DOI: https://doi.org/10.1186/s12943-023-01925-5
PMID: https://pubmed.ncbi.nlm.nih.gov/38195537
تاريخ النشر: 2024-01-09
مراجعة شاملة لتحرير الجينات المعتمد على CRISPR: الآليات والتحديات والتطبيقات في علاج السرطان
الملخص
نظام CRISPR هو أداة ثورية لتحرير الجينوم لديها القدرة على إحداث ثورة في مجال أبحاث وعلاج السرطان. إن القدرة على استهداف وتحرير الطفرات الجينية المحددة التي تدفع نمو وانتشار الأورام قد فتحت آفاقًا جديدة لتطوير علاجات سرطانية أكثر فعالية وشخصية. في هذه المراجعة، سنناقش الاستراتيجيات المختلفة المعتمدة على CRISPR التي تم اقتراحها لعلاج السرطان، بما في ذلك تعطيل الجينات التي تدفع نمو الأورام، وتعزيز الاستجابة المناعية لخلايا السرطان، وإصلاح الطفرات الجينية التي تسبب السرطان، وتوصيل جزيئات قاتلة للسرطان مباشرة إلى خلايا الورم. سنلخص أيضًا الحالة الحالية للدراسات ما قبل السريرية والتجارب السريرية لعلاج السرطان المعتمد على CRISPR، مع تسليط الضوء على أكثر النتائج الواعدة والتحديات التي لا تزال بحاجة إلى التغلب عليها. تعتبر السلامة والتوصيل أيضًا تحديات مهمة لكي يصبح علاج السرطان المعتمد على CRISPR خيارًا سريريًا قابلاً للتطبيق. سنناقش التحديات والقيود التي يجب التغلب عليها، مثل التأثيرات غير المستهدفة، والسلامة، والتوصيل إلى موقع الورم. أخيرًا، سنقدم نظرة عامة على التحديات والفرص الحالية في مجال علاج السرطان المعتمد على CRISPR ونناقش الاتجاهات المستقبلية للبحث والتطوير. لدى نظام CRISPR القدرة على تغيير مشهد أبحاث السرطان، وتهدف هذه المراجعة إلى تقديم نظرة عامة على الحالة الحالية للمجال والتحديات التي يجب التغلب عليها لتحقيق هذه الإمكانية.
المقدمة
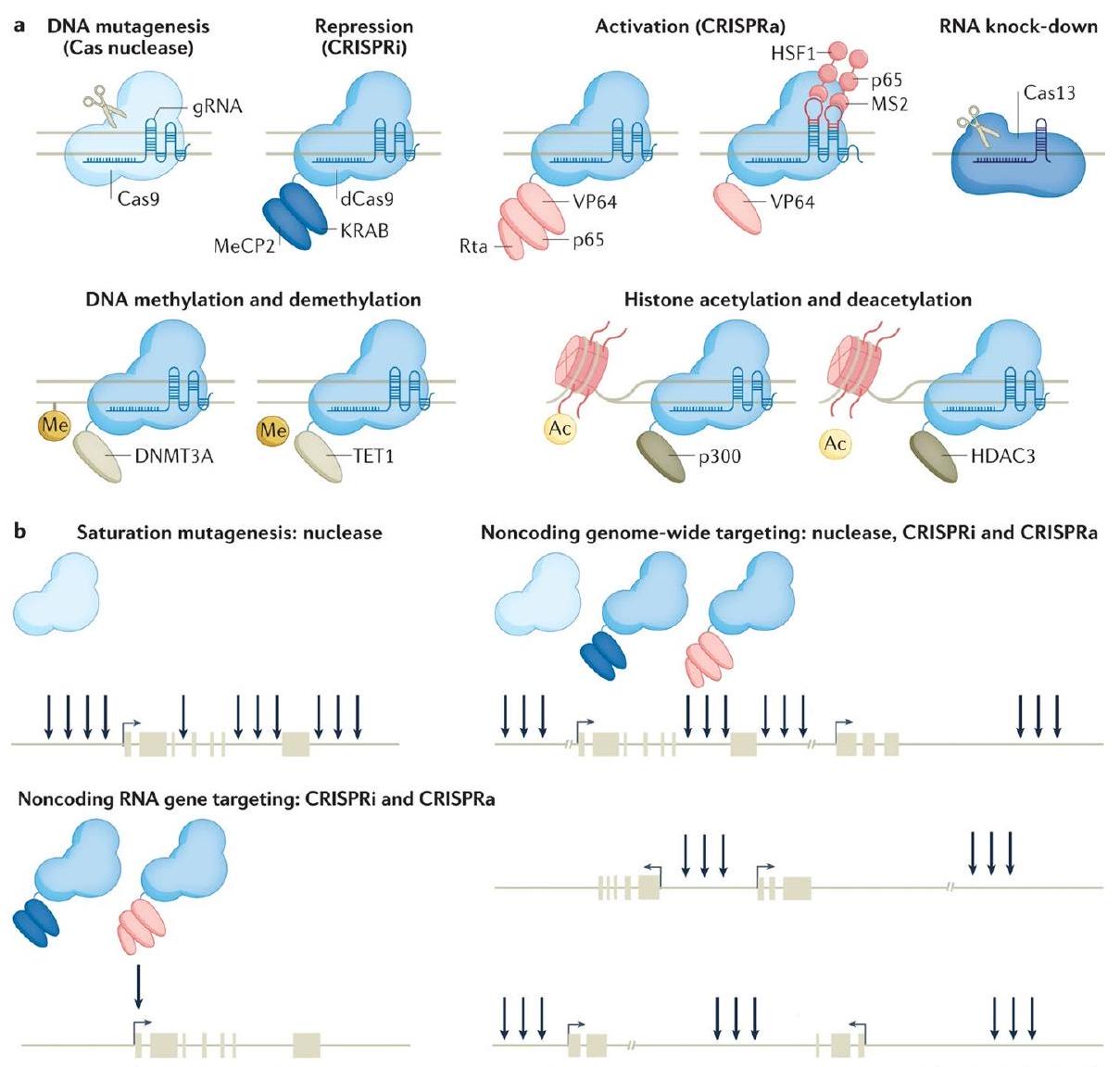
مجال تحرير الجينوم [4]. توضح الشكل 1 تطور أدوات CRISPR المستخدمة لاستكشاف بيولوجيا السرطان. يمكن برمجة الإنزيمات المرتبطة بـ CRISPR، مثل Cas9، لاستهداف تسلسلات DNA محددة، وعند دمجها مع RNAs الموجهة، يمكن استخدامها لقص أو تعديل أو حذف الجينات بطريقة دقيقة [4]. لقد تم استخدام هذه التقنية في مجموعة واسعة من التطبيقات، بما في ذلك الأبحاث الأساسية، وعلاج الجينات، والزراعة [1]. ومع ذلك، فإن تطبيقها المحتمل في أبحاث السرطان قد جذب اهتمامًا خاصًا بسبب القدرة على استهداف الطفرات الجينية التي تدفع نمو وانتشار الأورام [3]. هناك العديد من الاستراتيجيات المعتمدة على CRISPR التي تم اقتراحها لعلاج السرطان [4]. إحدى الطرق هي
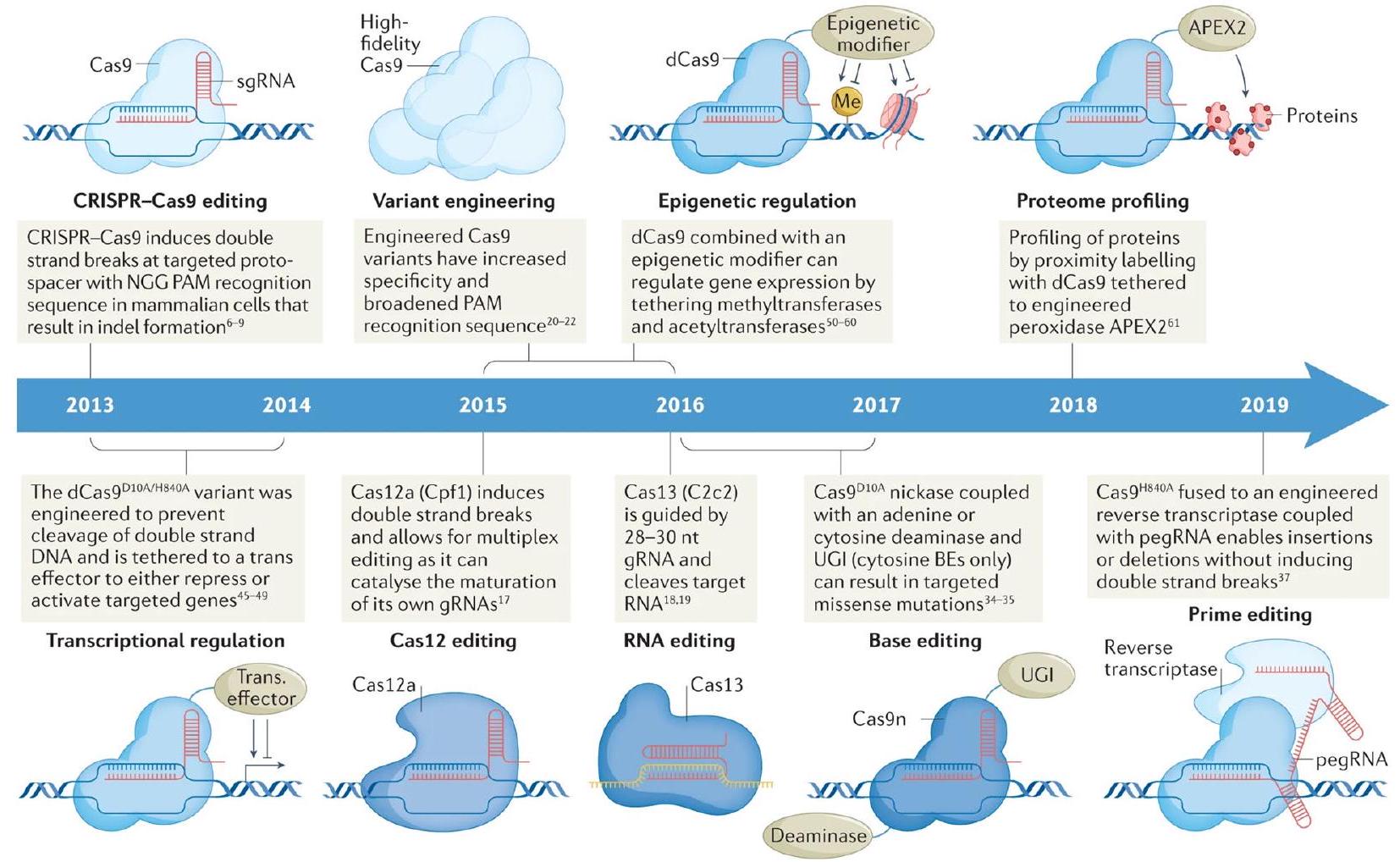
في هذه المقالة الاستعراضية، سنقدم لمحة عامة عن الحالة الحالية لمجال تحرير الجينات المعتمد على كريسبر في علاج السرطان، مع تسليط الضوء على النتائج الأكثر وعدًا والتحديات التي لا تزال بحاجة إلى التغلب عليها. سنصف الاستراتيجيات المختلفة المعتمدة على كريسبر التي تم اقتراحها لعلاج السرطان، ونلخص الحالة الحالية للدراسات قبل السريرية والتجارب السريرية، ونناقش التحديات والقيود التي يجب التغلب عليها لكي تصبح تقنية كريسبر لعلاج السرطان خيارًا سريريًا قابلاً للتطبيق. سنقدم أيضًا لمحة عامة عن الاتجاهات المستقبلية للبحث والتطوير ونناقش الآثار المحتملة لتقنية كريسبر لعلاج السرطان على مستقبل علاج السرطان والرعاية الصحية.
استراتيجيات كريسبر لعلاج السرطان
إلغاء تنشيط الجينات المسرطنة
(أ) فحوصات كريسبر: تحديد أهداف الأدوية
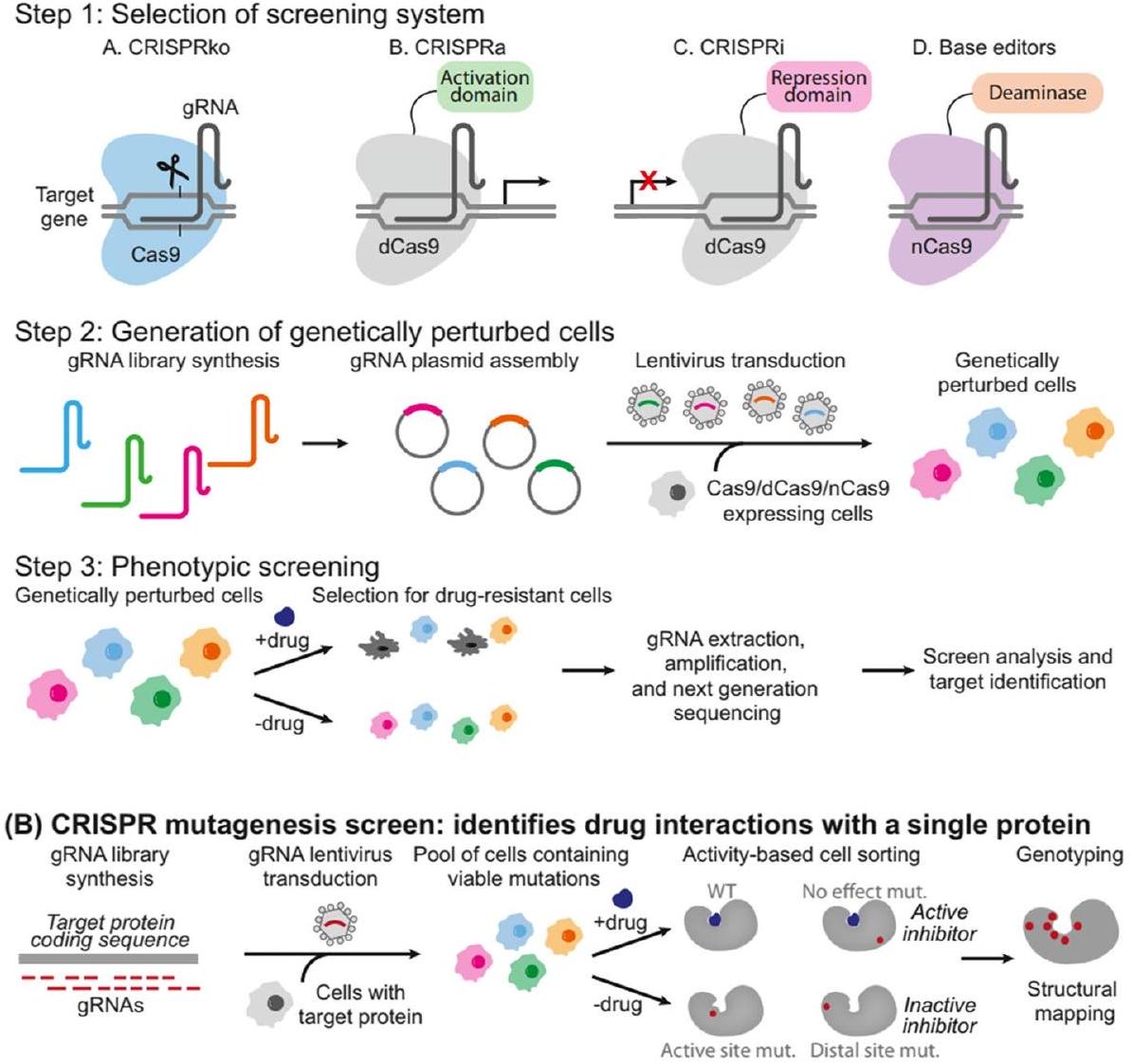
تمهيد الطريق لأساليب علاجية أكثر فعالية وتخصيصًا في مكافحة السرطان [27].
تعزيز الاستجابة المناعية
لنظام المناعة [23]. بالإضافة إلى ذلك، يمكن إدخال جينات مفيدة، مثل السيتوكينات أو وسطاء المناعة الآخرين، باستخدام CRISPR-Cas لتعزيز الاستجابة المناعية ضد مستضدات معينة [34]. يمكن أن يؤدي تحرير الجينات المعتمد على CRISPR أحيانًا إلى آثار غير محددة في المواقع، حيث تحدث تغييرات غير مقصودة في أجزاء أخرى من الجينوم. قد تؤدي هذه الآثار غير الانتقائية إلى تغييرات غير مرغوب فيها في وظيفة الجين وقد تشكل مخاوف تتعلق بالسلامة في سياق تعزيز المناعة [35، 36]. من الضروري تقييم هذه الآثار غير المرغوب فيها بدقة وتقليلها لضمان سلامة وفعالية استراتيجيات CRISPR المعتمدة [28]. يمكن استخدام CRISPR-Cas لتعديل خلايا المناعة، مثل خلايا T وخلايا NK، لتحسين وظيفتها وخصوصيتها في استهداف خلايا السرطان أو الخلايا المصابة. من خلال تحرير الجينات المسؤولة عن مستقبلات الخلايا ومسارات الإشارة، يمكن للباحثين تعزيز قدرة خلايا المناعة على التعرف على الأهداف المحددة وتدميرها [33]. فهم الآليات وتحسين البروتوكولات لهذه العملية في تحرير الجينات أمر حاسم لتطوير علاجات ناجحة تعتمد على خلايا المناعة [28]. بينما تظهر استراتيجيات CRISPR المعتمدة وعدًا كبيرًا في تعزيز الاستجابة المناعية، من الضروري التحقيق في آثارها طويلة المدى على جهاز المناعة لدى المضيف [30].
| استراتيجية | آلية العمل | المزايا | العيوب | نتائج ما قبل السريرية/السريرية | مرجع |
| تعطيل الجينات التي تحفز نمو الورم | استهداف وتعطيل الجينات المسرطنة وجينات مثبطات الورم لوقف نمو خلايا السرطان وتحفيز موت الخلايا المبرمج | استهداف دقيق: يمكن تصميم CRISPR لاستهداف جينات معينة أو طفرات تحفز نمو الورم، مما يزيد من الخصوصية ويقلل من الآثار الجانبية غير المستهدفة. فعالية عالية: أظهرت الدراسات أن تعطيل بعض الجينات باستخدام CRISPR يمكن أن يؤدي إلى تراجع الورم وزيادة البقاء في نماذج حيوانية | احتمالية حدوث آثار جانبية غير مستهدفة: بينما يوفر CRISPR خصوصية عالية، لا يزال هناك احتمال حدوث تغييرات غير مقصودة في الجينوم قد تسبب ضررًا للمريض. صعوبة استهداف جينات معينة أو توصيل العلاج إلى موقع الورم: قد يكون من الصعب استهداف بعض الأورام باستخدام طرق التوصيل الحالية | أظهرت الدراسة فعالية استخدام CRISPR لتعطيل الجين المسرطن KRAS في نموذج الفأر لسرطان الرئة. هذه خطوة رئيسية نظرًا لأن طفرات KRAS معروفة بصعوبتها في الاستهداف باستخدام علاجات أخرى | [16] |
| تعزيز الاستجابة المناعية ضد خلايا السرطان | تحرير خلايا المناعة للتعرف على خلايا السرطان وتدميرها، مثل تحرير خلايا T للتعبير عن مستقبلات مستضدات كيميرية (CARs) | يعزز الاستجابة المناعية الطبيعية للجسم: من خلال تحرير خلايا المناعة للتعرف على خلايا السرطان ومهاجمتها، يمكن للعلاج المناعي المعتمد على CRISPR تنشيط الاستجابة المناعية الطبيعية للجسم لمحاربة السرطان. يتجنب الآثار السامة للعلاج الكيميائي: على عكس العلاج الكيميائي، الذي يمكن أن يكون له آثار جانبية كبيرة، فإن العلاج المناعي باستخدام خلايا T المعدلة بواسطة CRISPR لديه القدرة على أن يكون نهج علاج أكثر استهدافًا وأقل سمية | احتمالية حدوث سمية أو رفض مناعي: هناك خطر من أن خلايا المناعة المعدلة بواسطة CRISPR قد تهاجم خلايا صحية أو يتم رفضها من قبل جهاز المناعة لدى المريض. توفر محدود من خلايا T معينة للتحرير: قد يكون من الصعب الحصول على عدد كافٍ من خلايا
|
أفادت الدراسة بتحقيق شفاء كامل في اثنين من ثلاثة مرضى تم علاجهم بخلايا T المعدلة بواسطة CRISPR في تجربة لعلاج اللمفومات المقاومة. هذه نتيجة واعدة، على الرغم من الحاجة إلى مزيد من البحث لتحديد سلامة وفعالية هذا النهج في مجموعات مرضى أكبر | [17] |
| إصلاح الطفرات الجينية التي تسبب السرطان | تصحيح الطفرات الجينية في جينات مثبطات الورم، جينات إصلاح DNA، أو جينات محركة أخرى | استهداف دقيق: يمكن استخدام CRISPR لتصحيح طفرات جينية معينة تسبب السرطان، مما قد يؤدي إلى فوائد طويلة الأمد. إمكانية تحقيق فوائد طويلة الأمد: قد يؤدي إصلاح الطفرات الجينية التي تسبب السرطان إلى تحقيق فوائد طويلة الأمد للمرضى | احتمالية حدوث آثار جانبية غير مستهدفة: كما هو الحال مع غيرها من الأساليب المعتمدة على CRISPR، هناك خطر حدوث تغييرات غير مقصودة في الجينوم قد تسبب ضررًا للمريض. صعوبة توصيل العلاج إلى موقع الورم: قد يكون من الصعب توصيل العلاج المعتمد على CRISPR مباشرة إلى موقع الورم | نتائج واعدة في الدراسات ما قبل السريرية، مثل استخدام CRISPR لتصحيح طفرات BRCA1 في خلايا سرطان المبيض | [18] |
| توصيل جزيئات قاتلة للسرطان مباشرة إلى خلايا الورم | استخدام CRISPR لتحرير الجينوم لفيروس أو بكتيريا لاستهداف خلايا السرطان بشكل محدد، وتوصيل جزيئات علاجية مثل السموم أو المعدلات المناعية مباشرة إلى خلايا الورم | استهداف دقيق: من خلال تعديل الجينوم لفيروس أو بكتيريا لاستهداف خلايا السرطان بشكل محدد، يمكن أن تقدم العلاجات المعتمدة على كريسبر توصيلًا مستهدفًا ودقيقًا للجزيئات العلاجية إلى خلايا الورم. فعالية عالية: أظهرت الدراسات أن توصيل الجزيئات القاتلة للسرطان بواسطة كريسبر يمكن أن يؤدي إلى تراجع الورم وزيادة البقاء في نماذج حيوانية. | احتمالية حدوث آثار غير مستهدفة: كما هو الحال مع approaches المعتمدة على كريسبر الأخرى، هناك خطر من تغييرات غير مقصودة في الجينوم قد تسبب ضررًا للمريض. توفر محدود للفيروسات أو البكتيريا المحددة للتعديل: قد يكون من الصعب الحصول على عدد كافٍ من الفيروسات أو البكتيريا المحددة للتعديل، مما قد يحد من الاستخدام الواسع النطاق لهذا النهج. | نتائج واعدة في الدراسات قبل السريرية، مثل استخدام كريسبر لتوصيل كريسبر لتفعيل الميكروRNAs المثبطة للورم في خلايا سرطان الكبد. تشمل أمثلة أخرى استخدام كريسبر لتوصيل حمولات سامة إلى خلايا الورم، مثل دراسة حيث تم استخدام كريسبر لتعديل البكتيريا لإنتاج سم يستهدف خلايا السرطان بشكل محدد في نموذج فأر لسرطان البنكرياس. | [17، 18] |
إصلاح الطفرات الجينية
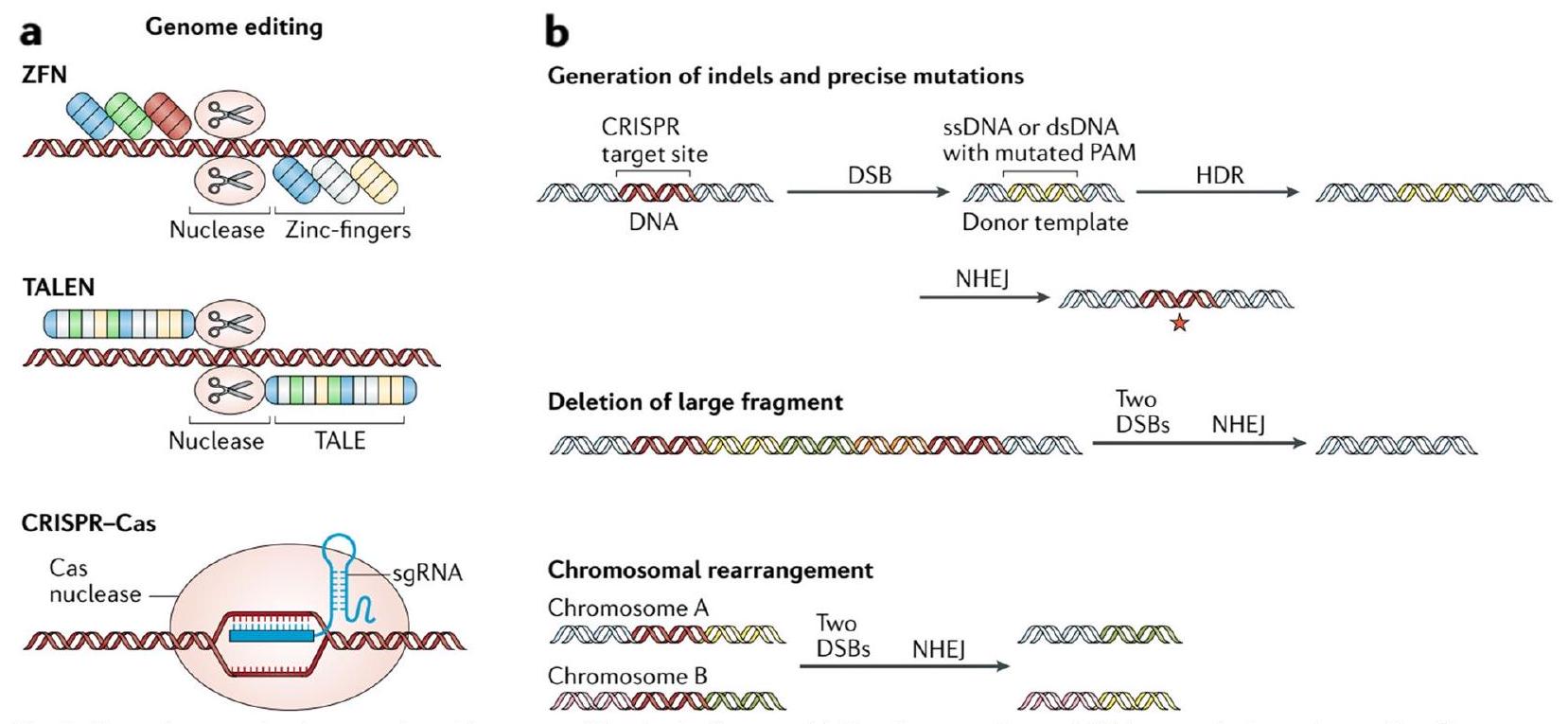
لطرق الإصلاح المعتمدة على كريسبر، مثل HDR وNHEJ، أمر حيوي. يمكن أن يقدم HDR التغييرات الجينية المرغوبة بدقة من خلال استخدام قالب مانح، ولكن كفاءته غالبًا ما تكون أقل مقارنة بـ NHEJ، الذي يمكن أن يؤدي إلى إدخالات أو حذف دون قالب [40]. سيساعد فهم التوازن بين الكفاءة والدقة في تحسين اختيار آلية الإصلاح للطفرات الجينية المحددة [41]. بينما أظهر كريسبر وعدًا كبيرًا، قد تكون هناك عواقب غير متوقعة من التلاعب بالجينيوم. قد تشمل هذه الطفرات غير الانتقائية أو إعادة ترتيب الجينوم على نطاق واسع، مما قد يقدم شذوذات جينية جديدة أو يسبب آثارًا غير مقصودة على تنظيم الجينات [42]. التقييم الدقيق والتقييم الشامل للنتائج غير المقصودة المحتملة أمر ضروري لضمان سلامة وموثوقية استراتيجيات كريسبر المعتمدة [37]. فهم استقرار الإصلاحات الجينية الناتجة عن كريسبر أمر حاسم لتقييم الجدوى طويلة الأمد للعلاجات المحتملة [40]. يجب أن تكون التعديلات الجينية مستقرة وتنتقل بدقة خلال انقسامات الخلايا لتوفير فوائد علاجية دائمة. سيوفر التحقيق في وراثة واستقرار الطفرات الجينية المصلحة الضوء على طول العمر وفعالية استراتيجيات كريسبر المعتمدة [41]. عند استخدام كريسبر-كاس9 للتطبيقات الحية، من الضروري تقييم الاستجابات المناعية المحتملة.
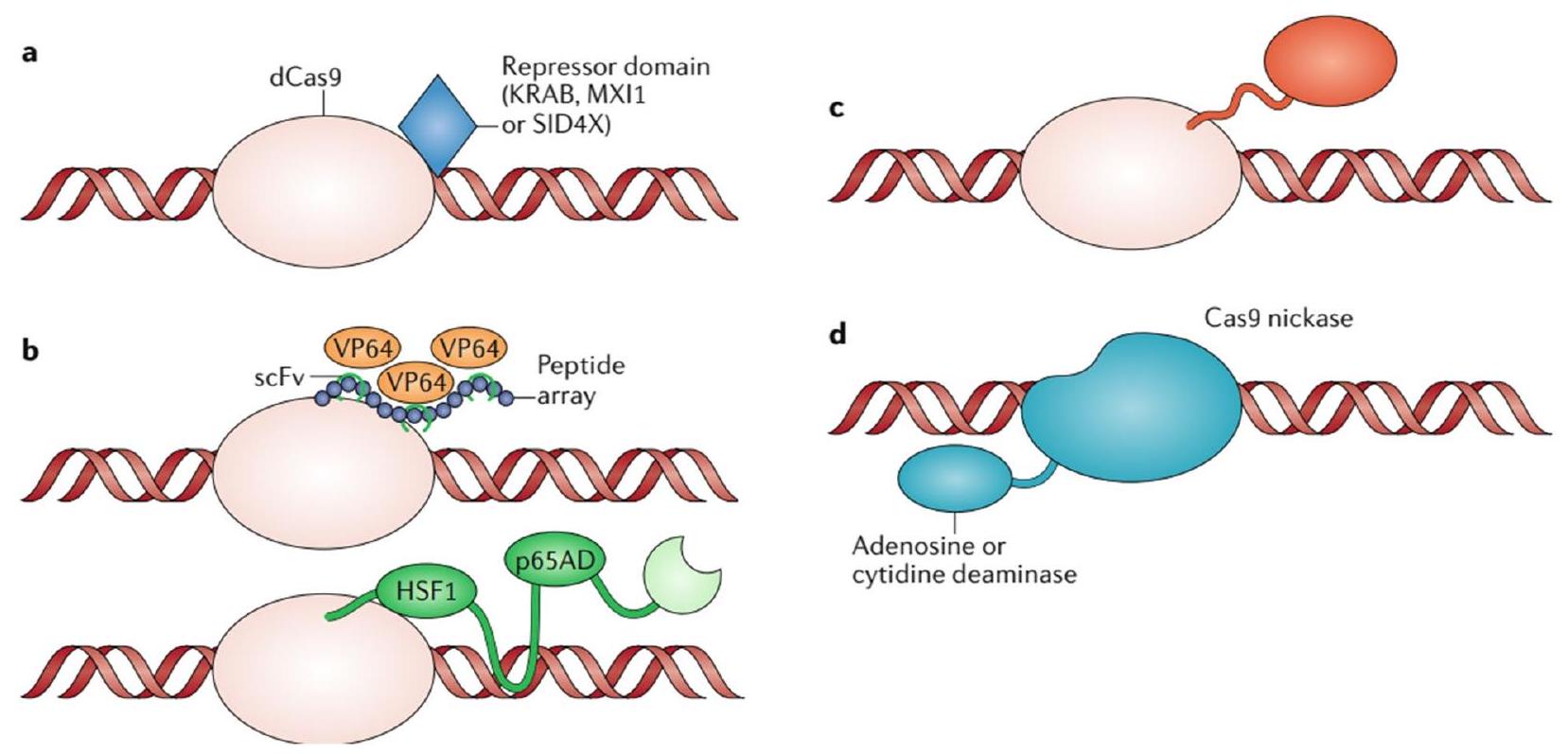
توصيل الجزيئات القاتلة للسرطان
يمكن أن تميز جزيئات RNA الدليل بشكل فعال بين الخلايا السرطانية والخلايا السليمة [50]. يعمل مركب Cas كحامل لجزيئات RNA الدليل. يشكل مركبًا مع RNA الدليل، مما ينشئ مركب CRISPR-Cas RNP [51]. يوفر بروتين Cas الآلات اللازمة للتعرف على RNA الدليل ويسهل ارتباطه بالحمض النووي المستهدف داخل الخلايا السرطانية. يعمل هذا المركب كقاطع جزيئي قوي، حيث يقطع ويعطل الجينات المسرطنة المسؤولة عن نمو السرطان [50]. يمكن إدخال مركب CRISPR-Cas RNP إلى جسم المريض من خلال طرق مختلفة. تتضمن إحدى الطرق الحقن المباشر في الأنسجة المستهدفة أو موقع الورم [49]. تتضمن طريقة أخرى هندسة خلايا المناعة، مثل خلايا T، للتعبير عن مركب CRISPR-Cas RNP. يمكن بعد ذلك إعادة إدخال هذه الخلايا المناعية المهندسة إلى مجرى دم المريض، حيث يمكنها استهداف الخلايا السرطانية ومهاجمتها بشكل محدد [51]. بمجرد دخولها إلى الخلايا السرطانية، يقوم مركب CRISPR-Cas RNP بتحديد تسلسلات الحمض النووي المستهدفة ويقطعها بدقة، مما يعطل الجينات المسرطنة المسؤولة عن الخباثة. يؤدي هذا التعطيل إلى إما بدء عملية الموت الخلوي (الموت الخلوي) في الخلايا السرطانية أو يجعلها أكثر عرضة للاستجابة المناعية للجسم، مما يؤدي إلى تدميرها [49]. تم تصميم جزيئات RNA الدليل لاستهداف الخلايا السرطانية بشكل محدد من خلال الارتباط بعلامات جينية فريدة أو طفرات موجودة في تلك الخلايا. من خلال استهداف الخلايا السرطانية بشكل انتقائي، ينجح مركب CRISPR-Cas RNP في تجنيب الخلايا السليمة من الضرر، مما يقلل من الآثار الجانبية المحتملة [52]. بالإضافة إلى ذلك، يسمح استخدام خلايا المناعة المهندسة بمزيد من التحديد في استهداف الخلايا السرطانية، مما يقلل من التأثير على الأنسجة السليمة [52، 53].
الدراسات ما قبل السريرية والتجارب السريرية لعلاج السرطان القائم على CRISPR
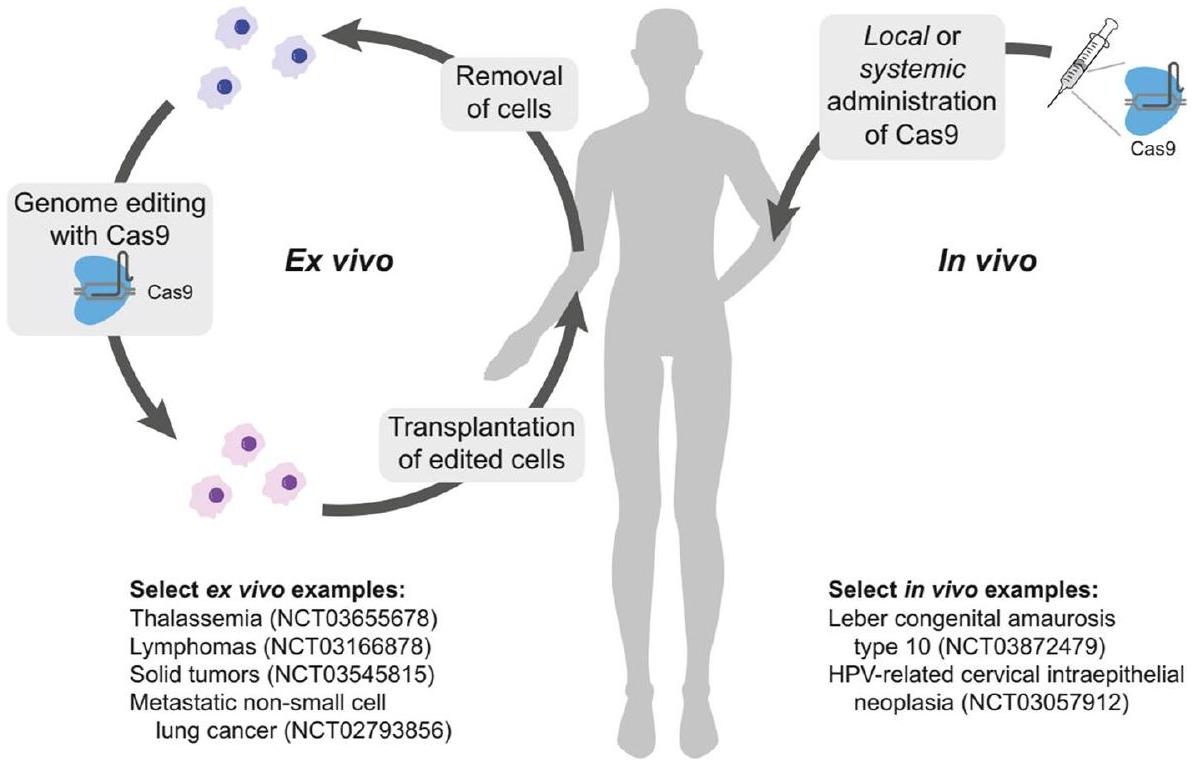
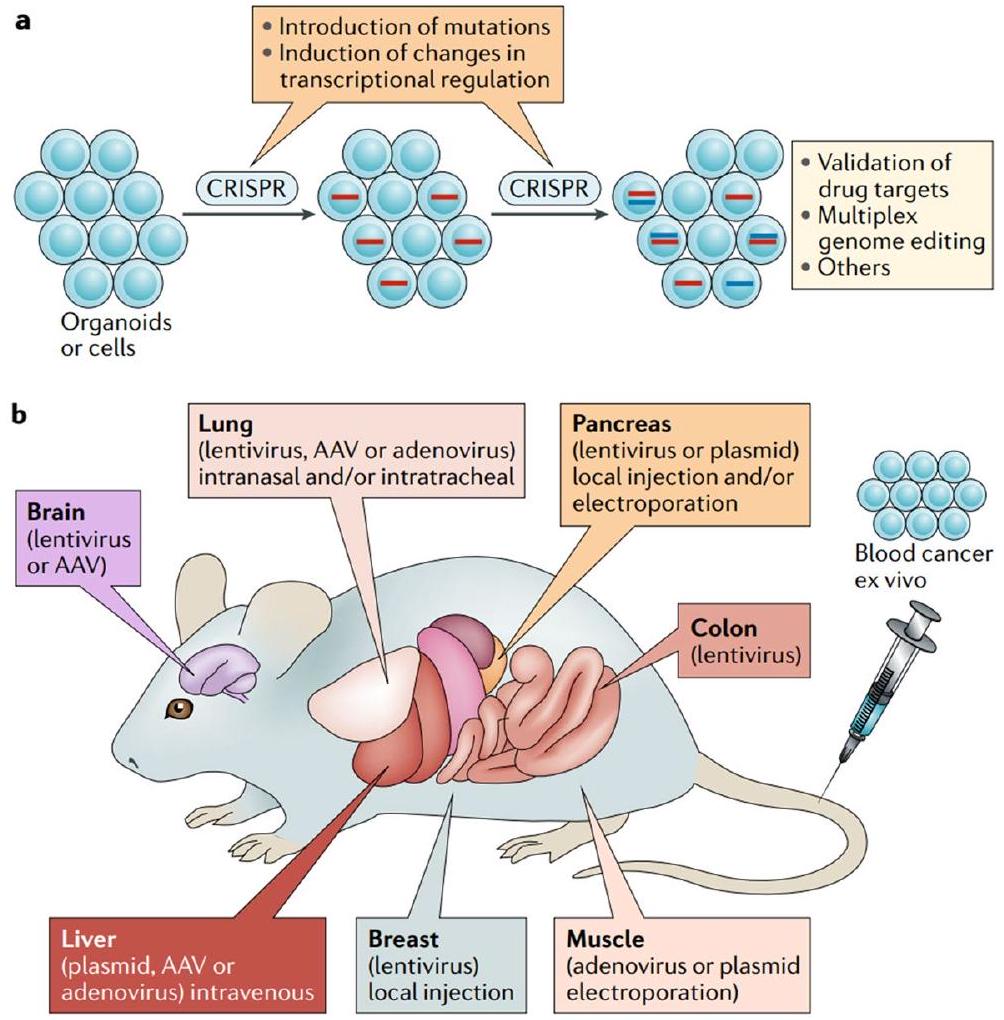
الدراسات ما قبل السريرية
P53 هو بروتين مثبط للورم حاسم مسؤول عن تنظيم تقدم دورة الخلية ومنع تكوين خلايا سرطانية [94]. في سياق تحرير الجينوم باستخدام CRISPR/Cas9، تصبح نشاط P53 مصدر قلق حاسم حيث أن استخدام هذه التقنية قد يؤدي إلى آثار جانبية غير مستهدفة، مما يسبب ضررًا غير مقصود للحمض النووي [95]. لذلك، من الضروري تقييم
أثر CRISPR/Cas9 على تعبير ووظيفة P53 لضمان سلامة وفعالية عملية التحرير [96]. يلعب P53 دورًا حاسمًا في مراقبة سلامة الحمض النووي للخلايا وتحفيز توقف دورة الخلية أو الموت الخلوي في حالة حدوث ضرر للحمض النووي [97]. للأسف، P53 عرضة للطفرات، مما يؤدي إلى تعطيله أو عدم وظيفته. هذه الطفرات شائعة في العديد من أنواع السرطان، بما في ذلك سرطان الرئة، وتساهم في نمو الخلايا غير المنضبط وتطور الورم [98]. يمكن أن يؤدي تعديل جين P53 إلى استعادة وظيفته، مما يؤدي إلى كبح نمو الخلايا السرطانية [94]. عندما يتم تنشيط جين P53 المعدل، فإنه يعزز إنتاج بروتين P21، وهو مثبط ورم معروف ينظم دورة الخلية. يؤدي زيادة تعبير P21 إلى توقف دورة الخلية، مما يمنع الخلايا السرطانية من التكاثر بشكل غير منضبط [96]. علاوة على ذلك، فإن تنشيط P21 يجعل الخلايا السرطانية أكثر عرضة للعلاج الكيميائي، حيث أن الخلايا التي تحتوي على بروتينات P21 نشطة تكون أكثر عرضة للموت الخلوي عند تعرضها لأدوية العلاج الكيميائي. يُعرف P21 أيضًا باسم مثبط كيناز المعتمد على السايلين 1A (CDKN1A)، وهو مثبط كيناز معتمد على السايلين يلعب دورًا محوريًا في تنظيم دورة الخلية وتعزيز توقف دورة الخلية. في سياق تحرير الجينوم باستخدام CRISPR/Cas9، قد يعمل P21 كسلاح ذو حدين [99]. من ناحية، قد يؤدي ارتفاع مستوياته استجابةً لضرر الحمض النووي الناجم عن CRISPR/Cas9 إلى توقف دورة الخلية، مما يمنع الخلايا من التكاثر وقد يهدد فعالية
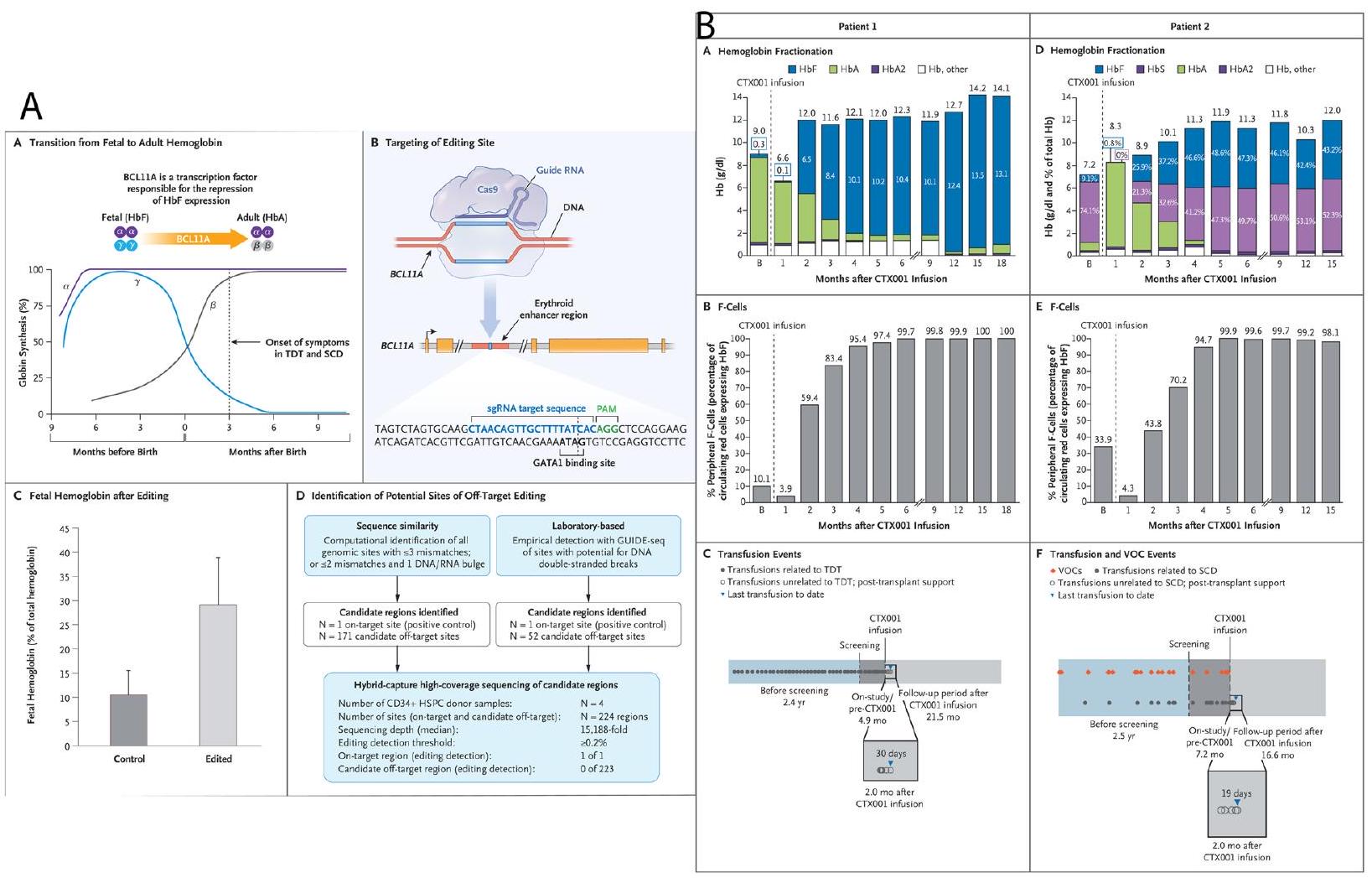
| نوع الخلية/النسيج | نهج كريسبر | النتائج | القيود/التحديات | مرجع |
| خلايا T من الفئران والبشر | إسكات PD-1 | زيادة نشاط خلايا T ضد خلايا السرطان، بقاء مطول في الفئران | مخاوف السلامة المتعلقة بإزالة جين PD-1 على المدى الطويل | [٥٩] |
| خلايا سرطان المبيض | إسكات الجين المسرطن | تقليل تكاثر الخلايا وتكوين المستعمرات | تقييم محدود للتأثيرات غير المستهدفة | [60] |
| خلايا سرطان الرئة | إسكات EGFR الطافر | انخفاض حيوية الخلايا ونمو الورم في الفئران | آثار غير مستهدفة في بعض الخلايا | [61] |
| خلايا سرطان الثدي الثلاثي السلبية | إسكات AXL | تقلص نمو الورم وزيادة الحساسية للعلاج الكيميائي | تقييم محدود للتأثيرات غير المستهدفة | [62] |
| خطوط خلايا السرطان المتنوعة | إسكات HIF-1α | تقليل نمو الورم وزيادة الحساسية للعلاج الإشعاعي | تقييم محدود للتأثيرات غير المستهدفة | [63] |
| خلايا الميلانوما | إسكات BRAF V600E | تقلص نمو الورم وزيادة الحساسية للعلاج المستهدف | آثار غير مستهدفة في بعض الخلايا | [64] |
| خلايا اللوكيميا | إسقاط MCL-1 | تحفيز موت الخلايا المبرمج وتقليل نمو الورم | تقييم محدود للتأثيرات غير المستهدفة | [65] |
| خلايا T البشرية | إسكات وتعبير مفرط عن NKG2D | تحسين التعرف على خلايا الورم وقتلها بواسطة خلايا CAR-T | المخاوف المتعلقة بالسلامة من الإفراط في التعبير عن NKG2D على المدى الطويل | [66,67] |
| الأورام الصلبة لدى الأطفال | إسكات الجينات المسرطنة الناتجة عن الاندماج | تقلص نمو الورم وزيادة الحساسية للعلاج الكيميائي | تقييم محدود للتأثيرات غير المستهدفة | [68] |
| خلايا سرطان البروستاتا | إسكات مستقبل الأندروجين | تقليل تكاثر الخلايا ونمو الورم في الفئران | آثار غير مستهدفة في بعض الخلايا | [69] |
| خلايا سرطان البنكرياس | إسكات TGF-
|
تقلص نمو الورم وزيادة الحساسية للعلاج الكيميائي | تقييم محدود للتأثيرات غير المستهدفة | [70] |
| خلايا سرطان الثدي | إسكات نهاية C من PAK1 | تقليل تكاثر الخلايا ونمو الورم في الفئران | آثار غير مستهدفة في بعض الخلايا | [71] |
| خلايا لمفوما بائية كبيرة منتشرة | إسكات BCL6 | تحفيز موت الخلايا المبرمج وتقليل نمو الورم | تقييم محدود للتأثيرات غير المستهدفة | [72] |
| خلايا الجليوبلاستوما | إسكات طفرات محفز TERT | تقليل تكاثر الخلايا ونمو الورم في الفئران | تقييم محدود للتأثيرات غير المستهدفة | [73] |
| خلايا اللوكيميا النقوية الحادة | إسكات جين GATA2 | تحفيز التمايز وتقليل نمو الورم | تقييم محدود للتأثيرات غير المستهدفة | [74] |
| خلايا الجذع الدموية البشرية | إسكات معزز BCL11A | تحفيز تعبير الهيموغلوبين الجنيني وتقليل أعراض فقر الدم المنجلي | تقييم محدود للتأثيرات غير المستهدفة | [75] |
| خلايا اللوكيميا اللمفاوية الحادة من نوع ب | إسكات CD19 | تحفيز موت الخلايا المبرمج وتقليل نمو الورم في الفئران | آثار غير مستهدفة في بعض الخلايا | [76] |
| خلايا لمفوما بائية كبيرة منتشرة | إسكات EZH2 | تقليل تكاثر الخلايا ونمو الورم في الفئران | تقييم محدود للتأثيرات غير المستهدفة | [77] |
| خلايا الورم الدبقي | إسكات IDH1 | انخفاض حيوية الخلايا ونمو الورم في الفئران | آثار غير مستهدفة في بعض الخلايا | [78] |
| خلايا الورم العصبي | إسكات MYCN | تحفيز موت الخلايا المبرمج وتقليل نمو الورم | تقييم محدود للتأثيرات غير المستهدفة | [79] |
| خلايا سرطان المبيض | إسكات MUC16 | تقليل تكاثر الخلايا والغزو | تقييم محدود للتأثيرات غير المستهدفة | [80] |
| خلايا ربيوميوساكروما الحويصلات الهوائية | إسكات PAX7-FOXO1 | تقليل تكاثر الخلايا ونمو الورم في الفئران | آثار غير مستهدفة في بعض الخلايا | [81] |
| خلايا سرطان المريء | إسكات SOX2 | تقليل تكاثر الخلايا وتكوين المستعمرات | تقييم محدود للتأثيرات غير المستهدفة | [82] |
| خلايا سرطان الكبد | إسكات TERT | تقليل تكاثر الخلايا ونمو الورم في الفئران | تقييم محدود للتأثيرات غير المستهدفة | [83] |
| خلايا سرطان القولون والمستقيم | إسكات Wnt/
|
تقليل تكاثر الخلايا ونمو الورم في الفئران | آثار غير مستهدفة في بعض الخلايا | [84] |
| نوع الخلية/النسيج | نهج كريسبر | النتائج | القيود/التحديات | مرجع |
| خلايا سرطان الثدي | إسكات P53 | زيادة تكاثر الخلايا وتكوين المستعمرات | آثار غير مستهدفة في بعض الخلايا | [85] |
| خلايا سرطان البنكرياس | إسكات KRAS | انخفاض حيوية الخلايا ونمو الورم في الفئران | تقييم محدود للتأثيرات غير المستهدفة | [86] |
| خلايا سرطان عنق الرحم | إسكات BIRC5 | تقليل تكاثر الخلايا ونمو الورم في الفئران | تقييم محدود للتأثيرات غير المستهدفة | [87] |
| خلايا سرطان البروستاتا | إسكات EZH2 | تقليل تكاثر الخلايا وتكوين المستعمرات | آثار غير مستهدفة في بعض الخلايا | [79] |
| خلايا الساركوما العظمية | إسكات HIF-1α | تقليل تكاثر الخلايا ونمو الورم في الفئران | تقييم محدود للتأثيرات غير المستهدفة | [88] |
| خلايا اللوكيميا اللمفاوية الحادة | إسكات MYB | تحفيز موت الخلايا المبرمج وتقليل نمو الورم | تقييم محدود للتأثيرات غير المستهدفة | [89] |
| خلايا سرطان الكلى | إسكات HIF-2α | تقليل تكاثر الخلايا ونمو الورم في الفئران | تقييم محدود للتأثيرات غير المستهدفة | [90] |
| خلايا سرطان الكبد | إسكات SALL4 | تقليل تكاثر الخلايا ونمو الورم في الفئران | تقييم محدود للتأثيرات غير المستهدفة | [91] |
| خلايا الميلانوما | إسكات CDK6 | تقليل تكاثر الخلايا ونمو الورم في الفئران | تقييم محدود للتأثيرات غير المستهدفة | [92] |
| خلايا اللوكيميا النقوية الحادة | إسكات ASXL1 | تحفيز التمايز وتقليل نمو الورم | تقييم محدود للتأثيرات غير المستهدفة | [93] |
من تطوير نماذج بديلة جديدة وذات صلة سريرية للدراسات الانتقالية [102].
خلايا السرطان أكثر عرضة للعلاج الكيميائي، حيث تميل الخلايا التي تم تنشيط P21 فيها إلى أن تكون أكثر استجابة لعلاج الكيميائي [106، 111]. يبرز بعض الباحثين إمكانية تحرير القاعدة المعتمد على CRISPR كمورد قيم للتقييم الوظيفي وإعادة تصنيف المتغيرات ذات الأهمية غير المؤكدة (VUSs) في جين BRCA1. علاوة على ذلك، تناولت هذه التحقيقات العقبات المرتبطة بتقييم الوظائف وتحديد pathogenicity للمتغيرات الجديدة في BRCA1، والتي من المعروف أنها تزيد بشكل كبير من خطر الإصابة بسرطان الثدي والمبيض وعادة ما يتم تحديدها من خلال الاختبارات الجينية السريرية. لتجاوز هذه العقبات، استخدم العلماء محرر قاعدة السايتوسين BE3 المعتمد على CRISPR للتحليل الوظيفي. قاموا بإجراء فحص شامل لتحرير القاعدة المعتمد على CRISPR باستخدام 745 RNA مرشد يستهدف جميع الإكسونات في BRCA1، مما أدى إلى تحديد عدة متغيرات غير معروفة سابقًا، بما في ذلك c.-97C > T، c.154C > T، c.3847C > T، c.5056C > T، و c.4986+5G>A. عرضت الدراسة بفعالية فائدة تحرير القاعدة المعتمد على CRISPR كأداة قوية لإعادة تقييم المتغيرات ذات الأهمية غير المؤكدة (VUSs) في BRCA1، مما يوفر رؤى قيمة للإدارة السريرية. يمكن أن يكون لهذا إعادة تصنيف VUSs في BRCA1 آثار كبيرة على المرضى ومقدمي الرعاية الصحية. يمكن للمرضى الذين تم توضيح تصنيفات المتغيرات أن يتلقوا تقييمات مخاطر أكثر دقة وخطط علاج فردية، قد تشمل زيادة المراقبة أو تدابير وقائية. بالنسبة لمقدمي الرعاية الصحية، تضمن تصنيف المتغيرات بدقة تقديم المشورة المناسبة والتواصل حول المخاطر للمرضى وعائلاتهم [112].
تعديلات غير مستهدفة. يمكن أن يقلل التصميم الدقيق لـ RNA المرشد، باستخدام خوارزميات متقدمة، والتحقق من المواقع غير المحددة المحتملة من خلال التسلسل من هذه الآثار [118]. لا يزال تسليم مكونات CRISPR-Cas9 إلى الخلايا المستهدفة يمثل تحديًا [119]. تم استكشاف طرق مثل الناقلات الفيروسية، والجسيمات النانوية الدهنية، والكهربة. لكل نهج مزايا وقيود من حيث الكفاءة، والسمية، والخصوصية [120]. يعزز تنشيط بروتين P21، وهو مثبط ورم معروف، إيقاف دورة الخلية عن طريق تثبيط كينازات معتمدة على السايلين التي تنظم انقسام الخلايا. من خلال إيقاف دورة الخلية، يتم تثبيط نمو خلايا السرطان [121]. يمكن أن يؤدي تنشيط بروتين P21 من خلال تحرير جين KRAS إلى زيادة حساسية خلايا السرطان للعلاج الكيميائي [122]. يؤدي زيادة تعبير P21 إلى إيقاف دورة الخلية، مما يسمح للأدوية الكيميائية باستهداف وقتل خلايا السرطان بشكل أكثر فعالية [123]. يؤدي تنشيط بروتين P21 من خلال تحرير جين KRAS إلى زيادة حساسية خلايا السرطان للعلاج الكيميائي. يسمح إيقاف دورة الخلية الناتج عن تنشيط P21 للأدوية الكيميائية باستهداف والقضاء على خلايا السرطان بشكل أكثر فعالية [115].
مع تشابه جزئي مع الموقع المستهدف [124]. لقد أدت الأبحاث المكثفة وتحسين تصميم RNA الموجه إلى تقليل الآثار الجانبية غير المرغوب فيها بشكل كبير [127]. لقد حسنت متغيرات Cas9 المتطورة، مثل Cas9 عالي الدقة ومحررات القواعد، من التخصص بشكل أكبر، مما يقلل من خطر التعديلات الجينية غير المقصودة [124]. تشمل التحديات المرتبطة بتحرير جين EGFR باستخدام CRISPR-Cas9 الآثار الجانبية غير المرغوب فيها، وكفاءة التسليم، والآثار المحتملة على المدى الطويل. تشمل الاعتبارات الأخلاقية الحاجة إلى الحصول على موافقة مستنيرة، وضمان الوصول العادل إلى التكنولوجيا، والاستخدام المسؤول لتجنب العواقب غير المقصودة أو إنشاء “أطفال مصممين”. التقييم الدقيق، والتنظيم، والإرشادات الأخلاقية ضرورية للتعامل مع هذه التحديات وضمان التطبيق المسؤول لتقنية CRISPR-Cas9 في تحرير جين EGFR أو أي جين آخر [128].
يمكن أن يلعب تحرير جين VEGF (عامل نمو الأوعية الدموية) باستخدام تقنية كريسبر-كاس9 دورًا حاسمًا في أنواع مختلفة من السرطان. VEGF هو بروتين يعزز نمو الأوعية الدموية الجديدة، وهي عملية تعرف باسم تكوين الأوعية، والتي تعتبر ضرورية لتطور الورم وانتشاره. من خلال استهداف وتعطيل جين VEGF باستخدام كريسبر-كاس9، يمكن للباحثين أن يعيقوا إنتاج VEGF وبالتالي يثبطوا تكوين الأوعية الدموية في الورم. قد يؤدي ذلك إلى تقليل نمو الورم وزيادة حساسية العلاجات الأخرى للسرطان. على الرغم من أن كريسبر-كاس9 محدد للغاية، إلا أن هناك احتمال حدوث آثار غير مستهدفة حيث تحدث تعديلات جينية غير مقصودة. في حالة تحرير جين VEGF، يجب على الباحثين تقييم المواقع المحتملة غير المستهدفة بعناية لضمان عدم تعديل أي جينات حيوية بشكل غير مقصود. لتقليل الآثار غير المرغوب فيها، يتم استخدام تحليلات المعلوماتية الحيوية الصارمة وطرق الفحص المتقدمة لاختيار RNA الموجه بأقل احتمال لنشاط المواقع غير المحددة. قد لا يكون تحرير جين VEGF القائم على كريسبر-كاس9 كافيًا بمفرده لعلاج السرطان بشكل كامل. على الرغم من أنه يمكن أن يعيق تكوين الأوعية الدموية في الورم، فإن استراتيجية علاج السرطان الشاملة عادة ما تتضمن دمج كريسبر-كاس9 مع علاجات أخرى مثل العلاج الكيميائي أو الإشعاعي أو المناعي. يمكن أن يؤدي دمج العلاجات إلى تأثير تآزري، يستهدف خلايا السرطان من خلال مسارات متعددة ويزيد من الفعالية العلاجية العامة. إن توصيل مكونات كريسبر-كاس9 إلى خلايا السرطان يمثل تحديًا كبيرًا. قد يحد الحجم الكبير لبروتين كاس9 ومجمع RNA الموجه من طرق التوصيل. يتم استكشاف طرق مختلفة، بما في ذلك الناقلات الفيروسية والجزيئات النانوية والليبوبومات، لضمان توصيل فعال وآمن إلى خلايا السرطان المستهدفة مع تجنب الإضرار بالأنسجة السليمة. إن ضمان كبت جين VEGF على المدى الطويل والثابت أمر ضروري لتحقيق تأثيرات علاجية مستدامة. يقوم الباحثون بالتحقيق في طرق لتحسين توصيل كريسبر-كاس9.
واستقرار داخل خلايا السرطان [129]. استراتيجيات مثل استخدام متغيرات كاس9 المعدلة أو دمج مكونات كريسبر في جينوم خلايا الهدف يمكن أن تعزز بشكل محتمل متانة تحرير جين VEGF وتأثيراته المضادة للسرطان [131].
إلى نمو الخلايا غير المنضبط، وهو سمة من سمات السرطان [137]. من خلال استخدام CRISPR-Cas9 لاستهداف وتحرير جين PTEN بدقة، يمكن للباحثين استعادة وظيفته كمثبط للأورام، مما يعيق نمو خلايا السرطان ويقلل من تطور الأورام [138]. يختلف احتمال استخدام تحرير جين PTEN بواسطة CRISPR-Cas9 كخيار علاجي بين أنواع السرطان المختلفة [136]. تظهر بعض أنواع السرطان طفرات في جين PTEN كمحرك رئيسي لتكوين الأورام، مما يجعلها أكثر قابلية لهذا النهج [135]. ومع ذلك، قد تعتمد فعالية هذه الاستراتيجية على السياق الجيني للسرطان، حيث قد تمتلك بعض الأورام آليات بديلة لتجاوز وظيفة PTEN [138]. هناك حاجة إلى دراسات قبل السريرية واسعة النطاق وتجارب سريرية لتحديد قابليتها وفعاليتها عبر أنواع السرطان المتنوعة [135]. تتعلق مخاوف السلامة في تحرير جين PTEN بواسطة CRISPR-Cas9 في علاج السرطان بالتأثيرات الجانبية غير المرغوب فيها المحتملة، حيث يمكن أن تحدث تغييرات جينية غير مقصودة في الخلايا غير السرطانية، مما يؤدي إلى عواقب سلبية [136]. بالإضافة إلى ذلك، يجب تقييم خطر إدخال طفرات جديدة أو تغيير جينات أساسية أخرى بعناية لتجنب الآثار الجانبية غير المرغوب فيها [138]. يعد الاختبار الدقيق في النماذج قبل السريرية والمراقبة الدقيقة خلال التجارب السريرية أمرًا حيويًا لضمان سلامة وقابلية هذا النهج العلاجي [135]. يستكشف الباحثون باستمرار استراتيجيات متنوعة لتحسين كفاءة تحرير جين CRISPR-Cas9 [136]. تتضمن إحدى الطرق تحسين نظام التسليم لضمان استهداف دقيق لخلايا السرطان. بالإضافة إلى ذلك، تقدم التطورات في تكنولوجيا CRISPR-Cas9، مثل استخدام محررات القواعد أو المحررات الرئيسية، تعديلات أكثر دقة وتقليل التأثيرات الجانبية غير المستهدفة [138]. علاوة على ذلك، قد يؤدي دمج CRISPR-Cas9 مع علاجات أخرى، مثل العلاجات المناعية أو العلاجات المستهدفة، إلى تعزيز الاستجابة العلاجية العامة، مما يسمح باستراتيجية علاجية أكثر شمولاً وفعالية. هناك العديد من التحديات التي يجب معالجتها عند استخدام CRISPR-Cas9 لتحرير جين PTEN في خلايا السرطان [135]. أولاً، يعد التسليم الفعال لمكونات CRISPR إلى خلايا السرطان المحددة أمرًا حيويًا لتجنب التأثيرات الجانبية غير المستهدفة [138]. ثانيًا، يعد ضمان التحرير الصحيح والدقيق لجين PTEN دون إدخال طفرات غير مقصودة أمرًا حيويًا لنجاح العلاج [135]. بالإضافة إلى ذلك، يجب تقييم الاستجابة المناعية لمكونات CRISPR وخطر الرفض المناعي المحتمل للخلايا المعدلة لتقييم قابليتها وسلامتها على المدى الطويل [138].
في نمو الأورام وتقدمها. يمكن استخدام CRISPR-Cas9 كأداة لتحرير الجينات لاستهداف وتعديل جين TERT في خلايا السرطان [140]. يتكون نظام CRISPR-Cas9 من RNA دليلي يوجه إنزيم Cas9 إلى الموقع الجيني المطلوب [141]. من خلال تصميم RNA دليلي محدد لتسلسل جين TERT، يمكن للباحثين توجيه Cas9 إلى جين TERT وإحداث كسر مزدوج في الموقع المستهدف [142]. ثم تقوم آلية إصلاح الحمض النووي في الخلية بإصلاح الكسر، غالبًا من خلال مسار NHEJ المعرض للأخطاء، والذي يقدم إدخالات أو حذف صغير (indels) تعطل وظيفة جين TERT [139]. بدلاً من ذلك، يمكن للباحثين استخدام CRISPR-Cas9 بالاشتراك مع قالب إصلاح لإدخال تعديلات محددة على تسلسل جين TERT، مثل حذف الجينات أو الطفرات النقطية [141]. يمكن أن يؤدي تحرير جين TERT باستخدام CRISPR-Cas9 إلى عدة نتائج. إحدى الاحتمالات هي تعطيل وظيفة جين TERT، مما يؤدي إلى انخفاض نشاط التيلوميراز في خلايا السرطان. يمكن أن يؤدي ذلك إلى تقصير التيلوميرات وشيخوخة الخلايا أو موتها، مما يعيق الإمكانية التكرارية غير المحدودة لخلايا السرطان [139]. نتيجة محتملة أخرى هي تعديل تعبير جين TERT، مثل تقليل مستوى تعبيره، مما يمكن أن يعيق نمو الورم [142]. بالإضافة إلى ذلك، قد يجعل تحرير جين TERT بواسطة CRISPR-Cas9 خلايا السرطان أكثر حساسية للعلاجات الأخرى، حيث يمكن أن يعزز تثبيط التيلوميراز فعالية العلاجات التقليدية مثل العلاج الكيميائي أو العلاج الإشعاعي [139].
المقاومة للعلاج الكيميائي والإشعاعي. من خلال تعطيل إشارة NF-kB من خلال تحرير CRISPR-Cas9، قد تصبح خلايا السرطان أكثر عرضة للعلاجات القياسية للسرطان، مما يحسن النتائج العلاجية العامة [145]. بينما يظهر تحرير جين NF-kB بواسطة CRISPR-Cas9 وعدًا، من المهم تقييم الآثار الجانبية المحتملة أو العواقب غير المقصودة [143]. التأثيرات الجانبية غير المرغوب فيها، حيث يقوم CRISPR-Cas9 بتحرير مواقع جينية غير مقصودة، يمكن أن تؤدي إلى عدم استقرار جيني أو تتداخل مع الوظائف الخلوية الطبيعية [145]. بالإضافة إلى ذلك، يجب تقييم الآثار طويلة المدى لتعطيل جين NF-kB على الاستجابة المناعية العامة والعمليات الالتهابية بشكل شامل [144]. يتطلب تحسين الإمكانات العلاجية لتحرير جين NF-kB بواسطة CRISPR-Cas9 مزيدًا من البحث والتطوير. يعد فهم الخصائص الجزيئية المحددة لأنواع السرطان المختلفة ومسارات إشارة NF-kB الخاصة بها أمرًا أساسيًا لتصميم استراتيجيات CRISPR-Cas9 دقيقة [145]. بالإضافة إلى ذلك، يمكن أن تعزز التطورات في أنظمة التسليم، مثل الناقلات الفيروسية أو الحوامل القائمة على الجسيمات النانوية، كفاءة وخصوصية تحرير جين NF-kB في خلايا السرطان [143].
وظائف مهمة في الخلايا الطبيعية، لذا فإن استهدافها قد يسبب عواقب غير مقصودة في الأنسجة غير السرطانية [149].
التجارب السريرية
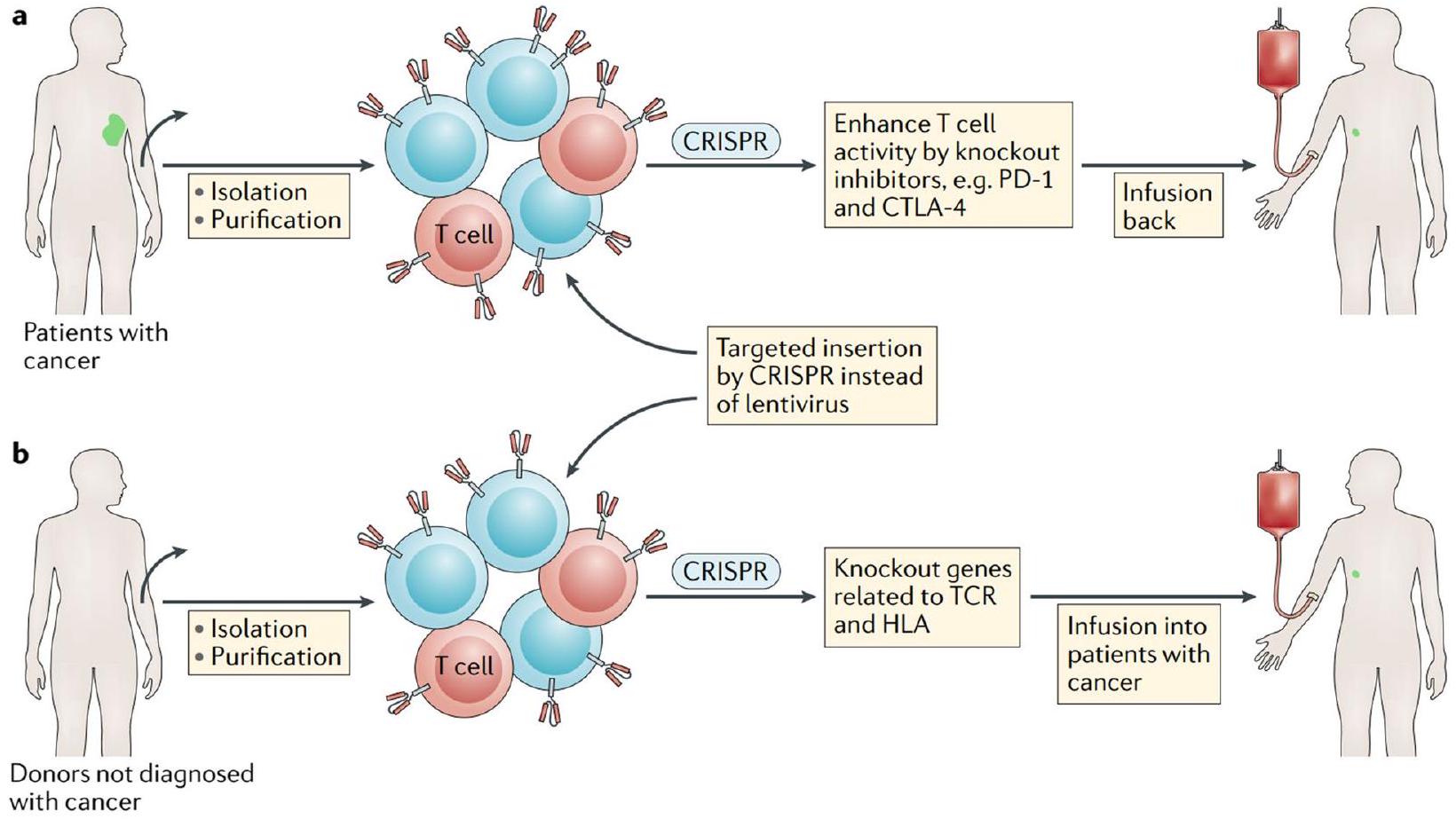
استهداف جينات أخرى لتحسين علاج خلايا CAR T. على سبيل المثال، تهدف التجربة السريرية NCT04037566 إلى تعزيز فعالية خلايا CAR T المستهدفة لـ CD19 من خلال تعديل الجين الداخلي HPK1 [157]. وبالمثل، تستخدم NCT04767308 حذف CD5 الداخلي في خلايا CAR T المستهدفة لـ CD5 خلال المرحلة الأولى من التجربة لتعزيز فعاليتها المحتملة
[158]. علاوة على ذلك، تستخدم NCT03166878 حذف كل من TCR و
تحديات السلامة والتوصيل
تحديات التوصيل وتدابير السلامة في تحرير الجينات المعتمد على CRISPR
المحررات الرئيسية إلى تقليل آثار المواقع غير الانتقائية، ولكن لا تزال الأبحاث الجارية والاعتبارات الأخلاقية ضرورية لاستغلال هذه التقنيات بشكل مسؤول للتطبيقات العلاجية وغيرها من التدخلات الجينية [215]. تشير آثار المواقع غير المستهدفة إلى التغييرات أو التعديلات غير المقصودة في الحمض النووي التي تحدث عند استخدام تقنيات تحرير الجينات مثل أنظمة CRISPR-Cas. يمكن أن تحدث هذه التعديلات غير المقصودة في مواقع أخرى غير الموقع المستهدف، مما قد يؤدي إلى تغييرات جينية غير متوقعة وغير مرغوب فيها [214]. المحررات الأساسية هي تقدم حديث في تكنولوجيا CRISPR يمكنها إجراء تعديلات كيميائية مستهدفة على قواعد الحمض النووي المحددة دون إنشاء كسور مزدوجة السلسلة مثل أنظمة CRISPR-Cas التقليدية. تقلل هذه الطريقة المستهدفة من خطر آثار المواقع غير المرغوب فيها من خلال تقليل الإمكانية للتعديلات العشوائية في الحمض النووي [215]. تقدم المحررات الرئيسية دقة محسنة في تحرير الجينات مقارنة بالمحررات الأساسية أو أنظمة CRISPR-Cas التقليدية. تجمع بين قدرات المحررات الأساسية والنكاسات، مما يسمح بالإدخال الدقيق والحذف والاستبدال لقاعدتين جينيتين معينتين داخل الجينوم. تزيد هذه الدقة من تقليل احتمال آثار المواقع غير المستهدفة [216]. يستخدم العلماء تقنيات متنوعة لتقييم آثار المواقع غير الانتقائية، مثل تسلسل الجينوم الكامل، والتسلسل عالي الإنتاجية، والتحليل الحسابي. تساعد هذه الطرق في تحديد التغييرات الجينية غير المقصودة وتحديد كفاءة وخصوصية تقنية تحرير الجينات المستخدمة [217]. على الرغم من التقدم في المحررات الأساسية والمحررات الرئيسية، تظل آثار المواقع غير المستهدفة مصدر قلق [217]. التحدي يكمن في تحقيق دقة مطلقة في استهداف مواقع جينومية معينة دون التأثير على المناطق المجاورة [215]. يعد التحسين المستمر لأدوات تحرير الجينات، جنبًا إلى جنب مع طرق التقييم والتحقق الدقيقة، أمرًا حاسمًا للتغلب على هذه التحديات [216]. لضمان سلامة استخدام المحررات الأساسية والمحررات الرئيسية في البيئات العلاجية، من الضروري إجراء دراسات قبل السريرية شاملة. تتضمن هذه الدراسات اختبارًا دقيقًا لأدوات تحرير الجينات على خطوط الخلايا والنماذج الحيوانية ذات الصلة لتقييم الآثار المحتملة للمواقع غير المستهدفة وضمان دقة التعديلات الجينية قبل التقدم إلى التجارب البشرية [215].
| نوع السرطان | نهج العلاج | فئة المرضى | الوصف | المزايا | العيوب | المرجع |
| الميلانوما النقيلي | علاج TCR/CAR-T المستهدف لـ NY-ESO-1 | المرضى الذين لديهم أورام إيجابية لـ NY-ESO-1 وفشلوا في العلاج السابق | تم تعديل خلايا T للتعبير عن TCR/CAR لـ NY-ESO-1، وتم ضخها مرة أخرى في المرضى | خصوصية الاستهداف، البقاء على المدى الطويل | احتمال آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [160] |
| لمفومة غير هودجكين | علاج CAR-T المستهدف لـ CD19 | المرضى الذين يعانون من لمفومة NHL المقاومة/الانتكاسية | تم تعديل خلايا T للتعبير عن CAR لـ CD19، وتم ضخها مرة أخرى في المرضى | معدل استجابة مرتفع، استجابة دائمة في بعض المرضى | متلازمة إطلاق السيتوكينات، سمية عصبية، احتمال هروب مستضد الورم | [161] |
| سرطان المثانة | حذف PD-1 عبر CRISPR/Cas9 | المرضى الذين يعانون من سرطان المثانة غير الغازي عالي الخطورة | تم ضخ خلايا T المعدلة بواسطة CRISPR في المرضى | إمكانية تعزيز الاستجابة المناعية المضادة للورم | آثار غير مستهدفة، احتمال حدوث أحداث سلبية مرتبطة بالمناعة | [162] |
| الساركومة | تحرير الجين المستهدف لجين الاندماج PAX3-FOXO1 | المرضى الذين يعانون من ساركومة نقيلي تعبر عن PAX3-FOXO1 | تم استخدام CRISPR/Cas9 لاستهداف جين الاندماج في خلايا الورم، تليها ضخ خلايا T المعدلة | استهداف محدد للطفرات المحفزة للسرطان | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [163] |
| الأورام الصلبة | نقل تكييفي لخلايا TCR/CAR-T المستهدفة للنيوانتجينات | المرضى الذين يعانون من أورام صلبة متقدمة | تم تعديل خلايا T للتعبير عن TCR/CAR المستهدف للنيوانتجينات الفريدة لكل ورم لدى المريض، وتم ضخها مرة أخرى في المرضى | خصوصية الاستهداف، إمكانية الاستجابة الدائمة | تباين النيوانتجينات الورمية، احتمال آثار غير مستهدفة | [164] |
| سرطان الخلايا الكلوية | حذف PD-1 عبر CRISPR/Cas9 | المرضى الذين يعانون من RCC المتقدم الذين فشلوا في العلاجات السابقة | تم ضخ خلايا T المعدلة بواسطة CRISPR في المرضى | إمكانية تعزيز الاستجابة المناعية المضادة للورم | آثار غير مستهدفة، احتمال حدوث أحداث سلبية مرتبطة بالمناعة | [165] |
| متعدد المايلوما | علاج CAR-T المستهدف لـ BCMA | المرضى الذين يعانون من متعدد المايلوما الانتكاسية/المقاومة | تم تعديل خلايا T للتعبير عن CAR لـ BCMA، وتم ضخها مرة أخرى في المرضى | معدل استجابة مرتفع، استجابة دائمة لدى بعض المرضى | متلازمة إطلاق السيتوكينات، السمية العصبية | [166] |
| ورم دبقي متعدد الأشكال | علاج CAR-T المستهدف لـ EGFRvIII | المرضى الذين يعانون من ورم دبقي متكرر يعبرون عن EGFRvIII | تم تعديل خلايا T للتعبير عن CAR EGFRvIII، وتم حقنها مرة أخرى في المرضى | خصوصية الهدف، الإمكانية للاستجابة المستدامة | تباين تعبير مستضدات الورم، الإمكانية للتأثيرات غير المستهدفة | [167] |
| سرطان المريء | تحرير الجين المستهدف لجين MUC1 | المرضى الذين يعانون من سرطان الخلايا الحرشفية المريئية MUC1 + | تم استخدام CRISPR/Cas9 لاستهداف جين MUC1 في خلايا الورم، تلاه ضخ خلايا T المعدلة. | استهداف محدد للطفرات المحفزة للسرطان | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [168] |
| سرطان البروستاتا | تحرير الجين المستهدف لجين مستقبل الأندروجين | المرضى الذين يعانون من سرطان البروستاتا المقاوم للإخصاء | تم استخدام CRISPR/Cas9 لاستهداف جين مستقبل الأندروجين في خلايا الورم، تلا ذلك ضخ خلايا T المعدلة. | استهداف محدد للطفرات المحفزة للسرطان | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [169] |
| سرطان الدم النخاعي الحاد | علاج CAR-T المستهدف لـ CD33 | المرضى الذين يعانون من سرطان الدم النخاعي الحاد المتكرر/المقاوم للعلاج | تم تعديل خلايا T للتعبير عن CAR CD33، ثم تم حقنها مرة أخرى في المرضى | خصوصية الهدف، إمكانية الاستجابة المستدامة | متلازمة إطلاق السيتوكينات، السمية العصبية | [170] |
| سرطان الرأس والعنق | تحرير الجين المستهدف لجين E6 لفيروس الورم الحليمي البشري 16 | المرضى الذين يعانون من سرطان الرأس والعنق المتكرر أو النقيلي إيجابي فيروس الورم الحليمي البشري 16 | تم استخدام CRISPR/Cas9 لاستهداف جين E6 لفيروس الورم الحليمي البشري 16 في خلايا الورم، تلا ذلك ضخ خلايا T المعدلة. | استهداف محدد للطفرات المحفزة للسرطان | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [171] |
| نوع السرطان | نهج العلاج | فئة المرضى | وصف | المزايا | العيوب | مرجع |
| سرطان الرئة | تحرير الجين المستهدف لجين KRAS | المرضى الذين يعانون من سرطان الرئة المتقدم الناتج عن طفرات KRAS | تم استخدام CRISPR/Cas9 لاستهداف جين KRAS في خلايا الورم، تلاه ضخ خلايا T المعدلة. | استهداف محدد للطفرات المحفزة للسرطان | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [172] |
| ورم الأرومة العصبية | تحرير الجين المستهدف لجين ALK | المرضى الذين يعانون من الورم العصبي النخاعي المتحور ALK | تم استخدام CRISPR/Cas9 لاستهداف جين ALK في خلايا الورم، تلاه ضخ خلايا T المعدلة. | استهداف محدد للطفرات المحفزة للسرطان | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [173] |
| لوكيميا اللمفاويات الحادة | علاج CAR-T المستهدف لـ CD19 | مرضى الأطفال الذين يعانون من اللوكيميا اللمفاوية الحادة المتكررة/المقاومة للعلاج | تم تعديل خلايا T للتعبير عن CAR CD19، ثم تم حقنها مرة أخرى في المرضى | معدل استجابة مرتفع، استجابة دائمة لدى بعض المرضى | متلازمة إطلاق السيتوكينات، السمية العصبية | [174] |
| الأورام الصلبة | تحرير الجين المستهدف لجين CCR4 | المرضى الذين يعانون من أورام صلبة متقدمة | تم استخدام CRISPR/Cas9 لاستهداف جين CCR4 في خلايا الورم، تلا ذلك ضخ خلايا T المعدلة. | استهداف محدد للطفرات المحفزة للسرطان | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [175] |
| الميلانوما | تحرير الجين المستهدف لجين NRAS | المرضى الذين يعانون من الميلانوما المتقدمة ذات الطفرة في جين NRAS | تم استخدام CRISPR/Cas9 لاستهداف جين NRAS في خلايا الورم، تلاه ضخ خلايا T المعدلة. | استهداف محدد للطفرات المحفزة للسرطان | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [176] |
| سرطان القناة الصفراوية | تحرير الجين المستهدف لجين IDH1 | المرضى الذين يعانون من سرطان القنوات الصفراوية المتقدم مع طفرات IDH1 | تم استخدام CRISPR/Cas9 لاستهداف جين IDH1 في خلايا الورم، تلا ذلك ضخ خلايا T المعدلة. | استهداف محدد للطفرات المحفزة للسرطان | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [177] |
| الأورام الصلبة | تحرير الجين المستهدف لجين TP53 | المرضى الذين يعانون من أورام صلبة متقدمة | تم استخدام CRISPR/Cas9 لاستهداف جين TP53 في خلايا الورم، تلاه ضخ خلايا T المعدلة. | استهداف محدد للطفرات المحفزة للسرطان | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [178] |
| الورم النقوي | علاج CAR-T المستهدف لـ CD19 | المرضى الذين يعانون من المايلوما المتكررة/المقاومة للعلاج | تم تعديل خلايا T للتعبير عن CAR CD19، ثم تم حقنها مرة أخرى في المرضى | معدل استجابة مرتفع، استجابة دائمة لدى بعض المرضى | متلازمة إطلاق السيتوكينات، السمية العصبية | [179] |
| الأورام الصلبة | تحرير الجين المستهدف لجين PD-1 | المرضى الذين يعانون من أورام صلبة متقدمة | تم استخدام CRISPR/Cas9 لاستهداف جين PD-1 في خلايا الورم، تلا ذلك ضخ خلايا T المعدلة. | يعزز المناعة المضادة للأورام من خلال تعطيل مسار نقاط التفتيش المناعية | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [180] |
| الأورام الصلبة | تحرير الجين المستهدف لجين MUC1 | المرضى الذين يعانون من أورام صلبة متقدمة MUC1 + | تم استخدام CRISPR/Cas9 لاستهداف جين MUC1 في خلايا الورم، تلاه ضخ خلايا T المعدلة. | استهداف محدد للطفرات المحفزة للسرطان | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [181] |
| أورام صلبة متعددة | تحرير الجين المستهدف لجين EGFR | المرضى الذين يعانون من أورام صلبة متقدمة تحمل طفرات في جين EGFR | تم استخدام CRISPR/Cas9 لاستهداف جين EGFR في خلايا الورم، تلاه ضخ خلايا T المعدلة. | استهداف محدد للطفرات المحفزة للسرطان | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [165] |
| نوع السرطان | نهج العلاج | فئة المرضى | وصف | المزايا | العيوب | مرجع |
| لوكيميا/ليمفوما | تحرير الجين المستهدف لجين CD22 | المرضى الذين يعانون من سرطان الدم/ اللمفوما CD22 + المتكرر/ المقاوم للعلاج | تم استخدام CRISPR/Cas9 لاستهداف جين CD22 في خلايا الورم، تلاه ضخ خلايا T المعدلة. | استهداف محدد للطفرات المحفزة للسرطان | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [182] |
| الأورام الصلبة | تحرير الجين المستهدف لجين PTEN | المرضى الذين يعانون من أورام صلبة متقدمة تفتقر إلى PTEN | تم استخدام CRISPR/Cas9 لاستهداف جين PTEN في خلايا الورم، تلاه ضخ خلايا T المعدلة. | استهداف محدد للطفرات المحفزة للسرطان | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [183] |
| أورام صلبة متعددة | تحرير الجين المستهدف لجين PDCD1 | المرضى الذين يعانون من أورام صلبة متقدمة | تم استخدام CRISPR/Cas9 لاستهداف جين PDCD1 في خلايا الورم، تلا ذلك ضخ خلايا T المعدلة. | يعزز المناعة المضادة للأورام من خلال تعطيل مسار نقاط التفتيش المناعية | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [184] |
| لوكيميا/ليمفوما | تحرير الجين المستهدف لجين TCR | المرضى الذين يعانون من سرطان الدم/اللمفوما المتكررة/المقاومة للعلاج | تم استخدام CRISPR/Cas9 لاستهداف جين TCR في خلايا T، تلا ذلك حقن خلايا T المعدلة. | استهداف محدد لجين TCR لتعزيز نشاط خلايا T | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [185] |
| لوكيميا/ليمفوما | علاج CAR-T المستهدف لـ CD19 | المرضى الذين يعانون من سرطان الدم/اللمفوما المتكررة/المقاومة للعلاج | تم تعديل خلايا T للتعبير عن CAR CD19، ثم تم حقنها مرة أخرى في المرضى | معدل استجابة مرتفع، استجابة دائمة لدى بعض المرضى | متلازمة إطلاق السيتوكينات، السمية العصبية | [186] |
| الأورام الصلبة | تحرير الجين المستهدف لـ HIF-1 ألفا | المرضى الذين يعانون من أورام صلبة متقدمة تعبر عن HIF-1a بشكل مفرط | تم استخدام CRISPR/Cas9 لاستهداف جين HIF-1 ألفا في خلايا الورم، تلاها ضخ خلايا T المعدلة. | استهداف محدد للطفرات المحفزة للسرطان | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [187] |
| سرطان الرئة غير صغير الخلايا | إزالة جين PD-1 بواسطة تقنية كريسبر/كاس9 | المرضى الذين يعانون من سرطان الرئة غير صغير الخلايا المتقدم إيجابي PD-L1 | تم استخدام CRISPR/Cas9 لإسكات جين PD-1 في خلايا T، تلا ذلك حقن خلايا T المعدلة. | يعيق مسار نقاط التفتيش المناعية | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [188] |
| أورام صلبة متعددة | إسكات TGF- بواسطة تقنية كريسبر/كاس9
|
المرضى الذين يعانون من TGF المتقدم
|
تم استخدام CRISPR/Cas9 لإسكات TGF-
|
استهداف محدد للطفرات المحفزة للسرطان | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [189] |
| الأورام الصلبة | إسكات DNMT1 بواسطة تقنية كريسبر/كاس9 | المرضى الذين يعانون من أورام صلبة متقدمة | تم استخدام CRISPR/Cas9 لإسكات جين DNMT1 في خلايا الورم، تلا ذلك ضخ خلايا T المعدلة. | استهداف محدد للمنظم الجيني الإيبيجيني | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [190] |
| الأورام الصلبة | إسكات LAP بواسطة تقنية كريسبر/كاس9 | المرضى الذين يعانون من أورام صلبة متقدمة تعبر عن LAP بشكل مفرط | تم استخدام CRISPR/Cas9 لإسكات جين LAP في خلايا الورم، تلا ذلك ضخ خلايا T المعدلة. | استهداف محدد للآلية المثبطة للمناعة | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [191] |
| نوع السرطان | نهج العلاج | فئة المرضى | وصف | المزايا | العيوب | مرجع |
| الأورام الصلبة | إسكات AXL بواسطة تقنية كريسبر/كاس9 | المرضى الذين يعانون من أورام صلبة متقدمة تعبر عن AXL بشكل مفرط | تم استخدام CRISPR/Cas9 لإسكات جين AXL في خلايا الورم، تلا ذلك ضخ خلايا T المعدلة. | استهداف محدد للطفرات المحفزة للسرطان | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [192] |
| الأورام الصلبة | إسكات HLA من الفئة الأولى بواسطة CRISPR/Cas9 | المرضى الذين يعانون من أورام صلبة متقدمة تعاني من نقص في فئة HLA I | تم استخدام CRISPR/Cas9 لإسكات جينات HLA من الفئة الأولى في خلايا الورم، تلاها ضخ خلايا T المعدلة. | استهداف محدد لآلية التهرب المناعي | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [193] |
| لوكيميا/ليمفوما | إسكات TCR و B2M بواسطة تقنية كريسبر/كاس9 | المرضى الذين يعانون من الأورام الخبيثة T-cell المتكررة/المقاومة للعلاج | تم استخدام CRISPR/Cas9 لإسكات جينات TCR وB2M في خلايا T، تلاها حقن خلايا T المعدلة. | تعطيل مستقبلات الخلايا التائية وتعبير فئة MHC I لمنع مرض الطعم ضد المضيف وتعزيز النشاط المضاد للورم | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [194] |
| الأورام الصلبة | إسكات بواسطة CRISPR/Cas9 لـ
|
المرضى الذين يعانون من مراحل متقدمة
|
تم استخدام CRISPR/Cas9 لإلغاء
|
استهداف محدد للطفرات المحفزة للسرطان | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [195] |
| لوكيميا/ليمفوما | إسكات CD7 بواسطة تقنية كريسبر/كاس9 | المرضى الذين يعانون من سرطان الدم/ اللمفوما CD7 + المتكرر/ المقاوم للعلاج | تم استخدام CRISPR/Cas9 لإسكات جين CD7 في خلايا T، تلا ذلك حقن خلايا T المعدلة. | استهداف محدد لمستضدات خلايا B | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [196] |
| الأورام الصلبة | إسكات PSCA بواسطة CRISPR/Cas9 | المرضى الذين يعانون من أورام صلبة تعبر عن PSCA المتقدمة | تم استخدام CRISPR/Cas9 لإسكات جين PSCA في خلايا الورم، تلا ذلك ضخ خلايا T المعدلة. | استهداف محدد لمستضدات مرتبطة بالورم | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [190] |
| الأورام الصلبة | إسكات APOBEC3B بواسطة تقنية كريسبر/كاس9 | المرضى الذين يعانون من أورام صلبة متقدمة تعبر عن APOBEC3B بشكل مفرط | تم استخدام CRISPR/Cas9 لإسكات جين APOBEC3B في خلايا الورم، تلا ذلك ضخ خلايا T المعدلة. | استهداف محدد للإنزيمات المسببة للطفرات | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [197] |
| الأورام الصلبة | إسقاط IL2RG بواسطة تقنية كريسبر/كاس9 | المرضى الذين يعانون من أورام صلبة متقدمة نقص IL2RG | تم استخدام CRISPR/Cas9 لإسكات جين IL2RG في خلايا T، تلا ذلك حقن خلايا T المعدلة. | استهداف محدد لجين نقص المناعة | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [198] |
| الأورام الصلبة | إسكات ARID1A بواسطة تقنية كريسبر/كاس9 | المرضى الذين يعانون من أورام صلبة متقدمة تحمل طفرات في جين ARID1A | تم استخدام CRISPR/Cas9 لإسكات جين ARID1A في خلايا الورم، تلا ذلك ضخ خلايا T المعدلة. | استهداف محدد للطفرات المحفزة للسرطان | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [199] |
| الأورام الصلبة | إسكات TRAC بواسطة تقنية كريسبر/كاس9 | المرضى الذين يعانون من أورام صلبة متقدمة ناقصة TRAC | تم استخدام CRISPR/Cas9 لإسكات جين TRAC في خلايا T، تلا ذلك حقن خلايا T المعدلة. | استهداف محدد لجين نقص المناعة | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [200] |
| نوع السرطان | نهج العلاج | فئة المرضى | وصف | المزايا | العيوب | مرجع |
| الأورام الصلبة | إسكات LAPTM4B بواسطة تقنية كريسبر/كاس9 | المرضى الذين يعانون من أورام صلبة متقدمة تعبر عن LAPTM4B بشكل مفرط | تم استخدام CRISPR/Cas9 لإسكات جين LAPTM4B في خلايا الورم، تلا ذلك ضخ خلايا T المعدلة. | استهداف محدد للجينات المسرطنة | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [201] |
| الأورام الصلبة | إسكات HPRT1 بواسطة تقنية كريسبر/كاس9 | المرضى الذين يعانون من أورام صلبة متقدمة تعبر عن HPRT1 بشكل مفرط | تم استخدام CRISPR/Cas9 لإسكات جين HPRT1 في خلايا الورم، تلاها ضخ خلايا T المعدلة. | استهداف محدد للجينات المسرطنة | آثار غير مستهدفة، فعالية محدودة في بعض المرضى | [202] |
| تحدي | النهج/ الاستراتيجيات الحالية | القيود | الاتجاهات المستقبلية | الاعتبارات الأخلاقية والتنظيمية | وصف | جدة | المزايا | العيوب | القيود / التحديات | مرجع |
| آثار غير مستهدفة | تحرير الجينوم المستهدف | – قد يكون من الصعب تحقيق دقة عالية | – تطوير طرق توصيل محسّنة وتقنيات مراقبة | – الحاجة إلى إرشادات واضحة ورقابة | يمكن أن يتسبب كريسبر في تغييرات غير مقصودة في الجينوم خارج المنطقة المستهدفة، مما قد يؤدي إلى آثار سلبية. | تحرير الجينوم بواسطة تقنية كريسبر هو نهج دقيق للغاية وموجه للعلاج الجيني | – يقلل من الضرر للخلايا السليمة | – إمكانية حدوث تغييرات غير مقصودة في الجينوم | يجب تحسين الخصوصية لتجنب التأثيرات غير المستهدفة | [٢٠٦، ٢٠٧] |
| التوصيل إلى موقع الورم | استخدام الناقلات الفيروسية | – نقص في التحديد، صعوبة في استهداف خلايا الورم | – تطوير أنظمة توصيل مستهدفة | – الحاجة إلى تقليل المخاطر على الأنسجة السليمة | تستخدم الناقلات الفيروسية عادةً لتوصيل العلاجات المعتمدة على كريسبر، ولكن قد يكون من الصعب استهدافها لخلايا الورم المحددة. | يمكن أن يقلل التوصيل المستهدف للعلاجات المعتمدة على كريسبر من الضرر الذي يلحق بالأنسجة السليمة | – يمكن استخدامه لاستهداف خلايا الورم المحددة | – يمكن أن يسبب ردود فعل مناعية وسمية | يجب تحسين خصوصية الاستهداف لتجنب الضرر للأنسجة السليمة | [٢٠٨، ٢٠٩] |
| السلامة | مراقبة التأثيرات غير المستهدفة | – حساسية ودقة محدودة لتقنيات المراقبة الحالية | – تطوير تقنيات مراقبة أكثر حساسية ودقة | – الحاجة إلى ضمان سلامة المرضى وتقليل الأذى المحتمل | رصد التأثيرات غير المستهدفة أمر ضروري لضمان سلامة العلاجات المعتمدة على كريسبر | يمكن أن توفر تقنيات المراقبة المحسّنة معلومات أكثر دقة حول تأثيرات تعديل CRISPR على الجينوم | – يمكن أن يساعد في اكتشاف التغيرات غير المقصودة في الجينوم | – التقنيات الحالية لديها حساسية ودقة محدودة | هناك حاجة إلى مزيد من البحث لتحسين تقنيات المراقبة | [210,211] |
| ردود الفعل المناعية | استخدام أنظمة كريسبر غير المناعية | – توفر محدود لأنظمة كريسبر غير المناعية | – تطوير أنظمة كريسبر غير المناعية | – الحاجة إلى تقليل مخاطر ردود الفعل المناعية والسمية | يمكن أن تؤدي العلاجات المعتمدة على كريسبر إلى تحفيز استجابات مناعية، مما قد يؤدي إلى آثار سلبية. | يتم تطوير أنظمة كريسبر غير المناعية لتقليل خطر ردود الفعل المناعية | يمكن أن تقلل أنظمة كريسبر غير المناعية من خطر ردود الفعل المناعية والسمية | – أنظمة كريسبر غير المناعية ليست متاحة على نطاق واسع | هناك حاجة إلى مزيد من البحث لتطوير أنظمة كريسبر غير المناعية | [212,213] |
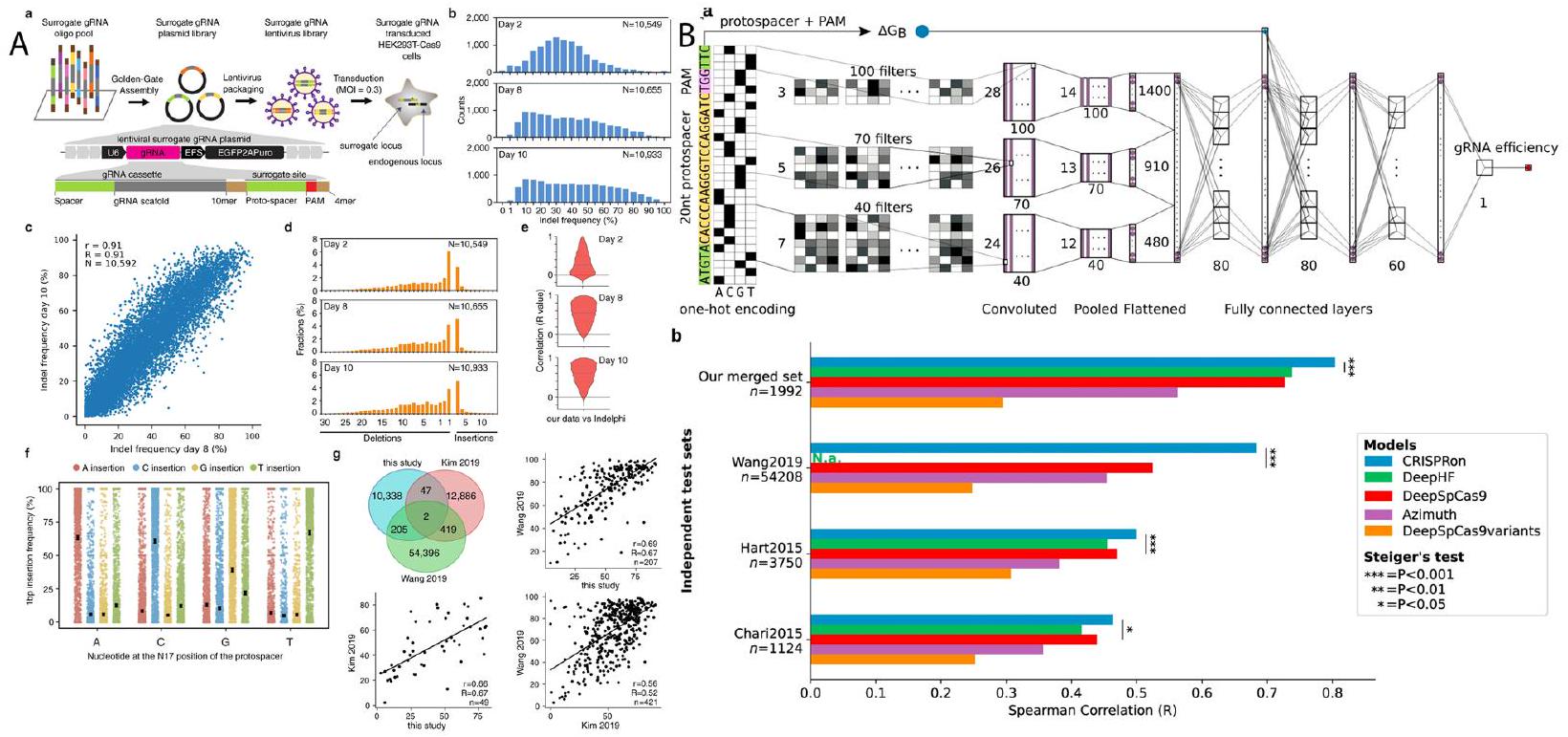
تنبؤات كفاءة الاستهداف. لتحقيق ذلك، جمع الباحثون بيانات نشاط gRNA عالية الجودة لـ 10,592 gRNA تستهدف إنزيم SpCas9. لتحسين توقعاتهم، دمج الباحثون هذه البيانات الجديدة مع البيانات التكميلية الموجودة من مصادر أخرى. ثم استخدموا نموذج تعلم عميق يسمى “CRISPR on”، الذي تم تدريبه على مجموعة بيانات مجمعة من 23,902 gRNA، بما في ذلك البيانات التي تم إنشاؤها حديثًا والبيانات المتاحة سابقًا. أظهرت نتائج دراستهم أن CRISPR on تفوق على الأدوات الحالية المستخدمة في توقع كفاءة gRNA. لوحظت الأداء المحسن عبر أربعة مجموعات بيانات اختبار لم تكن جزءًا من بيانات التدريب المستخدمة لتطوير أدوات التوقع الأخرى. وهذا يشير إلى أن توقعات CRISPR on كانت أكثر دقة وموثوقية مما هو متاح حاليًا. لجعل نتائجهم متاحة للمجتمع العلمي، طور الباحثون خادم ويب تفاعلي لتصميم gRNA بناءً على CRISPR on
الطفرات يمكن أن تعطل NFE2L2 وبالتالي تعزز نمو السرطان [139]. كان الغرض من استخدام CRISPR-Cas9 هو التحقيق في التأثير العلاجي المحتمل لاستهداف جين BRCA1/TP53/RAS، الذي يتعرض عادة للطفرات في سرطان الثدي والمبيض [231]. كانت العواقب غير المقصودة لاستخدام CRISPR-Cas9 على جين BRCA1/TP53/RAS هي طفرات غير مقصودة في عدة جينات غير مستهدفة، بما في ذلك RAD51D/MDM2/MAPK، على التوالي [232]. يمكن أن تكون الطفرات غير المقصودة في جينات RAD51D/MDM2/MAPK قد أضعفت التأثير العلاجي لاستهداف BRCA1/TP53/RAS حيث يمكن أن تنشط RAD51D/تعطل MDM2/تنشط MAPK، مما يؤدي إلى تعزيز نمو السرطان [233]. تسلط النتائج المتكررة للطفرات غير المقصودة في جينات غير مرغوب فيها في هذه الدراسات الضوء على الخطر الكبير للآثار الجانبية غير الانتقائية المرتبطة بتحرير الجينات باستخدام CRISPR-Cas9 [234]. يجب على البحث تحليل ومقارنة الآثار الجانبية غير المستهدفة التي لوحظت عند استهداف جينات مختلفة، مما قد يوفر رؤى حول التأثيرات الخاصة بالجنس من CRISPR-Cas9 [235، 236]. فهم الآثار الجانبية غير المستهدفة وتأثيرها المحتمل على نمو السرطان أمر بالغ الأهمية في تقييم سلامة وفعالية CRISPR-Cas9 كنهج علاجي لعلاج السرطان [232، 237].
السلامة
تحول الخلايا المعدلة إلى خلايا سرطانية [240]. يعتمد علاج السرطان القائم على كريسبر على القدرة على تحرير طفرات جينية محددة تدفع نمو الورم. ومع ذلك، إذا اكتسبت الخلايا المعدلة طفرات إضافية، فقد تصبح سرطانية [238]. يعمل الباحثون على فهم الآثار طويلة المدى لعلاج السرطان القائم على كريسبر وتطوير استراتيجيات لتقليل خطر تحول الخلايا المعدلة إلى خلايا سرطانية [241]. بالإضافة إلى هذه المخاوف، يثير علاج السرطان القائم على كريسبر عددًا من المخاوف الأخرى المتعلقة بالسلامة، بما في ذلك إمكانية حدوث تفاعلات مناعية تجاه الناقلات الفيروسية المستخدمة لتوصيل كريسبر، وخطر إنشاء طفرات جديدة تسبب السرطان. للتخفيف من هذه المخاوف، يعمل الباحثون على تطوير طرق توصيل أكثر أمانًا وتطوير استراتيجيات جديدة لتقليل خطر الآثار الجانبية غير المستهدفة [240]. يتم إجراء دراسات قبل السريرية والسريرية لتقييم سلامة وفعالية علاج السرطان القائم على كريسبر [238]. بالإضافة إلى ذلك، يعمل الباحثون على إيجاد طرق فعالة من حيث التكلفة وكفاءة لإنتاج أعداد كبيرة من الخلايا المعدلة بواسطة كريسبر، والتغلب على مشكلات القابلية للتوسع [240]. يمتلك تحرير الجينات القائم على كريسبر القدرة على إحداث ثورة في علاج السرطان، ولكن لا تزال هناك تحديات كبيرة تتعلق بالسلامة يجب معالجتها قبل أن يمكن استخدام هذا النهج بأمان وفعالية في العيادة [241]. البحث المستمر ضروري لفهم الآثار طويلة المدى لعلاج السرطان القائم على كريسبر بشكل أفضل، ولتطوير طرق توصيل أكثر أمانًا، وتقليل خطر الآثار الجانبية غير المستهدفة وغيرها من المخاوف المتعلقة بالسلامة [240]. جانب آخر من جوانب السلامة في علاج السرطان القائم على كريسبر هو طريقة التوصيل المستخدمة لتوصيل آلية كريسبر إلى خلايا الورم. واحدة من أكثر الطرق شيوعًا المستخدمة هي استخدام الناقلات الفيروسية، مثل الفيروسات الغدية أو الفيروسات القهقرية [241]. ومع ذلك، فإن هذه الناقلات لديها القدرة على التسبب في تفاعلات مناعية وآثار سلبية أخرى [238]. يعمل الباحثون على تطوير طرق توصيل غير فيروسية، مثل الجسيمات النانوية والإكسوزومات، كبديل للناقلات الفيروسية. هذه الطرق لديها القدرة على أن تكون أكثر أمانًا وفعالية، لكنها لا تزال في مراحل مبكرة من التطوير وتحتاج إلى مزيد من البحث لتحسين فعاليتها وسلامتها [241, 244]. بالإضافة إلى ذلك، فإن التصنيع والقابلية للتوسع لعلاج السرطان القائم على كريسبر هو مصدر قلق كبير آخر يتعلق بالسلامة. إنتاج كميات كبيرة من الخلايا المعدلة بواسطة كريسبر للاستخدام السريري يمثل تحديًا ومكلفًا [241]. يعمل الباحثون على إيجاد طرق فعالة من حيث التكلفة وكفاءة لإنتاج أعداد كبيرة من الخلايا المعدلة بواسطة كريسبر، والتغلب على مشكلات القابلية للتوسع. يشمل ذلك البحث عن طرق بديلة لإنتاج آلية كريسبر واستكشاف طرق لتحسين كفاءة عملية تحرير كريسبر [241]. السلامة هي مصدر قلق حاسم في تطوير علاج السرطان القائم على كريسبر [240]. يعمل الباحثون على معالجة هذه المخاوف من خلال تطوير
طرق توصيل أكثر أمانًا، وتطوير استراتيجيات جديدة لتقليل خطر الآثار الجانبية غير المستهدفة وغيرها من المخاوف المتعلقة بالسلامة، وإيجاد طرق فعالة من حيث التكلفة وكفاءة لإنتاج أعداد كبيرة من الخلايا المعدلة بواسطة كريسبر [238]. البحث المستمر ضروري لفهم الآثار طويلة المدى لعلاج السرطان القائم على كريسبر بشكل أفضل، ولضمان أن هذا النهج الجديد الواعد يمكن استخدامه بأمان وفعالية في العيادة. من المهم أن نلاحظ أنه بينما يعد علاج السرطان القائم على كريسبر نهجًا جديدًا واعدًا، إلا أنه لا يزال في مراحل مبكرة من التطوير. لا تزال العديد من المخاوف والتحديات المتعلقة بالسلامة التي تم مناقشتها أعلاه قيد الدراسة والتقييم في التجارب قبل السريرية والسريرية. لذلك، من المهم الاستمرار في مراقبة تقدم البحث في هذا المجال وتقييم سلامة وفعالية علاج السرطان القائم على كريسبر مع توفر المزيد من البيانات [240]. من الجدير بالذكر أيضًا أن المشهد التنظيمي لعلاج السرطان القائم على كريسبر لا يزال يتطور. لدى الدول والمناطق المختلفة لوائح وإرشادات مختلفة بشأن استخدام العلاجات القائمة على كريسبر في البشر [241]. سيحتاج الباحثون والشركات التي تطور علاجات السرطان القائمة على كريسبر إلى التنقل في هذه اللوائح والإرشادات من أجل طرح علاجاتهم في السوق [238]. يمتلك علاج السرطان القائم على كريسبر القدرة على إحداث ثورة في علاج السرطان، ولكن لا تزال هناك تحديات كبيرة تتعلق بالسلامة ومشكلات التوصيل التي يجب معالجتها. يعمل الباحثون على معالجة هذه المخاوف من خلال البحث والتطوير المستمر، ولكن سيستغرق الأمر وقتًا لفهم الآثار طويلة المدى وسلامة هذا النهج الجديد بشكل كامل. من المهم مراقبة تقدم البحث في هذا المجال وتقييم سلامة وفعالية علاج السرطان القائم على كريسبر مع توفر المزيد من البيانات [241].
التوصيل إلى موقع الورم
وتفعيل استجابة مناعية. يمكن أن يؤدي ذلك إلى التهاب وآثار سلبية أخرى، ويمكن أن يحد أيضًا من فعالية العلاج [250]. طريقة أخرى لتوصيل كريسبر إلى موقع الورم هي من خلال استخدام الجسيمات النانوية. هذه الجسيمات صغيرة بما يكفي لاختراق نسيج الورم بسهولة، ويمكن تصميمها لحمل معدات كريسبر [251]. يمكن أيضًا تصميم الجسيمات النانوية لاستهداف أنواع خلايا معينة، مثل خلايا السرطان، لزيادة كفاءة العلاج [252]. ومع ذلك، لا تزال فعالية الجسيمات النانوية في توصيل كريسبر إلى موقع الورم قيد التقييم، وهناك حاجة إلى مزيد من البحث لفهم سلامتها وفعاليتها [253]. تعتبر الإكسوزومات أيضًا وسيلة توصيل واعدة لكريسبر [254]. الإكسوزومات هي حويصلات صغيرة يتم إفرازها بشكل طبيعي بواسطة الخلايا ويمكن تصميمها لحمل معدات كريسبر [255]. تمتلك الإكسوزومات القدرة على عبور الحاجز الدموي الدماغي وتوصيل معدات كريسبر إلى موقع الورم. لكن هناك حاجة إلى مزيد من البحث لفهم سلامة وفعالية الإكسوزومات كوسيلة توصيل لكريسبر [256]. يقوم الباحثون بتطوير طرق توصيل كريسبر التي يمكن أن تستهدف خلايا السرطان بناءً على العلامات السطحية، مثل تعبير بروتينات معينة [257]. نهج آخر هو استهداف الطفرات الجينية التي تكون محددة لخلايا السرطان. يمكن القيام بذلك عن طريق تصميم معدات كريسبر للتعرف على واستهداف تسلسلات جينية معينة مرتبطة بالسرطان [255]. على سبيل المثال، قام الباحثون بتطوير علاجات قائمة على كريسبر تستهدف طفرات معينة في جينات مثل KRAS، والتي تتعرض للطفرات بشكل شائع في العديد من أنواع السرطان [256]. يمكن أيضًا استخدام مجموعة من هذه الاستراتيجيات لاستهداف أنواع خلايا معينة لتوصيل كريسبر إلى موقع الورم [248]. على سبيل المثال، يستكشف الباحثون استخدام الجسيمات النانوية المصممة لاستهداف علامات سطحية معينة على خلايا السرطان وتحمل أيضًا معدات كريسبر [245]. يعد توصيل كريسبر إلى موقع الورم خطوة حاسمة في تطوير علاج السرطان القائم على كريسبر [248]. يعمل الباحثون على تطوير طرق جديدة وفعالة لتوصيل كريسبر إلى موقع الورم، بما في ذلك الناقلات الفيروسية، والجسيمات النانوية، والإكسوزومات [251]. بالإضافة إلى ذلك، يمكن أن يزيد استهداف أنواع خلايا معينة، مثل خلايا السرطان، من كفاءة العلاج. بينما لا تزال هناك تحديات كبيرة يجب معالجتها، فإن إمكانيات علاج السرطان القائم على كريسبر في إحداث ثورة في علاج السرطان واضحة، وستساعد الأبحاث المستمرة في التغلب على هذه التحديات [255].
التصنيع وقابلية التوسع
النهج بأمان وفعالية في العيادة [200]. واحدة من التحديات الرئيسية هي التصنيع وقابلية التوسع للخلايا المعدلة بواسطة كريسبر للاستخدام السريري [259]. يعد تصنيع الخلايا المعدلة بواسطة كريسبر للاستخدام في علاج السرطان عملية معقدة ومكلفة. الخطوة الأولى هي الحصول على الخلايا التي سيتم تعديلها، والتي يمكن الحصول عليها من المريض أو من خط خلايا. بمجرد الحصول على الخلايا، يجب تعديلها باستخدام معدات كريسبر. يتضمن ذلك عادةً توصيل معدات كريسبر، بما في ذلك RNAs الموجهة وإنزيمات كاس، إلى الخلايا باستخدام ناقل فيروسي أو جسيم نانوي [260]. ومع ذلك، لم يتم تحسين هذه العملية بالكامل بعد وهناك حاجة إلى مزيد من البحث للعثور على طرق فعالة من حيث التكلفة لإنتاج أعداد كبيرة من الخلايا المعدلة بواسطة كريسبر [261]. تعتبر قابلية التوسع أيضًا تحديًا رئيسيًا في تصنيع الخلايا المعدلة بواسطة كريسبر لعلاج السرطان [262]. الطرق الحالية لإنتاج الخلايا المعدلة بواسطة كريسبر لا تزال غير قادرة على إنتاج الأعداد الكبيرة من الخلايا المطلوبة للاستخدام السريري [258]. على سبيل المثال، إذا تم إنتاج الخلايا باستخدام ناقل فيروسي، فإن العملية محدودة بعدد الخلايا التي يمكن أن تصاب في وقت واحد [263]. بالإضافة إلى ذلك، فإن الطرق الحالية لإنتاج الخلايا المعدلة بواسطة كريسبر لا تزال غير قادرة على إنتاج خلايا بكفاءة عالية بما يكفي لتكون ذات صلة سريرية [264]. هناك عدد من الحلول المحتملة لهذه التحديات. يعمل الباحثون على تطوير طرق أكثر كفاءة وفعالية من حيث التكلفة لإنتاج الخلايا المعدلة بواسطة كريسبر، مثل استخدام الإكسوزومات كوسيلة توصيل، وتحسين قابلية التوسع للعملية [265]. بالإضافة إلى ذلك، يعمل الباحثون على تحسين كفاءة عملية تحرير كريسبر وتقليل مخاطر الآثار الجانبية غير المستهدفة [200]. حل محتمل آخر لتحدي قابلية التوسع هو استخدام خطوط خلايا تم تعديلها جينيًا لإنتاج أعداد كبيرة من الخلايا المعدلة بواسطة كريسبر [259]. على سبيل المثال، قام الباحثون بتطوير خطوط خلايا تعبر بشكل مستقر عن إنزيمات كاس، والتي يمكن استخدامها لإنتاج أعداد كبيرة من الخلايا المعدلة بواسطة كريسبر [260]. بالإضافة إلى ذلك، يستكشف الباحثون استخدام خلايا جذعية كمصدر للخلايا المعدلة بواسطة كريسبر [264]. تمتلك الخلايا الجذعية القدرة على التجدد الذاتي والتمايز إلى مجموعة واسعة من أنواع الخلايا، مما يجعلها خيارًا جذابًا لإنتاج أعداد كبيرة من الخلايا المعدلة بواسطة كريسبر لعلاج السرطان. منطقة أخرى من البحث النشط هي تطوير منصات آلية لإنتاج الخلايا المعدلة بواسطة كريسبر. يمكن أن تقوم هذه المنصات بأتمتة العديد من الخطوات اليدوية المتضمنة في إنتاج الخلايا المعدلة بواسطة كريسبر، مما يجعل العملية أكثر كفاءة وفعالية من حيث التكلفة [200]. بالإضافة إلى ذلك، يمكن استخدام هذه المنصات لتحسين الظروف لإنتاج الخلايا المعدلة بواسطة كريسبر، مثل كمية إنزيمات كاس وRNAs الموجهة المستخدمة [259]. أخيرًا، يستكشف الباحثون استخدام توصيل كريسبر في الموقع.
الآلات، التي تسمح بتحرير الخلايا مباشرة في بيئة الورم الدقيقة [261]. تتجنب هذه الطريقة الحاجة إلى إنتاج وتوصيل أعداد كبيرة من الخلايا المعدلة باستخدام كريسبر، وقد تتغلب على تحديات القابلية للتوسع [264]. ومع ذلك، لا تزال هذه الطريقة في مراحلها الأولى من التطوير وتحتاج إلى مزيد من البحث لتحسين فعاليتها وسلامتها [260]. مثال واحد هو استخدام الإكسوزومات كطريقة توصيل لآلات كريسبر [264]. الإكسوزومات هي حويصلات صغيرة يتم إفرازها بشكل طبيعي بواسطة الخلايا ويمكن استخدامها لتوصيل مجموعة متنوعة من الجزيئات، بما في ذلك آلات كريسبر، إلى الخلايا المستهدفة [261]. أظهر الباحثون أن الإكسوزومات يمكن استخدامها لتوصيل آلات كريسبر إلى خلايا السرطان بكفاءة عالية وسمية منخفضة. يتم تطوير هذه الطريقة بشكل أكبر كحل محتمل لتحدي القابلية للتوسع. مثال آخر هو استخدام خطوط الخلايا التي تم تعديلها وراثيًا لإنتاج أعداد كبيرة من الخلايا المعدلة باستخدام كريسبر [264]. طور الباحثون خطوط خلايا تعبر بشكل مستقر عن إنزيمات كاس ويمكن استخدامها لإنتاج أعداد كبيرة من الخلايا المعدلة باستخدام كريسبر. يتم تطوير هذه الطريقة بشكل أكبر كحل محتمل لتحدي القابلية للتوسع. مثال آخر هو تطوير منصات آلية لإنتاج خلايا معدلة باستخدام كريسبر. تقوم هذه المنصات بأتمتة العديد من الخطوات اليدوية المتضمنة في إنتاج خلايا معدلة باستخدام كريسبر، مما يجعل العملية أكثر كفاءة وفعالية من حيث التكلفة. يتم تطوير هذه الطريقة بشكل أكبر كحل محتمل لتحدي القابلية للتوسع [200]. أخيرًا، هناك أمثلة على الأبحاث حول توصيل آلات كريسبر في الموقع [260]. طور الباحثون طرقًا لتوصيل آلات كريسبر مباشرة إلى خلايا السرطان في بيئة الورم الدقيقة [261]. تتجنب هذه الطريقة الحاجة إلى إنتاج وتوصيل أعداد كبيرة من الخلايا المعدلة باستخدام كريسبر، وقد تتغلب على تحديات القابلية للتوسع [264]. لا تزال هذه الطريقة في مراحلها الأولى من التطوير وتحتاج إلى مزيد من البحث لتحسين فعاليتها وسلامتها [259]. إن تصنيع وتوسيع نطاق الخلايا المعدلة باستخدام كريسبر للاستخدام السريري هو تحدٍ رئيسي يجب التغلب عليه لكي تصبح العلاجات القائمة على كريسبر خيارًا سريريًا قابلاً للتطبيق [261]. يعمل الباحثون على تطوير طرق أكثر كفاءة وفعالية من حيث التكلفة لإنتاج خلايا معدلة باستخدام كريسبر، مثل استخدام الإكسوزومات كطريقة توصيل، وتحسين القابلية للتوسع باستخدام خطوط خلايا معدلة وراثيًا أو خلايا جذعية، وتطوير منصات آلية وطرق توصيل في الموقع [200].
الخاتمة والاتجاهات المستقبلية
التعامل مع الخصائص المعقدة للسرطان من خلال التعديلات الجينية المستهدفة كبيرة. تشير الأساليب بما في ذلك تعطيل الجينات التي تعزز نمو الورم، وزيادة استجابة الجسم المناعية لخلايا السرطان، وتصحيح الشذوذات الجينية التي تؤدي إلى السرطان، ومهاجمة الأورام مباشرة باستخدام عوامل سامة، إلى مسارات واعدة في مجال العلاجات السرطانية. بدأت الأبحاث في مراحلها المبكرة والتجارب السريرية تكشف عن فعالية وإمكانات كريسبر التحويلية في سياق علاج السرطان. لم تسفر هذه التحقيقات عن نتائج واعدة فحسب، بل أوضحت أيضًا المسار الذي أمامنا. ومع ذلك، هناك عقبات ملحوظة يجب التغلب عليها. دقة كريسبر، وهي أكبر ميزة له، تثير القلق بشأن التأثيرات الجينية غير المقصودة، المعروفة باسم التأثيرات غير المستهدفة. تكمن الأهمية القصوى في ضمان أمان ودقة تدخلات كريسبر، مما يتطلب بحثًا مستمرًا لمعالجة هذه المخاوف. إن توصيل مكونات كريسبر إلى خلايا الورم يمثل تحديًا كبيرًا آخر. من الضروري تطوير طرق فعالة وآمنة لتوصيل هذه المكونات للاستخدام العملي لكريسبر في علاج السرطان. يتعزز هذا التحدي بسبب تنوع أنواع الأورام والتعقيد الكامن في البيولوجيا البشرية. ومع ذلك، فإن المستقبل المحتمل لكريسبر في علاج السرطان واعد بشكل استثنائي. مع التقدم المستمر في الأبحاث التي تتجاوز الحواجز الحالية، هناك إمكانية ملموسة لإنشاء علاجات أكثر كفاءة، مخصصة، وأقل تدخلاً لعلاج السرطان. يمكن أن تحول هذه التقدمات بشكل جذري النهج تجاه رعاية السرطان، من العلاج الكيميائي التقليدي والإشعاع إلى علاجات جينية محددة. تقدم منهجية كريسبر استراتيجية مبتكرة وقد تكون مغيرة لقواعد اللعبة في علاج السرطان. الرحلة المقبلة مليئة بالعقبات، مثل ضمان سلامة ودقة وكفاءة توصيل العلاجات. على الرغم من هذه التحديات، فإن التقدم المحرز حتى الآن واعد. إن التحقيق والتطوير المستمر في هذا المجال أمران حاسمان لاستغلال قدرات العلاجات القائمة على كريسبر بالكامل في مكافحة السرطان. من المهم مواجهة هذه التحديات مباشرة، مع التركيز على تحسين الأساليب، وتحسين أنظمة التوصيل، وإعطاء الأولوية لسلامة المرضى، من أجل استغلال الإمكانات الثورية لهذه التكنولوجيا في رعاية السرطان.
| أولويات البحث | التقنيات الناشئة | فرص النقل | الجهود التعاونية | الأثر الاجتماعي | مرجع |
| تطوير أنظمة توصيل أكثر دقة وكفاءة | توسيع أدوات تحرير الجينات لتتجاوز تعطيل الجينات وتصحيحها | تطوير مؤشرات حيوية للتنبؤ باستجابة العلاج | التطوير التعاوني لإرشادات أخلاقية للعلاج القائم على كريسبر | الوصول العادل إلى العلاج بالسرطان القائم على كريسبر | [268] |
| تطوير أدوات تحرير الجينات لاستهداف المناطق غير المشفرة من الجينوم | تطبيق شاشات كريسبر لتحديد أهداف علاجية جديدة | استخدام تحرير الجينات لتحسين استجابة العلاج المناعي | التعاون بين المؤسسات الأكاديمية والصناعة لتسريع تطوير الأدوية | التأثيرات على اقتصاديات الرعاية الصحية وتخصيص الموارد | [269] |
| استكشاف العلاجات المركبة التي تشمل تدخلات قائمة على كريسبر | دمج تكنولوجيا كريسبر مع تقنيات التصوير والاستشعار الناشئة | تطبيق تحرير الجينات باستخدام كريسبر لتطوير لقاحات سرطان مخصصة | التطوير التعاوني لأدوات تشخيصية قائمة على كريسبر | الآثار الأخلاقية لتحرير الجينات في السلالة الجرثومية للوقاية من السرطان وعلاجه | [271] |
| تطوير أساليب جديدة لتحسين دقة تحرير الجينات باستخدام كريسبر كاس9 | استكشاف استخدام تنظيم الجينات باستخدام كريسبر لتعديل التعبير الجيني | اختبار فعالية العلاج بالسرطان القائم على كريسبر بالاشتراك مع العلاجات القياسية الأخرى | تطوير أطر تنظيمية دولية لتكنولوجيا تحرير الجينات | التعليم والتوعية العامة لتعزيز الفهم والقبول لتكنولوجيا تحرير الجينات | [272] |
| دراسة تأثير تحرير الجينات باستخدام كريسبر على بيئة الورم والجهاز المناعي | تطوير أدوات قائمة على كريسبر للكشف عن السرطان ومراقبته بشكل غير تدخلي | دمج تحرير الجينات باستخدام كريسبر في تصنيف المرضى وتصميم التجارب السريرية | التعاون بين الباحثين لتعزيز فهم آليات كريسبر ودورها في بيولوجيا السرطان | معالجة قضايا التمييز الجيني واهتمامات الخصوصية المتعلقة بتكنولوجيا تحرير الجينات | [18] |
| تصميم أنظمة تحرير الجينات باستخدام كريسبر لعلاج السرطانات النادرة أو الصعبة العلاج | تقدم تكنولوجيا تحرير الجينات لاستهداف الشذوذات الجينية المعقدة في خلايا السرطان | إنشاء قواعد بيانات دولية لتعزيز تبادل بيانات وبروتوكولات العلاج المعتمد على كريسبر | تطوير إرشادات ومعايير لمراقبة الجودة وسلامة المنتج للعلاجات المعتمدة على كريسبر | ضمان الوصول العادل إلى العلاج المعتمد على كريسبر لمرضى السرطان من مختلف الفئات | [270] |
| تطوير استراتيجيات تحرير الجينات للتغلب على المقاومة للعلاجات القياسية للسرطان | دمج تقنية كريسبر مع تقنية النانو لتحسين توصيل واستهداف خلايا السرطان | إنشاء شراكات بين القطاعين العام والخاص لتسريع تطوير العلاجات المعتمدة على كريسبر للسرطان | التعاون بين الأطباء والباحثين الأساسيين لتحسين تصميم وتوصيل العلاجات المعتمدة على كريسبر للسرطان | تقييم السلامة والفعالية على المدى الطويل للعلاج المعتمد على كريسبر للسرطان | [273, 274] |
تطوير الأدوية وتعزيز فرص الترجمة الأكثر كفاءة [266]. علاوة على ذلك، فإن دمج التدخلات المعتمدة على كريسبر مع علاجات أخرى في نهج تآزري يستحق الاستكشاف، بينما ستساعد أدوات التشخيص المعتمدة على كريسبر في الكشف المبكر عن السرطان وتشخيصه [268]. يمكن الاستفادة من تقنية تحرير الجينات لتطوير لقاحات سرطان شخصية وتحسين استجابة العلاج المناعي. من حيث التأثير الاجتماعي
هناك حاجة إلى الوصول العادل إلى العلاج المعتمد على كريسبر للسرطان لضمان استفادة جميع المرضى من هذه التقدمات، بغض النظر عن خلفياتهم أو مواقعهم. يتطلب ذلك التعاون الدولي في إنشاء أطر تنظيمية لتقنية تحرير الجينات وتعزيز الفهم العام وقبول هذه العلاجات من خلال التعليم والتوعية [269]. تشمل أولويات البحث الأخرى دراسة آثار تحرير الجينات باستخدام كريسبر على الميكروبيئة الورمية والجهاز المناعي، ومعالجة التمييز الجيني ومخاوف الخصوصية المتعلقة بتحرير الجينات، وتقييم السلامة والفعالية على المدى الطويل للعلاج المعتمد على كريسبر للسرطان [18]. أخيرًا، فإن تطوير أنظمة تحرير الجينات باستخدام كريسبر للأورام النادرة أو الصعبة العلاج، واستغلال تقنية النانو لتوصيل كريسبر المستهدف، وإنشاء قواعد بيانات لتبادل البيانات أمر حاسم لدفع حدود العلاج المعتمد على كريسبر للسرطان [270]. تعتبر الشراكات بين القطاعين العام والخاص والتعاون بين الأطباء والباحثين أساسية في تحسين تصميم العلاج وتوصيله لتحقيق نتائج أفضل للمرضى [267].
تطوير طرق توصيل جديدة
سلامة وفعالية العلاجات المعتمدة على كريسبر باستخدام AAV [277]. توفر الجسيمات النانوية المصنوعة من البوليمرات أو المواد غير العضوية طرقًا بديلة لتوصيل مكونات كريسبر. يمكن تصميم هذه الجسيمات النانوية بخصائص فيزيائية كيميائية مختلفة، والتي قد تؤثر على امتصاص الخلايا وسرعة الإفراج [244]. بينما تتمتع الحويصلات الدهنية بمزايا في الاحتواء والتعديل، قد توفر الجسيمات النانوية الأخرى استقرارًا أفضل أو لديها قدرات فريدة للتوصيل المستهدف [282]. تمتلك الببتيدات المخترقة للخلايا والإكسوسومات القدرة على تحسين توصيل كريسبر [254]. يمكن للباحثين استكشاف التعديلات السطحية لهذه الأنظمة التوصيلية لزيادة ارتباطها المحدد للورم وامتصاصها [279]. بالإضافة إلى ذلك، فإن تحسين تحميل الحمولة وآليات الإفراج يمكن أن يعزز التحرير الدقيق للجينات المستهدفة مع تقليل الآثار غير المرغوب فيها [283].
العلاج المركب
استهداف جينات متعددة
يجب النظر في آثار المواقع غير الانتقائية، حيث يقوم CRISPR-Cas9 بتحرير جينات غير مقصودة، مما قد يؤدي إلى عواقب غير متوقعة. هناك حاجة إلى أبحاث واسعة وإجراءات أمان صارمة لتقليل هذه المخاطر وضمان التطبيق الآمن لتقنية CRISPR في علاجات السرطان. قد تختلف فعالية تحرير الجينات المعتمد على CRISPR في استهداف جينات متعددة بين أنواع السرطان المختلفة. يتميز كل نوع من أنواع السرطان بطفرات جينية فريدة، مما يتطلب نهجًا مخصصًا.
الطب الشخصي
الفتك الاصطناعي
الخلايا، مع الحفاظ على الخلايا الطبيعية [304]. في سياق تحرير الجينات المعتمد على كريسبر، تتضمن هذه الطريقة استخدام كريسبر-كاس9 لاستهداف جينين يتم تحويرهما بشكل متكرر في السرطان، مستغلين نقاط الضعف الجينية للسرطان [305]. تتمتع العلاجات السرطانية المعتمدة على القتل الاصطناعي بميزة استهداف خلايا السرطان بشكل انتقائي مع طفرات جينية محددة، مما يقلل من خطر إلحاق الضرر بالخلايا السليمة. يمكن أن تؤدي هذه الطريقة إلى علاجات أكثر فعالية ودقة مع آثار جانبية أقل من العلاجات التقليدية مثل العلاج الكيميائي والإشعاعي [304]. استهدف الباحثون تركيبات جينية مثل BRCA1 وBRCA2، التي توجد غالبًا في حالة تحور في سرطان الثدي والمبيض، وPARP1 وBRCA1، التي تتكرر فيها الطفرات في سرطان الثدي [303]. من خلال تعطيل هذه الأزواج الجينية في وقت واحد، فإنهم يحفزون القتل الاصطناعي في خلايا السرطان [302]. أظهرت الدراسات التجريبية إمكانيات القتل الاصطناعي المعتمد على كريسبر كاستراتيجية لعلاج السرطان [304]. من خلال استهداف تركيبات جينية محددة في خلايا السرطان، لاحظ الباحثون انخفاضات كبيرة في نمو الورم وحيوية الخلايا في النماذج قبل السريرية، مما يشير إلى إمكانيته كنهج علاجي واعد [303]. أحد التحديات الرئيسية هو توصيل مكونات كريسبر إلى موقع الورم بكفاءة [302]. كما أن ضمان الاستهداف الدقيق وآثار المواقع غير الانتقائية هي أيضًا مخاوف مهمة. بالإضافة إلى ذلك، فإن تحديد تركيبات جينية مناسبة لأنواع السرطان المحددة وضمان السلامة أثناء الترجمة السريرية هي اعتبارات حيوية [303].
علاج خلايا CAR-T
وورم ليمفوما غير هودجكين [310]. وقد أفادت التجارب السريرية بمعدلات استجابة عالية وحتى شفاء دائم في بعض المرضى [308]. ومع ذلك، فإن فعاليته في الأورام الصلبة لا تزال منطقة بحث وتطوير مهمة [307]. الآثار الجانبية طويلة الأمد لعلاج خلايا CAR-T لم تُفهم بالكامل بعد، حيث إن العلاج جديد نسبيًا [311]. ومع ذلك، تشمل بعض المخاوف المحتملة استمرار خلايا CAR-T في الجسم، والآثار الجانبية غير الانتقائية للتعديلات الجينية، وتأثيرها على وظيفة المناعة الطبيعية [312]. لمعالجة تحديات التصنيع، تُبذل جهود لتحسين وتبسيط عملية الإنتاج، بما في ذلك الأتمتة وتقليل الوقت والتكلفة المعنية [308]. علاوة على ذلك، يمكن أن يساعد إنشاء مراكز متخصصة مزودة بالخبرة والبنية التحتية في معالجة التحديات اللوجستية المرتبطة بعلاج خلايا CAR-T [307]. تشمل القيود الحالية التكلفة العالية للعلاج، والوصول المحدود بسبب المتطلبات المتخصصة، والحاجة إلى العلاج المخصص.
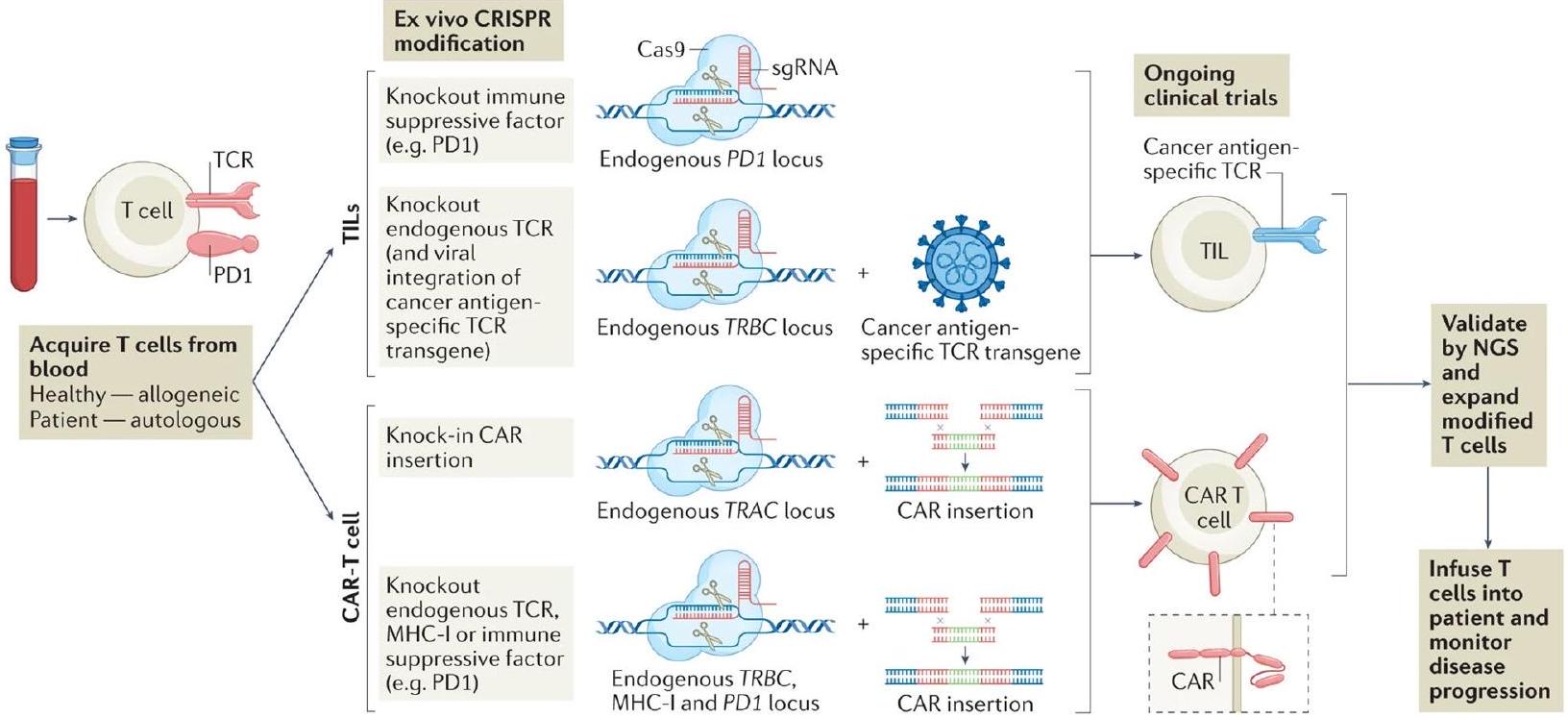
تصنيع لكل مريض [310]. بالإضافة إلى ذلك، يمكن أن تتأثر فعالية علاج خلايا CAR-T بعوامل مثل هروب المستضد، وتنوع الورم، والبيئة الدقيقة المثبطة للمناعة للورم [313]. توضح الشكل 11 التلاعب خارج الجسم باستخدام CRISPR لخلايا T البشرية لعلاج خلايا T بالتبني.
دمج تحرير الجينات المعتمد على كريسبر مع علاج الخلايا الجذعية
دمج تحرير الجينات المعتمد على كريسبر مع العلاج الجيني
تأثيرات [322]. بالإضافة إلى ذلك، قد يكون من الصعب الحفاظ على تنظيم طويل الأمد لنشاط الجينات عبر العلاج الجيني بسبب العمليات الخلوية التي قد تعيد هذه التغييرات مع مرور الوقت. يحتاج الباحثون إلى تطوير استراتيجيات للحفاظ على التعديلات الجينية المستقرة والقابلة للتوريث [323]. التباين الجيني، حيث تظهر خلايا مختلفة داخل ورم أو مرض طفرات جينية مميزة، يشكل تحديًا للعلاجات المستهدفة [256]. يسمح الجمع بين تقنية كريسبر والعلاج الجيني للباحثين باستهداف طفرات معينة مع تجاوز أخرى [324]. يمكن برمجة كريسبر للتعرف على وتحرير طفرات معينة، بينما يمكن للعلاج الجيني كبح نشاط جينات معينة متحورة، مما يؤدي إلى علاج أكثر شمولاً وفعالية [325]. يثير الجمع بين هذه التقنيات القوية أسئلة أخلاقية حول الآثار الجانبية المحتملة غير الانتقائية، والعواقب غير المقصودة، وتحرير الخط الجرثومي [325]. يجب على الباحثين وصانعي السياسات ضمان الالتزام الصارم ببروتوكولات السلامة والاستخدام المسؤول لمنع التغيرات الجينية غير المقصودة [326]. بالإضافة إلى ذلك، يجب معالجة الوصول العادل إلى مثل هذه العلاجات والفجوات المحتملة في الرعاية الصحية لتجنب تفاقم الفوارق الاجتماعية [327]. كما هو الحال مع أي تقنية ناشئة، هناك قيود يجب أخذها في الاعتبار. يمكن أن يكون توصيل مكونات كريسبر والأدوية الجينية إلى أنسجة أو أعضاء معينة تحديًا [328]. تركز الأبحاث الجارية على تحسين طرق التوصيل وزيادة كفاءة الاستهداف [325]. علاوة على ذلك، يبقى فهم العواقب طويلة الأمد للتعديلات الجينية والآثار الجانبية المحتملة أولوية لمزيد من التحقيق لضمان سلامة وفعالية هذه العلاج المركب [327].
تحديد أهداف دوائية جديدة
تعديل على بقاء الخلايا وتكاثرها [330، 333-336]. إذا كان فقدان أو تغيير جين ما يؤثر بشكل كبير على قدرة الخلايا على النمو والبقاء، فإن ذلك يشير إلى أن الجين يلعب دورًا حيويًا في دعم وظائف خلايا السرطان [332]. يوفر تحديد الجينات الحاسمة باستخدام تقنية كريسبر-كاس9 رؤى قيمة حول نقاط الضعف والاعتماديات في خلايا السرطان [48، 333، 335]. يمكن أن تُدرس الجينات التي وُجدت أنها ضرورية لنمو خلايا السرطان وبقائها كأهداف محتملة للأدوية [333]. قد يؤدي استهداف هذه الجينات بالأدوية إلى تعطيل العمليات الخلوية الحيوية، مما يؤدي إلى قتل أو قمع خلايا السرطان بشكل انتقائي مع تقليل الضرر على الخلايا الطبيعية [329، 331، 337، 338]. على الرغم من إمكانياتها، تواجه تقنية تحرير الجينات المعتمدة على كريسبر لتحديد أهداف الأدوية عدة تحديات [333]. يمكن أن تؤدي التأثيرات غير المستهدفة، وعدم اكتمال إلغاء الجين، والتكرار الوظيفي داخل المسارات الخلوية إلى تعقيد تفسير البيانات [333]. بالإضافة إلى ذلك، يتطلب تحويل النتائج المستندة إلى كريسبر إلى أهداف دوائية فعالة مزيدًا من التحقق من خلال الدراسات ما قبل السريرية والسريرية [48]. إن ضمان الخصوصية والفعالية والسلامة للأدوية التي تستهدف الجينات التي تم تحديدها حديثًا أمر حاسم للتنفيذ السريري الناجح [331، 333، 339].
شكر وتقدير
مساهمات المؤلفين
تمويل
توفر البيانات والمواد
الإعلانات
موافقة الأخلاقيات والموافقة على المشاركة
موافقة على النشر
المصالح المتنافسة
نُشر على الإنترنت: 09 يناير 2024
References
- De A, Biswas AR. Nanotechnology and Computational tool based study of CRISPR/Cas-9 research in Biomedical Engineering. J Nano Res Adv Mater Polym Sci. 2020;1:6-1.
- Hale CR, Majumdar S, Elmore J, Pfister N, Compton M, Olson S, et al. Essential features and rational design of CRISPR RNAs that function with the Cas RAMP module complex to cleave RNAs. Mol Cell. 2012;45(3):292-302.
- Ayanoğlu FB, Elçiln AE, Elçin YM. Bioethical issues in genome editing by CRISPR-Cas9 technology. Turkish J Biol. 2020;44(2):110-20.
- Feng Z, Zhang B, Ding W, Liu X, Yang D-L, Wei P, et al. Efficient genome editing in plants using a CRISPR/Cas system. Cell Res. 2013;23(10):1229-32.
- Shachaf CM, Kopelman AM, Arvanitis C, Karlsson Å, Beer S, Mandl S, et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004;431(7012):1112-7.
- Su S, Hu B, Shao J, Shen B, Du J, Du Y, et al. CRISPR-Cas9 mediated efficient PD-1 disruption on human primary T cells from cancer patients. Sci Rep. 2016;6. https://doi.org/10.1038/srep20070.
- Ye L, Wang J, Tan Y, Beyer AI, Xie F, Muench MO, et al. Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: An approach for treating sickle cell disease and
-thalassemia. Proc Natl Acad Sci. 2016;113(38):10661-5. - Walton J, Blagih J, Ennis D, Leung E, Dowson S, Farquharson M, et al. CRISPR/Cas9-mediated Trp53 and Brca2 knockout to generate improved murine models of ovarian high-grade serous carcinoma. Cancer Res. 2016;76(20):6118-29.
- Zhao R, Kaakati R, Liu X, Xu L, Lee AK, Bachelder R, et al. CRISPR/Cas9Mediated BRCA1 Knockdown Adipose Stem Cells Promote Breast Cancer Progression. Plast Reconstr Surg. 2019;143(3):747-56.
- Summary R, Doudna JA, Charpentier E. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346(6213). https://doi. org/10.1126/science. 1258096.
- Katti A, Diaz BJ, Caragine CM, Sanjana NE, Dow LE. CRISPR in cancer biology and therapy. Nat Rev Cancer. 2022;22(5):259-79.
- Awwad SW, Serrano-Benitez A, Thomas JC, Gupta V, Jackson SP. Revolutionizing DNA repair research and cancer therapy with CRISPRCas screens. Nat Rev Mol Cell Biol. 2023. https://doi.org/10.1038/ s41580-022-00571-x.
- Prakash G, Shokr A, Willemen N, Bashir SM, Shin SR, Hassan S. Microfluidic fabrication of lipid nanoparticles for the delivery of nucleic acids. Advanced Drug Delivery Reviews. 2022;184. https://doi.org/10.1016/j. addr.2022.114197.
- Modell AE, Lim D, Nguyen TM, Sreekanth V, Choudhary A. CRISPR-based therapeutics: current challenges and future applications. Trends Pharmacol Sci. 2022;43(2):151-61.
- Yin H, Xue W, Anderson DG. CRISPR-Cas: a tool for cancer research and therapeutics. Nat Rev Clin Oncol. 2019;16(5):281-95.
- Lakshmanan VK, Jindal S, Packirisamy G, Ojha S, Lian S, Kaushik A, et al. Nanomedicine-based cancer immunotherapy: recent trends and future perspectives. Cancer Gene Ther. 2021. https://doi.org/10.1038/ s41417-021-00299-4.
- Miliotou AN, Papadopoulou LC. CAR T-cell Therapy: A New Era in Cancer Immunotherapy. Curr Pharm Biotechnol. 2018. https://doi.org/10.2174/ 1389201019666180418095526.
- Huang D, Miller M, Ashok B, Jain S, Peppas NA. CRISPR/Cas systems to overcome challenges in developing the next generation of T cells for cancer therapy. Adv Drug Deliv Rev. 2020;158:17-35.
- Azangou-Khyavy M, Ghasemi M, Khanali J, Boroomand-Saboor M, Jamalkhah M, Soleimani M, et al. CRISPR/Cas: From Tumor Gene Editing to T Cell-Based Immunotherapy of Cancer. Front Immunol. 2020. https://doi.org/10.3389/fimmu.2020.02062.
- Shachaf CM, Felsher DW. Rehabilitation of cancer through oncogene inactivation. Trends Mol Med. 2005;11(7):316-21.
- Kennedy EM, Kornepati AVR, Goldstein M, Bogerd HP, Poling BC, Whisnant AW, et al. Inactivation of the Human Papillomavirus E6 or E7 Gene in Cervical Carcinoma Cells by Using a Bacterial CRISPR/Cas RNAGuided Endonuclease. J Virol. 2014;88(20):11965-72.
- Riedel M, Cai H, Stoltze IC, Vendelbo MH, Wagner EF, Bakiri L, et al. Targeting AP-1 transcription factors by CRISPR in the prostate. Oncotarget. 2021;12(19):1956-61.
- Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, et al. Evolution and classification of the CRISPR-Cas systems. Nature Reviews Microbiology. 2011;9(6):467-77.
- Hoellerbauer P, Kufeld M, Paddison PJ. Efficient Multi-Allelic Genome Editing of Primary Cell Cultures via CRISPR-Cas9 Ribonucleoprotein Nucleofection. Curr Protoc Stem Cell Biol. 2020. https://doi.org/10.1002/ cpsc. 126.
- Kim J, Jang G, Sim SH, Park IH, Kim K, Park C. Smarca4 depletion induces cisplatin resistance by activating yap1-mediated epithelial-to-mesenchymal transition in triple-negative breast cancer. Cancers (Basel). 2021;13(21). https://doi.org/10.3390/cancers13215474.
- Zeng K, Chen X, Hu X, Liu X, Xu T, Sun H, et al. LACTB, a novel epigenetic silenced tumor suppressor, inhibits colorectal cancer progression by attenuating MDM2-mediated p53 ubiquitination and degradation. Oncogene. 2018;37(41):5534-51.
- Estêvão D, Rios Costa N, Da Costa RG, Medeiros R. CRISPR-Cas9 therapies in experimental mouse models of cancer. Future Oncol. 2018;14(20):2083-95.
- Alves E, Taifour S, Dolcetti R, Chee J, Nowak AK, Gaudieri S, et al. Reprogramming the anti-tumor immune response via CRISPR genetic and epigenetic editing. Mol Ther Methods Clin Dev. 2021;21:592-606.
- Takata K, Chong LC, Ennishi D, Thakur A, Healy S, Viganò E, et al. The Tumor Associated Antigen PRAME Exhibits Dualistic Functions That Are Targetable in Diffuse Large B-Cell Lymphoma. Blood. 2020;136(Supplement 1):34-34.
- Zhi L, Su X, Yin M, Zhang Z, Lu H, Niu Z, et al. Genetical engineering for NK and T cell immunotherapy with CRISPR/Cas9 technology: Implications and challenges. Cell Immunol. 2021;369. https://doi.org/10.1016/j. cellimm.2021.104436.
- Afolabi LO, Afolabi MO, Sani MM, Okunowo WO, Yan D, Chen L, et al. Exploiting the CRISPR-Cas9 gene-editing system for human cancers and immunotherapy. Clin Transl Immunol. 2021;10(6). https://doi.org/ 10.1002/cti2.1286.
- Biggi AFB, Simioni PU. Inhibition of PD-1 protein by the CRISPR-Cas9 method as antitumor therapy of non-small cell lung cancers. Rev da Fac Ciências Médicas Sorocaba. 2019;21(1):2-7.
- Zarogoulidis P, Lampaki S, Yarmus L, Kioumis I, Pitsiou G, Katsikogiannis N , et al. Interleukin-7 and interleukin-15 for cancer. J Cancer. 2014. https://doi.org/10.7150/jca. 10471.
- Zhang Z, Kong X, Ligtenberg MA, van Hal-van Veen SE, Visser NL, de Bruijn B, et al. RNF31 inhibition sensitizes tumors to bystander killing by innate and adaptive immune cells. Cell Rep Med. 2022;3(6). https://doi. org/10.1016/j.xcrm.2022.100655.
- Wang L, Chen Y, Liu X, Li Z, Dai X. The Application of CRISPR/Cas9 Technology for Cancer Immunotherapy: Current Status and Problems. Front Oncol. 2022;11. https://doi.org/10.3389/fonc.2021.704999.
- Zhou XM, Li WQ, Wu YH, Han L, Cao XG, Yang XM, et al. Intrinsic expression of immune checkpoint molecule TIGIT could help tumor growth in vivoby suppressing the function of NK and CD8+T Cells. Front Immunol. 2018;9(NOV). https://doi.org/10.3389/fimmu.2018.02821.
- Reis A, Hornblower B, Robb B, Tzertzinis G. CRISPR/Cas9 and targeted genome editing: a new era in molecular biology. NEB expressions. 2014;1:3-6.
- Chavez M, Chen X, Finn PB, Qi LS. Advances in CRISPR therapeutics. Nat Rev Nephrol. 2023;19(1):9-22.
- Chehelgerdi
, Chehelgerdi . The use of RNA – based treatments in the field of cancer immunotherapy. 2023. BioMed Central. https://doi. org/10.1186/s12943-023-01807-w. - Lino CA, Harper JC, Carney JP, Timlin JA. Delivering crispr: A review of the challenges and approaches. Drug Delivery. 2018;25(1):1234-57.
- Hatada I, Morita S, Horii T. CRISPR/Cas9. Methods Mol Biol. 2023;2637:41-7.
- Pawluk A, Amrani N, Zhang Y, Garcia B, Hidalgo-Reyes Y, Lee J, et al. Naturally Occurring Off-Switches for CRISPR-Cas9. Cell. 2016. https:// doi.org/10.1016/j.cell.2016.11.017.
- Emily P, John H, Mitchell K, Margaret C, Bendzick L, Miller JS, et al. Enhancing Human NK Cell Function and Specificity for Cancer Immunotherapy. Blood. 2018;132(Supplement 1):2044-2044.
- Shmakov S, Smargon A, Scott D, Cox D, Pyzocha N, Yan W, et al. Diversity and evolution of class 2 CRISPR-Cas systems. Nat Rev Microbiol. 2017. https://doi.org/10.1038/nrmicro.2016.184.
- Biagioni A, Laurenzana A, Margheri F, Chillà A, Fibbi G, So M. Delivery systems of CRISPR/Cas9-based cancer gene therapy. J Biol Eng. 2018;12(1). https://doi.org/10.1186/s13036-018-0127-2.
- Sung B, Kim MH, Abelmann L. Magnetic microgels and nanogels: Physical mechanisms and biomedical applications. Bioeng Transl Med. 2021;6(1). https://doi.org/10.1002/btm2.10190.
- Jiang F, Doudna JA. CRISPR-Cas9 Structures and Mechanisms. Annu Rev Biophys. 2017;46:505-29.
- Zhang H, Qin C, An C, Zheng X, Wen S, Chen W, et al. Application of the CRISPR/Cas9-based gene editing technique in basic research, diagnosis, and therapy of cancer. Mol Cancer. 2021;20:1-22.
- Suzuki Y, Onuma H, Sato R, Sato Y, Hashiba A, Maeki M, et al. Lipid nanoparticles loaded with ribonucleoprotein-oligonucleotide complexes synthesized using a microfluidic device exhibit robust genome editing and hepatitis B virus inhibition. J Control Release. 2021;330:61-71.
- Wilbie D, Walther J, Mastrobattista E. Delivery Aspects of CRISPR/Cas for in Vivo Genome Editing. Acc Chem Res. 2019;52(6):1555-64.
- Künne T, Westra ER, Brouns SJJ. Electrophoretic mobility shift assay of DNA and CRISPR- Cas ribonucleoprotein complexes. Methods Mol Biol. 2015;1311:171-84.
- Bykonya AG, Lavrov AV, Smirnikhina SA. Methods for CRISPR-Cas as Ribonucleoprotein Complex Delivery In Vivo. Mol Biotechnol. 2023;65(2):181-95.
- Zhou Y, Bravo JPK, Taylor HN, Steens JA, Jackson RN, Staals RHJ, et al. Structure of a type IV CRISPR-Cas ribonucleoprotein complex. IScience. 2021;24(3). https://doi.org/10.1016/j.isci.2021.102201.
- Gregg JR, Thompson TC. Considering the potential for gene-based therapy in prostate cancer. Nat Rev Urol. 2021;18(3):170-84.
- Pomeroy E, Hunzeker J, Kluesner M, Crosby M, Bendzick L, Geller M, et al. Genetically engineered natural killer cells for cancer immunotherapy. Mol Ther. 2018;26(5):355-6.
- Li R, Wang Q, She K, Lu F, Yang Y. CRISPR/Cas systems usher in a new era of disease treatment and diagnosis. Molecular Biomedicine. 2022;3(1). https://doi.org/10.1186/s43556-022-00095-y.
- Martinez-Lage M, Torres-Ruiz R, Puig-Serra P, Moreno-Gaona P, Martin MC, Moya FJ, et al. In vivo CRISPR/Cas9 targeting of fusion oncogenes for selective elimination of cancer cells. Nat Commun. 2020;11(1). https://doi.org/10.1038/s41467-020-18875-x.
- Gao J, Luo T, Lin N, Zhang S, Wang J. A New Tool for CRISPR-Cas13aBased Cancer Gene Therapy. Mol Ther – Oncolytics. 2020;19:79-92.
- Chen J, López-Moyado IF, Seo H, Lio CWJ, Hempleman LJ, Sekiya T, et al. NR4A transcription factors limit CART cell function in solid tumours. Nature. 2019;567(7749):530-4.
- Bridges K, Yao HHC, Nicol B. Loss of Runx1 Induces Granulosa Cell Defects and Development of Ovarian Tumors in the Mouse. Int J Mol Sci. 2022;23(22). https://doi.org/10.3390/ijms232214442.
- Jung S, Kim DH, Choi YJ, Kim SY, Park H, Lee H, et al. Contribution of p53 in sensitivity to EGFR tyrosine kinase inhibitors in non-small cell lung cancer. Sci Rep. 2021;11(1). https://doi.org/10.1038/ s41598-021-99267-z.
- Wang L, Pearson N, Xiong Y, Renuse S, Cheng R, Carter JM, et al. Abstract 3913: Quantitative phosphoproteomic analysis of AXL signaling network in breast cancer. Cancer Res. 2022;82(12_Supplement):3913-3913.
- Hu X, Li L, Eid JE, Liu C, Yu J, Yue J, et al. IDH1 Mutation Induces HIF-1 a and Confers Angiogenic Properties in Chondrosarcoma JJ012 Cells. Dis Markers. 2022;2022. https://doi.org/10.1155/2022/7729968.
- Hui Goh CJ, Wong JH, El Farran C, Tan BX, Coffill CR, Loh YH, et al. Identification of pathways modulating vemurafenib resistance in melanoma cells via a genome-wide CRISPR/Cas9 screen. G3 Genes Genomes Genet. 2021;11(2). https://doi.org/10.1093/g3journal/jkaa069.
- Thus YJ, de Rooij MFM, Swier N, Beijersbergen RL, Guikema JEJ, Kersten MJ, et al. Inhibition of casein kinase 2 sensitizes mantle cell lymphoma to venetoclax through MCL-1 downregulation. Haematologica. 2023;108(3):797-810.
- Wrobel P, Shojaei H, Schittek B, Gieseler F, Wollenberg B, Kalthoff H, et al. Lysis of a broad range of epithelial tumour cells by human
cells: Involvement of NKG2D ligands and T-cell receptor- versus NKG2Ddependent recognition. Scand J Immunol. 2007;66(2-3):320-8. - Li Z, Chi Z, Ang WX, Chen C, Tay JCK, Ng YY, et al. Experimental treatment of colorectal cancer in mice with human
cells electroporated with NKG2D RNA CAR. Immunotherapy. 2020;12(10):733-48. - Mo F, Duan S, Jiang X, Yang X, Hou X, Shi W, et al. Nanobody-based chimeric antigen receptor T cells designed by CRISPR/Cas9 technology
for solid tumor immunotherapy. Signal Transduct Target Ther. 2021;6(1). https://doi.org/10.1038/s41392-021-00462-1. - Kuznik NC, Solozobova V, Lee II, Jung N, Yang L, Nienhaus K, et al. A chemical probe for BAG1 targets androgen receptor-positive prostate cancer through oxidative stress signaling pathway. iScience. 2022;25(5). https://doi.org/10.1016/j.isci.2022.104175.
- Ungefroren H, Otterbein H, Fiedler C, Mihara K, Hollenberg MD, Gieseler F, et al. RAC1B suppresses TGF-
-dependent cell migration in pancreatic carcinoma cells through inhibition of the TGF- type i receptor ALK5. Cancers (Basel). 2019;11(5). https://doi.org/10.3390/cancers110 50691. - Tishchenko A, Azorín DD, Vidal-Brime L, Muñoz MJ, Arenas PJ, Pearce C, et al. Cx43 and associated cell signaling pathways regulate tunneling nanotubes in breast cancer cells. Cancers (Basel). 2020;12(10):1-25.
- Cerchietti L, Melnick A. Targeting BCL6 in diffuse large B-cell lymphoma: What does this mean for the future treatment? Expert Rev Hematol. 2013;6(4):343-5.
- Aquilanti E, Kageler L, Watson J, Baird DM, Jones RE, Hodges M, et al. Telomerase inhibition is an effective therapeutic strategy in TERT promoter-mutant glioblastoma models with low tumor volume. Neuro Oncol. 2023. https://doi.org/10.1093/neuonc/noad024.
- Menendez-Gonzalez JB, Vukovic M, Abdelfattah A, Saleh L, Almotiri A, Thomas L anne, et al. Gata2 as a Crucial Regulator of Stem Cells in Adult Hematopoiesis and Acute Myeloid Leukemia. Stem Cell Rep. 2019;13(2):291-306.
- Chang KH, Smith SE, Sullivan T, Chen K, Zhou Q, West JA, et al. LongTerm Engraftment and Fetal Globin Induction upon BCL11A Gene Editing in Bone-Marrow-Derived CD34+ Hematopoietic Stem and Progenitor Cells. Mol Ther Methods Clin Dev. 2017;4:137-48.
- Hu Y, Zhou Y, Zhang M, Ge W, Li Y, Yang L, et al. CRISPR/Cas9-engineered universal CD19/CD22 dual-targeted CAR-T cell therapy for relapsed/ refractory B-cell acute lymphoblastic leukemia. Clin Cancer Res. 2021;27(10):2764-72.
- Tong KI, Yoon S, Isaev K, Bakhtiari M, Lackraj T, He MY, et al. Combined EZH2 inhibition and IKAROS degradation leads to enhanced antitumor activity in diffuse large B-cell lymphoma. Clin Cancer Res. 2021;27(19):5401-14.
- Yang Z, Hu N, Wang W, Hu W, Zhou S, Shi J, et al. Loss of FBXW7 Correlates with Increased IDH1 Expression in Glioma and Enhances IDH1-Mutant Cancer Cell Sensitivity to Radiation. Cancer Res. 2022;82(3):497-509.
- Chen L, Alexe G, Dharia NV, Ross L, Iniguez AB, Conway AS, et al. CRISPRCas9 screen reveals a MYCN-amplified neuroblastoma dependency on EZH2. J Clin Invest. 2018;128(1):446-62.
- Wang
, Chen , X, Wang , Zhu , Wu X. MUC16 impacts tumor proliferation and migration through cytoplasmic translocation of P120catenin in epithelial ovarian cancer cells: An original research. BMC Cancer. 2019;19(1). https://doi.org/10.1186/s12885-019-5371-4. - Liu J, Guzman MA, Pezanowski D, Patel D, Hauptman J, Keisling M, et al. FOXO1-FGFR1 fusion and amplification in a solid variant of alveolar rhabdomyosarcoma. Mod Pathol. 2011;24(10):1327-35.
- Calderon-Aparicio A, Yamamoto H, de Vitto H, Zhang T, Wang Q, Bode AM, et al. RCC2 promotes esophageal cancer growth by regulating activity and expression of the Sox2 transcription factor. Mol Cancer Res. 2020;18(11):1660-74.
- Kotiyal S, Evason KJ. Exploring the interplay of telomerase reverse transcriptase and
-catenin in hepatocellular carcinoma. Cancers. 2021;13(16). https://doi.org/10.3390/cancers13164202. - Jung YS, Jun S, Lee SH, Sharma A, Park J II. Wnt2 complements Wnt/
-catenin signaling in colorectal cancer. Oncotarget. 2015;6(35):37257-37268. - Blackburn AC, Jerry DJ. Knockout and transgenic mice of Trp53: What have we learned about p53 in breast cancer? Breast Cancer Res. 2002;4(3):101-11.
- Muzumdar MD, Chen PY, Dorans KJ, Chung KM, Bhutkar A, Hong E, et al. Survival of pancreatic cancer cells lacking KRAS function. Nat Commun. 2017;8(1). https://doi.org/10.1038/s41467-017-00942-5.
- Xu Y, Jin Y, Liu L, Zhang X, Chen Y, Wei J. Study of circulating IgG antibodies to peptide antigens derived from BIRC5 and MYC in cervical cancer. FEBS Open Bio. 2015;5:198-201.
- Ponnusamy L, Natarajan SR, Manoharan R. MARK2 potentiate aerobic glycolysis-mediated cell growth in breast cancer through regulating mTOR/HIF-1 a and p53 pathways. J Cell Biochem. 2022;123(4):759-71.
- Rahman S, Magnussen M, León TE, Farah N, Li Z, Abraham BJ, et al. Activation of the LMO2 oncogene through a somatically acquired neomorphic promoter in T-cell acute lymphoblastic leukemia. Blood. 2017;129(24):3221-6.
- Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, et al. HIF-2a drives an intrinsic vulnerability to ferroptosis in clear cell renal cell carcinoma. BioRxiv. 2018;9:388041.
- Chen T, Tsang JYS, Su XC, Li P, Sun WQ, Wong ILK, et al. SALL4 promotes tumor progression in breast cancer by targeting EMT. Mol Carcinog. 2020;59(10):1209-26.
- Kollmann K, Briand C, Bellutti F, Schicher N, Blunder S, Zojer M, et al. The interplay of CDK4 and CDK6 in melanoma. Oncotarget. 2019;10(14):1346-59.
- Gomez Limia CE, Devalle S, Reis M, Sochacki J, Carneiro M, Madeiro da Costa R, et al. Generation and characterization of a human induced pluripotent stem (iPS) cell line derived from an acute myeloid leukemia patient evolving from primary myelofibrosis carrying the CALR 52 bp deletion and the ASXL1 p.R693X mutation. Stem Cell Res. 2017;24:16-20.
- Ihry RJ, Worringer KA, Salick MR, Frias E, Ho D, Theriault K, et al. P53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat Med. 2018;24(7):939-46.
- Bowden AR, Morales-Juarez DA, Sczaniecka-Clift M, Agudo MM, Lukashchuk N, Thomas JC, et al. Parallel crispr-cas9 screens clarify impacts of p53 on screen performance. Elife. 2020;9:1.
- Zhan H, Xie H, Zhou Q, Liu Y, Huang W. Synthesizing a Genetic Sensor Based on CRISPR-Cas9 for Specifically Killing p53-Deficient Cancer Cells. ACS Synth Biol. 2018;7(7):1798-807.
- Tarangelo A, Magtanong L, Bieging-Rolett KT, Li Y, Ye J, Attardi LD, et al. p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep. 2018;22(3):569-75.
- Qiao C, Liu W, Jiang H, He M, Yang Q, Xing Y. Integrated analysis of miRNA and mRNA expression profiles in p53-edited PFF cells. Cell Cycle. 2020;19(8):949-59.
- D’Costa S, Rich MJ, Diekman BO. Engineered cartilage from human chondrocytes with homozygous knockout of cell cycle inhibitor p21. Tissue Eng Part A. 2020;26(7-8):441-9.
- Nicolae CM, O’connor MJ, Constantin D, Moldovan GL. NFkB regulates p21 expression and controls DNA damage-induced leukemic differentiation. Oncogene. 2018;37(27):3647-56.
- Fischietti M, Eckerdt F, Perez RE, Guillen Magaña JN, Mazewski C, Ho S, et al. SLFN11 Negatively Regulates Noncanonical NFkB Signaling to Promote Glioblastoma Progression. Cancer Res Commun. 2022;2(9):966-78.
- Hartmann O, Reissland M, Maier CR, Fischer T, Prieto-Garcia C, Baluapuri A, et al. Implementation of CRISPR/Cas9 Genome Editing to Generate Murine Lung Cancer Models That Depict the Mutational Landscape of Human Disease. Front Cell Dev Biol. 2021;9. https://doi.org/10.3389/ fcell.2021.641618.
- Walsh T, Casadei S, Munson KM, Eng M, Mandell JB, Gulsuner S, et al. CRISPR-Cas9/long-read sequencing approach to identify cryptic mutations in BRCA1 and other tumour suppressor genes. J Med Genet. 2021;58(12):850-2.
- Mintz RL, Lao YH, Chi CW, He S, Li M, Quek CH, et al. CRISPR/Cas9-mediated mutagenesis to validate the synergy between PARP1 inhibition and chemotherapy in BRCA1-mutated breast cancer cells. Bioeng Transl Med. 2020;5(1). https://doi.org/10.1002/btm2.10152.
- Walton JB, Farquharson M, Mason S, Port J, Kruspig B, Dowson S, et al. CRISPR/Cas9-derived models of ovarian high grade serous carcinoma targeting Brca1, Pten and Nf1, and correlation with platinum sensitivity. Sci Rep. 2017;7(1). https://doi.org/10.1038/s41598-017-17119-1.
- Tsujino T, Takai T, Hinohara K, Gui F, Tsutsumi T, Bai X, et al. CRISPR screens reveal genetic determinants of PARP inhibitor sensitivity and resistance in prostate cancer. Nat Commun. 2023;14(1). https://doi.org/ 10.1038/s41467-023-35880-y.
- Witz A, Dardare J, Husson M, Francois A, Merlin J-L, Gilson P, et al. 35P Increased sensitivity to olaparib by BRCA1/2 knockdown using a
108. Annunziato S, de Ruiter JR, Henneman L, Brambillasca CS, Lutz C, Vaillant F, et al. Comparative oncogenomics identifies combinations of driver genes and drug targets in BRCA1-mutated breast cancer. Nat Commun. 2019;10(1). https://doi.org/10.1038/s41467-019-08301-2.
109. Weber J, Öllinger R, Friedrich M, Ehmer U, Barenboim M, Steiger K, et al. CRISPR/Cas9 somatic multiplex-mutagenesis for high-Throughput functional cancer genomics in mice. Proc Natl Acad Sci U S A. 2015;112(45):13982-7.
110. Yoshimatsu S, Nakajima M, Qian E, Sanosaka T, Sato T, Okano H. Homologous Recombination-Enhancing Factors Identified by Comparative Transcriptomic Analyses of Pluripotent Stem Cell of Human and Common Marmoset. Cells. 2022;11 (3). https://doi.org/10.3390/cells 11030360.
111. Choudhury SR, Cui Y, Lubecka K, Stefanska B, Irudayaraj J. CRISPR-dCas9 mediated TET1 targeting for selective DNA demethylation at BRCA1 promoter. Oncotarget. 2016;7(29):46545-56.
112. Kweon J, Jang AH, Shin HR, See JE, Lee W, Lee JW, et al. A CRISPR-based base-editing screen for the functional assessment of BRCA1 variants. Oncogene. 2020;39(1):30-5.
113. Lee W, Lee JH, Jun S, Lee JH, Bang D. Selective targeting of KRAS oncogenic alleles by CRISPR/Cas9 inhibits proliferation of cancer cells. Sci Rep. 2018;8(1). https://doi.org/10.1038/s41598-018-30205-2.
114. Bender G, Fahrioglu Yamaci R, Taneri B. CRISPR and KRAS: a match yet to be made. J Biomed Sci. 2021;28(1). https://doi.org/10.1186/ s12929-021-00772-0.
115. Zhao X, Liu L, Lang J, Cheng K, Wang Y, Li X, et al. A CRISPR-Cas13a system for efficient and specific therapeutic targeting of mutant KRAS for pancreatic cancer treatment. Cancer Lett. 2018;431:171-81.
116. Burgess MR, Hwang E, Mroue R, Bielski CM, Wandler AM, Huang BJ, et al. KRAS Allelic Imbalance Enhances Fitness and Modulates MAP Kinase Dependence in Cancer. Cell. 2017;168(5):817-829.e15.
117. Wang B, Krall EB, Aguirre AJ, Kim M, Widlund HR, Doshi MB, et al. ATXN1L, CIC, and ETS Transcription Factors Modulate Sensitivity to MAPK Pathway Inhibition. Cell Rep. 2017;18(6):1543-57.
118. Sayed S, Sidorova OA, Hennig A, Augsburg M, Cortés Vesga CP, Abohawya M, et al. Efficient Correction of Oncogenic KRAS and TP53 Mutations through CRISPR Base Editing. Cancer Res. 2022;82(17):3002-15.
119. Lentsch E, Li L, Pfeffer S, Ekici AB, Taher L, Pilarsky C, et al. CRISPR/Cas9mediated knock-out of krasG12D mutated pancreatic cancer cell lines. Int J Mol Sci. 2019;20(22). https://doi.org/10.3390/ijms20225706.
120. Wang Z, Kang B, Gao Q, Huang L, Di J, Fan Y, et al. Quadrupleediting of the MAPK and PI3K pathways effectively blocks the progression of KRAS-mutated colorectal cancer cells. Cancer Sci. 2021;112(9):3895-910.
121. Yamada T, Amann JM, Tanimoto A, Taniguchi H, Shukuya T, Timmers C, et al. Histone deacetylase inhibition enhances the antitumor activity of a MEK inhibitor in lung cancer cells harboring RAS mutations. Mol Cancer Ther. 2018;17(1):17-25.
122. Thompson KN, Whipple RA, Yoon JR, Lipsky M, Charpentier MS, Boggs AE, et al. The combinatorial activation of the PI3K and Ras/ MAPK pathways is sufficient for aggressive tumor formation, while individual pathway activation supports cell persistence. Oncotarget. 2015;6(34):35231-46.
123. Maitra R, Seetharam R, Tesfa L, Augustine TA, Klampfer L, Coffey MC, et al. Oncolytic reovirus preferentially induces apoptosis in KRAS mutant colorectal cancer cells, and synergizes with irinotecan. Oncotarget. 2014;5(9):2807-19.
124. Yang YP, Ma H, Starchenko A, Huh WJ, Li W, Hickman FE, et al. A Chimeric Egfr Protein Reporter Mouse Reveals Egfr Localization and Trafficking In Vivo. Cell Rep. 2017;19(6):1257-67.
125. Koo T, Yoon AR, Cho HY, Bae S, Yun CO, Kim JS. Selective disruption of an oncogenic mutant allele by CRISPR/Cas9 induces efficient tumor regression. Nucleic Acids Res. 2017;45(13):7897-908.
126. Kiessling MK, Schuierer S, Stertz S, Beibel M, Bergling S, Knehr J, et al. Identification of oncogenic driver mutations by genome-wide CRISPRCas9 dropout screening. BMC Genomics. 2016;17(1). https://doi.org/10. 1186/s12864-016-3042-2.
127. Park JJ, Kim JE, Jeon Y, Lee MR, Choi JY, Song BR, et al. Deletion of NKX3.1 via CRISPR/Cas9 Induces Prostatic Intraepithelial Neoplasia in
128. Zhang BC, Luo BY, Zou JJ, Wu PY, Jiang JL, Le JQ, et al. Co-delivery of Sorafenib and CRISPR/Cas9 Based on Targeted Core-Shell Hollow Mesoporous Organosilica Nanoparticles for Synergistic HCC Therapy. ACS Appl Mater Interfaces. 2020;12(51):57362-72.
129. Chung SH, Frick SL, Yiu G. Targeting vascular endothelial growth factor using retinal gene therapy. Ann Transl Med. 2021;9(15):1277-1277.
130. Ameri H, Murat C, Arbabi A, Jiang W, Janga SR, Qin PZ, et al. Reduced expression of vegf-a in human retinal pigment epithelial cells and human muller cells following crispr-cas9 ribonucleoprotein-mediated gene disruption. Transl Vis Sci Technol. 2020;9(8). https://doi.org/10. 1167/TVST.9.8.23.
131. Hariprabu KNG, Sathya M, Vimalraj S. CRISPR/Cas9 in cancer therapy: A review with a special focus on tumor angiogenesis. Int J Biol Macromol. 2021;192:913-930.
132. Li XL, Li GH, Fu J, Fu YW, Zhang L, Chen W, et al. Highly efficient genome editing via CRISPR-Cas9 in human pluripotent stem cells is achieved by transient BCL-XL overexpression. Nucleic Acids Res. 2018;46(19):10195-215.
133. Abdul Rahman SF, Mohana-Kumaran N, Mohd Azzam MG, Mohd Norudin NA, Muniandy K. CRISPR/Cas9 Mediated BFL-1 Knock-out in Nasopharyngeal Carcinoma (NPC) Cell Lines. Front Pharmacol. 2018;9. https://doi.org/10.3389/conf.fphar.2018.63.00029.
134. Zhao W, Liu J, Wang D, Wang Y, Zhang F, Jin G, et al. Effect of silencing HIF-1 a gene on testicle spermatogenesis function in varicocele rats. Cell Tissue Res. 2019;378(3):543-54.
135. Fang N, Gu T, Wang Y, Wang S, Wang F, An Y, et al. Expression of PTENlong mediated by CRISPR/Cas9 can repress U87 cell proliferation. J Cell Mol Med. 2017;21(12):3337-46.
136. de Almeida Monteiro Melo Ferraz M, Nagashima JB, Venzac B, Le Gac S, Songsasen N. A dog oviduct-on-a-chip model of serous tubal intraepithelial carcinoma. Sci Rep. 2020;10(1). https://doi.org/10.1038/ s41598-020-58507-4.
137. Callif BL, Maunze B, Krueger NL, Simpson MT, Blackmore MG. The application of CRISPR technology to high content screening in primary neurons. Mol Cell Neurosci. 2017;80:170-9.
138. Heitink L, Whittle JR, Vaillant F, Capaldo BD, Dekkers JF, Dawson CA, et al. In vivo genome-editing screen identifies tumor suppressor genes that cooperate with Trp53 loss during mammary tumorigenesis. Mol Oncol. 2022;16(5):1119-31.
139. Xi L, Schmidt JC, Zaug AJ, Ascarrunz DR, Cech TR. A novel two-step genome editing strategy with CRISPR-Cas9 provides new insights into telomerase action and TERT gene expression. Genome Biol. 2015;16(1). https://doi.org/10.1186/s13059-015-0791-1.
140. James CD, Prabhakar AT, Otoa R, Evans MR, Wang X, Bristol ML, et al. SAMHD1 Regulates Human Papillomavirus 16-Induced Cell Proliferation and Viral Replication during Differentiation of Keratinocytes. mSphere. 2019;4(4). https://doi.org/10.1128/msphere.00448-19.
141. Zagorski JW, Maser TP, Liby KT, Rockwell CE. Nrf2-Dependent and -Independent Effects of tert-Butylhydroquinone, CDDO-Im, and H2O2 in Human Jurkat T Cells as Determined by CRISPR/Cas9 Gene Editing. J Pharmacol Exp Ther. 2017;361(2):259-67.
142. Cordeiro A, Deveau AP, Dhanraj S, Dror Y, Berman J. Characterizing Dyskeratosis Congenita Caused By Parn Mutations in the Zebrafish. Blood. 2019;134(Supplement_1):3744-3744.
143. Liu N, Xu S, Yao Q, Zhu Q, Kai Y, Hsu JY, et al. Transcription factor competition at the
144. Rui L. Abstract 3436: PRMT5 upregulation and its oncogenic cooperation with PI3K/AKT signaling in diffuse large B cell lymphoma. Cancer Res. 2019;79(13_Supplement):3436-3436.
145. Bonato A, Bomben R, Chakraborty S, Felician G, Martines C, Zucchetto A, et al. Chronic Lymphocytic Leukemia Cells with Mutated Nfkbie Are Positively Selected By Microenvironmental Signals and Display Reduced Sensitivity to Ibrutinib Treatment. Blood. 2021;138(Supplement 1):248-248.
146. Tagde A, Markert T, Rajabi H, Hiraki M, Alam M, Bouillez A, et al. Targeting MUC1-C suppresses polycomb repressive complex 1 in multiple myeloma. Oncotarget. 2017;8(41):69237-49.
147. Navarro-Guerrero E, Tay C, Whalley JP, Cowley SA, Davies B, Knight JC, et al. Genome-wide CRISPR/Cas9-knockout in human induced Pluripotent Stem Cell (iPSC)-derived macrophages. Sci Rep. 2021;11(1). https:// doi.org/10.1038/s41598-021-82137-z.
148. Hamada T, Akahane T, Yokoyama S, Higa N, Kirishima M, Matsuo K, et al. An oncogenic splice variant of PDGFRa in adult glioblastoma as a therapeutic target for selective CDK4/6 inhibitors. Sci Rep. 2022;12(1). https://doi.org/10.1038/s41598-022-05391-9.
149. Iacono D, Cinausero M, Gerratana L, Vitale MG, Basile D, Angione V, et al. Primary cutaneous melanoma in elderly patients: potential prognostic markers. Pigment Cell Melanoma Res. 2017;30(1):76-156.
150. Farrell J, Pietruska J, McRee S, Tsichlis P, Hinds P. Abstract PR14: Defining isoform-specific roles for AKTs in BRAFV600E-driven melanoma. Cancer Res. 2020;80(19_Supplement):PR14-PR14.
151. Kaulich M, Dowdy S. Abstract A10: A Cdk4-dependent phosphorylation threshold regulates the cell cycle entry decision. Mol Cancer Res. 2016;14(11_Supplement):A10-A10.
152. Frangoul H, Altshuler D, Cappellini MD, Chen Y-S, Domm J, Eustace BK, et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and
153. PD-1 Knockout Engineered T Cells for Advanced Esophageal Cancer – ClinicalTrials.gov. https://classic.clinicaltrials.gov/ct2/show/NCT03 081715. Accessed 22 Jul 2023.
154. PD-1 Knockout EBV-CTLs for Advanced Stage Epstein-Barr Virus (EBV) Associated Malignancies – ClinicalTrials.gov. https://classic.clinicaltrials. gov/ct2/show/NCT03044743. Accessed 22 Jul 2023.
155. TACE Combined With PD-1 Knockout Engineered T Cell in Advanced Hepatocellular Carcinoma – ClinicalTrials.gov. 2020. http://www.clini caltrials.gov/ct2/show/NCT04417764.
156. Study of CRISPR-Cas9 Mediated PD-1 and TCR Gene-knocked Out Mesothelin-directed CAR-T Cells in Patients With Mesothelin Positive Multiple Solid Tumors- ClinicalTrials.gov. 2020. http://www.clinicaltrials. gov/ct2/show/NCT03545815.
157. CRISPR (HPK1) Edited CD19-specific CAR-T Cells (XYF19 CAR-T Cells) for CD19+ Leukemia or Lymphoma- ClinicalTrials.gov. 2019. https://classic. clinicaltrials.gov/ct2/show/NCT04037566.
158. Safety and Efficacy of CT125A Cells for Treatment of Relapsed/Refractory CD5 + Hematopoietic Malignancies- ClinicalTrials.gov. 2021. http:// www.clinicaltrials.gov/ct2/show/NCT04767308.
159. A Safety and Efficacy Study Evaluating CTX130 in Subjects With Relapsed or Refractory T or B Cell Malignancies (COBALT-LYM). 2021. http://www.clinicaltrials.gov/ct2/show/NCT04502446.
160. Ishihara M, Nishida Y, Kitano S, Kawai A, Muraoka D, Momose F, et al. A phase 1 trial of NY-ESO-1-specific TCR-engineered T-cell therapy combined with a lymph node-targeting nanoparticulate peptide vaccine for the treatment of advanced soft tissue sarcoma. Int J Cancer. 2023;152(12):2554-66.
161. Kanwal B. Relapsed/Refractory Non-Hodgkin Lymphoma: Engineering T-Cells to Express Chimeric Antigen Receptors (CARs), a Salvage? Cureus. 2021. https://doi.org/10.7759/cureus.16307.
162. Fang C, Xiao G, Wang T, Song L, Peng B, Xu B, et al. Emerging Nano-/ Biotechnology Drives Oncolytic Virus-Activated and Combined Cancer Immunotherapy. Research. 2023;6. https://doi.org/10.34133/research. 0108.
163. Hölting TLB, Cidre-Aranaz F, Matzek D, Popper B, Jacobi SJ, Funk CM, et al. Neomorphic DNA-binding enables tumor-specific therapeutic gene expression in fusion-addicted childhood sarcoma. Mol Cancer. 2022;21(1). https://doi.org/10.1186/s12943-022-01641-6.
164. Malviya M, Aretz ZEH, Molvi Z, Lee J, Pierre S, Wallisch P, et al. Challenges and solutions for therapeutic TCR-based agents. Immunol Rev. 2023. https://doi.org/10.1111/imr.13233.
165. Chira S, Nutu A, Isacescu E, Bica C, Pop L, Ciocan C, et al. Genome Editing Approaches with CRISPR/Cas9 for Cancer Treatment: Critical Appraisal of Preclinical and Clinical Utility, Challenges, and Future Research. Cells. 2022;11(18). https://doi.org/10.3390/cells11182781.
166. Xu J, Chen LJ, Yang SS, Sun Y, Wu W, Liu YF, et al. Exploratory trial of a biepitopic CAR T-targeting B cell maturation antigen in relapsed/refractory multiple myeloma. Proc Natl Acad Sci U S A. 2019. https://doi.org/ 10.1073/pnas. 1819745116.
167. Feldman L, Brown C, Badie B. Chimeric Antigen Receptor T-Cell Therapy: Updates in Glioblastoma Treatment. Neurosurgery. 2021;88(6):1056-64.
168. Lin Y, Ying H, Shao L, Liu Q, Song M, Chen S. Safety and Efficacy of CRISPR/CAS9-Edited PD-1 Deficient CAR-T Cells in MUC1 Positive Advanced Esophageal Cancer. SSRN Electron J. 2023. https://doi.org/10. 2139/ssrn. 4316844.
169. Choi SYC, Ribeiro CF, Wang Y, Loda M, Plymate SR, Uo T. Druggable Metabolic Vulnerabilities Are Exposed and Masked during Progression to Castration Resistant Prostate Cancer. Biomolecules. 2022;12(11). https://doi.org/10.3390/biom12111590.
170. Acharya UH, Walter RB. Chimeric antigen receptor (Car)-modified immune effector cell therapy for acute myeloid leukemia (aml). Cancers. 2020;12(12):1-28.
171. Zhen S, Li X. Oncogenic Human Papillomavirus: Application of CRISPR/ Cas9 Therapeutic Strategies for Cervical Cancer. Cell Physiol Biochem. 2018;44(6):2455-66.
172. Uras IZ, Moll HP, Casanova E. Targeting KRAS mutant non-small-cell lung cancer: Past, present and future. Int J Mol Sci. 2020;21(12):1-30.
173. Trigg RM, Turner SD. ALK in neuroblastoma: Biological and therapeutic implications. Cancers. 2018;10(4). https://doi.org/10.3390/cancers100 40113.
174. Anagnostou T, Riaz IB, Hashmi SK, Murad MH, Kenderian SS. AntiCD19 chimeric antigen receptor T-cell therapy in acute lymphocytic leukaemia: a systematic review and meta-analysis. Lancet Haematol. 2020;7(11):e816-26.
175. Andrea AE, Chiron A, Mallah S, Bessoles S, Sarrabayrouse G, Hacein-Bey-Abina S. Advances in CAR-T Cell Genetic Engineering Strategies to Overcome Hurdles in Solid Tumors Treatment. Front Immunol. 2022;13. https://doi.org/10.3389/fimmu.2022.830292.
176. Li M, Sun J, Shi G. Application of CRISPR screen in mechanistic studies of tumor development, tumor drug resistance, and tumor immunotherapy. Front Cell Dev Biol. 2023;11. https://doi.org/10.3389/fcell.2023. 1220376.
177. Crispo F, Pietrafesa M, Condelli V, Maddalena F, Bruno G, Piscazzi A, et al. IDH1 Targeting as a New Potential Option for Intrahepatic Cholangiocarcinoma Treatment-Current State and Future Perspectives. Molecules. 2020;25(16). https://doi.org/10.3390/molecules25163754.
178. Mirgayazova R, Khadiullina R, Chasov V, Mingaleeva R, Miftakhova R, Rizvanov A, et al. Therapeutic editing of the TP53 gene: Is crispr/CAS9 an option? Genes. 2020;11(6):1-17.
179. Gagelmann N, Riecken K, Wolschke C, Berger C, Ayuk FA, Fehse B, et al. Development of CAR-T cell therapies for multiple myeloma. Leukemia. 2020;34(9):2317-32.
180. Wang Z, Li N, Feng K, Chen M, Zhang Y, Liu Y, et al. Phase I study of CAR-T cells with PD-1 and TCR disruption in mesothelin-positive solid tumors. Cell Mol Immunol. 2021;18(9):2188-98.
181. Bamdad CC, Yuan Y, Specht JM, Stewart AK, Smagghe BJ, Lin SC-M, et al. Phase I/II first-in-human CAR T-targeting MUC1 transmembrane cleavage product (MUC1*) in patients with metastatic breast cancer. J Clin Oncol. 2022;40(16_suppl):TPS1130-TPS1130.
182. Pennesi E, Michels N, Brivio E, van der Velden VHJ, Jiang Y, Thano A, et al. Inotuzumab ozogamicin as single agent in pediatric patients with relapsed and refractory acute lymphoblastic leukemia: results from a phase II trial. Leukemia. 2022;36(6):1516-24.
183. Swain SM, Shastry M, Hamilton E. Targeting HER2-positive breast cancer: advances and future directions. Nat Rev Drug Discovery. 2023;22(2):101-26.
184. Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex genome editing to generate universal CART cells resistant to PD1 inhibition. Clin Cancer Res. 2017;23(9):2255-66.
185. Liu J, Zhang Y, Guo R, Zhao Y, Sun R, Guo S, et al. Targeted CD7 CAR T-cells for treatment of T-Lymphocyte leukemia and lymphoma and acute myeloid leukemia: recent advances. Front Immunol. 2023;14. https://doi.org/10.3389/fimmu.2023.1170968.
186. Furqan F, Shah NN. Multispecific CART Cells Deprive Lymphomas of Escape via Antigen Loss. Annu Rev Med. 2023;74:279-91.
187. Li Q, Ni Y, Zhang L, Jiang R, Xu J, Yang H, et al. HIF-1 a-induced expression of m6A reader YTHDF1 drives hypoxia-induced autophagy and malignancy of hepatocellular carcinoma by promoting ATG2A and ATG14 translation. Signal Transduct Target Ther. 2021;6(1). https://doi. org/10.1038/s41392-020-00453-8.
188. Kumar R, Yu F, Zhen YH, Li B, Wang J, Yang Y, et al. PD-1 blockade restores impaired function of ex vivo expanded CD8+T cells and
enhances apoptosis in mismatch repair deficient EpCAM+PD-L1+ cancer cells. Onco Targets Ther. 2017;10:3453-65.
189. Haso W, Qin H, Zhang L, Orentas RJ, Fry TJ. CD22-Targeted Chimeric Antigen Receptor (CAR) T Cells Containing The 4-1BB Costimulatory Domain Demonstrate Enhanced Persistence and Superior Efficacy Against B-Cell Precursor Acute Lymphoblastic Leukemia (ALL) Compared To Those Containing CD28. Blood. 2013;122(21):1431-1431.
190. Dimitri A, Herbst F, Fraietta JA. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Molecular Cancer. 2022;21(1). https://doi.org/10.1186/s12943-022-01559-z.
191. Chen X, Kozhaya L, Tastan C, Placek L, Dogan M, Horne M, et al. Functional Interrogation of Primary Human T Cells via CRISPR Genetic Editing. J Immunol. 2018;201(5):1586-98.
192. Hedrich V, Breitenecker K, Ortmayr G, Pupp F, Huber H, Chen D, et al. PRAME Is a Novel Target of Tumor-Intrinsic Gas6/AxI Activation and Promotes Cancer Cell Invasion in Hepatocellular Carcinoma. Cancers (Basel). 2023;15(9). https://doi.org/10.3390/cancers15092415.
193. Kaligotla VSA, Jasti T, Kandra P. CRISPR/Cas9 in cancer: An attempt to the present trends and future prospects. Biotechnol Appl Biochem. 2022;69(3):1238-51.
194. Mehravar M, Roshandel E, Salimi M, Chegeni R, Gholizadeh M, Mohammadi MH, et al. Utilization of CRISPR/Cas9 gene editing in cellular therapies for lymphoid malignancies. Immunol Lett. 2020;226:71-82.
195. Zhang S, Zhang F, Chen Q, Wan C, Xiong J, Xu J. CRISPR/Cas9-mediated knockout of NSD1 suppresses the hepatocellular carcinoma development via the NSD1/H3/Wnt10b signaling pathway. J Exp Clin Cancer Res. 2019;38(1). https://doi.org/10.1186/s13046-019-1462-y.
196. Sürün D, von Melchner H, Schnütgen F. CRISPR/Cas9 genome engineering in hematopoietic cells. Drug Discov Today Technol. 2018;28:33-9.
197. Koh S, Kah J, Tham CYL, Yang N, Ceccarello E, Chia A, et al. Nonlytic Lymphocytes Engineered to Express Virus-Specific T-Cell Receptors Limit HBV Infection by Activating APOBEC3. Gastroenterology. 2018;155(1):180-193.e6.
198. Adigbli G, Ménoret S, Cross AR, Hester J, Issa F, Anegon I. Humanization of Immunodeficient Animals for the Modeling of Transplantation, Graft Versus Host Disease, and Regenerative Medicine. Transplantation. 2020;104(11):2290-306.
199. Stewart JR, Baxter J, Zatreanu D, Brough R, Song F, Konde A, et al. 3P Identification of novel biomarkers of response to ATR inhibitors in ARID1A mutant ovarian clear cell carcinoma. Ann Oncol. 2022;33:S383-4.
200. Kamali E, Rahbarizadeh F, Hojati Z, Frödin M. CRISPR/Cas9-mediated knockout of clinically relevant alloantigenes in human primary T cells. BMC Biotechnol. 2021;21(1). https://doi.org/10.1186/ s12896-020-00665-4.
201. Khan A, Zhang X. Function of the Long Noncoding RNAs in HepatocelIular Carcinoma: Classification, Molecular Mechanisms, and Significant Therapeutic Potentials. Bioengineering. 2022;9(8). https://doi.org/10. 3390/bioengineering9080406.
202. Kath J, Du W, Pruene A, Braun T, Thommandru B, Turk R, et al. Pharmacological interventions enhance virus-free generation of TRAC-replaced CART cells. Mol Ther – Methods Clin Dev. 2022;25:311-30.
203. Gao Q, Dong X, Xu Q, Zhu L, Wang F, Hou Y, et al. Therapeutic potential of CRISPR/Cas9 gene editing in engineered T-cell therapy. Cancer Med. 2019;8(9):4254-64.
204. Aghamiri S, Talaei S, Ghavidel AA, Zandsalimi F, Masoumi S, Hafshejani NH, et al. Nanoparticles-mediated CRISPR/Cas9 delivery: Recent advances in cancer treatment. J Drug Deliv Sci Technol. 2020;56. https://doi.org/10.1016/j.jddst.2020.101533.
205. Tong S, Moyo B, Lee CM, Leong K, Bao G. Engineered materials for in vivo delivery of genome-editing machinery. Nat Rev Mater. 2019;4(11):726-37.
206. Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNAGuided Human Genome Engineering via Cas9_Sup. Science. 2013;339(February):823-6.
207. Brouns SJJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJH, Snijders APL, et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321(5891):960-964.
208. Yin H, Kauffman KJ, Anderson DG. Delivery technologies for genome editing. Nat Rev Drug Discovery. 2017;16(6):387-99.
209. Chow MYT, Chang RYK, Chan HK. Inhalation delivery technology for genome-editing of respiratory diseases. Adv Drug Deliv Rev. 2021;168:217-28.
210. Sandhya D, Jogam P, Allini VR, Abbagani S, Alok A. The present and potential future methods for delivering CRISPR/Cas9 components in plants. J Genet Eng Biotechnol. 2020;18(1). https://doi.org/10.1186/ s43141-020-00036-8.
211. Taha EA, Lee J, Hotta A. Delivery of CRISPR-Cas tools for in vivo genome editing therapy: Trends and challenges. J Control Release. 2022;342:345-61.
212. Liu P, Liang SQ, Zheng C, Mintzer E, Zhao YG, Ponnienselvan K, et al. Improved prime editors enable pathogenic allele correction and cancer modelling in adult mice. Nat Commun. 2021;12(1). https://doi.org/10. 1038/s41467-021-22295-w.
213. Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, et al. CRISPRCas9 knockin mice for genome editing and cancer modeling. Cell. 2014;159(2):440-55.
214. Han HA, Pang JK, Soh BS. Mitigating off-target effects in CRISPR/Cas9mediated in vivo gene editing. J Mol Med. 2020;98(5):615-32.
215. Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, et al. Optimized sgRNA design to maximize activity and minimize offtarget effects of CRISPR-Cas9. Nat Biotechnol. 2016;34(2):184-91.
216. Burgess DJ. Characterizing CRISPR off-target effects. Nat Rev Genet. 2014;15(1):5-5.
217. Shen CC, Hsu MN, Chang CW, Lin MW, Hwu JR, Tu Y, et al. Synthetic switch to minimize CRISPR off-target effects by self-restricting Cas9 transcription and translation. Nucleic Acids Res. 2019;47(3). https://doi. org/10.1093/nar/gky1165.
218. Zhang S, Shen J, Li D, Cheng Y. Strategies in the delivery of Cas9 ribonucleoprotein for CRISPR/Cas9 genome editing. Theranostics. 2020. https://doi.org/10.7150/thno.47007.
219. Zhang D, Zhang Z, Unver T, Zhang B. CRISPR/Cas: A powerful tool for gene function study and crop improvement. J Adv Res. 2021;29:207-21.
220. Yin Y, Wang Q, Xiao L, Wang F, Song Z, Zhou C, et al. Advances in the engineering of the gene editing enzymes and the genomes: Understanding and handling the off-target effects of CRISPR/Cas9. J Biomed Nanotechnol. 2018. https://doi.org/10.1166/jbn.2018.2537.
221. Zhang X-H, Tee LY, Wang X-G, Huang Q-S, Yang S-H. Off-target Effects in CRISPR/Cas9-mediated Genome Engineering. Mol Ther Nucleic Acids. 2015;4: e264.
222. Li A, Lee CM, Hurley AE, Jarrett KE, De Giorgi M, Lu W, et al. A Self-Deleting AAV-CRISPR System for In Vivo Genome Editing. Mol Ther Methods Clin Dev. 2019. https://doi.org/10.1016/j.omtm.2018.11.009.
223. Xiang X, Corsi GI, Anthon C, Qu K, Pan X, Liang X, et al. Enhancing CRISPR-Cas9 gRNA efficiency prediction by data integration and deep learning. Nat Commun. 2021;12(1). https://doi.org/10.1038/ s41467-021-23576-0.
224. Kleinstiver BP, Prew MS, Tsai SQ, Topkar VV, Nguyen NT, Zheng Z, et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015. https://doi.org/10.1038/nature14592.
225. Shen B, Zhang W, Zhang J, Zhou J, Wang J, Chen L, et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat Methods. 2014. https://doi.org/10.1038/nmeth.2857.
226. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013. https:// doi.org/10.1038/nprot.2013.143.
227. Xie K, Yang Y. RNA-Guided Genome Editing in Plants Using a CRISPRCas System. Mol Plant. 2013;6(6):1975-83.
228. Izumi H, Wang Z, Goto Y, Ando T, Wu X, Zhang X, et al. Pathway-specific genome editing of PI3K/mTOR tumor suppressor genes reveals that PTEN loss contributes to cetuximab resistance in head and neck cancer. Mol Cancer Ther. 2020;19(7):1562-71.
229. Engstrom LD, Waters L, Gatto S, Fernandez-Banet J, Aranda R, Pavlicek A, et al. Abstract 5684: Drug-anchored in vitro and in vivo CRISPR screens to identify targetable vulnerabilities and modifiers of response to MRTX849 in KRASG12C-mutant models . Cancer Res. 2020;80(16_Supplement):5684-5684.
230. Li Y, Deutzmann A, Bell J, Ji H, Felsher D. Abstract PR02: Synthetic lethality screen identifies novel druggable targets in the MYC pathway. Mol Cancer Ther. 2017;16(10_Supplement):PR02-PR02.
231. Kolasa IK, Rembiszewska A, Janiec-Jankowska A, Dansonka-Mieszkowska A, Lewandowska AM, Konopka B, et al. PTEN mutation, expression and LOH at its locus in ovarian carcinomas. Relation to TP53, K-RAS and BRCA1 mutations. Gynecol Oncol. 2006;103(2):692-697.
232. Alenezi WM, Milano L, Fierheller CT, Serruya C, Revil T, Oros KK, et al. The Genetic and Molecular Analyses of RAD51C and RAD51D Identifies Rare Variants Implicated in Hereditary Ovarian Cancer from a Genetically Unique Population. Cancers (Basel). 2022;14(9). https://doi.org/10.3390/ cancers14092251.
233. Osher DJ, De Leeneer K, Michils G, Hamel N, Tomiak E, Poppe B, et al. Mutation analysis of RAD51D in non-BRCA1/2 ovarian and breast cancer families. Br J Cancer. 2012;106(8):1460-3.
234. Soukupová J, Lhotová K, Janatová M, Kleiblová P, Vočka M, Foretová L , et al. Germline mutations in RAD51C and RAD51D and hereditary predisposition to ovarian cancer. Klin Onkol. 2021;34(1):26-32.
235. Cummings S, Roman SS, Saam J, Bernhisel R, Brown K, Lancaster JM, et al. Age of ovarian cancer diagnosis among BRIP1, RAD51C, and RAD51D mutation carriers identified through multi-gene panel testing. J Ovarian Res. 2021;14(1). https://doi.org/10.1186/s13048-021-00809-w.
236. Loveday C, Turnbull C, Ramsay E, Hughes D, Ruark E, Frankum JR, et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat Genet. 2011;43(9):879-82.
237. Chen X, Li Y, Ouyang T, Li J, Wang T, Fan Z, et al. Associations between RAD51D germline mutations and breast cancer risk and survival in BRCA1/2-negative breast cancers. Ann Oncol. 2018;29(10):2046-51.
238. Tao J, Bauer DE, Chiarle R. Assessing and advancing the safety of CRISPR-Cas tools: from DNA to RNA editing. Nat Commun. 2023;14(1). https://doi.org/10.1038/s41467-023-35886-6.
239. Bier E, Nizet V. Driving to Safety: CRISPR-Based Genetic Approaches to Reducing Antibiotic Resistance. Trends Genet. 2021;37(8):745-57.
240. Cromer MK, Barsan VV., Jaeger E, Wang M, Hampton JP, Chen F, et al. Ultra-deep sequencing validates safety of CRISPR/Cas9 genome editing in human hematopoietic stem and progenitor cells. Nat Commun. 2022;13(1). https://doi.org/10.1038/s41467-022-32233-z.
241. Sun D. Design of time-delayed safety switches for CRISPR gene therapy. Sci Rep. 2021;11(1). https://doi.org/10.1038/s41598-021-96510-5.
242. Xu CF, Chen GJ, Luo YL, Zhang Y, Zhao G, Lu ZD, et al. Rational designs of in vivo CRISPR-Cas delivery systems. Adv Drug Deliv Rev. 2021;168:3-29.
243. Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, et al. CRISPR-engineered T cells in patients with refractory cancer. Science (80- ). 2020;367(6481). https://doi.org/10.1126/science.aba7365.
244. Wan F, Draz MS, Gu M, Yu W, Ruan Z, Luo Q. Novel strategy to combat antibiotic resistance: a sight into the combination of crispr/cas9 and nanoparticles. Pharmaceutics. 2021;13(3). https://doi.org/10.3390/ pharmaceutics13030352.
245. Tang N, Ning Q, Wang Z, Tao Y, Zhao X, Tang S. Tumor microenvironment based stimuli-responsive CRISPR/Cas delivery systems: A viable platform for interventional approaches. Colloids and Surfaces B: Biointerfaces. 2022;210. https://doi.org/10.1016/j.colsurfb.2021.112257.
246. Wang G, Chow RD, Bai Z, Zhu L, Errami Y, Dai X, et al. Multiplexed activation of endogenous genes by CRISPRa elicits potent antitumor immunity. Nat Immunol. 2019;20(11):1494-505.
247. Xu L, Lau YS, Gao Y, Li H, Han R. Life-Long AAV-Mediated CRISPR Genome Editing in Dystrophic Heart Improves Cardiomyopathy without Causing Serious Lesions in mdx Mice. Mol Ther. 2019;27(8):1407-14.
248. Wei S, Shao X, Liu Y, Xiong B, Cui P, Liu Z, et al. Genome editing of PD-L1 mediated by nucleobase-modified polyamidoamine for cancer immunotherapy. J Mater Chem B. 2022;10(8):1291-300.
249. Wu Y, Zheng J, Zeng Q, Zhang T, Xing D. Light-responsive charge-reversal nanovector for high-efficiency in vivo CRISPR/Cas9 gene editing with controllable location and time. Nano Res. 2020;13(9):2399-406.
250. Li C, Ding L, Sun CW, Wu LC, Zhou D, Pawlik KM, et al. Novel HDAd/EBV Reprogramming Vector and Highly Efficient Ad/CRISPR-Cas Sickle Cell Disease Gene Correction. Sci Rep. 2016;6. https://doi.org/10.1038/srep3 0422.
251. Preece R, Pavesi A, Gkazi SA, Stegmann KA, Georgiadis C, Tan ZM, et al. CRISPR-Mediated Base Conversion Allows Discriminatory Depletion of Endogenous T Cell Receptors for Enhanced Synthetic Immunity. Mol Ther Methods Clin Dev. 2020;19:149-61.
252. Liu J, Li G, Guo H, Ni C, Gao Y, Cao X, et al. Dual-Responsive Core-Shell Tecto Dendrimers Enable Efficient Gene Editing of Cancer Cells to Boost Immune Checkpoint Blockade Therapy. ACS Appl Mater Interfaces. 2023;15(10):12809-21.
253. Chen X, Chen Y, Xin H, Wan T, Ping Y. Near-infrared optogenetic engineering of photothermal nanoCRISPR for programmable genome editing. Proc Natl Acad Sci U S A. 2020;117(5):2395-405.
254. Villamizar O, Waters SA, Scott T, Saayman S, Grepo N, Urak R, et al. Targeted Activation of Cystic Fibrosis Transmembrane Conductance Regulator. Mol Ther. 2019;27(10):1737-48.
255. Pofali P, Mondal A, Londhe V. Exosome as a Natural Gene Delivery Vector for Cancer Treatment. Curr Cancer Drug Targets. 2020;20(11):821-30.
256. Ma SC, Zhang JQ, Yan TH, Miao MX, Cao YM, Cao YB, et al. Novel strategies to reverse chemoresistance in colorectal cancer. Cancer Med. 2023;12(10):11073-96.
257. Gong Z, Zhang X, Dai Z. Progress of nanoparticles inhibiting tumor metastasis. Kexue Tongbao/Chinese Sci Bull. 2018;63(15):1482-92.
258. Fix SM, Jazaeri AA, Hwu P. Applications of crispr genome editing to advance the next generation of adoptive cell therapies for cancer. Cancer Discov. 2021;11(3):560-74.
259. Choi E, Chang J-W, Krueger J, Lahr WS, Pomeroy E, Walsh M, et al. Engineering CD70-Directed CAR-NK Cells for the Treatment of Hematological and Solid Malignancies. Blood. 2021;138(Supplement 1):1691-1691.
260. Eyquem J, Mansilla-Soto J, Odak A, Sadelain M. 274. One-Step Generation of Universal CART Cells. Mol Ther. 2016;24:S109.
261. Kavanagh EW, Green JJ. Toward Gene Transfer Nanoparticles as Therapeutics. Advanced Healthcare Materials. 2022;11(7). https://doi.org/10. 1002/adhm. 202102145.
262. Wang X, Zabierowski S, Wu M, Del Casale C, Eyquem J, Mansilla-Soto J, et al. Establishing cGMP manufacturing of CRISPR/Cas9-edited human CART cells. Cytotherapy. 2020;22(5):S138-9.
263. Getahun YA, Ali DA, Taye BW, Alemayehu YA. Multidrug-Resistant Microbial Therapy Using Antimicrobial Peptides and the CRISPR/Cas9 System. Vet Med Res Reports. 2022;13:173-90.
264. Tay LS, Palmer N, Panwala R, Chew WL, Mali P. Translating CRISPR-Cas Therapeutics: Approaches and Challenges. CRISPR J. 2020;3(4):253-75.
265. Barrangou R, Horvath P. CRISPR: New horizons in phage resistance and strain identification. Annu Rev Food Sci Technol. 2012;3(1):143-62.
266. Xiu K, Saunders L, Wen L, Ruan J, Dong R, Song J, et al. Delivery of CRISPR/Cas9 Plasmid DNA by Hyperbranched Polymeric Nanoparticles Enables Efficient Gene Editing. Cells. 2023;12(1). https://doi.org/10. 3390/cells12010156.
267. Lau CH, Suh Y. In vivo genome editing in animals using AAV-CRISPR system: Applications to translational research of human disease. F1000Research. 2017;6. https://doi.org/10.12688/f1000research.11243.1.
268. Jiang C, Lin X, Zhao Z. Applications of CRISPR/Cas9 Technology in the Treatment of Lung Cancer. Trends Mol Med. 2019;25(11):1039-49.
269. Wang Z, Chen M, Zhang Y, Liu Y, Yang Q, Nie J, et al. Phase I study of CRISPR-engineered CAR-T cells with PD-1 inactivation in treating meso-thelin-positive solid tumors. J Clin Oncol. 2020;38(15_suppl):3038-3038.
270. Tratar UL, Horvat S, Cemazar M. Transgenic mouse models in cancer research. Front Oncol. 2018;8(JUL). https://doi.org/10.3389/fonc.2018. 00268.
271. Zhan T, Rindtorff N, Betge J, Ebert MP, Boutros M. CRISPR/Cas9 for cancer research and therapy. Semin Cancer Biol. 2019;55:106-19.
272. Ahmed
273. Zhao Z, Li C, Tong F, Deng J, Huang G, Sang Y. Review of applications of CRISPR-Cas9 gene-editing technology in cancer research. Biol Proced Online. 2021;23(1). https://doi.org/10.1186/s12575-021-00151-x.
274. Wang SW, Gao C, Zheng YM, Yi L, Lu JC, Huang XY, et al. Current applications and future perspective of CRISPR/Cas9 gene editing in cancer. Mol Cancer. 2022;21(1). https://doi.org/10.1186/s12943-022-01518-8.
275. Jo DH, Song DW, Cho CS, Kim UG, Lee KJ, Lee K, et al. CRISPR-Cas9mediated therapeutic editing of Rpe65 ameliorates the disease phenotypes in a mouse model of Leber congenital amaurosis. Sci Adv. 2019;5(10). https://doi.org/10.1126/sciadv.aax1210.
276. Liang SQ, Walkey CJ, Martinez AE, Su Q, Dickinson ME, Wang D, et al. AAV5 delivery of CRISPR-Cas9 supports effective genome editing in mouse lung airway. Mol Ther. 2022;30(1):238-43.
277. Yu W, Mookherjee S, Chaitankar V, Hiriyanna S, Kim JW, Brooks M, et al. Nrl knockdown by AAV-delivered CRISPR/Cas9 prevents retinal degeneration in mice. Nat Commun. 2017;8. https://doi.org/10.1038/ncomm s14716.
278. Zhen S, Liu Y, Lu J, Tuo X, Yang X, Chen H, et al. Human Papillomavirus Oncogene Manipulation Using Clustered Regularly Interspersed Short Palindromic Repeats/Cas9 Delivered by pH-Sensitive Cationic Liposomes. Hum Gene Ther. 2020;31(5-6):309-24.
279. Aksoy YA, Yang B, Chen W, Hung T, Kuchel RP, Zammit NW, et al. Spatial and Temporal Control of CRISPR-Cas9-Mediated Gene Editing Delivered via a Light-Triggered Liposome System. ACS Appl Mater Interfaces. 2020;12(47):52433-44.
280. Naeimi Kararoudi M, Likhite S, Elmas E, Yamamoto K, Schwartz M, Sorathia K, et al. Optimization and validation of CAR transduction into human primary NK cells using CRISPR and AAV. Cell Reports Methods. 2022;2(6). https://doi.org/10.1016/j.crmeth.2022.100236.
281. Fine EJ, Appleton CM, White DE, Brown MT, Deshmukh H, Kemp ML, et al. Trans-spliced Cas9 allows cleavage of HBB and CCR5 genes in human cells using compact expression cassettes. Sci Rep. 2015;5. https://doi.org/10.1038/srep10777.
282. Kazemian P, Yu SY, Thomson SB, Birkenshaw A, Leavitt BR, Ross CJD. Lipid-Nanoparticle-Based Delivery of CRISPR/Cas9 Genome-Editing Components. Mol Pharm. 2022;19(6):1669-86.
283. Osteikoetxea X, Silva A, Lázaro-Ibáñez E, Salmond N, Shatnyeva O, Stein J, et al. Engineered Cas9 extracellular vesicles as a novel gene editing tool. J Extracell Vesicles. 2022;11(5). https://doi.org/10.1002/jev2.12225.
284. Yang Z, Liang SQ, Yang H, Xu D, Bruggmann R, Gao Y, et al. CRISPRmediated kinome editing prioritizes a synergistic combination therapy for FGFR1-amplified lung cancer. Cancer Res. 2021;81(11):3121-33.
285. Chamberlain CA, Bennett EP, Kverneland AH, Svane IM, Donia M, Met Ö. Highly efficient PD-1-targeted CRISPR-Cas9 for tumor-infiltrating lymphocyte-based adoptive T cell therapy. Mol Ther Oncolytics. 2022;24:417-28.
286. Sajib AM, Agarwal P, Patton DJ, Nance RL, Stahr NA, Kretzschmar WP, et al. In vitro functional genetic modification of canine adenovirus type 2 genome by CRISPR/Cas9. Lab Investig. 2021;101(12):1627-36.
287. Sarkar E, Khan A. Erratic journey of CRISPR/Cas9 in oncology from bench-work to successful-clinical therapy. Cancer Treatment and Research Communications. 2021;27. https://doi.org/10.1016/j.ctarc. 2020.100289.
288. Michaels YS, Barnkob MB, Barbosa H, Baeumler TA, Thompson MK, Andre V , et al. Precise tuning of gene expression levels in mammalian cells. Nat Commun. 2019;10(1). https://doi.org/10.1038/ s41467-019-08777-y.
289. Lopez-Obando M, Hoffmann B, Géry C, Guyon-Debast A, Téoulé E, Rameau C, et al. Simple and efficient targeting of multiple genes through CRISPR-Cas9 in Physcomitrella patens. G3 Genes Genomes Genet. 2016;6(11):3647-3653.
290. Cao J, Wu L, Zhang SM, Lu M, Cheung WKC, Cai W, et al. An easy and efficient inducible CRISPR/Cas9 platform with improved specificity for multiple gene targeting. Nucleic Acids Res. 2016;44(19). https://doi.org/ 10.1093/nar/gkw660.
291. Ma X, Zhang Q, Zhu Q, Liu W, Chen Y, Qiu R, et al. A Robust CRISPR/Cas9 System for Convenient, High-Efficiency Multiplex Genome Editing in Monocot and Dicot Plants. Mol Plant. 2015;8(8):1274-84.
292. Wessels HH, Méndez-Mancilla A, Hao Y, Papalexi E, Mauck WM, Lu L, et al. Efficient combinatorial targeting of RNA transcripts in single cells with Cas13 RNA Perturb-seq. Nat Methods. 2023;20(1):86-94.
293. Hirata M, Wittayarat M, Namula Z, Le QA, Lin Q, Nguyen NT, et al. Evaluation of multiple gene targeting in porcine embryos by the CRISPR/Cas9 system using electroporation. Mol Biol Rep. 2020;47(7):5073-9.
294. Pan C, Wu X, Markel K, Malzahn AA, Kundagrami N, Sretenovic S, et al. CRISPR-Act3.0 for highly efficient multiplexed gene activation in plants. Nat Plants. 2021;7(7):942-953.
295. Selvakumar SC, Preethi KA, Ross K, Tusubira D, Khan MWA, Mani P, et al. CRISPR/Cas9 and next generation sequencing in the personalized treatment of Cancer. Mol Cancer. 2022;21(1). https://doi.org/10.1186/ s12943-022-01565-1.
296. Sioson VA, Kim M, Joo J. Challenges in delivery systems for CRISPRbased genome editing and opportunities of nanomedicine. Biomed Eng Lett. 2021;11(3):217-33.
297. Chandrasekaran AP, Karapurkar JK, Chung HY, Ramakrishna S. The role of the CRISPR-Cas system in cancer drug development: Mechanisms of action and therapy. Biotechnol J. 2022;17(7). https://doi.org/10.1002/ biot. 202100468.
298. Ma D, Liu F. Genome Editing and Its Applications in Model Organisms. Genomics Proteomics Bioinformatics. 2015;13(6):336-44.
299. Moses C, Kaur P. Applications of CRISPR systems in respiratory health: Entering a new’red pen’ era in genome editing. Respirology. 2019;24(7):628-37.
300. Kick L, Kirchner M, Schneider S. CRISPR-Cas9: From a bacterial immune system to genome-edited human cells in clinical trials. Bioengineered. 2017;8(3):280-6.
301. Leonova El, Reshetnikov VV, Sopova JV. CRISPR/Cas-edited pigs for personalized medicine: more than preclinical test-system. Res Results Pharmacol. 2022;8(3):87-98.
302. Castells-Roca L, Tejero E, Rodríguez-Santiago B, Surrallés J. Crispr screens in synthetic lethality and combinatorial therapies for cancer. Cancers. 2021;13(7). https://doi.org/10.3390/cancers13071591.
303. Huang A, Garraway LA, Ashworth A, Weber B. Synthetic lethality as an engine for cancer drug target discovery. Nat Rev Drug Discovery. 2020;19(1):23-38.
304. Dhanjal JK, Radhakrishnan N, Sundar D. Identifying synthetic lethal targets using CRISPR/Cas9 system. Methods. 2017;131:66-73.
305. Vit G, Duro J, Rajendraprasad G, Hertz EPT, Holland LKK, Weisser MB, et al. Chemogenetic profiling reveals PP2A-independent cytotoxicity of proposed PP2A activators iHAP1 and DT-061. EMBO J. 2022;41(14). https://doi.org/10.15252/embj.2022110611.
306. Li C, Mei H, Hu Y. Applications and explorations of CRISPR/Cas9 in CAR T-cell therapy. Brief Funct Genomics. 2020;19(3):175-82.
307. Hu Kjia, Yin ETS, Hu Y xian, Huang H. Combination of CRISPR/Cas9 System and CAR-T Cell Therapy: A New Era for Refractory and Relapsed Hematological Malignancies. Curr Med Sci. 2021;41(3):420-430.
308. Ajavavarakula T. CRISPR-edited CAR-T cells: Using CRISPR-Cas9 to Improve CAR-T Therapy. Highlights Sci Eng Technol. 2022;14:355-9.
309. Ye L, Park JJ, Peng L, Yang Q, Chow RD, Dong MB, et al. A genome-scale gain-of-function CRISPR screen in CD8 T cells identifies proline metabolism as a means to enhance CAR-T therapy. Cell Metab. 2022;34(4):595614.e14.
310. Zhou Z, Tao C, Li J, Tang JC on, Chan AS chi, Zhou Y. Chimeric antigen receptor T cells applied to solid tumors. Front Immunol. 2022;13. https://doi.org/10.3389/fimmu.2022.984864.
311. Cortese MJ, Sauter C. A ‘CRISPR’ non-viral manufacturing approach for CART cell therapies. Mol Ther. 2022;30(11):3338-40.
312. Yan X, Chen D, Ma X, Wang Y, Guo Y, Wei J, et al. CD58 loss in tumor cells confers functional impairment of CART cells. Blood Adv. 2022;6(22):5844-56.
313. Mollanoori H, Shahraki H, Rahmati Y, Teimourian S. CRISPR/Cas9 and CAR-T cell, collaboration of two revolutionary technologies in cancer immunotherapy, an instruction for successful cancer treatment. Hum Immunol. 2018;79(12):876-82.
314. Liu J, Wang X, Chen AT, Gao X, Himes BT, Zhang H, et al. ZNF117 regulates glioblastoma stem cell differentiation towards oligodendroglial lineage. Nat Commun. 2022;13(1). https://doi.org/10.1038/ s41467-022-29884-3.
315. Wilkinson AC, Dever DP, Baik R, Camarena J, Hsu I, Charlesworth CT, et al. Cas9-AAV6 gene correction of beta-globin in autologous HSCs improves sickle cell disease erythropoiesis in mice. Nat Commun. 2021;12(1). https://doi.org/10.1038/s41467-021-20909-x.
316. Smirnikhina SA, Kondrateva E V., Adilgereeva EP, Anuchina AA, Zaynitdinova MI, Slesarenko YS, et al. P.F508del editing in cells from cystic fibrosis patients. PLoS One. 2020;15(11 November). https://doi.org/10. 1371/journal.pone. 0242094.
317. Bonafont J, Mencía A, Chacón-Solano E, Srifa W, Vaidyanathan S, Romano R, et al. Correction of recessive dystrophic epidermolysis bullosa by homology-directed repair-mediated genome editing. Mol Ther. 2021;29(6):2008-18.
318. Valetdinova KR, Malankhanova TB, Zakian SM, Medvedev SP. The Cutting Edge of Disease Modeling: Synergy of Induced Pluripotent Stem Cell Technology and Genetically Encoded Biosensors. Biomedicines. 2021;9(8). https://doi.org/10.3390/biomedicines9080960.
319. Dessy-Rodriguez
320. Garcia-Bloj B, Moses C, Sgro A, Plani-Lam J, Arooj M, Duffy C, et al. Waking up dormant tumor suppressor genes with zinc fingers, TALEs and the CRISPR/dCas9 system. Oncotarget. 2016;7(37):60535-54.
321. Mazur PK, Herner A, Mello SS, Wirth M, Hausmann S, Sánchez-Rivera FJ, et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat Med. 2015;21(10):1163-71.
322. Ousterout DG, Gersbach CA. Genome Editing for Neuromuscular Diseases. Genome Editing: The Next Step in Gene Therapy. 2016:51-79.
323. Ansari I, Chaturvedi A, Chitkara D, Singh S. CRISPR/Cas mediated epigenome editing for cancer therapy. Semin Cancer Biol. 2022;83:570-83.
324. Zhang C, Quan R, Wang J. Development and application of CRISPR/Cas9 technologies in genomic editing. Hum Mol Genet. 2018;27(R2):R79-88.
325. Pickar-Oliver A, Gersbach CA. The next generation of CRISPR-Cas technologies and applications. Nat Rev Mol Cell Biol. 2019;20(8):490-507.
326. Rafii S, Tashkandi E, Bukhari N, Al-Shamsi HO. Current Status of CRISPR/ Cas9 Application in Clinical Cancer Research: Opportunities and Challenges. Cancers. 2022;14(4). https://doi.org/10.3390/cancers14040947.
327. Yang Y, Xu J, Ge S, Lai L. CRISPR/Cas: Advances, Limitations, and Applications for Precision Cancer Research. Front Med. 2021;8. https://doi.org/ 10.3389/fmed.2021.649896.
328. Moses C, Garcia-Bloj B, Harvey AR, Blancafort P. Hallmarks of cancer: The CRISPR generation. Eur J Cancer. 2018;93:10-8.
329. Behan FM, lorio F, Picco G, Gonçalves E, Beaver CM, Migliardi G, et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature. 2019;568(7753):511-6.
330. Kim D, Kim J, Hur JK, Been KW, Yoon SH, Kim JS. Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat Biotechnol. 2016;34(8):863-8.
331. Manghwar H, Lindsey K, Zhang X, Jin S. CRISPR/Cas System: Recent Advances and Future Prospects for Genome Editing. Trends Plant Sci. 2019. https://doi.org/10.1016/j.tplants.2019.09.006.
332. Doerflinger M, Forsyth W, Ebert G, Pellegrini M, Herold MJ. CRISPR/ Cas9-The ultimate weapon to battle infectious diseases? Cell Microbiol. 2017;19(2). https://doi.org/10.1111/cmi.12693.
333. Boos SL, Loevenich LP, Vosberg S, Engleitner T, Öllinger R, Kumbrink J, et al. Disease Modeling on Tumor Organoids Implicates AURKA as a Therapeutic Target in Liver Metastatic Colorectal Cancer. CMGH. 2022;13(2):517-40.
334. Manghwar H, Li B, Ding X, Hussain A, Lindsey K, Zhang X, et al. CRISPR/ Cas Systems in Genome Editing: Methodologies and Tools for sgRNA Design, Off-Target Evaluation, and Strategies to Mitigate Off-Target Effects. Adv Sci. 2020. https://doi.org/10.1002/advs.201902312.
335. Rayner E, Durin M-A, Thomas R, Moralli D, O’Cathail SM, Tomlinson I, et al. CRISPR-Cas9 Causes Chromosomal Instability and Rearrangements in Cancer Cell Lines, Detectable by Cytogenetic Methods. Cris J. 2019;2(6):406-16.
336. Chen
337. Teng F, Li J, Cui T, Xu K, Guo L, Gao Q, et al. Enhanced mammalian genome editing by new Cas12a orthologs with optimized crRNA scaffolds. Genome Biol. 2019;20(1). https://doi.org/10.1186/ s13059-019-1620-8.
338. Bikard D, Euler C, Jiang W, Nussenzweig PM, Goldberg GW, Duportet X , et al. Development of sequence-specific antimicrobials based on programmable CRISPR-Cas nucleases. Nat Biotechnol. 2014;32(11):1146.
339. Deltcheva E, Chylinski K, Sharma CM, Gonzales K. CRISPR RNA maturation by trans -encoded small RNA and host factor RNase III. Nature. 2011;471(7340):602-7.
ملاحظة الناشر
هل أنت مستعد لتقديم بحثك؟ اختر BMC واستفد من:
- تقديم سريع ومريح عبر الإنترنت
- مراجعة دقيقة من قبل باحثين ذوي خبرة في مجالك
- نشر سريع عند القبول
- دعم لبيانات البحث، بما في ذلك أنواع البيانات الكبيرة والمعقدة
- الوصول المفتوح الذهبي الذي يعزز التعاون الأوسع وزيادة الاقتباسات
- أقصى رؤية لبحثك: أكثر من 100 مليون مشاهدة للموقع سنويًا
تعرف على المزيد biomedcentral.com/submissions
DOI: https://doi.org/10.1186/s12943-023-01925-5
PMID: https://pubmed.ncbi.nlm.nih.gov/38195537
Publication Date: 2024-01-09
Comprehensive review of CRISPR-based gene editing: mechanisms, challenges, and applications in cancer therapy
Abstract
The CRISPR system is a revolutionary genome editing tool that has the potential to revolutionize the field of cancer research and therapy. The ability to precisely target and edit specific genetic mutations that drive the growth and spread of tumors has opened up new possibilities for the development of more effective and personalized cancer treatments. In this review, we will discuss the different CRISPR-based strategies that have been proposed for cancer therapy, including inactivating genes that drive tumor growth, enhancing the immune response to cancer cells, repairing genetic mutations that cause cancer, and delivering cancer-killing molecules directly to tumor cells. We will also summarize the current state of preclinical studies and clinical trials of CRISPR-based cancer therapy, highlighting the most promising results and the challenges that still need to be overcome. Safety and delivery are also important challenges for CRISPR-based cancer therapy to become a viable clinical option. We will discuss the challenges and limitations that need to be overcome, such as off-target effects, safety, and delivery to the tumor site. Finally, we will provide an overview of the current challenges and opportunities in the field of CRISPR-based cancer therapy and discuss future directions for research and development. The CRISPR system has the potential to change the landscape of cancer research, and this review aims to provide an overview of the current state of the field and the challenges that need to be overcome to realize this potential.
Introduction
the field of genome editing [4]. Figure 1 illustrates the evolution of CRISPR tools used for exploring cancer biology. CRISPR-associated enzymes, such as Cas9, can be programmed to target specific DNA sequences, and when combined with guide RNAs, can be used to cut, modify or delete genes in a precise manner [4]. This technology has been used in a wide range of applications, including basic research, gene therapy, and agriculture [1]. However, its potential application in cancer research has attracted particular interest due to the ability to target the genetic mutations that drive the growth and spread of tumors [3]. There are several different CRISPR-based strategies that have been proposed for cancer therapy [4]. One approach is
In this review article, we will provide an overview of the current state of the field of CRISPR-based gene editing in cancer therapy, highlighting the most promising results and the challenges that still need to be overcome. We will describe the different CRISPR-based strategies that have been proposed for cancer therapy, summarize the current state of preclinical studies and clinical trials, and discuss the challenges and limitations that need to be overcome for CRISPR-based cancer therapy to become a viable clinical option. We will also provide an overview of future directions for research, development and discuss the potential implications of CRISPR-based cancer therapy for the future of cancer treatment and healthcare.
CRISPR-based strategies for cancer therapy
Inactivation of oncogenes
(A) CRISPR screens: identifies drug targets
and pave the way for more effective and personalized treatment approaches in the fight against cancer [27].
Enhancement of immune response
activity of the immune system [23]. Additionally, beneficial genes, such as cytokines or other immune mediators, can be inserted using CRISPR-Cas to further enhance the immune response against specific antigens [34]. CRISPRbased gene editing can sometimes result in non-specific site effects, where unintended changes occur in other parts of the genome. These non-selective site effects may lead to unwanted alterations in gene function and could pose safety concerns in the context of immune enhancement [35, 36]. It is essential to thoroughly evaluate and minimize these undesirable site effects to ensure the safety and effectiveness of CRISPR-based strategies [28]. CRISPR-Cas can be utilized to modify immune cells, such as T-cells and NK cells, to improve their functionality and specificity in targeting cancer cells or infected cells. By editing the genes responsible for cell receptors and signaling pathways, researchers can enhance the ability of immune cells to recognize and destroy specific targets [33]. Understanding the mechanisms and optimizing the protocols for this gene editing process is crucial for developing successful immune cell-based therapies [28]. While CRISPR-based strategies show great promise in enhancing the immune response, it is essential to investigate their long-term effects on the host’s immune system [30]. Prolonged activation or manipulation of immune
| Strategy | Mechanism of Action | Advantages | Disadvantages | Preclinical/clinical results | Ref |
| Inactivating genes that drive tumor growth | Targeting and disrupting oncogenes and tumor suppressor genes to stop cancer cell growth and induce apoptosis | Precision targeting: CRISPR can be designed to target specific genes or mutations that drive tumor growth, increasing specificity and reducing off-target effects. High efficacy: Studies have shown that inactivating certain genes using CRISPR can result in tumor regression and increased survival in animal models | Potential for off-target effects: While CRISPR offers high specificity, there is still the potential for unintended changes in the genome that could cause harm to the patient. Difficulty targeting specific genes or delivering therapy to tumor site: Some tumors may be difficult to target using current delivery methods | Study showed efficacy of using CRISPR to inactivate KRAS oncogene in mouse model of lung cancer. This is a major milestone since KRAS mutations are notoriously difficult to target with other therapies | [16] |
| Enhancing immune response to cancer cells | Editing immune cells to recognize and destroy cancer cells, such as by editing T cells to express chimeric antigen receptors (CARs) | Enhances body’s natural immune response: By editing immune cells to recognize and attack cancer cells, CRISPR-based immunotherapy can activate the body’s natural immune response to fight the cancer. Avoids toxic effects of chemotherapy: Unlike chemotherapy, which can have significant side effects, immunotherapy using CRISPR-edited T cells has the potential to be a more targeted and less toxic treatment approach | Potential for toxicity or immune rejection: There is a risk that CRISPRedited immune cells could attack healthy cells or be rejected by the patient’s immune system. Limited availability of specific T cells for editing: It can be challenging to obtain a sufficient number of
|
Study reported complete remission in two out of three patients treated with CRISPR-edited T cells in a trial for refractory lymphomas. This is a promising result, although more research is needed to determine the safety and efficacy of this approach in larger patient populations | [17] |
| Repairing genetic mutations that cause cancer | Correcting genetic mutations in tumor suppressor genes, DNA repair genes, or other driver genes | Precision targeting: CRISPR can be used to correct specific genetic mutations that cause cancer, potentially leading to long-term benefits. Potential for long-term benefits: Repairing genetic mutations that cause cancer could potentially result in long-term benefits for patients | Potential for off-target effects: As with other CRISPR-based approaches, there is a risk of unintended changes to the genome that could cause harm to the patient. Difficulty delivering therapy to tumor site: It can be challenging to deliver CRISPR-based therapy directly to the tumor site | Promising results in preclinical studies, such as using CRISPR to correct BRCA1 mutations in ovarian cancer cells | [18] |
| Delivering cancer-killing molecules directly to tumor cells | Using CRISPR to edit the genome of a virus or bacteria to specifically target cancer cells, delivering therapeutic molecules such as toxins or immune modulators directly to tumor cells | Precision targeting: By editing the genome of a virus or bacteria to specifically target cancer cells, CRISPR-based therapy can offer highly targeted and specific delivery of therapeutic molecules to tumor cells. High efficacy: Studies have shown that CRISPR-mediated delivery of cancer-killing molecules can result in tumor regression and increased survival in animal models | Potential for off-target effects: As with other CRISPR-based approaches, there is a risk of unintended changes to the genome that could cause harm to the patient. Limited availability of specific viruses or bacteria for editing: It can be challenging to obtain a sufficient number of specific viruses or bacteria for editing, which could limit the widespread use of this approach | Promising results in preclinical studies, such as using CRISPR to deliver CRISPRa to activate tumor-suppressive microRNAs in liver cancer cells. Other examples include using CRISPR to deliver toxic payloads to tumor cells, such as in a study where CRISPR was used to engineer bacteria to produce a toxin that specifically targets cancer cells in a mouse model of pancreatic cancer | [17, 18] |
Repair of genetic mutations
of CRISPR-mediated repair methods, such as HDR and NHEJ, is vital. HDR can accurately introduce the desired genetic changes by utilizing a donor template, but its efficiency is often lower compared to NHEJ, which can result in insertions or deletions without a template [40]. Understanding the balance between efficiency and accuracy will help optimize the choice of repair mechanism for specific genetic mutations [41]. While CRISPR has shown great promise, there might be unforeseen consequences of manipulating the genome. These could include nonselective site mutations or large-scale genomic rearrangements, which may introduce new genetic abnormalities or cause unintended effects on gene regulation [42]. Careful evaluation and thorough assessment of potential unintended outcomes are essential to ensure the safety and reliability of CRISPR-based strategies [37]. Understanding the stability of CRISPR-induced genetic repairs is critical for assessing the long-term viability of potential treatments [40]. Genetic modifications must be stable and faithfully passed on during cell divisions to provide lasting therapeutic benefits. Investigating the heritability and stability of repaired genetic mutations will shed light on the longevity and efficacy of CRISPR-based strategies [41]. When using CRISPR-Cas9 for in vivo applications, it is crucial to evaluate potential immune responses to
Delivery of cancer-killing molecules
guide RNAs can effectively distinguish cancerous cells from healthy ones [50]. The Cas complex serves as a carrier for the guide RNA molecules. It forms a complex with the guide RNA, creating the CRISPR-Cas RNP complex [51]. The Cas protein provides the necessary machinery to recognize the guide RNA and facilitates its binding to the target DNA within cancer cells. This complex acts as a powerful molecular scissor, cutting and deactivating the oncogenes responsible for cancer growth [50]. The CRISPR-Cas RNP complex can be introduced into the patient’s body through different methods. One approach involves direct injection into the target tissue or tumor site [49]. Another method involves engineering immune cells, such as T-cells, to express the CRISPR-Cas RNP complex. These engineered immune cells can then be reintroduced into the patient’s bloodstream, where they can specifically target and attack cancer cells [51]. Once inside the cancer cells, the CRISPR-Cas RNP complex locates the targeted DNA sequences and precisely cuts them, deactivating the oncogenes responsible for the malignancy. This deactivation leads to either the initiation of apoptosis (cell death) in cancer cells or renders them more susceptible to the body’s immune response, resulting in their destruction [49]. The guide RNA molecules are designed to specifically target cancer cells by binding to unique genetic markers or mutations found in those cells. By selectively targeting cancerous cells, the CRISPR-Cas RNP complex effectively spares healthy cells from damage, minimizing potential side effects [52]. Additionally, the use of engineered immune cells allows for even greater specificity in targeting cancer cells, further reducing the impact on healthy tissues [52, 53].
Preclinical studies and clinical trials for CRISPR-based cancer therapy
Preclinical studies
P53 is a critical tumor suppressor protein responsible for regulating cell cycle progression and preventing the formation of cancerous cells [94]. In the context of CRISPR/Cas9 genome editing, the activity of P53 becomes a crucial concern as the use of this technology may lead to non-targeted site effects, causing unintended DNA damage [95]. Therefore, it is essential to evaluate
the impact of CRISPR/Cas9 on P53 expression and function to ensure the safety and efficacy of the editing process [96]. P53 plays a crucial role in monitoring the integrity of the cell’s DNA and inducing cell cycle arrest or apoptosis in case of DNA damage [97]. Unfortunately, P53 is susceptible to mutations, leading to its inactivation or dysfunction. These mutations are common in many cancer types, including lung cancer, and contribute to uncontrolled cell growth and tumor development [98]. Modifying the P53 gene can restore its function, leading to the suppression of cancer cell growth [94]. When the modified P53 gene is activated, it enhances the production of the P21 protein, a well-known tumor suppressor that regulates the cell cycle. Increased P21 expression induces cell cycle arrest, preventing cancer cells from proliferating uncontrollably [96]. Moreover, the activation of P21 also makes cancer cells more susceptible to chemotherapy, as cells with active P21 proteins are more prone to apoptosis when exposed to chemotherapy drugs. P21, also known as cyclin-dependent kinase inhibitor 1A (CDKN1A), is a cyclin-dependent kinase inhibitor that plays a pivotal role in regulating the cell cycle and promoting cell cycle arrest. In the context of CRISPR/Cas9 genome editing, P21 may act as a dou-ble-edged sword [99]. On one hand, its upregulation in response to DNA damage caused by CRISPR/Cas9 may induce cell cycle arrest, preventing cells from proliferating and potentially compromising the effectiveness of the
| Cell/Tissue Type | CRISPR Approach | Results | Limitations/Challenges | Ref |
| Mouse and Human T cells | Knockout of PD-1 | Enhanced T cell activity against cancer cells, prolonged survival in mice | Safety concerns with long-term PD-1 knockout | [59] |
| Ovarian Cancer Cells | Knockout of oncogene | Reduced cell proliferation and colony formation | Limited assessment of off-target effects | [60] |
| Lung Cancer Cells | Knockout of mutant EGFR | Reduced cell viability and tumor growth in mice | Off-target effects in some cells | [61] |
| Triple-Negative Breast Cancer Cells | Knockout of AXL | Reduced tumor growth and increased sensitivity to chemotherapy | Limited assessment of off-target effects | [62] |
| Various Cancer Cell Lines | Knockout of HIF-1a | Reduced tumor growth and increased sensitivity to radiation therapy | Limited assessment of off-target effects | [63] |
| Melanoma Cells | Knockout of BRAF V600E | Reduced tumor growth and increased sensitivity to targeted therapy | Off-target effects in some cells | [64] |
| Leukemia Cells | Knockout of MCL-1 | Induced apoptosis and reduced tumor growth | Limited assessment of off-target effects | [65] |
| Human T cells | Knockout and overexpression of NKG2D | Enhanced tumor cell recognition and killing by CAR-T cells | Safety concerns with long-term NKG2D overexpression | [66,67] |
| Pediatric Solid Tumors | Knockout of fusion oncogenes | Reduced tumor growth and increased sensitivity to chemotherapy | Limited assessment of off-target effects | [68] |
| Prostate Cancer Cells | Knockout of androgen receptor | Reduced cell proliferation and tumor growth in mice | Off-target effects in some cells | [69] |
| Pancreatic Cancer Cells | Knockout of TGF-
|
Reduced tumor growth and increased sensitivity to chemotherapy | Limited assessment of off-target effects | [70] |
| Breast Cancer Cells | Knockout of PAK1 C-terminus | Reduced cell proliferation and tumor growth in mice | Off-target effects in some cells | [71] |
| Diffuse Large B-cell Lymphoma Cells | Knockout of BCL6 | Induced apoptosis and reduced tumor growth | Limited assessment of off-target effects | [72] |
| Glioblastoma Cells | Knockout of TERT promoter mutations | Reduced cell proliferation and tumor growth in mice | Limited assessment of off-target effects | [73] |
| Acute Myeloid Leukemia Cells | Knockout of GATA2 | Induced differentiation and reduced tumor growth | Limited assessment of off-target effects | [74] |
| Human Hematopoietic Stem Cells | Knockout of BCL11A enhancer | Induced fetal hemoglobin expression and reduced sickle cell symptoms | Limited assessment of off-target effects | [75] |
| B Cell Acute Lymphoblastic Leukemia Cells | Knockout of CD19 | Induced apoptosis and reduced tumor growth in mice | Off-target effects in some cells | [76] |
| Diffuse Large B-cell Lymphoma Cells | Knockout of EZH2 | Reduced cell proliferation and tumor growth in mice | Limited assessment of off-target effects | [77] |
| Glioma Cells | Knockout of IDH1 | Reduced cell viability and tumor growth in mice | Off-target effects in some cells | [78] |
| Neuroblastoma Cells | Knockout of MYCN | Induced apoptosis and reduced tumor growth | Limited assessment of off-target effects | [79] |
| Ovarian Cancer Cells | Knockout of MUC16 | Reduced cell proliferation and invasion | Limited assessment of off-target effects | [80] |
| Alveolar Rhabdomyosarcoma Cells | Knockout of PAX7-FOXO1 | Reduced cell proliferation and tumor growth in mice | Off-target effects in some cells | [81] |
| Esophageal Cancer Cells | Knockout of SOX2 | Reduced cell proliferation and colony formation | Limited assessment of off-target effects | [82] |
| Hepatocellular Carcinoma Cells | Knockout of TERT | Reduced cell proliferation and tumor growth in mice | Limited assessment of off-target effects | [83] |
| Colorectal Cancer Cells | Knockout ofWnt/
|
Reduced cell proliferation and tumor growth in mice | Off-target effects in some cells | [84] |
| Cell/Tissue Type | CRISPR Approach | Results | Limitations/Challenges | Ref |
| Breast Cancer Cells | Knockout of P53 | Increased cell proliferation and colony formation | Off-target effects in some cells | [85] |
| Pancreatic Cancer Cells | Knockout of KRAS | Reduced cell viability and tumor growth in mice | Limited assessment of off-target effects | [86] |
| Cervical Cancer Cells | Knockout of BIRC5 | Reduced cell proliferation and tumor growth in mice | Limited assessment of off-target effects | [87] |
| Prostate Cancer Cells | Knockout of EZH2 | Reduced cell proliferation and colony formation | Off-target effects in some cells | [79] |
| Osteosarcoma Cells | Knockout of HIF-1a | Reduced cell proliferation and tumor growth in mice | Limited assessment of off-target effects | [88] |
| Acute Lymphoblastic Leukemia Cells | Knockout of MYB | Induced apoptosis and reduced tumor growth | Limited assessment of off-target effects | [89] |
| Renal Cell Carcinoma Cells | Knockout of HIF-2a | Reduced cell proliferation and tumor growth in mice | Limited assessment of off-target effects | [90] |
| Hepatocellular Carcinoma Cells | Knockout of SALL4 | Reduced cell proliferation and tumor growth in mice | Limited assessment of off-target effects | [91] |
| Melanoma Cells | Knockout of CDK6 | Reduced cell proliferation and tumor growth in mice | Limited assessment of off-target effects | [92] |
| Acute Myeloid Leukemia Cells | Knockout of ASXL1 | Induced differentiation and reduced tumor growth | Limited assessment of off-target effects | [93] |
to efficiently develop new and clinically relevant surrogate models for translational studies [102].
the cancer cells more susceptible to chemotherapy, as P21-activated cells tend to be more responsive to chemotherapy treatment [106, 111]. Some researchers highlight the potential of CRISPR-based base editing as a valuable resource for the functional evaluation and reclassification of variants of uncertain significance (VUSs) in the BRCA1 gene. Furthermore, this investigation tackled the obstacles associated with assessing functionality and determining the pathogenicity of new BRCA1 variants, which are known to substantially elevate the risk of breast and ovarian cancers and are typically identified through clinical genetic testing. To surmount these hurdles, the scientists employed CRISPR-mediated cytosine base editor BE3 for functional analysis. They carried out a comprehensive screening of CRISPR-mediated base editing using 745 guide RNAs targeting all exons in BRCA1, identifying several previously unidentified variants, including c.-97C > T, c.154C > T, c.3847C > T, c.5056C > T, and c.4986+5G>A. The study effectively showcased the utility of CRISPR-mediated base editing as a potent instrument for reevaluating variants of uncertain significance (VUSs) in BRCA1, offering valuable insights for clinical management. This reclassification of VUSs in BRCA1 can have substantial implications for patients and healthcare providers. Patients with clarified variant classifications can receive more precise risk assessments and individualized treatment plans, potentially involving heightened surveillance or preventative measures. For healthcare providers, accurate variant classification guarantees appropriate counseling and risk communication for patients and their families [112].
off-target edits. Careful gRNA design, utilizing advanced algorithms, and validation of potential non-specific sites through sequencing can minimize these effects [118]. Efficient delivery of CRISPR-Cas9 components to target cells remains a challenge [119]. Methods such as viral vectors, lipid nanoparticles, and electroporation have been explored. Each approach has advantages and limitations in terms of efficiency, toxicity, and specificity [120]. The activation of the P21 protein, a well-known tumor suppressor, promotes cell cycle arrest by inhibiting cyc-lin-dependent kinases that regulate cell division. By halting the cell cycle, the growth of cancer cells is inhibited [121]. Activating the P21 protein through editing the KRAS gene can sensitize cancer cells to chemotherapy [122]. The increased expression of P21 leads to cell cycle arrest, which allows the chemotherapy drugs to target and kill the cancer cells more effectively [123]. Activating the P21 protein through KRAS gene editing sensitizes cancer cells to chemotherapy. Cell cycle arrest caused by P21 activation allows chemotherapy drugs to more effectively target and eliminate cancer cells [115].
with partial similarity to the target site [124]. Extensive research and optimization of guide RNA design have significantly reduced undesirable site effects [127]. State-of-the-art Cas9 variants, such as high-fidelity Cas9 and base editors, have further improved specificity, minimizing the risk of unintended genetic modifications [124]. Challenges associated with CRISPR-Cas9 editing of the EGFR gene include undesirable site effects, delivery efficiency, and potential long-term effects. Ethical considerations include the need for informed consent, ensuring equitable access to the technology, and responsible use to avoid unintended consequences or the creation of “designer babies.” Rigorous evaluation, regulation, and ethical guidelines are essential to navigate these challenges and ensure the responsible application of CRISPR-Cas9 in editing the EGFR gene or any other gene [128].
CRISPR-Cas9 editing of the VEGF (Vascular Endothelial Growth Factor) gene can play a crucial role in various cancers. VEGF is a protein that promotes the growth of new blood vessels, a process known as angiogenesis, which is essential for tumor development and metastasis [129]. By targeting and disrupting the VEGF gene using CRISPR-Cas9, researchers can potentially hinder the production of VEGF and, consequently, inhibit tumor angiogenesis. This could lead to reduced tumor growth and increased sensitivity to other cancer treatments [130]. While CRISPR-Cas9 is highly specific, there is a possibility of off-target effects where unintended gene edits occur. In the case of VEGF gene editing, researchers must carefully assess potential off-target sites to ensure that no critical genes are unintentionally modified [131]. To minimize undesirable site effects, rigorous bioinformatics analyses and advanced screening methods are employed to select guide RNAs with the least likelihood of non-specific site activity. CRISPR-Cas9-based VEGF gene editing, on its own, may not be sufficient for complete cancer treatment [129]. While it can impede tumor angiogenesis, a comprehensive cancer treatment strategy usually involves combining CRISPR-Cas9 with other therapies like chemotherapy, radiation, or immunotherapy [130]. Combining treatments can lead to a synergistic effect, targeting cancer cells through multiple pathways and increasing the overall therapeutic efficacy [129]. Delivering CRISPR-Cas9 components to cancer cells poses a significant challenge [131]. The large size of the Cas9 protein and guide RNA complex may limit delivery methods [131]. Various approaches are being explored, including viral vectors, nanoparticles, and liposomes, to ensure efficient and safe delivery to target cancer cells while avoiding harm to healthy tissues [129]. Ensuring long-term and stable VEGF gene suppression is essential for sustained therapeutic effects [131]. Researchers are investigating methods to improve CRISPR-Cas9 delivery
and stability within cancer cells [129]. Strategies like utilizing modified Cas9 variants or integrating the CRISPR components into the genome of the target cells could potentially enhance the durability of VEGF gene editing and its anticancer effects [131].
to uncontrolled cell growth, a hallmark of cancer [137]. By using CRISPR-Cas9 to precisely target and edit the PTEN gene, researchers can potentially restore its function as a tumor suppressor, thereby inhibiting cancer cell growth and reducing tumor development [138]. The potential of CRISPR-Cas9-mediated PTEN gene editing as a treatment option varies among different cancer types [136]. Some cancers exhibit PTEN mutations as a dominant driver of tumorigenesis, making them more amenable to this approach [135]. However, the efficacy of this strategy may depend on the cancer’s genetic context, as some tumors may possess alternative mechanisms to bypass PTEN function [138]. Extensive preclinical studies and clinical trials are required to determine its applicability and effectiveness across diverse cancer types [135]. Safety concerns in CRISPR-Cas9 gene editing for PTEN in cancer therapy involve potential undesirable site effects, where unintended genetic changes could occur in noncancerous cells, leading to adverse consequences [136]. Additionally, the risk of introducing new mutations or altering other essential genes must be carefully evaluated to avoid unwanted side effects [138]. Rigorous testing in preclinical models and careful monitoring during clinical trials are crucial to ensure the safety and feasibility of this therapeutic approach [135]. Researchers are continuously exploring various strategies to improve CRISPR-Cas9 gene editing efficiency [136]. One approach involves optimizing the delivery system to ensure precise targeting of cancer cells. Additionally, advancements in CRISPR-Cas9 technology, such as using base editors or prime editors, offer more precise modifications and reduced non-targeted site effects [138]. Moreover, combining CRISPRCas9 with other therapies, such as immunotherapies or targeted therapies, may enhance the overall therapeutic response, allowing for a more comprehensive and effective treatment strategy. Several challenges need to be addressed when using CRISPR-Cas9 to edit the PTEN gene in cancer cells [135]. Firstly, efficient delivery of CRISPR components to specific cancer cells is crucial to avoid non-targeted site effects [138]. Secondly, ensuring the correct and precise editing of the PTEN gene without introducing unintended mutations is vital for therapeutic success [135]. Additionally, the immune response to the CRISPR components and potential immune rejection of edited cells must be evaluated to assess their long-term viability and safety [138].
to tumor growth and progression. CRISPR-Cas9 can be employed as a gene-editing tool to target and modify the TERT gene in cancer cells [140]. The CRISPR-Cas9 system consists of a guide RNA that directs the Cas9 nuclease to the desired genomic location [141]. By designing a guide RNA specific to the TERT gene sequence, researchers can guide Cas9 to the TERT gene and induce a DSB at the targeted site [142]. The cell’s DNA repair machinery then repairs the break, often through the error-prone NHEJ pathway, which introduces small insertions or deletions (indels) that disrupt the TERT gene’s function [139]. Alternatively, researchers can use CRISPR-Cas9 in combination with a repair template to introduce specific modifications to the TERT gene sequence, such as gene knockouts or point mutations [141]. Editing the TERT gene using CRISPR-Cas9 can lead to several outcomes. One possibility is the disruption of TERT gene function, resulting in decreased telomerase activity in cancer cells. This can lead to telomere shortening and cellular senescence or apoptosis, inhibiting the unlimited replicative potential of cancer cells [139]. Another potential outcome is the modification of TERT gene expression, such as reducing its expression level, which can hinder tumor growth [142]. Additionally, CRISPR-Cas9-mediated TERT gene editing may sensitize cancer cells to other therapies, as telomerase inhibition can enhance the effectiveness of conventional treatments like chemotherapy or radiation therapy [139].
resistance to chemotherapy and radiation. By disrupting NF-kB signaling through CRISPR-Cas9 editing, cancer cells may become more vulnerable to standard cancer treatments, improving overall treatment outcomes [145]. While CRISPR-Cas9 editing of the NF-kB gene shows promise, it is important to evaluate potential side effects or unintended consequences [143]. undesirable site effects, where CRISPR-Cas9 edits unintended genomic sites, could lead to genetic instability or interfere with normal cellular functions [145]. Additionally, the long-term effects of NF-kB gene disruption on overall immune response and inflammatory processes need to be thoroughly assessed [144]. Optimizing the therapeutic potential of CRISPR-Cas9 editing of the NF-kB gene requires further research and development. Understanding the specific molecular characteristics of different cancer types and their NF-kB signaling pathways is essential for designing precise CRISPR-Cas9 strategies [145]. Additionally, advancements in delivery systems, such as viral vectors or nanoparticle-based carriers, can enhance the efficiency and specificity of NF-kB gene editing in cancer cells [143].
important functions in normal cells, so targeting it may cause unintended consequences in non-cancerous tissues [149].
Clinical trials
on targeting other genes to improve CAR T cell therapy. For instance, the clinical trial NCT04037566 aims to enhance the effectiveness of CD19-targeting CAR T cells by editing the endogenous HPK1 gene [157]. Similarly, NCT04767308 utilizes endogenous CD5 knockout in allogeneic CD5-targeting CAR T cells during the early phase I trial to potentially enhance their efficacy
[158]. Moreover, NCT03166878 uses the knockout of both TCR and
Safety and delivery challenges
Delivery challenges and safety measures in CRISPR-based gene editing
prime editors aim to minimize non-selective site effects, but ongoing research and ethical considerations are necessary to harness these technologies responsibly for therapeutic applications and other genetic interventions [215]. Off-target effects refer to unintended changes or alterations in the DNA that occur when using gene editing technologies like CRISPR-Cas systems. These unintended modifications can happen at sites other than the targeted location, potentially leading to unpredictable and unwanted genetic changes [214]. Base editors are a recent advancement in CRISPR technology that can perform targeted chemical modifications to specific DNA bases without creating double-stranded breaks like traditional CRISPR-Cas systems. This targeted approach reduces the risk of undesirable site effects by minimizing the potential for random DNA alterations [215]. Prime editors offer enhanced precision in gene editing compared to base editors or traditional CRISPR-Cas systems. They combine the capabilities of base editors and nickases, allowing for accurate insertion, deletion, and substitution of particular genetic bases within the genome. This increased precision further reduces the likelihood of non-targeted site effects [216]. Scientists use various techniques to assess non-selective site effects, such as whole-genome sequencing, high-throughput sequencing, and computational analysis. These methods help identify unintended genetic changes and determine the efficiency and specificity of the gene-editing technology being used [217]. Despite the advancements in base editors and prime editors, off-target effects remain a concern [217]. The challenge lies in achieving absolute precision in targeting specific genomic sites without affecting nearby regions [215]. Continuous refinement of gene-editing tools, along with rigorous evaluation and validation methods, are crucial to overcoming these challenges [216]. To ensure the safety of using base editors and prime editors in therapeutic settings, comprehensive preclinical studies are necessary. These studies involve rigorous testing of the gene-editing tools on relevant cell lines and animal models to assess potential non-targeted site effects and ensure the accuracy of genetic modifications before progressing to human trials [215].
| Cancer Type | Treatment Approach | Patient Population | Description | Advantages | Disadvantages | Ref |
| Metastatic melanoma | TCR/CAR-T therapy targeting NY-ESO-1 | Patients with NY-ESO-1 + tumors who failed prior therapy | T cells were edited to express NY-ESO-1 TCR/CAR, infused back into patients | Target specificity, long-term persistence | Possible off-target effects, limited efficacy in some patients | [160] |
| Non-Hodgkin’s Lymphoma | CD19-targeting CAR-T therapy | Patients with refractory/ relapsed NHL | T cells were edited to express CD19 CAR, infused back into patients | High response rate, durable response in some patients | Cytokine release syndrome, neurotoxicity, potential for tumor antigen escape | [161] |
| Bladder Cancer | PD-1 knockout via CRISPR/ Cas9 | Patients with high-risk non-muscle-invasive bladder cancer | CRISPR-edited autologous T cells were infused into patients | Potential for enhanced antitumor immune response | Off-target effects, potential for immune-related adverse events | [162] |
| Sarcoma | Targeted genome editing of PAX3-FOXO1 fusion gene | Patients with metastatic sarcoma expressing PAX3-FOXO1 | CRISPR/Cas9 was used to target the fusion gene in tumor cells, followed by infusion of edited T cells | Specific targeting of oncogenic driver mutation | Off-target effects, limited efficacy in some patients | [163] |
| Solid Tumors | Adoptive transfer of TCR/ CAR-T cells targeting neoantigens | Patients with advanced solid tumors | T cells were edited to express TCR/CAR targeting neoantigens unique to each patient’s tumor, infused back into patients | Target specificity, potential for durable response | Heterogeneity of tumor neoantigens, potential for offtarget effects | [164] |
| Renal Cell Carcinoma | PD-1 knockout via CRISPR/ Cas9 | Patients with advanced RCC who failed prior therapies | CRISPR-edited autologous T cells were infused into patients | Potential for enhanced antitumor immune response | Off-target effects, potential for immune-related adverse events | [165] |
| Multiple Myeloma | BCMA-targeting CAR-T therapy | Patients with relapsed/refractory multiple myeloma | T cells were edited to express BCMA CAR, infused back into patients | High response rate, durable response in some patients | Cytokine release syndrome, neurotoxicity | [166] |
| Glioblastoma | EGFRvIII-targeting CAR-T therapy | Patients with recurrent GBM expressing EGFRvIII | T cells were edited to express EGFRvIII CAR, infused back into patients | Target specificity, potential for durable response | Heterogeneity of tumor antigen expression, potential for off-target effects | [167] |
| Esophageal Cancer | Targeted genome editing of MUC1 gene | Patients with MUC1 + esophageal squamous cell carcinoma | CRISPR/Cas9 was used to target the MUC1 gene in tumor cells, followed by infusion of edited T cells | Specific targeting of oncogenic driver mutation | Off-target effects, limited efficacy in some patients | [168] |
| Prostate Cancer | Targeted genome editing of androgen receptor gene | Patients with castration-resistant prostate cancer | CRISPR/Cas9 was used to target the androgen receptor gene in tumor cells, followed by infusion of edited T cells | Specific targeting of oncogenic driver mutation | Off-target effects, limited efficacy in some patients | [169] |
| Acute Myeloid Leukemia | CD33-targeting CAR-T therapy | Patients with relapsed/refractory AML | T cells were edited to express CD33 CAR, infused back into patients | Target specificity, potential for durable response | Cytokine release syndrome, neurotoxicity | [170] |
| Head and Neck Cancer | Targeted genome editing of HPV16 E6 gene | Patients with HPV16+ recurrent or metastatic head and neck cancer | CRISPR/Cas9 was used to target the HPV16 E6 gene in tumor cells, followed by infusion of edited T cells | Specific targeting of oncogenic driver mutation | Off-target effects, limited efficacy in some patients | [171] |
| Cancer Type | Treatment Approach | Patient Population | Description | Advantages | Disadvantages | Ref |
| Lung Cancer | Targeted genome editing of KRAS gene | Patients with advanced KRASmutant lung cancer | CRISPR/Cas9 was used to target the KRAS gene in tumor cells, followed by infusion of edited T cells | Specific targeting of oncogenic driver mutation | Off-target effects, limited efficacy in some patients | [172] |
| Neuroblastoma | Targeted genome editing of ALK gene | Patients with ALK-mutant neuroblastoma | CRISPR/Cas9 was used to target the ALK gene in tumor cells, followed by infusion of edited T cells | Specific targeting of oncogenic driver mutation | Off-target effects, limited efficacy in some patients | [173] |
| Acute Lymphoblastic Leukemia | CD19-targeting CAR-T therapy | Pediatric patients with relapsed/refractory ALL | T cells were edited to express CD19 CAR, infused back into patients | High response rate, durable response in some patients | Cytokine release syndrome, neurotoxicity | [174] |
| Solid Tumors | Targeted genome editing of CCR4 gene | Patients with advanced solid tumors | CRISPR/Cas9 was used to target the CCR4 gene in tumor cells, followed by infusion of edited T cells | Specific targeting of oncogenic driver mutation | Off-target effects, limited efficacy in some patients | [175] |
| Melanoma | Targeted genome editing of NRAS gene | Patients with advanced NRASmutant melanoma | CRISPR/Cas9 was used to target the NRAS gene in tumor cells, followed by infusion of edited T cells | Specific targeting of oncogenic driver mutation | Off-target effects, limited efficacy in some patients | [176] |
| Cholangiocarcinoma | Targeted genome editing of IDH1 gene | Patients with advanced IDH1mutant cholangiocarcinoma | CRISPR/Cas9 was used to target the IDH1 gene in tumor cells, followed by infusion of edited T cells | Specific targeting of oncogenic driver mutation | Off-target effects, limited efficacy in some patients | [177] |
| Solid Tumors | Targeted genome editing of TP53 gene | Patients with advanced solid tumors | CRISPR/Cas9 was used to target the TP53 gene in tumor cells, followed by infusion of edited T cells | Specific targeting of oncogenic driver mutation | Off-target effects, limited efficacy in some patients | [178] |
| Myeloma | CD19-targeting CAR-T therapy | Patients with relapsed/refractory myeloma | T cells were edited to express CD19 CAR, infused back into patients | High response rate, durable response in some patients | Cytokine release syndrome, neurotoxicity | [179] |
| Solid Tumors | Targeted genome editing of PD-1 gene | Patients with advanced solid tumors | CRISPR/Cas9 was used to target the PD-1 gene in tumor cells, followed by infusion of edited T cells | Enhances anti-tumor immunity by disrupting immune checkpoint pathway | Off-target effects, limited efficacy in some patients | [180] |
| Solid Tumors | Targeted genome editing of MUC1 gene | Patients with advanced MUC1 + solid tumors | CRISPR/Cas9 was used to target the MUC1 gene in tumor cells, followed by infusion of edited T cells | Specific targeting of oncogenic driver mutation | Off-target effects, limited efficacy in some patients | [181] |
| Multiple Solid Tumors | Targeted genome editing of EGFR gene | Patients with advanced EGFRmutant solid tumors | CRISPR/Cas9 was used to target the EGFR gene in tumor cells, followed by infusion of edited T cells | Specific targeting of oncogenic driver mutation | Off-target effects, limited efficacy in some patients | [165] |
| Cancer Type | Treatment Approach | Patient Population | Description | Advantages | Disadvantages | Ref |
| Leukemia/Lymphoma | Targeted genome editing of CD22 gene | Patients with relapsed/ refractory CD22 + leukemia/ lymphoma | CRISPR/Cas9 was used to target the CD22 gene in tumor cells, followed by infusion of edited T cells | Specific targeting of oncogenic driver mutation | Off-target effects, limited efficacy in some patients | [182] |
| Solid Tumors | Targeted genome editing of PTEN gene | Patients with advanced PTENdeficient solid tumors | CRISPR/Cas9 was used to target the PTEN gene in tumor cells, followed by infusion of edited T cells | Specific targeting of oncogenic driver mutation | Off-target effects, limited efficacy in some patients | [183] |
| Multiple Solid Tumors | Targeted genome editing of PDCD1 gene | Patients with advanced solid tumors | CRISPR/Cas9 was used to target the PDCD1 gene in tumor cells, followed by infusion of edited T cells | Enhances anti-tumor immunity by disrupting immune checkpoint pathway | Off-target effects, limited efficacy in some patients | [184] |
| Leukemia/Lymphoma | Targeted genome editing of TCR gene | Patients with relapsed/refractory leukemia/lymphoma | CRISPR/Cas9 was used to target the TCR gene in T cells, followed by infusion of edited T cells | Specific targeting of TCR gene for enhanced T-cell activity | Off-target effects, limited efficacy in some patients | [185] |
| Leukemia/Lymphoma | CD19-targeting CAR-T therapy | Patients with relapsed/refractory leukemia/lymphoma | T cells were edited to express CD19 CAR, infused back into patients | High response rate, durable response in some patients | Cytokine release syndrome, neurotoxicity | [186] |
| Solid Tumors | Targeted genome editing of HIF-1 a gene | Patients with advanced HIF-1a-overexpressing solid tumors | CRISPR/Cas9 was used to target the HIF-1 a gene in tumor cells, followed by infusion of edited T cells | Specific targeting of oncogenic driver mutation | Off-target effects, limited efficacy in some patients | [187] |
| Non-small cell lung cancer | CRISPR/Cas9-mediated PD-1 knockout | Patients with advanced PD-L1 + non-small cell lung cancer | CRISPR/Cas9 was used to knock out the PD-1 gene in T cells, followed by infusion of edited T cells | Disrupts immune checkpoint pathway | Off-target effects, limited efficacy in some patients | [188] |
| Multiple Solid Tumors | CRISPR/Cas9-mediated knockout of TGF-
|
Patients with advanced TGF-
|
CRISPR/Cas9 was used to knock out the TGF-
|
Specific targeting of oncogenic driver mutation | Off-target effects, limited efficacy in some patients | [189] |
| Solid Tumors | CRISPR/Cas9-mediated knockout of DNMT1 | Patients with advanced solid tumors | CRISPR/Cas9 was used to knock out the DNMT1 gene in tumor cells, followed by infusion of edited T cells | Specific targeting of epigenetic regulator | Off-target effects, limited efficacy in some patients | [190] |
| Solid Tumors | CRISPR/Cas9-mediated knockout of LAP | Patients with advanced LAPoverexpressing solid tumors | CRISPR/Cas9 was used to knock out the LAP gene in tumor cells, followed by infusion of edited T cells | Specific targeting of immunosuppressive mechanism | Off-target effects, limited efficacy in some patients | [191] |
| Cancer Type | Treatment Approach | Patient Population | Description | Advantages | Disadvantages | Ref |
| Solid Tumors | CRISPR/Cas9-mediated knockout of AXL | Patients with advanced AXLoverexpressing solid tumors | CRISPR/Cas9 was used to knock out the AXL gene in tumor cells, followed by infusion of edited T cells | Specific targeting of oncogenic driver mutation | Off-target effects, limited efficacy in some patients | [192] |
| Solid Tumors | CRISPR/Cas9-mediated knockout of HLA class I | Patients with advanced HLA class I-deficient solid tumors | CRISPR/Cas9 was used to knock out the HLA class I genes in tumor cells, followed by infusion of edited T cells | Specific targeting of immune evasion mechanism | Off-target effects, limited efficacy in some patients | [193] |
| Leukemia/Lymphoma | CRISPR/Cas9-mediated knockout of TCR and B2M | Patients with relapsed/refractory T-cell malignancies | CRISPR/Cas9 was used to knock out the TCR and B2M genes in T cells, followed by infusion of edited T cells | Disruption of T-cell receptor and MHC class I expression to prevent graft-versus-host disease and enhance antitumor activity | Off-target effects, limited efficacy in some patients | [194] |
| Solid Tumors | CRISPR/Cas9-mediated knockout of
|
Patients with advanced
|
CRISPR/Cas9 was used to knock out the
|
Specific targeting of oncogenic driver mutation | Off-target effects, limited efficacy in some patients | [195] |
| Leukemia/Lymphoma | CRISPR/Cas9-mediated knockout of CD7 | Patients with relapsed/ refractory CD7 + leukemia/ lymphoma | CRISPR/Cas9 was used to knock out the CD7 gene in T cells, followed by infusion of edited T cells | Specific targeting of B-cell antigen | Off-target effects, limited efficacy in some patients | [196] |
| Solid Tumors | CRISPR/Cas9-mediated knockout of PSCA | Patients with advanced PSCAexpressing solid tumors | CRISPR/Cas9 was used to knock out the PSCA gene in tumor cells, followed by infusion of edited T cells | Specific targeting of tumorassociated antigen | Off-target effects, limited efficacy in some patients | [190] |
| Solid Tumors | CRISPR/Cas9-mediated knockout of APOBEC3B | Patients with advanced APOBEC3B-overexpressing solid tumors | CRISPR/Cas9 was used to knock out the APOBEC3B gene in tumor cells, followed by infusion of edited T cells | Specific targeting of mutagenic enzyme | Off-target effects, limited efficacy in some patients | [197] |
| Solid Tumors | CRISPR/Cas9-mediated knockout of IL2RG | Patients with advanced IL2RGdeficient solid tumors | CRISPR/Cas9 was used to knock out the IL2RG gene in T cells, followed by infusion of edited T cells | Specific targeting of immunodeficiency gene | Off-target effects, limited efficacy in some patients | [198] |
| Solid Tumors | CRISPR/Cas9-mediated knockout of ARID1A | Patients with advanced ARID1A-mutant solid tumors | CRISPR/Cas9 was used to knock out the ARID1A gene in tumor cells, followed by infusion of edited T cells | Specific targeting of oncogenic driver mutation | Off-target effects, limited efficacy in some patients | [199] |
| Solid Tumors | CRISPR/Cas9-mediated knockout of TRAC | Patients with advanced TRACdeficient solid tumors | CRISPR/Cas9 was used to knock out the TRAC gene in T cells, followed by infusion of edited T cells | Specific targeting of immunodeficiency gene | Off-target effects, limited efficacy in some patients | [200] |
| Cancer Type | Treatment Approach | Patient Population | Description | Advantages | Disadvantages | Ref |
| Solid Tumors | CRISPR/Cas9-mediated knockout of LAPTM4B | Patients with advanced LAPTM4B-overexpressing solid tumors | CRISPR/Cas9 was used to knock out the LAPTM4B gene in tumor cells, followed by infusion of edited T cells | Specific targeting of oncogene | Off-target effects, limited efficacy in some patients | [201] |
| Solid Tumors | CRISPR/Cas9-mediated knockout of HPRT1 | Patients with advanced HPRT1-overexpressing solid tumors | CRISPR/Cas9 was used to knock out the HPRT1 gene in tumor cells, followed by infusion of edited T cells | Specific targeting of oncogene | Off-target effects, limited efficacy in some patients | [202] |
| Challenge | Current Approaches/ Strategies | Limitations | Future Directions | Ethical and Regulatory Considerations | Description | Novelty | Advantages | Disadvantages | Limitations/ Challenges | Ref |
| Off-Target Effects | Targeted Genome Editing | – Can be difficult to achieve high specificity | – Development of improved delivery methods and monitoring techniques | – Need for clear guidelines and oversight | CRISPR can cause unintended changes to the genome outside the target region, which can lead to adverse effects | CRISPR-mediated genome editing is a highly precise and targeted approach to gene therapy | – Minimizes damage to healthy cells | – Possibility of unintended changes to the genome | Specificity needs to be improved to avoid offtarget effects | [206, 207] |
| Delivery to Tumor Site | Use of Viral Vectors | – Lack of specificity, difficulty targeting tumor cells | – Development of targeted delivery systems | – Need to minimize risk to healthy tissues | Viral vectors are commonly used to deliver CRISPR-based therapies, but can be difficult to target to specific tumor cells | Targeted delivery of CRISPR-based therapies can minimize damage to healthy tissues | – Can be used to target specific tumor cells | – Can cause immune reactions and toxicity | Targeting specificity needs to be improved to avoid damage to healthy tissues | [208, 209] |
| Safety | Monitoring OffTarget Effects | – Limited sensitivity and accuracy of current monitoring techniques | – Development of more sensitive and accurate monitoring techniques | – Need to ensure patient safety and minimize potential harm | Monitoring of off-target effects is essential to ensure the safety of CRISPR-based therapies | Improved monitoring techniques can provide more accurate information about the effects of CRISPR editing on the genome | – Can help to detect unintended changes to the genome | – Current techniques have limited sensitivity and accuracy | More research is needed to improve monitoring techniques | [210,211] |
| Immune Reactions | Use of NonImmunogenic CRISPR Systems | – Limited availability of nonimmunogenic CRISPR systems | – Development of non-immunogenic CRISPR systems | – Need to minimize the risk of immune reactions and toxicity | CRISPR-based therapies can trigger immune responses, which can lead to adverse effects | Non-immunogenic CRISPR systems are being developed to minimize the risk of immune reactions | Non-immunogenic CRISPR systems can reduce the risk of immune reactions and toxicity | – Non-immunogenic CRISPR systems are not widely available | More research is needed to develop nonimmunogenic CRISPR systems | [212,213] |
predictions of on-target efficiency. To achieve this, the researchers gathered high-quality gRNA activity data for 10,592 gRNAs that target the SpCas9 enzyme. To further improve their predictions, the researchers integrated this new data with existing complementary data from other sources. They then employed a deep learning model called “CRISPR on,” which was trained on a combined dataset of 23,902 gRNAs, including both the newly generated data and the previously available data. The results of their study showed that CRISPR on outperformed existing tools used for gRNA efficiency prediction. The improved performance was observed across four test datasets that were not part of the training data used for developing other prediction tools. This suggests that CRISPR on’s predictions were more accurate and reliable than what was currently available. To make their findings accessible to the scientific community, the researchers developed an interactive webserver for gRNA design based on the CRISPR on
mutations could inactivate NFE2L2 and thus promote cancer growth [139]. The purpose of using CRISPR-Cas9 was to investigate the potential therapeutic effect of targeting the BRCA1/TP53/RAS gene, which is commonly mutated in breast and ovarian cancer/cancer [231]. The unintended consequences of using CRISPR-Cas9 on the BRCA1/TP53/RAS gene were unintended mutations in several off-target genes, including RAD51D/MDM2/ MAPK, respectively [232]. The unintended mutations in RAD51D/MDM2/MAPK genes could have compromised the therapeutic effect of targeting BRCA1/TP53/RAS as they could activate RAD51D/ inactivate MDM2/activate MAPK, leading to the promotion of cancer growth [233]. The frequent findings of unintended mutations in undesirable site genes in these studies highlight the significant risk of non-selective site effects associated with CRISPRCas9 gene editing [234]. The research should analyze and compare the off-target effects observed when targeting different genes, which could provide insights into the gene-specific effects of CRISPR-Cas9 [235, 236]. Understanding the off-target effects and their potential impact on cancer growth is crucial in assessing the safety and efficacy of CRISPR-Cas9 as a therapeutic approach for cancer treatment [232, 237].
Safety
of the edited cells becoming cancerous [240]. CRISPRbased cancer therapy relies on the ability to edit specific genetic mutations that drive tumor growth. However, if the edited cells acquire additional mutations, they could become cancerous [238]. Researchers are working to understand the long-term effects of CRISPR-based cancer therapy and to develop strategies to minimize the risk of the edited cells becoming cancerous [241]. In addition to these concerns, CRISPR-based cancer therapy raises a number of other safety concerns, including the potential for immune reactions to the viral vectors used to deliver CRISPR, and the risk of creating new cancer-causing mutations. To mitigate these concerns, researchers are developing safer delivery methods and developing new strategies to minimize the risk of non-targeted site effects [240]. Preclinical and clinical studies are being conducted to evaluate the safety and efficacy of CRISPR-based cancer therapy [238]. Additionally, researchers are working to find cost-effective and efficient methods for producing large numbers of CRISPR-edited cells, and to overcome scalability issues [240]. CRISPR-based gene editing has the potential to revolutionize cancer therapy, but significant safety challenges remain to be addressed before this approach can be safely and effectively used in the clinic [241]. Ongoing research is essential to better understand the long-term effects of CRISPR-based cancer therapy, to develop safer delivery methods, and to minimize the risk of non-targeted site effects and other safety concerns [240]. Another aspect of safety in CRISPR-based cancer therapy is the delivery method used to deliver the CRISPR machinery to the tumor cells. One of the most commonly used methods is the use of viral vectors, such as adenoviruses or lentiviruses [241]. However, these vectors have the potential to cause immune reactions and other adverse effects [238]. Researchers are working on developing non-viral delivery methods, such as nanoparticles and exosomes, as an alternative to viral vectors. These methods have the potential to be safer and more effective, but they are still in early stages of development and more research is needed to optimize their effectiveness and safety [241, 244]. Additionally, the manufacturing and scalability of CRISPR-based cancer therapy is another important safety concern. Producing large quantities of CRISPR-modified cells for clinical use is challenging and costly [241]. Researchers are working to find cost-effective and efficient methods for producing large numbers of CRISPR-edited cells, and to overcome scalability issues. This includes researching alternative methods of producing the CRISPR machinery and exploring ways to improve the efficiency of the CRISPR editing process [241]. Safety is a critical concern in the development of CRISPR-based cancer therapy [240]. Researchers are working to address these concerns by developing
safer delivery methods, developing new strategies to minimize the risk of non-targeted site effects and other safety concerns, and finding cost-effective and efficient methods for producing large numbers of CRISPR-edited cells [238]. Ongoing research is essential to better understand the long-term effects of CRISPR-based cancer therapy, and to ensure that this promising new approach can be safely and effectively used in the clinic. It’s important to note that while CRISPR-based cancer therapy is a promising new approach, it is still in the early stages of development. Many of the safety concerns and challenges discussed above are still being studied and evaluated in preclinical and clinical trials. Therefore, it is important to continue monitoring the progress of research in this field and to evaluate the safety and efficacy of CRISPRbased cancer therapy as more data becomes available [240]. It is also worth noting that the regulatory landscape for CRISPR-based cancer therapy is still evolving. Different countries and regions have different regulations and guidelines regarding the use of CRISPR-based therapies in humans [241]. Researchers and companies developing CRISPR-based cancer therapies will need to navigate these regulations and guidelines in order to bring their therapies to market [238]. The CRISPR-based cancer therapy has the potential to revolutionize cancer treatment, but significant safety challenges and delivery issues still need to be addressed. Researchers are working to address these concerns through ongoing research and development, but it will take time to fully understand the long-term effects and safety of this new approach. It is important to monitor the progress of research in this field and to evaluate the safety and efficacy of CRISPRbased cancer therapy as more data becomes available [241].
Delivery to the tumor site
and mount an immune response. This can lead to inflammation and other adverse effects, and can also limit the effectiveness of the therapy [250]. Another method to deliver CRISPR to the tumor site is through the use of nanoparticles. These particles are small enough to easily penetrate the tumor tissue, and can be engineered to carry the CRISPR machinery [251]. Nanoparticles can also be designed to target specific cell types, such as cancer cells, to increase the efficiency of the therapy [252]. However, the efficacy of nanoparticles in delivering CRISPR to the tumor site is still being evaluated, and more research is needed to understand their safety and effectiveness [253]. Exosomes are also considered as a promising delivery method for CRISPR [254]. Exosomes are small vesicles that are naturally released by cells and can be engineered to carry CRISPR machinery [255]. Exosomes have the ability to cross the blood-brain barrier and deliver the CRISPR machinery to the tumor site. But more research is needed to understand the safety and efficacy of exosomes as a delivery method for CRISPR [256]. Researchers are developing CRISPR delivery methods that can target cancer cells based on surface markers, such as the expression of specific proteins [257]. Another approach is to target genetic mutations that are specific to cancer cells. This can be done by engineering the CRISPR machinery to recognize and target specific genetic sequences associated with cancer [255]. For example, researchers have developed CRISPRbased therapies that target specific mutations in genes such as KRAS, which is commonly mutated in many types of cancer [256]. A combination of these strategies to target specific cell types can also be used to deliver CRISPR to the tumor site [248]. For example, researchers are exploring the use of nanoparticles that are designed to target specific surface markers on cancer cells and also carry the CRISPR machinery [245]. Delivery of CRISPR to the tumor site is a critical step in the development of CRISPR-based cancer therapy [248]. Researchers are working to develop new and efficient methods for delivering CRISPR to the tumor site, including viral vectors, nanoparticles, and exosomes [251]. Additionally, targeting specific cell types, such as cancer cells, can increase the efficiency of the therapy. While significant challenges remain to be addressed, the potential of CRISPR-based cancer therapy to revolutionize cancer treatment is clear, and ongoing research will help to overcome these challenges [255].
Manufacturing and scalability
approach can be safely and effectively used in the clinic [200]. One of the major challenges is the manufacturing and scalability of CRISPR-modified cells for clinical use [259]. Manufacturing CRISPR-edited cells for use in cancer therapy is a complex and costly process. The first step is to obtain the cells that will be edited, which can be obtained from the patient or from a cell line. Once the cells are obtained, they must be modified using the CRISPR machinery. This typically involves delivering the CRISPR machinery, including the guide RNAs and Cas enzymes, to the cells using a viral vector or nanoparticle [260]. However, this process is not yet fully optimized and more research is needed to find efficient and cost-effective methods for producing large numbers of CRISPR-edited cells [261]. Scalability is also a major challenge in the manufacture of CRISPR-edited cells for cancer therapy [262]. The current methods for producing CRISPR-edited cells are not yet able to produce the large numbers of cells required for clinical use [258]. For example, if the cells are produced using a viral vector, the process is limited by the number of cells that can be infected at one time [263]. Additionally, the current methods for producing CRISPR-edited cells are not yet able to produce cells with a high enough efficiency to be clinically relevant [264]. There are a number of potential solutions to these challenges. Researchers are working to develop more efficient and cost-effective methods for producing CRISPR-edited cells, such as using exosomes as a delivery method, and to improve the scalability of the process [265]. Additionally, researchers are working to improve the efficiency of the CRISPR-editing process and to minimize the risk of non-targeted site effects [200]. Another potential solution to the scalability challenge is the use of cell lines that have been genetically engineered to produce high numbers of CRISPR-edited cells [259]. For example, researchers have developed cell lines that stably express Cas enzymes, which can be used to produce large numbers of CRISPR-edited cells [260]. Additionally, researchers are exploring the use of stem cells as a source for CRISPR-edited cells [264]. Stem cells have the ability to self-renew and differentiate into a wide range of cell types, making them an attractive option for producing large numbers of CRISPR-edited cells for cancer therapy. Another area of active research is the development of automated platforms for producing CRISPR-edited cells. These platforms can automate many of the manual steps involved in the production of CRISPR-edited cells, making the process more efficient and cost-effective [200]. Additionally, these platforms can be used to optimize the conditions for producing CRISPR-edited cells, such as the amount of Cas enzymes and guide RNAs used [259]. Finally, researchers are exploring the use of in situ delivery of CRISPR
machinery, which allows the cells to be edited directly in the tumor microenvironment [261]. This approach avoids the need to produce and deliver large numbers of CRISPR-edited cells, and could potentially overcome the scalability challenges [264]. However, this approach is still in the early stages of development and more research is needed to optimize its effectiveness and safety [260]. One example is the use of exosomes as a delivery method for CRISPR machinery [264]. Exosomes are small vesicles that are naturally released by cells and can be used to deliver a variety of molecules, including CRISPR machinery, to target cells [261]. Researchers have shown that exosomes can be used to deliver CRISPR machinery to cancer cells with high efficiency and minimal toxicity. This approach is being further developed as a potential solution to the scalability challenge. Another example is the use of cell lines that have been genetically engineered to produce high numbers of CRISPR-edited cells [264]. Researchers have developed cell lines that stably express Cas enzymes and can be used to produce large numbers of CRISPR-edited cells. This approach is being further developed as a potential solution to the scalability challenge. Another example is the development of automated platforms for producing CRISPR-edited cells. These platforms automate many of the manual steps involved in the production of CRISPR-edited cells, making the process more efficient and cost-effective. This approach is being further developed as a potential solution to the scalability challenge [200]. Finally, there are examples of research on in situ delivery of CRISPR machinery [260]. Researchers have developed methods for delivering CRISPR machinery directly to cancer cells in the tumor microenvironment [261]. This approach avoids the need to produce and deliver large numbers of CRISPR-edited cells, and could potentially overcome the scalability challenges [264]. This approach is still in the early stages of development and more research is needed to optimize its effectiveness and safety [259]. The manufacturing and scalability of CRISPR-modified cells for clinical use is a major challenge that needs to be overcome for CRISPRbased cancer therapy to become a viable clinical option [261]. Researchers are working to develop more efficient and cost-effective methods for producing CRISPR-edited cells, such as using exosomes as a delivery method, and improving scalability by using genetically engineered cell lines or stem cells, and by developing automated platforms and in situ delivery method [200].
Conclusion and future directions
tackling the intricate characteristics of cancer via targeted genomic alterations are substantial. Approaches including deactivating genes that promote tumor growth, boosting the body’s immune reaction to cancer cells, correcting genetic anomalies that lead to cancer, and attacking tumors directly with toxic agents, have all indicated promising pathways in the realm of cancer therapeutics. Early-stage research and clinical experiments have started to reveal the effectiveness and transformative potential of CRISPR in the context of cancer treatment. These investigations have not only yielded hopeful outcomes but have also clarified the trajectory ahead. Yet, there are notable obstacles to overcome. The accuracy of CRISPR, its greatest advantage, raises concerns about unintended genetic impacts, known as off-target effects. The paramount importance lies in ensuring the security and precision of CRISPR interventions, necessitating continuous research to address these concerns. The delivery of CRISPR components to tumor cells presents another significant challenge. Developing methods that are both effective and safe for delivering these components is vital for the practical use of CRISPR in treating cancer. This challenge is heightened by the variability of tumor types and the complexity inherent in human biology. Nonetheless, the prospective future of CRISPR in cancer treatment is exceptionally promising. With ongoing advancements in research surmounting existing barriers, there is a tangible possibility for the creation of more efficient, individualized, and minimally invasive treatments for cancer. Such advancements could fundamentally transform the approach to cancer care, moving from traditional chemotherapy and radiation to specific genetic treatments. The CRISPR methodology presents an innovative and potentially game-changing strategy in cancer therapy. The journey ahead is laden with hurdles, such as ensuring the safety, accuracy, and efficient delivery of treatments. Despite these challenges, the progress achieved to date is promising. Continuous investigation and development in this area are crucial to fully harness the capabilities of CRISPR-based therapies in combating cancer. Looking forward, it is vital to confront these challenges directly, concentrating on refining methods, improving delivery systems, and prioritizing patient safety, in order to fully exploit the revolutionary potential of this technology in cancer care.
| Research Priorities | Emerging Technologies | Translational Opportunities | Collaborative Efforts | Societal Impact | Ref |
| Development of more precise and efficient delivery systems | Expansion of genome editing tools beyond gene knockout and correction | Development of biomarkers to predict treatment response | Collaborative development of ethical guidelines for CRISPR-based therapy | Equitable access to CRISPR-based cancer therapy | [268] |
| Development of gene editing tools to target non-coding regions of the genome | Application of CRISPR screens to identify new therapeutic targets | Use of gene editing to improve immunotherapy response | Collaboration between academic institutions and industry to accelerate drug development | Impacts on healthcare economics and resource allocation | [269] |
| Exploration of combination therapies that include CRISPR-based interventions | Integration of CRISPR technology with emerging imaging and sensing technologies | Application of CRISPR gene editing to develop personalized cancer vaccines | Collaborative development of CRISPR-based diagnostic tools | Ethical implications of germline editing for cancer prevention and treatment | [271] |
| Developing new approaches to improve specificity of CRISPRCas9 gene editing | Exploring the use of CRISPR gene regulation to modulate gene expression | Testing the efficacy of CRISPR-based cancer therapy in combination with other standard therapies | Development of international regulatory frameworks for gene editing technology | Education and public outreach to promote understanding and acceptance of gene editing technology | [272] |
| Studying the impact of CRISPR gene editing on tumor microenvironment and the immune system | Developing CRISPR-based tools for non-invasive cancer detection and monitoring | Incorporating CRISPR gene editing into patient stratification and clinical trial design | Collaboration among researchers to advance the understanding of CRISPR mechanisms and their role in cancer biology | Addressing issues of genetic discrimination and privacy concerns related to gene editing technology | [18] |
| Designing CRISPR gene editing systems for the treatment of rare or difficult-to-treat cancers | Advancing gene editing technology to target complex genetic abnormalities in cancer cells | Establishing international databases to promote sharing of CRISPR-based therapy data and protocols | Development of guidelines and standards for quality control and product safety for CRISPRbased therapies | Ensuring equitable access to CRISPR-based cancer therapy in diverse patient populations | [270] |
| Developing gene editing strategies to overcome resistance to standard cancer therapies | Integration of CRISPR technology with nanotechnology for enhanced delivery and targeting of cancer cells | Establishing public-private partnerships to accelerate the development of CRISPR-based cancer therapies | Collaboration between clinicians and basic researchers to optimize the design and delivery of CRISPRbased cancer therapies | Evaluation of the long-term safety and efficacy of CRISPR-based cancer therapy | [273, 274] |
drug development and foster more efficient translational opportunities [266]. Furthermore, combining CRISPRbased interventions with other therapies in a synergistic approach warrants exploration, while CRISPR-based diagnostic tools will aid in early cancer detection and diagnosis [268]. Gene editing technology can be leveraged to develop personalized cancer vaccines and improve immunotherapy response. In terms of societal
impact, there is a need for equitable access to CRISPRbased cancer therapy to ensure all patients benefit from these advancements, regardless of their background or location. This calls for international cooperation in establishing regulatory frameworks for gene editing technology and promoting public understanding and acceptance of these therapies through education and outreach [269]. Other research priorities involve studying the effects of CRISPR gene editing on the tumor microenvironment and immune system, addressing genetic discrimination and privacy concerns related to gene editing, and evaluating the long-term safety and efficacy of CRISPR-based cancer therapy [18]. Lastly, developing CRISPR gene editing systems for rare or difficult-to-treat cancers, harnessing nanotechnology for targeted delivery of CRISPR, and establishing databases for data sharing are critical for pushing the boundaries of CRISPR-based cancer therapy [270]. Public-private partnerships and collaboration between clinicians and researchers are instrumental in optimizing therapy design and delivery for better patient outcomes [267].
Development of new delivery methods
the safety and efficacy of AAV-based CRISPR therapies [277]. Nanoparticles made of polymers or inorganic materials offer alternative approaches to deliver CRISPR components. These nanoparticles can be designed with different physicochemical properties, which may influence cellular uptake and release kinetics [244]. While liposomes have advantages in encapsulation and modification, other nanoparticles may provide better stability or have unique capabilities for targeted delivery [282]. Cell-penetrating peptides and exosomes have the potential to improve CRISPR delivery [254]. Researchers can explore surface modifications of these delivery systems to increase their tumor-specific binding and uptake [279]. Additionally, optimizing cargo loading and release mechanisms could enhance the precise editing of target genes while minimizing unwanted effects [283].
Combination therapy
Targeting multiple genes
consider. non-selective site effects, where CRISPR-Cas9 inadvertently edits unintended genes, could result in unforeseen consequences [292]. Extensive research and stringent safety measures are necessary to minimize such risks and ensure the safe application of CRISPR technology in cancer treatments [293]. The CRISPR-based gene editing’s effectiveness in targeting multiple genes may vary between different types of cancer. Each cancer type is characterized by unique genetic mutations, necessitating tailored approaches [294].
Personalized medicine
Synthetic lethality
cells, while sparing normal cells [304]. In the context of CRISPR-based gene editing, this approach involves using CRISPR-Cas9 to simultaneously target two genes that are frequently mutated in cancer, exploiting the cancer’s genetic vulnerabilities [305]. Synthetic lethality-based cancer treatments have the advantage of selectively targeting cancer cells with specific gene mutations, reducing the risk of harming healthy cells. This approach can potentially lead to more effective and precise therapies with fewer side effects than conventional treatments like chemotherapy and radiation [304]. Researchers have targeted gene combinations such as BRCA1 and BRCA2, frequently found mutated in breast and ovarian cancer, and PARP1 and BRCA1, commonly mutated in breast cancer [303]. By disrupting these gene pairs simultaneously, they trigger synthetic lethality in cancer cells [302]. Experimental studies have demonstrated the potential of CRISPR-based synthetic lethality as a cancer treatment strategy [304]. By targeting specific gene combinations in cancer cells, researchers have observed significant reductions in tumor growth and cell viability in preclinical models, indicating its potential as a promising therapeutic approach [303]. One major challenge is the delivery of CRISPR components to the tumor site efficiently [302]. Ensuring precise targeting and non-selective site effects are also important concerns. Additionally, identifying suitable gene combinations for specific cancer types and ensuring safety during clinical translation are vital considerations [303].
CAR-T cell therapy
and non-Hodgkin lymphoma [310]. Clinical trials have reported high response rates and even durable remissions in some patients [308]. However, its effectiveness in solid tumors is still a significant area of research and development [307]. Long-term side effects of CAR-T cell therapy are not yet fully understood, as the therapy is relatively new [311]. However, some potential concerns include the persistence of CAR-T cells in the body, potential non-selective site effects of genetic modifications, and the impact on normal immune function [312]. To address manufacturing challenges, efforts are underway to optimize and streamline the production process, including automation and reducing the time and cost involved [308]. Furthermore, establishing specialized centers equipped with expertise and infrastructure can help address logistical challenges associated with CAR-T cell therapy [307]. Current limitations include the high cost of treatment, limited accessibility due to specialized requirements, and the need for personalized
manufacturing for each patient [310]. Additionally, the effectiveness of CAR-T cell therapy can be influenced by factors such as antigen escape, tumor heterogeneity, and the immunosuppressive tumor microenvironment [313]. Figure 11 illustrates the ex vivo CRISPR manipulation of human T cells for adoptive T cell therapy.
Combination of CRISPR-based gene editing with stem cell therapy
Combination of CRISPR-based gene editing with epigenetic therapy
effects [322]. Additionally, maintaining long-term regulation of gene activity via epigenetic therapy might be challenging due to cellular processes that could revert these changes over time. Researchers need to develop strategies to maintain stable and heritable epigenetic modifications [323]. Genetic heterogeneity, where different cells within a tumor or disease exhibit distinct genetic mutations, poses a challenge for targeted therapies [256]. The combination of CRISPR and epigenetic therapy allows researchers to target specific mutations while bypassing others [324]. CRISPR can be programmed to recognize and edit particular mutations, while epigenetic therapy can suppress the activity of specific mutated genes, leading to a more comprehensive and effective treatment [325]. The combination of these powerful technologies raises ethical questions about potential non-selective site effects, unintended consequences, and germline editing [325]. Researchers and policymakers must ensure strict adherence to safety protocols and responsible use to prevent unintended genetic alterations [326]. Additionally, equitable access to such therapies and potential disparities in healthcare must be addressed to avoid exacerbating social inequalities [327]. As with any emerging technology, there are limitations to consider. The delivery of CRISPR components and epigenetic drugs to specific tissues or organs can be challenging [328]. Ongoing research focuses on refining delivery methods and increasing targeting efficiency [325]. Moreover, understanding the long-term consequences of epigenetic modifications and potential off-target effects remains a priority for further investigation to ensure the safety and efficacy of this combination therapy [327].
Identification of new drug targets
modification on cell viability and proliferation [330, 333-336]. If the loss or alteration of a gene significantly impairs the cells’ ability to grow and survive, it suggests that the gene plays a vital role in supporting cancer cell functions [332]. The identification of crucial genes using CRISPR-Cas9 provides valuable insights into the vulnerabilities and dependencies of cancer cells [48, 333, 335]. Genes found to be essential for cancer cell growth and survival can be further investigated as potential drug targets [333]. Targeting these genes with drugs may disrupt critical cellular processes, leading to the selective killing or suppression of cancer cells while minimizing harm to normal cells [329, 331, 337, 338]. Despite its potential, CRISPR-based gene editing for drug target discovery faces several challenges [333]. Off-target effects, incomplete gene knockout, and functional redundancy within cellular pathways can complicate data interpretation [333]. Additionally, the translation of CRISPR-based findings into effective drug targets requires further validation through preclinical and clinical studies [48]. Ensuring the specificity, efficacy, and safety of drugs targeting newly identified genes is crucial for successful clinical implementation [331, 333, 339].
Acknowledgements
Authors’ contributions
Funding
Availability of data and materials
Declarations
Ethics approval and consent to participate
Consent for publication
Competing interests
Published online: 09 January 2024
References
- De A, Biswas AR. Nanotechnology and Computational tool based study of CRISPR/Cas-9 research in Biomedical Engineering. J Nano Res Adv Mater Polym Sci. 2020;1:6-1.
- Hale CR, Majumdar S, Elmore J, Pfister N, Compton M, Olson S, et al. Essential features and rational design of CRISPR RNAs that function with the Cas RAMP module complex to cleave RNAs. Mol Cell. 2012;45(3):292-302.
- Ayanoğlu FB, Elçiln AE, Elçin YM. Bioethical issues in genome editing by CRISPR-Cas9 technology. Turkish J Biol. 2020;44(2):110-20.
- Feng Z, Zhang B, Ding W, Liu X, Yang D-L, Wei P, et al. Efficient genome editing in plants using a CRISPR/Cas system. Cell Res. 2013;23(10):1229-32.
- Shachaf CM, Kopelman AM, Arvanitis C, Karlsson Å, Beer S, Mandl S, et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004;431(7012):1112-7.
- Su S, Hu B, Shao J, Shen B, Du J, Du Y, et al. CRISPR-Cas9 mediated efficient PD-1 disruption on human primary T cells from cancer patients. Sci Rep. 2016;6. https://doi.org/10.1038/srep20070.
- Ye L, Wang J, Tan Y, Beyer AI, Xie F, Muench MO, et al. Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: An approach for treating sickle cell disease and
-thalassemia. Proc Natl Acad Sci. 2016;113(38):10661-5. - Walton J, Blagih J, Ennis D, Leung E, Dowson S, Farquharson M, et al. CRISPR/Cas9-mediated Trp53 and Brca2 knockout to generate improved murine models of ovarian high-grade serous carcinoma. Cancer Res. 2016;76(20):6118-29.
- Zhao R, Kaakati R, Liu X, Xu L, Lee AK, Bachelder R, et al. CRISPR/Cas9Mediated BRCA1 Knockdown Adipose Stem Cells Promote Breast Cancer Progression. Plast Reconstr Surg. 2019;143(3):747-56.
- Summary R, Doudna JA, Charpentier E. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346(6213). https://doi. org/10.1126/science. 1258096.
- Katti A, Diaz BJ, Caragine CM, Sanjana NE, Dow LE. CRISPR in cancer biology and therapy. Nat Rev Cancer. 2022;22(5):259-79.
- Awwad SW, Serrano-Benitez A, Thomas JC, Gupta V, Jackson SP. Revolutionizing DNA repair research and cancer therapy with CRISPRCas screens. Nat Rev Mol Cell Biol. 2023. https://doi.org/10.1038/ s41580-022-00571-x.
- Prakash G, Shokr A, Willemen N, Bashir SM, Shin SR, Hassan S. Microfluidic fabrication of lipid nanoparticles for the delivery of nucleic acids. Advanced Drug Delivery Reviews. 2022;184. https://doi.org/10.1016/j. addr.2022.114197.
- Modell AE, Lim D, Nguyen TM, Sreekanth V, Choudhary A. CRISPR-based therapeutics: current challenges and future applications. Trends Pharmacol Sci. 2022;43(2):151-61.
- Yin H, Xue W, Anderson DG. CRISPR-Cas: a tool for cancer research and therapeutics. Nat Rev Clin Oncol. 2019;16(5):281-95.
- Lakshmanan VK, Jindal S, Packirisamy G, Ojha S, Lian S, Kaushik A, et al. Nanomedicine-based cancer immunotherapy: recent trends and future perspectives. Cancer Gene Ther. 2021. https://doi.org/10.1038/ s41417-021-00299-4.
- Miliotou AN, Papadopoulou LC. CAR T-cell Therapy: A New Era in Cancer Immunotherapy. Curr Pharm Biotechnol. 2018. https://doi.org/10.2174/ 1389201019666180418095526.
- Huang D, Miller M, Ashok B, Jain S, Peppas NA. CRISPR/Cas systems to overcome challenges in developing the next generation of T cells for cancer therapy. Adv Drug Deliv Rev. 2020;158:17-35.
- Azangou-Khyavy M, Ghasemi M, Khanali J, Boroomand-Saboor M, Jamalkhah M, Soleimani M, et al. CRISPR/Cas: From Tumor Gene Editing to T Cell-Based Immunotherapy of Cancer. Front Immunol. 2020. https://doi.org/10.3389/fimmu.2020.02062.
- Shachaf CM, Felsher DW. Rehabilitation of cancer through oncogene inactivation. Trends Mol Med. 2005;11(7):316-21.
- Kennedy EM, Kornepati AVR, Goldstein M, Bogerd HP, Poling BC, Whisnant AW, et al. Inactivation of the Human Papillomavirus E6 or E7 Gene in Cervical Carcinoma Cells by Using a Bacterial CRISPR/Cas RNAGuided Endonuclease. J Virol. 2014;88(20):11965-72.
- Riedel M, Cai H, Stoltze IC, Vendelbo MH, Wagner EF, Bakiri L, et al. Targeting AP-1 transcription factors by CRISPR in the prostate. Oncotarget. 2021;12(19):1956-61.
- Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, et al. Evolution and classification of the CRISPR-Cas systems. Nature Reviews Microbiology. 2011;9(6):467-77.
- Hoellerbauer P, Kufeld M, Paddison PJ. Efficient Multi-Allelic Genome Editing of Primary Cell Cultures via CRISPR-Cas9 Ribonucleoprotein Nucleofection. Curr Protoc Stem Cell Biol. 2020. https://doi.org/10.1002/ cpsc. 126.
- Kim J, Jang G, Sim SH, Park IH, Kim K, Park C. Smarca4 depletion induces cisplatin resistance by activating yap1-mediated epithelial-to-mesenchymal transition in triple-negative breast cancer. Cancers (Basel). 2021;13(21). https://doi.org/10.3390/cancers13215474.
- Zeng K, Chen X, Hu X, Liu X, Xu T, Sun H, et al. LACTB, a novel epigenetic silenced tumor suppressor, inhibits colorectal cancer progression by attenuating MDM2-mediated p53 ubiquitination and degradation. Oncogene. 2018;37(41):5534-51.
- Estêvão D, Rios Costa N, Da Costa RG, Medeiros R. CRISPR-Cas9 therapies in experimental mouse models of cancer. Future Oncol. 2018;14(20):2083-95.
- Alves E, Taifour S, Dolcetti R, Chee J, Nowak AK, Gaudieri S, et al. Reprogramming the anti-tumor immune response via CRISPR genetic and epigenetic editing. Mol Ther Methods Clin Dev. 2021;21:592-606.
- Takata K, Chong LC, Ennishi D, Thakur A, Healy S, Viganò E, et al. The Tumor Associated Antigen PRAME Exhibits Dualistic Functions That Are Targetable in Diffuse Large B-Cell Lymphoma. Blood. 2020;136(Supplement 1):34-34.
- Zhi L, Su X, Yin M, Zhang Z, Lu H, Niu Z, et al. Genetical engineering for NK and T cell immunotherapy with CRISPR/Cas9 technology: Implications and challenges. Cell Immunol. 2021;369. https://doi.org/10.1016/j. cellimm.2021.104436.
- Afolabi LO, Afolabi MO, Sani MM, Okunowo WO, Yan D, Chen L, et al. Exploiting the CRISPR-Cas9 gene-editing system for human cancers and immunotherapy. Clin Transl Immunol. 2021;10(6). https://doi.org/ 10.1002/cti2.1286.
- Biggi AFB, Simioni PU. Inhibition of PD-1 protein by the CRISPR-Cas9 method as antitumor therapy of non-small cell lung cancers. Rev da Fac Ciências Médicas Sorocaba. 2019;21(1):2-7.
- Zarogoulidis P, Lampaki S, Yarmus L, Kioumis I, Pitsiou G, Katsikogiannis N , et al. Interleukin-7 and interleukin-15 for cancer. J Cancer. 2014. https://doi.org/10.7150/jca. 10471.
- Zhang Z, Kong X, Ligtenberg MA, van Hal-van Veen SE, Visser NL, de Bruijn B, et al. RNF31 inhibition sensitizes tumors to bystander killing by innate and adaptive immune cells. Cell Rep Med. 2022;3(6). https://doi. org/10.1016/j.xcrm.2022.100655.
- Wang L, Chen Y, Liu X, Li Z, Dai X. The Application of CRISPR/Cas9 Technology for Cancer Immunotherapy: Current Status and Problems. Front Oncol. 2022;11. https://doi.org/10.3389/fonc.2021.704999.
- Zhou XM, Li WQ, Wu YH, Han L, Cao XG, Yang XM, et al. Intrinsic expression of immune checkpoint molecule TIGIT could help tumor growth in vivoby suppressing the function of NK and CD8+T Cells. Front Immunol. 2018;9(NOV). https://doi.org/10.3389/fimmu.2018.02821.
- Reis A, Hornblower B, Robb B, Tzertzinis G. CRISPR/Cas9 and targeted genome editing: a new era in molecular biology. NEB expressions. 2014;1:3-6.
- Chavez M, Chen X, Finn PB, Qi LS. Advances in CRISPR therapeutics. Nat Rev Nephrol. 2023;19(1):9-22.
- Chehelgerdi
, Chehelgerdi . The use of RNA – based treatments in the field of cancer immunotherapy. 2023. BioMed Central. https://doi. org/10.1186/s12943-023-01807-w. - Lino CA, Harper JC, Carney JP, Timlin JA. Delivering crispr: A review of the challenges and approaches. Drug Delivery. 2018;25(1):1234-57.
- Hatada I, Morita S, Horii T. CRISPR/Cas9. Methods Mol Biol. 2023;2637:41-7.
- Pawluk A, Amrani N, Zhang Y, Garcia B, Hidalgo-Reyes Y, Lee J, et al. Naturally Occurring Off-Switches for CRISPR-Cas9. Cell. 2016. https:// doi.org/10.1016/j.cell.2016.11.017.
- Emily P, John H, Mitchell K, Margaret C, Bendzick L, Miller JS, et al. Enhancing Human NK Cell Function and Specificity for Cancer Immunotherapy. Blood. 2018;132(Supplement 1):2044-2044.
- Shmakov S, Smargon A, Scott D, Cox D, Pyzocha N, Yan W, et al. Diversity and evolution of class 2 CRISPR-Cas systems. Nat Rev Microbiol. 2017. https://doi.org/10.1038/nrmicro.2016.184.
- Biagioni A, Laurenzana A, Margheri F, Chillà A, Fibbi G, So M. Delivery systems of CRISPR/Cas9-based cancer gene therapy. J Biol Eng. 2018;12(1). https://doi.org/10.1186/s13036-018-0127-2.
- Sung B, Kim MH, Abelmann L. Magnetic microgels and nanogels: Physical mechanisms and biomedical applications. Bioeng Transl Med. 2021;6(1). https://doi.org/10.1002/btm2.10190.
- Jiang F, Doudna JA. CRISPR-Cas9 Structures and Mechanisms. Annu Rev Biophys. 2017;46:505-29.
- Zhang H, Qin C, An C, Zheng X, Wen S, Chen W, et al. Application of the CRISPR/Cas9-based gene editing technique in basic research, diagnosis, and therapy of cancer. Mol Cancer. 2021;20:1-22.
- Suzuki Y, Onuma H, Sato R, Sato Y, Hashiba A, Maeki M, et al. Lipid nanoparticles loaded with ribonucleoprotein-oligonucleotide complexes synthesized using a microfluidic device exhibit robust genome editing and hepatitis B virus inhibition. J Control Release. 2021;330:61-71.
- Wilbie D, Walther J, Mastrobattista E. Delivery Aspects of CRISPR/Cas for in Vivo Genome Editing. Acc Chem Res. 2019;52(6):1555-64.
- Künne T, Westra ER, Brouns SJJ. Electrophoretic mobility shift assay of DNA and CRISPR- Cas ribonucleoprotein complexes. Methods Mol Biol. 2015;1311:171-84.
- Bykonya AG, Lavrov AV, Smirnikhina SA. Methods for CRISPR-Cas as Ribonucleoprotein Complex Delivery In Vivo. Mol Biotechnol. 2023;65(2):181-95.
- Zhou Y, Bravo JPK, Taylor HN, Steens JA, Jackson RN, Staals RHJ, et al. Structure of a type IV CRISPR-Cas ribonucleoprotein complex. IScience. 2021;24(3). https://doi.org/10.1016/j.isci.2021.102201.
- Gregg JR, Thompson TC. Considering the potential for gene-based therapy in prostate cancer. Nat Rev Urol. 2021;18(3):170-84.
- Pomeroy E, Hunzeker J, Kluesner M, Crosby M, Bendzick L, Geller M, et al. Genetically engineered natural killer cells for cancer immunotherapy. Mol Ther. 2018;26(5):355-6.
- Li R, Wang Q, She K, Lu F, Yang Y. CRISPR/Cas systems usher in a new era of disease treatment and diagnosis. Molecular Biomedicine. 2022;3(1). https://doi.org/10.1186/s43556-022-00095-y.
- Martinez-Lage M, Torres-Ruiz R, Puig-Serra P, Moreno-Gaona P, Martin MC, Moya FJ, et al. In vivo CRISPR/Cas9 targeting of fusion oncogenes for selective elimination of cancer cells. Nat Commun. 2020;11(1). https://doi.org/10.1038/s41467-020-18875-x.
- Gao J, Luo T, Lin N, Zhang S, Wang J. A New Tool for CRISPR-Cas13aBased Cancer Gene Therapy. Mol Ther – Oncolytics. 2020;19:79-92.
- Chen J, López-Moyado IF, Seo H, Lio CWJ, Hempleman LJ, Sekiya T, et al. NR4A transcription factors limit CART cell function in solid tumours. Nature. 2019;567(7749):530-4.
- Bridges K, Yao HHC, Nicol B. Loss of Runx1 Induces Granulosa Cell Defects and Development of Ovarian Tumors in the Mouse. Int J Mol Sci. 2022;23(22). https://doi.org/10.3390/ijms232214442.
- Jung S, Kim DH, Choi YJ, Kim SY, Park H, Lee H, et al. Contribution of p53 in sensitivity to EGFR tyrosine kinase inhibitors in non-small cell lung cancer. Sci Rep. 2021;11(1). https://doi.org/10.1038/ s41598-021-99267-z.
- Wang L, Pearson N, Xiong Y, Renuse S, Cheng R, Carter JM, et al. Abstract 3913: Quantitative phosphoproteomic analysis of AXL signaling network in breast cancer. Cancer Res. 2022;82(12_Supplement):3913-3913.
- Hu X, Li L, Eid JE, Liu C, Yu J, Yue J, et al. IDH1 Mutation Induces HIF-1 a and Confers Angiogenic Properties in Chondrosarcoma JJ012 Cells. Dis Markers. 2022;2022. https://doi.org/10.1155/2022/7729968.
- Hui Goh CJ, Wong JH, El Farran C, Tan BX, Coffill CR, Loh YH, et al. Identification of pathways modulating vemurafenib resistance in melanoma cells via a genome-wide CRISPR/Cas9 screen. G3 Genes Genomes Genet. 2021;11(2). https://doi.org/10.1093/g3journal/jkaa069.
- Thus YJ, de Rooij MFM, Swier N, Beijersbergen RL, Guikema JEJ, Kersten MJ, et al. Inhibition of casein kinase 2 sensitizes mantle cell lymphoma to venetoclax through MCL-1 downregulation. Haematologica. 2023;108(3):797-810.
- Wrobel P, Shojaei H, Schittek B, Gieseler F, Wollenberg B, Kalthoff H, et al. Lysis of a broad range of epithelial tumour cells by human
cells: Involvement of NKG2D ligands and T-cell receptor- versus NKG2Ddependent recognition. Scand J Immunol. 2007;66(2-3):320-8. - Li Z, Chi Z, Ang WX, Chen C, Tay JCK, Ng YY, et al. Experimental treatment of colorectal cancer in mice with human
cells electroporated with NKG2D RNA CAR. Immunotherapy. 2020;12(10):733-48. - Mo F, Duan S, Jiang X, Yang X, Hou X, Shi W, et al. Nanobody-based chimeric antigen receptor T cells designed by CRISPR/Cas9 technology
for solid tumor immunotherapy. Signal Transduct Target Ther. 2021;6(1). https://doi.org/10.1038/s41392-021-00462-1. - Kuznik NC, Solozobova V, Lee II, Jung N, Yang L, Nienhaus K, et al. A chemical probe for BAG1 targets androgen receptor-positive prostate cancer through oxidative stress signaling pathway. iScience. 2022;25(5). https://doi.org/10.1016/j.isci.2022.104175.
- Ungefroren H, Otterbein H, Fiedler C, Mihara K, Hollenberg MD, Gieseler F, et al. RAC1B suppresses TGF-
-dependent cell migration in pancreatic carcinoma cells through inhibition of the TGF- type i receptor ALK5. Cancers (Basel). 2019;11(5). https://doi.org/10.3390/cancers110 50691. - Tishchenko A, Azorín DD, Vidal-Brime L, Muñoz MJ, Arenas PJ, Pearce C, et al. Cx43 and associated cell signaling pathways regulate tunneling nanotubes in breast cancer cells. Cancers (Basel). 2020;12(10):1-25.
- Cerchietti L, Melnick A. Targeting BCL6 in diffuse large B-cell lymphoma: What does this mean for the future treatment? Expert Rev Hematol. 2013;6(4):343-5.
- Aquilanti E, Kageler L, Watson J, Baird DM, Jones RE, Hodges M, et al. Telomerase inhibition is an effective therapeutic strategy in TERT promoter-mutant glioblastoma models with low tumor volume. Neuro Oncol. 2023. https://doi.org/10.1093/neuonc/noad024.
- Menendez-Gonzalez JB, Vukovic M, Abdelfattah A, Saleh L, Almotiri A, Thomas L anne, et al. Gata2 as a Crucial Regulator of Stem Cells in Adult Hematopoiesis and Acute Myeloid Leukemia. Stem Cell Rep. 2019;13(2):291-306.
- Chang KH, Smith SE, Sullivan T, Chen K, Zhou Q, West JA, et al. LongTerm Engraftment and Fetal Globin Induction upon BCL11A Gene Editing in Bone-Marrow-Derived CD34+ Hematopoietic Stem and Progenitor Cells. Mol Ther Methods Clin Dev. 2017;4:137-48.
- Hu Y, Zhou Y, Zhang M, Ge W, Li Y, Yang L, et al. CRISPR/Cas9-engineered universal CD19/CD22 dual-targeted CAR-T cell therapy for relapsed/ refractory B-cell acute lymphoblastic leukemia. Clin Cancer Res. 2021;27(10):2764-72.
- Tong KI, Yoon S, Isaev K, Bakhtiari M, Lackraj T, He MY, et al. Combined EZH2 inhibition and IKAROS degradation leads to enhanced antitumor activity in diffuse large B-cell lymphoma. Clin Cancer Res. 2021;27(19):5401-14.
- Yang Z, Hu N, Wang W, Hu W, Zhou S, Shi J, et al. Loss of FBXW7 Correlates with Increased IDH1 Expression in Glioma and Enhances IDH1-Mutant Cancer Cell Sensitivity to Radiation. Cancer Res. 2022;82(3):497-509.
- Chen L, Alexe G, Dharia NV, Ross L, Iniguez AB, Conway AS, et al. CRISPRCas9 screen reveals a MYCN-amplified neuroblastoma dependency on EZH2. J Clin Invest. 2018;128(1):446-62.
- Wang
, Chen , X, Wang , Zhu , Wu X. MUC16 impacts tumor proliferation and migration through cytoplasmic translocation of P120catenin in epithelial ovarian cancer cells: An original research. BMC Cancer. 2019;19(1). https://doi.org/10.1186/s12885-019-5371-4. - Liu J, Guzman MA, Pezanowski D, Patel D, Hauptman J, Keisling M, et al. FOXO1-FGFR1 fusion and amplification in a solid variant of alveolar rhabdomyosarcoma. Mod Pathol. 2011;24(10):1327-35.
- Calderon-Aparicio A, Yamamoto H, de Vitto H, Zhang T, Wang Q, Bode AM, et al. RCC2 promotes esophageal cancer growth by regulating activity and expression of the Sox2 transcription factor. Mol Cancer Res. 2020;18(11):1660-74.
- Kotiyal S, Evason KJ. Exploring the interplay of telomerase reverse transcriptase and
-catenin in hepatocellular carcinoma. Cancers. 2021;13(16). https://doi.org/10.3390/cancers13164202. - Jung YS, Jun S, Lee SH, Sharma A, Park J II. Wnt2 complements Wnt/
-catenin signaling in colorectal cancer. Oncotarget. 2015;6(35):37257-37268. - Blackburn AC, Jerry DJ. Knockout and transgenic mice of Trp53: What have we learned about p53 in breast cancer? Breast Cancer Res. 2002;4(3):101-11.
- Muzumdar MD, Chen PY, Dorans KJ, Chung KM, Bhutkar A, Hong E, et al. Survival of pancreatic cancer cells lacking KRAS function. Nat Commun. 2017;8(1). https://doi.org/10.1038/s41467-017-00942-5.
- Xu Y, Jin Y, Liu L, Zhang X, Chen Y, Wei J. Study of circulating IgG antibodies to peptide antigens derived from BIRC5 and MYC in cervical cancer. FEBS Open Bio. 2015;5:198-201.
- Ponnusamy L, Natarajan SR, Manoharan R. MARK2 potentiate aerobic glycolysis-mediated cell growth in breast cancer through regulating mTOR/HIF-1 a and p53 pathways. J Cell Biochem. 2022;123(4):759-71.
- Rahman S, Magnussen M, León TE, Farah N, Li Z, Abraham BJ, et al. Activation of the LMO2 oncogene through a somatically acquired neomorphic promoter in T-cell acute lymphoblastic leukemia. Blood. 2017;129(24):3221-6.
- Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, et al. HIF-2a drives an intrinsic vulnerability to ferroptosis in clear cell renal cell carcinoma. BioRxiv. 2018;9:388041.
- Chen T, Tsang JYS, Su XC, Li P, Sun WQ, Wong ILK, et al. SALL4 promotes tumor progression in breast cancer by targeting EMT. Mol Carcinog. 2020;59(10):1209-26.
- Kollmann K, Briand C, Bellutti F, Schicher N, Blunder S, Zojer M, et al. The interplay of CDK4 and CDK6 in melanoma. Oncotarget. 2019;10(14):1346-59.
- Gomez Limia CE, Devalle S, Reis M, Sochacki J, Carneiro M, Madeiro da Costa R, et al. Generation and characterization of a human induced pluripotent stem (iPS) cell line derived from an acute myeloid leukemia patient evolving from primary myelofibrosis carrying the CALR 52 bp deletion and the ASXL1 p.R693X mutation. Stem Cell Res. 2017;24:16-20.
- Ihry RJ, Worringer KA, Salick MR, Frias E, Ho D, Theriault K, et al. P53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat Med. 2018;24(7):939-46.
- Bowden AR, Morales-Juarez DA, Sczaniecka-Clift M, Agudo MM, Lukashchuk N, Thomas JC, et al. Parallel crispr-cas9 screens clarify impacts of p53 on screen performance. Elife. 2020;9:1.
- Zhan H, Xie H, Zhou Q, Liu Y, Huang W. Synthesizing a Genetic Sensor Based on CRISPR-Cas9 for Specifically Killing p53-Deficient Cancer Cells. ACS Synth Biol. 2018;7(7):1798-807.
- Tarangelo A, Magtanong L, Bieging-Rolett KT, Li Y, Ye J, Attardi LD, et al. p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep. 2018;22(3):569-75.
- Qiao C, Liu W, Jiang H, He M, Yang Q, Xing Y. Integrated analysis of miRNA and mRNA expression profiles in p53-edited PFF cells. Cell Cycle. 2020;19(8):949-59.
- D’Costa S, Rich MJ, Diekman BO. Engineered cartilage from human chondrocytes with homozygous knockout of cell cycle inhibitor p21. Tissue Eng Part A. 2020;26(7-8):441-9.
- Nicolae CM, O’connor MJ, Constantin D, Moldovan GL. NFkB regulates p21 expression and controls DNA damage-induced leukemic differentiation. Oncogene. 2018;37(27):3647-56.
- Fischietti M, Eckerdt F, Perez RE, Guillen Magaña JN, Mazewski C, Ho S, et al. SLFN11 Negatively Regulates Noncanonical NFkB Signaling to Promote Glioblastoma Progression. Cancer Res Commun. 2022;2(9):966-78.
- Hartmann O, Reissland M, Maier CR, Fischer T, Prieto-Garcia C, Baluapuri A, et al. Implementation of CRISPR/Cas9 Genome Editing to Generate Murine Lung Cancer Models That Depict the Mutational Landscape of Human Disease. Front Cell Dev Biol. 2021;9. https://doi.org/10.3389/ fcell.2021.641618.
- Walsh T, Casadei S, Munson KM, Eng M, Mandell JB, Gulsuner S, et al. CRISPR-Cas9/long-read sequencing approach to identify cryptic mutations in BRCA1 and other tumour suppressor genes. J Med Genet. 2021;58(12):850-2.
- Mintz RL, Lao YH, Chi CW, He S, Li M, Quek CH, et al. CRISPR/Cas9-mediated mutagenesis to validate the synergy between PARP1 inhibition and chemotherapy in BRCA1-mutated breast cancer cells. Bioeng Transl Med. 2020;5(1). https://doi.org/10.1002/btm2.10152.
- Walton JB, Farquharson M, Mason S, Port J, Kruspig B, Dowson S, et al. CRISPR/Cas9-derived models of ovarian high grade serous carcinoma targeting Brca1, Pten and Nf1, and correlation with platinum sensitivity. Sci Rep. 2017;7(1). https://doi.org/10.1038/s41598-017-17119-1.
- Tsujino T, Takai T, Hinohara K, Gui F, Tsutsumi T, Bai X, et al. CRISPR screens reveal genetic determinants of PARP inhibitor sensitivity and resistance in prostate cancer. Nat Commun. 2023;14(1). https://doi.org/ 10.1038/s41467-023-35880-y.
- Witz A, Dardare J, Husson M, Francois A, Merlin J-L, Gilson P, et al. 35P Increased sensitivity to olaparib by BRCA1/2 knockdown using a
108. Annunziato S, de Ruiter JR, Henneman L, Brambillasca CS, Lutz C, Vaillant F, et al. Comparative oncogenomics identifies combinations of driver genes and drug targets in BRCA1-mutated breast cancer. Nat Commun. 2019;10(1). https://doi.org/10.1038/s41467-019-08301-2.
109. Weber J, Öllinger R, Friedrich M, Ehmer U, Barenboim M, Steiger K, et al. CRISPR/Cas9 somatic multiplex-mutagenesis for high-Throughput functional cancer genomics in mice. Proc Natl Acad Sci U S A. 2015;112(45):13982-7.
110. Yoshimatsu S, Nakajima M, Qian E, Sanosaka T, Sato T, Okano H. Homologous Recombination-Enhancing Factors Identified by Comparative Transcriptomic Analyses of Pluripotent Stem Cell of Human and Common Marmoset. Cells. 2022;11 (3). https://doi.org/10.3390/cells 11030360.
111. Choudhury SR, Cui Y, Lubecka K, Stefanska B, Irudayaraj J. CRISPR-dCas9 mediated TET1 targeting for selective DNA demethylation at BRCA1 promoter. Oncotarget. 2016;7(29):46545-56.
112. Kweon J, Jang AH, Shin HR, See JE, Lee W, Lee JW, et al. A CRISPR-based base-editing screen for the functional assessment of BRCA1 variants. Oncogene. 2020;39(1):30-5.
113. Lee W, Lee JH, Jun S, Lee JH, Bang D. Selective targeting of KRAS oncogenic alleles by CRISPR/Cas9 inhibits proliferation of cancer cells. Sci Rep. 2018;8(1). https://doi.org/10.1038/s41598-018-30205-2.
114. Bender G, Fahrioglu Yamaci R, Taneri B. CRISPR and KRAS: a match yet to be made. J Biomed Sci. 2021;28(1). https://doi.org/10.1186/ s12929-021-00772-0.
115. Zhao X, Liu L, Lang J, Cheng K, Wang Y, Li X, et al. A CRISPR-Cas13a system for efficient and specific therapeutic targeting of mutant KRAS for pancreatic cancer treatment. Cancer Lett. 2018;431:171-81.
116. Burgess MR, Hwang E, Mroue R, Bielski CM, Wandler AM, Huang BJ, et al. KRAS Allelic Imbalance Enhances Fitness and Modulates MAP Kinase Dependence in Cancer. Cell. 2017;168(5):817-829.e15.
117. Wang B, Krall EB, Aguirre AJ, Kim M, Widlund HR, Doshi MB, et al. ATXN1L, CIC, and ETS Transcription Factors Modulate Sensitivity to MAPK Pathway Inhibition. Cell Rep. 2017;18(6):1543-57.
118. Sayed S, Sidorova OA, Hennig A, Augsburg M, Cortés Vesga CP, Abohawya M, et al. Efficient Correction of Oncogenic KRAS and TP53 Mutations through CRISPR Base Editing. Cancer Res. 2022;82(17):3002-15.
119. Lentsch E, Li L, Pfeffer S, Ekici AB, Taher L, Pilarsky C, et al. CRISPR/Cas9mediated knock-out of krasG12D mutated pancreatic cancer cell lines. Int J Mol Sci. 2019;20(22). https://doi.org/10.3390/ijms20225706.
120. Wang Z, Kang B, Gao Q, Huang L, Di J, Fan Y, et al. Quadrupleediting of the MAPK and PI3K pathways effectively blocks the progression of KRAS-mutated colorectal cancer cells. Cancer Sci. 2021;112(9):3895-910.
121. Yamada T, Amann JM, Tanimoto A, Taniguchi H, Shukuya T, Timmers C, et al. Histone deacetylase inhibition enhances the antitumor activity of a MEK inhibitor in lung cancer cells harboring RAS mutations. Mol Cancer Ther. 2018;17(1):17-25.
122. Thompson KN, Whipple RA, Yoon JR, Lipsky M, Charpentier MS, Boggs AE, et al. The combinatorial activation of the PI3K and Ras/ MAPK pathways is sufficient for aggressive tumor formation, while individual pathway activation supports cell persistence. Oncotarget. 2015;6(34):35231-46.
123. Maitra R, Seetharam R, Tesfa L, Augustine TA, Klampfer L, Coffey MC, et al. Oncolytic reovirus preferentially induces apoptosis in KRAS mutant colorectal cancer cells, and synergizes with irinotecan. Oncotarget. 2014;5(9):2807-19.
124. Yang YP, Ma H, Starchenko A, Huh WJ, Li W, Hickman FE, et al. A Chimeric Egfr Protein Reporter Mouse Reveals Egfr Localization and Trafficking In Vivo. Cell Rep. 2017;19(6):1257-67.
125. Koo T, Yoon AR, Cho HY, Bae S, Yun CO, Kim JS. Selective disruption of an oncogenic mutant allele by CRISPR/Cas9 induces efficient tumor regression. Nucleic Acids Res. 2017;45(13):7897-908.
126. Kiessling MK, Schuierer S, Stertz S, Beibel M, Bergling S, Knehr J, et al. Identification of oncogenic driver mutations by genome-wide CRISPRCas9 dropout screening. BMC Genomics. 2016;17(1). https://doi.org/10. 1186/s12864-016-3042-2.
127. Park JJ, Kim JE, Jeon Y, Lee MR, Choi JY, Song BR, et al. Deletion of NKX3.1 via CRISPR/Cas9 Induces Prostatic Intraepithelial Neoplasia in
128. Zhang BC, Luo BY, Zou JJ, Wu PY, Jiang JL, Le JQ, et al. Co-delivery of Sorafenib and CRISPR/Cas9 Based on Targeted Core-Shell Hollow Mesoporous Organosilica Nanoparticles for Synergistic HCC Therapy. ACS Appl Mater Interfaces. 2020;12(51):57362-72.
129. Chung SH, Frick SL, Yiu G. Targeting vascular endothelial growth factor using retinal gene therapy. Ann Transl Med. 2021;9(15):1277-1277.
130. Ameri H, Murat C, Arbabi A, Jiang W, Janga SR, Qin PZ, et al. Reduced expression of vegf-a in human retinal pigment epithelial cells and human muller cells following crispr-cas9 ribonucleoprotein-mediated gene disruption. Transl Vis Sci Technol. 2020;9(8). https://doi.org/10. 1167/TVST.9.8.23.
131. Hariprabu KNG, Sathya M, Vimalraj S. CRISPR/Cas9 in cancer therapy: A review with a special focus on tumor angiogenesis. Int J Biol Macromol. 2021;192:913-930.
132. Li XL, Li GH, Fu J, Fu YW, Zhang L, Chen W, et al. Highly efficient genome editing via CRISPR-Cas9 in human pluripotent stem cells is achieved by transient BCL-XL overexpression. Nucleic Acids Res. 2018;46(19):10195-215.
133. Abdul Rahman SF, Mohana-Kumaran N, Mohd Azzam MG, Mohd Norudin NA, Muniandy K. CRISPR/Cas9 Mediated BFL-1 Knock-out in Nasopharyngeal Carcinoma (NPC) Cell Lines. Front Pharmacol. 2018;9. https://doi.org/10.3389/conf.fphar.2018.63.00029.
134. Zhao W, Liu J, Wang D, Wang Y, Zhang F, Jin G, et al. Effect of silencing HIF-1 a gene on testicle spermatogenesis function in varicocele rats. Cell Tissue Res. 2019;378(3):543-54.
135. Fang N, Gu T, Wang Y, Wang S, Wang F, An Y, et al. Expression of PTENlong mediated by CRISPR/Cas9 can repress U87 cell proliferation. J Cell Mol Med. 2017;21(12):3337-46.
136. de Almeida Monteiro Melo Ferraz M, Nagashima JB, Venzac B, Le Gac S, Songsasen N. A dog oviduct-on-a-chip model of serous tubal intraepithelial carcinoma. Sci Rep. 2020;10(1). https://doi.org/10.1038/ s41598-020-58507-4.
137. Callif BL, Maunze B, Krueger NL, Simpson MT, Blackmore MG. The application of CRISPR technology to high content screening in primary neurons. Mol Cell Neurosci. 2017;80:170-9.
138. Heitink L, Whittle JR, Vaillant F, Capaldo BD, Dekkers JF, Dawson CA, et al. In vivo genome-editing screen identifies tumor suppressor genes that cooperate with Trp53 loss during mammary tumorigenesis. Mol Oncol. 2022;16(5):1119-31.
139. Xi L, Schmidt JC, Zaug AJ, Ascarrunz DR, Cech TR. A novel two-step genome editing strategy with CRISPR-Cas9 provides new insights into telomerase action and TERT gene expression. Genome Biol. 2015;16(1). https://doi.org/10.1186/s13059-015-0791-1.
140. James CD, Prabhakar AT, Otoa R, Evans MR, Wang X, Bristol ML, et al. SAMHD1 Regulates Human Papillomavirus 16-Induced Cell Proliferation and Viral Replication during Differentiation of Keratinocytes. mSphere. 2019;4(4). https://doi.org/10.1128/msphere.00448-19.
141. Zagorski JW, Maser TP, Liby KT, Rockwell CE. Nrf2-Dependent and -Independent Effects of tert-Butylhydroquinone, CDDO-Im, and H2O2 in Human Jurkat T Cells as Determined by CRISPR/Cas9 Gene Editing. J Pharmacol Exp Ther. 2017;361(2):259-67.
142. Cordeiro A, Deveau AP, Dhanraj S, Dror Y, Berman J. Characterizing Dyskeratosis Congenita Caused By Parn Mutations in the Zebrafish. Blood. 2019;134(Supplement_1):3744-3744.
143. Liu N, Xu S, Yao Q, Zhu Q, Kai Y, Hsu JY, et al. Transcription factor competition at the
144. Rui L. Abstract 3436: PRMT5 upregulation and its oncogenic cooperation with PI3K/AKT signaling in diffuse large B cell lymphoma. Cancer Res. 2019;79(13_Supplement):3436-3436.
145. Bonato A, Bomben R, Chakraborty S, Felician G, Martines C, Zucchetto A, et al. Chronic Lymphocytic Leukemia Cells with Mutated Nfkbie Are Positively Selected By Microenvironmental Signals and Display Reduced Sensitivity to Ibrutinib Treatment. Blood. 2021;138(Supplement 1):248-248.
146. Tagde A, Markert T, Rajabi H, Hiraki M, Alam M, Bouillez A, et al. Targeting MUC1-C suppresses polycomb repressive complex 1 in multiple myeloma. Oncotarget. 2017;8(41):69237-49.
147. Navarro-Guerrero E, Tay C, Whalley JP, Cowley SA, Davies B, Knight JC, et al. Genome-wide CRISPR/Cas9-knockout in human induced Pluripotent Stem Cell (iPSC)-derived macrophages. Sci Rep. 2021;11(1). https:// doi.org/10.1038/s41598-021-82137-z.
148. Hamada T, Akahane T, Yokoyama S, Higa N, Kirishima M, Matsuo K, et al. An oncogenic splice variant of PDGFRa in adult glioblastoma as a therapeutic target for selective CDK4/6 inhibitors. Sci Rep. 2022;12(1). https://doi.org/10.1038/s41598-022-05391-9.
149. Iacono D, Cinausero M, Gerratana L, Vitale MG, Basile D, Angione V, et al. Primary cutaneous melanoma in elderly patients: potential prognostic markers. Pigment Cell Melanoma Res. 2017;30(1):76-156.
150. Farrell J, Pietruska J, McRee S, Tsichlis P, Hinds P. Abstract PR14: Defining isoform-specific roles for AKTs in BRAFV600E-driven melanoma. Cancer Res. 2020;80(19_Supplement):PR14-PR14.
151. Kaulich M, Dowdy S. Abstract A10: A Cdk4-dependent phosphorylation threshold regulates the cell cycle entry decision. Mol Cancer Res. 2016;14(11_Supplement):A10-A10.
152. Frangoul H, Altshuler D, Cappellini MD, Chen Y-S, Domm J, Eustace BK, et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and
153. PD-1 Knockout Engineered T Cells for Advanced Esophageal Cancer – ClinicalTrials.gov. https://classic.clinicaltrials.gov/ct2/show/NCT03 081715. Accessed 22 Jul 2023.
154. PD-1 Knockout EBV-CTLs for Advanced Stage Epstein-Barr Virus (EBV) Associated Malignancies – ClinicalTrials.gov. https://classic.clinicaltrials. gov/ct2/show/NCT03044743. Accessed 22 Jul 2023.
155. TACE Combined With PD-1 Knockout Engineered T Cell in Advanced Hepatocellular Carcinoma – ClinicalTrials.gov. 2020. http://www.clini caltrials.gov/ct2/show/NCT04417764.
156. Study of CRISPR-Cas9 Mediated PD-1 and TCR Gene-knocked Out Mesothelin-directed CAR-T Cells in Patients With Mesothelin Positive Multiple Solid Tumors- ClinicalTrials.gov. 2020. http://www.clinicaltrials. gov/ct2/show/NCT03545815.
157. CRISPR (HPK1) Edited CD19-specific CAR-T Cells (XYF19 CAR-T Cells) for CD19+ Leukemia or Lymphoma- ClinicalTrials.gov. 2019. https://classic. clinicaltrials.gov/ct2/show/NCT04037566.
158. Safety and Efficacy of CT125A Cells for Treatment of Relapsed/Refractory CD5 + Hematopoietic Malignancies- ClinicalTrials.gov. 2021. http:// www.clinicaltrials.gov/ct2/show/NCT04767308.
159. A Safety and Efficacy Study Evaluating CTX130 in Subjects With Relapsed or Refractory T or B Cell Malignancies (COBALT-LYM). 2021. http://www.clinicaltrials.gov/ct2/show/NCT04502446.
160. Ishihara M, Nishida Y, Kitano S, Kawai A, Muraoka D, Momose F, et al. A phase 1 trial of NY-ESO-1-specific TCR-engineered T-cell therapy combined with a lymph node-targeting nanoparticulate peptide vaccine for the treatment of advanced soft tissue sarcoma. Int J Cancer. 2023;152(12):2554-66.
161. Kanwal B. Relapsed/Refractory Non-Hodgkin Lymphoma: Engineering T-Cells to Express Chimeric Antigen Receptors (CARs), a Salvage? Cureus. 2021. https://doi.org/10.7759/cureus.16307.
162. Fang C, Xiao G, Wang T, Song L, Peng B, Xu B, et al. Emerging Nano-/ Biotechnology Drives Oncolytic Virus-Activated and Combined Cancer Immunotherapy. Research. 2023;6. https://doi.org/10.34133/research. 0108.
163. Hölting TLB, Cidre-Aranaz F, Matzek D, Popper B, Jacobi SJ, Funk CM, et al. Neomorphic DNA-binding enables tumor-specific therapeutic gene expression in fusion-addicted childhood sarcoma. Mol Cancer. 2022;21(1). https://doi.org/10.1186/s12943-022-01641-6.
164. Malviya M, Aretz ZEH, Molvi Z, Lee J, Pierre S, Wallisch P, et al. Challenges and solutions for therapeutic TCR-based agents. Immunol Rev. 2023. https://doi.org/10.1111/imr.13233.
165. Chira S, Nutu A, Isacescu E, Bica C, Pop L, Ciocan C, et al. Genome Editing Approaches with CRISPR/Cas9 for Cancer Treatment: Critical Appraisal of Preclinical and Clinical Utility, Challenges, and Future Research. Cells. 2022;11(18). https://doi.org/10.3390/cells11182781.
166. Xu J, Chen LJ, Yang SS, Sun Y, Wu W, Liu YF, et al. Exploratory trial of a biepitopic CAR T-targeting B cell maturation antigen in relapsed/refractory multiple myeloma. Proc Natl Acad Sci U S A. 2019. https://doi.org/ 10.1073/pnas. 1819745116.
167. Feldman L, Brown C, Badie B. Chimeric Antigen Receptor T-Cell Therapy: Updates in Glioblastoma Treatment. Neurosurgery. 2021;88(6):1056-64.
168. Lin Y, Ying H, Shao L, Liu Q, Song M, Chen S. Safety and Efficacy of CRISPR/CAS9-Edited PD-1 Deficient CAR-T Cells in MUC1 Positive Advanced Esophageal Cancer. SSRN Electron J. 2023. https://doi.org/10. 2139/ssrn. 4316844.
169. Choi SYC, Ribeiro CF, Wang Y, Loda M, Plymate SR, Uo T. Druggable Metabolic Vulnerabilities Are Exposed and Masked during Progression to Castration Resistant Prostate Cancer. Biomolecules. 2022;12(11). https://doi.org/10.3390/biom12111590.
170. Acharya UH, Walter RB. Chimeric antigen receptor (Car)-modified immune effector cell therapy for acute myeloid leukemia (aml). Cancers. 2020;12(12):1-28.
171. Zhen S, Li X. Oncogenic Human Papillomavirus: Application of CRISPR/ Cas9 Therapeutic Strategies for Cervical Cancer. Cell Physiol Biochem. 2018;44(6):2455-66.
172. Uras IZ, Moll HP, Casanova E. Targeting KRAS mutant non-small-cell lung cancer: Past, present and future. Int J Mol Sci. 2020;21(12):1-30.
173. Trigg RM, Turner SD. ALK in neuroblastoma: Biological and therapeutic implications. Cancers. 2018;10(4). https://doi.org/10.3390/cancers100 40113.
174. Anagnostou T, Riaz IB, Hashmi SK, Murad MH, Kenderian SS. AntiCD19 chimeric antigen receptor T-cell therapy in acute lymphocytic leukaemia: a systematic review and meta-analysis. Lancet Haematol. 2020;7(11):e816-26.
175. Andrea AE, Chiron A, Mallah S, Bessoles S, Sarrabayrouse G, Hacein-Bey-Abina S. Advances in CAR-T Cell Genetic Engineering Strategies to Overcome Hurdles in Solid Tumors Treatment. Front Immunol. 2022;13. https://doi.org/10.3389/fimmu.2022.830292.
176. Li M, Sun J, Shi G. Application of CRISPR screen in mechanistic studies of tumor development, tumor drug resistance, and tumor immunotherapy. Front Cell Dev Biol. 2023;11. https://doi.org/10.3389/fcell.2023. 1220376.
177. Crispo F, Pietrafesa M, Condelli V, Maddalena F, Bruno G, Piscazzi A, et al. IDH1 Targeting as a New Potential Option for Intrahepatic Cholangiocarcinoma Treatment-Current State and Future Perspectives. Molecules. 2020;25(16). https://doi.org/10.3390/molecules25163754.
178. Mirgayazova R, Khadiullina R, Chasov V, Mingaleeva R, Miftakhova R, Rizvanov A, et al. Therapeutic editing of the TP53 gene: Is crispr/CAS9 an option? Genes. 2020;11(6):1-17.
179. Gagelmann N, Riecken K, Wolschke C, Berger C, Ayuk FA, Fehse B, et al. Development of CAR-T cell therapies for multiple myeloma. Leukemia. 2020;34(9):2317-32.
180. Wang Z, Li N, Feng K, Chen M, Zhang Y, Liu Y, et al. Phase I study of CAR-T cells with PD-1 and TCR disruption in mesothelin-positive solid tumors. Cell Mol Immunol. 2021;18(9):2188-98.
181. Bamdad CC, Yuan Y, Specht JM, Stewart AK, Smagghe BJ, Lin SC-M, et al. Phase I/II first-in-human CAR T-targeting MUC1 transmembrane cleavage product (MUC1*) in patients with metastatic breast cancer. J Clin Oncol. 2022;40(16_suppl):TPS1130-TPS1130.
182. Pennesi E, Michels N, Brivio E, van der Velden VHJ, Jiang Y, Thano A, et al. Inotuzumab ozogamicin as single agent in pediatric patients with relapsed and refractory acute lymphoblastic leukemia: results from a phase II trial. Leukemia. 2022;36(6):1516-24.
183. Swain SM, Shastry M, Hamilton E. Targeting HER2-positive breast cancer: advances and future directions. Nat Rev Drug Discovery. 2023;22(2):101-26.
184. Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex genome editing to generate universal CART cells resistant to PD1 inhibition. Clin Cancer Res. 2017;23(9):2255-66.
185. Liu J, Zhang Y, Guo R, Zhao Y, Sun R, Guo S, et al. Targeted CD7 CAR T-cells for treatment of T-Lymphocyte leukemia and lymphoma and acute myeloid leukemia: recent advances. Front Immunol. 2023;14. https://doi.org/10.3389/fimmu.2023.1170968.
186. Furqan F, Shah NN. Multispecific CART Cells Deprive Lymphomas of Escape via Antigen Loss. Annu Rev Med. 2023;74:279-91.
187. Li Q, Ni Y, Zhang L, Jiang R, Xu J, Yang H, et al. HIF-1 a-induced expression of m6A reader YTHDF1 drives hypoxia-induced autophagy and malignancy of hepatocellular carcinoma by promoting ATG2A and ATG14 translation. Signal Transduct Target Ther. 2021;6(1). https://doi. org/10.1038/s41392-020-00453-8.
188. Kumar R, Yu F, Zhen YH, Li B, Wang J, Yang Y, et al. PD-1 blockade restores impaired function of ex vivo expanded CD8+T cells and
enhances apoptosis in mismatch repair deficient EpCAM+PD-L1+ cancer cells. Onco Targets Ther. 2017;10:3453-65.
189. Haso W, Qin H, Zhang L, Orentas RJ, Fry TJ. CD22-Targeted Chimeric Antigen Receptor (CAR) T Cells Containing The 4-1BB Costimulatory Domain Demonstrate Enhanced Persistence and Superior Efficacy Against B-Cell Precursor Acute Lymphoblastic Leukemia (ALL) Compared To Those Containing CD28. Blood. 2013;122(21):1431-1431.
190. Dimitri A, Herbst F, Fraietta JA. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Molecular Cancer. 2022;21(1). https://doi.org/10.1186/s12943-022-01559-z.
191. Chen X, Kozhaya L, Tastan C, Placek L, Dogan M, Horne M, et al. Functional Interrogation of Primary Human T Cells via CRISPR Genetic Editing. J Immunol. 2018;201(5):1586-98.
192. Hedrich V, Breitenecker K, Ortmayr G, Pupp F, Huber H, Chen D, et al. PRAME Is a Novel Target of Tumor-Intrinsic Gas6/AxI Activation and Promotes Cancer Cell Invasion in Hepatocellular Carcinoma. Cancers (Basel). 2023;15(9). https://doi.org/10.3390/cancers15092415.
193. Kaligotla VSA, Jasti T, Kandra P. CRISPR/Cas9 in cancer: An attempt to the present trends and future prospects. Biotechnol Appl Biochem. 2022;69(3):1238-51.
194. Mehravar M, Roshandel E, Salimi M, Chegeni R, Gholizadeh M, Mohammadi MH, et al. Utilization of CRISPR/Cas9 gene editing in cellular therapies for lymphoid malignancies. Immunol Lett. 2020;226:71-82.
195. Zhang S, Zhang F, Chen Q, Wan C, Xiong J, Xu J. CRISPR/Cas9-mediated knockout of NSD1 suppresses the hepatocellular carcinoma development via the NSD1/H3/Wnt10b signaling pathway. J Exp Clin Cancer Res. 2019;38(1). https://doi.org/10.1186/s13046-019-1462-y.
196. Sürün D, von Melchner H, Schnütgen F. CRISPR/Cas9 genome engineering in hematopoietic cells. Drug Discov Today Technol. 2018;28:33-9.
197. Koh S, Kah J, Tham CYL, Yang N, Ceccarello E, Chia A, et al. Nonlytic Lymphocytes Engineered to Express Virus-Specific T-Cell Receptors Limit HBV Infection by Activating APOBEC3. Gastroenterology. 2018;155(1):180-193.e6.
198. Adigbli G, Ménoret S, Cross AR, Hester J, Issa F, Anegon I. Humanization of Immunodeficient Animals for the Modeling of Transplantation, Graft Versus Host Disease, and Regenerative Medicine. Transplantation. 2020;104(11):2290-306.
199. Stewart JR, Baxter J, Zatreanu D, Brough R, Song F, Konde A, et al. 3P Identification of novel biomarkers of response to ATR inhibitors in ARID1A mutant ovarian clear cell carcinoma. Ann Oncol. 2022;33:S383-4.
200. Kamali E, Rahbarizadeh F, Hojati Z, Frödin M. CRISPR/Cas9-mediated knockout of clinically relevant alloantigenes in human primary T cells. BMC Biotechnol. 2021;21(1). https://doi.org/10.1186/ s12896-020-00665-4.
201. Khan A, Zhang X. Function of the Long Noncoding RNAs in HepatocelIular Carcinoma: Classification, Molecular Mechanisms, and Significant Therapeutic Potentials. Bioengineering. 2022;9(8). https://doi.org/10. 3390/bioengineering9080406.
202. Kath J, Du W, Pruene A, Braun T, Thommandru B, Turk R, et al. Pharmacological interventions enhance virus-free generation of TRAC-replaced CART cells. Mol Ther – Methods Clin Dev. 2022;25:311-30.
203. Gao Q, Dong X, Xu Q, Zhu L, Wang F, Hou Y, et al. Therapeutic potential of CRISPR/Cas9 gene editing in engineered T-cell therapy. Cancer Med. 2019;8(9):4254-64.
204. Aghamiri S, Talaei S, Ghavidel AA, Zandsalimi F, Masoumi S, Hafshejani NH, et al. Nanoparticles-mediated CRISPR/Cas9 delivery: Recent advances in cancer treatment. J Drug Deliv Sci Technol. 2020;56. https://doi.org/10.1016/j.jddst.2020.101533.
205. Tong S, Moyo B, Lee CM, Leong K, Bao G. Engineered materials for in vivo delivery of genome-editing machinery. Nat Rev Mater. 2019;4(11):726-37.
206. Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNAGuided Human Genome Engineering via Cas9_Sup. Science. 2013;339(February):823-6.
207. Brouns SJJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJH, Snijders APL, et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321(5891):960-964.
208. Yin H, Kauffman KJ, Anderson DG. Delivery technologies for genome editing. Nat Rev Drug Discovery. 2017;16(6):387-99.
209. Chow MYT, Chang RYK, Chan HK. Inhalation delivery technology for genome-editing of respiratory diseases. Adv Drug Deliv Rev. 2021;168:217-28.
210. Sandhya D, Jogam P, Allini VR, Abbagani S, Alok A. The present and potential future methods for delivering CRISPR/Cas9 components in plants. J Genet Eng Biotechnol. 2020;18(1). https://doi.org/10.1186/ s43141-020-00036-8.
211. Taha EA, Lee J, Hotta A. Delivery of CRISPR-Cas tools for in vivo genome editing therapy: Trends and challenges. J Control Release. 2022;342:345-61.
212. Liu P, Liang SQ, Zheng C, Mintzer E, Zhao YG, Ponnienselvan K, et al. Improved prime editors enable pathogenic allele correction and cancer modelling in adult mice. Nat Commun. 2021;12(1). https://doi.org/10. 1038/s41467-021-22295-w.
213. Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, et al. CRISPRCas9 knockin mice for genome editing and cancer modeling. Cell. 2014;159(2):440-55.
214. Han HA, Pang JK, Soh BS. Mitigating off-target effects in CRISPR/Cas9mediated in vivo gene editing. J Mol Med. 2020;98(5):615-32.
215. Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, et al. Optimized sgRNA design to maximize activity and minimize offtarget effects of CRISPR-Cas9. Nat Biotechnol. 2016;34(2):184-91.
216. Burgess DJ. Characterizing CRISPR off-target effects. Nat Rev Genet. 2014;15(1):5-5.
217. Shen CC, Hsu MN, Chang CW, Lin MW, Hwu JR, Tu Y, et al. Synthetic switch to minimize CRISPR off-target effects by self-restricting Cas9 transcription and translation. Nucleic Acids Res. 2019;47(3). https://doi. org/10.1093/nar/gky1165.
218. Zhang S, Shen J, Li D, Cheng Y. Strategies in the delivery of Cas9 ribonucleoprotein for CRISPR/Cas9 genome editing. Theranostics. 2020. https://doi.org/10.7150/thno.47007.
219. Zhang D, Zhang Z, Unver T, Zhang B. CRISPR/Cas: A powerful tool for gene function study and crop improvement. J Adv Res. 2021;29:207-21.
220. Yin Y, Wang Q, Xiao L, Wang F, Song Z, Zhou C, et al. Advances in the engineering of the gene editing enzymes and the genomes: Understanding and handling the off-target effects of CRISPR/Cas9. J Biomed Nanotechnol. 2018. https://doi.org/10.1166/jbn.2018.2537.
221. Zhang X-H, Tee LY, Wang X-G, Huang Q-S, Yang S-H. Off-target Effects in CRISPR/Cas9-mediated Genome Engineering. Mol Ther Nucleic Acids. 2015;4: e264.
222. Li A, Lee CM, Hurley AE, Jarrett KE, De Giorgi M, Lu W, et al. A Self-Deleting AAV-CRISPR System for In Vivo Genome Editing. Mol Ther Methods Clin Dev. 2019. https://doi.org/10.1016/j.omtm.2018.11.009.
223. Xiang X, Corsi GI, Anthon C, Qu K, Pan X, Liang X, et al. Enhancing CRISPR-Cas9 gRNA efficiency prediction by data integration and deep learning. Nat Commun. 2021;12(1). https://doi.org/10.1038/ s41467-021-23576-0.
224. Kleinstiver BP, Prew MS, Tsai SQ, Topkar VV, Nguyen NT, Zheng Z, et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015. https://doi.org/10.1038/nature14592.
225. Shen B, Zhang W, Zhang J, Zhou J, Wang J, Chen L, et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat Methods. 2014. https://doi.org/10.1038/nmeth.2857.
226. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013. https:// doi.org/10.1038/nprot.2013.143.
227. Xie K, Yang Y. RNA-Guided Genome Editing in Plants Using a CRISPRCas System. Mol Plant. 2013;6(6):1975-83.
228. Izumi H, Wang Z, Goto Y, Ando T, Wu X, Zhang X, et al. Pathway-specific genome editing of PI3K/mTOR tumor suppressor genes reveals that PTEN loss contributes to cetuximab resistance in head and neck cancer. Mol Cancer Ther. 2020;19(7):1562-71.
229. Engstrom LD, Waters L, Gatto S, Fernandez-Banet J, Aranda R, Pavlicek A, et al. Abstract 5684: Drug-anchored in vitro and in vivo CRISPR screens to identify targetable vulnerabilities and modifiers of response to MRTX849 in KRASG12C-mutant models . Cancer Res. 2020;80(16_Supplement):5684-5684.
230. Li Y, Deutzmann A, Bell J, Ji H, Felsher D. Abstract PR02: Synthetic lethality screen identifies novel druggable targets in the MYC pathway. Mol Cancer Ther. 2017;16(10_Supplement):PR02-PR02.
231. Kolasa IK, Rembiszewska A, Janiec-Jankowska A, Dansonka-Mieszkowska A, Lewandowska AM, Konopka B, et al. PTEN mutation, expression and LOH at its locus in ovarian carcinomas. Relation to TP53, K-RAS and BRCA1 mutations. Gynecol Oncol. 2006;103(2):692-697.
232. Alenezi WM, Milano L, Fierheller CT, Serruya C, Revil T, Oros KK, et al. The Genetic and Molecular Analyses of RAD51C and RAD51D Identifies Rare Variants Implicated in Hereditary Ovarian Cancer from a Genetically Unique Population. Cancers (Basel). 2022;14(9). https://doi.org/10.3390/ cancers14092251.
233. Osher DJ, De Leeneer K, Michils G, Hamel N, Tomiak E, Poppe B, et al. Mutation analysis of RAD51D in non-BRCA1/2 ovarian and breast cancer families. Br J Cancer. 2012;106(8):1460-3.
234. Soukupová J, Lhotová K, Janatová M, Kleiblová P, Vočka M, Foretová L , et al. Germline mutations in RAD51C and RAD51D and hereditary predisposition to ovarian cancer. Klin Onkol. 2021;34(1):26-32.
235. Cummings S, Roman SS, Saam J, Bernhisel R, Brown K, Lancaster JM, et al. Age of ovarian cancer diagnosis among BRIP1, RAD51C, and RAD51D mutation carriers identified through multi-gene panel testing. J Ovarian Res. 2021;14(1). https://doi.org/10.1186/s13048-021-00809-w.
236. Loveday C, Turnbull C, Ramsay E, Hughes D, Ruark E, Frankum JR, et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat Genet. 2011;43(9):879-82.
237. Chen X, Li Y, Ouyang T, Li J, Wang T, Fan Z, et al. Associations between RAD51D germline mutations and breast cancer risk and survival in BRCA1/2-negative breast cancers. Ann Oncol. 2018;29(10):2046-51.
238. Tao J, Bauer DE, Chiarle R. Assessing and advancing the safety of CRISPR-Cas tools: from DNA to RNA editing. Nat Commun. 2023;14(1). https://doi.org/10.1038/s41467-023-35886-6.
239. Bier E, Nizet V. Driving to Safety: CRISPR-Based Genetic Approaches to Reducing Antibiotic Resistance. Trends Genet. 2021;37(8):745-57.
240. Cromer MK, Barsan VV., Jaeger E, Wang M, Hampton JP, Chen F, et al. Ultra-deep sequencing validates safety of CRISPR/Cas9 genome editing in human hematopoietic stem and progenitor cells. Nat Commun. 2022;13(1). https://doi.org/10.1038/s41467-022-32233-z.
241. Sun D. Design of time-delayed safety switches for CRISPR gene therapy. Sci Rep. 2021;11(1). https://doi.org/10.1038/s41598-021-96510-5.
242. Xu CF, Chen GJ, Luo YL, Zhang Y, Zhao G, Lu ZD, et al. Rational designs of in vivo CRISPR-Cas delivery systems. Adv Drug Deliv Rev. 2021;168:3-29.
243. Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, et al. CRISPR-engineered T cells in patients with refractory cancer. Science (80- ). 2020;367(6481). https://doi.org/10.1126/science.aba7365.
244. Wan F, Draz MS, Gu M, Yu W, Ruan Z, Luo Q. Novel strategy to combat antibiotic resistance: a sight into the combination of crispr/cas9 and nanoparticles. Pharmaceutics. 2021;13(3). https://doi.org/10.3390/ pharmaceutics13030352.
245. Tang N, Ning Q, Wang Z, Tao Y, Zhao X, Tang S. Tumor microenvironment based stimuli-responsive CRISPR/Cas delivery systems: A viable platform for interventional approaches. Colloids and Surfaces B: Biointerfaces. 2022;210. https://doi.org/10.1016/j.colsurfb.2021.112257.
246. Wang G, Chow RD, Bai Z, Zhu L, Errami Y, Dai X, et al. Multiplexed activation of endogenous genes by CRISPRa elicits potent antitumor immunity. Nat Immunol. 2019;20(11):1494-505.
247. Xu L, Lau YS, Gao Y, Li H, Han R. Life-Long AAV-Mediated CRISPR Genome Editing in Dystrophic Heart Improves Cardiomyopathy without Causing Serious Lesions in mdx Mice. Mol Ther. 2019;27(8):1407-14.
248. Wei S, Shao X, Liu Y, Xiong B, Cui P, Liu Z, et al. Genome editing of PD-L1 mediated by nucleobase-modified polyamidoamine for cancer immunotherapy. J Mater Chem B. 2022;10(8):1291-300.
249. Wu Y, Zheng J, Zeng Q, Zhang T, Xing D. Light-responsive charge-reversal nanovector for high-efficiency in vivo CRISPR/Cas9 gene editing with controllable location and time. Nano Res. 2020;13(9):2399-406.
250. Li C, Ding L, Sun CW, Wu LC, Zhou D, Pawlik KM, et al. Novel HDAd/EBV Reprogramming Vector and Highly Efficient Ad/CRISPR-Cas Sickle Cell Disease Gene Correction. Sci Rep. 2016;6. https://doi.org/10.1038/srep3 0422.
251. Preece R, Pavesi A, Gkazi SA, Stegmann KA, Georgiadis C, Tan ZM, et al. CRISPR-Mediated Base Conversion Allows Discriminatory Depletion of Endogenous T Cell Receptors for Enhanced Synthetic Immunity. Mol Ther Methods Clin Dev. 2020;19:149-61.
252. Liu J, Li G, Guo H, Ni C, Gao Y, Cao X, et al. Dual-Responsive Core-Shell Tecto Dendrimers Enable Efficient Gene Editing of Cancer Cells to Boost Immune Checkpoint Blockade Therapy. ACS Appl Mater Interfaces. 2023;15(10):12809-21.
253. Chen X, Chen Y, Xin H, Wan T, Ping Y. Near-infrared optogenetic engineering of photothermal nanoCRISPR for programmable genome editing. Proc Natl Acad Sci U S A. 2020;117(5):2395-405.
254. Villamizar O, Waters SA, Scott T, Saayman S, Grepo N, Urak R, et al. Targeted Activation of Cystic Fibrosis Transmembrane Conductance Regulator. Mol Ther. 2019;27(10):1737-48.
255. Pofali P, Mondal A, Londhe V. Exosome as a Natural Gene Delivery Vector for Cancer Treatment. Curr Cancer Drug Targets. 2020;20(11):821-30.
256. Ma SC, Zhang JQ, Yan TH, Miao MX, Cao YM, Cao YB, et al. Novel strategies to reverse chemoresistance in colorectal cancer. Cancer Med. 2023;12(10):11073-96.
257. Gong Z, Zhang X, Dai Z. Progress of nanoparticles inhibiting tumor metastasis. Kexue Tongbao/Chinese Sci Bull. 2018;63(15):1482-92.
258. Fix SM, Jazaeri AA, Hwu P. Applications of crispr genome editing to advance the next generation of adoptive cell therapies for cancer. Cancer Discov. 2021;11(3):560-74.
259. Choi E, Chang J-W, Krueger J, Lahr WS, Pomeroy E, Walsh M, et al. Engineering CD70-Directed CAR-NK Cells for the Treatment of Hematological and Solid Malignancies. Blood. 2021;138(Supplement 1):1691-1691.
260. Eyquem J, Mansilla-Soto J, Odak A, Sadelain M. 274. One-Step Generation of Universal CART Cells. Mol Ther. 2016;24:S109.
261. Kavanagh EW, Green JJ. Toward Gene Transfer Nanoparticles as Therapeutics. Advanced Healthcare Materials. 2022;11(7). https://doi.org/10. 1002/adhm. 202102145.
262. Wang X, Zabierowski S, Wu M, Del Casale C, Eyquem J, Mansilla-Soto J, et al. Establishing cGMP manufacturing of CRISPR/Cas9-edited human CART cells. Cytotherapy. 2020;22(5):S138-9.
263. Getahun YA, Ali DA, Taye BW, Alemayehu YA. Multidrug-Resistant Microbial Therapy Using Antimicrobial Peptides and the CRISPR/Cas9 System. Vet Med Res Reports. 2022;13:173-90.
264. Tay LS, Palmer N, Panwala R, Chew WL, Mali P. Translating CRISPR-Cas Therapeutics: Approaches and Challenges. CRISPR J. 2020;3(4):253-75.
265. Barrangou R, Horvath P. CRISPR: New horizons in phage resistance and strain identification. Annu Rev Food Sci Technol. 2012;3(1):143-62.
266. Xiu K, Saunders L, Wen L, Ruan J, Dong R, Song J, et al. Delivery of CRISPR/Cas9 Plasmid DNA by Hyperbranched Polymeric Nanoparticles Enables Efficient Gene Editing. Cells. 2023;12(1). https://doi.org/10. 3390/cells12010156.
267. Lau CH, Suh Y. In vivo genome editing in animals using AAV-CRISPR system: Applications to translational research of human disease. F1000Research. 2017;6. https://doi.org/10.12688/f1000research.11243.1.
268. Jiang C, Lin X, Zhao Z. Applications of CRISPR/Cas9 Technology in the Treatment of Lung Cancer. Trends Mol Med. 2019;25(11):1039-49.
269. Wang Z, Chen M, Zhang Y, Liu Y, Yang Q, Nie J, et al. Phase I study of CRISPR-engineered CAR-T cells with PD-1 inactivation in treating meso-thelin-positive solid tumors. J Clin Oncol. 2020;38(15_suppl):3038-3038.
270. Tratar UL, Horvat S, Cemazar M. Transgenic mouse models in cancer research. Front Oncol. 2018;8(JUL). https://doi.org/10.3389/fonc.2018. 00268.
271. Zhan T, Rindtorff N, Betge J, Ebert MP, Boutros M. CRISPR/Cas9 for cancer research and therapy. Semin Cancer Biol. 2019;55:106-19.
272. Ahmed
273. Zhao Z, Li C, Tong F, Deng J, Huang G, Sang Y. Review of applications of CRISPR-Cas9 gene-editing technology in cancer research. Biol Proced Online. 2021;23(1). https://doi.org/10.1186/s12575-021-00151-x.
274. Wang SW, Gao C, Zheng YM, Yi L, Lu JC, Huang XY, et al. Current applications and future perspective of CRISPR/Cas9 gene editing in cancer. Mol Cancer. 2022;21(1). https://doi.org/10.1186/s12943-022-01518-8.
275. Jo DH, Song DW, Cho CS, Kim UG, Lee KJ, Lee K, et al. CRISPR-Cas9mediated therapeutic editing of Rpe65 ameliorates the disease phenotypes in a mouse model of Leber congenital amaurosis. Sci Adv. 2019;5(10). https://doi.org/10.1126/sciadv.aax1210.
276. Liang SQ, Walkey CJ, Martinez AE, Su Q, Dickinson ME, Wang D, et al. AAV5 delivery of CRISPR-Cas9 supports effective genome editing in mouse lung airway. Mol Ther. 2022;30(1):238-43.
277. Yu W, Mookherjee S, Chaitankar V, Hiriyanna S, Kim JW, Brooks M, et al. Nrl knockdown by AAV-delivered CRISPR/Cas9 prevents retinal degeneration in mice. Nat Commun. 2017;8. https://doi.org/10.1038/ncomm s14716.
278. Zhen S, Liu Y, Lu J, Tuo X, Yang X, Chen H, et al. Human Papillomavirus Oncogene Manipulation Using Clustered Regularly Interspersed Short Palindromic Repeats/Cas9 Delivered by pH-Sensitive Cationic Liposomes. Hum Gene Ther. 2020;31(5-6):309-24.
279. Aksoy YA, Yang B, Chen W, Hung T, Kuchel RP, Zammit NW, et al. Spatial and Temporal Control of CRISPR-Cas9-Mediated Gene Editing Delivered via a Light-Triggered Liposome System. ACS Appl Mater Interfaces. 2020;12(47):52433-44.
280. Naeimi Kararoudi M, Likhite S, Elmas E, Yamamoto K, Schwartz M, Sorathia K, et al. Optimization and validation of CAR transduction into human primary NK cells using CRISPR and AAV. Cell Reports Methods. 2022;2(6). https://doi.org/10.1016/j.crmeth.2022.100236.
281. Fine EJ, Appleton CM, White DE, Brown MT, Deshmukh H, Kemp ML, et al. Trans-spliced Cas9 allows cleavage of HBB and CCR5 genes in human cells using compact expression cassettes. Sci Rep. 2015;5. https://doi.org/10.1038/srep10777.
282. Kazemian P, Yu SY, Thomson SB, Birkenshaw A, Leavitt BR, Ross CJD. Lipid-Nanoparticle-Based Delivery of CRISPR/Cas9 Genome-Editing Components. Mol Pharm. 2022;19(6):1669-86.
283. Osteikoetxea X, Silva A, Lázaro-Ibáñez E, Salmond N, Shatnyeva O, Stein J, et al. Engineered Cas9 extracellular vesicles as a novel gene editing tool. J Extracell Vesicles. 2022;11(5). https://doi.org/10.1002/jev2.12225.
284. Yang Z, Liang SQ, Yang H, Xu D, Bruggmann R, Gao Y, et al. CRISPRmediated kinome editing prioritizes a synergistic combination therapy for FGFR1-amplified lung cancer. Cancer Res. 2021;81(11):3121-33.
285. Chamberlain CA, Bennett EP, Kverneland AH, Svane IM, Donia M, Met Ö. Highly efficient PD-1-targeted CRISPR-Cas9 for tumor-infiltrating lymphocyte-based adoptive T cell therapy. Mol Ther Oncolytics. 2022;24:417-28.
286. Sajib AM, Agarwal P, Patton DJ, Nance RL, Stahr NA, Kretzschmar WP, et al. In vitro functional genetic modification of canine adenovirus type 2 genome by CRISPR/Cas9. Lab Investig. 2021;101(12):1627-36.
287. Sarkar E, Khan A. Erratic journey of CRISPR/Cas9 in oncology from bench-work to successful-clinical therapy. Cancer Treatment and Research Communications. 2021;27. https://doi.org/10.1016/j.ctarc. 2020.100289.
288. Michaels YS, Barnkob MB, Barbosa H, Baeumler TA, Thompson MK, Andre V , et al. Precise tuning of gene expression levels in mammalian cells. Nat Commun. 2019;10(1). https://doi.org/10.1038/ s41467-019-08777-y.
289. Lopez-Obando M, Hoffmann B, Géry C, Guyon-Debast A, Téoulé E, Rameau C, et al. Simple and efficient targeting of multiple genes through CRISPR-Cas9 in Physcomitrella patens. G3 Genes Genomes Genet. 2016;6(11):3647-3653.
290. Cao J, Wu L, Zhang SM, Lu M, Cheung WKC, Cai W, et al. An easy and efficient inducible CRISPR/Cas9 platform with improved specificity for multiple gene targeting. Nucleic Acids Res. 2016;44(19). https://doi.org/ 10.1093/nar/gkw660.
291. Ma X, Zhang Q, Zhu Q, Liu W, Chen Y, Qiu R, et al. A Robust CRISPR/Cas9 System for Convenient, High-Efficiency Multiplex Genome Editing in Monocot and Dicot Plants. Mol Plant. 2015;8(8):1274-84.
292. Wessels HH, Méndez-Mancilla A, Hao Y, Papalexi E, Mauck WM, Lu L, et al. Efficient combinatorial targeting of RNA transcripts in single cells with Cas13 RNA Perturb-seq. Nat Methods. 2023;20(1):86-94.
293. Hirata M, Wittayarat M, Namula Z, Le QA, Lin Q, Nguyen NT, et al. Evaluation of multiple gene targeting in porcine embryos by the CRISPR/Cas9 system using electroporation. Mol Biol Rep. 2020;47(7):5073-9.
294. Pan C, Wu X, Markel K, Malzahn AA, Kundagrami N, Sretenovic S, et al. CRISPR-Act3.0 for highly efficient multiplexed gene activation in plants. Nat Plants. 2021;7(7):942-953.
295. Selvakumar SC, Preethi KA, Ross K, Tusubira D, Khan MWA, Mani P, et al. CRISPR/Cas9 and next generation sequencing in the personalized treatment of Cancer. Mol Cancer. 2022;21(1). https://doi.org/10.1186/ s12943-022-01565-1.
296. Sioson VA, Kim M, Joo J. Challenges in delivery systems for CRISPRbased genome editing and opportunities of nanomedicine. Biomed Eng Lett. 2021;11(3):217-33.
297. Chandrasekaran AP, Karapurkar JK, Chung HY, Ramakrishna S. The role of the CRISPR-Cas system in cancer drug development: Mechanisms of action and therapy. Biotechnol J. 2022;17(7). https://doi.org/10.1002/ biot. 202100468.
298. Ma D, Liu F. Genome Editing and Its Applications in Model Organisms. Genomics Proteomics Bioinformatics. 2015;13(6):336-44.
299. Moses C, Kaur P. Applications of CRISPR systems in respiratory health: Entering a new’red pen’ era in genome editing. Respirology. 2019;24(7):628-37.
300. Kick L, Kirchner M, Schneider S. CRISPR-Cas9: From a bacterial immune system to genome-edited human cells in clinical trials. Bioengineered. 2017;8(3):280-6.
301. Leonova El, Reshetnikov VV, Sopova JV. CRISPR/Cas-edited pigs for personalized medicine: more than preclinical test-system. Res Results Pharmacol. 2022;8(3):87-98.
302. Castells-Roca L, Tejero E, Rodríguez-Santiago B, Surrallés J. Crispr screens in synthetic lethality and combinatorial therapies for cancer. Cancers. 2021;13(7). https://doi.org/10.3390/cancers13071591.
303. Huang A, Garraway LA, Ashworth A, Weber B. Synthetic lethality as an engine for cancer drug target discovery. Nat Rev Drug Discovery. 2020;19(1):23-38.
304. Dhanjal JK, Radhakrishnan N, Sundar D. Identifying synthetic lethal targets using CRISPR/Cas9 system. Methods. 2017;131:66-73.
305. Vit G, Duro J, Rajendraprasad G, Hertz EPT, Holland LKK, Weisser MB, et al. Chemogenetic profiling reveals PP2A-independent cytotoxicity of proposed PP2A activators iHAP1 and DT-061. EMBO J. 2022;41(14). https://doi.org/10.15252/embj.2022110611.
306. Li C, Mei H, Hu Y. Applications and explorations of CRISPR/Cas9 in CAR T-cell therapy. Brief Funct Genomics. 2020;19(3):175-82.
307. Hu Kjia, Yin ETS, Hu Y xian, Huang H. Combination of CRISPR/Cas9 System and CAR-T Cell Therapy: A New Era for Refractory and Relapsed Hematological Malignancies. Curr Med Sci. 2021;41(3):420-430.
308. Ajavavarakula T. CRISPR-edited CAR-T cells: Using CRISPR-Cas9 to Improve CAR-T Therapy. Highlights Sci Eng Technol. 2022;14:355-9.
309. Ye L, Park JJ, Peng L, Yang Q, Chow RD, Dong MB, et al. A genome-scale gain-of-function CRISPR screen in CD8 T cells identifies proline metabolism as a means to enhance CAR-T therapy. Cell Metab. 2022;34(4):595614.e14.
310. Zhou Z, Tao C, Li J, Tang JC on, Chan AS chi, Zhou Y. Chimeric antigen receptor T cells applied to solid tumors. Front Immunol. 2022;13. https://doi.org/10.3389/fimmu.2022.984864.
311. Cortese MJ, Sauter C. A ‘CRISPR’ non-viral manufacturing approach for CART cell therapies. Mol Ther. 2022;30(11):3338-40.
312. Yan X, Chen D, Ma X, Wang Y, Guo Y, Wei J, et al. CD58 loss in tumor cells confers functional impairment of CART cells. Blood Adv. 2022;6(22):5844-56.
313. Mollanoori H, Shahraki H, Rahmati Y, Teimourian S. CRISPR/Cas9 and CAR-T cell, collaboration of two revolutionary technologies in cancer immunotherapy, an instruction for successful cancer treatment. Hum Immunol. 2018;79(12):876-82.
314. Liu J, Wang X, Chen AT, Gao X, Himes BT, Zhang H, et al. ZNF117 regulates glioblastoma stem cell differentiation towards oligodendroglial lineage. Nat Commun. 2022;13(1). https://doi.org/10.1038/ s41467-022-29884-3.
315. Wilkinson AC, Dever DP, Baik R, Camarena J, Hsu I, Charlesworth CT, et al. Cas9-AAV6 gene correction of beta-globin in autologous HSCs improves sickle cell disease erythropoiesis in mice. Nat Commun. 2021;12(1). https://doi.org/10.1038/s41467-021-20909-x.
316. Smirnikhina SA, Kondrateva E V., Adilgereeva EP, Anuchina AA, Zaynitdinova MI, Slesarenko YS, et al. P.F508del editing in cells from cystic fibrosis patients. PLoS One. 2020;15(11 November). https://doi.org/10. 1371/journal.pone. 0242094.
317. Bonafont J, Mencía A, Chacón-Solano E, Srifa W, Vaidyanathan S, Romano R, et al. Correction of recessive dystrophic epidermolysis bullosa by homology-directed repair-mediated genome editing. Mol Ther. 2021;29(6):2008-18.
318. Valetdinova KR, Malankhanova TB, Zakian SM, Medvedev SP. The Cutting Edge of Disease Modeling: Synergy of Induced Pluripotent Stem Cell Technology and Genetically Encoded Biosensors. Biomedicines. 2021;9(8). https://doi.org/10.3390/biomedicines9080960.
319. Dessy-Rodriguez
320. Garcia-Bloj B, Moses C, Sgro A, Plani-Lam J, Arooj M, Duffy C, et al. Waking up dormant tumor suppressor genes with zinc fingers, TALEs and the CRISPR/dCas9 system. Oncotarget. 2016;7(37):60535-54.
321. Mazur PK, Herner A, Mello SS, Wirth M, Hausmann S, Sánchez-Rivera FJ, et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat Med. 2015;21(10):1163-71.
322. Ousterout DG, Gersbach CA. Genome Editing for Neuromuscular Diseases. Genome Editing: The Next Step in Gene Therapy. 2016:51-79.
323. Ansari I, Chaturvedi A, Chitkara D, Singh S. CRISPR/Cas mediated epigenome editing for cancer therapy. Semin Cancer Biol. 2022;83:570-83.
324. Zhang C, Quan R, Wang J. Development and application of CRISPR/Cas9 technologies in genomic editing. Hum Mol Genet. 2018;27(R2):R79-88.
325. Pickar-Oliver A, Gersbach CA. The next generation of CRISPR-Cas technologies and applications. Nat Rev Mol Cell Biol. 2019;20(8):490-507.
326. Rafii S, Tashkandi E, Bukhari N, Al-Shamsi HO. Current Status of CRISPR/ Cas9 Application in Clinical Cancer Research: Opportunities and Challenges. Cancers. 2022;14(4). https://doi.org/10.3390/cancers14040947.
327. Yang Y, Xu J, Ge S, Lai L. CRISPR/Cas: Advances, Limitations, and Applications for Precision Cancer Research. Front Med. 2021;8. https://doi.org/ 10.3389/fmed.2021.649896.
328. Moses C, Garcia-Bloj B, Harvey AR, Blancafort P. Hallmarks of cancer: The CRISPR generation. Eur J Cancer. 2018;93:10-8.
329. Behan FM, lorio F, Picco G, Gonçalves E, Beaver CM, Migliardi G, et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature. 2019;568(7753):511-6.
330. Kim D, Kim J, Hur JK, Been KW, Yoon SH, Kim JS. Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat Biotechnol. 2016;34(8):863-8.
331. Manghwar H, Lindsey K, Zhang X, Jin S. CRISPR/Cas System: Recent Advances and Future Prospects for Genome Editing. Trends Plant Sci. 2019. https://doi.org/10.1016/j.tplants.2019.09.006.
332. Doerflinger M, Forsyth W, Ebert G, Pellegrini M, Herold MJ. CRISPR/ Cas9-The ultimate weapon to battle infectious diseases? Cell Microbiol. 2017;19(2). https://doi.org/10.1111/cmi.12693.
333. Boos SL, Loevenich LP, Vosberg S, Engleitner T, Öllinger R, Kumbrink J, et al. Disease Modeling on Tumor Organoids Implicates AURKA as a Therapeutic Target in Liver Metastatic Colorectal Cancer. CMGH. 2022;13(2):517-40.
334. Manghwar H, Li B, Ding X, Hussain A, Lindsey K, Zhang X, et al. CRISPR/ Cas Systems in Genome Editing: Methodologies and Tools for sgRNA Design, Off-Target Evaluation, and Strategies to Mitigate Off-Target Effects. Adv Sci. 2020. https://doi.org/10.1002/advs.201902312.
335. Rayner E, Durin M-A, Thomas R, Moralli D, O’Cathail SM, Tomlinson I, et al. CRISPR-Cas9 Causes Chromosomal Instability and Rearrangements in Cancer Cell Lines, Detectable by Cytogenetic Methods. Cris J. 2019;2(6):406-16.
336. Chen
337. Teng F, Li J, Cui T, Xu K, Guo L, Gao Q, et al. Enhanced mammalian genome editing by new Cas12a orthologs with optimized crRNA scaffolds. Genome Biol. 2019;20(1). https://doi.org/10.1186/ s13059-019-1620-8.
338. Bikard D, Euler C, Jiang W, Nussenzweig PM, Goldberg GW, Duportet X , et al. Development of sequence-specific antimicrobials based on programmable CRISPR-Cas nucleases. Nat Biotechnol. 2014;32(11):1146.
339. Deltcheva E, Chylinski K, Sharma CM, Gonzales K. CRISPR RNA maturation by trans -encoded small RNA and host factor RNase III. Nature. 2011;471(7340):602-7.
Publisher’s Note
Ready to submit your research? Choose BMC and benefit from:
- fast, convenient online submission
- thorough peer review by experienced researchers in your field
- rapid publication on acceptance
- support for research data, including large and complex data types
- gold Open Access which fosters wider collaboration and increased citations
- maximum visibility for your research: over 100 M website views per year
Learn more biomedcentral.com/submissions