DOI: https://doi.org/10.1038/s41467-024-45106-4
PMID: https://pubmed.ncbi.nlm.nih.gov/38296948
تاريخ النشر: 2024-01-31
مسار أوليغوميرازا مدفوع بالنانوحبس لإزالة فعالة للملوثات الفينولية عبر تفاعل شبيه بفنتون
تم القبول: 15 يناير 2024
نُشر على الإنترنت: 31 يناير 2024
(أ) التحقق من التحديثات
الملخص
تمثل تفاعل فينتون غير المتجانس واحدة من أكثر التقنيات موثوقية لضمان سلامة المياه، ولكنها تواجه حالياً تحديات تتمثل في بطء اختزال Fe(III)، والإدخال المفرط للمواد الكيميائية لتعدين المواد العضوية، وانبعاث الكربون غير المرغوب فيه. تركز الجهود الحالية لتحسين الأداء التحفيزي لتفاعل فينتون في الغالب على كيفية تسريع اختزال Fe(III)، بينما يتم تجاهل خطوة تحلل الملوثات عادةً. هنا، نبلغ عن استراتيجية نانو احتجاز باستخدام هلام الجرافين (GA) لدعم UiO-66-
النتائج
توصيف المحفزات

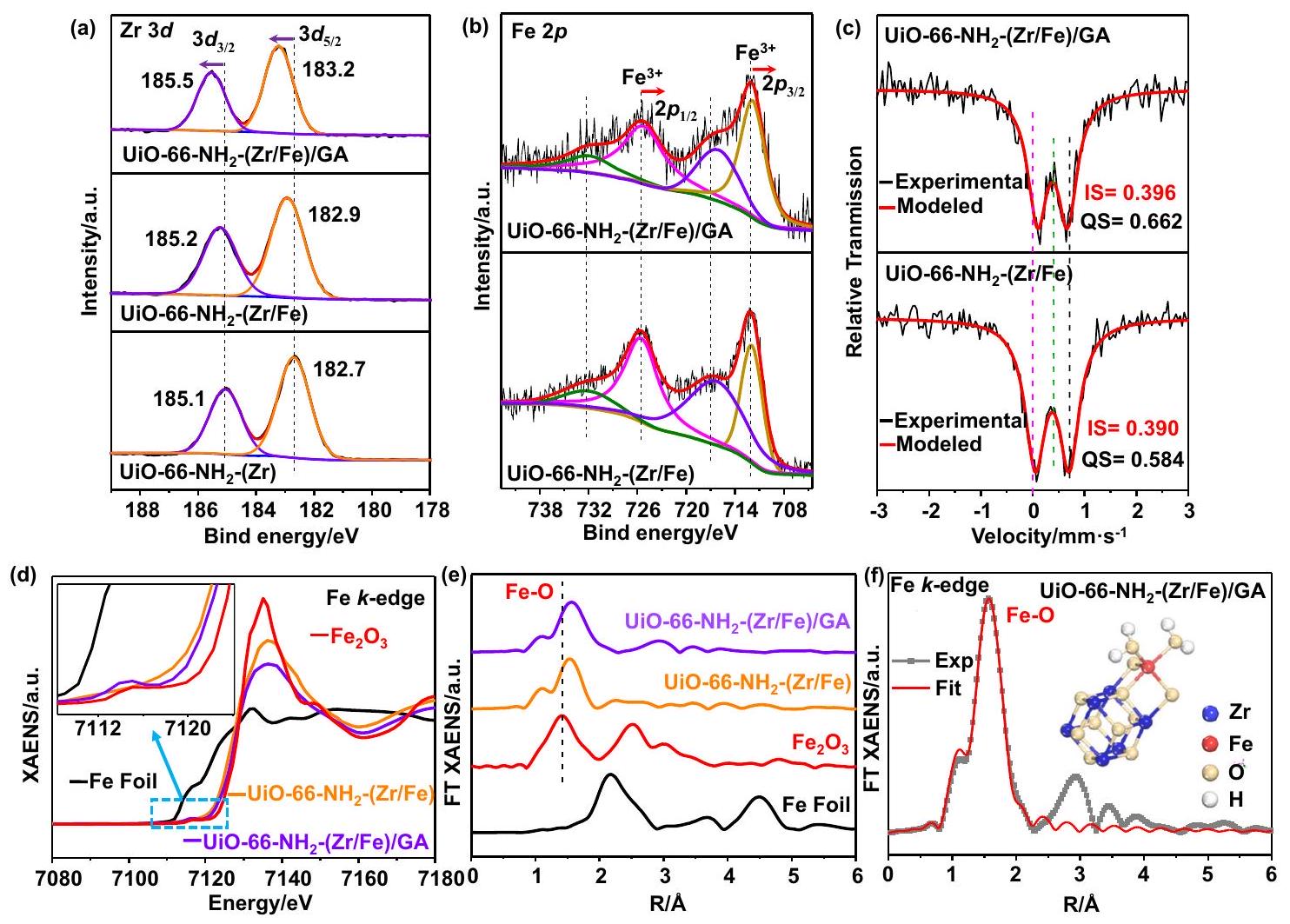
طيف) و (e) FT-EXAFS الحديد
تم توضيح الهيكل والشكل الخارجي لـ GA بمفرده في الشكل التوضيحي الإضافي 4.
بدون
مواقع حافة امتصاص الحديد في كلا العينتين قريبة جدًا من تلك الخاصة بـ
عدد ذرات الحديد يقل قليلاً من 4.9 في
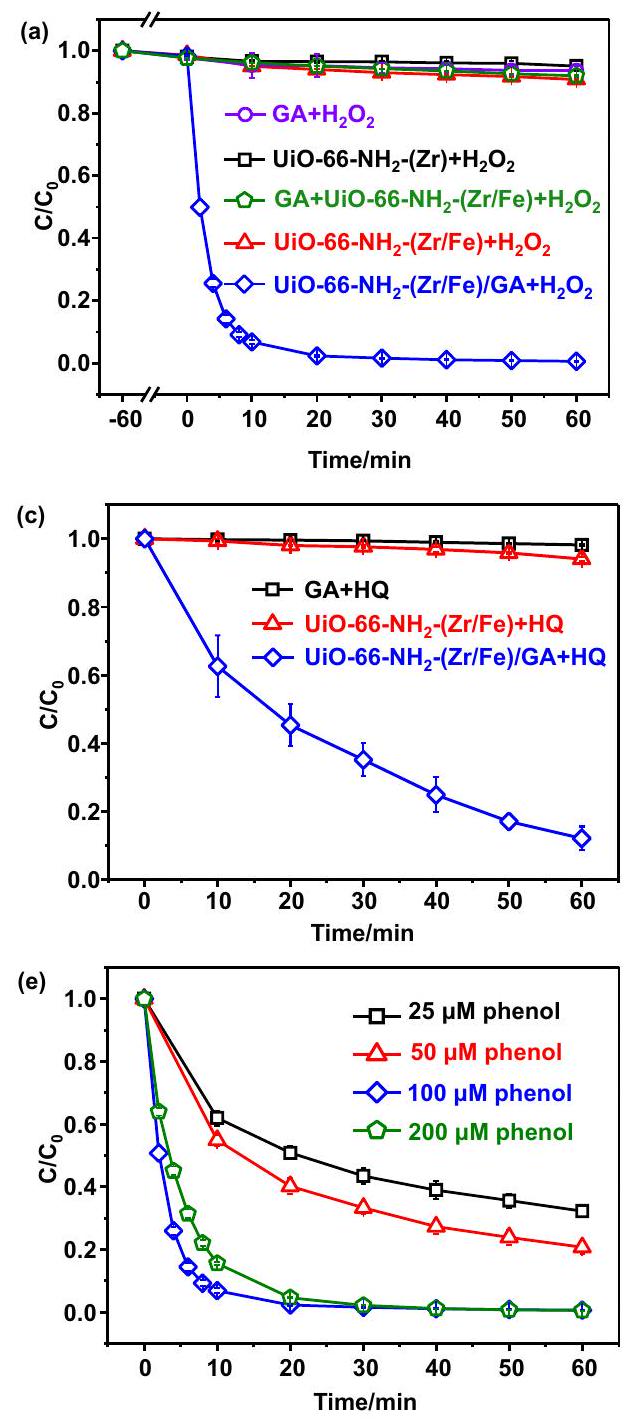
تفاعلات تحفيزية شبيهة بفينتون لإزالة الملوثات
(
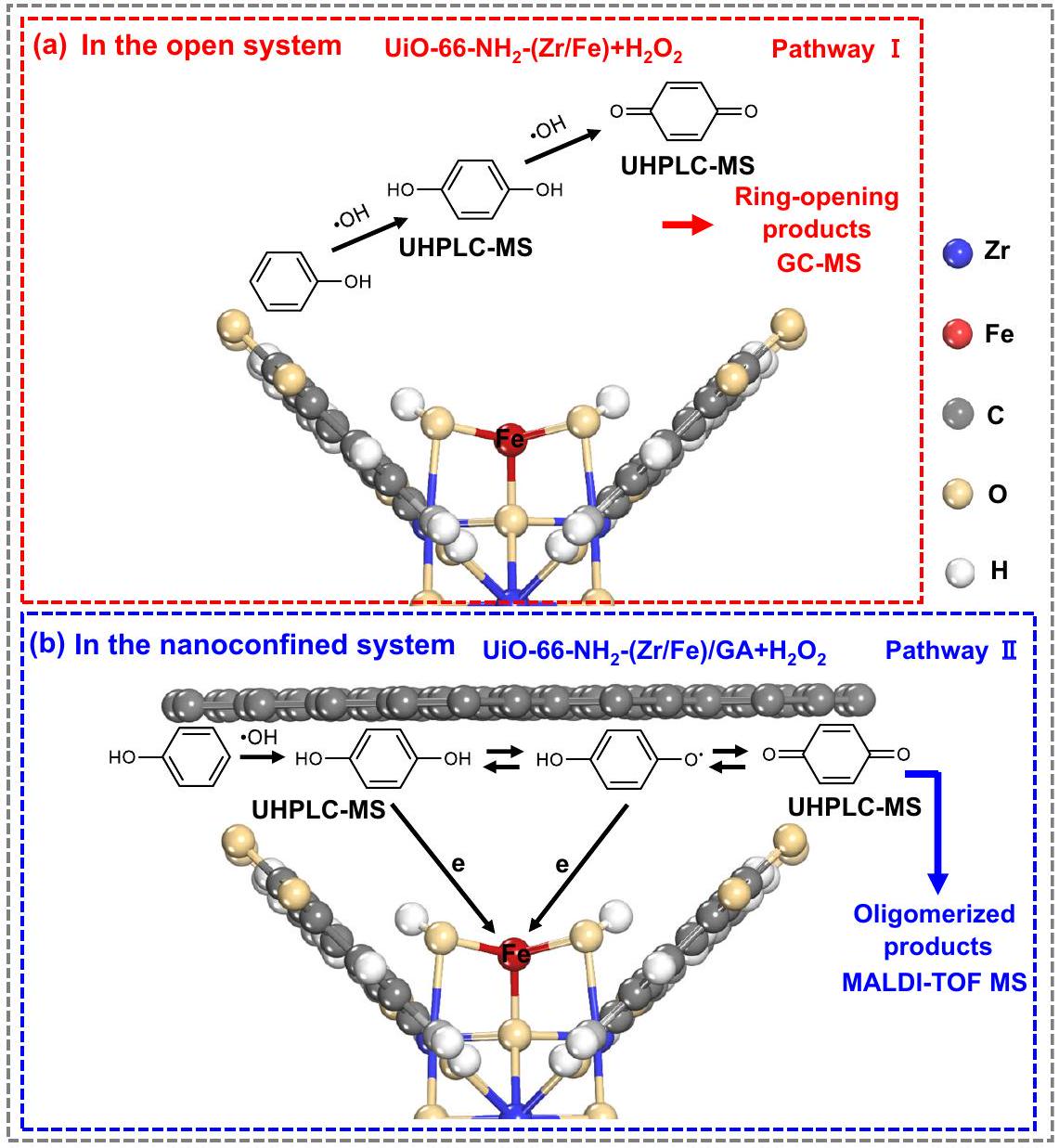
التحقيق الميكانيكي أثناء إزالة الملوثات
( MeOH ) كـ
نقاش
mولارية
طرق
المواد الكيميائية
الخصائص
تحضير المحفزات
غسل المنتج بالإيثانول والماء ثلاث مرات، تلتها عملية التجفيف بالتجميد لمدة 24 ساعة لإنتاج UiO-66-
فحص المحفزات
توفر البيانات
References
- Tang, Z. M. et al. Biomedicine meets Fenton chemistry. Chem. Rev. 121, 1981-2019 (2021).
- Liu, Q. et al. A Fenton reaction at the endoplasmic reticulum is involved in the redox control of hypoxia-inducible gene expression. Proc. Natl Acad. Sci. USA 101, 4302-4307 (2004).
- Liu, L. et al.
sensitized 1530 nm to 1180 nm second near-infrared window upconversion nanocrystals for in vivo biosensing. Angew. Chem. Int. Ed. 57, 7518-7522 (2018). - Feng, G. D. et al. Accelerated crystallization of zeolites via hydroxyl free radicals. Science 351, 1188-1191 (2016).
- Dai, X. C. et al. Sustainable co-synthesis of glycolic acid, formamides and formates from 1, 3-dihydroxyacetone by a
catalyst with a single active site. Angew. Chem. Int. Ed. 58, 5251-5255 (2019). - Liu, T. C. et al. Water decontamination via nonradical process by nanoconfined Fenton-like catalysts. Nat. Commun. 14, 2881 (2023).
- Xiao, F. et al. Selective electrocatalytic reduction of oxygen to hydroxyl radicals via 3-electron pathway with FeCo alloy encapsulated carbon aerogel for fast and complete removing pollutants. Angew. Chem. Int. Ed. 60, 10375-10383 (2021).
- Zhang, S. et al. Mechanism of heterogeneous Fenton reaction kinetics enhancement under nanoscale spatial confinement. Environ. Sci. Technol. 54, 10868-10875 (2020).
- Hodges, B. C., Cates, E. L. & Kim, J. H. Challenges and prospects of advanced oxidation water treatment processes using catalytic nanomaterials. Nat. Nanotechnol. 13, 642-650 (2018).
- Yang, Z. et al. The Fenton reaction in water assisted by picolinic acid: Accelerated iron cycling and co-generation of a selective Febased oxidant. Environ. Sci. Technol. 55, 8299-8308 (2021).
- Chen, Y., Miller, C. J. & Waite, T. D. Heterogeneous Fenton chemistry revisited: Mechanistic insights from ferrihydrite-mediated oxidation of formate and oxalate. Environ. Sci. Technol. 55, 14414-14425 (2021).
- Yang, X. J. et al. Iron oxychloride (FeOCl): an efficient Fenton-like catalyst for producing hydroxyl radicals in degradation of organic contaminants. J. Am. Chem. Soc. 135, 16058-16061 (2013).
- Hou, X. et al. Hydroxylamine promoted goethite surface Fenton degradation of organic pollutants. Environ. Sci. Technol. 51, 5118-5126 (2017).
- Su, L. et al. Regulating local electron density of iron single sites by introducing nitrogen vacancies for efficient photo-Fenton process. Angew. Chem. Int. Ed. 60, 21261-21266 (2021).
- Mao, Y. et al. Accelerating Fe
-aqua complex reduction in an efficient solid-liquid-interfacial Fenton reaction over the Mn-CNH co-catalyst at near-neutral pH. Environ. Sci. Technol. 55, 13326-13334 (2021). - Yan, Q. et al. Constructing an acidic microenvironment by
in heterogeneous Fenton reaction for pollutant control. Angew. Chem. Int. Ed. 60, 17155-17163 (2021). - Ling, C. et al. Atomic-layered
nanoclusters on with dual catalytic sites for efficient and selective activation. Angew. Chem. Int. Ed. 61, e202200670 (2022). - Yang, Z. et al. Singlet oxygen mediated iron-based Fenton-like catalysis under nanoconfinement. Proc. Natl Acad. Sci. USA 116, 6659-6664 (2019).
- Wang, J. et al. Interlayer structure manipulation of iron oxychloride by potassium cation intercalation to steer
activation pathway. J. Am. Chem. Soc. 144, 4294-4299 (2022). - Henzler-Wildman, K. A. et al. Intrinsic motions along an enzymatic reaction trajectory. Nature 450, 838-844 (2007).
- Huang, Y., Ren, J. & Qu, X. Nanozymes: classification, catalytic mechanisms, activity regulation, and applications. Chem. Rev. 119, 4357-4412 (2019).
- Kapil, V. et al. The first-principles phase diagram of monolayer nanoconfined water. Nature 609, 512-516 (2022).
- Grommt, A. B., Feller, M. & Klajn, R. Chemical reactivity under nanoconfinement. Nat. Nanotechnol. 15, 256-271 (2020).
- Li, H. et al. Confined catalysis under two-dimensional materials. Proc. Natl Acad. Sci. USA 114, 5930-5934 (2017).
- Wang, Q. Q. et al. Self-assembled nanospheres with multiple endohedral binding sites pre-organize catalysts and substrates for highly efficient reactions. Nat. Chem. 8, 225-230 (2016).
- Jin, Z. et al. Hydrophobic zeolite modification for in situ peroxide formation in methane oxidation to methanol. Science 367, 193-197 (2020).
- Smit, B. & Maesen, T. L. M. Towards a molecular understanding of shape selectivity. Nature 451, 671-678 (2008).
- Zhang, Q., Gao, S. Q. & Yu, J. H. Metal sites in zeolites: synthesis, characterization, and catalysis. Chem. Rev. 123, 6039-6106 (2023).
- Dusselier, M. et al. Shape-selective zeolite catalysis for bioplastics production. Science 349, 78-80 (2015).
- Riscoe, A. R. et al. Transition state and product diffusion control by polymer-nanocrystal hybrid catalysts. Nat. Catal. 2, 852-863 (2019).
- Zhuang, T. T. et al. Copper nanocavities confine intermediates for efficient electrosynthesis of C3 alcohol fuels from carbon monoxide. Nat. Catal. 1, 946-951 (2018).
- Zang, Y. et al. Selective
electroreduction to ethanol over a carbon-coated catalyst. Angew. Chem. Int. Ed. 61, e202209629 (2022). - Manna, K. et al. Chemoselective single-site earth-abundant metal catalysts at metal-organic framework nodes. Nat. Commun. 7, 12610 (2016).
- Abdel-Mageed, A. M. et al. Highly Active and stable single-atom Cu catalysts supported by a metal-organic framework. J. Am. Chem. Soc. 141, 5201-5210 (2019).
- Ma, X. et al. Modulating Coordination environment of single-atom catalysts and their proximity to photosensitive units for boosting MOF photocatalysis. J. Am. Chem. Soc. 143, 12220-12229 (2021).
- Ferrighi, L. et al. Catalysis under cover: enhanced reactivity at the interface between (doped) graphene and anatase
. J. Am. Chem. Soc. 138, 7365-7376 (2016). - Virmani, E. et al. On-surface synthesis of highly oriented thin metalorganic framework films through vapor-assisted conversion. J. Am. Chem. Soc. 140, 4812-4819 (2018).
- Xu, C. et al. Turning on visible-light photocatalytic C-H oxidation over metal-organic frameworks by introducing metal-to-cluster charge transfer. J. Am. Chem. Soc. 141, 19110-19117 (2019).
- Vleet Van, M. J. et al. In situ, time-resolved, and mechanistic studies of metal-organic framework nucleation and growth. Chem. Rev. 118, 3681-3721 (2018).
- Li, C. X. et al. Ultralight covalent organic framework/graphene aerogels with hierarchical porosity. Nat. Commun. 11, 4712 (2020).
- Zhang, Y. et al. Weakly hydrophobic nanoconfinement by graphene aerogels greatly enhances the reactivity and ambient stability of reactivity of MIL-101-Fe in Fenton-like reaction. Nano Res. 14, 2383-2389 (2021).
- Ye, G. et al. In situ implanting of single tungsten sites in to defective UiO-66(Zr) by solvent-free route for efficient oxidative desulfurizationat at room temperature. Angew. Chem. Int. Ed. 60, 20318-20324 (2021).
- Pan, J. B. et al. Activity and stability boosting of an oxygen-vacancyrich
photoanode by NiFe-MOFs thin layer for water oxidation. Angew. Chem. Int. Ed. 60, 1433-1440 (2021). - Tang, J. & Wang, J. Metal organic framework with coordinatively unsaturated sites as efficient Fenton-like catalyst for enhanced degradation of sulfamethazine. Environ. Sci. Technol. 52, 5367-5377 (2018).
- Yang, C. et al. A semiconducting layered metal-organic framework magnet. Nat. Commun. 10, 3260 (2019).
- Bao, Y. et al. Generating high-valent iron-oxo
complexes in neutral microenvironments through peroxymonosulfate activation by Zn -Fe layered double hydroxides. Angew. Chem. Int. Ed. 61, e202209542 (2022). - Zhang, W. et al. Redox-active metal-organic composites for highly selective oxygen separation applications. Adv. Mater. 28, 3572-3577 (2016).
- Zhao, W. et al. Fe-O clusters anchored on nodes of metal-organic frameworks for direct methane oxidation. Angew. Chem. Int. Ed. 60, 5811-5815 (2021).
- Feng,
. et al. Creation of exclusive artificial cluster defects by selective metal removal in the ( ) mixed-metal UiO-66. J. Am. Chem. Soc. 143, 21511-21518 (2021). - Yang, Y. et al. Which micropollutants in water environments deserve more attention globally? Environ. Sci. Technol. 56, 13-29 (2022).
- Zazo, J. A. et al. Chemical pathway and kinetics of phenol oxidation by Fenton’s reagent. Environ. Sci. Technol. 39, 9295-9302 (2005).
- Chen, L. et al. Interaction between organic compounds and catalyst steers the oxidation pathway and mechanism in the iron oxidebased heterogeneous Fenton system. Environ. Sci. Technol. 56, 14059-14068 (2022).
- Chen, R. Z. & Pignatello, J. J. Role of quinone intermediates as electron shuttles in Fenton and photoassisted Fenton oxidations of aromatic compounds. Environ. Sci. Technol. 31, 2399-2406 (1997).
- Duesterberg, C. K. & Waite, T. D. Kinetic modeling of the oxidation of p-hydroxybenzoic acid by Fenton’s reagent: implications of the role of quinones in the redox cycling of iron. Environ. Sci. Technol. 41, 4103-4110 (2007).
- Li, Y. et al. Interactive enhancements of ascorbic acid and iron in hydroxyl radical generation in quinone redox cycling. Environ. Sci. Technol. 46, 10302-10309 (2012).
- Yun, E. T. et al. Exploring the role of persulfate in the activation process: radical precursor versus electron acceptor. Environ. Sci. Technol. 51, 10090-10099 (2017).
- Zhang, A. J. et al. Oxidative polymerization of hydroquinone using deoxycholic acid supramolecular template. Sci. China Chem. 55, 830-835 (2012).
- Razimov, A. Z., Bekmashi, F. T. & Liogon’kii, B. I. Thermal polymerization of p-benzoquinone. Polym. Sci. U. S. S. R. 17, 3164-3170 (1975).
- Qiu, X. et al. Cleaving arene rings for acyclic alkenylnitrile synthesis. Nature 597, 64-69 (2021).
- Shearer, G. C. et al. Defect engineering: tuning the porosity and composition of the metal-organic framework UiO-66 via modulated synthesis. Chem. Mater. 28, 3749-3761 (2016).
الشكر والتقدير
مساهمات المؤلفين
المصالح المتنافسة
معلومات إضافية
المواد التكميلية المتاحة على
https://doi.org/10.1038/s41467-024-45106-4.
http://www.nature.com/reprints
© المؤلفون 2024
مختبر جيانغسو الرئيسي للتحكم في تلوث المواد الكيميائية وإعادة استخدام الموارد، كلية الهندسة البيئية والبيولوجية، جامعة نانجينغ للعلوم والتكنولوجيا، نانجينغ 210094، الصين. مركز أبحاث تكنولوجيا النانو البيئية (ReCENT)، المختبر الرئيسي للدولة للتحكم في التلوث وإعادة استخدام الموارد، كلية البيئة، جامعة نانجينغ، نانجينغ 210023، الصين. مختبر أكاديمي رئيسي لعلوم وهندسة تحويل الكربون المنخفض، معهد شنغهاي للأبحاث المتقدمة، الأكاديمية الصينية للعلوم، شنغهاي 201210، الصين. كلية الهندسة البيئية، جامعة ووشي، جيانغسو 214105، جمهورية الصين الشعبية. ساهم هؤلاء المؤلفون بالتساوي: شيانغ تشانغ، جينغجينغ تانغ، لينغ لينغ وانغ. البريد الإلكتروني:qianjieshu@foxmail.com; bcpan@nju.edu.cn - صور لـ UiO-66-
. صور (e) SEM و (f) TEM التمثيلية لـ UiO- . صورة HRTEM (g)، وأنماط SAED (h)، وصور رسم خرائط العناصر HAADF و EDS (i) لـ UiO-66- .
DOI: https://doi.org/10.1038/s41467-024-45106-4
PMID: https://pubmed.ncbi.nlm.nih.gov/38296948
Publication Date: 2024-01-31
Nanoconfinement-triggered oligomerization pathway for efficient removal of phenolic pollutants via a Fenton-like reaction
Accepted: 15 January 2024
Published online: 31 January 2024
(A) Check for updates
Abstract
Heterogeneous Fenton reaction represents one of the most reliable technologies to ensure water safety, but is currently challenged by the sluggish Fe(III) reduction, excessive input of chemicals for organic mineralization, and undesirable carbon emission. Current endeavors to improve the catalytic performance of Fenton reaction are mostly focused on how to accelerate Fe(III) reduction, while the pollutant degradation step is habitually overlooked. Here, we report a nanoconfinement strategy by using graphene aerogel (GA) to support UiO-66-
Results
Characterization of catalysts
spectra) and (e) FT-EXAFS Fe
structure and morphology of the GA alone are illustrated in Supplementary Fig. 4.
without
positions of the Fe absorption edge in both samples are very close to that of
number of Fe atom reduces slightly from 4.9 in
Fenton-like catalytic reactions for pollutant removal
(
Mechanistic investigation during pollutant removal
( MeOH ) as the typical
Discussion
molar mass
Methods
Reagents
Characterizations
Preparation of Catalysts
washed by ethanol and water three times, followed by freeze drying for 24 h to generate UiO-66-
Examination of catalysts
Data availability
References
- Tang, Z. M. et al. Biomedicine meets Fenton chemistry. Chem. Rev. 121, 1981-2019 (2021).
- Liu, Q. et al. A Fenton reaction at the endoplasmic reticulum is involved in the redox control of hypoxia-inducible gene expression. Proc. Natl Acad. Sci. USA 101, 4302-4307 (2004).
- Liu, L. et al.
sensitized 1530 nm to 1180 nm second near-infrared window upconversion nanocrystals for in vivo biosensing. Angew. Chem. Int. Ed. 57, 7518-7522 (2018). - Feng, G. D. et al. Accelerated crystallization of zeolites via hydroxyl free radicals. Science 351, 1188-1191 (2016).
- Dai, X. C. et al. Sustainable co-synthesis of glycolic acid, formamides and formates from 1, 3-dihydroxyacetone by a
catalyst with a single active site. Angew. Chem. Int. Ed. 58, 5251-5255 (2019). - Liu, T. C. et al. Water decontamination via nonradical process by nanoconfined Fenton-like catalysts. Nat. Commun. 14, 2881 (2023).
- Xiao, F. et al. Selective electrocatalytic reduction of oxygen to hydroxyl radicals via 3-electron pathway with FeCo alloy encapsulated carbon aerogel for fast and complete removing pollutants. Angew. Chem. Int. Ed. 60, 10375-10383 (2021).
- Zhang, S. et al. Mechanism of heterogeneous Fenton reaction kinetics enhancement under nanoscale spatial confinement. Environ. Sci. Technol. 54, 10868-10875 (2020).
- Hodges, B. C., Cates, E. L. & Kim, J. H. Challenges and prospects of advanced oxidation water treatment processes using catalytic nanomaterials. Nat. Nanotechnol. 13, 642-650 (2018).
- Yang, Z. et al. The Fenton reaction in water assisted by picolinic acid: Accelerated iron cycling and co-generation of a selective Febased oxidant. Environ. Sci. Technol. 55, 8299-8308 (2021).
- Chen, Y., Miller, C. J. & Waite, T. D. Heterogeneous Fenton chemistry revisited: Mechanistic insights from ferrihydrite-mediated oxidation of formate and oxalate. Environ. Sci. Technol. 55, 14414-14425 (2021).
- Yang, X. J. et al. Iron oxychloride (FeOCl): an efficient Fenton-like catalyst for producing hydroxyl radicals in degradation of organic contaminants. J. Am. Chem. Soc. 135, 16058-16061 (2013).
- Hou, X. et al. Hydroxylamine promoted goethite surface Fenton degradation of organic pollutants. Environ. Sci. Technol. 51, 5118-5126 (2017).
- Su, L. et al. Regulating local electron density of iron single sites by introducing nitrogen vacancies for efficient photo-Fenton process. Angew. Chem. Int. Ed. 60, 21261-21266 (2021).
- Mao, Y. et al. Accelerating Fe
-aqua complex reduction in an efficient solid-liquid-interfacial Fenton reaction over the Mn-CNH co-catalyst at near-neutral pH. Environ. Sci. Technol. 55, 13326-13334 (2021). - Yan, Q. et al. Constructing an acidic microenvironment by
in heterogeneous Fenton reaction for pollutant control. Angew. Chem. Int. Ed. 60, 17155-17163 (2021). - Ling, C. et al. Atomic-layered
nanoclusters on with dual catalytic sites for efficient and selective activation. Angew. Chem. Int. Ed. 61, e202200670 (2022). - Yang, Z. et al. Singlet oxygen mediated iron-based Fenton-like catalysis under nanoconfinement. Proc. Natl Acad. Sci. USA 116, 6659-6664 (2019).
- Wang, J. et al. Interlayer structure manipulation of iron oxychloride by potassium cation intercalation to steer
activation pathway. J. Am. Chem. Soc. 144, 4294-4299 (2022). - Henzler-Wildman, K. A. et al. Intrinsic motions along an enzymatic reaction trajectory. Nature 450, 838-844 (2007).
- Huang, Y., Ren, J. & Qu, X. Nanozymes: classification, catalytic mechanisms, activity regulation, and applications. Chem. Rev. 119, 4357-4412 (2019).
- Kapil, V. et al. The first-principles phase diagram of monolayer nanoconfined water. Nature 609, 512-516 (2022).
- Grommt, A. B., Feller, M. & Klajn, R. Chemical reactivity under nanoconfinement. Nat. Nanotechnol. 15, 256-271 (2020).
- Li, H. et al. Confined catalysis under two-dimensional materials. Proc. Natl Acad. Sci. USA 114, 5930-5934 (2017).
- Wang, Q. Q. et al. Self-assembled nanospheres with multiple endohedral binding sites pre-organize catalysts and substrates for highly efficient reactions. Nat. Chem. 8, 225-230 (2016).
- Jin, Z. et al. Hydrophobic zeolite modification for in situ peroxide formation in methane oxidation to methanol. Science 367, 193-197 (2020).
- Smit, B. & Maesen, T. L. M. Towards a molecular understanding of shape selectivity. Nature 451, 671-678 (2008).
- Zhang, Q., Gao, S. Q. & Yu, J. H. Metal sites in zeolites: synthesis, characterization, and catalysis. Chem. Rev. 123, 6039-6106 (2023).
- Dusselier, M. et al. Shape-selective zeolite catalysis for bioplastics production. Science 349, 78-80 (2015).
- Riscoe, A. R. et al. Transition state and product diffusion control by polymer-nanocrystal hybrid catalysts. Nat. Catal. 2, 852-863 (2019).
- Zhuang, T. T. et al. Copper nanocavities confine intermediates for efficient electrosynthesis of C3 alcohol fuels from carbon monoxide. Nat. Catal. 1, 946-951 (2018).
- Zang, Y. et al. Selective
electroreduction to ethanol over a carbon-coated catalyst. Angew. Chem. Int. Ed. 61, e202209629 (2022). - Manna, K. et al. Chemoselective single-site earth-abundant metal catalysts at metal-organic framework nodes. Nat. Commun. 7, 12610 (2016).
- Abdel-Mageed, A. M. et al. Highly Active and stable single-atom Cu catalysts supported by a metal-organic framework. J. Am. Chem. Soc. 141, 5201-5210 (2019).
- Ma, X. et al. Modulating Coordination environment of single-atom catalysts and their proximity to photosensitive units for boosting MOF photocatalysis. J. Am. Chem. Soc. 143, 12220-12229 (2021).
- Ferrighi, L. et al. Catalysis under cover: enhanced reactivity at the interface between (doped) graphene and anatase
. J. Am. Chem. Soc. 138, 7365-7376 (2016). - Virmani, E. et al. On-surface synthesis of highly oriented thin metalorganic framework films through vapor-assisted conversion. J. Am. Chem. Soc. 140, 4812-4819 (2018).
- Xu, C. et al. Turning on visible-light photocatalytic C-H oxidation over metal-organic frameworks by introducing metal-to-cluster charge transfer. J. Am. Chem. Soc. 141, 19110-19117 (2019).
- Vleet Van, M. J. et al. In situ, time-resolved, and mechanistic studies of metal-organic framework nucleation and growth. Chem. Rev. 118, 3681-3721 (2018).
- Li, C. X. et al. Ultralight covalent organic framework/graphene aerogels with hierarchical porosity. Nat. Commun. 11, 4712 (2020).
- Zhang, Y. et al. Weakly hydrophobic nanoconfinement by graphene aerogels greatly enhances the reactivity and ambient stability of reactivity of MIL-101-Fe in Fenton-like reaction. Nano Res. 14, 2383-2389 (2021).
- Ye, G. et al. In situ implanting of single tungsten sites in to defective UiO-66(Zr) by solvent-free route for efficient oxidative desulfurizationat at room temperature. Angew. Chem. Int. Ed. 60, 20318-20324 (2021).
- Pan, J. B. et al. Activity and stability boosting of an oxygen-vacancyrich
photoanode by NiFe-MOFs thin layer for water oxidation. Angew. Chem. Int. Ed. 60, 1433-1440 (2021). - Tang, J. & Wang, J. Metal organic framework with coordinatively unsaturated sites as efficient Fenton-like catalyst for enhanced degradation of sulfamethazine. Environ. Sci. Technol. 52, 5367-5377 (2018).
- Yang, C. et al. A semiconducting layered metal-organic framework magnet. Nat. Commun. 10, 3260 (2019).
- Bao, Y. et al. Generating high-valent iron-oxo
complexes in neutral microenvironments through peroxymonosulfate activation by Zn -Fe layered double hydroxides. Angew. Chem. Int. Ed. 61, e202209542 (2022). - Zhang, W. et al. Redox-active metal-organic composites for highly selective oxygen separation applications. Adv. Mater. 28, 3572-3577 (2016).
- Zhao, W. et al. Fe-O clusters anchored on nodes of metal-organic frameworks for direct methane oxidation. Angew. Chem. Int. Ed. 60, 5811-5815 (2021).
- Feng,
. et al. Creation of exclusive artificial cluster defects by selective metal removal in the ( ) mixed-metal UiO-66. J. Am. Chem. Soc. 143, 21511-21518 (2021). - Yang, Y. et al. Which micropollutants in water environments deserve more attention globally? Environ. Sci. Technol. 56, 13-29 (2022).
- Zazo, J. A. et al. Chemical pathway and kinetics of phenol oxidation by Fenton’s reagent. Environ. Sci. Technol. 39, 9295-9302 (2005).
- Chen, L. et al. Interaction between organic compounds and catalyst steers the oxidation pathway and mechanism in the iron oxidebased heterogeneous Fenton system. Environ. Sci. Technol. 56, 14059-14068 (2022).
- Chen, R. Z. & Pignatello, J. J. Role of quinone intermediates as electron shuttles in Fenton and photoassisted Fenton oxidations of aromatic compounds. Environ. Sci. Technol. 31, 2399-2406 (1997).
- Duesterberg, C. K. & Waite, T. D. Kinetic modeling of the oxidation of p-hydroxybenzoic acid by Fenton’s reagent: implications of the role of quinones in the redox cycling of iron. Environ. Sci. Technol. 41, 4103-4110 (2007).
- Li, Y. et al. Interactive enhancements of ascorbic acid and iron in hydroxyl radical generation in quinone redox cycling. Environ. Sci. Technol. 46, 10302-10309 (2012).
- Yun, E. T. et al. Exploring the role of persulfate in the activation process: radical precursor versus electron acceptor. Environ. Sci. Technol. 51, 10090-10099 (2017).
- Zhang, A. J. et al. Oxidative polymerization of hydroquinone using deoxycholic acid supramolecular template. Sci. China Chem. 55, 830-835 (2012).
- Razimov, A. Z., Bekmashi, F. T. & Liogon’kii, B. I. Thermal polymerization of p-benzoquinone. Polym. Sci. U. S. S. R. 17, 3164-3170 (1975).
- Qiu, X. et al. Cleaving arene rings for acyclic alkenylnitrile synthesis. Nature 597, 64-69 (2021).
- Shearer, G. C. et al. Defect engineering: tuning the porosity and composition of the metal-organic framework UiO-66 via modulated synthesis. Chem. Mater. 28, 3749-3761 (2016).
Acknowledgements
Author contributions
Competing interests
Additional information
supplementary material available at
https://doi.org/10.1038/s41467-024-45106-4.
http://www.nature.com/reprints
© The Author(s) 2024
Jiangsu Key Laboratory of Chemical Pollution Control and Resources Reuse, School of Environmental and Biological Engineering, Nanjing University of Science and Technology, Nanjing 210094, China. Research Center for Environmental Nanotechnology (ReCENT), State Key Laboratory of Pollution Control and Resources Reuse, School of Environment, Nanjing University, Nanjing 210023, China. CAS key Laboratory of Low-carbon Conversion Science and Engineering, Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai 201210, China. School of Environmental Engineering, Wuxi University, Jiangsu 214105, P. R. China. These authors contributed equally: Xiang Zhang, Jingjing Tang, Lingling Wang. e-mail: qianjieshu@foxmail.com; bcpan@nju.edu.cn - images of UiO-66-
. Representative (e) SEM and (f) TEM images of UiO- . g HRTEM image, (h) SAED patterns, and (i) High-angle annular dark field (HAADF) and EDS elemental mapping images of UiO-66- .