محفزات كهربائية Pd/NiMoO4/NF للتخليق الفعال والفائق الاستقرار واستخراج الجلايكولات بمساعدة الإلكتروليت Pd/NiMoO4/NF electrocatalysts for the efficient and ultra-stable synthesis and electrolyte-assisted extraction of glycolate
تاريخ الاستلام: 26 سبتمبر 2023 تم القبول: 18 مارس 2024 نُشر على الإنترنت: 04 أبريل 2024 (أ) التحقق من التحديثات
كاي شيدي سيشيوي تنغليسونغ تشينجيانلين شي (ب
التحويل الكهروكيميائي للجزيئات العضوية الصغيرة هو تقنية واعدة لإنتاج مواد كيميائية ذات قيمة مضافة، ولكنها تعاني من استهلاك مرتفع للمعادن الثمينة، وضعف استقرار المحفزات الكهروكيميائية، وصعوبة فصل المنتجات. هنا، تم استخدام محفز كهروكيميائي Pd/NiMoO4/NF مع كمية منخفضة جداً من Pd.تم تطوير ( ) من أجل تخليق الجلايكولات بكفاءة اقتصادية واستقرار فائق، حيث يظهر كفاءة فاراداي عالية (98.9%)، وعائد (98.8%)، واستقرار فائق (1500 ساعة) تجاه أكسدة الإيثيلين جلايكول الكهروكيميائية. علاوة على ذلك، تم تحويل حمض الجلايك إلى جلايكولات الصوديوم ذو القيمة المضافة من خلال تفاعل الحمض والقاعدة في إلكتروليت NaOH، والذي يتمتع بكفاءة ذرية ولا يحتاج إلى إضافة حمض إضافي لفصل المنتج. علاوة على ذلك، تلعب الامتزاز الضعيف لجلايكولات الصوديوم على سطح المحفز دورًا كبيرًا في تجنب الأكسدة المفرطة وتحقيق انتقائية عالية. قد توفر هذه الدراسة إرشادات لتصميم المحفز الكهروكيميائي وكذلك فصل المنتج لتحويلات الكحول الكهروكيميائية.
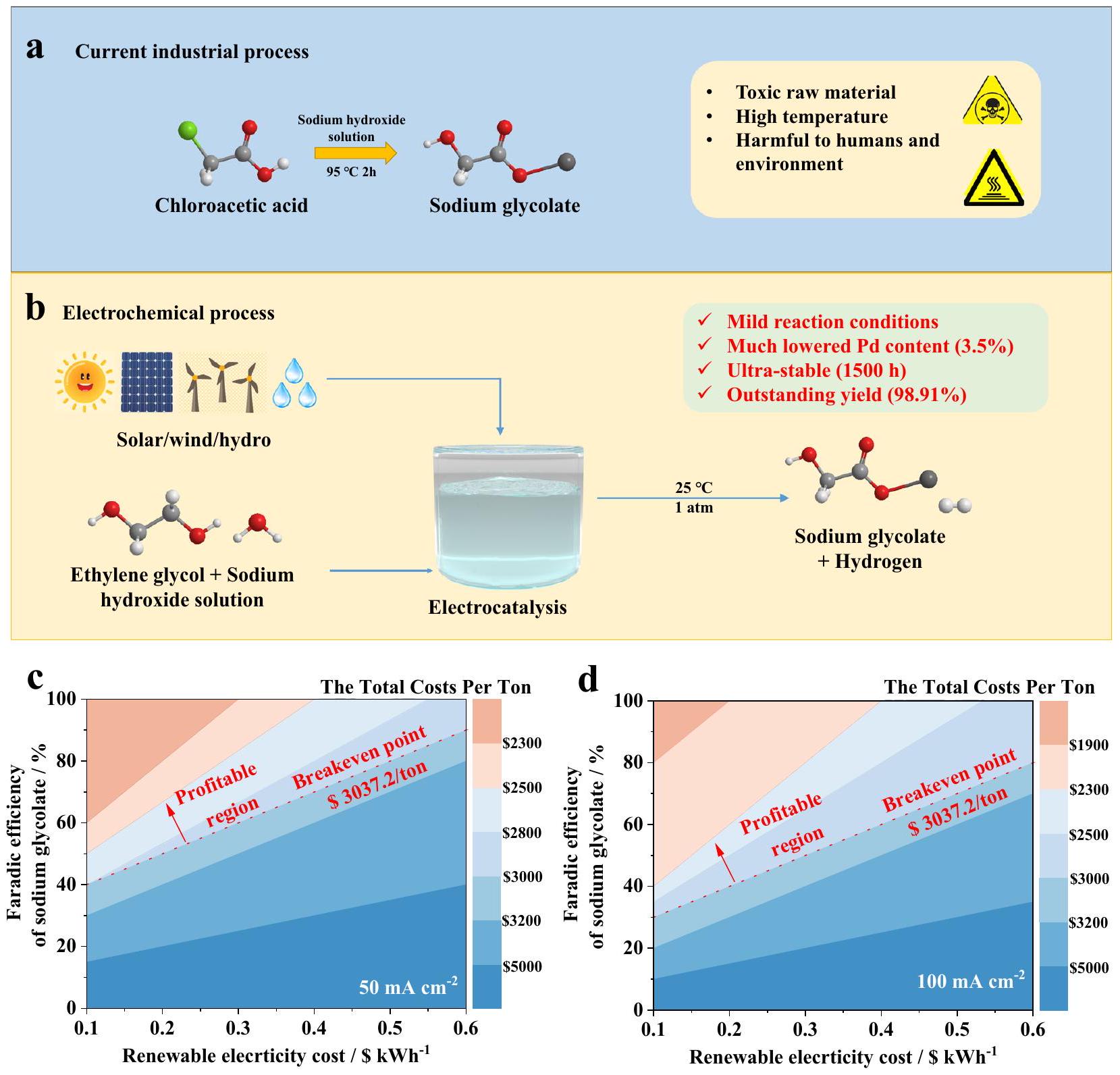
غليكولات الصوديوم، وهو مادة كيميائية أساسية، يُستخدم على نطاق واسع كوسيط مهم في تقنيات إنتاج مجموعة متنوعة من المواد الكيميائية الهامة. يصل الإنتاج العالمي من غليكولات الصوديوم إلى حوالي 0.1 مليون طن سنويًا، وسعر السوق لغليكولات الصوديوم حوالي 3.0 آلاف دولار أمريكي.في الوقت الحاضر، يتم إنتاج جلايكولات الصوديوم من خلال عملية كثيفة الطاقة تحت درجات حرارة عالية. عادةً ما يتفاعل حمض الكلورو أسيتيك مع هيدروكسيد الصوديوم في تفاعل استبدال لإنتاج جلايكولات الصوديوم وكلوريد الصوديوم، يتبع ذلك عملية فصل معقدة للحصول على المنتج النهائي. بالإضافة إلى التكلفة العالية واستهلاك الطاقة للتكنولوجيا المذكورة، فإن حمض الكلورو أسيتيك الخام عادةً ما يكون سامًا وقابلًا للتآكل، مما يضر بالبيئة وصحة الإنسان. في هذا الصدد، فإن تطوير طرق فعالة وموفرة للطاقة وغير سامة وغير ضارة تحت ظروف محيطية لإنتاج جلايكولات الصوديوم يعد أمرًا بالغ الأهمية..
مع التطور السريع لأساليب توليد الكهرباء المتجددة، أصبحت التخليق الكهربائي شائعًا كوسيلة فعالة أداة التخليق تحت الظروف المحيطةعلى سبيل المثال، تم الإبلاغ عن الأكسدة الكهروكيميائية للجزيئات العضوية الصغيرة (الميثانول، الإيثانول، الإيثيلين غليكول، الجلسرين، 5 هيدروكسي ميثيل فورفورال، وما إلى ذلك) إلى مواد كيميائية قيمة مثل حمض الفورميك، حمض الجليكوليك، وحمض 2,5-فوران ثنائي الكربوكسيليك.. تم استكشاف عدة أقطاب كهربائية من المعادن النبيلة سابقًا لأكسدة الإيثيلين غليكول؛ ومع ذلك، فإنها تنتج فقط جزيئات C 1، مثل وقد تم الحصول على الشكل والصيغة كأهم المنتجات في معظم التقارير، كما أن ندرة وارتفاع تكلفة المعادن النبيلة قد حد من تطبيقها على نطاق واسع.مؤخراً، قام الباحثون بتحميل المعادن النبيلة على الركائز من الأكاسيد أو الهيدروكسيدات للحصول على منتجات C2. على سبيل المثال، أبلغت مجموعتنا عن الأكسدة الكهروكيميائية للإيثيلين غليكول (EG) إلى حمض الغليكوليك (GA) بواسطة PdAg/NF، مما جذب اهتماماً واسعاً.على الرغم من أن العديد من الباحثين قد قدموا مساهمات في هذا المجال، لا تزال هناك مشاكل قائمة.من ناحية، فإن الكمية العالية من المعادن الثمينة في المحفزات الكهربائية وتوقفها السريع (لا يتجاوز 200 ساعة) لا يمكن أن تلبي المتطلبات.
الشكل 1 | توضيح تخطيطي لإنتاج جلايكولات الصوديوم. أ الطريق الصناعي الحالي.الطريقة الكهروكيميائية المقترحة.نتائج اختبار TEA فينتائج اختبار TEA في.
تُستخدم محلول هيدروكسيد البوتاسيوم بتركيز عالٍ كإلكتروليت للحفاظ على انتقائية التفاعل العالية، والتي تحتاج إلى أن تُعادل قبل فصل المنتج، وبالتالي يتم إهدار كمية كبيرة من القلويات والأحماض بشكل لا مفر منه.
لحل المشكلات المذكورة أعلاه، نقترح استراتيجية تفاعل الحمض والقاعدة في الموقع لاستخراج جليكولات الصوديوم من الإلكتروليت. على عكس الحالات في التقارير السابقة، يمكن الحصول على جليكولات الصوديوم من التفاعل في الموقع بين إلكتروليت هيدروكسيد الصوديوم وGA الناتج من أكسدة EG (الشكل 1b)، مما لا يتيح فقط الاستفادة الكاملة من الإلكتروليتات لتحسين معدل استخدام الذرات، بل يقلل أيضًا من هدر الحمض.
لإظهار الإمكانات الاقتصادية لهذا النهج، تم تقديم تحليل تقني اقتصادي (TEA) (الملاحظة التكميلية 1 والشكل التكميلية 1)تشير دراسة الجدوى الأولية (الشكل 1c و d والشكل التوضيحي 2) إلى أن الأكسدة الانتقائية للإيثيلين الجليكول إلى صوديوم جليكولات (الانتقائية) عند كثافة تيار مرتفعة نسبياً ( ) مربح.
في هذا العمل،محفز كهربائي بمحتوى Pd منخفض جداًتم تطويره من أجل تخليق الجليكولات بكفاءة واقتصادية واستقرار فائق. يظهر كفاءة فاراداي عالية بشكل معقول (98.9%) وعائد (98.8%)، وثبات فائق (1500 ساعة) تجاه أكسدة الإيثيلين غليكول الكهروكيميائية. علاوة على ذلك، تم تحويل حمض الغليكوليك الناتج إلى غليكولات الصوديوم ذات القيمة المضافة من خلال تفاعل حمض-قاعدة في الموقع مع إلكتروليت NaOH، مما يجعله فعالاً من حيث الذرات ولا حاجة لإضافة حمض إضافي لفصل المنتج. تكشف نتائج التجربة أن i) الامتصاص الضعيف لغليكولات الصوديوم على سطح المحفز يلعب دورًا كبيرًا في تجنب الأكسدة المفرطة وتحقيق انتقائية عالية؛ ii) انتقال الإلكترون بين و وانخفاض مستوى النطاق في مركز d لـ Pd يعزز امتصاصوزيادة النشاط التحفيزي والاستقرار.
النتائج
تركيب وتوصيف المحفزات
تم تخليقه من خلال عملية من خطوتين كما هو موضح في الشكل 2a (يمكن العثور على التفاصيل التجريبية في قسم الطرق في SI). في الخطوة الأولى، تم تنمية هيدروكسيد ثنائي المعدن على رغوة النيكل بواسطة طريقة الهيدروحرارية، تلتها عملية تلدين بالغاز الأرجون للحصول على الشكل المتبلور.أنابيب نانوية مدعومة على رغوة النيكل ) . في الخطوة الثانية، تم ترسيب رقائق النانو من البالاديوم على بواسطةنهج التخفيض للحصول على المحفز النهائيعينات التحكم و تم الحصول عليها أيضًا بطرق مماثلة.
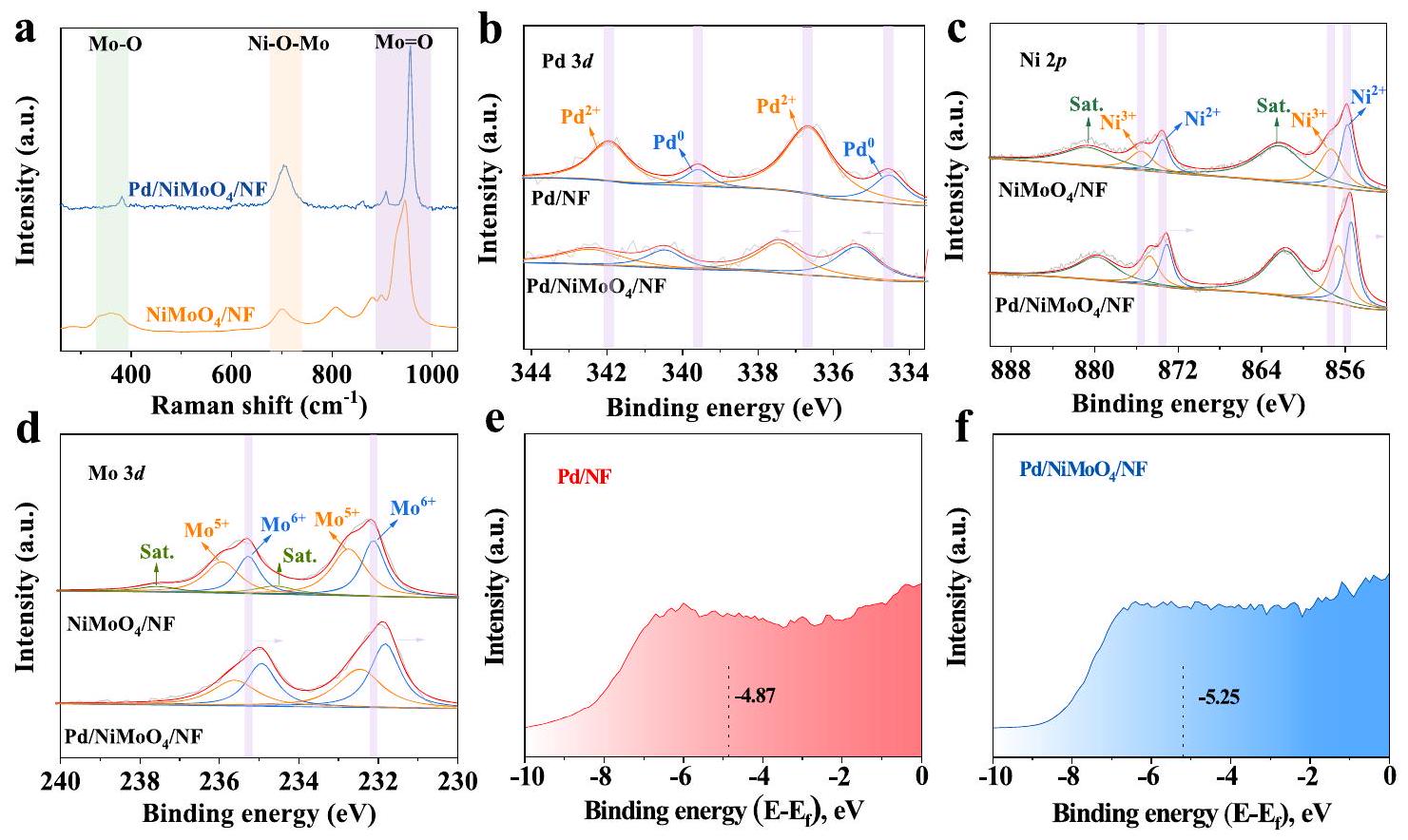
أولاً، أنماط حيود الأشعة السينية (XRD) لـبالإضافة إلى عوامل التحفيز الأخرى التي تم الحصول عليها لإظهار التخليق الناجح لهذه المحفزات الكهربائية. كما هو موضح في الشكل 2b، فإن هناك فقط قمم حيود مميزة تنتمي إلى الشبكة المكعبة المركزية الوجه المميزة لـ Pd والبنية الرباعية.على التوالي، دون العثور على قمم حيود أخرى. عادةً، الانعكاسات الموجودة في و تتوافق مع (111) (200) و (220) من طائرات Pd (رقم JCPDS 46-1043)، بينما تلك الموجودة في، و تتوافق مع مستويات (110) و(112) و(220) و(022) (رقم JCPDS 33-0948)، مما يدل على النجاح في تخليق الـ، و تم التحقق من الميكروهياكل لهذه المحفزات الكهربائية التي تم الحصول عليها بواسطة المجهر الإلكتروني الماسح (SEM). تؤكد الصور النموذجية للمجهر الإلكتروني الماسح أن مصفوفات نانوورقة Pd المتراصة بشكل جيد قد نمت بنجاح على سطح الـالنانو القضبان (الشكل 2ج، د). صور المجهر الإلكتروني الماسح للنموذج الناتج، و NF معروضة في الأشكال التكميلية 3-5. بمزيد من التفصيل، هيكل تم تأكيد ذلك بشكل أكبر من خلال تصوير المجهر الإلكتروني الناقل (TEM) (الشكل 2e). من خلال تسليط الضوء على الحواف الشبكية المميزة لـ و في تباعد الشبكة، الذي يتوافق مع مستوى (111) من البالاديوم ومستوى (312) منعلى التوالي، تؤكد صورة TEM عالية الدقة (HRTEM) بشكل إضافي على البلورات Pd/ (الشكل 2f، g). صور TEM للعينات التي تم الحصول عليها و مُعروضة في الشكل التكميلية 6. تشير تحليل خريطة العناصر باستخدام التحليل الطيفي للطاقة المشتتة (EDS) إلى
الشكل 2 | خصائص الشكل والهياكل للعوامل الحفازة. أ رسم تخطيطي لعملية تخليقأنماط حيود الأشعة السينية، ، وصورة SEM لـ Pd/NiMoO4/NF. e صور TEM و f، g صور HRTEM لـ Pd/NiMoO4/NF.تخطيط SEM-EDS لـ.
ذلكو O موزعة بشكل متجانس في جميع أنحاء (الشكل 2h). بالإضافة إلى ذلك، تم تحليل كمية تحميل البالاديوم الدقيقة بواسطة مطيافية الانبعاث البلازمي المقترن بالتحريض (ICP-OES)، ونسبة وزن البالاديوم منخفضة تصل إلى ، وهو أقل بكثير من تلك المبلغ عنها حتى الآن (الشكل التوضيحي 7).
هيكل وخصائص إلكترونية للعوامل المساعدة
للتعمق أكثر في التركيب الكيميائي لـتم إجراء قياسات طيفية باستخدام مطياف رامان وتحويل فورييه للأشعة تحت الحمراء (FT-IR). كما هو موضح في الشكل 3a، فإن الحزم الموجودة في و تُنسب إلى التمددات المتماثلة وغير المتماثلة للنهاياتبينما الفرقة فيمخصصة لوضع الشد لرابطة Ni-O-Mo. بالإضافة إلى ذلك، فإن الأطياف عندينتمي إلى وضع الانحناء لـيمكن أيضًا الحصول على معلومات مماثلة من منحنيات FT-IR (الشكل التوضيحي 8)، والتي تظهر منها القمم المميزة الواضحة عند و يمكن أن يُعزى إلىالتمدد المتماثل وتمدد Ni-Mo-O المتماثل، على التوالي. بالإضافة إلى ذلك، القمم المميزة عند و ينتمي إلى تراكب اهتزاز الشد لـ و مجموعاتمن الواضح أن الانزياح الأحمر لكل قمة يمكن ملاحظته في منحنيات الطيف Raman و FT-IR، مما يدل على التحميل الناجح للـ Pd على.
علاوة على ذلك، تم استخدام مطيافية الإلكترونات الضوئية بالأشعة السينية (XPS) لتحليل الحالات الإلكترونية للبلاتين (Pd) وتركيب العناصر السطحية لـطيف الاستطلاع (الشكل التوضيحي 9) يؤكد وجود ، مو، وأو، والتي تتوافق جيدًا مع نتائج EDS. كما هو موضح في الشكل 3ب، طيف Pd 3d عالي الدقة لـتتكون من أربع قمم. تقع القمم على ارتفاع حوالي و ) ينتمي إلى المعدن Pd؛ بينما القمم الموجودة عند حوالي و يتوافق معذروة Pd 3 d فييتحول في اتجاه إيجابي بمقدار 0.9 إلكترون فولت مقارنةً بـ، مما يوحي بتفاعلات كهربائية بين Pd ويمكن رؤية ستة قمم بوضوح في طيف XPS عالي الدقة لـ Ni 2p (الشكل 3c)، حيث تُعزى القمم عند 855.8 و 873.6 eV إلى و من، على التوالي، بينما تُنسب القمم عند 857.3 و 875.5 إلكترون فولت إلى و من“، على التوالي، وتُنسب القيم عند 862.4 و 880.8 إلكترون فولت إلى قمرين مصاحبين.يعرض نسبة عالية منالذي يرتبط بالهيدروكسلة السطحية. بالنسبة لطيف XPS ثلاثي الأبعاد عالي الدقة لمعدن الموليبدينوم، يتم ملاحظة ثلاث مجموعات من قمم الزوجية بين 230 و 240 إلكترون فولت (الشكل 3d)، والتي يمكن أن تُعزى إلى قمم الزوجية لـوقممهم التابعةطاقة الربط لـ Ni 2p و Mo 3d لـم shifted سلبياً ( ) مقارنةً مع تلك الخاصة بـ . بالإضافة إلى ذلك، كما هو موضح في الشكل التوضيحي التكميلي 10، تشير نتائج فرق كثافة الشحنة إلى أن 1.63 إلكترونًا تنتقل من البالاديوم إلى الركيزة، التي تتفق جيدًا مع نتائج XPS. تكشف النتائج أعلاه عن انتقال الإلكترونات من Pd إلى، مما يؤكد المزيد من التفاعل الإلكتروني القوي بين Pd و.
من أجل استكشاف تأثير التفاعل بين الواجهات على قوة ارتباط الممتصات بشكل أعمق، تم استخدام طيف الانبعاث الضوئي لفرقة التكافؤ السطحية لتقييم مركز حزمة d- و كما هو موضح في الشكل 3e و f والشكل التكميلي 11، فإن مركز نطاق d لـينخفض إلى -5.25 إلكترون فولت مقارنةً بـ، على الرغم من أن مركز نطاق d لـ هو الأدنى، لا يظهر المحفز أي نشاط أكسدة EG واضح عند الجهد المنخفض، لذا لن يتم مناقشته. علاوة على ذلك، كما هو موضح في الشكل التكميلي 12، فإن مركز نطاق d لـ و تم حسابها لتكون -1.83 eV و -2.45 eV، على التوالي، والتي تتوافق جيدًا مع نتائج طيفية الفوتوإلكترون للطبقة السطحية. يُعزى الانخفاض في مركز نطاق d إلى إزالة الوسائط الكربونية (مثل حمض الجليكولات) لتحقيق استقرار طويل الأمد وانتقائية عالية للمنتجات المستهدفة..
أداءات التحفيز الكهربائي لأكسدة الإيثيلين غليكول
لتحقيق أداء التحفيز الكهربائي للعينات المحضرةبالإضافة إلى عينات التحكم لأكسدة الإيثيلين جلايكول عند القطب الموجب، تم إجراء سلسلة من القياسات الكهروكيميائية في إعداد ثلاثي الأقطاب. الشكل التكميلي 13 يقدم منحنيات الفولتمترية ذات المسح الخطي (LSV) لـفي 1.0 م NaOH مع أو بدون 1 م إيثيلين جلايكول. في غياب الإيثيلين
الشكل 3 | هيكل وخصائص إلكترونية للعوامل المساعدة. أ طيف رامان لـ و طيف XPS عالي الدقة لـ و (د) مو 3د من و طيف نطاق التكافؤ XPS لـ و .
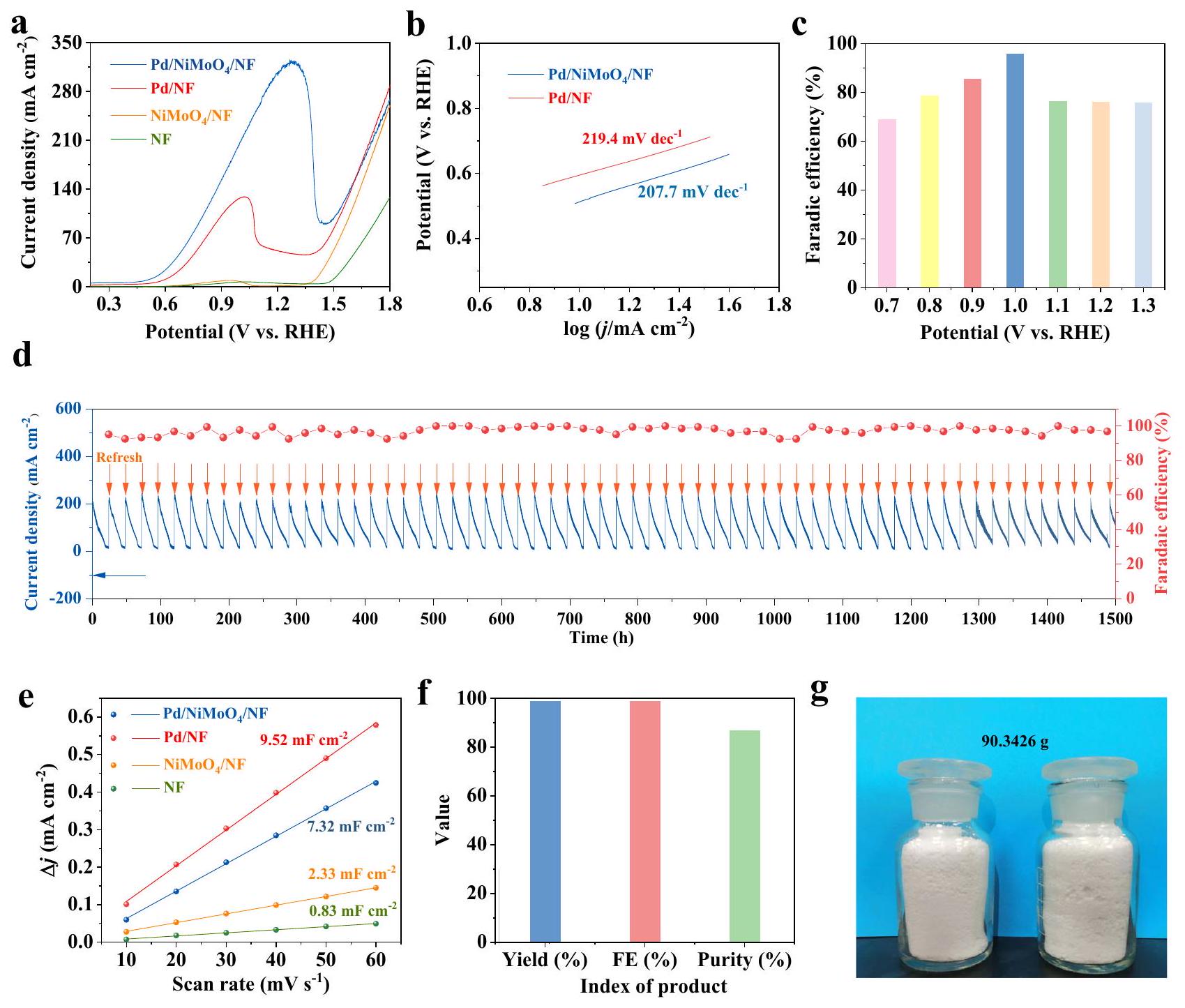
الجليكول، يظهر القطب نشاطًا معتدلاً في تفاعل الأكسدة، والذي يصل إلى كثافة التيار الأنودي لـ عند جهد 1.61 فولت مقابل القطب الهيدروجيني القابل للعكس (vs. RHE). عند إضافة 1 م من الإيثيلين غليكول، يزداد كثافة التيار بشكل ملحوظ، وينخفض الجهد الأنودي بشكل لافت إلى 0.79 فولت مقابل RHE عند ، وهو أقل بمقدار 820 مللي فولت من ذلك الخاص بتفاعل الأكسدة الكهربائي. أيضًا، لتقديم مقارنة أكثر تفصيلًا مع تفاعل الأكسدة الكهربائي، تعرض الشكل التكميلي 14 أن الجهود الأنودية في محلول الإيثيلين غليكول قد انخفضت بمقدار لا يقل عن 820 مللي فولت عند كثافة التيار ، و بشكل ملحوظ، يظهر أداءً أعلى بكثير من تلك الخاصة بـ ، ومراجع NF النقية لأكسدة الإيثيلين غليكول الكهروكيميائية (الشكل 4أ)، مما يشير إلى أن التفاعل بين Pd وتلعب دورًا حاسمًا في تعزيز نشاط EGOR. تم أيضًا دراسة تأثير تركيز NaOH والإيثيلين جلايكول في الإلكتروليت على أداء الأكسدة للإيثيلين جلايكول عند القطب الموجب، كما هو موضح في الشكل التكميلي 15. كانت أعلى نشاط أكسدة للإيثيلين جلايكول ( ) يتم تحقيقه في محلول 9 م من NaOH يحتوي على 1 م من EG. ومع ذلك، لتجنب الهدر المفرط لـ NaOH، نحن اختر محلول 1 M NaOH المضاف إليه 1 M EG. علاوة على ذلك، يتم مناقشة أداء المحفزات بمحتوى Pd مختلف وركائز أكسيد بشكل أعمق (الأشكال التكميلية 16-19). كما هو موضح في الشكل 4b،يظهر انحدار تافل منخفض جداً (207.7 مللي فولت مقارنةً بذلك من ، مما يشير إلى سرعة حركية التفاعل الأسرع لـ عند الأنود. بالإضافة إلى ذلك، طيف impedancia الكهروكيميائية (EIS) لـ ، وتم الحصول على NF عند جهد الدائرة المفتوحة.يوفر مقاومة نقل شحنة أصغر (Rct) منو NF النقي، (الشكل التوضيحي التكميلي 20)، مما يشير إلى الديناميات السريعة لنقل الشحنة.
لفهم أصل الأداء المرتفع بشكل ملحوظ لـ EGORسعة الطبقة الثنائية الكهروكيميائيةتم الحصول على ) لحساب المساحة السطحية النشطة كهربائياً (ECSA) (الشكل 4e والشكل التكميلي 21).يظهر مشابهًا قيمة ( ) مع ( )، والتي يبدو أنها أعلى من تلك الخاصة بـ ( ) و NF ( تم الحصول أيضًا على أنشطة EGOR المعيرة بواسطة ECSA، والتي تكون في حدود
الشكل 4 | أداءات التحفيز الكهربائي لـ نحو أكسدة الإيثيلين جلايكول عند الأنود. أ منحنيات LSV للمواد الكهروكيميائية المستخلصة لأكسدة الإيثيلين جلايكول عند الأنود. ب انحدارات تافل لـ و . كفاءات فاراداي (FEs) لـلإنتاج غليكولات الصوديوم لمدة ساعتين باستخدام الكرونوأمبرو متري عند إمكانيات متنوعة. كثافة التيار وFEs لـ Pd/NiMoO4/
NF لإنتاج جلايكولات الصوديوم لدورات التحليل الكهربائي لمدة 1500 ساعة. السعة الثنائية الطبقة (Cdl) لـو NF النقي.العائد، FE والنقاء لـ GA المستخرج من EGOR. الصورة البصرية لمنتج GA المستخرج. (الشكل التوضيحي 22)، مما يوضح بشكل أكبر النشاط المرتفع بشكل كبير لـ EGOR محفز
بينما يعتبر تحقيق نشاط EGOR العالي أمرًا مهمًا، قد تلعب الاستقرار الجيد دورًا أكثر أهمية في EGOR للتطبيقات الكبيرة النطاق في المستقبل. بعد ذلك، تم إجراء اختبار كرونوأمبيرومترية طويل الأمد باستخدامتم إجراء التجربة كعامل كهربائي عند مقابل RHE لتحديد منتجات أكسدة الإيثيلين جلايكول التي تم الحصول عليها عند الأنود بواسطة الرنين المغناطيسي النوويطيف الرنين المغناطيسي النووي (الأشكال التكميلية 23، 24). كما هو موضح فيطيف NMR (الشكل التكميلي 23)، يعتبر جلايكولات الصوديوم (3.89 جزء في المليون) هو المنتج الرئيسي لعملية أكسدة الإيثيلين غليكول. بالإضافة إلى ذلك، تم الكشف عن كميات صغيرة فقط من الفورمات (FA). أعلى كفاءة فاراداي (FE) لـتم الحصول عليه عند 1.0 فولت مقابل RHE، لذلك تم اختيار هذا الجهد كأقصى جهد (الشكل 4c والشكل التكميلية 25). مع تغير FEs في نطاقلإنتاج حمض الجليكوليك وتم الحفاظ على الشحنات الإجمالية لمدة 1500 ساعة من الكرونوأمبرو متقطعة (انظر التفاصيل في الطرق) (الشكل 4d والشكل التكميلي 26)لا يمكن ملاحظة أي انخفاضات كبيرة في الشحنات المستهلكة و FE خلال اختبارات الاستقرار التي استمرت 1500 ساعة، مما يوضح المتانة القوية لـالمحفز الكهربائي. بقدر ما نعلم، هذا هو أطول اختبار استقرار بين التقارير الأدبية. (الشكل التكميلية 27). في المقابل، خلال التحليل الكهربائي الدوري، المحفز يظهر فعالية أقل بكثير تجاه حمض الجليكوليك (87-88%)، مصحوبًا بتدهورات سريعة في كثافة التيار، مما يشير إلى الدور الحاسم لـفي تعزيز الانتقائية والاستقرار تجاه EGOR (الشكل التوضيحي التكميلي 28).
تطوير أساليب الفصل الاقتصادي أمر ضروري للحصول على المنتجات ذات القيمة المضافة والتطبيقات الصناعية للأكسدة الكهروكيميائية. تتطلب طرق الفصل المبلغ عنها سابقًا كمية كبيرة من الأحماض القوية لتحييد القواعد القوية الزائدة المحتفظ بها في الإلكتروليت، مما يؤدي حتمًا إلى هدر كل من الأحماض والقواعد. هنا، استنادًا إلى مكونات EG وNaOH في الإلكتروليت قبل التفاعل وتفاعل الحمض والقاعدة في الموقع، يصل الرقم الهيدروجيني للإلكتروليت بعد اختبار الدورة إلى حوالي 10.08 (الشكل التكميلي 29) بسبب التحلل المائي لغلوتينات الصوديوم، مما يشير إلى احتفاظ ضئيل بالقلويات في الإلكتروليت. لذلك، لا حاجة لحمض إضافي لفصل المنتج، مما يتيح الاستخدام الكامل للإلكتروليت ويتجنب هدر الحمض والقاعدة. بعد 1500 ساعة من التحليل الكهربائي الدوري، تم عزل 90.34 جرام من غلوتينات الصوديوم بنجاح ب purity منوعائد قدره (الشكل 4f و g والشكل التوضيحي 30). بالإضافة إلى ذلك، تمت دراسة القطب بعد أكسدة الإيثيلين جلايكول لمدة 1500 ساعة باستخدام XRD و SEM و EDS و TEM و HRTEM لتقييم استقرار المحفز. لم يتم العثور على تغييرات ملحوظة في نمط XRD للمحفز (الشكل التكميلي 31). علاوة على ذلك، فإن صورة SEM لـيظهر الحفاظ على الشكل الأصلي بعد اختبارات استقرار أكسدة الإيثيلين جلايكول (الشكل التكميلي 32)، مما يبرز القوة الهيكلية الفائقة. تحليل رسم الخرائط العنصرية SEMEDS يظهر أنو O موزعة بشكل متجانس في جميع أنحاء (الشكل التوضيحي 33). بالإضافة إلى ذلك، كل من Pd ويمكن العثور على الشبكة في صورة HRTEM (الشكل التوضيحي 34). تشير هذه النتائج جميعها إلى الاستقرار الممتاز للمحفز الذي تم الحصول عليه. علاوة على ذلك، فإن المنتجتم تحديد الكمية بواسطة كروماتوغرافيا الغاز (GC). الشكل التوضيحي الإضافي 35 يظهر المنحنى القياسي لـالإنتاج المقاس بواسطة GC. كما هو موضح في الشكل التوضيحي التكميلي 36، فإن الكفاءة فاراداي.تم حساب الرسوم المستهلكة المتنوعة لتكون قريبة منونقاء هو .
فهم الآلية
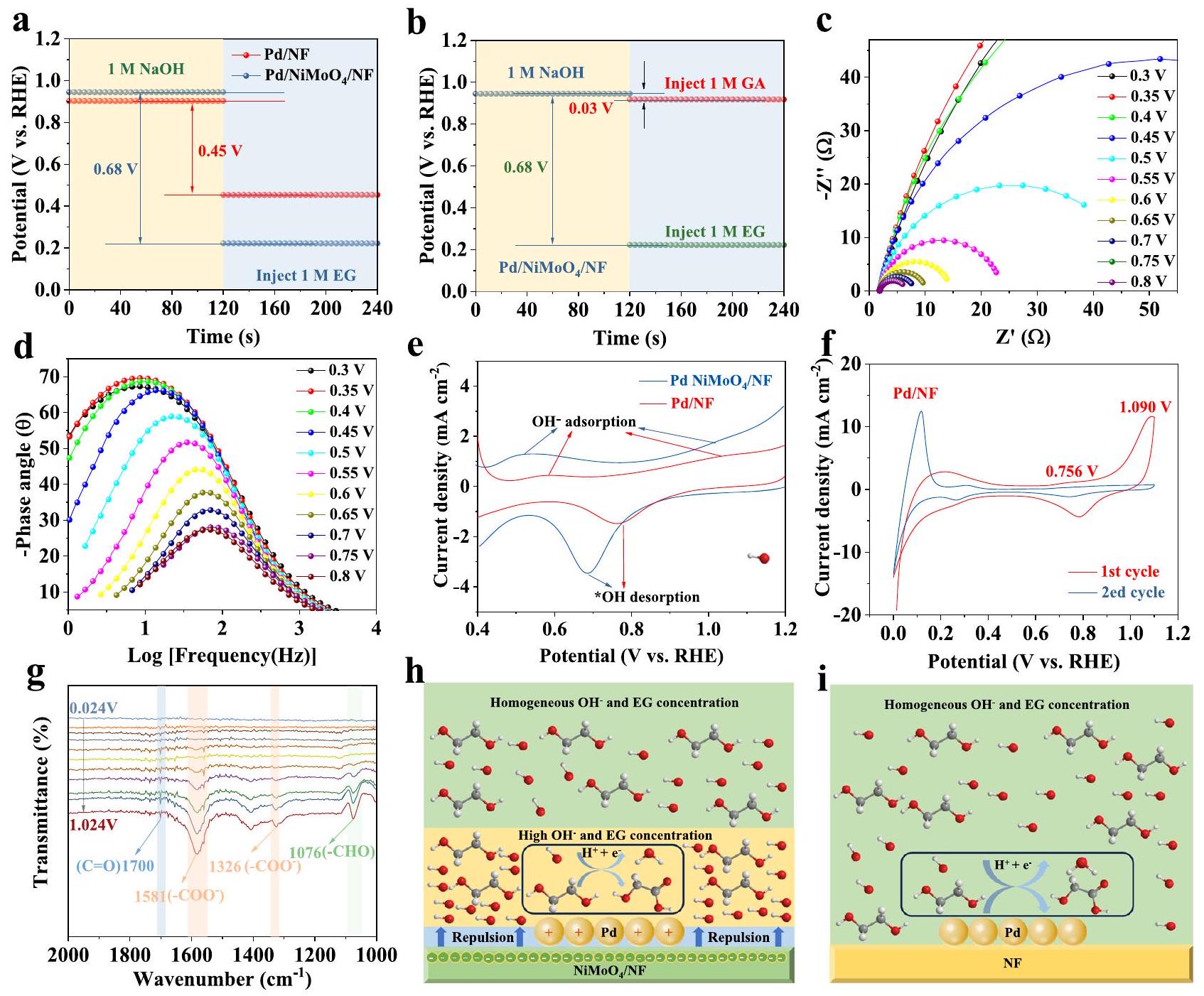
إن امتصاص أنواع EG مهم لعملية الأكسدة الكهروكيميائية لـ EG في الإلكتروليت القلوي. توضح الشكل 5a والشكل التكميلي 37 نتائج قياسات الجهد المفتوح (OCP)، والتي يمكن استخدامها لإظهار تأثيرات الممتزات العضوية على طبقة هلمهولتز الداخليةعند إضافة 1 م من EG، يتم تقليل OCP بشكل ملحوظ لـمقارنة مع و ، مما يشير إلى أن EG يتم امتصاصه بشكل أسهل علىمن و السطح. في الوقت نفسه، فإن الامتزاز الضعيف للجلايكولات هو المفتاح لتجنب الأكسدة المفرطة. كما هو موضح في الشكل 5ب، عند إضافة 1 م من GA، فإن تغير OCP يكون ضئيلاً ( ) مقارنةً مع 1 م EG ( )، مما يشير إلى أن GA يتم امتصاصه بشكل ضعيف في طبقة هيلمولتز الداخلية لمنع أكسدته المفرطة. علاوة على ذلك، تم تطبيق قياسات EIS في الموقع لاستكشاف الحركيات الحفازة وخصائص واجهة القطب/الالكتروليت عند إمكانيات مختلفة.مخططات نيكويست وبودي لـفي 1 م NaOH الذي يحتوي على 1 م EG كما هو موضح في الشكل 5c و d. يمكن ملاحظة تحول واضح في ذروة الطور عند 0.45 فولت (بالنسبة إلى RHE) في الترددات المنخفضة، مما يشير إلى بداية التفاعل عند هذه الجهد. مع زيادة الجهد، تضعف ذروات زاوية الطور وتتحول نحو ترددات أعلى، مما يدل على أن المزيد والمزيد من EG الممتص يتم أكسدته بسرعة بمعدل نقل شحنات واجهي سريع. كما هو موضح في الشكل التكميلي 38، يظهر أكسدة GA اتجاهًا معاكسا لذلك الخاص بـ EG، مما يشير أيضًا إلى امتصاص أضعف لـ GA علىالسطح. تظهر النتائج أعلاه نشاطًا تحفيزيًا ضعيفًا لـنحو تفاعل أكسدة GA. بالإضافة إلى ذلك، للتحقق من النتائج المذكورة أعلاه، تم استكشاف نشاط أكسدة GA في 1 م NaOH، والذي يعتبر ضئيلاً مقارنةً بنشاط EG في نفس المحلول (الشكل التكميلي 39). ستضمن الامتصاص الأضعف بشكل ملحوظ لـ GA مقارنةً بـ EG على Pd/NiMoO4/NF منع الأكسدة الزائدة وزيادة الانتقائية لـ GA مقارنةً بـ EG.
وبالمثل، فإن امتصاصالنوع مهم أيضًا لعملية الأكسدة الكهروكيميائية لـ EG في الإلكتروليت القلوي. الشكل 5e يوضح منحنيات الامتصاص والانفصال التمثيلية لـ OH على Pd في و . كما هو موضح في الشكل.يعرض نطاقات امتصاص/إطلاق OH عند جهد بدء أقل بكثير (0.5 فولت مقابل RHE) من (0.6 فولت مقابل RHE)، والذي يُعزى إلى الأنواع النيكل المحبة للأكسجين المدخلة في، التي يمكن أن تؤكسد بكفاءةإلى * OH الممتص على أسطح Pd عند جهد منخفض نسبيًا. في الوقت نفسه، بالاقتران مع نتائج XPS، فإن الركيزة الغنية بالإلكتروناتلا يمكن أن يمتصالأنيونات على سطحه، بينما يمكن لأوراق النانو Pd الموجبة الشحنة أن تلتقطلتسهيل الأكسدة الإضافية لـالأدوار المهمة لـتم استكشاف الأنواع بشكل أعمق من خلال قياسات الفولتمترية الدورية مع إزالة ثاني أكسيد الكربون (CV)يظهر ذروة مميزة لأكسدة أول أكسيد الكربون و في المسح الأنودي الأول عند ذروات الجهد عند 1.090 فولت مقابل RHE و0.785 فولت مقابل RHE (الأشكال التكميلية 40، 41). بعد إدخالتنخفض إمكانيات الذروة لأكسدة أول أكسيد الكربون إلى جهد منخفض قدره 1.026 فولت مقابل RHE (الشكل 5f) لـ، مما يشير إلى أن أول أكسيد الكربون يتم امتصاصه بشكل ضعيف على السطح ويمكن إزالته بسهولة عن طريق الأكسدة بسبب انخفاض مركز نطاق d للبلاتين، مما يساهم في الاستقرار المتفوق الملحوظ لتفاعل أكسدة الإيثيلين.
علاوة على ذلك، تم استخدام مطيافية تحويل فورييه بالأشعة تحت الحمراء الكهروكيميائية في الموقع لفهم أصل الأداء العالي لـ EGOR بشكل أفضل.تم وضع المحفزات الكهربية على السطح المسطح لبرواز نصف أسطواني من السيليكون وضغطها بواسطة إلكترود من الكربون الزجاجي. تم تسجيل طيف FTIR في محلول مائي 1 م من NaOH مع 1 م من EG عند جهد يتراوح من -0.9 إلى 0.1 فولت مقابلكما هو موضح في الشكل التكميلية 42، هناك قمة عندالمتوافق مع الروابط المتعددةيمكن ملاحظته جيدًا على؛ ومع ذلك، لا يمكن اكتشاف الإشارة بسهولة لـ NF على مدى كامل النطاق المحتمل، مما يوضح المزيد من تحمل CO العالي لـ، وهو ما يتماشى مع نتائج تجارب إزالة أول أكسيد الكربون. من الواضح أن هناك نطاق تعزيز نحو الأسفل عنديمكن ملاحظته، والذي يُعزى إلى اهتزاز الشد للألدهيد، مما يشير إلى أن EG يتم أكسدته أولاً إلى نوع جلايكوالدهيد. علاوة على ذلك، القمم في و تنتمي إلى نطاقات التمدد المتماثلة وغير المتماثلة لـ
الشكل 5 | فهم الآلية. أ الجهود المفتوحة للدائرة (OCPs) لـ Pd/ و في محلول 1 م NaOH قبل وبعد إضافة EG.أقراص منع الحملفي محلول 1 م من NaOH مع إضافة EG أو GA.مخططات نايكويست و (د) مخططات طور بودي المقابلة لـالقطب الكهربائي عند مستويات مختلفة الجهود في 1 م NaOH مع 1 م EG. منحنيات CV لـ و في 1 متجارب التجريد منطيف FTIR الكهروكيميائي في الموقع لـ EGOR المحفز بواسطة. رسم توضيحي تخطيطي لآلية EGOR المقترحة على و (أنا) . مجموعة الكربوكسيل من GA، على التوالي، تشير إلى الأكسدة الإضافية للجليكوالدهيد إلىبالإضافة إلى ذلك، فإن قمم الاهتزاز المميزة عند 1700 و تم اكتشافها على و على التوالي، والتي يمكن أن تُنسب إلى اهتزاز الشد لمجموعة الكربونيل ( )، مما يشير إلى تشكيل 2-هيدروكسي أسيتيل ( * المتوسطات (الشكل 5 ج).
علاوة على ذلك، تم استخدام حسابات نظرية الكثافة (DFT) أيضًا للتحقيق في آلية التفاعل. تم عرض النماذج المحسّنة لأكسدة EG على محفزات Pd/NiMoO4 وPd في الأشكال التكميلية 43 و44. بالإضافة إلى ذلك، تم حساب ملف الطاقة الحرة غيبس المقابل عند 0 فولت مقابل RHE. كما هو موضح في الشكل التكميلية 45، نجد بوضوح أن إضافةلتشكيل الامتزاز المشترك لـ *OH وهو الخطوة المحددة لمعدل التفاعل (RDS) في التفاعل الكامل من الإيثيلين جلايكول إلى حمض الجليكوليك ومحفزات Pd. على وجه الخصوص، تغيير الطاقة الحرة لخطوة تحديد المعدل (RDS) على أقل بكثير من 0.86 إلكترون فولت من البالاديوم، مما يشير إلى أنيمكن أن تمتص المحفزات OH عند جهد بدء نسبي أقل، وهو ما يتماشى مع نتائج CV.
علاوة على ذلك، كما هو موضح في الشكل التوضيحي التكميلي 10، تشير نتائج فرق كثافة الشحنة إلى أن 1.63 إلكترونًا تنتقل من البالاديوم إلىالركيزة، مما يشير إلى أن البالاديوم إيجابي مشحون والمادة السطحية مشحونة سلبًا. يمكن أن يجذب البالاديوم المشحون إيجابيًا الأنيونات OH لتعزيز عملية سحب الهيدروجين من الإيثيلين غليكول. تم حساب طاقات الامتزاز لـ OH على أسطحهم، و 0.28 إلكترون فولت في مواقع امتصاص Pd و Ni و Mo، على التوالي (الشكل التكميلية 46). تشير النتائج أعلاه إلى أن إضافة OH تحدث في موقع Pd. بالإضافة إلى ذلك، كما هو موضح في الشكل التكميلية 47، فإن طاقات الامتصاص المحسوبة لـ EG على Pd (111) وكانت -0.57 و -0.98 إلكترون فولت، على التوالي، مما يشير إلى أن EG يتم امتصاصه بسهولة أكبر على، وفقًا لنتائج قياسات OCP. بالإضافة إلى ذلك، فإن طاقات الامتزاز المحسوبة لـ EG على أسطحهم، و -0.36 إلكترون فولت عند ومواقع امتصاص Mo، على التوالي (الشكل التكميلي 48)، مما يشير إلى أن EG أكثر ملاءمة للامتصاص عند موقع Pd. علاوة على ذلك، كما هو موضح في الشكل التكميلي 28، فإن المنتجات الرئيسية لـ EGOR المحفز بواسطة Pd/NF هي صوديوم جلايكولات؛ ومع ذلك، فإنلا توجد نشاط أكسدة EG واضح عند الجهد المنخفض. تشير النتائج أعلاه إلى أن تجريد الهيدروجين يحدث أيضًا في موقع البالاديوم.
وفقًا للنتائج المذكورة أعلاه، تم اقتراح آلية التفاعل ومسار EG كما هو موضح في الشكل 5h والشكل التكميلي 49. سطحيصبح مشحونًا سلبًا بسبب نقل الإلكترونات من Pd إلى، مما يمكن أن يعزز هجرة الهيدروكسيلات بعيدًا عن السطح وتؤدي إلى زيادة تركيز الهيدروكسيل المحلي حول سطح البالاديوم. على النقيض من ذلك، فإن الهيدروكسيلات على سطح البالاديوم موزعة بشكل موحد علىالمحفزات (الشكل 5i). يمكن أن تعزز التركيزات المحلية الأعلى من الهيدروكسيل حول سطح البالاديوم التحويل الأسرع للإيثيلين غليكول وتنشيط أول أكسيد الكربون، مما يعزز نشاط واستقرار تفاعل أكسدة الإيثيلين غليكول. بالإضافة إلى ذلك، فإن إدخاليمكن أن يعزز أيضًا امتصاص EG، والتركيز المحلي الأعلى يسهل تحويله بشكل أسرع.
أداء إلكتروليزر التدفق بدون غشاء
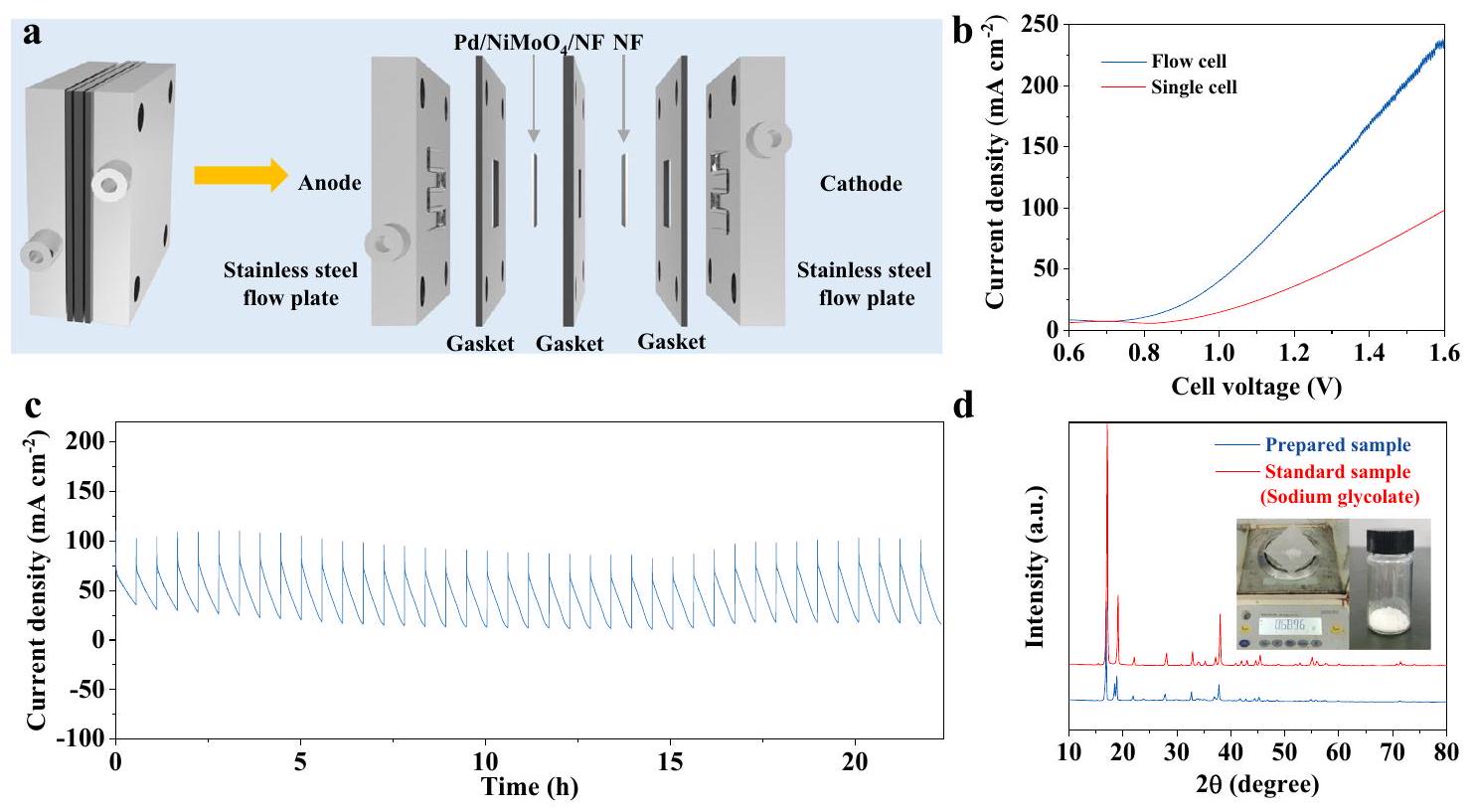
لإظهار جدواها الاقتصادية في نظام ذو قطبين، تم تجميع جهاز إلكتروليزر تدفق بدون غشاء محلي الصنع باستخدامالمحفز كأنود ورغوة النيكل المعالجة مسبقًا ككاثود في منطقة عمل (الشكل 6أ). تم تدوير إلكتروليت متجانس (1 م NaOH يحتوي على 1 م من الإيثيلين جلايكول) في مفاعل التدفق باستخدام مضخة بيرستالتية بمعدل تدفق قدره تظهر منحنيات LSV (الشكل 6ب) أن الأكسدة الكهربائية للإيثيلين جلايكول تحدث منويصل إلى كثافة التيار الحاليةعند 1.2 فولت في خلية التدفق، وهو أعلى من ذلك في خلية الثلاثة أقطاب المقابلة، مما يظهر إمكانيات تطبيق واعدة لأكسدة الإيثيلين جلايكول في خلية التدفق.
ثم قمنا بإجراء اختبارات جهد متقطع لتقييم أداء إنتاج جلايكولات الصوديوم في الخلايا الكهروكيميائية.بعد 23 ساعة من اختبار الاستقرار (الشكل 6c)، تصل نسبة تحويل الإيثيلين جلايكول إلى، وخصائص التفاعل والانتقائية لحمض الصوديوم الجليكوليك هي و وأخيرًا، تم الحصول على بلورة جلايكولات الصوديوم كما تم تأكيده بواسطة حيود الأشعة السينية (الشكل 6d).
الاستنتاجات
باختصار، محفز كهربائيمع تحميل Pd منخفض نسبيًا (تم الحصول على ( ) من أجل أكسدة الإيثيلين غليكول الكهروكيميائية بكفاءة، والتي تتميز بالكفاءة الفارادية، وعائد صوديوم غليكولات، واستقرار يصل إلى، و1500 ساعة، على التوالي. والأهم من ذلك، تم تحويل حمض الجليكوليك الناتج إلى غليكولات الصوديوم ذو القيمة المضافة من خلال تفاعل الحمض والقاعدة في الموقع مع إلكتروليت NaOH، مما يعزز من استخدام الذرات وفصل غليكولات الصوديوم. تلعب الامتزاز الضعيف لغليكولات الصوديوم على سطح المحفز دورًا كبيرًا في تجنب الأكسدة المفرطة وتحقيق انتقائية عالية. يحدث نقل الإلكترون بين البالاديوم ووقد تم اقتراح أن الانخفاض في مركز نطاق d للبلاتين (Pd) يعزز الامتصاص لـوإزالة الأكسدة لثاني أكسيد الكربون، مما يؤدي إلى زيادة النشاط والاستقرار لـ.
طرق
المواد الكيميائية
نيترات النيكل سداسي الماء ) ، موليبدات الصوديوم ثنائي الهيدرات ( ) ، وبوروهيدريد الصوديوم ( ، 99.99%) تم شراؤها من مجموعة الصين الوطنية للأدوية المحدودة. تيتراكلوريد البالاديوم الصوديوم ( ) تم شراؤه من شركة شنغهاي تيتان للتكنولوجيا المحدودة. هيدروكسيد الصوديوم ( ) كانت من ماكلين. تم شراء رغوة النيكل (NF، بسماكة 0.15 مم) من شركة تيانجين غاوشي رويليان للتكنولوجيا الضوئية المحدودة. حمض الماليك ( ، الدرجة) وماء الديوتيريوم (تم شراء ( ) من شركة أداماس للمواد الكيميائية المحدودة. تم شراء الإيثيلين جلايكول (AR) من مواد كيميائية صينية.
تم استخدام جميع المواد الكيميائية كما هي دون أي تنقية إضافية. تم استخدام الماء المقطر (DIW) في جميع التجارب.
تركيب الـأنابيب نانوية/ NF
تم تحضيره كمادة سابقة باستخدام التخليق الهيدروحراري. أولاً،تم تنظيفه بالموجات فوق الصوتية في 3 M HCl، والإيثانول، والماء المقطر لإزالة أي شوائب سطحية. بعد ذلك، Ni و تم إذابتها في ماء منزوع الأيونات (30 مل) وتم تحريكها لمدة 10 دقائق. بعد ذلك، تم وضع محلول السلفيكتور الناتج وقطعة من NF في وعاء ضغط من الفولاذ المقاوم للصدأ مبطن بتفلون وتم الاحتفاظ بها عندلمدة 6 ساعات. بعد أن تم تبريد الأوتوكلاف إلى درجة حرارة الغرفة، تم إخراج NF مع رواسب خضراء فاتحة على السطح وغسله بالماء المقطر والإيثانول على التوالي لإزالة أي بقايا غير متفاعلة قبل أن يتم تجفيفه بالكامل عندبين عشية وضحاها تحت الفراغ. من أجل الحصول على بلوراتتم حرق النانوهياكل، الركائز الموصلة ذات الهياكل الهرمية الناتجة في حالتها الأصلية عندلمدة ساعتين بمعدل تسخين للحرارةفي جو من الأرجون.
الشكل 6 | خلية تدفق بدون غشاء لأكسدة الإيثيلين غليكول. أ إعداد MEA لتفاعل HER المزدوج (-) // أكسدة الإيثيلين غليكول (+). ب منحنيات LSV لـفي 1 م NaOH مع 1 م EG في خلية مفردة أو خلية تدفق. ج. اختبارات الاستقرار لـنحو EGOR. نمط XRD لحمض الصوديوم الجليكولات الذي تم تحضيره ذاتيًا.
تركيب Pd/NiMoO4/NF
ال و تم تفريقها في 30 مل من الماء وتم تحريكها لمدة 12 ساعة. تم الحصول على العينة السوداء بعد إضافة 1 مل منوتم التحريك لمدة 4 ساعات. بعد ذلك، تم غسل العينة الناتجة بالماء المنزوع الأيونات والإيثانول، ثم تم تجفيفها بشكل إضافي عندلمدة 10 ساعات في فراغ للحصول علىمحفزات.تمت عملية التخليق بطريقة مشابهة باستخدام NF المعالج مسبقًا ( ) كالسلف.
توصيف المواد
تم تسجيل أنماط حيود الأشعة السينية (XRD) على جهاز حيود Rigaku D/MAX 2550 عند 35 كيلوفولت و25 مللي أمبير باستخدام إشعاع النحاس Ka.تم الحصول على صور المجهر الإلكتروني الماسح (SEM) وصور رسم الخرائط العنصرية الموزعة بالطاقة (EDS) باستخدام جهاز ZEISS Gemini 450. تم الحصول على صور المجهر الإلكتروني الناقل (TEM) على جهاز JEM-2100F عند 200 كيلوفولت. تم إجراء قياس الطيف الضوئي للأشعة السينية (XPS) على جهاز AXIS SUPRA معمصدر الإشعاع ). تم استخدام موضع قمة C 1 s عند 284.8 eV كمرجع للمعايرة لتحديد الطاقة الربط الدقيقة ( تم تسجيل تحليل طيف الانبعاث بالتحليل الطيفي للبلازما المقترنة بالحث (ICP-OES) على جهاز Agilent 700 Series. تم إجراء قياسات FTIR الكهروكيميائية في الموقع على خلية Linglu ECIR-II المثبتة على Pike Veemax III ATR باستخدام بلورة سيليكون أحادية مترددة. تم الإشارة إلى قيم طاقة الربط باستخدام موضع قمة C 1 s عند 284.8 eV. تم تقييم مراكز نطاق d من خلال تطبيق الصيغة التالية.
القياسات الكهروكيميائية
تم إجراء جميع القياسات لتفاعل OER وأكسدة الإيثيلين جلايكول على جهاز العمل الكهربائي BioLogic VSP-300 في خلية مفردة ذات ثلاثة أقطاب في درجة حرارة الغرفة معمن المواد المصنوعة كما هي كالكاثود العامل، وقطعة من البلاتين كالكاثود المضاد، و ( 1.0 م NaOH ) كإلكترود مرجعي، على التوالي. تم معايرة القطب المرجعي (1 م NaOH) بالنسبة إلى RHE في إلكتروليتات 1 م NaOH المشبعة بالهيدروجين عالي النقاء، حيث تم استخدام قطبي سلك من البلاتين كقطب مضاد وقطب عمل في هذه القياسات، على التوالي. صيغة التحويل لـالقطب المرجعي هو:تم الحصول على منحنيات الاستقطاب باستخدام LSV بمعدل مسحفي منطقة (ضد تم قياس الفولتمترية الدورية (CV) باستخدام مسح CV من -0.1 فولت إلى 1.2 فولت مقابل RHE بمعدل مسحتم إجراء اختبار المتانة بواسطة الكرونوأمبيرومترية عند 1.0 فولت مقابل RHE. تم إجراء قياسات EIS في نطاق التردد من 100 كيلو هرتز إلى 10 ميغا هرتز بجهد تيار متردد قدره 5 مللي فولت. سعات الطبقة المزدوجة الكهروكيميائية (تم تأكيد ) من عينات مختلفة بواسطة CV في المنطقة المحتملة دون عملية فارادائية لحساب ECSA. تم إجراء اختبارات EGOR في محلول 1 م NaOH يحتوي على 1 م من الإيثيلين جلايكول. كانت مساحة القطب العامل في الإلكتروليت ثابتة عندوتم تطبيع جميع كثافات التيار إلى المساحة الهندسية للقطب. تم استخدام جميع المنحنيات دون تعويض IR.
تجارب الفولتمترية لانتزاع أول أكسيد الكربون
تم إجراء منحنيات إزالة ثاني أكسيد الكربون في محلول 1 م NaOH. قبل الاختبارات، تم إزالة الهواء من محلول 1 م NaOH أولاً باستخدام نقاء عالٍ.لمدة 30 دقيقة. ثم، تم تمرير غاز CO النقي في المحلول لمدة 30 دقيقة بينما تم الحفاظ على جهد القطب العامل عند جهد ثابت قدره 0.1 فولت مقابل RHE لضمان فعالية CO. الامتزاز على سطح القطب. ثم تم طرد جزيئات CO غير الممتزة عن طريق الفقاعاتالغاز في المحلول لمدة 20 دقيقة. بعد ذلك، تم بدء منحنيات إزالة ثاني أكسيد الكربون منإلى 1.1 فولت مقابل RHE في الاتجاه الأنودي عند معدل مسحلمدة دورتين متتاليتين على الأقل.
تحديد كمية المنتج
تم إجراء اختبارات الكرونوأمبرو متري عند 1.0 فولت مقابل RHE (لنظام الثلاثة أقطاب) أو عند جهد الخلية 1.2 فولت (لنظام القطبين). بعد ساعتين من التحليل الكهربائي، تم تحليل المنتجات وقياسها بواسطة و الرنين المغناطيسي النووي و NMR). و تم تسجيل طيف NMR على جهاز Avance II 300 (بروكير).تم جمع الإلكتروليت وتخفيفه بـمنتم استخدام حمض الماليك كمعيار داخلي. تم استخدام جهاز كروماتوغرافيا الغاز Ramin GC2060 مع عمود محشو وكاشف موصلية حرارية لت quantifying الناتج.المولد عند الكاثود خلال التحليل الكهربائي. المنحنى القياسي لـتم عرضه في الشكل التوضيحي التكميلي 31. كمية ما تم توليده نظريًاتم حسابه على أنه ( هو الشحنة الكلية التي مرت عبر الأقطاب، هو الحجم المولي للغاز، هو عدد الإلكترونات اللازمة لإنتاج جزيء من هو ثابت فاراداي). الصيغة المستخدمة لحساب نقاء جليكولات الصوديوم هي: النقاء = m1/m2 m 1 تشير إلى كتلة جليكولات الصوديوم التي تم فصلها فعليًا، بينما m 2 تشير إلى الكتلة الإجمالية للمنتج المعزول.
يمكن تحديد العائد (%) والانتقائية (%) لتكوين حمض الجليكوليك بواسطة المعادلتين (1) و (2) على التوالي.
الانتقائية
كفاءة فاراداي (%) لحمض الجليكوليك ويمكن تحديد الإنتاج بواسطة المعادلتين (3) و (4) على التوالي
أين و هي الشحنة الكلية المارة عبر الأقطاب، هو عدد الإلكترونات التي تشكل مولًا من حمض الجليكوليك، هو عدد الإلكترونات التي تنتج جزيئًا من هو ثابت فاراداي.
توفر البيانات
جميع البيانات متاحة من المؤلفين عند الطلب.
References
Tang, W. et al. Efficient Conversion of Biomass to Formic Acid Coupled with Low Energy Consumption Hydrogen Production from Water Electrolysis. Angew. Chem. Int. Ed. 62, e202305843 (2023).
Gao, P., Zhong, L., Han, B., He, M. & Sun, Y. Green Carbon Science: Keeping the Pace in Practice. Angew. Chem. Int Ed. Engl. 61, e202210095 (2022).
Fan, L. et al. Selective production of ethylene glycol at high rate via cascade catalysis. Nat. Catal. 6, 2520-1158 (2023).
Lum, Y. et al. Tuning OH binding energy enables selective electrochemical oxidation of ethylene to ethylene glycol. Nat. Catal. 3, 14-22 (2020).
Xu, H., Chen, L. & Shi, J. Advanced electrocatalytic systems for enhanced atom/electron utilization. Energy Environ. Sci. 16, 1334-1363 (2023).
Si, D. et al. Hydrogen anode/cathode co-productions-coupled anode alcohol selective oxidation and distinctive H/e transfer pathways. Appl. Catal. B: Environ. 331, 122664 (2023).
Radwan, A., Jin, H., He, D. & Mu, S. Design Engineering, Synthesis Protocols, and Energy Applications of MOF-Derived Electrocatalysts. Nano-Micro Lett. 13, 132 (2021).
Lin, H. et al. Two-dimensional titanium carbide MXenes as efficient non-noble metal electrocatalysts for oxygen reduction reaction. Sci. China Mater. 62, 662-670 (2018).
Li, G. et al. Dual hydrogen production from electrocatalytic water reduction coupled with formaldehyde oxidation via a copper-silver electrocatalyst. Nat. Commun. 14, 525 (2023).
Wang, T. et al. Combined anodic and cathodic hydrogen production from aldehyde oxidation and hydrogen evolution reaction. Nat. Catal. 5, 66-73 (2021).
Fan, L. et al. High Entropy Alloy Electrocatalytic Electrode toward Alkaline Glycerol Valorization Coupling with Acidic Hydrogen Production. J. Am. Chem. Soc. 144, 7224-7235 (2022).
Huang, H. et al. Ni, Co hydroxide triggers electrocatalytic production of high-purity benzoic acid over . Energy Environ. Sci. 13, 4990-4999 (2020).
Zhu, Y. et al. Single-Atom In-Doped Subnanometer Pt Nanowires for Simultaneous Hydrogen Generation and Biomass Upgrading. Adv. Funct. Mater. 30, 2004310 (2020).
Wei, X., Li, Y., Chen, L. & Shi, J. Formic Acid Electro-Synthesis by Concurrent Cathodic Reduction and Anodic Oxidation. Angew. Chem. Int. Ed. 60, 3148-3155 (2021).
Zhou, P. et al. Heterogeneous-Interface-Enhanced Adsorption of Organic and Hydroxyl for Biomass Electrooxidation. Adv. Mater. 34, 2204089 (2022).
Zhang, W., Xia, G.-J. & Wang, Y.-G. Mechanistic insight into methanol electro-oxidation catalyzed by PtCu alloy. Chin. J. Catal. 43, 167-176 (2022).
Shi, K., Si, D., Teng, X., Chen, L. & Shi, J. Enhanced electrocatalytic glycerol oxidation on at significantly reduced potentials. Chin. J. Catal. 53, 143-152 (2023).
Peng, T. et al. Electrocatalytic valorization of lignocellulose-derived aromatics at industrial-scale current densities. Nat. Commun. 14, 7229 (2023).
Qin, Y. et al. Extraordinary p-d Hybridization Interaction in Heterostructural Pd-PdSe Nanosheets Boosts C-C Bond Cleavage of Ethylene Glycol Electrooxidation. Angew. Chem. Int Ed. Engl. 61, e202200899 (2022).
Wang, Y. et al. Implanting Mo Atoms into Surface Lattice of Alloys Enclosed by High-Indexed Facets: Promoting Highly Active Sites for Ethylene Glycol Oxidation. ACS Catal. 9, 442-455 (2018).
Yang, X. et al. Interface-Rich Three-Dimensional Au-Doped PtBi Intermetallics as Highly Effective Anode Catalysts for Application in Alkaline Ethylene Glycol Fuel Cells. Adv. Funct. Mater. 31, 2103671 (2021).
Zhang, S. et al. Highly Strained Au-Ag-Pd Alloy Nanowires for Boosted Electrooxidation of Biomass-Derived Alcohols. Nano Lett. 21, 1074-1082 (2021).
Bai, S., Xu, Y., Cao, K. & Huang, X. Selective Ethanol Oxidation Reaction at the Interface. Adv. Mater. 33, e2005767 (2021).
Zhu, X.-Y. et al. Hollow nanotube bundles with high electrocatalytic performances for hydrogen evolution and ethylene
glycol oxidation reactions. J. Colloid Interface Sci. 532, 571-578 (2018).
Qi, J. et al. Energy-saving and product-oriented hydrogen peroxide electrosynthesis enabled by electrochemistry pairing and product engineering. Nat. Commun. 14, 6263 (2023).
Si, D., Xiong, B., Chen, L. & Shi, J. Highly selective and efficient electrocatalytic synthesis of glycolic acid in coupling with hydrogen evolution. Chem. Catal. 1, 941-955 (2021).
Yan, Y. et al. Electrocatalytic Upcycling of Biomass and Plastic Wastes to Biodegradable Polymer Monomers and Hydrogen Fuel at High. Curr. Densities. J. Am. Chem. Soc. 145, 6144-6155 (2023).
Liu, F. et al. Concerted and Selective Electrooxidation of Polyethylene-Terephthalate-Derived Alcohol to Glycolic Acid at an Industry-Level Current Density over a Pd-Ni(OH)2 Catalyst. Angew. Chem. Int. Ed. 62, 202300094 (2023).
Zhao, B.-H. et al. Economically viable electrocatalytic ethylene production with high yield and selectivity. Nat. Sustainability 6, 827-837 (2023).
Wang, M. et al. Pd Nanoparticles Coupled to Nanorods for Enhanced Electrocatalytic Ethanol Oxidation. ACS Appl. Mater. Interfaces 13, 53777-53786 (2021).
Liu, X. C. et al. Reaction Mechanism and Selectivity Tuning of Propene Oxidation at the Electrochemical Interface. J. Am. Chem. Soc. 144, 20895-20902 (2022).
An, L. et al. Epitaxial Heterogeneous Interfaces on Nanowires/Nanosheets to Boost Hydrogen and Oxygen Production for Overall Water Splitting. Adv. Funct. Mater. 29, 1805298 (2019).
Huang, B. et al. Hydrangea-like NiMoO4-Ag/rGO as Battery-type electrode for hybrid supercapacitors with superior stability. J. Colloid Interface Sci. 606, 1652-1661 (2022).
Ge, R. et al. Selective Electrooxidation of Biomass-Derived Alcohols to Aldehydes in a Neutral Medium: Promoted Water Dissociation over a Nickel-Oxide-Supported Ruthenium Single-Atom Catalyst. Angew. Chem. Int. Ed. 61, e202200211 (2022).
Li, Y., Wei, X., Chen, L., Shi, J. & He, M. Nickel-molybdenum nitride nanoplate electrocatalysts for concurrent electrolytic hydrogen and formate productions. Nat. Commun. 10, 5335 (2019).
Li, Z. et al. Alcohols electrooxidation coupled with production at high current densities promoted by a cooperative catalyst. Nat. Commun. 13, 147 (2022).
Huang, W. et al. Highly active and durable methanol oxidation electrocatalyst based on the synergy of platinum-nickel hydroxide-graphene. Nat. Commun. 6, 10035 (2015).
Liu, F. et al. Concerted and Selective Electrooxidation of Poly-ethylene-Terephthalate-Derived Alcohol to Glycolic Acid at an Industry-Level Current Density over a Pd-Ni(OH)2 Catalyst. Angew. Chem. Int. Ed. 62, 202300094 (2023).
Zhu, J. et al. Ultrahigh Stable Methanol Oxidation Enabled by a High Hydroxyl Concentration on Pt Clusters/MXene Interfaces. J. Am. Chem. Soc. 144, 15529-15538 (2022).
Chen, Y. et al. Trimetallic PtRhCo petal-assembled alloyed nanoflowers as efficient and stable bifunctional electrocatalyst for ethylene glycol oxidation and hydrogen evolution reactions. J. Colloid Interface Sci. 559, 206-214 (2020).
Marinho, V. L. et al. Ethylene glycol oxidation on carbon supported binary PtM ( an Ni ) electrocatalysts in alkaline media. J. Electroanalytical Chem. 880, 114859 (2021).
Li, S., Lai, J., Luque, R. & Xu, G. Designed multimetallic Pd nanosponges with enhanced electrocatalytic activity for ethylene glycol and glycerol oxidation. Energy Environ. Sci. 9, 3097-3102 (2016).
Arjona, N., Espinosa-Magaña, F., Bañuelos, J. A., Álvarez-Contreras, L. & Guerra-Balcázar, M. Manganese Oxides as
Co-catalysts in Pd-Based Nanomaterials for the Ethylene Glycol Electro-Oxidation. ChemElectroChem 9, 202200015 (2022).
44. Qin, Y. et al. Extraordinary p-d Hybridization Interaction in Heterostructural Pd-PdSe Nanosheets Boosts C-C Bond Cleavage of Ethylene Glycol Electrooxidation. Angew. Chem. Int. Ed. 61, 202200899 (2022).
45. Qian, Q. et al. Electrochemical Biomass Upgrading Coupled with Hydrogen Production under Industrial-level Current Density. Adv. Mater. 35, 2300935 (2023).
46. Wang, H.-Y. et al. In Operando Identification of Geometrical-SiteDependent Water Oxidation Activity of Spinel . J. Am. Chem. Soc. 138, 36-39 (2015).
47. Luo, H. et al. Amorphous with high oxophilicity interfaced with PtMo alloy nanoparticles boosts anti-CO hydrogen electrocatalysis. Adv. Mater. 35, e2211854 (2023).
شكر وتقدير
تم دعم هذا العمل من قبل البرنامج الوطني الرئيسي للبحث والتطوير في الصين (2022YFB4002700)، وبرنامج النجوم الصاعدة من لجنة العلوم والتكنولوجيا في شنغهاي (22QA1403400) ومؤسسة العلوم الطبيعية في شنغهاي (21ZR1418700). يود المؤلفون أن يشكروا منصة ECNU متعددة الوظائف للابتكار على دعمها في تحديد خصائص TEM (004). ويعبر المؤلفون عن امتنانهم لمختبر Shiyanjia (www.شينغيانجيا.كومللمساعدة القيمة في تحليل SEM.
مساهمات المؤلفين
قاد المشروع J.S. و L.C. صمم K.S. وأجرى التجارب؛ ساهم X.T. و D.S. بشكل كبير في مراجعة المخطوطة. ناقش جميع المؤلفين النتائج وعلقوا على المخطوطة.
المصالح المتنافسة
يعلن المؤلفون عدم وجود مصالح متنافسة.
معلومات إضافية
معلومات إضافية النسخة الإلكترونية تحتوي على المواد التكميلية متاحة على https://doi.org/10.1038/s41467-024-47179-7. يجب توجيه المراسلات والطلبات للحصول على المواد إلى ليسونغ تشين.
معلومات مراجعة الأقران تشكر مجلة Nature Communications المراجعين المجهولين على مساهمتهم في مراجعة هذا العمل. يتوفر ملف مراجعة الأقران.
معلومات إعادة الطباعة والتصاريح متاحة على http://www.nature.com/reprints ملاحظة الناشر: تظل شركة سبرينغر ناتشر محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
الوصول المفتوح هذه المقالة مرخصة بموجب رخصة المشاع الإبداعي النسب 4.0 الدولية، التي تسمح بالاستخدام والمشاركة والتكيف والتوزيع وإعادة الإنتاج بأي وسيلة أو صيغة، طالما أنك تعطي الائتمان المناسب للمؤلفين الأصليين والمصدر، وتوفر رابطًا لرخصة المشاع الإبداعي، وتوضح ما إذا تم إجراء تغييرات. الصور أو المواد الأخرى من طرف ثالث في هذه المقالة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة، ما لم يُشار إلى خلاف ذلك في سطر الائتمان للمادة. إذا لم تكن المادة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة وكان استخدامك المقصود غير مسموح به بموجب اللوائح القانونية أو يتجاوز الاستخدام المسموح به، فستحتاج إلى الحصول على إذن مباشرة من صاحب حقوق الطبع والنشر. لعرض نسخة من هذه الرخصة، قم بزيارةhttp://creativecommons.org/رخصة/بواسطة/4.0/. (ج) المؤلف(ون) 2024
المختبر الوطني الرئيسي لهندسة الجزيئات والعمليات البترولية، المختبر الرئيسي لعلوم الكيمياء الخضراء والعمليات الكيميائية، كلية الكيمياء والهندسة الجزيئية، جامعة شرق الصين العادية، شنغهاي 200062، الصين.معهد إيكو تشونغ مينغ، شنغهاي 202162، الصين. معهد شنغهاي للسيراميك، الأكاديمية الصينية للعلوم، شنغهاي 200050، جمهورية الصين الشعبية.البريد الإلكتروني: Ischen@chem.ecnu.edu.cn
Pd/NiMoO4/NF electrocatalysts for the efficient and ultra-stable synthesis and electrolyte-assisted extraction of glycolate
Received: 26 September 2023
Accepted: 18 March 2024
Published online: 04 April 2024
(A) Check for updates
Kai Shi , Di Si , Xue Teng , Lisong Chen Jianlin Shi (B
Electrocatalytic conversion of organic small molecules is a promising technique for value-added chemical productions but suffers from high precious metal consumption, poor stability of electrocatalysts and tedious product separation. Here, a Pd/NiMoO4/NF electrocatalyst with much lowered Pd loading amount ( ) has been developed for efficient, economic, and ultra-stable glycolate synthesis, which shows high Faradaic efficiency (98.9%), yield (98.8%), and ultrahigh stability ( 1500 h ) towards electrocatalytic ethylene glycol oxidation. Moreover, the obtained glycolic acid has been converted to value-added sodium glycolate by in-situ acid-base reaction in the NaOH electrolyte, which is atomic efficient and needs no additional acid addition for product separation. Moreover, the weak adsorption of sodium glycolate on the catalyst surface plays a significant role in avoiding excessive oxidation and achieving high selectivity. This work may provide instructions for the electrocatalyst design as well as product separation for the electrocatalytic conversions of alcohols.
Sodium glycolate, an essential commodity chemical, is widely employed as an important intermediate in the production techniques of various important chemicals. The global production of sodium glycolate reaches around 0.1 million tons per year and the market price of sodium glycolate is around 3.0 thousand US dollars . At present, sodium glycolate is produced through a highly energy-intensive process under high temperatures. Typically, chloroacetic acid and sodium hydroxide undergo a substitution reaction to produce sodium glycolate and sodium chloride followed by a complex separation process to obtain the final product. In addition to the high cost and energy consumption of above technology, the raw chloroacetic is usually acidpoisonous and corrosive, which is detrimental to the environment and human health (Fig. 1a). In this regard, developing efficient, energysaving, non-toxic and harmless ways under ambient conditions to produce sodium glycolate is of great importance .
With the rapid development of renewable electricity generation approaches, electrosynthesis is becoming popular as an effective
synthesis tool under ambient conditions . For example, the electrochemical oxidations of organic small molecules (methanol, ethanol, ethylene glycol, glycerol, 5 hydroxymethyl furfural, and so on) to valuable chemicals such as formic acid, glycolic acid, and 2,5-furan dicarboxylic acid, have been reported . Previously, several noblemetal electrodes have been explored for ethylene glycol oxidation; however, only C 1 molecules, such as and formate, have been obtained as the main products in most reports, and the scarcity and high cost of noble metals have also limited their large-scale application . Recently, researchers load the noble metal on oxides or hydroxide substrates to obtain C2 products. For example, our group has reported the electrocatalytic oxidation of ethylene glycol (EG) to glycolic acid (GA) by PdAg/NF, which has attracted wide attention . Although many researchers have made contributions to this field, there are still problems remaining . On the one hand, the high loading amount of precious metal in the electrocatalysts and its rapid deactivation (no longer than 200 h ) can’t meet the requirements
Fig. 1 | Schematic illustration of the production of sodium glycolate. a Current industrial route. Proposed electrochemical route. TEA results at TEA results at .
of large-scale industrial applications; on the other hand, excess of high concentration potassium hydroxide solution is used as the electrolyte to keep the high reaction selectivity, which need to be neutralized before product separation, therefore a large amount of alkaline and acid are inevitably wasted.
In order to solve the above problems, we propose a strategy of insitu acid-base reaction for sodium glycolate extraction from the electrolyte. Unlike the situations in previous reports, in the electrolytic process of this report, sodium glycolate can be obtained from the insitu reaction between the sodium hydroxide electrolyte and GA produced from EG oxidation (Fig. 1b), which not only can make full use of electrolytes to improve the utilization rate of atoms, but also minimize the waste of acid.
To demonstrate the economic potential of this approach, a techno-economic analysis (TEA) has been provided (Supplementary Note 1 and Supplementary Fig. 1) . Preliminary TEA (Fig. 1c, d and Supplementary Fig. 2) indicates that the selective oxidation of ethylene
glycol to sodium glycolate ( selectivity) at a rather high current density ( ) is profitable.
In this work, a electrocatalyst with much lowered Pd content ( ) has been developed for efficient, economic, and ultra-stable glycolate synthesis. shows reasonably high Faradaic efficiency (98.9%) and yield (98.8%), and ultrahigh stability ( 1500 h ) towards electrocatalytic ethylene glycol oxidation. Moreover, the obtained glycolic acid has been converted to valueadded sodium glycolate by in-situ acid-base reaction with the NaOH electrolyte, which is atomically efficient and no additional acid addition is needed for product separation. The experiment results reveal that i) the weak adsorption of sodium glycolate on the catalyst surface plays a significant role in avoiding excessive oxidation and achieving high selectivity; ii) the electron transfer between and and the downshift in the d band-center of Pd promote the adsorption of and the enhancement of catalytic activity and stability.
Results
Synthesis and characterization of catalysts
was synthesized by a two-step process as shown in Fig. 2a (Experimental details can be found in the methods part in SI). In the first step, bi-metal hydroxide was grown on nickel foam by a hydrothermal method, followed by Ar annealing to obtain the crystallized nanorods supported on nickel foam ( ) . In the second step, palladium nanosheets were deposited on the by a reduction approach to obtain the final catalyst . Control samples and have also been obtained by similar methods.
Firstly, X-ray diffraction (XRD) patterns of as well as other catalysts have been obtained to demonstrate the successful synthesis of these electrocatalysts. As shown in Fig. 2b, only characteristic diffraction peaks belonging to the distinct face-centered cubic lattice of Pd and the tetragonal structure of , respectively, without other diffraction peaks being found . Typically, the reflections located at and correspond to the (111),
(200), and (220) planes of Pd (JCPDS no. 46-1043), while the those located at , and correspond to the (110), (112), (220) and (022) planes of (JCPDS no. 33-0948), indicating the successful synthesis of the , and . The microstructures of these obtained electrocatalysts were verified by scanning electron microscopy (SEM). Typical SEM images confirm that well-aligned Pd nanosheet arrays have been grown successfully on the surface of the nanorods (Fig. 2c, d). The SEM images of the as-obtained , and NF are displayed in Supplementary Figs. 3-5. In more detail, the structure of was further confirmed by the transmission electron microscopic (TEM) imaging (Fig. 2e). By highlighting the distinct lattice fringes of and in the lattice d-spacing, which correspond to the (111) plane of the Pd and the ( 312 ) plane of the respectively, the highresolution TEM (HRTEM) image further confirms the crystalline Pd/ (Fig. 2f, g). The TEM images of the as-obtained and are displayed in Supplementary Fig. 6. The energydispersive spectroscopy (EDS) elemental mapping analysis indicates
Fig. 2 | Morphology and structures characterizations of catalysts. a Schematic illustration for the synthesis of XRD patterns of , , and pure NF. c, d SEM images of Pd/NiMoO4/NF. e TEM and f, g HRTEM images of Pd/NiMoO4/NF. SEM-EDS mapping of .
that , and O are homogeneously distributed throughout the (Fig. 2h). In addition, the precise Pd loading amount has been analyzed by inductively coupled plasma mission spectroscopy (ICP-OES), and the weight percentage of Pd is as low as , which is much lower than those reported so far (Supplementary Fig. 7).
Structure and electronic properties of catalysts
To further probe the chemical structure of , Raman and Fourier transform infrared (FT-IR) spectroscopic measurements were carried out. As shown in Fig. 3a, the bands located at and are attributed to the symmetric and asymmetric stretches of terminal , while the band at is assigned to the stretching mode of the Ni-O-Mo bond. Besides, the bands at belongs to the bending mode of . Similar information can also be obtained from the FT-IR curves (Supplementary Fig. 8), from which the obvious characteristic peaks at and can be attributed to the symmetric stretching and the Ni-Mo-O symmetrically stretching, respectively. Besides, the characteristic peaks at and belongs to the superposition of stretching vibration of and groups . Obviously, the red shift of each peak can be observed in Raman and FT-IR spectroscopic curves, which indicates the successful loading of Pd on .
Furthermore, X-ray photoelectron spectroscopy (XPS) was adopted to analyze the electronic states of Pd and the surface element composition of the . The survey spectrum of (Supplementary Fig. 9) confirms the presence of , Mo, and O, which agrees well with the EDS results. As shown in Fig. 3b, the high-resolution Pd 3 d spectrum of consists of four peaks. The peaks located at about and ) belongs to the metallic Pd; while the peaks located at about and corresponds to the . The peak of Pd 3 d in shifts in a positive direction by 0.9 eV compared to that of the , implying electric interactions between Pd and . Six peaks can be obviously seen in the highresolution Ni 2p XPS spectrum (Fig. 3c), among which peaks at 855.8 and 873.6 eV are assigned to and of , respectively, while peaks at 857.3 and 875.5 eV are attributed to and
of , respectively, and those at 862.4 and 880.8 eV are ascribed to two accompanying satellites. The exhibits a high proportion of , which is associated with surface hydroxylation . For highresolution Mo 3d XPS spectra, three sets of doublet peaks are observed between 230 and 240 eV (Fig. 3d), which can be attributed to the doublet peaks of , and their satellite peaks . The binding energy of Ni 2 p and Mo 3 d for is negatively shifted ( ) compared with those of . Besides, as shown in Supplementary Fig. 10, the charge density difference results indicate that 1.63 electrons transfer from the Pd to the substrate, which agree well the results of the XPS. The above results reveal the transferring of electrons from Pd to , which further confirms the strong electronic interaction between Pd and .
In order to further explore the effect of interfacial interaction on the binding strength of absorbates, the surface valence band photoemission spectroscopy was employed to evaluate the d-band center of and . As shown in Fig. 3e, f and Supplementary Fig. 11, the d-band center of the drops to -5.25 eV compared with that of , although the d-band center of the is the lowest, the catalyst has no obvious EG oxidation activity at low potential, so it will not be discussed. Furthermore, as shown in Supplementary Fig. 12, the d-band center of and are calculated to be -1.83 eV and -2.45 eV , respectively, which agree well the results of the surface valance band photoemission spectroscopy. The downshift of d-band center is contributed to the desorption of carbonyl intermediates (such as glycolate acid) to achieve long-term stability and high selectivity of the target products .
Electrocatalytic performances for ethylene glycol oxidation
To investigate the electrocatalytic performance of as-prepared as well as control samples for ethylene glycol oxidation at the anode, a series of electrochemical measurements were performed in a three-electrode setup. Supplementary Fig. 13 presents the linear sweep voltammetry (LSV) curves of in 1.0 M NaOH with or without 1 M ethylene glycol. In the absence of ethylene
Fig. 3 | Structure and electronic properties of catalysts. a Raman spectras of and . b High-resolution XPS spectra of and (d) Mo 3d of and . e XPS valence band spectra of and .
glycol, the electrode shows a moderate OER activity, which reaches the anodic current density of at the potential of 1.61 V versus reversible hydrogen electrode (vs. RHE). When introducing 1 M ethylene glycol, the current density increases markedly, and the anodic potential strikingly decreases to 0.79 V vs. RHE at , which is 820 mV lower than that of OER. Also, to give a more detailed comparison with OER, Supplementary Fig. 14 displays that the anodic potentials in ethylene glycol solution are reduced by at least 820 mV at the current density of , and . Noticeably, shows significantly higher performance than those of the , and pure NF references for electrocatalytic ethylene glycol oxidation (Fig. 4a), which indicates that the interaction between Pd and plays a crucial role in enhancing the EGOR activity. The effects of the concentration of NaOH and ethylene glycol in the electrolyte on the oxidation performance of ethylene glycol at the anode were also studied, as shown in Supplementary Fig. 15. The highest EG oxidation activity ( ) is achieved in 9 M NaOH solution containing 1 M EG. Nevertheless, in order to avoid excessive waste of NaOH , we
choose 1 M NaOH solution added with 1 M EG. What’s more, the performance of catalysts with different Pd content and oxide substrates is further discussed (Supplementary Figs. 16-19). As shown in Fig. 4b, shows much lowered Tafel slop ( 207.7 mV ) compared to that of , indicating the faster reaction kinetics of at the anode. Besides, electrochemical impedance spectra (EIS) of , and NF at the open-circuit voltage were obtained. delivers a smaller charge transfer resistance (Rct) than and pure NF, (Supplementary Fig. 20), which implies the rapid charge transfer kinetics of .
To better understand the origin of the markedly high EGOR performance of , the electrochemical double-layer capacitance ( ) has been obtained to calculate the electrochemically active surface area (ECSA) (Fig. 4e and Supplementary Fig. 21). shows a similar value ( ) with ( ), which is apparently higher than those of ( ) and NF ( ). The ECSA-normalized EGOR activities have also been obtained, which are in the order of
Fig. 4 | Electrocatalytic performances of toward ethylene glycol oxidation at the anode. a LSV curves of obtained electrocatalysts for ethylene glycol anodic oxidation. b Tafel slopes of and . c Faradaic efficiencies (FEs) of for sodium glycolate production for 2 h chronoamperometry at varied potentials. d FEs and current density of Pd/NiMoO4/
NF for sodium glycolate production for 1500 h electrolysis cycles. e Double layer capacitance (Cdl) of , and pure NF. The yield, FE and purity of GA obtained from EGOR. g The optical image of obtained GA product. (Supplementary Fig. 22), further demonstrating largely elevated EGOR activity of the catalyst.
While achieving high EGOR activity is important, good stability may play an even more important role in EGOR for further large-scale applications. Subsequently, a long-term chronoamperometry test using as the electrocatalyst was carried out at vs. RHE to determine the obtained ethylene glycol oxidation products at the anode by NMR and NMR spectroscopy (Supplementary Figs. 23, 24). As shown in the NMR spectra (Supplementary Fig. 23), sodium glycolate ( 3.89 ppm ) is the primary EG oxidation product. Besides, only small amounts of formate (FA) were detected. The highest faradaic efficiency (FE) of was obtained at 1.0 V vs RHE, therefore this voltage was chosen as the optimum potential (Fig. 4c and Supplementary Fig. 25). With The FEs varies in the range of for glycolic acid production and the total charges have been maintained for 1500 h of intermittent chronoamperometry (see details in Methods) (Fig. 4d and Supplementary Fig. 26) . No significant decreases in FE and consumed charges can be observed during the 1500 h stability tests, elucidating the robust durability of the electrocatalyst. As far as we know, this is the longest stability test among literature reports. (Supplementary Fig. 27) . In contrast, during cyclic electrolysis, catalyst shows a much lower FE toward glycolic acid (87-88%), accompanied by quick decays of the current density, indicating the critical role of in enhancing the selectivity and stability towards EGOR (Supplementary Fig. 28).
The development of economic separation approaches is essential to obtaining the value-added products and the industrial applications of electrocatalytic oxidations. The previously reported separation methods necessitate a large amount of strong acids to neutralize excess strong bases retained in the electrolyte, which inevitably leads to the wastes of both acids and bases. Here, based on the stoichiometric EG and NaOH components in the electrolyte before reaction and in-situ acid-base reaction, the pH of the electrolyte after a cycle test reaches about 10.08 (Supplementary Fig. 29) due to the hydrolysis of sodium glycolate, which implies negligible alkaline retained in the electrolyte. Therefore, no additional acid is needed for the product separation, which enables full use of electrolyte and avoids the waste of acid and base. After 1500 hours of cyclic electrolysis, 90.34 g sodium glycolate was successfully isolated at a purity of and a yield of (Fig. 4f, g and Supplementary Fig. 30). Besides, the electrode after ethylene glycol electro-oxidation for 1500 h was further characterized by XRD, SEM, EDS, TEM and HRTEM to evaluate the stability of the catalyst. No significant changes in the XRD pattern of the catalyst can be found (Supplementary Fig. 31). Moreover, the SEM image of shows maintained original morphology after ethylene glycol oxidation stability tests (Supplementary Fig. 32), highlighting the superior structural robustness. SEMEDS elemental mapping analysis displays that the and O are homogeneously distributed throughout the (Supplementary Fig. 33). In addition to this, both the Pd and lattice can be found in the HRTEM image (Supplementary Fig. 34). These results all suggest the excellent stability of the obtained catalyst. Furthermore, the produced amount was quantified by gas chromatography (GC). Supplementary Fig. 35 shows the standard curve of production measured by GC. As exhibited in Supplementary Fig. 36, the Faradaic efficiency for at varied consumed charges have been calculated to be close to and the purity of is .
Understanding the mechanism
The adsorption of EG species is important for the electrocatalytic EG oxidation in alkaline electrolyte. Figure 5a and Supplementary Fig. 37 shows the open circuit potential (OCP) measurement results, which can be applied to show the influences of organic absorbates on the
inner Helmholtz layer . Upon adding 1 M EG, the OCP is significantly decreased for compared with and , indicating that EG is more easily adsorbed on the than on and surface. At the same time, the weak adsorption of glycolate is the key to avoid over-oxidation. As shown in Fig. 5b, upon the addition of 1 M GA, the OCP changes is negligible ( ) compared with that of 1 M EG ( ), which indicates that GA is weakly absorbed in the inner Helmholtz layer so as to prevent its over-oxidation. Furthermore, the in-situ EIS measurements were applied to explore the catalytic kinetics and the electrode/electrolyte interface properties at varied potentials . The Nyquist and Bode plots of in 1 M NaOH containing 1 M EG are shown in Fig. 5c, d. A phase peak shift at 0.45 V (vs. RHE) in the low-frequency can be clearly observed, indicating the beginning of the reaction at this potential. As the potential increases, the phase angle peaks become weakened and shift towards higher frequency, demonstrating that more and more adsorbed EG are rapidly oxidized at a fast interfacial charge transferring rate. As shown in Supplementary Fig. 38, the oxidation of GA shows an opposite trend to that of EG, which also implies a weaker adsorption of GA on the surface. The above results demonstrate a poor catalytic activity of toward GA oxidation reaction. Besides, to further verify the above results, the oxidation activity of GA in 1 M NaOH has also been explored, which is even negligible compared with that of EG in the same solution (Supplementary Fig. 39). The markedly weaker adsorption of GA than that of EG on Pd/NiMoO4/NF will ensure the over-oxidation prevention and selectivity enhancement of GA in comparison with EG.
Similarly, the adsorption of species is also crucial for the electrocatalytic EG oxidation in alkaline electrolyte. Figure 5e shows the representative OH adsorption and desorption curves on Pd in and . As shown in Fig. exhibits OH adsorption/desorption bands at significantly lower onset potential ( 0.5 V vs RHE) than ( 0.6 V vs RHE), which is attributed to the introduced oxyphilic Ni species in , which can efficiently oxidize to * OH adsorbed on Pd surfaces at a relatively low potential. At the same time, combined with the XPS results, the electron-rich substrate can absorb no anions on its surface, while the positively charged Pd nanosheets can capture to facilitate the further oxidization of . The important roles of species were further explored by CO-stripping Cyclic Voltammetry (CV) measurements . A distinct CO oxidation peak appears for and in the first anodic scan at the peak potentials at 1.090 V vs. RHE and 0.785 V vs. RHE (Supplementary Figs. 40, 41). After introducing , the peak potentials of CO oxidation decrease to a lowered potential 1.026 V vs RHE (Fig. 5f) for , which indicates that CO is weakly adsorbed on the surface and can be easily oxidative-removed on account of the downshift of the d-band center of Pd, eventually contributing to the observed superior EGOR stability.
Moreover, in-situ electrochemical Fourier Transform Infrared (FTIR) spectroscopy has been employed to further understand the origin of the high EGOR performance of . The electrocatalysts were placed on the flat plane of a Si hemicylindrical prism and pressed by a glass carbon electrode. The FTIR spectra were recorded in 1 M NaOH aqueous solution with 1 M EG at the potential of -0.9 to 0.1 V vs . As displayed in Supplementary Fig. 42, a peak at corresponding to the multiple bonded can be well observed on ; however, signal can be hardly detected for NF over the whole potential range, further demonstrating high CO tolerance of , which is consistent with the CO stripping experimental results. Apparently, a downward enhancement band at can be observed, which is attributed to the stretching vibration of aldehyde, indicating that EG is firstly oxidized to glycolaldehyde species . Further, the peaks at and , belonging to symmetric and antisymmetric stretching bands of
Fig. 5 | Understanding the mechanism. a Open circuit potentials (OCPs) of Pd/ and in 1 M NaOH solution before and after EG was added. OCPs of in 1 M NaOH solution with EG or GA addition. The Nyquist plots and (d) corresponding Bode phase plots of electrode at varied
potentials in 1 M NaOH with 1 M EG. e CV curves of and in 1 M stripping experiments of In-situ electrochemical FTIR spectra of EGOR catalyzed by . Schematic illustration of the proposed EGOR mechanism on and (i) .
carboxyl group of GA, respectively, indicates the further oxidation of glycolaldehyde to . Besides, the distinct vibration peaks at 1700 and have been detected on and , respectively, which can be assigned to the stretching vibration of carbonyl group ( ), indicating the formation of 2 -hydroxyacetyl ( * ) intermediates (Fig. 5 g ).
Furthermore, the Density functional theory (DFT) calculation is also implied to investigate the reaction mechanism. The optimized models of EG oxidation on the Pd/NiMoO4 and Pd catalysts are shown in Supplementary Figs. 43, 44. Besides, the corresponding Gibbs free energy profile at 0 V vs RHE were calculated. As shown in Supplementary Fig. 45, obviously, we find that the addition of to form the co-adsorption of * OH and is the rate-determining step (RDS) in the whole reaction from EG to the glycolic acid on and Pd catalysts. In particular, the free-energy change of the RDS on is much lower than 0.86 eV of Pd , suggesting that the catalysts could adsorb OH at a relatively lower onset potential, which is in accordance with the results of CV .
What’s more, As shown in Supplementary Fig. 10, the charge density difference results indicate that 1.63 electrons transfer from the Pd to the substrate, which suggests that the Pd is positively
charged and the substrate is negatively charged. The positively charged Pd could attract anions OH to promote the hydrogen abstraction of EG. The calculated adsorption energies of OH on the surfaces of are , and 0.28 eV at Pd , Ni , and Mo adsorption sites, respectively (Supplementary Fig. 46). The above results indicate that the addition of OH is at the Pd site. Besides, as shown in Supplementary Fig. 47, the calculated adsorption energies of EG on Pd (111) and were -0.57 and -0.98 eV , respectively, which indicates that the EG is more easily adsorbed on the , in accordance with the results of OCP measurements. Besides, the calculated adsorption energies of EG on the surfaces of are , and -0.36 eV at and Mo adsorption sites, respectively (Supplementary Fig. 48), indicating that the EG is more favorable for adsorption at Pd site. Furthermore, as shown in Supplementary Fig. 28, the main products of EGOR catalyzed by Pd/NF is sodium glycolate; however, the has no obvious EG oxidation activity at low potential. The above results indicate that the abstraction of hydrogen is also at the Pd site.
According to above results, the reaction mechanism and path of EG has been proposed as following (Fig. 5h and Supplementary Fig. 49). The surface of becomes negatively charged due to
the transferring of electrons from Pd to , which can promote migration of hydroxyls away from the surface and result in the enhanced local hydroxyl concentration around the Pd surface. In contrast, the hydroxyls on the Pd surface are uniformly distributed on catalysts (Fig. 5i). The higher local hydroxyl concentration around the Pd surface can promote the faster conversion of ethylene glycol and activation of CO, thus favoring the enhancement of EGOR activity and stability. In addition, the introduction of can also promote the adsorption of EG, and the higher local concentration facilitates its faster conversion.
Membrane-free flow electrolyzer performance
To demonstrate its economic feasibility in a two-electrode system, a homemade membrane-free flow electrolyzer has been assembled by using catalyst as the anode and pre-treated nickel foam as the cathode at a working area of (Fig. 6a). A homogenous electrolyte ( 1 M NaOH containing 1 M ethylene glycol) was circulated in the flow reactor by using a peristaltic pump at a flow rate of . The LSV curves (Fig. 6b) shows that electrooxidation of ethylene glycol occurs from and reaches the current density of at 1.2 V in flow cell, which is higher than that in the corresponding three-electrode cell, demonstrating priomising application potentials of ethylene glycol electrooxidation in flow cell.
We then performed intermittent potential tests to evaluate the performance of sodium glycolate production in the electrolyzer . After 23 hours of stability test (Fig. 6c), the conversion of ethylene glycol reaches , and the FE and selectivity of sodium glycolate are and , respectively. Finally, the sodium glycolate crystal was obtained as confirmed by XRD (Fig. 6d).
Conclusions
In summary, an electrocatalyst with rather low Pd loading ( ) has been obtained for efficient electrocatalytic ethylene glycol oxidation, which features the faradaic efficiency, yield of sodium glycolate and stability of as high as , and 1500 h , respectively. More importantly, the obtained glycolic acid has been converted to value-added sodium glycolate by in-situ acid-base reaction with NaOH electrolyte, which favors the enhancement of
atom utilization and the separation of sodium glycolate separation. The weak adsorption of sodium glycolate on the catalyst surface plays a significant role in avoiding excessive oxidation and achieving high selectivity. The electron transfer between Pd and and the downshift of the d band-center of Pd have been proposed to promote the adsorption of and the oxidation removal of CO , thereby leading to the enhanced activity and stability of .
Methods
Chemicals
Nickel nitrate hexahydrate ( ), Sodium molybdate dihydrate ( ), and Sodium borohydride ( , 99.99%) were purchased from China National Pharmaceutical Group Co., Ltd. Sodium tetrachloropalladate ( ) was purchased from Shanghai Titan Technology Co. Ltd. Sodium hydroxide ( ) were from Macklin. Nickel Foam (NF, 0.15 mm thick) was purchased from Tianjin Gaoshi Ruilian Photoelectric Technology Co., Ltd. Maleic acid ( , grade) and deuterated water ( ) were bought from Adamas Reagent Co., Ltd. Ethylene glycol (AR) was bought from Chinese medicine reagent.
All chemicals were used as received without any further purification. Deionized water (DIW) was used in all experiments.
Synthesis of the nanorods/NF
was prepared as a precursor using hydrothermal synthesis. First, was ultrasonically cleaned in 3 M HCl , ethanol, and deionized water to remove any surface impurities. Thereafter, Ni and were dissolved in deionized water ( 30 mL ) and stirred for 10 min . Subsequently, the asobtained precursor solution and a piece of NF were placed in a Teflonlined stainless-steel autoclave and maintained at for 6 h . After the autoclave was cooled to room temperature, the NF with light green precipitates on the surface was taken out and washed with deionized water and ethanol respectively to remove any unreacted residues before being fully dried at overnight under vacuum. In order to obtain crystallized nanostructures, the conductive substrates with as-grown precursor hierarchical structures were calcined at for 2 h with a temperature ramp rate of in an argon atmosphere.
Fig. 6 | Membrane-free flow cell for EG oxidation. a The MEA setup for paired HER (-) // EG oxidation (+). b LSV curves of in 1 M NaOH with 1 M EG in single cell or flow cell. c Stability tests of toward EGOR. d XRD pattern of self-prepared sodium glycolate.
Synthesis of the Pd/NiMoO4/NF
The and were dispersed in 30 mL of water and stirred for 12 h . The black sample was generated after adding 1 mL of and stirring for 4 h . Subsequently, the resulting sample was washed via deionized water and ethanol and further dried at for 10 h in a vacuum to obtain catalysts. was synthesized in a similar manner using the pretreated NF ( ) as the precursor.
Materials characterization
X-ray diffraction (XRD) patterns were recorded on a Rigaku D/MAX 2550 diffractometer at 35 kV and 25 mA using Cu Ka radiation ( ). Scanning electron microscope (SEM) images and energydispersive elemental mapping (EDS) images were acquired using a ZEISS Gemini 450. Transmission electron microscopy (TEM) images were acquired on a JEM-2100F at 200 kV . X-ray photoelectron spectroscopy (XPS) was performed on AXIS SUPRA with an radiation source ( ). The position of the C 1 s peak at 284.8 eV was utilized as a calibration reference for determining the precise binding energy ( ). Inductively coupled plasma emission spectroscopic (ICP-OES) analysis was recorded on Agilent 700 Series instrument. Electrochemical in situ FTIR measurements were performed on a Linglu instruments ECIR-II cell mounted on a Pike Veemax III ATR using a single crystal bouncing silicon crystal. The binding energy values were referenced using the C 1 s peak position at 284.8 eV . The d-band centers were evaluated by applying the following formula.
Electrochemical measurements
All measurements for OER and ethylene glycol oxidation were conducted on a BioLogic VSP-300 electrochemical workstation in a threeelectrode single cell at room temperature with of the as-made materials as the working electrode, a Pt plate as the counter electrode, and ( 1.0 M NaOH ) as the reference electrode, respectively. The ( 1 M NaOH ) reference electrode was calibrated with respect to RHE in high-purity hydrogen-saturated 1 M NaOH electrolytes, two platinum wire electrodes were used as the counter electrode and the working electrode in this measurement, respectively. The conversion formula of the reference electrode is: . Polarization curves were obtained using LSV with a scanning rate of in the region of (vs ). Cyclic Voltammetry (CV) was measured using CV scans from -0.1 V to 1.2 V vs. RHE at a scan rate of . The durability test was performed by chronoamperometry at 1.0 V vs. RHE. EIS measurements were performed in the frequency range of 100 kHz to 10 MHz with an alternating current voltage of 5 mV . The electrochemical double layer capacitances ( ) of various samples were confirmed by CV in the potential region without faradaic process to calculate the ECSA. EGOR tests were conducted in a 1 M NaOH solution containing 1 M ethylene glycol. The area of the working electrode in the electrolyte was fixed at and all current densities were normalized to the geometrical area of the electrode. All the curves were used without IR compensation.
CO stripping voltammetry experiments
The CO stripping curves were carried out in a 1 M NaOH solution. Prior to the tests, the 1 M NaOH solution was first deaerated with high-purity for 30 min . Then, pure CO gas was bubbled into the solution for 30 min while the potential of the working electrode was held at a constant potential of 0.1 V vs. RHE to ensure effective CO
adsorption on the electrode surface. Nonadsorbed CO molecules were then repelled by bubbling gas in the solution for 20 min . Subsequently, CO stripping curves were initiated from to 1.1 V vs. RHE in the anodic direction at a scan rate of for at least two consecutive cycles.
Product quantification
The chronoamperometry tests were conducted at 1.0 V vs. RHE (for the three-electrode system) or at the cell voltage of 1.2 V (for the two-electrode system). After 2 h of electrolysis, the products were analyzed and quantified by and nuclear magnetic resonance ( and NMR). and NMR spectra were recorded on an Avance II 300 instrument (Bruker). electrolyte was collected and diluted with of . Maleic acid was used as an internal standard. A Ramin GC2060 gas chromatograph with a packed column and a thermal conductivity detector was used to quantify the generated generated at the cathode during electrolysis. The standard curve of was exhibited in Supplementary Fig. 31. The amount of theoretically generated was calculated as ( is the total charge passed through the electrodes, is the molar volume of gas, is the number of electrons needed to produce a molecule of is the Faraday constant). The formula used to calculate the purity of sodium glycolate is:
Purity = m1/m2
m 1 refers to the mass of sodium glycolate is actually separated, m 2 refers to the total mass of the isolated product.
The yield (%) and selectivity (%) of glycolic acid formation can be determined by the following Eqs. (1) and (2), respectively
Selectivity
The Faraday efficiency (%) of the glycolic acid and production can be determined by the following Eqs. (3) and (4), respectively
Where and are the total charge passed through the electrodes, is the number of electrons that form a mole of glycolic acid, is the number of electrons that produce a molecule of is the Faraday constant .
Data availability
All data is available from the authors upon request.
References
Tang, W. et al. Efficient Conversion of Biomass to Formic Acid Coupled with Low Energy Consumption Hydrogen Production from Water Electrolysis. Angew. Chem. Int. Ed. 62, e202305843 (2023).
Gao, P., Zhong, L., Han, B., He, M. & Sun, Y. Green Carbon Science: Keeping the Pace in Practice. Angew. Chem. Int Ed. Engl. 61, e202210095 (2022).
Fan, L. et al. Selective production of ethylene glycol at high rate via cascade catalysis. Nat. Catal. 6, 2520-1158 (2023).
Lum, Y. et al. Tuning OH binding energy enables selective electrochemical oxidation of ethylene to ethylene glycol. Nat. Catal. 3, 14-22 (2020).
Xu, H., Chen, L. & Shi, J. Advanced electrocatalytic systems for enhanced atom/electron utilization. Energy Environ. Sci. 16, 1334-1363 (2023).
Si, D. et al. Hydrogen anode/cathode co-productions-coupled anode alcohol selective oxidation and distinctive H/e transfer pathways. Appl. Catal. B: Environ. 331, 122664 (2023).
Radwan, A., Jin, H., He, D. & Mu, S. Design Engineering, Synthesis Protocols, and Energy Applications of MOF-Derived Electrocatalysts. Nano-Micro Lett. 13, 132 (2021).
Lin, H. et al. Two-dimensional titanium carbide MXenes as efficient non-noble metal electrocatalysts for oxygen reduction reaction. Sci. China Mater. 62, 662-670 (2018).
Li, G. et al. Dual hydrogen production from electrocatalytic water reduction coupled with formaldehyde oxidation via a copper-silver electrocatalyst. Nat. Commun. 14, 525 (2023).
Wang, T. et al. Combined anodic and cathodic hydrogen production from aldehyde oxidation and hydrogen evolution reaction. Nat. Catal. 5, 66-73 (2021).
Fan, L. et al. High Entropy Alloy Electrocatalytic Electrode toward Alkaline Glycerol Valorization Coupling with Acidic Hydrogen Production. J. Am. Chem. Soc. 144, 7224-7235 (2022).
Huang, H. et al. Ni, Co hydroxide triggers electrocatalytic production of high-purity benzoic acid over . Energy Environ. Sci. 13, 4990-4999 (2020).
Zhu, Y. et al. Single-Atom In-Doped Subnanometer Pt Nanowires for Simultaneous Hydrogen Generation and Biomass Upgrading. Adv. Funct. Mater. 30, 2004310 (2020).
Wei, X., Li, Y., Chen, L. & Shi, J. Formic Acid Electro-Synthesis by Concurrent Cathodic Reduction and Anodic Oxidation. Angew. Chem. Int. Ed. 60, 3148-3155 (2021).
Zhou, P. et al. Heterogeneous-Interface-Enhanced Adsorption of Organic and Hydroxyl for Biomass Electrooxidation. Adv. Mater. 34, 2204089 (2022).
Zhang, W., Xia, G.-J. & Wang, Y.-G. Mechanistic insight into methanol electro-oxidation catalyzed by PtCu alloy. Chin. J. Catal. 43, 167-176 (2022).
Shi, K., Si, D., Teng, X., Chen, L. & Shi, J. Enhanced electrocatalytic glycerol oxidation on at significantly reduced potentials. Chin. J. Catal. 53, 143-152 (2023).
Peng, T. et al. Electrocatalytic valorization of lignocellulose-derived aromatics at industrial-scale current densities. Nat. Commun. 14, 7229 (2023).
Qin, Y. et al. Extraordinary p-d Hybridization Interaction in Heterostructural Pd-PdSe Nanosheets Boosts C-C Bond Cleavage of Ethylene Glycol Electrooxidation. Angew. Chem. Int Ed. Engl. 61, e202200899 (2022).
Wang, Y. et al. Implanting Mo Atoms into Surface Lattice of Alloys Enclosed by High-Indexed Facets: Promoting Highly Active Sites for Ethylene Glycol Oxidation. ACS Catal. 9, 442-455 (2018).
Yang, X. et al. Interface-Rich Three-Dimensional Au-Doped PtBi Intermetallics as Highly Effective Anode Catalysts for Application in Alkaline Ethylene Glycol Fuel Cells. Adv. Funct. Mater. 31, 2103671 (2021).
Zhang, S. et al. Highly Strained Au-Ag-Pd Alloy Nanowires for Boosted Electrooxidation of Biomass-Derived Alcohols. Nano Lett. 21, 1074-1082 (2021).
Bai, S., Xu, Y., Cao, K. & Huang, X. Selective Ethanol Oxidation Reaction at the Interface. Adv. Mater. 33, e2005767 (2021).
Zhu, X.-Y. et al. Hollow nanotube bundles with high electrocatalytic performances for hydrogen evolution and ethylene
glycol oxidation reactions. J. Colloid Interface Sci. 532, 571-578 (2018).
Qi, J. et al. Energy-saving and product-oriented hydrogen peroxide electrosynthesis enabled by electrochemistry pairing and product engineering. Nat. Commun. 14, 6263 (2023).
Si, D., Xiong, B., Chen, L. & Shi, J. Highly selective and efficient electrocatalytic synthesis of glycolic acid in coupling with hydrogen evolution. Chem. Catal. 1, 941-955 (2021).
Yan, Y. et al. Electrocatalytic Upcycling of Biomass and Plastic Wastes to Biodegradable Polymer Monomers and Hydrogen Fuel at High. Curr. Densities. J. Am. Chem. Soc. 145, 6144-6155 (2023).
Liu, F. et al. Concerted and Selective Electrooxidation of Polyethylene-Terephthalate-Derived Alcohol to Glycolic Acid at an Industry-Level Current Density over a Pd-Ni(OH)2 Catalyst. Angew. Chem. Int. Ed. 62, 202300094 (2023).
Zhao, B.-H. et al. Economically viable electrocatalytic ethylene production with high yield and selectivity. Nat. Sustainability 6, 827-837 (2023).
Wang, M. et al. Pd Nanoparticles Coupled to Nanorods for Enhanced Electrocatalytic Ethanol Oxidation. ACS Appl. Mater. Interfaces 13, 53777-53786 (2021).
Liu, X. C. et al. Reaction Mechanism and Selectivity Tuning of Propene Oxidation at the Electrochemical Interface. J. Am. Chem. Soc. 144, 20895-20902 (2022).
An, L. et al. Epitaxial Heterogeneous Interfaces on Nanowires/Nanosheets to Boost Hydrogen and Oxygen Production for Overall Water Splitting. Adv. Funct. Mater. 29, 1805298 (2019).
Huang, B. et al. Hydrangea-like NiMoO4-Ag/rGO as Battery-type electrode for hybrid supercapacitors with superior stability. J. Colloid Interface Sci. 606, 1652-1661 (2022).
Ge, R. et al. Selective Electrooxidation of Biomass-Derived Alcohols to Aldehydes in a Neutral Medium: Promoted Water Dissociation over a Nickel-Oxide-Supported Ruthenium Single-Atom Catalyst. Angew. Chem. Int. Ed. 61, e202200211 (2022).
Li, Y., Wei, X., Chen, L., Shi, J. & He, M. Nickel-molybdenum nitride nanoplate electrocatalysts for concurrent electrolytic hydrogen and formate productions. Nat. Commun. 10, 5335 (2019).
Li, Z. et al. Alcohols electrooxidation coupled with production at high current densities promoted by a cooperative catalyst. Nat. Commun. 13, 147 (2022).
Huang, W. et al. Highly active and durable methanol oxidation electrocatalyst based on the synergy of platinum-nickel hydroxide-graphene. Nat. Commun. 6, 10035 (2015).
Liu, F. et al. Concerted and Selective Electrooxidation of Poly-ethylene-Terephthalate-Derived Alcohol to Glycolic Acid at an Industry-Level Current Density over a Pd-Ni(OH)2 Catalyst. Angew. Chem. Int. Ed. 62, 202300094 (2023).
Zhu, J. et al. Ultrahigh Stable Methanol Oxidation Enabled by a High Hydroxyl Concentration on Pt Clusters/MXene Interfaces. J. Am. Chem. Soc. 144, 15529-15538 (2022).
Chen, Y. et al. Trimetallic PtRhCo petal-assembled alloyed nanoflowers as efficient and stable bifunctional electrocatalyst for ethylene glycol oxidation and hydrogen evolution reactions. J. Colloid Interface Sci. 559, 206-214 (2020).
Marinho, V. L. et al. Ethylene glycol oxidation on carbon supported binary PtM ( an Ni ) electrocatalysts in alkaline media. J. Electroanalytical Chem. 880, 114859 (2021).
Li, S., Lai, J., Luque, R. & Xu, G. Designed multimetallic Pd nanosponges with enhanced electrocatalytic activity for ethylene glycol and glycerol oxidation. Energy Environ. Sci. 9, 3097-3102 (2016).
Arjona, N., Espinosa-Magaña, F., Bañuelos, J. A., Álvarez-Contreras, L. & Guerra-Balcázar, M. Manganese Oxides as
Co-catalysts in Pd-Based Nanomaterials for the Ethylene Glycol Electro-Oxidation. ChemElectroChem 9, 202200015 (2022).
44. Qin, Y. et al. Extraordinary p-d Hybridization Interaction in Heterostructural Pd-PdSe Nanosheets Boosts C-C Bond Cleavage of Ethylene Glycol Electrooxidation. Angew. Chem. Int. Ed. 61, 202200899 (2022).
45. Qian, Q. et al. Electrochemical Biomass Upgrading Coupled with Hydrogen Production under Industrial-level Current Density. Adv. Mater. 35, 2300935 (2023).
46. Wang, H.-Y. et al. In Operando Identification of Geometrical-SiteDependent Water Oxidation Activity of Spinel . J. Am. Chem. Soc. 138, 36-39 (2015).
47. Luo, H. et al. Amorphous with high oxophilicity interfaced with PtMo alloy nanoparticles boosts anti-CO hydrogen electrocatalysis. Adv. Mater. 35, e2211854 (2023).
Acknowledgements
This work was supported by National Key R&D Program of China (2022YFB4002700), Shanghai Science and Technology Committee Rising-Star Program (22QA1403400) and the Natural Science Foundation of Shanghai (21ZR1418700). The authors would like to thank ECNU Multifunctional Platform for Innovation for support of TEM characterizations (004). The authors extend their gratitude to Shiyanjia Lab (www. shiyanjia.com) for providing in valuable assistance with the SEM analysis.
Author contributions
J.S. and L.C. led the project. K.S. designed and performed the experiments; X.T. and D.S. contributed considerably to the revision of the manuscript. All authors discussed the results and commented on the manuscript.
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information The online version contains
supplementary material available at https://doi.org/10.1038/s41467-024-47179-7.
Correspondence and requests for materials should be addressed to Lisong Chen.
Peer review information Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Reprints and permissions information is available at http://www.nature.com/reprints
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/ licenses/by/4.0/.
(c) The Author(s) 2024
State Key Laboratory of Petroleum Molecular & Process engineering, Shanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China. Institute of Eco-Chongming, Shanghai 202162, China. Shanghai Institute of Ceramics, Chinese Academy of Sciences, Shanghai 200050, P. R. China. e-mail: Ischen@chem.ecnu.edu.cn