في هذه الدراسة، نقدم محفزًا أحادي الذرة غير معدني فعال للغاية (SAC)، يُشار إليه باسم I-NC، والذي يتم احتجازه استراتيجيًا داخل هيكل كربوني مخدر بالنيتروجين (NC). تتميز هذه التهيئة بتنسيق C-I مميز يحسن الهيكل الإلكتروني لمواقع الكربون المجاورة للنيتروجين. ونتيجة لذلك، تعزز هذه الترتيبات نقل الإلكترون من بيروكسيمونوكبريتات (PMS) إلى المواقع النشطة، وخاصة الكربون الناقص الإلكترون. يتبع هذا النقل الإلكتروني عملية إزالة بروتون تنتج الجذري بيروكسيمونوكبريتات (). بعد ذلك، يخضع الجذري () لتفاعل عدم التناسب، مما يؤدي إلى إنتاج الأكسجين الأحادي (). علاوة على ذلك، فإن حاجز الطاقة للخطوة المحددة للسرعة في توليد () في I-NC أقل بكثير عند 1.45 eV، مقارنة بـ 1.65 eV في هيكل NC. هذا الانخفاض في حاجز الطاقة يتغلب بشكل فعال على العقبات الحركية، مما يسهل توليدًا معززًا لـ (). وبالتالي، يظهر محفز I-NC كفاءة تحفيزية ملحوظة وتفاعل لا مثيل له لتفعيل PMS. وهذا يؤدي إلى تسريع كبير في تحلل الملوثات، كما يتضح من ثابت معدل الحركية الملحوظ نسبيًا () مقارنةً بمحفزات SAC المعدنية الأخرى. تقدم هذه الدراسة رؤى قيمة في التصميم العقلاني لمحفزات SAC غير المعدنية الفعالة، مما يبرز إمكاناتها الواعدة لتفاعلات شبيهة بفنتون في تطبيقات معالجة المياه.
نظرًا للقدرة على إنتاج أنواع الأكسجين التفاعلية (ROS) عالية التفاعل على مدى واسع من قيم pH، ظهرت عمليات الأكسدة المتقدمة المعتمدة على PMS (PMSAOPs) التي تتميز بسهولة التعامل والتخزين والنقل للأكسيد كتقنيات واعدة لتنقية المياه. يبدأ تنشيط PMS عادةً بامتصاص جزيء PMS على سطح المحفز ()، والذي يحفز بعد ذلك تكوين أنواع تفاعلية مختلفة، بما في ذلك الجذور (مثل، جذور الهيدروكسيل، جذور الكبريتات، وجذور فوق أكسيد) وغير الجذور
(مثل، وأنواع المعادن عالية التكافؤ) . ومع ذلك، فإن فعالية الأنواع الجذرية تتأثر بتداخل الأنيونات والمواد العضوية الموجودة في المياه، مما يقلل بشكل كبير من استخدام PMS وقد يؤدي إلى تكوين منتجات ثانوية غير مرغوب فيها من الهاليدات . بالمقابل، توفر المسارات غير الجذرية آليات أكسدة انتقائية وقوية ، خاصة في حالة التي تسمح بالتحلل الانتقائي للملوثات بسبب خصوصيتها وطول عمرها ومقاومتها لمكونات المصفوفة .
من خلال الاستفادة من فوائد SACs، مثل الكفاءة الذرية القصوى، والمواقع النشطة المحددة، والهياكل الإلكترونية القابلة للتعديل، والنشاط المعزز، فقد حظيت باهتمام كبير في مجالات متعددة، بما في ذلك التحفيز الكهربائي، والتخليق الكيميائي، وإزالة التلوث البيئي . من الجدير بالذكر أن الدور الحاسم لـ SACs في توضيح العلاقة بين الهيكل والنشاط على المستوى الذري يبرز الإمكانات في PMS-AOPs . تميزت SACs المعدنية (M-SACs) بطبيعة المواقع النشطة، وقد أظهرت وعدًا في تحويل PMS إلى لتحلل الملوثات الدقيقة . ومع ذلك، فإن التكاليف العالية للإنتاج، وتعقيد التخليق، وخطر التلوث الثانوي للمحفزات المعدنية تشجعنا على تصميم SACs غير المعدنية (NM-SACs) لمعالجة المياه، والتي تعتبر أكثر جدوى اقتصاديًا وبيئيًا.
في الوقت الحاضر، لا تزال التحقيقات المتعلقة بـ NM-SACs في مراحلها الأولى، ويرجع ذلك أساسًا إلى الصعوبات في تحديد المواقع النشطة وتوضيح مسارات التحفيز. ومع ذلك، تكشف الأعمال الرائدة حول NM-SACs عن إمكاناتها في تعديل الهياكل الإلكترونية لتعزيز التفاعل الشبيه بفنتون. طور يانغ وآخرون NM-SAC يتكون من ذرات فوسفور معزولة عالية الكثافة مثبتة على حافة الجرافين كمحفز كهربائي قوي لـ الاختزال . ومع ذلك، بسبب القدرة المحدودة على زيادة النشاط الجوهري للمحفزات، فإن المحفزات المخدرة بالفوسفور تُستخدم بشكل أقل في PMSAOPs . عمومًا، هناك العديد من العناصر غير المعدنية في هذه الفئة، بما في ذلك الفلور، والكلور، والبروم، واليود (I)، التي يمكن أن تعمل كمرشحين فعالين لـ PMS-AOPs. من بينها، تمتلك ذرة I عدة مزايا ملحوظة. أولاً، الهيكل الذري لـ I يختلف بوضوح عن ذلك الخاص بالكربون، ويمكن أن يؤدي دمج I إلى تعديل الهيكل الإلكتروني لمصفوفة الكربون المجاورة بشكل فعال. بعد ذلك، يمكن أن يعزز إضافة I من موصلية الدعم، مما يسهل نقل الإلكترون بكفاءة بين المحفز وPMS . علاوة على ذلك، سيؤدي دمج ذرة I إلى المزيد من العيوب بسبب نصف قطرها الذري الأكبر من الكربون، مما يكشف عن المزيد من المواقع النشطة ويعزز نقل الكتلة لجزيئات PMS بشكل فعال . أخيرًا، يسمح الوزن الذري الأكبر لذرة I بتحسين تحديد مواقعها النشطة على المستوى الذري باستخدام تقنيات متقدمة مثل مجهر الإلكترون الناقل بتصحيح التشوهات ومطيافية الامتصاص بالأشعة السينية، مما يسهل إنشاء علاقة الهيكل والنشاط للمحفز بشكل أكثر شمولاً . بسبب خصائصه الفريدة، يظهر I كمرشح قوي لتعزيز النشاط الشبيه بفنتون للمواد القائمة على الكربون.
تقدم هذه الدراسة محفزًا قويًا من النيتروجين والكربون المخدر باليود (المشار إليه باسم I-NC)، تم تصنيعه من خلال ترسيب البخار الكيميائي، والذي يظهر فعالية في تنشيط PMS لتطبيقات معالجة المياه. من الجدير بالذكر أن دمج I يعزز بشكل كبير الهيكل الإلكتروني للمواد القائمة على الكربون، مما يحسن كفاءة تحويل PMS في مواقع الكربون الناقصة الإلكترون ويعزز أدائها التحفيزي الشبيه بفنتون. يبرز الوجود السائد لـ () في تفاعلات الأكسدة، التي تم التحقق منها من خلال التوصيفات الشاملة والتحليلات النظرية، الآلية الأساسية وفعالية المحفز. إن الأداء المذهل والمتانة الملحوظة لـ I-NC يضعه كخيار رائد في التحفيز أحادي الذرة لمعالجة المياه العملية، مما يحدد معيارًا لتصميم المحفزات وتطبيقها في PMS-AOPs.
النتائج والمناقشة
تصنيع محفز I-NC وتوصيف الهيكل
تم تصنيع محفز I-NC مع I الموزع ذريًا باستخدام تقنية ترسيب البخار الكيميائي (CVD). في البداية، تم إخضاع هياكل ZIF-8، التي تشكلت من تجميع 2-ميثيل إيميدازول ونترات الزنك، للتفكك الحراري في جو خامل لإنتاج ركائز كربون مخدر بالنيتروجين (NC). تم تأكيد نجاح تصنيع هذه الركائز من خلال حيود الأشعة السينية (XRD)
أنماط، كما هو موضح في الشكل التكميلي 1. بعد ذلك، تم وضع ركيزة NC في فرن أنبوبي لبدء عملية CVD، باستخدام كمادة مؤكسدة ومصدر I (الشكل 1a). على وجه التحديد، تم وضع قارب خزفي يحتوي على NC في المركز داخل الفرن الأنبوبي، مع وضع في الطرف العلوي لضمان التعرض لتدفق الغاز المتطور. تم إجراء عملية التلدين في بيئة خاملة، مما أدى إلى تحلل وإطلاق الأمونيا ، والهيدروجين اليودي ، وبخار اليود ، الذي يتفاعل مع NC. يعزز هذا التفاعل تكوين محفزات أحادية الذرة I-NC مع مجموعات كربون-يود (C-I)، مما يزيد من النشاط الجوهري .
كما هو موضح في الشكل التكميلي 2، تكشف تحليل المجهر الإلكتروني الماسح أن محفزات I-NC تحتفظ إلى حد كبير بالشكل الأصلي للمعين ذو الاثني عشر وجهًا المميز لـ ZIF-8، مع حجم جزيئي متوسط يتراوح بين 300 و 500 نانومتر وتوزيع حجم متسق. تشير صور المجهر الإلكتروني الناقل (TEM)، المعروضة في الشكل التكميلي 3a، b، إلى أن سطح I-NC يبدو أكثر خشونة بشكل ملحوظ مقارنة بـ NC. تشير هذه الملاحظة إلى أنوغازات HI المنبعثة خلاليمكن أن يؤدي التحلل إلى نقش موحد لمصفوفة الكربون لتعزيز تثبيت الذرة الفردية I. تؤكد تحقيقات مجهر الإلكترون الناقل عالي الدقة (HRTEM) (الشكل التكميلي 3) غياب الشبكات البلورية الملحوظة، مما يشير إلى عدم وجود جزيئات نانوية كبيرة في إطار I-NC. لتحديد توزيع الذرات على المستوى الذري لـ I داخل I-NC، تم استخدام مجهر الإلكترون الناقل بتصحيح الشذوذ والمجال المظلم الحلقي عالي الزاوية (HAADF-STEM). كما هو موضح في الشكل 1b وc، أظهرت ذرات I تباينًا أكثر سطوعًا بسبب عددها الذري الأعلى مقارنة بذرات Zn، وهي موزعة بشكل موحد عبر سطح I-NC. علاوة على ذلك، باستخدام تقنية رسم الخرائط المتقدمة لتناسب دالة غاوسية ثلاثية الأبعاد (3D) التي تتداخل فيها الذرات، يمكن إعادة بناء المنطقة المميزة بمربع أرجواني في الشكل 1c إلى الشكل 1d، حيث يتم تمييز ذرات I غير المعدنية المعزولة بوضوح داخل مصفوفة محفز I-NC. تؤكد خرائط التحليل الطيفي بالأشعة السينية المشتتة للطاقة (EDS) التكميلية، المعروضة في الشكل 1e، التوزيع المتجانس لـ I والنيتروجين (N) عبر الركيزة الكربونية. بشكل جماعي، تؤكد هذه النتائج النجاح في إدخال وتوزيع الذرات لـ I على الركائز الكربونية المدعمة بالنيتروجين.
نمط حيود مسحوق الأشعة السينية (XRD) لـ I-NC (الشكل التوضيحي 4) يظهر قمتين بارزتين عندحوالي و . تم تخصيص هذه القمم للطائرات (002) و(101) للكربون الجرافيتي، على التوالي، وتظهر درجة عالية من التشابه مع القمم المقابلة في NC. يشير هذا التشابه إلى أن الهيكل الجرافيتي الأساسي محفوظ في I-NC. لاستكشاف الخصائص الميكروهيكلية لـ NC و I-NC بشكل أكبر، تم إجراء مطيافية رامان ونتائجها مقدمة في الشكل التكميلية 5. نحن نحدد قمتين متميزتين عند حوالي 1349 و“، والتي تُنسب إلى نطاق D ونطاق G للمواد القائمة على الكربون، على التوالي. ومن الجدير بالذكر أن نسبة شدة نطاق D إلى نطاق G ( ) بالنسبة لسجلات I-NC تسجل زيادة طفيفة مقارنة بـ NC. هذه الزيادة تشير إلى درجة أقل من الجرافيتية، مما يتماشى مع الملاحظات المتعلقة بالنقش على سطح الكربون التي كشفتها تحليل TEM. بشكل جماعي، تدعم هذه الرؤى الطيفية التعديلات الهيكلية التي أحدثتها العلاج وتوفير فهم مجهرى للخصائص المتغيرة لـ I-NC.
طيف الرنين المغناطيسي الإلكتروني (EPR) لـ I-NC، الموضح في الشكل 2a، يظهر ذروة بارزة عند قيمة g تبلغ 2.005، مما يدل على وجود فراغات كربونية معززة داخل الهيكل الكتلي لـ NC.من المتوقع أن تعزز هذه العيوب الفراغية نقل الكتلة لجزيئات المتفاعلات وتعزز دمج المزيد من ذرات اليود. تم إجراء مزيد من تحليل هيكل المسام والمساحة السطحية من خلال قياس إيزوثيرم الامتصاص-الامتزاز للنيتروجين باستخدام منهجية بروناوير-إيميت-تيلر (BET) (الشكل التوضيحي التكميلي 6). يظهر I-NC إيزوثيرم من النوع الأول، مما يشير إلى هيكل يهيمن عليه المسام الدقيقة، بما يتماشى مع توزيع حجم المسام.
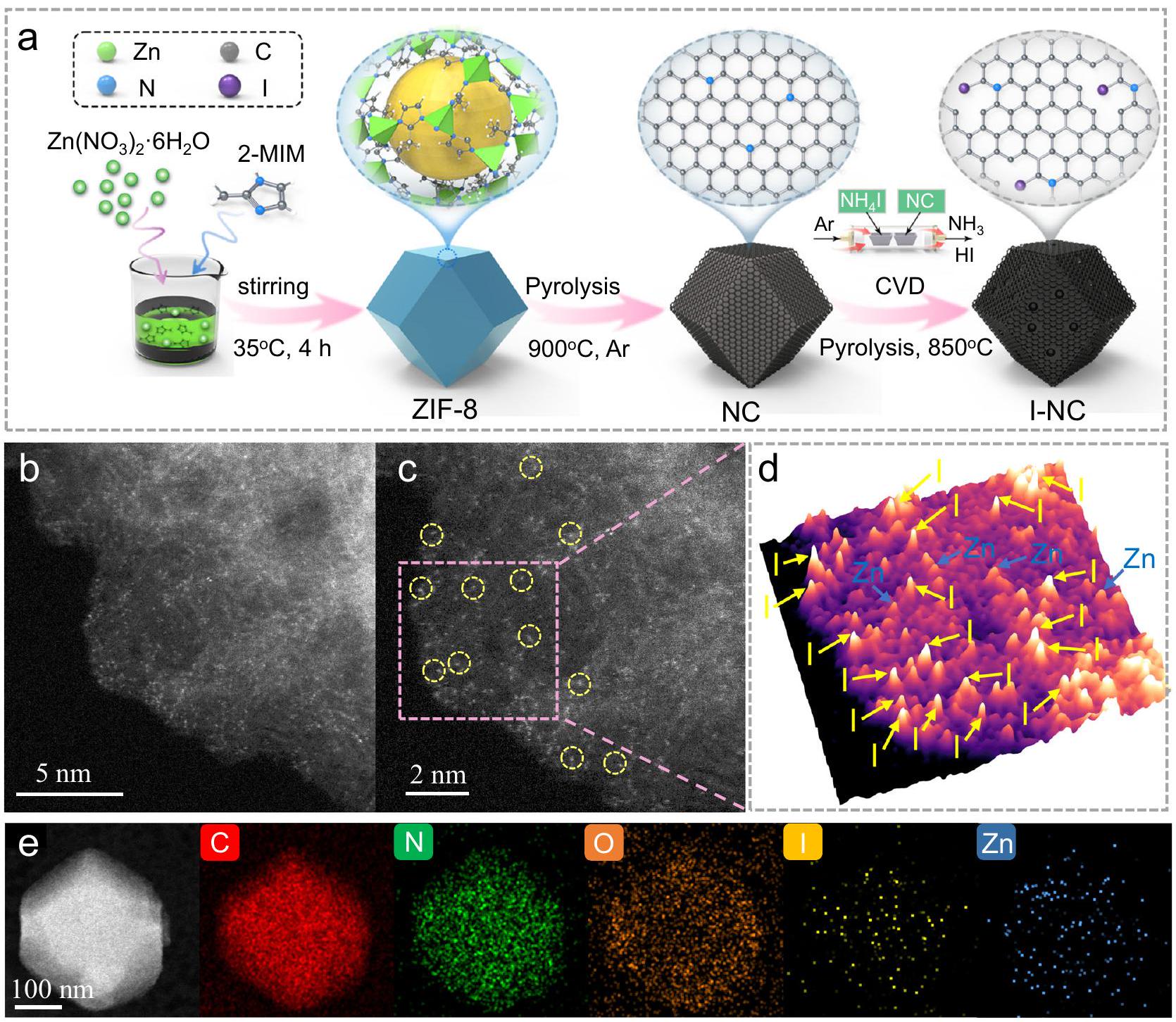
الشكل 1 | التحضير والتوصيف الهيكلي لمحفز I-NC. أ رسم تخطيطي لتحضير محفز الكربون النيتروجيني المدعوم باليود الموزع ذريًا (المشار إليه بـ I-NC). تمثل الكرة الصفراء أكبر كرة فان دير فالس يمكن أن تناسب في التجويف. ب، ج صورة عالية الدقة من الزاوية العالية. صورة مجهر الإلكترون الناقل الماسح عالي الزاوية (HAADF-STEM) لـ I-NC. الدوائر الصفراء تشير إلى ذرات اليود الفردية. د خريطة تركيب دالة غاوسي ثلاثية الأبعاد متداخلة لمنطقة في الإطار (ج). هـ خرائط العناصر بواسطة مطيافية الأشعة السينية المشتتة للطاقة (EDS)وزنك (Zn) من I-NC. النتائج. من الجدير بالذكر أن ميزات I-NC تحسن بشكل ملحوظ في مساحة السطح BET ( ) وحجم المسام ( ) عند مقارنتها مع NC ( و تعمل هذه التحسينات على تحسين تعرض المواقع الحفازة وتعزيز انتشار جزيئات المتفاعلات (مثل، ROS، CIP).
تمت دراسة الهياكل التركيبية والإلكترونية لـ NC و I-NC بشكل شامل باستخدام مطيافية الأشعة السينية للأشعة السطحية (XPS)، مما يوفر رؤى حول التركيب العنصري السطحي والحالات الكيميائية. كشفت طيف المسح XPS (الشكل التوضيحي 7) عن مستويات متقاربة من الكربون (C) والنيتروجين (N) والأكسجين (O) عبر كلا المحفزين. دقة عالية Nيمكن تحليل الأطياف لـ NC و I-NC إلى أربعة قمم متميزة تتوافق مع تكوينات النيتروجين المختلفة: نيتروجين بيريديني (عندن (فين (في ) وأنواع النيتروجين المؤكسدة ( )، كما هو موضح في الشكل 2b، والشكل التكميلي 8 ومُلخص في الجدول التكميلي 2 والجدول 3. تم ملاحظة تحسين ملحوظ في محتوى النيتروجين الجرافيتي لـ ، التسجيل في ، زيادة من NC عند تُبرز هذه الهيمنة على النيتروجين الجرافيتي في I-NC، خاصة عند مقارنتها بأنواع النيتروجين الأخرى، دورها المحتمل في تعزيز الأنشطة التحفيزية الشبيهة بفنتون.
علاوة على ذلك، يتميز طيف I-NC بوجود قمة جديدة عند حوالي 620 إلكترون فولت، تعود إلى عنصر I. التحليل التفصيلي لـ Iالمنطقة في طيف XPS الخاص بـ I-NC (الشكل 2c) تكشف عن تحليل تفكيكي إلى قمتين عند 618.58 و620.46 إلكترون فولت، مما يتوافق مع الأيونية ( ) وأنواع اليود التساهمي (C-I) على التوالي. تدعم هذه الملاحظة دمج تكوينات رابطة C-I، التي تم تأكيدها من خلال قمة محددة عند 285.6 eV في طيف XPS C 1s (الشكل التكميلي 9)، مما يؤكد النجاح في إدخال والتفاعل الكيميائي لليود داخل هيكل I-NC.
تم توضيح التركيب الإلكتروني والتكوين الذري لـ I-NC بشكل أكبر باستخدام مطيافية امتصاص الأشعة السينية عند حافة I K (XAFS). في الشكل 2d، تكشف ملفات هيكل الامتصاص القريب من الحافة للأشعة السينية (XANES) لـ I-NC والمراجع عن تشابه ملحوظ في حافة الامتصاص لـ I داخل I-NC مع ما لوحظ لـ iohexol مع روابط نموذجية C-I (الشكل التكميلي 10). يشير هذا التشابه إلى أن حالة الأكسدة لـ I في I-NC تقع بين اليود العنصري و iohexol، مما يشير إلى وجود حالة مختلطة من I. تم الحصول على مزيد من الرؤى من خلال تحليل هيكل الامتصاص الدقيق الممتد للأشعة السينية المحولة فورييه (EXAFS)، المفصل في الشكل 2e، f والشكل التكميلي 11. تسلط هذه النتائج الضوء على غياب القمم المميزة.
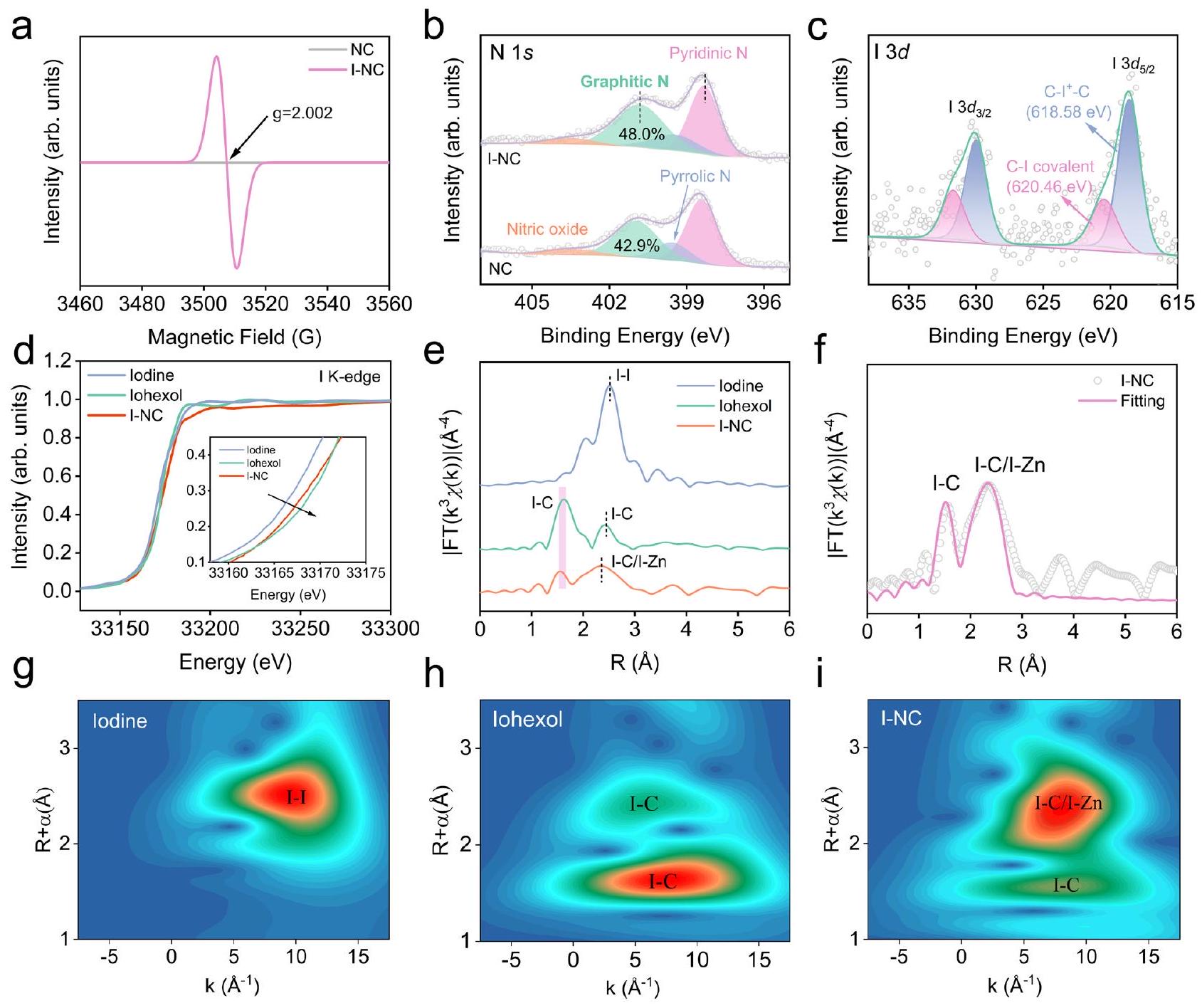
الشكل 2 | توصيف الحالة الإلكترونية والبنية الذرية للمحفزات المُعدة. أ طيف الرنين المغناطيسي الإلكتروني (EPR) لـ NC و I-NC. ب طيف الأشعة السينية للأشعة السينية عالية الدقة (XPS) لـ N 1s لـ NC و I-NC. ج XPS عالية الدقة لـ Iطيف I-NC. المعلمات المناسبة لتناسب XPS و مدرجة في الجدول التكميلي 3. د أشعة إكس عند حافة K المعايرة طيف هيكل الامتصاص بالقرب من الحافة (XANES) لليود، إيوهكسول، وI-NC. e طيف هيكل الامتصاص الممتد بالأشعة السينية المحوّل فوريًا (EXAFS) لليود، إيوهكسول، وI-NC. المنطقة البنفسجية تشير إلى القمم المميزة لـ I-C سواء في NC أو I-NC. f تحليل ملاءمة EXAFS لحافة I K لـ I-NC. g-i مخططات تحويل الموجات EXAFS لليود، إيوهكسول، وI-NC. يتوافق مع رابطة I-I عند“، مؤكدًا الفرضية بأن ذرات I مدمجة داخل مصفوفة I-NC ككيانات معزولة. تدعم هذه العزلة التأكيد على تنسيق الذرات الفردية، مما يتماشى مع التحسين المتوقع للنشاط التحفيزي ويقدم تفسيرًا منطقيًا للخصائص الفيزيائية والكيميائية الملحوظة لـ I-NC. يتطلب الفحص الإضافي لبيانات XAFS لـ I-NC، وخاصة الذروة الملحوظة عنديؤكد وجود تنسيق C-I، الذي يشبه ما تم تحديده في الإيوهكسول. يمكن أن يُعزى ذروة إضافية ملحوظة في طيف I-NC إلى مسارات التشتت الخلفي C-I وZn-I. تشير اكتشافات تفاعلات Zn-I إلى احتمال وجود تنسيق مباشر بين I وZn داخل هيكل NC أو تشكيل يوديد الزنك.كنتيجة لنقش بخار اليود، وهي فرضية مدعومة بغياب خطوة الغسيل بعد النقش. ومع ذلك، يجب ملاحظة أن ميزة Zn-I ليست سائدة، مما يتماشى مع الاستنتاج بأن I داخل المادة يوجد بشكل أساسي في حالة مشحونة إيجابياً، وتم تقديم المزيد من الأدلة لاحقاً. كما تم تقديم مزيد من الدعم من خلال تحليل تحويل الموجات EXAFS الموضح في الشكل 2g-i، مما يؤكد عدم وجود روابط I-I داخل I-NC. هذه النتيجة تتماشى مع الملاحظات من تحقيقات HAADF-STEM. علاوة على ذلك، EXAFS تشير نتائج التركيب إلى أن متوسط عدد التنسيق حول C-I في I-NC يعكس ما تم ملاحظته في الإيوهكسول، مما يعزز الطبيعة الموزعة ذريًا لمواقع I داخل المحفز (الشكل التكميلي 12 والجدول 4).
الأداء التحفيزي وتحديد أنواع الأكسجين التفاعلية
تم تقييم الأداء التحفيزي الشبيه بفنتون لـ I-NC من خلال تنشيط PMS لتفكيك السيبروفلوكساسين (CIP). في البداية، تم إجراء فحص لتأثير معلمات التشغيل المختلفة، كما هو موضح في الأشكال التكميلية 13-15. على وجه التحديد، تم تحديد التركيز الأمثل لـ PMS ليكون 1 مللي مول بينما تم اختبار جرعة المحفز عندتم العثور على أن أنسب درجة حرارة للتسخين الثانوي في عملية CVD لتصنيع I-NC هيتظهر النتائج المعروضة في الشكل 3أ أن CIP تم تحليله تقريبًا بالكامل بواسطة نظام I-NC/PMS في غضون 10 دقائق. بالمقابل، فقط حواليتمت ملاحظة تحلل CIP مع نظام NC/PMS. هذه الفروق الكبيرة تبرز النشاط التحفيزي الفائق لنظام I-NC على غرار فنتون. كما أكدت كروماتوغرافيا السائل-مطيافية الكتلة (LC-MS) لكروماتوغرامات CIP قبل وبعد التفاعل في نظام I-NC/PMS أن CIP تم القضاء عليه تقريبًا خلال 10 دقائق (المكملات
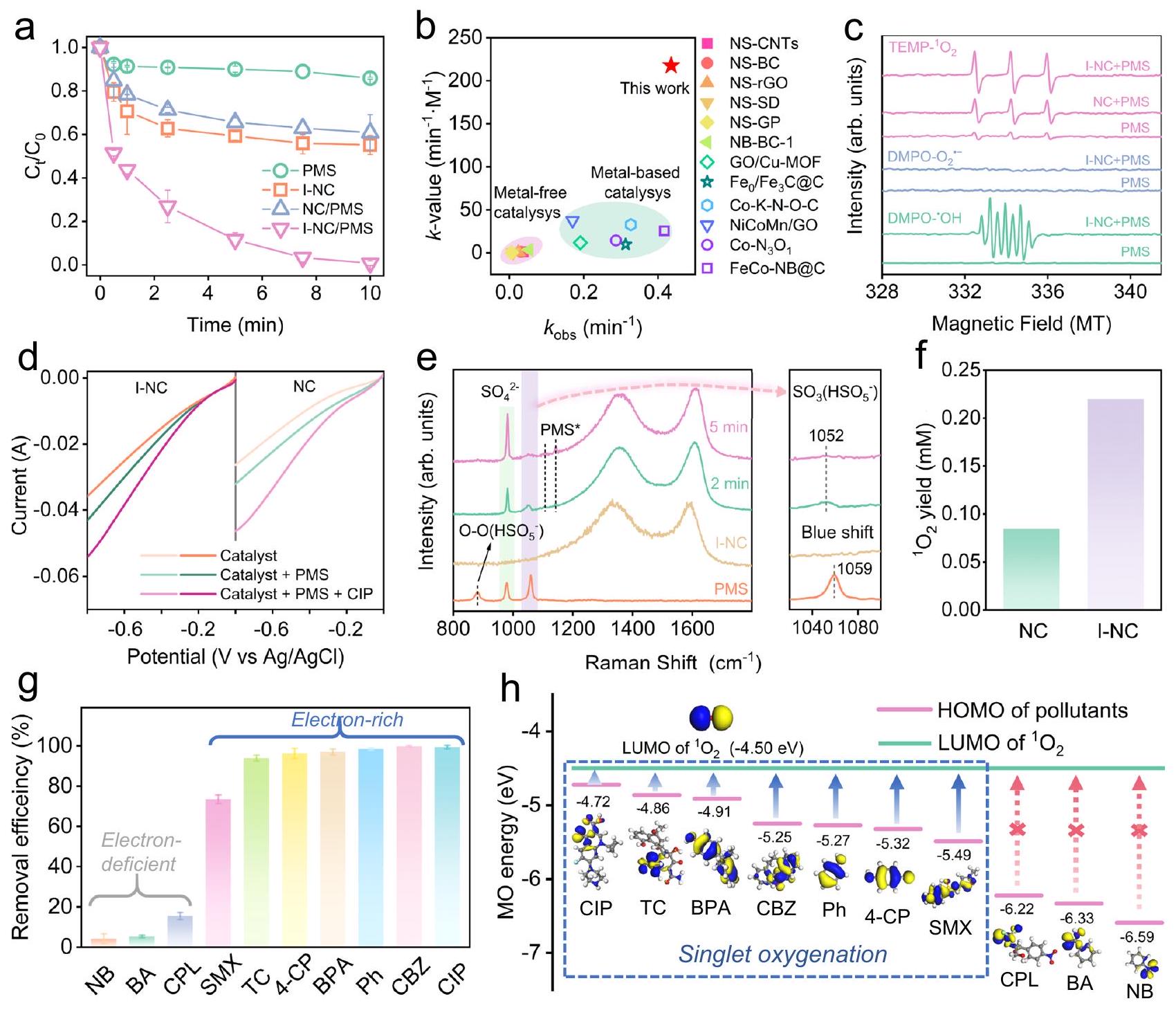
الشكل 3 | أداء تحفيزي شبيه بفنتون، تحديد الأنواع التفاعلية وتوصيف عملية التفاعل. أ تدهور السيبروفلوكساسين (CIP) في أنظمة PMS المفعلة بواسطة محفزات مختلفة. ب مقارنة بين I-NC والمحفات المبلغ عنها في تفاعل شبيه بفنتون. المراجع ذات الصلة مدرجة في الجدول التكميلي 5. ج طيف EPR باستخدام 5,5-ثنائي ميثيل-1-بيرولين-N-أكسيد (DMPO) و2,2,6,6-تترا ميثيل بيبيريدين (TEMP) كعوامل حبس. د منحنيات الفولتميترية الخطية (LSV) لـ NC و I-NC تحت ظروف مختلفة. هـ طيف رامان في الموقع لـ PMS و I-NC وتفاعلهما. تمثل المناطق السماوية، الأرجوانية والصفراء القمم المميزة لـو متلازمة ما قبل الحيض*، على التوالي. fالعائد في أنظمة المحفزات المختلفة / PMS / CIP. ج تدهور الملوثات المتعددة في نظام I-NC / PMS. يتم سرد اسم الهيكل الكيميائي للملوثات الدقيقة ذات الصلة في الشكل التكميلية 33.آلية الأكسدةلعدة ملوثات دقيقة. تمثل الأسطح الزرقاء والصفراء السطح الإيجابي والسطح السلبي، على التوالي. تمثل الكرات البيضاء والرمادية والزرقاء والحمراء والصفراء والسماوية ذرات الهيدروجين والكربون والنيتروجين والأكسجين والكبريت والفلور، على التوالي. الشروط: [المحفز]الملوثات50 مليمول (إذا كان هناك أي). تشير أشرطة الخطأ إلى الانحراف المعياري للتجارب التي أُجريت ثلاث مرات. يتم توفير بيانات المصدر كملف بيانات مصدر.
الشكل 16). بالإضافة إلى ذلك، فإن PMS وحده لم يتدهور إلا أقل من من CIP، وحول تمت إزالة CIP بواسطة I-NC من خلال الامتزاز، مما يشير إلى أن الأكسدة الحفزية لعبت دورًا رئيسيًا في الإزالة السريعة لـ CIP. ومن الجدير بالذكر أن تسربأيونات اليود من نظام I-NC/PMS ساهمت في حواليمعدل تحلل CIP، مما يشير إلى أن أيونات اليود المستخرجة كان لها تأثير ضئيل على عملية التحلل (الشكل التوضيحي 17).
يمكن وصف تحلل CIP بشكل جيد بواسطة حركيات من الدرجة الأولى الزائفة. ثابت الحركية ( ) من I-NC/PMS ( كان أعلى بـ 10.1 مرات من NC/PMS )، و أضعاف أكبر من تلك الخاصة بالمواد الكربونية التجارية. ومن الجدير بالذكر أنلقد تفوقت I-NC على معظم المحفزات المتطورة من حيث تفعيل PMS الفعال في تحلل CIP (الشكل التكميلي 18). بالإضافة إلى ذلك، نظرًا لأنقد يختلف مع ظروف التفاعل ونوع المواد العضوية، نموذج حركي موحد ( – تم استخدام هذا النموذج لضبط القيمة.عن طريق قسمته على تركيز PMS وجرعة المحفز، وضربه في تركيز الملوث، لتقييم معدلات الإزالة بشكل موحد عبر أنظمة فينتون غير المتجانسة المختلفة.كما هو موضح في الشكل 3ب والموجز في الجدول التكميلي 5،-قيمة لـ I-NC (تجاوزت تلك الخاصة بجميع المحفزات الخالية من المعادن المقارنة (0.23 إلىمن حيث تفعيل PMS لتدهور CIP وكان أعلى من معظم المحفزات المعدنية الرائدة، على سبيل المثال،لـ.
أظهرت النتائج أن I-NC يتفوق بشكل كبير على NC في النشاط التحفيزي الشبيه بفينتون، مما يبرز فعالية الـطريقة النقش. كما هو موضح في الشكل التكميلي 19a، هناك علاقة مباشرة واضحة بين النشاط التحفيزي الشبيه بفنتون والعوامل مثل المساحة السطحية، وحجم المسام، ونسبة. تشير هذه العلاقة إلى أنالنقش عدل المسام بنجاح هيكل NC، الذي زاد من عدد المواقع النشطة وحسن من وصولها. بشكل ملحوظ، تم تحقيق زيادة بمقدار 6.8 مرة في المساحة السطحية المحددة المعدلة قيمة ( لكل وحدة سطحية) لا يزال محققًا، مما يدل على نشاط تحفيزي داخلي أكثر فائدة لـ I-NC (الشكل التكميلي 19b). من المثير للاهتمام أن خصائص التحلل لـ I-NC لم تتماشى مع التغيرات في تركيز الزنك، كما هو موضح في الشكل التكميلي 20. وهذا يشير إلى أن الزنك يلعب دورًا ضئيلًا في فعالية نظام I-NC/PMS. بالإضافة إلى ذلك، فإن الكلسنة الثانوية لـ NC في جو خامل بدون “، المشار إليه باسم NC-فارغ، لم يعزز بشكل كبير أداء تحلل CIP. وهذا يشير إلى أن عملية التكليس الثانوية وحدها لا تؤثر بشكل كبير على المواقع النشطة الجوهرية لـ NC، كما تدعمه البيانات في الشكل التوضيحي 21. الدراسات السابقةلقد أظهرت المواد القائمة على الكربون أنها يمكن أن تخضع لنقش فعال لإدخال العيوب من خلالمعالجة حرارية. وبالتالي، تم اختيار اليوريا كـمصدر لعلاج NC. أظهرت النتائج التجريبية أن كفاءة تحلل CIP في نظام NC-يوريا/PMS أظهرت تحسنًا طفيفًا فقط مقارنةً بـ NC بمفرده. علاوة على ذلك، على الرغم من إضافة أيونات اليود الإضافية، لم يُلاحظ تحسن كبير في نظام NC-يوريا وأيونات اليود/PMS، مما يوضح مرة أخرى التأثير المحدود لأيونات اليود المذابة في التفاعلات الحفزية التي تتوسطها PMS (الشكل التوضيحي 21). تسلط هذه النتيجة الضوء على الدور الحاسم لذرة اليود المفردة في تعزيز تنشيط PMS وتسهيل تحلل CIP.
أنواع تفاعلية متنوعة، بما في ذلك، و تم تحديدها في عمليات المحفزات القائمة على الكربون / PMS. للتحقيق في الأنواع التفاعلية المحددة الناتجة أثناء تنشيط PMS مع NC و I-NC، تم إجراء تجارب امتصاص باستخدام الميثانول (MeOH) والكحول التيرت-بيوتيلي (TBA) كمواد امتصاص للجذور الحرة. على وجه التحديد، تم استخدام MeOH و TBA كمواد امتصاص للجذور الحرة من و/أووجود كميات زائدة من الميثانول (MeOH) وTBA بالكاد أعاق تحلل CIP (الشكل التوضيحي 22)، مما يشير إلى أن و من المحتمل أن لا تكون الأنواع النشطة الرئيسية في NC/PMS و I-NC/PMS. حيث إن الميثانول (MeOH) و TBA لا يمكنهما إخماد الجذور الحرة المرتبطة بالسطح، تم استخدام ثنائي ميثيل سلفوكسيد (DMSO) و NaF لاستكشاف مساهمة الجذور الحرة على سطح I-NC.كما هو موضح في الشكل التوضيحي 23، كان لمادة DMSO و NaF تأثير محدد على تحلل CIP في نظام I-NC/PMS، مما يشير إلى أن الجذور الحرة المرتبطة بالسطح تساهم قليلاً في تحلل CIP. بالإضافة إلى ذلك، فإن إدخال الكلوروفورم ( )، مع ثابت معدل مرتفع للتفاعل مع من ، تسبب فقط في انخفاض طفيف في تحلل CIP. تشير هذه النتيجة إلى أن لم تلعب أيضًا دورًا كبيرًا في تحلل CIP. بالإضافة إلى ذلك، تم استخدام ثنائي كلوريد نيتروبلاو تيترازوليوم (NBT) ككاشف لـ ، كما هو موضح في الشكل التوضيحي 24، فإن التأثير الضئيل لـ NBT يشير إلى الحد الأدنى من مشاركة في نظام I-NC/PMS. على النقيض من ذلك، أدى إدخال كحول الفرفوريل، مع ثابت معدل معروف مع من ، إلى إعاقة كبيرة لتحلل CIP. تشير هذه المثبطات الملحوظة إلى دور حاسم لـ في عملية التحلل، مما يتماشى مع سلوك الأكسدة الانتقائية الملحوظ تجاه مختلف الملوثات الدقيقة في I-NC/PMS. كما هو موضح في الشكل التوضيحي 25، فإن التأثيرات المماثلة في التثبيط التي لوحظت في كل من NC و I-NC عند استخدام L-histidine، و 2،2،6،6-tetramethylpiperidine (TEMP) كعوامل تثبيط تشير إلى أن هذه المحفزات تشترك في آلية شائعة لتفعيل PMS وتحلل الملوثات الدقيقة. وهذا يبرز بشكل أكبر هيمنة في هذه التفاعلات .
نظرًا لأن المواد القائمة على الكربون يمكن أن تعمل كوسائط إلكترونية لتسهيل نقل الإلكترون من الملوثات إلى PMS، وبالتالي أكسدة الملوثات باستقلالية ROS. للتحقق من مساهمة نقل الإلكترون في تحلل CIP، تم إجراء سلسلة من التجارب بما في ذلك القياس الكهروكيميائي، تجربة الخلط المسبق، وتجربة تحلل PMS. كما هو موضح في الشكل التوضيحي 26، زاد الجهد المفتوح للدائرة (OCP) للأقطاب الكربونية الزجاجية المطلية بـ NC أو I-NC على الفور بعد إضافة PMS، مما يشير إلى أن المركبات النشطة لـ PMS التي تتشكل على سطح المحفز يمكن أن ترفع الجهد . ومع ذلك، لم يكن هناك انخفاض ملحوظ في الجهد مع إضافة CIP، مما يشير إلى أن عملية نقل الإلكترون التي يتم تحفيزها بواسطة المحفز للملوثات إلى مركبات المحفز-PMS ضئيلة . علاوة على ذلك، للتحقق من وجود عملية نقل الإلكترون التي يتم تحفيزها بواسطة المحفز، تم خلط المحفز و PMS في المحلول الفارغ، وتم إضافة محلول يحتوي على CIP في ، وبعد 9 دقائق لاحقًا. كما هو موضح في الشكل التوضيحي 27، لاحظنا أن زيادة وقت الخلط المسبق أدت إلى انخفاض أكبر في كفاءة تحلل CIP، مع تحلل فقط من CIP مع من بعد فترة خلط مسبق في نظام I-NC/PMS. تشير هذه النتيجة إلى أن عملية نقل الإلكترون قد لا تلعب دورًا كبيرًا في تحلل CIP في ظل هذه الظروف. بالإضافة إلى ذلك، تشير الدراسات الحالية إلى أن تحلل PMS يعتمد غالبًا على الركائز العضوية التي تعمل كمانحات للإلكترون في آلية نقل الإلكترون . ومع ذلك، عندما تم إدخال CIP كمانح محتمل للإلكترون، لم يتم ملاحظة تسريع في استهلاك PMS، كما هو موضح في الشكل التوضيحي 28. تشير هذه النتيجة إلى أن CIP لا يسهل تحلل PMS عبر نقل الإلكترون، مما يتناقض مع السلوك المتوقع إذا كانت عملية نقل الإلكترون هي المسار الرئيسي للتحلل في I-NC/PMS.
توفر الدراسات المرجعية أدلة على أن عمليات نقل الإلكترون التي يتم تحفيزها بواسطة المحفز ليست مساهمات كبيرة في نشاط I-NC/ PMS. لتعزيز هذا الاستنتاج، استخدمت الأبحاث 9،10-diphenylanthracene (DPA) ككاشف محدد للكشف عن وجود في النظام . يتفاعل DPA مع ، ويخضع للأكسدة إلى 9،10-diphenylanthracene dioxide (DPAO2)، والتي تترافق مع انخفاض في شدة قمة الامتصاص حول 380 نانومتر. يؤكد الانخفاض الملحوظ في هذه القمة، كما هو موضح في الشكل التوضيحي 29، على توليد ومشاركة في عملية تحلل CIP. بالإضافة إلى ذلك، استخدمت الدراسة 5،5-dimethyl-1-pyrroline-Noxide (DMPO) و TEMP كعوامل حبس للكشف عن أنواع تفاعلية أخرى، على وجه التحديد و و ، على التوالي. كانت استخدام هذه العوامل الحبس في الشكل 3c تهدف إلى تمييز الأنواع التفاعلية المعنية وتأكيد الدور السائد لـ في آلية التحلل، على عكس مشاركة أنواع الجذور الأخرى في I-NC/PMS. علاوة على ذلك، كشفت تجارب EPR عن رؤى مهمة حول الأنواع التفاعلية المسؤولة عن تحلل CIP داخل I-NC/PMS. أظهرت طيف EPR بوضوح قمة مميزة تعود إلى 5،5-dimethylpyrroline-N-oxide (DMPOX)، مما يشير إلى أن DMPO يخضع لأكسدة عميقة، ربما بسبب الإنتاج السريع للجذور الهيدروكسيلية، أو الأنواع المعدنية عالية التكافؤ-أوكسي، أو . نظرًا لاستبعاد الجذور الحرة كأنواع نشطة سائدة والطبيعة الخالية من المعادن المتعددة للمحفز، يمكننا التكهن بأن مسؤولة عن تشكيل DMPOX. علاوة على ذلك، كشفت التجربة التي استخدمت TEMP كعامل حبس عن إشارة مميزة لـ -TEMP المركبات. كانت هذه الإشارة أكثر كثافة بشكل ملحوظ في نظام I-NC/PMS مقارنة بنظام NC/PMS، مما يعزز فكرة أن تلعب دورًا حاسمًا في التحلل التحفيزي لـ CIP بواسطة I-NC. توفر وجود هذه -TEMP المركبات دليلًا قويًا على أن هي نوع مؤكسد سائد في نظام I-NC/PMS، تشارك بفعالية في عملية التحلل. تشير مجموعة الأدلة التجريبية إلى أن تلعب دورًا محوريًا في INC/PMS و NC/PMS، مع عرض I-NC أداءً تحفيزيًا متفوقًا مقارنة بـ NC/PMS. يعزز الانخفاض الكبير في شدة قمة -TEMP المركب عند إدخال CIP في نظام I-NC/PMS الاستنتاج بأن تكون أساسية في تحلل CIP (الشكل التوضيحي 30). بالإضافة إلى ذلك، تدعم النتائج من تجربة التثبيط لـ وكاشف NBT، جنبًا إلى جنب مع تجربة EPR الحبس TEMP، الاستنتاج بأن في النظام لا تنبع من . تشير الملاحظة بأن تهوية الغاز بالأرجون (Ar) والأكسجين () لا تؤثر على إزالة CIP (الشكل التوضيحي 31)، مما يشير إلى أن الأكسجين المذاب ليس مقدمة لـ التي تتولد في هذه التفاعلات. ومن ثم، فإن الاستنتاج المستمد هو أن تنتج من خلال التفاعل بين جزيئات PMS ومواقع المحفز النشطة.
توصيف عملية التفاعل لنظام شبيه بفنتون نظرًا للدور المزدوج لـ PMS كعامل مؤكسد ومختزل، فإن مسارات نقل الإلكترون بين PMS والمحفز ضرورية لتوليد . عندما يتم نقل الإلكترونات من PMS إلى المحفز، فإن هذه العملية تحفز تفكك ، مما يؤدي إلى تشكيل (Eq. 1). يؤدي هذا الجذر بعد ذلك إلى توليد من خلال تفاعل التباين (Eq. 2) . على العكس من ذلك، عندما يتم نقل الإلكترونات من المحفز إلى PMS، فإن هذا يبدأ في كسر رابطة لإنتاج *OH، والتي تتفكك بعد ذلك إلى * O، والتي تتحد بعد ذلك مع وسيط آخر لتوليد . لتمييز عملية نقل الإلكترون بين I-NC و PMS، تم إجراء اختبارات كهروكيميائية. في تحليلات الفولتامترية ذات المسح الخطي (الشكل 3d)، زادت كثافة التيار لكل من القطب I-NC و القطب NC بشكل كبير في وجود PMS، مع زيادة الأولى أكبر بكثير من الثانية، مما يظهر نقل إلكترون معزز من PMS إلى I-NC مقارنة بـ NC . علاوة على ذلك، زاد التيار الملحوظ في النظام بشكل كبير بعد إضافة CIP، مما يشير إلى تفاعل مواتٍ بين PMS و CIP و المحفزات. بالإضافة إلى ذلك، أظهر OCP للقطب الكربوني الزجاجي المطلي بمحفزات NC و I-NC زيادة فورية عند إضافة PMS. من الجدير بالذكر أن الزيادة في OCP لمحفز I-NC كانت 0.54 فولت، والتي كانت أكبر بكثير من الزيادة البالغة 0.45 فولت التي لوحظت مع محفز NC (الشكل التوضيحي 26). تدعم هذه الفروق فكرة نقل الإلكترون الفعال من PMS إلى المواقع النشطة على المحفزات .
للمزيد من التحقيق في عملية نقل الإلكترون بين I-NC و PMS، تم إجراء التحليل الطيفي رامان (الشكل 3e). كشفت الأطياف عن ثلاث قمم مميزة في محلول PMS (، الموجودة عند 880، 980، و )، والتي تتوافق مع اهتزازات الشد لـ ، و ، على التوالي. ومن الجدير بالذكر أن القمم المرتبطة بـ و أظهرت القمم انخفاضًا ملحوظًا عند إضافة I-NC مع مرور الوقت، مما يشير إلى تحويل سريع لـ PMS إلىبعد تنشيطها بواسطة I-NC. بالإضافة إلى ذلك، فإن امتصاص PMS على المحفزات أمر حاسم لتنشيطها. يمكن أن يؤدي ارتباط جزيئات PMS بسطح المحفز إلى تكوين معقدات غير مستقرة، يُشار إليها بـ *PMS. وقد تم إثبات هذه العملية من خلال ظهور قمم جديدة عند 1110 وفي طيف رامان، مما يشير إلى وجود مركب *PMSمن الجدير بالذكر أن الاهتزاز المميز لـظهر انزياح أزرق ملحوظالتحول الأزرق عادة ما ينشأ من انخفاض كثافة الإلكترونات، مما يشير إلى تبرع الإلكترونات من PMS إلى المحفزات.. بعد ذلك، يتم تحويل PMS إلى ، مما يؤدي بعد ذلك إلى توليد . تشير هذه النتائج إلى أن نقل الإلكترون من PMS إلى المواقع النشطة يحدث أولاً، يليه إزالة البروتون لتكوين. ثم يخضع هذا الجذر لعملية تفكك غير متناسب، مما يؤدي إلى إنتاج . علاوة على ذلك، فإن دمج I يُحسن التكوين الإلكتروني المحلي لـ NC، مما يسهل نقل الإلكترون من PMS إلى المحفزات وبالتالي يعزز تشكيل .
علاوة على ذلك، تم مراقبة التغيرات في درجة الحموضة قبل وبعد عملية التفاعل، كما هو موضح في الشكل التكميلي 32. إن الانخفاض التدريجي في قيمة pH للمحلول يؤكد على الكفاءة في توليدم coupled with إطلاق البروتون (المعادلة 3). هذه الملاحظة تستبعد بشكل أكبر الدور السائد لمسار الجذور الحرة الذي ينتجالحفاظ على الرقم الهيدروجيني أو توليدبالاقتران مع زيادة في الرقم الهيدروجيني. من الجدير بالذكر أننا يمكننا حساب القيمة النظرية لـالجيل باستخدام المعادلة 3 والتغيرات الملحوظة في الرقم الهيدروجيني.تم العثور على التركيز الناتج عن نظام إدارة الطاقة المنشط I-NC ليكون 0.22 مللي مول، وهو أعلى بمقدار 2.59 مرة من التركيز الناتج عن NC (الشكل 3f). تشير هذه النتيجة أيضًا إلى أن إضافة ذرات I تعدل بشكل فعال الهيكل الإلكتروني للمادة، مما يعزز نشاطها التحفيزي الفطري و تحسين الكفاءة لـكفاءة التوليد.
يعمل كعامل إلكتروني، ويشارك بسهولة في الإضافة الإلكترونية تجاه الملوثات الغنية بالإلكترونات. وبالتالي، يمكن لنظام تنشيط PMS الذي يحفزه I-NC القضاء بشكل انتقائي على مجموعة واسعة من الملوثات. على وجه الخصوص، تجاوزت معدلات تحلل المركبات الغنية بالإلكترونات مثل CIP، وكاربامازيبين (CBZ)، والفينول (Ph)، وبيسفينول أ (BPA)، و4-كلوروفينول (4-CP) والتتراسيكلين (TC)داخلمدة، باستثناء السلفاميثوكسازول (SMX)، الذي حقق تقريبًا. على العكس من ذلك، أظهرت المواد المحتوية على نقص في الإلكترونات، بما في ذلك النيتروبنزين (NB) وحمض البنزويك (BA) والكلورامفينيكول (CPL) كفاءات تحلل أقل بكثير، حيث كانت جميعها أقل من 20% (الشكل 3g). تبرز هذه التفاعلية الانتقائية فعالية نظام PMS المنشط بواسطة I-NC في استهداف أنواع معينة من الملوثات، كما هو موضح في الشكل التكميلي 33. الفرق في تم استكشاف قدرة الأكسدة تجاه مجموعة مختارة من المواد العضوية من خلال منظور مسار نقل الشحنة، كما هو موضح في الشكل 3h. فجوة الطاقة الأصغر بين أدنى مدار جزيئي غير مشغول (LUMO) لـوأعلى مدار جزيئي مشغول (HOMO) للملوثات يمثل انتقال الإلكترونات من الملوثات إلىيمكن أن يبدأ بشكل أكثر سهولة. هذا يسهل ويعجل عملية الأكسدة المستهدفة للملوثات المحددة. وبالتالي، فإن كفاءةفي أكسدة المركبات العضوية المختلفة يرتبط ارتباطًا وثيقًا بالتوافق الطاقي لأوربيتالاتها الجزيئيةلقد لوحظ بوضوح أن مستوى الطاقة الأعلى المشغول (HOMO) للملوثات الغنية بالإلكترونات مثل CIP (-4.72 eV) و TC و SMX ( -5.49 eV ) كانت أقل قليلاً من LUMO لـ ( -4.50 eV ). تشير محاذاة الطاقة هذه إلى أن يمكن أن تسحب الإلكترونات بسهولة من HOMO لهذه الملوثات الغنية بالإلكترونات، مما يسهل تحللها. وعلى العكس، فإن LUMO لـكان أعلى بكثير من مستويات طاقة HOMO لـ CPLبكالوريوس و ملاحظة . هذه الفجوة الكبيرة في الطاقة توضح أنه من الصعب على لإجراء تفاعلات الأكسدة على هذه المركبات الفقيرة بالإلكترونات، مما يبرز المزيد من التفاعل الانتقائي لـاستنادًا إلى التركيب الإلكتروني للملوثات. باختصار، يخضع CIP لفعالية تدهور سريعة في نظام I-NC/PMS مقارنةً بـ 10 ملوثات تم فحصها، بسبب الفجوة الضيقة بين LUMO لـ و HOMO من CIP، مما يسهل نقل الإلكترونات بشكل أكثر كفاءة والأكسدة اللاحقة. تم تحديد الوسائط المتحللة لـ CIP بواسطة LC-MS كما هو موضح في الشكل التكميلية 34. على وجه التحديد، ظهر الذروة المقابلة لـ CIP عند زمن احتباس قدره 7.5 دقيقة. بالإضافة إلى ذلك، تم تحديد ستة وسائط تحلل، والتي تشير معًا إلى مسار تحلل محتمل لـ CIP، كما هو موضح في الشكل التوضيحي 35. يوفر هذا المسار رؤى حول تحول CIP في نظام I-NC/PMS، مما يبرز تعقيد وفعالية عملية التحلل.
دراسة نظرية لآلية التحفيز
كما تم التحقق منه من خلال النتائج السابقةتظهر الأنواع المختلفة من النيتروجين آليات تنشيط مختلفة في عملية تنشيط PMS التي تتوسطها مواقع نشطة غير معدنية، وتعتبر المحفزات الكربونية المدعمة بالنيتروجين الجرافيتي ملائمة لتنشيط PMS لإنتاج. هنا وجدنا محتوى أعلى من النيتروجين الجرافيتي مقارنة بالنيتروجين البيريديني والنيتروجين البيرولي في المحفزات، مما يرتبط بسطوة كنوع نشط في كل من NC/PMS و I-NC/PMS. وهذا يشير إلى دور حاسم للنيتروجين الجرافيتي في عملية تنشيط PMS التي تهيمن عليها
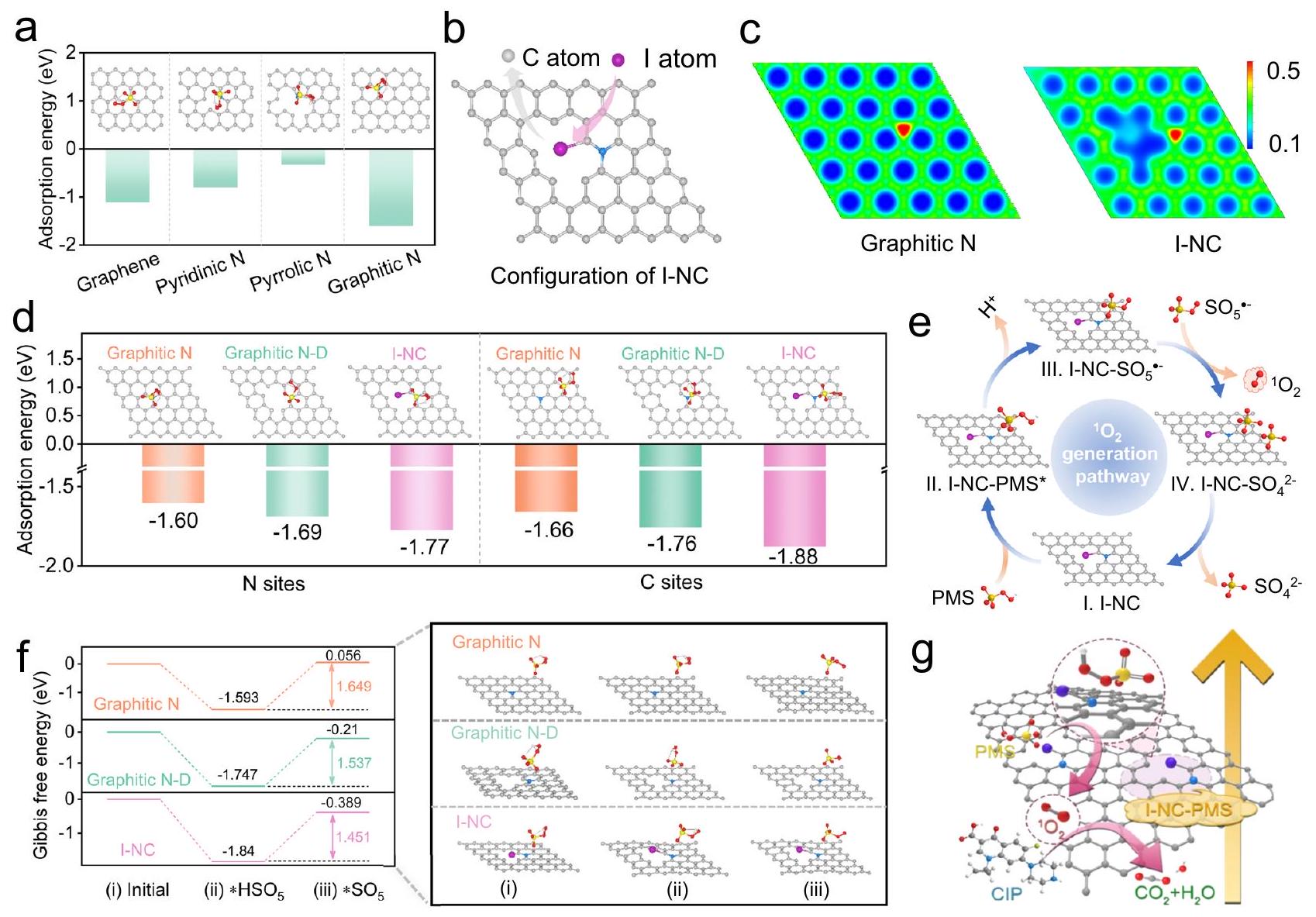
الشكل 4 | حسابات DFT لتوضيح الآليات الأساسية. أ طاقة الامتزاز لـ PMS على الجرافين النقي، النيتروجين البيريديني، النيتروجين البيرولي، والنيتروجين الجرافيتي. ب مخطط تخطيطي لتكوين I-NC. ج صور توزيع كثافة الشحنة لنماذج الكربون المضافة بالنيتروجين مع تكوينات مختلفة من النيتروجين الجرافيتي، I-NC. د طاقة الامتزاز لـ PMS على مواقع النيتروجين ومواقع الكربون في نماذج النيتروجين الجرافيتي، النيتروجين الجرافيتي-D، وI-NC. هـ عملية التفاعل المقترحة لأكسدة PMS إلىعلى I-NC العوامل المساعدة.ملفات الطاقة الكامنة لمسار التفاعل لتوليد الأنواع. نموذج N-D الجرافيتي يمثل النيتروجين الجرافيتي مع عيب. الكرات الرمادية والزرقاء والبنفسجية والحمراء والصفراء والبيضاء تمثل ذرات الكربون والنيتروجين واليود والأكسجين والكبريت والهيدروجين، على التوالي.تمثيل بياني لآلية تحلل CIP في نظام I-NC/PMS. تشير المنطقة البنفسجية والمنطقة المتقطعة في الشكل 4g إلى الموقع النشط لـ I-NC ومعقد I-NC-PMS، على التوالي. تم توفير بيانات المصدر كملف بيانات مصدر. مسار غير جذري. لدعم هذه الفرضية بشكل أكبر، تم إنشاء نماذج حسابية لأشكال الكربون المختلفة، بما في ذلك الجرافين النقي وتكوينات مشوبة بالنيتروجين (النيتروجين الجرافيتي، النيتروجين البيريديني، والنيتروجين البيرولي)، وتُعرض تكويناتها الهندسية المثلى في الشكل التكميلية 36. أشارت حسابات DFT إلى أن طاقة الامتزاز ( ) لمواقع النيتروجين الرسوبي على الجرافين هو أكثر سلبية بشكل ملحوظ ( -1.60 eV ) مقارنة بالجرافين النقي ( -1.11 eV )، البيريديني أو بيروليك. تؤكد هذه النتيجة على الكثافة الإلكترونية العالية والتفاعل الأفضل لمواقع النيتروجين الجرافيتية، مما يجعلها مفضلة للتفاعل مع PMS، كما هو موضح في الشكل.نتيجة لذلك، تم اختيار هيكل الكربون المضاف إليه النيتروجين على شكل جرافيت كنموذج نظري لفهم وتحسين نشاط محفز NC.
قمنا أيضًا بإجراء حسابات DFT لتوضيح أساس الأداء الممتاز لـ I-NC، والتي تهدف إلى استكشاف تأثير تشويب I على الهياكل الإلكترونية للإطار الكربوني، بالإضافة إلى تأثيره على الخصائص الحفازة المتعلقة بتفعيل PMS. استنادًا إلى التوصيف الهيكلي، تم بناء نماذج قابلة للتطبيق لـ N الجرافيتي مع عيب (N-D الجرافيتي) و I-NC مع عيب. تم تصوير التكوينات الهندسية المثلى لهذه النماذج في الشكل 4b والشكل التكميلي 37a، مما يوفر تمثيلًا بصريًا للهياكل الجزيئية التي تسهل فهم الآلية الحفازة المعززة. علاوة على ذلك، تكشف صور توزيع كثافة الشحنة (الشكل 4c والشكل التكميلي 37b) أن إدخال العيوب والتشويب بـ I إعادة تكوين الهيكل الإلكتروني لإطار الكربون بشكل كبير. تم إجراء هذا التعديل للتأثير بشكل كبير على تنشيط PMS. الدراسات الحديثةلقد أظهرت أن ذرات الكربون ذات العجز الإلكتروني يمكن أن تعمل كمواقع نشطة لتنشيط PMS، مما يؤدي إلى تكوينجذري. هذه الخطوة هي خطوة حاسمة محدودة المعدل ومحورية لتوليد. وبالتالي، تم تحليل امتصاص PMS على مواقع الكربون ذات أقل كثافة إلكترونية (الشكل التكميلي 38) وعلى مواقع النيتروجين ذات أعلى كثافة إلكترونية. وأشارت النتائج إلى أن في مواقع الكربون على المحفزات أكثر سلبية مقارنة بتلك الموجودة في مواقع النيتروجين (الشكل 4d). وهذا يشير إلى أن PMS من المرجح أن تمتص على ذرات الكربون الناقصة الإلكترونات، وهو ما يتماشى مع النتائج التجريبية السابقة حول عملية نقل الإلكترون بين المحفز وPMS. علاوة على ذلك،قيمة لامتصاص PMS على I-NCأكثر سلبية من N-D الجرافيتي (-1.76 eV) والجرافيتي. هذا أكد أن التلاعب يمكن أن يؤدي إلى توزيع شحنات موضعية، وكسر تماثل توزيع الإلكترونات على السطح، وخلق منطقة ناقصة الإلكترونات، وزيادة نقل الإلكترونات من PMS إلى مواقع الكربون الناقصة الإلكترونات، وبالتالي زيادة الإنتاج من وزيادة النشاط التحفيزي الشبيه بفنتون.
لذلك، فإن عملية أكسدة PMS لتوليدتم اقتراح أن يتم تحفيز I-NC، كما هو موضح في الشكل 4e. في البداية، يميل جزيء PMS إلى الامتصاص بشكل تفضيلي على المواقع التي تحتوي على كربون ناقص الإلكترونات، مما يؤدي إلى تشكيل. بعد ذلك، تم امتصاص
الشكل 5 | الإمكانيات التطبيقية لنظام I-NC/PMS لتنقية المياه. أ كفاءة الإزالة لأنظمة PMS المنشطة بـ I-NC في وجود أيونات مختلفة، وتركيزات مختلفة من الحمض الهيومي (HA)، ودرجة حموضة ابتدائية مختلفة، ومياه طبيعية مختلفة. الشروط: [المحفز]، (إذا كان هناك أي). كل نقطة بيانات في الشكل 5أ مستقلة عن الأخرى. ب تقليل CIP في خمسة متتالية دورات في نظام I-NC/PMS. تركيز CIP وTOC في المياه الناتجة من مفاعل التدفق المستمر الذي يتكون من عمود مملوء بـ I-NC، الشروط: جرعة المحفزمعدل التدفقتشير أشرطة الخطأ إلى الانحراف المعياري للتجارب التي أُجريت ثلاث مرات. يتم توفير بيانات المصدر كملف بيانات مصدر.
يخضع PMS للتفعيل وإزالة البروتون، تليها تفكك الـرابطة لتوليدالخطوة النهائية تتضمن التباين في النسب، مما يمكّن في النهاية من إنتاج . علاوة على ذلك، من أجل التحقق من صحة هذا الرأي، فإن الحواجز الطاقية المرتبطة بالمسارات لتوليدفي شكل جرافيتيتم حساب PMS و N-D/PMS الجرافيتي و I-NC/PMS (الشكل 4f). ومن الجدير بالذكر أن PMS يتم تمثيله أولاً بالشحنة السلبية.، وبالتالي، تم اعتبار الشحنة معلمة خلفية، دون إضافة شحنة إضافية عند بناء النموذج الذي يحتوي على إما أو باختصار، يمتص PMS على مواقع الكربون الناقصة من الإلكترونات، مكونًا * وإطلاق الطاقة في هذه العملية. أطلق I-NC طاقة أكبر (1.840 إلكترون فولت) مقارنةً بالرسوم البيانية الكربونية. و N-D الجرافيتي (1.747 eV)، مما يشير إلى أن امتصاص PMS على I-NC من المرجح أن يحدث بطريقة طاردة للحرارة وعفوية. علاوة على ذلك، فإن تشكيل حالة الانتقال الوسيطة لـ * (الخطوة II) تمثل الخطوة ذات أعلى حاجز طاقة. تعتبر هذه الخطوة هي الخطوة المحددة لمعدل التفاعل للأنظمة التي تشمل النيتروجين الجرافيتي، والنيتروجين الجرافيتي-D، وI-NC على حد سواء. بالإضافة إلى ذلك، تم ملاحظة حاجز الطاقة المنخفض للـ I-NC.مقارنةً بـ N الجرافيتيو N-D الجرافيتياقترح أن التلاعب يسهل مسارًا بحاجز طاقة منخفض نسبيًا لكل من الامتزاز وإزالة البروتون من PMS أثناء التوليد لـلتلخيص، فإن دمج I في مواقع العيوب يعدل بشكل فعال الهيكل الإلكتروني لمواقع الكربون المجاورة للنيتروجين الجرافيتي. يؤدي هذا التعديل إلى خفض حاجز الطاقة لتوليدويعزز حركية تفاعل فنتون الشبيه. وبالتالي، يظهر تكوين I-NC نشاطًا تحفيزيًا فنتونيًا جوهريًا أعلى بكثير مقارنةً بـ NC (الشكل 4 ج).
الجدوى المحتملة لـ I-NC
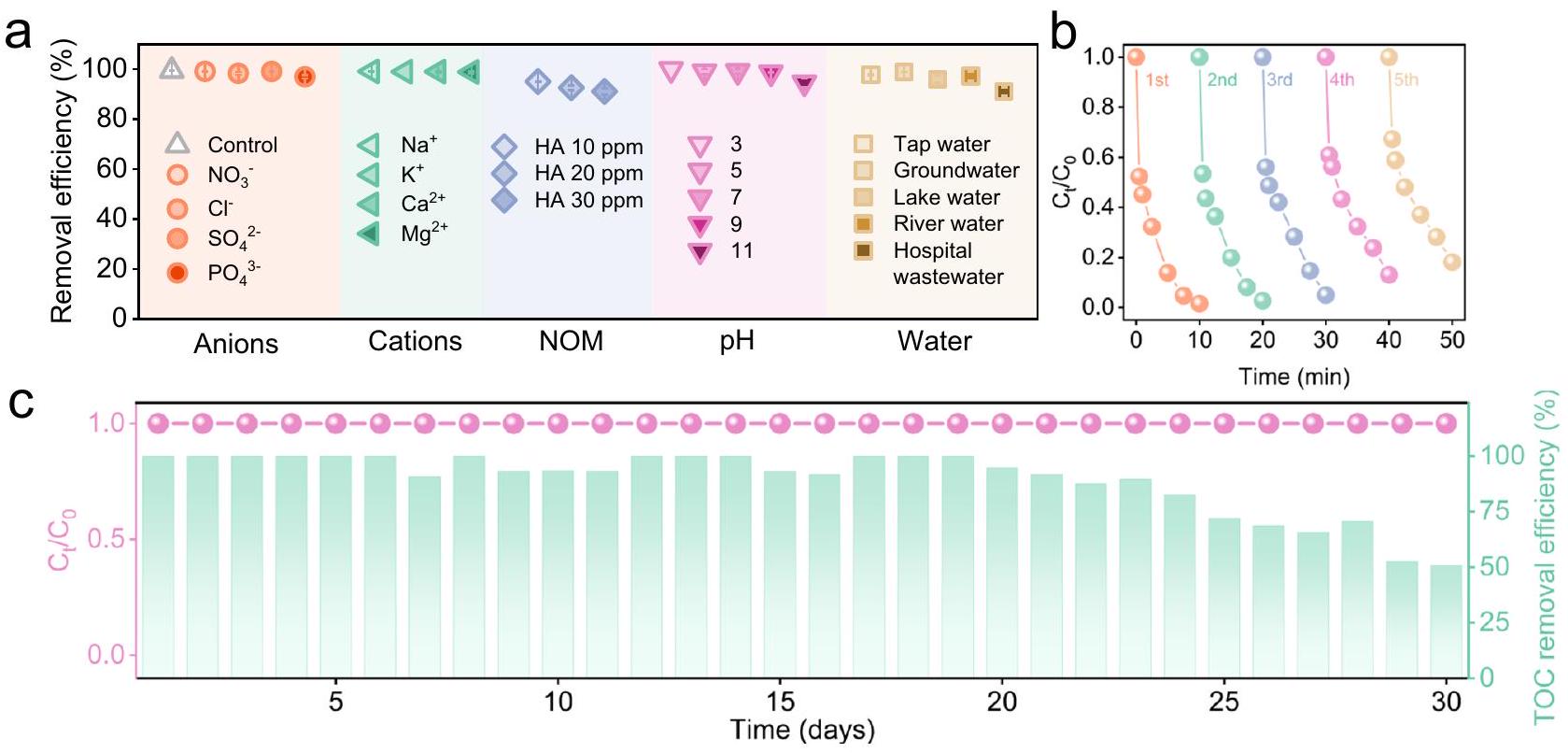
التطبيق العملي للعوامل المساعدة أمر حاسم لمعالجة مشكلة تلوث المياه. ومع ذلك، فإن الانتقال بالعوامل المساعدة الشبيهة بفنتون من البيئات المخبرية إلى المصانع الصناعية يمثل تحديًا بسبب وجود أيونات غير عضوية معقدة ومكونات عضوية في الطبيعة. المسطحات المائية. قد تتنافس هذه المواد مع الملوثات المستهدفة لاستهلاك ROS. ومن الجدير بالذكر أن الشكل 5a يوضح أن كفاءة إزالة CIP بواسطة I-NC/PMS تأثرت بشكل طفيف فقط بالأنيونات النموذجية (أي،، و )، الكاتيونات (أي، ، و ) و DOM (أي، HA). هذه الملاحظة تشير إلى أن نظام التحلل الذي يتم بوساطة – يظهر مقاومة عالية لتأثير الأيونات غير العضوية والملوثات التي تفتقر إلى الإلكترونات. علاوة على ذلك، أظهر نظام INC/PMS أداءً ممتازًا عبر نطاق واسع من الرقم الهيدروجيني الابتدائي (من 3.1 إلى 10.9)، محققًا أكثر منإزالة CIP (الشكل 5 أ والشكل التكميلي 39). تؤكد هذه النتيجة أن كل من I-NC وتتمتع الأنواع النشطة بقدرة قوية على التحمل لتغيرات في درجة حموضة المحلول. الحركيات السريعة (يمكن أن يُعزى الإزالة خلال 30 ثانية التي لوحظت في إزالة CIP عند درجة حموضة 10.9 إلى تنشيط PMS تحت ظروف قلوية عالية (أي، درجة حموضة أكبر من 10.0).تم تقييم جدوى I-NC/PMS بشكل إضافي عبر مصفوفات مائية مختلفة، مع توفير بيانات جودة المياه في الجدول التكميلية 6. أظهر العملية معدلات إزالة متفوقة لـ CIP في أنواع مختلفة من المياه الطبيعية. تحقيق حواليإزالة في مياه الصنبور، والمياه الجوفية، ومياه البحيرات، ومياه الأنهار. بالإضافة إلى ذلك، أظهرت فعالية كبيرة في مياه الصرف الصحي الحقيقية، بحواليتم تحقيق الإزالة في مياه الصرف الصحي بالمستشفى. وهذا يبرز تحسين المقاومة لمواجهة الأنيونات الشائعة الموجودة غالبًا في المياه. علاوة على ذلك، حافظ النظام على نشاط عالي في إزالة التلوث خلال خمس دورات تشغيل متتالية (الشكل 5ب)، على الرغم من حدوث انخفاض طفيف في الأداء خلال التشغيل المطول بسبب تفعيل المحفز بواسطة وسائط الأكسدة. كما هو موضح في الشكل التكميلي 40. تم تطوير مفاعل تدفق مستمر لاحقًا لإظهار الإمكانات الكبيرة للتطبيق العملي لـ I-NC في التشغيل طويل الأمد (معدل التدفق لـ ). بشكل مثير للإعجاب، أظهرت I-NC قدرة مثيرة للإعجاب في المعالجة المستمرة لـ CIP على مدى فترة 30 يومًا من التشغيل المتواصل. على وجه التحديد، تم الحفاظ على كفاءة إزالة CIP عند حوالي كما هو موضح في الشكل 5c. ومن الجدير بالذكر أنه بسبب النشاط التحفيزي القوي لـ I-NC ووجود مواقع امتصاص النيتروجين الغنية في الجرافيت، حدثت معدنة ملحوظة. تم تحقيق الكفاءة في إزالة CIP، مع أكثر منإزالة TOC خلال أول 20 يومًا. الانخفاض الملحوظ في الكفاءة، مع أكثر منيمكن أن يُعزى إزالة TOC في الأيام العشرة التالية إلى تعطيل بعض المواقع النشطة بواسطة وسائط CIP. علاوة على ذلك، أجرينا تحليلًا شاملاً للمحفزات قبل وبعد خمس تجارب دورية، بما في ذلك طيف XPS وخصائص HAADF-STEM. كما هو موضح في الأشكال التكميلية 41 و42، لم تُظهر أطياف XPS لمحفز I-NC أي تغييرات ملحوظة قبل وبعد الاستخدام. بالإضافة إلى ذلك، أكدت خرائط HAADF-STEM وEDS المرتبطة أنه لم يحدث أي تكتل سواء لمواقع N أو I، مما يوضح المزيد من الاستقرار الهيكلي الممتاز لمحفز I-NC. باختصار، تؤكد النشاط القوي والقدرة على التحمل لمحفز I-NC على إمكانياته الواسعة في تطبيقات الترميم البيئي في العالم الحقيقي.
باختصار، عملية CVD معتم تطوير النقش لتوليف ذرة مفردة مميزة وموزعة بشكل جيد مثبتة على ركائز كربونية مشوبة بالنيتروجين. كشفت حسابات DFT أن الهيكل الإلكتروني للنيتروجين الجرافيتي المجاور للكربون يمكن تحسينه من خلال بناء تنسيق C-I في الذرة المفردة غير المعدنية I. تسهل هذه التهيئة التنسيقية الفريدة نقل الإلكترون من PMS إلى المواقع النشطة وتقلل من حاجز الطاقة لتكوين.، مما يعزز النشاط الشبيه بفنتون لعنصر I-NC. علاوة على ذلك، يظهر محفز I-NC كفاءة عالية في الأكسدة الشبيهة بفنتون من حيث الانتقائية العالية للأكسدة وقدرته على مقاومة التداخل في تحلل الملوثات الدقيقة. لقد أظهرت تجارب العمود الإمكانيات الكبيرة لـ I-NC للتطبيقات الواقعية، حيث أظهرت قدرته على إزالة CIP وTOC بشكل مستمر وفعال على مدى 30 يومًا. يفتح عملنا طريقًا لتصميم وتخليق محفزات قائمة على الكربون مدعومة بذرات أحادية غير معدنية لتفاعلات شبيهة بفنتون عالية الكفاءة.
طرق
تحضير المحفزات
تم تفصيل المواد الكيميائية والمواد المحددة المستخدمة في الملاحظة التكميلية 1 في المعلومات الداعمة. تم توضيح إجراءات التخليق لـ ZIF-8 و NC في الملاحظة التكميلية 2. لتحضير I-NC، تم استخدام المنهجية التالية: خضعت NC لعملية تلدين ثانوية حيث تم وضع قارب خزفي يحتوي على NC في مركز فرن أنبوبي. تم تسخين هذا الإعداد إلى وتم الاحتفاظ بها لمدة ساعة واحدة تحت جو من الأرجون (Ar). في الوقت نفسه، كانت هناك قارب خزفي آخر يحتوي على يوديد الأمونيوم، الذي يعمل كعامل حفر ومصدر I، وُضع في الطرف العلوي من فرن الأنبوب، حيث كانت درجة الحرارة تقريبًا. بعد عملية التلدين الثانوية ومرحلة التبريد اللاحقة، تم الحصول على منتج I-NC ولم يتطلب أي معالجة إضافية. في حالة عينة التحكم NC-blank، كانت تحضيرها مشابهًا لتحضير I-NC مع الاستثناء أن تم حذفها خلال مرحلة التلدين الثانوي. وبالمثل، تم إعداد عينة التحكم NC-يوريا باستخدام بروتوكول مشابه لذلك المستخدم في I-NC، باستثناء أنتم استبداله باليوريا خلال خطوة التلدين الثانوية. بالإضافة إلى ذلك، كانت تحضيرات عينات التحكم I-NC-1000 و I-NC-acid متطابقة مع عينة I-NC. تم تحقيق تخليق NC عن طريق تغيير درجة حرارة التكليس (أي، التلدين عند ) وغسل الحمض، على التوالي.
تجارب الانحلال
لتقييم الكفاءة التحفيزية، تم إجراء تجارب دفعة باستخدام قوارير مخروطية سعة 100 مل، تحتوي كل منها على 50 مل من محلول CIP.تم ضبط مستويات pH للمعلقات بدقة لتتراوح بين 3.1 و 10.9 باستخداممحاليل HCl. لعملية التحلل،تم إدخال المحفز I-NC في محلول CIP، ثم تم تفعيله بإضافة PMS (1 مللي مول) تحت تحريك ميكانيكي مستمر عند
400 دورة في الدقيقة. في فترات زمنية محددة مسبقًا، تم استخراج عينات بحجم حوالي 0.5 مل، وتم تثبيتها على الفور بحوالي 1 مل من محلول الميثانول، ثم تم تصفيتها من خلالمرشح غشاء متوافق مع الماء. تم تحليل العينات المفلترة باستخدام كروماتوغرافيا السائل عالية الأداء (طراز LC-20 AT، شيمادزو). تم الحصول على عينات مياه الصنبور من مختبرنا. تم الحصول على عينات المياه الجوفية من نبع بايخه، جبل يويلو. تم أخذ عينات مياه البحيرة من بحيرة تاوزي. تم أخذ عينات مياه الصرف الصحي من مستشفى في تشانغشا، الصين. تم قياس الكربون العضوي الكلي في عينات المياه باستخدام جهاز تحليل الكربون العضوي الكلي Elementar liquiTOC II. يتم الكشف عن تركيز الأنيونات في عينات المياه بواسطة كروماتوغرافيا الأنيونات (Dionex Integrion HPIC، الولايات المتحدة الأمريكية).
توفر البيانات
البيانات التي تدعم نتائج الدراسة متاحة ضمن الورقة والمعلومات التكميلية. يتم توفير بيانات المصدر مع هذه الورقة.
References
Wu, Q.-Y., Yang, Z.-W., Wang, Z.-W. & Wang, W.-L. Oxygen doping of cobalt-single-atom coordination enhances peroxymonosulfate activation and high-valent cobalt-oxo species formation. Proc. Natl. Acad. Sci. USA 120, e2219923120 (2023).
Wang, C. et al. Metal-organic frameworks and their derived materials: emerging catalysts for a sulfate radicals-based advanced oxidation process in water purification. Small 15, 1900744 (2019).
Guo, Z.-Y. et al. Crystallinity engineering for overcoming the activity-stability tradeoff of spinel oxide in Fenton-like catalysis. Proc. Natl. Acad. Sci. USA 120, e2220608120 (2023).
Zhang, D. et al. Dynamic active-site induced by host-guest interactions boost the Fenton-like reaction for organic wastewater treatment. Nat. Commun. 14, 3538 (2023).
Miao, J. et al. Spin-state-dependent peroxymonosulfate activation of single-atom M-N moieties via a radical-free pathway. ACS Catal. 11, 9569-9577 (2021).
Zhou, Q. et al. Generating dual-active species by triple-atom sites through peroxymonosulfate activation for treating micropollutants in complex water. Proc. Natl. Acad. Sci. USA 120, e2300085120 (2023).
Yu, X. et al. A green edge-hosted zinc single-site heterogeneous catalyst for superior Fenton-like activity. Proc. Natl. Acad. Sci. 120, e2221228120 (2023).
Huang, B. et al. Modulating electronic structure engineering of atomically dispersed cobalt catalyst in Fenton-like reaction for efficient degradation of organic pollutants. Environ. Sci. Technol. 57, 14071-14081 (2023).
Yang, M. et al. Unveiling the origins of selective oxidation in singleatom catalysis via Co-N4-C intensified radical and nonradical pathways. Environ. Sci. Technol. 56, 11635-11645 (2022).
Song, J. et al. Asymmetrically coordinated CoB1N3 moieties for selective generation of high-valence Co-Oxo species via coupled electron-proton transfer in Fenton-like reactions. Adv. Mater. 35, 2209552 (2023).
Shao, P. et al. Revisiting the graphitized nanodiamond-mediated activation of peroxymonosulfate: singlet oxygenation versus electron transfer. Environ. Sci. Technol. 55, 16078-16087 (2021).
Yang, Z. et al. Toward selective oxidation of contaminants in aqueous systems. Environ. Sci. Technol. 55, 14494-14514 (2021).
Qian, K. et al. Single-atom Fe catalyst outperforms its homogeneous counterpart for activating peroxymonosulfate to achieve effective degradation of organic contaminants. Environ. Sci. Technol. 55, 7034-7043 (2021).
Yao, Y. et al. Rational regulation of Co-N-C coordination for highefficiency generation of 102 toward Nearly 100% selective
degradation of organic pollutants. Environ. Sci. Technol. 56, 8833-8843 (2022).
. et al. diatomite actuates peroxymonosulfate activation process: mechanism for active species transformation and pesticide degradation. Water Res. 235, 119843 (2023).
Yang, P., Long, Y., Huang, W. & Liu, D. Single-atom copper embedded in two-dimensional MXene toward peroxymonosulfate activation to generate singlet oxygen with nearly selectivity for enhanced Fenton-like reactions. Appl. Catal. B: Environ. 324, 122245 (2023).
Wu, Z.-Y. et al. Electrochemical ammonia synthesis via nitrate reduction on Fe single atom catalyst. Nat. Commun. 12, 2870 (2021).
Li, J. et al. Metal-organic framework-derived graphene mesh: a robust scaffold for highly exposed Fe-N4 Active sites toward an excellent oxygen reduction catalyst in acid media. J. Am. Chem. Soc. 144, 9280-9291 (2022).
Mehmood, A. et al. High loading of single atomic iron sites in Fe-NC oxygen reduction catalysts for proton exchange membrane fuel cells. Nat. Catal. 5, 311-323 (2022).
Zhou, X. et al. Identification of Fenton-like active Cu sites by heteroatom modulation of electronic density. Proc. Natl. Acad. Sci. USA 119, e2119492119 (2022).
Liu, D., He, Q., Ding, S. & Song, L. Structural regulation and support coupling effect of single-atom catalysts for heterogeneous catalysis. Adv. Energy Mater. 10, 2001482 (2020).
Shan J. et al. Metal-metal interactions in correlated single-atom catalysts. Sci. Adv. 8, eabo0762 (2022).
Tang, W. et al. Ru single atom catalyst with dual reaction sites for efficient Fenton-like degradation of organic contaminants. Appl. Catal. B: Environ. 320, 121952 (2023).
Gao, Y. et al. Activity trends and mechanisms in peroxymonosulfate-assisted catalytic production of singlet oxygen over atomic Metal-N-C catalysts. Angew. Chem. Int. Ed. 60, 22513-22521 (2021).
Gao, J. et al. Cobalt single-atom catalysts for domino reductive amination and amidation of levulinic acid and related molecules to N-heterocycles. Chem. Catal. 2, 178-194 (2022).
Cao, D. et al. Volcano-type relationship between oxidation states and catalytic activity of single-atom catalysts towards hydrogen evolution. Nat. Commun. 13, 5843 (2022).
Shang, Y., Xu, X., Gao, B., Wang, S. & Duan, X. Single-atom catalysis in advanced oxidation processes for environmental remediation. Chem. Soc. Rev. 50, 5281-5322 (2021).
Bin Yang, H. et al. Identification of non-metal single atomic phosphorus active sites for the CO2 reduction reaction. EES Catal. 1, 774-783 (2023).
Duan, X., Indrawirawan, S., Sun, H. & Wang, S. Effects of nitrogen-, boron-, and phosphorus-doping or codoping on metal-free graphene catalysis. Catal. Today 249, 184-191 (2015).
Singh, K. P., Song, M. Y. & Yu, J.-S. lodine-treated heteroatomdoped carbon: conductivity driven electrocatalytic activity. J. Mater. Chem. A 2, 18115-18124 (2014).
Jeon, I.-Y. et al. Facile, scalable synthesis of edge-halogenated graphene nanoplatelets as efficient metal-free eletrocatalysts for oxygen reduction reaction. Sci. Rep. 3, 1810 (2013).
Liu, J. et al. lodine-doping-induced electronic structure tuning of atomic cobalt for enhanced hydrogen evolution electrocatalysis. ACS Nano 15, 18125-18134 (2021).
Tian, H. et al. High durability of Fe-N-C single-atom catalysts with carbon vacancies toward the oxygen reduction reaction in alkaline media. Adv. Mater. 35, 2210714 (2023).
Wang, B. et al. A site distance effect induced by reactant molecule matchup in single-atom catalysts for fenton-like reactions. Angew. Chem. 134, e202207268 (2022).
Dong, Y. et al. Ammonia thermal treatment toward topological defects in porous carbon for enhanced carbon dioxide electroreduction. Adv. Mater. 32, 2001300 (2020).
Liu, S. et al. Atomically dispersed iron sites with a nitrogen-carbon coating as highly active and durable oxygen reduction catalysts for fuel cells. Nat. Energy 7, 652-663 (2022).
Gao, Y., Chen, Z., Zhu, Y., Li, T. & Hu, C. New insights into the generation of singlet oxygen in the metal-free peroxymonosulfate activation process: important role of electron-deficient carbon atoms. Environ. Sci. Technol. 54, 1232-1241 (2020).
Sun, B. et al. Polyaniline: a new metal-free catalyst for peroxymonosulfate activation with highly efficient and durable removal of organic pollutants. Environ. Sci. Technol. 53, 9771-9780 (2019).
Li, F., Lu, Z., Li, T., Zhang, P. & Hu, C. Origin of the excellent activity and selectivity of a single-atom copper catalyst with unsaturated Cu-N2 Sites via peroxydisulfate activation: III as a dominant oxidizing species. Environ. Sci. Technol. 56, 8765-8775 (2022).
Guo, C. et al. Ball-milled layer double hydroxide as persulfate activator for efficient degradation of organic: alkaline sites-triggered non-radical mechanism. J. Hazard. Mater. 461, 132219 (2024).
Li, M. et al. Single cobalt atoms anchored on Ti3C2Tx with dual reaction sites for efficient adsorption-degradation of antibiotic resistance genes. Proc. Natl. Acad. Sci. USA 120, e2305705120 (2023).
Wu, Z. et al. Facilely tuning the first-shell coordination microenvironment in iron single-atom for Fenton-like chemistry toward highly efficient Wastewater purification. Environ. Sci. Technol. 57, 14046-14057 (2023).
Pan, Y., Su, H., Zhu, Y., Vafaei Molamahmood, H. & Long, M. CaO2 based Fenton-like reaction at neutral pH : accelerated reduction of ferric species and production of superoxide radicals. Water Res. 145, 731-740 (2018).
Bokare, A. D. & Choi, W. Singlet-oxygen generation in alkaline periodate solution. Environ. Sci. Technol. 49, 14392-14400 (2015).
Lei, Y. et al. Assessing the use of probes and quenchers for understanding the reactive species in advanced oxidation processes. Environ. Sci. Technol. 57, 5433-5444 (2023).
Chen, F. et al. Single-atom iron anchored tubular g-C3N4 catalysts for ultrafast Fenton-like reaction: roles of high-valency iron-oxo species and organic radicals. Adv. Mater. 34, 2202891 (2022).
Li, S. et al. Performance enhancement and mechanism of electroenhanced peroxymonosulfate activation by single-atom Fe catalyst modified electrodes. Proc. Natl. Acad. Sci. USA 121, e2404965121 (2024).
Meng, Y. et al. Nanoconfinement steers nonradical pathway transition in single atom Fenton-like catalysis for improving oxidant utilization. Nat. Commun. 15, 5314 (2024).
Wu, Z. et al. Long-range interactions driving neighboring Fe-N4 sites in Fenton-like reactions for sustainable water decontamination. Nat. Commun. 15, 7775 (2024).
Yun, E.-T., Lee, J. H., Kim, J., Park, H.-D. & Lee, J. Identifying the nonradical mechanism in the peroxymonosulfate activation process: singlet oxygenation versus mediated electron transfer. Environ. Sci. Technol. 52, 7032-7042 (2018).
Li, H., Yuan, N., Qian, J. & Pan, B. Mn2O3 as an Electron shuttle between peroxymonosulfate and organic pollutants: the dominant role of surface reactive species. Environ. Sci. Technol. 56, 4498-4506 (2022).
Wang, Z. et al. Cobalt single atoms anchored on oxygen-doped tubular carbon nitride for efficient peroxymonosulfate activation: simultaneous coordination structure and morphology modulation. Angew. Chem. Int. Ed. 61, e202202338 (2022).
Xu P. et al. A nanoconfined FeCo 2 O 4 -embedded ceramic membrane regulates electron transfer in peroxymonosulfate activation
to selectively generate singlet oxygen for water decontamination. Environ. Sci. Technol. 58, 17464-17474 (2024).
Jiang, X. et al. Precise coordination of high-loading Fe single atoms with sulfur boosts selective generation of nonradicals. Proc. Natl. Acad. Sci. USA 121, e2309102121 (2024).
Gu, C.-H. et al. Tuning electronic structure of metal-free dual-site catalyst enables exclusive singlet oxygen production and in-situ utilization. Nat. Commun. 15, 5771 (2024).
Mo, F. et al. The optimized Fenton-like activity of Fe single-atom sites by Fe atomic clusters-mediated electronic configuration modulation. Proc. Natl. Acad. Sci. USA 120, e2300281120 (2023).
Lan, M.-Y. et al. Multi-channel electron transfer induced by polyvanadate in metal-organic framework for boosted peroxymonosulfate activation. Nat. Commun. 15, 7208 (2024).
Liu, S. et al. Carbonized polyaniline activated peroxymonosulfate (PMS) for phenol degradation: role of PMS adsorption and singlet oxygen generation. Appl. Catal. B: Environ. 286, 119921 (2021).
Zhang, J. et al. Melamine-cyanurate supramolecule induced graphitic N -rich graphene for singlet oxygen-dominated peroxymonosulfate activation to efficiently degrade organic pollutants. Sep. Purif. Technol. 265, 118474 (2021).
Qin, J. et al. Rational design of efficient metal-free catalysts for peroxymonosulfate activation: selective degradation of organic contaminants via a dual nonradical reaction pathway. J. Hazard. Mater. 398, 122808 (2020).
Liang, X. et al. Coordination number dependent catalytic activity of single-atom cobalt catalysts for Fenton-like reaction. Adv. Funct. Mater. 32, 2203001 (2022).
Zhang, X. et al. Metal-free catalysts with local fluorination regulation for peroxymonosulfate activation: nearly singlet oxygen production for selective degradation of aqueous organic pollutants. Chem. Eng. J. 480, 148026 (2024).
Wu, Z. et al. Active center size-dependent Fenton-like chemistry for sustainable water decontamination. Environ. Sci. Technol. 57, 21416-21427 (2023).
Zhen, J. et al. M-N3 configuration on boron nitride boosts singlet oxygen generation via peroxymonosulfate activation for selective oxidation. Angew. Chem. Int. Ed. 63, e202402669 (2024).
Hu, P. et al. Selective degradation of organic pollutants using an efficient metal-free catalyst derived from carbonized polypyrrole via peroxymonosulfate activation. Environ. Sci. Technol. 51, 11288 (2017).
شكر وتقدير
تم دعم العمل من قبل المؤسسة الوطنية للعلوم الطبيعية في الصين (22176124، ج.ل.)، وتمويل بدء البحث من جامعة شنغهاي جياو تونغ (WH22O416002، ج.ل.). كما يشكر المؤلفون المساعدة من خدمة أبحاث سيسيجو.www.ceshigo.com) للحسابات النظرية. الآراء والأفكار المعبر عنها هنا هي فقط هؤلاء من المؤلفين ولا تمثل أفكارهم أي شكل من أشكال وكالات التمويل.
مساهمات المؤلفين
قام J.P. و J.Luo بتصميم البحث. قام J.P. و J.Liu و K.F. و Y.F. بتخليق المواد وإجراء التجارب. ساهم J.P. و K.Y. و J.Luo في تفسير النتائج. كتب J.P. المخطوطة. قام K.Y. و S.L. و D.Y. و M.X. و J.Luo بمراجعة المخطوطة. قدم جميع المؤلفين ملاحظات حاسمة وساعدوا في تشكيل البحث والتحليل والمخطوطة.
يجب توجيه المراسلات والطلبات للحصول على المواد إلى جينمينغ لو.
معلومات مراجعة الأقران تشكر مجلة Nature Communications شياوكيانغ آن، ليجوان كاي، والمراجعين الآخرين المجهولين على مساهمتهم في مراجعة هذا العمل. يتوفر ملف مراجعة الأقران.
ملاحظة الناشر: تظل شركة سبرينغر ناتشر محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
الوصول المفتوح هذه المقالة مرخصة بموجب رخصة المشاع الإبداعي النسبية-غير التجارية-بدون مشتقات 4.0 الدولية، والتي تسمح بأي استخدام غير تجاري، ومشاركة، وتوزيع، وإعادة إنتاج في أي وسيلة أو صيغة، طالما أنك تعطي الائتمان المناسب للمؤلفين الأصليين والمصدر، وتوفر رابطًا لرخصة المشاع الإبداعي، وتوضح إذا قمت بتعديل المادة المرخصة. ليس لديك إذن بموجب هذه الرخصة لمشاركة المواد المعدلة المشتقة من هذه المقالة أو أجزاء منها. الصور أو المواد الأخرى من طرف ثالث في هذه المقالة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة، ما لم يُشار إلى خلاف ذلك في سطر الائتمان للمادة. إذا لم تكن المادة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة وكان استخدامك المقصود غير مسموح به بموجب اللوائح القانونية أو يتجاوز الاستخدام المسموح به، فستحتاج إلى الحصول على إذن مباشرة من صاحب حقوق الطبع والنشر. لعرض نسخة من هذه الرخصة، قم بزيارة http:// creativecommons.org/licenses/by-nc-nd/4.0/. (ج) المؤلف(ون) 2025
¹كلية علوم البيئة والهندسة، جامعة هونان، تشانغشا، جمهورية الصين الشعبية.المختبر الرئيسي لحماية البيئة للدولة لتقييم تأثير الصحة البيئية للملوثات الناشئة، كلية علوم البيئة والهندسة، جامعة جياو تونغ بشنغهاي، شنغهاي، جمهورية الصين الشعبية.المختبر الوطني الرئيسي للكيمياء/الاستشعار الحيوي والكيميوميتري، جامعة هونان، تشانغشا، جمهورية الصين الشعبية.مركز أبحاث الهندسة لصباغة وتجميل الأقمشة البيئية (وزارة التعليم)، جامعة تشجيانغ للعلوم والتكنولوجيا، هانغتشو، جمهورية الصين الشعبية.مدرسة الكيمياء والهندسة الجزيئية، جامعة شرق الصين للعلوم والتكنولوجيا، شنغهاي، جمهورية الصين الشعبية. البريد الإلكتروني:جينمينغ.لو@sjtu.edu.cn
Non-metallic iodine single-atom catalysts with optimized electronic structures for efficient Fenton-like reactions
Received: 27 September 2024
Accepted: January 2025
Published online: 18 January 2025
Check for updates
Junjun Pei , Jianbin Liu , Kaixing Fu , Yukui Fu , Kai Yin , Shenglian Luo , Deyou Yu® , Mingyang Xing & Jinming Luo →
In this study, we introduce a highly effective non-metallic iodine single-atom catalyst (SAC), referred to as I-NC, which is strategically confined within a nitrogen-doped carbon (NC) scaffold. This configuration features a distinctive C-I coordination that optimizes the electronic structure of the nitrogenadjacent carbon sites. As a result, this arrangement enhances electron transfer from peroxymonosulfate (PMS) to the active sites, particularly the electrondeficient carbon. This electron transfer is followed by a deprotonation process that generates the peroxymonosulfate radical ( ). Subsequently, the radical undergoes a disproportionation reaction, leading to the production of singlet oxygen ( ). Furthermore, the energy barrier for the rate-limiting step of generation in I-NC is significantly lower at 1.45 eV , compared to 1.65 eV in the NC scaffold. This reduction in energy barrier effectively overcomes kinetic obstacles, thereby facilitating an enhanced generation of . Consequently, the I-NC catalyst exhibits remarkable catalytic efficiency and unmatched reactivity for PMS activation. This leads to a significantly accelerated degradation of pollutants, evidenced by a relatively high observed kinetic rate constant ( ) compared to other metallic SACs. This study offers valuable insights into the rational design of effective non-metallic SACs, showcasing their promising potential for Fenton-like reactions in water treatment applications.
Given the capacity to produce highly reactive oxygen species (ROS) over a wide pH range, PMS-based advanced oxidation processes (PMSAOPs) featuring easy handling, storage, and transport of oxidant have emerged as promising technologies for water decontamination . The activation of PMS typically commences with the adsorption of a PMS molecule onto a catalyst’s surface , which subsequently catalyzes the formation of various reactive species, including radicals (e.g., hydroxyl radicals, sulfate radicals, and superoxide radicals) and nonradicals
(e.g., and high-valent metal-oxo species) . However, the efficacy of radical species is compromised by the interference of anions and organic matter presenting in waters, which largely reduces PMS utilization and may lead to the formation of undesirable halide byproducts . In contrast, nonradical pathways offer selective and robust oxidation mechanisms , especially in the case of that allows for selective degradation of contaminants due to its specificity, longevity, and resistance to matrix components .
Leveraging the benefits of SACs, such as maximal atomic efficiency, defined active sites, tunable electronic structures, and heightened activity, they have gained great attention in various domains, including electrocatalysis, chemical synthesis, and environmental remediation . Notably, the critical role of SACs in elucidating the structure-activity relationship at an atomic level underscores the potential in PMS-AOPs . Distinguished by the nature of the active sites, metallic SACs (M-SACs) have shown promise in transforming PMS to for micro-pollutant degradation . However, the high production costs, complexity in synthesis, and risk of secondary contamination of metallic SACs encourage us to design the nonmetallic SACs (NM-SACs) for water treatment, which are economically and environmentally more viable.
At present, the investigation regarding NM-SACs is nascent, primarily due to the difficulties in ascertaining active sites and elucidating catalytic pathways. Yet, pioneering works on NM-SACs unveil their potential in modulating electronic structures for the enhancement of Fenton-like reactivity. Yang et al. developed a NM-SAC composed of high-density, isolated phosphorus atoms anchored at the edge of graphene as a robust electrocatalyst for the reduction . Nevertheless, because of the limited ability to increase the catalysts’ intrinsic activity, phosphorus atom-doped catalysts are less used in PMSAOPs . Generally, there are several non-metallic elements in this category, including fluorine, chlorine, bromine and iodine (I), that can perform as effective candidates for PMS-AOPs. Among them, the I atom possesses several notable advantages. Primarily, the atomic structure of I is clearly distinct from that of carbon, and the incorporation of I can effectively modulate the electronic structure of the neighboring carbon matrix. Subsequently, the addition of I can enhance the conductivity of the support, thereby facilitating efficient electron transfer between the catalyst and PMS . Moreover, the incorporation of I atom will lead to more defects due to its larger atomic radius than carbon, thereby exposing more active sites and enhancing the mass transfer of PMS molecules effectively . Finally, the larger atomic mass of I atom allows for the improved identification of its active sites at the atomic scale using advanced techniques such as aberration-corrected high-angle annular dark-field scanning transmission electron microscopy and X-ray absorption fine spectroscopy, thereby facilitating a more thorough establishment of the structureactivity relationship of the catalyst . Due to its unique properties, I emerges as a strong candidate for enhancing the Fenton-like activity of carbon-based materials.
This study presents a potent atomically dispersed I-doped nitro-gen-carbon catalyst (referred to as I-NC), synthesized through chemical vapor deposition, which demonstrates effectiveness in activating PMS for water treatment applications. Notably, the incorporation of I significantly enhances the electronic structure of the carbon-based materials, thereby improving the conversion efficiency of PMS at the active electron-deficient carbon sites and boosting its Fenton-like catalytic performance. The predominant presence of species in oxidation reactions, validated through comprehensive characterizations and theoretical analyses, highlights the catalyst’s underlying mechanism and efficacy. The impressive performance and remarkable durability of I-NC position it as a leading option in single-atom catalysis for practical water treatment, setting a benchmark for catalyst design and application in PMS-AOPs.
Results and discussion
Fabrication of the I-NC catalyst and structure characterization
The I-NC catalyst with atomically dispersed I was synthesized using a chemical vapor deposition (CVD) technique. Initially, ZIF-8 frameworks, formed by the assembly of 2-methylimidazole and zinc nitrate, were subjected to pyrolysis in an inert atmosphere to produce nitrogen-doped carbon (NC) substrates. The successful synthesis of these substrates was confirmed through X-ray diffraction (XRD)
patterns, as illustrated in Supplementary Fig. 1. Subsequently, the NC substrate placed in a tube furnace to initiate the CVD process, utilizing as both an etchant and an I source (Fig. 1a). Specifically, a porcelain boat containing NC was situated centrally within the tube furnace, with positioned at the upstream end to ensure exposure to the evolving gas flow. The annealing was conducted in an inert environment, leading to the decomposition of and the release of ammonia , hydrogen iodide , and iodine vapor , which interact with the NC. This interaction promotes the formation of I-NC single-atom catalysts with carbon-iodine (C-I) moieties, thereby augmenting the intrinsic activity .
As depicted in Supplementary Fig. 2, the scanning electron microscopy analysis reveals that the I-NC catalysts largely retain the original rhombic dodecahedral shape characteristic of ZIF-8, with an average particle size ranging between 300 and 500 nm and a consistent size distribution. Transmission electron microscopy (TEM) images, shown in Supplementary Fig. 3a, b, indicate that the surface of I-NC appears notably rougher compared to NC. This observation suggests that the and HI gases released during decomposition can uniformly etch the carbon matrix to promote the anchor of I single-atom. High-resolution TEM (HRTEM) investigations (Supplementary Fig. 3) confirm the absence of discernible crystal lattices, indicating that no large nanoparticles are presented in the I-NC framework. To delineate the atomic-level dispersion of I within I-NC, aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) was employed. As shown in Fig. 1b, c, the I atoms exhibited a brighter contrast due to their higher atomic number compared to Zn atoms and are uniformly scattered across the I-NC surface. Furthermore, using the advanced threedimensional (3D) atom-overlapping Gaussian-function fitting mapping technique, the area marked by a purple square in Fig. 1c can be reconstructed into Fig. 1d, where the isolated non-metallic I atoms are clearly distinguished within the matrix of the I-NC catalyst. Complementary energy-dispersive X-ray spectroscopy (EDS) mapping, showcased in Fig. 1e, corroborates the homogeneous distribution of I and nitrogen (N) across the carbon substrate. Collectively, these findings affirm the successful introduction and atomic dispersion of I on the N -doped carbon substrates.
The X-ray powder diffraction (XRD) pattern of I-NC (Supplementary Fig. 4) displays two pronounced peaks at the of approximately and . These peaks are assigned to the (002) and (101) planes of graphitic carbon, respectively, and show a high degree of similarity to the corresponding peaks in NC. This resemblance suggests that the fundamental graphitic structure is preserved in I-NC. To further probe the microstructural attributes of NC and I-NC, Raman spectroscopy was conducted and the results are presented in Supplementary Fig. 5. We identify two distinct peaks at roughly 1349 and , which are attributed to the D-band and G-band of carbon-based materials, respectively. Notably, the intensity ratio of the D-band to G-band ( ) for I-NC registers a marginal increase compared to NC. This increment is indicative of a reduced degree of graphitization, aligning with the observations of etching on the carbon surface revealed through TEM analysis. Collectively, these spectroscopic insights corroborate the structural modifications induced by treatment and provide a microscopic understanding of the altered properties of I-NC.
The electron paramagnetic resonance (EPR) spectrum for I-NC, shown in Fig. 2a, displays a pronounced peak signal at a g-value of 2.005, indicative of enhanced carbon vacancies within the NC bulk structure . These vacancy defects are expected to enhance the mass transfer of reactant molecules and promote further I atoms incorporation. Further pore structure and surface area analysis were conducted by measuring nitrogen adsorption-desorption isotherms via Brunauer-Emmett-Teller (BET) methodology (Supplementary Fig. 6). I-NC exhibits a typical type-I isotherm, suggesting a microporousdominant structure, in alignment with the pore size distribution
Fig. 1 | Preparation and structural characterization of I-NC catalyst. a Schematic illustration of the preparation of atomically dispersed I-doped nitrogen-carbon catalyst (denoted as I-NC). The yellow sphere represents the largest van der Waals sphere that would fit in the cavity. b, c A high-resolution high-angle annular dark-
field scanning transmission electron microscopy (HAADF-STEM) image of I-NC. Yellow circles indicate single iodine atoms. d 3D atom-overlapping Gaussianfunction fitting map of a region in inset (c). e Energy-dispersive X-ray spectroscopy (EDS) elemental mappings ( , and Zn) of I-NC.
outcomes. Notably, I-NC features significantly improvement in BET surface area ( ) and pore volume ( ) when compared with NC ( and ). These enhancements presumably optimize the exposure of catalytic sites and promote the diffusion of reactant molecules (e.g., ROS, CIP).
The compositional and electronic structures of NC and I-NC were thoroughly investigated using X-ray photoelectron spectroscopy (XPS), providing insights into the surface elemental compositions and chemical states. The XPS survey spectra (Supplementary Fig. 7) revealed comparable levels of carbon ( C ), N , and oxygen ( O ) across both catalysts. High-resolution N spectra for NC and I-NC could be deconvolved into four distinct peaks corresponding to various N configurations: pyridinic N (at ), pyrrolic N (at ), graphitic N (at ), and oxidized N species ( ), as displayed in Fig. 2b, and Supplementary Fig. 8 and summarized in Supplementary Table 2 and Table 3. A notable enhancement in the graphitic N content was observed for , registering at , an increase from NC at . This predominance of graphitic N in I-NC, especially when compared to other nitrogen species, underscores its potential role in enhancing Fenton-like catalytic activities.
Furthermore, the I-NC spectrum features a new peak at approximately 620 eV , attributing to I element. The detailed analysis of the I region in I-NC’s XPS spectrum (Fig. 2c) reveals deconvolution into two peaks at 618.58 and 620.46 eV , corresponding to ionic ( ) and covalent (C-I) iodine species, respectively. This observation supports the integration of C-I bonding configurations, corroborated by a specific peak at 285.6 eV in the XPS C 1s spectrum (Supplementary Fig. 9), affirming the successful introduction and chemical interaction of I within the I-NC structure.
The electronic structure and atomic configuration of I-NC were further elucidated using I K-edge X-ray absorption fine spectroscopy (XAFS). In Fig. 2d, the X-ray absorption near edge structure (XANES) profiles of I-NC and references reveal a notable resemblance in the absorption edge of I within I-NC to that observed for iohexol with model C-I bonds (Supplementary Fig. 10). This similarity indicates that the oxidation state of I in I-NC is between elemental iodine and iohexol, suggesting a mixed valence of I. Further insights were obtained through the Fourier-transformed extended X-ray absorption fine structure (EXAFS) analysis, detailed in Fig. 2e, f and Supplementary Fig. 11. These results highlight the absence of distinct peaks
Fig. 2 | Electronic state and atomic structure characterization of the prepared catalysts. a The electron paramagnetic resonance (EPR) spectra of NC and I-NC. b High-resolution X-ray photoelectron spectroscopy (XPS) N 1s spectra of NC and I-NC. c High-resolution XPS I spectra of I-NC. The relevant XPS fitting parameters of and are listed in Supplementary Table 3. d Normalized I K-edge X-ray
absorption near edge structure (XANES) spectra of Iodine, Iohexol, and I-NC. e I K-edge Fourier-transformed extended X-ray absorption fine structure (EXAFS) spectra for Iodine, Iohexol, and I-NC. Purple region denotes the characterized peaks of I-C both in NC and I-NC. fI K-edge EXAFS fitting analysis for I-NC. g-i Wavelet transform EXAFS plots of iodine, iohexol, and I-NC.
corresponding to I-I bonding at , affirming the premise that I atoms are integrated within the I-NC matrix as isolated entities. This isolation supports the assertion of single-atom coordination, aligning with the anticipated enhancement of catalytic activity and providing a cogent explanation for the observed physicochemical properties of I-NC. Additional examination of the XAFS data for I-NC, particularly the peak observed at , corroborates the presence of C-I coordination, which is akin to that identified in iohexol. An additional discernible peak in the I-NC spectrum can be ascribed to the C-I and Zn-I backscattering pathways. The detection of Zn-I interactions suggests potential direct coordination between I and Zn within the NC structure or the formation of zinc iodide as a consequence of iodine vapor etching, a hypothesis supported by the absence of a washing step postetching. However, it should be noted that the Zn-I feature is not predominant, which aligns with the inference that I within the material predominantly exists in a positively charged state, more evidences were provided later. Further substantiation is provided by the EXAFS wavelet transform (WT) analysis depicted in Fig. 2g-i, affirming the nonexistence of I-I bonding within I-NC. This finding is congruent with observations from HAADF-STEM investigations. Moreover, EXAFS
fitting results suggest the mean coordination number around C-I in I-NC mirrors observed in iohexol, reinforcing the atomically dispersed nature of I sites within the catalyst (Supplementary Fig. 12 and Table 4).
Catalytic Performance and Identification of ROS
The Fenton-like catalytic performance of I-NC was evaluated by activating PMS for the degradation of ciprofloxacin (CIP). Initially, an examination of the impact of various operating parameters was carried out, illustrated in Supplementary Figs. 13-15. Specifically, the optimal concentration of PMS was determined to be 1 mM while the catalyst dosage was screened at . The most effective secondary annealing temperature in the CVD process for fabricating I-NC was found to be . The results presented in Fig. 3a demonstrate that CIP was nearly completely degraded by the I-NC/PMS system within 10 min . In contrast, only about of CIP degradation was observed with the NC/PMS system. This significant difference underscores the superior Fenton-like catalytic activity of the I-NC system. The liquid chromatography-mass spectrometry (LC-MS) chromatograms of CIP before and after the reaction in the I-NC/PMS system further confirmed that CIP was virtually eliminated within 10 min (Supplementary
Fig. 3 | Fenton-like catalytic performance, reactive species identification and reaction process characterization. a The degradation of ciprofloxacin (CIP) in different catalysts-activated PMS systems. b Comparison of I-NC and reported catalysts in a Fenton-like reaction. The relevant references are listed in Supplementary Table 5. c EPR spectra using 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) and 2,2,6,6-tetramethylpiperidine (TEMP) as trapping agents. d Linear sweep voltammetry (LSV) curves of NC and I-NC under different conditions. e In situ Raman spectra of PMS, I-NC and their reaction. The cyan, purple and yellow regions represent the characteristic peaks of and PMS*, respectively.
f The yield in different catalyst/PMS/CIP systems. g Degradation of multiple pollutants in the I-NC/PMS system. The name and chemical structure of relevant micropollutants are listed in Supplementary Fig. 33. The oxidation mechanism of for multiple micropollutants. The blue and yellow surfaces represent positive isosurface and negative isosurface, respectively. white, Grey, blue, red, yellow, and cyan spheres represent H, C, N, O, S, and F atoms, respectively. Conditions: [catalyst] , [pollutants] 50 mM (if any). Error bars denote standard deviation of the experiments performed in triplicate. Source data are provided as a Source Data file.
Fig. 16). In addition, PMS alone only degraded less than of CIP, and around of CIP was removed by I-NC through adsorption, indicating that catalytic oxidation played a major role in the rapid elimination of CIP. Notably, the leaching of of iodine ions from the I-NC/PMS system contributed to about degradation rate of CIP, indicating that the leached iodine ions had a minimal impact on the degradation process (Supplementary Fig. 17).
CIP degradation can be well described by pseudo-first-order kinetics. The kinetic constant ( ) of I-NC/PMS ( ) was 10.1 times higher than that of NC/PMS ( ), and times greater than those of commercial carbonaceous materials. Notably, the of I-NC outperformed most of the state-of-the-art catalysts with effective PMS activation on CIP degradation (Supplementary Fig. 18). Additionally, given that may vary with reaction conditions and the type of organic substances, a normalized kinetic model ( –
value) was employed. This model adjusts the by dividing it by the concentration of PMS and the catalyst dosage, and multiplying by the concentration of the pollutant, to uniformly assess the removal rates across different heterogeneous Fenton-like systems . As illustrated in Fig. 3b and summarized in Supplementary Table 5, the -value for I-NC ( ) surpassed those of all compared metal-free catalysts ( 0.23 to ) in terms of PMS activation for CIP degradation and was higher than most leading-edge metallic catalysts, for example, for .
The results underscored that I-NC significantly outperforms NC in Fenton-like catalytic activity, highlighting the effectiveness of the etching method. As depicted in Supplementary Fig. 19a, a direct relationship is evident between the Fenton-like catalytic activity and factors such as surface area, pore volume, and the ratio. This correlation suggests that etching successfully modified the pore
structure of NC, which enhanced the number of active sites and improved their accessibility. Remarkably, a 6.8 -fold enhancement in the specific surface area normalized value ( per surface area) was still attained, indicating a more advantageous intrinsic catalytic activity of I-NC (Supplementary Fig. 19b). Intriguingly, the degradation characteristics of I-NC did not align with the variations in Zn concentration, as illustrated in Supplementary Fig. 20. This indicates that Zn plays a negligible role in the effectiveness of the I-NC/PMS system. Additionally, the secondary calcination of NC in an inert atmosphere without , referred to as NC-blank, did not significantly enhance CIP degradation performance. This indicates that the secondary calcination process alone does not substantially affect the intrinsic active sites of NC, as supported by the data in Supplementary Fig. 21. Previous studies have shown that carbon-based materials can undergo effective etching to introduce defects through heat treatment. Consequently, urea was chosen as the source for treating NC. The experimental results revealed that the CIP degradation efficiency in the NC-urea/PMS system exhibited only a slight improvement over that of NC alone. Moreover, despite the addition of extra iodine ions, minimal improvement was observed in the NC-urea and iodine ions/PMS system, further demonstrating the limited influence of dissolved iodine ions in PMS-mediated catalytic reactions (Supplementary Fig. 21). This finding highlights the critical role of iodine single-atom in enhancing PMS activation and facilitating the degradation of CIP.
Various reactive species, including , and , have been identified in carbon-based catalyst/PMS processes . To investigate the specific reactive species generated during PMS activation with NC and I-NC, scavenging experiments were conducted using methanol (MeOH) and tert-butyl alcohol (TBA) as radical scavengers. Specifically, MeOH and TBA were employed as radical scavengers of and/or . The presence of overdosed MeOH and TBA scarcely hindered CIP degradation (Supplementary Fig. 22), suggesting that and were likely not the primary active species in the NC/PMS and I-NC/PMS. As MeOH and TBA cannot quench surface-bound radicals, dimethyl sulfoxide (DMSO) and NaF were employed to explore the contribution of radicals on the surface of I-NC . As illustrated in Supplementary Fig. 23, DMSO and NaF had a limiting effect on the degradation of CIP in the I-NC/PMS system, suggesting that surfacebound radicals contribute little to the degradation of CIP. Additionally, the introduction of chloroform ( ), with a high-rate constant for reacting with of , caused only a slight decrease in CIP degradation. This result indicates that also did not play a significant role in degrading CIP. Additionally, nitroblue tetrazolium dichloride (NBT) was employed as a probe for , as shown in Supplementary Fig. 24, the negligible influence of NBT suggested the minimal involvement of in the I-NC/PMS system. In stark contrast, the introduction of furfuryl alcohol, with a known rate constant with of , significantly impeded the degradation of CIP. This remarkable inhibition points to a critical role of in the degradation process, aligning with the observed selective oxidation behavior towards various micropollutants in the I-NC/PMS. As depicted in Supplementary Fig. 25, the similar quenching effects observed in both NC and I-NC when using L-histidine, and 2,2,6,6-tetramethylpiperidine (TEMP) as quenchers suggest that these catalysts share a common mechanism for PMS activation and micropollutant degradation. This further emphasizes the dominance of in these reactions .
Considering that carbon-based materials can act as electronic mediators to facilitate electron transfer from pollutants to PMS, thereby oxidizing pollutants with the independence of ROS. To validate the contribution of electron transfer to CIP degradation, a series of experiments including electrochemical measurement, premixing experiment, and PMS decomposition experiment were conducted. As shown in Supplementary Fig. 26, the open circuit potential (OCP) of glassy carbon electrodes coated with NC or I-NC increased
immediately after the addition of PMS, indicating that the active PMS complexes formed on the catalyst surface can elevate the potential . Nevertheless, there was no discernible drop in potential with the addition of CIP, suggesting that the catalyst-mediated electron transfer process for pollutants to the catalyst-PMS complexes is negligible . Furthermore, to further verify the existence of the catalyst-mediated electron transfer process, the catalyst and PMS were mixed in the blank solution, and the CIP-containing solution was added at , and 9 min subsequently. As depicted in Supplementary Fig. 27, we observed an extended premixing time resulted in a more significant decrease in CIP degradation efficiency, with only of CIP being degraded with of after a premixing period in the I-NC/PMS system. This outcome further suggests that the electron transfer process might not play a significant role in CIP degradation under these conditions. Additionally, existing studies indicate that the decomposition of PMS often depends on organic substrates acting as electron donors in the electron transfer mechanism . However, when CIP was introduced as a potential electron donor, there was no observed acceleration in PMS consumption, as illustrated in Supplementary Fig. 28. This lack of impact indicates that CIP does not facilitate the PMS decomposition via electron transfer, contrasting with the expected behavior if electron transfer were a predominant degradation pathway in the I-NC/PMS.
The referenced studies provide evidence that catalyst-mediated electron transfer processes are not significant contributors to the I-NC/ PMS activity. To reinforce this conclusion, the research utilized 9,10diphenylanthracene (DPA) as a specific probe to detect the presence of in the system . DPA interacts with , undergoing oxidation to 9,10-diphenylanthracene dioxide (DPAO2), which is accompanied by a decrease in the absorption peak intensity around 380 nm . The observed reduction in this peak, as shown in Supplementary Fig. 29, confirms the generation and involvement of in the CIP degradation process. Additionally, the study employed 5,5-dimethyl-1-pyrroline-Noxide (DMPO) and TEMP as trapping agents to probe for other reactive species, specifically and , and , respectively. The utilization of these trapping agents in Fig. 3c aimed to differentiate the reactive species involved and further ascertain the dominant role of in the degradation mechanism, unlike the involvement of other radical species in I-NC/PMS. Furthermore, the EPR experiments revealed significant insights into the reactive species responsible for CIP degradation within the I-NC/PMS. The EPR spectra clearly showed a distinctive peak attributed to 5,5-dimethylpyrroline-N-oxide (DMPOX), indicating that DMPO undergoes deep oxidation, potentially caused by the rapid production of hydroxyl radicals, high-valent metal-oxo species, or . Given the exclusion of free radicals as the dominant active species and the catalyst’s multivalent metal-free nature, we can speculate that is responsible for the formation of DMPOX. Moreover, the experiment utilizing TEMP as a trapping agent revealed a distinct signal for -TEMP adducts. This signal was significantly more intense in the I-NC/PMS system compared to the NC/PMS system, reinforcing the idea that plays a crucial role in the catalytic degradation of CIP by I-NC. The presence of this -TEMP adduct provides strong evidence that is a dominant oxidative species in the I-NC/PMS system, effectively participating in the degradation process. The accumulation of experimental evidence indicates that plays a pivotal role in the INC/PMS and NC/PMS, with I-NC exhibiting superior catalytic performance compared to NC/PMS. The significant reduction in the intensity of the -TEMP adduct peak upon introducing CIP into the I-NC/PMS system reinforces the conclusion that is instrumental in degrading CIP (Supplementary Fig. 30). Additionally, the results from the quenching experiment and the NBT probe, along with the TEMPtrapping EPR experiment, support the conclusion that in the system does not stem from . The observation that gas aeration with argon (Ar) and oxygen ( ) does not influence CIP elimination (Supplementary Fig. 31), suggesting that dissolved oxygen is not a
precursor to the generated in these reactions. Hence, the derived inference is that is produced through the interaction between PMS molecules and the catalyst’s active sites.
The reaction process characterization of the Fenton-like system Considering PMS’s dual role as both an oxidant and reductant, the electron transfer pathways between PMS and the catalyst are essential for the generation of . When electrons are transferred from PMS to the catalyst, this process triggers the dissociation of the , resulting in the formation of the (Eq. 1). This radical subsequently leads to the generation of through disproportionation reaction (Eq. 2) . Conversely, when electrons are transferred from the catalyst to PMS, this initiates the cleavage of the bond to produce *OH, subsequently dissociated into * O , which then combine with another intermediate to ultimately generate . To discriminate the electron transfer process between I-NC and PMS, electrochemical tests were carried out. In linear sweep voltammetry analyses (Fig. 3d), the current density of both the I-NC electrode and NC electrode increased significantly in PMS presence, with the increment of the former being much greater than that of the latter, showcasing an enhanced electron transfer from PMS to I-NC compare to NC . Moreover, the current observed in the system increased significantly following the addition of CIP, indicating a favorable reaction among PMS, CIP, and the catalysts. Additionally, the OCP of glassy carbon electrode coated with NC and I-NC catalysts exhibited an immediate increase upon the addition of PMS. Notably, the increase in OCP for the I-NC catalyst was 0.54 V , which was substantially greater than the 0.45 V increase observed with the NC catalyst (Supplementary Fig. 26). This difference further supports the notion of effective electron transfer from PMS to the active sites on the catalysts .
To further investigate the electron transfer process between I-NC and PMS, the Raman spectroscopy was conducted (Fig. 3e). The spectra revealed three distinct peaks in the PMS solution ( , located at 880,980 , and ), corresponding to the stretching vibrations of , and , respectively. Notably, the peaks associated with the and peaks showed a marked decrease upon the addition of I-NC over time, indicating a rapid conversion of PMS to following activation by I-NC. In addition, the adsorption of PMS on catalysts is crucial for its activation. The attachment of PMS molecules to the catalyst surface can lead to the formation of a metastable complexes, denoted as *PMS. This process was evidenced by the appearance of new peaks at 1110 and in the Raman spectra, suggesting the presence of the *PMS complex . Notably, the characteristic vibration of appeared significant blue shift ( ). The blue shift typically derives from the decrease in electron density, suggesting the donation of electrons from PMS to catalysts . Subsequently, PMS is converted into the , which then leads to the generation of . These findings suggest that the electron transfer from PMS to the active sites occurs first, followed by deprotonation to form . This radical then undergoes a disproportionation process, resulting in the production of . Furthermore, the incorporation of I optimizes the local electronic configuration of NC, facilitating the electron transfer from PMS to catalysts and thereby enhancing the forming of .
Moreover, changes in pH were monitored before and after reaction process, as depicted in Supplementary Fig. 32. The gradual decline in the solution’s pH value reaffirms the efficient generation of coupled with proton release (Eq. 3). This observation further rules out the predominant role of free radical pathway that produces maintaining pH or generates in conjunction with an increasing pH . Notably, we can calculate the theoretical value of generation using Eq. 3 and the observed variations in pH . The generated by I-NC activated PMS was found to be 0.22 mM , which is 2.59 times higher than that generated by NC (Fig. 3f). This result further indicates that the addition of I atoms effectively modifies the electronic structure of the material, enhancing its inherent catalytic activity and
improving the efficiency of generation efficiency.
serves as an electrophilic agent, readily participating in electrophilic addition toward electron-rich pollutants. Consequently, the PMS activation system catalyzed by I-NC can selectively eliminate a wide range of pollutants. In particular, the degradation rates of electron-rich compounds such as CIP, carbamazepine (CBZ), phenol (Ph), bisphenol A (BPA), 4-chlorophenol (4-CP) and tetracycline (TC) exceeded within a duration, with the exception of sulfamethoxazole (SMX), which achieved approximately . Conversely, electron-deficient containments, including nitrobenzene (NB), benzoic acid (BA) and chloramphenicol (CPL) exhibited significantly lower degradation efficiencies, all falling below 20% (Fig. 3g). This selective reactivity highlights the effectiveness of the I-NC activated PMS system in targeting specific types of pollutants, as illustrated in Supplementary Fig. 33. The difference of oxidation ability toward different selected organics were further explored through the perspective of the charge transfer pathway, as depicted in Fig. 3h. A smaller energy gap between lowest unoccupied molecular orbital (LUMO) of and highest occupied molecular orbital (HOMO) of the pollutants represents that electrons migration from the pollutants to can be more readily initiated. This facilitates and accelerates the oxidation process targeted at the specific pollutants. Thus, the efficiency of in oxidizing various organic compounds is closely linked to the energetic compatibility of their molecular orbitals . It was clearly observed that the HOMO of electron-rich contaminants such as CIP ( -4.72 eV ), TC and SMX ( -5.49 eV ) were slightly lower than the LUMO of ( -4.50 eV ). This energy alignment indicates that can readily withdraw electrons from HOMO of these electron-rich contaminants, facilitating their degradation. Conversely, the LUMO of was substantially higher than the HOMO energy levels of CPL , BA and NB . This substantial energy difference demonstrates that it is challenging for to perform oxidation reactions on these electron-deficient compounds, further highlighting the selective reactivity of based on the electronic structure of the pollutants. In summary, CIP undergoes a rapid degrading effectiveness in the I-NC/PMS system compared to the 10 representative pollutants examined, due to a narrower gap between LUMO of and HOMO of CIP, facilitating more efficient electron transfer and subsequent oxidation. The degradation intermediates of CIP were identified by LC-MS as shown in Supplementary Fig. 34. Specifically, the peak corresponding to CIP appeared at a retention time of 7.5 min . Additionally, six degradation intermediates were identified, which together suggest a possible degradation pathway for CIP, as shown in Supplementary Fig. 35. This pathway provides insights into the transformation of CIP in the I-NC/PMS system, highlighting the complexity and effectiveness of the degradation process.
Theoretical study of the catalytic mechanism
As validated by previous findings , the different types of N species exhibit different activation mechanisms in non-metal active sitemediated PMS activation process, and graphitic N-doped carbonaceous catalysts are favorable for PMS activation to generate . Here we found a higher content of graphitic N compared to pyridinic and pyrrolic N in the catalysts, associating this with the dominance of as the active species in both NC/PMS and I-NC/PMS. This suggests a crucial role of graphitic N in the PMS activation process dominated by
Fig. 4 | DFT calculations for elucidating the underlying mechanisms. a The adsorption energy of PMS on pristine graphene, pyridinic N, pyrrolic N and graphitic N. b Schematic diagram of I-NC configuration. c Charge density distribution images of N -doped carbon models with different configurations of graphitic N , I-NC. d The adsorption energy of PMS on N sites and C sites of graphitic N, graphitic N-D, and I-NC models. e Proposed reaction process for PMS oxidation to on I-NC
catalysts. Potential energy profiles of the reaction pathway for generating the species. Graphitic N-D model represent graphitic N with defect. Grey, blue, purple, red, yellow, and white spheres represent C, N, I, O, S, and H atoms, respectively. Diagrammatic representation of the CIP degradation mechanism in the I-NC/PMS system. The purple region and dashed area in Fig. 4g denote I-NC’s active site and I-NC-PMS complex, respectively. Source data are provided as a Source Data file.
non-radical pathway. To further substantiate this conjecture, computational models of various carbon forms, including pristine graphene and N -doped configurations (graphitic N , pyridinic N , and pyrrolic N ), were created, and their optimal geometric configurations are presented in Supplementary Fig. 36. DFT calculations indicated that the adsorption energy ( ) of PMS on graphitic N sites is significantly more negative ( -1.60 eV ) than on pristine graphene ( -1.11 eV ), pyridinic , or pyrrolic . This finding underscores the superior electron density and reactivity of graphitic N sites, establishing them as preferential for PMS interaction, as depicted in Fig. . As a result, the graphitic N -doped carbon structure was chosen as the theoretical model for understanding and optimizing the NC catalyst’s activity.
We further performed the DFT calculations to elucidate the foundation of the I-NC excellent performance, which aimed to explore the impact of I doping on the electronic structures of the carbon framework, as well as its influence on the catalytic characteristics relevant to PMS activation. Drawing on structural characterization, feasible models for graphitic N with defect (graphitic N-D) and I-NC with defect were constructed. The optimal geometrical configurations of these models are depicted in Fig. 4b and Supplementary Fig. 37a, providing a visual representation of the molecular structures that facilitate an understanding of the enhanced catalytic mechanism. Furthermore, the charge density distribution images (Fig. 4c and Supplementary Fig. 37b) reveal that the introduction of defects and the doping with I
substantially reconfigure the electronic structure of the carbon framework. This modification is made to significantly influence the activation of PMS. Recent studies have highlighted that carbon atoms with electronic deficiency can serve as active sites for the activation of PMS, leading to the formation of the radical. This step is a crucial rate-limited step and pivotal for the generation of . Thus, the adsorption of PMS on carbon sites with the lowest electron density (Supplementary Fig. 38) and on nitrogen sites with the highest electron density were analyzed. The results indicated that the at carbon sites on the catalysts is more negative compared to those on nitrogen sites (Fig. 4d). This suggests that PMS is more likely to adsorb onto electrondeficient carbon atoms which is in accordance with previous experimental results on the electron transfer process between the catalyst and PMS. Moreover, the value for PMS adsorption on I-NC is more negative than on graphitic N-D ( -1.76 eV ) and graphitic . This confirmed that I doping can led to localized charge distributions, break the symmetry of surface electron distribution, create an electron-deficient region, further enhance the electron transfer of PMS to electron-deficient carbon sites, and consequently increase the generation of and enhance the Fenton-like catalytic activity.
Therefore, the process of PMS oxidation to generate triggered by I-NC was proposed, as depicted in Fig. 4e. Initially, the PMS molecule preferentially adsorbs onto the sites with electron-deficient carbon, leading to the formation of . Following this, the adsorbed
Fig. 5 | Application potential of I-NC/PMS system for water purification. a The removal efficiency of I-NC activated PMS systems in the presence of different ions, different humic acid (HA) concentrations, different initial pH, and different natural waters. Conditions: [catalyst] , (if any). Each data point in Fig. 5a is independent of the other. b CIP abatement in five consecutive
cycles in the I-NC/PMS system. c CIP concentration and TOC in the effluent from a continuous-flow reactor consisting of the I-NC-filled column, conditions: catalyst dosage , flow rate . Error bars denote standard deviation of the experiments performed in triplicate. Source data are provided as a Source Data file.
PMS undergoes activation and deprotonation, followed by the dissociation of the bond to generate . The final step involves the disproportionation of , which ultimately enables the production of . Furthermore, in order to validate this view, the energy barriers associated with the pathways for the generation of in graphitic PMS, graphitic N-D/PMS, and I-NC/PMS were calculated (Fig. 4f). Notably, the PMS is first represented by the negatively charged , and therefore, the charge was considered a background parameter, with no additional charge added when constructing the model containing either or . Briefly, PMS adsorbs onto electrondeficient carbon sites, forming * and releasing energy in the process. The I-NC released more energy ( 1.840 eV ) compared to the graphitic and graphitic N-D ( 1.747 eV ), indicating that the adsorption of PMS onto I-NC is more likely to occur in an exothermic and spontaneous manner. Furthermore, the formation of the intermediate transition state of * (step II) represents the step with the highest energy barrier. This step is considered the rate-determining step for the systems involving graphitic N, graphitic N-D, and I-NC alike. In addition, the observed lower energy barrier for the I-NC compared to graphitic N and graphitic N-D suggested that I doping facilitates a pathway with a relatively low energy barrier for both the adsorption and deprotonation of PMS during the generation of . To summarize, the incorporation of I into defect sites effectively modulates the electronic structure of carbon sites adjacent to graphitic N. This modulation leads to a lower energy barrier for the generation of and enhances the kinetics of the Fenton-like reaction. Consequently, the I-NC configuration demonstrates significantly higher intrinsic Fenton-like catalytic activity compared to the NC (Fig. 4 g ).
Potential practicability of I-NC
The practical application of catalysts is crucial for addressing the issue of water pollution. However, transitioning Fenton-like catalysts from laboratory settings to industrial plants is challenging due to the presence of complex inorganic ions and organic components in natural
water bodies. These substances may compete with the target pollutants for consuming ROS. Notably, Fig. 5a illustrates that the removal efficiency of CIP by I-NC/PMS was only marginally affected by typical anions (i.e., , and ), cations (i.e., , and ) and DOM (i.e., HA). This observation suggests that the -mediated degradation system exhibits a high resistance to the influence of inorganic ions and electron-deficient pollutants. Furthermore, the INC/PMS demonstrated excellent performance across a broad initial pH range (from 3.1 to 10.9), achieving more than removal of CIP (Fig. 5a and Supplementary Fig. 39). This result confirms that both the I-NC and the active species possess a strong tolerance to variations in the solution pH . The rapid kinetics ( elimination within 30 s ) observed in the removal of CIP at a pH of 10.9 can be attributed to the activation of PMS under highly alkaline conditions (i.e., pH greater than 10.0. The viability of the I-NC/PMS was further evaluated across various water matrices, with water quality data provided in Supplementary Table 6. The process demonstrated superior removal rates of CIP in different types of natural waters. Achieving about removal in tap water, groundwater, lake water, and river water. Additionally, it showed significant effectiveness in real wastewater, with approximately removal achieved in hospital wastewater. This highlights the improvement of resistance to counteract common anions frequently found in water. Furthermore, the system maintained high decontamination activity during five consecutive running cycles (Fig. 5b), although a slight performance decline occurred during prolonged operation due to catalyst passivation by the oxidation intermediates. As illustrated in Supplementary Fig. 40. a continuous-flow reactor was subsequently developed to showcase the significant potential for the practical application of the I-NC in long-term operation (flow rate of ). Impressively, I-NC demonstrated impressive capability in the continuous treatment of CIP over a period of 30 days of consecutive operation. Specifically, the removal efficiency of CIP was maintained at approximately , as shown in Fig. 5c. Notably, due to the potent catalytic activity of I-NC and the abundant graphite N adsorption sites, a remarkable mineralization
efficiency was achieved in the removal of CIP, with over TOC removal within the first 20 days. The observed decrease in efficiency, with more than TOC removal in the subsequent 10 days, could be attributed to the inactivation of some active sites by the CIP intermediates. Furthermore, we conducted a comprehensive analysis of the catalysts before and after five cyclic tests, including XPS spectra and HAADF-STEM characterizations. As shown in Supplementary Figs. 41 and 42, the XPS spectra of I-NC catalyst exhibited no significant changes before and after use. Additionally, the HAADF-STEM and associated EDS mapping confirmed that no aggregation occurred for either N or I sites, further demonstrating the excellent structural stability of I-NC catalyst. In summary, the I-NC’s powerful activity and durability underscore its vast potential for real-world environmental remediation applications.
In summary, the CVD process with etching was developed to synthesize a distinctive well-dispersed I single-atom anchored onto N -doped carbon substrates. DFT calculations revealed that the electronic structure of the graphitic N adjacent to carbon could be optimized by the construction of C-I coordination in the non-metallic I single-atom. This unique coordination configuration facilitates the electron transfer from PMS to active sites and reduces the energy barrier for the formation of , thereby enhancing the Fenton-like activity of I-NC catalyst. Moreover, the I-NC catalyst exhibits great Fenton-like oxidation efficiency in terms of high oxidation selectivity and anti-interference capability for the degradation of micropollutants. Column experiments have demonstrated the great potential of I-NC for real-world applications, showcasing its ability to continuously and effectively remove CIP and TOC over a 30-day period. Our work opens an avenue for the design and synthesis of nonmetallic single atom-doped carbon-based catalysts for highly efficient Fenton-like reactions.
Methods
Preparation of catalysts
The specific chemicals and materials utilized are detailed in Supplementary Note 1 in the Supporting Information. The synthesis procedures for ZIF-8 and NC are elaborated in Supplementary Note 2. For preparing I-NC, the following methodology was employed: NC underwent a secondary annealing process wherein a porcelain boat containing NC was positioned at the center of a tube furnace. This setup was heated to and maintained for 1 h under an argon (Ar) atmosphere. Concurrently, another porcelain boat containing ammonium iodide , serving both as an etchant and an I source, was placed at the tube furnace’s upstream end, where the temperature was approximately . Following the secondary annealing and subsequent cooling phases, the I-NC product was obtained and required no further processing. In the case of the NC-blank control sample, its preparation mirrored that of I-NC with the exception that was omitted during the secondary annealing phase. Similarly, the NC-urea control sample was prepared using a protocol akin to that for I-NC, except that was substituted with urea during the secondary annealing step. Additionally, the preparations of the I-NC-1000 and I-NC-acid control samples were identical to that of the I-NC sample. The NC synthesis was accomplished by altering the calcination temperature (i.e., annealing at ) and acid washing, respectively.
Degradation experiments
To evaluate catalytic efficiency, batch experiments were conducted using 100 mL conical flasks, each containing 50 mL of CIP solution ( ). The pH levels of the suspensions were meticulously adjusted to range from 3.1 to 10.9 utilizing and HCl solutions. For the degradation process, of the I-NC catalyst was introduced into the CIP solution, and subsequently activated by the addition of PMS ( 1 mM ) under consistent mechanical stirring at
400 rpm . At predetermined time intervals, approximately 0.5 mL samples were extracted, promptly stabilized with roughly 1 mL of methanol solution, and subsequently filtered through a watercompatible membrane filter. The filtered samples were analyzed using high-performance liquid chromatography (model LC-20 AT, Shimadzu). Tap water samples were obtained from our laboratory. Groundwater samples were obtained from Baihe Spring, Yuelu Mountain. Lake water samples were taken from the Taozi Lake. Hospital wastewater samples were taken from a hospital in changsha, China. TOC in the water samples was measured on an Elementar liquiTOC II Total Organic Carbon analyzer. Anion concentration in water samples is detected by anion chromatography (Dionex Integrion HPIC, USA).
Data availability
The data supporting the findings of the study are available within the paper and its Supplementary Information. Source data are provided with this paper.
References
Wu, Q.-Y., Yang, Z.-W., Wang, Z.-W. & Wang, W.-L. Oxygen doping of cobalt-single-atom coordination enhances peroxymonosulfate activation and high-valent cobalt-oxo species formation. Proc. Natl. Acad. Sci. USA 120, e2219923120 (2023).
Wang, C. et al. Metal-organic frameworks and their derived materials: emerging catalysts for a sulfate radicals-based advanced oxidation process in water purification. Small 15, 1900744 (2019).
Guo, Z.-Y. et al. Crystallinity engineering for overcoming the activity-stability tradeoff of spinel oxide in Fenton-like catalysis. Proc. Natl. Acad. Sci. USA 120, e2220608120 (2023).
Zhang, D. et al. Dynamic active-site induced by host-guest interactions boost the Fenton-like reaction for organic wastewater treatment. Nat. Commun. 14, 3538 (2023).
Miao, J. et al. Spin-state-dependent peroxymonosulfate activation of single-atom M-N moieties via a radical-free pathway. ACS Catal. 11, 9569-9577 (2021).
Zhou, Q. et al. Generating dual-active species by triple-atom sites through peroxymonosulfate activation for treating micropollutants in complex water. Proc. Natl. Acad. Sci. USA 120, e2300085120 (2023).
Yu, X. et al. A green edge-hosted zinc single-site heterogeneous catalyst for superior Fenton-like activity. Proc. Natl. Acad. Sci. 120, e2221228120 (2023).
Huang, B. et al. Modulating electronic structure engineering of atomically dispersed cobalt catalyst in Fenton-like reaction for efficient degradation of organic pollutants. Environ. Sci. Technol. 57, 14071-14081 (2023).
Yang, M. et al. Unveiling the origins of selective oxidation in singleatom catalysis via Co-N4-C intensified radical and nonradical pathways. Environ. Sci. Technol. 56, 11635-11645 (2022).
Song, J. et al. Asymmetrically coordinated CoB1N3 moieties for selective generation of high-valence Co-Oxo species via coupled electron-proton transfer in Fenton-like reactions. Adv. Mater. 35, 2209552 (2023).
Shao, P. et al. Revisiting the graphitized nanodiamond-mediated activation of peroxymonosulfate: singlet oxygenation versus electron transfer. Environ. Sci. Technol. 55, 16078-16087 (2021).
Yang, Z. et al. Toward selective oxidation of contaminants in aqueous systems. Environ. Sci. Technol. 55, 14494-14514 (2021).
Qian, K. et al. Single-atom Fe catalyst outperforms its homogeneous counterpart for activating peroxymonosulfate to achieve effective degradation of organic contaminants. Environ. Sci. Technol. 55, 7034-7043 (2021).
Yao, Y. et al. Rational regulation of Co-N-C coordination for highefficiency generation of 102 toward Nearly 100% selective
degradation of organic pollutants. Environ. Sci. Technol. 56, 8833-8843 (2022).
. et al. diatomite actuates peroxymonosulfate activation process: mechanism for active species transformation and pesticide degradation. Water Res. 235, 119843 (2023).
Yang, P., Long, Y., Huang, W. & Liu, D. Single-atom copper embedded in two-dimensional MXene toward peroxymonosulfate activation to generate singlet oxygen with nearly selectivity for enhanced Fenton-like reactions. Appl. Catal. B: Environ. 324, 122245 (2023).
Wu, Z.-Y. et al. Electrochemical ammonia synthesis via nitrate reduction on Fe single atom catalyst. Nat. Commun. 12, 2870 (2021).
Li, J. et al. Metal-organic framework-derived graphene mesh: a robust scaffold for highly exposed Fe-N4 Active sites toward an excellent oxygen reduction catalyst in acid media. J. Am. Chem. Soc. 144, 9280-9291 (2022).
Mehmood, A. et al. High loading of single atomic iron sites in Fe-NC oxygen reduction catalysts for proton exchange membrane fuel cells. Nat. Catal. 5, 311-323 (2022).
Zhou, X. et al. Identification of Fenton-like active Cu sites by heteroatom modulation of electronic density. Proc. Natl. Acad. Sci. USA 119, e2119492119 (2022).
Liu, D., He, Q., Ding, S. & Song, L. Structural regulation and support coupling effect of single-atom catalysts for heterogeneous catalysis. Adv. Energy Mater. 10, 2001482 (2020).
Shan J. et al. Metal-metal interactions in correlated single-atom catalysts. Sci. Adv. 8, eabo0762 (2022).
Tang, W. et al. Ru single atom catalyst with dual reaction sites for efficient Fenton-like degradation of organic contaminants. Appl. Catal. B: Environ. 320, 121952 (2023).
Gao, Y. et al. Activity trends and mechanisms in peroxymonosulfate-assisted catalytic production of singlet oxygen over atomic Metal-N-C catalysts. Angew. Chem. Int. Ed. 60, 22513-22521 (2021).
Gao, J. et al. Cobalt single-atom catalysts for domino reductive amination and amidation of levulinic acid and related molecules to N-heterocycles. Chem. Catal. 2, 178-194 (2022).
Cao, D. et al. Volcano-type relationship between oxidation states and catalytic activity of single-atom catalysts towards hydrogen evolution. Nat. Commun. 13, 5843 (2022).
Shang, Y., Xu, X., Gao, B., Wang, S. & Duan, X. Single-atom catalysis in advanced oxidation processes for environmental remediation. Chem. Soc. Rev. 50, 5281-5322 (2021).
Bin Yang, H. et al. Identification of non-metal single atomic phosphorus active sites for the CO2 reduction reaction. EES Catal. 1, 774-783 (2023).
Duan, X., Indrawirawan, S., Sun, H. & Wang, S. Effects of nitrogen-, boron-, and phosphorus-doping or codoping on metal-free graphene catalysis. Catal. Today 249, 184-191 (2015).
Singh, K. P., Song, M. Y. & Yu, J.-S. lodine-treated heteroatomdoped carbon: conductivity driven electrocatalytic activity. J. Mater. Chem. A 2, 18115-18124 (2014).
Jeon, I.-Y. et al. Facile, scalable synthesis of edge-halogenated graphene nanoplatelets as efficient metal-free eletrocatalysts for oxygen reduction reaction. Sci. Rep. 3, 1810 (2013).
Liu, J. et al. lodine-doping-induced electronic structure tuning of atomic cobalt for enhanced hydrogen evolution electrocatalysis. ACS Nano 15, 18125-18134 (2021).
Tian, H. et al. High durability of Fe-N-C single-atom catalysts with carbon vacancies toward the oxygen reduction reaction in alkaline media. Adv. Mater. 35, 2210714 (2023).
Wang, B. et al. A site distance effect induced by reactant molecule matchup in single-atom catalysts for fenton-like reactions. Angew. Chem. 134, e202207268 (2022).
Dong, Y. et al. Ammonia thermal treatment toward topological defects in porous carbon for enhanced carbon dioxide electroreduction. Adv. Mater. 32, 2001300 (2020).
Liu, S. et al. Atomically dispersed iron sites with a nitrogen-carbon coating as highly active and durable oxygen reduction catalysts for fuel cells. Nat. Energy 7, 652-663 (2022).
Gao, Y., Chen, Z., Zhu, Y., Li, T. & Hu, C. New insights into the generation of singlet oxygen in the metal-free peroxymonosulfate activation process: important role of electron-deficient carbon atoms. Environ. Sci. Technol. 54, 1232-1241 (2020).
Sun, B. et al. Polyaniline: a new metal-free catalyst for peroxymonosulfate activation with highly efficient and durable removal of organic pollutants. Environ. Sci. Technol. 53, 9771-9780 (2019).
Li, F., Lu, Z., Li, T., Zhang, P. & Hu, C. Origin of the excellent activity and selectivity of a single-atom copper catalyst with unsaturated Cu-N2 Sites via peroxydisulfate activation: III as a dominant oxidizing species. Environ. Sci. Technol. 56, 8765-8775 (2022).
Guo, C. et al. Ball-milled layer double hydroxide as persulfate activator for efficient degradation of organic: alkaline sites-triggered non-radical mechanism. J. Hazard. Mater. 461, 132219 (2024).
Li, M. et al. Single cobalt atoms anchored on Ti3C2Tx with dual reaction sites for efficient adsorption-degradation of antibiotic resistance genes. Proc. Natl. Acad. Sci. USA 120, e2305705120 (2023).
Wu, Z. et al. Facilely tuning the first-shell coordination microenvironment in iron single-atom for Fenton-like chemistry toward highly efficient Wastewater purification. Environ. Sci. Technol. 57, 14046-14057 (2023).
Pan, Y., Su, H., Zhu, Y., Vafaei Molamahmood, H. & Long, M. CaO2 based Fenton-like reaction at neutral pH : accelerated reduction of ferric species and production of superoxide radicals. Water Res. 145, 731-740 (2018).
Bokare, A. D. & Choi, W. Singlet-oxygen generation in alkaline periodate solution. Environ. Sci. Technol. 49, 14392-14400 (2015).
Lei, Y. et al. Assessing the use of probes and quenchers for understanding the reactive species in advanced oxidation processes. Environ. Sci. Technol. 57, 5433-5444 (2023).
Chen, F. et al. Single-atom iron anchored tubular g-C3N4 catalysts for ultrafast Fenton-like reaction: roles of high-valency iron-oxo species and organic radicals. Adv. Mater. 34, 2202891 (2022).
Li, S. et al. Performance enhancement and mechanism of electroenhanced peroxymonosulfate activation by single-atom Fe catalyst modified electrodes. Proc. Natl. Acad. Sci. USA 121, e2404965121 (2024).
Meng, Y. et al. Nanoconfinement steers nonradical pathway transition in single atom Fenton-like catalysis for improving oxidant utilization. Nat. Commun. 15, 5314 (2024).
Wu, Z. et al. Long-range interactions driving neighboring Fe-N4 sites in Fenton-like reactions for sustainable water decontamination. Nat. Commun. 15, 7775 (2024).
Yun, E.-T., Lee, J. H., Kim, J., Park, H.-D. & Lee, J. Identifying the nonradical mechanism in the peroxymonosulfate activation process: singlet oxygenation versus mediated electron transfer. Environ. Sci. Technol. 52, 7032-7042 (2018).
Li, H., Yuan, N., Qian, J. & Pan, B. Mn2O3 as an Electron shuttle between peroxymonosulfate and organic pollutants: the dominant role of surface reactive species. Environ. Sci. Technol. 56, 4498-4506 (2022).
Wang, Z. et al. Cobalt single atoms anchored on oxygen-doped tubular carbon nitride for efficient peroxymonosulfate activation: simultaneous coordination structure and morphology modulation. Angew. Chem. Int. Ed. 61, e202202338 (2022).
Xu P. et al. A nanoconfined FeCo 2 O 4 -embedded ceramic membrane regulates electron transfer in peroxymonosulfate activation
to selectively generate singlet oxygen for water decontamination. Environ. Sci. Technol. 58, 17464-17474 (2024).
Jiang, X. et al. Precise coordination of high-loading Fe single atoms with sulfur boosts selective generation of nonradicals. Proc. Natl. Acad. Sci. USA 121, e2309102121 (2024).
Gu, C.-H. et al. Tuning electronic structure of metal-free dual-site catalyst enables exclusive singlet oxygen production and in-situ utilization. Nat. Commun. 15, 5771 (2024).
Mo, F. et al. The optimized Fenton-like activity of Fe single-atom sites by Fe atomic clusters-mediated electronic configuration modulation. Proc. Natl. Acad. Sci. USA 120, e2300281120 (2023).
Lan, M.-Y. et al. Multi-channel electron transfer induced by polyvanadate in metal-organic framework for boosted peroxymonosulfate activation. Nat. Commun. 15, 7208 (2024).
Liu, S. et al. Carbonized polyaniline activated peroxymonosulfate (PMS) for phenol degradation: role of PMS adsorption and singlet oxygen generation. Appl. Catal. B: Environ. 286, 119921 (2021).
Zhang, J. et al. Melamine-cyanurate supramolecule induced graphitic N -rich graphene for singlet oxygen-dominated peroxymonosulfate activation to efficiently degrade organic pollutants. Sep. Purif. Technol. 265, 118474 (2021).
Qin, J. et al. Rational design of efficient metal-free catalysts for peroxymonosulfate activation: selective degradation of organic contaminants via a dual nonradical reaction pathway. J. Hazard. Mater. 398, 122808 (2020).
Liang, X. et al. Coordination number dependent catalytic activity of single-atom cobalt catalysts for Fenton-like reaction. Adv. Funct. Mater. 32, 2203001 (2022).
Zhang, X. et al. Metal-free catalysts with local fluorination regulation for peroxymonosulfate activation: nearly singlet oxygen production for selective degradation of aqueous organic pollutants. Chem. Eng. J. 480, 148026 (2024).
Wu, Z. et al. Active center size-dependent Fenton-like chemistry for sustainable water decontamination. Environ. Sci. Technol. 57, 21416-21427 (2023).
Zhen, J. et al. M-N3 configuration on boron nitride boosts singlet oxygen generation via peroxymonosulfate activation for selective oxidation. Angew. Chem. Int. Ed. 63, e202402669 (2024).
Hu, P. et al. Selective degradation of organic pollutants using an efficient metal-free catalyst derived from carbonized polypyrrole via peroxymonosulfate activation. Environ. Sci. Technol. 51, 11288 (2017).
Acknowledgements
The work was supported by the National Natural Science Foundation of China (22176124, J.L.), and the Research Start-up Funding from Shanghai Jiao Tong University (WH22O416002, J.L.). The authors also thank the help from Ceshigo Research Service (www.ceshigo.com) for the theoretical calculation. The views and ideas expressed herein are solely
those of the authors and do not represent the ideas of the funding agencies in any form.
Author contributions
J.P. and J.Luo designed the research. J.P., J.Liu, K.F., and Y.F. synthesized the materials and carried out the experiments. J.P., K.Y., and J.Luo contributed to the interpretation of the results. J.P. wrote the manuscript. K.Y., S.L., D.Y., M.X., and J.Luo revised the manuscript. All authors provided critical feedback and helped shape the research, analysis, and manuscript.
Correspondence and requests for materials should be addressed to Jinming Luo.
Peer review information Nature Communications thanks Xiaoqiang An, Lejuan Cai, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http:// creativecommons.org/licenses/by-nc-nd/4.0/.
(c) The Author(s) 2025
¹College of Environmental Science and Engineering, Hunan University, Changsha, P.R. China. State Environmental Protection Key Laboratory of Environmental Health Impact Assessment of Emerging Contaminants, School of Environmental Science and Engineering, Shanghai Jiao Tong University, Shanghai, P.R. China. State Key Laboratory of Chemo/Biosensing and Chemometrics, Hunan University, Changsha, P.R. China. Engineering Research Center for Eco-Dyeing and Finishing of Textiles (Ministry of Education), Zhejiang Sci-Tech University, Hangzhou, P. R. China. School of Chemistry and Molecular Engineering, East China University of Science and Technology, Shanghai, P.R. China. e-mail: jinming.luo@sjtu.edu.cn