DOI: https://doi.org/10.1172/jci176345
PMID: https://pubmed.ncbi.nlm.nih.gov/38299591
تاريخ النشر: 2024-02-01
مرض الكبد المرتبط بالكحول
الملخص
مرض الكبد المرتبط بالكحول (ALD) هو سبب رئيسي لمرض الكبد المزمن في جميع أنحاء العالم، ويشمل طيفًا من عدة اضطرابات مختلفة، بما في ذلك التدهور الدهني البسيط، التهاب الكبد الدهني، تليف الكبد، وسرطان الكبد الخلوي المتداخل. على الرغم من التقدم الكبير الذي تم إحرازه في مجال ALD على مدار العشرين عامًا الماضية، لا تزال آلية حدوث ALD غامضة، ولا توجد حاليًا أدوية معتمدة من إدارة الغذاء والدواء الأمريكية لعلاج ALD. في هذه المراجعة، نناقش رؤى جديدة حول آلية حدوث ALD والأهداف العلاجية، مستفيدين من دراسة متعدد الأوميات وغيرها من الأساليب المتطورة. يتم مناقشة إمكانية ترجمة هذه الدراسات إلى الممارسة السريرية والعلاج. كما نناقش النماذج قبل السريرية لـ ALD، وتفاعل ALD مع الخلل الأيضي، وسرطان الكبد المرتبط بالكحول، وتنوع ALD، وبعض آفاق البحث الترجمية المحتملة لـ ALD.
مقدمة
آلية المرض والأهداف العلاجية لمرض الكبد الدهني الكحولي
كان يُعتقد أنه يعزز تجديد الكبد وتم اختباره في التجارب السريرية لالتهاب الكبد الكحولي الحاد. ومع ذلك، فإن الأدلة على تحفيز G-CSF لتجديد الكبد غير كافية، وكانت نتائج التجارب السريرية لـ G-CSF مثيرة للجدل في حالات الفشل الكبدي الحاد على المزمن بما في ذلك التهاب الكبد الكحولي، ولم يوفر G-CSF أي فائدة للبقاء على قيد الحياة بعد 90 يومًا في الأفراد الذين يعانون من التهاب الكبد الكحولي الحاد، مما يشير إلى أن المزيد من الأدلة مطلوبة لمزيد من التحقيقات السريرية حول G-CSF. أخيرًا، قد يكون IL-22 هدفًا استثنائيًا يحمي بشكل خاص من موت خلايا الكبد ويعزز تكاثر خلايا الكبد دون التأثير على الخلايا المناعية نظرًا للتعبير المحدود عن IL-22.
| أنواع | الأهداف/الأدوية | الوظائف البيولوجية | حالة | |||
| استهداف موت وتجديد الخلايا الكبدية | منشط IL-22 (F-562) |
|
نتائج واعدة من تجربة المرحلة الثانية ب (25) تجارب المرحلة الثانية ب الجارية على فشل الكبد الحاد (CTR20212657) | |||
| مضاد الالتهاب | مضاد IL-1R (أناكينرا) |
|
فوائد مشابهة لعلاج الستيرويد من تجربة المرحلة الثانية (34) | |||
| استهداف تجديد الكبد؟ | جي-سي إس إف (بيغفيلغراستيم) |
|
أفادت تجربة المرحلة الثانية الأخيرة بوجود مخاوف من ارتفاع عدد كريات الدم البيضاء وعدم وجود فوائد (21) | |||
| مضاد الالتهابات | مثبطات مستقبلات CXCR1 و CXCR2 |
|
نتائج واعدة في نماذج مرض أديسون في الفئران
|
|||
| مضاد الالتهابات | مضاد LPS (اللبأ البقري عالي المناعة الغني بـ IgG) |
|
تجربة سريرية مستمرة المرحلة الثانية (NCT01968382) | |||
| استهداف موت الخلايا | مثبط الكاسباز إيمريكسين (IDN-6556) |
|
لا فوائد من تجربة المرحلة الثانية (NCT01912404) | |||
| مضاد الالتهاب | مثبطات CCR2/5 المزدوجة (سينيكريفيروك) |
|
نتائج واعدة في نماذج ALD الفأرية (37) | |||
| مضاد الالتهاب | TNF-
|
|
زيادة في الوفيات (33) | |||
| مضاد الالتهاب | TNF-
|
|
يحسن من معدل الوفيات على المدى القصير (35) | |||
| استهداف موت الخلايا المبرمج | مثبط ASK-1 (سيلونسرطيب GS-4997) |
|
لا فوائد من تجربة المرحلة الثانية (NCTO2854631) | |||
| مضاد الأكسدة | مضاد ROS (N-acetylcysteine) |
|
دراسة سريرية عشوائية مزدوجة التعمية في مرضى السكتة الدماغية الحادة
|
|||
| مضاد أكسدة؛ مثبط استرات الأحماض الدهنية | مضاد ROS (ميتادوكسيين) |
|
أبلغت تجربة سريرية عشوائية عن فائدة بقاء قصيرة الأمد (88) | |||
| استهداف موت الخلايا وتجديدها | DNMT1 (مثبط فرط الميثلة للحمض النووي لارسوكوستيرول) (DUR-928) |
|
نتائج واعدة من تجربة سريرية من المرحلة الثانية (85) | |||
| عوامل تعمل على الميكروبيوم ومحور الأمعاء والكبد | زراعة ميكروبيوم البراز من متبرع صحي (FMT) | تصحيح اختلال التوازن الميكروبي في مرض الكبد الدهني الكحولي | أظهر تجربة سريرية عشوائية فائدة في البقاء على قيد الحياة بعد 90 يومًا في المرضى الذين يعانون من sAH (62) |
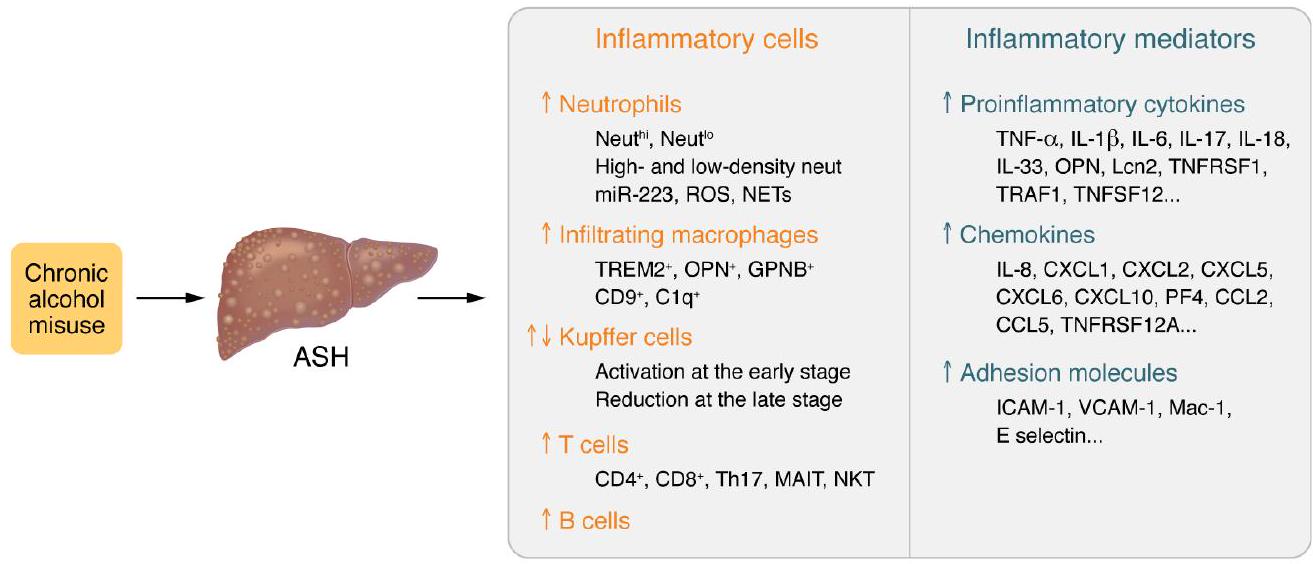
تسلل كبير لخلايا T يُلاحظ أيضًا في مرض الكبد الدهني الكحولي، خاصة في تليف الكبد المرتبط بالكحول، لكن أدوارها الدقيقة لم تُحدد بشكل جيد في المرضى الذين يعانون من مرض الكبد الدهني الكحولي. تشير الأدلة الناشئة إلى أن خلايا T لها أدوار مهمة في تعزيز التليف في مرض الكبد الدهني المرتبط بالخلل الأيضي، لذا سيكون من المهم فحص ما إذا كانت خلايا T تساهم أيضًا في تكوين الألياف في مرض الكبد الدهني الكحولي. من المثير للاهتمام أن هناك علاقة سلبية بين العدلات داخل الكبد وداخل الكبد.
زيادة نفاذية الأمعاء، تقليل إنتاج الجزيئات المضادة للميكروبات، زيادة سمك المخاط، انخفاض ملحوظ في خلايا المناعة المخاطية، وتغيرات مرتبطة بميكروبيوم الأمعاء. بشكل عام، يعتبر تقليل خلايا المناعة في الأمعاء سمة فريدة من نوعها لمرض الأمعاء المرتبط بالكحول، والذي يختلف عن الأمراض المعوية الأخرى (مثل مرض السيلياك، مرض الأمعاء الالتهابي) التي تتميز بالالتهاب المعوي. يؤدي تقليل خلايا المناعة المعوية الناتج عن الكحول إلى خلل في المناعة المعوية ويساهم بعد ذلك في تدمير حاجز الأمعاء. ومع ذلك، لا يزال غير واضح كيف يؤثر استهلاك الكحول المزمن بالضبط على مجموعات خلايا المناعة المؤيدة والمعادية للالتهابات في أجزاء الأمعاء المختلفة. علاوة على ذلك، يرتبط إساءة استخدام الكحول ومرض الكبد الكحولي بزيادة نمو البكتيريا في الأمعاء الدقيقة، وتغيرات في ميكروبيوتا الأمعاء (“خلل الميكروبيوم”)، وانتقال البكتيريا.
حتى وقت قريب. أظهر مختبر دينغ أن استهلاك الكحول يقلل من بروتين دينامين المرتبط بالكبد 1 (DRP1)، وهو بروتين يشارك في انقسام الميتوكوندريا، ويؤدي إلى تكوين ميتوكوندريا ضخمة في الخلايا ونموذج الفأر لمرض الكبد الكحولي (ALD) (80). المرضى الذين يعانون من السمنة الكبدية الحادة (sAH) لديهم انخفاض في DRP1 الكبدي مرتبط بزيادة تراكم الميتوكوندريا الضخمة في الكبد، والحذف الجيني لـ
تطبيق التقنيات المتطورة في ALD
| المؤلفون، السنة (المرجع) | نوع التحليل، العينات، النماذج | رقم الانضمام |
| دياسيك وآخرون 2004 (100) | الميكروأري، الكبد، نموذج الإيثانول داخل المعدة | |
| شو وآخرون 2015 (102) | الميكروأري، الكبد، نموذج NIAAA | GSE67546 |
| كيربيتش وآخرون. 2016 (191) | الميتابولوميات، عينات البراز، الإيثانول المزمن | |
| إيغوتشي وآخرون. 2017 (118) | تسلسل RNA صغير، مصل، إيثانول داخل المعدة + نظام غذائي عالي الدهون، فئران | |
| خانوفا وآخرون. 2018 (107) | تسلسل RNA والبروتيوميات، الكبد، الإيثانول داخل المعدة + نظام غذائي عالي الدهون + تناول مفرط | GSE97234 |
| ليو وآخرون 2020 (104) | RNA-Seq، خلايا النجمية الكبدية البشرية/الفأرية المنشطة بالكحول | GSE141100 |
| جيانغ وآخرون. 2020 (105) | تسلسل RNA، الكبد والأمعاء، نموذج NIAAA | مشروع البيولوجيا PRJNA597350 |
| يانغ وآخرون 2021 (103) | تسلسل RNA، الميتابولوميات، والليبيدوميات؛ الكبد؛ نموذج NIAAA | GSE137059 |
| بالوج وآخرون 2022 (120) | تسلسل RNA أحادي الخلية، خلايا تعبر عن Colla1، إيثانول داخل المعدة + نظام غذائي عالي الدهون + نوبات تناول الطعام | |
| كاو وآخرون. 2023 (106) | تسلسل RNA أحادي الخلية، الكبد، تناول الإيثانول المتعدد على المدى القصير | |
| نموذج المعهد الوطني لسوء استخدام الكحول وإدمانه (NIAAA)، تغذية الإيثانول المزمنة مع نوبات الشرب. | ||
| المؤلفون، السنة (المرجع) | نوع التحليل، الجين أو العينة، المرض | رقم الانضمام |
| تيان وآخرون 2010 (97) | دراسات الارتباط الجينومي الواسعة، PNPLA3، مرض الكبد الدهني الكحولي | |
| ترابو وآخرون 2011 (95) | دراسات الارتباط الجينومي الواسعة، PNPLA3/أديبونوترين، مرض الكبد الدهني الكحولي | |
| بوش وآخرون 2015 (93) | دراسات الارتباط على مستوى الجينوم؛ PNPLA3، TM6SF2، وMBOAT7؛ AC | |
| أبو الحسن وآخرون 2018 (96) | دراسة الارتباط الجيني، بروتين HSD17B13 المقطوع، الحماية ضد مرض الكبد الدهني الكحولي | |
| بودوان وآخرون. 2017 (98) | دراسات الارتباط على مستوى الجينوم، AH | |
| إنيس وآخرون 2020 (94) | GWAS، MARC1 و HNRNPUL1، AC | |
| أفو وآخرون 2013 (101) | الميكروأري، الكبد، المرضى الذين يعانون من التهاب الكبد الكحولي | GSE28619 |
| راشاكندا وآخرون 2014 (112) | الميتابولوميات، المصل، المرضى الذين يعانون من ارتفاع ضغط الدم | |
| يانغ وآخرون. 2017 (111) | مصفوفة LncRNA، مصل، مرضى مع AC | |
| ترابو وآخرون 2018 (119) | الميكروأري، الكبد، المرضى الذين يعانون من السAH | GSE94417، E-MTAB-2664 |
| أرجيمي وآخرون 2019 (108) | تسلسل RNA، الميثيلوميات، ودراسات الارتباط الجينومي الواسعة؛ الكبد؛ مرضى يعانون من ASH المبكر، AH، sAH | dbGAP (phs001807.v1.p1.) |
| راما تشاندرا، وآخرون. 2019 (121)
|
تسلسل RNA أحادي الخلية، الكبد، المرضى الذين يعانون من AC | |
| هيون وآخرون. 2020 (125) | تسلسل RNA، الكبد، المرضى الذين يعانون من sAH | GSE143318 |
| ويشسلباوم وآخرون 2020 (123) | تسلسل RNA للوحيدات وATAC-Seq، المرضى الذين يعانون من sAH | GSE135286 |
| ماسي، وآخرون. 2021 (113) | تسلسل RNA، الميتابولوميات، وتسلسل CHIP؛ الكبد؛ المرضى الذين يعانون من AH أو AC | GSE142530 |
| ليو وآخرون. 2021 (124) | تسلسل RNA و CHIP-Seq، الكبد، المرضى الذين يعانون من AH | GSE155926، GSE166564 |
| بو صالح وآخرون 2021 (122) | تسلسل RNA لعينة YAP المقطعة ميكروسكوبياً
|
phs001807.v1.p1., GSE167308 |
| هاردستي وآخرون 2022 (117) | البروتيوم والفوسفوبروتيوم، الكبد، المرضى الذين يعانون من AH أو AC | مستودع ماسيف MSV000089168 |
| هاريس وآخرون 2022 (116) | البروتيوميات، الكبد، المرضى الذين يعانون من التهاب الكبد الكحولي | |
| لوثر وآخرون 2022 (114) | بروتيوميات المصل، المرضى الذين يعانون من اضطراب استخدام الكحول أو التهاب الكبد الخفيف/المعتدل/الشديد | |
| أرجيمي وآخرون 2022 (110) | البروتيوميات، البلازما، المرضى الذين يعانون من AH أو AC | الانضمام الضخم MSV000084528 |
| ليستوباد وآخرون 2022 (115) | RNA-Seq للـ PBMC والكبد، المرضى الذين يعانون من AH أو AC | phs001807.v1.p1., GSE142530 |
| نيو وآخرون 2022 (109) | بروتيوميات البلازما والكبد، الكبد، المرضى الذين يعانون من مرض الكبد الدهني | |
| ما وآخرون 2022 (30) | تسلسل RNA، الكبد، المرضى الذين يعانون من AH، نموذج NIAAA | GSE143318 |
| أحمدي وآخرون 2023 (31) | مصفوفة الأجسام المضادة للبروتيوم، الكبد، المرضى الذين يعانون من sAH | |
| تليف الكبد المرتبط بالكحول (AC)؛ نموذج NIAAA، تغذية الإيثانول المزمنة مع نوبات.
|
||
| جين | تعدد الأشكال | التأثير على ALD | المراجع. | ||
| PNPLA3 (بروتين يحتوي على مجال الفوسفوليباز الشبيه بالباتاتين 3) | rs738408، rs738409 |
|
93، 95، 97، 161، 163، 192 | ||
| MBOAT7 (نطاق نقل الأحماض الدهنية المرتبط بالغشاء 7) | rs626283، rs641738 |
|
93 | ||
| TM6SF2/SUGP1 (عضو في عائلة البروتينات الغشائية العابرة 6) | rs10401969، r558542926 |
|
93، 161، 163 | ||
| MARC1 (مكون تقليل الأميودوكسيم الميتوكوندري) | rs2642438 |
|
94 | ||
| HNRNPUL1 (بروتين ريبونوكليوتيد نووي غير متجانس U مثل 1) | rs15052 |
|
94 | ||
| HSD17B13 (هيدروكسيستيرويد 17-
|
rs72613567 |
|
96 | ||
| SERPINA1 (عضو من عائلة السيربين A 1) | rs28929474 |
|
٩٦,٩٩ | ||
| WNT3A-WNT9A (عضو عائلة Wnt 3A/9A) | rs708113 |
|
161 | ||
| ترانسكريبتاز العكسية للتيلوميراز (TERT) | rs2242652 |
|
172 | ||
| APOE (أبوليبوبروتين E) | rs429358 |
|
171 | ||
| LPL (ليبوبروتين ليباز) | rs13702 |
|
170 | ||
| FAF2 (عضو عائلة العامل المرتبط بفاس 2) | rs374702773 |
|
99 |
وجد أن بروتين تنظيم التقطيع الظهاري 2 يلعب دورًا مهمًا في التحكم في إعادة برمجة الخلايا الكبدية في التهاب الكبد الكحولي (125). وأفادت دراسة أخرى أن بروتين كيناز الغني بالسيرين والأرجينين 2، وهو كيناز رئيسي يتحكم في التقطيع البديل، يتم تنشيطه في الخلايا الكبدية استجابةً للكحول ويعزز مرض الكبد الكحولي في الفئران (126).
نماذج ما قبل السريرية لمرض الكبد الدهني الكحولي
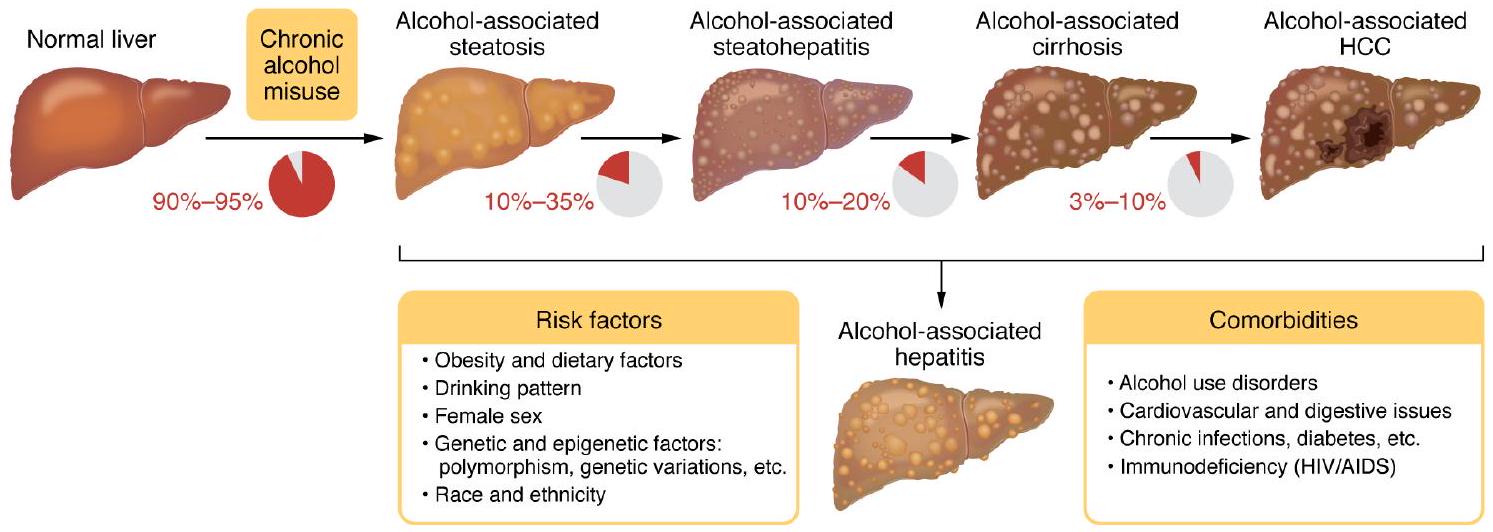
| النماذج (المراجع) | خصائص | آليات | الميزات التي تمثل مرض ALD البشري | ||||||||||||
| نموذج تغذية الإيثانول المفرطة
|
|
|
يمثل إصابة كبدية حادة خفيفة ناتجة عن الكحول لدى البشر. | ||||||||||||
| تغذية الإيثانول المزمنة حسب الرغبة، والمعروفة أيضًا بنموذج ليبر-ديكارلي (131) |
|
|
يمثل المراحل المبكرة من مرض الكبد الدهني المزمن البشري الخفيف. دراسة تنكس الكبد الدهني وتنشيط البلعميات. | ||||||||||||
| تغذية الكحول المزمنة داخل المعدة، والمعروفة أيضًا بنموذج تسوكاموتو-فرينش (132) |
|
آليات مشابهة لتغذية الإيثانول المزمنة | يمثل مرض الكبد الدهني الكحولي المزمن المعتدل لدى البشر: تنكس دهني، تنشيط البلعميات، تليف خفيف. | ||||||||||||
| نموذج التغذية المزمنة مع الإفراط في الأكل، المعروف أيضًا باسم NIAAA
|
|
|
يمثل المراحل المبكرة من التهاب الكبد الدهني البشري الخفيف. دراسة التهاب الكبد الدهني، تسلل العدلات، الاستجابة الليفية. | ||||||||||||
|
|
|
يمثل نموذجًا لدراسة التفاعل بين السمنة وشرب الكحول بشكل مفرط على التهاب الكبد الدهني الحاد. | ||||||||||||
|
|
|
|
||||||||||||
| نموذج “الضربة الثانية” أو “الضربات المتعددة” (136) |
|
تغذية الكحول المزمنة تزيد من قابلية الكبد للإصابة بأضرار والتهابات ناتجة عن ضربة ثانية أو متعددة. | تزيد تغذية الإيثانول المزمنة من قابلية الكبد للإصابة بالضرر والالتهاب الناتج عن الضربة الثانية أو الضربات المتعددة. |
تفاعل ALD والخلل الأيضي
تليف الكبد المرتبط بالكحول
سرطان الكبد المرتبط بالكحول
في السكان الذين يعانون من مرض الكبد الكحولي (ALD) (155). من الجدير بالذكر أنه مقارنة بالسكان العامين، كان الخطر النسبي (RR) لسرطان الكبد (HCC) 2.4 لمرضى اضطراب استخدام الكحول (AUD) فقط، وزادت وجود التليف الكبدي من الخطر النسبي لتطور سرطان الكبد بين الأشخاص الذين يعانون من AUD إلى 22.4 (156). التليف الكبدي هو خطوة وسيطة ضرورية لتطور سرطان الكبد المرتبط بالكحول (A-HCC) ويعزز الخطر العام للتسرطن لدى المرضى الذين يعانون من ALD (157، 158). أظهرت تحليل أسباب الوفاة للمرضى الدنماركيين الذين يعانون من ALD أن الغالبية العظمى من الوفيات ناتجة عن مرض الكبد نفسه في السنوات الخمس التي تلي التشخيص، وبعد ذلك تصبح الوفيات الناتجة عن أمراض القلب والأوعية الدموية خارج الكبد، والسرطان، والوفيات المرتبطة بـ AUD أكثر شيوعًا، بينما تعتبر السرطانات الفردية، بما في ذلك A-HCC، مساهمات صغيرة في الوفيات المرتبطة بـ ALD (159). ومع ذلك، من المحتمل أن تؤدي تحسينات العلاجات لـ ALD في المستقبل إلى زيادة طول عمر المرضى الذين يعانون من ALD، مما قد يؤدي إلى زيادة عدد المرضى الذين يعانون من A-HCC.
تباين مرض الكبد الدهني الكحولي
متغيرات خطية في TERT ومتغيرات لعدة جينات مرتبطة بتمثيل الدهون، بما في ذلك MARC1 rs2642438، APOE rs429358، HSD17B13 rs72613567، و
| عوامل/تقنيات | غرض الدراسات |
| موت الخلايا الكبدية، التجديد | حدد النوع (الأنواع) السائدة من موت الخلايا وآلياته (مثل، إجهاد الشبكة الإندوبلازمية والإجهاد التأكسدي، الالتهام الذاتي، إلخ)، والتي قد تساعد في اكتشاف أهداف علاجية أفضل لحماية الكبد. |
| التهاب | حدد المحفزات وميز الالتهاب في مرض الكبد الدهني الكحولي، مما سيساعد في اكتشاف أهداف علاجية أكثر تحديدًا للالتهاب. |
| الأطباء | ترتبط DRs بشدة ALD، وتحدد الآليات الكامنة وراء DRs وكيفية الوقاية من DRs، وتحويل DRs إلى خلايا كبد ناضجة. |
| scRNA، snRNA-Seq | حدد مجموعات فرعية من البلعميات، العدلات، خلايا T، إلخ، التي قد توفر أهداف علاجية أكثر تحديدًا لخلايا المناعة في مرض الكبد الدهني الكحولي. |
| تلطيخ متعدد | توصيف النمط الظاهري للتنوع الخلوي الكامل في مرض الكبد الدهني غير الكحولي مع السياق المكاني، مما سيساعد في فهم التفاعل بين الخلايا في مرض الكبد الدهني غير الكحولي. |
| الترانسكريبتوميات المكانية | حدد السياق الموضعي للنشاط النسخي للمناطق أو الخلايا الفردية في ALD، على سبيل المثال، حدد الفروق في DRs في المناطق الخلوية والمناطق الليفية، والتي قد تساعد في الكشف عن الآليات المختلفة الكامنة وراء DRs. |
| الجهاز المناعي المعوي | حدد كيف يؤثر استهلاك الكحول على نظام المناعة في الأمعاء وكيف تؤثر عواقبه على ميكروبيوم الأمعاء والالتهاب في مرض الكبد الدهني الكحولي. |
| العوامل الغذائية | حدد العوامل الغذائية التي تؤثر على مرض الكبد الدهني الكحولي، مثل الأحماض الدهنية، الكوليسترول، السكر، الملح، الألياف، وغيرها، في النماذج التجريبية. |
| ALD في الأفراد الذين لديهم ALDH2 غير نشط | وصف مرض الكبد الدهني الكحولي (ALD) في الفئران والبشر الذين لديهم نشاط غير نشط لإنزيم ALDH2 و/أو نشاط أعلى لإنزيم ADH، وتحديد ما إذا كانت هناك حاجة لإرشادات تشخيصية جديدة وعلاج لهذه الفئة. |
| سرطان الكبد | دراسة الآليات الكامنة وراء سرطان الكبد المرتبط بالكحول وتأثيرات الكحول على بيئة الورم الدقيقة. |
| نماذج تجريبية | النماذج الحالية لمرض الكبد الدهني الكحولي تسبب إصابة كبدية خفيفة إلى متوسطة، والتهاب، وتليف. يجب اختبار الجمع بين تغذية الكحول مع عوامل غذائية مختلفة وتعديلات جينية مختلفة في المستقبل. |
| علامات تشخيص المصل | تطبيق تقنيات الأوميكس المتعددة في تحديد علامات مصلية يمكن استخدامها لتشخيص مرض الكبد الدهني الكحولي المبكر. |
الاستنتاجات وآفاق البحث الانتقالي
تم اختبارها لمرض الكبد الدهني الكحولي. بالإضافة إلى ذلك، يجب اختبار الجمع بين الكحول وأنظمة غذائية مختلفة في النماذج ما قبل السريرية، مما قد يساعد في تحديد العوامل الغذائية التي تلعب دورًا مهمًا في مرض الكبد الدهني الكحولي. بالإضافة إلى ذلك، من المحتمل أن تساعد التقدمات الأخيرة في التحرير المتعدد السريع في الجسم للكبد البالغ للفأر باستخدام تقنية كريسبر/كاسبي 9 في تحديد كيفية تفاعل الأنظمة المختلفة في مرض الكبد الدهني الكحولي في النماذج ما قبل السريرية. أخيرًا، فإن تطوير مؤشرات حيوية لمرض الكبد الدهني الكحولي أمر ضروري أيضًا للتشخيص المبكر لمرض الكبد الدهني الكحولي “الصامت” سريريًا، مما يسمح بالتدخل المبكر مع علاج اضطراب استخدام الكحول لتقليل استهلاك الكحول وإمكانية عكس مرض الكبد الدهني الكحولي في بعض المرضى.
مساهمات المؤلفين
شكر وتقدير
6. باتالر آر، وآخرون. التهاب الكبد المرتبط بالكحول. نيو إنجلاند جورنال أوف ميديسين. 2022؛ 387(26): 2436-2448.
7. غيّو A، وآخرون. استهداف إنزيم الألدهايد ديهيدروجيناز-2 في الكبد يمنع شرب الكحول بكميات كبيرة ولكن ليس المعتدلة. إجراءات الأكاديمية الوطنية للعلوم في الولايات المتحدة الأمريكية. 2019؛ 116(51): 25974-25981.
8. رامشان داني VA، وآخرون. التقدم في أبحاث استقلاب الإيثانول. علم الأمراض البيولوجي (باريس). 2001؛49(9):676-682.
9. كليمنتي-سانشيز أ، وآخرون. التهاب الكبد الكحولي المعتدل. أمراض الكبد السريرية. 2021؛ 25(3): 537-555.
10. فاتساليا V، وآخرون. التأثيرات المفيدة لعلاج لاكتوباسيلس GG على الكبد وتقييمات الشرب لدى المرضى الذين يعانون من التهاب الكبد المرتبط بالكحول المعتدل. مجلة أمريكان جاستروإنترولوجي. 2023؛118(8):1457-1460.
11. لوفيه أ، وآخرون. زراعة الكبد المبكرة لالتهاب الكبد الشديد المرتبط بالكحول الذي لا يستجيب للعلاج الطبي: دراسة مستقبلية محكومة. لانسيت لأمراض الجهاز الهضمي والكبد. 2022؛7(5):416-425.
12. كرايب DW، وآخرون. تشخيص وعلاج أمراض الكبد المرتبطة بالكحول: إرشادات الممارسة لعام 2019 من الجمعية الأمريكية لدراسة أمراض الكبد. علم الكبد. 2020؛ 71(1): 306-333.
13. روجال س، وآخرون. تأثير علاج اضطراب استخدام الكحول على النتائج السريرية بين المرضى الذين يعانون من تليف الكبد. علم الكبد. 2020;71(6):2080-2092.
14. فانييه AGL، وآخرون. حدوث وتقدم مرض الكبد المرتبط بالكحول بعد العلاج الطبي لاضطراب استخدام الكحول. JAMA Netw Open. 2022;5(5):e2213014.
15. سيتز إتش كيه، وآخرون. مرض الكبد الكحولي. مراجعات الطبيعة لمبادئ الأمراض. 2018؛4(1):16.
16. أفيلّا م. أ، وآخرون. التقدمات الحديثة في مرض الكبد المرتبط بالكحول (ALD): ملخص لاجتماع طاولة مستديرة في مجلة غوت. غوت. 2020؛69(4):764-780.
17. قاو ب، وآخرون. مسارات الالتهاب في التهاب الكبد الدهني الكحولي. مجلة الكبد. 2019;70(2):249-259.
18. وو إكس، وآخرون. التقدمات الحديثة في فهم آلية حدوث مرض الكبد المرتبط بالكحول. مراجعة سنوية لعلم الأمراض. 2023؛ 18: 411-438.
19. سبahr ل، وآخرون. عامل تحفيز مستعمرات العدلات يحفز تكاثر الخلايا الجذعية الكبدية في التهاب الكبد الدهني الكحولي: تجربة عشوائية. علم الكبد. 2008؛ 48(1): 221-229.
20. إنجلمان سي، وآخرون. عامل تحفيز مستعمرات العدلات (G-CSF) لعلاج الفشل الكبدي الحاد على المزمن: تجربة عشوائية متعددة المراكز (دراسة GRAFT). مجلة الكبد. 2021؛75(6):1346-1354.
21. تايك JA، وآخرون. تجربة عشوائية مفتوحة متعددة المراكز من المرحلة الثانية للبيغ فيلجرستيم للمرضى الذين يعانون من التهاب الكبد المرتبط بالكحول. EClinicalMedicine. 2022;54:101689.
22. راداييفا س، وآخرون. يلعب الإنترلوكين 22 (IL-22) دورًا وقائيًا في التهاب الكبد الفيروسي المعتمد على الخلايا التائية: IL-22 هو عامل بقاء للخلايا الكبدية عبر تنشيط STAT3. علم الكبد. 2004؛39(5):1332-1342.
23. كي ش، وآخرون. علاج الإنترلوكين-22 يحسن من إصابة الكبد الكحولية في نموذج فئري لتغذية الإيثانول المزمنة المتقطعة: دور ناقل الإشارة ومفعل النسخ 3. علم الكبد.
24. شيانغ
25. عرب جي بي، وآخرون. دراسة مفتوحة، تصعيد الجرعة لتقييم سلامة وفعالية المحفز IL-22 F-652 في المرضى الذين يعانون من التهاب الكبد المرتبط بالكحول. علم الكبد. 2020؛72(2):441-453.
26. سابو جي، ماندريكار بي. منظور حديث حول الكحول، المناعة، والدفاع المضيف. ألكحول كلين إكسب ريس. 2009؛33(2):220-232.
27. خان آر إس، وآخرون. دور العدلات في التهاب الكبد المرتبط بالكحول. مجلة الكبد. 2023;79(4):1037-1048.
28. تشو ي، وآخرون. تساهم الفخاخ خارج الخلوية للعدلات في تلف الكبد وتزيد من العدلات منخفضة الكثافة المعيبة في التهاب الكبد المرتبط بالكحول. مجلة الكبد. 2023؛78(1):28-44.
29. ميهال و. آليات تليف الكبد في متلازمة الأيض. إي غاستروإنتيرولوجي. 2023;1(1):e100015.
30. ما ج، وآخرون. أنماط هيستوباثولوجية مميزة لالتهاب الكبد الكحولي الشديد تشير إلى آليات مختلفة تؤدي إلى إصابة الكبد والفشل. ج Clin Invest. 2022;132(14):e157780.
31. أحمدي AR، وآخرون. اكتشاف وتوصيف الأجسام المضادة داخل الكبد المتفاعلة بشكل متقاطع في التهاب الكبد الكحولي الشديد. Elife. 2023;12:RP86678.
32. لوفيه أ، وآخرون. الكورتيكوستيرويدات تقلل من خطر الوفاة خلال 28 يومًا للمرضى الذين يعانون من التهاب الكبد الكحولي الشديد، مقارنةً بالبينتوكسيفيلين أو الدواء الوهمي – تحليل تلوي للبيانات الفردية من التجارب المضبوطة. أمراض الجهاز الهضمي. 2018؛155(2):458-468.
33. بوتيشر NC، وآخرون. تجربة عشوائية مزدوجة التعمية، خاضعة للرقابة الوهمية متعددة المراكز لاستخدام الإيتانرسبت في علاج التهاب الكبد الكحولي. أمراض الجهاز الهضمي. 2008؛ 135(6): 1953-1960.
34. سابو جي، وآخرون. مضاد مستقبلات IL-1 بالإضافة إلى البنتوكسيفيلين والزنك لعلاج التهاب الكبد الشديد المرتبط بالكحول. علم الكبد. 2022;76(4):1058-1068.
35. أكرِيفياديس إ، وآخرون. البنتوكسيفيلين يحسن البقاء على قيد الحياة على المدى القصير في التهاب الكبد الكحولي الحاد الشديد: تجربة مزدوجة التعمية، خاضعة للرقابة مع دواء وهمي. أمراض الجهاز الهضمي. 2000؛119(6):1637-1648.
36. كرينكل O، وآخرون. التثبيط العلاجي لتجنيد وحيدات النوى الالتهابية يقلل من التهاب الكبد الدهني وتليف الكبد. علم الكبد. 2018؛67(4):1270-1283.
37. أمباد أ، وآخرون. التثبيط الدوائي لإشارات CCR2/5 يمنع ويعكس تلف الكبد الناتج عن الكحول، وتراكم الدهون، والالتهاب في الفئران. علم الكبد. 2019؛69(3):1105-1121.
38. فنغ دي، وآخرون. تساهم البلعميات المشتقة من وحيدات النوى في تنسيق تفاعلات متعددة بين أنواع الخلايا لإصلاح آفات الكبد النخرية في نماذج الأمراض. ج Clin Invest. 2023;133(15):e166954.
39. غيّو A، وتاكي F. الأبعاد المكانية لتنوع البلعميات في أمراض الكبد. eGastroenterology. 2023;1(1):e000003.
40. لي م، وآخرون. الميكرو RNA-223 يحسن إصابة الكبد الناتجة عن الكحول من خلال تثبيط IL-6-p47
41. بيرتولا أ، وآخرون. التغذية المزمنة مع تناول الإيثانول بشكل مفرط تحفز بشكل متزامن تسلل العدلات وإصابة الكبد في الفئران: دور حاسم لـ E-selectin. علم الكبد. 2013؛58(5):1814-1823.
42. ويسر V، وآخرون. عكس التهاب الكبد الدهني الكحولي في الفئران بواسطة حجب وظيفي قائم على البيبدوسين لمستقبلات الإنترلوكين-8. غوت. 2017؛ 66(5): 930-938.
43. فرنسي SW، وآخرون. دور مسار إشارات IL-8 في تسلل الكريات البيضاء إلى كبد المرضى الذين يعانون من التهاب الكبد الكحولي. Exp Mol Pathol. 2017;103(2):137-140.
44. ماسيوني ل، وآخرون. مرض الأمعاء المرتبط بالكحول: رؤى جديدة في الآلية المرضية. الإغاثة الهضمية. 2023؛ 1(1): e100013.
45. تشانغ جي تي. الفيزيولوجيا المرضية لأمراض الأمعاء الالتهابية. نيو إنجلاند جورنال أوف ميديسين. 2020;383(27):2652-2664.
46. تريباتي أ، وآخرون. محور الأمعاء والكبد والتقاطع مع الميكروبيوم. مراجعة الطبيعة لأمراض الجهاز الهضمي والكبد. 2018؛15(7):397-411.
47. كاسارد إيه إم، سيكان د. الميكروبيوتا، لاعب رئيسي في مرض الكبد الكحولي. كلين مول هيباتول. 2018؛ 24(2): 100-107.
48. موتلو إي، وآخرون. اختلال ميكروبات الأمعاء: آلية محتملة للإندوتوكسيميا الناتجة عن الكحول والتهاب الكبد الدهني الكحولي في الجرذان. أبحاث الكحول السريرية والتجريبية. 2009؛ 33(10): 1836-1846.
49. يان AW، وآخرون. اختلال ميكروبات الأمعاء المرتبط بنموذج الفأر لمرض الكبد الكحولي. علم الكبد. 2011؛ 53(1): 96-105.
50. دوان ي، وآخرون. استهداف الفيروسات البكتيرية للبكتيريا المعوية يخفف من مرض الكبد الكحولي. الطبيعة. 2019؛ 575(7783): 505-511.
51. هارتمن ب، شنابل ب. العدوى الفطرية والميكروبيوم الفطري في الاضطرابات الكبدية الصفراوية. مجلة الكبد. 2023;78(4):836-851.
52. جيانغ ل، وآخرون. الفيروسات المعوية في مرضى التهاب الكبد الكحولي. علم الكبد. 2020;72(6):2182-2196.
53. أوهاشي و، وآخرون. الحفاظ على توازن الظهارة المعوية بواسطة ناقلات الزنك. علوم الهضم والمرض. 2019؛64(9):2404-2415.
54. إيريتاني س، وآخرون. المؤشرات المفيدة لنقص الزنك لإدارة مرض الكبد المزمن. مجلة أمراض الجهاز الهضمي. 2022؛57(4):322-332.
55. تشونغ و، وآخرون. خلل وظيفة خلايا بانث يؤدي إلى التهاب الكبد الدهني المرتبط بالكحول من خلال تعزيز انتقال البكتيريا في الفئران: دور نقص الزنك. علم الكبد. 2020؛ 71(5): 1575-1591.
56. باركس أو بي، وآخرون. إشارات الإنترلوكين-22 في تنظيم صحة الأمعاء والمرض. فرونت سيل ديف بيول. 2016؛3:85.
57. هندريكس تي، وآخرون. بكتيريا مُهندسة لإنتاج IL-22 في الأمعاء تحفز تعبير REG3G لتقليل مرض الكبد الناتج عن الإيثانول في الفئران. غوت. 2019؛68(8):1504-1515.
58. كونو تي، وآخرون. البكتيريا المهندسة التي تنتج محفزات مستقبلات الهيدروكربون العطرية تحمي من مرض الكبد الناتج عن الإيثانول في الفئران. ألكحول كلين إكسب ريس (هوبوكين). 2023؛ 47(5): 856-867.
59. تشيان م، وآخرون. نقص مستقبلات الهيدروكربون العطرية في خلايا الظهارة المعوية يزيد من حدة مرض الكبد المرتبط بالكحول. خلية مول جستروإنتيرول هيباتول. 2022؛ 13(1): 233-256.
60. ورزوسك ل، وآخرون. استقلاب التربتوفان بواسطة الميكروبيوتا يحفز تنشيط مستقبلات الهيدروكربونات العطرية ويحسن إصابة الكبد الناتجة عن الكحول. غوت. 2021؛70(7):1299-1308.
61. رنجبرين T، شنابل B. العلاجات المتمحورة حول ميكروبيوم الأمعاء لمرض الكبد المرتبط بالكحول. سيمين ليفر ديس. 2023;43(3):311-322.
62. باندي أ، وآخرون. زراعة ميكروبات البراز
مقارنة مع البريدنيزولون في مرضى التهاب الكبد الكحولي الشديد: تجربة عشوائية. هباتول إنترناشيونال. 2023;17(1):249-261.
63. باجاج جي إس، وآخرون. زراعة ميكروبات البراز من متبرع براز عقلاني تحسن من اعتلال الدماغ الكبدي: تجربة سريرية عشوائية. أمراض الكبد. 2017;66(6):1727-1738.
64. بلوم بي بي، وآخرون. زراعة ميكروبات البراز تحسن الإدراك في الاعتلال الدماغي الكبدي وتأثيرها يختلف حسب المتبرع والمستقبل. نيراطول كوميونيكشن. 2022؛6(8):2079-2089.
65. شاناهان ف، وآخرون. الميكروبيوم الصحي – ما هو تعريف الميكروبيوم المعوي الصحي؟ أمراض الجهاز الهضمي. 2021؛ 160(2): 483-494.
66. ساتو ك، وآخرون. رد فعل القنوات في أمراض الكبد: الآليات المرضية والأهمية الانتقالية. علم الكبد. 2019;69(1):420-430.
67. تشاو إكس، وآخرون. تنشيط مستمر لـ mTORC1 بسبب فقدان معقد التصلب الحدبي الكبدي 1 يعزز إصابة الكبد في التهاب الكبد الكحولي. علم الكبد. 2023;78(2):503-517.
68. Elßner C، وآخرون. الانتقال النووي لـ RELB يزداد في الكبد البشري المريض ويعزز رد الفعل القنوي والتليف الصفراوي في الفئران. علم الجهاز الهضمي. 2019؛ 156(4): 1190-1205.
69. أغيلار-برابو ب، وآخرون. خلايا التفاعل القنوي تظهر ملفًا التهابيًا وتستقطب العدلات في التهاب الكبد الكحولي. علم الكبد. 2019؛69(5):2180-2195.
70. أغيلار-برابو ب، وآخرون. تحديد ملف تعريف تمايز الخلايا الكبدية في مرض الكبد المرتبط بالكحول يحدد CXCR4 كمحرك لإعادة برمجة الخلايا. مجلة الكبد. 2023؛79(3):728-740.
71. جيلوت أ، وآخرون. رسم خريطة المشهد المناعي الكبدي يحدد البلعميات الوحيدة كعوامل رئيسية في تقدم التهاب الكبد الدهني ومرض القنوات الصفراوية. علم الكبد. 2023؛78(1):150-166.
72. أرينو س، وآخرون. الخلايا المتعادلة المرتبطة بتفاعل القنوات تعزز تكاثر الظهارة الصفراوية في أمراض الكبد المزمنة. مجلة الكبد. 2023؛79(4):1025-1036.
73. تشاو إكس، وآخرون. بروتين p62 في الكبد يثبط التفاعل القنوي وتكون الأورام في كبد الفئران مع تنشيط mTORC1 وخلل في البلعمة الذاتية. مجلة الكبد. 2022؛76(3):639-651.
74. نافارو-كوركويرا أ، وآخرون. RNA غير المشفر الطويل ACTA2-AS1 يعزز التفاعل القنوي من خلال التفاعل مع مركب p300/ELK1. مجلة الكبد. 2022؛76(4):921-933.
75. زانغ ز، وآخرون. نيك الصفراوي يعزز رد الفعل القنوي وإصابة الكبد والتليف في الفئران. نات كوميونيك. 2022;13(1):5111.
76. ماكويك ب، قاو ب. تنشيط mTORC1 في خلايا القنوات الصفراوية يحفز رد الفعل القنوي الناتج عن الكحول. علم الكبد. 2023;78(2):378-381.
77. نصير ف، إبداح JA. دور الميتوكوندريا في مرض الكبد الكحولي. مجلة العالم لأمراض الجهاز الهضمي. 2014;20(9):2136-2142.
78. ما إكس، وآخرون. دور وآليات الميتوفاجي في أمراض الكبد. خلايا. 2020؛ 9(4): 837.
79. برغويرا م، وآخرون. الميتوكوندريا العملاقة في الخلايا الكبدية: تلميح تشخيصي لمرض الكبد الكحولي. أمراض الجهاز الهضمي. 1977؛73(6):1383-1387.
80. ما إكس، وآخرون. فقدان DRP1 الكبدي يزيد من حدة التهاب الكبد الكحولي من خلال تحفيز الميتوكوندريا الضخمة وسوء التكيف الميتوكوندري. علم الكبد. 2023;77(1):159-175.
81. هاو ل، وآخرون. تنشيط ATF4 يعزز الكبد
خلل الميتوكوندريا عن طريق كبت إشارة NRF1TFAM في التهاب الكبد الدهني الكحولي. غوت. 2021;70(10):1933-1945.
82. باركر ر، وآخرون. الكحول، الأنسجة الدهنية وأمراض الكبد: الروابط الميكانيكية والاعتبارات السريرية. مراجعة الطبيعة لأمراض الجهاز الهضمي والكبد. 2018؛15(1):50-59.
83. ماثور م، وآخرون. تحلل الدهون في الأنسجة الدهنية مهم لتمكين الإيثانول من التسبب في الكبد الدهني في نموذج الفئران لمعهد الوطني لإساءة استخدام الكحول وإدمان الكحول للتغذية المزمنة والمفرطة بالإيثانول. علم الكبد. 2023؛77(5):1688-1701.
84. سينغال AK، وآخرون. الأهداف الناشئة للعلاج في ALD: دروس من NASH [نُشر على الإنترنت في 21 مارس 2023]. أمراض الكبد.https://doi. org/10.1097/hep.0000000000000381.
85. حسنين ت، وآخرون. السلامة، الديناميكا الدوائية، وإشارات الفعالية للارسوكوستيرول (DUR-928) في التهاب الكبد المرتبط بالكحول [نُشر على الإنترنت في 8 مايو 2023]. المجلة الأمريكية لأمراض الجهاز الهضمي.https://doi. org/10.14309/ajg.0000000000002275.
86. نغوين-خاك إي، وآخرون. الجلوكوكورتيكويدات بالإضافة إلى N-أسيتيل سيستين في التهاب الكبد الكحولي الشديد. نيو إنجلاند جورنال أوف ميديسين. 2011؛ 365(19): 1781-1789.
87. ستيوارت س، وآخرون. تجربة عشوائية للعلاج بمضادات الأكسدة بمفردها أو مع الكورتيكوستيرويدات في التهاب الكبد الكحولي الحاد. مجلة الكبد. 2007؛47(2):277-283.
88. هيغيرا-دي لا تيخيرا ف، وآخرون. العلاج بالميتادوكسيين وتأثيره على الوفيات المبكرة لدى المرضى الذين يعانون من التهاب الكبد الكحولي الشديد. آن هباتول. 2014؛ 13(3): 343-352.
89. ويليامز JA، دينغ WX. دور الالتهام الذاتي في إصابة الكبد الناتجة عن الكحول والمخدرات. كيمياء الغذاء والسموم. 2020؛136:111075.
90. تشاو إكس، دينغ WX. دور وآليات البلعمة الذاتية في إصابة الكبد الناتجة عن الكحول.
91. تشيان إتش، وآخرون. البلعمة الذاتية في أمراض الكبد: مراجعة. الجوانب الجزيئية للطب. 2021؛82:100973.
92. وانغ إكس، وآخرون. الميكرو RNA كمنظمات، وعلامات حيوية، وأهداف علاجية في أمراض الكبد. غوت. 2021؛ 70(4): 784-795.
93. Buch S، وآخرون. دراسة ارتباط على مستوى الجينوم تؤكد PNPLA3 وتحدد TM6SF2 و MBOAT7 كمواقع خطر لتشمع الكبد المرتبط بالكحول. نات جينت. 2015؛ 47(12): 1443-1448.
94. إينيس إتش، وآخرون. دراسة ارتباط على مستوى الجينوم لتشمع الكبد المرتبط بالكحول تحدد مواقع الخطر في MARC1 و HNRNPUL1. أمراض الجهاز الهضمي. 2020;159(4):1276-1289.
95. ترابو إي، وآخرون. تعدد الأشكال الشائع في جين PNPLA3/adiponutrin يزيد من خطر الإصابة بالتشمع وتلف الكبد في مرض الكبد الكحولي. مجلة الكبد. 2011؛55(4):906-912.
96. أبو الحسن NS، وآخرون. متغير HSD17B13 الذي يسبب تقصير البروتين والحماية من مرض الكبد المزمن. نيو إنجلاند جورنال أوف ميديسين. 2018؛378(12):1096-1106.
97. تيان سي، وآخرون. الطفرة في PNPLA3 مرتبطة بمرض الكبد الكحولي. الطبيعة الجينية. 2010؛ 42(1): 21-23.
98. بودوان جي جي، وآخرون. تحليل جيني استكشافي على مستوى الجينوم لمخاطر الجينات لالتهاب الكبد الكحولي. مجلة العلوم الهضمية الاسكندنافية. 2017؛52(11):1263-1269.
99. شوانتس-آن تي إتش، وآخرون. دراسة ارتباط جينومي شاملة وتحليل ميتا حول تليف الكبد المرتبط بالكحول يحدد عوامل الخطر الجينية. علم الكبد. 2021؛ 73(5): 1920-1931.
100. دياسيك الرابع، وآخرون. تحليل جيني واسع النطاق لـ
الكبد في نموذج الفأر لحقن الإيثانول المزمن داخل المعدة. مجلة الكبد. 2004;40(2):219-227.
101. أفو
102. شو، م-ج وآخرون. البروتين الدهني المحدد 27/ CIDEC يعزز تطور التهاب الكبد الدهني الكحولي في الفئران والبشر. أمراض الجهاز الهضمي. 2015;149(4):1030-1041.
103. يانغ ز، وآخرون. دور SHP/REV-ERB
104. ليو إكس، وآخرون. خلايا النجم الكبدية البشرية والفأرية المنشطة بالكحول الأولي تشترك في تشابهات في ملفات التعبير الجيني. اتصالات الكبد. 2020؛ 4(4): 606-626.
105. جيانغ ل، وآخرون. تحديد ملف التعبير الجيني يكتشف جينات كبدية ومعوية جديدة بعد التغذية المزمنة مع تناول الكحول بشكل مفرط في الفئران. علوم الهضم والمرض. 2020؛65(12):3592-3604.
106. كاو إل، وآخرون. تحليل نسخ RNA على مستوى الخلية الواحدة لخلايا الكبد في إصابة الكبد الكحولية قصيرة الأمد في الفئران. المجلة الدولية للعلوم الجزيئية. 2023;24(5):4344.
107. خانوفا إي، وآخرون. الموت الخلوي بالنار عبر مسار كاسبيز 11/4-غاز-ديرمين-D في التهاب الكبد الكحولي في الفئران والمرضى. علم الكبد. 2018;67(5):1737-1753.
108. أرجيمي ج، وآخرون. تعبير الجينات المعتمد على HNF4alpha المعيب كعامل مؤثر في فشل الكبد الخلوي في التهاب الكبد الكحولي. نات كوم. 2019؛10(1):3126.
109. نيو ل، وآخرون. علامات بروتينية غير جراحية للأمراض الكبدية المرتبطة بالكحول. نات ميد. 2022؛ 28(6): 1277-1287.
110. أرجيمي ج، وآخرون. تحليل متكامل للتعبير الجيني والبروتيني يحدد علامات حيوية في البلازما لفشل الكبد في التهاب الكبد المرتبط بالكحول. مجلة الأمراض الباثولوجية الأمريكية. 2022؛192(12):1658-1669.
111. يانغ ز، وآخرون. LncRNA AK054921 و AK128652 هما مؤشرات محتملة في المصل ومتنبئان ببقاء المرضى الذين يعانون من تليف الكبد الكحولي. هيباتول سوتمن. 2017؛ 1(6): 513-523.
112. راشاكندا V، وآخرون. تحليل الميتابولوم في مصل الدم في التهاب الكبد الكحولي الحاد يحدد مسارات متعددة غير منظمة. PLoS One. 2014;9(12):e113860.
113. ماسي V، وآخرون. تكامل المولتيومكس يكشف إعادة برمجة استخدام الجلوكوز ويحدد هيكسوكيناز جديد في التهاب الكبد الكحولي. أمراض الجهاز الهضمي. 2021؛ 160(5): 1725-1740.
114. لوثر ج، وآخرون. التوقيع البروتيني المتداول لمرض الكبد المرتبط بالكحول. JCI Insight. 2022;7(14):e159775.
115. Listopad S، وآخرون. التمييز بين أمراض الكبد من خلال تطبيق أساليب التعلم الآلي متعددة الفئات على بيانات النسخ الجيني لنسج الكبد أو عينات الدم. JHEP Rep. 2022;4(10):100560.
116. هاريس بي إس، وآخرون. التحليل البروتيني لالتهاب الكبد المرتبط بالكحول يكشف عن الجليكوبروتين NMB (GPNMB) كعلامة حيوية جديدة للكبد والمصل. الكحول. 2022؛99:35-48.
117. هاردستي ج، وآخرون. بصمات البروتينات والبروتينات الفوسفاتية الكبدية لالتهاب الكبد والتشمع المرتبط بالكحول. مجلة الطب الشرعي الأمريكية. 2022؛192(7):1066-1082.
118. إيغوتشي أ، وآخرون. الحويصلات خارج الخلوية التي تطلقها خلايا الكبد من نموذج التسريب المعدي
تحتوي أمراض الكبد الكحولي على شيفرة ميكرو RNA يمكن اكتشافها في الدم. علم الكبد. 2017;65(2):475-490.
119. ترابو إي، وآخرون. دمج توقيع التعبير الجيني ونموذج درجة مرض الكبد في مراحله النهائية يتنبأ ببقاء المرضى الذين يعانون من التهاب الكبد الكحولي الشديد. أمراض الجهاز الهضمي. 2018؛ 154(4): 965-975.
120. بالوغ س، وآخرون. ظهور مجموعة فرعية من خلايا الكبد الجذعية Lrat+Fbln2+ ذات خصائص عالية التليف والتهاب في التهاب الكبد الكحولي. علم الكبد. 2023;78(1):212-224.
121. راماشاندران ب، وآخرون. حل الفضاء الليفي لتشمع الكبد البشري على مستوى الخلية الواحدة. ناتشر. 2019;575(7783):512-518.
122. بوعصالح م، وآخرون. فقدان هوية الخلايا الكبدية بعد تنشيط YAP غير الطبيعي: آلية رئيسية في التهاب الكبد الكحولي. مجلة الكبد. 2021;75(4):912-923.
123. ويشيلباوم ل، وآخرون. الأساس الوراثي اللاجيني لخلل أحادية النواة في المرضى الذين يعانون من التهاب الكبد الكحولي الشديد. مجلة الكبد. 2020؛73(2):303-314.
124. ليو م، وآخرون. تنظيم المعززات الفائقة لإنتاج الكيموكينات المستحثة بواسطة السيتوكينات في التهاب الكبد الكحولي. نات كوم. 2021؛ 12(1): 4560.
125. هيون ج، وآخرون. بروتين تنظيم التقطيع الظهاري 2 الذي يتوسط التقطيع البديل يعيد برمجة الخلايا الكبدية في التهاب الكبد الكحولي الشديد. استثمار سريري. 2020؛130(4):2129-2145.
126. لي جي، وآخرون. استهداف كيناز البروتين سيرين-أرجينين الكبدي 2 يحسن مرض الكبد المرتبط بالكحول من خلال التحكم في الانقسام البديل لعملية تصنيع الدهون. علم الكبد. 2023؛ 78(5): 1506-1524.
127. غيّو A، وآخرون. فك شفرة البيئة المناعية في قسم نسيج محفوظ في الفورمالين ومثبت بالشمع من خلال بروتوكول تلوين مناعي متعدد الفلورسنت يمكن تنفيذه على الفور. السرطانات (بازل). 2020؛ 12(9): 2449.
128. جيلوت أ، وآخرون. تروج البلعوم النشطة بواسطة الأحماض الصفراوية لتكاثر خلايا الظهارة الصفراوية من خلال الإنتغرين
129. تسوكاموتو إتش، وآخرون. نموذج تسريب الإيثانول داخل المعدة في القوارض. طرق البيولوجيا الجزيئية. 2008؛447:33-48.
130. براندون-وارنر إي، وآخرون. نماذج القوارض لمرض الكبد الكحولي: من الفئران إلى البشر. الكحول. 2012؛46(8):715-725.
131. ليبر سي إس، وآخرون. طرق تجريبية لإدارة الإيثانول. علم الكبد. 1989؛ 10(4): 501-510.
132. أونو أ، وآخرون. نموذج التسريب داخل المعدة في الفئران (iG). بروتوكولات الطبيعة. 2012؛7(4):771-781.
133. بيرتولا أ، وآخرون. نموذج الفأر للتغذية المزمنة والمفرطة بالإيثانول (نموذج NIAAA). بروتوكولات الطبيعة. 2013؛ 8(3): 627-637.
134. وانغ هـ، وآخرون. نماذج تجريبية لأمراض الكبد الدهني: الحالة والتقييم. اتصالات الكبد. 2023؛ 7(7): e00200.
135. قاو ب، وآخرون. نماذج حيوانية لمرض الكبد الكحولي: الآلية المرضية والأهمية السريرية. تعبير الجينات. 2017؛ 17(3): 173-186.
136. تسوكاموتو إتش، وآخرون. نماذج “الضربة الثانية” لمرض الكبد الكحولي. مجلة أمراض الكبد. 2009؛29(2):178-187.
137. زو ز، وآخرون. تفاعلات العدلات مع خلايا النجم الكبدية تعزز التليف في التهاب الكبد الدهني التجريبي. خلية مول جستروإنتيرول هيباتول. 2018؛ 5(3): 399-413.
138. تشانغ ب، وآخرون. تغذية على نظام غذائي عالي الدهون على المدى القصير أو الطويل بالإضافة إلى تناول كميات كبيرة من الإيثانول بشكل حاد تحفز بشكل تآزري إصابة الكبد الحادة في الفئران: دور مهم لـ CXCL1. علم الكبد. 2015؛62(4):1070-1085.
139. يي ي-تي، وآخرون. تناول إيثانول واحد يسبب إصابة شديدة في الكبد في الفئران التي تتبع نظام غذائي غربي. اتصالات الكبد. 2023؛ 7(7): e00174.
140. لازارو ر، وآخرون. نقص الأوستيو بونتين لا يمنع ولكن يعزز التهاب الكبد النيوتروفيل الكحولي في الفئران. علم الكبد. 2015؛61(1):129-140.
141. شونفيلد م، وآخرون. نظام غذائي غربي مع الكحول في مياه الشرب يعيد تجسيد ميزات مرض الكبد المرتبط بالكحول في الفئران. ألكحول كلين إكسب ريس. 2021؛ 45(10): 1980-1993.
142. Buyco DG، وآخرون. النظام الغذائي الغربي المتزامن مع الإفراط في تناول الكحول يخل بتوازن الأيض الكبدي. PLoS One. 2023;18(5):e0281954.
143. رينلا م. إ., وآخرون. بيان توافق دلفي متعدد المجتمعات حول تسميات جديدة لمرض الكبد الدهني. مجلة الكبد. 2023;79(6):1542-1556.
144. إسرائيلسن م، وآخرون. MetALD: فرص جديدة لفهم دور الكحول في مرض الكبد الدهني. لانسيت لأمراض الجهاز الهضمي والكبد. 2023؛ 8(10): 866-868.
145. دياز LA، وآخرون. التقاطع بين مرض الكبد المرتبط بالكحول ومرض الكبد الدهني غير الكحولي. مراجعة الطبيعة لأمراض الجهاز الهضمي والكبد. 2023؛20(12):764-783.
146. فوس تي، وآخرون. العبء العالمي لـ 369 مرضًا وإصابة في 204 دول وأقاليم، 1990-2019: تحليل منهجي لدراسة العبء العالمي للمرض 2019. لانسيت. 2020؛ 396(10258): 1204-1222.
147. تابير إي بي، بارك ن دي. الوفيات بسبب تليف الكبد وسرطان الكبد في الولايات المتحدة، 1999-2016: دراسة رصدية. BMJ. 2018;362:k2817.
148. تابير إي بي، بارك ن دي. تشخيص وإدارة تليف الكبد ومضاعفاته: مراجعة. مجلة الجمعية الطبية الأمريكية. 2023;329(18):1589-1602.
149. هوانغ دي كيو، وآخرون. التغير في وبائيات سرطان الكبد العالمية من 2010 إلى 2019: التهاب الكبد الدهني غير الكحولي هو أسرع الأسباب نمواً لسرطان الكبد. ميتابوليزم الخلايا. 2022؛34(7):969-977.
150. بايك جي إم، وآخرون. التغيرات في العبء العالمي للأمراض الكبدية المزمنة من 2012 إلى 2017: التأثير المتزايد لمرض الكبد الدهني غير الكحولي. علم الكبد. 2020؛ 72(5): 1605-1616.
151. هوانغ دي كيو، وآخرون. الوبائيات العالمية للتشمع الكبدي المرتبط بالكحول وسرطان الكبد: الاتجاهات، التوقعات وعوامل الخطر. مراجعة طبيعية لأمراض الجهاز الهضمي والكبد. 2023؛ 20(1): 37-49.
152. هاجستروم إتش، وآخرون. خطر الإصابة بالسرطان في مرض الكبد المرتبط بالكحول المثبت بواسطة الخزعة: دراسة قائمة على السكان تضم 3410 أشخاص. مجلة أمراض الجهاز الهضمي والكبد السريرية. 2022؛ 20(4): 918-929.
153. بوشي ل، وآخرون. مقارنة بين سرطان الكبد الخلوي المرتبط بالكحول وفيروس التهاب الكبد C: العرض السريري، العلاج والنتيجة. أليمنت فارماكول ثير. 2016؛ 43(3): 385-399.
154. كوستنتين سي إي، وآخرون. يتم تشخيص سرطان الكبد الخلوي في مرحلة متأخرة لدى المرضى المدمنين على الكحول: نتائج دراسة مستقبلية على مستوى البلاد. السرطان. 2018؛ 124(9): 1964-1972.
155. كوستنتين سي إي، وآخرون. الفوارق الجغرافية في نتائج سرطان الكبد الخلوي في فرنسا: العبء الأكبر للكحول مقارنةً بالتهاب الكبد C. علوم الهضم والمرض. 2020؛ 65(1): 301-311.
156. كوبر هـ، وآخرون. خطر الإصابة بسرطان الكبد والقنوات الصفراوية
في المرضى الذين يعانون من التهاب الكبد الفيروسي المزمن، أو إدمان الكحول، أو تليف الكبد. علم الكبد. 2001؛34(4 الجزء 1):714-718.
157. مانسيو A، وآخرون. incidence السنوي لسرطان الكبد الخلوي بين المرضى الذين يعانون من تليف الكبد الكحولي وتحديد مجموعات المخاطر. مجلة الطب الباطني وأمراض الكبد. 2013؛ 11(1): 95-101.
158. ماريرو JA، وآخرون. تشخيص وتصنيف وإدارة سرطان الكبد الخلوي: إرشادات الممارسة لعام 2018 من الجمعية الأمريكية لدراسة أمراض الكبد. علم الكبد. 2018؛68(2):723-750.
159. Kann AE، وآخرون. الوفيات المحددة بسبب الأسباب في المرضى الذين يعانون من مرض الكبد المرتبط بالكحول في الدنمارك: دراسة قائمة على السكان. لانسيت لأمراض الجهاز الهضمي والكبد. 2023؛ 8(11): 1028-1034.
160. براندون-وارنر إي، وآخرون. التغذية المزمنة بالإيثانول تسرع من تقدم سرطان الكبد الخلوي بطريقة تعتمد على الجنس في نموذج الفأر لسرطان الكبد. ألكحول كلينيكال إكسب ريس. 2012؛ 36(4): 641-653.
161. Trepo E، وآخرون. التباين الجيني الشائع في سرطان الكبد المرتبط بالكحول: دراسة حالة-شاهد على مستوى الجينوم. لانسيت أونكول. 2022؛ 23(1): 161-171.
162. ميروني م، وآخرون. العوامل الوراثية والوراثية المعدلة لمرض الكبد الكحولي. المجلة الدولية للعلوم الجزيئية. 2018؛19(12):3857.
163. ستكل F، وآخرون. المتغيرات الجينية في PNPLA3 و TM6SF2 تزيد من خطر تطور سرطان الكبد الخلوي لدى الأفراد الذين يعانون من تليف الكبد المرتبط بالكحول. المجلة الأمريكية لأمراض الجهاز الهضمي. 2018؛113(10):1475-1483.
164. مانسينا RM، وآخرون. المتغير MBOAT7-TMC4 rs641738 يزيد من خطر الإصابة بمرض الكبد الدهني غير الكحولي لدى الأفراد من أصل أوروبي. أمراض الجهاز الهضمي. 2016؛150(5):1219-1230.
165. سبليوتس EK، وآخرون. تحليل الارتباط على مستوى الجينوم يحدد المتغيرات المرتبطة بمرض الكبد الدهني غير الكحولي التي لها تأثيرات مميزة على الصفات الأيضية. PLoS Genet. 2011;7(3):e1001324.
166. نيشالك إتش دي، وآخرون. تعدد أشكال شائع في جين NCAN مرتبط بسرطان الكبد الخلوي في مرض الكبد الكحولي. مجلة الكبد. 2014؛ 61(5): 1073-1079.
167. ساكاموتو تي، وآخرون. تأثير استهلاك الكحول وتعدد أشكال الجينات ADH2 و ALDH2 على سرطان الكبد الخلوي في السكان اليابانيين. المجلة الدولية للسرطان. 2006؛ 118(6): 1501-1507.
168. أبي إتش، وآخرون. تعدد أشكال إنزيم الألدهايد ديهيدروجيناز 2 لتطور سرطان الكبد الخلوي في تليف الكبد الكحولي في شرق آسيا. مجلة أمراض الجهاز الهضمي والكبد. 2015؛30(9):1376-1383.
169. هومان ن، وآخرون. إنزيم الكحول ديهيدروجيناز
170. شملز ف، وآخرون. قد يحمي النوع عالي الإنتاج من ليبوبروتين ليباز من سرطان الكبد الخلوي في تليف الكبد المرتبط بالكحول. تقارير JHEP. 2023؛ 5(4): 100684.
171. إينيس إتش، وآخرون. موقع rs429358 في بروتين الأبوليبوبروتين E مرتبط بسرطان الكبد الخلوي في المرضى الذين يعانون من تليف الكبد. اتصالات الكبد. 2022؛ 6(5): 1213-1226.
172. Buch S، وآخرون. التباين الجيني في TERT يعدل خطر الإصابة بسرطان الكبد الخلوي في تليف الكبد المرتبط بالكحول: نتائج من دراسة حالة-شاهد على مستوى الجينوم. غوت. 2023؛72(2):381-391.
173. ستكل F، وآخرون. وراثيات الاعتماد على الكحول وأمراض الكبد المرتبطة بالكحول. مجلة الكبد. 2017؛ 66(1): 195-211.
174. بارك ش، وآخرون. الإيثانول ومستقلباته غير المؤكسدة تعزز الإصابة الحادة للكبد من خلال تحفيز إجهاد الشبكة الإندوبلازمية، وموت الخلايا الدهنية، وتحلل الدهون. خلية مول جاستروإنترول هيباتول. 2023؛ 15(2): 281-306.
175. وانغ و، وآخرون. إنزيم الألدهايد ديهيدروجيناز، أمراض الكبد والسرطان. المجلة الدولية للعلوم البيولوجية. 2020؛16(6):921-934.
176. بوليماني آر، جيليرنتر ج. ADH1B: من إدمان الكحول، الانتقاء الطبيعي، والسرطان إلى الفينوم البشري. المجلة الأمريكية لعلم الوراثة الطبية B علم الوراثة النفسية العصبية. 2018؛177(2):113-125.
177. شاراتشارونويتثايا ب، وآخرون. مرض الكبد المرتبط بالكحول: الشرق مقابل الغرب. أمراض الكبد السريرية (هوبوكين). 2020؛ 16(6): 231-235.
178. تشانغ جي إس، وآخرون. تعدد أشكال ALDH2 وسرطانات الكحول في الآسيويين: منظور الصحة العامة. مجلة العلوم الطبية الحيوية. 2017;24(1):19.
179. تشين YC، وآخرون. الأساس الدوائي الحركي والدوائي الديناميكي للتغلب على ردود الفعل السلبية الناتجة عن الأسيتالديهيد في المدمنين على الكحول من الآسيويين، الذين يحملون الأليل المتغاير ALDH2*2. علم الوراثة الدوائية والجينومية. 2009؛19(8):588-599.
180. كوان إتش جي، وآخرون. نقص إنزيم الألدهايد ديهيدروجيناز 2 يحسن الكبد الدهني الكحولي ولكنه يزيد من التهاب الكبد والتليف في الفئران. علم الكبد. 2014؛60(1):146-157.
181. قاو ي، وآخرون. الكحول يثبط استقلاب الجلوكوز في خلايا T والتهاب الكبد في نقص ALDH2
الفئران والبشر: أدوار الأسيتالديهيد والجلوكوكورتيكويدات. الأمعاء. 2019;68(7):1311-1322.
182. سابو ج. النساء ومرض الكبد الكحولي تحذير من خطر صامت. مراجعة طبيعية لأمراض الجهاز الهضمي والكبد. 2018;15(5):253-254.
183. ديلكوت سي، وآخرون. نموذج لتحديد الشاربين بكثرة المعرضين لخطر تقدم مرض الكبد. مجلة الطب الباطني وأمراض الكبد. 2020؛ 18(10): 2315-2323.
184. سنغال إيه كيه، ماثورين ب. تشخيص وعلاج مرض الكبد المرتبط بالكحول: مراجعة. جاما. 2021;326(2):165-176.
185. بيمماساني ج، وآخرون. الفروق الجنسية في الخصائص السريرية والنتائج المرتبطة بالتهاب الكبد الكحولي. المجلة الأوروبية لأمراض الجهاز الهضمي والكبد. 2023؛ 35(10): 1192-1196.
186. إلياس ف، وآخرون. ارتفاع معدلات الوفيات المرتبطة بأمراض الكبد الناتجة عن الكحول في الولايات المتحدة من 1999 إلى 2022. اتصالات الكبد. 2023؛ 7(7): e00180.
187. رهم ج، وآخرون. الكحول كعامل خطر لتليف الكبد: مراجعة منهجية وتحليل تلوي. مراجعة المخدرات والكحول. 2010؛29(4):437-445.
188. أنوتي أ، ميلينجر JL. تغير وبائيات مرض الكبد المرتبط بالكحول: الجنس، العرق، وعوامل الخطر. سيمين ليفر ديس. 2023؛ 43(1): 50-59.
189. هوانغ س، وآخرون. الإنترلوكين-22 يحسن التهاب الكبد الدهني غير الكحولي المدفوع بالعدلات من خلال أهداف متعددة. علم الكبد. 2020;72(2):412-429.
190. كاتسودا ت، وآخرون. تحرير متعدد سريع في الجسم (RIME) لكبد الفأر البالغ. علم الكبد. 2023;78(2):486-502.
191. كيربيتش IA، وآخرون. الدهون الغذائية المشبعة وغير المشبعة تعدل بشكل مختلف التغيرات الناتجة عن الإيثانول في الميكروبيوم المعوي والميتابولوم في نموذج الفأر لمرض الكبد الكحولي. المجلة الأمريكية لعلم الأمراض. 2016؛186(4):765-776.
192. كيم إتش إس، وآخرون. الجمع التآزري لمتغير PNPLA3 I148M، تناول الكحول، والسمنة مع خطر تليف الكبد، وسرطان الكبد، والوفيات. مجلة JAMA Netw Open. 2022؛ 5(10): e2234221.
193. شوكلا إس دي، وآخرون. الإيثانول المفرط والكبد: تطورات جزيئية جديدة. أبحاث الكحول السريرية والتجريبية. 2013؛37(4):550-557.
194. وو إكس، وآخرون. MLKL المشتق من البلعميات في مرض الكبد المرتبط بالكحول: تنظيم البلعمة. أمراض الكبد. 2023؛ 77(3): 902-919.
195. وانغ ل، وآخرون. الالتهاب الذي يتوسطه STING يساهم في نموذج تغذية الإيثانول المفرط في غاو. مجلة فسيولوجيا الخلايا. 2022؛237(2):1471-1485.
196. تشيان إتش، وآخرون. فقدان SQSTM1/p62 يؤدي إلى السمنة ويزيد من إصابة الكبد الناتجة عن الكحول في الفئران المسنّة. خلية مول جستروإنتيرول هيباتول. 2023؛ 15(5): 1027-1049.
197. وو م، وآخرون. الحذف المحدد للخلايا الكبدية لمثبط الجينات المحفزة بواسطة E1A 1 يزيد من تفاقم إصابة الكبد الناتجة عن الكحول من خلال تنشيط كينازات الإجهاد. المجلة الدولية للعلوم البيولوجية. 2022؛18(4):1612-1626.
- ملاحظة المؤلف: ساهم BM و YF بالتساوي في هذا العمل.
تعارض المصالح: أعلن المؤلفون أنه لا يوجد تعارض في المصالح.
حقوق الطبع والنشر: © 2024، ماكويك وآخرون. هذه مقالة مفتوحة الوصول نُشرت بموجب شروط ترخيص المشاع الإبداعي النسبة 4.0 الدولية.
معلومات مرجعية: / Clin Invest. 2024;134(3):e176345.
https://doi.org/10.1172/JCI176345. - جوليان ج، وآخرون. الانتشار المتوقع والوفيات المرتبطة بأمراض الكبد الناتجة عن الكحول في الولايات المتحدة الأمريكية، 2019-2040: دراسة نمذجة. لانسيت للصحة العامة. 2020؛ 5(6): e316-e323.
- عصراني إس ك، وآخرون. تقليل العبء العالمي لمرض الكبد المرتبط بالكحول: خطة عمل. أمراض الكبد. 2021؛ 73(5): 2039-2050.
- ديفاربهافي إتش، وآخرون. العبء العالمي لأمراض الكبد-
سهولة: تحديث 2023. مجلة الكبد. 2023;79(2):516-537. - غاو ب، باتالر ر. مرض الكبد الكحولي: الآلية المرضية وأهداف علاجية جديدة. أمراض الجهاز الهضمي. 2011؛141(5):1572-1585.
DOI: https://doi.org/10.1172/jci176345
PMID: https://pubmed.ncbi.nlm.nih.gov/38299591
Publication Date: 2024-02-01
Alcohol-associated liver disease
Abstract
Alcohol-associated liver disease (ALD) is a major cause of chronic liver disease worldwide, and comprises a spectrum of several different disorders, including simple steatosis, steatohepatitis, cirrhosis, and superimposed hepatocellular carcinoma. Although tremendous progress has been made in the field of ALD over the last 20 years, the pathogenesis of ALD remains obscure, and there are currently no FDA-approved drugs for the treatment of ALD. In this Review, we discuss new insights into the pathogenesis and therapeutic targets of ALD, utilizing the study of multiomics and other cutting-edge approaches. The potential translation of these studies into clinical practice and therapy is deliberated. We also discuss preclinical models of ALD, interplay of ALD and metabolic dysfunction, alcohol-associated liver cancer, the heterogeneity of ALD, and some potential translational research prospects for ALD.
Introduction
Pathogenesis and therapeutic targets of ALD
was thought to promote liver regeneration and was tested in clinical trials for sAH. However, the evidence for G-CSF stimulation of liver regeneration is insufficient (19), the clinical trial results for G-CSF were controversial in acute-on-chronic liver failure including AH, and G-CSF provided no survival benefit at 90 days in individuals with sAH , indicating that more evidence is required for further clinical investigation of G-CSF (20, 21). Finally, IL-22 may be an exceptional target that specifically protects against hepatocyte death and promotes hepatocyte proliferation without affecting immune cells owing to the restricted expression of IL-22
| Types | Targets/drugs | Biological functions | Status | |||
| Targeting hepatocyte death and regeneration | IL-22 agonist (F-562) |
|
Promising results from a phase Ilb trial (25) Ongoing phase Ilb trials on ACLF (CTR20212657) | |||
| Antiinflammation | IL-1R antagonist (anakinra) |
|
Similar benefits to steroid therapy from a phase II trial (34) | |||
| Targeting liver regeneration? | G-CSF (pegfilgrastim) |
|
A recent phase II trial reported concerns with elevation of WBC and no benefits (21) | |||
| Antiinflammation | CXCR1 and CXCR2 receptor inhibitors |
|
Promising results in mouse ALD models
|
|||
| Antiinflammation | Anti-LPS (hyperimmune bovine colostrum enriched with IgG) |
|
Ongoing phase lla clinical trial (NCT01968382) | |||
| Targeting cell death | Caspase inhibitor emricasan (IDN-6556) |
|
No benefits from a phase lla trial (NCT01912404) | |||
| Antiinflammation | Dual CCR2/5 blockers (cenicriviroc) |
|
Promising results in mouse ALD models (37) | |||
| Antiinflammation | TNF-
|
|
Increased mortality (33) | |||
| Antiinflammation | TNF-
|
|
Improves short-term mortality (35) | |||
| Targeting apoptosis | ASK-1 inhibitor (selonsertib GS-4997) |
|
No benefits from a phase lla trial (NCTO2854631) | |||
| Antioxidant | Anti-ROS (N-acetylcysteine) |
|
Double-blind randomized controlled clinical trial in patients with sAH
|
|||
| Antioxidant; fatty acid esters inhibitor | Anti-ROS (metadoxine) |
|
A randomized clinical trial reported short-term survival benefit (88) | |||
| Targeting cell death and regeneration | DNMT1 (DNA hypermethylation inhibitor Larsucosterol) (DUR-928) |
|
Promising results from a phase lla clinical trial (85) | |||
| Agents acting on microbiome and gut-liver axis | Healthy donor fecal microbiota transplantation (FMT) | Correct dysbiosis in ALD | A randomized clinical trial revealed survival benefit at 90 days in patients with sAH (62) |
ty, etc. (17, 18). Significant infiltration of T cells is also observed in ALD, especially in the alcohol-associated cirrhosis, but their exact roles have not been well characterized in patients with ALD (17). Emerging evidence suggests that T cells have important profibrotic roles in metabolic dysfunction-associated steatotic liver disease (MASLD) (29), so it will be important to examine whether T cells also contribute to liver fibrogenesis in ALD. Interestingly, a negative correlation of intrahepatic neutrophils with intrahepatic
num, increased intestinal permeability, reduced production of antimicrobial molecules, increased mucus thickness, a striking diminution of mucosal immune cells, and gut microbiomerelated changes (44). In general, reduction of immune cells in the intestine is a unique feature of alcohol-associated bowel disease, which is different from other intestinal diseases (e.g., celiac disease, inflammatory bowel disease) characterized by intestinal inflammation (45). Alcohol-mediated reduction of intestinal immune cells results in intestinal immune dysfunction and subsequently contributes to gut barrier disruption (44). However, how chronic alcohol consumption exactly affects different pro- and antiinflammatory immune cell populations in different intestinal tracts still remains unclear. Moreover, alcohol misuse and ALD are associated with small intestinal bacterial overgrowth, alterations of gut microbiota (“dysbiosis”), and bacterial translocation
until recently. The Ding lab demonstrated that alcohol consumption decreased hepatic dynamin-related protein 1 (DRP1), a protein involved in mitochondrial fission, and induced megamitochondria in cells and a mouse model of ALD (80). Patients with sAH have decreased hepatic DRP1 that is associated with increased accumulation of megamitochondria in the liver, and genetic deletion of the
Application of cutting-edge technologies in ALD
| Authors, year (ref.) | Type of analysis, samples, models | Accession no. |
| Deaciuc, et al. 2004 (100) | Microarray, liver, intragastric ethanol model | |
| Xu, et al. 2015 (102) | Microarray, liver, NIAAA model | GSE67546 |
| Kirpich, et al. 2016 (191) | Metabolomics, fecal samples, chronic ethanol | |
| Eguchi, et al. 2017 (118) | Small RNA-Seq, serum, intragastric ethanol+HFD, mice | |
| Khanova, et al. 2018 (107) | RNA-Seq and proteomics, liver, intragastric ethanol+HFD+binge | GSE97234 |
| Liu, et al. 2020 (104) | RNA-Seq, alcohol-activated human/mouse hepatic stellate cells | GSE141100 |
| Jiang, et al. 2020 (105) | RNA-Seq, liver and intestine, NIAAA model | BioProject PRJNA597350 |
| Yang, et al. 2021 (103) | RNA-Seq, metabolomics, and lipidomics; liver; NIAAA model | GSE137059 |
| Balog, et al. 2022 (120) | scRNA-Seq, Colla1-expressing cells, intragastric ethanol+HFD+binge | |
| Cao, et al. 2023 (106) | scRNA-Seq, liver, short-term multiple binge ethanol | |
| National Institute on Alcohol Abuse and Alcoholism (NIAAA) model, chronic-plus-binge ethanol feeding. | ||
| Authors, year (ref.) | Type of analysis, gene or sample, disease | Accession no. |
| Tian, et al. 2010 (97) | GWAS, PNPLA3, ALD | |
| Trépo, et al. 2011 (95) | GWAS, PNPLA3/adiponutrin, ALD | |
| Buch, et al. 2015 (93) | GWAS; PNPLA3, TM6SF2, and MBOAT7; AC | |
| Abul-Husn, et al. 2018 (96) | GWAS, a protein-truncating HSD17B13, protection against ALD | |
| Beaudoin, et al. 2017 (98) | GWAS, AH | |
| Innes, et al. 2020 (94) | GWAS, MARC1 and HNRNPUL1, AC | |
| Affo, et al. 2013 (101) | Microarray, liver, patients with AH | GSE28619 |
| Rachakonda, et al. 2014 (112) | Metabolomics, serum, patients with AH | |
| Yang, et al. 2017 (111) | LncRNA array, serum, patients with AC | |
| Trépo, et al. 2018 (119) | Microarray, liver, patients with sAH | GSE94417, E-MTAB-2664 |
| Argemi, et al. 2019 (108) | RNA-Seq, methylomics, and GWAS; liver; patients with early ASH, AH, sAH | dbGAP (phs001807.v1.p1.) |
| Ramachandran, et al. 2019 (121)
|
scRNA-Seq, liver, patients with AC | |
| Hyun, et al. 2020 (125) | RNA-Seq, liver, patients with sAH | GSE143318 |
| Weichselbaum, et al. 2020 (123) | Monocyte RNA-Seq and ATAC-Seq, patients with sAH | GSE135286 |
| Massey, et al. 2021 (113) | RNA-Seq, metabolomics, and CHIP-Seq; liver; patients with AH or AC | GSE142530 |
| Liu, et al. 2021 (124) | RNA-Seq and CHIP-Seq, liver, patients with AH | GSE155926, GSE166564 |
| Bou Saleh, et al. 2021 (122) | RNA-Seq of microdissected YAP
|
phs001807.v1.p1., GSE167308 |
| Hardesty, et al. 2022 (117) | Proteome and phosphoproteome, liver, patients with AH or AC | MassIVE repository MSV000089168 |
| Harris, et al. 2022 (116) | Proteomics, liver, patients with AH | |
| Luther, et al. 2022 (114) | Serum proteomics, patients with AUD or mild/moderate/severe AH | |
| Argemi, et al 2022 (110) | Proteomics, plasma, patients with AH or AC | MassIVE Accession MSV000084528 |
| Listopad, et al. 2022 (115) | PBMC and liver RNA-Seq, patients with AH or AC | phs001807.v1.p1., GSE142530 |
| Niu, et al. 2022 (109) | Plasma and liver proteomics, liver, patients with ALD | |
| Ma, et al. 2022 (30) | RNA-Seq, liver, patients with AH, NIAAA model | GSE143318 |
| Ahmadi, et al. 2023 (31) | Proteome antibody array, liver, patients with sAH | |
| AC, alcohol-associated cirrhosis; NIAAA model, chronic-plus-binge ethanol feeding.
|
||
| Gene | Polymorphism | Effect on ALD | Refs. | ||
| PNPLA3 (patatin-like phospholipase domain-containing 3) | rs738408, rs738409 |
|
93, 95, 97, 161, 163, 192 | ||
| MBOAT7 (membrane-bound 0-acyltransferase domain containing 7) | rs626283, rs641738 |
|
93 | ||
| TM6SF2/SUGP1 (transmembrane 6 superfamily member 2) | rs10401969, r558542926 |
|
93, 161, 163 | ||
| MARC1 (mitochondrial amidoxime reducing component 1) | rs2642438 |
|
94 | ||
| HNRNPUL1 (heterogeneous nuclear ribonucleoprotein U like 1) | rs15052 |
|
94 | ||
| HSD17B13 (hydroxysteroid 17-
|
rs72613567 |
|
96 | ||
| SERPINA1 (serpin family A member 1) | rs28929474 |
|
96,99 | ||
| WNT3A-WNT9A (Wnt family member 3A/9A) | rs708113 |
|
161 | ||
| TERT (telomerase reverse transcriptase) | rs2242652 |
|
172 | ||
| APOE (apolipoprotein E) | rs429358 |
|
171 | ||
| LPL (lipoprotein lipase) | rs13702 |
|
170 | ||
| FAF2 (Fas-associated factor family member 2) | rs374702773 |
|
99 |
found that epithelial splicing regulatory protein 2 plays an important role in controlling hepatocyte reprogramming in AH (125). Another study reported that serine-arginine-rich protein kinase 2, a key kinase controlling alternative splicing, is activated in hepatocytes in response to alcohol and promotes ALD in mice (126).
Preclinical models of ALD
| Models (Refs.) | Characteristics | Mechanisms | Features that are representative of human ALD | ||||||||||||
| Binge ethanol feeding model
|
|
|
Represents mild acute human alcohol-induced liver injury. | ||||||||||||
| Chronic ad libitum ethanol feeding, also known as Lieber-DeCarli model (131) |
|
|
Represents the early stages of mild chronic human ALD. Study of ALD steatosis and macrophage activation. | ||||||||||||
| Intragastric chronic ethanol feeding, also known as Tsukamoto-French model (132) |
|
Similar mechanisms as chronic ethanol feeding | Represents moderate chronic human ALD: ALD steatosis, macrophage activation, mild fibrosis. | ||||||||||||
| Chronic-plus-binge feeding model, also known as NIAAA
|
|
|
Represents early stages of mild human AH. Study AH steatosis, neutrophil infiltration, fibrogenic response. | ||||||||||||
|
|
|
Represents a model to study of the interaction between obesity and binge drinking on acute steatohepatitis. | ||||||||||||
|
|
|
|
||||||||||||
| “Second hit” or “multiple hits” model (136) |
|
Chronic ethanol feeding increases susceptibility of livers to second or multiple hit(s)-induced liver injury and inflammation | Chronic ethanol feeding increases the susceptibility of livers to second or multiple hit(s)-induced liver injury and inflammation. |
Interplay of ALD and metabolic dysfunction
Alcohol-associated cirrhosis
Alcohol-associated liver cancer
ing in populations with ALD (155). Of note, compared with the general population, the relative risk (RR) of HCC was 2.4 for AUD alone, and the presence of cirrhosis increased the RR of developing HCC among people with AUD to 22.4 (156). Cirrhosis is a necessary intermediate step for A-HCC development and amplifies the overall risk for carcinogenesis in patients with ALD (157, 158). Analysis of causes of death for Danish patients with ALD revealed that the majority of deaths are due to the liver disease itself in the 5 years after diagnosis, after which extrahepatic cardiovascular, cancer, AUD-related deaths become more common, while individual cancers, including A-HCC, are minor contributors to ALD-related mortality (159). However, improved ALD treatments in the future will likely increase longevity of patients with ALD, which may lead to an increase in a number of patients with A-HCC.
Heterogeneity of ALD
line variant in TERT and variants of several lipid metabolism-related genes, including MARC1 rs2642438, APOE rs429358, HSD17B13 rs72613567, and
| Factors/techniques | Purpose of the studies |
| Hepatocyte death, regeneration | Identify the predominant type(s) of cell death and its mechanisms (e.g., ER and oxidative stress, autophagy, etc.,), which may help discover the better therapeutic targets for hepatoprotection. |
| Inflammation | Identify the triggers of and characterize the inflammation in ALD, which will help discover more specific therapeutic targets for inflammation. |
| DRs | DRs correlate with the severity of ALD, identify the mechanisms underlying DRs and how to prevent DRs, and convert DRs to mature hepatocytes. |
| scRNA, snRNA-Seq | Identify subsets of macrophage, neutrophils, T cells, etc., which may provide more specific therapeutic targets for immune cells in ALD. |
| Multiplex staining | Phenotype the full cellular diversity of ALD with spatial context, which will help understand the cell-cell interaction in ALD. |
| Spatial transcriptomics | Identify the positional context of transcriptional activity for regions or single cells in ALD, e.g., identify the differences of DRs in parenchymal and fibrotic regions, which may help reveal the different mechanisms underlying DRs. |
| Intestinal immune system | Characterize how alcohol consumption modulates gut immune system and its consequences affect gut microbiome and inflammation in ALD. |
| Dietary factors | Identify the dietary factors that modulate ALD, such as fatty acid, cholesterol, sugar, salt, fiber, etc., in experimental models. |
| ALD in individuals with inactive ALDH2 | Characterize ALD in mice and humans with inactive ALDH2 and/or greater ADH activity and define whether new diagnosis guideline and therapy are needed for this population. |
| Liver cancer | Study the mechanisms underlying alcohol-associated liver cancer and the effects of alcohol on tumor microenvironment. |
| Experimental models | Current ALD models generate mild-to-moderate liver injury, inflammation, and fibrosis. Combination of alcohol feeding with different dietary factors and different genetic modification should be tested in the future. |
| Serum diagnosis markers | The application of multiple omics in identifying serum markers that can be used for the diagnosis of early ALD. |
Conclusions and translational research prospects
tested for ALD. In addition, combination of alcohol with different diets in preclinical models should be tested, which may identify dietary factors that play an important role in ALD. In addition, the recent advances in rapid in vivo multiplexed editing of the adult mouse liver using CRISPR/caspase-9 will likely help to identify how different systems interact in ALD in preclinical models (190). Finally, development of ALD biomarkers is also essential for the early diagnosis of clinically “silent” ALD, allowing early intervention with AUD therapy to decrease alcohol consumption and potentially reverse ALD in some patients.
Author contributions
Acknowledgments
6. Bataller R, et al. Alcohol-associated hepatitis. N Engl J Med. 2022;387(26):2436-2448.
7. Guillot A, et al. Targeting liver aldehyde dehy-drogenase-2 prevents heavy but not moderate alcohol drinking. Proc Natl Acad Sci USA. 2019;116(51):25974-25981.
8. Ramchandani VA, et al. Research advances in ethanol metabolism. Pathol Biol (Paris). 2001;49(9):676-682.
9. Clemente-Sanchez A, et al. Moderate alcoholic hepatitis. Clin Liver Dis. 2021;25(3):537-555.
10. Vatsalya V, et al. The beneficial effects of lactobacillus GG therapy on liver and drinking assessments in patients with moderate alco-hol-associated hepatitis. Am J Gastroenterol. 2023;118(8):1457-1460.
11. Louvet A, et al. Early liver transplantation for severe alcohol-related hepatitis not responding to medical treatment: a prospective controlled study. Lancet Gastroenterol Hepatol. 2022;7(5):416-425.
12. Crabb DW, et al. Diagnosis and treatment of alcohol-associated liver diseases: 2019 Practice Guidance From the American Association for the Study of Liver Diseases. Hepatology. 2020;71(1):306-333.
13. Rogal S, et al. Impact of alcohol use disorder treatment on clinical outcomes among patients with cirrhosis. Hepatology. 2020;71(6):2080-2092.
14. Vannier AGL, et al. Incidence and progression of alcohol-associated liver disease after medical therapy for alcohol use disorder. JAMA Netw Open. 2022;5(5):e2213014.
15. Seitz HK, et al. Alcoholic liver disease. Nat Rev Dis Primers. 2018;4(1):16.
16. Avila MA, et al. Recent advances in alcohol-related liver disease (ALD): summary of a Gut round table meeting. Gut. 2020;69(4):764-780.
17. Gao B, et al. Inflammatory pathways in alcoholic steatohepatitis. J Hepatol. 2019;70(2):249-259.
18. Wu X, et al. Recent advances in understanding of pathogenesis of alcohol-associated liver disease. Апnu Rev Pathol. 2023;18:411-438.
19. Spahr L, et al. Granulocyte-colony stimulating factor induces proliferation of hepatic progenitors in alcoholic steatohepatitis: a randomized trial. Hepatology. 2008;48(1):221-229.
20. Engelmann C, et al. Granulocyte-colony stimulating factor (G-CSF) to treat acute-on-chronic liver failure: A multicenter randomized trial (GRAFT study). J Hepatol. 2021;75(6):1346-1354.
21. Tayek JA, et al. A phase II, multicenter, open-label, randomized trial of pegfilgrastim for patients with alcohol-associated hepatitis. EClinicalMedicine. 2022;54:101689.
22. Radaeva S, et al. Interleukin 22 (IL-22) plays a protective role in T cell-mediated murine hepatitis: IL-22 is a survival factor for hepatocytes via STAT3 activation. Hepatology. 2004;39(5):1332-1342.
23. Ki SH , et al. Interleukin-22 treatment ameliorates alcoholic liver injury in a murine model of chron-ic-binge ethanol feeding: role of signal transducer and activator of transcription 3. Hepatology.
24. Xiang
25. Arab JP, et al. An open-label, dose-escalation study to assess the safety and efficacy of IL-22 Agonist F-652 in patients with alcohol-associated hepatitis. Hepatology. 2020;72(2):441-453.
26. Szabo G, Mandrekar P. A recent perspective on alcohol, immunity, and host defense. Alcohol Clin Exp Res. 2009;33(2):220-232.
27. Khan RS, et al. The role of neutrophils in alcoholrelated hepatitis. J Hepatol. 2023;79(4):1037-1048.
28. Cho Y, et al. Neutrophil extracellular traps contribute to liver damage and increase defective low-density neutrophils in alcohol-associated hepatitis. J Hepatol. 2023;78(1):28-44.
29. Mehal W. Mechanisms of liver fibrosis in metabolic syndrome. eGastroenterology. 2023;1(1):e100015.
30. Ma J, et al. Distinct histopathological phenotypes of severe alcoholic hepatitis suggest different mechanisms driving liver injury and failure. J Clin Invest. 2022;132(14):e157780.
31. Ahmadi AR, et al. Discovery and characterization of cross-reactive intrahepatic antibodies in severe alcoholic hepatitis. Elife. 2023;12:RP86678.
32. Louvet A, et al. Corticosteroids reduce risk of death within 28 days for patients with severe alcoholic hepatitis, compared with pentoxifylline or placebo-a meta-analysis of individual data from controlled trials. Gastroenterology. 2018;155(2):458-468.
33. Boetticher NC, et al. A randomized, dou-ble-blinded, placebo-controlled multicenter trial of etanercept in the treatment of alcoholic hepatitis. Gastroenterology. 2008;135(6):1953-1960.
34. Szabo G, et al. IL-1 receptor antagonist plus pentoxifylline and zinc for severe alcohol-associated hepatitis. Hepatology. 2022;76(4):1058-1068.
35. Akriviadis E, et al. Pentoxifylline improves shortterm survival in severe acute alcoholic hepatitis: a double-blind, placebo-controlled trial. Gastroenterology. 2000;119(6):1637-1648.
36. Krenkel O, et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology. 2018;67(4):1270-1283.
37. Ambade A, et al. Pharmacological inhibition of CCR2/5 signaling prevents and reverses alcoholinduced liver damage, steatosis, and inflammation in mice. Hepatology. 2019;69(3):1105-1121.
38. Feng D, et al. Monocyte-derived macrophages orchestrate multiple cell-type interactions to repair necrotic liver lesions in disease models. J Clin Invest. 2023;133(15):e166954.
39. Guillot A, and Tacke F. Spatial dimension of macrophage heterogeneity in liver diseases. eGastroenterology. 2023;1(1):e000003.
40. Li M, et al. MicroRNA-223 ameliorates alcoholic liver injury by inhibiting the IL-6-p47
41. Bertola A, et al. Chronic plus binge ethanol feeding synergistically induces neutrophil infiltration and liver injury in mice: a critical role for E-selectin. Hepatology. 2013;58(5):1814-1823.
42. Wieser V, et al. Reversal of murine alcoholic steatohepatitis by pepducin-based functional blockade of interleukin- 8 receptors. Gut. 2017;66(5):930-938.
43. French SW, et al. The role of the IL-8 signaling pathway in the infiltration of granulocytes into the livers of patients with alcoholic hepatitis. Exp Mol Pathol. 2017;103(2):137-140.
44. Maccioni L, et al. Alcohol-associated bowel disease: new insights into pathogenesis. eGastroenterology. 2023;1(1):e100013.
45. Chang JT. Pathophysiology of inflammatory bowel diseases. N Engl J Med. 2020;383(27):2652-2664.
46. Tripathi A , et al. The gut-liver axis and the intersection with the microbiome. Nat Rev Gastroenterol Hepatol. 2018;15(7):397-411.
47. Cassard AM, Ciocan D. Microbiota, a key player in alcoholic liver disease. Clin Mol Hepatol. 2018;24(2):100-107.
48. Mutlu E, et al. Intestinal dysbiosis: a possible mechanism of alcohol-induced endotoxemia and alcoholic steatohepatitis in rats. Alcohol Clin Exp Res. 2009;33(10):1836-1846.
49. Yan AW, et al. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology. 2011;53(1):96-105.
50. Duan Y, et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature. 2019;575(7783):505-511.
51. Hartmann P, Schnabl B. Fungal infections and the fungal microbiome in hepatobiliary disorders. J Hepatol. 2023;78(4):836-851.
52. Jiang L, et al. Intestinal virome in patients with alcoholic hepatitis. Hepatology. 2020;72(6):2182-2196.
53. Ohashi W, et al. Maintenance of intestinal epithelial homeostasis by zinc transporters. Dig Dis Sci. 2019;64(9):2404-2415.
54. Iritani S, et al. The useful predictors of zinc deficiency for the management of chronic liver disease. J Gastroenterol. 2022;57(4):322-332.
55. Zhong W, et al. Paneth cell dysfunction mediates alcohol-related steatohepatitis through promoting bacterial translocation in mice: role of zinc deficiency. Hepatology. 2020;71(5):1575-1591.
56. Parks OB, et al. Interleukin-22 signaling in the regulation of intestinal health and disease. Front Cell Dev Biol. 2016;3:85.
57. Hendrikx T, et al. Bacteria engineered to produce IL-22 in intestine induce expression of REG3G to reduce ethanol-induced liver disease in mice. Gut. 2019;68(8):1504-1515.
58. Kouno T, et al. Engineered bacteria producing aryl-hydrocarbon receptor agonists protect against ethanol-induced liver disease in mice. Alcohol Clin Exp Res (Hoboken). 2023;47(5):856-867.
59. Qian M, et al. Aryl hydrocarbon receptor deficiency in intestinal epithelial cells aggravates alcohol-related liver disease. Cell Mol Gastroenterol Hepatol. 2022;13(1):233-256.
60. Wrzosek L, et al. Microbiota tryptophan metabolism induces aryl hydrocarbon receptor activation and improves alcohol-induced liver injury. Gut. 2021;70(7):1299-1308.
61. Ranjbarian T, Schnabl B. Gut microbiome-centered therapies for alcohol-associated liver disease. Semin Liver Dis. 2023;43(3):311-322.
62. Pande A, et al. Fecal microbiota transplantation
compared with prednisolone in severe alcoholic hepatitis patients: a randomized trial. Hepatol Int. 2023;17(1):249-261.
63. Bajaj JS, et al. Fecal microbiota transplant from a rational stool donor improves hepatic encephalopathy: A randomized clinical trial. Hepatology. 2017;66(6):1727-1738.
64. Bloom PP, et al. Fecal microbiota transplant improves cognition in hepatic encephalopathy and its effect varies by donor and recipient. Нераtol Commun. 2022;6(8):2079-2089.
65. Shanahan F, et al. The healthy microbiome-what is the definition of a healthy gut microbiome? Gastroenterology. 2021;160(2):483-494.
66. Sato K, et al. Ductular reaction in liver diseases: pathological mechanisms and translational significances. Hepatology. 2019;69(1):420-430.
67. Chao X, et al. Persistent mTORC1 activation due to loss of liver tuberous sclerosis complex 1 promotes liver injury in alcoholic hepatitis. Hepatology. 2023;78(2):503-517.
68. Elßner C, et al. Nuclear translocation of RELB is increased in diseased human liver and promotes ductular reaction and biliary fibrosis in mice. Gastroenterology. 2019;156(4):1190-1205.
69. Aguilar-Bravo B, et al. Ductular reaction cells display an inflammatory profile and recruit neutrophils in alcoholic hepatitis. Hepatology. 2019;69(5):2180-2195.
70. Aguilar-Bravo B, et al. Hepatocyte dedifferentiation profiling in alcohol-related liver disease identifies CXCR4 as a driver of cell reprogramming. J Hepatol. 2023;79(3):728-740.
71. Guillot A, et al. Mapping the hepatic immune landscape identifies monocytic macrophages as key drivers of steatohepatitis and cholangiopathy progression. Hepatology. 2023;78(1):150-166.
72. Ariño S, et al. Ductular reaction-associated neutrophils promote biliary epithelium proliferation in chronic liver disease. J Hepatol. 2023;79(4):1025-1036.
73. Chao X, et al. Hepatocytic p62 suppresses ductular reaction and tumorigenesis in mouse livers with mTORC1 activation and defective autophagy. J Hepatol. 2022;76(3):639-651.
74. Navarro-Corcuera A, et al. Long non-coding RNA ACTA2-AS1 promotes ductular reaction by interacting with the p300/ELK1 complex. J Hepatol. 2022;76(4):921-933.
75. Zhang Z, et al. Biliary NIK promotes ductular reaction and liver injury and fibrosis in mice. Nat Commun. 2022;13(1):5111.
76. Mackowiak B, Gao B. Activation of cholangiocyte mTORC1 drives alcohol-induced ductular reaction. Hepatology. 2023;78(2):378-381.
77. Nassir F, Ibdah JA. Role of mitochondria in alcoholic liver disease. World J Gastroenterol. 2014;20(9):2136-2142.
78. Ma X, et al. Role and mechanisms of mitophagy in liver diseases. Cells. 2020;9(4):837.
79. Bruguera M, et al. Giant mitochondria in hepatocytes: a diagnostic hint for alcoholic liver disease. Gastroenterology. 1977;73(6):1383-1387.
80. Ma X, et al. Loss of hepatic DRP1 exacerbates alcoholic hepatitis by inducing megamitochondria and mitochondrial maladaptation. Hepatology. 2023;77(1):159-175.
81. Hao L, et al. ATF4 activation promotes hepatic
mitochondrial dysfunction by repressing NRF1TFAM signalling in alcoholic steatohepatitis. Gut. 2021;70(10):1933-1945.
82. Parker R, et al. Alcohol, adipose tissue and liver disease: mechanistic links and clinical considerations. Nat Rev Gastroenterol Hepatol. 2018;15(1):50-59.
83. Mathur M, et al. Adipose lipolysis is important for ethanol to induce fatty liver in the National Institute on Alcohol Abuse and Alcoholism murine model of chronic and binge ethanol feeding. Hepatology. 2023;77(5):1688-1701.
84. Singal AK, et al. Emerging targets for therapy in ALD: Lessons from NASH [published online March 21, 2023]. Hepatology. https://doi. org/10.1097/hep.0000000000000381.
85. Hassanein T, et al. Safety, pharmacokinetics, and efficacy signals of larsucosterol (DUR-928) in alcohol-associated hepatitis [published online May 8, 2023]. Am J Gastroenterol. https://doi. org/10.14309/ajg.0000000000002275.
86. Nguyen-Khac E, et al. Glucocorticoids plus N -acetylcysteine in severe alcoholic hepatitis. N Engl J Med. 2011;365(19):1781-1789.
87. Stewart S, et al. A randomized trial of antioxidant therapy alone or with corticosteroids in acute alcoholic hepatitis. J Hepatol. 2007;47(2):277-283.
88. Higuera-de la Tijera F, et al. Treatment with metadoxine and its impact on early mortality in patients with severe alcoholic hepatitis. Ann Hepatol. 2014;13(3):343-352.
89. Williams JA, Ding WX. Role of autophagy in alcohol and drug-induced liver injury. Food Chem Toxicol. 2020;136:111075.
90. Chao X, Ding WX. Role and mechanisms of autophagy in alcohol-induced liver injury.
91. Qian H, et al. Autophagy in liver diseases: A review. Mol Aspects Med. 2021;82:100973.
92. Wang X, et al. MicroRNAs as regulators, biomarkers and therapeutic targets in liver diseases. Gut. 2021;70(4):784-795.
93. Buch S, et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat Genet. 2015;47(12):1443-1448.
94. Innes H, et al. Genome-wide association study for alcohol-related cirrhosis identifies risk loci in MARC1 and HNRNPUL1. Gastroenterology. 2020;159(4):1276-1289.
95. Trépo E, et al. Common polymorphism in the PNPLA3/adiponutrin gene confers higher risk of cirrhosis and liver damage in alcoholic liver disease. J Hepatol. 2011;55(4):906-912.
96. Abul-Husn NS, et al. A protein-truncating HSD17B13 variant and protection from chronic liver disease. NEngl JMed. 2018;378(12):1096-1106.
97. Tian C, et al. Variant in PNPLA3 is associated with alcoholic liver disease. Nat Genet. 2010;42(1):21-23.
98. Beaudoin JJ, et al. An exploratory genome-wide analysis of genetic risk for alcoholic hepatitis. Scand J Gastroenterol. 2017;52(11):1263-1269.
99. Schwantes-An TH, et al. Genome-wide association study and meta-analysis on alcohol-associated liver cirrhosis identifies genetic risk factors. Hepatology. 2021;73(5):1920-1931.
100.Deaciuc IV, et al. Large-scale gene profiling of the
liver in a mouse model of chronic, intragastric ethanol infusion. J Hepatol. 2004;40(2):219-227.
101. Affò
102. Xu M-J, et al. Fat-specific protein 27/CIDEC promotes development of alcoholic steatohepatitis in mice and humans. Gastroenterology. 2015;149(4):1030-1041.
103. Yang Z, et al. The role of SHP/REV-ERB
104. Liu X, et al. Primary alcohol-activated human and mouse hepatic stellate cells share similarities in gene-expression profiles. Hepatol Commun. 2020;4(4):606-626.
105. Jiang L, et al. Transcriptomic profiling identifies novel hepatic and intestinal genes following chronic plus binge ethanol feeding in mice. Dig Dis Sci. 2020;65(12):3592-3604.
106. Cao L, et al. Single-cell RNA transcriptome profiling of liver cells of short-term alcoholic liver injury in mice. Int J Mol Sci. 2023;24(5):4344.
107. Khanova E, et al. Pyroptosis by caspase11/4-gas-dermin-D pathway in alcoholic hepatitis in mice and patients. Hepatology. 2018;67(5):1737-1753.
108. Argemi J, et al. Defective HNF4alpha-dependent gene expression as a driver of hepatocellular failure in alcoholic hepatitis. Nat Commun. 2019;10(1):3126.
109. Niu L, et al. Noninvasive proteomic biomarkers for alcohol-related liver disease. Nat Med. 2022;28(6):1277-1287.
110.Argemi J, et al. Integrated transcriptomic and proteomic analysis identifies plasma biomarkers of hepatocellular failure in alcohol-associated hepatitis. Am J Pathol. 2022;192(12):1658-1669.
111. Yang Z, et al. LncRNA AK054921 and AK128652 are potential serum biomarkers and predictors of patient survival with alcoholic cirrhosis. Hepatol Сотmun. 2017;1(6):513-523.
112. Rachakonda V, et al. Serum metabolomic profiling in acute alcoholic hepatitis identifies multiple dysregulated pathways. PLoS One. 2014;9(12):e113860.
113. Massey V, et al. Integrated multiomics reveals glucose use reprogramming and identifies a novel hexokinase in alcoholic hepatitis. Gastroenterology. 2021;160(5):1725-1740.
114. Luther J, et al. The circulating proteomic signature of alcohol-associated liver disease. JCI Insight. 2022;7(14):e159775.
115. Listopad S, et al. Differentiating between liver diseases by applying multiclass machine learning approaches to transcriptomics of liver tissue or blood-based samples. JHEP Rep. 2022;4(10):100560.
116. Harris PS, et al. Proteomic analysis of alcohol-associated hepatitis reveals glycoprotein NMB (GPNMB) as a novel hepatic and serum biomarker. Alcohol. 2022;99:35-48.
117. Hardesty J, et al. Hepatic protein and phosphoprotein signatures of alcohol-associated cirrhosis and hepatitis. Am J Pathol. 2022;192(7):1066-1082.
118. Eguchi A, et al. Extracellular vesicles released by hepatocytes from gastric infusion model of
alcoholic liver disease contain a MicroRNA barcode that can be detected in blood. Hepatology. 2017;65(2):475-490.
119. Trépo E, et al. Combination of gene expression signature and model for end-stage liver disease score predicts survival of patients with severe alcoholic hepatitis. Gastroenterology. 2018;154(4):965-975.
120. Balog S, et al. Emergence of highly profibrotic and proinflammatory Lrat+Fbln2+ HSC subpopulation in alcoholic hepatitis. Hepatology. 2023;78(1):212-224.
121. Ramachandran P , et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature. 2019;575(7783):512-518.
122. Bou Saleh M, et al. Loss of hepatocyte identity following aberrant YAP activation: A key mechanism in alcoholic hepatitis. J Hepatol. 2021;75(4):912-923.
123. Weichselbaum L, et al. Epigenetic basis for monocyte dysfunction in patients with severe alcoholic hepatitis. J Hepatol. 2020;73(2):303-314.
124. Liu M, et al. Super enhancer regulation of cyto-kine-induced chemokine production in alcoholic hepatitis. Nat Commun. 2021;12(1):4560.
125. Hyun J, et al. Epithelial splicing regulatory protein 2-mediated alternative splicing reprograms hepatocytes in severe alcoholic hepatitis. J Clin Invest. 2020;130(4):2129-2145.
126. Li G, et al. Targeting hepatic serine-arginine protein kinase 2 ameliorates alcohol-associated liver disease by alternative splicing control of lipogenesis. Hepatology. 2023;78(5):1506-1524.
127. Guillot A , et al. Deciphering the immune microenvironment on a single archival formalin-fixed paraffin-embedded tissue section by an immediately implementable multiplex fluorescence immunostaining protocol. Cancers (Basel). 2020;12(9):2449.
128. Guillot A, et al. Bile acid-activated macrophages promote biliary epithelial cell proliferation through integrin
129. Tsukamoto H, et al. Intragastric ethanol infusion model in rodents. Methods Mol Biol. 2008;447:33-48.
130. Brandon-Warner E, et al. Rodent models of alcoholic liver disease: of mice and men. Alcohol. 2012;46(8):715-725.
131. Lieber CS, et al. Experimental methods of ethanol administration. Hepatology. 1989;10(4):501-510.
132. Ueno A, et al. Mouse intragastric infusion (iG) model. Nat Protoc. 2012;7(4):771-781.
133. Bertola A, et al. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc. 2013;8(3):627-637.
134. Wang H, et al. Experimental models of fatty liver diseases: Status and appraisal. Hepatol Commun. 2023;7(7):e00200.
135. Gao B, et al. Animal models of alcoholic liver disease: pathogenesis and clinical relevance. Gene Expr. 2017;17(3):173-186.
136. Tsukamoto H, et al. “Second hit” models of alcoholic liver disease. Semin Liver Dis. 2009;29(2):178-187.
137. Zhou Z, et al. Neutrophil-hepatic stellate cell interactions promote fibrosis in experimental steatohepatitis. Cell Mol Gastroenterol Hepatol. 2018;5(3):399-413.
138. Chang B, et al. Short- or long-term high-fat diet feeding plus acute ethanol binge synergistically induce acute liver injury in mice: an important role for CXCL1. Hepatology. 2015;62(4):1070-1085.
139. Yeh Y-T, et al. Single ethanol binge causes severe liver injury in mice fed Western diet. Hepatol Commun. 2023;7(7):e00174.
140. Lazaro R, et al. Osteopontin deficiency does not prevent but promotes alcoholic neutrophilic hepatitis in mice. Hepatology. 2015;61(1):129-140.
141. Schonfeld M, et al. A Western diet with alcohol in drinking water recapitulates features of alco-hol-associated liver disease in mice. Alcohol Clin Exp Res. 2021;45(10):1980-1993.
142. Buyco DG, et al. Concomitant Western diet and chronic-binge alcohol dysregulate hepatic metabolism. PLoS One. 2023;18(5):e0281954.
143. Rinella ME, et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. J Hepatol. 2023;79(6):1542-1556.
144.Israelsen M, et al. MetALD: new opportunities to understand the role of alcohol in steatotic liver disease. Lancet Gastroenterol Hepatol. 2023;8(10):866-868.
145. Díaz LA, et al. The intersection between alco-hol-related liver disease and nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. 2023;20(12):764-783.
146.Vos T, et al. Global burden of 369 diseases and injuries in 204 countries and territories, 1990-2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet. 2020;396(10258):1204-1222.
147. Tapper EB, Parikh ND. Mortality due to cirrhosis and liver cancer in the United States, 1999-2016: observational study. BMJ. 2018;362:k2817.
148. Tapper EB, Parikh ND. Diagnosis and management of cirrhosis and its complications: a review. JAMA. 2023;329(18):1589-1602.
149. Huang DQ, et al. Changing global epidemiology of liver cancer from 2010 to 2019: NASH is the fastest growing cause of liver cancer. Cell Metab. 2022;34(7):969-977.
150. Paik JM, et al. Changes in the global burden of chronic liver diseases from 2012 to 2017: the growing impact of NAFLD. Hepatology. 2020;72(5):1605-1616.
151. Huang DQ, et al. Global epidemiology of alco-hol-associated cirrhosis and HCC: trends, projections and risk factors. Nat Rev Gastroenterol Hepatol. 2023;20(1):37-49.
152. Hagstrom H, et al. Risk of cancer in biopsy-proven alcohol-related liver disease: a popula-tion-based cohort study of 3410 Persons. Clin Gastroenterol Hepatol. 2022;20(4):918-929.
153. Bucci L, et al. Comparison between alcohol- and hepatitis C virus-related hepatocellular carcinoma: clinical presentation, treatment and outcome. Aliment Pharmacol Ther. 2016;43(3):385-399.
154. Costentin CE, et al. Hepatocellular carcinoma is diagnosed at a later stage in alcoholic patients: Results of a prospective, nationwide study. Cancer. 2018;124(9):1964-1972.
155. Costentin CE, et al. Geographical disparities of outcomes of hepatocellular carcinoma in france: the heavier burden of alcohol compared to hepatitis C. Dig Dis Sci. 2020;65(1):301-311.
156. Kuper H, et al. The risk of liver and bile duct cancer
in patients with chronic viral hepatitis, alcoholism, or cirrhosis. Hepatology. 2001;34(4 pt 1):714-718.
157. Mancebo A, et al. Annual incidence of hepatocellular carcinoma among patients with alcoholic cirrhosis and identification of risk groups. Clin Gastroenterol Hepatol. 2013;11(1):95-101.
158. Marrero JA, et al. Diagnosis, staging, and management of hepatocellular carcinoma: 2018 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology. 2018;68(2):723-750.
159. Kann AE, et al. Cause-specific mortality in patients with alcohol-related liver disease in Denmark: a population-based study. Lancet Gastroenterol Hepatol. 2023;8(11):1028-1034.
160. Brandon-Warner E, et al. Chronic ethanol feeding accelerates hepatocellular carcinoma progression in a sex-dependent manner in a mouse model of hepatocarcinogenesis. Alcohol Clin Exp Res. 2012;36(4):641-653.
161. Trepo E, et al. Common genetic variation in alcohol-related hepatocellular carcinoma: a case-control genome-wide association study. Lancet Oncol. 2022;23(1):161-171.
162. Meroni M, et al. Genetic and epigenetic modifiers of alcoholic liver disease. Int J Mol Sci. 2018;19(12):3857.
163. Stickel F, et al. Genetic variants in PNPLA3 and TM6SF2 predispose to the development of hepatocellular carcinoma in individuals with alcohol-related cirrhosis. Am J Gastroenterol. 2018;113(10):1475-1483.
164. Mancina RM, et al. The MBOAT7-TMC4 variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of European descent. Gastroenterology. 2016;150(5):1219-1230.
165. Speliotes EK, et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011;7(3):e1001324.
166. Nischalke HD, et al. A common polymorphism in the NCAN gene is associated with hepatocellular carcinoma in alcoholic liver disease. J Hepatol. 2014;61(5):1073-1079.
167. Sakamoto T, et al. Influence of alcohol consumption and gene polymorphisms of ADH2 and ALDH2 on hepatocellular carcinoma in a Japanese population. Int J Cancer. 2006;118(6):1501-1507.
168. Abe H, et al. Aldehyde dehydrogenase 2 polymorphism for development to hepatocellular carcinoma in East Asian alcoholic liver cirrhosis. J Gastroenterol Hepatol. 2015;30(9):1376-1383.
169. Homann N , et al. Alcohol dehydrogenase
170.Schmalz F, et al. High producer variant of lipoprotein lipase may protect from hepatocellular carcinoma in alcohol-associated cirrhosis. JHEP Rep. 2023;5(4):100684.
171. Innes H, et al. The rs429358 locus in apolipoprotein E is associated with hepatocellular carcinoma in patients with cirrhosis. Hepatol Commun. 2022;6(5):1213-1226.
172. Buch S, et al. Genetic variation in TERT modifies the risk of hepatocellular carcinoma in alco-hol-related cirrhosis: results from a genome-wide case-control study. Gut. 2023;72(2):381-391.
173. Stickel F, et al. The genetics of alcohol dependence and alcohol-related liver disease. J Hepatol. 2017;66(1):195-211.
174. Park SH, et al. Ethanol and its nonoxidative metabolites promote acute liver injury by inducing ER Stress, adipocyte death, and lipolysis. Cell Mol Gastroenterol Hepatol. 2023;15(2):281-306.
175. Wang W, et al. Aldehyde dehydrogenase, liver disease and cancer. Int J Biol Sci. 2020;16(6):921-934.
176. Polimanti R, Gelernter J. ADH1B: From alcoholism, natural selection, and cancer to the human phenome. Am J Med Genet B Neuropsychiatr Genet. 2018;177(2):113-125.
177. Charatcharoenwitthaya P, et al. Alcohol-Associated Liver Disease: East Versus West. Clin Liver Dis (Hoboken). 2020;16(6):231-235.
178. Chang JS, et al. ALDH2 polymorphism and alco-hol-related cancers in Asians: a public health perspective. J Biomed Sci. 2017;24(1):19.
179. Chen YC, et al. Pharmacokinetic and pharmacodynamic basis for overcoming acetaldehyde-induced adverse reaction in Asian alcoholics, heterozygous for the variant ALDH2*2 gene allele. Pharmacogenet Genomics. 2009;19(8):588-599.
180. Kwon HJ, et al. Aldehyde dehydrogenase 2 deficiency ameliorates alcoholic fatty liver but worsens liver inflammation and fibrosis in mice. Hepatology. 2014;60(1):146-157.
181. Gao Y, et al. Alcohol inhibits T-cell glucose metabolism and hepatitis in ALDH2-deficient
mice and humans: roles of acetaldehyde and glucocorticoids. Gut. 2019;68(7):1311-1322.
182.Szabo G. Women and alcoholic liver disease warning of a silent danger. Nat Rev Gastroenterol Hepatol. 2018;15(5):253-254.
183. Delacote C, et al. A model to identify heavy drinkers at high risk for liver disease progression. Clin Gastroenterol Hepatol. 2020;18(10):2315-2323.
184.Singal AK, Mathurin P. Diagnosis and treatment of alcohol-associated liver disease: a review. JAMA. 2021;326(2):165-176.
185. Pemmasani G , et al. Sex differences in clinical characteristics and outcomes associated with alcoholic hepatitis. Eur J Gastroenterol Hepatol. 2023;35(10):1192-1196.
186.Ilyas F, et al. Rising alcohol-associated liver disease-related mortality rates in the United States from 1999 to 2022. Hepatol Commun. 2023;7(7):e00180.
187. Rehm J, et al. Alcohol as a risk factor for liver cirrhosis: a systematic review and meta-analysis. Drug Alcohol Rev. 2010;29(4):437-445.
188. Anouti A, Mellinger JL. The changing epidemiology of alcohol-associated liver disease: gender, race, and risk factors. Semin Liver Dis. 2023;43(1):50-59.
189. Hwang S, et al. Interleukin-22 ameliorates neutrophil-driven nonalcoholic steatohepatitis through multiple targets. Hepatology. 2020;72(2):412-429.
190.Katsuda T, et al. Rapid in vivo multiplexed editing (RIME) of the adult mouse liver. Hepatology. 2023;78(2):486-502.
191. Kirpich IA, et al. Saturated and unsaturated dietary fats differentially modulate ethanol-induced changes in gut microbiome and metabolome in a mouse model of alcoholic liver disease. Am J Pathol. 2016;186(4):765-776.
192. Kim HS, et al. Synergistic associations of PNPLA3 I148M variant, alcohol intake, and obesity with risk of cirrhosis, hepatocellular carcinoma, and mortality. JAMA Netw Open. 2022;5(10):e2234221.
193. Shukla SD, et al. Binge ethanol and liver: new molecular developments. Alcohol Clin Exp Res. 2013;37(4):550-557.
194. Wu X, et al. Macrophage-derived MLKL in alcohol-associated liver disease: Regulation of phagocytosis. Hepatology. 2023;77(3):902-919.
195. Wang L, et al. STING-mediated inflammation contributes to Gao binge ethanol feeding model. J Cell Physiol. 2022;237(2):1471-1485.
196. Qian H, et al. Loss of SQSTM1/p62 induces obesity and exacerbates alcohol-induced liver injury in aged mice. Cell Mol Gastroenterol Hepatol. 2023;15(5):1027-1049.
197. Wu M, et al. Hepatocyte-specific deletion of cellular repressor of E1A-stimulated genes 1 exacerbates alcohol-induced liver injury by activating stress kinases. Int J Biol Sci. 2022;18(4):1612-1626.
- Authorship note: BM and YF contributed equally to this work.
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2024, Mackowiak et al. This is an open access article published under the terms of the Creative Commons Attribution 4.0 International License.
Reference information: / Clin Invest. 2024;134(3):e176345.
https://doi.org/10.1172/JCI176345. - Julien J, et al. Projected prevalence and mortality associated with alcohol-related liver disease in the USA, 2019-40: a modelling study. Lancet Public Health. 2020;5(6):e316-e323.
- Asrani SK, et al. Reducing the global burden of alcohol-associated liver disease: a blueprint for action. Hepatology. 2021;73(5):2039-2050.
- Devarbhavi H, et al. Global burden of liver dis-
ease: 2023 update. J Hepatol. 2023;79(2):516-537. - Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology. 2011;141(5):1572-1585.