محللات المياه الكهربائية ذات كفاءة عالية باستخدام أغشية تبادل الأنيونات مع محفزات كهربائية غير متبلورة مشوبة بالكروم Highly efficient anion exchange membrane water electrolyzers via chromium-doped amorphous electrocatalysts
فهم عميق وتعديل التركيب الإلكتروني لمواقع المعادن النشطة أمر حاسم لتعزيز نشاطها الجوهري في تفاعل تطور الأكسجين الكهروكيميائي (OER) تجاه إلكتروليز المياه باستخدام أغشية تبادل الأنيونات (AEMWEs). هنا، نوضح سلسلة من المحفزات المعدنية غير المتبلورة. و مع أداء عالٍ لمواد AEMWE بواسطة مادة الكروم عالية التكافؤ. نكتشف أن التأثير الإيجابي للانتقال من التكافؤ المنخفض إلى التكافؤ العالي لموقع الكوبالت على طاقة الامتزاز للوسط والحاجز الأكسيدي المنخفض هو العامل الرئيسي لزيادة نشاطه بواسطة تقنيات الإشعاع السنكروتروني في الموقع. بشكل خاص،يحقق محفز الأنود كثافة تيار عالية تبلغعند 2.1 فولت ويستمر لأكثر من 120 ساعة مع ت attenuation أقل منفي اختبار AEMWE. إن هذا الأداء الاستثنائي يُظهر آفاقًا واعدة للتطبيق الصناعي ويوفر إرشادات عامة لتصميم أنظمة AEMWE عالية الكفاءة.
تعتبر عملية التحليل الكهربائي باستخدام غشاء تبادل الأنيونات (AEM) تقنية محورية للاقتصاد الطاقي المستدام.. لديها مزايا على تقنية غشاء تبادل البروتون (PEM) مع أكسيد المعادن النبيلة ( ) الأنودات. تشمل هذه المزايا القدرة على استخدام محفزات غير مكلفة من المعادن غير الثمينة (PGM) على جانب الأنود، وأغشية تبادل الأيونات الأرخص، الخالية من البوليمرات القائمة على الفلور الضارة بالبيئة، وعدم الحاجة إلى مواد تكديس مقاومة للأحماض، مما يؤدي إلى خفض التكاليف الإجمالية للجهاز.ومع ذلك، فإن تقنية AEMWE هي تقنية جديدة نسبياً وتواجه العديد من القضايا التي يجب حلها قبل تحقيق إمكاناتها الكاملة، حيث تعتبر المحفزات عاملاً حاسماً.هناك تحديان رئيسيان في تطوير محفزات الأكسدة الكهربائية (OER) ذات الأنود غير النبيل ذات الأداء العالي. الأول هو أن طرق التخليق الحالية لهذه المحفزات ليست محسّنة للتطبيقات الصناعية. لذلك، في السيناريوهات العملية، من الضروري أخذ في الاعتبار طرق تخليق بسيطة وسهلة الوصول. طرق يمكن أن تنتج كميات كبيرة من المحفزات. والآخر هو أنه على الرغم منتمت دراسة المواد القائمة على – بشكل مكثف كعوامل حفازة لتفاعل الأكسدة، لكن استقرارها ضعيف ونادراً ما تُستخدم في أبحاث التحليل الكهربائي باستخدام أغشية تبادل الأنيونات.الاستقرار والنشاط هما عاملان حاسمان، خاصة في تطبيقات التحليل الكهربائي، والتي يجب أن تحظى بمزيد من الاهتمام.
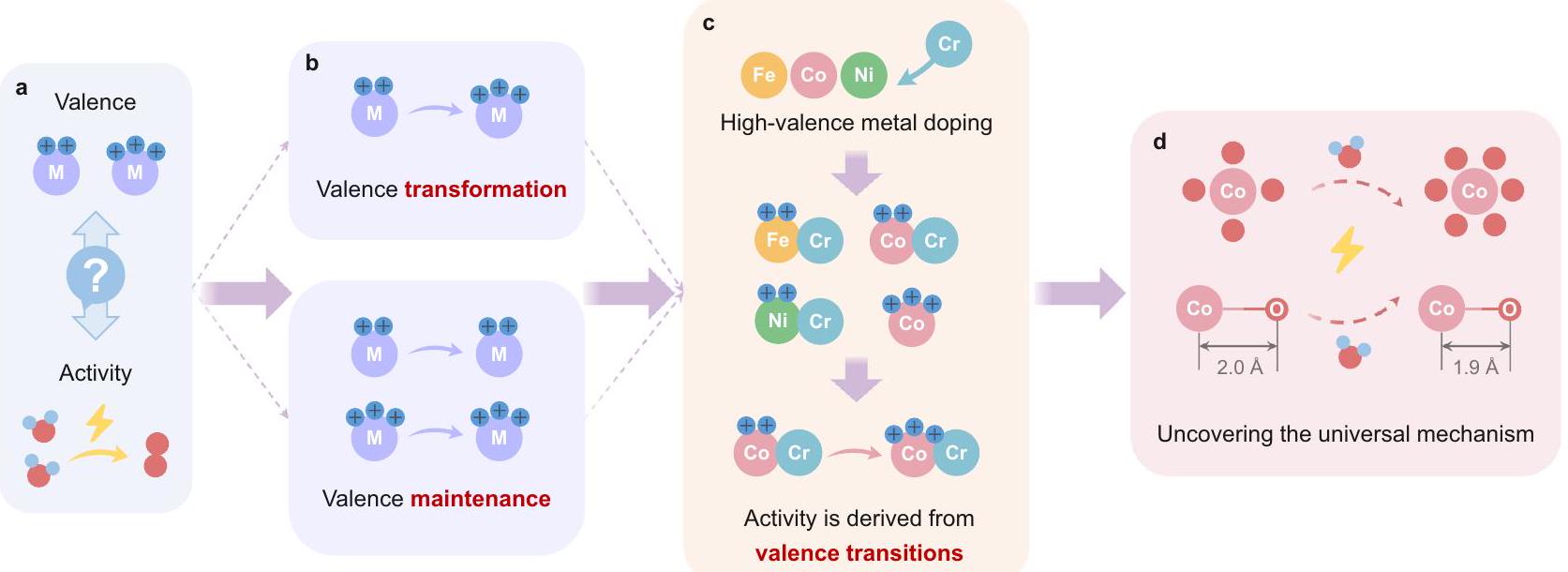
على جانب الأنود من خلايا التحليل الكهربائي للمياه القلوية، تعتبر عملية تطور الأكسجين عملية معقدة تتضمن نقل عدة إلكترونات. مع نقل الإلكترونات، يتغير حالة التكافؤ للمعادن النشطة، والتي يُعتقد أنها مصدر النشاط الجوهري لتطور الأكسجين.. ومن ثم، هناك نقاش مستمر بشأن العلاقة بين حالة التكافؤ ونشاط الموقع النشط في المحفزات القائمة على المعادن الانتقالية (الشكل 1أ). وقد كشفت العديد من الدراسات أن مركبات المعادن الانتقالية ذات مواقع المعادن عالية حالة الأكسدة تظهر نشاطًا كبيرًا في تفاعل تطور الأكسجين، بينما تلك التي تحتوي على مواقع المعادن منخفضة الأكسدة تتمتع بنشاط منخفض نسبيًا.
الشكل 1 | مخطط توضيحي لأصل أفكار البحث والتصميم. أ النقاش الجوهري بين قيمة الموقع النشط والنشاط التحفيزي.أنواع مختلفة من التغيرات في التكافؤ خلال عملية OER. ج سلسلة من الأفكار المدروسة بعناية و تم إجراء تجارب مصممة باستخدام الكروم كعامل تشويب عالي التكافؤ. d كشف التغيرات في التكافؤ والآليات العالمية التي تؤثر على الأداء التحفيزي. نشاط. ومع ذلك، فقد تحدت الأبحاث الحديثة هذه الفكرة، مشيرة إلى أن مواقع المعادن ذات الأكسدة المنخفضة يمكن أن تلعب أيضًا دورًا مهمًا في تحديد نشاط تفاعل تطور الأكسجين (OER) استنادًا إلى هذه النظرية، قدم العلماء عيوبًا غنية بالأكسجين والمعادن عالية التكافؤ المخدرة (مثل و ) لخفض حالة التكافؤ للمعادن النشطة. على الرغم من هذه الجهود، لا تزال العلاقة بين النشاط التحفيزي وتغيرات التكافؤ خلال التفاعل غير واضحة، بسبب نقص تحليل التطور الديناميكي لمواقع المعادن النشطة خلال ظروف OER (الشكل 1b). لا يزال الفهم العميق وتنظيم الهيكل الإلكتروني لمواقع المعادن النشطة في المحفزات، بالإضافة إلى تطبيقها في خلايا AEMWE العملية لفهم الآلية، يمثل تحديًا كبيرًا. توفر تقنيات الإشعاع السنكروتروني في الموقع وسيلة عملية لمعالجة هذه القضية من خلال مراقبة ديناميات حالة التكافؤ وتوضيح الأسباب الرئيسية لتأثيرها على الأداء التحفيزي.
في هذا العمل، اخترنا الكروم منخفض التكلفة والوفير ذو الشحنة التكافؤية العالية كعامل مضاف معدني واستخدمنا طريقة اختزال الطور السائل ذات الخطوة الواحدة لتخليق سلسلة من المحفزات الفعالة من أكاسيد المعادن غير المتبلورة لأجهزة التحليل الكهربائي للمياه القلوية (الشكل 1c). من بينها،قدم المحفز نشاطًا واستقرارًا متميزين. استخدمنا طيف انبعاث الأشعة السينية من القلب إلى التكافؤ (vtc-XES) في النظام الكهروكيميائي للمرة الأولى لتحديد روابط الكوبالت بدقة، و-تعزيز قوة التفاعل لـ Co-O خلال عملية OER. سمحت لنا مجموعة من تجارب مطيافية الامتصاص بالأشعة السينية في الموقع (XAS) والأشعة تحت الحمراء من إشعاع السنكروترون (SR-IR) وتجارب أخرى، بالإضافة إلى الحسابات النظرية، بالتعرف على تطور الهيكل الذري والإلكتروني في موقع Co الذي يحسن من طاقة الامتصاص للوسط الأكسجيني (الشكل 1d)، مما يؤدي إلى انخفاض حاجز طاقة الأكسدة الذي يعد حاسماً للنشاط التحفيزي العالي. من المثير للإعجاب، تم تجميع AEMWE باستخدامككاثود أنودي حقق كثافة تيار عالية منعند 2.1 فولت وأظهرت متانة طويلة الأمد رائعة للعمل لأكثر من 120 ساعة فيمما يوضح المزيد من إمكانياته الكبيرة في تطبيق صناعة التحليل الكهربائي للماء.
النتائج
التركيب والتوصيف الهيكلي
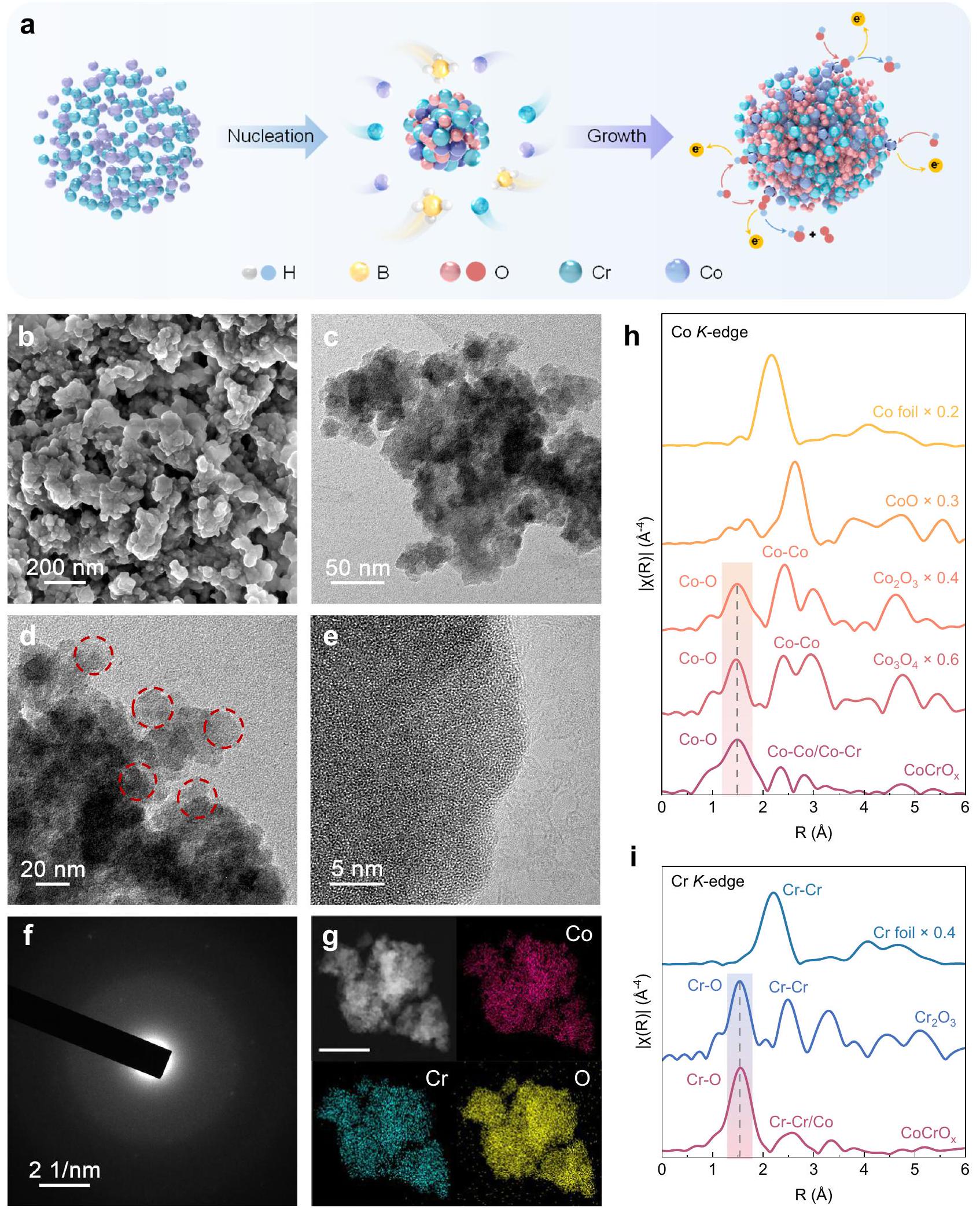
يوضح الشكل 2a مخطط طريقة تصنيع المحفزات. تم تحضير المحفزات بواسطة طريقة اختزال سائلة بسيطة من خطوة واحدة عن طريق خلطمع حل مختلف كلوريدات المعادن تحت التحريك، تليه الغسيل والتجفيف في درجة حرارة الغرفة. وجودسهلت التخفيض المتزامن للمواد الأولية المعدنية خلال مراحل التكوين والنمو.
سمح هذا العملية التخفيضية أيضًا بالتكوين الفوري لشبكات ثلاثية الأبعاد من خلال اندماج نوى المعادن.، مما يتيح التحضير السهل لمحفزات عالية الأداء. تم ملاحظة شكل المحفزات المحضرة باستخدام المجهر الإلكتروني الماسح (SEM)، مما يكشف عن الخصائص المسامية لـ و (الشكل 2ب والشكل التكميلي 1). علاوة على ذلك، تم التحقيق في الميزات الهيكلية بواسطة مجهر الإلكترون الناقل (TEM)، الذي أشار إلى أن المحفزات المحضرة لها بنية ذرية غير مرتبة (الشكل 2ج)، وتمت ملاحظة جزيئات نانوية كروية بمتوسط حجم حوالي 20 نانومتر (الشكل 2د). أظهر مجهر الإلكترون الناقل عالي الدقة (HRTEM) ومجهر الإلكترون الناقل الماسح ذو الحقل المظلم عالي الزاوية المصحح للانحراف (AC HAADF-STEM) بشكل أكبر أنه لا توجد خطوط شبكية واضحة (الشكل 2هـ والشكل التكميلي 2)، مما يدل على طبيعتها غير المتبلورة. أظهر نمط حيود الأشعة السينية (XRD) (الشكل التكميلي 3) قمم حيود واسعة وضعيفة جداً، مما يدل على ميزة الاضطراب على المدى الطويل. تم تأكيد الطابع غير المتبلور بشكل أكبر من خلال حلقات الحيود التي تظهرها حيود الإلكترون في المنطقة المختارة (SAED) (الشكل، مع الحلقات الداخلية اللامعة التي تشير إلى ميزة الترتيب قصير المدىالمورفولوجيا والبنية و وُجد أنها مشابهة لـ (الأشكال التكميلية 4-7). بالإضافة إلى ذلك، أظهر تحليل خرائط طيف الأشعة السينية المشتتة للطاقة (EDS) أن، وكان O موزعًا بشكل موحد في جميع أنحاء الهيكل (الشكل 2g)، مما يدل على تكوين أكاسيد CoCr بدلاً من الهياكل المختلطة المنفصلة عن بعضها. وقد لوحظ نفس الظاهرة لـ و (الشكل التوضيحي 8)، بينما يُعزى ارتفاع محتوى الأكسجين إلى سهولة أكسدة المعادن الانتقالية. تم استخدام مطياف الانبعاث الذري المتصل بلاسما التحريض (ICP-AES) لتحديد النسب العنصرية للمحفزات المُعدة (الجدول التوضيحي 1). أظهرت النتائج أن النسب الذرية لـ، كانت Ni و Cr قريبتين من، مما يدل على التحكم الدقيق في الطريقة الاصطناعية المعتمدة في هذه الدراسة. علاوة على ذلك، تم استخدام طيف الامتصاص بالأشعة السينية الممتد (EXAFS) لتأكيد أن و (الشكل 2h و i والأشكال التكميلية 9-12) كانت المحفزات لها هياكل محلية وبيئات تنسيق مشابهة، مما يشير إلى أن كل منها يتشكل بشكل مشابه من الناحية الهيكليةو نيالأكاسيد، التي تشكل الأساس لمزيد من التحليل.
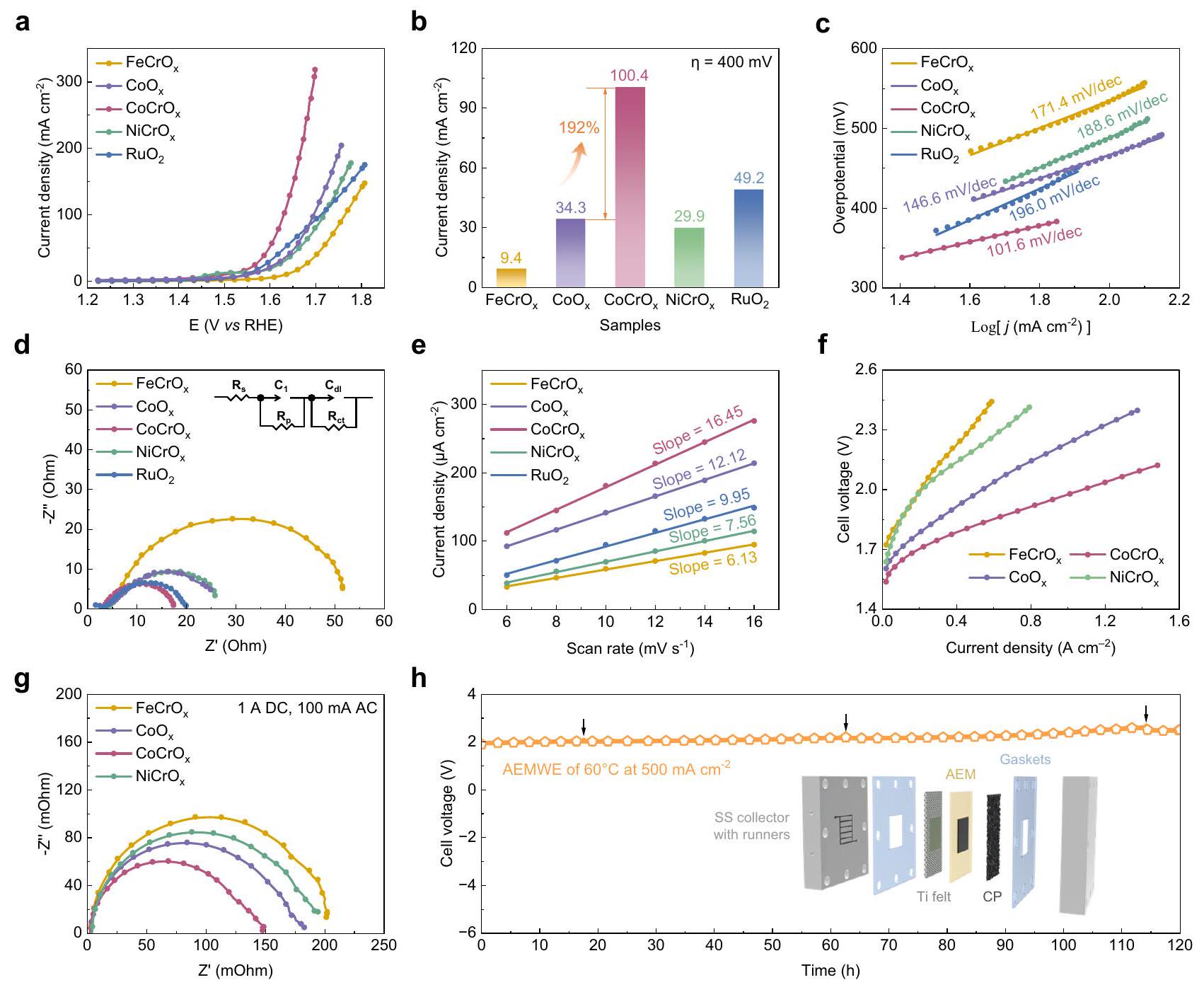
تم إجراء قياسات نشاط OER للمواد الحفازة فيمحلول KOH بتركيز 1 م مشبع باستخدام نظام ثلاثي الأقطاب القياسي. كانت الأداء التحفيزي الأمثل للعناصر
الشكل 2 | التحضير والخصائص الهيكلية والشكلية لـ. توضيح لإجراء التحضير، (ب) SEM، (ج، د) TEM، (هـ) صور HRTEM. (و) أنماط SAED. (ز) خرائط EDS تظهر توزيع Co (وردي)، Cr (أزرق- الأخضر) و O (الأصفر)، على التوالي. مقياس الرسم: 100 نانومتر. طيف EXAFS لـ ( ) شركة -حافة و (أنا)-حافة لـوعينات مرجعية. تحقق عندما تكون النسبة الذرية لـكان 1:1، كما تم تحديده من خلال الفحص القائم على الأداء (الشكل التكميلي 13). من أجل الوضوح، فإن النسبة الذرية لـإلى كر لـ، و العوامل المساعدةما لم يُذكر خلاف ذلك. لتقييم تأثير إضافة الكروم، قمنا بتخليق مادة غير مضافة بالكرومالعامل المساعد وأيضًا شمل المعيارمحفز للمقارنة. كما هو موضح في منحنيات LSV المصححة بواسطة iR (الشكل 3أ)،أظهرت أداءً متفوقًا في OER، مما يتطلب فقط جهدًا زائدًا قدره
الشكل 3 | النشاط الكهروتحفيزي والثبات. أ منحنيات LSV لـ، و المحفزات في 1 م KOH عند معدل المسحكثافة التيار الحالية لمختلف المحفزات المسجلة عند 400 مللي فولت. ج. مخططات تافل. د. مخططات نايكويست المكتسبة عند 1.57 فولت (مقابل RHE) في نصف الخلية. الإدراجات في اللوحات د تظهر الدائرة المعادلة لتفاعل الأكسدة. هـ. الفروق في كثافات التيار المرسومة مقابل معدلات المسح إلى
حدد ECSA.منحنيات I-V لأجهزة AEMWEs المحفزات المعدة كأنودات. g مخططات نايكويست لأجهزة AEMWEs التي تطبق 1 أمبير من التيار المباشر (DC) و100 مللي أمبير من الجذر التربيعي لمتوسط التيار المتردد (AC).منحنى الكرونوپوتنشيومترمحفز عند كثافة تيار ثابتة منفي AEMWEs فيتمثل المواقع المشار إليها بالسهم الأسود استبدال الإلكتروليت.
268 مللي فولت لتحقيق كثافة التيار . كانت الأداء يتفوق على أداء المحفزات الأخرى المعدة والمقارنة محفز، متجاوزًا حتى التجاري. وقد اقترح أن التأثير المفيد لتطعيم الكروم في تعزيز نشاط تفاعل تطهير الأكسجين. حيث تم تثبيت الجهد الزائد عند 400 مللي فولت، كانت كثافة التيار لـتجاوزت بشكل كبير المحفزات الأخرى، حيث كانت 10.7 مرات، و3.4 مرات، ومرتين أعلى من، و على التوالي. ومن المهم أن نقول إن إضافة الكروم زادت أيضًا من كثافة التيار بـبالنسبة إلى (الشكل 3ب). بالإضافة إلى ذلك، من أجل التحقيق بشكل أعمق في النشاط الجوهري للمحفزات، تم الحصول على ميل تافل من منحنيات LSV المقابلة (الشكل 3ج). مع ميل تافل قدره ، الـتفوقت بشكل ملحوظ على المحفزات الأخرى، بما في ذلك المحفزات غير المضافة بالكروممحفز (146.6 مللي فولت ) و ، مما يدل على حركيات OER الأكثر ملاءمة. كشفت طيفيات impedancia الكهروكيميائية (EIS) (الشكل 3d) أنأظهرت مقاومة نقل الشحنة أقلفي منطقة التردد العالي مقارنة بـ ( ) ، ، و ، مما يشير إلى توصيل كهربائي متفوق وعملية نقل شحن أسرع. ويدعم هذا الاتجاه تحليل مقارن للسطح النشط كهربائياً (ECSA) للمواد الحفازة (الشكل 3e والمكمل
تم إجراء الشكل 14) من خلال قياس سعة الطبقة المزدوجة في منطقة الجهد غير الفارادي.لـتم حسابه ليكونأعلى من، و من الجدير بالذكر أن كل من تحليلات EIS والحركية تتماشى مع النشاط الممتاز لـتمت ملاحظته في منحنيات LSV. الاستقرار هو أيضًا معيار رئيسي لتقييم الأداء التحفيزي طويل الأمد للمواد الكهروكيميائية (الشكل التكميلي 15).لم يظهر أي فقدان ملحوظ في النشاط خلال 50 ساعة من التحليل الكهربائي في نصف خلية عند جهد ثابت قدره 1.56 فولتRHE)، حيث تزداد كثافة التيار أولاً ثم تستقر. يمكن أن يُعزى الزيادة في كثافة التيار إلى تكوين المزيد من المادة النشطة واختراق الإلكتروليت تدريجياً في الهيكل المسامي..
من أجل استغلال الإمكانيات الصناعية لـالمحفزات الكهربائية، قمنا بإنشاء نظام إلكتروليزر AEM باستخدام العينات المحضرة كمحفز أنودي ومادة Pt/C التجارية كمحفز كاثودي لتقييم أدائها التحفيزي تحت ظروف صناعية محاكاة. ). الجهد-التيار ( تظهر المنحنيات المميزة في الشكل 3f أنيعرض المحفز أداءً متفوقًا، محققًا كثافة تيار عالية تبلغعند 2.1 فولت. كما قمنا بدراسة تأثير أقطاب الغشاء المختلفة طرق التحضير على أداء المحلل الكهربائي (الشكل التكميلي 16) ورسم المخططات التخطيطية لتجمعات المحلل الكهربائي للعمليات الثلاث (الشكل التكميلي 17). لتقديم توضيح للأداء المتميز لـالعوامل المساعدة في AEMWE، تكشف مخططات نايكويست للخلية الكهروكيميائية التي تم الحصول عليها من خلال EIS عن ثلاثة مقاومات مختلفة في AEMWEs: المقاومة الأومية (OR)، مقاومة نقل الشحنة (CTR) ومقاومة نقل الكتلة (MTR) (الشكل 3g). تمثل التقاطعات في منطقة التردد العالي المقاومة الأومية، بينما تشير أقواس مناطق التردد العالي والمنخفض إلى مقاومة نقل الشحنة المرتبطة بالتفاعلات الكهروكيميائية ومقاومة نقل الكتلة المرتبطة بنقل المتفاعلات والمنتجات، على التوالي. من الواضح أن المقاومة الأومية للعينات متطابقة تقريبًا، معيظهر أدنى معدل نقر (CTR) ومعدل نقل الكتلة (MTR)، مما يشير إلى أن نشاطه المتفوق ناتج عن تسريع نقل الشحنة وتقليل مقاومة نقل الكتلة. توضح الشكل التوضيحي الإضافي 18 أنه تم استخدام قياس الطيف الكهربائي في الحالة التشغيلية (EIS) عند تيارات مختلفة لتحديد مقاومة نقل الشحنة لكل محفز. أظهرت النتائج التي تم الحصول عليها انخفاضًا ملحوظًا في المقاومة مع زيادة التيار، مما يعني نقل أسرع للإلكترونات وكفاءة أعلى في تحويل الطاقة أثناء التحليل الكهربائي عند تيارات مرتفعة. كما قمنا بتقييم استقرار الـالعامل المساعد تحت وظروف درجة حرارة الغرفة عند كثافة تيار عالية من لتقييم إمكانيته للتطبيقات الصناعية بشكل أكبر (الشكل 3h والشكل التكميلي 19). كما هو موضح في الشكل 3h، أظهر المحفز استقرارًا كبيرًا خلال فترات التحليل الكهربائي التي استمرت 120 ساعة، مما يثبت قابليته للتطبيق في العالم الحقيقي. يعرض الرسم البياني المقدم في الشكل 3h تمثيلًا تخطيطيًا لـ AEMWE، والذي يتضمن جامعَين من الفولاذ المقاوم للصدأ (SS) مع عدائين، وحشوات من بولي تترافلوروإيثيلين (PTFE)، وأقمشة من التيتانيوم، وAEM، وورق الكربون (CP). تم تجميع البيانات الكهروكيميائية المختلفة في الجدول التكميلي 2 لمقارنة أكثر وضوحًا بين أداء كل محفز من حيث الكهروكيمياء.
تحليل تطور التكافؤ والبنية المحلية
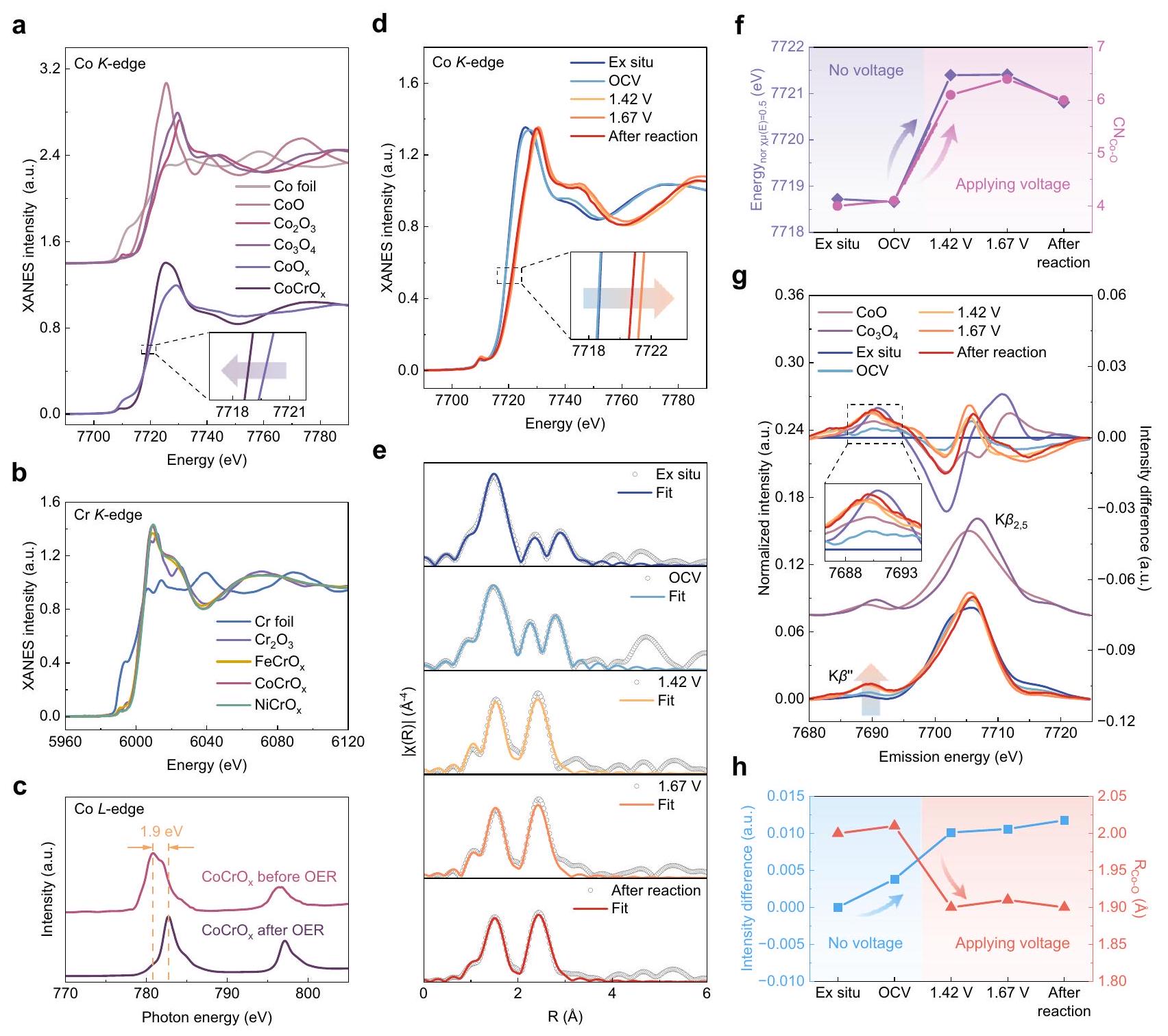
من أجل مقارنة والتحقيق في الأسباب الرئيسية للنشاط الممتاز لـيتم تحديد الحالة الأولية لجهد العناصر النشطة بشكل متزامن. من خلال مطيافية الامتصاص الهيكلي القريب من حافة الأشعة السينية (XANES)، تمكنا من تحديد حالات الجهد للعناصر النشطة بدقة. بالنسبة لعناصر المعادن الانتقالية، يمثل التحول نحو اليمين لحافة الامتصاص القريبة حالة أكسدة مرتفعة. مقارنة موضع حافة الامتصاص مع العينات القياسية تكشف أن يقع بين CoO و (الشكل 4أ). بالإضافة إلى ذلك، فإن الانزياح نحو اليسار لحافة الامتصاص لـبالنسبة إلىتشير إلى أن تشويب الكروم يقلل من متوسط حالة التكافؤ للكوبالت، وهو ما يتوافق مع ذروة الامتصاص ذات التكافؤ المنخفض الأقوى في مطيافية الامتصاص بالأشعة السينية الناعمة (sXAS) (الشكل التكميلي 20). كما أن التعايش بين كل من حالات التكافؤ +2 و +3 لكل عنصر معدني نشط في مطيافية الإلكترون بالأشعة السينية (XPS) يؤكد الاستنتاج أعلاه (الشكل التكميلي 21a-f والجدول التكميلي 3). وبالمثل، تم تثبيت حالات التكافؤ للحديد والنيكل المشوبين بالكروم (الشكل التكميلي 22) بين التكافؤ +2 و +3. يوضح الشكل 4 ب أن-حافة جميع المحفزات تقريبًا تتداخل مع، مشيرًا إلى أن حالة التكافؤ للكروم هي +3، وهو ما يتوافق بشكل كبير مع النتائج في sXAS (الشكل التوضيحي 23) و XPS (الشكل التوضيحي ). إن التناسق بين التكافؤ والبنية يعزز موثوقية التحليل الإضافي. لتوضيح سبب الاختلاف في النشاط التحفيزي الجوهري للمنشطات الثلاثة، قمنا بفحص التغيرات في التكافؤ في العناصر النشطة لـ و قبل وبعد التفاعل باستخدام sXAS (الشكل 4c والشكل التكميلي 24). اكتشفنا تغييرًا ملحوظًا في الكوبالت فقط خلال التفاعل، حيث انزاح ذروة الامتصاص بمقدار 1.9 إلكترون فولت نحو الطاقة الأعلى، مما يمثل زيادة كبيرة في حالة الأكسدة للكوبالت، وهو ما تدعمه أيضًا نتائج XPS (الشكل التكميلي 25). نظرًا لأن طاقة الربط لـأعلى من ذلك لـإن التحول في قمة طاقة الربط نحو الطاقة المنخفضة يؤكد على ارتفاع التكافؤ ولاية. من الواضح أيضًا أن الذروة المميزة للأقمار الصناعية لـيكاد يختفي، مما يثبت أكثر أنتقريبًا يتم أكسدته بالكامل إلى حالة التكافؤ الأعلى خلال عملية OER.
لكشف المزيد عن التطور الديناميكي للبنية الإلكترونية والبيئة المحلية الذرية لمواقع الكوبالت النشطة خلال عملية التحفيز الكهربائي لتفاعل الأكسدة، تم إجراء قياسات XAFS وvtc-XES في الموقع باستخدام خلية كيميائية كهربائية مبنية في المختبر. كما هو موضح في الشكل 4d، فإن الكوبالتتشير طيفيات XANES عند الحافة إلى أن مواقع حافة الامتصاص للكوبيك تحت ظروف خارج الموقع وجهد الدائرة المفتوحة (OCV) متشابهة تقريبًا. عند تطبيق الجهد، تكون حافة الامتصاص القريبة من الحافة يتم تحريكه بشكل إيجابي، مما يعني ارتفاع الحالة الأكسيدية المتوسطة للكوبيليوم خلال عملية OER، وهو ما يتماشى مع نتائج sXAS و XPS. النتائج الملائمة للكوبيليومتشير طيفيات EXAFS عند حافة الطاقة إلى أن عدد التنسيق للطبقة الأولى من الغلافيزداد بشكل كبير من 4 إلى أكثر من 6 عند تطبيق الجهد (الشكل 4e، الشكل التكميلية 26 والجدول التكميلية 4)، وقد يكون الزيادة في عدد التنسيق هي السبب في ارتفاع حالة الفالنس لعنصر الكوبالت.الرسم البياني موضح بشكل بديهي في الشكل.يوضح وجود ارتباط إيجابي كبير بين موضع حافة الامتصاص وعدد تنسيق الكوبالت-أكسجين. بالإضافة إلى ذلك، لاحظنا أن التركيب المحلي للكوبالت تغير خلال التفاعل، مما قد يشير إلى إعادة تكوين ديناميكية.. يعود ذلك إلى الذوبان الكهربائي الجزئي للكروم تحت ظروف قلوية، وهو ما تدعمه تقليل محتوى الكروم الذي كشفت عنه خرائط EDS بعد التفاعل (الشكل التوضيحي 27). تشير نتائج HRTEM (الشكل التوضيحي 28a) إلى أن المحفز حافظ بشكل أساسي على شكله الأصلي، ولكن ظهرت خطوط شبكية قصيرة وفتحات نانوية، وقد توفر هذه الفتحات مساحة سطح محددة أكبر وسهولة الوصول إلى المواقع النشطة، بالإضافة إلى نقل الكتلة السريع، مما من شأنه تعزيز حركية تفاعل الأكسدة. قد يكون الهيكل غير المتبلور قد تعرض لضرر طفيف خلال عملية التحليل الكهربائي، كما يتضح من وجود عدة بقع ساطعة في نمط SAED بعد التفاعل. ومع ذلك، تم الحفاظ على الحالة غير المتبلورة الأصلية إلى حد كبير (الشكل التوضيحي 28b).
على الرغم من أن EXAFS يمكن أن يوفر أعداد التنسيق بالإضافة إلى أطوال روابط المعدن-الليغاند، إلا أنه يفشل في التمييز بين الليغاندات ذات الأرقام الذرية المتشابهة (على سبيل المثال، ) أو حالات بروتونية مختلفة للرابطة . لذلك، تم استخدام تقنية vtc-XES في الموقع (الشكل 4 ج) لتحديد الليغاند وبيئة التنسيق للكوبيك، مما يؤكد نوع الليغاند وتغير قوة الرابطة.ذروة حوالي 7690 إلكترون فولت هي دليل مهم على تفاعل ligand-metal من خلالالتفاعلات، ويُعترف بهذا القمة كقمة مميزة لـتنسيق. مكثفالقمة المميزة تشير إلى أن تفاعل Co-O يقوى مع الجهد خلال عملية OER. هذه الملاحظة تدعم انكماش مسافة رابطة Co-O كما هو موضح في طيف EXAFS المحول فورييه (FT-EXAFS).تم توضيح الأطياف المخصومة vtc-XES في أعلى الشكل 4 g لتسليط الضوء على الفروقات. من الملحوظ أنه بعد تطبيق الجهد، فإن شدة القمة تشبه إلى حد كبير تلك الخاصة بـعينة مرجعية ( ). علاوة على ذلك، تُظهر الشكل 4 ح العلاقة السلبية الموثوقة بين شدة طيف vtc-XES المخصوم و أطوال الروابط، مما يؤكد موثوقية التحليل. يمكن أن توفر روابط Co-O الأقصر طاقة ربط محسّنة لامتصاص وسيط الأكسجين، مما يسهل عملية كيمياء الأكسدة المعجلة..
تحقيق الآلية والمحاكاة
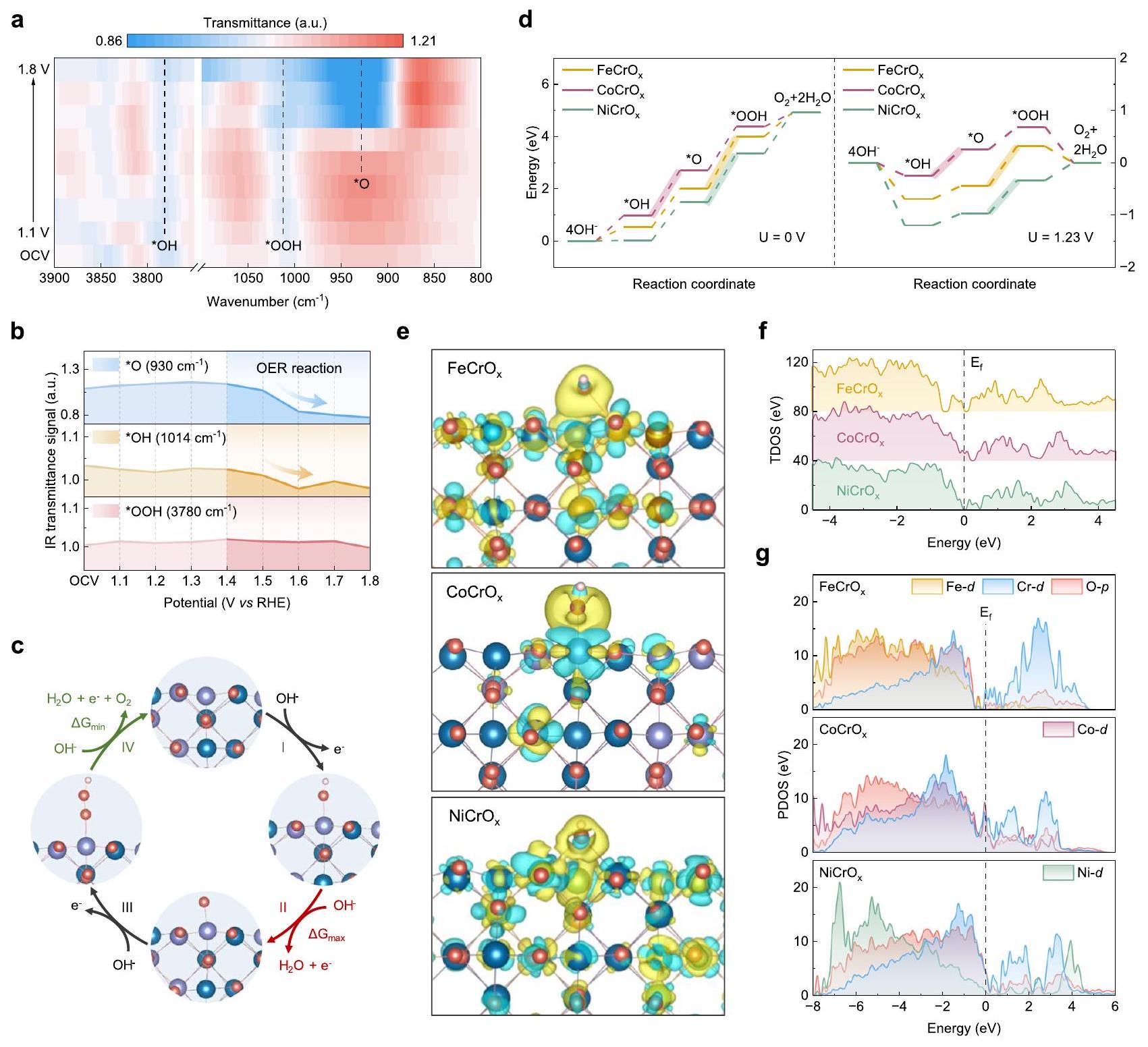
من أجل الحصول على فهم أعمق لسلوكيات الامتزاز وآليات التفاعل لمختلف المحفزات، قمنا بإجراء قياسات SR-IR في الموقع وحسابات نظرية الكثافة الوظيفية (DFT) لـ و كما هو موضح في الشكل 5أ، خريطة كثافة الانعكاس الناتجة عن طيف الأشعة تحت الحمراء السريعةحدد الثلاثة وسائط التفاعلية الحاسمة لتفاعل الأكسدة، وهي *OH ( )، *OOH ( ) و * . من المثير للاهتمام أن قمم امتصاص *O تظهر فقط عند إمكانيات أعلى. عندما
الشكل 4 | حالة التكافؤ، تغيير في خصائص المحفز المسجلة عند فولتية مطبقة مختلفة خلال عملية التحفيز الكهربائي لتفاعل الأكسدة. أ طيف XANES لـ Co-حافةوعينات المرجع.طيف XANES لـ-حافة، وعينات المرجع.طيف sXAS المُعَدلحافةقبل وبعد OER. طيف XANES في الموقع لـ . منحنيات التناسب لـ -طيف FT-EXAFS الموزون بـ-حافة لـ. العلاقة بين طاقة حافة الامتصاص المعيارية و عدد التنسيق خلال عملية OER.طيف VTC-XES في الموقعوعينات المرجع. يتم عرض طيف vtc-XES المخصوم (كل طيف ناقص الطيف الخارجي) في الأعلى لتسليط الضوء على الفروقات.العلاقة بين اختلافات الشدة في طيف vtc-XES في الموقع وأطوال روابط Co-O خلال عملية OER. يصل الجهد إلى منطقة OER ( )، تزداد شدة قمم *OH و *O بشكل ملحوظ. وهذا يشير إلى وجود علاقة إيجابية بين امتصاص الوسطاء وتطبيق الجهد. ومع ذلك، فإن شدة قمة *OOH تضعف بشكل متناقض أو حتى تختفي (الشكل 5b والشكل التكميلي 29b). بالمقابل، لم تُلاحظ أي قمم لامتصاص *O لـ و في جميع الظروف المحتملة، مما يشير إلى أنلديه طاقة امتصاص متوسطة محسّنة (الشكل التكميلي 29a، c). هذه النتيجة تدعم أكثر أن * O يتم إنتاجه وامتصاصه على مواقع الكوبالت خلال التفاعل، مما أدى إلى زيادة في عدد الليغاندات وتغير التكافؤ للكوبالت، مما يسهل بدوره عملية OER.
لتوضيح المنطق وراء هذه الظاهرة، تم افتراض أن آلية OER ذات الأربعة إلكترونات تسير من خلال *OH و *O و (النجوم تشير إلى مواقع الامتزاز) (الشكل 5ج) تم حساب الطاقة الحرة لجيبس وحاجز التفاعل المحدد لكل خطوة أساسية في تفاعل الأكسدة، والتي تعتمد على الطاقة الحرة لـ خطوة تحديد المعدل (RDS) (الشكل 5d). تم اكتشاف أنه بالنسبة لـ، فإن عملية الأكسدة من *OH إلى *O قد تكون تفسيرًا معقولًا لكون *O يتم توليده فقط عند إمكانيات أعلى. بينما يتم نزع الهيدروجين من *OOH لتشكيليعرض أدنى حاجز طاقة تفاعل مقارنة بالخطوات الثلاث الأخرى. ونتيجة لذلك، يتم استهلاك *OOH الوسيطة بسرعة عند الجهود العالية، مما قد يفسر الاختفاء القريب لقمة *OOH. من المهم أن نذكر أنأظهر أقل قيمة لـ RDS بين ثلاثة محفزات، حيث بلغت فقط 0.50 إلكترون فولت عند. إن RDSs لأكسدة *O إلى *OOH في و كانت 0.76 إلكترون فولت و0.63 إلكترون فولت، على التوالي. علاوة على ذلك، تكشف كثافات الشحن التفاضلية في الشكل 5e أن نقل الشحنة لـإلى * OH أقل بكثير والتفاعلات أضعف مقارنة بـ و . هذا يتوافق مع الامتصاص الأضعف نسبيًا لمتوسطات *OH في الـمخطط الطاقة الحرة. يسمح الامتصاص المحسن لـ *OH بحدوث الخطوات اللاحقة عند إمكانيات أقل، مما يسهل حركية تفاعل الأكسدة.. في هذه الأثناء، الكثافة الإجمالية لـ
الشكل 5 | مراقبة الوسائط التفاعلية وحسابات DFT. أ قياسات SRIR في الموقع في نطاق و تحت إمكانيات مختلفة لـخلال عملية OER.إشارات نفاذية الأشعة تحت الحمراء مقابل إمكانيات *O و *OH و *OOH الوسيطة. ج الآلية المقترحة لتفاعل الأكسدة في القلويات.ألوان النموذج هي Co (أرجواني)، Cr (أزرق)، O (أحمر) وH مخططات الطاقة الحرة OER عند 0 فولت (يسار) و1.23 فولت (يمين) لـ، و . كثافات الشحن التفاضلية (تشير المناطق الزرقاء والصفراء إلى نقص الإلكترونات وتراكمها، على التوالي). ألوان النموذج هي Fe (بني-أصفر)، Co (أرجواني)، Ni (أخضر)، Cr (أزرق)، O (أحمر) و H (وردي فاتح). f TDOS و (g) PDOS من و . حسابات حالات (TDOS) تظهر أنيمتلك كثافة حالات أكبر عند مستوى طاقة فيرمي، مما يشير إلى تحسين الموصلية ويشرح النشاط الاستثنائي لـ (الشكل 5f). توضح كثافة الحالة المتوقعة (PDOS) أن – و تساهم الإلكترونات المدارية بشكل رئيسي في نطاق التكافؤ، بينما يتكون نطاق التوصيل بشكل أساسي-الإلكترونات المدارية (الشكل 5 ج). مزيد من التحليل لـمدارات المعدن النشط تكشف أن و الإسقاطات المدارية لـ، وNi تساهم بشكل رئيسي تحت مستوى فيرمي ولها تفاعلات مدارية قوية مع المدارات، مما يدل على أنلديه قدرة ربط مستقرة مع ذرات الأكسجين، مما يسهل تعزيز امتصاص ذرات الأكسجين النشطة وحركية تفاعل الأكسدة (الأشكال التكميلية 30، 31). بالإضافة إلى ذلك، فإن كثافة الإلكترون بالقرب من مستوى طاقة فيرمي للعناصر النشطة تساهم بشكل أساسي فيالمدارات. ومع ذلك، فإن وفرةالإلكترونات القريبة من مستوى فيرمي، بدلاً من أن تكون بعيدة عنه بشكل كبير، تسهل نقل الشحنة من المعادن الانتقالية إلى المواد الماصة المحتوية على الأكسجين خلال عملية OER. هذا واضح بشكل خاص في حالة ، وهو ما يزيد بشكل كبير عن و . هذا يعزز أكثر سبب نشاطه الاستثنائي.
نقاش
لتلخيص، قمنا بتفصيل وتجميع سلسلة من المحفزات المعدنية غير المتبلورة المخدومة بالكروم عالية التكافؤ باستخدام طريقة اختزال سائلة بسيطة من خطوة واحدة.حقق المحفز كثافة تيار عالية منعند 2.1 فولت وعمل عند كثافة تيارلأكثر من 120 ساعة في AEMWE. تشير القياسات في الموقع باستخدام السنكروترون والحسابات النظرية إلى أن تحسين امتصاص *O، وقصر أطوال روابط Co-O، وزيادة أعداد التنسيق أدت إلى تغييرات في حالة الفلز Co وزيادة نشاط OER. وبالتالي، فإن انخفاض حاجز طاقة الأكسدة للفلز النشط هو الهدف النهائي لتعديل الهيكل الإلكتروني، وهو المفتاح. عامل للنشاط الأكبر للمعادن الانتقالية القابلة للأكسدة بسهولة. هذه النتائج توسع الطريق لتعزيز نشاط التحفيز لتفاعل الأكسدة الكهربائية للأكسجين وأداء خلايا الماء القلوية، بينما تشير تطبيقات الطريقة الجديدة في الموقع إلى العديد من الاحتمالات لمعالجة المشكلات ذات الصلة في مجال تفاعلات التحفيز الطاقي.
طرق
المواد الكيميائية
كلوريد الحديد سداسي الماء ) ، كلوريد النيكل سداسي الماء ( ) ، كلوريد الكوبالت سداسي الهيدرات ( كلوريد الكروم سداسي الهيدرات ( ) ، بوروهيدريد الصوديوم ( ) والإيثانول تم شراؤهما من شركة سينوفارم للمواد الكيميائية. هيدروكسيد البوتاسيوم (KOH، درجة إلكترونية، 99.999% أساس المعادن، باستثناء الصوديوم) تم شراؤه من شركة شنغهاي ألالدين للتكنولوجيا الحيوية. ثنائي أكسيد الروثينيوم (99.9% أساس المعادن النادرة) ونافيوونتم شراء المحلول (5%) من سيغما-ألدريش. الكربون الأسود (ECP600JD، كيتجن بلاكتم الحصول على ) لقياسات الكهروكيميائية من شركة سوزو سينيرو تكنولوجي المحدودة، وتم الحصول على مسحوق الجرافيت (XF009 7782-42-5) لتوصيف XAS من شركة XFNANO لمواد التكنولوجيا المحدودة. يمكن استخدام جميع المواد الكيميائية دون الحاجة إلى تنقية إضافية.
تركيب المحفزات
قم بإذابة 11.90 جرام من و 13.32 جرام من في 25 مل من الماء المقطر (DI) لصنع محلول كلوريد المعدن بتركيز 2 م ولتتركه جانبًا. خذ 2 مل من المحلول المحضر أعلاه و المحاليل على التوالي وخلطها بشكل متساوٍ. حضر محلولًا جديدًا من بوروهيدريد الصوديوم عن طريق إذابة 3 جرام منفي 20 مل من الماء المقطر، ثم أضف بالتنقيط إلى خليط كلوريدات أيونات المعادن المذكورة أعلاه، حرك ميكانيكياً لمدة 15 دقيقة، اتركه ليبرد، ثم اتركه طوال الليل وجففه بالتجميد. بعد التجفيف بالتجميد، تم غسل المحفز عدة مرات باستخدام الماء المقطر والإيثانول، وجفف تحت الفراغ للحصول علىمحفز و تم الحصول على المحفزات بنفس الإجراء، باستثناء أنه تم استبدال محلول كلوريد المعدن بآخر.
القياسات الكهروكيميائية
تم إجراء جميع الاختبارات الكهروكيميائية في نظام ثلاثي الأقطاب نموذجي على محطة عمل كهروكيميائية (CHI760E، تشين هوا).تم استخدام قطب مرجعي (1 م KOH، تجيدة) كقطب مرجعي، وقطب من رقائق البلاتين ( ) أو قضيب الجرافيت كالكاثود وتم طلاء المحفز على ورق الكربون (HESEN, HCPO20N) لصنع قطب كهربائي عامل. 6 ملغ من المحفز، 3 ملغ من الكربون الأسود الموصل (ECP600JD، كيتجن بلاكتم خلطه بشكل متجانس معمن ماء DI، من الإيثانول و مننافيوون وتم صوته لجعل الحبر متجانساً.من ماء DI، من الإيثانول و منتم خلط نافيون لصنعمحلول نافيون. ثمتم إسقاط حبر المحفز على ورق الكربون )، مجفف ومنتم إضافة نافيون قطرة قطرة وتجفيفه في درجة حرارة الغرفة واستخدامه للقياسات الكهروكيميائية.
تم إجراء الفولتمترية الماسحة الخطية (LSV) في محلول 1 م كوه مشبع بالأكسجين عند درجة حرارة الغرفة. ) باستخدام معدل مسح مقاومة المحلولتم تحديده من ملاءمة مخطط نايكويست الناتج واستخدم لتصحيح السقوط الأومي بناءً علىأين هو الجهد التصحيحي و هو الجهد المقاس، على التوالي). تم قياس طيف EIS على مدى مجموعة من مع سعة 0.005 فولت. تم ضبط الجهد عند 1.57 فولت (مقابل RHE) وتمت معالجة بيانات EIS بواسطة برنامج ZView. في مخطط الدائرة المعادلة (في الجزء العلوي من الشكل 3d)،يمثل مقاومة الحل، و هي المقاومات لنقل الشحنة، و و تُستخدم لوصف سعة الطبقة المزدوجة. تم الحصول على المساحة السطحية النشطة كهربائياً (ECSA) من خلال قياس سعة الطبقة المزدوجة في منطقة الجهد غير الفارادي مع نطاق قياس الجهد من 0.875 إلى 0.975 فولت.RHE).
تصنيع MEAs وتقييم AEMWEs
تمت عملية تصنيع MEAs وتقييم AEMWEs في شركة زونغك إنثالبي (أنhui) لتكنولوجيا الطاقة الجديدة المحدودة. بالنسبة للأنود، تم تحضير المحفزات ( و تم خلط المحفزات مع الأيونومر (PiperION-A5-HCO3) في محلول مائي (1:3 ماء وإيزوبروبانول). بالنسبة للقطب السالب، (تانكا، تم خلط ) مع الأيونومر في محلول مائي (1:5 ماء وإيزوبروبانول). تم رش حبر المحفز على AEM (PiperION-A60-HCO3) أو شعيرات مغطاة بالبلاتين والتيتانيوم (Sti0.25Pt0.5) لصنع أقطاب انتشار الغاز (GDEs)، مع تحميل معدني تقريبي منللأنود وللكاثود. تم غمر AEM في 0.5 M KOH لمدة ساعة، ثم تم استبداله بمحلول KOH جديد وغمره لمدة ساعة أخرى. ثم تم شطف الأنيون البيكربونات بالماء المقطر لتحويل الأنيون البيكربونات إلى الأنيون الهيدروكسيد. تم تحميل MEA في جهاز تثبيت وتم تثبيته بعزم دورانتم اختبار الخلية بواسطة جهاز قياس الجهد الثابت مع إلكتروليت متدفق بتركيز 1 مولي من هيدروكسيد البوتاسيوم، وتم اختبار خلايا AEMWEs لقياس المقاومة الداخلية باستخدام مقياس المقاومة الداخلية.لـلـلـ و لـ ). تم حساب الفقد الأومى بناءً على تصحيح لتعويض المقاومة الداخلية. تم إجراء قياسات EIS لأجهزة AEMWEs بواسطة محلل impedancia (DH7007، DongHua Analytical) على مدى amplitودات ، وتيارات (التيار المتردد RMS المطبق هو من DC المقابل ومساحة MEA الفعالة هي). تم إجراء اختبارات الأداء واختبارات EIS عند درجة حرارة، بينما تم إجراء اختبارات المتانة عند كل منودرجة حرارة الغرفة.
توصيف المواد
تم الحصول على أنماط XRD باستخدام جهاز قياس الحيود Philips X’Pert ProSuper معإشعاع (). تم فحص الشكل بواسطة SEM باستخدام ZEISS GeminiSEM 500. تم إجراء صور TEM وHRTEM على مجهر إلكتروني ميداني JEM-2100F بجهد تسريع قدره 200 kV. تم إجراء HAADF-STEM على جهاز JEOL JEM-ARF200F HRTEM مع مصحح انحراف كروي عند جهد 200 kV. تم الحصول على خرائط العناصر EDS على جهاز JEOL JEM-F200. تم إذابة جميع المحفزات للتخليق في ماء الملكية وتم تحديد محتوياتوCr في المحفزات المعدة مسبقًا بواسطة ICP-AES على جهاز PerkinElmer Optima 7300 DV ICP-AES. تم تسجيل XPS على جهاز Thermo ESCALAB 250Xi مع مصدر إثارة من Al Ka () وطاقة مرور قدرها 30 eV. تم معايرة قيم طاقات الربط مع قمة C 1 s للكربون الملوث عند 284.80 eV.
قياسات X-ray الناعمة XANES
تم إجراء قياسات XANES عند حافةوCr في محطة الإشعاع الضوئي في BL12B من مختبر الإشعاع السنكروتروني الوطني (NSRL) في الصين. تم توصيل مغناطيس انحناء بخط الشعاع، الذي كان مزودًا بثلاثة شبكات تغطي طاقات الفوتون من 100 إلى 1000 eV بدقة طاقة قدرها. تم تسجيل البيانات في وضع العائد الكلي للإلكترونات عن طريق جمع تيار تصريف العينة. كانت قوة الحل لمصفوفة الشبكة عادةً، وكان تدفق الفوتونفوتون في الثانية.
قياسات XAFS
تم قياس طيف XAFS في خط الشعاع BL14W1 من منشأة الإشعاع السنكروتروني في شنغهاي (SSRF) وخط الشعاع 1W1B من منشأة الإشعاع السنكروتروني في بكين (BSRF) في الصين. تم تشغيل حلقة تخزين SSRF عند 3.5 GeV وBSRF عند 2.5 GeV، وكلاهما بحد أقصى لتيار الإلكترون قدره 250 mA. تم خلط حوالي 64 ملغ من العينة المناسبة بشكل متجانس مع الجرافيت وضغطها في كريات دائرية بقطر 8 مم. تم تسجيل طيف
لحافةول
جميع العينات في وضع النقل، وتمت معايرة موضع حافة الامتصاص ( ) بواسطة ورقة العنصر المقابلة، على التوالي.تم قياس طيف XAFS في الموقع في خط الشعاع BL14W1 من SSRF، الصين. تم إجراء قياسات XAFS في الموقع باستخدام ورق كربون مغطى بالمحفز باستخدام خلية كهربائية مصنوعة في المنزل، وتم جمع طيفها في وضع الفلورية. تم توزيع مسحوق المحفز بشكل متجانس في الماء والإيثانول (
) لتشكيل حبر، الذي تم إسقاطه على ورق الكربون كقطب عمل. للحصول على معلومات تطور مواقع Co تحت ظروف عمل OER، تم تحليل طيف XAFS لسلسلة من الظروف التمثيلية (ex situ، OCV، 1.42 V مقابل RHE و1.67 V مقابل RHE، بعد التفاعل).
تحليل بيانات XAFS
تم معالجة بيانات EXAFS المكتسبة وفقًا للإجراءات القياسية باستخدام وحدة ATHENA المدمجة في حزمة برامج IFEFFIT
. تم تحويل بيانات-weightedفي-space إلى الفضاء الحقيقي (R) باستخدام نوافذ هانينغ (Åفي R-space باستخدام وحدة ARTEMIS من IFEFFIT. تم حساب سعة الانعكاس الفعالةوانزياح الطورلكل مسار ملاءمة باستخدام كود ab initio FEFF8.0. بالنسبة للعينات في الموقع المختلفة، تم استخدام نطاق k منÅمنÅمنÅمنÅالمختار لـ-weightedلوظيفة ملاءمة المنحنى، يتم إعطاء عدد النقاط المستقلة بواسطةأما بالنسبة للعينات ex-situ وOCV:
بالنسبة لـ
وبعد العينات التفاعلية:أثناء ملاءمة المنحنى، تم تثبيت عامل تقليل الكفاءة السعوية
عند قيمة 0.71 لعينات Co التي تم تحديدها عن طريق ملاءمة بيانات ورق Co، مع عدد تنسيق Co-Co مضبوط على 12.بالنسبة للعينات ex-situ وOCV، أظهرت منحنيات FT ثلاثة قمم بالقرب من
ÅÅوالتنسيق، على التوالي. بعد ذلك، تم استخدام نموذج هيكلي ثلاثي القذائف يتضمنوومسار تشتت لملاءمة بيانات EXAFS للعينات في الموقع. في كلا العينتين، تم تعيين عوامل Debye-Waller () لتكون متساوية، وتم تعيينلأول طبقة قذيفة () من عينة OCV كمعلمة حرة. في الوقت نفسه، تم تعيينللعينة ex-situ إلى أفضل قيمة ملاءمة لعينة OCV. تم تعيين أعداد التنسيق (CN) والمسافات بين الذراتوانزياحات الطاقة () كمعلمات حرة لكلتا العينتين.لذلك، فإن عدد المعلمات القابلة للتعديل لعينة ex-situ هو
بالنسبة لعينة OCV، فإن عدد المعلمات القابلة للتعديل هو
بالنسبة لـ
وبعد العينات التفاعلية، أظهرت منحنيات FT قمتين بالقرب منÅÅوالتنسيق، والتي تم ملاءمتها باستخدام نموذج هيكلي ثنائي القذائف يتضمن واحدًاوواحدًامسار تشتت. بالنظر إلى هذه العينات الثلاث، يجب تعيينلتكون متسقة ووتم تعيينها كمعلمات حرة.لذا، فإن عدد المعلمات القابلة للتعديل للعينات الثلاث هو
كانت جميع عوامل R الناتجة لجميع العينات لا تزيد عن 0.020، مما يشير إلى النمذجة المناسبة، وإعدادات المعلمات المعقولة وبالتالي، جودة الملاءمة الجيدة.
قياسات Vtc-XES
تم قياس قياسات Co vtc-XES في خط الشعاع BL20U (E-line) من SSRF، الصين. تم تعديل الأشعة السينية الساقطة إلى 7706 eV بواسطة مقياس الطيف Si (111) مزدوج البلورة. تم استخدام خمسة بلورات كروية Si (440) وكاشف عد الفوتونات Pilatus3 لجمع وتحليل فلورية Co
وبشكل أكثر كفاءة. لتقليل امتصاص الفلورية بواسطة الهواء، تم وضع كيس غاز الهيليوم بين العينة والبلورات الكروية والكاشف. كانت الإعدادات في الموقع مماثلة لقياسات XAFS في الموقع. تم تصحيح طيف vtcXES باستخدام خط مستقيم وتم تطبيعه على المساحة الكلية بين 7624.6 و7680 eV، ثم تم تنعيم البيانات باستخدام طريقة Savitzky-Golay.
قياسات SR-IR في الموقع
تم إجراء قياسات SR-IR في الموقع في خط الشعاع تحت الحمراء BL01B من NSRL باستخدام إعداد خلية انعكاسية محلية الصنع مع بلورة ZnSe كنافذة نقل تحت حمراء. تم ضغط قطب المحفز بإحكام ضد نافذة بلورة ZnSe بفجوة ميكرونية لتقليل فقد الضوء تحت الأحمر. لضمان طيف SR-IR عالي الجودة، استخدم الجهاز وضع الانعكاس مع سقوط عمودي للضوء تحت الأحمر. تم الحصول على الطيف تحت الأحمر عن طريق متوسط 128 مسح بدقة
. قبل كل قياس OER نظامي، تم الحصول على الطيف الخلفي لقطب المحفز عند جهد دائرة مفتوحة. تم قياس نطاقات جهد OER بينمقابل RHE بفاصل 0.1 V. تمت معالجة بيانات الأشعة تحت الحمراء وتنعيمها باستخدام برنامج OPUS.
حسابات DFT
تم إجراء الحسابات ضمن إطار نظرية الوظيفة الكثافة (DFT) المدمجة في حزمة محاكاة فيينا ab initio (VASP)
. تم وصف التفاعل بين الأيونات والإلكترونات باستخدام طريقة الموجة المعززة بالمشاريع (PAW). تم وصف طاقة تبادل الإلكترون والتفاعل باستخدام الوظيفة المعتمدة على التقريب العام للتدرج Perdew-Burke-Erzenhorf (PBE). تم إجراء تصحيحات الانحراف شبه التجريبية من Grimme وزملائه (DFT-D3) لحساب التفاعلات بين الممتصات والألواح. تم تطبيق تصحيح Hubbard-U لوصف أفضل للإلكترونات d المحلية لـوNi. اخترنا قيمة فعالةقدرها 3.0 eV لـلذرات Co و5.5 eV لذرات Ni. تم بناء نماذج هيكلية سطحية لـ(110)،و(110) و(110) تم بناء منطقة فراغ كبيرة بما فيه الكفاية منتم استخدامه لجميع النماذج لضمان فصل الصور الدورية بشكل جيد. تم إجراء تكاملات منطقة بريل باستخدام شبكات نقاط خاصة من نوع مونكهورست-باك. تمركزت النقطة عند نقطة غاما (تم استخدام شبكة نقاط k لنظام السوبرسيل. للحصول على الهيكل الدقيق، تم ضبط طاقة قطع الموجة المسطحة على 500 إلكترون فولت.
تم تحديد تقارب القوة ليكون، وتم تحديد تقارب الطاقة الكلي ليكونتم حساب الطاقة الحرة للحالة الممتصة على النحو التالي استنادًا إلى طاقة الامتزاز:
أين هو طاقة الامتزاز المستخرجة مباشرة من حسابات DFT، هي طاقة النقطة الصفرية، هو طاقة تصحيح سعة الحرارة، و T هي درجة الحرارة ( ) ، هو التغير في الإنتروبيا. هنا، يتم تصحيح طاقة غيبس باستخدام كود VASPKIT .
توفر البيانات
البيانات المصدرية التي تستند إليها الأشكال 2-5، والأشكال التكميلية 3، 9-16، 18-26، 29-31، وحسابات الهيكل الإلكتروني التي تم إنشاؤها في هذه الدراسة متاحة كملف بيانات مصدرية. يتم تقديم البيانات المصدرية مع هذه الورقة.
References
Abbasi, R. et al. A roadmap to low-cost hydrogen with hydroxide exchange membrane electrolyzers. Adv. Mater 31, 1805876 (2019).
Xu, D. et al. Earth-abundant oxygen electrocatalysts for alkaline anion-exchange-membrane water electrolysis: effects of catalyst conductivity and comparison with performance in three-electrode cells. ACS Catal 9, 7-15 (2018).
Li, D. et al. Highly quaternized polystyrene ionomers for high performance anion exchange membrane water electrolysers. Nat. Energy 5, 378-385 (2020).
Seh, Z. W. et al. Combining theory and experiment in electrocatalysis: insights into materials design. Science 355, eaad4998 (2017).
Li, Q. et al. Anion exchange membrane water electrolysis: the future of green hydrogen. J. Phys. Chem. C 127, 7901-7912 (2023).
Cousins, I. T. et al. The concept of essential use for determining when uses of PFASs can be phased out. Environ. Sci. Processes Impacts 21, 1803-1815 (2019).
Santoro, C. et al. What is next in anion-exchange membrane water electrolyzers? Bottlenecks, benefits, and future. ChemSusChem 15, e202200027 (2022).
Li, C. & Baek, J.-B. The promise of hydrogen production from alkaline anion exchange membrane electrolyzers. Nano Energy 87, 106162 (2021).
. et al. Carbon-based bifunctional electrocatalysts for oxygen reduction and oxygen evolution reactions: optimization strategies and mechanistic analysis. J. Energy Chem. 71, 234-265 (2022).
Yao, N. et al. Intermolecular energy gap-induced formation of highvalent cobalt species in CoOOH surface layer on cobalt sulfides for efficient water oxidation. Angew Chem. Int. Ed. 61, e202117178 (2022).
Zheng, X. et al. Theory-driven design of high-valence metal sites for water oxidation confirmed using in situ soft X-ray absorption. Nat. Chem. 10, 149-154 (2018).
Zhang, B. et al. High-valence metals improve oxygen evolution reaction performance by modulating 3d metal oxidation cycle energetics. Nat. Catal 3, 985-992 (2020).
Li, N. et al. Influence of iron doping on tetravalent nickel content in catalytic oxygen evolving films. Proc. Natl Acad. Sci. USA 114, 1486-1491 (2017).
Xiao, Z. et al. Operando identification of the dynamic behavior of oxygen vacancy-rich for oxygen evolution reaction. J. Am. Chem. Soc. 142, 12087-12095 (2020).
Novák, M. et al. Primary oxide minerals in the system and their breakdown products from the pegmatite No. 3 at Dolni Bory-Hatě, Czech Republic. Eur. J. Mineral 20, 487-499 (2008).
Kuepper, K. et al. Electronic and magnetic properties of highly ordered . Phys. Status Solidi A 201, 3252-3256 (2004).
Wang, M. H. et al. Operando high-valence Cr-modified NiFe hydroxides for water oxidation. Small 18, e2200303 (2022).
Krishna, K. S., Sandeep, C. S. S., Philip, R. & Eswaramoorthy, M. Mixing does the magic: a rapid synthesis of high surface area noble metal nanosponges showing broadband nonlinear optical response. ACS Nano 4, 2681-2688 (2010).
Kim, J. J., Choi, Y., Suresh, S. & Argon, A. S. Nanocrystallization during nanoindentation of a bulk amorphous metal alloy at room temperature. Science 295, 654-657 (2002).
Wu, G. et al. A general synthesis approach for amorphous noble metal nanosheets. Nat. Commun. 10, 4855 (2019).
Cai, W. et al. Amorphous versus crystalline in water oxidation catalysis: a case study of NiFe alloy. Nano Lett. 20, 4278-4285 (2020).
Liu, J., Yang, Y., Ni, B., Li, H. & Wang, X. Fullerene-like Nickel Oxysulfide hollow nanospheres as bifunctional electrocatalysts for water splitting. Small 13, 1602637 (2017).
Zhang, R. et al. Synthesis and conductivity properties of double perovskite by sol-gel combustion. J. Mater Sci: Mater Electron. 26, 9941-9948 (2015).
Qin, C. et al. Increasing cerium dispersion favours lattice oxygen activity of cobalt oxides for CO catalytic combustion. Combust Flame 260, 113219 (2024).
Wang, J. et al. Redirecting dynamic surface restructuring of a layered transition metal oxide catalyst for superior water oxidation. Nat. Catal 4, 212-222 (2021).
Zhou, J. et al. Electrochemically accessing ultrathin Co (oxy)hydroxide nanosheets and operando identifying their active phase for the oxygen evolution reaction. Energy Environ. Sci. 12, 739-746 (2019).
Lv, L. et al. Coordinating the edge defects of bismuth with sulfur for enhanced electroreduction to formate. Angew Chem. Int. Ed. 62, e202303117 (2023).
Zhu, J. et al. Surface passivation for highly active, selective, stable, and scalable electroreduction. Nat. Commun. 14, 4670 (2023).
Xu, D. et al. The role of Cr doping in Ni Fe oxide/(oxy)hydroxide electrocatalysts for oxygen evolution. Electrochim Acta 265, 10-18 (2018).
Bo, X., Li, Y., Hocking, R. K. & Zhao, C. NiFeCr hydroxide holey nanosheet as advanced electrocatalyst for water oxidation. ACS Appl. Mater Interfaces 9, 41239-41245 (2017).
Gallo, E. & Glatzel, P. Valence to core X-ray emission spectroscopy. Adv. Mater 26, 7730-7746 (2014).
Pollock, C. J. & DeBeer, S. Valence-to-Core X-ray emission spectroscopy: a sensitive probe of the nature of a bound ligand. J. Am. Chem. Soc. 133, 5594-5601 (2011).
Kuhn, T. J., Hormes, J., Matoussevitch, N., Bonnemann, H. & Glatzel, P. Site-selective high-resolution X-ray absorption spectroscopy and high-resolution X-ray emission spectroscopy of cobalt nanoparticles. Inorg Chem. 53, 8367-8375 (2014).
Pollock, C. J. & DeBeer, S. Insights into the geometric and electronic structure of transition metal centers from valence-to-core X-ray emission spectroscopy. Acc. Chem. Res. 48, 2967-2975 (2015).
Zhang, S. et al. Spontaneous delithiation under operando condition triggers formation of an amorphous active layer in spinel cobalt oxides electrocatalyst toward oxygen evolution. ACS Catal 9, 7389-7397 (2019).
Haase, F. T. et al. Size effects and active state formation of cobalt oxide nanoparticles during the oxygen evolution reaction. Nat. Energy 7, 765-773 (2022).
Su, H. et al. Dynamic evolution of solid-liquid electrochemical interfaces over single-atom active sites. J. Am. Chem. Soc. 142, 12306-12313 (2020).
Su, H. et al. In-situ spectroscopic observation of dynamic-coupling oxygen on atomically dispersed iridium electrocatalyst for acidic water oxidation. Nat. Commun. 12, 6118 (2021).
Lin, C. et al. In-situ reconstructed Ru atom array on with enhanced performance for acidic water oxidation. Nat. Catal 4, 1012-1023 (2021).
Zhou, Z. et al. Cation-vacancy-enriched Nickel phosphide for efficient electrosynthesis of hydrogen peroxides. Adv. Mater 34, 2106541 (2022).
Ullman, A. M., Brodsky, C. N., Li, N., Zheng, S. L. & Nocera, D. G. Probing edge site reactivity of oxidic cobalt water oxidation catalysts. J. Am. Chem. Soc. 138, 4229-4236 (2016).
Zhang, L. et al. Fe-doped and sulfur-enriched nanowires with enhanced reaction kinetics for boosting water oxidation. Green Chem. Eng. 3, 367-373 (2022).
Zhao, C. et al. D-Orbital manipulated Ru nanoclusters for highefficiency overall water splitting at industrial-level current densities. Adv. Funct. Mater 34, 2307917 (2024).
Newville, M. IFEFFIT: interactive XAFS analysis and FEFF fitting. J. Synchrotron. Radiat. 8, 322-324 (2001).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron. Radiat. 12, 537-541 (2005).
Ankudinov, A. L., Ravel, B., Rehr, J. J. & Conradson, S. D. Real-space multiple-scattering calculation and interpretation of x-rayabsorption near-edge structure. Phys. Rev. B 58, 7565-7576 (1998).
Kresse, G. av JF. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Phys. Rev. B 54, 11169-11186 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953-17979 (1994).
Perdew, John P., Burke, Kieron & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865-3868 (1996).
Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 21, 1787-1799 (2006).
Conesa, J. C. Electronic structure of the (Undoped and Fe-Doped) evolution electrocatalyst. J. Phys. Chem. C 120, 18999-19010 (2016).
Tkalych, A. J., Zhuang, H. L. & Carter, E. A. A density functional + U assessment of oxygen evolution reaction mechanisms on – NiOOH . ACS Catal 7, 5329-5339 (2017).
Wang, V., Xu, N., Liu, J.-C., Tang, G. & Geng, W.-T. VASPKIT: a userfriendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 267, 108033 (2021).
شكر وتقدير
تم دعم هذا العمل من قبل البرنامج الوطني الرئيسي للبحث والتطوير في الصين (2021YFA1600800)، ومؤسسة العلوم الطبيعية الوطنية في الصين (12025505، 22179125، و 12205304)، ومؤسسة غوانغدونغ للبحث الأساسي والبحث التطبيقي (2022A1515011828)، وبرنامج البحث الاستراتيجي الأولوي للأكاديمية الصينية للعلوم (XDB0450200)، وبرنامج الابتكار الجامعي في مقاطعة آنهوي (GXXT-2020-053)، وصناديق البحث الأساسية للجامعات المركزية. (WK2060000038، WK2310000113)، جمعية تعزيز الابتكار الشبابي CAS (2022458). نشكر NSRL و BSRF و SSRF على وقت شعاع السنكروترون ونظام الحوسبة الفائقة في مركز الحوسبة الفائقة بجامعة العلوم والتكنولوجيا في الصين على الحسابات. تم تنفيذ هذا العمل جزئيًا في مركز أدوات العلوم الفيزيائية، جامعة العلوم والتكنولوجيا في الصين.
مساهمات المؤلفين
ت.ي. تصور الفكرة وأشرف على العمل. س.ل.، و.ز. و ت.د. خططوا وأجروا تخليق المحفز. س.ل.، م.و. و و.ز. قاموا بإجراء قياسات AEMWEs. ت.ل.، ج.ك. و ق.ل. نفذوا حسابات DFT. ح.ز. و إكس.ل. قاموا بإجراء قياسات XAFS وساعدوا في تحليل البيانات. د.ل.، ل.ز.، ي.ج.، س.ج.، ج.ك. و ح.و. قاموا بإجراء قياسات TEM و HRTEM و EDS-mapping، وساعدوا في تحليل البيانات. ت.ي.، س.ل.، و.ز. و ت.د. شاركوا في كتابة وتحسين وتنقيح المخطوطة. ناقش جميع المؤلفين النتائج وقدموا تعليقات على المخطوطة.
يجب توجيه المراسلات والطلبات للحصول على المواد إلى وي زانغ، تاو دينغ أو تاو ياو.
معلومات مراجعة الأقران تشكر مجلة Nature Communications جونغانغ هو والمراجعين الآخرين المجهولين على مساهمتهم في مراجعة هذا العمل. يتوفر ملف مراجعة الأقران.
ملاحظة الناشر: تظل شركة سبرينجر ناتشر محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
الوصول المفتوح هذه المقالة مرخصة بموجب رخصة المشاع الإبداعي النسب 4.0 الدولية، التي تسمح بالاستخدام والمشاركة والتكيف والتوزيع وإعادة الإنتاج بأي وسيلة أو صيغة، طالما أنك تعطي الائتمان المناسب للمؤلفين الأصليين والمصدر، وتوفر رابطًا لرخصة المشاع الإبداعي، وتوضح إذا ما تم إجراء تغييرات. الصور أو المواد الأخرى من طرف ثالث في هذه المقالة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة، ما لم يُشار إلى خلاف ذلك في سطر الائتمان للمواد. إذا لم تكن المادة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة وكان استخدامك المقصود غير مسموح به بموجب اللوائح القانونية أو يتجاوز الاستخدام المسموح به، فسيتعين عليك الحصول على إذن مباشرة من صاحب حقوق الطبع والنشر. لعرض نسخة من هذه الرخصة، قم بزيارةhttp://creativecommons.org/رخص/بواسطة/4.0/. (ج) المؤلف(ون) 2024
مدرسة العلوم والتكنولوجيا النووية، المختبر الرئيسي للكيمياء الدقيقة والذكية، المركز الوطني للبحوث الفيزيائية على المقياس المجهري في هيفي، مختبر الإشعاع السنكروتروني الوطني، جامعة العلوم والتكنولوجيا في الصين، هيفي، جمهورية الصين الشعبية.شركة تشونغكه إنثالبي (أنhui) لتكنولوجيا الطاقة الجديدة المحدودة، هيفي، جمهورية الصين الشعبية.المركز التجريبي للهندسة وعلوم المواد، جامعة العلوم والتكنولوجيا في الصين، هيفي، جمهورية الصين الشعبية.معاهد العلوم الفيزيائية وتكنولوجيا المعلومات، جامعة آنهوي، هيفي، جمهورية الصين الشعبية.هؤلاء المؤلفون ساهموا بالتساوي: سيتشينغ لي، تونغ ليو. البريد الإلكتروني:zhangw94@ustc.edu.cn; dingtao@ustc.edu.cn; ياوت@ustc.edu.cn
In-depth comprehension and modulation of the electronic structure of the active metal sites is crucial to enhance their intrinsic activity of electrocatalytic oxygen evolution reaction (OER) toward anion exchange membrane water electrolyzers (AEMWEs). Here, we elaborate a series of amorphous metal oxide catalysts and with high performance AEMWEs by high-valent chromium dopant. We discover that the positive effect of the transition from low to high valence of the Co site on the adsorption energy of the intermediate and the lower oxidation barrier is the key factor for its increased activity by synchrotron radiation in-situ techniques. Particularly, the anode catalyst achieves the high current density of at 2.1 V and maintains for over 120 h with attenuation less than in AEMWE testing. Such exceptional performance demonstrates a promising prospect for industrial application and providing general guidelines for the design of highefficiency AEMWEs systems.
Anion exchange membrane (AEM) electrolysis is considered a pivotal technology for the sustainable energy economy . It has advantages over proton exchange membrane (PEM) technology with noble metal oxide ( ) anodes. These advantages include the ability to use less expensive non-platinum group metal (PGM) catalysts on the anode side, cheaper ion exchange membranes, free of environmentally harmful fluorine-based polymers, and no need for acid-resistant stacking materials, resulting in lower overall device costs . Nevertheless, AEMWE technology is a relatively new technology and faces many issues that have to be solved before fulfilling its full potential, with catalysts being a crucial factor . There are two major challenges in developing higher performance non-noble metal anode OER electrocatalysts. One is that current synthesis methods for these catalysts are not optimized for industrial applications. Therefore, in practical scenarios, it is essential to consider simple and easily accessible synthesis
methods that can produce large quantities of catalysts. The other is that although -based materials have been extensively studied as OER catalysts, their stability is poor and they are rarely used in AEM electrolysis research . The stability and activity are both critical factors especially in the electrolysis applications, which should receive more attention.
On the anode side of AEMWEs, the OER is a complex process involving the transfer of multiple electrons. As the electrons are transferred, the valence state of the active metal changes, which is believed to be the source of the OER intrinsic activity . Hence, there is ongoing debate regarding the relationship between the valence state and activity of the active site in transition-metal-based catalysts (Fig. 1a). Numerous studies have revealed that transition metal compounds with high oxidation state metal sites exhibit great OER activity, while those with low oxidation metal sites have comparatively low
Fig. 1 | Schematic diagram of the origin of the research and design ideas. a The inherent debate between active site valence and catalytic activity. Different types of valence changes during the OER. c A series of meticulously conceived and
designed experiments were conducted using Cr as the high-valence dopant. d Uncovering valence changes and universal mechanisms impacting catalytic performance.
activity . However, recent research has challenged this notion, suggesting that low oxidation metal sites can also play a significant role in determine OER activity . Based on this theory, scientists have introduced oxygen-rich defects and doped high valence metals (such as and ) to lower the valence state of the active metal. Despite these efforts, the relationship between catalytic activity and valence changes during the reaction remains unclear, owing to the lack of the dynamic evolution analysis of active metal sites during the OER condition (Fig. 1b). In-depth understanding and regulation of the electronic structure of the active metal sites in catalysts, as well as their application in practical AEMWEs for mechanistic comprehension, remains a significant challenge. Synchrotron radiation in-situ techniques provide a practical way to address this issue by monitoring the dynamics of the valence state and elucidating the key reasons for its impact on catalytic performance.
In this work, we chose low-cost, abundant Cr with high valence charge as the metal dopant and utilized a one-step liquid phase reduction method to synthesize a series of efficient amorphous metal oxide catalysts for AEMWEs (Fig. 1c). Among them, the catalyst delivered outstanding activity and stability. We used in-situ valence to core X-ray emission spectroscopy (vtc-XES) for the first time in the electrocatalytic system to precisely identify the ligands of Co , and the -interaction strength enhancement of Co-O during the OER process. The combination of in-situ X-ray absorption spectroscopy (XAS), synchrotron radiation infrared (SR-IR) and other experiments along with theoretical calculations allowed us to discern the atomic and electronic structure evolution at the Co site optimizes the adsorption energy of the oxygen intermediate (Fig. 1d), resulting in the low oxidation energy barrier that are crucial for high catalytic activity. Impressively, the AEMWE was assembled by using as anode catalyst achieved the high current density of at 2.1 V and exhibited outstanding long-term durability for operating over 120 h at , further demonstrating its great potential in the application of water electrolysis industry.
Results
Synthesis and structural characterization
A diagram of the catalysts synthetic method is shown in Fig. 2a. The catalysts were prepared by a simple one-step liquid-phase reduction method by mixing with the solution of different metal chlorides under stirring, following by washing and drying at room temperature. The presence of facilitated the simultaneous reduction of the metal precursors during the nucleation and growth phases.
This reduction process also allowed for the instantaneous formation of three-dimensional networks through the fusion of metal nuclei , enabling the facile preparation of high performance catalysts. The morphology of the prepared catalysts was observed using the scanning electron microscopy (SEM), revealing the porous characteristics of and (Fig. 2b and Supplementary Fig. 1). Moreover, the structural features were investigated by transmission electron microscopy (TEM), which indicated that the prepared catalysts as the disordered atomic structure (Fig. 2c), and spherical nanoparticles with an average size of about 20 nm were observed (Fig. 2d). The high-resolution TEM (HRTEM) and aberration-corrected highangle annular dark-field scanning transmission electron microscopy (AC HAADF-STEM) further showed that there were no obvious lattice striations (Fig. 2e and Supplementary Fig. 2), indicating its amorphous nature. X-ray diffraction (XRD) pattern (Supplementary Fig. 3) showed very broad and weak diffraction peaks, indicating the long-range disorder feature. The amorphous character is further confirmed by the diffraction rings shown by selected area electron diffraction (SAED) (Fig. , with the bright inner rings suggesting the short-range order feature . The morphology and structure of and were found to be similar to (Supplementary Figs. 4-7). In addition, energy-dispersive X-ray spectroscopy (EDS) mapping analysis showed that , and O were uniformly distributed throughout the architecture (Fig. 2g), demonstrating the formation of CoCr oxides rather than phase-separated mixed structures. The same phenomenon was observed for and (Supplementary Fig. 8), while the higher O content is attributed to the ease of oxidation of the transition metals. The inductively coupled plasma-atomic emission spectrometer (ICP-AES) was used to further determine the elemental ratios of the prepared catalysts (Supplementary Table 1). The results showed that the atomic ratios of , Ni to Cr were close to , indicating the precise control of the synthetic method adopted in this study. Furthermore, the extended X-ray absorption fine structure (EXAFS) spectra was used to confirm that the and (Fig. 2h, i and Supplementary Figs. 9-12) catalysts had similar local structures and coordination environments, suggesting that each form structurally similar and Ni oxides, which lays the basis for further analysis.
Electrocatalytic OER and AEMWEs performance
The OER activity measurements of the catalysts were conducted in saturated 1 M KOH solution by using a standard three-electrode system. The optimal catalytic performance of the samples was
Fig. 2 | Preparation, structural and morphological characteristics of . a lllustration of the preparation procedure, (b) SEM, (c, d) TEM, (e) HRTEM images. f SAED patterns. g EDS mappings showing the dispersion of Co (pink), Cr (blue-
green) and O (yellow), respectively. Scale bar: 100 nm . EXAFS spectra of ( ) Co -edge and (i) -edge for and reference samples.
achieved when the atomic ratio of was 1:1, as determined through performance-based screening (Supplementary Fig. 13). For the sake of clarity, the atomic ratio of to Cr for the , and catalysts is unless otherwise specified. To assess
the impact of Cr doping, we synthesized a non- Cr doped catalyst and also included the benchmark catalyst for comparison. As shown in the iR-corrected LSV curves (Fig. 3a), the exhibited superior OER performance, necessitating a mere overpotential of
Fig. 3 | Electrocatalytic activity and stability. a LSV curves of , and catalysts in 1 M KOH at the scan rate of The current density of different catalysts recorded at 400 mV . c Tafel plots. d Nyquist plots acquired at 1.57 V (vs RHE) in half cell. Inserts in panels d show equivalent circuit of the OER. e Differences in current densities plotted against scan rates to
determine the ECSA. I-V curves of AEMWEs the prepared catalysts as anodes. g Nyquist plots of AEMWEs applying 1 A direct current (DC) and 100 mA root mean square (RMS) of alternating current (AC). Chronopotentiometry curve of catalyst at constant current density of in the AEMWEs at . Positions indicated by the black arrows represent that the replacement of electrolyte.
268 mV to achieve the current density of . The performance outmatched that of other prepared catalysts and the comparative catalyst , surpassing even the commercial . It suggested that the beneficial impact of Cr doping on enhancing the OER activity. As the overpotential was fixed at 400 mV , the current density of considerably exceeded other catalysts, being 10.7 times, 3.4 times, and twice as high as , and , respectively. Significantly, Cr doping also enhanced the current density by relative to (Fig. 3b). Additionally, in order to further investigate the intrinsic activity of the catalysts, the Tafel slope was further obtained from the corresponding LSV curves (Fig. 3c). With a Tafel slope of , the significantly outperformed other catalysts, including the non- Cr doped catalyst ( 146.6 mV ) and , indicative of the more favorable OER kinetics. The electrochemical impedance spectra (EIS) (Fig. 3d) revealed that exhibited a lower charge transfer resistance in the high-frequency region ( ) compared to ( ), , and , suggesting superior electrical conductivity and expedited charge transfer process. Corroborating this trend, a comparative analysis of the electrochemically active surface area (ECSA) of the catalysts (Fig. 3e and Supplementary
Fig. 14) was undertaken, obtained by measuring the double-layer capacitance in the non-faradaic potential region. The for was calculated to be , higher than , and . Notably, both EIS and kinetic analyses are in accordance with the excellent activity of observed in the LSV curves. Stability is also a key criterion to evaluate the long-term catalytic performance of electrocatalysts (Supplementary Fig. 15). exhibited no significant loss of activity during 50 h of electrolysis in a half-cell at the constant voltage of 1.56 V ( RHE), with the current density first increasing and then stabilizing. The increase in current density could be attributed to the formation of more active material and the gradual penetration of the electrolyte into the porous structure .
In order to capture the industrial potential of electrocatalysts, we constructed an AEM electrolyzer system with the prepared samples as an anode catalyst and commercial Pt/C as a cathode catalyst to evaluate its catalytic performance under simulated industrial conditions ( ). The current-voltage ( ) characteristic curves in Fig. 3f demonstrate that the catalyst displays superior performances, achieving the high current density of at 2.1 V . We also investigated the impact of different membrane electrode
preparation methods on electrolyzer performance (Supplementary Fig. 16) and plotted the schematic diagrams of the electrolyzer assemblies for the three processes (Supplementary Fig. 17). To provide clarification for the outstanding performance of catalysts in AEMWE, the Nyquist plots of the electrolyzer obtained through EIS reveal three different resistances in AEMWEs: ohmic resistance (OR), charge-transfer resistance (CTR) and mass transport resistance (MTR) (Fig. 3g). The intercepts in the high-frequency region represents OR, while the high- and low-frequency regions arcs indicate the CTR associated with electrochemical reactions and MTR corresponding to the transportation of reactants and products, respectively. It is evident that the OR of the samples are nearly identical, with exhibiting the lowest CTR and MTR, indicating its superior activity derives from expedited charge transport and reduced mass transfer resistance. Supplementary Fig. 18 demonstrates that the operando EIS was employed at various currents to determine the charge transfer resistance of each catalyst. The obtained results indicated a notable decline in resistance with increased current, implying faster electron transfer and higher energy conversion efficiency during electrolysis at elevated currents. We additionally assessed the stability of the catalyst under and room temperature conditions at a high current density of to further evaluate its potential for industrial applications (Fig. 3h and Supplementary Fig. 19). As shown in Fig. 3h, the catalyst displayed considerable stability during electrolysis durations of 120 h , verifying their practicability for real-world deployment. The diagram presented in Fig. 3h displays a diagrammatic representation of the AEMWE, which includes two stainless steel (SS) collectors with runners, two polytetrafluoroethylene (PTFE) gaskets, titanium felts, AEM, and carbon paper (CP). The various electrochemical data are compiled in Supplementary Table 2 for a cleaner comparison and contrast of each catalyst’s electrochemical performance.
Valence and local structure evolution analysis
In order to compare and investigate the key reasons for the excellent activity of , the initial valence state of the active elements in unison is determined. By X-ray near-edge structural absorption spectroscopy (XANES), we were able to precisely determine the valence states of the active elements. For transition metal elements, the rightward shift of the nearside absorption edge represents an elevated oxidation state. Comparing the position of the absorption edge to the standard samples reveals that is located between CoO and (Fig. 4a). In addition, the leftward shift of the absorption edge of relative to indicates that the Cr doping decreases the average valence state of Co , which matches the stronger lower valence absorption peak in the soft XAS (sXAS) (Supplementary Fig. 20). The coexistence of both +2 and +3 valences for each active metal element in X-ray photoelectron spectroscopy (XPS) also confirms the above conclusion (Supplementary Fig. 21a-f and Supplementary Table 3). Correspondingly, the valence states of Cr -doped Fe and Ni (Supplementary Fig. 22) were similarly stabilized between +2 and +3 valence. Figure 4 b shows that the -edge of all catalysts almost overlaps with , pointing to the valence state of Cr is +3 , which is in high agreement with the results in sXAS (Supplementary Fig. 23) and XPS (Supplementary Fig. ). The consistency between valence and structure bolsters the reliability of further analysis. To further elucidate the reason for the difference in the intrinsic catalytic activity of the three catalysts, we examined the valence alterations in the active elements of and before and after the reaction using sXAS (Fig. 4c and Supplementary Fig. 24). We discovered a conspicuous change in Co alone during the reaction, its absorption peak shifted by 1.9 eV towards the higher energy, representing a significant increase in the oxidation state of Co , which is also supported by the XPS results (Supplementary Fig. 25). Given that the binding energy of is higher than that of , the shift in the binding energy peak to the lower energy corroborates the higher valence
state . It is also evident that the characteristic satellite peak of almost disappears, further proving that is almost completely oxidized to the higher valence state during the OER process.
To further uncover the dynamic evolution of the electronic structure and atomic local environment of the Co active sites during the electrocatalytic OER, the in-situ XAFS and vtc-XES measurements were performed using a lab-built electrochemical cell. As shown in Fig. 4d, the Co -edge XANES spectra indicate that the positions of the absorption edge of Co under ex-situ and open-circuit voltage (OCV) conditions are almost the same. When the voltage is applied, the nearedge absorption edge of is positively shifted, implying the elevation of the average oxidation state of Co during the OER process, which is consistent with the sXAS and XPS results. The fitted results of the Co -edge EXAFS spectra indicate that the coordination number of the first shell layer ( ) increases significantly from 4 to above 6 as the voltage is applied (Fig. 4e, Supplementary Fig. 26 and Supplementary Table 4), and the increase in coordination number may be the reason for the elevated Co valence state . The graph depicted intuitively in Fig. illustrates a significant positive correlation between the absorption edge position and the Co-O coordination number. In addition, we noted that the local structure of Co changed during the reaction, which may indicate the dynamic reconfiguration of . This is due to the partial electrolytic dissolution of Cr under alkaline conditions, which is supported by the reduction of Cr content revealed EDS mappings after reaction (Supplementary Fig. 27). The HRTEM results (Supplementary Fig. 28a) indicate that the catalyst essentially maintained its original morphology, but short lattice streaks and nanopores appeared, and these formed pores may provide a larger specific surface area and easy access to the active sites, as well as rapid mass transfer, which would further promote the OER kinetics . The amorphous structure may have been slightly damaged during the electrolysis process, as evidenced by several bright spots in the SAED pattern after reaction. However, the original amorphous state was largely preserved (Supplementary Fig. 28b).
Although EXAFS can provide the coordination numbers as well as the metal-ligand bond lengths, it fails to distinguish between ligands with similar atomic numbers (e.g., ) or different protonation states of the ligands . Therefore, the in-situ vtc-XES (Fig. 4 g ) was employed to identify the ligand and coordination environment of Co, thereby further confirm the ligand type and the variation in bond strength. The peak around 7690 eV is significant evidence of ligandmetal interaction through interactions , and this peak is recognized as the characteristic peak of coordination . An intensified peak characterized implied that the Co-O interaction strengthens with voltage during the OER. This observation corroborates the contraction of the Co-O bond distance as depicted in the Fourier-transformed EXAFS (FT-EXAFS) spectra . The subtracted spectra vtc-XES is illustrated on the top of Fig. 4 g to highlight the differences. It is observable that post-voltage application, the peak intensity closely resembles that of the reference sample ( ). Furthermore, Fig. 4 h showcases the dependable negative connection between the intensity of the subtracted vtc-XES spectra and the bond lengths, thereby corroborating the reliability of the analysis. Shorter Co-O bonds can provide optimized binding energy for the adsorption of oxygen intermediate, thereby facilitates the accelerated OER kinetic process .
Mechanism investigation and simulation
In order to gain deeper understanding of the adsorption behaviors and reaction mechanisms of various catalysts, we conducted in-situ SR-IR measurements and density functional theory (DFT) calculations for and . As presented in Fig. 5a, the reflectance intensity heatmap obtained from SR-IR spectroscopy of indicate the three crucial reactive intermediates of OER, namely *OH ( ), *OOH ( ) and * . Interestingly, the *O adsorption peaks only appear at higher potentials. When the
Fig. 4 | Valence state, change in catalyst properties recorded at different applied voltages during electrocatalytic OER. a XANES spectra of Co -edge of and the reference samples. XANES spectra of -edge of , and the reference samples. Normalized sXAS spectra of edge of before and after OER. d In-situ XANES spectra of . e The fitting curves of -weighted FT-EXAFS spectra of -edge for . Relationship between normalized absorption edge energy and coordination number during the OER process. In-situ vtc-XES spectra of and the reference samples. The subtracted vtc-XES spectra (each spectrum minus ex-situ spectrum) is shown at the top highlight the differences. Relationship between intensity differences in in-situ vtc-XES spectra and Co-O bond lengths during the OER process.
potential reaches the OER region ( ), the intensities of the *OH and *O peaks markedly increase. This indicates a positive correlation between intermediate adsorption and voltage application. Nevertheless, the intensity of the *OOH peak paradoxically weakens or even vanishes (Fig. 5b and Supplementary Fig. 29b). In contrast, no *O adsorption peaks were observed for and at all potential conditions, suggesting that has an optimized intermediate adsorption energy (Supplementary Fig. 29a, c). This finding further corroborates that * O is produced and adsorbed on the Co sites during the reaction of , resulting in an increase in the number of ligands and the Co valence change, which in turn facilitates the OER process .
To elucidate the rationale behind these occurrence, a fourelectron OER mechanism was assumed to proceed through *OH, *O and (asterisks denote adsorption sites) (Fig. 5c) . The Gibbs free energy and the limiting reaction barrier were also calculated for each elementary step in the OER, which based on the free energy of the
rate-determining step (RDS) (Fig. 5d). It was discovered that for , the RDS is the oxidation of *OH to *O, which may be a plausible explanation for the fact that *O is generated only at higher potentials. Whereas the *OOH dehydrogenation to form exhibits the lowest reaction energy barrier in comparison to the other three steps. As a result, the *OOH intermediates are consumed rapidly at high potentials, potentially explaining the near disappearance of the *OOH peak. It is pertinent to mention that exhibited the lowest RDS among three catalysts, measuring only 0.50 eV at . The RDSs for the oxidation of *O to *OOH in and were 0.76 eV and 0.63 eV , respectively. Furthermore, the differential charge densities in Fig. 5e reveals that the charge transfer of to * OH is significantly less and the interactions are weaker compared to and . This corresponds to the relatively weaker adsorption of *OH intermediates in the free energy diagram. The optimized *OH adsorption allows the subsequent steps to occur at lower potentials, thus facilitating the OER kinetics . Meanwhile, the total density of
Fig. 5 | Monitoring of reaction intermediates and DFT calculations. a In-situ SRIR measurements in the range of and under various potentials for during the OER process. Infrared transmittance signals versus potentials of * O , * OH and *OOH intermediates. c The proposed alkaline OER mechanism on . The model colors are Co (purple), Cr (blue), O (red) and H
(pale pink). d OER free-energy diagrams at 0 V (left) and 1.23 V (right) for , and . e Differential charge densities (blue and yellow regions indicate electron depletion and accumulation, respectively). The model colors are Fe (yellow-brown), Co (purple), Ni (green), Cr (blue), O (red) and H (pale pink). f TDOS and (g) PDOS of and .
states (TDOS) calculation shows that has greater density of states at the Fermi energy level, suggesting improved conductivity and explaining the exceptional activity of (Fig. 5f). The projected density of state (PDOS) illustrates that – and -orbital electrons contributions are mainly in the valence band, whereas the conduction band is composed predominantly -orbital electrons (Fig. 5 g ). Further analysis of the orbitals of the active metal reveals that the and orbital projections of , and Ni mainly contribute below the Fermi level and have strong orbital interactions with the orbitals, indicating that has stable bonding ability with O atoms, which is conducive to promoting adsorption of active O atoms and OER kinetics (Supplementary Figs. 30, 31). In addition, the electron density near the Fermi energy level of the active elements is primarily contributed by the orbitals. However, the abundance of electrons near the Fermi level, rather than significantly below it, facilitates the charge transfer from
transition metals to oxygen-containing adsorbates during the OER process . This is particularly evident in the case of , which is significantly higher than and . This further corroborates the reason for its exceptional activity.
Discussion
To summarize, we elaborated and synthesized a series of high-valent Cr-doped amorphous metal oxide catalysts using a facile one-step liquid phase reduction method. The catalyst achieved a high current density of at 2.1 V and operated at a current density of for over 120 h in the AEMWE. In-situ synchrotron measurements and theoretical calculations indicate that optimized *O adsorption, shorter Co-O bond lengths, and elevated coordination numbers resulted in Co valence changes and improved OER activity. Hence, the low oxidation energy barrier of the active metal is the ultimate goal of electronic structure modulation, which is the key
factor for the greater activity of the easily oxidized transition metals. These findings broaden the pathway to enhance OER catalytic activity and practical AEMWEs performance, while the application of new insitu method suggests numerous possibilities for addressing relevant problems in the field of energy catalytic reactions.
Methods
Chemicals
Iron chloride hexahydrate ( ), nickel chloride hexahydrate ( ), cobalt chloride hexahydrate ( ), chromium chloride hexahydrate ( ), sodium borohydride ( ) and ethanol were purchased from Sinopharm Chemical Reagent Co., Ltd. Potassium hydroxide (KOH, electronic grade, 99.999% metals basis, except sodium) was purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. Ruthenium dioxide (99.9% trace metals basis) and Nafion solution (5%) were purchased from Sigma-Aldrich. Carbon black (ECP600JD, Ketjenblack ) for electrochemical measurements was obtained from Suzhou Sinero Technology Co., Ltd. and graphite powder (XF009 7782-42-5) for XAS characterization was obtained from XFNANO Materials Tech Co., Ltd. All chemicals can be used without further purification.
Synthesis of catalysts
Dissolve 11.90 g of and 13.32 g of in 25 mL of deionized (DI) water to make a 2 M metal chloride solution and set aside. Take 2 mL of the above prepared and solutions respectively and mix them evenly. Prepare a fresh solution of sodium borohydride by dissolving 3 g of in 20 mL of DI water, then add dropwise to the above mixture of metal ion chlorides, stir mechanically for 15 min , allow to cool, then leave overnight and freeze dry. After freeze-drying, the catalyst was repeatedly washed using DI water and ethanol, and dried under vacuum to obtain the catalyst. and catalysts were obtained by the same procedure, except that the metal chloride solution was replaced with another.
Electrochemical measurements
All electrochemical tests were performed in a typical three-electrode system on an electrochemical workstation (CHI760E, Chen Hua). A electrode ( 1 M KOH , Tjaida) was used as the reference electrode, a platinum foil electrode ( ) or graphite rod as the counter electrode and the catalyst was coated on carbon paper (HESEN, HCPO20N) to make a working electrode. 6 mg of catalyst, 3 mg of conductive carbon black (ECP600JD, Ketjenblack ) was homogenously mixed with of DI water, of ethanol and of Nafion and sonicated to make a homogeneous ink. of DI water, of ethanol and of Nafion were mixed to make Nafion solution. Then of catalyst ink was dropped onto carbon paper ( ), dried and of Nafion was added dropwise and dried at room temperature and used for electrochemical measurements.
Linear scanning voltammetry (LSV) was performed in oxygensaturated 1 M KOH at room temperature ( ) using a scan rate of . The solution resistance was determined from the resulting Nyquist plot fit and used for ohmic drop correction based on where is the correction potential and is the measured potential, respectively). EIS spectra were measured over a range of with an amplitude of 0.005 V . The voltage was set at 1.57 V (vs RHE) and the EIS data was fitted by ZView software. In the equivalent circuit diagram (inset of Fig. 3d), represents the solution resistance, and are the resistances for charge transfer, and and are used to describe the double-layer capacitance. Electrochemical active surface area (ECSA) was obtained by measuring the double-layer capacitance in the nonfaradaic potential region with a voltage measurement range of 0.875 to 0.975 V ( RHE).
MEAs fabrication and AEMWEs evaluation
The MEAs fabrication and AEMWEs evaluation were performed at Zhongke Enthalpy (Anhui) New Energy Technology Co, Ltd. For the anode, the prepared catalysts ( and ) catalysts were mixed with the ionomer (PiperION-A5-HCO3) in an aqueous solution (1:3 water and isopropanol). For the cathode, (TANAKA, ) was mixed with the ionomer in an aqueous solution (1:5 water and isopropanol). The catalyst ink was sprayed onto AEM (PiperION-A60-HCO3) or platinum-titanium-coated felt (Sti0.25Pt0.5) to make gas diffusion electrodes (GDEs), with an approximate metal loading of for the anode and for the cathode. The AEM was immersed in 0.5 M KOH for 1 h , then replaced with fresh KOH solution and immersed for another 1 h . The bicarbonate anion was then rinsed with deionized water to convert the bicarbonate anion to hydroxide anion. The MEA was loaded into a fixture and clamped with a torque of . The cell was tested by a constant-potentiostat with a flowing electrolyte of 1 M KOH , and the AEMWEs were tested for internal resistance using an internal resistance meter ( for for for and for ). The ohmic drop was calculated based on the correction for iR compensation. EIS measurements of the AEMWEs were performed by an impedance analyzer (DH7007, DongHua Analytical) over the range of , amplitudes of , and currents of (The applied RMS AC is of the corresponding DC and the effective area of the MEA is ). The performance and EIS tests were conducted at a temperature of , while the durability tests were conducted at both and room temperature.
Material characterizations
XRD patterns were obtained by using a Philips X’Pert ProSuper diffractometer with radiation ( ). The morphology was examined by SEM using the ZEISS GeminiSEM 500. TEM and HRTEM images were undertaken on a JEM-2100F field-emission electron microscope with an accelerating voltage of 200 kV . HAADF-STEM was performed on a JEOL JEM-ARF200F HRTEM with a spherical aberration corrector at voltage of 200 kV . EDS elemental mappings were obtained on JEOL JEM-F200 instrument. All catalysts for synthesis were dissolved in aqua regia and obtain the contents of and Cr in asprepared catalysts were determined by ICP-AES on a PerkinElmer Optima 7300 DV ICP-AES instrument. The XPS were recorded on a Thermo ESCALAB 250Xi spectrometer with an excitation source of monochromatized Al Ka ( ) and a pass energy of 30 eV . The values of binding energies were calibrated with the C 1 s peak of contaminant carbon at 284.80 eV .
Soft X-ray XANES measurements
The -edge XANES measurements of and Cr were performed at the photoemission endstation at BL12B beamlines of the National Synchrotron Radiation Laboratory (NSRL), China. A bending magnet was connected to the beamline, which was equipped with three gratings covering photon energies from 100 to 1000 eV with an energy resolution of . The data were recorded in the total electron yield mode by collecting the sample drain current. The resolving power of the grating was typically , and the photon flux was photons per second.
XAFS measurements
The XAFS spectra were measured at the BL14W1 beamline of the Shanghai Synchrotron Radiation Facility (SSRF) and the 1W1B beamline of the Beijing Synchrotron Radiation Facility (BSRF), China. The SSRF storage ring was operated at 3.5 GeV and the BSRF at 2.5 GeV , both with a maximum electron current of 250 mA . A total of about 64 mg of the appropriate sample was homogeneously mixed with graphite and pressed into round pellets of 8 mm diameter. The spectra of the -edge of and for
all samples were recorded in transmission mode, and the position of the absorption edge ( ) was calibrated by the corresponding element foil, respectively.
The in-situ XAFS spectra were measured at the BL14W1 beamline of the SSRF, China. The in-situ XAFS measurements were performed with catalyst-coated carbon paper using a home-built electrolytic cell, the spectra of which were collected in fluorescent mode. The catalyst powders were homogeneously dispersed in water and ethanol ( ) to form an ink, which was dropcast onto the carbon paper as a working electrode. To obtain the evolution information of the Co sites under OER working conditions, XAFS spectra were analyzed for a series of presentative conditions (ex situ, OCV, 1.42 V vs RHE and 1.67 V vs RHE, after reaction).
XAFS data analysis
The acquired EXAFS data were processed according to standard procedures using the ATHENA module implemented in the IFEFFIT software package . The -weighted data in the -space were Fourier-transformed into real ( R ) space using a hanning windows ( ) to separate the EXAFS contributions from different coordination shells. To obtain detailed structural parameters around the Co atoms in the as-prepared samples, quantitative curve fitting was performed on for the Fourier-transformed in R-space using the ARTEMIS module of IFEFFIT . The effective backscattering amplitude and the phase shift of all fitting paths were calculated using the ab initio code FEFF8.0 . For different in-situ samples, the k range of and the range of were used for ex-situ and OCV samples. For 1.42 V vs RHE, 1.67 V vs RHE and after reaction samples, the range of and the range of were chosen. For the selected range for -weighted for the curve fitting function, the number of independent points is given by
As for ex-situ and OCV samples:
For and after reaction samples:
During the curve fitting, the amplitude-efficient reduction factor was fixed at the value of 0.71 for Co samples determined by fitting the data of Co foil, with the coordination number of Co-Co set to 12.
For the ex-situ and OCV samples, the FT curves showed three peaks near and , which were assigned to and coordination, respectively. Subsequently, a three-shell structure model including a , a and a scattering path was used to fit the EXAFS data of the in-situ samples. In both samples, the Debye-Waller factors ( ) set to be the same, the of the first shell layer ( ) of the OCV sample was set as a free parameter. Meanwhile, the of the ex-situ sample was set to the best fitting value for the OCV sample. The coordination numbers (CN), interatomic distances , and energy shifts ( ) were all set as free parameters for both samples.
Therefore, the number of adjustable parameters for the ex-situ sample is
For the OCV sample, the number of adjustable parameters is
For the and after reaction samples, the FT curves showed two peaks near and assigned to and coordination, which were fitted using a two-shell structure model including one and one scattering path. Considering these three samples, should be set to be consistent and and were set as free parameters.
Hence, the number of adjustable parameters for the three samples is
All yielded R-factors for all samples were no larger than 0.020, indicating the appropriate modeling, rational parameter settings and thus, the good fitting qualities.
Vtc-XES measurements
The Co vtc-XES measurements were measured at the BL20U beamline (E-line) of SSRF, China. The incident X-ray was monochromatized to 7706 eV by Si (111) double-crystal monochromator. Five Si (440) spherical crystals and a Pilatus3 photon counting detector were used to collect and analysis Co and fluorescence more efficiently. To reduce the absorption of fluorescence by air, a helium gas bag was placed between the sample, spherical crystals, and the detector. The in-situ setup was identical to the in-situ XAFS measurements. The vtcXES spectra were baseline-corrected using a straight line and normalized to the total area between 7624.6 and 7680 eV , and then the data were smoothed using the Savitzky-Golay method.
In-situ SR-IR measurements
In-situ SR-IR measurements were conducted at the infrared beamline BL01B of NSRL using a homemade top-plate cell reflection IR setup with a ZnSe crystal as the infrared transmission window. The catalyst electrode was pressed tightly against the ZnSe crystal window with a micron-scale gap to minimize the loss of infrared light. To ensure high-quality SR-IR spectra, the apparatus utilized a reflection mode with a vertical incidence of infrared light. The infrared spectrum was obtained by averaging 128 scans at a resolution of . Prior to each systemic OER measurement, the background spectrum of the catalyst electrode was obtained at an open-circuit voltage. The OER potential ranges were measured between vs RHE with an interval of 0.1 V . The infrared data was processed and smoothed using OPUS software.
DFT calculations
The calculations were performed within the Density Functional Theory (DFT) framework implanted in Vienna ab initio Simulation Package (VASP) . The interaction between ions and electrons was described in the Projector Augmented Wave (PAW) Method . The electron exchange and correlation energy were described using the generalized gradient approximation-based Perdew-Burke-Erzenhorf (PBE) functional . The semi-empirical London dispersion corrections of Grimme and colleagues (DFT-D3) were conducted to calculate the interactions between absorbers and slabs . The Hubbard-U correction was applied for better description of the localized d-electrons of and Ni . We chose an effective value of 3.0 eV for for Co and 5.5 eV for Ni atoms . The surface structure models of (110), , (110) and (110) were built. A sufficiently large vacuum region of was used for all the models to ensure the periodic images were well separated. The Brillouin-zone integrations were carried out using Monkhorst-Pack grids of special points. A gammacentered ( ) k-point grid were used for supercell. To obtain the accurate structure, the plane-wave cutoff energy was set up to 500 eV .
The force convergence was set to be , and the total energy convergence was set to be . The free energy of the adsorbed state was calculated as follows based on the adsorption energy:
where is the adsorption energy directly obtained from DFT calculations, is the zero-point energy, is the heat capacity correction energy, and T is the temperature ( ), is the change in entropy. Herein, the Gibbs energy is corrected by using the VASPKIT code .
Data availability
The source data underlying Figs. 2-5, Supplementary Figs. 3, 9-16, 18-26, 29-31 and the electronic structure calculations generated in this study are provided as a Source Data file. Source data are provided with this paper.
References
Abbasi, R. et al. A roadmap to low-cost hydrogen with hydroxide exchange membrane electrolyzers. Adv. Mater 31, 1805876 (2019).
Xu, D. et al. Earth-abundant oxygen electrocatalysts for alkaline anion-exchange-membrane water electrolysis: effects of catalyst conductivity and comparison with performance in three-electrode cells. ACS Catal 9, 7-15 (2018).
Li, D. et al. Highly quaternized polystyrene ionomers for high performance anion exchange membrane water electrolysers. Nat. Energy 5, 378-385 (2020).
Seh, Z. W. et al. Combining theory and experiment in electrocatalysis: insights into materials design. Science 355, eaad4998 (2017).
Li, Q. et al. Anion exchange membrane water electrolysis: the future of green hydrogen. J. Phys. Chem. C 127, 7901-7912 (2023).
Cousins, I. T. et al. The concept of essential use for determining when uses of PFASs can be phased out. Environ. Sci. Processes Impacts 21, 1803-1815 (2019).
Santoro, C. et al. What is next in anion-exchange membrane water electrolyzers? Bottlenecks, benefits, and future. ChemSusChem 15, e202200027 (2022).
Li, C. & Baek, J.-B. The promise of hydrogen production from alkaline anion exchange membrane electrolyzers. Nano Energy 87, 106162 (2021).
. et al. Carbon-based bifunctional electrocatalysts for oxygen reduction and oxygen evolution reactions: optimization strategies and mechanistic analysis. J. Energy Chem. 71, 234-265 (2022).
Yao, N. et al. Intermolecular energy gap-induced formation of highvalent cobalt species in CoOOH surface layer on cobalt sulfides for efficient water oxidation. Angew Chem. Int. Ed. 61, e202117178 (2022).
Zheng, X. et al. Theory-driven design of high-valence metal sites for water oxidation confirmed using in situ soft X-ray absorption. Nat. Chem. 10, 149-154 (2018).
Zhang, B. et al. High-valence metals improve oxygen evolution reaction performance by modulating 3d metal oxidation cycle energetics. Nat. Catal 3, 985-992 (2020).
Li, N. et al. Influence of iron doping on tetravalent nickel content in catalytic oxygen evolving films. Proc. Natl Acad. Sci. USA 114, 1486-1491 (2017).
Xiao, Z. et al. Operando identification of the dynamic behavior of oxygen vacancy-rich for oxygen evolution reaction. J. Am. Chem. Soc. 142, 12087-12095 (2020).
Novák, M. et al. Primary oxide minerals in the system and their breakdown products from the pegmatite No. 3 at Dolni Bory-Hatě, Czech Republic. Eur. J. Mineral 20, 487-499 (2008).
Kuepper, K. et al. Electronic and magnetic properties of highly ordered . Phys. Status Solidi A 201, 3252-3256 (2004).
Wang, M. H. et al. Operando high-valence Cr-modified NiFe hydroxides for water oxidation. Small 18, e2200303 (2022).
Krishna, K. S., Sandeep, C. S. S., Philip, R. & Eswaramoorthy, M. Mixing does the magic: a rapid synthesis of high surface area noble metal nanosponges showing broadband nonlinear optical response. ACS Nano 4, 2681-2688 (2010).
Kim, J. J., Choi, Y., Suresh, S. & Argon, A. S. Nanocrystallization during nanoindentation of a bulk amorphous metal alloy at room temperature. Science 295, 654-657 (2002).
Wu, G. et al. A general synthesis approach for amorphous noble metal nanosheets. Nat. Commun. 10, 4855 (2019).
Cai, W. et al. Amorphous versus crystalline in water oxidation catalysis: a case study of NiFe alloy. Nano Lett. 20, 4278-4285 (2020).
Liu, J., Yang, Y., Ni, B., Li, H. & Wang, X. Fullerene-like Nickel Oxysulfide hollow nanospheres as bifunctional electrocatalysts for water splitting. Small 13, 1602637 (2017).
Zhang, R. et al. Synthesis and conductivity properties of double perovskite by sol-gel combustion. J. Mater Sci: Mater Electron. 26, 9941-9948 (2015).
Qin, C. et al. Increasing cerium dispersion favours lattice oxygen activity of cobalt oxides for CO catalytic combustion. Combust Flame 260, 113219 (2024).
Wang, J. et al. Redirecting dynamic surface restructuring of a layered transition metal oxide catalyst for superior water oxidation. Nat. Catal 4, 212-222 (2021).
Zhou, J. et al. Electrochemically accessing ultrathin Co (oxy)hydroxide nanosheets and operando identifying their active phase for the oxygen evolution reaction. Energy Environ. Sci. 12, 739-746 (2019).
Lv, L. et al. Coordinating the edge defects of bismuth with sulfur for enhanced electroreduction to formate. Angew Chem. Int. Ed. 62, e202303117 (2023).
Zhu, J. et al. Surface passivation for highly active, selective, stable, and scalable electroreduction. Nat. Commun. 14, 4670 (2023).
Xu, D. et al. The role of Cr doping in Ni Fe oxide/(oxy)hydroxide electrocatalysts for oxygen evolution. Electrochim Acta 265, 10-18 (2018).
Bo, X., Li, Y., Hocking, R. K. & Zhao, C. NiFeCr hydroxide holey nanosheet as advanced electrocatalyst for water oxidation. ACS Appl. Mater Interfaces 9, 41239-41245 (2017).
Gallo, E. & Glatzel, P. Valence to core X-ray emission spectroscopy. Adv. Mater 26, 7730-7746 (2014).
Pollock, C. J. & DeBeer, S. Valence-to-Core X-ray emission spectroscopy: a sensitive probe of the nature of a bound ligand. J. Am. Chem. Soc. 133, 5594-5601 (2011).
Kuhn, T. J., Hormes, J., Matoussevitch, N., Bonnemann, H. & Glatzel, P. Site-selective high-resolution X-ray absorption spectroscopy and high-resolution X-ray emission spectroscopy of cobalt nanoparticles. Inorg Chem. 53, 8367-8375 (2014).
Pollock, C. J. & DeBeer, S. Insights into the geometric and electronic structure of transition metal centers from valence-to-core X-ray emission spectroscopy. Acc. Chem. Res. 48, 2967-2975 (2015).
Zhang, S. et al. Spontaneous delithiation under operando condition triggers formation of an amorphous active layer in spinel cobalt oxides electrocatalyst toward oxygen evolution. ACS Catal 9, 7389-7397 (2019).
Haase, F. T. et al. Size effects and active state formation of cobalt oxide nanoparticles during the oxygen evolution reaction. Nat. Energy 7, 765-773 (2022).
Su, H. et al. Dynamic evolution of solid-liquid electrochemical interfaces over single-atom active sites. J. Am. Chem. Soc. 142, 12306-12313 (2020).
Su, H. et al. In-situ spectroscopic observation of dynamic-coupling oxygen on atomically dispersed iridium electrocatalyst for acidic water oxidation. Nat. Commun. 12, 6118 (2021).
Lin, C. et al. In-situ reconstructed Ru atom array on with enhanced performance for acidic water oxidation. Nat. Catal 4, 1012-1023 (2021).
Zhou, Z. et al. Cation-vacancy-enriched Nickel phosphide for efficient electrosynthesis of hydrogen peroxides. Adv. Mater 34, 2106541 (2022).
Ullman, A. M., Brodsky, C. N., Li, N., Zheng, S. L. & Nocera, D. G. Probing edge site reactivity of oxidic cobalt water oxidation catalysts. J. Am. Chem. Soc. 138, 4229-4236 (2016).
Zhang, L. et al. Fe-doped and sulfur-enriched nanowires with enhanced reaction kinetics for boosting water oxidation. Green Chem. Eng. 3, 367-373 (2022).
Zhao, C. et al. D-Orbital manipulated Ru nanoclusters for highefficiency overall water splitting at industrial-level current densities. Adv. Funct. Mater 34, 2307917 (2024).
Newville, M. IFEFFIT: interactive XAFS analysis and FEFF fitting. J. Synchrotron. Radiat. 8, 322-324 (2001).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron. Radiat. 12, 537-541 (2005).
Ankudinov, A. L., Ravel, B., Rehr, J. J. & Conradson, S. D. Real-space multiple-scattering calculation and interpretation of x-rayabsorption near-edge structure. Phys. Rev. B 58, 7565-7576 (1998).
Kresse, G. av JF. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Phys. Rev. B 54, 11169-11186 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953-17979 (1994).
Perdew, John P., Burke, Kieron & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865-3868 (1996).
Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 21, 1787-1799 (2006).
Conesa, J. C. Electronic structure of the (Undoped and Fe-Doped) evolution electrocatalyst. J. Phys. Chem. C 120, 18999-19010 (2016).
Tkalych, A. J., Zhuang, H. L. & Carter, E. A. A density functional + U assessment of oxygen evolution reaction mechanisms on – NiOOH . ACS Catal 7, 5329-5339 (2017).
Wang, V., Xu, N., Liu, J.-C., Tang, G. & Geng, W.-T. VASPKIT: a userfriendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 267, 108033 (2021).
Acknowledgements
This work was supported by the National Key R&D Program of China (2021YFA1600800), the National Natural Science Foundation of China (12025505, 22179125, and 12205304), the Guangdong Basic and Applied Basic Research Foundation (2022A1515011828), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0450200), the University of China Innovation Program of Anhui Province (GXXT-2020-053), the Fundamental Research Funds for the Central Universities
(WK2060000038, WK2310000113), the Youth Innovation Promotion Association CAS (2022458). We would thank NSRL, BSRF and SSRF for the synchrotron beam time and the supercomputing system in the Supercomputing Center of University of Science and Technology of China for the calculations. This work was partially carried out at the Instruments Center for Physical Science, University of Science and Technology of China.
Author contributions
T.Y. conceived the idea and supervised the work. S.L., W.Z. and T.D. planned and performed the catalyst synthesis. S.L., M.W. and W.Z. performed the AEMWEs measurement. T.L., C.Q. and Q.L. carried out the DFT calculations. H.Z. and X.L. performed the XAFS measurements and assisted in analyzing the data. D.L., L.Z., Y.C., S.J., C.Q. and H.W. performed TEM, HRTEM, EDS-mapping measurements, and assisted in data analysis. T.Y., S.L., W.Z. and T.D. co-wrote, optimized and revised the manuscript. All authors discussed the results and provided comments on the manuscript.
Correspondence and requests for materials should be addressed to Wei Zhang, Tao Ding or Tao Yao.
Peer review information Nature Communications thanks Jungang Hou and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/ licenses/by/4.0/.
(c) The Author(s) 2024
School of Nuclear Science and Technology, Key Laboratory of Precision and Intelligent Chemistry, Hefei National Research Center for Physical Sciences at the Microscale, National Synchrotron Radiation Laboratory, University of Science and Technology of China, Hefei, P.R. China. Zhongke Enthalpy (Anhui) New Energy Technology Co. Ltd, Hefei, P.R. China. Experimental Center of Engineering and Materials Science, University of Science and Technology of China, Hefei, P.R. China. Institutes of Physical Science and Information Technology, Anhui University, Hefei, P.R. China. These authors contributed equally: Sicheng Li, Tong Liu. e-mail: zhangw94@ustc.edu.cn; dingtao@ustc.edu.cn; yaot@ustc.edu.cn