DOI: https://doi.org/10.1038/s41420-024-01803-z
PMID: https://pubmed.ncbi.nlm.nih.gov/38225241
تاريخ النشر: 2024-01-15
علم الوراثة اللاجينية للسرطان: من الدراسات المخبرية والتجارب السريرية إلى الطب الدقيق
الملخص
الخلل التنظيمي الجيني هو سمة شائعة للعديد من الأمراض البشرية، وخاصة السرطان. أصبح تحديد العيوب الجينية المرتبطة بالأورام الخبيثة محورًا لبحوث السرطان مما أدى إلى توضيح تدريجي لتنظيم الجينات في خلايا السرطان. في الواقع، تصاحب معظم مراحل تقدم الورم، بما في ذلك تكوين الورم، والترويج، والتقدم، والانتكاس، تغييرات جينية، يمكن عكس بعضها بواسطة أدوية جينية. الهدف الرئيسي من العلاج الجيني في عصر الطب الدقيق المخصص هو اكتشاف علامات السرطان الحيوية لتحسين تقييم المخاطر، والتشخيص، والتدخلات العلاجية المستهدفة. لقد دفعت التقدمات التكنولوجية السريعة التي تسهل توصيف التغيرات الجينية الجزيئية المرتبطة بالسرطانات أبحاث وتطوير الأدوية الجينية. تلخص هذه المراجعة الآليات الرئيسية للخلل التنظيمي الجيني وتناقش أمثلة سابقة وحالية من مثبطات الجينات في تشخيص وعلاج السرطان، مع التركيز على تطوير مثبطات أو أدوية الإنزيمات الجينية. في الجزء الأخير، يتم النظر في آفاق التشخيص والعلاج الدقيق بناءً على فهم أفضل للاختلالات الجينية في السرطان.
حقائق
- تشمل الآليات التنظيمية الوراثية البيئية بيولوجيا السرطان، وخاصة الميثلة الحمض النووي، والأسيتيل الهستوني، والميكرو RNA.
- تعبير الجينات المرتبطة بالأورام مرتبط ارتباطًا وثيقًا بالعملية التنظيمية الجينية للأورام.
- تُحدث العلاجات الجينية مثل DNMTi و HDACis و BETis وغيرها من العلاجات الجينية باستمرار وتُستخدم في العيادات.
- العلاج المركب المعتمد على علم الوراثة اللاجيني هو اتجاه واعد.
- الأنظمة المتعددة، والعلاج الجيني، والذكاء الاصطناعي هي انتقالات مواتية من العلاج الجيني إلى الطب الدقيق.
أسئلة مفتوحة
- هل يمكن تحسين الفعالية العلاجية للأورام الصلبة من خلال دمج العلاجات التي تستهدف علامات جينية مختلفة؟
- ما هي التغيرات المقاسة في تشخيص وعلاج السرطان الدقيق من خلال علم الوراثة اللاجيني التي تعتبر مؤقتة، وأيها تعتبر علامات حيوية حقيقية للأورام؟
- كيف يمكن مقارنة النتائج في العلاج المخصص عندما تكون استنتاجات الدراسات المخبرية والدراسات السريرية متناقضة؟
مقدمة
آليات التنظيم الجيني غير الطبيعي في السرطان
ميثيلation الحمض النووي
يساهم في زيادة تعبير الجينات المسرطنة [24، 25]. يمكن أن تؤدي الطفرات وتفعيل جينات كابحة الورم إلى تلف الحمض النووي أو نمو الخلايا غير المنضبط، مما يعزز تقدم السرطان [26]. بالإضافة إلى ذلك، فإن القابلية المحتملة لعكس نشاط الميثيل ترانسفيراز تجعلها هدفًا جذابًا للتدخلات العلاجية، على عكس التغيرات الجينية.
ميثيلات الهيستون
أسيتيلation الهيستون
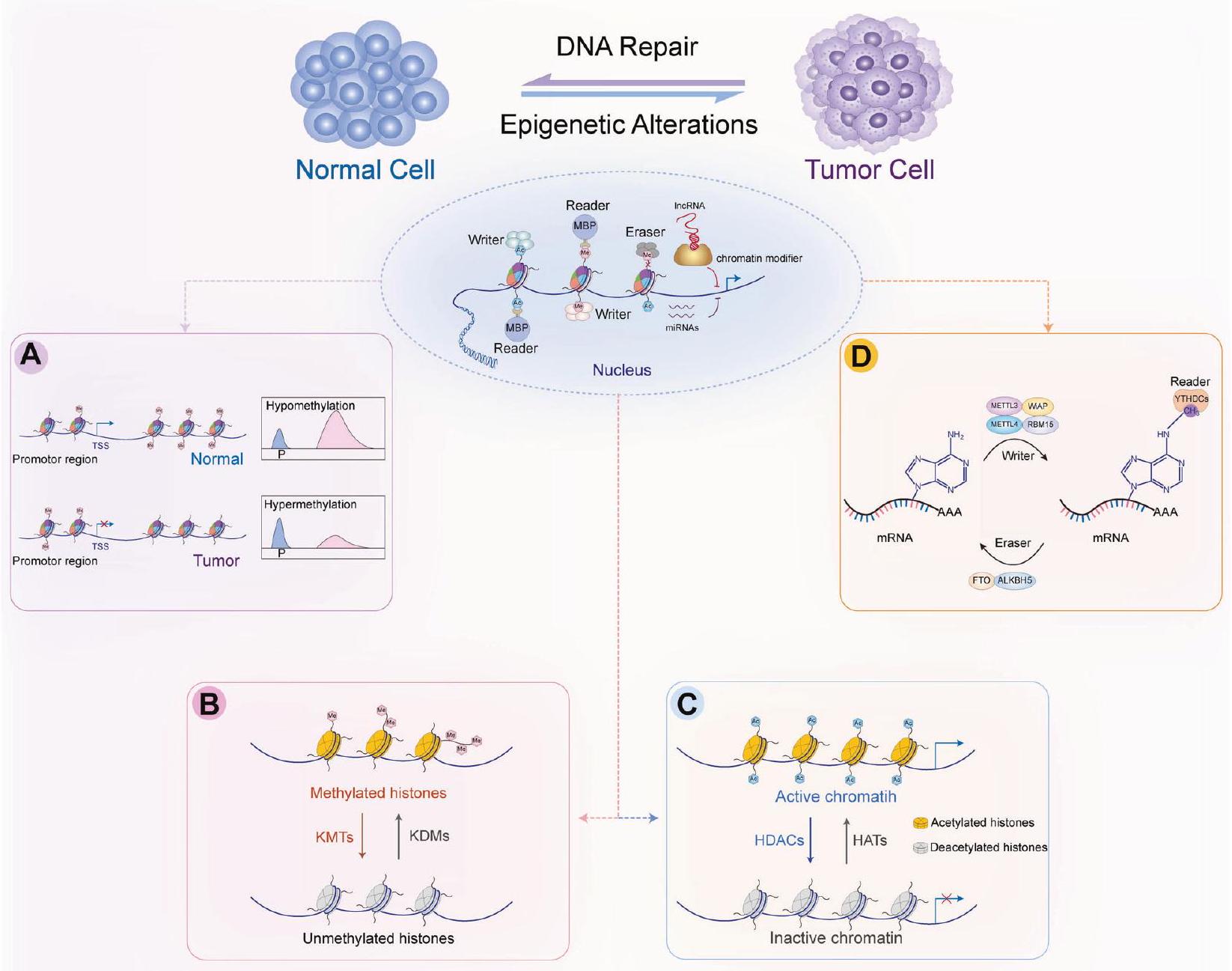
(بما في ذلك HDAC1 و HDAC2 و HDAC3 و HDAC8) التي تتواجد بشكل أساسي في النواة وعادة ما يتم التعبير عنها في الجينوم البشري. تتواجد HDACs من الفئة الثانية في النواة والسيتوبلازم وتظهر أنماط تعبير محددة حسب الأنسجة ونشاط إزالة الأسيتيل غير الهيستوني. وهي مقسمة إلى فئتين فرعيتين، Ila (HDAC4 و 5 و 7 و 9) و Ilb (HDAC6 و 10) [44]. تتكون HDACs من الفئة الثالثة من بروتينات شبيهة بـ SIR2 (بما في ذلك SIRT1 و SIRT2 و SIRT3 و SIRT4 و SIRT5 و SIRT6 و SIRT7) التي تشارك في تنظيم عمليات خلوية متعددة مثل البقاء، الشيخوخة، استجابة الإجهاد، والتمثيل الغذائي [43]. تتكون HDACs من الفئة الرابعة فقط من HDAC11، التي تشترك في تشابه جزئي مع HDACs من الفئة الأولى والثانية وتعمل كإزالة الأسيتيل للأحماض الدهنية طويلة السلسلة [45]. تعتمد HDACs من الفئات الأولى والثانية والرابعة على الركائز المرتبطة بالزنك و
علم الوراثة اللاجينية للـ RNA
سرطان الثدي وmiR-124a في الأورام القولونية مقارنة بالأنسجة الطبيعية [64، 65]. قد يعكس كتم الجينات غير الوراثية للـmiRNAs خصوصية الأنسجة. يؤثر التعبير الخاص بالأنسجة ونوع الخلايا للـmiRNAs بشكل واسع على تمايز الخلايا، ودوراتها، وشيخوختها، والتمثيل الغذائي [66]. ومن ثم، يمكن أن تساعد تحليل تعبير الـmiRNA في أنسجة الأورام أو خزعات السوائل في توجيه تشخيص السرطان، وتوقع تشخيص المرضى، وتحديد الأهداف العلاجية المحتملة.
استراتيجيات مضادة للسرطان على مستوى الإبيجينيتك
مثبطات DNMT
مثبطات HDAC
إكس. يو وآخرون.
الخلايا في الدراسات ما قبل السريرية. وهذا يؤدي إلى الموافقة على عدة أدوية لعلاج بعض الأورام الدموية.
مثبطات عائلة BET
| هدف | مثبط | سرطان مرتبط | الحالة السريرية | مرجع |
| دي إن إم تي | أزاستيدين | الأورام النخاعية المرتبطة بالعلاج | يحقق استجابة للمرض | [71] |
| ديستابين | مكافحة غسل الأموال | يحقق استجابة للمرض وبقاءً عامًا أفضل | [72] | |
| SGI-110 | مكافحة غسل الأموال | يحقق استجابة للمرض | [73] | |
| CP-4200 | مكافحة غسل الأموال | يسبب إعادة تنشيط فعالة للجينات المثبطة للورم التي تم كبتها إبيجينياً | [74] | |
| MG98 | سرطان الخلايا الكلوية | يعيق تكاثر خلايا السرطان النامية | [75] | |
| نانوميسين أ | سرطان الخلايا الكبدية | تظهر تأثيرات مضادة للتكاثر | [76] | |
| HDAC | فورينوسيت (SAHA) | لمفومة الخلايا التائية | يحدد معدلات الاستجابة الكاملة والجزئية | [80] |
| روميديبسين (ديبسيبيبتيد) | CTCL | يحقق استجابة للمرض | [81] | |
| بيلينوسات | لمفومة الخلايا التائية المحيطية | يحقق استجابة للمرض | [82] | |
| بانوبينوسات | MM | يحقق استجابة للمرض | [83] | |
| TSA | سرطان الثدي | لديه خصوصية أكبر للسرطان مقابل الخلايا الطبيعية | [84] | |
| توباسين | الميلانوما، سرطان القولون والمستقيم | يزيد من الإفراز خارج الخلية لمؤشر خلايا السرطان الجذعية | [85] | |
| MC1568، MC1575 | سرطان الثدي، الميلانوما | تظهر تأثيرات مضادة للتكاثر | [٨٦، ٨٧] | |
| ريكولينوسات (ACY1215) | جي بي إم | يعيق نمو خلايا الورم | [88] | |
| IN-2001 | سرطان الثدي | يُثبِّط نمو الورم | [89] | |
| AR-42 | ورم السحايا | يزيد من تعبير الجينات المؤيدة للاستماتة ويقلل من مستويات البروتينات المعاكسة للاستماتة | [90] | |
| جيفينوسات (ITF2357) | بي سي بي – الكل | يعيق التكاثر ويحفز موت الخلايا المبرمج | [91] | |
| رهان | JQ1 | التهاب النخاع الشوكي، اللوكيميا الحادة، سرطان الغدد الليمفاوية الكبير B، سرطان البروستاتا، سرطان الثدي | يُنتج تأثيرًا مضادًا للتكاثر قويًا مرتبطًا بتوقف دورة الخلية والشيخوخة الخلوية، والتمايز النخاعي النهائي، والقضاء على خلايا جذعية اللوكيميا | [94-98] |
| OTX015 (MK-8628) | لمفوما الخلايا البائية، ورم الأرومة العصبية | يعيق تكاثر خلايا السرطان؛ يقلل من مستوى c-Myc وMYCN وغيرها من الجينات المسرطنة المرتبطة بالمُعزِّزات الفائقة. | [٩٩، ١٠٠] | |
| MS645 | سرطان الثدي الثلاثي السلبي | يعيق تكاثر خلايا السرطان | [101] | |
| ABBV-075 | AML، لمفوما غير هودجكين، MM | يحفز موت الخلايا المبرمج | [102] | |
| ABBV-744 | سرطان البروستاتا | يزيح BRD4 من المعززات الفائقة المحتوية على AR ويثبط النسخ المعتمد على AR | [103] | |
| I-BET151 | جي بي إم | يعيق تكاثر خلايا الورم الدبقي | [105] | |
| CC-90011 | الأورام الصلبة | يحقق استجابة كاملة أو استجابة جزئية؛ يطيل المرض المستقر | [104] | |
| آي-بيت 762 | سرطان البنكرياس | يعيق مسارات متعددة مرتبطة بنمو الخلايا | [107] | |
| KDM | ORY-1001 | مكافحة غسل الأموال | يقلل من نمو خلايا السرطان | [108] |
| كيمت | بي إكس – 01294 | DLBCL، ورم العصبي الدبقي | يعيق تكاثر الخلايا ويحفز موت الخلايا المبرمج في خلايا السرطان | [١٠٩، ١١٠] |
| UNC0638 | غير صغير الخلايا سرطان الرئة | يعيق نمو الخلايا ويحفز موت الخلايا المبرمج | [111] | |
| بينوميتوستات | MLL | يعيق تكاثر خطوط خلايا اللوكيميا الحاملة لـ MLL-r ويؤدي إلى تراجع مستدام | [112] | |
| EPZ004777 | MLL | القتل الانتقائي للخلايا الحاملة لانتقال جين MLL | [113] | |
| GSK126 | خلايا مثبطة مشتقة من النخاع الشوكي | يعيق نمو خلايا الورم | [114] |
سرطان الغدد اللمفاوية، وخلايا MM [102]، بينما يستهدف ABBV-744 بشكل انتقائي مجال BD2، ويزيح BRD4 من المحسنات الفائقة المحتوية على مستقبل الأندروجين (AR)، ويثبط النسخ المعتمد على AR. يظهر هذا نشاطًا مضادًا للورم أفضل من ABBV075 في نموذج زراعة الفئران باستخدام خلايا سرطان البروستاتا البشرية [103]. CC-90010 هو مثبط BET من الجيل التالي مع
نشاط مضاد للورم مشجع في المرضى الذين يعانون من أورام صلبة متقدمة [104]. تزداد وفرة BRD2 و BRD4 بشكل كبير في GBM. لذلك، فإن العلاج بمثبط بروتين BET (I-BET151) يثبط تكاثر خلايا GBM [105]. علاوة على ذلك، يظهر OTX015 تأثيرًا مضادًا للتكاثر أعلى من نظيره (JQ1) في خطوط خلايا GBM [106]. في الوقت نفسه، يعمل JQ1 و I-BET 762 بشكل فعال
| أشكال التركيب | العلاجات المركبة | السرطان المرتبط | المزايا | المرجع |
| DNMTi+HDACi | أزا سيتيدين + فوري نوسات | MDS؛ CMML | أكثر فعالية من العلاج الأحادي | [116] |
| أزا سيتيدين + إنتينوسات | CRC | تحسين النشاط المضاد للورم | [117] | |
| دواء إبي + دواء مستهدف | ACY-1215 + بورتزوميب | MM | تأخير نمو الورم؛ إطالة البقاء | [121] |
| TSA + جزيئات نانوية من البالاديوم | سرطان عنق الرحم | زيادة الإمكانية لعلاج ناجح | [122] | |
| JQ1
|
T-ALL | تصدت لمقاومة
|
[123] | |
| دواء إبي + مناعيات | بانوبينستات + بورتزوميب + ديكساميثازون | MM | تحسين البقاء بدون تقدم | [126] |
| حاصرات PD-1 + ديسيتابين | CRC | تثبيط نمو الورم؛ إطالة البقاء | [127] | |
| أزا سيتيدين + بيمبروليزوماب | MDS | آمن مع سمية قابلة للتحكم | [128] |
تتداخل مع مسارات متعددة مرتبطة بنمو الخلايا في سرطان البنكرياس [107]. بشكل عام، يظهر مثبط BRD4 التقليدي JQ1 ومثبطات BET المطورة حديثًا نتائج واعدة في أنواع مختلفة من السرطانات البشرية.
مثبطات KMT و KDM
استراتيجيات العلاج المركب
يجمع بين JQ1 و
وجهات نظر في الطب الدقيق
التقنيات التي يمكن أن تحدد الجينوميات، والترنسكريبتوميات، والجينوميات ثلاثية الأبعاد وتوضح التفاعلات بين التغيرات الجينية والوراثية في بيولوجيا السرطان، فعالة في إدارة المرض بدقة والتنبؤ به. يمكن لتسلسل الجينوم الكامل وتسلسل الإكسوم الكامل تتبع التغيرات الجينومية في الأورام المختلفة. في الوقت نفسه، يمكن لتقنية RNA-seq التقاط ملفات النسخ الميكروية وتحليل تباين الأورام. حاليًا، يُعتبر تسلسل البيسلفيت للجينوم الكامل هو الطريقة القياسية الذهبية لتحليل بيانات ميثيل الحمض النووي على نطاق الجينوم بدقة قاعدة واحدة. بالإضافة إلى ذلك، تؤثر إمكانية الوصول إلى الكروماتين على ارتباط الحمض النووي بعوامل النسخ والعناصر التنظيمية. وهذا يوفر رؤى مهمة حول الآليات التي يتم من خلالها تنشيط الجينومات السرطانية وإسكاتها. يمكن لاختبار الكروماتين القابل للوصول بواسطة الترانسبوزاز باستخدام التسلسل (ATAC-seq) تقييم مشهد إمكانية الوصول إلى الكروماتين في السرطانات البشرية الأولية. في الوقت نفسه، يمكن أن يؤدي التدخل في الهيكل ثلاثي الأبعاد للجينوم إلى تنشيط الأورام الغريبة من خلال التفاعل مع المعززات القريبة أو البعيدة ومناطق المحفزات التي تبدأ نسخ الأورام. وبالتالي، يمكن استخدام تقنية ترسيب المناعة للكروماتين والتسلسل (ChIP-seq) لتحديد نشاط المعززات والمعززات الفائقة على نطاق الجينوم بناءً على علامات الهيستون H3K4me1 وH3K27ac، على التوالي. بالإضافة إلى ذلك، يمكن أن تحدد تحليل التقاط الكروماتين المكاني على نطاق الجينوم (HiC) التفاعلات غير الطبيعية بين المعززات والمحفزات في جميع أنحاء جينوم الورم. إن التطور السريع لهذه التقنيات يسهم في تحديد بانوراما الإبيجينوم للسرطان ويسهل المزيد من دمج مؤشرات الإبيجينوم في التطبيقات السريرية. في الواقع، ستوفر مجموعة تقنيات الإبيجينetics مع المنصات الأكثر استخدامًا (مثل تسلسل الجينوم الكامل وRNA-seq) رؤى شاملة بشأن الشذوذات الجينومية والإبيجينومية لدى المرضى المصابين بالسرطان. بالإضافة إلى ذلك، ستعمل تطبيقات الأساليب متعددة الأوميات لتحديد العلامات البيولوجية التشخيصية على تحسين التدخلات العلاجية من خلال توفير بيانات للعلاج الشخصي.
نموذج التعلم الآلي، أفوكادو، الذي هو مزيج من تحليل الموترات والشبكات العصبية العميقة التي تضغط البيانات الإبيجينية إلى تمثيلات كثيفة وغنية بالمعلومات. يتفوق أفوكادو على النماذج المدربة مباشرة على البيانات الإبيجينية في مجموعة متنوعة من المهام الجينومية ويحقق توقعات دقيقة عالية لتعبير الجينات وتفاعلات المحفزات والمعززات، بما في ذلك المناطق التي تتفاعل بشكل متكرر في بيانات HiC، ووقت النسخ، وبنية الكروماتين ثلاثية الأبعاد. يمكن للذكاء الاصطناعي اكتشاف كميات صغيرة من العلامات الحيوية، مما يحسن بشكل كبير من تشخيص السرطان المبكر ويخصص الرعاية السريرية بينما يساعد في اكتشاف أدوية جديدة لمكافحة السرطان. يوفر نضوج التحليل الإبيجيني فرصًا تآزرية لجهود التصوير المعتمدة على الذكاء الاصطناعي. بشكل عام، فإن تطبيق الذكاء الاصطناعي في تشخيص السرطان الدقيق وعلاجه يقدم آفاقًا واسعة، مما يؤدي إلى تطوير علاجات ذكية للسرطان.
الخاتمة
ملخص التقرير
REFERENCES
- Quina AS, Buschbeck M, Di Croce L. Chromatin structure and epigenetics. Biochem Pharm. 2006;72:1563-9.
- Peng A, Peng W, Wang R, Zhao H, Yu X, Sun Y. Regulation of 3D organization and its role in cancer biology. Front Cell Dev Biol. 2022;10:879465.
- Li E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat Rev Genet. 2002;3:662-73.
- Mohammad HP, Barbash O, Creasy CL. Targeting epigenetic modifications in cancer therapy: erasing the roadmap to cancer. Nat Med. 2019;25:403-18.
- Rutten BP, Mill J. Epigenetic mediation of environmental influences in major psychotic disorders. Schizophr Bull. 2009;35:1045-56.
- Kanwal R, Gupta K, Gupta S. Cancer epigenetics: an introduction. Methods Mol Biol. 2015;1238:3-25.
- Chen Y, Hong T, Wang S, Mo J, Tian T, Zhou X. Epigenetic modification of nucleic acids: from basic studies to medical applications. Chem Soc Rev. 2017;46:2844-72.
- Singh M, Kumar V, Sehrawat N, Yadav M, Chaudhary M, Upadhyay SK, et al. Current paradigms in epigenetic anticancer therapeutics and future challenges. Semin Cancer Biol. 2022;83:422-40.
- Biswas S, Rao CM. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur J Pharm. 2018;837:8-24.
- Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology 2013;38:23-38.
- Robert MF, Morin S, Beaulieu N, Gauthier F, Chute IC, Barsalou A, et al. DNMT1 is required to maintain CpG methylation and aberrant gene silencing in human cancer cells. Nat Genet. 2003;33:61-5.
- Foulks JM, Parnell KM, Nix RN, Chau S, Swierczek K, Saunders M, et al. Epigenetic drug discovery: targeting DNA methyltransferases. J Biomol Screen. 2012;17:2-17.
- Chen T, Hevi S, Gay F, Tsujimoto N, He T, Zhang B, et al. Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nat Genet. 2007;39:391-6.
- Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999;99:247-57.
- Simo-Riudalbas L, Melo SA, Esteller M. DNMT3B gene amplification predicts resistance to DNA demethylating drugs. Genes Chromosomes Cancer. 2011;50:527-34.
- Stewart ML, Tamayo P, Wilson AJ, Wang S, Chang YM, Kim JW, et al. KRAS genomic status predicts the sensitivity of ovarian cancer cells to Decitabine. Cancer Res. 2015;75:2897-906.
- Baylin SB. DNA methylation and gene silencing in cancer. Nat Clin Pr Oncol. 2005;2:S4-11.
- Unoki M, Brunet J, Mousli M. Drug discovery targeting epigenetic codes: the great potential of UHRF1, which links DNA methylation and histone modifications, as a drug target in cancers and toxoplasmosis. Biochem Pharm. 2009;78:1279-88.
- Yamashita K, Dai T, Dai Y, Yamamoto F, Perucho M. Genetics supersedes epigenetics in colon cancer phenotype. Cancer Cell. 2003;4:121-31.
- Laux DE, Curran EM, Welshons WV, Lubahn DB, Huang TH. Hypermethylation of the Wilms’ tumor suppressor gene CpG island in human breast carcinomas. Breast Cancer Res Treat. 1999;56:35-43.
- Arechederra M, Daian F, Yim A, Bazai SK, Richelme S, Dono R, et al. Hypermethylation of gene body CpG islands predicts high dosage of functional oncogenes in liver cancer. Nat Commun. 2018;9:3164.
- Yegnasubramanian S, Kowalski J, Gonzalgo ML, Zahurak M, Piantadosi S, Walsh PC, et al. Hypermethylation of CpG islands in primary and metastatic human prostate cancer. Cancer Res. 2004;64:1975-86.
- Abbosh PH, Wang M, Eble JN, Lopez-Beltran A, Maclennan GT, Montironi R, et al. Hypermethylation of tumor-suppressor gene CpG islands in small-cell carcinoma of the urinary bladder. Mod Pathol. 2008;21:355-62.
- Ando M, Saito Y, Xu G, Bui NQ, Medetgul-Ernar K, Pu M, et al. Chromatin dysregulation and DNA methylation at transcription start sites associated with transcriptional repression in cancers. Nat Commun. 2019;10:2188.
- Timp W, Feinberg AP. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat Rev Cancer. 2013;13:497-510.
- Corradini P, Inghirami G, Astolfi M, Ladetto M, Voena C, Ballerini P, et al. Inactivation of tumor suppressor genes, p53 and Rb1, in plasma cell dyscrasias. Leukemia 1994;8:758-67.
- Hyun K, Jeon J, Park K, Kim J. Writing, erasing and reading histone lysine methylations. Exp Mol Med. 2017;49:e324.
- Husmann D, Gozani O. Histone lysine methyltransferases in biology and disease. Nat Struct Mol Biol. 2019;26:880-9.
- Sterling J, Menezes SV, Abbassi RH, Munoz L Histone lysine demethylases and their functions in cancer. Int J Cancer. 2020;148:2375-88.
- Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell. 2012;48:491-507.
- Alam H, Tang M, Maitituoheti M, Dhar SS, Kumar M, Han CY, et al. KMT2D deficiency impairs super-enhancers to confer a glycolytic vulnerability in lung cancer. Cancer Cell. 2020;37:599-617.e7.
- Duan R, Du W, Guo W. EZH2: a novel target for cancer treatment. J Hematol Oncol. 2020;13:104.
- Nguyen AT, Zhang Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011;25:1345-58.
- Kurani H, Razavipour SF, Harikumar KB, Dunworth M, Ewald AJ, Nasir A, et al. DOT1L is a novel cancer stem cell target for triple-negative breast cancer. Clin Cancer Res. 2022;28:1948-65.
- Griffin GK, Wu J, Iracheta-Vellve A, Patti JC, Hsu J, Davis T, et al. Epigenetic silencing by SETDB1 suppresses tumour intrinsic immunogenicity. Nature 2021;595:309-14.
- Nachiyappan A, Gupta N, Taneja R. EHMT1/EHMT2 in EMT, cancer stemness and drug resistance: emerging evidence and mechanisms. FEBS
. 2022;289:1329-51. - Vicioso-Mantis M, Aguirre S, Martinez-Balbas MA. JmjC family of Histone Demethylases form nuclear condensates. Int J Mol Sci. 2022;23:7664.
- Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, et al. LSD1 ablation stimulates anti-tumor immunity and enables checkpoint blockade. Cell 2018;174:549-63.e19.
- Shen DD, Pang JR, Bi YP, Zhao LF, Li YR, Zhao L, et al. LSD1 deletion decreases exosomal PD-L1 and restores T-cell response in gastric cancer. Mol Cancer. 2022;21:75.
- Musella M, Guarracino A, Manduca N, Galassi C, Ruggiero E, Potenza A, et al. Type I IFNs promote cancer cell stemness by triggering the epigenetic regulator KDM1B. Nat Immunol. 2022;23:1379-92.
- Goswami S, Raychaudhuri D, Singh P, Natarajan SM, Chen Y, Poon C, et al. Myeloid-specific KDM6B inhibition sensitizes glioblastoma to PD1 blockade. Nat Cancer. 2023;4:1455-73.
- Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381-95.
- Li Y, Seto E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb Perspect Med. 2016;6:a026831.
- Seto E, Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol. 2014;6:a018713.
- Nunez-Alvarez Y, Suelves M. HDAC11: a multifaceted histone deacetylase with proficient fatty deacylase activity and its roles in physiological processes. FEBS J. 2022;289:2771-92.
- Zheng W. The Zinc-dependent HDACs: Non-histone substrates and catalytic deacylation beyond deacetylation. Mini Rev Med Chem. 2022;22:2478-85.
- Marks PA, Richon VM, Rifkind RA. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J Natl Cancer Inst. 2000;92:1210-6.
- Wu CC, Jin LW, Wang IF, Wei WY, Ho PC, Liu YC, et al. HDAC1 dysregulation induces aberrant cell cycle and DNA damage in progress of TDP-43 proteinopathies. EMBO Mol Med. 2020;12:e10622.
- Kim MS, Kwon HJ, Lee YM, Baek JH, Jang JE, Lee SW, et al. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat Med. 2001;7:437-43.
- Hanigan CL, Van Engeland M, De Bruine AP, Wouters KA, Weijenberg MP, Eshleman JR, et al. An inactivating mutation in HDAC2 leads to dysregulation of apoptosis mediated by APAF1. Gastroenterology 2008;135:1654-64.e2.
- Barbieri I, Kouzarides T. Role of RNA modifications in cancer. Nat Rev Cancer. 2020;20:303-22.
- Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA modifications in gene expression regulation. Cell 2017;169:1187-200.
- Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, et al. m(6)A RNA methylation regulates the self-renewal and Tumorigenesis of Glioblastoma stem cells. Cell Rep. 2017;18:2622-34.
- Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, et al. FTO plays an oncogenic role in acute myeloid leukemia as a N(6)-Methyladenosine RNA Demethylase. Cancer Cell. 2017;31:127-41.
- Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5mediated m(6)A-demethylation of NANOG mRNA. Proc Natl Acad Sci USA. 2016;113:E2047-56.
- Statello L, Guo CJ, Chen LL, Huarte M. Gene regulation by long non-coding RNAs and its biological functions. Nat Rev Mol Cell Biol. 2021;22:96-118.
- Battistelli C, Garbo S, Riccioni V, Montaldo C, Santangelo L, Vandelli A, et al. Design and functional validation of a mutant variant of the LncRNA HOTAIR to counteract snail function in epithelial-to-mesenchymal transition. Cancer Res. 2021;81:103-13.
- Wurm AA, Pina C. Long non-coding RNAs as functional and structural chromatin modulators in acute myeloid leukemia. Front Oncol. 2019;9:899.
- Zhu S, Wang JZ, Chen D, He YT, Meng N, Chen M, et al. An oncopeptide regulates m (6)A recognition by the m (6)A reader IGF2BP1 and tumorigenesis. Nat Commun. 2020;11:1685.
- Saito Y, Suzuki H, Tsugawa H, Nakagawa I, Matsuzaki J, Kanai Y, et al. Chromatin remodeling at Alu repeats by epigenetic treatment activates silenced microRNA-512-5p with downregulation of Mcl-1 in human gastric cancer cells. Oncogene. 2009;28:2738-44.
- Garzon R, Liu S, Fabbri M, Liu Z, Heaphy CE, Callegari E, et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood. 2009;113:6411-8.
- Iorio MV, Piovan C, Croce CM. Interplay between microRNAs and the epigenetic machinery: an intricate network. Biochim Biophys Acta. 2010;1799:694-701.
- Lee YS, Dutta A. MicroRNAs in cancer. Annu Rev Pathol. 2009;4:199-227.
- Lehmann U, Hasemeier B, Christgen M, Muller M, Romermann D, Langer F, et al. Epigenetic inactivation of microRNA gene hsa-mir-9-1 in human breast cancer. J Pathol. 2008;214:17-24.
- Lujambio A, Ropero S, Ballestar E, Fraga MF, Cerrato C, Setien F, et al. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 2007;67:1424-9.
- Fasihi-Ramandi M, Moridnia A, Najafi A, Sharifi M. Inducing cell proliferative prevention in human acute promyelocytic leukemia by miR-182 inhibition through modulation of CASP9 expression. Biomed Pharmacother. 2017;89:1152-8.
- Cheng Y, He C, Wang M, Ma X, Mo F, Yang S, et al. Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Signal Transduct Target Ther. 2019;4:62.
- Schuh AC, Dohner H, Pleyer L, Seymour JF, Fenaux P, Dombret H. Azacitidine in adult patients with acute myeloid leukemia. Crit Rev Oncol Hematol. 2017;116:159-77.
- Klimek VM, Dolezal EK, Tees MT, Devlin SM, Stein K, Romero A, et al. Efficacy of hypomethylating agents in therapy-related myelodysplastic syndromes. Leuk Res. 2012;36:1093-7.
- Sato T, Cesaroni M, Chung W, Panjarian S, Tran A, Madzo J, et al. Transcriptional selectivity of epigenetic therapy in cancer. Cancer Res. 2017;77:470-81.
- Fianchi L, Criscuolo M, Lunghi M, Gaidano G, Breccia M, Levis A, et al. Outcome of therapy-related myeloid neoplasms treated with azacitidine. J Hematol Oncol. 2012;5:44.
- Fili C, Candoni A, Zannier ME, Olivieri J, Imbergamo S, Caizzi M, et al. Efficacy and toxicity of Decitabine in patients with acute myeloid leukemia (AML): A multicenter real-world experience. Leuk Res. 2019;76:33-8.
- Kantarjian HM, Roboz GJ, Kropf PL, Yee KWL, O’Connell CL, Tibes R, et al. Guadecitabine (SGI-110) in treatment-naive patients with acute myeloid leukaemia: phase 2 results from a multicentre, randomised, phase
trial. Lancet Oncol. 2017;18:1317-26. - Brueckner B, Rius M, Markelova MR, Fichtner I, Hals PA, Sandvold ML, et al. Delivery of 5-azacytidine to human cancer cells by elaidic acid esterification increases therapeutic drug efficacy. Mol Cancer Ther. 2010;9:1256-64.
- Amato RJ, Stephenson J, Hotte S, Nemunaitis J, Belanger K, Reid G, et al. MG98, a second-generation DNMT1 inhibitor, in the treatment of advanced renal cell carcinoma. Cancer Invest. 2012;30:415-21.
- Lai SC, Su YT, Chi CC, Kuo YC, Lee KF, Wu YC, et al. Correction to: DNMT3b/OCT4 expression confers sorafenib resistance and poor prognosis of hepatocellular carcinoma through IL-6/STAT3 regulation. J Exp Clin Cancer Res. 2020;39:10.
- Kazanets A, Shorstova T, Hilmi K, Marques M, Witcher M. Epigenetic silencing of tumor suppressor genes: Paradigms, puzzles, and potential. Biochim Biophys Acta. 2016;1865:275-88.
- Matthews GM, Newbold A, Johnstone RW. Intrinsic and extrinsic apoptotic pathway signaling as determinants of histone deacetylase inhibitor antitumor activity. Adv Cancer Res. 2012;116:165-97.
- Yang H, Sun B, Xu K, He Y, Zhang T, Hall SRR, et al. Pharmaco-transcriptomic correlation analysis reveals novel responsive signatures to HDAC inhibitors and identifies Dasatinib as a synergistic interactor in small-cell lung cancer. EBioMedicine. 2021;69:103457.
- Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2007;109:31-9.
- Whittaker SJ, Demierre MF, Kim EJ, Rook AH, Lerner A, Duvic M, et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol. 2010;28:4485-91.
- Lee HZ, Kwitkowski VE, Del Valle PL, Ricci MS, Saber H, Habtemariam BA, et al. FDA Approval: Belinostat for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma. Clin Cancer Res. 2015;21:2666-70.
- Garnock-Jones KP. Panobinostat: first global approval. Drugs 2015;75:695-704.
- Chang J, Varghese DS, Gillam MC, Peyton M, Modi B, Schiltz RL, et al. Differential response of cancer cells to HDAC inhibitors trichostatin A and depsipeptide. Br J Cancer. 2012;106:116-25.
- Chao OS, Chang TC, Di Bella MA, Alessandro R, Anzanello F, Rappa G, et al. The HDAC6 inhibitor tubacin induces release of CD133(+) extracellular vesicles from cancer cells. J Cell Biochem. 2017;118:4414-24.
- Duong V, Bret C, Altucci L, Mai A, Duraffourd C, Loubersac J, et al. Specific activity of class II histone deacetylases in human breast cancer cells. Mol Cancer Res. 2008;6:1908-19.
- Venza I, Visalli M, Oteri R, Cucinotta M, Teti D, Venza M. Class II-specific histone deacetylase inhibitors MC1568 and MC1575 suppress IL-8 expression in human melanoma cells. Pigment Cell Melanoma Res. 2013;26:193-204.
- Li S, Liu X, Chen X, Zhang L, Wang X. Histone deacetylase 6 promotes growth of glioblastoma through inhibition of SMAD2 signaling. Tumour Biol. 2015;36:9661-5.
- Joung KE, Min KN, An JY, Kim DK, Kong G, Sheen YY. Potent in vivo anti-breast cancer activity of IN-2001, a novel inhibitor of histone deacetylase, in MMTV/cNeu mice. Cancer Res. 2006;66:5394-402.
- Burns SS, Akhmametyeva EM, Oblinger JL, Bush ML, Huang J, Senner V, et al. Histone deacetylase inhibitor AR-42 differentially affects cell-cycle transit in meningeal and meningioma cells, potently inhibiting NF2-deficient meningioma growth. Cancer Res. 2013;73:792-803.
- Savino AM, Sarno J, Trentin L, Vieri M, Fazio G, Bardini M, et al. The histone deacetylase inhibitor givinostat (ITF2357) exhibits potent anti-tumor activity against CRLF2-rearranged BCP-ALL. Leukemia 2017;31:2365-75.
- Gilan O, Rioja I, Knezevic K, Bell MJ, Yeung MM, Harker NR, et al. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immunoinflammation. Science 2020;368:387-94.
- Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell. 2014;54:728-36.
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011;146:904-17.
- Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, et al. RNAi screen identifies Brd 4 as a therapeutic target in acute myeloid leukaemia. Nature 2011;478:524-8.
- Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell. 2013;24:777-90.
- Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014;510:278-82.
- Lu L, Chen Z, Lin X, Tian L, Su Q, An P, et al. Inhibition of BRD4 suppresses the malignancy of breast cancer cells via regulation of Snail. Cell Death Differ. 2020;27:255-68.
- Boi M, Gaudio E, Bonetti P, Kwee I, Bernasconi E, Tarantelli C, et al. The BET Bromodomain Inhibitor OTX015 Affects Pathogenetic Pathways in Preclinical B-cell Tumor Models and Synergizes with Targeted Drugs. Clin Cancer Res. 2015;21:1628-38.
- Henssen A, Althoff K, Odersky A, Beckers A, Koche R, Speleman F, et al. Targeting MYCN-driven transcription by BET-Bromodomain inhibition. Clin Cancer Res. 2016;22:2470-81.
- Ren C, Zhang G, Han F, Fu S, Cao Y, Zhang F, et al. Spatially constrained tandem bromodomain inhibition bolsters sustained repression of BRD4 transcriptional activity for TNBC cell growth. Proc Natl Acad Sci USA. 2018;115:7949-54.
- Bui MH, Lin X, Albert DH, Li L, Lam LT, Faivre EJ, et al. Preclinical characterization of BET family Bromodomain inhibitor ABBV-075 suggests combination therapeutic strategies. Cancer Res. 2017;77:2976-89.
- Faivre EJ, McDaniel KF, Albert DH, Mantena SR, Plotnik JP, Wilcox D, et al. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature 2020;578:306-10.
- Moreno V, Sepulveda JM, Vieito M, Hernandez-Guerrero T, Doger B, Saavedra O, et al. Phase I study of CC-90010, a reversible, oral BET inhibitor in patients with advanced solid tumors and relapsed/refractory non-Hodgkin’s lymphoma. Ann Oncol. 2020;31:780-8.
- Pastori C, Daniel M, Penas C, Volmar CH, Johnstone AL, Brothers SP, et al. BET bromodomain proteins are required for glioblastoma cell proliferation. Epigenetics 2014;9:611-20.
- Berenguer-Daize C, Astorgues-Xerri L, Odore E, Cayol M, Cvitkovic E, Noel K, et al. OTX015 (MK-8628), a novel BET inhibitor, displays in vitro and in vivo antitumor effects alone and in combination with conventional therapies in glioblastoma models. Int J Cancer. 2016;139:2047-55.
- Leal AS, Williams CR, Royce DB, Pioli PA, Sporn MB, Liby KT. Bromodomain inhibitors, JQ1 and I-BET 762, as potential therapies for pancreatic cancer. Cancer Lett. 2017;394:76-87.
- Maes T, Mascaro C, Tirapu I, Estiarte A, Ciceri F, Lunardi S, et al. ORY-1001, a potent and selective covalent KDM1A inhibitor, for the treatment of acute leukemia. Cancer Cell. 2018;33:495-511.e12.
- Xu L, Gao X, Yang P, Sang W, Jiao J, Niu M, et al. EHMT2 inhibitor BIX-01294 induces endoplasmic reticulum stress mediated apoptosis and autophagy in diffuse large B-cell lymphoma cells. J Cancer. 2021;12:1011-22.
- Lu Z, Tian Y, Salwen HR, Chlenski A, Godley LA, Raj JU, et al. Histone-lysine methyltransferase EHMT2 is involved in proliferation, apoptosis, cell invasion, and DNA methylation of human neuroblastoma cells. Anticancer Drugs. 2013;24:484-93.
- Wang L, Dong X, Ren Y, Luo J, Liu P, Su D, et al. Targeting EHMT2 reverses EGFRTKI resistance in NSCLC by epigenetically regulating the PTEN/AKT signaling pathway. Cell Death Dis. 2018;9:129.
- Stein EM, Garcia-Manero G, Rizzieri DA, Tibes R, Berdeja JG, Savona MR, et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 2018;131:2661-9.
- Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20:53-65.
- Huang S, Wang Z, Zhou J, Huang J, Zhou L, Luo J, et al. EZH2 inhibitor GSK126 suppresses antitumor immunity by driving production of myeloidderived suppressor cells. Cancer Res. 2019;79:2009-20.
- Mohammad F, Weissmann S, Leblanc B, Pandey DP, Hojfeldt JW, Comet I, et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat Med. 2017;23:483-92.
- Sekeres MA, Othus M, List AF, Odenike O, Stone RM, Gore SD, et al. Randomized Phase II study of azacitidine alone or in combination with lenalidomide or with vorinostat in higher-risk myelodysplastic syndromes and chronic myelomonocytic leukemia: North American Intergroup Study SWOG S1117. J Clin Oncol. 2017;35:2745-53.
- Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci USA. 2014;111:11774-9.
- Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet. 1999;21:103-7.
- Kalac M, Scotto L, Marchi E, Amengual J, Seshan VE, Bhagat G, et al. HDAC inhibitors and decitabine are highly synergistic and associated with unique gene-expression and epigenetic profiles in models of DLBCL. Blood 2011;118:5506-16.
- Bruyer A, Maes K, Herviou L, Kassambara A, Seckinger A, Cartron G, et al. DNMTi/ HDACi combined epigenetic targeted treatment induces reprogramming of myeloma cells in the direction of normal plasma cells. Br J Cancer. 2018;118:1062-73.
- Santo L, Hideshima T, Kung AL, Tseng JC, Tamang D, Yang M, et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 2012;119:2579-89.
- Zhang XF, Yan Q, Shen W, Gurunathan S. Trichostatin A enhances the apoptotic potential of palladium nanoparticles in human cervical cancer cells. Int J Mol Sci. 2016;17:1354.
- Knoechel B, Roderick JE, Williamson KE, Zhu J, Lohr JG, Cotton MJ, et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat Genet. 2014;46:364-70.
- Kuang C, Park Y, Augustin RC, Lin Y, Hartman DJ, Seigh L, et al. Pembrolizumab plus azacitidine in patients with chemotherapy refractory metastatic colorectal cancer: a single-arm phase 2 trial and correlative biomarker analysis. Clin Epigenet. 2022;14:3.
- Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 2015;162:974-86.
- San-Miguel JF, Hungria VT, Yoon SS, Beksac M, Dimopoulos MA, Elghandour A, et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014;15:1195-206.
- Yu G, Wu Y, Wang W, Xu J, Lv X, Cao X, et al. Low-dose decitabine enhances the effect of PD-1 blockade in colorectal cancer with microsatellite stability by remodulating the tumor microenvironment. Cell Mol Immunol. 2019;16:401-9.
- Chien KS, Kim K, Nogueras-Gonzalez GM, Borthakur G, Naqvi K, Daver NG, et al. Phase II study of azacitidine with pembrolizumab in patients with intermediate1 or higher-risk myelodysplastic syndrome. Br J Haematol. 2021;195:378-87.
- Yuan Z, Chen S, Gao C, Dai Q, Zhang C, Sun Q, et al. Development of a versatile DNMT and HDAC inhibitor CO2S modulating multiple cancer hallmarks for breast cancer therapy. Bioorg Chem. 2019;87:200-8.
- Nepali K, Liou JP. Recent developments in epigenetic cancer therapeutics: clinical advancement and emerging trends. J Biomed Sci. 2021;28:27.
- Romanelli A, Stazi G, Fioravanti R, Zwergel C, Di Bello E, Pomella S, et al. Design of first-in-class dual EZH2/HDAC inhibitor: biochemical activity and biological evaluation in cancer cells. ACS Med Chem Lett. 2020;11:977-83.
- Konig IR, Fuchs O, Hansen G, von Mutius E, Kopp MV. What is precision medicine? Eur Respir J. 2017;50:1700391.
- Balloux F, Bronstad Brynildsrud O, van Dorp L, Shaw LP, Chen H, Harris KA, et al. From theory to practice: translating Whole-Genome Sequencing (WGS) into the clinic. Trends Microbiol. 2018;26:1035-48.
- Nicot C. RNA-seq reveals novel CircRNAs involved in breast cancer progression and patient therapy response. Mol Cancer. 2020;19:76.
- Nikanjam M, Kato S, Kurzrock R. Liquid biopsy: current technology and clinical applications. J Hematol Oncol. 2022;15:131.
- Corces MR, Granja JM, Shams S, Louie BH, Seoane JA, Zhou W, et al. The chromatin accessibility landscape of primary human cancers. Science 2018;362:eaav1898.
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10:1213-8.
- Pott S, Lieb JD. What are super-enhancers? Nat Genet. 2015;47:8-12.
- Ren B, Yang J, Wang C, Yang G, Wang H, Chen Y, et al. High-resolution Hi-C maps highlight multiscale 3D epigenome reprogramming during pancreatic cancer metastasis. J Hematol Oncol. 2021;14:120.
- Bi WL, Hosny A, Schabath MB, Giger ML, Birkbak NJ, Mehrtash A, et al. Artificial intelligence in cancer imaging: Clinical challenges and applications. CA Cancer J Clin. 2019;69:127-57.
- Gur D, Sumkin JH, Rockette HE, Ganott M, Hakim C, Hardesty L, et al. Changes in breast cancer detection and mammography recall rates after the introduction of a computer-aided detection system. J Natl Cancer Inst. 2004;96:185-90.
- Nishikawa RM, Gur D. CADe for early detection of breast cancer-current status and why we need to continue to explore new approaches. Acad Radio. 2014;21:1320-1.
- Campanella G, Hanna MG, Geneslaw L, Miraflor A, Werneck Krauss Silva V, Busam KJ, et al. Clinical-grade computational pathology using weakly supervised deep learning on whole slide images. Nat Med. 2019;25:1301-9.
- Coudray N, Ocampo PS, Sakellaropoulos T, Narula N, Snuderl M, Fenyo D, et al. Classification and mutation prediction from non-small cell lung cancer histopathology images using deep learning. Nat Med. 2018;24:1559-67.
- Sun Y, Zhu S, Ma K, Liu W, Yue Y, Hu G, et al. Identification of 12 cancer types through genome deep learning. Sci Rep. 2019;9:17256.
- Libbrecht MW, Noble WS. Machine learning applications in genetics and genomics. Nat Rev Genet. 2015;16:321-32.
- Ernst J, Kellis M. Large-scale imputation of epigenomic datasets for systematic annotation of diverse human tissues. Nat Biotechnol. 2015;33:364-76.
- Durham TJ, Libbrecht MW, Howbert JJ, Bilmes J, Noble WS. PREDICTD PaRallel Epigenomics Data Imputation with Cloud-based Tensor Decomposition. Nat Commun. 2018;9:1402.
- Schreiber J, Durham T, Bilmes J, Noble WS. Avocado: a multi-scale deep tensor factorization method learns a latent representation of the human epigenome. Genome Biol. 2020;21:81.
- Bhinder B, Gilvary C, Madhukar NS, Elemento O. Artificial intelligence in cancer research and precision medicine. Cancer Discov. 2021;11:900-15.
شكر وتقدير
مساهمات المؤلفين
الجدول والأشكال. قام أنغوي بينغ بمراجعة المخطوطة وأخيرًا وافق على النسخة التي ستُنشر.
تمويل
المصالح المتنافسة
معلومات إضافية
© المؤلف(ون) 2024
مختبر قوانغدونغ الإقليمي الرئيسي لتشخيص وعلاج التدخل الورمي، معهد تشجيع الطب التحويلي في تشوهاي، (مستشفى الشعب في تشوهاي، كلية الطب السريرية بجامعة جينان)، تشوهاي 519000، الصين. قسم جراحة العمود الفقري، مستشفى ييتشانغ المركزي الشعبي التابع لجامعة الصين ثلاث شلالات، ييتشانغ، هوبي 443000، الصين. قسم الصيدلة، مستشفى الشعب في تشوهاي، مستشفى الشعب في تشوهاي (كلية الطب السريري في جامعة جينان)، تشوهاي، قوانغدونغ 519000، الصين. ساهم هؤلاء المؤلفون بالتساوي: شين يانغ يو، هاو تشاو. البريد الإلكتروني:syh@alumni.tongji.edu.cn; m13387383303@163.com
DOI: https://doi.org/10.1038/s41420-024-01803-z
PMID: https://pubmed.ncbi.nlm.nih.gov/38225241
Publication Date: 2024-01-15
Cancer epigenetics: from laboratory studies and clinical trials to precision medicine
Abstract
Epigenetic dysregulation is a common feature of a myriad of human diseases, particularly cancer. Defining the epigenetic defects associated with malignant tumors has become a focus of cancer research resulting in the gradual elucidation of cancer cell epigenetic regulation. In fact, most stages of tumor progression, including tumorigenesis, promotion, progression, and recurrence are accompanied by epigenetic alterations, some of which can be reversed by epigenetic drugs. The main objective of epigenetic therapy in the era of personalized precision medicine is to detect cancer biomarkers to improve risk assessment, diagnosis, and targeted treatment interventions. Rapid technological advancements streamlining the characterization of molecular epigenetic changes associated with cancers have propelled epigenetic drug research and development. This review summarizes the main mechanisms of epigenetic dysregulation and discusses past and present examples of epigenetic inhibitors in cancer diagnosis and treatment, with an emphasis on the development of epigenetic enzyme inhibitors or drugs. In the final part, the prospect of precise diagnosis and treatment is considered based on a better understanding of epigenetic abnormalities in cancer.
FACTS
- Epigenetic regulatory mechanisms involve cancer biology, especially DNA methylation, histone acetylation, and miRNAs.
- The expression of tumor-related genes is closely related to the epigenetic regulatory process of tumors.
- DNMTi, HDACis, BETis and other epigenetic therapies are constantly being updated and used in the clinic.
- Epigenetic combination therapy is a promising direction.
- Multi-omics, gene therapy, and AI are favorable transitions from epigenetic therapy to precision medicine.
OPEN QUESTIONS
- Can the therapeutic efficacy of solid tumors be improved by combining therapies targeting different epigenetic markers?
- Which changes measured in epigenetic cancer precision diagnosis and treatment are temporary, and which are true tumor biomarkers?
- How to compare the results in personalized treatment when the conclusions of laboratory studies and clinical studies are contradictory?
INTRODUCTION
MECHANISMS OF EPIGENETIC DYSREGULATION IN CANCER
DNA methylation
contributes to the increased expression of oncogenes [24, 25]. Mutation and inactivation of tumor suppressor genes can lead to DNA damage or uncontrolled cell growth, thereby promoting cancer progression [26]. Additionally, the potential reversibility of methyltransferase activity makes it an attractive target for therapeutic interventions, unlike genetic changes.
Histone methylations
Histone acetylation
(including HDAC1, HDAC2, HDAC3, and HDAC8) that are primarily localized in the nucleus and are generally expressed in the human genome. Class II HDACs are in the nucleus and cytoplasm and exhibit tissue-specific expression patterns and non-histone deacetylation activity. They are further divided into two subclasses, Ila (HDAC4, 5, 7, and 9) and Ilb (HDAC6 and 10) [44]. Class III HDACs are comprised of SIR2-like proteins (including SIRT1, SIRT2, SIRT3, SIRT4, SIRT5, SIRT6, and SIRT7) that are involved in regulating multiple cellular processes such as survival, aging, stress response, and metabolism [43]. Class IV HDACs only comprise HDAC11, which shares partial homology with class I and II HDACs and acts as a long-chain fatty acid deacylase [45]. Class I, II, and IV HDACs rely on zinc-binding substrates and
RNA epigenetics
breast cancer and miR-124a in colorectal tumors relative to normal tissues [64, 65]. Epigenetic silencing of miRNAs may reflect tissue specificity. Tissue- and cell-type-specific expression of miRNAs widely affect cell differentiation, cycling, aging, and metabolism [66]. Hence, the analysis of miRNA expression in tumor tissues or liquid biopsies can help direct cancer diagnosis, predict patient prognosis, and identify potential therapeutic targets.
EPIGENETIC ANTICANCER STRATEGIES
DNMT Inhibitors
HDAC inhibitors
X. Yu et al.
cells in preclinical studies. This results in the approval of several drugs to treat certain hematologic malignancies.
BET family inhibitors
| Target | Inhibitor | Associated cancer | Clinical status | Reference |
| DNMT | Azacitidine | Therapy-related myeloid neoplasms | Achieves disease response | [71] |
| Decitabine | AML | Achieves disease response and better overall survival | [72] | |
| SGI-110 | AML | Achieves disease response | [73] | |
| CP-4200 | AML | Causes efficient reactivation of epigenetically silenced tumor suppressor genes | [74] | |
| MG98 | Renal cell carcinoma | Inhibits the proliferation of growing cancer cells | [75] | |
| Nanaomycin A | Hepatocellular carcinoma | Exhibits antiproliferative effects | [76] | |
| HDAC | Vorinostat (SAHA) | T-cell lymphoma | Determines complete and partial response rates | [80] |
| Romidepsin (depsipeptide) | CTCL | Achieves disease response | [81] | |
| Belinostat | Peripheral T-cell lymphoma | Achieves disease response | [82] | |
| Panobinostat | MM | Achieves disease response | [83] | |
| TSA | Breast cancer | Has greater specificity for cancer vs normal cells | [84] | |
| Tubacin | Melanoma, CRC | Increases the extracellular release of a cancer stem cell marker | [85] | |
| MC1568, MC1575 | Breast cancer, melanoma | Exhibits antiproliferative effects | [86, 87] | |
| Ricolinostat (ACY1215) | GBM | Inhibits tumor cell growth | [88] | |
| IN-2001 | Breast cancer | Suppresses tumor growth | [89] | |
| AR-42 | Meningioma | Increases proapoptotic gene expression and decreases antiapoptotic protein levels | [90] | |
| Givinostat (ITF2357) | BCP-ALL | Inhibits the proliferation and induces apoptosis | [91] | |
| BET | JQ1 | MM, AML, DLBCL, prostate cancer, breast cancer | Produces a potent antiproliferative effect associated with cell-cycle arrest and cellular senescence, terminal myeloid differentiation, and elimination of leukemia stem cells | [94-98] |
| OTX015 (MK-8628) | B-cell lymphoma, neuroblastoma | Inhibits the proliferation of cancer cells; downregulates c-Myc, MYCN, and other oncogenes associated with superenhancers | [99, 100] | |
| MS645 | Triple-negative breast cancer | Inhibits cancer cell proliferation | [101] | |
| ABBV-075 | AML, non-Hodgkin lymphoma, MM | Triggers apoptosis | [102] | |
| ABBV-744 | Prostate cancer | Displaces BRD4 from AR-containing super-enhancers and Inhibits AR-dependent transcription | [103] | |
| I-BET151 | GBM | Inhibits GBM cell proliferation | [105] | |
| CC-90011 | Solid tumors | Achieves complete response or partial response; prolongs stable disease | [104] | |
| I-BET 762 | Pancreatic cancer | Hinders multiple pathways associated with cell growth | [107] | |
| KDM | ORY-1001 | AML | Reduces the growth of cancer cells | [108] |
| KMT | BIX-01294 | DLBCL, neuroblastoma | Inhibits cell proliferation and induces apoptosis of cancer cells | [109, 110] |
| UNC0638 | non-SCLC | Inhibits cell growth and induces apoptosis | [111] | |
| Pinometostat | MLL | Inhibits the proliferation of leukemia cell lines harboring MLL-r and induced sustained regressions | [112] | |
| EPZ004777 | MLL | Selective kills cells bearing the MLL gene translocation | [113] | |
| GSK126 | Myeloid-derived suppressor cells | Inhibits the growth of tumor cells | [114] |
lymphoma, and MM cells [102], whereas ABBV-744 selectively targets the BD2 domain, displaces BRD4 from androgen receptor (AR)-containing super-enhancers, and inhibits AR-dependent transcription. This shows better antitumor activity than ABBV075 in a mouse xenograft model using human prostate cancer cells [103]. CC-90010 is another next-generation BET inhibitor with
encouraging antitumor activity in patients with advanced solid tumors [104]. The abundances of BRD2 and BRD4 are significantly increased in GBM. Therefore, treatment with BET protein inhibitor (I-BET151) inhibits GBM cell proliferation [105]. Moreover, OTX015 exhibits a higher antiproliferative effect than its analog (JQ1) in GBM cell lines [106]. Meanwhile, JQ1 and I-BET 762 effectively
| Combination forms | Combination therapeutics | Associated cancer | Advantages | Reference |
| DNMTi+HDACi | azacytidine + vorinostat | MDS; CMML | more effective than monotherapy | [116] |
| azacytidine + entinostat | CRC | improved antitumor activity | [117] | |
| Epi-drug + targeted drug | ACY-1215 + bortezomib | MM | delayed tumor growth; prolonged survival | [121] |
| TSA + palladium nanoparticles | cervical cancer | increased the potential for successful treatment | [122] | |
| JQ1
|
T-ALL | countered the resistance of
|
[123] | |
| Epi-drug + immunomodulators | Panobinostat + bortezomib+dexamethasone | MM | improved progression-free survival | [126] |
| PD-1 blockers +Decitabine | CRC | inhibited tumor growth; prolonged survival | [127] | |
| Azacytidine+pembrolizumab | MDS | safe with controllable toxicity | [128] |
interfere with multiple pathways associated with cell growth in pancreatic cancer [107]. Overall, the traditional small-molecule BRD4 inhibitor JQ1 and the newly developed BET inhibitors show promising results in various human cancers.
KMT & KDM inhibitors
Combination therapy strategies
combination of JQ1 and
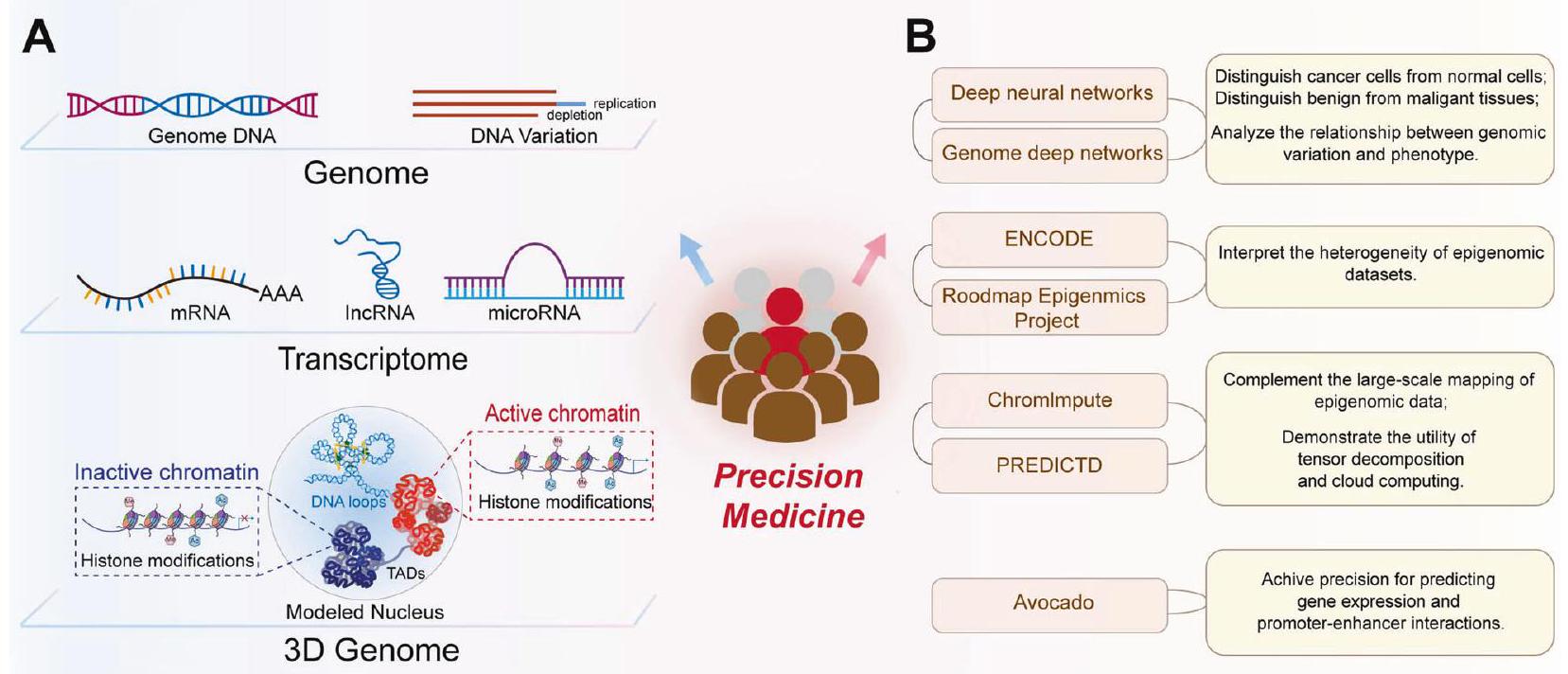
PERSPECTIVES IN PRECISION MEDICINE
technologies that can profile genomics, transcriptomics, and 3D genomics and elucidate the interactions between genetic and epigenetic changes in cancer biology, are effective in precise disease management and prediction. Whole-genome and wholeexome sequencing can trace genomic variations in different tumors [133]. Meanwhile, RNA-seq can capture microarray transcription profiles and dissect tumor heterogeneity [134]. Currently, whole-genome bisulfite sequencing is the gold standard method for analyzing DNA methylation data on a genome-wide scale at single-base resolution [135]. Additionally, chromatin accessibility affects the DNA binding to transcription factors and regulatory elements. This provides important insights into the mechanisms by which cancer genomes are activated and silenced [136]. The assay for transposase-accessible chromatin using sequencing (ATAC-seq) can assess the chromatin accessibility landscape of primary human cancers [137]. Meanwhile, interference with the 3D structure of the genome can lead to ectopic oncogene activation by interacting with proximal or distant enhancers and promoter regions that initiate oncogene transcription. Thus, chromatin immunoprecipitation and sequencing (ChIP-seq) technology can be employed to identify activity enhancers and super-enhancers on a genome-wide scale based on H3K4me1 and H3K27ac histone markers, respectively [138]. Additionally, high-throughput genome-wide chromatin spatial capture (HiC) analysis can identify abnormal enhancer-promoter interactions throughout the tumor genome [139]. The rapid development of these technologies is conducive to defining the epigenomic panorama of cancer and facilitates further integration of epigenomic indicators in clinical applications. Indeed, the combination of epigenetic techniques with more commonly used platforms (such as whole-genome sequencing and RNA-seq) will provide comprehensive insights regarding genomic and epigenomic abnormalities in patients with cancer. Additionally, the application of multi-omics approaches to identify diagnostic biomarkers will improve therapeutic interventions by providing data for personalized treatment.
machine learning model, Avocado, which is a combination of tensor factorization and deep neural networks that compresses epigenomic data into dense, information-rich representations [149]. Avocado outperforms models trained directly on epigenomic data in a variety of genomic tasks and achieves high precision predictions of gene expression and promoter-enhancer interactions, including frequently interacting regions in HiC data, replication time, and 3D chromatin structure. Al can detect small amounts of biomarkers, significantly improve early cancer diagnosis and personalize clinical care while aiding in the discovery of new anticancer drugs. The maturation of epigenomic analysis provides synergistic opportunities for Al-based imaging efforts [150]. In general, the application of AI in precision cancer diagnosis and treatment presents broad prospects, resulting in the development of intelligent cancer treatments.
CONCLUSION
Reporting summary
REFERENCES
- Quina AS, Buschbeck M, Di Croce L. Chromatin structure and epigenetics. Biochem Pharm. 2006;72:1563-9.
- Peng A, Peng W, Wang R, Zhao H, Yu X, Sun Y. Regulation of 3D organization and its role in cancer biology. Front Cell Dev Biol. 2022;10:879465.
- Li E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat Rev Genet. 2002;3:662-73.
- Mohammad HP, Barbash O, Creasy CL. Targeting epigenetic modifications in cancer therapy: erasing the roadmap to cancer. Nat Med. 2019;25:403-18.
- Rutten BP, Mill J. Epigenetic mediation of environmental influences in major psychotic disorders. Schizophr Bull. 2009;35:1045-56.
- Kanwal R, Gupta K, Gupta S. Cancer epigenetics: an introduction. Methods Mol Biol. 2015;1238:3-25.
- Chen Y, Hong T, Wang S, Mo J, Tian T, Zhou X. Epigenetic modification of nucleic acids: from basic studies to medical applications. Chem Soc Rev. 2017;46:2844-72.
- Singh M, Kumar V, Sehrawat N, Yadav M, Chaudhary M, Upadhyay SK, et al. Current paradigms in epigenetic anticancer therapeutics and future challenges. Semin Cancer Biol. 2022;83:422-40.
- Biswas S, Rao CM. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur J Pharm. 2018;837:8-24.
- Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology 2013;38:23-38.
- Robert MF, Morin S, Beaulieu N, Gauthier F, Chute IC, Barsalou A, et al. DNMT1 is required to maintain CpG methylation and aberrant gene silencing in human cancer cells. Nat Genet. 2003;33:61-5.
- Foulks JM, Parnell KM, Nix RN, Chau S, Swierczek K, Saunders M, et al. Epigenetic drug discovery: targeting DNA methyltransferases. J Biomol Screen. 2012;17:2-17.
- Chen T, Hevi S, Gay F, Tsujimoto N, He T, Zhang B, et al. Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nat Genet. 2007;39:391-6.
- Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999;99:247-57.
- Simo-Riudalbas L, Melo SA, Esteller M. DNMT3B gene amplification predicts resistance to DNA demethylating drugs. Genes Chromosomes Cancer. 2011;50:527-34.
- Stewart ML, Tamayo P, Wilson AJ, Wang S, Chang YM, Kim JW, et al. KRAS genomic status predicts the sensitivity of ovarian cancer cells to Decitabine. Cancer Res. 2015;75:2897-906.
- Baylin SB. DNA methylation and gene silencing in cancer. Nat Clin Pr Oncol. 2005;2:S4-11.
- Unoki M, Brunet J, Mousli M. Drug discovery targeting epigenetic codes: the great potential of UHRF1, which links DNA methylation and histone modifications, as a drug target in cancers and toxoplasmosis. Biochem Pharm. 2009;78:1279-88.
- Yamashita K, Dai T, Dai Y, Yamamoto F, Perucho M. Genetics supersedes epigenetics in colon cancer phenotype. Cancer Cell. 2003;4:121-31.
- Laux DE, Curran EM, Welshons WV, Lubahn DB, Huang TH. Hypermethylation of the Wilms’ tumor suppressor gene CpG island in human breast carcinomas. Breast Cancer Res Treat. 1999;56:35-43.
- Arechederra M, Daian F, Yim A, Bazai SK, Richelme S, Dono R, et al. Hypermethylation of gene body CpG islands predicts high dosage of functional oncogenes in liver cancer. Nat Commun. 2018;9:3164.
- Yegnasubramanian S, Kowalski J, Gonzalgo ML, Zahurak M, Piantadosi S, Walsh PC, et al. Hypermethylation of CpG islands in primary and metastatic human prostate cancer. Cancer Res. 2004;64:1975-86.
- Abbosh PH, Wang M, Eble JN, Lopez-Beltran A, Maclennan GT, Montironi R, et al. Hypermethylation of tumor-suppressor gene CpG islands in small-cell carcinoma of the urinary bladder. Mod Pathol. 2008;21:355-62.
- Ando M, Saito Y, Xu G, Bui NQ, Medetgul-Ernar K, Pu M, et al. Chromatin dysregulation and DNA methylation at transcription start sites associated with transcriptional repression in cancers. Nat Commun. 2019;10:2188.
- Timp W, Feinberg AP. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat Rev Cancer. 2013;13:497-510.
- Corradini P, Inghirami G, Astolfi M, Ladetto M, Voena C, Ballerini P, et al. Inactivation of tumor suppressor genes, p53 and Rb1, in plasma cell dyscrasias. Leukemia 1994;8:758-67.
- Hyun K, Jeon J, Park K, Kim J. Writing, erasing and reading histone lysine methylations. Exp Mol Med. 2017;49:e324.
- Husmann D, Gozani O. Histone lysine methyltransferases in biology and disease. Nat Struct Mol Biol. 2019;26:880-9.
- Sterling J, Menezes SV, Abbassi RH, Munoz L Histone lysine demethylases and their functions in cancer. Int J Cancer. 2020;148:2375-88.
- Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell. 2012;48:491-507.
- Alam H, Tang M, Maitituoheti M, Dhar SS, Kumar M, Han CY, et al. KMT2D deficiency impairs super-enhancers to confer a glycolytic vulnerability in lung cancer. Cancer Cell. 2020;37:599-617.e7.
- Duan R, Du W, Guo W. EZH2: a novel target for cancer treatment. J Hematol Oncol. 2020;13:104.
- Nguyen AT, Zhang Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011;25:1345-58.
- Kurani H, Razavipour SF, Harikumar KB, Dunworth M, Ewald AJ, Nasir A, et al. DOT1L is a novel cancer stem cell target for triple-negative breast cancer. Clin Cancer Res. 2022;28:1948-65.
- Griffin GK, Wu J, Iracheta-Vellve A, Patti JC, Hsu J, Davis T, et al. Epigenetic silencing by SETDB1 suppresses tumour intrinsic immunogenicity. Nature 2021;595:309-14.
- Nachiyappan A, Gupta N, Taneja R. EHMT1/EHMT2 in EMT, cancer stemness and drug resistance: emerging evidence and mechanisms. FEBS
. 2022;289:1329-51. - Vicioso-Mantis M, Aguirre S, Martinez-Balbas MA. JmjC family of Histone Demethylases form nuclear condensates. Int J Mol Sci. 2022;23:7664.
- Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, et al. LSD1 ablation stimulates anti-tumor immunity and enables checkpoint blockade. Cell 2018;174:549-63.e19.
- Shen DD, Pang JR, Bi YP, Zhao LF, Li YR, Zhao L, et al. LSD1 deletion decreases exosomal PD-L1 and restores T-cell response in gastric cancer. Mol Cancer. 2022;21:75.
- Musella M, Guarracino A, Manduca N, Galassi C, Ruggiero E, Potenza A, et al. Type I IFNs promote cancer cell stemness by triggering the epigenetic regulator KDM1B. Nat Immunol. 2022;23:1379-92.
- Goswami S, Raychaudhuri D, Singh P, Natarajan SM, Chen Y, Poon C, et al. Myeloid-specific KDM6B inhibition sensitizes glioblastoma to PD1 blockade. Nat Cancer. 2023;4:1455-73.
- Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381-95.
- Li Y, Seto E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb Perspect Med. 2016;6:a026831.
- Seto E, Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol. 2014;6:a018713.
- Nunez-Alvarez Y, Suelves M. HDAC11: a multifaceted histone deacetylase with proficient fatty deacylase activity and its roles in physiological processes. FEBS J. 2022;289:2771-92.
- Zheng W. The Zinc-dependent HDACs: Non-histone substrates and catalytic deacylation beyond deacetylation. Mini Rev Med Chem. 2022;22:2478-85.
- Marks PA, Richon VM, Rifkind RA. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J Natl Cancer Inst. 2000;92:1210-6.
- Wu CC, Jin LW, Wang IF, Wei WY, Ho PC, Liu YC, et al. HDAC1 dysregulation induces aberrant cell cycle and DNA damage in progress of TDP-43 proteinopathies. EMBO Mol Med. 2020;12:e10622.
- Kim MS, Kwon HJ, Lee YM, Baek JH, Jang JE, Lee SW, et al. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat Med. 2001;7:437-43.
- Hanigan CL, Van Engeland M, De Bruine AP, Wouters KA, Weijenberg MP, Eshleman JR, et al. An inactivating mutation in HDAC2 leads to dysregulation of apoptosis mediated by APAF1. Gastroenterology 2008;135:1654-64.e2.
- Barbieri I, Kouzarides T. Role of RNA modifications in cancer. Nat Rev Cancer. 2020;20:303-22.
- Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA modifications in gene expression regulation. Cell 2017;169:1187-200.
- Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, et al. m(6)A RNA methylation regulates the self-renewal and Tumorigenesis of Glioblastoma stem cells. Cell Rep. 2017;18:2622-34.
- Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, et al. FTO plays an oncogenic role in acute myeloid leukemia as a N(6)-Methyladenosine RNA Demethylase. Cancer Cell. 2017;31:127-41.
- Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5mediated m(6)A-demethylation of NANOG mRNA. Proc Natl Acad Sci USA. 2016;113:E2047-56.
- Statello L, Guo CJ, Chen LL, Huarte M. Gene regulation by long non-coding RNAs and its biological functions. Nat Rev Mol Cell Biol. 2021;22:96-118.
- Battistelli C, Garbo S, Riccioni V, Montaldo C, Santangelo L, Vandelli A, et al. Design and functional validation of a mutant variant of the LncRNA HOTAIR to counteract snail function in epithelial-to-mesenchymal transition. Cancer Res. 2021;81:103-13.
- Wurm AA, Pina C. Long non-coding RNAs as functional and structural chromatin modulators in acute myeloid leukemia. Front Oncol. 2019;9:899.
- Zhu S, Wang JZ, Chen D, He YT, Meng N, Chen M, et al. An oncopeptide regulates m (6)A recognition by the m (6)A reader IGF2BP1 and tumorigenesis. Nat Commun. 2020;11:1685.
- Saito Y, Suzuki H, Tsugawa H, Nakagawa I, Matsuzaki J, Kanai Y, et al. Chromatin remodeling at Alu repeats by epigenetic treatment activates silenced microRNA-512-5p with downregulation of Mcl-1 in human gastric cancer cells. Oncogene. 2009;28:2738-44.
- Garzon R, Liu S, Fabbri M, Liu Z, Heaphy CE, Callegari E, et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood. 2009;113:6411-8.
- Iorio MV, Piovan C, Croce CM. Interplay between microRNAs and the epigenetic machinery: an intricate network. Biochim Biophys Acta. 2010;1799:694-701.
- Lee YS, Dutta A. MicroRNAs in cancer. Annu Rev Pathol. 2009;4:199-227.
- Lehmann U, Hasemeier B, Christgen M, Muller M, Romermann D, Langer F, et al. Epigenetic inactivation of microRNA gene hsa-mir-9-1 in human breast cancer. J Pathol. 2008;214:17-24.
- Lujambio A, Ropero S, Ballestar E, Fraga MF, Cerrato C, Setien F, et al. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 2007;67:1424-9.
- Fasihi-Ramandi M, Moridnia A, Najafi A, Sharifi M. Inducing cell proliferative prevention in human acute promyelocytic leukemia by miR-182 inhibition through modulation of CASP9 expression. Biomed Pharmacother. 2017;89:1152-8.
- Cheng Y, He C, Wang M, Ma X, Mo F, Yang S, et al. Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Signal Transduct Target Ther. 2019;4:62.
- Schuh AC, Dohner H, Pleyer L, Seymour JF, Fenaux P, Dombret H. Azacitidine in adult patients with acute myeloid leukemia. Crit Rev Oncol Hematol. 2017;116:159-77.
- Klimek VM, Dolezal EK, Tees MT, Devlin SM, Stein K, Romero A, et al. Efficacy of hypomethylating agents in therapy-related myelodysplastic syndromes. Leuk Res. 2012;36:1093-7.
- Sato T, Cesaroni M, Chung W, Panjarian S, Tran A, Madzo J, et al. Transcriptional selectivity of epigenetic therapy in cancer. Cancer Res. 2017;77:470-81.
- Fianchi L, Criscuolo M, Lunghi M, Gaidano G, Breccia M, Levis A, et al. Outcome of therapy-related myeloid neoplasms treated with azacitidine. J Hematol Oncol. 2012;5:44.
- Fili C, Candoni A, Zannier ME, Olivieri J, Imbergamo S, Caizzi M, et al. Efficacy and toxicity of Decitabine in patients with acute myeloid leukemia (AML): A multicenter real-world experience. Leuk Res. 2019;76:33-8.
- Kantarjian HM, Roboz GJ, Kropf PL, Yee KWL, O’Connell CL, Tibes R, et al. Guadecitabine (SGI-110) in treatment-naive patients with acute myeloid leukaemia: phase 2 results from a multicentre, randomised, phase
trial. Lancet Oncol. 2017;18:1317-26. - Brueckner B, Rius M, Markelova MR, Fichtner I, Hals PA, Sandvold ML, et al. Delivery of 5-azacytidine to human cancer cells by elaidic acid esterification increases therapeutic drug efficacy. Mol Cancer Ther. 2010;9:1256-64.
- Amato RJ, Stephenson J, Hotte S, Nemunaitis J, Belanger K, Reid G, et al. MG98, a second-generation DNMT1 inhibitor, in the treatment of advanced renal cell carcinoma. Cancer Invest. 2012;30:415-21.
- Lai SC, Su YT, Chi CC, Kuo YC, Lee KF, Wu YC, et al. Correction to: DNMT3b/OCT4 expression confers sorafenib resistance and poor prognosis of hepatocellular carcinoma through IL-6/STAT3 regulation. J Exp Clin Cancer Res. 2020;39:10.
- Kazanets A, Shorstova T, Hilmi K, Marques M, Witcher M. Epigenetic silencing of tumor suppressor genes: Paradigms, puzzles, and potential. Biochim Biophys Acta. 2016;1865:275-88.
- Matthews GM, Newbold A, Johnstone RW. Intrinsic and extrinsic apoptotic pathway signaling as determinants of histone deacetylase inhibitor antitumor activity. Adv Cancer Res. 2012;116:165-97.
- Yang H, Sun B, Xu K, He Y, Zhang T, Hall SRR, et al. Pharmaco-transcriptomic correlation analysis reveals novel responsive signatures to HDAC inhibitors and identifies Dasatinib as a synergistic interactor in small-cell lung cancer. EBioMedicine. 2021;69:103457.
- Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2007;109:31-9.
- Whittaker SJ, Demierre MF, Kim EJ, Rook AH, Lerner A, Duvic M, et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol. 2010;28:4485-91.
- Lee HZ, Kwitkowski VE, Del Valle PL, Ricci MS, Saber H, Habtemariam BA, et al. FDA Approval: Belinostat for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma. Clin Cancer Res. 2015;21:2666-70.
- Garnock-Jones KP. Panobinostat: first global approval. Drugs 2015;75:695-704.
- Chang J, Varghese DS, Gillam MC, Peyton M, Modi B, Schiltz RL, et al. Differential response of cancer cells to HDAC inhibitors trichostatin A and depsipeptide. Br J Cancer. 2012;106:116-25.
- Chao OS, Chang TC, Di Bella MA, Alessandro R, Anzanello F, Rappa G, et al. The HDAC6 inhibitor tubacin induces release of CD133(+) extracellular vesicles from cancer cells. J Cell Biochem. 2017;118:4414-24.
- Duong V, Bret C, Altucci L, Mai A, Duraffourd C, Loubersac J, et al. Specific activity of class II histone deacetylases in human breast cancer cells. Mol Cancer Res. 2008;6:1908-19.
- Venza I, Visalli M, Oteri R, Cucinotta M, Teti D, Venza M. Class II-specific histone deacetylase inhibitors MC1568 and MC1575 suppress IL-8 expression in human melanoma cells. Pigment Cell Melanoma Res. 2013;26:193-204.
- Li S, Liu X, Chen X, Zhang L, Wang X. Histone deacetylase 6 promotes growth of glioblastoma through inhibition of SMAD2 signaling. Tumour Biol. 2015;36:9661-5.
- Joung KE, Min KN, An JY, Kim DK, Kong G, Sheen YY. Potent in vivo anti-breast cancer activity of IN-2001, a novel inhibitor of histone deacetylase, in MMTV/cNeu mice. Cancer Res. 2006;66:5394-402.
- Burns SS, Akhmametyeva EM, Oblinger JL, Bush ML, Huang J, Senner V, et al. Histone deacetylase inhibitor AR-42 differentially affects cell-cycle transit in meningeal and meningioma cells, potently inhibiting NF2-deficient meningioma growth. Cancer Res. 2013;73:792-803.
- Savino AM, Sarno J, Trentin L, Vieri M, Fazio G, Bardini M, et al. The histone deacetylase inhibitor givinostat (ITF2357) exhibits potent anti-tumor activity against CRLF2-rearranged BCP-ALL. Leukemia 2017;31:2365-75.
- Gilan O, Rioja I, Knezevic K, Bell MJ, Yeung MM, Harker NR, et al. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immunoinflammation. Science 2020;368:387-94.
- Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell. 2014;54:728-36.
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011;146:904-17.
- Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, et al. RNAi screen identifies Brd 4 as a therapeutic target in acute myeloid leukaemia. Nature 2011;478:524-8.
- Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell. 2013;24:777-90.
- Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014;510:278-82.
- Lu L, Chen Z, Lin X, Tian L, Su Q, An P, et al. Inhibition of BRD4 suppresses the malignancy of breast cancer cells via regulation of Snail. Cell Death Differ. 2020;27:255-68.
- Boi M, Gaudio E, Bonetti P, Kwee I, Bernasconi E, Tarantelli C, et al. The BET Bromodomain Inhibitor OTX015 Affects Pathogenetic Pathways in Preclinical B-cell Tumor Models and Synergizes with Targeted Drugs. Clin Cancer Res. 2015;21:1628-38.
- Henssen A, Althoff K, Odersky A, Beckers A, Koche R, Speleman F, et al. Targeting MYCN-driven transcription by BET-Bromodomain inhibition. Clin Cancer Res. 2016;22:2470-81.
- Ren C, Zhang G, Han F, Fu S, Cao Y, Zhang F, et al. Spatially constrained tandem bromodomain inhibition bolsters sustained repression of BRD4 transcriptional activity for TNBC cell growth. Proc Natl Acad Sci USA. 2018;115:7949-54.
- Bui MH, Lin X, Albert DH, Li L, Lam LT, Faivre EJ, et al. Preclinical characterization of BET family Bromodomain inhibitor ABBV-075 suggests combination therapeutic strategies. Cancer Res. 2017;77:2976-89.
- Faivre EJ, McDaniel KF, Albert DH, Mantena SR, Plotnik JP, Wilcox D, et al. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature 2020;578:306-10.
- Moreno V, Sepulveda JM, Vieito M, Hernandez-Guerrero T, Doger B, Saavedra O, et al. Phase I study of CC-90010, a reversible, oral BET inhibitor in patients with advanced solid tumors and relapsed/refractory non-Hodgkin’s lymphoma. Ann Oncol. 2020;31:780-8.
- Pastori C, Daniel M, Penas C, Volmar CH, Johnstone AL, Brothers SP, et al. BET bromodomain proteins are required for glioblastoma cell proliferation. Epigenetics 2014;9:611-20.
- Berenguer-Daize C, Astorgues-Xerri L, Odore E, Cayol M, Cvitkovic E, Noel K, et al. OTX015 (MK-8628), a novel BET inhibitor, displays in vitro and in vivo antitumor effects alone and in combination with conventional therapies in glioblastoma models. Int J Cancer. 2016;139:2047-55.
- Leal AS, Williams CR, Royce DB, Pioli PA, Sporn MB, Liby KT. Bromodomain inhibitors, JQ1 and I-BET 762, as potential therapies for pancreatic cancer. Cancer Lett. 2017;394:76-87.
- Maes T, Mascaro C, Tirapu I, Estiarte A, Ciceri F, Lunardi S, et al. ORY-1001, a potent and selective covalent KDM1A inhibitor, for the treatment of acute leukemia. Cancer Cell. 2018;33:495-511.e12.
- Xu L, Gao X, Yang P, Sang W, Jiao J, Niu M, et al. EHMT2 inhibitor BIX-01294 induces endoplasmic reticulum stress mediated apoptosis and autophagy in diffuse large B-cell lymphoma cells. J Cancer. 2021;12:1011-22.
- Lu Z, Tian Y, Salwen HR, Chlenski A, Godley LA, Raj JU, et al. Histone-lysine methyltransferase EHMT2 is involved in proliferation, apoptosis, cell invasion, and DNA methylation of human neuroblastoma cells. Anticancer Drugs. 2013;24:484-93.
- Wang L, Dong X, Ren Y, Luo J, Liu P, Su D, et al. Targeting EHMT2 reverses EGFRTKI resistance in NSCLC by epigenetically regulating the PTEN/AKT signaling pathway. Cell Death Dis. 2018;9:129.
- Stein EM, Garcia-Manero G, Rizzieri DA, Tibes R, Berdeja JG, Savona MR, et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 2018;131:2661-9.
- Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20:53-65.
- Huang S, Wang Z, Zhou J, Huang J, Zhou L, Luo J, et al. EZH2 inhibitor GSK126 suppresses antitumor immunity by driving production of myeloidderived suppressor cells. Cancer Res. 2019;79:2009-20.
- Mohammad F, Weissmann S, Leblanc B, Pandey DP, Hojfeldt JW, Comet I, et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat Med. 2017;23:483-92.
- Sekeres MA, Othus M, List AF, Odenike O, Stone RM, Gore SD, et al. Randomized Phase II study of azacitidine alone or in combination with lenalidomide or with vorinostat in higher-risk myelodysplastic syndromes and chronic myelomonocytic leukemia: North American Intergroup Study SWOG S1117. J Clin Oncol. 2017;35:2745-53.
- Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci USA. 2014;111:11774-9.
- Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet. 1999;21:103-7.
- Kalac M, Scotto L, Marchi E, Amengual J, Seshan VE, Bhagat G, et al. HDAC inhibitors and decitabine are highly synergistic and associated with unique gene-expression and epigenetic profiles in models of DLBCL. Blood 2011;118:5506-16.
- Bruyer A, Maes K, Herviou L, Kassambara A, Seckinger A, Cartron G, et al. DNMTi/ HDACi combined epigenetic targeted treatment induces reprogramming of myeloma cells in the direction of normal plasma cells. Br J Cancer. 2018;118:1062-73.
- Santo L, Hideshima T, Kung AL, Tseng JC, Tamang D, Yang M, et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 2012;119:2579-89.
- Zhang XF, Yan Q, Shen W, Gurunathan S. Trichostatin A enhances the apoptotic potential of palladium nanoparticles in human cervical cancer cells. Int J Mol Sci. 2016;17:1354.
- Knoechel B, Roderick JE, Williamson KE, Zhu J, Lohr JG, Cotton MJ, et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat Genet. 2014;46:364-70.
- Kuang C, Park Y, Augustin RC, Lin Y, Hartman DJ, Seigh L, et al. Pembrolizumab plus azacitidine in patients with chemotherapy refractory metastatic colorectal cancer: a single-arm phase 2 trial and correlative biomarker analysis. Clin Epigenet. 2022;14:3.
- Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 2015;162:974-86.
- San-Miguel JF, Hungria VT, Yoon SS, Beksac M, Dimopoulos MA, Elghandour A, et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014;15:1195-206.
- Yu G, Wu Y, Wang W, Xu J, Lv X, Cao X, et al. Low-dose decitabine enhances the effect of PD-1 blockade in colorectal cancer with microsatellite stability by remodulating the tumor microenvironment. Cell Mol Immunol. 2019;16:401-9.
- Chien KS, Kim K, Nogueras-Gonzalez GM, Borthakur G, Naqvi K, Daver NG, et al. Phase II study of azacitidine with pembrolizumab in patients with intermediate1 or higher-risk myelodysplastic syndrome. Br J Haematol. 2021;195:378-87.
- Yuan Z, Chen S, Gao C, Dai Q, Zhang C, Sun Q, et al. Development of a versatile DNMT and HDAC inhibitor CO2S modulating multiple cancer hallmarks for breast cancer therapy. Bioorg Chem. 2019;87:200-8.
- Nepali K, Liou JP. Recent developments in epigenetic cancer therapeutics: clinical advancement and emerging trends. J Biomed Sci. 2021;28:27.
- Romanelli A, Stazi G, Fioravanti R, Zwergel C, Di Bello E, Pomella S, et al. Design of first-in-class dual EZH2/HDAC inhibitor: biochemical activity and biological evaluation in cancer cells. ACS Med Chem Lett. 2020;11:977-83.
- Konig IR, Fuchs O, Hansen G, von Mutius E, Kopp MV. What is precision medicine? Eur Respir J. 2017;50:1700391.
- Balloux F, Bronstad Brynildsrud O, van Dorp L, Shaw LP, Chen H, Harris KA, et al. From theory to practice: translating Whole-Genome Sequencing (WGS) into the clinic. Trends Microbiol. 2018;26:1035-48.
- Nicot C. RNA-seq reveals novel CircRNAs involved in breast cancer progression and patient therapy response. Mol Cancer. 2020;19:76.
- Nikanjam M, Kato S, Kurzrock R. Liquid biopsy: current technology and clinical applications. J Hematol Oncol. 2022;15:131.
- Corces MR, Granja JM, Shams S, Louie BH, Seoane JA, Zhou W, et al. The chromatin accessibility landscape of primary human cancers. Science 2018;362:eaav1898.
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10:1213-8.
- Pott S, Lieb JD. What are super-enhancers? Nat Genet. 2015;47:8-12.
- Ren B, Yang J, Wang C, Yang G, Wang H, Chen Y, et al. High-resolution Hi-C maps highlight multiscale 3D epigenome reprogramming during pancreatic cancer metastasis. J Hematol Oncol. 2021;14:120.
- Bi WL, Hosny A, Schabath MB, Giger ML, Birkbak NJ, Mehrtash A, et al. Artificial intelligence in cancer imaging: Clinical challenges and applications. CA Cancer J Clin. 2019;69:127-57.
- Gur D, Sumkin JH, Rockette HE, Ganott M, Hakim C, Hardesty L, et al. Changes in breast cancer detection and mammography recall rates after the introduction of a computer-aided detection system. J Natl Cancer Inst. 2004;96:185-90.
- Nishikawa RM, Gur D. CADe for early detection of breast cancer-current status and why we need to continue to explore new approaches. Acad Radio. 2014;21:1320-1.
- Campanella G, Hanna MG, Geneslaw L, Miraflor A, Werneck Krauss Silva V, Busam KJ, et al. Clinical-grade computational pathology using weakly supervised deep learning on whole slide images. Nat Med. 2019;25:1301-9.
- Coudray N, Ocampo PS, Sakellaropoulos T, Narula N, Snuderl M, Fenyo D, et al. Classification and mutation prediction from non-small cell lung cancer histopathology images using deep learning. Nat Med. 2018;24:1559-67.
- Sun Y, Zhu S, Ma K, Liu W, Yue Y, Hu G, et al. Identification of 12 cancer types through genome deep learning. Sci Rep. 2019;9:17256.
- Libbrecht MW, Noble WS. Machine learning applications in genetics and genomics. Nat Rev Genet. 2015;16:321-32.
- Ernst J, Kellis M. Large-scale imputation of epigenomic datasets for systematic annotation of diverse human tissues. Nat Biotechnol. 2015;33:364-76.
- Durham TJ, Libbrecht MW, Howbert JJ, Bilmes J, Noble WS. PREDICTD PaRallel Epigenomics Data Imputation with Cloud-based Tensor Decomposition. Nat Commun. 2018;9:1402.
- Schreiber J, Durham T, Bilmes J, Noble WS. Avocado: a multi-scale deep tensor factorization method learns a latent representation of the human epigenome. Genome Biol. 2020;21:81.
- Bhinder B, Gilvary C, Madhukar NS, Elemento O. Artificial intelligence in cancer research and precision medicine. Cancer Discov. 2021;11:900-15.
ACKNOWLEDGEMENTS
AUTHOR CONTRIBUTIONS
table and figures. Anghui Peng reviewed the manuscript and finally approved the version to be published.
FUNDING
COMPETING INTERESTS
ADDITIONAL INFORMATION
© The Author(s) 2024
Guangdong Provincial Key Laboratory of Tumor Interventional Diagnosis and Treatment, Zhuhai Institute of Translational Medicine, (Zhuhai People’s Hospital Zhuhai Clinical Medical College of Jinan University), Zhuhai 519000, China. Department of Spinal Surgery, Yichang Central People’s Hospital Affiliated with China Three Gorges University, Yichang, Hubei 443000, China. Department of Pharmacy, Zhuhai People’s Hospital, Zhuhai People’s Hospital (Zhuhai Clinical Medical College of Jinan University), Zhuhai, Guangdong 519000, China. These authors contributed equally: Xinyang Yu, Hao Zhao. email: syh@alumni.tongji.edu.cn; m13387383303@163.com