية المقابلة لـ على طول ( ) [110] و ( ) [001] محاور المنطقة، شريط القياس: 2 نانومتر. رسم توضيحي تخطيطي لـ. كما هو موضح في الأشكال التكميلية 22-24، فإنها تتوافق بشكل جيد مع توقعات DFT. كما هو موضح في الجداول التكميلية 6 و 7، فإن كل من النشاط والاستقرار لـكانت أعلى من تلك الخاصة بالعوامل المحفزة الكهربائية المعتمدة على الروثينيوم التي تم الإبلاغ عنها مؤخرًا، مما يؤكد الكفاءة الاقتصادية لـ.
لتحقيق إمكانيات التطبيق لـلإجراء التحليل الكهربائي للماء، قمنا بتصميم جهاز PEMWE (غشاء نافيوني 117) باستخداموتجاري كعامل حفاز للأنود والكاثود، على التوالي (الشكل 3e). منحنيات التيار-الجهد (بدونالتعويض) في الشكل 3f توضح بوضوح الأداء المتفوق لتحليل الماء الكهربائي لـPEMWE القائم على المقارنة مع التجاريجهاز PEMWE. تحديداً، Er-RuOالتحليل الكهربائي القائم علىمطلوب فقط، و 1.837 فولت للوصول إلى كثافة تيار صناعية قدرها 1.2، وعلى التوالي، متفوقين على PEMWE باستخدام التقنيات الحديثة الأخرىالمحفزات القائمة على – (الجدول التكميلي 8). علاوة على ذلك، فإن PEMWE الذي يستخدم المحفز يحقق كفاءة تبلغ حواليفي (الشكل التوضيحي 26)، بتكلفة تقدر بحوالي 0.85 دولار أمريكي لكل كيلوجرام من ، وهو أقل بكثير من هدف وزارة الطاقة الأمريكية (DOE) البالغ 2 دولار أمريكي لكل كيلوغرام مناستقرار الإربيومتم تقييم جهاز PEMWE عند 200 و 500 وعلى التوالي، ولم يُلاحظ أي تدهور ملحوظ في النشاط على الجهاز بعداختبار الاستقرار لكل حالة (الشكل 3 ج). معدل التدهور عندمجرد، مما يوضح الإمكانية التطبيقية لـلإنتاج الهيدروجين الأخضر.
أصل الأداء الممتاز لـ
تم إجراء مطيافية الإلكترونات الضوئية بالأشعة السينية (XPS) لتحديد التركيب الكيميائي وحالات التكافؤ للمواد المحفزة التي تم إعدادها. في الروديومطيف XPS (الشكل التوضيحي 27)، يمكن أن تُنسب القمم عند 463.7 و 463.1 إلكترون فولت إلى و على التوالي. بالمقارنة مع ، الـنسبة فيأقل من ذلك فيمما يدل على حالة أكسدة أقل للروثينيوم فيلاستكشاف تفاعل الإربيوم والروثينيوم، تم استخدام مطيافية امتصاص الأشعة السينية (XAS) لاستكشاف البيئة الذرية والتنسيقية. تكشف طيف هيكل الامتصاص القريب من حافة الأشعة السينية (XANES) لحافة الروثينيوم (الشكل 4a) أن موضع عتبة الامتصاص لـأعلى من تلك الخاصة بورق الروثينيوم ولكن أقل من تلك الخاصة بـحالة الأكسدة المحسوبة للروثينيوم في هو 3.80 (الشكل التوضيحي التكميلي 28)، متسق مع نتائج XPS وشحنة بادر. علاوة على ذلك، فإن طيف تحويل فورييه لحد Ru K لهيكل الامتصاص الدقيق للأشعة السينية الممتدة (FT-EXAFS) لـ Er-RuO (الشكل 4ب) يعرض قمة تقع عند ، المخصصة لروابط رو-أو. يظهر هذا الذروة انزياحًا إيجابيًا نحو مسافات ذرية أطول مقارنةً بتلك الخاصة بـبسبب الضعف الطفيفالتكافؤ (الجدول التكميلية 9)، يتطابق جيدًا مع توقعات الحسابات النظرية. كما أن تحليل تحويل الموجات يظهر أنالتنسيق فييشبه ذلك لـبينما لايتم اكتشاف الرابطة في، متماشياً مع نتائج FT-EXAFS (الشكل. ).
ثم قمنا بإجراء قياس طيف رامان في الموقع (الشكل التوضيحي 29) لتوضيح تطور الهيكل خلال
الشكل 3 | الأداء الكهروتحفيزي للمحفزات المُعدة حديثًا. أمنحنيات الاستقطاب المصنوعة في المنزلوتجاريمع مصحح )، حيث تم قياسه ليكون. رسومات جدولية منزليةوتجاري . منحنيات الاستقطاب وتجاريقبل (خط مستمر) وبعد (خط منقط) دورات. منحنيات CP لـ Er-RuOوتجاريفيرسم تخطيطي لمحول كهربائي من نوع PEMWE.منحنيات الاستقطاب لـوتجاري-المعتمد على PEMWE في. منحنيات CP لـ Er-التحليل الكهربائي PEMWE القائم على – يعمل عند 200 و 500 و، على التوالي.
الطيف الرامان يظهر الطبيعة النانوية البلورية لـ و (الشكل 4ج، د)، مع قمتين رئيسيتين تقعان في و ، الذي يتوافق مع و على التوالي. مع زيادة الجهد من جهد الدائرة المفتوحة (OCP) إلى 1.8 فولت مقابل RHE،يحافظ على انزياح رامان شبه ثابت، مما يدل على الاستقرار لـالتكوينات. بالمقابل،يتم ملاحظة الانزياح الأحمر في، مما يشير إلى الانكماش فيطول الرابطة أثناء عملية أكسدة الأكسجين.
علاوة على ذلك، XANES عند حافة Ru K لـ و تم جمعها مع زيادة الجهد المطبق من 1.3 إلى 1.7 فولت مقابل RHE للتحقيق في التغيرات في الهيكل الذري المحلي للروثينيوم والتنسيق الكيميائي. كما هو موضح في الشكل 4e و f والشكل التكميلي 30، و تظهر اختلافات ملحوظة في تباين حالات أكسدة الروثينيوم، خاصة عند الجهد العالي. بالتفصيل، عندما يكون الجهد المطبق 1.3 فولت، تكون حالة أكسدة الروثينيوم فييزداد من 3.80 إلى 4.16، تليه تغيرات ضئيلة عندما زادت الإمكانية إلى 1.7 فولت. بالمقابل، حالات الأكسدة للروثينيوم فييزداد بشكل مستمر إلى 4.36 مع تغير الجهد المطبق من 1.3 إلى 1.7 فولت، دون إظهار أي علامة على الاستقرار. تشير النتيجة إلى أن الهيكل المستقر لـيمكن الحفاظ عليها عند إمكانيات أكسدة عالية، بينمايخضع لتطور هيكلي كبير. قمة Ru 3d في Er-RuOطيف XPS بعد اختبار الاستقرار يظهر انزياحًا طفيفًا نحو الإيجابية (الشكل التكميلي 31)، مما يشير إلى أن سطحيتأكسد إلى حالة تكافؤ أعلى، متوافقة مع نتائج XANES. تؤكد صور TEM بعد اختبار الاستقرار أن الهيكل البلوري لـ (الشكل التوضيحي 32) محفوظ بشكل جيد، مما يشير إلى عدم حدوث تدهور هيكلي كبير تحت التأثير الأكسيدي القوي.
تحليل سلوك الامتزاز لـمع وسائط الأكسجين
لتبرير الأداء المحسن لمعدل الأكسدةامتصاصات و تم تقييم الوسائط المتوسطة للأكسجين. كما هو موضح في الشكل 5أ،يعرض مركز نطاق d أقل للروثينيوممن ذلك النقي، مما يشير إلى أن تغيير الهجين المداري قليلاً البيئة الإلكترونية للروثينيومالمدارات. علاوة على ذلك،يمكن دمج المدارات في و معالوسطاء. الروثينيوم-مستوى الطاقة في الحالةمنتتحول إلى مستوى فيرمي بالنسبة لذلك من (الشكل 5ب)، مما يؤدي إلى تقليل الشغل الإلكتروني في حالات المضاد للرابطة وزيادة امتصاص *OH (الشكل 5ج). كما هو موضح في الشكل 5د، فإن تراكم الشحنة نحو *OH يؤكد سلوك نقل الإلكترون الأقوى بين و ، مما يؤكد تعزيز امتصاص *OH.
تم التحقق تجريبيًا من تعزيز امتصاص *OH باستخدام الميثانول كأداة. تفاعل أكسدة الميثانول
الشكل 4 | التوصيف الهيكلي لـ و . طيف XANES لحافة Ru K من رقائق الروثينيوم، ، وإر-رو. طيف EXAFS لرقائق الروثينيوم، ، و طيف رامان في الموقع الذي تم الحصول عليه تحت إمكانيات مختلفة على (ج) و (د)
إر-رو. طيف XANES لحافة K من إيريديوم-روثينيوممع انحياز مطبق مختلف.اتجاه تباين حالات أكسدة الروثينيوم في و تحت إمكانيات مختلفة. WT-EXAFS لـ (g) رقائق الروثينيوم، (h)، و (ط) . (MOR) يتبع آلية راسخة، حيث تميل جزيئات الميثانول إلى الهجوم النيوكليوفيلي على *OH الكهربية. ونتيجة لذلك، فإن MOR أكثر نشاطًا على الأسطح التي تتمتع بامتصاص أقوى لـ *OH.. عندما تم إدخال 1.0 ميثانول في حل، كثافات التيار لـ و أظهر زيادة كبيرة مقارنة بتلك التي كانت قبل إضافة الميثانول، والتي تعزى إلى أكسدة الميثانول الكهربية (الشكل 5e، f). تم قياس الفرق في كثافات التيار الناتجة عن MOR، والتي كانت تتناسب طردياً مع عدد الشحنات المنقولة، من خلال حساب المساحة المملوءة بين المنحنيات. كان الفرق الأكبر في التيار الملحوظ بين MOR و OER علىمن ذلكاقترح رد فعل أقوى لمنافسة MOR، مما يحقق تعزيز امتصاص *OH على (الشكل 5 ج).
للحصول على معلومات حول وسائط الأكسجين لفهم آلي شامل، تم إجراء تقنية الانعكاس الكلي المخفف – مطيافية الامتصاص بالأشعة تحت الحمراء المعززة بالسطح (ATR-SEIRAS) باستخدام خلية كهروكيميائية مصنوعة في المنزل. تم إجراء ATR-SEIRAS لكل من و تم قياسها بين 1000 ومن 1.3 فولت إلى 1.7 فولت مقابل RHE. كما هو موضح في الشكل 5 ح، قمة تعتمد على الجهد عندلـيتوافق مع اهتزاز الشد لـنوعأصبح أكثر بروزًا مع زيادة الجهد من 1.5 فولت إلى 1.7 فولت.ظهر ذروة *OOH عند 1.4 فولت (الشكل 5i)، بما يتماشى مع انخفاض PDS لـ *O إلى *OOH (الشكل 1d). في الوقت نفسه، أشارت ظهور أنواع *OOH إلى أن آلية تطور الامتصاص كانت هي السائدة.جيل فوق.
نقاش
باختصار، نوضح أن الضبط الدقيق للتعاون التساهمي بين الروثينيوم والأكسجين فييمكن تحقيقه من خلال إدخال عناصر Ln عبرالهجينة المدارية. الاستفادة من تحسين التساهمية بين الروثينيوم والأكسجين،يتم استبعاده ويظهر استقرارًا تحفيزيًا عاليًا، متفوقًا بشكل ملحوظ على الحالة النقيةبأوامر من حيث الحجم. تشير التوصيفات في الظروف التشغيلية إلى أن حالة الأكسدة للروثينيوم فييزداد في البداية مع زيادة الجهد المطبق ثم يبقى تقريبًا ثابتًا. بالمقابل، حالة الأكسدة للروثينيوم في يرتفع باستمرار دون أي علامة على الاستقرار. تؤكد حسابات DFT وتجارب مجس الميثانول على امتصاص *OH الأقوى على بالنسبة لذلك على، مما أدى إلى تعزيز نشاط OER. تستخدم PEMWE (غشاء نافيوني 117) حيث أن محفز الأنود يتطلب فقط 1.837 فولت للوصول إلىوتظهر استقرارًا طويل الأمد فيلمدة 100 ساعة بمعدل تدهور بسيطتوفر هذه الدراسة إطار تصميم محفزات مبني على مبادئ للتحكم الدقيق في التساهمية بين الروثينيوم والأكسجين، مما يوجه تطوير محفزات قائمة على الروثينيوم مناسبة للتطبيق العملي في أنظمة PEMWE.
طرق
المواد
كلوريد الروثينيومتم شراءه من شركة بكين إنوكيم للعلوم والتكنولوجيا المحدودة. نترات الهولميوم خماسية الماءنترات الإربيوم خماسية الماء، ) و نترات الثوليوم سداسي الماء كان
الشكل 5|تحليل سلوك الامتزاز. أ تم حساب PDOS للروثينيومو مدارات Er f. الـ PDOS لـمدار و . مخطط تخطيطي لتهجين المدارات بينمدار و ومدارات الربط *OH. فرق كثافة الشحنة ل *OH-الممتص على Er-RuO (يسار) و (يمين). تمثل الأسطح المعزولة الصفراء والزرقاء تراكم الشحنة و نقص، على التوالي. منحنيات الاستقطاب لـ (هـ) و (و) في 0.5 مالحل مع (الخطوط المتقطعة) وبدون (الخطوط الصلبة) 1 ميثانول. ج الفرق في التيار بين منحنيات الاستقطاب فيمحلول مع وبدون 1.0 ميثانول لـ و . تحليل ATR-SEIRAS لـ (h) التجارية و (أنا) . تم الشراء من شركة شانغهاي ماكلين للكيماويات الحيوية المحدودة. تم الحصول على اليوريا (AR) والجلوكوز (AR) من شركة بكين تونغ جوانغ للكيماويات الدقيقة. Pt/C التجارية (تم الحصول على ( ) من شركة جونسون ماثي. تم الحصول على غشاء نافيون 117 (سمك: 0.18 مم) من شركة دو بونت. تم استخدام جميع المواد الكيميائية دون مزيد من التنقية.
تركيب و
في التخليق النموذجي لـوكمية معينة منتمت إضافتها إلى 5 مل من الماء المقطر مع 1 جرام من اليوريا و5 جرام من الجلوكوز (مع الحفاظ على النسبة الذرية لـفي ). تم تحريك الخليط حتى تم الحصول على محلول متجانس. بعد ذلك، تم تعريضه للتسخين عند لمدة 8 ساعات في فرن لتشكيل رغوة مسامية، ثم تم تلدينها عند لمدة 10 ساعات في الهواء للحصول على. للمقارنة، النقيتم تحضيره أيضًا دون إضافة.
القياسات الكهروكيميائية
تم إجراء جميع القياسات الكهروكيميائية باستخدام جهاز العمل الكهروكيميائي CHI 760E مع خلية ثلاثية الأقطاب في درجة حرارة الغرفة. تم توزيع المحفزات المحضرة (2 ملغ) في خليط منإيزوبروبانول ونافيو D-521 ) الحل. بعد الموجات فوق الصوتية لمدة ساعة واحدة، تم إسقاط الحبر المتجانس بعناية على ورق الكربون ( ) للحصول على القطب العامل بتحميل مرغوب فيه . الـ كان القطب المرجعيتم معايرته في نظام ثلاثي الأقطاب حيث كانت أسلاك البلاتين تعمل ككلا من القطب العامل والقطب المضاد، ومشبعتم استخدام المحلول كإلكتروليت. بعد ذلك، تم قياس CV بمعدل مسحتم تحديد الجهد المتوسط الذي يعبر فيه التيار عن الصفر كجهد حراري نسبي لـ. تم معايرة جميع الإمكانيات إلى RHE بواسطة المعادلة: E (بالنسبة إلى RHE مقابل تم استخدام قضيب الجرافيت كقطب مضاد. تم إجراء منحنيات الاستقطاب بمعدل مسحفيحل0.3). جميع منحنيات الفولتمترية المسح الخطي (LSV) المقاسة في خلية ثلاثية الأقطاب كانت-مصحح ) ما لم يُذكر خلاف ذلك، حيث تم قياسه ليكونخلال تحضير الإلكتروليت، تم إضافة 13.6 مل من حمض الكبريتيك (98%) إلى دورق يحتوي على حجم مناسب من الماء المقطر، تلاه ضبط الحجم إلى 500 مل في قنينة حجمية واهتزاز لضمان الخلط الجيد. يتم تحضير الإلكتروليت حديثًا واستخدامه على الفور. تم إجراء مطيافية الامتزاز الكهروكيميائي عند 1.485 فولت مقابل RHE في نطاق التردد منإلى. السير الذاتية بمعدلات مسح مختلفة (أي، ، و تم إجراء ( ) لحساب المساحة السطحية النشطة كهربائياً (ECSA)، والتي كانت متناسبة مع سعة الطبقة المزدوجة.باستخدام فرضية أن السعة النوعية لسطح مستو كانتلـمن المساحة السطحية الحقيقية، ثم تم تقدير ECSA كالتالي: ECSA.
اختبارات PEMWE
تم تجميع PEMWE معأو تجاريكأنود، مع تحميل قدرهتجاريتم استخدامه كعامل حفاز للقطب السالبتم استخدام ألياف التيتانيوم المطلية بالبلاتين كطبقات لتوزيع الغاز. تم استخدام نافيون 117 كغشاء لتبادل البروتونات (PEM)، الذي تم معالجته بشكل متتابع بـ و في لمدة ساعة واحدة. تم تحضير حبر المحفز للأنود والكاثود بشكل منفصل عن طريق توزيع المحفز في خليط من نافيون (5%)، وإيزوبروبانول، والماء المقطر. تم رش جميع حبر الكاثود على غشاء تبادل البروتون. أما بالنسبة للأنود، فقد تم رش نصف حبر المحفز على غشاء تبادل البروتون، بينما تم رش النصف المتبقي على سطح طبقة انتشار الغاز. ثم تم ضغط غشاء تبادل البروتون المحمّل بالمحفز وطبقات انتشار الغاز معًا تحت حرارة.لمدة دقيقتين تحت ضغط 2 ميجا باسكال لتصنيع مجموعة أقطاب الغشاء (MEA)، والتي تم وضعها بين لوحين ثنائي القطب من التيتانيوم لإكمال جهاز PEMWE. كان لكل لوح ثنائي القطب من التيتانيوم قناة تدفق متعرجة معمنطقة تفاعلية 1 سم. تم تشغيل PEMWEs عنداستخدام الماء المقطر كإلكتروليت، الذي تم توصيله إلى الأنود بواسطة مضخة بيرستالتية. تم تسجيل جميع القياسات في خلايا PEMWE دون تصحيح iR.
قياسات ATR-SEIRAS في الموقع
تم إجراء قياس ATR-SEIRAS في الموقع على جهاز مطياف الأشعة تحت الحمراء بتقنية تحويل فورييه (FTIR) من نوع Bruker 70 V. تميز القياس بدقة طيفية قدرها، مع دمج 64 صورة تداخلية لكل طيف. تضمنت إعداد الأقطاب الكهربائية العاملة خطوتين: أولاً، تم ترسيب فيلم رقيق جداً من الذهب كيميائياً على بلورة السيليكون لتعزيز إشارة الأشعة تحت الحمراء وتسهيل توصيل الإلكترونات؛ ثانياً، تم إسقاط حبر المحفز على فيلم الذهب بتحميل قدره تم وضع بلورة السيليكون المحملة بالمواد الحفازة في خلية ثلاثية الأقطاب للطيف الكهروكيميائي. كانت قطب Ag/AgCl و سلك Pt يعملان كأقطاب مرجعية وأقطاب مضادة، على التوالي.تم استخدام المحلول كإلكتروليت. تم الحصول على جميع طيف SEIRAS خلال اختبار LSV.
حسابات DFT
تم إجراء الحسابات باستخدام نظرية الكثافة الوظيفية كما تم تنفيذها في حزمة المحاكاة الأولية فيينا (VASP). تم وصف دالة التبادل-الارتباط باستخدام تقريب التدرج العام (GGA) مُعَامَل بواسطة نموذج بيردو-بورك-إرنزرهوف (PBE). تم تحديد طاقة القطع لقاعدة الموجة المسطحة إلىتم نمذجة السطح (الشكل التوضيحي 1) بواسطةسوبرسيل (مع 55 ذرة أكسجين و24 ذرة روديوم)، حيث يُسمح للطبقات الثلاث العليا من الذرات بالاسترخاء. أما بالنسبة لـذرتان من الروديوم فيتم استبدال النموذج بذرات Ln (الأشكال التكميلية 2-4 والبيانات التكميلية 1)، مما أدى إلى تركيبة كيميائية بنسبة 8.3٪ من Ln و91.7٪ من Ru. تم استخدام فراغ فراغي منتم تعيينه على طول اتجاه z لمنع التفاعل بين اللوح ونمطه الدوري. إنهاء و يحتوي على جميع مواقع الروديوم مليئة بالأكسجين باستثناء موقع واحد غير مشبع تنسيقياً غير ممتلئ، وهو الموقع النشط لامتصاص الوسائط. تم استخدام طريقة مونكهورست-باك لأخذ عينات من منطقة بريل.الشبكة. كانت معايير استرخاء الهندسة والتقارب للهيكل الإلكتروني و ، على التوالي. تم حساب الطاقات الحرة لخطوات التفاعل باستخدام المعادلة:، حيث هو طاقة الامتزاز للوسائط، و هو درجة الحرارة. و تمثل تباين الطاقة في طاقة النقطة الصفرية والإنتروبيا، على التوالي. عند حساب طاقة تشكيل فراغات الروديوم، فإن ذرات الروديوم المفقودة تنشأ من مواقع التنسيق السطحية غير المشبعة، باستمرار من نفس الموقع عبر جميع النماذج. في حساب طاقة تشكيل فراغات الأكسجين، كلاهما و فقدان ذرات الأكسجين المحيطة بمواقع تنسيق الروثينيوم غير المشبعة على السطح. تمثل قيم ICOHP وشحنة بادر القيم عند الموقع النشط.
توفر البيانات
البيانات التي تدعم استنتاجات هذه الدراسة متاحة ضمن الورقة والمعلومات التكميلية. يتم توفير بيانات المصدر مع هذه الورقة.
References
Chong, L. et al. La- and Mn-doped cobalt spinel oxygen evolution catalyst for proton exchange membrane electrolysis. Science 380, 609-616 (2023).
Seh, Z. W. et al. Combining theory and experiment in electrocatalysis: insights into materials design. Science 355, eaad4998 (2017).
Guo, J. et al. Direct seawater electrolysis by adjusting the local reaction environment of a catalyst. Nat. Energy 8, 264-272 (2023).
Li, F. & Baek, J.-B. Active site engineering accelerates water electrolysis. Nat. Synth. 3, 558-559 (2024).
Shi, Z. et al. Enhanced acidic water oxidation by dynamic migration of oxygen species at the catalyst/support interfaces. Angew. Chem. Int. Ed. 61, e202212341 (2022).
Shi, Z. et al. Customized reaction route for ruthenium oxide towards stabilized water oxidation in high-performance PEM electrolyzers. Nat. Commun. 14, 843 (2023).
Liu, H. et al. Eliminating over-oxidation of ruthenium oxides by niobium for highly stable electrocatalytic oxygen evolution in acidic media. Joule 7, 558-573 (2023).
Wang, Q. et al. Long-term stability challenges and opportunities in acidic oxygen evolution electrocatalysis. Angew. Chem. Int. Ed. 62, e202216645 (2023).
Ge, S. et al. A robust chromium-iridium oxide catalyst for high-current-density acidic oxygen evolution in proton exchange membrane electrolyzers. Energy Environ. Sci. 16, 3734-3742 (2023).
Liu, R.-T. et al. Recent advances in proton exchange membrane water electrolysis. Chem. Soc. Rev. 52, 5652-5683 (2023).
Chen, F.-Y., Wu, Z.-Y., Adler, Z. & Wang, H. Stability challenges of electrocatalytic oxygen evolution reaction: from mechanistic understanding to reactor design. Joule 5, 1704-1731 (2021).
Dang, Q. et al. Iridium metallene oxide for acidic oxygen evolution catalysis. Nat. Commun. 12, 6007 (2021).
Chen, S. et al. Reconstructed Ir-O-Mo species with strong Brønsted acidity for acidic water oxidation. Nat. Commun. 14, 4127 (2023).
Gao, J., Tao, H. & Liu, B. Progress of nonprecious-metal-based electrocatalysts for oxygen evolution in acidic media. Adv. Mater. 33, 2003786 (2021).
Li, L. et al. Electrochemically modifying the electronic structure of nanoparticles for overall electrochemical water splitting with extensive adaptability. Adv. Energy Mater. 10, 2001600 (2020).
Cao, L. et al. Dynamic oxygen adsorption on single-atomic ruthenium catalyst with high performance for acidic oxygen evolution reaction. Nat. Commun. 10, 4849 (2019).
Yao, Y. et al. Engineering the electronic structure of single atom Ru sites via compressive strain boosts acidic water oxidation electrocatalysis. Nat. Catal. 2, 304-313 (2019).
Zagalskaya, A. & Alexandrov, V. Role of defects in the interplay between adsorbate evolving and lattice oxygen mechanisms of the oxygen evolution reaction in and . ACS Catal. 10, 3650-3657 (2020).
Wu, Z.-Y. et al. Non-iridium-based electrocatalyst for durable acidic oxygen evolution reaction in proton exchange membrane water electrolysis. Nat. Mater. 22, 100-108 (2023).
Wang, Y. et al. Unraveling oxygen vacancy site mechanism of Rhdoped catalyst for long-lasting acidic water oxidation. Nat. Commun. 14, 1412 (2023).
Galyamin, D. et al. Active and durable pyrochlores with low Ru content for acidic oxygen evolution. Nat. Commun. 14, 2010 (2023).
Jin, H. et al. Safeguarding the phase against lattice oxygen oxidation during acidic water electrooxidation. Energy Environ. Sci. 15, 1119-1130 (2022).
Miao, X. et al. Quadruple perovskite ruthenate as a highly efficient catalyst for acidic water oxidation. Nat. Commun. 10, 3809 (2019).
Zhu, W. et al. Direct dioxygen radical coupling driven by octahedral ruthenium-oxygen-cobalt collaborative coordination for acidic oxygen evolution reaction. J. Am. Chem. Soc. 145, 17995-18006 (2023).
Li, L. et al. Spin-polarization strategy for enhanced acidic oxygen evolution activity. Adv. Mater. 35, 2302966 (2023).
Li, L. et al. Compensating electronic effect enables fast site-to-site electron transfer over ultrathin RuMn nanosheet branches toward highly electroactive and stable water splitting. Adv. Mater. 33, 2105308 (2021).
Zheng, X. et al. Ru-Co pair sites catalyst boosts the energetics for the oxygen evolution reaction. Angew. Chem. Int. Ed. 61, e202205946 (2022).
Su, J. et al. Assembling ultrasmall copper-doped ruthenium oxide nanocrystals into hollow porous polyhedra: highly robust electrocatalysts for oxygen evolution in acidic media. Adv. Mater. 30, 1801351 (2018).
Zhang, D. et al. Construction of Zn -doped nanowires for efficient and stable water oxidation in acidic media. Nat. Commun. 14, 2517 (2023).
Wang, X. et al. Engineering 3d-2p-4f gradient orbital coupling to enhance electrocatalytic oxygen reduction. Adv. Mater. 34, 2206540 (2022).
Wang, X. et al. Spin-selective coupling in Mott-Schottky boosts electrocatalytic oxygen reduction. Small Methods 7, 2300100 (2023).
Li, L. et al. Optimizing the electronic structure of ruthenium oxide by neodymium doping for enhanced acidic oxygen evolution catalysis. Adv. Funct. Mater. 33, 2213304 (2023).
Bender, G. & Dinh, H. N. HydroGEN: Low-Temperature Electrolysis (LTE) and LTE/Hybrid Supernode (No. NREL/PR-5900-659). (National Renewable Energy Lab.(NREL), 2020).
Hu, Y. et al. Single Ru atoms stabilized by hybrid amorphous/crystalline FeCoNi layered double hydroxide for ultraefficient oxygen evolution. Adv. Energy Mater. 11, 2002816 (2021).
Tao, H. B. et al. A general method to probe oxygen evolution intermediates at operating conditions. Joule 3, 1498-1509 (2019).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
شكر وتقدير
يقر S.J.G. بالتمويلات من المؤسسة الوطنية للعلوم الطبيعية في الصين (الأرقام 52333010، 52025133، 22309004، 52261135633، 52303363، 52302207، 22205010، 22305010، 22105007)، البرنامج الوطني الرئيسي للبحث والتطوير في الصين (رقم 2022YFE0128500)، مشروع التعاون الاستراتيجي بين شركة النفط الوطنية الصينية وجامعة بكين للبحث الأساسي، ومؤسسة بكين للعلوم الطبيعية.
(رقم Z220020)، مؤسسة كورنرستون للعلوم الجديدة من خلال جائزة XPLORER، صندوق الابتكار CNPC (رقم 2021DQ02-1002)، البرنامج الوطني الصيني لما بعد الدكتوراه للمواهب المبتكرة (رقم BX20220009)، مؤسسة العلوم لما بعد الدكتوراه في الصين (الأرقام 2022M720225، 2023M730029، 2022M710187، 2023M730051، 2020M670018) ومشاريع البحث الأساسية في يونان (رقم المنحة 202401ATO7O37O). تم تنفيذ هذا العمل بدعم من خط شعاع 1W1B في منشأة الإشعاع المتزامن في بكين. يشكر المؤلفون محطات الإشعاع الضوئي BL14W1 في منشأة الإشعاع المتزامن في شنغهاي (SSRF) على المساعدة في التوصيفات.
مساهمات المؤلفين
S.J.G. تصور وأشرف على المشروع. M.C.L. قاد وأشرف على البحث بالكامل. L.L. و G.W.Z. نفذا التجارب، وجمعا وحللا البيانات. شارك C.H.Z. و F.L. و H.L. و D.W.W. في اختبارات PEMWE. ساعد Y.J.T. و Y.H. و Y.X.L. و C.S.S. في قياسات Operando ATR-SEIRAS. شارك L.Y.Z. و Q.Z.H. و R.J.Z. و N.Y. في جزء من التجارب الأساسية. كتبت L.L. المخطوطة. شارك جميع المؤلفين في مناقشة البيانات وقدموا تعليقات على المخطوطة.
ملاحظة الناشر تظل Springer Nature محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
الوصول المفتوح هذه المقالة مرخصة بموجب رخصة المشاع الإبداعي للاستخدام والمشاركة والتكيف والتوزيع وإعادة الإنتاج في أي وسيلة أو صيغة، طالما أنك تعطي الائتمان المناسب للمؤلفين الأصليين والمصدر، وتوفر رابطًا لرخصة المشاع الإبداعي، وتوضح ما إذا كانت هناك تغييرات قد أجريت. الصور أو المواد الأخرى من طرف ثالث في هذه المقالة مشمولة في رخصة المشاع الإبداعي للمقالة، ما لم يُذكر خلاف ذلك في سطر الائتمان للمادة. إذا لم تكن المادة مشمولة في رخصة المشاع الإبداعي للمقالة واستخدامك المقصود غير مسموح به بموجب اللوائح القانونية أو يتجاوز الاستخدام المسموح به، ستحتاج إلى الحصول على إذن مباشرة من صاحب حقوق الطبع والنشر. لعرض نسخة من هذه الرخصة، قم بزيارة http://creativecommons.org/licenses/by/4.0/.
(ج) المؤلفون 2024
مدرسة علوم المواد والهندسة، جامعة بكين، بكين، الصين.كلية الهندسة المعدنية والطاقة، جامعة كونمينغ للعلوم والتكنولوجيا، كونمينغ، يونان، الصين.ساهم هؤلاء المؤلفون بالتساوي: لو لي، قنغ وي زانغ.البريد الإلكتروني: guosj@pku.edu.cn
Precisely modulating the Ru-O covalency in for enhanced stability in proton exchange membrane water electrolysis is highly desired. However, transition metals with -valence electrons, which were doped into or alloyed with , are inherently susceptible to the influence of coordination environment, making it challenging to modulate the Ru-O covalency in a precise and continuous manner. Here, we first deduce that the introduction of lanthanide with gradually changing electronic configurations can continuously modulate the Ru-O covalency owing to the shielding effect of orbitals. Theoretical calculations confirm that the durability of following a volcanic trend as a function of Ru-O covalency. Among various is identified as the optimal catalyst and possesses a stability 35.5 times higher than that of . Particularly, the -based device requires only 1.837 V to reach and shows a long-term stability at for 100 h with a degradation rate of mere .
Renewable-driven water electrolysis is widely recognized as a promising and sustainable route to scalable production of green hydrogen . Proton exchange membrane water electrolysis (PEMWE) technology shows great potential on account of its high intermittent compatibility, low Ohmic resistance, high current density, low operating pressure, and limited side reactions . However, the widespread application of PEMWE is obstructed by the lack of efficient and costaffordable electrocatalysts for acidic oxygen evolution reaction . OER with four proton-coupled electron transfer necessitates a high overpotential due to its sluggish kinetics, thereby decreasing the operating efficiency of PEMWE . Besides, strong oxidative bias and extremely acidic corrosion collectively challenge the durability of available OER electrocatalysts for high-current-density operation .
Currently, iridium (Ir)-based catalysts, e.g. , remain the only practical choice for anode electrocatalysts in PEMWE due to their well balance in durability and activity . However, the spreading of PEMWE is drastically limited by the availability of Ir, which eventually leads to severe Ir shortage . In this context, numerous efforts have
been devoted to searching for alternatives to Ir, of which the most promising one is identified to be Ru-based oxides for their intrinsic high activity. Nevertheless, Ru-based OER catalysts always suffer from insufficient stability at industrial current density, which hampers their deployment in PEMWE for green hydrogen industry . The instability of Ru-based anode originates from the over-oxidation of Ru species, generating soluble under OER potentials . Despite considerable efforts on stabilizing Ru for OER electrocatalysis, a significant performance gap remains to meet the industrial requirements . To fill this gap, it would be more efficient to establish a fundamental and manageable strategy for stabilizing Ru-based OER electrocatalysts, instead of the conventional trial-and-error approach.
The stability of nanocrystalline -based catalysts is closely tied to the covalency of bonds . Weakening the bond covalency can localize and Ru orbitals below Fermi level, inhibiting lattice oxygen’s participation in OER and the formation of oxygen vacancies, thereby preventing excessive overoxidation of Ru species into soluble during . Meanwhile, excessively low
covalency is detrimental as it makes Ru be easily leached, leading to the direct demetallation of surface Ru and the subsequent structural degradation of . Prior studies focused on modulating the electronic structure of using , and metal substrates/ dopants . However, as orbital locates the outermost of transition metals, it is susceptible to external influences of crystal field and coordination environment . For instance, the introduction of consecutive elements such as , and into results in the transformation of their valence electrons into , , and , respectively . Such external influences render it be extremely challenging to modulate the Ru-O covalency in a precise and continuous manner.
Herein, we reason that lanthanide (Ln)-group elements with the orbital buried under can minimize external influences and consequently enable precise and continuous tuning of Ru-O covalency for durable OER electrocatalysis. Density functional theory (DFT) calculations were conducted on systems, showing that the resultant hybridization induces a continuously varying Ru-O covalency for dictating OER performance. Benefiting from an optimal covalency, was screened out as it demonstrated the largest formation energy of the lattice oxygen and Ru vacancy. The operando characterizations confirm the critical role of Er dopants in stabilizing the Ru-O structure for improved OER durability. Moreover, the up-shifted -state energy level ( -0.855 eV ) of , relevant to the benchmark , results in less electronic occupancy in the antibonding states and a stronger *OH adsorption, as validated by methanol molecular probe experiments, thereby significantly boosting the catalytic activity of . This work validates a novel regulating approach for precisely and continuously modulating Ru-O covalency, aiding to more economic affordability of PEMWE in a hydrogen economy scenario.
Results
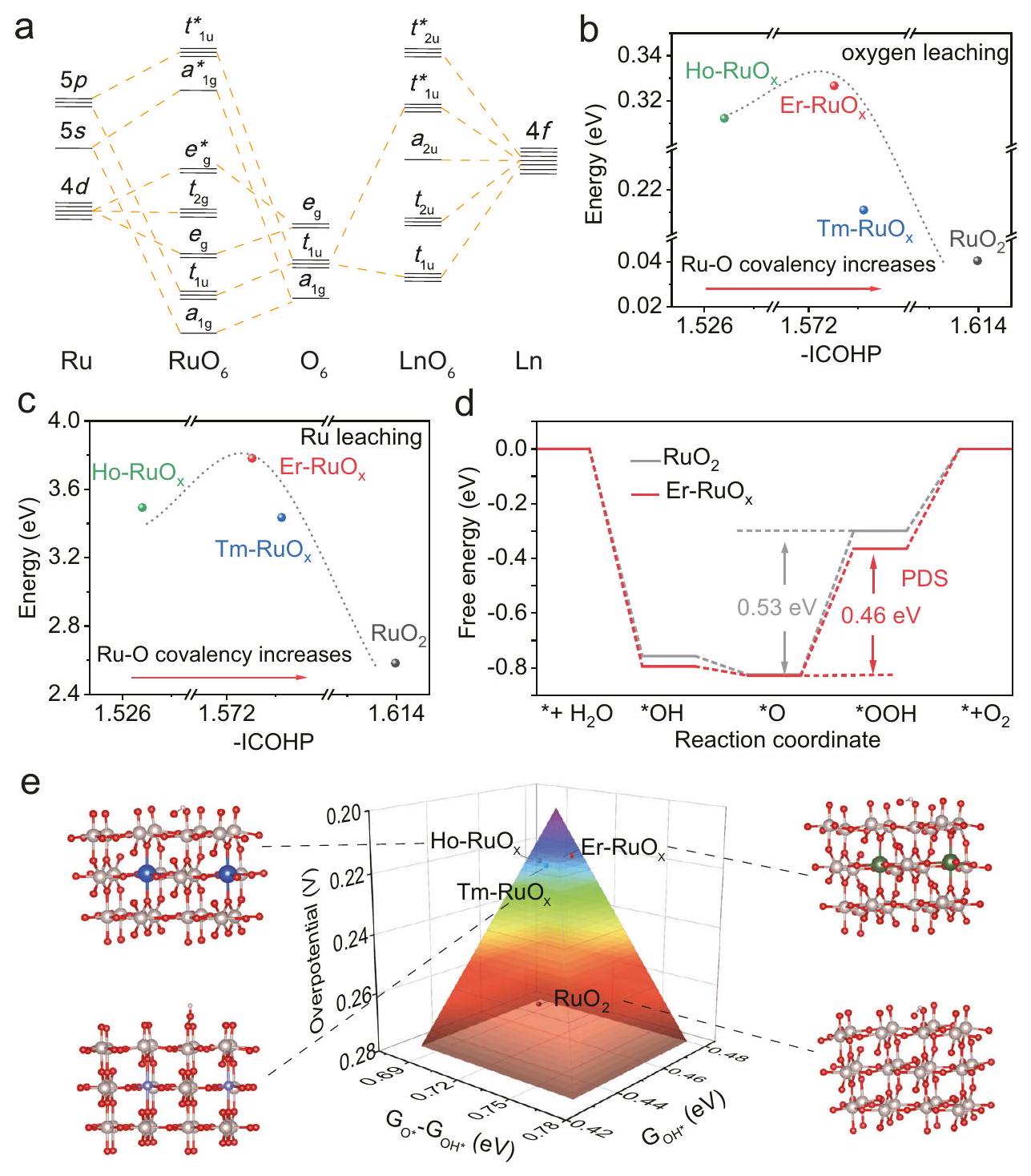
Lanthanide-regulating Ru-O covalency as OER descriptor
Ln elements offer a flexible avenue for fine-tuning and optimizing the electrocatalytic performance of catalysts due to the shielding effect of orbitals, gradient-filled orbital electron configuration, rich electronic energy levels, and the ability to accommodate various coordination numbers. The gradient orbital coupling of Ru, O , and Ln shows promise for enhancing the OER performance based on group theory-directed symmetric analysis . For , the orbital coupling of valence , and orbitals with -mixing orbitals of coordinated O atoms produces metal-oxygen (M-O) bonding states composed of , with their corresponding (MO )* antibonding states , and non-bonding terms. For [ ], the orbital coupling between and is contributed by and with their antibonding and terms, and nonbonding terms. Thanks to the conjunction with orbitals, the gradient orbital coupling of Ru-O-Ln can be formed (Fig. 1a), resulting in more flexible electronic interactions for electrocatalytic adaptation.
We first carried out the DFT calculations with Ru-O-Ln configuration constructed based on the rutile (Supplementary Figs. 1-4). The Bader charge analysis shows electron transfers from Ln to Ru , reducing the charge of Ru sequentially from to 1.50 , and (Supplementary Fig. 5), which confirms the orbital hybridization in . Through the analysis of bonding and antibonding orbital filling, the crystal orbital Hamilton population (COHP) and integrated COHP (ICOHP) calculations results demonstrate that the introduction of can weaken the bonding state occupancy from to -1.523 eV , and (Supplementary Fig. 6). The fine tuning of Ru-O covalency originates from the shielding effect of orbitals for Ln elements with gradient-filled orbital electron configuration.
To evaluate the stability of lattice oxygen and Ru, we calculated the formation energy of the lattice oxygen ( vacancy) and Ru vacancy ( ), which are utilized together to assess the stability of the electrocatalysts. As Ln dissolution would take place during OER process , defective structures containing Ln vacancies were constructed. In the presence of Ln vacancies, exhibits the highest , surpassing , and by 0.29 , 0.02 , and 0.12 eV , respectively (Fig. 1b). Specifically, the regulation of Ru-O covalency leads to a modified vacancy, increasing from 2.58 eV in to , and 3.44 eV for , , and , respectively (Fig. 1c). Considering the vacancy and vacancy in combination, the stability of follows the volcanic-like trend as a function of Ru-O covalency. This in turn verifies our proposed design principle that the fine tuning of Ru-O covalency by Ln regulates stability.
To further investigate the mechanism by which regulated the OER activity, we calculated the Gibbs free energy of oxygen intermediates during OER, yielding the theoretical overpotential for Ho, and . As shown in Fig. 1d and Supplementary Figs. 7 and 8, the potential determining step (PDS) for is the evolution from *O to *OOH, with a calculated overpotential of 0.53 V , while the overpotential decreases to , and 0.50 V for , and , respectively. The theoretical overpotential of various electrocatalysts are depicted in Fig. 1e employing a three-dimensional volcano-shaped plot that delineates the free energy difference between *O and *OH intermediates. Benefitting from the optimized covalency, demonstrates an enhanced binding strength (Supplementary Fig. 9) and provides near-optimal free energies for each intermediate, thus leading to the low theoretical OER overpotential. For the samples with both lower ) and higher ( ) Ru-O covalency, the free energy increase of PDS is observed. Ru-O covalency of the catalysts reflects the Ru-*OH bonding interaction, suggesting an optimal Ru-O covalency that is neither too weak nor too strong is favorable for OER.
Overall, the incorporation of Ln can continuously optimize the Ru-O covalency within a narrow range, thereby controlling the dissolution kinetics of -based catalysts and influencing the energy barriers of key reaction steps.
Materials synthesis and characterization
As theoretically possesses high OER activity and stability, it is selected for further investigation. To experimentally verify the predictions above, the catalyst was prepared by a one-pot glu-cose-blowing method. Pure was also synthesized employing the same method for comparison. The scanning electron microscopy (SEM) images (Supplementary Fig. 10) manifest that possesses a porous sheet-like structure with a specific surface area ( ) of (Supplementary Fig. 11). Furthermore, the porous sheets are composed of a large number of small nanoparticles (Supplementary Fig. 12). The X-ray diffraction (XRD) patterns reveal that possesses a rutile-type structure with no distinct diffraction peaks related to (Supplementary Fig. 13), suggesting the successful incorporation of Er atoms into . High-resolution transmission electron microscopy (HRTEM) image shows nanosheets consist of small nanoparticles with sizes around , which are composed of the (110) facet-dominated (Supplementary Fig. 14).
Furthermore, we utilized the aberration-corrected transmission electron microscopy (AC-TEM) to reveal the fine atomic-scale structure of the catalyst. The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images (Fig. 2a, d) show the clear edge of the Er-RuO grain. Figure 2 b and Supplementary Fig. 15 display the surface plots of , allowing the depiction of the intensity of the atomic columns. The atomic configuration of Er can be clearly seen from the atomic HAADF-STEM images along the [001] and [110] zone axes (Fig. 2c, e), consistent with that of pure
Fig. 1 | Prediction of the OER performance utilizing DFT calculations.
a The qualitative molecular orbital diagram obtained from and .
b The and (c) as a function of -ICOHP for . The
upshift values of ICOHP indicate lower Ru-O covalency. d The reaction paths on Er and at 1.23 V . e Volcano plot for different electrocatalysts and corresponding structures. . The energy-dispersive spectra (EDS) line scan in Fig. 2f was recorded from the HAADF-STEM image of (Fig. 2d, the green line ). The , and Er signals are detected and recorded in the line scan profile. Especially, the signals resulting from Er only appears in the regions of Ru signal, further indicating that Er was successfully introduced into . The relatively weak signal intensity of Er element in contrast to that of Ru reveals the low Er content, matching with the result of EDS spectra (Supplementary Fig. 16). The element mapping images further demonstrate the coexistence and atomic-scale distribution of Ru and Er atoms (Fig. ). Er atoms are distributed within the catalyst, with a preference for surface localization, which contributes to the electrocatalytic performance as the Ru-O covalency can be regulated as discussed above.
Electrocatalytic performance in three-electrode configurations and PEMWE devices
The OER performance of and the control samples were measured in solution in a three-electrode system. As exhibited in Fig. 3a, Er-RuO represents superior OER activity to commercial and home-made . Excitingly, the required overpotential to reach current density of is only on
and lower than those of commercial and home-made , respectively. Furthermore, when normalized to the electrochemical active area, the catalytic activity of remains better than that of commercial (Supplementary Fig. 19). The catalyst also demonstrates a decreased Tafel slope of compared to commercial (Fig. 3b), suggesting the boosted reaction kinetics.
The stability of the as-prepared catalysts was investigated by cyclic voltammetry (CV) between 1 V and 1.45 V vs. RHE. demonstrates a much smaller attenuation than commercial after 30,000 cycling tests (Fig. 3c). The concentration of dissolved Ru after cycles for was measured to be 13.7 ppb , which was much lower than that of commercial . These results suggest that the incorporation of Er suppresses the dissolution of . In addition, the catalytic durability was evaluated by chronopotentiometry (CP) at (Fig. 3d), demonstrating a more pronounced stability advantage of over commercial . In detail, the required overpotential of commercial increased by 674 mV after 73 h stability test at , which was times higher than that of , verifying the beneficial role of Er on catalytic stability. The OER performance of and was
Fig. 2 | Atomic-scale structure characterization of Er-RuO . a HAADF-STEM and b surface plot of along [001] zone axes, scale bar: 2 nm . c Atomic STEM image along [001] zone axes and structural model (inset) of Er-RuO , scale bar: . d HAADF-STEM (scale bar: 2 nm ) and (e) atomic STEM images (scale bar: ) of
Er-RuO along [110] zone axes. f EDS line scan of , and Er signal recorded from the green line in (d). HAADF-STEM image and corresponding element mapping images of along the ( ) [110] and ( ) [001] zone axes, scale bar: 2 nm . q Schematic illustration of .
shown in Supplementary Figs. 22-24, being in good agreement with DFT prediction. As shown in Supplementary Tables 6 and 7, both the activity and stability of were higher than those of the recently reported Ru-based electrocatalysts , confirming the economic efficiency of .
To investigate the application potential of for water electrolysis, we constructed a PEMWE device (Nafion 117 membrane) using and commercial as the anode and cathode catalyst, respectively (Fig. 3e). The current-voltage curves (without compensation) in Fig. 3f clearly demonstrate the superior water electrolysis performance of the -based PEMWE in comparison to the commercial PEMWE device. Specifically, the Er-RuO -based electrolyzer (at ) required only , and 1.837 V to reach an industrial current density of 1,2 , and , respectively, outperforming the PEMWE using the other state-of-the-art -based catalysts (Supplementary Table 8). Moreover, the PEMWE employing the catalyst achieves an efficiency of approximately at (Supplementary Fig. 26), with an estimated cost of about US$ 0.85 per kg of , which is significantly below the US Department of Energy (DOE)’s target of US$ 2 per kg of . The stability of the Er PEMWE device was evaluated at 200 , 500 , and , respectively, and no significant activity decay was observed over the device after a stability test for each condition (Fig. 3 g ). The degradation rate at is mere , demonstrating the application potential of for green hydrogen production.
Origin of the excellent performance of
X-ray photoelectron spectroscopy (XPS) was conducted to determine the chemical composition and valence states of the as-prepared catalysts. In the Ru XPS spectrum (Supplementary Fig. 27), the peaks at 463.7 and 463.1 eV can be attributed to and , respectively . Compared with , the ratio in is lower than that in , indicating a lower oxidation state of Ru in . To explore the interaction of Er and Ru, X-ray absorption spectroscopy (XAS) was utilized to probe the atomic and coordination environment. The X-ray absorption near-edge structure spectra (XANES) of Ru K-edge (Fig. 4a) reveal that the absorption threshold position of is higher than that of the Ru foil but lower than that of . The calculated oxidation state of Ru in is 3.80 (Supplementary Fig. 28), consistent with the results of XPS and Bader charge. Moreover, the Ru K-edge Fourier transform extended X-ray absorption fine structure (FT-EXAFS) spectrum of Er-RuO (Fig. 4b) displays a peak located at , assigned to the Ru-O bonds. This peak exhibits a positive shift towards longer interatomic distances in comparison to that of because of the slightly weakened covalency (Supplementary Table 9), matching well with theoretical calculation predictions. The Wavelet transform analysis also demonstrates that coordination in is similar to that of , while no bond is detected in , aligning with the FT-EXAFS results (Fig. ).
We then conducted the in situ Raman spectroscopy measurement (Supplementary Fig. 29) to elucidate the structure evolution during
Fig. 3 | Electrocatalytic performance of the as-prepared catalysts. a polarization curves of home-made and commercial with corrected ( ), where was measured to be . b Tafel plots of homemade and commercial . c Polarization curves of and commercial before (solid line) and after (dash line)
cycles. d The CP curves of Er-RuO and commercial at . e Schematic diagram of the PEMWE electrolyzer. Polarization curves of the and commercial -based PEMWE at . g The CP curves of Er--based PEMWE electrolyzer operated at 200,500 , and , respectively.
OER. Raman spectra shows the nanocrystalline nature of and (Fig. 4c, d), with two major peaks located at and , corresponding to the and , respectively. As the potential increases from open circuit potential (OCP) to 1.8 V vs. RHE, maintains a nearly constant Raman shift, indicating the stability of configurations. In contrast, red shift is observed in , suggesting the shrinkage in bonding length during OER.
Furthermore, Ru K -edge XANES of and were collected with applied potential rise from 1.3 to 1.7 V vs. RHE to investigate the changes in local Ru atomic structure and chemical coordination. As presented in Fig. 4e, f and Supplementary Fig. 30, and exhibit significant differences in the variation of Ru oxidation sates, especially at high bias. In detail, when the applied voltage is 1.3 V , the oxidation state of Ru in increases from 3.80 to 4.16, followed by a negligible change when potential further increased to 1.7 V . In contrast, the oxidation states of Ru in steadily increases to 4.36 as the applied bias varies from 1.3 to 1.7 V , showing no sign of stabilization. The result suggests that the stable structure of can be maintained at high oxidation potentials, whereas undergoes significant structural evolution. The Ru 3d peak in Er-RuO XPS spectrum after stability test is slightly positive-shift (Supplementary Fig. 31), indicating that the surface of is oxidized
to a higher valence state, consistent with the XANES results. TEM images after the stability test confirm that the crystalline structure of (Supplementary Fig. 32) is well preserved, indicating no significant structure deterioration occurred under the strong oxidative bias.
Adsorption behavior analysis of with oxygen intermediates
To rationalize the improved OER performance on , the adsorptions of and to oxygen intermediates were evaluated. As shown in Fig. 5a, exhibits a lower d-band center of Ru than that of the pure , indicating that the orbital hybridization slightly changes the electronic environment of Ru orbitals. Further, the orbitals can be hybridized into and with intermediates. The Ru -state energy level of up-shifts to the Fermi level relative to that of the (Fig. 5b), thereby resulting in less electronic occupancy in the antibonding states and stronger *OH adsorption (Fig. 5c). As shown in Fig. 5d, more charge accumulation toward *OH verifies a stronger electron transfer behavior between and , confirming the strengthened *OH adsorption.
The strengthened * OH adsorption was also experimentally verified using methanol as a probe. The methanol oxidation reaction
Fig. 4 | Structural characterization of and . a Ru K-edge XANES spectra of Ru foil, , and Er-RuO . b EXAFS spectra of Ru foil, , and . In situ Raman spectra obtained under various applied potential on (c) and (d)
Er-RuO . e Ru K-edge XANES spectra of Er-RuO with different applied bias. The variation trend of Ru oxidation states in and under different potentials. WT-EXAFS of (g) Ru foil, (h) , and (i) .
(MOR) follows a well-established mechanism, in which methanol molecules tend to nucleophilically attack the electrophilic *OH. As a result, MOR is more active on surfaces with stronger *OH adsorption . When 1.0 M methanol was introduced into the solution, the current densities of and showed a substantial increase compared to those before the addition of methanol, attributed to the methanol electrooxidation (Fig. 5e, f). The difference in current densities induced by MOR, which was directly proportional to the number of charges transferred, was quantified by calculating the filled area between the curves. The bigger current difference observed between the MOR and OER over than that of suggested its stronger MOR competition reaction, verifying the enhanced *OH adsorption on (Fig. 5 g ).
To acquire the information of oxygen intermediates for a more comprehensive mechanistic understanding, the in situ attenuated total reflection-surface enhanced infrared absorption spectroscopy (ATR-SEIRAS) was performed using a home-made electrochemical cell. The ATR-SEIRAS of both and were measured between 1000 and from 1.3 V to 1.7 V vs. RHE. As shown in Fig. 5 h , a potential-dependent peak at for , corresponding to the stretching vibration of species , became more prominent as the potential increased from 1.5 V to 1.7 V . For , the peak of *OOH species was emerged at 1.4 V (Fig. 5i), in accordance with the lower PDS of *O to *OOH (Fig. 1d). Meanwhile, the emergence of *OOH species indicated that the adsorbate evolution mechanism pathway dominated generation over .
Discussion
In summary, we demonstrate that precisely tuning of the Ru-O covalency in can be achieved by introducing Ln elements through orbital hybridization. Benefiting from the optimized Ru-O covalency, is screened out and exhibits the high catalytic stability, significantly outperforming pristine by orders of magnitude. The operando characterizations indicate that the oxidation state of Ru in initially increases as the applied potential increases and then remains nearly constant. In contrast, the oxidation state of Ru in continuously rises with no sign of stabilization. DFT calculations and methanol molecular probe experiments validate the stronger *OH adsorption on relative to that on , thus leading to the enhanced OER activity. The PEMWE (Nafion 117 membrane) employing as the anode catalyst requires only 1.837 V to reach and exhibit long-term stability at for 100 h with a degradation rate of mere . This study provides a principled catalyst design framework for the precise Ru-O covalency control, thereby guiding the development of ruthenium-based catalysts suitable for practical implementation in PEMWE systems.
Methods
Materials
Ruthenium chloride ( ) was purchased from Beijing InnoChem Science & Technology Co., Ltd. Holmium nitrate pentahydrate , erbium nitrate pentahydrate , ) and thulium nitrate hexahydrate was
Fig. 5|Analysis of adsorption behavior. a The calculated PDOS of Ru , and Er f orbitals of . The PDOS of orbital of and . Schematic diagram of orbital hybridization between orbital of and and the *OH bonding orbitals. d Charge density difference of *OH-adsorbed Er-RuO (left) and (right). Yellow and blue iso-surfaces represent charge accumulation and
depletion, respectively. Polarization curves of (e) and (f) in 0.5 M solution with (dashed lines) and without (solid lines) 1 M methanol. g Current difference between the polarization curves in solution with and without 1.0 M methanol for and . ATR-SEIRAS analysis of (h) commercial and (i) .
purchased from Shanghai Macklin Biochemical Co., Ltd. Urea (AR) and glucose (AR) were obtained from Beijing Tong Guang Fine Chemicals Company. Commercial Pt/C ( ) was obtained from Johnson Matthey Company. Nafion 117 membrane (thickness: 0.18 mm ) was obtained from DuPont Co. All reagents were used without further purification.
Synthesis of and
In the typical synthesis of and a certain amount of were added into 5 mL deionized water with 1 g urea and 5 g glucose (keeping the atomic ratio of at ). The mixture was stirred until a homogeneous solution was attained. Subsequently, it was subjected to heating at for 8 h in an oven to form a porous foam, and then annealed at for 10 h in the air to obtain . For comparison, pure was also prepared without the addition of .
Electrochemical measurements
All electrochemical measurements were conducted with the CHI 760E electrochemical workstation employing a three-electrode cell at room temperature. The as-prepared catalysts ( 2 mg ) were dispersed in a mixture of isopropanol and Nafion D-521 ( ) solution. After ultrasonication for 1 h , the homogeneous ink was carefully dropped onto the carbon paper ( ) to obtain the working electrode with a desirable loading of . The
reference electrode was , calibrated in a three-electrode system in which Pt wires served as both working electrode and counter electrode, and -saturated solution was employed as the electrolyte. Subsequently, CV was measured at a scan rate of . The average potential at which the current crosses zero was determined as the thermodynamic potential relative to . All potentials were calibrated to RHE by the equation: E (vs. RHE vs. . The graphite rod was used as the counter electrode. The polarization curves were performed at a scan rate of in solution ( 0.3). All linear sweep voltammetry (LSV) curves measured in threeelectrode cell were -corrected ( ) unless otherwise stated, where was measured to be . During electrolyte preparation, 13.6 mL sulfuric acid (98%) was added to a beaker containing a suitable volume of deionized water, followed by adjustment of the volume to 500 mL in a volumetric flask and shake to ensure thorough mixing. The electrolyte is freshly prepared and promptly utilized. Electrochemical impedance spectroscopy was performed at 1.485 V vs. RHE in the frequency range from to . CVs at various scan rates (namely, , and ) were performed to calculate the electrochemical active surface area (ECSA), which was proportional to the double layer capacitance . Assuming that the specific capacitance of a flat surface was for of real surface area, then the ECSA was estimated as: ECSA .
PEMWE tests
The PEMWE was assembled with or commercial as anode, with a loading of . Commercial was utilized as the cathode catalyst . Pt-coated Ti fiber was used as gas diffusion layers. Nafion 117 was used as the proton exchange membrane (PEM), which was sequentially treated with and at for 1 h . The anode and cathode catalyst ink were separately prepared by dispersing the catalyst in a mixture of Nafion (5%), isopropanol, and distilled water. All the cathode ink was sprayed onto the PEM. As for anode, half of catalyst ink was sprayed onto the PEM, while the remaining half was sprayed onto the surface of gas diffusion layer. The catalyst-loaded PEM and gas diffusion layers were then hot-pressed together at for 2 min under a pressure of 2 MPa to fabricate the membrane electrode assembly (MEA), which was sandwiched by two Ti bipolar plates to complete a PEMWE device. Each Ti bipolar plate featured a serpentine flow channel with 1 cm reactive area. The PEMWEs were operated at utilizing distilled water as the electrolyte, which was delivered to the anode by a peristaltic pump. All measurements in PEMWEs were recorded without iR-correction.
In situ ATR-SEIRAS measurements
The in situ ATR-SEIRAS measurement was carried out on Bruker 70 V Fourier-transform infrared (FTIR) spectrometer. The measurement featured a spectral resolution of , with 64 interferograms coadded for each spectrum. The preparation of working electrodes comprised of two steps: firstly, an ultra-thin Au film was chemical deposited on the Si crystal to enhance the IR signal and facilitate electron conduction; secondly, the catalyst ink was dropped onto the Au film with a loading of . The Si crystal loaded with catalyst was placed onto a spectro-electrochemical three-electrode cell. Ag/ AgCl electrode and Pt wire served as the reference and counter electrodes, respectively. The solution was used as the electrolyte. All SEIRAS spectra were obtained during the LSV test.
DFT calculations
The calculations were performed employing the density functional theory as implemented in the Vienna ab initio simulation package (VASP) . The exchange-correlation function was described using the generalized gradient approximation (GGA) parameterized by the Perdew-Burke-Ernzerhof (PBE). The cut-off energy for the plane wave basis was set to surface (Supplementary Fig. 1) was modeled by a supercell (with 55 O atoms and 24 Ru atoms), in which the top three atomic layers are allowed to relax. As for , two Ru atoms in the model was substituted by Ln atoms (Supplementary Figs. 2-4 and Supplementary Data 1), leading to a chemical composition of 8.3 at percentage Ln and 91.7 at percentage Ru. A vacuum spacing of was set along the z -direction to prevent the interaction between the slab and its periodic motif. The termination of and has all Ru filled with oxygen but one coordinatively unsaturated site Ru unfilled, which is the active site for intermediates adsorption. The Monkhorst-Pack method was used for sampling the Brillouin zone with a mesh. The geometry relaxation and convergence criteria for the electronic structure were and , respectively. The free energies of the reaction steps were calculated by the equation: , where is the adsorption energy of intermediates, and is temperature. and represent the energy variance in zero-point energy and entropy, respectively. When calculating the formation energy of Ru vacancies, the lost Ru atoms originate from unsaturated surface coordination sites, consistently from the same position across all models. In the calculation of oxygen vacancy formation energy, both and lose oxygen atoms surrounding unsaturated Ru coordination sites on the surface. ICOHP and Bader charge represent the values at active site.
Data availability
The data that support the conclusions of this study are available within the paper and Supplementary information. Source data are provided with this paper.
References
Chong, L. et al. La- and Mn-doped cobalt spinel oxygen evolution catalyst for proton exchange membrane electrolysis. Science 380, 609-616 (2023).
Seh, Z. W. et al. Combining theory and experiment in electrocatalysis: insights into materials design. Science 355, eaad4998 (2017).
Guo, J. et al. Direct seawater electrolysis by adjusting the local reaction environment of a catalyst. Nat. Energy 8, 264-272 (2023).
Li, F. & Baek, J.-B. Active site engineering accelerates water electrolysis. Nat. Synth. 3, 558-559 (2024).
Shi, Z. et al. Enhanced acidic water oxidation by dynamic migration of oxygen species at the catalyst/support interfaces. Angew. Chem. Int. Ed. 61, e202212341 (2022).
Shi, Z. et al. Customized reaction route for ruthenium oxide towards stabilized water oxidation in high-performance PEM electrolyzers. Nat. Commun. 14, 843 (2023).
Liu, H. et al. Eliminating over-oxidation of ruthenium oxides by niobium for highly stable electrocatalytic oxygen evolution in acidic media. Joule 7, 558-573 (2023).
Wang, Q. et al. Long-term stability challenges and opportunities in acidic oxygen evolution electrocatalysis. Angew. Chem. Int. Ed. 62, e202216645 (2023).
Ge, S. et al. A robust chromium-iridium oxide catalyst for high-current-density acidic oxygen evolution in proton exchange membrane electrolyzers. Energy Environ. Sci. 16, 3734-3742 (2023).
Liu, R.-T. et al. Recent advances in proton exchange membrane water electrolysis. Chem. Soc. Rev. 52, 5652-5683 (2023).
Chen, F.-Y., Wu, Z.-Y., Adler, Z. & Wang, H. Stability challenges of electrocatalytic oxygen evolution reaction: from mechanistic understanding to reactor design. Joule 5, 1704-1731 (2021).
Dang, Q. et al. Iridium metallene oxide for acidic oxygen evolution catalysis. Nat. Commun. 12, 6007 (2021).
Chen, S. et al. Reconstructed Ir-O-Mo species with strong Brønsted acidity for acidic water oxidation. Nat. Commun. 14, 4127 (2023).
Gao, J., Tao, H. & Liu, B. Progress of nonprecious-metal-based electrocatalysts for oxygen evolution in acidic media. Adv. Mater. 33, 2003786 (2021).
Li, L. et al. Electrochemically modifying the electronic structure of nanoparticles for overall electrochemical water splitting with extensive adaptability. Adv. Energy Mater. 10, 2001600 (2020).
Cao, L. et al. Dynamic oxygen adsorption on single-atomic ruthenium catalyst with high performance for acidic oxygen evolution reaction. Nat. Commun. 10, 4849 (2019).
Yao, Y. et al. Engineering the electronic structure of single atom Ru sites via compressive strain boosts acidic water oxidation electrocatalysis. Nat. Catal. 2, 304-313 (2019).
Zagalskaya, A. & Alexandrov, V. Role of defects in the interplay between adsorbate evolving and lattice oxygen mechanisms of the oxygen evolution reaction in and . ACS Catal. 10, 3650-3657 (2020).
Wu, Z.-Y. et al. Non-iridium-based electrocatalyst for durable acidic oxygen evolution reaction in proton exchange membrane water electrolysis. Nat. Mater. 22, 100-108 (2023).
Wang, Y. et al. Unraveling oxygen vacancy site mechanism of Rhdoped catalyst for long-lasting acidic water oxidation. Nat. Commun. 14, 1412 (2023).
Galyamin, D. et al. Active and durable pyrochlores with low Ru content for acidic oxygen evolution. Nat. Commun. 14, 2010 (2023).
Jin, H. et al. Safeguarding the phase against lattice oxygen oxidation during acidic water electrooxidation. Energy Environ. Sci. 15, 1119-1130 (2022).
Miao, X. et al. Quadruple perovskite ruthenate as a highly efficient catalyst for acidic water oxidation. Nat. Commun. 10, 3809 (2019).
Zhu, W. et al. Direct dioxygen radical coupling driven by octahedral ruthenium-oxygen-cobalt collaborative coordination for acidic oxygen evolution reaction. J. Am. Chem. Soc. 145, 17995-18006 (2023).
Li, L. et al. Spin-polarization strategy for enhanced acidic oxygen evolution activity. Adv. Mater. 35, 2302966 (2023).
Li, L. et al. Compensating electronic effect enables fast site-to-site electron transfer over ultrathin RuMn nanosheet branches toward highly electroactive and stable water splitting. Adv. Mater. 33, 2105308 (2021).
Zheng, X. et al. Ru-Co pair sites catalyst boosts the energetics for the oxygen evolution reaction. Angew. Chem. Int. Ed. 61, e202205946 (2022).
Su, J. et al. Assembling ultrasmall copper-doped ruthenium oxide nanocrystals into hollow porous polyhedra: highly robust electrocatalysts for oxygen evolution in acidic media. Adv. Mater. 30, 1801351 (2018).
Zhang, D. et al. Construction of Zn -doped nanowires for efficient and stable water oxidation in acidic media. Nat. Commun. 14, 2517 (2023).
Wang, X. et al. Engineering 3d-2p-4f gradient orbital coupling to enhance electrocatalytic oxygen reduction. Adv. Mater. 34, 2206540 (2022).
Wang, X. et al. Spin-selective coupling in Mott-Schottky boosts electrocatalytic oxygen reduction. Small Methods 7, 2300100 (2023).
Li, L. et al. Optimizing the electronic structure of ruthenium oxide by neodymium doping for enhanced acidic oxygen evolution catalysis. Adv. Funct. Mater. 33, 2213304 (2023).
Bender, G. & Dinh, H. N. HydroGEN: Low-Temperature Electrolysis (LTE) and LTE/Hybrid Supernode (No. NREL/PR-5900-659). (National Renewable Energy Lab.(NREL), 2020).
Hu, Y. et al. Single Ru atoms stabilized by hybrid amorphous/crystalline FeCoNi layered double hydroxide for ultraefficient oxygen evolution. Adv. Energy Mater. 11, 2002816 (2021).
Tao, H. B. et al. A general method to probe oxygen evolution intermediates at operating conditions. Joule 3, 1498-1509 (2019).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Acknowledgements
S.J.G. acknowledge the fundings from National Natural Science Foundation of China (Nos. 52333010, 52025133, 22309004, 52261135633, 52303363, 52302207, 22205010, 22305010, 22105007), National Key R&D Program of China (No. 2022YFE0128500), China National Petroleum Corporation-Peking University Strategic Cooperation Project of Fundamental Research, the Beijing Natural Science Foundation
(No. Z220020), New Cornerstone Science Foundation through the XPLORER PRIZE, CNPC Innovation Found (No. 2021DQ02-1002), China National Postdoctoral Program for Innovative Talents (No. BX20220009), China Postdoctoral Science Foundation (Nos. 2022M720225, 2023M730029, 2022M710187, 2023M730051, 2020M670018) and Yunnan Fundamental Research Projects (grant NO. 202401ATO7O37O). This work was carried out with the support of 1W1B beamline at Beijing Synchrotron Radiation Facility. The authors thank the photoemission photoendstations BL14W1 in the Shanghai Synchrotron Radiation Facility (SSRF) for the help with characterizations.
Author contributions
S.J.G. conceived and supervised the project. M.C.L. guided and supervised the whole research. L.L. and G.W.Z. performed the experiments, collected and analyzed the data. C.H.Z., F.L., H.L. and D.W.W. participated in the PEMWE tests. Y.J.T., Y.H., Y.X.L. and C.S.S. helped with the Operando ATR-SEIRAS measurements. L.Y.Z., Q.Z.H., R.J.Z. and N.Y. participated in part of basic experiments. L.L. wrote the manuscript. All authors took part in the discussion of data and gave comments on the manuscript.
Correspondence and requests for materials should be addressed to Shaojun Guo.
Peer review information Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/ licenses/by/4.0/.
(c) The Author(s) 2024
School of Materials Science and Engineering, Peking University, Beijing, China. Faculty of Metallurgical and Energy Engineering, Kunming University of Science and Technology, Kunming, Yunnan, China. These authors contributed equally: Lu Li, Gengwei Zhang. e-mail: guosj@pku.edu.cn