سبائك عالية الانتروبيا تحفز التحول البوليمري للملوثات المائية مع تحسين ملحوظ في كفاءة استخدام الإلكترونات High-entropy alloys catalyzing polymeric transformation of water pollutants with remarkably improved electron utilization efficiency
تظهر جزيئات نانوية من سبائك عالية الانتروبيا (HEA-NPs) خصائص ملائمة في العمليات التحفيزية، حيث تضمن مواقعها متعددة المعادن كلاً من النشاط الجوهري العالي والكفاءة الذرية. ومع ذلك، لا يزال من الصعب التحكم في تخليق مجموعات متعددة المعادن بشكل موحد على المستوى الذري. تنجح هذه الدراسة في تحميل HEA-NPs على حامل كربوني مخدر بالنيتروجين (HEAs) وتبتكر تطبيقًا في تنشيط بيروكسيمونوكبريتات (PMS) لدفع الأكسدة على غرار فنتون. يحقق نظام HEAs-PMS إزالة سريعة للغاية للملوثات عبر نطاق واسع من الرقم الهيدروجيني مع مقاومة قوية للتداخلات المائية في العالم الحقيقي. علاوة على ذلك، يقوم نظام HEAs-PMS غير الجذري بتحويل الفينولات بشكل انتقائي إلى منتجات ذات وزن جزيئي عالٍ عبر مسار بلمرة. يقلل النظام الفريد غير المعدني بشكل ملحوظ من استهلاك PMS ويحقق كفاءة استخدام إلكترون عالية تصل إلىتظهر المزيد من حسابات DFT والتحليل التجريبي أن الحديد والكوبالت في HEA-NPs يعملان كمواقع تحفيزية رئيسية للتعقيد مع PMS للتفعيل، بينما، وتعمل Pd كوسائط شحن لتسهيل نقل الإلكترونات. تمتلك المركبات الناتجة PMS* على HEAs جهد أكسدة مرتفع، مما يدفع أكسدة الفينول المكانية المنفصلة على دعم الجرافين المضاف إليه النيتروجين لتكوين جذور الفينوكسي، مما يؤدي بعد ذلك إلى تحفيز تكوين منتجات بوليمرية عالية الجزيئية عبر تفاعلات البلمرة. تقدم هذه الدراسة محفزات HEAs مصممة لمعالجة المياه مع استهلاك منخفض للأكسدة وانبعاثات.
إن معالجة المياه العادمة العضوية هي قضية تزداد أهمية في التنمية طويلة الأمد للمجتمع، مما يستلزم تطوير تقنيات تنقية المياه الصديقة للبيئة والفعالة من حيث التكلفة والكفاءة.تعتبر عمليات الأكسدة المتقدمة (AOPs) حيوية في تكسير وتعدين الملوثات العضوية إلى التخفيف من التلوث البيئيلتلبية الطلبات المتزايدة بسرعة، فإن تطوير المحفزات عالية الأداء ذات النشاط العالي والانتقائية والاستقرار في التفاعلات الحفزية أمر ضروري. وقد أسفرت الأبحاث المكثفة عن إنتاج مجموعة متنوعة من المحفزات، مثل الذرات الفردية، والذرات الفردية ثنائية المعدن، وأكاسيد/كبريتيدات المعادن، وغيرها..
هناك إجماع على أن التركيب العنصري والبنية البلورية للعوامل الحفازة تؤثر بشكل كبير على أدائها الحفازي. لذلك، يبدو أن تطوير عوامل حفازة أكثر تقدمًا وشيك، مما يوفر فرصًا واسعة لضبط الهيكل الدقيق للميكروإلكترونيات وتحسين الأداء..
سبائك عالية الانتروبيا، وهي فئة مميزة من سبائك متعددة المعادن تتكون عادة من خمسة معادن أو أكثر، قد حازت على اهتمام بحثي واسع في السنوات الأخيرة ووجدت تطبيقات واسعة في تحويل الطاقة والتخليق الأخضر.تقدم المرحلة غير المرتبة لعناصرها مزايا فطرية، بما في ذلك تنوع العناصر واستقرار الهيكل، مما يؤدي إلى زيادة كبيرة في الجهد الكيميائي وخصائص إلكترونية سطحية مختلفة مقارنةً بنظائرها التقليدية من المعادن الفردية/المختلطة.في التحفيز، يؤثر امتصاص الجزيئات والمواد الوسيطة على سطح المحفز على النشاط التحفيزي. مقارنة بالعناصر النقية، يمكن تنظيم طاقات الامتصاص هذه من خلال السبائك لتعزيز النشاط التحفيزي.نتيجة لذلك، تم استكشاف السبائك عالية الانتشار (HEAs) بشكل واسع في تفاعلات تحفيزية متنوعة مثل تطور الهيدروجين، وتطور الأكسجين، وأكسدة الكحول، وأكسدة الأمونيا، حيث تظهر هذه السبائك أداءً متفوقًا مقارنةً بالمواد المحفزة التقليدية.ومع ذلك، فإن التحديات الرئيسية المرتبطة بالسبائك عالية الانتروبيا هي التركيب الدقيق والتوصيف، بالإضافة إلى آليات التفاعل المعقدة بسبب الهياكل متعددة المكونات.
في هذا السياق، لتعزيز التركيب الإلكتروني للسبائك عالية الانتروبيا من أجل الامتزاز الوسيط، نبلغ عن تخليق جزيئات نانوية من سبيكة CuPdFeCoNi محملة على مواد كربونية تحتوي على النيتروجين باستخدام مزيج من الكيمياء الرطبة وطرق التحلل الحراري المسيطر عليه. تكشف النتائج أن التركيب الإلكتروني المعدل يسرع من تنشيط بيروكسيمونوكبريتات (PMS) مقارنةً بـ NG لإزالة الملوثات العضوية من الماء. يعمل نظام HEAsPMS من خلال مسار نقل إلكتروني غير جذري (ETP) بكفاءة عالية تحت ظروف pH قاسية ومقاومة للتداخل من عوامل خلفية الماء. علاوة على ذلك، تم إجراء تحليل شامل للهيكل والتركيب للسبائك عالية الانتروبيا. تم التحقيق في الآلية المرتبطة باستخدام الرنين المغناطيسي الإلكتروني (EPR) وتجارب الاستكشاف، مدعومة بالتوصيف الكهروكيميائي. ولدهشتنا، أسفر النظام غير الجذري الناتج عن HEA عن كمية كبيرة من منتجات البلمرة على سطح المحفز ضد التمعدن، مما أدى إلى كفاءة استخدام إلكترونية استثنائية تصل إلىيُنسب ذلك إلى الإمكانيات العالية لمركب HEAs-PMS* وسرعة نقل الإلكترون (ديناميكية أكسدة الملوثات السريعة)، المنسقة بواسطة العناصر المتعددة الموزعة بشكل موحد في HEAs، مما ينتج عنه منتجات ذات درجة بوليمرة عالية. تم إثبات الاستقرار على المدى الطويل والجدوى العملية لـ HEAs من خلال تجربة إزالة المياه الملوثة المستمرة لمدة 50 يومًا. بشكل عام، تقدم هذه الدراسة حلاً لتطوير محفزات غير متجانسة متقدمة مناسبة لمعالجة المياه مع تقليل استهلاك المؤكسدات وإعادة تدوير ملوثات المياه.
النتائج
تركيب وتوصيف المواد عالية التشتت
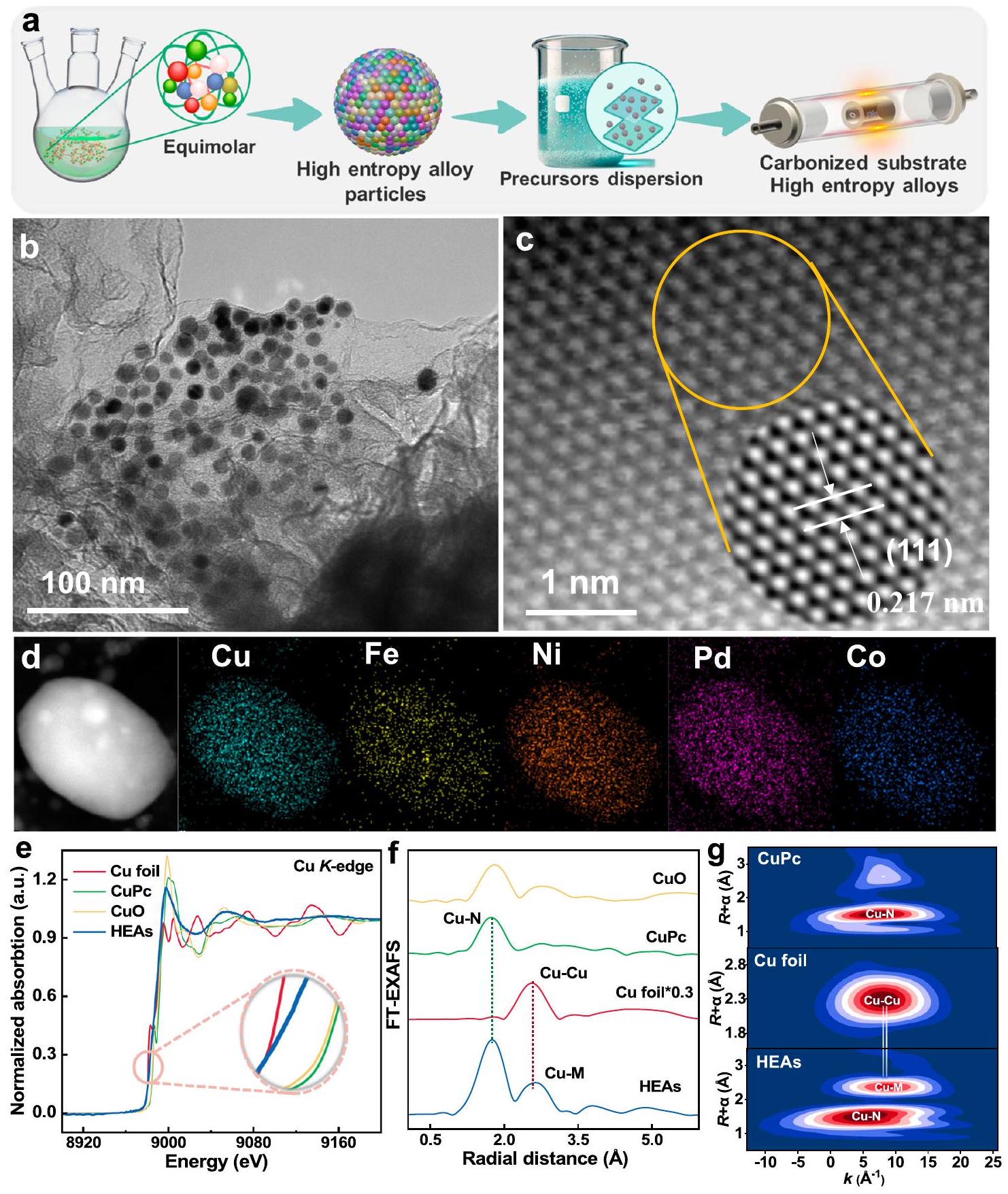
تم اعتماد نهج بسيط لتخليق الطور الزيتي في وعاء واحد عن طريق تسخين خمسة من سوائل أملاح المعادن.، وني ) في لمدة ساعتين، تليها التحلل الحراري مع وجود سلف كربوني للحصول على HEAsتم تحديد التركيب المحسوب للسبائك عالية التداخل (HEAs) التي تم الحصول عليها على أنهمن خلال التحليل الطيفي الانبعاثي البصري المتصل بالتحليل الطيفي البلازمي (ICP-OES) والتحليل العنصري (الجدولان التكميليان 1 و 2). نجح إجراء التخليق، الموضح في الشكل 1a والمفصل في قسم التجارب، في إنتاج مواد متجانسة.جزيئات نانوية بقطركما هو موضح في صور المجهر الإلكتروني الناقل (TEM) (الشكل 1b والأشكال التكميلية 1 و2). أكد نمط حيود الأشعة السينية للمواد المسحوقة الهيكل البلوري المكعب المتمركز على الوجوه (fcc) لسبائك CuPd (رقم JCPDS 48-1551) NPs، مع الطور الرئيسي المحدد على أنه سبيكة CuPd من خلال القمم الموجودة حول و ، التي تتوافق مع الوجوه (111) و (200) (الشكل التكميلي 3)“. بالإضافة إلى ذلك، فإن التحول الملحوظ في موضع قمة الانكسار العريضة، على عكس قمم الانكسار للمعادن النقية (Pd،، وCu)، يشير إلى تغيير كبير. يشير التحول الطفيف في القمة لسبيكة CuPd إلى دمج Fe وNi وCo في شبكة CuPd، مما يؤدي إلى إنشاء هيكل سبيكة موحد مع انكماش متزامن في الشبكة.صورة TEM الماسح ذات الزاوية العالية والحقل المظلم الحلقي (AC-HAADF-STEM) في الشكل 1c تظهر تباعد الشبكة بمقدار 0.217 نانومتر، مما يتوافق مع وجه (111) منتظهر خرائط العناصر باستخدام AC-HAADF-STEM-EDS أن كل عنصر موزع بشكل موحد، مما يوضح أن المعادن الخمسة (Pd وFe وCo وNi وCu) موزعة بالتساوي على سطح جزيئات HEA (الشكل 1d).. هذه التوصيفات الشاملة تدعم التخليق الناجح والتحميل لـNPs على الجرافين المخصب بالنيتروجين (NG).
لتحديد الهياكل الإلكترونية والتنسيقية لذرات النحاس في السبائك عالية الانتشار (HEAs)، تم استخدام مطيافية امتصاص الأشعة السينية بالقرب من الحافة (XANES) وهيكل الامتصاص الدقيق الممتد للأشعة السينية (EXAFS). يكشف منحنى XANES أن شدة الخط الأبيض أعلى من رقائق النحاس ولكنها أقل بكثير من تلك الخاصة بـ CuO وCuPc، مما يؤكد أن أنواع النحاس في HEAs موجودة بشكل رئيسي في الحالة المعدنية (الشكل 1e).. علاوة على ذلك، يقع مركز ما قبل الحافة للسبائك عالية الانتقال (HEAs) بين رقائق النحاس (Cu) وCuPc، مما يشير إلى احتمال التنسيق بين ذرات النحاس (Cu) والنيتروجين (N) في NG. يعرض طيف هيكل الامتصاص الدقيق للأشعة السينية الممتد المحول فورييه (FT-EXAFS) للسبائك عالية الانتقال (HEAs) في فضاء R قمتين رئيسيتين تقعان تقريبًا عند لـرابطة وانحراف ذروة عند، الذي يتوافق مع التفاعل. يشير هذا الإزاحة إلى وجودالتفاعلات ) داخل السبائك عالية التشتت (HEAs)، والتي من المحتمل أن تكون ناتجة عن التفاعل المتبادل لعدة عناصر في السبائك عالية التشتت (الشكل 1f). بالإضافة إلى ذلك، تحويلات الموجات (WT) لـ طيف EXAFS الموزون للسبائك عالية التشتت يكشف أيضًا عن ذروتين مهيمنتين تصلان إلى الحد الأقصى عندنُسِبَ إلىرابطة والتفاعلات (الشكل 1g). قوية و يمكن ملاحظة التنسيق في مخططات كونتور WT لرقائق النحاس وCuPc، وهو ما يتوافق مع النتائج المستخلصة من طيف EXAFS. بالإضافة إلى ذلك، فإن طيف امتصاص الأشعة السينية لذرات النيكل في HEAs يعطي أيضًا استنتاجات مشابهة، على الرغم من التفاعلات السائدة لنيكل M (الشكل التكميلي 5). وهذا يعزز النتائج السابقة للتوصيف، مؤكداً دمج النيكل في سبيكة CuPd.
كفاءة تحفيزية متفوقة للسبائك عالية الانتشار
تمت دراسة القدرة التحفيزية للسبائك عالية التشتت (HEAs) باستخدام بيروكسيد الهيدروجين كمادة سابقة، وتم تقييم النشاط الشبيه بفنتون من خلال استخدام الفينول كملوث عضوي نموذجي. كما هو موضح في الشكل 2a، حدثت امتصاصية ضئيلة في أنظمة المحفز فقط، حيث حقق نظام HEAs إزالة 100% من الفينول في 10 دقائق مع إضافة PMS، بينما أظهر نظام NG أقل منإزالة الفينول. من الجدير بالذكر أن معدل التحلل للـ HEAs تجاوز معدل التحلل للـ NG بأكثر من 7 مرات (الشكل التوضيحي التكميلي 6)، مما يشير إلى أن دمج مواقع جزيئات السبيكة في الـ NG قد يكون مواقع نشطة حاسمة لتفعيل الـ PMS.. بالإضافة إلى ذلك، أظهرت تحسين تركيز PMS أن التحلل الكامل كان ممكنًا مع جرعة PMS تبلغ 0.25 مللي مول، بعد ارتباط إيجابي لسرعة الأكسدة مع تركيز PMS، على الأرجح بسبب تأثيراته الإيجابية على تنشيط PMS (الشكل التوضيحي 7). انخفضت معدلات الإزالة مع زيادة تركيز الفينول الابتدائي، على الرغم من أنه تم تحقيق إزالة كاملة للفينول خلال 10 دقائق عند تركيز 0.1 مللي مول (الشكل التوضيحي 8). ومع ذلك، عندما تجاوز تركيز الفينول الابتدائي ما يمكن أن تقبله معادلات الإلكترون من PMSتم تقليل كفاءة الإزالة بشكل كبير.
تم اكتشاف أن أيونات الكوبالت والحديد والمعادن الأخرى تظهر أداءً تحفيزياً كمنشطات كلاسيكية في تفاعلات شبيهة بفنتون، والتي تُستخدم على نطاق واسع لتفعيل PMS. ومع ذلك، فإن كفاءة
الشكل 1 | تخليق وتوصيف المواد عالية التشتت. أ الرسم التخطيطي لعملية تخليق المواد عالية التشتت.صور TEM للسبائك عالية الانتشار.صورة HAADFSTEM المصححة للانحراف ونمط FFT العكسي المقابل لـ HEAs (المميزة في الدائرة الصفراء). د رسم الخرائط العنصرية للسبائك عالية الانتقال (مقياس، 5 نانومتر). هـ XANES عند حافة النحاس L.طيف FT-EXAFS في فضاء R.WTs لطيفيات EXAFS للسبائك عالية التشتت،رقائق، وCuPc. المحفزات غير المتجانسة مقارنة بأيونات المعادن مثليبقى محدودًالتحقيق الأداء الاستثنائي للسبائك عالية التشتت في تحلل الفينول، تم إجراء تجارب باستخدام تركيزات متساوية من أيونات المعادن.، و ) كعوامل تحفيز مقارنة بناءً على محتوى المعادن في HEAs (الشكل التكميلي 9 والجدول 3). لوحظ أنه عند تركيز PMS يبلغ 0.25 مللي مول، فقط أظهرت أداءً متدهورًا بشكل ملحوظ. تم إجراء تجارب تدهور إضافية عند تركيزات أعلى باستخدام أيونات الكوبالت ومزيج من 5 أيونات معدنية أخرى. أظهرت النتائج في الشكل التوضيحي 10 أنه حتى مع زيادة بمقدار 40 مرة فيوزيادة بمقدار 20 ضعف في الأيونات المختلطة، لا تزال كفاءة إزالة الفينول لا تستطيع المنافسة مع نظام HEAs-PMS. كانت الثابتة الحركية الظاهرة لنظام HEAs-PMS 5.3 مرات. و2.7 مرة أعلى من ذلك الخاص بالمتجانسأنظمة PMS وأيونات مختلطة-PMS، على التوالي. تدعم هذه النتائج المزيد من مزايا المحفزات المحملة بجزيئات سبائك عالية الانتروبيا من حيث النشاط الجوهري العالي وانخفاض تسرب المعادن في معالجة التلوث البيئي. علاوة على ذلك، اقترح مراقبة معدل استهلاك PMS من خلال تفاعل اختزال اليود أن الأداء الاستثنائي للسبائك عالية الانتروبيا يمكن أن يُعزى إلى تفعيل PMS المحسن بشكل ملحوظ على سطح جزيئات السبائك، محققًا معدلًا أعلى بـ 12 مرة من NG (الشكل التكميلي 11)..
تأثيرات المواد العضوية المذابة المتواجدة معًا (DOM، الممثلة بحمض الهيوميك) وأنواع الأنيونات النموذجية (مثل،، ، و ) على إزالة الفينول بواسطة HEAs و NG تم التحقيق فيها بشكل أكبر. كما هو موضح في الشكل 2b، كانت المادة العضوية الذائبة بتركيز
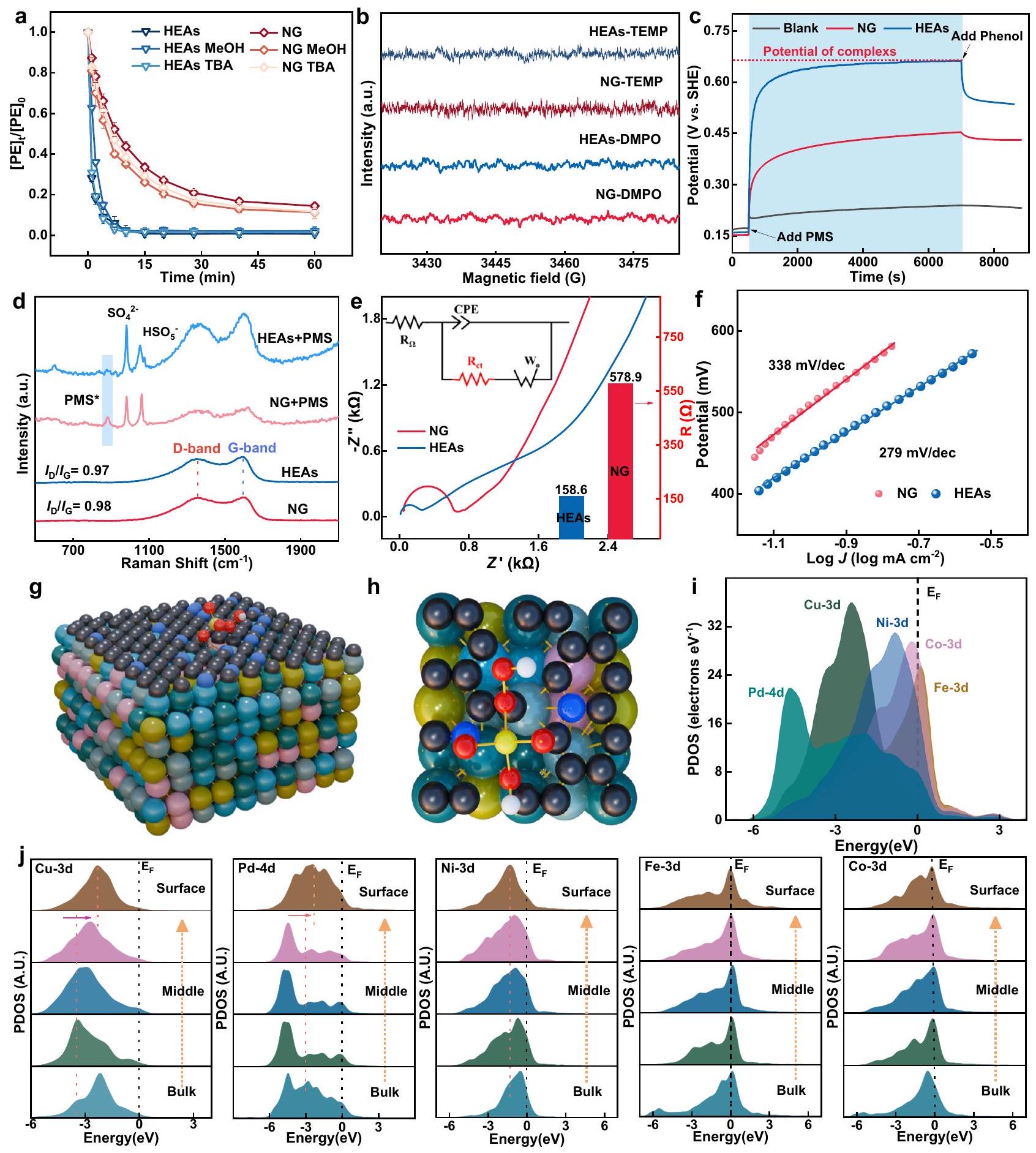
الشكل 3 | تحديد الأنواع النشطة. أ تأثيرات الإخماد بواسطة مختلف الماسحات. تمثل أشرطة الخطأ الانحرافات المعيارية من الاختبارات الثلاثية. الجرعة: [الفينول]، PMS: 0.25 مللي مول ، المنظفات: 2.5 مللي مول ، محلول التفاعل: 50 مل ، المحفز: . تم إجراء اختبارات EPR على مركبات DMPO وTEMP ضمن نظام HEAs-PMS أثناء تنشيط PMS. ج الجهد المفتوح على أقطاب HEAs-GCE وNG-GCE. د طيف رامان في الموقع لنظام HEAs-PMS وNG-PMS في محلول سائل. رسومات نيكويست وحسابات الملاءمة للمقاومة الداخلية للسبائك عالية الانتشار (HEAs) والمواد الجديدة (NG).منحدر تافل على أقطاب HEAs-GCE و NG-GCE في محاليل الفينول.المنظر الجانبي للتكوين الهيكلي لـ PMS على HEAs.المنظر العلوي للتكوين الهيكلي لـ PMS على HEAs. i دوال الكثافة المحلية لحالات HEAs. j دوال الكثافة المحلية لحالات تعتمد على الموقع، و Co في HEAs. تيتراميثيلبيبيريدين (TEMP) كـلم يؤثر المثبط على كفاءة إزالة الفينول، مما يدل على التأثير الضئيل لـعلى الأداء التحفيزي (الشكل التكميلي 19). في الوقت نفسه، استبداللأن الماء المنزوع الأيونات في المحلول لم يسهل حركيات الأكسدة، ولم تؤدِ التجارب التي أجريت باستخدام 9,10-ثنائي فينيل أنثراسين (DPA) كأداة استكشاف إلى إنتاج المنتج المميز منالأكسدة (الأشكال التكميلية 20 و21)³4. هذه النتائج أشار إلى أنلم يتم تقديمه في أنظمة HEAs-PMS و NG-PMS. تشير التأثيرات المثبطة الضئيلة لثنائي ميثيل سلفوكسيد ككاشف محدد إلى غياب الأنواع المعدنية عالية التكافؤ من الأكسو في نظام HEAs-PMS (الشكل التوضيحي 22).. تستبعد هذه النتائج الجماعية مساهمات المتطرفين ( أو )، وأنواع المعادن الأكسو ذات الشحنة العالية، مما يوحي بأن ETP التي يتم تحفيزها بواسطة PMS من المحتمل أن تكون متورطة في إزالة الفينول بطريقة غير جذرية.
تم استخدام تقنيات كيميائية كهربائية متنوعة لتقييم أكسدة الفينول التي تتم بوساطة ETP في أنظمة HEAs-PMS و NG-PMS، بما في ذلك OCPT و LSV و CA و EIS و منحنى تافل وتحليل ECSA.كما هو موضح في الشكل 3c، بعد حقن PMS، زادت OCPT بسرعة لـ HEAs-GCE و NG-GCE، مما يدل على تكوين معقد PMS المنشط على السطح (PMS*) الذي رفع من قدرة المحفز. ومن الجدير بالذكر أن قدرة HEAs-PMS* تجاوزت تلك الخاصة بـ NG-PMS*، مما يشير إلى قدرة أكسدة أعلى. عند إضافة الفينول، أشار الانخفاض اللاحق في OCPT إلى حدوث تفاعل ETP بين HEAs/NG-PMS* والفينول. علاوة على ذلك، أثبتت مطيافية رامان في الموقع في الشكل 3d مباشرة وجود معقد PMS* التفاعلي على أسطح HEAs و NG.
أظهر المزيد من المراقبة لمحتوى PMS خلال تجربة OCPT التفاعل الأكثر كفاءة لجزيئات السبيكة مع PMS، مما أدى إلى الإمكانية الأعلى الملحوظة لقطب HEAs-GCE (الشكل التكميلي 23). أظهرت تجربة CV كثافة تيار أعلى بكثير لأكسدة الفينول بواسطة HEAs مقارنة بـ NG، مما يؤكد القدرة الفائقة على نقل الإلكترونات لجزيئات السبيكة من الملوث إلى PMS (الشكل التكميلي 24). تم دعم الاقتراح أيضًا بواسطة قياسات CA ووقت التيار وECSA (الأشكال التكملية 25-27). تم تقدير الموصلية والديناميكا الكهربائية لـ HEAs باستخدام EIS وميل Tafel، مما يحدد الخطوة المحددة للسرعة (الشكل 3e والشكل التكميلي 28). أشارت نتائج تركيب الدائرة إلى أن مقاومة HEAs () كانت أقل بكثير من تلك الخاصة بـ NG ()، بسبب وجود HEAs المعدنية، التي تفيد عمليات هجرة الشحن. تم الحصول على ميول Tafel من خلال تركيب بيانات LSV على منحنى Tafel، مما أسفر عن حساب ميول Tafel قدرها 279 و لـ HEAs و NG، على التوالي (الشكل 3f). تشير هذه الفروقات إلى أن HEAs تظهر ديناميات أكسدة أسرع وقدرة أعلى على نقل الإلكترونات عند إمكانيات عالية مقارنة بـ NG. تم إجراء تجارب المفاعل الجلفاني لتحليل آلية تنشيط PMS بواسطة HEAs/NG كما هو موضح في الشكل التكميلي 29. كان جهد القطب داخل غرفة PMS أعلى بكثير من ذلك في غرفة الفينول، مما يشير إلى أن غرفة PMS كانت تعمل كغرفة كاثود، بينما كانت محلول الفينول يعمل كغرفة أنود (الشكل التكميلي 30). وبالتالي، تم أكسدة الفينول مباشرة من خلال ETP المرتبط بالبروتون في تفاعل الخلية الجلفانية وتوسط تدفق الإلكترونات بشكل متزامن من الخلية العضوية إلى PMS المنشط على الطرف الآخر. أظهرت التغيرات المسجلة في التيار خلال التفاعلات الجلفانية أن HEAs يمكن أن تولد تيارًا أكبر بكثير مقارنة بـ NG، مما يبرز تنشيط PMS الفائق (قبول الإلكترون) وديناميات أكسدة الفينول (تقديم الشحنة) على HEAsNPs عبر نظام ETP .
لكشف النشاط الفائق وقدرة نقل الإلكترونات لـ HEAs، تم حساب كثافة الحالة المتوقعة (PDOS) لامتصاص PMS على HEAs. في البداية، تم مقارنة PDOS لثلاثة HEAs مع اختلافات ستوكيومترية طفيفة. كما هو موضح في الشكل التكميلي 31، عرضت نماذج المقارنة هياكل إلكترونية متشابهة للغاية مع تغييرات طفيفة في مواقع الذروة وأنماط المدارات d لكل عنصر. لذلك، من غير المحتمل أن تؤثر التغيرات الطفيفة في ستوكيومترية HEAs بشكل كبير على الهيكل الإلكتروني في نتائج PDOS . بعد ذلك، تم اختيار هيكل HEAs الأكثر استقرارًا كنموذج شبكي (الشكل 3g وh). لفهم الهيكل الإلكتروني بشكل أفضل، يوضح الشكل 3i PDOS لكل عنصر في HEAs. من الجدير بالذكر أن مدارات Pd-4d احتلت أعمق موقع بالقرب من نطاق التكافؤ () عند -4.6 eV، مما يعمل كخزان للإلكترونات. عرضت مدارات Ni-3d ذروة عند Ev -1.0 eV، بينما أظهرت مدارات Co-3d تقاطعًا بارزًا بكثافة إلكترونية عالية عند مستوى فيرمي ()، مما يدعم النشاط الكهربائي العالي لمواقع Ni وCo لتنسيق تفاعل الأكسدة السطحية. بالإضافة إلى ذلك، أشار التداخل الكبير بين مدارات d المختلفة إلى وجود روابط قوية وتواصل إلكتروني بين العناصر المعدنية. مكنت هذه التفاعلات القوية من اقتران المتفاعلات (PMS) مع مدارات 3d لمواقع Fe وCo، مما يسهل
تعديل الإلكترونات المرنة وتسريع نقل الإلكترونات على سطح HEAs NPs. وهذا يدل على أن مدارات 3d لـ Cu وNi وCo وFe تعمل أيضًا كوسائط جسرية، مما يساعد على تخفيف حاجز الطاقة لـ ETP خلال عمليات الأكسدة .
لتحديد التعديلات الإلكترونية بشكل أكبر، تم توفير PDOSs المعتمدة على الموقع لكل عنصر في HEAs (الشكل 3j). من المواقع الكتلية إلى السطح، لوحظ تحول واضح نحو الأسفل لـ ومدارات Pd-4d، بينما أظهرت مواقع Pd تخفيفًا لتأثير الانقسام ، مما يدعم كفاءة نقل الإلكترونات المحسنة. بالنسبة لمواقع Ni الأقرب إلى السطح، أظهرت مدارات 3d تحولًا طفيفًا لدعم نشاط نقل الإلكترونات لـ HEAs. عرضت كل من مواقع Fe وCo مركز نطاق d مستقر نسبيًا، يُعزى إلى تأثير تثبيت مزدوج من تأثير الحماية للمدارات المعدنية المجاورة. ساعد ذلك في الحفاظ على مركز تعزيز الإلكترونات وسهل الاقتران مع PMS، مما يظهر القدرة القوية على تقديم الإلكترونات لعملية الأكسدة. نظرًا لوجود حواجز منخفضة لنضوب الإلكترونات، تم تحديد مواقع , وNi كمواقع عازلة، تحمي المواقع المعدنية والنشطة كهربائيًا Cu وPd داخل HEAs NPs من الأكسدة. وهذا يضمن أداء إزالة فائق واستقرار HEAs في أكسدة ETP. بناءً على النتائج المذكورة أعلاه، استنتجنا أن Fe وCo يعملان كمواقع تفاعلية رئيسية للارتباط مع PMS، وفي الوقت نفسه، Ni وCu وPd هم وسطاء يسرعون نقل الإلكترونات إلى الفينول الممتص على سطح الجرافيت المدعوم بالنيتروجين، مما يعزز ديناميات أكسدة الفينول.
دراسة آلية البوليمرية في إزالة الملوثات
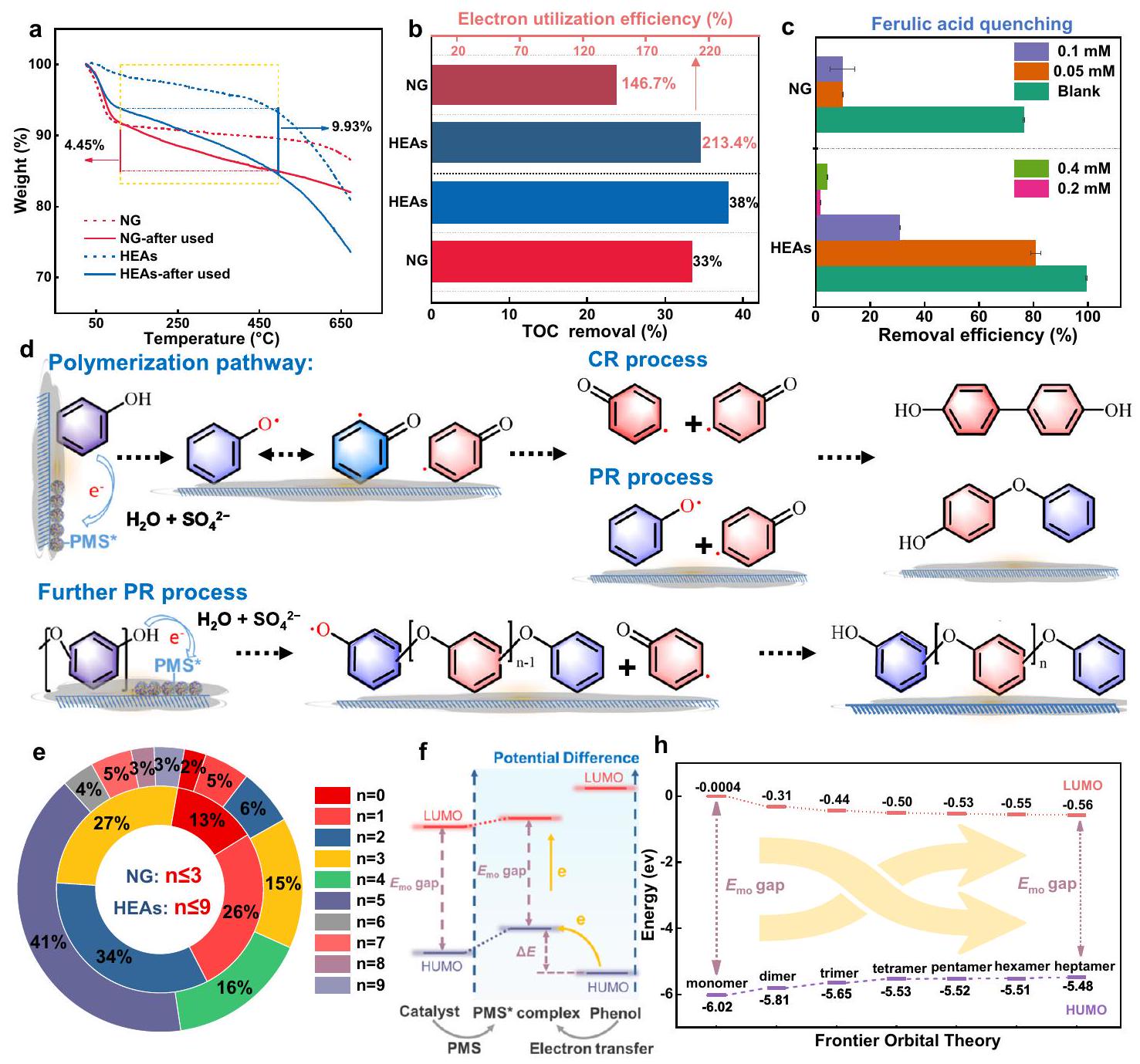
مؤخراً، تم الإبلاغ عن أنظمة تحفيزية تعتمد على أكاسيد المعادن والمواد الكربونية لتحفيز بوليمرية ملوثات عطرية محددة (فينولات وأنيلينات) مع وجود بيرسلفات . من خلال مسار البوليمرية، يتم تحويل الملوثات العضوية من المحاليل المائية إلى منتجات قابلة لإعادة التدوير ترسب على سطح المحفز، مما يغير مسار AOPs من التمعدن إلى البوليمرية. من شأن هذا الانتقال في النظام أن يقلل بشكل ملحوظ من مدخلات البيروكسيد مع كفاءة عالية في إزالة التلوث ويقلل من انبعاثات الكربون في AOPs. هنا، تم إجراء تحليل الوزن الحراري (TGA) لفحص وجود منتجات بوليمرية من الفينول على أسطح هذه الأنظمة. كشفت نتائج TGA عن تحلل حراري أكبر للمنتجات البوليمرية لـ HEAs بعد معالجة AOPs (15.6%) مقارنة بـ HEAs الأصلية وNG، مما يظهر كخسائر في الكتلة قدرها 9.9% و، على التوالي (الشكل 4a والشكل التكميلي 32). اقترحت هذه الملاحظة أن HEAs أظهرت قدرة فائقة على تعزيز التحول البوليمري للملوثات، ناتجة عن الإمكانية الأكثر ملاءمة لمجمع PMS* وقدرة نقل الإلكترونات الأسرع لتحويل الملوثات العضوية بسرعة إلى جذر أحادي عبر سحب الإلكترون لبدء تفاعل سلسلة البوليمرية.
بالإضافة إلى ذلك، تم تحليل كفاءة إزالة الكربون العضوي الكلي (TOC) لأنظمة HEAs-PMS وNG-PMS لتسليط الضوء على التباين في مكافئات الإلكترون الملاحظة خلال تمعدن الفينول (الجدول التكميلي 6). كشفت النتائج أنه بينما كانت المكافئات النظرية القصوى للإلكترون التي حصلت عليها PMS (0.5 مليمول) أقل بكثير من المكافئات الفعلية المفقودة خلال تمعدن الفينول . يمكن أن يُعزى هذا التباين إلى الأكسدة الانتقائية للفينول عبر نقل إلكترون واحد في مسار البوليمرية (PMS:فينول=1:2)، بدلاً من عملية التمعدن، التي تتطلب استهلاك PMS نظري أعلى بكثير (PMS:فينول = 14:1). مستفيدًا من هذه المسار الأكسدي، أظهرت نظام HEAs-PMS () كفاءة أعلى في استخدام الإلكترونات مقارنة بنظام NG-PMS ()، مما يقلل بشكل كبير من استهلاك المؤكسدات في التطبيقات العملية مقارنة بتحفيز التمعدن المتجانس (الشكل 4b).
بعد ذلك، استخدمنا BHT وحمض الفيروليك (FA) كمثبط للبوليمرية لقمع نشاط البوليمرية عبر حجب الجذر الفينوكسي الوسيط. أدى إضافة BHT(2,4,6-tri-tertbutylphenol) إلى تقليل ملحوظ في إزالة الفينول،
الشكل 4 | خصائص منتجات البلمرة. أ منحنيات TGA لـ HEAs و NG قبل وبعد التفاعل. ب كفاءة إزالة TOC النهائية لأنظمة المحاليل المائية HEAs-PMS و NG-PMS، واستخدام الإلكترون. ج تأثير تركيزات مختلفة من FA على كفاءة إزالة الفينول في أنظمة NG-PMS و HEAs-PMS (تمثل أشرطة الخطأ الانحرافات المعيارية من اختبارات ثلاثية). د مسارات التفاعل المقترحة للربط الأكسيدي و بلمرة الفينول على سطح المحفز. توزيع منتجات درجات بلمرة الوحدات المختلفة على سطح محفزات NG و HEAs. رسم تخطيطي لنظرية المدارات الحدودية لبلمرة الفينول الحفازة على سطح المحفز. الرسوم البيانية للمدارات الحدودية المحسوبة لوحدات بلمرة الفينول بدرجات مختلفة. الجرعة: [الفينول]محلول التفاعل: 50 مل، العامل المساعد:. تظهر وجود عمليات البلمرة في كل من أنظمة HEAs-PMS و NG-PMS (الشكل التكميلي 33). يمكن لـ FA تحويل الجذور الفينوكسي إلى سلف الفينول غير النشط من حيث الأكسدة والاختزال، مما يجعله مخلصًا أكثر دقة لجذور الفينوكسي (الشكل 4c والشكل التكميلي 34). تظهر النتائج أن حوالي 0.05 مللي مول من FA تمنع تمامًا إزالة الفينول في نظام NG-PMS، بينما في نظام HEAs-PMS، زادت درجة التثبيط مع زيادة تركيز FA، مما أدى في النهاية إلى تثبيط كامل (أعلى من 0.2 مللي مول). وبالتالي، تدعم هذه النتائج بقوة أن الإمكانية الأعلى لمركب PMS* في نظام HEAs-PMS (مقارنةً بـ NG-PMS) ستؤدي إلى توليد أسرع وتراكم لجذور الفينوكسي، مما يسرع عملية البلمرة.
تم إجراء تحليل لل intermediates / المنتجات للتحقيق في مسارات تحويل الفينول في كلا النظامين. تم استخدام جهاز الكروماتوغرافيا السائلة عالية الأداء – مطياف الكتلة (UPLC-QTOF-MS) لفحص المنتجات الوسيطة للفينول في الطور المائي والممتصة على أسطح المحفز.استخدمنا النوكليوفيل الأسيتونيتريل كعامل انتزاع، مما استبعد عملية نقل الإلكترون المزدوج التي تولد أيون الفينوكسيونيوم كوسيط (الشكل التوضيحي التكميلي 35). وبالتالي، الفينول في المحلول يتبرع أولاً بإلكترون واحد إلىمعقد لتشكيل جذر الفينوكسي مع حالات رنين مختلفة (الشكل 4d). تعمل هذه الجذور الفينوكسي كمواد سابقة للمنتجات الأوليغومرية في تفاعلات السلسلة الجذرية، وتستمر في الخضوع لـالاقتران لتوليد منتجات ثنائية مع تنسيق مختلف (نفس ).
من الجدير بالذكر أن الديمرات ومنتجات أخرى قد تفقد أيضًا إلكترونات لتشكيل جذر عضوي أكبر، مما يزيد من درجة البلمرة من خلال اقتران جذر الديمر أو جذر بوليمر آخر لتشكيل بوليمرات عالية الجزيئية. تشارك البوليمرات الناتجة بشكل متكرر في تفاعلات الاقتران المؤكسد، مما يوفر فرصًا لاقتران أي جذرين بوليمريين معًا، وبالتالي زيادة درجة البلمرة مما يؤدي إلى كمية كبيرة من البوليمرات عالية الوزن الجزيئي الملتصقة بسطح المحفز.في الأشكال التكميلية 36 و37، كشفت التحليلات الإضافية لمنتجات البلمرة على سطح المحفز في أنظمة HEAs-PMS وNG-PMS عن وجود منتجات بوليفينول بدرجة بلمرة أعلى على سطح HEAs. )، بينما تم الكشف عن منتجات بلمرة ذات وزن جزيئي منخفض فقط على سطح NG ( ، ). مزيد من التحليل لاستخراج وتركيز المحلول الكتلي
الشكل 5 | تقييم قابلية التكيف والاستقرار للسبائك عالية الانتشار. أ. حيوية الخلايا لـ 16 HBE تحت تركيزات مختلفة من السبائك عالية الانتشار ووقت زراعة الخلايا: 48 ساعة. تمت ملاحظة خلايا في وسط الثقافة تحت المجهر. معالجة الفينول في مصفوفات المياه الحقيقية. طيف EEM لمياه الصرف الصحي من صناعة الفحم الكيميائية المجمعة قبل (د) وبعد المعالجة (هـ) بواسطة نظام HEAs-PMS. رسم تخطيطي لـ نظام التدفق المستمر لعمود السرير الثابت.كفاءة إزالة الفينول من HEAs في التجارب الدورية. كفاءة إزالة الفينول وتسرب المعادن من السبائك عالية الأداء في نظام تدفق مستمر. تمثل أعمدة الخطأ الانحراف المعياري، الذي تم الحصول عليه من تكرار التجربة ثلاث مرات. الجرعة: [الفينول] : 0.1 مللي مول ، PMS: 0.25 مللي مول ، محلول التفاعل: 50 مل ، المحفز: . من أنظمة HEAs-PMS و NG-PMS كشفت عن اكتشاف منتجات تتراوح بين ثنائيات إلى رباعيات. )، بالإضافة إلى المنتجات الناتجة عن فتح الحلقة المؤكسدة جزئيًا، مما يشير إلى أن المنتجات ذات درجة البوليمرة المنخفضة قد ذابت في المحلول (الأشكال التكميلية 38-44).
علاوة على ذلك، عند استنفاد مصدر الإلكترون (PMS)، يمكن أن يؤدي الربط غير الكافي للجذور البوليمرية إلى إنهاء تفاعلات البلمرة. هذه النتيجة تتماشى مع نتائج تجربة TGA التي أظهرت أن HEAs تتمتع بكفاءة أعلى في تحويل الملوثات إلى بوليمر. في الشكل 4e، أظهر تحليل توزيع درجة البلمرة على أسطح NG وHEAs أن التترايمرات ( ) و خماسيات ( ) تُلاحظ بشكل رئيسي على سطح NG، بينما معظم السداسيات ( ) إلى نونامرات ( ) موجودة على سطح HEAs. بالمقابل، يتم إذابة المنتجات ذات درجة البوليمرة المنخفضة في الطور الحلولي. وبالتالي، فإن الجهد الكهروكيميائي الأعلى لـ HEAs-PMS* وقدرة نقل الإلكترون الأسرع يمكن أن تحفيز التوليد السريع وتراكم الجذور الحرة الفينوكسي، مما يعزز حركية تحويل الفينول إلى بوليمر لإنتاج منتجات بوليفينول أكبر.
تم إجراء تحقق إضافي من الآلية المذكورة أعلاه من خلال نظرية المدارات الحدودية، كما هو موضح في الشكل 5f و h والشكل التكميلي 45)، حيث تتناقص الفجوات الطاقية بين أعلى مدار جزيئي مشغول (HOMO) وأدنى مدار جزيئي غير مشغول للبوليفينول مع أحجام جزيئية مختلفة تدريجياً مع زيادة درجة البوليمرة. وهذا يشير إلى أن المتفاعلات ذات درجات البوليمرة الأعلى لديها HOMO أعلى وفجوة طاقية أقل. ) لفقدان الإلكترونات، مما يجعلها أكثر سهولة في الأكسدة إلى جذر الفينوكسي وتشكيل المنتجات البوليمرية المقابلة. بالإضافة إلى ذلك، تشير الحواجز الطاقية لتكوين الجذور العضوية في المنتجات ذات درجات البلمرة المختلفة إلى أنه مع زيادة درجة البلمرة، تصبح التفاعل أكثر ملاءمة من الناحية الديناميكية الحرارية (الشكل التوضيحي 46).
لفهم أفضل لماذا حققت HEAs درجات أعلى من البلمرة، تم بلمرة منتجات الفينول البوليمرية بمقادير مختلفة على أسطح HEAs وNG باستخدام تفاعل بلمرة محفز بواسطة PMS. كما هو موضح في الشكل التكميلي 47، بعد إدخال كميات مختلفة من منتجات بلمرة الفينول على HEAs، استمرت هذه الأسطح في تنشيط PMS بسرعة، مما أدى إلى مزيد من بلمرة الفينول وإنتاج منتجات ذات وزن جزيئي عالٍ. في المقابل، فقدت NG بسرعة قدرتها على تنشيط PMS. هذه الظاهرة توضح بشكل أكبر أن التركيب العطري المترافق والتفاعلات بين جزيئات فان دير فالز في الجرافيت المدعوم بالنيتروجين تمتص وتغني منتجات البلمرة بشكل تفضيلي، والتي تتشكل بشكل أساسي على سطح الجرافيت. في الوقت نفسه، يسمح فصل المواقع النشطة بتنشيط PMS على جزيئات HEA، مما يدعم عملية البلمرة التي تتوسطها ETP. على النقيض من ذلك، غطت منتجات البلمرة في NG بشكل عشوائي المواقع التفاعلية لتنشيط PMS، مما أدى إلى فقدان سريع للنشاط التحفيزي. في الوقت نفسه، أظهرت تجارب OCPT في الشكل 3c أن HEAs حافظت على إمكانات معقدة أعلى من NG مع وجود الفينول، مما يسمح باستمرار عملية البلمرة. تشير هذه الاستنتاجات بوضوح إلى أن فصل المواقع النشطة لـ PMS والملوثات العضوية في HEAs يمكّن من تنشيط PMS المستمر، مما يولد إمكانات أعلى ويحافظ على عملية البلمرة التي تتوسطها ETP. في الوقت نفسه، يمتص واجهة الجرافيت باستمرار، ويتركز، ويستقر البوليمرات، مما يؤدي إلى تشكيل منتجات بوليفينول أكبر مع درجة أعلى من البلمرة مقارنة بـ NG.
اختبار شمولية السمية البيولوجية واستقرار التطبيق
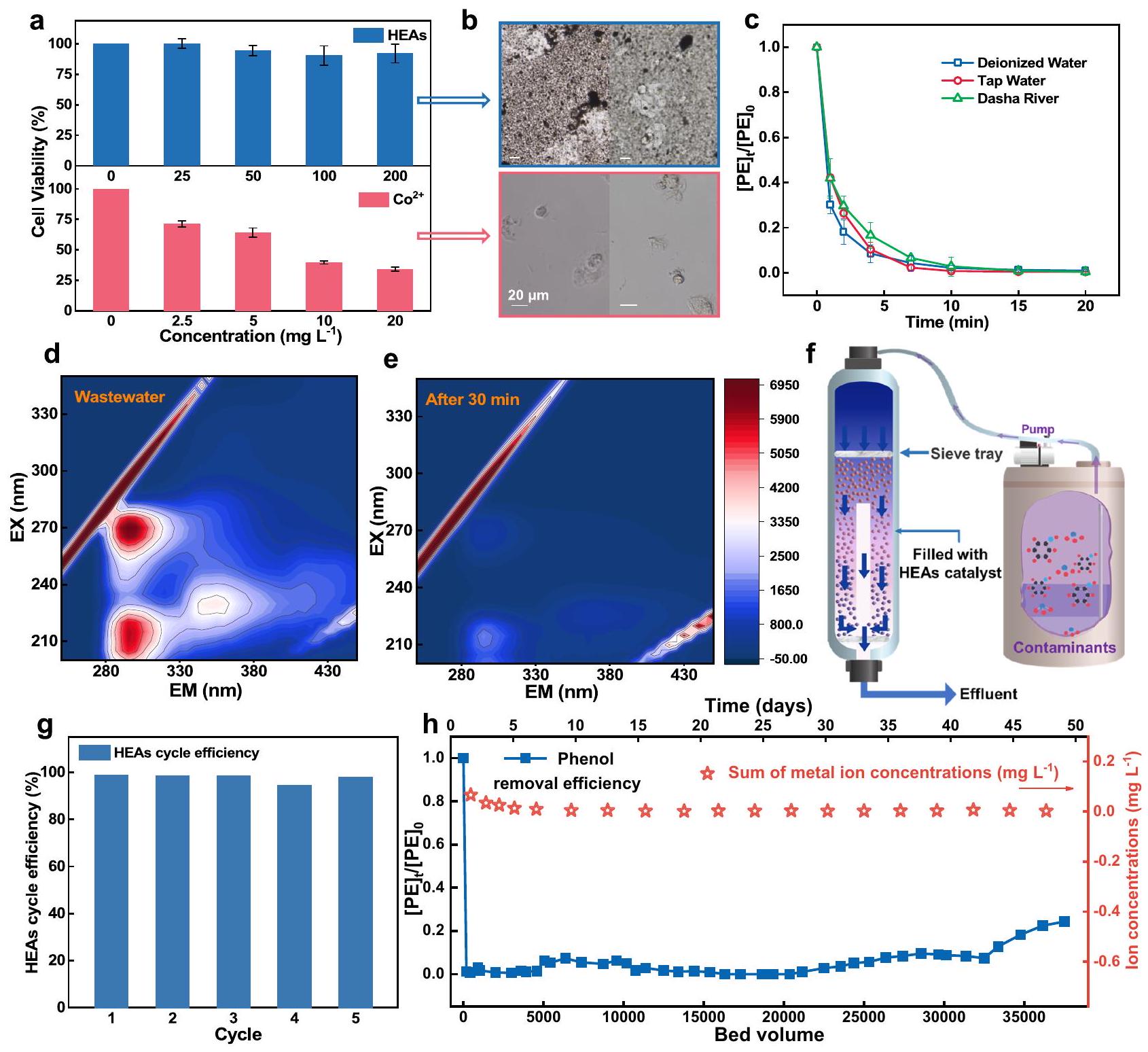
عند تقييم ملاءمة المواد عالية الكفاءة كعوامل حفازة لمعالجة المياه، فإن فهم سمّيتها البيولوجية أمر بالغ الأهمية. لتقييم خطر التعرض أثناء الاستخدام العملي للعوامل الحفازة، قمنا بإجراء تجارب سمّية خلوية في المختبر على خلايا الظهارة الشعب الهوائية البشرية.كما هو موضح في الشكل 5a، تكشف تجارب اختبار CTL عن سمية بيولوجية تعتمد على الجرعة لـوأيونات مختلطة، مع انخفاض قابلية الخلايا للحياة إلى ما دون و عند تركيزات من لـ و لتركيز الأيونات المختلطة (ما يعادل تقريبًا أداء HEAs) (الشكل التكميلي 48). ومن الجدير بالذكر أن إضافة HEAs أسفرت عن نتائج واعدة، حيث حافظت على بقاء خط خلايا 16 HBE فوقحتى مع ضعف التركيز. تم التأكيد على هذا التباين بين HEAs والأيونات بشكل أكبر في الشكل 5b، حيث كان هناك فرق ملحوظ في عدد الخلايا وشكل البقاء في المجال المجهري. بالإضافة إلى ذلك، تم تقييم سمية المنتجات البوليمرية باستخدام أداة تقدير السمية، التي أظهرت زيادة طفيفة في السمية مع زيادة درجة البلمرة. تسلط هذه النتيجة الضوء على أهمية تطوير تقنيات استرداد فعالة لتقليل الآثار البيئية المحتملة وتعزيز إعادة استخدام المنتجات البوليمرية المعاد تدويرها. (الشكل التكميلي 49)
علاوة على ذلك، في تجارب مصفوفات المياه العملية، يظهر نظام HEAs-PMS قدرة ممتازة على مقاومة التداخل في البيئات ذات مصفوفات المياه الأكثر تعقيدًا (الشكل 5c). تم استخدام مياه الصرف الثانوية المجمعة من صناعة الكيماويات الفحمية لتقييم جدوى نظام HEAs-PMS. تشير طيف مصفوفة انبعاث الإثارة (EEM) الموضحة في الشكل 5d و e إلى أن المنتجات الثانوية الميكروبية القابلة للذوبان والمركبات العضوية الشبيهة بحمض الهيوميك الفينولي في عينات مياه الصرف كانت سهلة الإزالة، كما يتضح من الانخفاض الكبير في شدة الفلورية عند القمم المكتشفة.
كما أظهر المحفز استقرارًا ممتازًا في الإزالة الدورية للفينول، حيث حقق إزالة بنسبة 95% من الفينول في خمس دورات (الشكل 5g والشكل التكميلي 50). بالمقابل، قدمت تجارب التشغيل المستمر على المدى الطويل تقييمًا أكثر شمولاً لأداء المحفز في مفاعل السرير الثابت لتفكيك الملوثات العضوية (الشكل 5f).. نتائج تجربة التحلل على المدى الطويل التي استمرت 50 يومًا في ظل ظروف أظهر الفينول أن أداء التحلل لنظام HEAs-PMS انخفض تدريجياً فقط بعد معالجة محلول الفينول بحجم سرير يبلغ 30,000 (الشكل 5h والشكل التكميلي 51). بالإضافة إلى ذلك، فإن تسرب أيونات المعادن المكتشفة يكاد يكون صفراً (الجدول التكميلي 7)، مما يثبت بشكل أكبر الاستقرار الهيكلي القوي لـ HEAs وأداء الإزالة المستدام في التشغيل على المدى الطويل..
نقاش
باختصار، قمنا بتخليق وتطبيق HEAs بنجاح لإزالة الملوثات المائية في نظام PMS-AOPs. بالمقارنة مع NG، فإن HEAs المدعومة أكثر فعالية في تحفيز تنشيط PMS لتحقيق إزالة سريعة للفينول (PMS:فينول=2.5:1)، حيث تظهر استقرارًا استثنائيًا حتى في ظروف pH القاسية ومناعة للتداخل من المواد الأيونية في خلفية الماء. أظهرت HEAs نشاطًا تحفيزيًا مستقرًا وطويل الأمد في تجارب طويلة الأمد استمرت 50 يومًا دون أي تسرب ملحوظ للمعادن. حققت جزيئات HEAs نانو كفاءة عالية في تنشيط PMS وتوصيلية ممتازة، مما أدى إلى تعزيز إمكانيات مركب PMS* وزيادة معدلات نقل الإلكترون. نتيجة لذلك، أدى نظام ETP غير الجذري إلى تحفيز إزالة الفينول غير المعدني من خلال أكسدة إلكترون واحد وحقق كفاءة استخدام إلكترون عالية تصل إلى، مما يؤدي إلى الإنتاج المستدام لمنتجات بوليفينولية عالية الوزن الجزيئي. بالإضافة إلى ذلك، نظرًا لكون HEAs صديقة للبيئة وذات سمية حيوية منخفضة، فإن هذه الدراسة تقدم منظورًا لتصميم محفزات غير متجانسة بدقة ذرية لمعالجة مياه الصرف الصناعي مع استهلاك منخفض للأكسدة وانبعاثات كربونية.
طرق
تحضير المحفزات
في تخليق شائع للسبائك عالية الانتشار،، الجلوكوز، وتم إذابة CTAC (50 ملغ) في 10 مل من أوليامين، تلا ذلك معالجة بالموجات فوق الصوتية لمدة 30 دقيقة. ثم تم تسخين المزيج في حمام زيت عندلمدة ساعتين. تم جمع المنتج المبرد عن طريق الطرد المركزي وغسله ثلاث مرات بـنسبة حجم خليط الإيثانول/السيكلوهكسان. أخيرًا، تم توزيع المنتجات الغروية السوداء في السيكلوهكسان،تمت الإضافة، تلتها عملية الصوتنة لمدة 60 دقيقة، ثم الطرد المركزي، وغسل ثلاث مرات بمزيج من الإيثانول/السيكلوهكسان. بعد التجفيف تحت الفراغ عندلمدة 12 ساعة، تم إضافة 500 ملغ من الجلوكوز وطحنه جيدًا حتى تم تحقيق تشتت متجانس. ثم تم نقل الخليط إلى فرن أنبوبي وتم تلدينه عندلمدة ساعتين بمعدل تسخين قدرهتحت جو من الأرجون الحامي. بعد ذلك، تم إزالته بسرعة وتركه ليبرد إلى درجة حرارة الغرفة للحصول على السبائك عالية الانتشار. بالنسبة لتخليق NC، فإن جميع الظروف مشابهة لتلك الخاصة بالسبائك عالية الانتشار باستثناء غياب المنتجات الغروية السوداء خلال عملية التلدين.
توصيف المحفز
تم استخدام تقنية المجهر الإلكتروني الناقل الميداني ذو الزاوية العالية والحقل المظلم (HAADF-STEM) باستخدام مجهر إلكتروني Titan Themis 80-200 (شركة FEI، هولندا) لمراقبة الشكل والبنية للمحفزات ورسم الخرائط العنصرية. تم تحديد الأطوار البلورية للمحفزات من خلال أنماط حيود الأشعة السينية باستخدام إشعاع CuK-α (SmartLab 3kw، شركة Rigaku). تم إجراء التقاط الإلكترونات بواسطة هيدروكلوريد 2،2،6،6-تترا ميثيل-4-بيبيريدون (TEMP) واحتجاز الجذور بواسطة 5،5-ثنائي ميثيل-1-بيرولين N-أكسيد (DMPO) على مطياف ESR5000 (شركة Bruker). تم إجراء تجارب مطيافية الانبعاث الضوئي للبلازما المقترنة بالحث (ICP-OES) باستخدام جهاز Avio 550 Max (شركة Perkin Elmer) لتحديد محتوى المعادن في المحفزات وتسرب أيونات المعادن أثناء التحلل. تم تقييم كفاءة تحلل الفينول باستخدام محلل الكربون العضوي الكلي (TOC) (Multi N/C 3100، شركة Analytik Jena)، بينما تم تحديد وسائط الفينول من خلال الكروماتوغرافيا السائلة عالية الأداء للغاية مع رباعي القطب. تم قياس الكتلة الأولية للرحلة باستخدام مطياف الكتلة عالي الدقة (UPLC-QTOF-MS، Waters Xevo G2XS QTOF). علاوة على ذلك، تم تحديد تركيز PMS باستخدام مطياف الأشعة فوق البنفسجية والمرئية (AQ8100، Thermo Orion Co.) من خلال طريقة اليوديد البوتاسيوم.
اختبارات التحليل الكهروكيميائي
تم عرض طرق التحليل الكهروكيميائي مثل الفولتمترية ذات المسح الخطي (LSV)، والفولتمترية الدورية (CV)، والكرونوأمبيرومترية (CA)، وطيف الامتزاز الكهروكيميائي (EIS)، والمساحة السطحية النشطة كهروكيميائيًا (ECSA)، وإمكانات الدائرة المفتوحة (OCPT) في المعلومات التكميلية بواسطة محطة عمل كهروكيميائية (CHI 760E).
قياسات XAFS وتحليل EXAFS
تم تحديد الأنواع الكيميائية للنحاس والنيكل بواسطة XANES عند حافة K. تم الحصول على طيف XANES للنحاس والنيكل في مسحوق HEAs باستخدام نموذج النقل من خلال خط الشعاع MEX-1 في منشأة الإشعاع السنكروتروني الأسترالية. تم اختيار رقائق النحاس، CuO، CuPc، رقائق النيكل، NiO، وNiPc كمرجع. تم خلط عينات المرجع المقابلة مع السليلوز وقياسها في وضع النقل. تم استخدام رقاقة الحديد للمعايرة. تم معالجة بيانات EXAFS المكتسبة وفقًا للإجراءات القياسية باستخدام وحدة ATHENA (الإصدار 0.9.26) المدمجة في حزم برامج IFEFFIT للمعايرة الخلفية، وخط ما قبل الحافة، وخط ما بعد الحافة. بعد ذلك،-مرجحالبيانات في نطاق R، تمتد من 0 إلىتم تحويلها باستخدام تحويل فورييه إلى الفضاء الحقيقي (R) باستخدام معلمات دالة مورلي كابا مورلي وسيغما مورليت ، بمساعدة hama_fortran. تم إجراء هذا التحويل لتمييز مساهمات EXAFS من قذائف التنسيق المختلفة.
دراسات الحسابات النظرية
تستخدم هذه الحسابات وحدة حزمة الطاقة الكلية التسلسلية من كامبريدج ضمن برنامج MaterialsStudio 20.1. الدالة المستخدمة لتبادل وتفاعل الإلكترونات هي دالة بيردو-بورك-إرنزرهوف تحت التقريب العام للمتدرجات. يتم استخدام طريقة المجال الذاتي المتسق (SCF) لحل معادلات كوهين-شام. تم تعيين معيار التقارب لطاقة SCF إلى /ذرة. خطأ الطاقة الكلي في النظام ضمن الذرة، انحراف الإجهاد أقل من 0.02 جيجا باسكال، ومعيار التقارب لقوى الذرات هو تُستخدم شبكات مونكهورست-باك لمنطقة بريلوان-نقطة أخذ العينات، معتم اختيار شبكة من النقاط لإجراء الحسابات. تم تعيين طاقة القطع لمجموعة الأساس الموجي المسطح إلى 440 إلكترون فولت. يتم استخدام الزيف الناعم لوصف التفاعل بين النوى الأيونية والإلكترونات التكافؤية.
ملخص التقرير
معلومات إضافية حول تصميم البحث متاحة في ملخص تقارير مجموعة نيتشر المرتبط بهذه المقالة.
توفر البيانات
البيانات التي تدعم نتائج الدراسة مدرجة في النص الرئيسي وملفات المعلومات التكميلية. البيانات الإضافية متاحة من المؤلف المراسل عند الطلب. يتم توفير بيانات المصدر مع هذه الورقة.
References
Dong, C. et al. Dual-functional single-atomic Mo/Fe clustersdecorated via three electron-pathway in oxygen reduction reaction for tandemly removing contaminants from water. Proc. Natl. Acad. Sci. USA 120, e2305883120 (2023).
Yu, F. et al. Rapid self-heating synthesis of Fe-based nanomaterial catalyst for advanced oxidation. Nat. Commun. 14, 4975 (2023).
Gao, Y. et al. Subtle tuning of nanodefects actuates highly efficient electrocatalytic oxidation. Nat. Commun. 14, 2059 (2023).
Liang, X. et al. Coordination number dependent catalytic activity of single-atom cobalt catalysts for fenton-like reaction. Adv. Funct. Mater. 32, 2203001 (2022).
Chen, F. et al. Single-atom iron anchored tubular catalysts for ultrafast fenton-like reaction: roles of high-valency iron-oxo species and organic radicals. Adv. Mater. 34, 2202891 (2022).
Zhao, Z. et al. Improved electronic structure from spin-state reconstruction of a heteronuclear diatomic pair to boost the fenton-like reaction. Environ. Sci. Technol. 57, 4556-4567 (2023).
Chen, Z. et al. Single-atom Mo-Co catalyst with low biotoxicity for sustainable degradation of high-ionization-potential organic pollutants. Proc. Natl. Acad. Sci. USA 120, e2305933120 (2023).
Jiang, Y. et al. In situ turning defects of exfoliated MXene into Fenton-like catalytic active sites. Proc. Natl. Acad. Sci. USA 120, e2210211120 (2023).
Guo, Z.-Y. et al. Electron delocalization triggers nonradical Fentonlike catalysis over spinel oxides. Proc. Natl. Acad. Sci. USA 119, e2201607119 (2022).
He, Z. et al. perovskite for efficient sulfafurazole degradation via peroxymonosulfate activation: catalytic mechanism of interfacial structure. Appl. Catal. B Environ. 335, 122883 (2023).
Sun, Y., Li, M., Duan, J., Antonietti, M. & Chen, S. Entropy-driven direct air electrofixation. Angew. Chem. Int. Ed. 63, e202402678 (2024).
Wang, J., Zhang, J., Hu, Y., Jiang, H. & Li, C. Activating multisite highentropy alloy nanocrystals via enriching M-pyridinic N-C bonds for superior electrocatalytic hydrogen evolution. Sci. Bull. 67, 1890-1897 (2022).
Ren, J.-T., Chen, L., Wang, H.-Y. & Yuan, Z.-Y. High-entropy alloys in electrocatalysis: from fundamentals to applications. Chem. Soc. Rev. 52, 8319-8373 (2023).
Wu, D. et al. Noble-metal high-entropy-alloy nanoparticles: atomiclevel insight into the electronic structure. J. Am. Chem. Soc. 144, 3365-3369 (2022).
Kang, Y. et al. Mesoporous multimetallic nanospheres with exposed highly entropic alloy sites. Nat. Commun. 14, 4182 (2023).
Men, Y. et al. Understanding alkaline hydrogen oxidation reaction on PdNiRulrRh high-entropy-alloy by machine learning potential. Angew. Chem. Int. Ed. 62, e202217976 (2023).
Li, K. & Chen, W. Recent progress in high-entropy alloys for catalysts: synthesis, applications, and prospects. Mater. Today Energy 20, 100638 (2021).
Zou, X. et al. High-entropy engineering with regulated defect structure and electron interaction tuning active sites for trifunctional electrocatalysis. Proc. Natl. Acad. Sci. USA 121, e2313239121 (2024).
Cui, X. et al. Rapid high-temperature liquid shock synthesis of highentropy alloys for hydrogen evolution reaction. ACS Nano 18, 2948-2957 (2024).
Li, B. et al. Boosting catalytic decomposition by the synergistic effect of multiple elements in cobalt-based high-entropy oxides. Environ. Sci. Technol. 58, 2153-2161 (2024).
Wang, Y. et al. Supported high-entropy alloys for electrooxidation of benzyl alcohol assisted water electrolysis. Adv. Funct. Mater. 34, 2311611 (2024).
Li, H. et al. Fast site-to-site electron transfer of high-entropy alloy nanocatalyst driving redox electrocatalysis. Nat. Commun. 11, 5437 (2020).
Li, M. et al. Potential-dependent selectivity for the efficient capture of gold from E-waste acid leachate using sulfhydryl-functionalized carbon. Sci. Bull. 68, 1095-1099 (2023).
Zhan, C. et al. Medium/high-entropy amalgamated core/shell nanoplate achieves efficient formic acid catalysis for direct formic acid fuel cell. Angew. Chem. Int. Ed. 62, e202213783 (2023).
Chen, W. et al. High-entropy intermetallic PtRhBiSnSb nanoplates for highly efficient alcohol oxidation electrocatalysis. Adv. Mater. 34, 2206276 (2022).
Li, M. et al. Deep separation between In(III) and Fe(III) lons by regulating the lewis basicity of adsorption sites on electrospun fibers. Adv. Funct. Mater. 34, 2313443 (2024).
Shao, P. et al. Revisiting the graphitized nanodiamond-mediated activation of peroxymonosulfate: singlet oxygenation versus electron transfer. Environ. Sci. Technol. 55, 16078-16087 (2021).
Ren, W. et al. Origins of electron-transfer regime in persulfatebased nonradical oxidation processes. Environ. Sci. Technol. 56, 78-97 (2022).
Zhao, Z. et al. Construction of dual active sites on diatomic metal (FeCo-N/C-x) catalysts for enhanced Fenton-like catalysis. Appl. Catal. B Environ. 309, 121256 (2022).
Wang, Z. et al. Cobalt single atoms anchored on oxygen-doped tubular carbon nitride for efficient peroxymonosulfate activation: simultaneous coordination structure and morphology modulation. Angew. Chem. Int. Ed. 61, e202202338 (2022).
Yang, J. et al. Insights into the role of dual reaction sites for single Ni atom Fenton-like catalyst towards degradation of various organic contaminants. J. Hazard. Mater. 430, 128463 (2022).
Shao, H. et al. Naproxen as a turn-on chemiluminescent probe for real-time quantification of sulfate radicals. Environ. Sci. Technol. 57, 8818-8827 (2023).
Gong, Y. et al. Whose oxygen atom is transferred to the products? A case study of peracetic acid activation via complexed for organic contaminant degradation. Environ. Sci. Technol. 57, 6723-6732 (2023).
Zhang, Q. et al. Mineralization versus polymerization pathways in heterogeneous Fenton-like reactions. Water Res. 249, 120931 (2024).
Deng, G. et al. Ferryl ion in the photo-fenton process at acidic pH: occurrence, fate, and implications. Environ. Sci. Technol. 57, 18586-18596 (2023).
Zhang, B., Li, X., Akiyama, K., Bingham, P. A. & Kubuki, S. Elucidating the mechanistic origin of a spin state-dependent Catalyst toward organic contaminant oxidation via peroxymonosulfate activation. Environ. Sci. Technol. 56, 1321-1330 (2022).
Liu, H.-Z. et al. Tailoring d-band center of high-valent metal-oxo species for pollutant removal via complete polymerization. Nat. Commun. 15, 2327 (2024).
Zhang, Y.-J. et al. Simultaneous nanocatalytic surface activation of pollutants and oxidants for highly efficient water decontamination. Nat. Commun. 13, 3005 (2022).
Chen, Y. et al. Transformative removal of aqueous micropollutants into polymeric products by advanced oxidation processes. Environ. Sci. Technol. 58, 4844-4851 (2024).
Peng, J. et al. Removal of phenols by highly active periodate on carbon nanotubes: a mechanistic investigation. Environ. Sci. Technol. 57, 10804-10815 (2023).
Zhang, Y.-J. et al. Distinguishing homogeneous advanced oxidation processes in bulk water from heterogeneous surface reactions in organic oxidation. Proc. Natl. Acad. Sci. USA 120, e2302407120 (2023).
Yao, Z. et al. Thiol-rich, porous carbon for the efficient capture of silver: understanding the relationship between the surface groups and transformation pathways of silver. Chem. Eng. J. 427, 131470 (2022).
Shang, Y., Xu, X., Gao, B., Wang, S. & Duan, X. Single-atom catalysis in advanced oxidation processes for environmental remediation. Chem. Soc. Rev. 50, 5281-5322 (2021).
شكر وتقدير
نحن نعترف بالدعم المالي المقدم من المؤسسة الوطنية للعلوم الطبيعية في الصين (رقم 52321005)، وزمالة برنامج البحث والتطوير في المناطق الرئيسية لمقاطعة قوانغدونغ (رقم 2023B0101200004)، وبرنامج الابتكار العلمي والتكنولوجي في شنتشن (رقم GXWD2O231129122140001، KQTD20190929172630447)، والمشروع المفتوح للمختبر الوطني الرئيسي لموارد المياه الحضرية والبيئة (معهد هاربين للتكنولوجيا) (رقم QA202440)، ومؤسسة العلوم لمركز البحث الهندسي الوطني للتخلص الآمن واستعادة الموارد من الحمأة (معهد هاربين للتكنولوجيا، منحة رقم K2024B001). كما يشكر X.D. الدعم المالي من مجلس الأبحاث الأسترالي عبر زمالة ARC المستقبلية (FT230100526).
مساهمات المؤلفين
صمم Z.Y. وY.C. وN.R. وX.D. البحث؛ قام Z.Y. بإجراء البحث؛ ساهم Z.Y. وK.H. وZ.J. وZ.S. وS.R. في توفير مواد جديدة/أدوات تحليلية؛ قام Z.Y. وS.R. وX.W. بتحليل البيانات؛ كتب Z.Y. وY.C. وX.D. الورقة.
ملاحظة الناشر: تظل شركة سبرينجر ناتشر محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
الوصول المفتوح هذه المقالة مرخصة بموجب رخصة المشاع الإبداعي النسب 4.0 الدولية، التي تسمح بالاستخدام والمشاركة والتكيف والتوزيع وإعادة الإنتاج بأي وسيلة أو صيغة، طالما أنك تعطي الائتمان المناسب للمؤلفين الأصليين والمصدر، وتوفر رابطًا لرخصة المشاع الإبداعي، وتوضح إذا ما تم إجراء تغييرات. الصور أو المواد الأخرى من طرف ثالث في هذه المقالة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة، ما لم يُشار إلى خلاف ذلك في سطر الائتمان للمواد. إذا لم تكن المادة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة وكان استخدامك المقصود غير مسموح به بموجب اللوائح القانونية أو يتجاوز الاستخدام المسموح به، فسيتعين عليك الحصول على إذن مباشرة من صاحب حقوق الطبع والنشر. لعرض نسخة من هذه الرخصة، قم بزيارةhttp://creativecommons.org/رخصة/بواسطة/4.0/. (ج) التاج 2024
المختبر الوطني الرئيسي لموارد المياه الحضرية والبيئة، مختبر شنتشن الرئيسي للوقاية من التلوث العضوي ومكافحته، كلية الهندسة المدنية والبيئية، معهد هاربين للتكنولوجيا، شنتشن، جمهورية الصين الشعبية.مدرسة الهندسة الكيميائية، جامعة أديلايد، أديلايد، سا، أستراليا.كلية المواد والهندسة البيئية، جامعة شنتشن بوليتكنيك، شنتشن، جمهورية الصين الشعبية. البريد الإلكتروني: chenyidi@hit.edu.cn; xiaoguang.duan@adelaide.edu.au
High-entropy alloys catalyzing polymeric transformation of water pollutants with remarkably improved electron utilization efficiency
Received: 18 September 2024
Accepted: 18 December 2024
Published online: 02 January 2025
Check for updates
Ziwei Yao , Yidi Chen ® , Xiaodan Wang , Kunsheng Hu , Shiying Ren , Jinqiang Zhang , Zhao Song , Nanqi Ren & Xiaoguang Duan
High-entropy alloy nanoparticles (HEA-NPs) exhibit favorable properties in catalytic processes, as their multi-metallic sites ensure both high intrinsic activity and atomic efficiency. However, controlled synthesis of uniform multimetallic ensembles at the atomic level remains challenging. This study successfully loads HEA-NPs onto a nitrogen-doped carbon carrier (HEAs) and pioneers the application in peroxymonosulfate (PMS) activation to drive Fenton-like oxidation. The HEAs-PMS system achieves ultrafast pollutant removal across a wide pH range with strong resistance to real-world water interferences. Furthermore, the nonradical HEAs-PMS system selectively transforms phenolics into high-molecular-weight products via a polymerization pathway. The unique non-mineralization regime remarkably reduces PMS consumption and achieves a high electron utilization efficiency of up to . Further DFT calculations and experimental analysis reveal that Fe and Co in HEA-NPs act as the primary catalytic sites to complex with PMS for activation, while , and Pd serve as charge mediators to facilitate electron transfer. The resulting PMS* complexes on HEAs possess a high redox potential, which drives spatially separated phenol oxidation on nitrogendoped graphene support to form phenoxyl radicals, subsequently triggering the formation of high-molecule polymeric products via polymerization reactions. This study offers engineered HEAs catalysts for water treatment with low oxidant consumption and emissions.
The treatment of organic wastewater is an increasingly critical issue in the long-term development of society, necessitating the development of environmentally friendly, cost-effective, and efficient water purification technologies . Advanced oxidation processes (AOPs) are pivotal in breaking down and mineralizing organic pollutants to
mitigate environmental contamination . To meet the rapidly growing demands, the development of high-performance catalysts with high activity, selectivity, and stability of catalytic reactions is essential. Extensive research has produced a variety of catalysts, such as single atoms, bimetallic single atoms, metal oxides/sulfides, and others .
There is a consensus that the elemental composition and crystal structure of catalysts significantly influence their catalytic performance. Therefore, the development of more advanced catalysts appears imminent, offering extensive opportunities for microelectronic structure fine-tuning and performance optimization .
High-entropy alloys, a distinctive class of multi-metal alloys typically composed of five or more metals, have garnered widespread research interest in recent years and have found extensive applications in energy conversions and green synthesis . The disordered phase of their elements provides inherent advantages, including elemental diversity and structural stability, leading to significantly higher chemical potential and different surface electronic properties compared to traditional single/mixed metal counterparts . In catalysis, the adsorption of molecules and intermediate substances on the catalyst surface influences catalytic activity. Compared to pure elements, these adsorption energies can be regulated through alloying to enhance catalytic activity . Consequently, HEAs have been extensively explored in diverse catalytic reactions such as hydrogen evolution, oxygen evolution, alcohol oxidation, and ammonia oxidation, where these alloys demonstrate superior performance compared to traditional catalysts . However, the primary challenges associated with highentropy alloys are the precise synthesis and characterization, as well as complex reaction mechanisms due to the multicomponent structures.
Herein, to enhance the electronic structure of high-entropy alloys for intermediate adsorption, we report the synthesis of CuPdFeCoNi alloy nanoparticles (NPs) loaded onto nitrogen-containing carbon materials using a combination of wet chemistry and controlled pyrolysis methods. The findings reveal that the modified electronic structure accelerates the activation of peroxymonosulfate (PMS) compared to NG for organic pollutant removal from water. The HEAsPMS system operates through a non-radical electron-transfer pathway (ETP) with high efficiency under harsh pH conditions and resistance to interference from water background factors. Furthermore, a comprehensive analysis of the structure and composition of HEAs was conducted. The associated mechanism was investigated using electron paramagnetic resonance (EPR) and probe experiments, substantiated with electrochemical characterization. To our delight, the HEAinduced non-radical regime yielded a substantial amount of polymerization products on the catalyst surface against mineralization, resulting in an exceptionally high electron utilization efficiency of up to . This is attributed to the higher HEAs-PMS* composite potential and faster electron transfer (fast pollutant oxidation kinetics), coordinated by the uniformly distributed multi-elements in HEAs, producing high-polymerization-degree products. The long-term stability and practicality of HEAs were demonstrated through a 50-day continuous flow and actual wastewater removal efficiency experiment. Overall, this study presents a solution for developing advanced heterogeneous catalysts suitable for water remediation with reduced oxidant consumption and upcycled water pollutants.
Results
Synthesis and characterization of HEAs
A straightforward one-pot oil-phase synthesis approach was adopted by heating five metal salt precursors ( , and Ni ) at for 2 h , followed by thermal decomposition along with a carbon precursor to obtain HEAs . The calculated composition of the obtained HEAs was determined to be through inductively coupled plasma-optical emission spectrometry (ICP-OES) and elemental analysis (Supplementary Tables 1 and 2). The synthesis procedure, depicted in Fig. 1a and detailed in the Experimental section, successfully yielded uniform nanoparticles with a diameter of , as shown in transmission electron microscopy (TEM) images (Fig. 1b and Supplementary Figs. 1 and 2). The powder X-ray diffraction pattern confirmed the face-centered cubic (fcc) crystalline structure of CuPd alloy (JCPDS No. 48-1551) NPs, with
the main phase identified as CuPd alloy by the peaks at around and , corresponding to the (111) and (200) facets (Supplementary Fig. 3) . Additionally, the pronounced shift in the position of the broad diffraction peak, in contrast to the diffraction peaks of pure metals (Pd, , and Cu ), indicates a significant alteration. The subtle peak shift for the CuPd alloy further suggests the incorporation of Fe , Ni , and Co into the CuPd lattice, leading to the creation of a uniform alloy structure with concurrent lattice contraction . High-angle annular dark-field scanning TEM (AC-HAADF-STEM) image in Fig. 1c shows a lattice spacing of 0.217 nm , corresponding to the (111) facet of NPs (Supplementary Fig. 4). Moreover, AC-HAADF-STEM-EDS elemental mappings show that each element is uniformly distributed, demonstrating that the five metals (Pd, Fe, Co, Ni , and Cu ) are evenly positioned on the surface of HEA NPs (Fig. 1d) . These comprehensive characterizations substantiate the successful synthesis and loading of NPs onto N-doped graphene (NG).
To determine the electronic and coordination structures of Cu atoms in HEAs, X-ray absorption near-edge spectroscopy (XANES) and extended X-ray absorption fine structure (EXAFS) were utilized. XANES curve reveals that the white line intensity is higher than Cu foil but much lower than that of CuO and CuPc , confirming that the Cu species in HEAs are mainly present in the metallic state (Fig. 1e) . Moreover, the pre-edge centroid of HEAs is situated between the Cu foil and CuPc , indicating potential coordination between Cu atoms and N in NG. The Fourier-transformed extended X-ray absorption fine structure (FT-EXAFS) spectrum of HEAs in R space displays two dominant peaks located at approximately for the bond and a peak offset at , corresponding to the interaction. This offset indicates the presence of interactions ( ) within the HEAs, which is likely caused by the mutual interaction of multiple elements for HEAs (Fig. 1f). Besides, the wavelet transforms (WT) of the weighted EXAFS spectra of HEAs also reveal two dominating peaks maximum at ascribed to bonding and interactions (Fig. 1g). Strong and coordination can be observed in the WT contour plots of Cu foil and CuPc , which agrees with the results from EXAFS spectra. Additionally, X-ray absorption spectroscopy of Ni atoms in HEAs also yields similar conclusions, despite the dominant Ni M interactions (Supplementary Fig. 5). This further corroborates the earlier characterization findings, confirming the incorporation of Ni into the CuPd alloy.
Superior catalytic efficiency of HEAs
The catalytic capability of HEAs was investigated by using PMS as a peroxide precursor, and the Fenton-like activity was assessed by employing phenol as a model organic contaminant. As shown in Fig. 2a, minimal adsorption occurred in the catalyst-only systems, with the HEAs system achieving 100% phenol removal in 10 min with PMS addition, while the NG system exhibited less than phenol removal. Notably, the degradation rate of HEAs exceeded that of NG by over 7 times (Supplementary Fig. 6), suggesting that the integration of alloy NPs sites into NG could be crucial active sites for PMS activation . Additionally, the optimization of PMS concentration demonstrated that complete degradation was achievable with a PMS dose of 0.25 mM , following a positive correlation of oxidation kinetics with PMS concentration, likely due to its positive feedback effects on PMS activation (Supplementary Fig. 7). The removal rates decreased with increasing initial phenol concentrations, though complete phenol removal was still achieved within 10 min at a concentration of 0.1 mM (Supplementary Fig. 8). However, when the initial phenol concentration exceeded the electron equivalents that PMS could accept , the removal efficiency was significantly reduced.
Cobalt, iron, and other metal ions have been discovered to exhibit catalytic performance as classic activators in Fenton-like reactions, widely utilized for PMS activation. However, the efficiency of
Fig. 1 | Synthesis and characterization of HEAs. a The schematic diagram for synthesis process of HEAs. TEM images of HEAs. Aberration-corrected HAADFSTEM image and the corresponding inverse FFT pattern of HEAs (highlighted in the
yellow circle). d Elemental mapping for HEAs (scale bar, 5 nm ). e XANES at Cu L-edge. FT-EXAFS spectra in R space. WTs of EXAFS spectra of HEAs, foil, and CuPc.
heterogeneous catalysts in comparison to metal ions such as remains limited . To investigate the exceptional performance of HEAs in phenol degradation, experiments were conducted using equimolar concentrations of metal ions ( , and ) as comparative catalysts based on the metal content in HEAs (Supplementary Fig. 9 and Table 3). It was observed that at a PMS concentration of 0.25 mM , only exhibited a significant degradation performance. Further degradation experiments were conducted at higher concentrations using cobalt ions and a mixture of 5 other metal ions. The results in Supplementary Fig. 10 showed that even with a 40 -fold increase in and a 20-fold increase in mixed ions, the phenol removal efficiency still cannot compete with the HEAs-PMS system. The apparent kinetic constant of the HEAs-PMS system was 5.3 times
and 2.7 times higher than that of the homogeneous PMS and mixed ions-PMS systems, respectively. These results further support the advantages of high-entropy alloy NPs-loaded catalysts in terms of high intrinsic activity and low metal leaching in addressing environmental pollution. Furthermore, monitoring the PMS consumption rate via the iodine reduction reaction suggested that the exceptional performance of HEAs could be attributed to remarkably enhanced PMS activation on the alloy NPs surface, achieving a rate 12 times higher than NG (Supplementary Fig. 11) .
The impacts of the co-existing dissolved organic matter (DOM, represented by humic acid) and typical anions species (e.g., , , and ) on phenol removal by HEAs and NG were further investigated. As illustrated in Fig. 2b, DOM at the concentration
Fig. 3 | Identification of active species. a Quenching effects by various scavengers. The error bars represent the standard deviations from triplicate tests. Dosage: [Phenol] , PMS: 0.25 mM , scavengers: 2.5 mM , reaction solution: 50 mL , catalyst: . EPR tests were performed on DMPO and TEMP adducts within the HEAs-PMS system during PMS activation. c The open circuit potential on HEAs -GCE and NG-GCE electrode. d In situ Raman spectra of HEAs-PMS and NG-PMS in
the liquid solution. e Nyquist plots and fitting calculations of internal resistance for HEAs and NG. Tafel slope on the HEAs-GCE and NG-GCE electrodes in phenol solutions. The side view of the structural configuration of PMS on HEAs. The top view of the structural configuration of PMS on HEAs. i The PDOSs of the HEAs. j The site-dependent PDOSs of , and Co in HEAs.
tetramethylpiperidine (TEMP) as a quencher did not affect phenol removal efficiency, signifying the negligible influence of on the catalytic performance (Supplementary Fig. 19). Simultaneously, substituting for deionized water in the solution did not facilitate the oxidation kinetics, and conducting experiments with 9,10-Diphenylanthracene (DPA) as a probe did not generate the featured product from oxidation (Supplementary Figs. 20 and 21)³4. These results
indicated that did not present in the HEAs-PMS and NG-PMS systems. The negligible inhibitive impact of dimethyl sulfoxide as a specific scavenger suggests the absence of high-valence metal-oxo species in the HEAs-PMS system (Supplementary Fig. 22) . These collective findings exclude the contributions of radicals ( or ), , and high-valence metal-oxo species, implying that PMS-triggered ETP are likely involved in phenol removal in a non-radical manner.
Various electrochemical techniques were employed to assess ETPmediated phenol oxidation in the HEAs-PMS and NG-PMS systems, including OCPT, LSV, CA, EIS, Tafel curve, and ECSA analysis . As shown in Fig. 3c, after the injection of PMS, OCPT promptly increased for HEAs-GCE and NG-GCE, indicating the formation of surfaceactivated PMS complex (PMS*) which elevated the catalyst potential. Notably, the potential of HEAs-PMS* exceeded that of NG-PMS*, implying a higher oxidation capacity. Upon phenol addition, a subsequent decrease in OCPT indicated ETP occurs between HEAs/NGPMS* and phenol. Moreover, in situ Raman spectroscopy in Fig. 3d directly evidenced the existence of the PMS* reactive complex on HEAs and NG surfaces.
Further monitoring of PMS content during the OCPT experiment revealed the more efficient interaction of alloy NPs with PMS, giving rise to the higher potential observed for the HEAs-GCE electrode (Supplementary Fig. 23). The CV experiment showed significantly higher current density for phenol oxidation by HEAs than NG, confirming the superior electron transfer capability of alloy NPs from the pollutant to PMS (Supplementary Fig. 24). The proposition was also supported by the CA, current-time and ECSA measurement (Supplementary Figs. 25-27). The conductivity and electrochemical kinetics of the HEAs were further estimated using EIS and the Tafel slope, defining the rate-limiting step (Fig. 3e and Supplementary Fig. 28). The circuit fitting results indicated that the impedance of HEAs ( ) was significantly lower than that of NG ( ), due to the presence of metallic HEAs, which benefit the charge migration processes. Tafel slopes were obtained by fitting LSV data to the Tafel curve, resulting in calculated Tafel slopes of 279 and for HEAs and NG, respectively (Fig. 3f). These disparities suggest that HEAs exhibit faster oxidation kinetics and higher electron transfer capacity at high potentials compared to NG. Galvanic reactor experiments were performed to analyze the mechanism of PMS activation by HEAs/NG as shown in Supplementary Fig. 29. The electrode potential within the PMS chamber was notably higher than that in the phenol chamber, indicating that the PMS chamber served as the cathode chamber, while the phenol solution acted as the anode chamber (Supplementary Fig. 30). Consequently, phenol was directly oxidized through protoncoupled ETP in the galvanic cell reaction and synchronously mediated electron flow from the organic cell to the surface-activated PMS on the other end. The recorded current changes during the galvanic reactions demonstrated that HEAs could generate a significantly larger current compared to NG, underscoring the superior PMS activation (electron acceptance) and phenol oxidation (charge donation) kinetics on HEAsNPs via an ETP regime .
To unravel the superior activity and electron transfer capability of HEAs, the projected density of state (PDOS) for PMS adsorption on HEAs was calculated. Initially, the PDOS of three HEAs with slight stoichiometric differences was compared. As shown in Supplementary Fig. 31, the comparison models displayed highly similar electronic structures with minimal changes in the peak positions and patterns of d orbitals for each element. Therefore, slight variations in HEAs stoichiometry are unlikely to significantly affect the electronic structure in the PDOS results . Subsequently, the HEAs structure with the highest stability was selected as the lattice model (Fig. 3g and h). To further understand the electronic structure, Fig. 3i has illustrated the PDOS of each element in the HEAs. Notably, the Pd-4d orbitals occupied the deepest position near the valence band ( ) at -4.6 eV , serving as an electron reservoir. The Ni-3d orbitals exhibited a peak at Ev -1.0 eV , while the Co-3d orbitals showed a pronounced intersection with high electron density at the Fermi level ( ), supporting the high electroactivity of Ni and Co sites to coordinate surface redox reaction. Additionally, the significant overlap between different d orbitals indicated strong bonding and electronic communication between the metal elements. These strong interactions enabled the coupling of reactants (PMS) with the 3d orbitals of Fe and Co sites, facilitating
flexible electron modulation and accelerating electron transfer on the surface of the HEAs NPs. This signified that the 3d orbitals of Cu, Ni, Co, and Fe also acted as bridging intermediaries, helping to alleviate the energy barrier for ETP during oxidation processes .
To further identify the electronic modulations, the site-dependent PDOSs of each element were supplied in HEAs (Fig. 3j). From bulk to surface sites, an evident downward shift was observed for and Pd-4d orbitals, while Pd sites displayed an alleviation of the splitting effect, supporting an enhanced electron transfer efficiency. For Ni sites closer to the surface, the 3d orbitals showed a slight shift to support the electron transfer activity of the HEAs. Both Fe and Co sites exhibited a relatively stable d-band center, attributed to a dual anchoring effect from the shielding effect of neighboring metal orbitals. This helped maintain the electron boosting center and facilitated coupling with PMS, exhibiting the strong electron-donating capability for the oxidation process. Due to low barriers for electron depletion, , and Ni sites were identified as buffer sites, protecting the metallic and electroactive Cu and Pd sites within HEAs NPs from oxidation. This ensures superior removal performance and stability of HEAs in ETP oxidation. Based the aforementioned results, we inferred that Fe and Co act as the primary reactive sites for binding with PMS, and meanwhile, Ni, Cu, and Pd are mediators that accelerate electron transfer to phenol adsorbed on the surface of nitrogen-doped graphite, thereby enhancing the kinetics of phenol oxidation.
Mechanistic study of polymerization in pollutant removal
Recently, catalytic systems based on metal oxides and carbonaceous materials have been reported to induce polymerization of specific aromatic pollutants (phenolics and anilines) with the presence of persulfates . Through the polymerization pathway, organic pollutants are converted from aqueous solutions into recyclable products deposited onto the catalyst surface, thus altering the AOPs pathway from mineralization to polymerization. Such regime transition would remarkably reduce the peroxide inputs with high decontamination efficiency and minimize the carbon emission in AOPs. Here, thermogravimetric analysis (TGA) was conducted to examine the presence of phenol polymerization products on the surfaces of these systems. The TGA results revealed more significant pyrolysis decomposition of polymeric products for HEAs after the AOPs treatment (15.6%) compared to the pristine HEAs and NG, manifesting as mass losses of 9.9% and , respectively (Fig. 4a and Supplementary Fig. 32). This observation suggested that HEAs exhibited a superior potential to promote polymeric transformation of pollutants, stemming from the more favorable PMS* complex potential and faster electron transfer capability to rapidly convert organic pollutants into monomer radicals via electron abstraction to initiate the polymer chain reaction.
Additionally, the total organic carbon (TOC) removal efficiency of the HEAs-PMS and NG-PMS systems was analyzed to shed light on the discrepancy in electron equivalents observed during phenol mineralization (Supplementary Table 6). The results revealed that while the theoretical maximum electron equivalents obtained by PMS ( 0.5 mM ) were significantly lower than the actual electron equivalents lost during phenol mineralization . This disparity could be attributed to the selective oxidation of phenol via one-electron transfer in the polymerization pathway (PMS:phenol=1:2), rather than the mineralization process, which requires much higher theoretical PMS consumption (PMS:phenol = 14:1). Benefiting from this oxidative pathway, the HEAs-PMS system ( ) demonstrated higher electron utilization efficiency compared to the NG-PMS system ( ), significantly reducing the consumption of oxidants in practical applications compared to homogeneous mineralization catalysis (Fig. 4b).
Subsequently, we use BHT and Ferulic acid (FA) as a polymerization inhibitor to suppress polymerization activity via blocking the phenoxyl radical intermediate. The addition of BHT(2,4,6-tri-tertbutylphenol) resulted in a notable reduction in phenol removal,
Fig. 4 | Characteristics of the polymerization products. a TGA curves of HEAs and NG before and after the reaction. b HEAs-PMS and NG-PMS aqueous solution systems final TOC removal efficiency, and electron utilization. c The effect of different concentrations of FA on the phenol removal efficiency in the NG-PMS and HEAs-PMS systems (The error bars represent the standard deviations from triplicate tests). d Proposed reaction pathways of the oxidative coupling and
polymerization of phenol on the catalyst surface. e The distribution of different units polymerization-degree products on the surface of NG and HEAs catalysts. f Schematic diagram of the frontier orbital theory for catalytic polymerization of phenol on the catalyst surface. h Calculated frontier orbital diagrams for different units polymerization-degree phenol. Dosage: [Phenol] , reaction solution: 50 mL , catalyst: .
demonstrating the presence of polymerization processes in both HEAs-PMS and NG-PMS systems (Supplementary Fig. 33). FA can convert phenoxyl radicals back to redox-inert phenol precursor, making it a more precise phenoxyl radical scavenger (Fig. 4c and Supplementary Fig. 34). The results show that ca. 0.05 mM FA completely inhibit phenol removal in the NG-PMS system, while in the HEAs-PMS system, the inhibition degree increased with FA concentration, eventually reaching complete inhibition (above 0.2 mM ). Thus, these results firmly support that the higher potential of the PMS* complex in the HEAs-PMS system (compared to NG-PMS) would induce faster generation and accumulation of phenoxyl radicals, thereby accelerating the polymerization process.
Analysis of the intermediates/products was conducted to investigate the phenol transformation pathways in both systems. The ultrahigh performance liquid chromatograph-mass spectrometer (UPLC-QTOF-MS) was employed to examine the intermediate products of phenol in the aqueous phase and adsorbed on the catalyst surfaces . We used the nucleophile acetonitrile as a scavenger, which ruled out the double-electron-transfer process generating the phenoxonium ion as the intermediate (Supplementary Fig. 35). Thus,
phenol in the solution first donates one electron to the complex to form phenoxyl radicals with various resonance states (Fig. 4d). These phenoxyl radicals serve as precursors for oligomeric products in the radical chain reactions, further undergoing coupling to generate dimeric products with different coordination (same ).
Notably, dimers and other products may also lose electrons to form larger organic radicals, increasing the polymerization degree via coupling dimer radicals or other polymer radicals to form highmolecule polymers. The generated polymers repeatedly participate in oxidative coupling reactions, providing opportunities for pairing any two polymer radicals together, thereby increasing the polymerization degree and leading to a large quantity of high-molecular-weight polymers adhering to the catalyst surface . In Supplementary Figs. 36 and 37, further analysis of the polymerization products on the catalyst surface in the HEAs-PMS and NG-PMS systems revealed the presence of polyphenol products with a higher polymerization degree on the surface of HEAs ( ), while only lower-molecular polymerization products were detected on the NG surface ( , ). Further analysis of extraction and concentrated bulk solution
Fig. 5 | Assessment of the adaptability and stability of HEAs. a Cell viability of 16 HBE under different concentrations of HEAs and . Cell culture time: 48 h .
b Cells in the culture medium were observed under a microscope. c Treatment of phenol in real water matrices. d, e EEM spectra of collected coal chemical industry wastewater before (d) and after treatment (e) by HEAs-PMS system. f Schematic of
the fixed-bed column continuous flow system. The phenol removal efficiency of HEAs in cyclic experiments. The efficiency of phenol removal and the metal leakage of HEAs in a continuous flow system. Error bars represent the standard deviation, obtained by repeating the experiment three times. Dosage: [Phenol] : 0.1 mM , PMS: 0.25 mM , reaction solution: 50 mL , catalyst: .
from the HEAs-PMS and NG-PMS systems revealed the detection of products ranging from dimers to tetramers ( ), as well as partially oxidized ring-opening products, indicating that the lower-polymerization-degree products were dissolved into the solution (Supplementary Figs. 38-44).
Moreover, upon the depletion of the electron source (PMS), the insufficient coupling of polymer radicals can lead to the termination of polymerization reactions. This result is consistent with the findings of the TGA experiment that HEAs exhibit higher pollutant-to-polymer transformation efficiency. In Fig. 4e, polymerization degree distribution analysis on the surfaces of NG and HEAs revealed that tetramers ( ) and pentamers ( ) are predominantly observed on the NG surface, while most hexamers ( ) to nonamers ( ) are present on the HEAs surface. In contrast, lower-polymerization-degree products are dissolved in the solution phase. Thus, the higher redox potential of HEAs-PMS* and faster electron transfer capability can
trigger the rapid generation and accumulation of phenoxyl radicals, thereby enhancing the phenol-to-polymer transformation kinetics to yield larger polyphenol products.
Further verification of the above mechanism was conducted through the frontier orbital theory, as shown in Fig. 5f, h and Supplementary Fig. 45), the energy gaps between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital of polyphenol with different molecular sizes decrease gradually with the increased polymerization degree. This suggests that reactants with higher degrees of polymerization have a higher HOMO and lower energy gap ( ) for losing electrons, making them more easily oxidized into phenoxy radicals and form the corresponding polymeric products. Additionally, the energy barriers for the formation of organic radicals in products with different degrees of polymerization indicate that as the degree of polymerization increases, the reaction becomes more thermodynamically favorable (Supplementary Fig. 46).
To better understand why HEAs achieved higher degrees of polymerization, phenol polymeric products in different equivalents were prepolymerized on the surfaces of HEAs and NG using a PMStriggered polymerization reaction. As shown in Supplementary Fig. 47, after introducing different amounts of phenol polymerization products onto HEAs, these surfaces continued to rapidly activate PMS, resulting in further polymerization of phenol and the production of high-molecular-weight products. In contrast, NG quickly lost its ability to activate PMS. This phenomenon further demonstrates that the conjugated aromatic structure and van der Waals interactions of nitrogen-doped graphite preferentially adsorb and enrich polymerization products, which predominantly form on the graphite surface. Meanwhile, the separation of active sites enables PMS activation on the HEA NPs, sustaining the ETP-mediated polymerization process. In contrast, the polymerization products in NG indiscriminately covered the reactive sites for PMS activation, leading to a rapid loss of catalytic activity. Simultaneously, OCPT experiments in Fig. 3c showed that HEAs maintained a higher complex potential than NG with the presence of phenol, allowing for the continued polymerization process. These conclusions clearly indicate that the separation of active sites for PMS and organic pollutants in HEAs enables continuous PMS activation, generating a higher potential and maintaining the ETPmediated polymerization process. At the same time, the graphite interface consistently adsorbs, concentrates, and stabilizes the polymers, leading to the formation of larger polyphenol products with a higher degree of polymerization compared to NG.
Biotoxicity universality and application stability test
In assessing the suitability of HEAs as catalysts for water treatment, understanding their biotoxicity is crucial. To assess the exposure risk during practical catalyst use, we conducted in the vitro cytotoxicity experiments on human bronchial epithelial cells . As represented by Fig. 5a, CTL assay experiments reveal dose-dependent biological toxicity of and mixed ions, with cell viability dropping below and at concentrations of for and for the mixed ion concentration (approximately equivalent to HEAs performance) (Supplementary Fig. 48). Notably, the addition of HEAs yielded promising results, maintaining 16 HBE cell line viability above even with twice the concentration. This contrast between HEAs and ions was further emphasized in Fig. 5b, where a noticeable difference in cell count and survival morphology in the microscopic field. In addition, the toxicity of the polymeric products was assessed using the Toxicity Estimation Software Tool, which showed a slight increase in toxicity as the polymerization degree increased. This finding highlights the importance of developing efficient recovery technologies to reduce potential environmental impacts and promote the reutilization the upcycled polymeric products. (Supplementary Fig. 49)
Furthermore, in practical water matrices experiments, the HEAsPMS system demonstrates excellent anti-interference capability in environments with more complex water matrices (Fig. 5c). The secondary wastewater collected from the coal chemical industry was utilized to assess the practicality of HEAs-PMS system. The excitation emission matrix (EEM) spectra shown in Fig. 5d, e indicated that soluble microbial byproducts and phenolic humic acid-like organic compounds in the wastewater samples were easily removed, as evidenced by the significant reduction in fluorescence intensity at the detected peaks.
The catalyst also exhibited excellent stability in the cyclic removal of phenol, achieving a 95% removal of phenol in five cycles (Fig. 5g and Supplementary Fig. 50). In contrast, continuous-flow long-term running experiments provided a more comprehensive evaluation of the catalyst’s performance in a fixed-bed reactor for degrading organic pollutants (Fig. 5f) . The results of the 50-day long-term degradation experiment under conditions of phenol demonstrated that
the degradation performance of the HEAs-PMS system gradually declined only after treating a phenol solution with a bed volume of 30,000 (Fig. 5h and Supplementary Fig. 51). Additionally, the detected leakage of metal ions is almost zero (Supplementary Table 7), further proving the robust structural stability of HEAs and sustained removal performance in long-term operation .
Discussion
In summary, we successfully synthesized and applied HEAs for aqueous pollutant removals in the PMS-AOPs system. Compared to NG, the supported HEAs are more effective in catalyzing PMS activation to achieve rapid phenol removal (PMS:phenol=2.5:1), exhibiting exceptional stability even under harsh pH conditions and immunity to interference from ionic substances in the water background. HEAs exhibited stable and long-lasting catalytic activity in the 50-day longterm experiments with no observable metal leaching. The HEAs NPs secured both high PMS activation activity and excellent conductivity, resulting in enhanced PMS* composite potentials and accelerated electron transfer rates. As a result, the nonradical ETP regime trigger non-mineralization phenol removal via one-electron oxidation and achieved a high electron utilization efficiency of up to , leading to the sustained production of high-molecular-weight polyphenolic products. Additionally, due to the environmental friendlies and low biotoxicity of HEAs, this study provides a perspective for designing atom-precision heterogeneous catalysts for the treatment of industrial wastewater with low oxidant consumption and carbon emission.
Methods
Preparation of catalysts
In a typical synthesis of HEAs, , , glucose , and CTAC ( 50 mg ) were dissolved in 10 mL of oleylamine, followed by ultrasound treatment for 30 min . Then, the mixture was heated in an oil bath at for 2 h . The cooled product was collected by centrifugation and washed three times with a volume ratio ethanol/cyclohexane mixture. Finally, the black colloidal products were dispersed in cyclohexane, was added, followed by sonication for 60 min , centrifugation, and washing three times with an ethanol/cyclohexane mixture. After vacuum drying at for 12 h , 500 mg of glucose was added and finely ground until a homogeneous dispersion was achieved. The mixture was then transferred into a tubular furnace and annealed at for 2 h at a heating rate of under a protective Ar atmosphere. Subsequently, it was promptly removed and allowed to cool to room temperature to obtain HEAs. For the synthesis of NC, all the conditions are similar to those of HEAs except for the absence of black colloidal products during the annealing process.
Catalyst characterization
The high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) technique using a Titan Themis 80-200 electron microscope (FEI Company, Netherlands) was used to observe the morphology and structure of the catalysts and elemental mapping. The crystal phases of the catalysts were characterized by X-ray diffraction patterns with CuK-α radiation (SmartLab 3kw, Rigaku Co.). Electron capture by 2,2,6,6-Tetramethyl-4-piperidone hydrochloride (TEMP) and radical trapping by 5,5-dimethyl-1-pyrrolineN-oxide (DMPO) were conducted on an ESR5000 spectrometer (Bruker Co.). Inductively coupled plasma optical emission spectroscopy (ICP-OES) experiments were performed using an Avio 550 Max (Perkin Elmer Co.) to determine the content of metals in catalysts and leakage of metal ions during degradation. The efficiency of phenol mineralization was assessed using a total organic carbon (TOC) analyzer (Multi N/C 3100, Analytik Jena), while the phenol intermediates were identified through ultrahigh performance liquid chromatography with quadrupole time-
of-flight initial mass spectrometry (UPLC-QTOF-MS, Waters Xevo G2XS QTOF). Furthermore, the concentration of PMS was quantified using a UV-visible spectrophotometer (AQ8100, Thermo Orion Co.) through the potassium iodide method.
Electrochemical analysis tests
The electrochemical analysis methods linear sweep voltammetry (LSV), Cyclic Voltammetry (CV), Chronoamperometry (CA), Electrochemical impedance spectroscopy (EIS), Electrochemical active surface area (ECSA), and open circuit potential (OCPT) were shown in Supplementary Information by an electrochemical workstation (CHI 760E).
XAFS measurements and EXAFS analysis
Chemical speciations of Cu and Ni were determined by K-edge XANES. XANES spectra of Cu and Ni in HEAs powder were obtained in the transmission model using the beamline of MEX-1 in the Australian Synchrotron Radiation Facility. Cu foil, CuO, CuPc, Ni foil, NiO, and NiPc were selected as references. The corresponding reference samples were mixed with cellulose and measured in transmission mode. Fe foil was employed for the calibration. The acquired EXAFS data were processed following standard procedures using the ATHENA (version 0.9.26) module implemented in IFEFFIT software packages for background, pre-edge line, and post-edge line calibrations. Subsequently, the -weighted data in the R range, spanning from 0 to , were Fourier transformed to real (R) space using the Morlet function parameters kappaMorlet and sigmaMorlet , with the assistance of hama_fortran. This transformation was carried out to distinguish the EXAFS contributions from various coordination shells.
Theoretical calculation studies
This calculation utilizes the Cambridge Serial Total Energy Package module within the MaterialsStudio 20.1 software. The exchangecorrelation functional chosen for electron-electron interactions is the Perdew-Burke-Ernzerhof functional under the Generalized Gradient Approximation. The self-consistent field (SCF) method is employed to solve the Kohn-Sham equations. The convergence criterion for the SCF energy is set to /atom. The total energy error of the system is within atom, the stress deviation is less than 0.02 GPa , and the convergence criterion for atomic forces is . Monkhorst-Pack grids are used for Brillouin zone -point sampling, with a -point mesh chosen for calculations. The cutoff energy for the plane-wave basis set is set to 440 eV . Ultra-soft pseudopotentials are employed to describe the interaction between ionic cores and valence electrons.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings of the study are included in the main text and supplementary information files. Additional data are available from the corresponding author upon request. Source data are provided with this paper.
References
Dong, C. et al. Dual-functional single-atomic Mo/Fe clustersdecorated via three electron-pathway in oxygen reduction reaction for tandemly removing contaminants from water. Proc. Natl. Acad. Sci. USA 120, e2305883120 (2023).
Yu, F. et al. Rapid self-heating synthesis of Fe-based nanomaterial catalyst for advanced oxidation. Nat. Commun. 14, 4975 (2023).
Gao, Y. et al. Subtle tuning of nanodefects actuates highly efficient electrocatalytic oxidation. Nat. Commun. 14, 2059 (2023).
Liang, X. et al. Coordination number dependent catalytic activity of single-atom cobalt catalysts for fenton-like reaction. Adv. Funct. Mater. 32, 2203001 (2022).
Chen, F. et al. Single-atom iron anchored tubular catalysts for ultrafast fenton-like reaction: roles of high-valency iron-oxo species and organic radicals. Adv. Mater. 34, 2202891 (2022).
Zhao, Z. et al. Improved electronic structure from spin-state reconstruction of a heteronuclear diatomic pair to boost the fenton-like reaction. Environ. Sci. Technol. 57, 4556-4567 (2023).
Chen, Z. et al. Single-atom Mo-Co catalyst with low biotoxicity for sustainable degradation of high-ionization-potential organic pollutants. Proc. Natl. Acad. Sci. USA 120, e2305933120 (2023).
Jiang, Y. et al. In situ turning defects of exfoliated MXene into Fenton-like catalytic active sites. Proc. Natl. Acad. Sci. USA 120, e2210211120 (2023).
Guo, Z.-Y. et al. Electron delocalization triggers nonradical Fentonlike catalysis over spinel oxides. Proc. Natl. Acad. Sci. USA 119, e2201607119 (2022).
He, Z. et al. perovskite for efficient sulfafurazole degradation via peroxymonosulfate activation: catalytic mechanism of interfacial structure. Appl. Catal. B Environ. 335, 122883 (2023).
Sun, Y., Li, M., Duan, J., Antonietti, M. & Chen, S. Entropy-driven direct air electrofixation. Angew. Chem. Int. Ed. 63, e202402678 (2024).
Wang, J., Zhang, J., Hu, Y., Jiang, H. & Li, C. Activating multisite highentropy alloy nanocrystals via enriching M-pyridinic N-C bonds for superior electrocatalytic hydrogen evolution. Sci. Bull. 67, 1890-1897 (2022).
Ren, J.-T., Chen, L., Wang, H.-Y. & Yuan, Z.-Y. High-entropy alloys in electrocatalysis: from fundamentals to applications. Chem. Soc. Rev. 52, 8319-8373 (2023).
Wu, D. et al. Noble-metal high-entropy-alloy nanoparticles: atomiclevel insight into the electronic structure. J. Am. Chem. Soc. 144, 3365-3369 (2022).
Kang, Y. et al. Mesoporous multimetallic nanospheres with exposed highly entropic alloy sites. Nat. Commun. 14, 4182 (2023).
Men, Y. et al. Understanding alkaline hydrogen oxidation reaction on PdNiRulrRh high-entropy-alloy by machine learning potential. Angew. Chem. Int. Ed. 62, e202217976 (2023).
Li, K. & Chen, W. Recent progress in high-entropy alloys for catalysts: synthesis, applications, and prospects. Mater. Today Energy 20, 100638 (2021).
Zou, X. et al. High-entropy engineering with regulated defect structure and electron interaction tuning active sites for trifunctional electrocatalysis. Proc. Natl. Acad. Sci. USA 121, e2313239121 (2024).
Cui, X. et al. Rapid high-temperature liquid shock synthesis of highentropy alloys for hydrogen evolution reaction. ACS Nano 18, 2948-2957 (2024).
Li, B. et al. Boosting catalytic decomposition by the synergistic effect of multiple elements in cobalt-based high-entropy oxides. Environ. Sci. Technol. 58, 2153-2161 (2024).
Wang, Y. et al. Supported high-entropy alloys for electrooxidation of benzyl alcohol assisted water electrolysis. Adv. Funct. Mater. 34, 2311611 (2024).
Li, H. et al. Fast site-to-site electron transfer of high-entropy alloy nanocatalyst driving redox electrocatalysis. Nat. Commun. 11, 5437 (2020).
Li, M. et al. Potential-dependent selectivity for the efficient capture of gold from E-waste acid leachate using sulfhydryl-functionalized carbon. Sci. Bull. 68, 1095-1099 (2023).
Zhan, C. et al. Medium/high-entropy amalgamated core/shell nanoplate achieves efficient formic acid catalysis for direct formic acid fuel cell. Angew. Chem. Int. Ed. 62, e202213783 (2023).
Chen, W. et al. High-entropy intermetallic PtRhBiSnSb nanoplates for highly efficient alcohol oxidation electrocatalysis. Adv. Mater. 34, 2206276 (2022).
Li, M. et al. Deep separation between In(III) and Fe(III) lons by regulating the lewis basicity of adsorption sites on electrospun fibers. Adv. Funct. Mater. 34, 2313443 (2024).
Shao, P. et al. Revisiting the graphitized nanodiamond-mediated activation of peroxymonosulfate: singlet oxygenation versus electron transfer. Environ. Sci. Technol. 55, 16078-16087 (2021).
Ren, W. et al. Origins of electron-transfer regime in persulfatebased nonradical oxidation processes. Environ. Sci. Technol. 56, 78-97 (2022).
Zhao, Z. et al. Construction of dual active sites on diatomic metal (FeCo-N/C-x) catalysts for enhanced Fenton-like catalysis. Appl. Catal. B Environ. 309, 121256 (2022).
Wang, Z. et al. Cobalt single atoms anchored on oxygen-doped tubular carbon nitride for efficient peroxymonosulfate activation: simultaneous coordination structure and morphology modulation. Angew. Chem. Int. Ed. 61, e202202338 (2022).
Yang, J. et al. Insights into the role of dual reaction sites for single Ni atom Fenton-like catalyst towards degradation of various organic contaminants. J. Hazard. Mater. 430, 128463 (2022).
Shao, H. et al. Naproxen as a turn-on chemiluminescent probe for real-time quantification of sulfate radicals. Environ. Sci. Technol. 57, 8818-8827 (2023).
Gong, Y. et al. Whose oxygen atom is transferred to the products? A case study of peracetic acid activation via complexed for organic contaminant degradation. Environ. Sci. Technol. 57, 6723-6732 (2023).
Zhang, Q. et al. Mineralization versus polymerization pathways in heterogeneous Fenton-like reactions. Water Res. 249, 120931 (2024).
Deng, G. et al. Ferryl ion in the photo-fenton process at acidic pH: occurrence, fate, and implications. Environ. Sci. Technol. 57, 18586-18596 (2023).
Zhang, B., Li, X., Akiyama, K., Bingham, P. A. & Kubuki, S. Elucidating the mechanistic origin of a spin state-dependent Catalyst toward organic contaminant oxidation via peroxymonosulfate activation. Environ. Sci. Technol. 56, 1321-1330 (2022).
Liu, H.-Z. et al. Tailoring d-band center of high-valent metal-oxo species for pollutant removal via complete polymerization. Nat. Commun. 15, 2327 (2024).
Zhang, Y.-J. et al. Simultaneous nanocatalytic surface activation of pollutants and oxidants for highly efficient water decontamination. Nat. Commun. 13, 3005 (2022).
Chen, Y. et al. Transformative removal of aqueous micropollutants into polymeric products by advanced oxidation processes. Environ. Sci. Technol. 58, 4844-4851 (2024).
Peng, J. et al. Removal of phenols by highly active periodate on carbon nanotubes: a mechanistic investigation. Environ. Sci. Technol. 57, 10804-10815 (2023).
Zhang, Y.-J. et al. Distinguishing homogeneous advanced oxidation processes in bulk water from heterogeneous surface reactions in organic oxidation. Proc. Natl. Acad. Sci. USA 120, e2302407120 (2023).
Yao, Z. et al. Thiol-rich, porous carbon for the efficient capture of silver: understanding the relationship between the surface groups and transformation pathways of silver. Chem. Eng. J. 427, 131470 (2022).
Shang, Y., Xu, X., Gao, B., Wang, S. & Duan, X. Single-atom catalysis in advanced oxidation processes for environmental remediation. Chem. Soc. Rev. 50, 5281-5322 (2021).
Acknowledgements
We acknowledge the financial support provided by the National Natural Science Foundation of China (No. 52321005), the fellowship of the KeyArea Research and Development Program of Guangdong Province (No. 2023B0101200004), the Shenzhen Science and Technology Innovation Program (No. GXWD2O231129122140001, KQTD20190929172630447), the Open Project of State Key Laboratory of Urban Water Resource and Environment (Harbin Institute of Technology) (No. QA202440), the Science Foundation of National Engineering Research Center for Safe Disposal and Resources Recovery of Sludge (Harbin Institute of Technology, Grant No K2024B001). X.D. acknowledges the financial support from Australian Research Council via ARC Future Fellowship (FT230100526).
Author contributions
Z.Y., Y.C., N.R., and X.D. designed research; Z.Y. performed research; Z.Y., K.H., Z.J., Z.S., and S.R. contributed new reagents/analytic tools; Z.Y., S.R., and X.W. analyzed data; Z.Y., Y.C., and X.D. wrote the paper.
Correspondence and requests for materials should be addressed to Yidi Chen or Xiaoguang Duan.
Peer review information Nature Communications thanks Lei Wu and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/ licenses/by/4.0/.
(c) Crown 2024
State Key Laboratory of Urban Water Resource and Environment, Shenzhen Key Laboratory of Organic Pollution Prevention and Control, School of Civil and Environmental Engineering, Harbin Institute of Technology, Shenzhen, Shenzhen, P. R. China. School of Chemical Engineering, The University of Adelaide, Adelaide, SA, Australia. School of Materials and Environmental Engineering, Shenzhen Polytechnic University, Shenzhen, P. R. China.
↩e-mail: chenyidi@hit.edu.cn; xiaoguang.duan@adelaide.edu.au