رؤى لعلم الأورام الدقيق من دمج البيانات الجينومية والسريرية لـ 13,880 ورم من برنامج جينومات السرطان 100,000 Insights for precision oncology from the integration of genomic and clinical data of 13,880 tumors from the 100,000 Genomes Cancer Programme
رؤى حول علم الأورام الدقيق من دمج البيانات الجينومية والسريرية لـ 13,880 ورم من برنامج جينومات السرطان
تاريخ الاستلام: 19 ديسمبر 2022
تاريخ القبول: 2 نوفمبر 2023
تاريخ النشر على الإنترنت: 11 يناير 2024
(ط) تحقق من التحديثات
تظهر قائمة المؤلفين وانتماءاتهم في نهاية الورقة
كان برنامج السرطان لمشروع الجينومات الـ 100,000 مبادرة لتوفير تسلسل الجينوم الكامل (WGS) للمرضى المصابين بالسرطان، وتقييم الفرص لرعاية السرطان الدقيقة ضمن نظام الرعاية الصحية الوطني في المملكة المتحدة (NHS). قامت جينومكس إنجلترا، جنبًا إلى جنب مع NHS إنجلترا، بتحليل بيانات WGS من 13,880 ورم صلب تغطي 33 نوعًا من السرطان، ودمج البيانات الجينومية مع بيانات العلاج والنتائج في العالم الحقيقي، ضمن بيئة بحثية آمنة. اختلفت نسبة الطفرات الجسدية في الجينات الموصى بها للاختبار القياسي عبر أنواع السرطان. على سبيل المثال، في الورم الدبقي متعدد الأشكال، كانت المتغيرات الصغيرة موجودة في 94% من الحالات وخلل عدد النسخ في جين واحد على الأقل في 58% من الحالات، بينما أظهر الساركوما أعلى حدوث للمتغيرات الهيكلية القابلة للتنفيذ (13%). تم تحديد نقص إعادة التركيب المتماثل في 40% من حالات سرطان المبيض عالي الدرجة مع المرتبطة بالطفرات الجينية المسببة للأمراض، مما يبرز قيمة التحليل المشترك للجسيمات والجينات الجرثومية. سمح ربط بيانات WGS وبيانات الحياة السريرية الطولية بتقييم نتائج العلاج للمرضى المصنفين وفقًا للعلامات الجينومية الشاملة. تظهر نتائجنا فائدة ربط البيانات الجينومية والسريرية في العالم الحقيقي لتمكين تحليل البقاء لتحديد جينات السرطان التي تؤثر على التشخيص وتعزيز فهمنا لكيفية تأثير علم جينوم السرطان على نتائج المرضى.
على مدار العقد الماضي، زادت نسبة الإصابة بالسرطان في المملكة المتحدة بنحو 4% (مرجع 1)، مما زاد الحاجة إلى الاختبارات الجزيئية للسرطان، بما في ذلك الاختبارات الجرثومية لجينات الاستعداد للسرطان وعلامات الأدوية الجينية . كان مشروع الجينومات الـ 100,000، وهو مبادرة تحويلية من الحكومة البريطانية أجريت ضمن خدمة الصحة الوطنية (NHS) في إنجلترا، يهدف إلى إنشاء تسلسل جينوم كامل موحد عالي الإنتاجية (WGS) للمرضى المصابين بالسرطان والأمراض النادرة عبر خط أنابيب معلومات حيوية معتمد من منظمة دولية
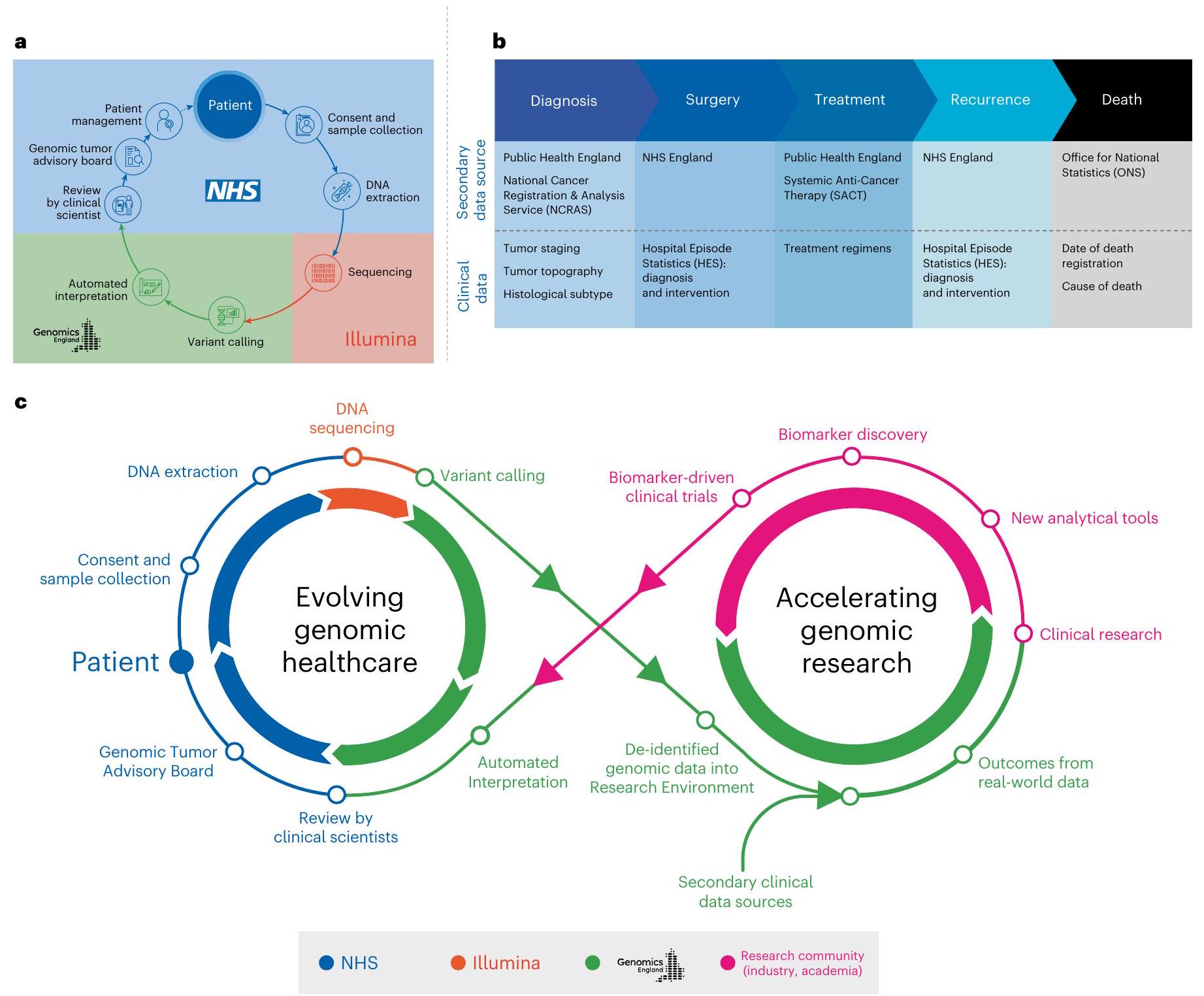
للتوحيد – معتمد من منظمة التوحيد الدولية (يوفر استدعاء المتغيرات المعتمدة سريريًا وترتيب المتغيرات) . تم تقييم دور WGS على نطاق واسع للمرضى المصابين بالسرطان في NHS ضمن برنامج السرطان لمشروع الجينومات الـ 100,000 (الشكل 1أ). قدم المشاركون موافقة مستنيرة مكتوبة لربط بياناتهم الجينومية بسجلات صحية طولية مجهولة الهوية ومشاركتها مع الباحثين في بيئة بحثية آمنة (www.genomic-sengland.co.uk/research/research-environment) لدفع معرفتنا
عبر أنواع السرطان المختلفة . ثم تم استخدام البيانات الناتجة لإنشاء منصة بيانات جزيئية وطنية (مكتبة البحث الجينومي الوطنية) مع روابط آمنة لبيانات العالم الحقيقي الطولية في بيئة البحث (الشكل 1ب). تشمل مجموعات البيانات السريرية الوطنية مجموعة بيانات تسجيل وتحليل السرطان الوطنية (NCRAS) التي تتكون من بيانات تسجيل السرطان ومجموعة بيانات العلاج الكيميائي المضاد للسرطان (SACT)، بالإضافة إلى حلقات السرطان اللاحقة، بما في ذلك إحصاءات حلقات المستشفى (HES) وبيانات الوفيات من مكتب الإحصاءات الوطنية (ONS) (الشكل 1ب). تتيح هذه الطريقة إجراء الأبحاث الجينومية والاكتشافات لتغذية الرعاية الصحية الجينومية (الشكل 1ج).
كان الهدف على المدى الطويل هو تسريع تقديم الاختبارات الجزيئية، بما في ذلك WGS، في رعاية السرطان السريرية NHS . بناءً على المعرفة المتطورة من مشروع الجينومات الـ 100,000 وتوفير الاختبارات الجزيئية الحالية ضمن NHS، تم إطلاق خدمة الطب الجينومي NHS (GMS) في أكتوبر 2018 لتقديم الاختبارات الجينومية والرعاية السريرية والتفسير للأمراض النادرة والسرطان في جميع أنحاء إنجلترا، باستخدام دليل اختبار جينومي وطني موحد , بما في ذلك لوحات جينات كبيرة مستهدفة وWGS، لتمكين الوصول العادل والاختبار الجينومي الشامل. يهدف دليل الاختبار الجينومي الوطني إلى توفير اتساق في منهجيات الاختبار، وأهداف الجينات ومعايير الأهلية عبر المؤشرات السريرية من خلال شبكة موحدة من سبعة مختبرات جينومية إقليمية NHS إنجلترا (NHSE) . يحدد الاختبارات الجينومية التي يتم تكليفها وبالتالي تمويلها من قبل NHS في إنجلترا كجزء من ملف تعريف الجزيئات القياسي الذهبي في مؤشرات السرطان السريرية المختلفة ويوفر فرصًا للمرضى للمشاركة في الأبحاث؟
لقد قامت دراسات التسلسل على نطاق واسع مثل التحالف الدولي لجينوم السرطان (ICGC) و أطلس جينوم السرطان (TCGA) بتوثيق طيف الطفرات الجسدية عبر أنواع السرطان من مجموعة استعادية من 2,658 عينة ورم أولية . وقد أبلغت مبادرات أكثر حداثة، مثل مؤسسة هارتويغ الطبية، عن نتائج ذات صلة سريريًا لـ 4,784 عينة ورم صلب بالغ نقيلة ودعمت التوظيف في تجربة بروتوكول إعادة اكتشاف الأدوية (DRUP) . تمثل هذه المبادرات، حتى الآن، أكبر مجموعتين من بيانات WGS المتاحة للبحث. في هذه المقالة، نقدم تحليلنا لبيانات WGS من 13,880 ورم صلب، مع التركيز على الجينات القابلة للتنفيذ سريريًا والعلامات الجينومية الشاملة، المرتبطة ببيانات العلاج والنجاة الطولية في العالم الحقيقي لتسليط الضوء على الدروس المستفادة من برنامج السرطان والآثار على الرعاية السريرية الحالية.
النتائج
الخصائص الديموغرافية للمجموعة
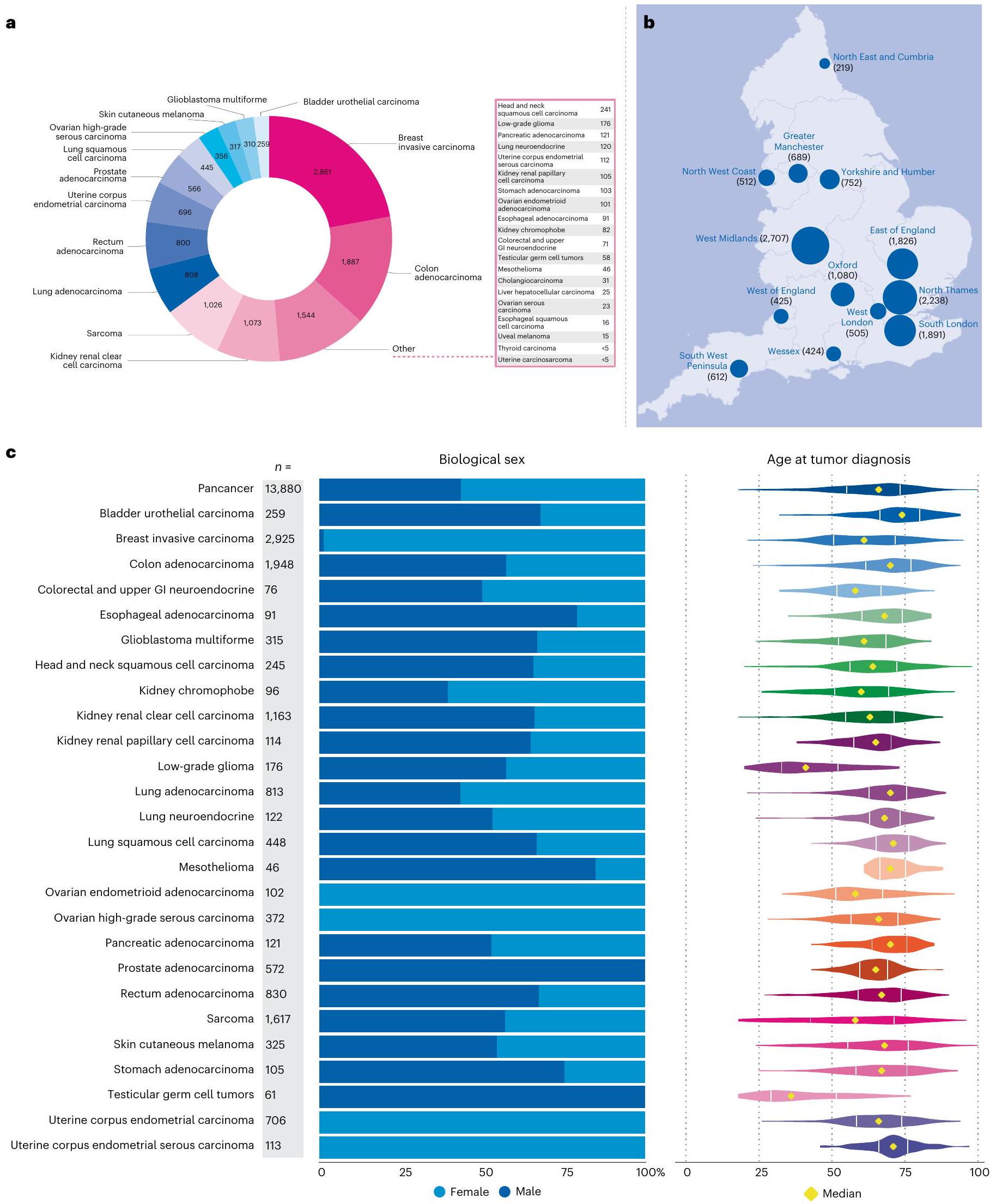
قمنا بتسلسل 16,358 زوجًا من عينات الورم الطبيعي من 15,241 مريضًا تم تشخيصهم بالسرطان ضمن NHS الذين تم تجنيدهم في برنامج السرطان لمشروع الجينومات الـ 100,000 بين عامي 2015 و2019، مع تجنيد ما يقرب من نصف المرضى في عام 2018 والباقي في هذا المشروع تم تجنيدهم من خلال ذراع الأمراض النادرة. غطت تحليلاتنا الجينومية الكاملة (WGA) 33 نوعًا من الأورام (الشكل 2أ) من 13,880 عينة ورم، تتكون من 13,311 عينة مجمدة طازجة () و569 عينة ورم مثبتة في الفورمالين (4.1%). تضمنت عينات الدم الطبيعية (الجرثومية) 13,493 (99.1%) مأخوذة من الدم، و100 (0.7%) من الأنسجة الطبيعية و23 (0.2%) من عينات اللعاب. تم تسلسل عينات الورم إلى تغطية وعينات الدم الطبيعية إلى لضمان حساسية عالية لاستدعاء المتغيرات (الطرق) في الإعدادات السريرية (مقارنة بـ و في مجموعة TCGA). تم استبعاد الجينومات من الأورام الدموية () والسرطانات الأطفال () والسرطانات ذات الأصل غير المعروف () والأورام التي لم ترتبط بمجموعات بيانات خارجية () من هذا التحليل. تم تأكيد التشخيص المقدم عند جمع العينات من خلال ربط بيانات الجينوم مع مجموعات بيانات NCRAS وHES. تضمنت أنواع الأورام التي تم تسلسل أكثر من 1,000 جينوم ورم الثدي الغازي () والقولون الغدي () والساركوما () وسرطان الكلى الخلوي الواضح (). يوضح الشكل 2ب التجنيد عبر 13 مركز جينومي NHS (تشمل أكثر من
80 مؤسسة مستشفى) في إنجلترا. يتم عرض توزيع الجنس البيولوجي والعمر عبر أنواع الأورام في الشكل 2ج. لوحظ ظهور مبكر (متوسط العمر أقل من 50 عامًا) لورم الدبقي منخفض الدرجة وأورام الخلايا الجرثومية الخصوية بما يتماشى مع إحصائيات الإصابة .
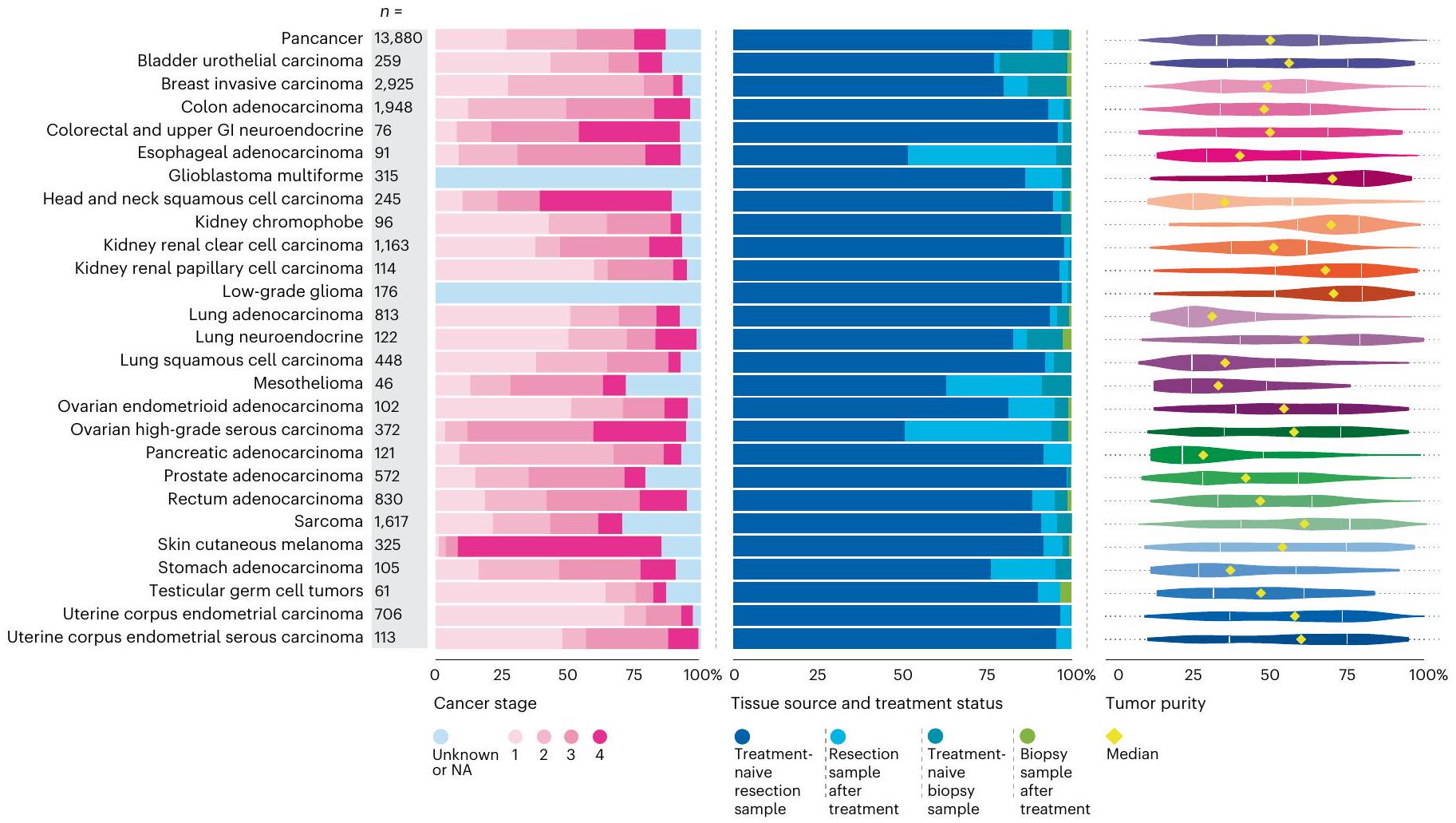
كانت معلومات المرحلة متاحة في مجموعة بيانات NCRAS لـ 12,040 (86.7%) ورم. يتم عرض توزيع المراحل المختلفة لأنواع الأورام التي تم تسلسلها في الشكل 3؛ من 13,880) من المرضى كانوا يعانون من سرطان المرحلة 4 (مرض نقيل متقدم) مع الحصول على عينات من مواقع نقيلية تشمل الكبد، والعقد اللمفاوية، والرئة والدماغ. أظهر سرطان المبيض عالي الدرجة وسرطان الجلد الميلانيني انتشارًا أعلى للمرض المتقدم (المراحل 3 و4)، بينما كانت سرطانات الثدي الغازية لها انتشار أعلى للمرحلة المبكرة (المراحل 1 و2) بسبب تحيزات أخذ العينات في تحديد الأنسجة. نشأت عينات الورم بشكل رئيسي من الاستئصال الجراحي ()، بما في ذلك حالات غير معالجة و حالات بعد العلاج المساعد. فقط 5.5% (جاءت من خزعات نقيلية أو تشخيصية، بنسبة 10.9% ( ) بعد العلاج (الشكل 3). توضح نقاء الورم الموضح في الشكل 3 التحديات في الحصول على عينات تحتوي على محتوى ورمي كافٍ (أكثر من ) في أنواع معينة من السرطانات، مثل سرطان الرئة وسرطان البنكرياس الغدي، وهو ما يتماشى مع المنشورات السابقة .
العمل السريري من خلال تسلسل الجينوم الكامل
يمكن أن يسهل اختبار واحد مثل تسلسل الجينوم الكامل (WGS)، الذي يتضمن تسلسل الورم الطبيعي المزدوج، الكشف المتزامن عن المتغيرات الصغيرة الجسدية بما في ذلك المتغيرات أحادية النوكليوتيد (SNVs) والإضافات والحذوفات (indels)، والانحرافات في عدد النسخ (CNAs) والمتغيرات الهيكلية (SVs)، بما في ذلك اندماجات الجينات. بالإضافة إلى ذلك، مكنت النتائج الجينية، مثل المتغيرات في جينات القابلية للإصابة بالسرطان والنتائج الصيدلانية الجينية (المتغيرات التي تؤثر على استقلاب العوامل العلاجية المستخدمة لعلاج السرطانات)، من تحقيق عائد أكبر من النتائج السريرية ذات الصلة. قدم برنامج السرطان نتائج WGA موحدة، تم إنشاؤها في خط أنابيب معلومات حيوية آلي، وعادت إلى مختبرات NHS GMC. تمت مراجعة النتائج القابلة للتنفيذ المحتمل في البداية من قبل العلماء السريريين ومن ثم في مجالس الأورام الجزيئية متعددة التخصصات، المشار إليها باسم مجالس استشارات الأورام الجينومية (GTABs). تظهر أمثلة على نتائج WGA في المعلومات التكميلية؛ وتوصف التفاصيل الكاملة للتحليل والتفسير في الطرق، مما يظهر فائدة WGS في التقاط التغيرات الجينومية المختلفة ذات الصلة السريرية من خلال اختبار واحد.
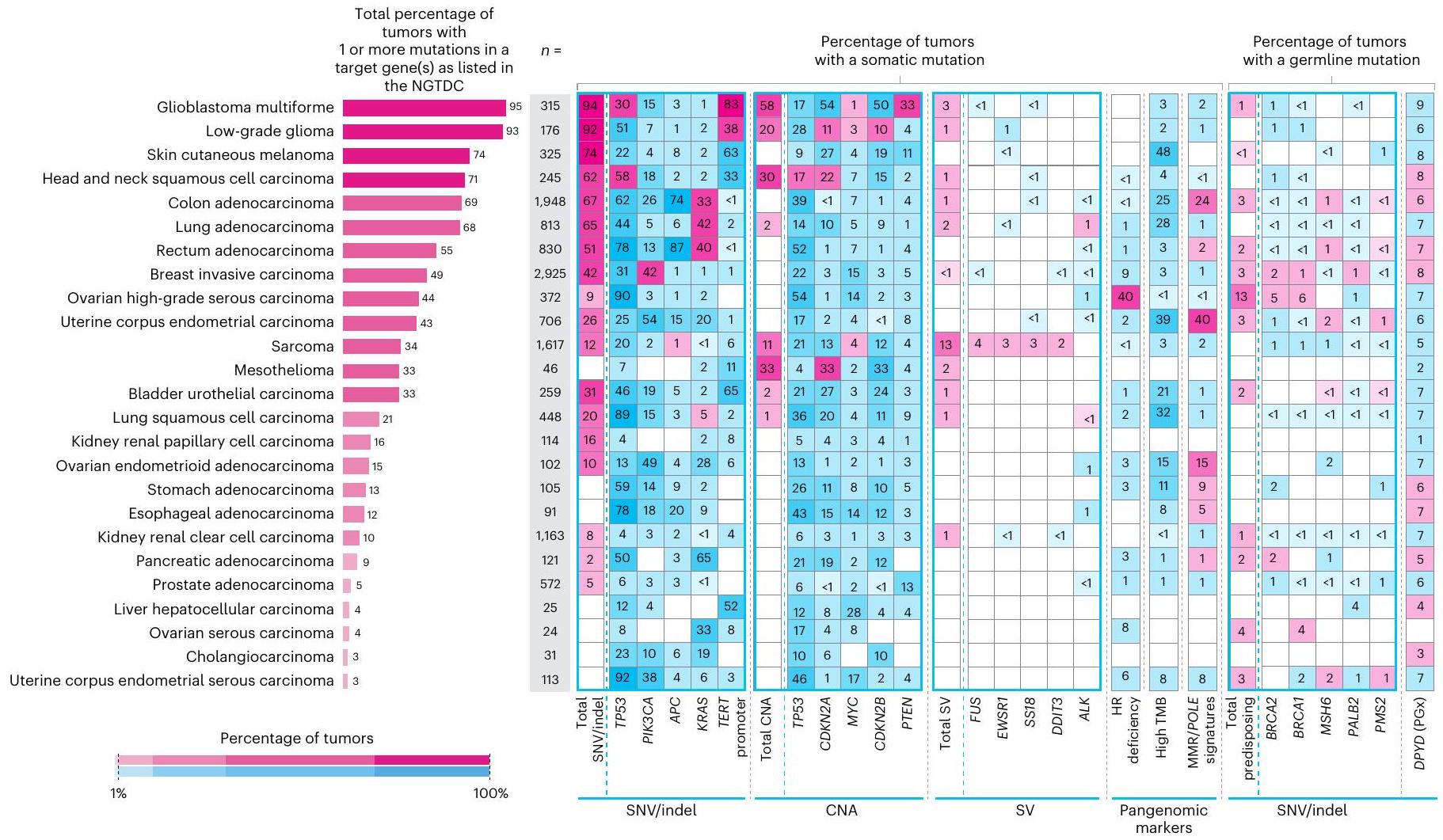
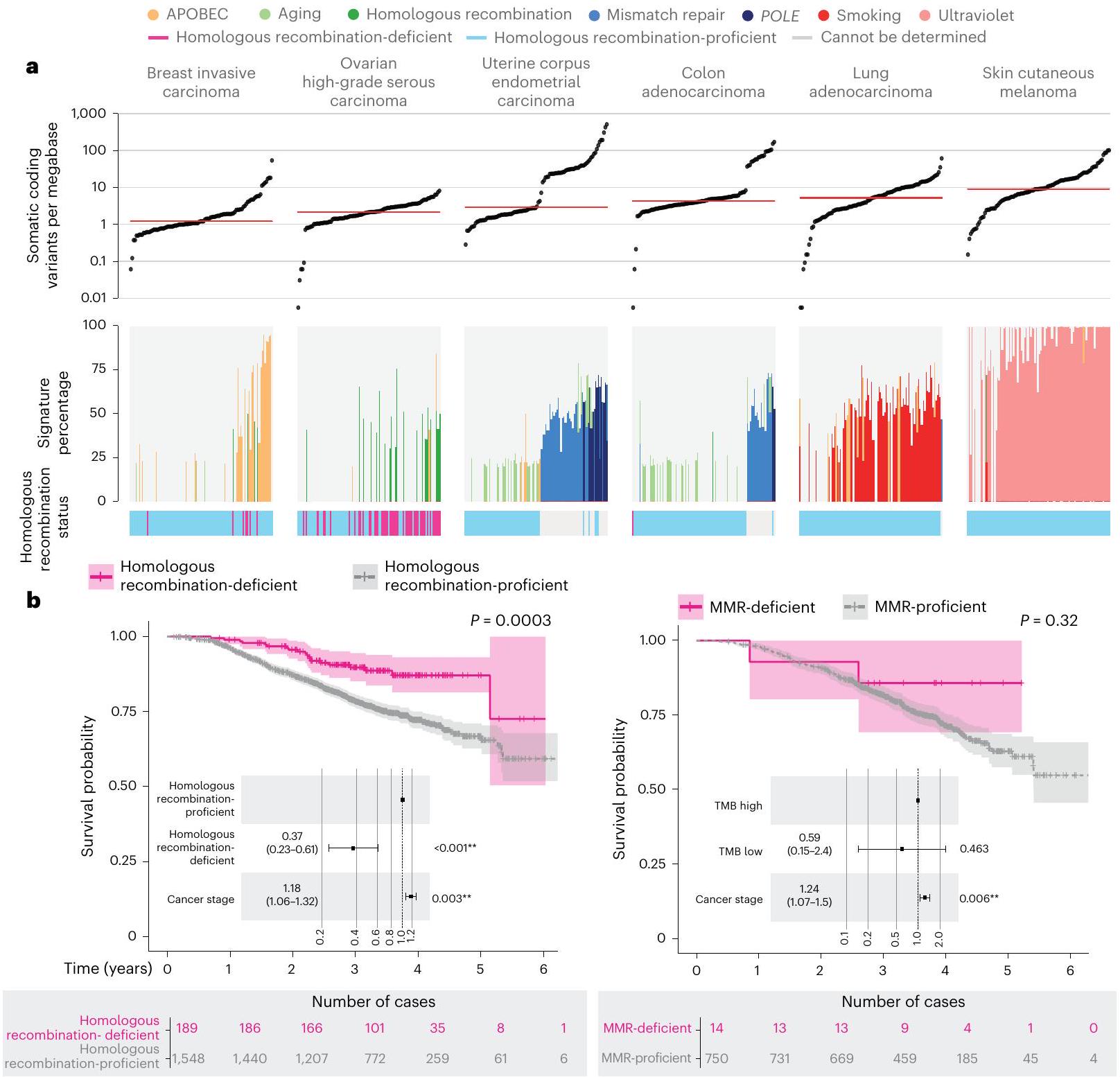
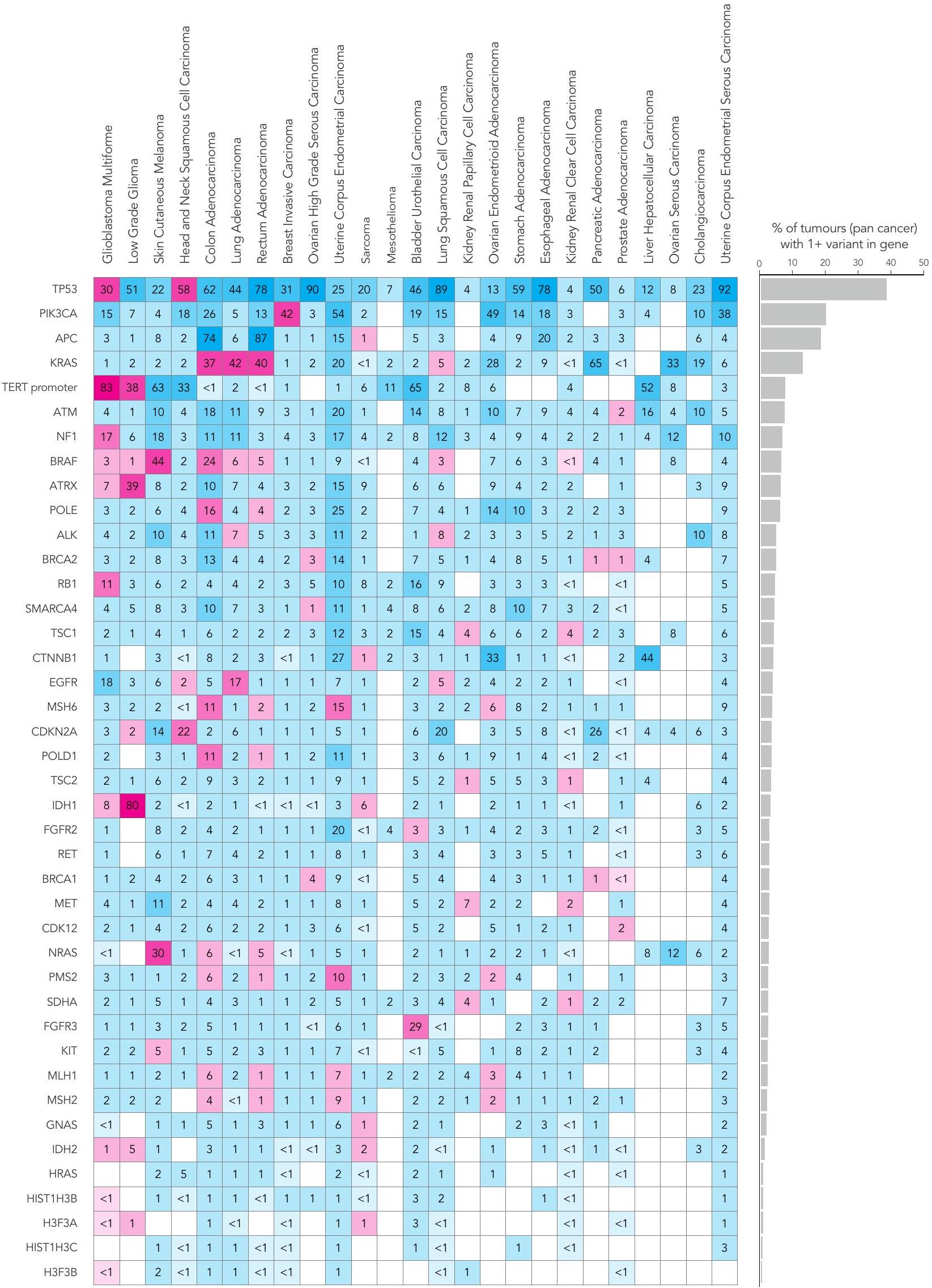
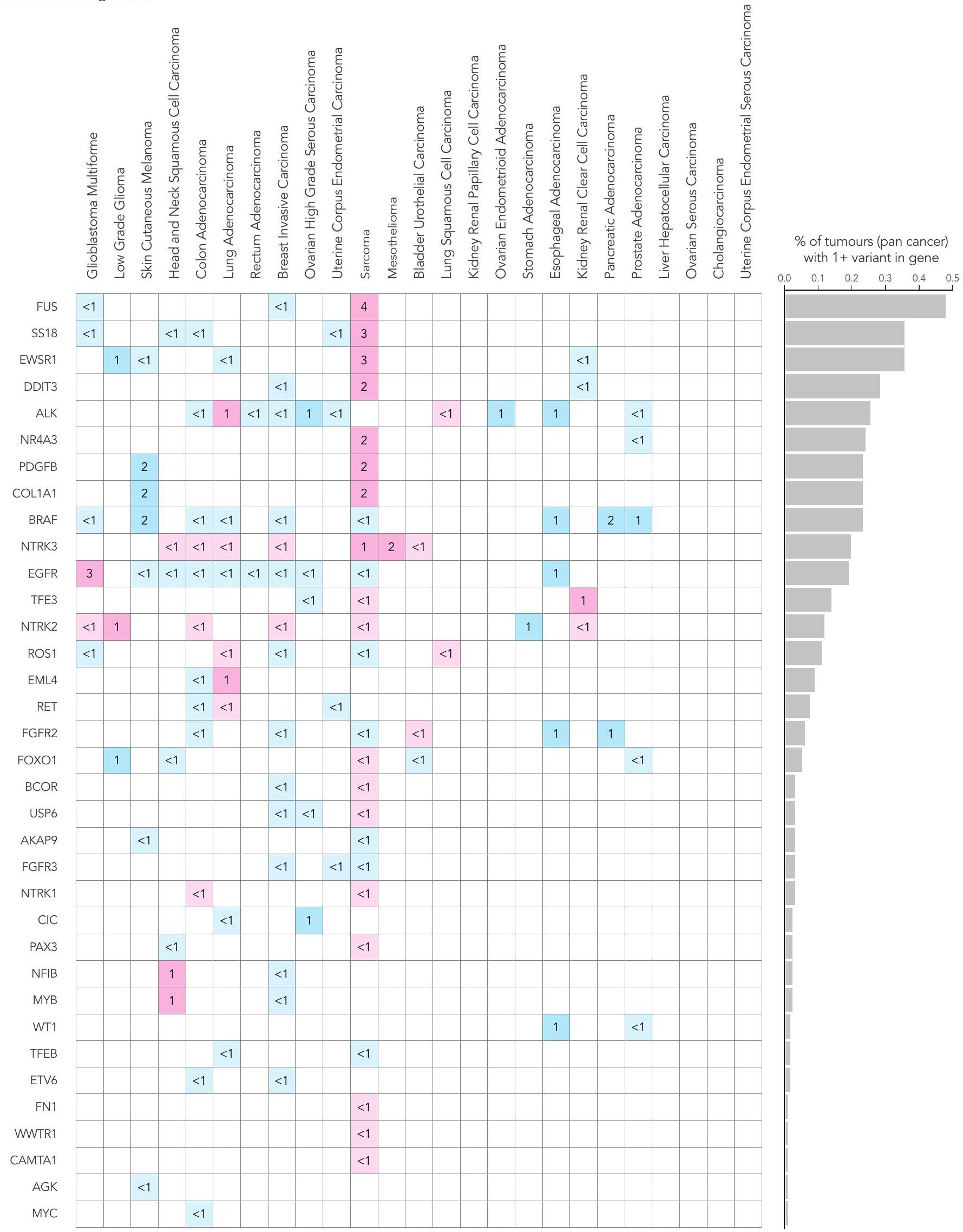
قمنا بتحليل بيانات مجمعة من 13,880 جينوم كامل في سياق الدليل الوطني الحالي للاختبارات الجينومية للسرطان (NGTDC) الإصدار 6.0 المحدث في 3 أبريل 2023 (المرجع 7)؛ تم اكتشاف عدة أنواع من الطفرات المتعلقة بالأهداف المحددة في NGTDC، بما في ذلك المتغيرات الصغيرة، والتغيرات العددية في الكروموسومات، والاندماجات، بالإضافة إلى المتغيرات الجرثومية المرتبطة بمخاطر السرطان الوراثي ونتائج الصيدلة الجينومية (انظر الطرق عبر الإنترنت لمزيد من التفاصيل). كانت نسبة الحالات التي تحتوي على طفرة جسدية واحدة أو أكثر في الجينات المشار إليها في NGTDC لنوع السرطان المعني مرتفعة، على الرغم من أنها متغيرة (الشكل 4). على سبيل المثال، أكثر منمن الأورام التي تحتوي على طفرة واحدة أو أكثر تم العثور عليها في الجينات المحددة للاختبار في NGTDC في الورم الدبقي متعدد الأشكال، الورم الدبقي منخفض الدرجة، الميلانوما الجلدية، سرطان الخلايا الحرشفية في الرأس والعنق، أدينوكارسينوما القولون والمستقيم، وأدينوكارسينوما الرئة (الشكل 4). تم العثور على طفرات ذات صلة سريرية في 20-49% من حالات سرطان الثدي الغازي، سرطان المبيض عالي الدرجة، بطانة الرحم، الساركوما، الميزوثليوما، سرطان الخلايا الانتقالية في المثانة وسرطان الخلايا الحرشفية في الرئة، بينما في أنواع السرطان الأخرى مثل أدينوكارسينوما البنكرياس، البروستاتا، المريء والمعدة، كانت النسبة أقل منمن الحالات التي تمتلك طفرات في الجينات الموجودة في NGTDC (الشكل 4). نلاحظ أن القابلية السريرية للعمل على هذه الطفرات ستعتمد على الحالة الفردية والظروف السريرية، مثل مرحلة الورم والأمراض المصاحبة للمشارك. وهذا يبرز الحاجة إلى التفسير السريري والنقاش حيثما كان ذلك مناسبًا سريريًا ضمن GTAB.
الشكل 1| نظرة عامة علىبرنامج جينومات السرطان. أ، رحلة جينوم المريض. قدم المرضى موافقة خطية مستنيرة لتحليل تسلسل الجينوم الكامل للورم والعينة الطبيعية (الجرثومية). تم استخراج الحمض النووي من عينات الورم والعينة الطبيعية (الدم) باستخدام بروتوكولات موحدة وتم تقديم العينات لتحليل تسلسل الجينوم الكامل، الذي تم إجراؤه على جهاز تسلسل إيلومينا. تم إنشاء خط أنابيب آلي لمراقبة جودة التسلسل، والمحاذاة، واستدعاء المتغيرات و تفسير، مع نتائج تُعاد إلى 13 مركزًا للطب الجيني في NHS للمراجعة في GTABs الإقليمية. ب، مجموعات بيانات جينية وسريرية مرتبطة بالعالم الحقيقي. في مشروع 100,000 جينوم، يتم متابعة المشاركين على مدار حياتهم باستخدام السجلات الصحية الإلكترونية (جميع حلقات المستشفى، إدخالات تسجيل السرطان، العلاجات المضادة للسرطان النظامية وسبب الوفاة). ج، حلقة لانهائية تمثل الرابط بين الرعاية الصحية والبحث في علم الجينوم.
قمنا بتقييم الطفرات المدرجة في NGTDC في أنواع السرطان الأخرى التي لا يُشار حاليًا إلى اختبار تلك الجينات أو الطفرات لها (الشكل 4 وفي الشكل الإضافي 1a-d). يتم الإشارة إلى هذه المتغيرات باللون الأزرق في الشكل 4 وقد تشير إلى نتائج قابلة للتنفيذ قد تمكن من التوظيف في التجارب السريرية أو تحفز مراجعة إضافية ضمن GTAB. على سبيل المثال، تم تحديد SNVs في PIK3CA وعبر أنواع السرطان المختلفة، وكذلك العلامات الجينومية الشاملة، مثل نقص إعادة التركيب المتماثل (HRD) وعبء الطفرات الورمية (TMB)، والتي قد تكون متاحة لها تجارب سريرية. مع تزايد الأدلة من التجارب المدفوعة بالعلامات الحيوية، من المتوقع أن تتوسع مؤشرات NGTDC، لتشمل جينات جديدة وعلامات حيوية عبر عدة أنواع من السرطان.
منظر المتغيرات الصغيرة الجسدية
كان الجين الأكثر تكرارًا في الطفرات هو TP53 (5,411 من 13,880، 39.0% من المرضى؛ الشكل 4 والطرق عبر الإنترنت). ضمن أنواع السرطان الفردية، كانت وتيرة طفرات TP53 متغيرة ولكنها كانت الأعلى في سرطان بطانة الرحم الغدي المسامي، وسرطان المبيض عالي الدرجة المسامي، وسرطان الخلايا الحرشفية في الرئة، وسرطان الغدة الدرقية في المستقيم، وسرطان الغدة الدرقية في المريء وسرطان الخلايا الحرشفية في المريء (أكثر من 70% من الحالات). من بين الأفراد الذين لديهم على الأقل طفرة واحدة في TP53،من 5,411) كانت تحتوي على واحد أو أكثر من المتغيرات المتوقعة أن تكون مقلصة للبروتين أو مغيرة للوصلات و (3,544 من 5,411) يحملون واحدًا أو أكثر من المتغيرات غير المعنية (207 فردًا يحملون كلا نوعي المتغيرات)، مع كون التغييرات البروتينية الخمسة الأكثر شيوعًا هي R175H (5.3%)، R273C (3.2%)، R248Q (3.2%)، R273H (3.2%) و R282W (2.7%) (الجدول التكميلي 1). كانت PIK3CA الجين الثاني الأكثر تغييرًا، مع وجود طفرات فيه من المرضى (2,750 من 13,880)، تحدث بشكل متكرر في سرطان بطانة الرحم (53.5%)، سرطان الغدة الكظرية المبيضية (49.0%)، سرطان الثدي الغازي (42.2%)، سرطان بطانة الرحم المسال (38.1%) وسرطان القولون الغدي (26.5%). الأكثر
الشكل 2 | نظرة عامة علىالخصائص السكانية لمجموعة برنامج جينومات السرطان. أ، توزيع 12,948 حالة تمثل 33 نوعًا من الأورام (تم احتساب الحالات التي تحتوي على أكثر من عينة واحدة لكل ورم مرة واحدة فقط). ب، قامت ثلاثة عشر مركزًا طبيًا تابعًا للخدمة الصحية الوطنية بتجنيد مرضى تم تشخيصهم بالسرطان في جميع أنحاء إنجلترا. مساحة الرسم البياني الدائري تتناسب مع عدد المرضى المجندين؛ عدد المشاركين الإجمالي المجندين لكل GMC موضح بين قوسين. مصدر الخريطة: مكتب الإحصاءات الوطنية مرخص بموجب ترخيص الحكومة المفتوحة v.3.0. ج، تحليل الجنس البيولوجي والعمر عند التشخيص وفقًا للمرض. يوضح مخطط العمر النطاق الربعي (IQR) والقيم الوسيطة.
الشكل 3 | نظرة عامة على خصائص العينة لـبرنامج جينومات السرطان. تحليل وفقًا لمرحلة المرض (يسار) (NA، غير متوفر أو غير قابل للتطبيق في سياق الورم الدبقي متعدد الأشكال والورم الدبقي منخفض الدرجة)، نوع العينة المأخوذة (وسط) ونقاء الورم (يمين) لكل نوع من أنواع الأورام؛ يتم عرض قيم النطاق الربعي والقيم الوسيطة.
كانت الكودونات المتحورة بشكل شائع في PIK3CA هي E545 و H1047. وُجد أن أكثر من 69.9% من جميع الطفرات في هذا الجين كانت موجودة في النقاط الساخنة الخمس المعروفة جيدًا.. بينما يُشار حاليًا إلى الاختبار في سرطان الثدي الغازي فقط، كانت طفرات PIK3CA موجودة عبر أنواع متعددة من الأورام، مما يشير إلى أنه يمكن النظر في التجارب السريرية مع مثبطات PIK3CA في المستقبل، إذا كان ذلك مناسبًا سريريًا. كانت جينات أخرى مثل APC وKRAS وVHL وIDH1 غنية جدًا بالطفرات في نوع أو نوعين فقط من الأورام. تحليلنا الشامل للسرطان يتوافق مع جهود التسلسل واسعة النطاق الأخرى.مثل ICGC و TCGA، على الرغم من وجود اختلافات بسبب نسب أنواع السرطان، والتي تعكسها النسبة الأعلى من أدينوكارسينوما القولون والمستقيم، والساركوما في مجموعتنا (الشكل 2أ). تسلسل عدد كبير من عينات أورام المبيض (سمح بتصنيف فرعي إضافي، مع وجود انتشار مرتفع لمتغيرات TP53 التي تم تحديدها في السرطان الغدي الحليمي عالي الدرجة (89.8% من الحالات)، ومتغيرات PIK3CA في السرطان الغدي البطاني المبيضي (49.0%) وطفرات في سرطان المبيض الحليمي منخفض الدرجة (33.3%).
الاندماجات والتغيرات العددية في الكروموسومات
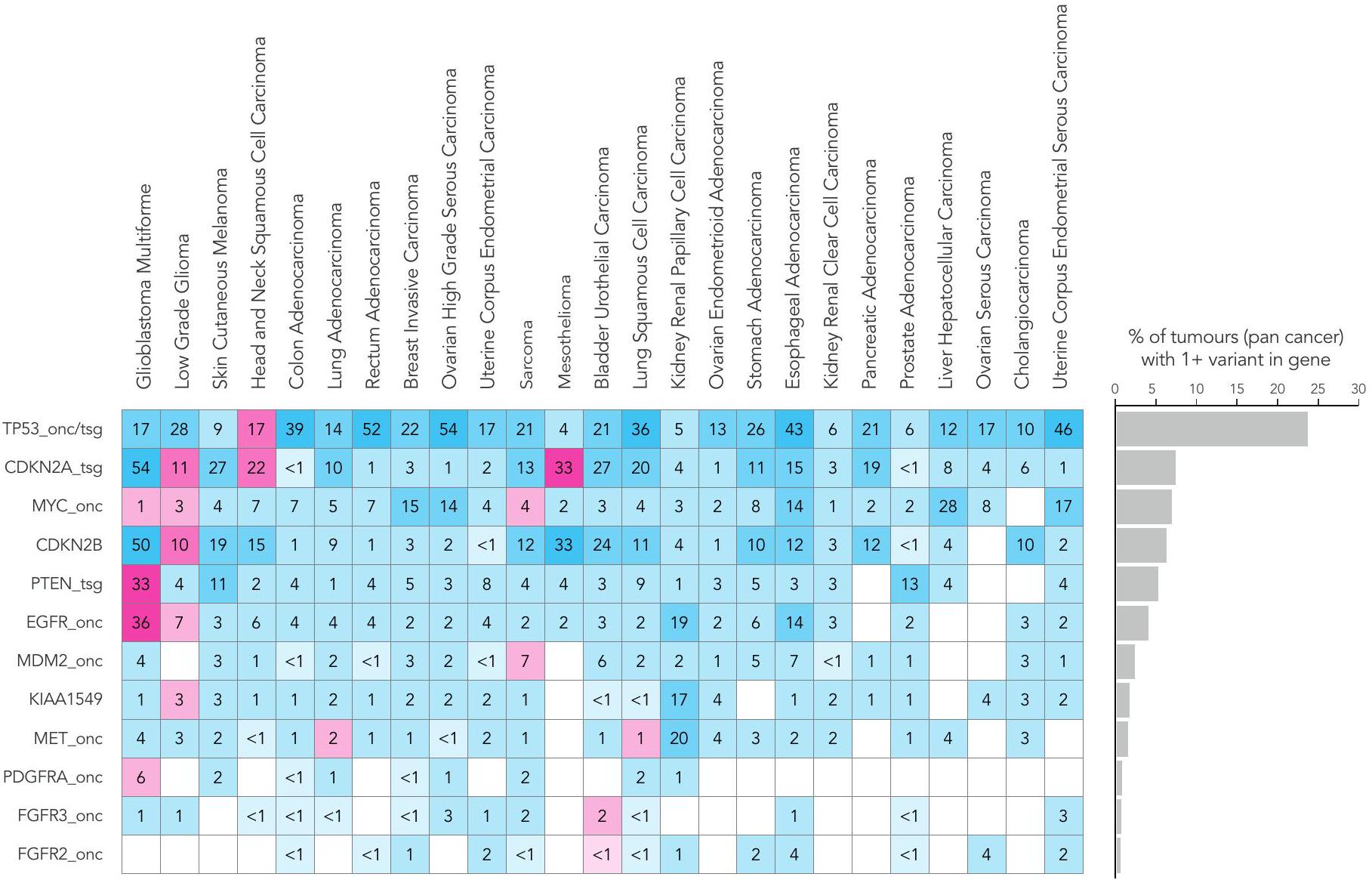
تم العثور على انتشار مرتفع من التضخيمات أو الخسائر في TP53 و CDKN2A و MYC و CDKN2B و PTEN عبر جميع أنواع السرطان (الشكل 4). أظهرت الأورام الدبقية متعددة الأشكال، والأورام الدبقية منخفضة الدرجة، وسرطان الخلايا الحرشفية في الرأس والعنق، والميزوثليوما، والساركوما (الشكل 4 والشكل الإضافي 1b) أعلى عدد من التغيرات العددية الكروموسومية ذات الصلة السريرية. مع زيادة العلاجات المستهدفة، أصبحت الاختبارات الجزيئية لأنواع الطفرات المختلفة، بما في ذلك الاندماجات، معيار الرعاية.على سبيل المثال، اندماجات NTRK (عبر جميع أنواع السرطان) ولكن أيضًا اندماجات كيناز أخرى (على سبيل المثال، و لسرطانات الرئة)، تم تضمينها الآن في NGTDC. على الرغم من أن نسبة صغيرة فقط من المرضى تظهر نتائج إيجابية لاندماجات محددة، إلا أن وجود طفرة يمكن أن يكون حاسمًا لتصنيف المرض. مثال بارز على ذلك يوجد في الساركوما الغضروفية الميزنشيمية، حيث تكون اندماجات HEY1-NCOA2 حصرية لذلك. النوع الفرعي. في الواقع، كانت الساركوما هي الأكثر انتشارًا بين الأورام (13%) مع نتائج SV ذات الصلة السريرية. (الشكل 4 والشكل الإضافي 1c).
نتائج السلالة الجرثومية
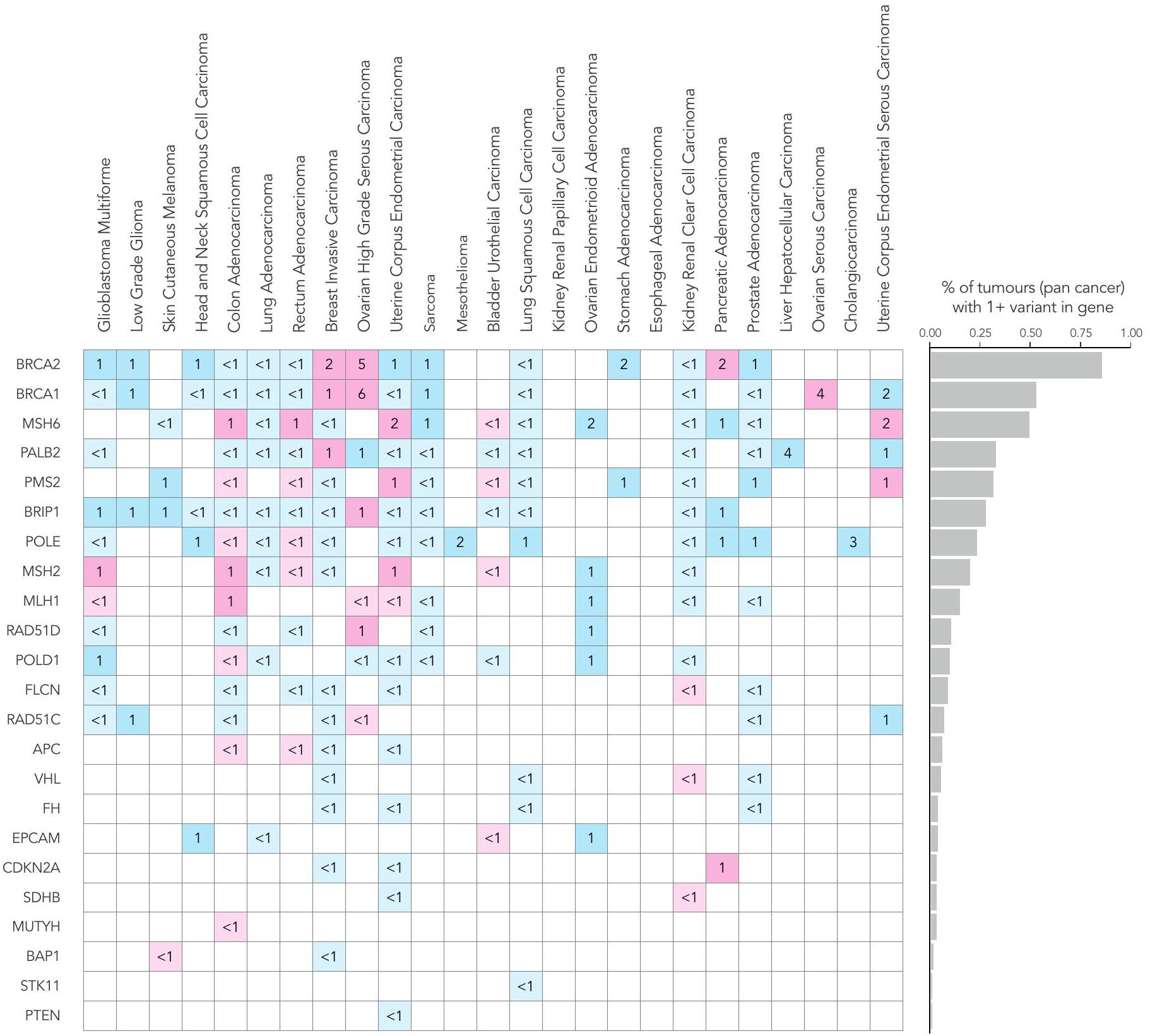
على عكس اختبارات الألواح المستهدفة التي تُجرى بشكل متكرر على عينات الأورام فقط، يسمح تسلسل الجينوم الكامل المقارن بين الورم والأنسجة الطبيعية بالكشف عن المتغيرات الجسدية والوراثية معًا. يمكن أن يكون لدرجة اليقين حول أصل المتغير تأثيرات على إدارة المريض، مثل اختبار الجينات العائلية أو الأهلية للعلاج. كان لدى المرضى المصابين بسرطان المبيض عالي الدرجة من النوع السيروسي أعلى معدل لوجود نتائج وراثية قابلة للتنفيذ بالنسبة للمتغيرات الفردية (SNVs) والإدخالات (indels)، حيث كان 13% من المرضى يحملون متغيرات في جين BRCA1 والجينات (الشكل 4 والشكل الإضافي 1d؛ تم الإبلاغ عن المتغيرات الصغيرة المسببة للتوقف أو الطفرات غير المعنية ذات التصنيف المرضي في Clinvar؛ لمزيد من التفاصيل، انظر الطرق عبر الإنترنت). يتم عرض متوسط عمر تشخيص الورم في الشكل 2c؛ كما هو متوقع، كان هناك متوسط عمر أصغر عند تشخيص الورم في أولئك المرضى الذين لديهم نتائج جينية مهيئة (الجدول الإضافي 1). من الجدير بالذكر أن المرضى الذين لديهم متغيرات جينية في جينات إصلاح عدم التطابق (MMR) أظهروا عمراً مبكراً بشكل ملحوظ عند ظهور أدينوكارسينومات القولون، بينما أظهر المرضى الذين لديهم متغيرات جينية في جينات إصلاح إعادة التركيب المتماثل بداية مبكرة بشكل ملحوظ في السرطانات المصلية عالية الدرجة في المبيضين والسرطانات الغازية في الثدي. وقد لوحظ ذلك أيضًا في سرطان الخلايا الكلوية الواضحة مع متغيرات جينية تتركز بشكل رئيسي في جين VHL.كانت المتغيرات المرتبطة بسمية الفلوروبيريميدين موجودة في 5-10% من المشاركين، مما يوجه التوصيات بشأن حذف الجرعة أو تعديلها في علاج سرطانات الثدي الغازية وسرطانات القولون والمستقيم وسرطانات الغدة البنكرياسية وسرطانات الخلايا الحرشفية في الرأس والعنق كما هو موصى به في NGTDC.
علامات بانجينومية وتوقيعات الطفرات
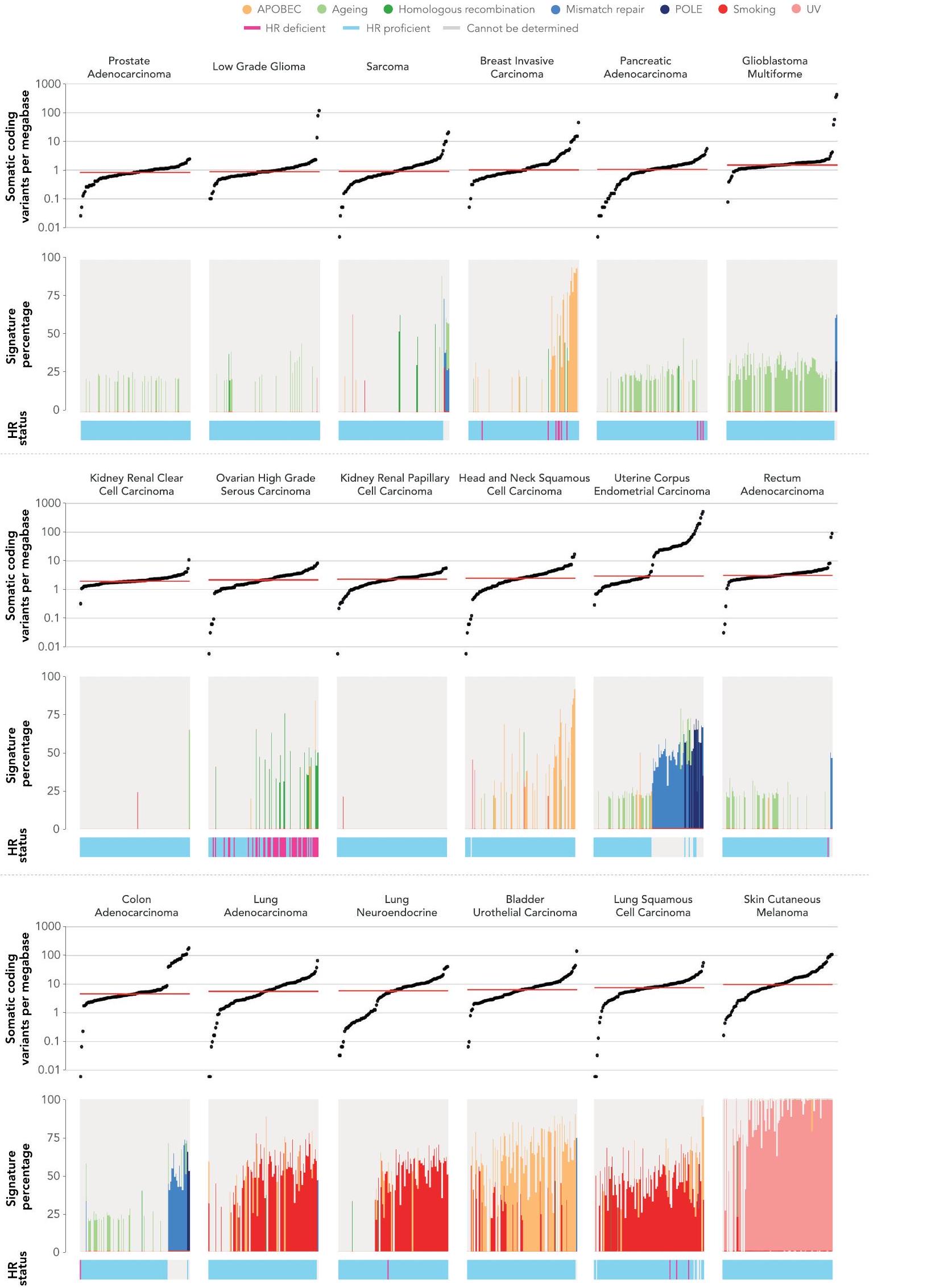
تم الإشارة إلى TMB كعلامة حيوية محتملةوفي هذه المجموعة من البيانات، لاحظنا تباينًا كبيرًا عبر أنواع السرطان وداخلها. وفقًا لـ
الشكل 4 | التغيرات الجسدية والجرثومية عبر أنواع الأورام الشائعة. انتشار أنواع مختلفة من الطفرات التي تم تحديدها باستخدام تسلسل الجينوم الكامل في الجينات المشار إليها للاختبار في NGTDC. تشير اللوحة الأكثر يسارًا إلى النسبة المئوية الإجمالية للحالات التي تحتوي على تغيير جيني واحد أو أكثر ذي صلة سريرية كما هو مدرج في NGTDC (حيث يكون عدد الأورام التي تم تسلسلها عشرة أو أكثر). في اللوحات التالية، تتكون المتغيرات الجسدية (من اليسار إلى اليمين) من متغيرات صغيرة (SNVs، indels)، CNAs، SVs، HRD، توقيعات MMR وTMB بالإضافة إلى المتغيرات الجرثومية المتعلقة بمخاطر السرطان الوراثية (المهيئة النتائج الجينية ونتائج علم الأدوية الجينومي (PGx) المرتبطة بالسميةتظهر (المتغيرات). يتم عرض أعلى خمسة جينات ذات أعلى معدلات الطفرات لكل نوع من أنواع الطفرات (انظر الشكل 1 في البيانات الموسعة للتحليل الكامل). يتم عرض نسبة الأورام التي تحتوي على نوع محدد من الطفرة في الجين (أو الجينات) المشار إليها للاختبار وفقًا لنوع الورم في NGTDC باللون الأرجواني. يتم عرض حدوث الطفرات (كنسبة مئوية) في أنواع الأورام الأخرى، التي لم يتم الإشارة إليها حاليًا في NGTDC، باللون الأزرق. تعكس تدرجات الألوان نسبة الحالات المتأثرة. مع التقارير السابقةوجدنا أن الميلانوما الجلدية وسرطان الرئة الغدي كان لهما أعلى متوسط لمعدل التحولات الطفيلية (TMB) (الشكل 5أ). أظهر سرطان القولون الغدي وسرطان بطانة الرحم تباينًا في وجود أو عدم وجود عدم استقرار الميكروساتلايت أو فرط الطفرة الناتج عن طفرات POLE (انظر التوافق مع التوقيعات الطفرية المقابلة).
عند فحص توقيعات الطفرات (COSMIC v.3) ذات الأسباب الموصوفة جيدًا، لاحظنا ترددات متوقعة ضمن أنواع معينة من السرطان. (الشكل 5 أ والشكل البياني الموسع 2). كما هو متوقع، كانت توقيعات APOBEC 2 و 13 مرتبطة بسرطان الثدي الغازي، وسرطان الخلايا الحرشفية في الرأس والعنق، وسرطان الظهارة البولية في المثانة وسرطان الغدد الرئوية؛ وكانت توقيعات التدخين 4 و 92 مرتبطة بسرطانات الرئة (سرطان الغدد الرئوية، وسرطان الغدد الصماء العصبية في الرئة وسرطان الخلايا الحرشفية في الرئة)؛ وتوقيع الأشعة فوق البنفسجية (التوقيعات ) مع الميلانوما الجلدية. توقيعات إصلاح الحمض النووي MMR 21 و 26 و 44 كانت غنية في سرطان الغدد الكولونية عالي عدم استقرار الميكروساتلايت وسرطان بطانة الرحم (الشكل 5أ).
تم تعريف حالة HRD بواسطة مصنفات بانكانسر المستندة إلى ندبات الطفرات على مستوى الجينوم، CHORDو HRDetect. أظهرت الخوارزميتان توافقًا بنسبة 99.2% في عينة الدراسة لدينا (الطرق). أظهر سرطان المبيض عالي الدرجة من النوع الحليمي أعلى انتشار لعيوب إصلاح الحمض النووي (HRD) (بينما تُستخدم مثبطات PARP حاليًا فقط في الأورام المبيضية التي تحتوي على عيوب في إصلاح الحمض النووي، تم اكتشاف عيوب في إصلاح الحمض النووي أيضًا بنسب منخفضة في أنواع أخرى من السرطان التي يمكن أن تصل إلى مثبطات PARP من خلال التجارب السريرية أو مسارات الوصول الرحيم.
الفائدة السريرية للتسلسل الجيني الكامل
بشكل عام، تُظهر هذه النتائج قدرة بيانات تسلسل الجينوم الكامل على وصف المشهد الجينومي السريري للورم بشكل كامل. يمكن لاختبار واحد أن يُبلغ عن SNVs جسدية، واندماجات جينية، وفقدان عدد النسخ (CNAs)، بالإضافة إلى طفرات جينية محتملة pathogenic، وعلامات بانجينية مثل توقيعات الطفرات وTMB (الشكل 4). في المعلومات التكميلية، نقدم أمثلة على نتائج WGA كما تم تقديمها إلى مختبرات NHS GMC. على سبيل المثال، في مريض مصاب بسرطان المبيض عالي الدرجة، تم تحديد SNV جسدي TP53، متوافق مع التشخيص، بالإضافة إلى متغير BRCA1 جيني و فقدان عدد النسخ BRCA1 (CN) الجسدي الذي يقود HRD، والذي تم دعمه لاحقًا من خلال تحليل HRD. وبالمثل، في حالة أخرى، في مريض مصاب بسرطان بطانة الرحم، تم تحديد توقيعات نقص MMR بالاقتران مع TMB مرتفع، بالإضافة إلى متغير BRCA2 جيني pathogenic، وطفرات فقدان بداية PMS2 الجسدية ومتغير دوائي (جيني) في الـالجين (المتعلق بالسمية للفوروبيريميدينات). توضح هذه الأمثلة حالات محددة حيث كانت تحديد أنواع مختلفة من الطفرات والعلامات الجينومية الشاملة ذات صلة سريرية.
علامات بانجينومية والنتائج من البيانات الواقعية
من خلال ربط بيانات WGS مع بيانات السجل السريري على مدى الحياة (SACT وONS)، قمنا بتقييم نتائج العلاج للمرضى المصنفين وفقًا لمؤشرات الجينوم الشامل (الشكل 5b والجدول التكميلي 2). كما هو موضح في الشكل 5b، في المرضى الذين تم علاجهم بعلاجات البلاتين، توقعت HRD نتائج أفضل.، )، بشكل أساسي في المرضى الذين يعانون من سرطان الثدي الغازي ( ) وسرطانات المبيض عالية الدرجة من النوع الحليمي (، نتائج العلاج المناعي في الحالات التي تعاني من نقص MMRكانت النتائج غير حاسمة بسبب الأعداد الصغيرة. ثم قمنا بتقييم TMB كعلامة تنبؤية.وفارق كبير في البقاء، ) لوحظت لدى هؤلاء المرضى الذين لديهم TMB في الربع الأدنى (وسيط 3.8 متغيرات صغيرة غير مرادفة لكل ميغابايت) مقارنة بالربع الأعلى (وسيط 20.98 متغيرات صغيرة غير مرادفة لكل ميغابايت) لدى الذين تم تشخيصهم بسرطان الجلد الميلانوما (الشكل 5c والجدول التكميلي 2). من المثير للاهتمام أنه لم يتم ملاحظة فرق كبير في سرطان الرئة الغدي ( )، حيث كانت قيم TMB الوسيطة في الربع الأدنى والأعلى 2.2 و10.5 متغيرات صغيرة غير مرادفة لكل ميغابايت، على التوالي. قد يشير هذا إلى أن مستوى TMB ذو صلة بالتنبؤ ويدعم الحاجة إلى مزيد من تحسين العلامات البيولوجية الجينومية كعلامات تنبؤية واستباقية لاستجابة العلاج المناعي، كما تم تسليط الضوء عليه في الدراسات السابقة .
تزامن المتغيرات الصغيرة وCNA
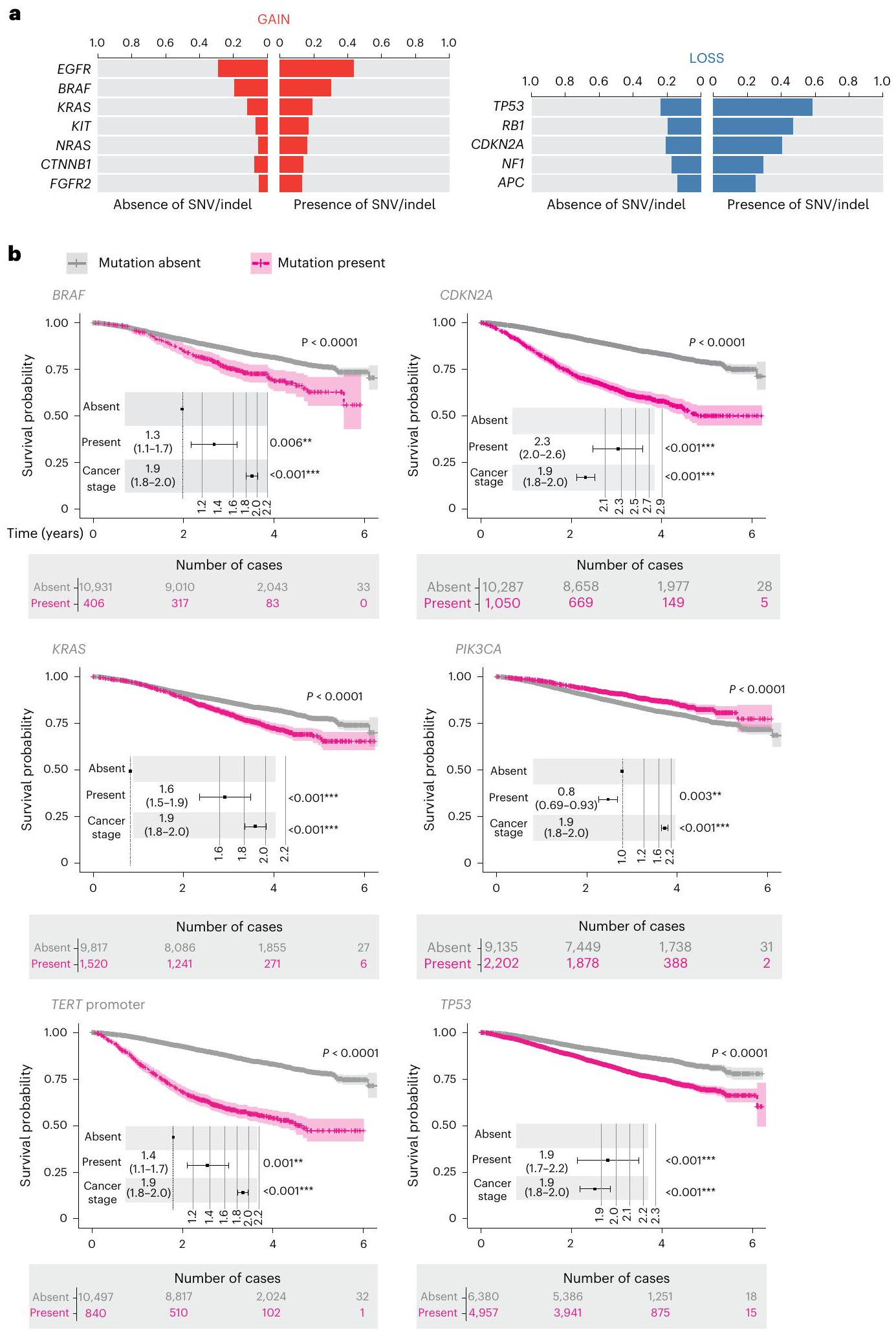
تزامن SNVs وindels وCNA موثق جيدًا . مع WGS، تمكنا من استكشاف تزامن CNA والمتغيرات الصغيرة الجسدية التي تؤثر على جينات السرطان في NGTDC. قسمنا الحالات إلى تلك التي تحتوي على متغيرات صغيرة وتلك التي لا تحتوي عليها لكل جين ثم قارننا تكرار CNA لكل جين عبر هاتين المجموعتين (الشكل 6a والجدول التكميلي 3). بعد تصحيح الاختبارات المتعددة، وجدنا أن 12 جينًا أظهرت فرقًا كبيرًا في تكرار التغيرات النسخية. أكدنا النتائج السابقة، وهي أن و، في أنواع السرطان المحددة، كانت تميل إلى أن تكون مضاعفة عندما كان SNV تنشيطي محتمل موجودًا. لقد تم مناقشة دور مكاسب النسخ على بعض الأورام السرطانية لفترة طويلة ووجد تحليلنا أن هناك تزامنًا كبيرًا للمكاسب في وجود متغيرات صغيرة تؤثر على و. كما وجدنا أن خمسة جينات مثبطة للورم أو ذات دور مزدوج كانت لديها تكرارات أعلى بشكل كبير لفقدان النسخ في وجود متغيرات صغيرة جسدية، بما في ذلك أمثلة مثبتة مثل TP53 (مرجع 28)، RB1 (مرجع 29)، و، مما يبرز قيمة تفسير أنواع مختلفة من المتغيرات بشكل متزامن.
تحليل البقاء باستخدام بيانات العالم الحقيقي
قمنا بعد ذلك بتقييم البقاء العام في جميع 33 نوعًا فرعيًا من السرطان مقسمة وفقًا لوجود أو عدم وجود طفرات في 40 جينًا محددًا من NGTDC (تم تضمين المتغيرات الصغيرة التي تغير البروتين (SNVs وindels) بالإضافة إلى الحذف المتماثل في جينات مثبطة الورم). قدمت البيانات السريرية من مصادر بيانات ثانوية مثل HES وONS بيانات البقاء. تم إجراء تحليلات Kaplan-Meier وCox proportional-hazards على مجموعة مرضانا الشاملة. بعد تصحيح المرحلة والاختبارات المتعددة، أثر 15 جينًا على البقاء العام (الشكل 6b والشكل التكميلي 3). الجين الذي أثر على نتائج المرضى بشكل أكثر حدة كان CDKN2A (, )، والذي يتوافق مع ارتباطه بالمرض عالي الدرجة وسوء التنبؤ في بعض أنواع السرطان، مثل الورم الدبقي وساركوما الأنسجة الرخوة . تتفق نتائجنا مع الارتباطات التنبؤية المبلغ عنها سابقًا لأنواع الأورام المحددة، على سبيل المثال، سوء
التنبؤ لطفرات KRAS في سرطان القولون وسرطان الرئة غير صغير الخلايا أو طفرات TP53 في سرطان الرئة غير صغير الخلايا . كانت الطفرات في PIK3CA مرتبطة بنتائج إيجابية، بما يتماشى مع التقارير في الأدبيات .
نقاش
أنشأ مشروع الجينوم 100,000 البنية التحتية والموارد لربط البيانات الجينومية والبيانات السريرية الطولية. ساعدت نتائجنا من برنامج السرطان في اختيار الأهداف الجينومية في دليل الاختبار الجينومي الوطني NHS. قدم تقييم بيانات WGS دعمًا لتكليف WGS السريري لسرطان الساركوما، والورم الدبقي، وسرطان المبيض عالي الدرجة، وسرطان الثدي الثلاثي السلبي، لاكتشاف أنواع مختلفة من الطفرات، بما في ذلك العلامات الجينومية، من خلال اختبار واحد لإبلاغ الرعاية السريرية. تم دمج البنية التحتية الناتجة عن مشروع الجينوم 100,000 في نظام GMS NHS لتمكين التوصيف الجزيئي القياسي للأورام ولتوسيع الفائدة السريرية للتوصيف الجزيئي الاستباقي لمزيد من المرضى المصابين بالسرطان. يتماشى ذلك مع الدراسات السابقة نحن نبلغ عن انتشار مرتفع للمتغيرات الجينية المستخدمة لتصنيف المرضى نحو العلاجات المعتمدة والتجارب السريرية عبر أنواع السرطان المختلفة. يتماشى نهجنا مع برامج مماثلة في دول أخرى، مثل مستشفى أبحاث الأطفال St.Jude في الولايات المتحدة، BC Cancer في كندا ، برنامج سرطان الطفولة Zero في أستراليا ، فرنسا Médecine Génomique وGenomic Medicine Sweden . هذه المبادرات إما جارية ولم تنشر بعد عن مجموعتها أو تمثل مجموعة أصغر من سرطانات الطفولة.
شمل دراستنا بيانات WGS فقط، وعلى الرغم من أن الجينوميات قد توفر نقطة انطلاق قيمة لتصنيف السرطان الجزيئي، فمن المحتمل أن تكون هناك طرق أخرى، مثل الحمض النووي الخالي من الخلايا، تسلسل RNA، الميثيل، وتحليل التعبير الجيني، البروتيوميات، التسلسل طويل القراءة، وتسلسل الخلايا المفردة ستتطور نحو الاستخدام السريري. على هذا النحو، نتوقع تضمين بيانات متعددة الأوميات جنبًا إلى جنب مع بيانات الحياة الطولية ودمج البيانات الجزيئية والسريرية متعددة الوسائط، بما في ذلك علم الأمراض الرقمي والأشعة، لتعظيم فائدة رعاية السرطان الدقيقة للمرضى .
مع انتشار اختبار الجينوم، من الضروري دمج هذه البيانات مع بيانات العلاج والسريريات من العالم الحقيقي. هذا الدمج ضروري لتعزيز فهمنا للتأثير طويل الأمد للجينوميات السريرية للسرطان على نتائج المرضى. في هذه الدراسة، أظهرنا قيمة البيانات المرتبطة بالعالم الحقيقي في تقييم النتائج ومطابقة العلامات الجزيئية السلبية من التجارب السريرية. إن تراكم البيانات الجينومية جنبًا إلى جنب مع البيانات الصحية الإلكترونية المضمنة في سجلات السرطان، مثل التصنيف، وعلم الأمراض، والعلاج، والنتائج، يغني مجموعة البيانات وقد يساهم في تحسين اختيار العلامات البيولوجية. من المحتمل أن يعزز تزامن المتغيرات في نفس الجين، أو التعايش للطفرات في جينات مختلفة، القيمة التنبؤية والاستباقية لاختيار العلامات البيولوجية وقد يكشف عن إشارات كامنة طويلة الأمد من الفائدة أو الضرر ويساعد في اتخاذ القرارات السريرية والتنظيمية . الآثار العلاجية المرتبطة بتزامن CNA والمتغيرات الصغيرة الجسدية غير واضحة، وقد لا تكون هذه المستوى من المعلومات الجينومية متاحة بسهولة من بيانات لوحات السرطان الكبيرة . نقدم تحليل بقاء واسع على مستوى الجين؛ مع توسع مجموعة البيانات، سيكون من الممكن
الشكل 5|القيمة التنبؤية للعلامات الجينومية المستمدة من بيانات WGS. أ، توزيع TMB وتوقيعات الطفرات عبر ستة أنواع من الأورام. (تم استبعاد العينات التي خضعت لتكبير PCR أثناء إعداد المكتبة وتم تقليل مجموعة البيانات لكل نوع ورم إلى 100 عينة.) تشير الشريط الأحمر الأفقي إلى الوسيط TMB لكل نوع سرطان. تعريفات الأسباب بناءً على توقيعات استبدال القاعدة الواحدة COSMIC (v.3): نشاط APOBEC، التوقيعات 2 و13؛ الشيخوخة، التوقيع 1؛ HRD، التوقيع 3؛ نقص MMR، التوقيعات 6،15،20،21،26 و44؛ طفرات POLE، التوقيعات 10a، 10b و14؛ التدخين، التوقيعات 4 و92؛ التعرض للأشعة فوق البنفسجية، التوقيعات . تظهر فقط التوقيعات التي لديها أكثر من مساهمة. يتم الإشارة إلى حالة إعادة التركيب المتماثل في الأشرطة أسفل التوقيع
الرسوم البيانية. ب، ج، تقديرات Kaplan-Meier للبقاء العام مع القيم المحسوبة باستخدام اختبار log-rank المصنف. يتم الإشارة إلى أعداد المرضى المعرضين للخطر في نقاط زمنية مختلفة أدناه منحنيات البقاء. تشير النقاط وأشرطة الخطأ في الرسوم البيانية المدمجة إلى نسب المخاطر (HRs) مع فترات الثقة (CIs) بنسبة 95%، على التوالي. تم حساب HRs وCIs و القيم من نماذج Cox proportional-hazards المصححة وفقًا لمرحلة السرطان. تم تصنيف المرضى وفقًا لحالة HRD في السرطانات المعالجة بعلاج كيميائي بالبلاتين (، اليسار، )؛ وفقًا لتوقيعات MMR في السرطانات المعالجة بالعلاج المناعي (، اليمين، ب)؛ وفقًا لـ TMB العالي والمنخفض في سرطان الجلد الميلانوما (، اليسار، ج)؛ ووفقًا لسرطان الرئة الغدي (، اليمين، ). يمكن العثور على القيم الدقيقة لـ في الجدول التكميلي 2.
C
لفحص هذه البيانات بشكل أعمق لتحديد الآثار التنبؤية والتنبؤية للمتغيرات المحددة، كما لوحظ مع متغيرات KRASومع ذلك، لا تزال هناك تحديات في تنفيذ تسلسل الجينوم الكامل السريري في هيئة الخدمات الصحية الوطنية في إنجلترا، ليس أقلها بسبب التكلفة الإجمالية مقارنةً باختبارات الألواح الجينية الكبيرة. يتطلب تقديم خدمة جينوميات متطورة في المملكة المتحدة ليس فقط البنية التحتية للتسلسل والتحليل، ولكن أيضًا مراعاة المتطلبات التشغيلية (مثل تحسين مسارات الأنسجة وأوقات الاستجابة لإبلاغ اتخاذ القرارات السريرية) جنبًا إلى جنب مع تحويل المسارات المحلية وتطوير المعرفة والمهارات للقوى العاملة متعددة التخصصات التي تدعم رعاية السرطان.
تتم مناقشة نتائج تسلسل الجينوم الكامل في مجالس الأورام الجزيئية متعددة التخصصات أو GTABs لتقييم المتغيرات الجسدية والوراثية، وتحديد القابلية للعمل السريري وتقديم التوصيات السريرية. تلعب GTABs دورًا حيويًا في ضمان توصيل النتائج القابلة للتنفيذ إلى فرق العلاج والأطباء، بينما تستكشف أيضًا الأهلية للعلاجات المعتمدة والتجارب السريرية.يلعب GTAB مصمم بشكل جيد ومنظم بشكل جيد دورًا رئيسيًا في التفسير السريري لاختبارات الجينوم السرطاني، حيث يوجه الأطباء في اتخاذ القرارات من خلال التوصيات، ويسهل تسجيل المرضى في التجارب السريرية وقد يعزز النتائج.. تتماشى هذه المقاربة مع تجارب السلة التكيفية مثل DETERMINEالذي تم إنشاؤه لتقييم العلاجات المرخصة في مؤشرات غير مرخصة مشابهة لتجربة DRUPالهدف هو تمكين اختبارات جزيئية أكثر عدلاً وشمولية ضمن خدمة الصحة الوطنية (NHS) وتحسين رعاية السرطان من خلال تحديد جميع الطفرات ذات الصلة السريرية لسرطان معين (كما هو موضح في الشكل 4) وعلاقتها بالأدوية الدقيقة المعتمدة، ولكن أيضًا لضمان أن يتم أخذ المرضى بعين الاعتبار بالكامل للبحث السريري والتجارب بسبب هذا الاختبار الجينومي واستكشاف خيارات التجارب السريرية، بما في ذلك استخدام الأدوية المعروفة والمميزة المعاد توجيهها.
بيئة البحث، وهي منصة أنشأتها جينومكس إنجلاند وNHSE، تتيح للباحثين المعتمدين الوصول الآمن إلى البيانات الجينومية والبيانات الصحية المرتبطة بها. وقد سمحت بتقدم في الأبحاث الأساسية، مثل اكتشاف جينات محركات السرطان.توقيعات الطفراتأو التغييرات في الممارسة السريرية المدفوعة بتوافر اختبار تسلسل الجينوم الكامل.
تؤكد نتائجنا على الإمكانية التي توفرها هذه البيانات لتقديم رؤى تنبؤية إضافية بناءً على غياب أو وجود طفرات محددة. مع تراكم البيانات داخل بيئة البحث مع ربط البيانات الجينومية والسريرية وبيانات النتائج، يمكن إجراء تحليلات أكثر دقة باستخدام بيانات العالم الحقيقي، مدعومة بتوصيف أكثر شمولاً للأورام. سيمكن ذلك من مزيد من تحسين العلامات الجزيئية التنبؤية والتنبؤية، ليس فقط من خلال تركيبات من تغييرات جينومية مختلفة، ولكن أيضًا بما يتجاوز الجينوميات، بما في ذلك التقنيات الناشئة لتوسيع نطاق علم الأورام الدقيق لتحسين نتائج السرطان.
المحتوى عبر الإنترنت
أي طرق، مراجع إضافية، ملخصات تقارير Nature Portfolio، بيانات المصدر، بيانات موسعة، معلومات تكميلية، شكر وتقدير، معلومات مراجعة الأقران؛ تفاصيل مساهمات المؤلفين والمصالح المتنافسة؛ وبيانات توفر البيانات والرموز متاحة علىhttps://doi.org/10.1038/s41591-023-02682-0.
Zehir, A. et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 23, 703-713 (2017).
Smedley, D. et al. 100,000 Genomes Pilot on Rare Disease Diagnosis in Health Care-preliminary report. N. Engl. J. Med. 385, 1868-1880 (2021).
Turnbull, C. et al. The 100000 Genomes Project: bringing whole genome sequencing to the NHS. BMJ 361, k1687 (2018).
Turnbull, C. Introducing whole-genome sequencing into routine cancer care: the Genomics England 100000 Genomes Project. Ann. Oncol. 29, 784-787 (2018).
National Genomic Test Directory. NHS England www.england.nhs. uk/publication/national-genomic-test-directories (2023).
NHS England. Board Paper (2017); www.england.nhs.uk/ wp-content/uploads/2017/03/board-paper-300317-item-6.pdf
Berner, A. M., Morrissey, G. J. & Murugaesu, N. Clinical analysis of whole genome sequencing in cancer patients. Curr. Genet. Med. Rep. 7, 136-143 (2019).
Aaltonen, L. A. et al. Pan-cancer analysis of whole genomes. Nature 578, 82-93 (2020).
Martínez-Jiménez, F. et al. Pan-cancer whole-genome comparison of primary and metastatic solid tumours. Nature 618, 333-341 (2023).
van der Velden, D. L. et al. The Drug Rediscovery protocol facilitates the expanded use of existing anticancer drugs. Nature 574, 127-131 (2019).
Aran, D., Sirota, M. & Butte, A. J. Systematic pan-cancer analysis of tumour purity. Nat. Commun. 6, 8971 (2015).
Zhong, L. et al. Small molecules in targeted cancer therapy: advances, challenges, and future perspectives. Signal Transduct. Target Ther. 6, 201 (2021).
Lanic, M.-D. et al. Detection of sarcoma fusions by a nextgeneration sequencing based-ligation-dependent multiplex RT-PCR assay. Mod. Pathol. 35, 649-663 (2022).
Chan, T. A. et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann. Oncol. 30, 44-56 (2019).
Lawrence, M. S. et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495-501 (2014).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415-421 (2013).
Nguyen, L., Martens, J. W. M., Van Hoeck, A. & Cuppen, E. Pan-cancer landscape of homologous recombination deficiency. Nat. Commun. 11, 5584 (2020).
Davies, H. et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 23, 517-525 (2017).
Xiao, D. et al. Analysis of ultra-deep targeted sequencing reveals mutation burden is associated with gender and clinical outcome in lung adenocarcinoma. Oncotarget 7, 22857-22864 (2016).
Klempner, S. J. et al. Tumor mutational burden as a predictive biomarker for response to immune checkpoint inhibitors: a review of current evidence. Oncologist 25, e147-e159 (2020).
McGrail, D. J. et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann. Oncol. 32, 661-672 (2021).
Inoue, K. & Fry, E. A. Haploinsufficient tumor suppressor genes. Adv. Med. Biol. 118, 83-122 (2017).
Sigismund, S., Avanzato, D. & Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 12, 3-20 (2018).
Cheng, L. et al. KIT gene mutation and amplification in dysgerminoma of the ovary. Cancer 117, 2096-2103 (2011).
Shetzer, Y. et al. The onset of p53 loss of heterozygosity is differentially induced in various stem cell types and may involve the loss of either allele. Cell Death Differ. 21, 1419-1431 (2014).
Latil, A. et al. Loss of heterozygosity at chromosome arm and RB1 status in human prostate cancer. Hum. Pathol. 30, 809-815 (1999).
Foulkes, W. D., Flanders, T. Y., Pollock, P. M. & Hayward, N. K. The CDKN2A (p16) gene and human cancer. Mol. Med. 3, 5-20 (1997).
Horbinski, C., Berger, T., Packer, R. J. & Wen, P. Y. Clinical implications of the 2021 edition of the WHO classification of central nervous system tumours. Nat. Rev. Neurol. 18, 515-529 (2022).
Bui, N. Q. et al. A clinico-genomic analysis of soft tissue sarcoma patients reveals CDKN2A deletion as a biomarker for poor prognosis. Clin. Sarcoma Res. 9, 12 (2019).
Ozer, M. et al. Age-dependent prognostic value of mutation in metastatic colorectal cancer. Future Oncol. 17, 4883-4893 (2021).
Aredo, J. V. et al. Impact of mutation subtype and concurrent pathogenic mutations on non-small cell lung cancer outcomes. Lung Cancer 133, 144-150 (2019).
Jiao, X.-D., Qin, B.-D., You, P., Cai, J. & Zang, Y.-S. The prognostic value of TP53 and its correlation with EGFR mutation in advanced non-small cell lung cancer, an analysis based on cBioPortal data base. Lung Cancer 123, 70-75 (2018).
Kalinsky, K. et al. PIK3CA mutation associates with improved outcome in breast cancer. Clin. Cancer Res. 15, 5049-5059 (2009).
Priestley, P. et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 575, 210-216 (2019).
Ma, X. et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 555, 371-376 (2018).
Pleasance, E. et al. Whole-genome and transcriptome analysis enhances precision cancer treatment options. Ann. Oncol. 33, 939-949 (2022).
Wong, M. et al. Whole genome, transcriptome and methylome profiling enhances actionable target discovery in high-risk pediatric cancer. Nat. Med. 26, 1742-1753 (2020).
Préindications d’Accès au Séquençage Génomique. France Medecine Genomique 2025 https://pfmg2025.aviesan.fr/le-plan/ indications-dacces-au-sequencage-genomique/ (undated).
Sequencing of 7,000 Genomes in Swedish Clinical Practice in 2021 for Better Diagnosis and Treatment. Genomic Medicine Sweden https://genomicmedicine.se/en/2022/04/11/ sequencing-of-7000-genomes-in-swedish-clinical-practice-2021-for-better-diagnosis-and-treatment (2022).
Donoghue, M. T. A., Schram, A. M., Hyman, D. M. & Taylor, B. S. Discovery through clinical sequencing in oncology. Nat. Cancer 1, 774-783 (2020).
Nogrady, B. How cancer genomics is transforming diagnosis and treatment. Nature 579, S10-S11 (2020).
Chandramohan, R. et al. A validation framework for somatic copy number detection in targeted sequencing panels. J. Mol. Diagn. 24, 760-774 (2022).
Huang, L., Guo, Z., Wang, F. & Fu, L. KRAS mutation: from undruggable to druggable in cancer. Signal Transduct. Target Ther. 6, 386 (2021).
van de Haar, J. et al. Codon-specific KRAS mutations predict survival benefit of trifluridine/tipiracil in metastatic colorectal cancer. Nat. Med. 29, 605-614 (2023).
Academy of Medical Royal Colleges. Principles for the Implementation of Genomic Medicine (2019); www.aomrc.org. uk/wp-content/uploads/2019/10/Principles_implementation_ genomic_medicine_011019.pdf
Malone, E. R., Oliva, M., Sabatini, P. J. B., Stockley, T. L. & Siu, L. L. Molecular profiling for precision cancer therapies. Genome Med. 12, 8 (2020).
Kato, S. et al. Real-world data from a molecular tumor board demonstrates improved outcomes with a precision N-of-One strategy. Nat. Commun. 11, 4965 (2020).
Cornish, A. J. et al. Whole genome sequencing of 2,023 colorectal cancers reveals mutational landscapes, new driver genes and immune interactions. Preprint at bioRxiv https://doi. org/10.1101/2022.11.16.515599 (2022).
Degasperi, A. et al. Substitution mutational signatures in whole-genome-sequenced cancers in the UK population. Science 376, science.abl9283 (2022).
Prendergast, S. C. et al. Sarcoma and the 100,000 Genomes Project: our experience and changes to practice. J. Pathol. Clin. Res. 6, 297-307 (2020).
Trotman, J. et al. The NHS England 100,000 Genomes Project: feasibility and utility of centralised genome sequencing for children with cancer. Br. J. Cancer 127, 137-144 (2022).
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
متطلبات جمع العينات واستخراج الحمض النووي موصوفة في إرشادات التعامل مع العينات (الإصدار 4.0) المتاحة علىhttps://files. genomicsengland.co.uk/forms/Sample-Handling-Guidance-v4.0.pdf. إجمالي حمض نووي جرثومي و على الأقلكان من الضروري الحصول على الحمض النووي للورم لإجراء تحضير مكتبة Illumina TruSeq الخالية من PCR. تم استخدام تحضير المكتبة القائم على PCR عندما لم يكن من الممكن الحصول على كمية كافية من الحمض النووي للتسلسل الخالي من PCR، مع حد أدنى مطلوب قدره 500 نانوغرام. تم السماح باستخدام أنسجة الورم المثبتة بالفورمالين لإجراء التسلسل الجيني الكامل في ظروف استثنائية، حيث كانت حجم الورم يحد من توفر الأنسجة الطازجة، أو إذا لم يكن هناك ورم موجود في العينة المجمدة الطازجة.
جودة بيانات التسلسل. تم تسلسل جميع العينات على منصة HiSeq بمتوسط تغطية قدرهللورم وللعينات الطبيعية. تم تنفيذ الفحوصات التالية لضمان جودة العينة: كانت العينات الطبيعية تحتوي على أكثر من 85 جيجابايت وكانت عينات الورم تحتوي على أكثر من 210 جيجابايت من بيانات التسلسل عالية الجودة (جودة القاعدة أكبر من 30، تمت إزالة القراءات المكررة)؛ كانت العينات الطبيعية تحتوي على أكثر من 95% من الجينوم الجسمي مغطاة عندأو أكثر بعد إزالة القراءات ذات جودة التعيين أقل من 10؛ كانت العينات الطبيعية تحتوي على تلوث عبر المرضى أقل من 3% كما تم تقييمه باستخدام VerifyBamID؛ كانت عينات الورم تحتوي على تلوث عبر المرضى أقل من 2.5% وزوج عينات الورم الطبيعي originating من نفس المريض كما تم تقييمه باستخدام ConPair؛ تم مراقبة جودة بيانات التسلسل باستخدام تحليل المكونات الرئيسية استنادًا إلى المقاييس التالية: نسبة القراءات الموجهة إلى الجينوم المرجعي، نسبة قطع الحمض النووي الشيميري، حجم القطعة الوسيطة، عدم تساوي تغطية الجينوم المحلي ونسبة القراءات المفقودة من المناطق الجينومية الغنية بـ AT أو GC (انخفاض AT وGC).
تخطيط واستدعاء المتغيرات. تم استخدام خط أنابيب إلومينا نورث ستار (الإصدار 2.6.53.23) للتحليل الأساسي لتسلسل الجينوم الكامل. تم إجراء محاذاة القراءة ضد الجينوم المرجعي البشري GRCh38 + الدخيل + فيروس إبشتاين-بار باستخدام ISAAC (الإصدار iSAAC-03.16.02.19). نحن نعترف بنقص في برنامج محاذاة ISAAC لتقديرات دقيقة لتردد الأليل المتغير.ولتحليل تطور الورم، نلاحظ أن جميع الجينومات من مشروع الجينومات الـ 100,000 قد تم إعادة محاذاتها مؤخرًا باستخدام منصة إيلومينا دراجن (البيانات متاحة في بيئة البحث). تم إجراء استدعاء المتغيرات الصغيرة في السلالة الجرثومية باستخدام ستارلينغ (الإصدار 2.4.7) وتم إجراء استدعاء المتغيرات الصغيرة في السلالة الجسدية باستخدام ستريلكا (الإصدار 2.4.7). بالإضافة إلى الفلاتر الافتراضية لستريلكا، تم تطبيق الفلاتر الإضافية التالية لتقليل معدل الإيجابيات الكاذبة في مجموعة المتغيرات الجسدية المستخدمة كمدخلات في حساب TMB وتوقيعات الطفرات: (1) المتغيرات التي لديها تردد أليل سلالة جرثومية في السكان يزيد عن 1% في مجموعات بيانات جينومكس إنجلاند أو gnomAD؛ (2) المتغيرات الجسدية المتكررة التي لديها تردد يزيد عنفي مجموعة بيانات جينومكس إنجلاند؛ (3) المتغيرات التي تتداخل مع التكرارات البسيطة كما هو محدد بواسطة أداة العثور على التكرارات المتتالية؛ (4) التغيرات الصغيرة في المناطق ذات مستويات عالية من ضوضاء التسلسل حيث على الأقلتم تصفية مكالمات القاعدة في نافذة تمتد 50 قاعدة على كل جانب من مكالمة indel بواسطة Strelka بسبب الجودة الضعيفة؛ (5) SNVs الناتجة عن عيوب منهجية في التخطيط والمكالمات مع درجة Phred لاختبار فيشر أقل من 50. تم تنفيذ تصنيف التخطيط والمكالمات المنهجية من خلال اختبار ما إذا كانت نسبة عمق الأليل في الورم في كل موقع SNV جسمي تختلف بشكل كبير عن نسبة عمق الأليل في هذا الموقع في مجموعة من الأفراد الطبيعيين. كانت مجموعة الأفراد الطبيعيين تتكون من مجموعة من 7000 جينوم غير ورمي من Genomics England. في مجموعة البيانات؛ تم تضمين الأفراد الذين لا يحملون الأليل البديل المعني فقط في حساب أعماق الأليلات في كل موقع جيني. لم يتم إزالة المتغيرات التي تم وضع علامة عليها بأي من الفلاتر الداخلية المذكورة أعلاه من نتائج WGA للمتغيرات القابلة للتنفيذ سريرياً، ولكن تم وضع علامة عليها في المخرجات المشتركة مع العلماء السريريين.
تم تحديد CNAs باستخدام Canvas v.1.3.1. تم استخدام Manta (v.0.28.0) لاستدعاء SVs والانحرافات الطويلة (أكثر من 50 نقطة أساسية)، مع دمج الأدلة من القراءة المزدوجة والمقسمة لاكتشاف SV وتسجيله.
تم إنتاج تقديرات دقة استدعاء المتغيرات الجسدية في خط أنابيب مشروع الجينومات 100,000 كمتطلب للاعتماد بموجب معيار المنظمة الدولية للتوحيد القياسي رقم 15189. لقد قدمنا ‘تحقق من خط أنابيب المعلومات الحيوية. تقرير السرطان، سبتمبر 2018’ كمعلومات تكميلية وقمنا بتلخيص النتائج في الجدول التكميلية 4. سيتم تقديم التحقق الشامل والتحسينات الوظيفية لخط الأنابيب لخدمات الجينوم في NHS في منشور منفصل.
تمت معالجة التعليقات والإبلاغ عن القابلية للعمل. تم محاذاة SNVs والـ small indels إلى اليسار، وتم تقليمها، وتم تفكيك المتغيرات متعددة الأليلات، قبل التعليق باستخدام Cellbase، مع الاستعانة بقواعد بيانات Ensembl (الإصدار 90/GRCh38) وCOSMIC (الإصدار 86/GRCh38) وClinVar (إصدار أكتوبر 2018). تم إجراء تعليق أنواع العواقب بواسطة مُعَلِّم متغيرات عالي الأداء ضمن Cellbase؛ وتم الإبلاغ فقط عن المتغيرات التي تم التعليق عليها بمجموعة مُنَسَّقة من أنواع العواقب (توقف مكتسب أو مفقود، فقدان البداية، متغير إزاحة الإطار، إدخال أو حذف ضمن الإطار، متغير غير معني، متغير مستقبل أو مانح للقطع، متغير منطقة القطع) في النسخ الكنسية.
تم أخذ تفسير CNAs في الاعتبار نمط عمل الجينات كما هو محدد في إحصاء جينات السرطان COSMIC (أي، جين ورمي أو جين مثبط للورم). حيث كان للجين دور غامض أو غير معروف في السرطان، تم تضمينه في فئتي الجينات الورمية والمثبطة للورم. تم الإبلاغ عن الزيادات في الجينات الورمية إذا كان عدد النسخ على الأقل مرتين أعلى من العدد الكلي للكروموسومات كما هو محدد بواسطة Canvas. تم الإبلاغ عن السيناريوهات التالية كخسائر في جينات المثبطات الورمية: (1) الحذف المتماثل الذي تم تحديده بواسطة Canvas ( ); (2) فقدان التغايرية (LOH) الذي تم الإبلاغ عنه بواسطة Canvas ( ) أو LOH محايد نسبيًا، بالاشتراك مع متغير صغير جسمي غير مرمز؛ و (3) SVs من Manta التي لديها القدرة على تعطيل منطقة ترميز الجين بالاشتراك مع متغير صغير جسمي غير مرمز. فقط العينات التي تحتوي على نقاء الورم أكبر من تم تضمينها في تحليل قابلية العمل لـ CNA. بالنسبة لتحليل التزامن بين المتغيرات الصغيرة الجسدية و CNAs في الشكل 6a، تم اعتبار اكتساب نسخة واحدة على الأقل من الجينات المسرطنة أو فقدان نسخة واحدة على الأقل من جينات كابحة الورم كحدث CNA.
تم تقييم مكالمات مانتا (نهاية الكسر، الحذف، التكرار أو الانعكاس) بشكل إضافي من حيث القدرة على توليد اندماجات منتجة باستخدام نهج داخلي يعتمد على اتجاه النسخ وموثوقية إطار القراءة عبر نقطة كسر SV. تم التخلص من SVs التي تم تحديدها على أنها خارج الإطار أو غير مكتوبة. تم الإبلاغ عن الاندماجات المحتملة في الإطار والأحداث الغامضة التي تحتوي على نقطة كسر في الإكسون المشفر أو في 5-‘UTR للشركاء اللاحقين.
تم الإبلاغ عن المتغيرات الجينية في السلالة الجرثومية المدرجة في ClinVar على أنها مسببة للأمراض أو ربما مسببة للأمراض بتصنيف لا يقل عن نجمتين والمتغيرات التي تؤدي إلى تقصير البروتين في الجينات التي كان آلية المسببة للأمراض فيها هي فقدان الوظيفة (توقف مكتسب أو مفقود، بدء مفقود، متغير إزاحة الإطار، متغير مستقبل أو مانح للقطع) لمجموعة فرعية من جينات الاستعداد للسرطان المحددة للاختبار الجيني في NGTD.
في سياق برنامج السرطان لمشروع الجينومات المئة ألف، تم مراجعة جميع المتغيرات المعادة إلى مراكز الجينوم الطبية ضمن لجان تقييم الجينوم لتصنيف المتغيرات بشكل إضافي إذا كانت مسببة للأمراض أو ربما مسببة للأمراض (خطية) أو مسرطنة أو ربما مسرطنة (جسدية) وتقديم التوصيات السريرية حيثما كان ذلك مناسبًا.
التوقيعات وTMB. لكل عينة ورم، تم حساب الترددات عبر جميع سياقات ثلاثي النوكليوتيدات SNV باستخدام ملفات VCF التي تم تصفيتها للمتغيرات المحتملة الإيجابية الكاذبة (انظر استدعاء المتغيرات) تم اشتقاق مساهمة كل من توقيعات استبدال القاعدة الواحدة COSMIC (الإصدار 3) في العبء الطفري الكلي الملحوظ في الورم باستخدام تحليل التفكيك بواسطة مجموعة أدوات SigProfiler.تمت بناء تعريفات الأسباب على أساس مجموعات التوقيع التالية: نشاط APOBEC، التوقيعات 2 و 13؛ الشيخوخة، التوقيع 1؛ نقص MMR، التوقيعات 6 و 15 و 20 و 21 و 26 و 44؛ طفرات POLE، التوقيعات 10a و 10b و 14؛ التدخين، التوقيعات 4 و 92؛ التعرض للأشعة فوق البنفسجية، التوقيعات 7a-d. لم يتم تضمين التوقيع 14 (المبلغ عنه مع السبب ‘طفرة بوليميراز إبسيلون المتزامنة وعيب MMR في الحمض النووي’) في مجموعة نقص MMR لتجنب العد المزدوج في مجموعتي MMR و POLE. إن تضمين SBS14 في مجموعة MMR سيغير حالة MMR لـ 9 من 13,880 ورمًا وسيزيد فقط من عددالأورام في مجموعتنا بواسطة. بالنسبة لسبب معين، إذا كانت التوقيعات المجمعة النهائية أقل من تم تعيين التوقيع إلى ‘آخر’. تم تصنيف الأورام على أنها تعاني من نقص في MMR إذا كانت المساهمة الإجمالية لتوقيعات MMR أكثر من. HRDetect هو مصنف انحدار لوجستي يحسب درجة احتمال HRD استنادًا إلى حذف الميكروهومولوجيا، وتوقيعات الطفرات SNV وSV، ودرجة LOH. تم استرجاع حالة HRD باستخدام HRDetect من منشور سابق.خوارزمية CHORD هي مصنف يعتمد على الغابات العشوائية يتضمن عدداً من أنواع المتغيرات المختلفة كمدخلات (تغيرات أحادية النوكليوتيد، حذف الميكروهومولوجيا، والتغيرات الهيكلية) ولا تتطلب خطوة استخراج توقيع طفرات وسيطة.تم تدريب HRDetect و CHORD على مجموعات بيانات ICGC ومؤسسة هارتويغ الطبية، على التوالي. أعادت الخوارزميتان نتائج متوافقة لـ 99.2% من العينات في مجموعتنا (10,764 من 10,854) وتم استخدام نتائج CHORD للرسوم البيانية. تم حساب TMB كإجمالي عدد المتغيرات الصغيرة الجسدية غير المتناظرة عالية الثقة لكل ميغاباز من تسلسل الترميز (انظر قسم استدعاء المتغيرات لطريقة التصفية المستخدمة).
وصف موارد البيانات السريرية
تم جمع مجموعة بسيطة من بيانات المرضى والعينات من مراكز الرعاية الصحية العامة في وقت تقديم عينة الحمض النووي من خلال OpenClinica v.3.4، على سبيل المثال، نوع الورم، سنة الميلاد، مصدر الأنسجة، الجنس المبلغ عنه ذاتيًا. لأغراض التحليل، تم التحقق من صحة الجنس المبلغ عنه ذاتيًا مع الجنس البيولوجي المستنتج باستخدام نسبة متوسط تغطية التسلسل لكروموسومات الجنس ومتوسط تغطية التسلسل للكروموسومات الجسدية. تم استخدام الجنس البيولوجي المعين في خط أنابيب المعلوماتية الحيوية كمدخل لاستدعاء المتغيرات. تم جمع معلومات سريرية ثانوية من NHSE و Public Health England (PHE)/ NCRAS. من NHSE، تم استخدام بيانات HES للحصول على تفاصيل جميع الأنشطة المفوضة خلال admissions؛ تم الحصول على معلومات الوفيات من بيانات سجل ONS لتسجيلات السرطان والوفيات داخل وخارج المستشفيات. من PHE/NCRAS، تم استخدام جدول av_tumor للحصول على تاريخ تشخيص الورم، مع رموز النسج والشكل. قدم جدول SACT معلومات عن تاريخ وأنواع العلاج. تم الوصول إلى جميع مجموعات البيانات عبر مكتبة الأبحاث الجينومية الوطنية باستخدام LabKey.
ربط البيانات الجينومية بمصادر البيانات الثانوية
تم اعتبار الأورام الدموية، والأورام لدى الأطفال، والسرطانات ذات الأصل غير المعروف خارج نطاق الدراسة وتم إزالتها قبل اختيار الأورام. تم استخدام البيانات الثانوية من كتالوج الأورام PHE/NCRAS (av_tumor) وبيانات NHS Digital HES لتأكيد البيانات السريرية المقدمة من قبل GMCs.
تم ربط مجموعة بيانات av_tumor بالبيانات الجينومية بناءً على معرف المشارك. تم إزالة الأورام المصنفة على أنها حميدة أو في الموقع من عملية الاختيار، مما ترك فقط الأورام الخبيثة أو غير المعروفة أو NA (الأخيرة كانت حالة لمشاركي Genomics England غير الموجودين في مجموعة بيانات av_tumor). حيثما كانت بيانات av_tumor متاحة لمشارك، تم استخدامها لتأكيد نوع الورم المقدم من قبل GMC. في الحالات التي لم تتطابق فيها بيانات av_tumor مع تقديم GMC، أو لم تكن البيانات موجودة، تم استخدام بيانات رعاية المرضى المقبولين من HES لاختيار أقرب مستشفى ذي صلة. موعد يتضمن تشخيصًا أوليًا للسرطان (استنادًا إلى التصنيف الإحصائي الدولي للأمراض والمشاكل الصحية ذات الصلة، الإصدار العاشر (ICD-10) الرمز) إلى عينة العيادة الزمنية المقدمة من قبل GMC. إذا كان رمز ICD-10 لذلك الموعد يعتبر مطابقًا لنوع الورم المقدم من قبل GMC، فإن بيانات HES تعتبر تأكيدًا لتقديم GMC.
حيث لم تتطابق بيانات HES مع نوع الورم المقدم من GMC، تم استخدام ثلاثة نهج إضافية: (1) بالنسبة للأورام الأولية، تم استخدام مجموعة من رموز عمليات HES المنسقة لمطابقة نوع الورم المقدم من GMC وبيانات HES إذا كانت تاريخ العملية يتطابق تمامًا مع تاريخ أخذ عينة الورم المقدم إلى Genomics England؛ (2) بالنسبة للأورام غير الأولية التي تم تحديدها على أنها قولونية من قبل بيانات av_tumor، وكما كانت إما كبدية-بنكرياسية-صفراوية، أو سرطان بطانة الرحم أو رئة في تقديم GMC، تم السماح بمطابقة أكثر مرونة لرموز ICD-10 من HES بشرط أن يكون الفرق في التاريخ بين تاريخ موعد HES وتاريخ أخذ عينة الورم المقدم من GMC أقل من 7 أيام؛ (3) بالنسبة لعدد قليل من العينات المتبقية، تم استخدام بيانات ICD-10 والمورفولوجيا المقدمة من GMC لتأكيد نوع الورم.
تم الحصول على مرحلة الورم من مجموعة بيانات NCRAS. حيث كانت stage_best موجودة في av_tumor وكان التاريخ في عمود قاعدة بيانات التشخيص أقل من 365 يومًا من وقت العينة السريرية المقدمة من GMC، تم استخدام stage_best (مبسطة إلى المراحل 1 و2 و3 و4) (11,618 منتم تصنيف الأورام المقدمة كأورام نقيلية على أنها من المرحلة 4 بشكل افتراضي. تم استخدام مرحلة FIGO (الاتحاد الدولي لأمراض النساء والتوليد) لمؤشرات السريرية المتعلقة بالمبيض والرحم، وتم استخدام تصنيف دوك لسرطانات الغدد الكولونية والمستقيم (كلاهما تم الحصول عليه من جدول av_tumor). في المجموع، تم الحصول على معلومات المرحلة لـ 12,040 من 13,880 (86.7%) من الأورام.
تحليل البقاء
تم إجراء جميع تحليلات البقاء في R باستخدام مكتبات survminer و survival. على وجه التحديد، تم استخدام دوال survfit و ggsurvplot لإنشاء مخططات كابلان-ماير، و coxph لنماذج المخاطر النسبية لكوكس. تم استخدام دالة ggforest لإنشاء مخططات الغابة. تم الحصول على تاريخ الوفاة من بيانات ONS. حيث لم يتم تسجيل وفاة لفرد، تم الحصول على تواريخ العلاج وحدث العملية من مجموعة بيانات HES واستخدمت لتحديد آخر تاريخ تم فيه رؤية الفرد لتمكين التقطيع الصحيح للبيانات.
الأخلاق
البحث الموصوف في هذه المخطوطة يتوافق مع جميع اللوائح الأخلاقية ذات الصلة. تم الحصول على الموافقة للمشروع من لجنة الأخلاقيات البحثية في شرق إنجلترا – كامبريدج الجنوبية (مرجع لجنة الأخلاقيات البحثية 14/EE/1112، معرف نظام تقديم الطلبات البحثية المتكاملة: 166046)تم اختيار المشاركين بناءً على تحديدهم من قبل المتخصصين في الرعاية الصحية والباحثين داخل خدمة الصحة الوطنية (NHS) على أنهم مصابون بتشخيص السرطان. تم تجنيد المشاركين من 13 مركزًا طبيًا تابعًا لـ NHS وتم الحصول على موافقة خطية مستنيرة من المشاركين.
ملخص التقرير
معلومات إضافية حول تصميم البحث متاحة في ملخص تقارير مجموعة نيتشر المرتبط بهذه المقالة.
توفر البيانات
البيانات التي تدعم نتائج هذه الدراسة متاحة ضمن بيئة البحث، وهي مساحة عمل سحابية آمنة. يمكن العثور على تفاصيل كيفية الوصول إلى البيانات لهذه المنشورة فيhttps://re-docs. genomicsengland.co.uk/pan_cancer_pub/يمكن العثور على بيانات مجمعة إضافية معالجة تم استخدامها لتوليد الأرقام في الجداول التكميلية 5-20. للوصول إلى البيانات الجينومية والسريرية داخل هذا البيئة البحثية، يجب على الباحثين أولاً التقدم ليصبحوا أعضاء في شبكة أبحاث جينوميات إنجلترا. (المعروف سابقًا بشراكة التفسير السريري لجينوميات إنجلترا، GECIP) (www.genomicsengland.co.uk/research/شريك صناعي في منتدى الاكتشاف (أكاديمي)www.genomic-sengland.co.uk/research/research-environment). يتم وصف عملية الانضمام إلى شبكة أبحاث جينومكس إنجلاند في www.genomicsengland.co.uk/research/academic/join-gecipويتكون من الخطوات التالية: (1) إذا لم تكن مؤسستك تشارك بالفعل، فسيتعين عليها توقيع اتفاقية المشاركة المتاحة فيhttps://files.genomicsengland.co.uk/documents/اتفاقية المشاركة Genomics-England-GeCIP-v2.0.pdf وقم بإرسال النسخة الموقعة إلىgecip-help@genomicsengland.co.uk; (2) بمجرد أن تؤكد أن مؤسستك مسجلة وقد وجدت مجال اهتمام، يمكنك التقديم من خلال النموذج الإلكتروني على www.genomicsengland.co.uk/research/academic/join-gecipبمجرد إنشاء حسابك في بوابة البحث، ستتمكن من تسجيل الدخول وتتبع طلبك؛ (3) سيتم مراجعة طلبك خلال عشرة أيام عمل؛ (4) ستقوم مؤسستك بالتحقق من انتمائك؛ و (5) ستكمل تدريبنا عبر الإنترنت حول حوكمة المعلومات وسيتم منحك الوصول إلى بيئة البحث خلال ساعتين من اجتياز التدريب عبر الإنترنت. البيانات المتاحة للمستخدمين المسجلين تشمل: المحاذاة بتنسيق BAM أو CRAM؛ استدعاءات المتغيرات المعلّقة بتنسيق VCF؛ تعيين التوقيع؛ عبء الطفرات الورمية؛ مقاييس جودة التسلسل؛ ملخص النتائج المشتركة مع مختبرات الجينوم؛ وبيانات سريرية ثانوية كما هو موضح في هذه الورقة. يمكن العثور على مزيد من التفاصيل حول أنواع البيانات المتاحة (على سبيل المثال، بيانات الوفيات، إحصائيات حلقات المستشفى وبيانات العلاج) فيhttps://re-docs.genomicsengland.co.uk/data_overview/.Germlineيمكن استكشاف المتغيرات باستخدام متصفح تحليل المتغيرات التفاعلي (https://re-docs.genomicsengland.co.uk/iva_variant/). يمكن استكشاف مجموعة المرضى المصابين بالسرطان والمعلومات السريرية الطولية حول العلاج والوفيات باستخدام مستكشف المشاركين (https://re-docs.genomicsengland.co.uk/pxa/).
نشكر المشاركين الذين جعلوا هذا العمل ممكنًا. تم تمكين هذا البحث من خلال الوصول إلى البيانات والنتائج التي تم توليدها بواسطة مشروع الجينوم 100,000. يتم إدارة مشروع الجينوم 100,000 بواسطة شركة جينومكس إنجلاند المحدودة، وهي شركة مملوكة بالكامل لوزارة الصحة والرعاية الاجتماعية. يتم تمويل مشروع الجينوم 100,000 من قبل المعهد الوطني للبحوث الصحية وNHS إنجلترا. كما قامت مؤسسة ويلكوم، وبحوث السرطان في المملكة المتحدة، ومجلس البحوث الطبية بتمويل بنية البحث التحتية. يستخدم مشروع الجينوم 100,000 البيانات تم توفيره من قبل المرضى وجمعه من قبل NHS كجزء من رعايتهم ودعمهم. نحن ممتنون للدعم من السيدة S. Hill والفريق في NHS إنجلترا لتأسيس وتمويل 13 مركزًا للطب الجينومي. نشكر جميع مراكز الطب الجينومي في NHS عبر إنجلترا: شرق إنجلترا؛ كلية إمبريال وغرب لندن؛ مانشستر الكبرى؛ شمال ثايمز؛ الساحل الشمالي الغربي؛ شمال شرق وإنجلترا؛ أكسفورد؛ جنوب ثايمز؛ جنوب غرب؛ غرب إنجلترا؛ ويسكس؛ غرب ميدلاندز؛ ويوركشاير وهامبر. لقد مكنوا مساهمة NHS، بما في ذلك الإرجاع السريري للنتائج ضمن NHS بتنسيق موحد ومُعتمد. M.C. هو خريج باحث كبير في المعهد الوطني للبحوث الصحية والرعاية (NIHR). هذا العمل هو جزء من محفظة البحث الانتقالي في مراكز البحث البيولوجي NIHR في بارتس، مستشفيات جامعة كامبريدج NHS، مؤسسة مستشفى غريت أورموند ستريت NHS، مؤسسة مستشفى جامعة مانشستر NHS، مؤسسة مستشفى نيوكاسل NHS، مؤسسة مستشفى جامعة أكسفورد NHS، مؤسسة غايز وسانت توماس NHS ومؤسسة جامعة كوليدج لندن NHS. نشكر مختبرات الجينوم NHS وأولئك الذين عملوا على إنشاء الدليل الوطني للاختبارات الجينومية وإجراء مراجعته السنوية، و NHS GMS. تم تحقيق هذا العمل بفضل كرم مرضى NHS ويستخدم بيانات سريرية من NHS و NHS Digital. نشكر جميع الموظفين في جينومكس إنجلترا المحدودة وأعضاء اتحاد أبحاث جينومكس إنجلترا. نشكر فريق خدمات مختبر إيلومينا في هينكستون على نصائحهم ولقيامهم بإجراء تسلسل الجينوم الكامل (WGS). نشكر جامعة كوليدج لندن، تكنولوجيا أبحاث السرطان وفريق TRACERx لتوفير عينات الأورام الرئوية التي تم استخدامها في التحقق من خط أنابيب WGS. نشكر J. Chalker، مستشفى غريت أورموند ستريت للأطفال NHS، و A. Wallace، مركز مانشستر للطب الجينومي لمشاركة البيانات من الاختبارات الجينومية المتوازية التي تم استخدامها في التحقق من خط أنابيب WGS. تم تمويل D.C. بالكامل من خلال منحة من أبحاث السرطان في المملكة المتحدة (رقم C1298/A8362) الممنوحة لـ R. Houlston في معهد أبحاث السرطان في المملكة المتحدة. تم تمويل B.N. من خلال منحة مركز أبحاث السرطان في المملكة المتحدة في برمنغهام رقم C17422/A25154.
مساهمات المؤلفين
قام كل من س.هـ، ز.د، س.ت، س. هندرسون، ل.ج، أ.هـ، ن.م، ت.ف، م.ج وس. هينغ بالإشراف على تنفيذ مشروع الجينوم 100,000 في هيئة الخدمات الصحية الوطنية. أشرف د.ب وس.ك على شراكة مع إيلومينا لتسلسل الجينوم الكامل. قام ج.ب، أ. صديق، ت.ز وت.س بتنفيذ عمليات تدفق العينات. قام ج.أ، ب.أ، ج.س وم.ب.ب بتنظيم البيانات الواقعية الطولية وربطها ببيانات التسلسل. ساهم أ.ر.-م، أ. سوسينسكي، د.ب.-ج، ج.ل، ج.م، د.س، ب.ن، ن.ف وأ.ر في تطوير خط أنابيب التحليل البيوانفورماتيكي. صمم أ. سوسينسكي، ن.م، م.ج، ج.أ و و.س الدراسة. قام ج.أ، و.س، د.ب وأ. سوسينسكي بإجراء التحليل وتفسير البيانات. قام ج.أ، و.س، ك.ب وأ.ي بإجراء تصور البيانات. أشرف ن.م، أ. سوسينسكي وم.ج على الدراسة. كتب ن.م، أ. سوسينسكي، س.و، س.ت، ج.أ و و.س المخطوطة بمشاركة جميع المؤلفين. اتخذ أ. سوسينسكي، م.ج ون.م القرار بتقديم المخطوطة للنشر.
المصالح المتنافسة
جينومكس إنجلاند هي شركة مملوكة بالكامل لوزارة الصحة والرعاية الاجتماعية في المملكة المتحدة، وقد تم إنشاؤها في عام 2013 لإدخال تسلسل الجينوم الكامل في الرعاية الصحية بالتعاون مع هيئة الخدمات الصحية الوطنية في إنجلترا. جميع المؤلفين المرتبطين بجينومكس إنجلاند (A. Sosinsky، J.A.، C.T.، S. Henderson، L.J.، A.H.، P.A.، G.C.، J.M.، S.W.، K.B.، D.P.، M.B.P.، N.V.، A.R.-M.، D.P.-G.، J.L.، J.P.، A. Siddiq، T.Z.، T.C.، O.Y.، T.F.، A.R.، M.C. و N.M.) هم، أو كانوا، يتقاضون رواتب من أو تم إلحاقهم بجينومكس إنجلاند. D.B. و C.K. هما موظفان بدوام كامل ومساهمان في شركة إلومينا. A.H. قد حصل على أتعاب متحدث من جلياد، روش، فايزر، جاز، أبفي، إنسايت وأستيلس. N.M. قدمت استشارات ودعم استشاري لفايزر، غاردانت، سيجن وجانسن، وتلقت أتعاب متحدث من نوفارتس، فايزر وسيرفييه. خارج العمل المقدم. يعلن المؤلفون المتبقون عدم وجود مصالح متنافسة.
يجب توجيه المراسلات والطلبات للحصول على المواد إلى مارك كولفيلد أو نيروبا موروغيسو.
معلومات مراجعة الأقران تشكر مجلة ناتشر ميديسن جو لين روكيتا، مارك روبين وستيفن ج. تشانوك على مساهمتهم في مراجعة هذا العمل. المحرر الرئيسي: آنا ماريا رانزوني، بالتعاون مع فريق ناتشر ميديسن.
الشكل 1D من البيانات الموسعة (الخطوط الجينية المسببة للسرطان)
الشكل البياني الموسع 1 | انتشار (كنسبة مئوية) لأنواع مختلفة من الطفرات التي تم تحديدها بواسطة تسلسل الجينوم الكامل في الجينات المشار إليها للاختبار في الدليل الوطني للاختبارات الجينومية للسرطان (NGTDC). (أ) الطفرات الصغيرة الجسدية (طفرات النوكليوتيد المفردة (SNVs)، الإدخالات والحذف). (ب) الشذوذات في عدد النسخ (CNAs)؛ onc = جين ورمي، tsg = جين مثبط للورم. (ج) المتغيرات الهيكلية (SVs). (د) المتغيرات الصغيرة المرتبطة بالوراثة خطر الإصابة بالسرطان (الجينات المهيئة). يتم عرض نسبة الأورام التي تحتوي على نوع محدد من الطفرات في الجين (أو الجينات) المشار إليها للاختبار حسب نوع الورم في NGTDC باللون الأرجواني. يتم عرض حدوث الطفرات (كنسبة مئوية) في أنواع الأورام الأخرى، التي لم يتم الإشارة إليها حاليًا في NGTDC، باللون الأزرق. تعكس تدرجات الألوان نسبة الحالات المتأثرة.
الشكل البياني الموسع 2 | توزيع عبء الطفرات الورمية (TMB) والتوقيعات الطفرية عبر أنواع الأورام. تتطابق تخصيص التوقيعات للأسباب المعروفة مع الشكل 3.
الشكل البياني الممتد 3 | تقديرات كابلان-ماير للبقاء العام مع-القيم المحسوبة باستخدام اختبار لوغاريتمي مصنف. أعداد المرضى المعرضين للخطر في نقاط زمنية مختلفة موضحة أسفل منحنيات البقاء. النقاط وأشرطة الخطأ في الرسوم البيانية المدمجة تشير إلى نسب المخاطر (HR) مع 95%
من نماذج المخاطر النسبية لكوكس المصححة حسب مرحلة السرطان. المرضى هم مُصنَّف حسب حالة الطفرات للجينات المحددة للاختبار في NGTDC عبر جميع أنواع السرطانيمكن العثور على قيم p الدقيقة في الجدول التكميلي S2. فترات الثقة (CI) ، على التوالي. يتم حساب HR و CI وقيم p. من نماذج المخاطر النسبية لكوكس المصححة حسب مرحلة السرطان. يتم تصنيف المرضى حسب حالة الطفرات للجينات المحددة للاختبار في NGTDC عبر جميع أنواع السرطان (يمكن العثور على قيم p الدقيقة في الجدول التكميلي S2.
الجدول البياني الموسع 1 | العمر الوسيط ونطاق الربعين (IQR) عند التشخيص في غياب ووجود النتائج الجينية ذات الصلة
نوع الورم
متغير خط الجرثومة
ن
الوسيط (المدى interquartile)
قيمة P المصححة
مستوى الدلالة
سرطان urothelial في المثانة
غائب
255
73 (67-80)
0.474
NS
حاضر
<5
82.5 (73.75-87.5)
سرطان الثدي الغازي
غائب
٢٨٣٩
61 (51-72)
***
حاضر
86
51.5 (46-63.75)
سرطان الغدة الدرقية في القولون
غائب
1893
70 (62-77)
٧.٣٨ × ١٠^-٦
***
حاضر
٥٥
61 (48-70)
الورم الدبقي متعدد الأشكال
غائب
٣١٢
61 (52-69)
0.474
NS
حاضر
<5
٥٨ (٥٤-٥٨.٥)
سرطان المبيض عالي الدرجة من النوع الحليمي
غائب
٣٢٢
67 (58-73)
***
حاضر
50
٥٩ (٤٩.٥-٦٣.٧٥)
سرطان الخلايا الكلوية الواضحة
غائب
١١٥٤
63 (55-71)
*
حاضر
9
٤٠ (٢٨-٥٦)
سرطان الغدة البنكرياسية
غائب
١١٨
70 (65-76)
0.474
NS
حاضر
<5
70 (63-70.5)
سرطان الغدة المخاطية في المستقيم
غائب
815
67 (60-74)
0.101
NS
حاضر
15
61 (49.5-62.5)
الميلانوما الجلدية
غائب
٣٢٤
68 (56-76.25)
0.474
NS
حاضر
<5
76 (76-76)
سرطان بطانة الرحم
غائب
682
67 (59-74)
0.343
NS
حاضر
٢٤
٥٩.٥ (٥٥-٦٩.٥)
سرطان بطانة الرحم الغدي المسامي
غائب
١١٠
71 (67-75)
0.474
NS
حاضر
<5
60 (59-72.5)
محفظة الطبيعة
مارك كولفيلد المؤلف(المؤلفون) المراسلون: نيروبا موروغيسو آخر تحديث من المؤلف(ين): 15 أكتوبر 2023
ملخص التقرير
تتمنى Nature Portfolio تحسين قابلية إعادة إنتاج العمل الذي ننشره. يوفر هذا النموذج هيكلًا للاتساق والشفافية في التقرير. لمزيد من المعلومات حول سياسات Nature Portfolio، يرجى الاطلاع على سياسات التحرير وقائمة مراجعة سياسة التحرير.
الإحصائيات
لجميع التحليلات الإحصائية، تأكد من أن العناصر التالية موجودة في أسطورة الشكل، أسطورة الجدول، النص الرئيسي، أو قسم الطرق.
مؤكد حجم العينة بالضبط ( ) لكل مجموعة/شرط تجريبي، معطاة كرقم منفصل ووحدة قياس
بيان حول ما إذا كانت القياسات قد أُخذت من عينات متميزة أو ما إذا كانت نفس العينة قد تم قياسها عدة مرات X اختبار(ات) الإحصاء المستخدمة وما إذا كانت أحادية الجانب أو ثنائية الجانب يجب أن تُوصف الاختبارات الشائعة فقط بالاسم؛ واصفًا التقنيات الأكثر تعقيدًا في قسم الطرق. وصف لجميع المتغيرات المرافقة التي تم اختبارها وصف لأي افتراضات أو تصحيحات، مثل اختبارات الطبيعية والتعديل للمقارنات المتعددة “凶” translates to “شر” in Arabic. وصف كامل للمعلمات الإحصائية بما في ذلك الاتجاه المركزي (مثل المتوسطات) أو تقديرات أساسية أخرى (مثل معامل الانحدار) والتباين (مثل الانحراف المعياري) أو تقديرات عدم اليقين المرتبطة (مثل فترات الثقة)
لاختبار الفرضية الصفرية، إحصائية الاختبار (على سبيل المثال، ) مع فترات الثقة، أحجام التأثير، درجات الحرية وقيمة ملحوظة أعطِالقيم كقيم دقيقة كلما كان ذلك مناسبًا. لتحليل بايزي، معلومات حول اختيار القيم الأولية وإعدادات سلسلة ماركوف مونت كارلو للتصاميم الهرمية والمعقدة، تحديد المستوى المناسب للاختبارات والتقارير الكاملة عن النتائج تقديرات أحجام التأثير (مثل حجم تأثير كوهين)بيرسون )، مما يشير إلى كيفية حسابها تحتوي مجموعتنا على الإنترنت حول الإحصائيات لعلماء الأحياء على مقالات تتناول العديد من النقاط المذكورة أعلاه.
البرمجيات والشيفرة
معلومات السياسة حول توفر كود الكمبيوتر
جمع البيانات
تحليل البيانات
OpenClinica v3.4
جودة بيانات التسلسل، التعيين واستدعاء المتغيرات نسخة 2.6.53.23 من خط أنابيب نورث ستار إصدار آيزاك iSAAC-03.16.02.19 ستارلينغ الإصدار 2.4.7 ستريلكا النسخة 2.4.7 إصدار كانفاس 1.3.1 مانتا الإصدار 0.28.0 إصدار سام تولز 1.9 حزم R المستخدمة في هذا التحليل R4.0.3 سيرفماينر_0.4.9 البقاء_3.2-7 dplyr_1.0.7 بور_0.3.4 tidyr_1.1.2 تبل_3.0.3 ggplot2_3.3.2 tidyverse_1.3.0
بالنسبة للمخطوطات التي تستخدم خوارزميات أو برامج مخصصة تكون مركزية في البحث ولكن لم يتم وصفها بعد في الأدبيات المنشورة، يجب أن تكون البرمجيات متاحة للمحررين والمراجعين. نحن نشجع بشدة على إيداع الشيفرة في مستودع مجتمعي (مثل GitHub). راجع إرشادات مجموعة Nature لتقديم الشيفرة والبرمجيات لمزيد من المعلومات.
بيانات
معلومات السياسة حول توفر البيانات
يجب أن تتضمن جميع المخطوطات بيانًا حول توفر البيانات. يجب أن يوفر هذا البيان المعلومات التالية، حيثما ينطبق:
رموز الانضمام، معرفات فريدة، أو روابط ويب لمجموعات البيانات المتاحة للجمهور
وصف لأي قيود على توفر البيانات
بالنسبة لمجموعات البيانات السريرية أو بيانات الطرف الثالث، يرجى التأكد من أن البيان يتماشى مع سياستنا
مجموعات البيانات العامة التي تم استخدامها لتوصيف المتغيرات: إصدار إنسيبل 90/GRCh38 نسخة كوزميك v86 إصدار كلينفار أكتوبر 2018 توقيعات كوزميك v3 البيانات التي تدعم نتائج هذه الدراسة متاحة ضمن بيئة البحث، وهي مساحة عمل سحابية آمنة. يمكن العثور على تفاصيل كيفية الوصول إلى البيانات لهذه المنشورة فيhttps://re-docs.genomicsengland.co.uk/pan_cancer_pub/يمكن العثور على بيانات مجمعة إضافية معالجة تم استخدامها لتوليد الأرقام في الجداول التكميلية S5-S20.
بمجرد أن تؤكد أن مؤسستك مسجلة وقد وجدت مجال GECIP الذي يهمك، يمكنك التقديم من خلال النموذج الإلكتروني على https://www.genomicsengland.co.uk/research/academic/join-gecipبمجرد إنشاء حسابك في بوابة البحث، ستتمكن من تسجيل الدخول وتتبع طلبك.
سيقوم قائد المجال بمراجعة طلبك خلال 10 أيام عمل.
ستقوم مؤسستك بالتحقق من انتمائك.
ستكمل تدريبنا عبر الإنترنت حول حوكمة المعلومات وسيتم منحك الوصول إلى بيئة البحث خلال ساعتين من اجتياز التدريب عبر الإنترنت.
البيانات المتاحة للمستخدمين المسجلين تشمل: المحاذاة بتنسيق BAM أو CRAM، استدعاءات المتغيرات المعلّمة بتنسيق VCF، تعيين التوقيعات، عبء الطفرات الورمية، مقاييس جودة التسلسل، ملخص النتائج الذي يتم مشاركته مع مختبرات الجينوم، البيانات السريرية الثانوية كما هو موصوف في هذه الورقة. يمكن العثور على مزيد من التفاصيل حول أنواع البيانات المتاحة (على سبيل المثال، بيانات الوفيات، إحصائيات حلقات المستشفى وبيانات العلاج) فيhttps://redocs.genomicsengland.co.uk/data_overview/يمكن استكشاف المتغيرات الجينية في متصفح تحليل المتغيرات التفاعلي (انظر الوصف فيhttps://redocs.genomicsengland.co.uk/iva_variant/). يمكن استكشاف مجموعة مرضى السرطان والمعلومات السريرية الطولية حول العلاج والوفيات باستخدام مستكشف المشاركين (انظر الوصف في https://re-docs.genomicsengland.co.uk/pxa/).
البحث الذي يتضمن مشاركين بشريين، بياناتهم، أو مواد بيولوجية
معلومات السياسة حول الدراسات التي تشمل مشاركين بشريين أو بيانات بشرية. انظر أيضًا معلومات السياسة حول الجنس، الهوية/التقديم الجنسي، والتوجه الجنسي والعرق، والاثنية والعنصرية.
التقارير عن الجنس والنوع
تم استنتاج الجنس البيولوجي باستخدام نسبة متوسط تغطية التسلسل لكروموسومات الجنس ومتوسط تغطية التسلسل للكروموسومات الجسدية. في تحليلنا، لم يتم تصنيف المرضى حسب الجنس لزيادة قوة المجموعة. قدم المرضى موافقة مستنيرة لتحليل تسلسل الجينوم الكامل (WGS) للأورام الطبيعية (الجينوم الجرثومي) المزدوج. كما أعطى المشاركون موافقتهم لربط بياناتهم الجينومية بسجلات صحية طولية مجهولة الهوية ومشاركتها مع الباحثين في بيئة بحث آمنة.
التقارير عن العرق أو الإثنية أو غيرها من المجموعات الاجتماعية ذات الصلة
خصائص السكان
لم يتم استخدام متغيرات التصنيف ذات الصلة الاجتماعية في هذه الدراسة.
15,241 مريضًا تم تشخيصهم بالسرطان ضمن NHS وتم تجنيدهم في برنامج السرطان لمشروع الجينوم 100,000 بين عامي 2015 و2019. أنواع الأورام التي تم تسلسل أكثر من 1,000 جينوم ورمي لها تشمل سرطان الثدي الغازي.سرطان الغدة الدرقية في القولون (الساركوما ) وسرطان الخلايا الكلوية الواضحة ( ). ( ) من المرضى كانوا يعانون من سرطان المرحلة الرابعة (مرض متقدم نقيل). تم ملاحظة بداية مبكرة (متوسط العمر <50 سنة) للأورام الدبقية منخفضة الدرجة وأورام الخلايا الجرثومية الخصوية بما يتماشى مع إحصائيات الحدوث. كانت عينات الورم تأتي بشكل رئيسي من الاستئصال الجراحي ( )، بما في ذلك حالات لم تتلقَ العلاجعلاج ما بعد العلاج المساعد. فقطجاءت من خزعات نقيلية أو تشخيصية، معبعد العلاج.
تم اختيار المشاركين بناءً على تحديدهم من قبل المتخصصين في الرعاية الصحية والباحثين داخل خدمة الصحة الوطنية (NHS) على أنهم مصابون بالسرطان. تم تجنيد المشاركين من 13 مركزًا للطب الجيني التابع لـ NHS وتم الحصول على موافقة خطية مستنيرة من المشاركين.
رقابة الأخلاقيات
البحث الموصوف في هذه المخطوطة يتوافق مع جميع اللوائح الأخلاقية ذات الصلة. تم الحصول على الموافقة للمشروع من لجنة الأخلاقيات البحثية في شرق إنجلترا – كامبريدج الجنوبية (مرجع REC 14/EE/1112، معرف IRAS 166046)
يرجى ملاحظة أنه يجب أيضًا تقديم معلومات كاملة حول الموافقة على بروتوكول الدراسة في المخطوطة.
التقارير الخاصة بالمجالات
يرجى اختيار الخيار أدناه الذي يناسب بحثك بشكل أفضل. إذا لم تكن متأكدًا، اقرأ الأقسام المناسبة قبل اتخاذ قرارك.
علوم الحياة العلوم السلوكية والاجتماعية العلوم البيئية والتطورية والبيئية
لنسخة مرجعية من الوثيقة مع جميع الأقسام، انظر nature.com/documents/nr-reporting-summary-flat.pdf
تصميم دراسة علوم الحياة
يجب على جميع الدراسات الإفصاح عن هذه النقاط حتى عندما يكون الإفصاح سلبيًا.
حجم العينة
النتائج المعروضة هنا ليست نتيجة لإعداد تجريبي. نحن نصف الملاحظات لمجموعة من 13,880 مريض سرطان تم تجنيدهم لبرنامج 100,000 جينوم. حساب حجم العينة غير ذي صلة لهذه الدراسة.
استبعاد البيانات
تم استبعاد السرطانات الأطفال، والأورام الدموية، والسرطانات ذات المصدر غير المعروف والعينات التي لم تحتوي على معلومات سريرية من مصادر ثانوية كما هو مذكور في المخطوطة.
التكرار
التكرار غير ذي صلة للسبب الموضح أعلاه
العشوائية
العشوائية غير ذات صلة للسبب الموضح أعلاه
التعمية
التعمية غير ذات صلة للسبب الموضح أعلاه
التقارير للمواد والأنظمة والأساليب المحددة
نحن نطلب معلومات من المؤلفين حول بعض أنواع المواد والأنظمة التجريبية والأساليب المستخدمة في العديد من الدراسات. هنا، حدد ما إذا كانت كل مادة أو نظام أو طريقة مدرجة ذات صلة بدراستك. إذا لم تكن متأكدًا مما إذا كان عنصر القائمة ينطبق على بحثك، اقرأ القسم المناسب قبل اختيار رد.
تم حساب الأهمية الإحصائية للاكتشافات الجينية ذات الصلة لبداية الورم المبكر بواسطة اختبار ويلكوكسون لمجموع الرتب مع اختبار بنجاميني-هوشبرغ المتعدد. ***P<0.0001، *P<0.05.
تم استبعاد أنواع الأورام التي لم يتم اختبارها لمتغيرات الجينوم في NGTDC.
Insights for precision oncology from the integration of genomic and clinical data of 13,880 tumors from the Genomes Cancer Programme
Received: 19 December 2022
Accepted: 2 November 2023
Published online: 11 January 2024
(i) Check for updates
A list of authors and their affiliations appears at the end of the paper
The Cancer Programme of the 100,000 Genomes Project was an initiative to provide whole-genome sequencing (WGS) for patients with cancer, evaluating opportunities for precision cancer care within the UK National Healthcare System (NHS). Genomics England, alongside NHS England, analyzed WGS data from 13,880 solid tumors spanning 33 cancer types, integrating genomic data with real-world treatment and outcome data, within a secure Research Environment. Incidence of somatic mutations in genes recommended for standard-of-care testing varied across cancer types. For instance, in glioblastoma multiforme, small variants were present in 94% of cases and copy number aberrations in at least one gene in 58% of cases, while sarcoma demonstrated the highest occurrence of actionable structural variants (13%). Homologous recombination deficiency was identified in 40% of high-grade serous ovarian cancer cases with linked to pathogenic germline variants, highlighting the value of combined somatic and germline analysis. The linkage of WGS and longitudinal life course clinical data allowed the assessment of treatment outcomes for patients stratified according to pangenomic markers. Our findings demonstrate the utility of linking genomic and real-world clinical data to enable survival analysis to identify cancer genes that affect prognosis and advance our understanding of how cancer genomics impacts patient outcomes.
Over the last decade, UK cancer incidence has increased by approximately 4% (ref. 1), driving the need for molecular cancer testing, including germline testing of cancer predisposition genes and pharmacogenomic markers . The 100,000 Genomes Project, a transformational UK Government initiative conducted within the National Health Service (NHS) in England, aimed to establish standardized high-throughput whole-genome sequencing (WGS) for patients with cancer and rare diseases via an automated, International Organization
for Standardization-accredited bioinformatics pipeline (providing clinically accredited variant calling and variant prioritization) . The role of WGS at scale for patients with cancer in the NHS was evaluated within the Cancer Programme of the 100,000 Genomes Project (Fig. 1a). Participants gave written informed consent for their genomic data to be linked to anonymized longitudinal health records and shared with researchers in a secure Research Environment (www.genomic-sengland.co.uk/research/research-environment) to drive forward our
knowledge across different cancers . The data generated were then used to establish a national molecular data platform (National Genomic Research Library) with secure links to longitudinal real-world data in the Research Environment (Fig.1b). The national clinical datasets include the National Cancer Registration and Analysis Service (NCRAS) dataset consisting of cancer registration data and the Systemic Anti-Cancer Therapy (SACT) dataset, as well as subsequent cancer episodes, including Hospital Episode Statistics (HES) and mortality data from the Office for National Statistics (ONS) (Fig. 1b). This approach enables genomic research and discovery to be fed back into genomic healthcare (Fig.1c).
A longer-term objective was to accelerate the delivery of molecular testing, including WGS, in NHS clinical cancer care . Building on evolving knowledge from the 100,000 Genomes Project and the existing molecular testing provision within the NHS, the NHS Genomic Medicine Service (GMS) was launched in October 2018 to deliver genomic testing, clinical care and interpretation for rare diseases and cancer across England, using a standardized National Genomic Test Directory , including targeted large gene panels and WGS, to enable equitable access and comprehensive genomic testing. The National Genomic Test Directory aims to provide consistency of test methodologies, gene targets and eligibility criteria across clinical indications via a consolidated network of seven NHS England (NHSE) Regional Genomic Laboratory Hubs . It specifies the genomic tests that are commissioned and thereby funded by the NHS in England as part of gold standard molecular profiling in different cancer clinical indications and provides opportunities for patients to participate in research?
Large-scale sequencing studies such as the International Cancer Genome Consortium (ICGC) and The Cancer Genome Atlas (TCGA) have extensively cataloged the spectra of somatic mutations across cancer types from a retrospective cohort of 2,658 primary tumor samples . More recent initiatives, such as The Hartwig Medical Foundation reported clinically relevant findings for 4,784 metastatic adult solid tumor samples and supported recruitment to the Drug Rediscovery Protocol (DRUP) trial . These initiatives represent, to date, the two largest WGS cohorts available for research. In this article, we present our analysis of WGS data from 13,880 solid tumors, focused on clinically actionable genes and pangenomic markers, linked to real-world longitudinal, life course clinical, treatment and long-term survival data to highlight the learnings from the Cancer Programme and the implications for current clinical care.
Results
Cohort demographics
We sequenced 16,358 tumor-normal sample pairs from 15,241 patients diagnosed with cancer within the NHS who were recruited to the Cancer Programme of the 100,000 Genomes Project between 2015 and 2019, with almost half of the patients being recruited in 2018 and the remainder in this Project being recruited through the Rare Disease arm. Our integrative whole-genome analysis (WGA) covered 33 tumor types (Fig. 2a) of 13,880 tumor samples, consisting of 13,311 fresh-frozen ( ) and 569 formalin-fixed paraffin-embedded tumor samples (4.1%). Matched normal (germline) samples included 13,493 (99.1%) blood-derived, 100 (0.7%) from normal tissue and 23 (0.2%) from saliva samples. Tumor samples were sequenced to coverage and normal samples to to ensure high sensitivity of variant calling (Methods) in clinical settings (compared with and in the TCGA cohort). Genomes from hematological tumors ( ), pediatric cancers ( ), carcinomas of unknown primary ( ) and tumors that were not linked to external datasets ( ) were excluded from this analysis. The diagnosis submitted at sample collection was confirmed by linking genomics data with the NCRAS and HES datasets. Tumor types with more than 1,000 sequenced tumor genomes included breast invasive carcinoma ( ), colon adenocarcinoma ( ), sarcoma ( ) and kidney renal clear cell carcinoma ( ). Figure 2b illustrates recruitment across 13 NHS GMCs (comprising over
80 hospital trusts) in England. The distribution of biological sex and age across tumor types is shown in Fig. 2c. Early onset (median age less than 50 years) was observed for low-grade glioma and testicular germ cell tumors in agreement with incidence statistics .
Staging information was available in the NCRAS dataset for 12,040 (86.7%) tumors. The breakdown of the different stages for the tumor types sequenced is shown in Fig. 3; of 13,880) of patients had stage 4 cancer (advanced metastatic disease) with samples obtained from metastatic sites including the liver, lymph nodes, lung and brain. Ovarian high-grade serous carcinoma and skin cutaneous melanoma exhibited higher prevalence of advanced (stages 3 and 4) disease, whereas invasive breast cancers had a higher prevalence of early-stage (stages 1 and 2) disease due to sampling biases in tissue ascertainment. Tumor samples mainly originated from surgical resections ( ), including treatment-naive cases and cases after neoadjuvant treatment. Only 5.5% ( ) came from metastatic or diagnostic biopsies, with 10.9% ( ) being after treatment (Fig. 3). The tumor purity depicted in Fig. 3 highlights challenges in obtaining samples with adequate tumor content (more than ) in specific cancers, such as lung and pancreatic adenocarcinomas, which is consistent with previous publications .
Clinical actionability through WGS
A single test such as WGS, comprising paired tumor-normal sequencing, can facilitate the concurrent detection of somatic small variants including single-nucleotide variants (SNVs) and insertions and deletions (indels), copy number aberrations (CNAs) and structural variants (SVs), including gene fusions. In addition, germline findings, such as variants in cancer susceptibility genes and pharmacogenomic findings (variants affecting the metabolism of therapeutic agents used to treat cancers), enabled a greater yield of clinically relevant findings. The Cancer Programme delivered standardized WGA results, generated in an automated bioinformatics pipeline, returned to NHS GMC Laboratories. Potentially actionable findings were reviewed initially by clinical scientists and subsequently at multidisciplinary Molecular Tumor Boards, referred to as Genomic Tumor Advisory Boards (GTABs). Examples of WGA results are shown in the Supplementary Information; full details of the analysis and interpretation are described in the Methods, showing the utility of WGS to capture various genomic alterations of clinical relevance with a single test.
We analyzed aggregated data from 13,880 whole genomes in the context of the current National Genomic Test Directory for Cancer (NGTDC) v.6.0 updated on 3 April 2023 (ref. 7); several types of mutations relating to targets specified in the NGTDC were detected, including small variants, CNAs and fusions, along with germline variants associated with inherited cancer risk and pharmacogenomic findings (see the online Methods for details). The percentage of cases with one or more somatic mutations present in genes indicated in the NGTDC for the applicable cancer type was high, although variable (Fig. 4). For example, over of tumors harbored one or more mutations found in genes indicated for testing in the NGTDC in glioblastoma multiforme, low-grade glioma, skin cutaneous melanoma, head and neck squamous cell carcinoma, colon and rectal adenocarcinoma, and lung adenocarcinoma (Fig. 4). Clinically relevant mutations were found in 20-49% of breast invasive carcinoma, ovarian high-grade serous carcinoma, uterine endometrial, sarcoma, mesothelioma, bladder urothelial carcinoma and lung squamous cell carcinoma cases, while in other cancer types such as pancreatic, prostate, esophageal and stomach adenocarcinomas, less than of cases possessed mutations in genes present in the NGTDC (Fig. 4). We note that the clinical actionability of these mutations will be dependent on the individual case and clinical circumstances, such as the stage of the tumor and associated comorbidities of the participant. This highlights the need for clinical interpretation and discussion where clinically appropriate within a GTAB.
Fig. 1| Overview of the Genomes Cancer Programme. a, Journey of the patient’s genome. Patients provided written informed consent for paired tumor and normal (germline) WGS analysis. DNA was extracted from tumor and normal (blood) samples using standardized protocols and samples were submitted for WGS, which was performed on an Illumina sequencer. An automated pipeline was constructed for sequence quality control, alignment, variant calling and
interpretation, with results returned to the 13 NHS Genomic Medicine Centers for review in regional GTABs. b, Linked genomic and real-world clinical datasets. In the 100,000 Genomes Project, participants are followed over their life course using electronic health records (all hospital episodes, cancer registration entries, systemic anticancer therapies and cause of death). c, Infinity loop representing the link between healthcare and research in genomics.
We assessed the mutations listed in the NGTDC in other cancer types for which testing of that gene or mutation is not currently indicated (Fig. 4 and in Extended Data Fig.1a-d). These variants are denoted in blue in Fig. 4 and could indicate potentially actionable findings that may enable recruitment into clinical trials or prompt further review within a GTAB. For example, SNVs were identified in PIK3CA and across different cancer types and similarly pangenomic markers, such as homologous recombination deficiency (HRD) and tumor mutational burden (TMB), for which clinical trials may be available. As biomarker-driven trial evidence grows, NGTDC indications are expected to expand, incorporating new genes and biomarkers across several cancer types.
Landscape of somatic small variants
The most frequently mutated gene was TP53 (5,411 of 13,880,39.0% of patients; Fig. 4 and online Methods). Within individual cancer types,
the frequency of TP53 mutations was variable but highest in uterine corpus endometrial serous carcinoma, ovarian high-grade serous carcinoma, lung squamous cell carcinoma, rectum adenocarcinoma, esophageal adenocarcinoma and esophageal squamous cell carcinoma (more than 70% of cases). Of the individuals with at least one TP53 mutation, of 5,411) harbored one or more variant predicted to be protein-truncating or splice-altering and (3,544 of 5,411) carried one or more missense variant (207 individuals carried both variant types), with the five most common protein changes being R175H (5.3%), R273C (3.2%), R248Q (3.2%), R273H (3.2%) and R282W (2.7%) (Supplementary Table 1). PIK3CA was the second most frequently altered gene, with mutations found in of patients ( 2,750 of 13,880 ), occurring most frequently in uterine corpus endometrial carcinoma (53.5%), ovarian endometrioid adenocarcinoma (49.0%), breast invasive carcinoma (42.2%), uterine corpus endometrial serous carcinoma (38.1%) and colon adenocarcinoma (26.5%). The most
Fig. 2 | Overview of the Genomes Cancer Programme cohort demographics. a, Distribution of 12,948 cases represented by 33 tumor types (cases with more than one sample per tumor were only counted once). b, Thirteen NHS GMCs recruited patients diagnosed with cancer across England. The area of the pie chart is proportional to the number of patients recruited;
the total number of participants recruited per GMC is indicated in parentheses. Map source: Office for National Statistics licensed under the Open Government Licence v.3.0. c, Breakdown of biological sex and age at diagnosis according to disease. The age plot shows the interquartile range (IQR) and median values.
Fig. 3 | Overview of the sample characteristics for the Genomes Cancer Programme cohort. Breakdown according to the stage of the disease (left) (NA, not available or not applicable in the context of glioblastoma multiforme and low-grade glioma), type of sample obtained (middle) and tumor purity (right) for each tumor type; the IQR and median values are shown.
commonly mutated codons in PIK3CA were E545 and H1047. Over 69.9% of all mutations in this gene were found in the five well-characterized hotspots . While currently indicated for testing in breast invasive carcinoma only, PIK3CA mutations were present across multiple tumor types, suggesting that clinical trials with PIK3CA inhibitors could be considered in the future, if clinically appropriate. Other genes such as APC, KRAS, VHL and IDH1 were highly enriched for mutations in only one or two tumor types. Our pancancer analysis is concordant with other large-scale sequencing endeavors such as ICGC and TCGA, albeit with variations due to cancer type proportions, reflected by a higher proportion of colon and rectum adenocarcinoma, and sarcoma in our cohort (Fig. 2a). The sequencing of a large number of ovarian tumor samples ( ) allowed further subtype classification, with a high prevalence of TP53 variants being identified in high-grade serous carcinoma (89.8% of cases), PIK3CA variants in ovarian endometrioid adenocarcinoma (49.0%) and variants in low-grade ovarian serous carcinoma (33.3%).
Fusions and CNAs
A high prevalence of amplifications or losses was found in TP53, CDKN2A, MYC, CDKN2B and PTEN across all cancer types (Fig. 4). Glioblastoma multiforme, low-grade glioma, head and neck squamous cell carcinoma, mesothelioma and sarcoma (Fig. 4 and Extended Data Fig. 1b) demonstrated the highest number of clinically relevant CNAs. With increased targeted therapies, molecular tests for different mutation types, including fusions, have become standard of care . For instance, NTRK fusions (across all cancer types) but also other kinase fusions (for example, and for lung cancers), are now included in the NGTDC. Although only a small percentage of patients test positive for specific fusion, the presence of a mutation can be critical for disease classification. A prime example is found in mesenchymal chondrosarcomas, where HEY1-NCOA2 fusions are exclusive to that
subtype. Indeed, sarcomas had the highest prevalence of tumors (13%) with clinically relevant SV findings (Fig. 4 and Extended Data Fig.1c).
Germline findings
Unlike targeted panel tests that are frequently performed on tumor-only samples, paired tumor and normal WGS allows somatic and germline variants to be detected together. The certainty of origin for a variant can have implications on patient management, such as family genetic testing or eligibility for treatment. Patients with ovarian high-grade serous carcinoma had the highest prevalence of actionable germline findings for SNVs and indels, with 13% of patients harboring variants in the BRCA1 and genes (Fig. 4 and Extended Data Fig. 1d; predicted truncating small variants or missense mutations with pathogenic classification in Clinvar are reported; for details, see the online Methods). Median age at tumor diagnosis is shown in Fig. 2c; as expected, there was a younger median age at tumor diagnosis in those patients with predisposing germline findings (Extended Data Table1). Notably, patients with germline variants in mismatch repair (MMR) genes showed significantly earlier age at onset of colon adenocarcinomas, while patients with germline variants in homologous recombination repair genes showed significantly earlier onset in ovarian high-grade serous carcinomas and breast invasive carcinomas. This was also observed in kidney renal clear cell carcinoma with germline variants predominantly in the VHL gene. variants, linked to fluoropyrimidine toxicity, were present in 5-10% of participants, guiding the recommendations for dose omission or adjustment in the treatment of breast invasive carcinomas, colon, rectum, pancreatic adenocarcinomas and head and neck squamous cell carcinomas as recommended in the NGTDC.
Pangenomic markers and mutational signatures
TMB has been cited as a potential biomarker and in this dataset we observed significant variation across and within cancer types. In line
Fig. 4 | Somatic and germline alterations across common tumor types. Prevalence of different types of mutations identified using WGS in genes indicated for testing in the NGTDC. The leftmost panel indicates the total percentage of cases harboring one or more genomic alterations of clinical relevance as listed in the NGTDC (where the number of cancers sequenced is ten or more). In the subsequent panels, somatic variants (from left to right) consisting of small variants (SNVs, indels), CNAs, SVs, HRD, MMR signatures and TMB along with germline variants related to inherited cancer risk (predisposing
genes) and pharmacogenomic (PGx) findings (toxicity-associated variants) are shown. The top five genes with the most prevalent mutation rates for each mutation type are shown (see Extended Data Fig. 1 for the full analysis). The percentage of tumors harboring a specific type of mutation in the gene(s) indicated for testing according to tumor type in the NGTDC are shown in magenta. Mutation incidence (as a percentage) in other tumor types, not currently indicated in the NGTDC, is shown in blue. Color gradation reflects the percentage of affected cases.
with previous reports , we found that skin cutaneous melanoma and lung adenocarcinoma had the highest average TMB (Fig. 5a). Colon adenocarcinoma and uterine corpus endometrial carcinoma showed variability in the presence or absence of microsatellite instability or hypermutation caused by POLE mutations (see alignment with corresponding mutational signatures).
When examining mutational signatures (COSMIC v.3) with well-described etiologies, we observed expected frequencies within certain cancer types (Fig. 5a and Extended Data Fig. 2). As expected, APOBEC signatures 2 and 13 were associated with breast invasive carcinoma, head and neck squamous cell carcinoma, bladder urothelial carcinoma and lung adenocarcinoma; smoking signatures 4 and 92 with lung cancers (lung adenocarcinoma, lung neuroendocrine and lung squamous cell carcinoma); and ultraviolet signature (signatures ) with skin cutaneous melanoma. DNA MMR signatures , 21, 26 and 44 were enriched in microsatellite instability-high colon adenocarcinoma and uterine corpus endometrial carcinoma (Fig. 5a).
HRD status was defined by two genome-wide mutational scar-based pancancer classifiers, CHORD and HRDetect . The two algorithms demonstrated 99.2% concordance in our sample cohort (Methods). Ovarian high-grade serous carcinoma showed the highest prevalence of HRD ( ). While PARP inhibitors are currently only indicated for use in ovarian tumors with HRD, HRD was also detected at low prevalence in other cancers that could potentially access PARP inhibitors via clinical trials or compassionate access pathways.
Clinical utility of WGS
Overall, these findings demonstrate the ability of WGS data to fully characterize the clinical genomic landscape of a tumor. A single test can report somatic SNVs, gene fusions and CNAs, along with potentially pathogenic germline mutations, and pangenomic markers such as mutational signatures and TMB (Fig. 4). In the Supplementary Information, we provide examples of WGA results as provided to NHS GMC Laboratories. For example, in a patient with ovarian high-grade serous carcinoma, a somatic TP53 SNV was identified, consistent with the diagnosis, along with a germline BRCA1 variant and somatic BRCA1 copy number (CN) loss driving HRD, which was subsequently supported by the HRD analysis. Similarly, in another case, in a patient with endometrial cancer, MMR deficiency signatures were identified in combination with high TMB, along with a PMS2 pathogenic germline variant, a somatic PMS2 start-loss mutation and a pharmacogenomic (germline) variant in the gene (associated with toxicity to fluoropyrimidines). These examples demonstrate specific instances where the identification of different types of mutations and pangenomic markers were clinically relevant.
Pangenomic markers and outcomes from real-world data
Through the link of the WGS data with longitudinal life course clinical data (SACT and ONS), we assessed treatment outcomes for patients stratified according to pangenomic markers (Fig. 5b and Supplementary Table 2). As shown in Fig. 5b, in patients treated with platinum therapies, HRD predicted better outcome ( , ), primarily in patients with invasive breast carcinomas ( ) and ovarian high-grade serous carcinomas ( , ). Immunotherapy outcomes in MMR-deficient cases ( ) were inconclusive because of small numbers. We then evaluated TMB as a prognostic marker and a significant difference in survival ( , ) was observed for those patients with TMB in the lowest quartile (median of 3.8 nonsynonymous small variants per Mb ) compared with the highest quartile (median of 20.98 nonsynonymous small variants per Mb ) in those diagnosed with skin cutaneous melanoma (Fig. 5c and Supplementary Table 2). Interestingly, a significant difference was not observed in lung adenocarcinoma ( ), where the lowest and highest quartile median TMB values were 2.2 and 10.5 nonsynonymous small variants per Mb , respectively. This may indicate that the level of TMB is relevant in prognosis and supports the need for further refining of pangenomic biomarkers as both prognostic and predictive for immunotherapy response, as highlighted in previous studies .
Co-occurrence of small variants and CNAs
The co-occurrence of SNVs, indels and CNAs is well documented . With WGS, we were able to explore the co-occurrence of CNAs and somatic small variants impacting cancer genes in the NGTDC. We divided cases into those with and without small variants for each gene and then compared the frequency of CNAs for each gene across these two groups (Fig. 6a and Supplementary Table 3). After multiple-testing correction, we found that 12 genes displayed a significant difference in the frequency of copy alterations. We confirmed previous findings, namely, that and , in specific cancer types, tended to be amplified when a putative activating SNV was present. The role of copy gains on certain oncogenes has long been debated and our analysis found that there was a significant co-occurrence of gains in the presence of small variants affecting and . We also found that five tumor suppressor or dual-role genes had significantly higher frequencies of copy loss in the presence of somatic small variants, including established examples such as TP53 (ref. 28), RB1 (ref. 29), and , further emphasizing the value of interpreting different types of variants concurrently.
Survival analysis using real-world data
We next assessed overall survival in all 33 cancer subtypes stratified according to the presence or absence of mutations in 40 NGTDC-indicated genes (protein-altering small variants (SNVs and indels) as well as homozygous deletions in tumor suppressor genes were included). Clinical data from secondary data sources such as HES and ONS provided survival data. Kaplan-Meier and Cox proportional-hazards analyses were performed on our pancancer cohort. After correcting for stage and multiple testing, 15 genes affected overall survival (Fig. 6b and Extended Data Fig. 3). The gene that affected patient outcome most severely was CDKN2A ( , ), which corresponds to its association with high-grade disease and poor prognosis in some cancer subtypes, such as glioma and soft-tissue sarcoma . Our results agree with previously reported prognostic associations for specific tumor types, for example, poor
prognosis for KRAS mutants in colorectal cancer and non-small cell lung cancer or TP53 mutations in non-small cell lung cancer . Mutations in PIK3CA were associated with favorable outcomes, in keeping with reports in the literature .
Discussion
The 100,000 Genomes Project established the infrastructure and resources for linking genomic and longitudinal clinical life course data. Our findings from the Cancer Programme aided the selection of genomic targets in the NHS National Genomic Test Directory. Evaluation of WGS data provided support for the commissioning of clinical WGS for sarcoma, glioblastoma, ovarian high-grade serous carcinoma and triple-negative breast cancers, to detect different types of mutations, including pangenomic markers, with a single test to inform clinical care. The infrastructure generated from the 100,000 Genomes Project has been incorporated into the NHS GMS to enable standardized molecular characterization of tumors and to extend the clinical benefit of prospective molecular characterization to more patients with cancer. Consistent with previous studies we report a high prevalence of genetic variants used to stratify patients toward approved therapies and clinical trials across different cancer types. Our approach aligns with similar programs in other countries, such as St.Jude Children’s Research Hospital in the USA, BC Cancer in Canada , Zero Childhood Cancer Program in Australia , France Médecine Génomique and Genomic Medicine Sweden . These initiatives are either ongoing and have yet to publish on their cohort or represent a smaller cohort of childhood cancers.
Our study only included WGS data and while genomics may provide a valuable starting point for molecular stratification of cancer, it is likely that other modalities, such as cell-free DNA, RNA sequencing, methylation and gene expression profiling, proteomics, long-read sequencing and single-cell sequencing will mature toward clinical use. As such, we envisage the inclusion of multi-omics data alongside longitudinal life course data and the integration of multimodal molecular and clinical data, including digital pathology and radiology, to maximize the benefit of precision cancer care for patients .
As genomic testing becomes more widespread, it is essential to combine these data with real-world clinical and treatment data. This integration is crucial to advancing our understanding of the long-term impact of clinical cancer genomics on patient outcomes. In this study, we demonstrated the value of linked real-world data in evaluating outcomes and mirroring adverse molecular markers from clinical trials. The accumulation of genomic data alongside electronic health data included in cancer registries, such as staging, pathology and treatment, and outcomes, enriches the dataset and may further refine the selection of biomarkers. The co-occurrence of variants in the same gene, or the coexistence of mutations in different genes, are likely to enhance the prognostic and predictive value of biomarker selection and may detect longer-term latent signals of benefit or harm and aid clinical and regulatory decision-making . The therapeutic implications associated with the co-occurrence of CNAs and somatic small variants are unclear, and this level of genomic information may not readily be available from large cancer panel data . We present a broad survival analysis at the gene level; as the dataset expands, it will be possible
Fig. 5|Predictive value of pangenomic markers derived from WGS data. a, Distribution of TMB and mutational signatures across six tumor types. (Samples that underwent PCR amplification during library preparation were excluded and the dataset for each tumor type was downsampled to 100 samples.) The horizontal red bar indicates the median TMB for each cancer type. Etiology definitions based on COSMIC (v.3) single-base substitution signatures: APOBEC activity, signatures 2 and 13; aging, signature 1; HRD, signature 3; MMR deficiency, signatures 6,15,20,21,26 and 44;POLE mutations, signatures 10a, 10b and 14; smoking, signatures 4 and 92; ultraviolet exposure, signatures . Only signatures with more than contribution are shown. Homologous recombination status is indicated in the bars below the signature
plots. b,c, Kaplan-Meier estimates of overall survival with values calculated using a stratified log-rank test. The numbers of patients at risk at different time points are indicated below the survival curves. The points and error bars on the embedded forest plots indicate the hazard ratios (HRs) with 95% confidence intervals (CIs), correspondingly. HRs, CIs and values were calculated from Cox proportional-hazards models corrected according to cancer stage. Patients were stratified according to HRD status in cancers treated with platinum chemotherapy ( , left, ); according to MMR signatures in cancers treated with immunotherapies ( , right, b); according to high and low TMB in skin cutaneous melanoma ( , left, c); and according to lung adenocarcinoma ( , right, ). Exact values can be found in Supplementary Table 2.
C
to examine these data further to establish prognostic and predictive implications for specific variants, as observed with KRAS variants . Yet, challenges remain in implementing clinical WGS in the NHS in England not least because of the overall cost compared to large gene panel testing. Providing a cutting-edge UK genomics service requires not only the sequencing and analytical infrastructure, but the consideration of operational requirements (such as improvements in tissue pathways and turnaround times to inform clinical decision-making) together with local pathway transformation and the development of knowledge and skills of the multiprofessional workforce supporting cancer care.
WGS results are discussed at multidisciplinary Molecular Tumor Boards or GTABs to evaluate somatic and germline variants, determine clinical actionability and provide clinical recommendations. GTABs have a vital role in ensuring that actionable results are communicated to treating teams and clinicians, while also exploring eligibility for approved therapies and clinical trials . A well-designed, well-structured GTAB has a key role in the clinical interpretation of cancer genomic testing, guiding clinicians in decision-making through recommendations, facilitating clinical trial enrollment and potentially enhancing outcomes . This approach aligns with adaptive basket trials such as DETERMINE , which has been established to evaluate licensed treatments in unlicensed indications similar to the DRUP trial . The aim is to enable more equitable and comprehensive molecular testing within the NHS and to optimize cancer care by identifying all clinically relevant mutations for a specific cancer (as shown in Fig. 4) and their relationship to approved precision medicines, but also to ensure that patients are fully considered for clinical research and trials because of this genomic testing and to explore clinical trial options, including the use of repurposed well-known and well-characterized drugs.
The Research Environment, a platform built by Genomics England and NHSE, allows approved researchers secure access to genomic data and associated health data. It has allowed advances in fundamental research, such as the discovery of cancer driver genes , mutational signatures or changes in clinical practice driven by availability of WGS testing .
Our findings underscore the potential for these data to provide additional prognostic insights based on the absence or presence of specific mutations. As data accumulate within the Research Environment with linkage of genomic, clinical and outcome data, more refined analyses using real-world data can take place, aided by more comprehensive tumor profiling. This will enable further refinement of prognostic and predictive molecular markers, not only with combinations of different genomic alterations, but beyond genomics, including emerging technologies to expand the reach of precision oncology to improve cancer outcomes.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41591-023-02682-0.
Zehir, A. et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 23, 703-713 (2017).
Smedley, D. et al. 100,000 Genomes Pilot on Rare Disease Diagnosis in Health Care-preliminary report. N. Engl. J. Med. 385, 1868-1880 (2021).
Turnbull, C. et al. The 100000 Genomes Project: bringing whole genome sequencing to the NHS. BMJ 361, k1687 (2018).
Turnbull, C. Introducing whole-genome sequencing into routine cancer care: the Genomics England 100000 Genomes Project. Ann. Oncol. 29, 784-787 (2018).
National Genomic Test Directory. NHS England www.england.nhs. uk/publication/national-genomic-test-directories (2023).
NHS England. Board Paper (2017); www.england.nhs.uk/ wp-content/uploads/2017/03/board-paper-300317-item-6.pdf
Berner, A. M., Morrissey, G. J. & Murugaesu, N. Clinical analysis of whole genome sequencing in cancer patients. Curr. Genet. Med. Rep. 7, 136-143 (2019).
Aaltonen, L. A. et al. Pan-cancer analysis of whole genomes. Nature 578, 82-93 (2020).
Martínez-Jiménez, F. et al. Pan-cancer whole-genome comparison of primary and metastatic solid tumours. Nature 618, 333-341 (2023).
van der Velden, D. L. et al. The Drug Rediscovery protocol facilitates the expanded use of existing anticancer drugs. Nature 574, 127-131 (2019).
Aran, D., Sirota, M. & Butte, A. J. Systematic pan-cancer analysis of tumour purity. Nat. Commun. 6, 8971 (2015).
Zhong, L. et al. Small molecules in targeted cancer therapy: advances, challenges, and future perspectives. Signal Transduct. Target Ther. 6, 201 (2021).
Lanic, M.-D. et al. Detection of sarcoma fusions by a nextgeneration sequencing based-ligation-dependent multiplex RT-PCR assay. Mod. Pathol. 35, 649-663 (2022).
Chan, T. A. et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann. Oncol. 30, 44-56 (2019).
Lawrence, M. S. et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495-501 (2014).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415-421 (2013).
Nguyen, L., Martens, J. W. M., Van Hoeck, A. & Cuppen, E. Pan-cancer landscape of homologous recombination deficiency. Nat. Commun. 11, 5584 (2020).
Davies, H. et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 23, 517-525 (2017).
Xiao, D. et al. Analysis of ultra-deep targeted sequencing reveals mutation burden is associated with gender and clinical outcome in lung adenocarcinoma. Oncotarget 7, 22857-22864 (2016).
Klempner, S. J. et al. Tumor mutational burden as a predictive biomarker for response to immune checkpoint inhibitors: a review of current evidence. Oncologist 25, e147-e159 (2020).
McGrail, D. J. et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann. Oncol. 32, 661-672 (2021).
Inoue, K. & Fry, E. A. Haploinsufficient tumor suppressor genes. Adv. Med. Biol. 118, 83-122 (2017).
Sigismund, S., Avanzato, D. & Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 12, 3-20 (2018).
Cheng, L. et al. KIT gene mutation and amplification in dysgerminoma of the ovary. Cancer 117, 2096-2103 (2011).
Shetzer, Y. et al. The onset of p53 loss of heterozygosity is differentially induced in various stem cell types and may involve the loss of either allele. Cell Death Differ. 21, 1419-1431 (2014).
Latil, A. et al. Loss of heterozygosity at chromosome arm and RB1 status in human prostate cancer. Hum. Pathol. 30, 809-815 (1999).
Foulkes, W. D., Flanders, T. Y., Pollock, P. M. & Hayward, N. K. The CDKN2A (p16) gene and human cancer. Mol. Med. 3, 5-20 (1997).
Horbinski, C., Berger, T., Packer, R. J. & Wen, P. Y. Clinical implications of the 2021 edition of the WHO classification of central nervous system tumours. Nat. Rev. Neurol. 18, 515-529 (2022).
Bui, N. Q. et al. A clinico-genomic analysis of soft tissue sarcoma patients reveals CDKN2A deletion as a biomarker for poor prognosis. Clin. Sarcoma Res. 9, 12 (2019).
Ozer, M. et al. Age-dependent prognostic value of mutation in metastatic colorectal cancer. Future Oncol. 17, 4883-4893 (2021).
Aredo, J. V. et al. Impact of mutation subtype and concurrent pathogenic mutations on non-small cell lung cancer outcomes. Lung Cancer 133, 144-150 (2019).
Jiao, X.-D., Qin, B.-D., You, P., Cai, J. & Zang, Y.-S. The prognostic value of TP53 and its correlation with EGFR mutation in advanced non-small cell lung cancer, an analysis based on cBioPortal data base. Lung Cancer 123, 70-75 (2018).
Kalinsky, K. et al. PIK3CA mutation associates with improved outcome in breast cancer. Clin. Cancer Res. 15, 5049-5059 (2009).
Priestley, P. et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 575, 210-216 (2019).
Ma, X. et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 555, 371-376 (2018).
Pleasance, E. et al. Whole-genome and transcriptome analysis enhances precision cancer treatment options. Ann. Oncol. 33, 939-949 (2022).
Wong, M. et al. Whole genome, transcriptome and methylome profiling enhances actionable target discovery in high-risk pediatric cancer. Nat. Med. 26, 1742-1753 (2020).
Préindications d’Accès au Séquençage Génomique. France Medecine Genomique 2025 https://pfmg2025.aviesan.fr/le-plan/ indications-dacces-au-sequencage-genomique/ (undated).
Sequencing of 7,000 Genomes in Swedish Clinical Practice in 2021 for Better Diagnosis and Treatment. Genomic Medicine Sweden https://genomicmedicine.se/en/2022/04/11/ sequencing-of-7000-genomes-in-swedish-clinical-practice-2021-for-better-diagnosis-and-treatment (2022).
Donoghue, M. T. A., Schram, A. M., Hyman, D. M. & Taylor, B. S. Discovery through clinical sequencing in oncology. Nat. Cancer 1, 774-783 (2020).
Nogrady, B. How cancer genomics is transforming diagnosis and treatment. Nature 579, S10-S11 (2020).
Chandramohan, R. et al. A validation framework for somatic copy number detection in targeted sequencing panels. J. Mol. Diagn. 24, 760-774 (2022).
Huang, L., Guo, Z., Wang, F. & Fu, L. KRAS mutation: from undruggable to druggable in cancer. Signal Transduct. Target Ther. 6, 386 (2021).
van de Haar, J. et al. Codon-specific KRAS mutations predict survival benefit of trifluridine/tipiracil in metastatic colorectal cancer. Nat. Med. 29, 605-614 (2023).
Academy of Medical Royal Colleges. Principles for the Implementation of Genomic Medicine (2019); www.aomrc.org. uk/wp-content/uploads/2019/10/Principles_implementation_ genomic_medicine_011019.pdf
Malone, E. R., Oliva, M., Sabatini, P. J. B., Stockley, T. L. & Siu, L. L. Molecular profiling for precision cancer therapies. Genome Med. 12, 8 (2020).
Kato, S. et al. Real-world data from a molecular tumor board demonstrates improved outcomes with a precision N-of-One strategy. Nat. Commun. 11, 4965 (2020).
Cornish, A. J. et al. Whole genome sequencing of 2,023 colorectal cancers reveals mutational landscapes, new driver genes and immune interactions. Preprint at bioRxiv https://doi. org/10.1101/2022.11.16.515599 (2022).
Degasperi, A. et al. Substitution mutational signatures in whole-genome-sequenced cancers in the UK population. Science 376, science.abl9283 (2022).
Prendergast, S. C. et al. Sarcoma and the 100,000 Genomes Project: our experience and changes to practice. J. Pathol. Clin. Res. 6, 297-307 (2020).
Trotman, J. et al. The NHS England 100,000 Genomes Project: feasibility and utility of centralised genome sequencing for children with cancer. Br. J. Cancer 127, 137-144 (2022).
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The sample collection and DNA extraction requirements are described in the Sample Handling Guidance (v.4.0) available at https://files. genomicsengland.co.uk/forms/Sample-Handling-Guidance-v4.0. pdf. A total of germline DNA and at least tumor DNA were required for Illumina TruSeq PCR-free library preparation to be performed. PCR-based library preparation was used when insufficient DNA could be obtained for PCR-free sequencing, with a minimum requirement of 500 ng . Optimized formalin-fixed tumor tissue was allowed for WGS under exceptional circumstances, where tumor size limited availability of fresh tissue, or if no tumor was present in the fresh-frozen sample.
Quality of sequencing data. All samples were sequenced on the HiSeq platform to an average coverage of for tumor and for normal. The following checks were implemented to ensure sample quality: normal samples had more than 85 Gb and tumor samples had more than 210 Gb of high-quality sequencing data (base quality greater than 30, duplicated reads removed); normal samples had more than 95% of the autosomal genome covered at or more after removing reads with mapping quality lower than 10 ; normal samples had cross-patient contamination lower than 3% as assessed using VerifyBamID; tumor samples had cross-patient contamination lower than 2.5% and normal tumor sample pair originating from the same patient as assessed using ConPair; the quality of the sequencing data was monitored using principal component analysis based on the following metrics: percentage of reads mapped to the reference genome, proportion of chimeric DNA fragments, median fragment size, unevenness of local genome coverage and percentage of reads missing from AT-rich or GC-rich genomic regions (AT and GC drop).
Mapping and variant calling. The Illumina North Star pipeline (v.2.6.53.23) was used for the primary WGS analysis. Read alignment against the human reference genome GRCh38 + decoy + Epstein-Barr virus was performed with ISAAC (v.iSAAC-03.16.02.19). We acknowledge deficiencies in the ISAAC alignment software for precise variant allele frequency estimates and for tumor evolution analysis and note that all genomes from the 100,000 Genomes Project were recently realigned with the Illumina Dragen platform (data available in the Research Environment). Germline small variant calling was performed using Starling (v.2.4.7) and somatic small variant calling was performed using Strelka (v.2.4.7). In addition to default Strelka filters, the following additional filters were applied to reduce the false positive rate in the set of somatic variants used as an input into the calculation of TMB and mutational signatures: (1) variants with a population germline allele frequency above 1% in the Genomics England or gnomAD datasets; (2) recurrent somatic variants with a frequency above in the Genomics England dataset; (3) variants overlapping simple repeats as defined by Tandem Repeats Finder; (4) small indels in regions with high levels of sequencing noise where at least of the base calls in a window extending 50 bases to either side of the indel call were filtered out by Strelka because of poor quality; (5) SNVs resulting from systematic mapping and calling artifacts with a Fisher’s exact test Phred score lower than 50. The flagging of systematic mapping and calling was performed by testing whether the ratio of tumor allele depths at each somatic SNV site were significantly different to the ratio of allele depths at this site in a panel of normals. The panel of normals consisted of a cohort of 7,000 non-tumor genomes from the Genomics England
dataset; at each genomic site only individuals not carrying the relevant alternate allele were included in the count of allele depths. Variants flagged with any of the above internal filters were not removed from the WGA results of clinically actionable variants but were labeled in the output shared with clinical scientists.
CNAs were identified with Canvas v.1.3.1. Manta (v.0.28.0) was used to call SVs and long indels (more than 50 bp), combining paired and split-read evidence for SV discovery and scoring.
Estimates of the accuracy of somatic variant calling in the 100,000 Genomes Project pipeline were produced as a requirement for accreditation under International Organization for Standardization no. 15189. We have provided ‘Bioinformatics Pipeline Validation. Cancer Report, September 2018’ as Supplementary Information and have summarized the findings in Supplementary Table 4. Extensive validation and functional improvements of the pipeline for the NHS GMS will be presented in a separate publication.
Annotation and reporting actionability. SNVs and small indels were left-aligned, trimmed, and multi-allelic variants decomposed, before annotation with Cellbase, using the Ensembl (v.90/GRCh38), COSMIC (v.v86/GRCh38) and ClinVar (October 2018 release) databases. Annotation of consequence types was carried out by a high-performance variant annotator within Cellbase; only variants annotated with a curated set of consequence types (stop gained or lost, start lost, frameshift variant, inframe insertion or deletion, missense variant, splice acceptor or donor variant, splice region variant) in canonical transcripts were reported.
Interpretation of CNAs took into account gene mode of action as defined in the COSMIC Cancer Gene Census (that is, oncogene or tumor suppressor gene). Where a gene had an ambiguous or unknown role in cancer, it was included in both oncogene and tumor suppressor categories. Gains in oncogenes were reported if CN was at least twice higher than the overall ploidy as defined by Canvas. The following scenarios were reported as losses in tumor suppressor genes: (1) homozygous deletions called by Canvas ( ); (2) loss of heterozygosity ( LOH ) called by Canvas ( ) or copy-neutral LOH , in combination with a nonsynonymous somatic small variant; and (3) Manta SVs with the potential to disrupt the gene coding region in combination with a nonsynonymous somatic small variant. Only samples with tumor purity greater than were included in the CNA actionability analysis. For the co-occurrence of somatic small variants and CNAs analysis in Fig. 6a, gain of at least one copy for oncogenes or loss of at least one copy for tumor suppressor genes was counted as a CNA event.
Manta calls (break end, deletion, duplication or inversion) were further assessed for the potential to generate productive fusions using an in-house approach based on transcript orientation and consistency of reading frame across the SV breakpoint. SVs that were identified as out of frame or untranscribed were discarded. Potential inframe fusions and ambiguous events with a breakpoint in the coding exon or in the 5-‘UTR of downstream partners were reported.
Germline variants listed in ClinVar as pathogenic or probably pathogenic with a rating of at least two stars and predicted protein-truncating variants in genes for which the mechanism of pathogenicity was loss of function (stop gained or lost, start lost, frameshift variant, splice acceptor or donor variant) were reported for a subset of cancer predisposition genes indicated for germline testing in NGTD.
Within the context of the 100,000 Genomes Project Cancer Programme, all variants returned to GMCs were reviewed within GTABs to classify further if variants were pathogenic or probably pathogenic (germline) or oncogenic or probably oncogenic (somatic) and to provide clinical recommendations where appropriate.
Signatures and TMB. For each tumor sample, frequencies across all SNV trinucleotide contexts were calculated using VCF files that were filtered for potential false positive variants (see the variant calling
section) and the contribution of each of the COSMIC (v.3) single-base substitution signatures to the overall mutational burden observed in the tumor was derived using decomposition by the SigProfiler suite of tools . Etiology definitions were based on the following signature combinations: APOBEC activity, signatures 2 and 13; aging, signature 1; MMR deficiency, signatures 6,15,20,21,26 and 44;POLE mutations, signatures 10a, 10b and 14; smoking, signatures 4 and 92; ultraviolet exposure, signatures 7a-d. Signature 14 (reported with the etiology ‘concurrent polymerase epsilon mutation and defective DNA MMR’) was not included in the MMR deficiency group to avoid double counting in the MMR and POLE groups. Including SBS14 in the MMR group would change MMR status for 9 of 13,880 tumors and would only increase the number of tumors in our cohort by . For a given etiology, if the final combined signatures summed to less than , the signature was assigned to ‘other’. Tumors were classified with MMR deficiency if the total contribution of MMR signatures was more than . HRDetect is a logistic regression classifier that computes a probability score of HRD based on microhomology deletions, SNV and SV mutational signatures, and LOH score. HRD status using HRDetect was retrieved from a previous publication . The CHORD algorithm is a random forest-based classifier that incorporates counts of different variant types as input (SNVs, microhomology deletions and SVs) and does not require an intermediate mutational signature extraction step) . HRDetect and CHORD were trained on the ICGC and Hartwig Medical Foundation cohorts, respectively. The two algorithms returned concordant results for 99.2% of samples in our cohort ( 10,764 of 10,854) and CHORD results were used for the figures. TMB was calculated as the total number of nonsynonymous high-confidence somatic small variants per megabase of coding sequence (see the variant calling section for the filtering method used).
Description of clinical data resources
A minimal set of patient and sample data was collected from GMCs at the time of DNA sample submission through OpenClinica v.3.4, for example, tumor type, year of birth, tissue source, self-reported gender. For the purposes of the analysis, self-reported gender was cross-validated with biological sex inferred using the ratio of mean sequencing coverage of sex chromosomes and mean sequencing coverage of autosomes. Assigned biological sex was used in the bioinformatics pipeline as an input for variant calling. Secondary clinical information was gathered from NHSE and Public Health England (PHE)/ NCRAS. From NHSE, HES data were used to obtain details of all commissioned activity during admissions; mortality information was obtained from the ONS registry data for cancer registrations and deaths inside and outside of hospitals. From PHE/NCRAS, the av_tumor table was used to obtain tumor date of diagnosis, together with histology and morphology codes. The SACT table provided information on the date and types of treatment. All datasets were accessed via the National Genomics Research Library using LabKey.
Linking genomic data with secondary data sources
Hematological tumors, pediatric tumors and carcinomas of unknown primary origin were considered to be outside the scope of the study and were removed before tumor selection. Secondary data from the PHE/NCRAS tumor catalog (av_tumor), and NHS Digital HES data were used to corroborate the clinical data submitted by the GMCs.
The av_tumor dataset was linked to genomic data on the basis of the participant identifier. Tumors labeled as either benign or in situ were removed from the selection process, leaving only malignant, unknown or NA (the latter being the case for Genomics England participants not present in the av_tumor dataset). Where av_tumor data were available for a participant, they were used to confirm the tumor type submitted by the GMC. For cases where the av_tumor data did not match the GMC submission, or data were not present, HES Admitted Patient Care data were used to select the closest relevant hospital
appointment involving a primary diagnosis of cancer (based on International Statistical Classification of Diseases and Related Health Problems,10th Revision (ICD-10) code) to the clinical sample time submitted by the GMC. If the ICD-10 code for that appointment was considered a match to the tumor type submitted by the GMC, the HES data were deemed as corroborating the GMC submission.
Where HES data did not corroborate the tumor type submitted by the GMC, three additional approaches were used: (1) for primary tumors, a curated set of HES operation codes was used to match the tumor type submitted by the GMC and the HES data if the operation date exactly matched the sampling date of the tumor submitted to Genomics England; (2) for non-primary tumors that were identified as colorectal by the av_tumor data, and as either hepato-pancreatobiliary, endometrial carcinoma or lung in the GMC submission, more flexible HES ICD-10 matching was allowed provided the date difference between the HES appointment date and tumor sampling date submitted by the GMC was fewer than 7 days; (3) for a small number of remaining samples, ICD-10 and morphology data submitted by the GMC were used to corroborate tumor type.
Tumor stage was obtained from the NCRAS dataset. Where stage_ best was present in av_tumor and the date in the diagnosis database column was fewer than 365 days from the clinical sample time submitted by the GMC, stage_best was used (simplified to stages 1,2,3 and 4) ( 11,618 of ). Tumors submitted as metastatic were assigned stage 4 by default. FIGO (Fédération Internationale de Gynécologie et d’Obstétrique) stage was used for ovarian- and endometrium-related clinical indications and Dukes’ staging was used for colon and rectum adenocarcinomas (both obtained from the av_tumor table). In total, stage information was obtained for 12,040 of 13,880 (86.7%) tumors.
Survival analysis
All survival analyses were performed in R using the survminer and survival libraries. Specifically, the survfit and ggsurvplot functions were used to create the Kaplan-Meier plots, and coxph for the Cox proportional-hazards models. The ggforest function was used to create the forest plots. Date of death was obtained from the ONS data. Where a death was not recorded for an individual, treatment and operation event dates were obtained from the HES dataset and used to determine the last date an individual was seen to enable right-censoring of the data.
Ethics
The research described in this manuscript complies with all relevant ethical regulations. Approval for the project was obtained from the East of England-Cambridge South Research Ethics Committee (Research Ethics Committee reference 14/EE/1112, Integrated Research Application System ID: 166046) . Participants were selected on the basis of having been identified by healthcare professionals and researchers within the NHS as having a cancer diagnosis. Participants were recruited across 13 NHS GMCs and written informed consent was obtained from participants.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings of this study are available within the Research Environment, a secure cloud workspace. Details on how to access data for this publication can be found at https://re-docs. genomicsengland.co.uk/pan_cancer_pub/. Additional processed aggregated data used to generate figures can be found in Supplementary Tables 5-20. To access the genomic and clinical data within this Research Environment, researchers must first apply to become a member of either the Genomics England Research Network
(previously known as the Genomics England Clinical Interpretation Partnership, GECIP) (www.genomicsengland.co.uk/research/ academic) or a Discovery Forum industry partner (www.genomic-sengland.co.uk/research/research-environment). The process for joining the Genomics England Research Network is described at www.genomicsengland.co.uk/research/academic/join-gecip and consists of the following steps: (1) If it is not already participating, your institution will need to sign a participation agreement available at https://files.genomicsengland.co.uk/documents/ Genomics-England-GeCIP-Participation-Agreement-v2.0.pdf and email the signed version to gecip-help@genomicsengland.co.uk; (2) once you have confirmed your institution is registered and have found a domain of interest, you can apply through the online form at www.genomicsengland.co.uk/research/academic/join-gecip. Once your Research Portal account is created you will be able to log in and track your application; (3) your application will be reviewed within ten working days; (4) your institution will validate your affiliation; and (5) you will complete our online Information Governance training and will be granted access to the Research Environment within 2 h of passing the online training. Data that have been made available to registered users include: alignments in BAM or CRAM format; annotated variant calls in VCF format; signature assignment; tumor mutational burden; sequencing quality metrics; summary of findings shared with the Genomic Lab Hubs; and secondary clinical data as described in this paper. Further details of the types of data available (for example, mortality, hospital episode statistics and treatment data) can be found at https://re-docs.genomicsengland.co.uk/data_overview/.Germline variants can be explored using the Interactive Variant Analysis Browser (https://re-docs.genomicsengland.co.uk/iva_variant/). The cohort of patients with cancer and longitudinal clinical information on treatment and mortality can be explored with Participant Explorer (https://re-docs.genomicsengland.co.uk/pxa/).
We thank the participants, who made this work possible. This research was made possible through access to the data and findings generated by the 100,000 Genomes Project. The 100,000 Genomes Project is managed by Genomics England Limited, a wholly owned company of the Department of Health and Social Care (DHSC). The 100,000 Genomes Project is funded by the National Institute for Health Research and NHS England. The Wellcome Trust, Cancer Research UK and the Medical Research Council also funded the research infrastructure. The 100,000 Genomes Project uses data
provided by patients and collected by the NHS as part of their care and support. We are grateful for the support from Dame S. Hill and the team at NHS England for establishing and funding the 13 Genomic Medicine Centres. We thank all of the NHS Genomic Medicine Centres across England: East of England; Imperial College and West London; Greater Manchester; North Thames; Northwest Coast; North East and Cumbria; Oxford; South Thames; South West; West of England; Wessex; West Midlands; and Yorks and Humber. They enabled the NHS contribution, including the clinical return of results within the NHS in a standardized and validated format. M.C. is a National Institute for Health and Care Research (NIHR) Senior Investigator Alumnus. This work is part of the portfolio of translational research at the NIHR Biomedical Research Centres at Barts, Cambridge University Hospitals NHS Foundation Trust, Great Ormond Street Foundation NHS Trust, Manchester University NHS Foundation Trust, Newcastle Hospitals NHS Foundation Trust, Oxford University Hospitals NHS Foundation Trust, Guys and St Thomas’ NHS Foundation Trust and University College London NHS Foundation Trust. We thank the NHS Genomic Laboratory Hubs and those who worked to create the National Genomic Test Directory and undertake its annual review, and the NHS GMS. This work was made possible through the generosity of NHS patients and uses clinical data from the NHS and NHS Digital. We thank all the staff at Genomics England Ltd and the Genomics England Research Consortium members. We thank the Illumina Laboratory Services team at Hinxton for their advice and for undertaking the WGS. We thank University College London, Cancer Research Technology and the TRACERx Team for providing the lung tumor samples that were used in the WGS pipeline validation. We thank J. Chalker, Great Ormond Street Hospital for Children NHS Foundation Trust, and A. Wallace, Manchester Centre for Genomic Medicine for sharing data from the orthogonal genomic tests that were used in the WGS pipeline validation. D.C. was solely funded by a Cancer Research UK grant (no. C1298/A8362) awarded to R. Houlston at The Institute of Cancer Research UK. B.N. was funded through the Cancer Research UK Birmingham Centre award no. C17422/A25154.
Author contributions
S.H., Z.D., C.T., S. Henderson, L.J., A.H., N.M., T.F., M.C. and S. Hing supervised the implementation of the 100,000 Genomes Project at the NHS. D.B. and C.K. supervised a partnership with Illumina for WGS. J.P., A. Siddiq, T.Z. and T.C. delivered the sample flow operations. J.A., P.A., G.C. and M.B.P. curated the longitudinal real-world data and linked them to the sequencing data. A.R.-M., A. Sosinsky, D.P.-G., J.L., J.M., D.C., B.N., N.V. and A.R. contributed to the development of the analytical bioinformatics pipeline. A. Sosinsky, N.M., M.C., J.A. and W.C. designed the study. J.A., W.C., D.P. and A. Sosinsky performed the analysis and interpreted the data. J.A., W.C., K.B. and O.Y. performed the data visualization. N.M., A. Sosinsky and M.C. supervised the study. N.M., A. Sosinsky, S.W., C.T., J.A. and W.C. wrote the manuscript with input from all authors. A. Sosinsky, M.C. and N.M. made the decision to submit the manuscript for publication.
Competing interests
Genomics England is a company wholly owned by the UK DHSC and was created in 2013 to introduce WGS into healthcare in conjunction with NHS England. All authors affiliated with Genomics England (A. Sosinsky, J.A., C.T., S. Henderson, L.J., A.H., P.A., G.C., J.M., S.W., K.B., D.P., M.B.P., N.V., A.R.-M., D.P.-G., J.L., J.P., A. Siddiq, T.Z., T.C., O.Y., T.F., A.R., M.C. and N.M.) are, or were, salaried by or seconded to Genomics England. D.B. and C.K. are full-time employees and shareholders of Illumina. A.H. has received speaker fees from Gilead, Roche, Pfizer, Jazz, AbbVie, Incyte and Astellas. N.M. has provided consulting and advisory support for Pfizer, Guardant, Seagen and Janssen, and received speaker fees from Novartis, Pfizer and Servier
outside of the submitted work. The remaining authors declare no competing interests.
Correspondence and requests for materials should be addressed to Mark Caulfield or Nirupa Murugaesu.
Peer review information Nature Medicine thanks Jo Lynne Rokita, Mark Rubin and Stephen J. Chanock for their contribution to the peer review of this work. Primary Handling Editor: Anna Maria Ranzoni, in collaboration with the Nature Medicine team.
Extended Data Fig. 1D (Cancer-predisposing germline)
Extended Data Fig. 1 | Prevalence (as percentage) of different types of mutations identified by WGS in genes indicated for testing in the National Genomic Test Directory for Cancer (NGTDC). (A) Somatic small variants (single nucleotide variants (SNVs), insertions and deletions). (B) Copy-number aberrations (CNAs); onc = oncogene, tsg = tumour suppressor gene.
(C) Structural variants (SVs). (D) Germline small variants related to inherited
cancer risk (predisposing genes). The percentage of tumours harbouring a specific type of mutation in gene(s) indicated for testing by tumour type in the NGTDC are shown in magenta. The incidence of mutations (as a percentage) in other tumour types, not currently indicated in the NGTDC, are shown in blue. Colour gradation reflects the percentage of affected cases.
Extended Data Fig. 2 | Distribution of tumor mutation burden (TMB) and mutational signatures across tumor types. Assignment of signatures to known etiologies matches Fig. 3.
Extended Data Fig. 3 | Kaplan-Meier estimates of overall survival with -values calculated using a stratified log-rank test. Numbers of patients at risk at different time points are indicated below the survival curves. Points and error bars on the embedded forest plots indicate hazard ratios (HR) with 95%
from cox proportional hazards models corrected by cancer stage. Patients are
stratified by mutational status of genes indicated for testing in NGTDC across all
cancer types . Exact p -values can be found in Supplementary Table S2. confidence intervals (CI), correspondingly. HR, CI and p-values are calculated
from cox proportional hazards models corrected by cancer stage. Patients are stratified by mutational status of genes indicated for testing in NGTDC across all
cancer types ( ). Exact p -values can be found in Supplementary Table S2.
Extended Data Table 1 | Median age and interquartile range (IQR) at diagnosis in the absence and presence of pertinent germline findings
Tumour Type
Germline Variant
N
Median (IQR)
Corrected P-value
Significance Level
Bladder Urothelial Carcinoma
absent
255
73 (67-80)
0.474
NS
present
<5
82.5 (73.75-87.5)
Breast Invasive Carcinoma
absent
2839
61 (51-72)
***
present
86
51.5 (46-63.75)
Colon Adenocarcinoma
absent
1893
70 (62-77)
7.38E-06
***
present
55
61 (48-70)
Glioblastoma Multiforme
absent
312
61 (52-69)
0.474
NS
present
<5
58 (54-58.5)
Ovarian High Grade Serous Carcinoma
absent
322
67 (58-73)
***
present
50
59 (49.5-63.75)
Kidney Renal Clear Cell Carcinoma
absent
1154
63 (55-71)
*
present
9
40 (28-56)
Pancreatic Adenocarcinoma
absent
118
70 (65-76)
0.474
NS
present
<5
70 (63-70.5)
Rectum Adenocarcinoma
absent
815
67 (60-74)
0.101
NS
present
15
61 (49.5-62.5)
Skin Cutaneous Melanoma
absent
324
68 (56-76.25)
0.474
NS
present
<5
76 (76-76)
Uterine Corpus Endometrial Carcinoma
absent
682
67 (59-74)
0.343
NS
present
24
59.5 (55-69.5)
Uterine Corpus Endometrial Serous Carcinoma
absent
110
71 (67-75)
0.474
NS
present
<5
60 (59-72.5)
natureportfolio
Mark Caulfield
Corresponding author(s):
Nirupa Murugaesu
Last updated by author(s): Oct 15,2023
Reporting Summary
Nature Portfolio wishes to improve the reproducibility of the work that we publish. This form provides structure for consistency and transparency in reporting. For further information on Nature Portfolio policies, see our Editorial Policies and the Editorial Policy Checklist.
Statistics
For all statistical analyses, confirm that the following items are present in the figure legend, table legend, main text, or Methods section.
Confirmed
X The exact sample size ( ) for each experimental group/condition, given as a discrete number and unit of measurement
A statement on whether measurements were taken from distinct samples or whether the same sample was measured repeatedly
X
The statistical test(s) used AND whether they are one- or two-sided
Only common tests should be described solely by name; describe more complex techniques in the Methods section.
A description of all covariates tested
A description of any assumptions or corrections, such as tests of normality and adjustment for multiple comparisons
凶
A full description of the statistical parameters including central tendency (e.g. means) or other basic estimates (e.g. regression coefficient) AND variation (e.g. standard deviation) or associated estimates of uncertainty (e.g. confidence intervals)
For null hypothesis testing, the test statistic (e.g. ) with confidence intervals, effect sizes, degrees of freedom and value noted Give values as exact values whenever suitable.
For Bayesian analysis, information on the choice of priors and Markov chain Monte Carlo settings
For hierarchical and complex designs, identification of the appropriate level for tests and full reporting of outcomes
Estimates of effect sizes (e.g. Cohen’s , Pearson’s ), indicating how they were calculated
Our web collection on statistics for biologists contains articles on many of the points above.
Software and code
Policy information about availability of computer code
Data collection
Data analysis
OpenClinica v3.4
SEquencing data QC, Mapping and variant calling
North Star pipeline version 2.6.53.23
ISAAC version iSAAC-03.16.02.19
Starling version 2.4.7
Strelka version 2.4.7
Canvas version 1.3.1
Manta version 0.28.0
samtools version 1.9
R packages used in this analysis
R4.0.3
survminer_0.4.9
survival_3.2-7
dplyr_1.0.7
purrr_0.3.4
tidyr_1.1.2
tibble_3.0.3
ggplot2_3.3.2
tidyverse_1.3.0
For manuscripts utilizing custom algorithms or software that are central to the research but not yet described in published literature, software must be made available to editors and reviewers. We strongly encourage code deposition in a community repository (e.g. GitHub). See the Nature Portfolio guidelines for submitting code & software for further information.
Data
Policy information about availability of data
All manuscripts must include a data availability statement. This statement should provide the following information, where applicable:
Accession codes, unique identifiers, or web links for publicly available datasets
A description of any restrictions on data availability
For clinical datasets or third party data, please ensure that the statement adheres to our policy
Public datasets that were used for variant annotation:
Ensembl version 90/GRCh38
COSMIC version v86
ClinVar October 2018 release
COSMIC signatures v3
The data supporting the findings of this study are available within the Research Environment, a secure cloud workspace. Details on how to access data for this publication can be found at https://re-docs.genomicsengland.co.uk/pan_cancer_pub/. Additional processed aggregated data used to generate figures can be found in Supplementary Tables S5-S20.
Once you have confirmed your institution is registered and have found a GECIP domain of interest, you can apply through the online form at https:// www.genomicsengland.co.uk/research/academic/join-gecip. Once your Research Portal account is created you will be able to log in and track your application.
The domain lead will review your application within 10 working days.
Your institution will validate your affiliation.
You will complete our online Information Governance training and will be granted access to the Research Environment within 2 hours of passing the online training.
Data that has been made available to registered users include: alignments in BAM or CRAM format, annotated variant calls in VCF format, signatures assignment, tumour mutation burden, sequencing quality metrics, summary of findings that is shared with Genomic Lab Hubs, secondary clinical data as described in this paper. Further details of the types of data available (for example, mortality, hospital episode statistics and treatment data) can be found at https://redocs.genomicsengland.co.uk/data_overview/. Germline variants can be explored in Interactive Variant Analysis Browser (see description at https://redocs.genomicsengland.co.uk/iva_variant/). Cancer patients cohort and longitudinal clinical information on treatment and mortality can be explored with Participant Explorer (see description at https://re-docs.genomicsengland.co.uk/pxa/).
Research involving human participants, their data, or biological material
Policy information about studies with human participants or human data. See also policy information about sex, gender (identity/presentation), and sexual orientation and race, ethnicity and racism.
Reporting on sex and gender
Biological sex that was inferred using the ratio of mean sequencing coverage of sex chromosomes and mean sequencing coverage of autosomes. In our analysis, patients were not stratified by sex to maximize the power of the cohort. Patients provided informed consent for paired tumour and normal (germline) whole genome sequencing (WGS) analysis. Participants also gave consent for their genomic data to be linked to anonymised longitudinal health records and shared with researchers in a secure Research Environment.
Reporting on race, ethnicity, or other socially relevant groupings
Population characteristics
Socially relevant categorization variables were not used in this study.
15,241 patients diagnosed with cancer within the NHS that were recruited to the Cancer Programme of the 100,000 Genomes Project between 2015 and 2019. Tumour types with more than 1,000 sequenced tumour genomes included breast invasive carcinoma ( ), colon adenocarcinoma ( ), sarcoma ( ) and kidney renal clear cell carcinoma ( ). ( ) of patients had stage 4 cancer (advanced metastatic disease). Early onset (median age <50 years) was observed for low grade glioma and testicular germ cell tumours in agreement with incidence statistics. Tumour samples mainly originated from surgical resections ( ), including treatment-naive cases and postneoadjuvant treatment. Only came from metastatic or diagnostic biopsies, with being posttreatment.
Participants were selected on the basis of having been identified by health care professionals and researchers within the NHS as having a cancer diagnosis. The participants were recruited across 13 NHS Genomic Medicine Centres and written informed consent was obtained from the participants.
Ethics oversight
Research described in this manuscript complies with all relevant ethical regulations. Approval for the project was obtained from the East of England – Cambridge South Research Ethics Committee (REC reference 14/EE/1112, IRAS ID 166046)
Note that full information on the approval of the study protocol must also be provided in the manuscript.
Field-specific reporting
Please select the one below that is the best fit for your research. If you are not sure, read the appropriate sections before making your selection.
Life sciences Behavioural & social sciences Ecological, evolutionary & environmental sciences
For a reference copy of the document with all sections, see nature.com/documents/nr-reporting-summary-flat.pdf
Life sciences study design
All studies must disclose on these points even when the disclosure is negative.
Sample size
The results shown here are not the result of an experimental set up. We are describing observations for a cohort of 13,880 cancer patients recruited for 100,000 Genomes Programm. Sample size calculation is not relevant for this study.
Data exclusions
Pediatric cancers, hematological malignancies, cancers of unknown primary ans samples that didn’t have clinical information from secondary sources were excluded as stated in the manuscript.
Replication
Replication is not relevant for the reason explained above
Randomization
Randomization is not relevant for the reason explained above
Blinding
Blinding is not relevant for the reason explained above
Reporting for specific materials, systems and methods
We require information from authors about some types of materials, experimental systems and methods used in many studies. Here, indicate whether each material, system or method listed is relevant to your study. If you are not sure if a list item applies to your research, read the appropriate section before selecting a response.
Statistical significance of pertinent germline finding for early tumor onset was calculated by Wilcoxon rank-sum test with Benjamini-Hochberg multiple testing. ***P<0.0001, *P<0.05.
Tumor types without germline variant testing indicated in the NGTDC were excluded.