مواقع أزواج الأيونات لاستخراج اليورانيوم بكفاءة من المياه النووية العادمة الحقيقية Ion pair sites for efficient electrochemical extraction of uranium in real nuclear wastewater
تمثل عملية استخراج اليورانيوم الكهروكيميائي من مياه الصرف النووي استراتيجية ناشئة لإعادة تدوير موارد اليورانيوم. ومع ذلك، في إنتاج الوقود النووي الذي ينتج غالبية مياه الصرف النووي المحتوية على اليورانيوم، أيون الفلوريد () يتواجد مع اليورانيوم ( )، مما أدى إلى الأنواع المعقدة من وبالتالي تقليل كفاءة الاستخراج. هنا، نقوم ببناءمواقع استخراج أزواج الأيونات فيلاستخراج اليورانيوم الكهربائي الفعال من مياه الصرف الصحي الناتجة عن إنتاج الوقود النووي. ترتبط هذه المواقع بشكل انتقائي معمن خلال التيتانيوم-فلوريد المركب والمتعدد الروابط. في استخراج اليورانيوم، تمر أنواع اليورانيوم بانتقال بلوري من إلى. في مياه الصرف النووي الحقيقية، يتم استخراج اليورانيوم كهربائياً بكفاءة عالية من وأخيرًا تم تنقيته كمسحوق أكسيد اليورانيوم، مما يتوافق مع قدرة الاستخراج لـدون تشبع. يمهد هذا العمل طريقًا فعالًا لإعادة تدوير اليورانيوم الكهروكيميائي في مياه الصرف الصحي الحقيقية الناتجة عن الإنتاج النووي.
اليورانيوم هو المورد الأساسي الرئيسي في صناعة النوويةمع تطور صناعة النووية، ستنفد موارد اليورانيوم في خام الأرض خلال أقل من قرن، مصحوبة بتوليد هائل لمياه الصرف النووي المحتوية على اليورانيوم.. في الحالات العملية، يتم إنتاج الغالبية العظمى من مياه الصرف النووي المحتوية على اليورانيوم من إجراءات إنتاج الوقود، مثل تخصيب اليورانيوم، وتحويل اليورانيوم، وتصنيع عناصر الوقود، والتي تتطلب الاستخدام الواسع لفلوريد اليورانيوم.“. بعد التحلل المائي لفلوريد اليورانيوم، يوجد اليورانيوم في مياه الصرف النووي عادةً في شكل يورانيوم ( )، جنبًا إلى جنب مع التركيز العالي المتزامن لأيون الفلورايد . وبالتالي، استخراج اليورانيوم تحت تأثير تركيزات عالية من هو قضية مهمة لحماية البيئة وإعادة تدوير موارد اليورانيوم. بالنسبة لمياه الصرف النووي هذه، تتطلب طريقة الامتزاز التقليدية أو تبادل الأيونات عادةً الترسيب المسبق لـبواسطة، مما أدى إلى تشكيل مواد تحتوي على اليورانيوم كنفايات صلبة مشعةوبناءً عليه، البحث عن تقنيات أخرى لاستخراج اليورانيوم مطلوب بشدة تحت تأثير التركيزات العالية من.
كأحد التقنيات الناشئة، جذبت عملية استخراج اليورانيوم الكهروكيميائية اهتمامًا متزايدًا بسبب السرعة العالية للتفاعلات، وزيادة سعة الاستخراج، ومقاومتها لتداخل الأنيونات.في الوقت الحاضر، تم تطبيق استخراج اليورانيوم الكهربائي في الأنظمة المائية دون. على سبيل المثال، الـذرات مفردة مع مجموعات أميدوكسيتم الإبلاغ عن تحقيق قدرة استخراج قدرهامن مياه البحر الحقيقية على مدى 24 ساعة. ومع ذلك، على الرغم من التقدم الكبير، فإن استخراج اليورانيوم الكهربائي في مياه الصرف النووي الحقيقية مع وجودلا يزال التحدي قائمًا، بسبب الأنواع المعقدة نسبيًا من اليورانيوم نتيجة تأثير التنسيق.. على وجه التحديد، اليورانيوم السداسي التكافؤ المسيطر (تتواجد الأنواع عمومًا على سلسلة من فلوريد اليورانيوممثل، و بدلاً من العاري. في هذه الحالة، تعاني مواقع استخراج اليورانيوم التقليدية من التنافس في تنسيق اليورانيوم و، مما يؤدي إلى انخفاض كفاءة الاستخراج
من اليورانيوم تحت تأثير تركيزات عالية من. لذلك، فإن إنشاء مواقع محددة لربط هو شرط أساسي لتحقيق استخراج اليورانيوم الكهربائي الفعال في مياه الصرف النووي الحقيقية ذات التركيزات العالية من .
في هذا، نطور الـمعمواقع استخراج أزواج الأيونات للربط الانتقائي لـواستخراج اليورانيوم الكهروكيميائي الفعال تحت تأثير تركيز عالٍ منكل من بنية الامتصاص الدقيق لأشعة السنكروترون السينية (XAFS) والمحاكاة النظرية تظهر أنمواقع استخراج أزواج الأيونات مرتبطة بقوة معمن خلال التفاعلات المشتركة لـ و . خلال استخراج اليورانيوم، الـيتم تقليصه إلىكترسب رمادي يتبعه أكسدة إضافية وتبلور. في 400 مل من المياه النووية الحقيقية الناتجة عن إنتاج الوقود، يحقق كفاءة استخراج كهروكيميائية منلليورانيوم خلال 7 ساعات، مع قدرة استخراج قدرهادون تشبع. يتم جمع المسحوق المحتوي على اليورانيوم بنجاح من مياه الصرف النووي الحقيقية، معنسبة اليورانيوم بين العناصر المعدنية في المسحوق.
النتائج
بناءمع مواقع أزواج الأيونات
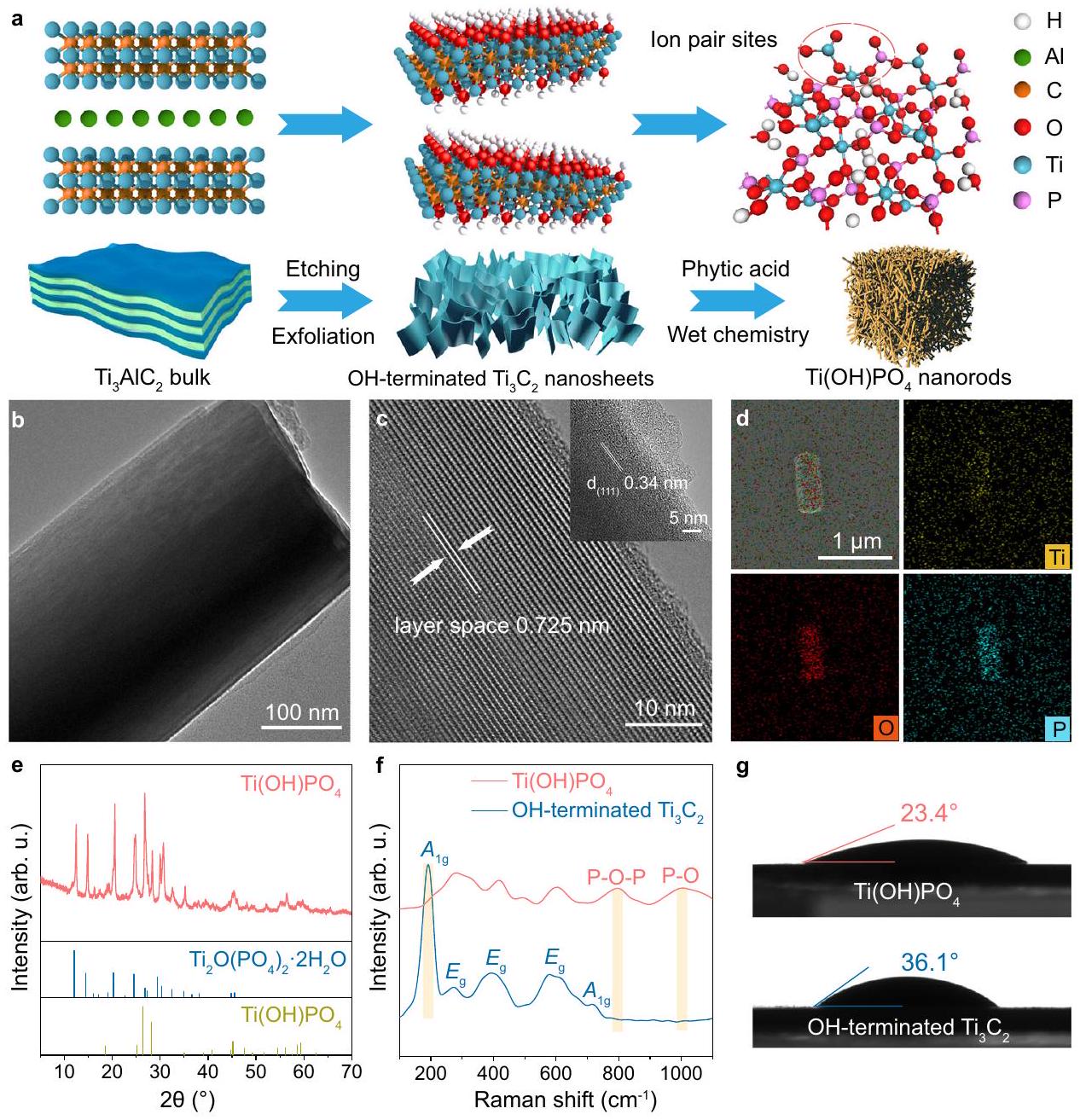
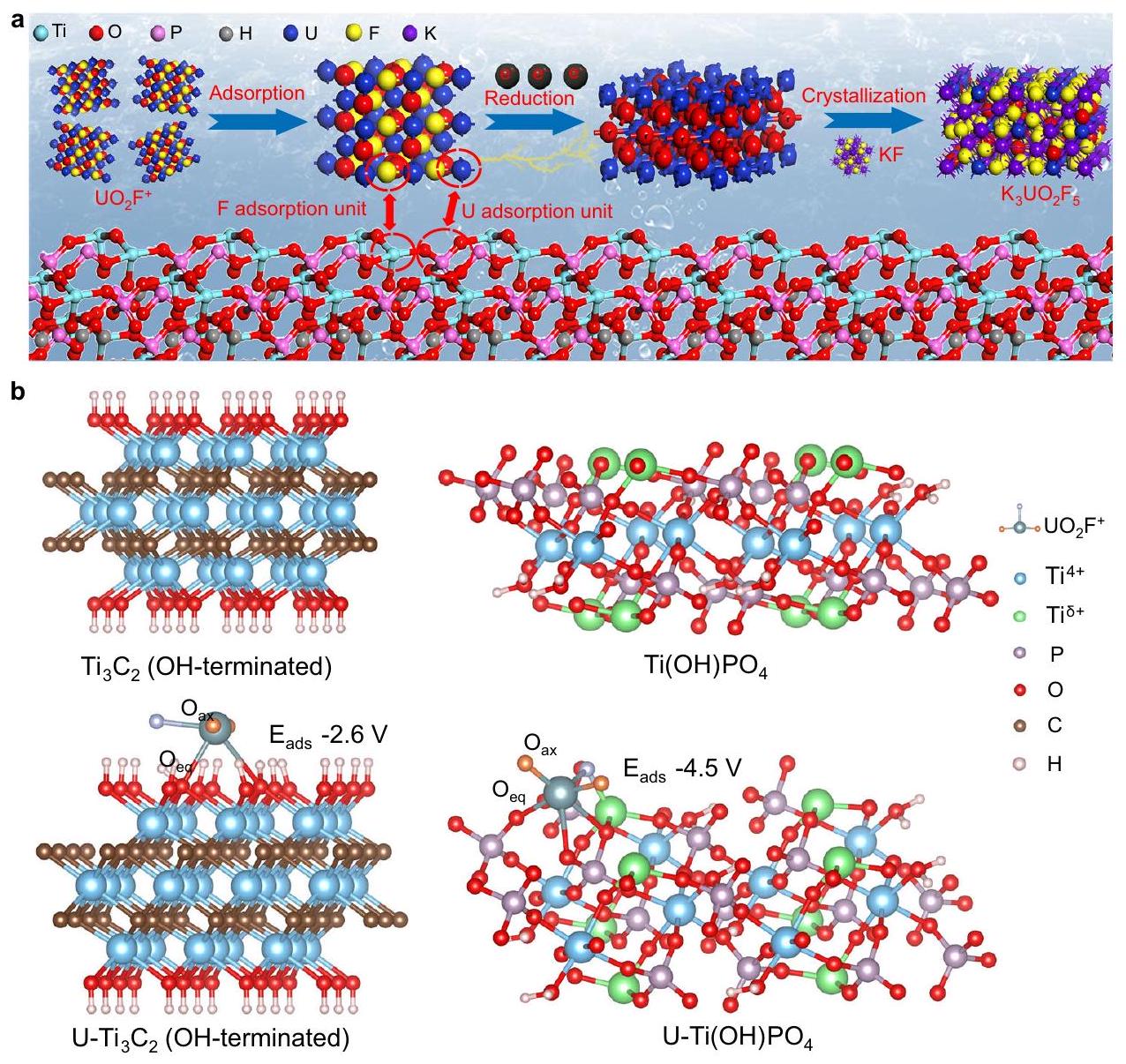
نشأت بناء مواقع أزواج الأيونات من إعادة بناء OH المنتهية اللاميلية.كما هو موضح في الشكل 1أ. في البداية، الألمنيوم طبقات من البضائع التجارية بالجملةتم نقشها لتشكيل كومة على شكل أكورديون من F-المُنهيةالأغشية النانوية. تليها عملية التقشير تحتالحماية في المحلول المائي، المتراص ذو النهاية F-تم تحويل النانوصفائح إلى طبقات نهائية مغطاة بـ OHالأغشية النانوية (الشكل التكميلي 1). الطبقات المرنة المنفصلة المنتهية بـ OH-تم تأكيد وجود الأوراق النانوية من خلال صور المجهر الإلكتروني الناقل عالي الدقة (HRTEM) ونمط حيود الأشعة السينية (XRD) (الشكل التوضيحي 2). كما هو موضح من خلال قياس طيف الأشعة تحت الحمراء باستخدام تحويل فورييه (FT-IR)، فإن الطبقات المتقشرة المنتهية بـ OH كانت الأوراق النانوية تفتقر إلى ذروة تمدد C-F عندبالنسبة إلى المكدس الشبيه بالأكورديونالأغشية النانوية، مما يظهر استبدال نهايات -F بنهايات -OH خلال عملية التقشير (الشكل التكميلي 3). بعد ذلك، تم تطبيق معالجة كيميائية رطبة لتحويل الطبقات المقشرة المنتهية بـ OH-إلى، الذي احتوى على زوج من الأيونات و كما هو موضح في صور المجهر الإلكتروني الناقل (TEM)، أظهر Ti(OH)PO4 شكلًا يشبه القضبان النانوية (الشكل 1b). تحليل المجهر القوة الذرية (AFM) والرسم البياني ثلاثي الأبعاد المقابل لـأشارت القضبان النانوية إلى توزيع الارتفاع الضيق بسبب التكديس المنتظم (الشكل التكميلي 4). أظهر الشكل 1c صورة HRTEM لهيكل قائم بذاته.نانوكريستال. الأظهرت القضبان النانوية هيكلًا دوريًا بطبقات متتالية مع
الشكل 1 | إنشاء مواقع أزواج الأيونات. أ الرسم التخطيطي لتحضير الـمع مواقع أزواج الأيونات.صورة TEM لـأنابيب نانوية. صورة HRTEM لـأنابيب نانوية. الصورة المرفقة: صورة HRTEM لحدودأنابيب نانوية. تحليل EDS لـأنابيب نانوية. نمط XRD، طيف رامان، وزاوية الاتصال الثابتةالأنابيب النانوية والطبقات
منتهي بـ OHالأغشية النانوية. المناطق المميزة في الشكل. إذا أظهرتذروة الطرف OHوقمم P-O-P و P-O، على التوالي. الحقوق: أ (نماذج تخطيطية) حقوق الطبع والنشر لشركة هانغتشو SPHERE للتكنولوجيا المحدودة. يتم توفير بيانات المصدر كملف بيانات مصدر.
الشكل 2 | الترابط والبنية الإلكترونية لمواقع أزواج الأيونات. أ طيف FT-IR، ب طيف ESR، ج Tiطيف XPS، وطيف XANES منأنابيب نانوية وOH نهائية طبقيةالأغشية النانوية. مخطط الطاقة لـ تقسيم المدارات لـذروة. و تمثل مستويين فرعيين مختلفين فيغلاف ذرة التيتانيوم، على التوالي. و تمثل انقسام مستويات الطاقة لـالمدارات في مجال التنسيق. يتم توفير بيانات المصدر كملف بيانات المصدر. فاصل يبلغ 0.725 نانومتر، وهو أكبر من قطر اليورانيوم أو فلوريد اليورانيوم ( ). كانت الصورة المرفقة هي منطقة الحافة لهيكل القضيب، الذي عرض حافة الشبكة مع تباعد الطبقات البينية بمقدار 0.34 نانومتر وأشارت إلى وجوه (111) من تم تحديده بواسطة مطيافية الأشعة السينية المشتتة للطاقة (EDS)، حيث ملأت عناصر التيتانيوم والأكسجين والفوسفور حدود المنطقة المختارة، مما يشير إلى التوزيع المتجانس للمكونات العنصرية فينانورود (الشكل 1د).
المرحلة الفيزيائية لـتم التحقق من القضبان النانوية بشكل إضافي. ظهرت مجموعتان من قمم الحيود في نمط XRD لـالنانو القضبان (الشكل 1e). تم تحديد مجموعتين من القمم المميزة على أنها المونوكلاين القياسي المرحلة (JCPDS #360697) و (JCPDS #52-1529). من الجدير بالذكر أن كان تفاعل الديمرة لـبعد عملية إزالة الماء، مما يدل على الهيدروكسيل القابل للتحويل فيالشكل 1f يوضح طيف رامان لـو OH المنتهي اللاملمي. بعد إعادة بناء OH -المُنهيةإلى، الـذروة OH-المُنتهية النقيةاختفى، مصحوبًا بالق peaks المميزة لـالسندات وسندات. علاوة على ذلك، عرضت زاوية تماس ثابتة مع الماء قدرها، والذي كان أقل من ذلك في الطبقات النقيةمُنهى (الشكل 1g). نتج عن هذه الظاهرة دمج الهيدروكسيل في الشبكة مع تعزيز الروابط الهيدروجينية في، مما يوضح بشكل أكبر وجود شكل من .
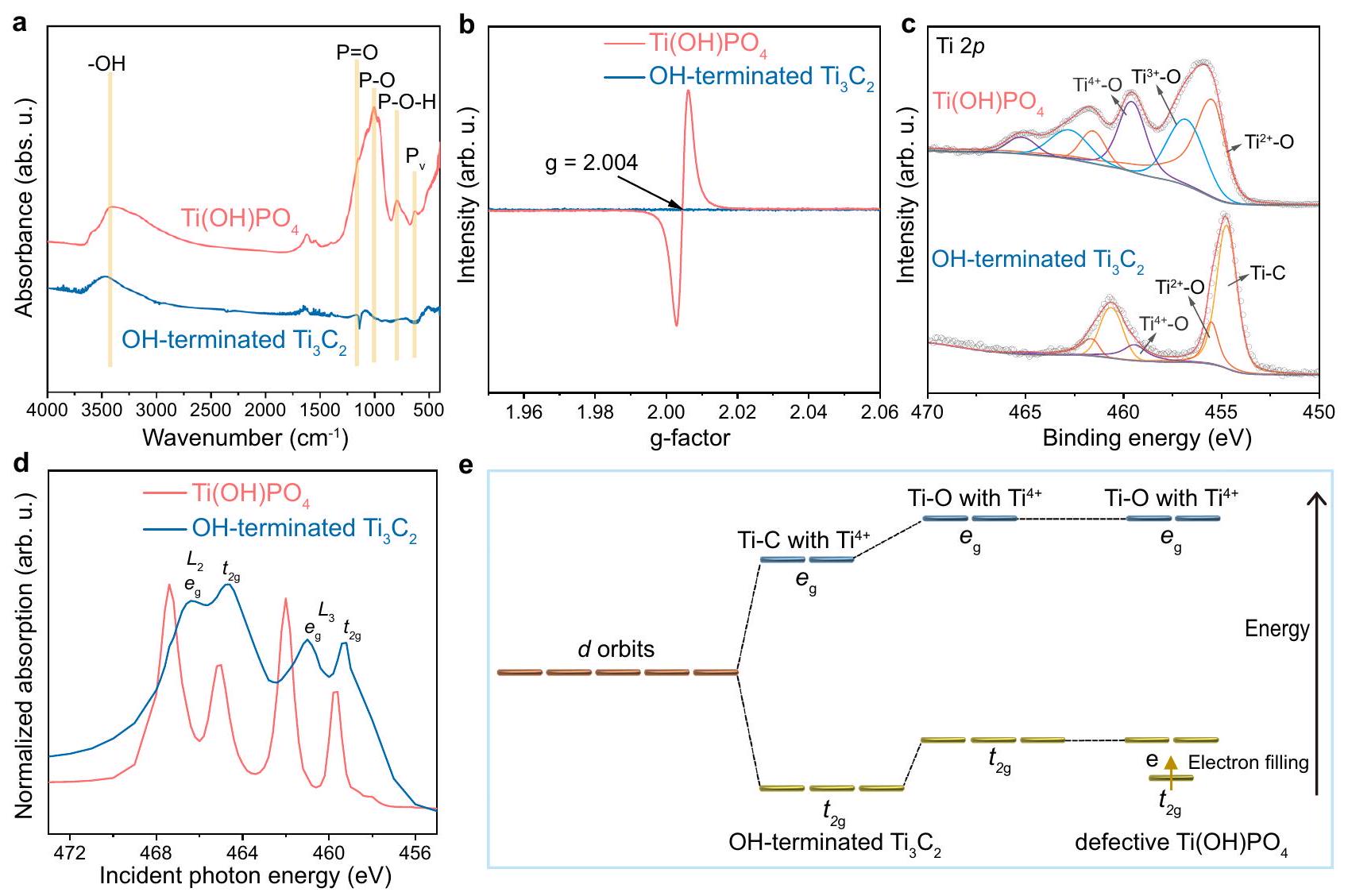
مواقع أزواج الأيونات فيتم التحقيق فيها من خلال تحليل الروابط والبنية الإلكترونية لـتم تحديدها بواسطة قياس FT-IR (الشكل 2أ)، اهتزازات الشد لـ، و P-O-H في تمت ملاحظة المجموعة في طيفالذي اختلف عن ذلك الخاص بـ OH -المُنهية. في دوران الإلكترون طيف الرنين (ESR) (الشكل 2ب)،أظهر إشارة رنين حادة عند عامل g يبلغ 2.004، مما يدل على وجود فراغات أكسجين ضخمة فيعلاوة على ذلك، تم إجراء تحليل طيف الإلكترون السيني (XPS) للتحقق من الروابط الكيميائية لـ، الذي عرض ، وإشارات O في طيف المسح (الشكل التوضيحي 5a). الحالة الأكسدية للـ P في الـتم تأكيد المجموعة بواسطةطيف من (الشكل التوضيحي الإضافي 5ب). في تم ملاحظة ذروة الأكسجين في نطاق الطيف بالقرب من فراغ الأكسجين عند 531.2 إلكترون فولت، مما يوضح الطبيعة المعيبة لـ (الشكل التوضيحي التكميلي 5c). في هذه الأثناء، امتلكت الإشارة السائدة لرابطة Ti-C، مع الإشارة الطفيفة لـ و الروابط، التي تم نسبها إلى إنهاء OH (الشكل 2c). بالمقارنة، فإن Ti طيف منأظهرت ثلاث مجموعات من القمم، التي تم تعيينها على أنها، و الكتلة الكبيرة ذات القيمة المنخفضة ( ) تشير إلى الطبيعة المعيبة لـ التي تم إنشاؤها بواسطة فراغات الأكسجين خلال التحول منمُنهىإلى.
لإظهار التركيب الإلكتروني للتيتانيوم فيالنانو القضبان، قمنا باختبارطيف الامتصاص بالأشعة السينية بالقرب من الحافة (XANES) المنتهي بالهيدروكسيل و . كما هو موضح في الشكل 2d، كلا من حافة وحافةأظهرت القضبان النانوية انزياحًا طفيفًاإلىانتقال وتحول دراماتيكي لـإلىالانتقال بالنسبة إلى OH -المُنهى النقي. تم توضيح هذه النتيجة من خلال مخطط الطاقة لمدارات الانقسام لـ Tiذروة (الشكل 2e). عادةً، كان التيتانيوم يمتلك خمسة مدارات، التي يمكن تقسيمها إلى اثنينمدارات وثلاثةمداراتطاقة الربط لـ و كانت المدارات مرتبطة بعاملين، بما في ذلك بيئة التنسيق وملء الإلكترونات لـ Ti. بالنسبة لبيئة التنسيق، فإن التحول منفي
الشكل 3 | استخراج اليورانيوم الكهربائي من مواقع أزواج الأيونات. أ اختبارات LSV على-منتهيوأقطاب من شعيرات الجرافيت العارية.يمثل الإمكانية. ب كفاءة الاستخراج الكهروكيميائي لـعلىمُنهى“، وأقطاب مصنوعة من شعيرات الجرافيت العارية. الإدراج: خطوط ترسيب ماكروسكوبية من اليورانيوم البلوري. استقرار الاستخراج الكهروكيميائي لـ U(VI) علىإلكترودتحت ظروف pH مختلفة،مع مختلف الـــ الأنواع الأيونية الموجودة.التغيير في تركيز اليورانيوم بعد استخراج اليورانيوم الكهروكيميائي تحت المحاليل الكهربائية المختلفةتركيزات (تتراوح منإلى ). كفاءة الاستخراج الكهروكيميائي لـعلىكم concentration الأولية لـتفاوتت منإلىتمثل أشرطة الخطأ الانحراف المعياري لثلاث قياسات. يتم توفير بيانات المصدر كملف بيانات مصدر.
منتهي بـ OHإلى Ti-O فيأدى إلى التحول الإيجابي لكل من و مدارات في التيتانيوم-edge XANES، بسبب الكهربية السالبة الأعلى للأكسجين مقارنة بالكربون. علاوة على ذلك، بالنسبة لعامل ملء الإلكترون،ممتلك فارغ و مدارات، في حين أن العيوبيمتلك وفرة و التي كان لديها على التوالي إلكترون واحد وإلكترونين فيمدارات. بسبب الطاقة المنخفضة لـمدارات منالمدارات، الإلكترونات الإضافية فيكانت تقع فيالمدارات، مما يؤدي إلى الانخفاض الدراماتيكي في الطاقة لـمدارات. كنتيجة شاملة لبيئة التنسيق وملء الإلكترونات لـ Ti،ذروة في أظهر تحولًا واضحًا، في حين أن أظهر الذروة تحولاً ضئيلاً بالنسبة إلى المجموعات المنتهية بـ OHفي-edge XANES (الجدول التكميلي 1). الـ و الـ مجموعة فيتم إنشاء مواقع أزواج الأيونات، التي كانت الأنواع الرئيسية لاستخراج اليورانيوم تحت تأثير التركيز العالي من.
استخراج اليورانيوم الكهربائي الكيميائي
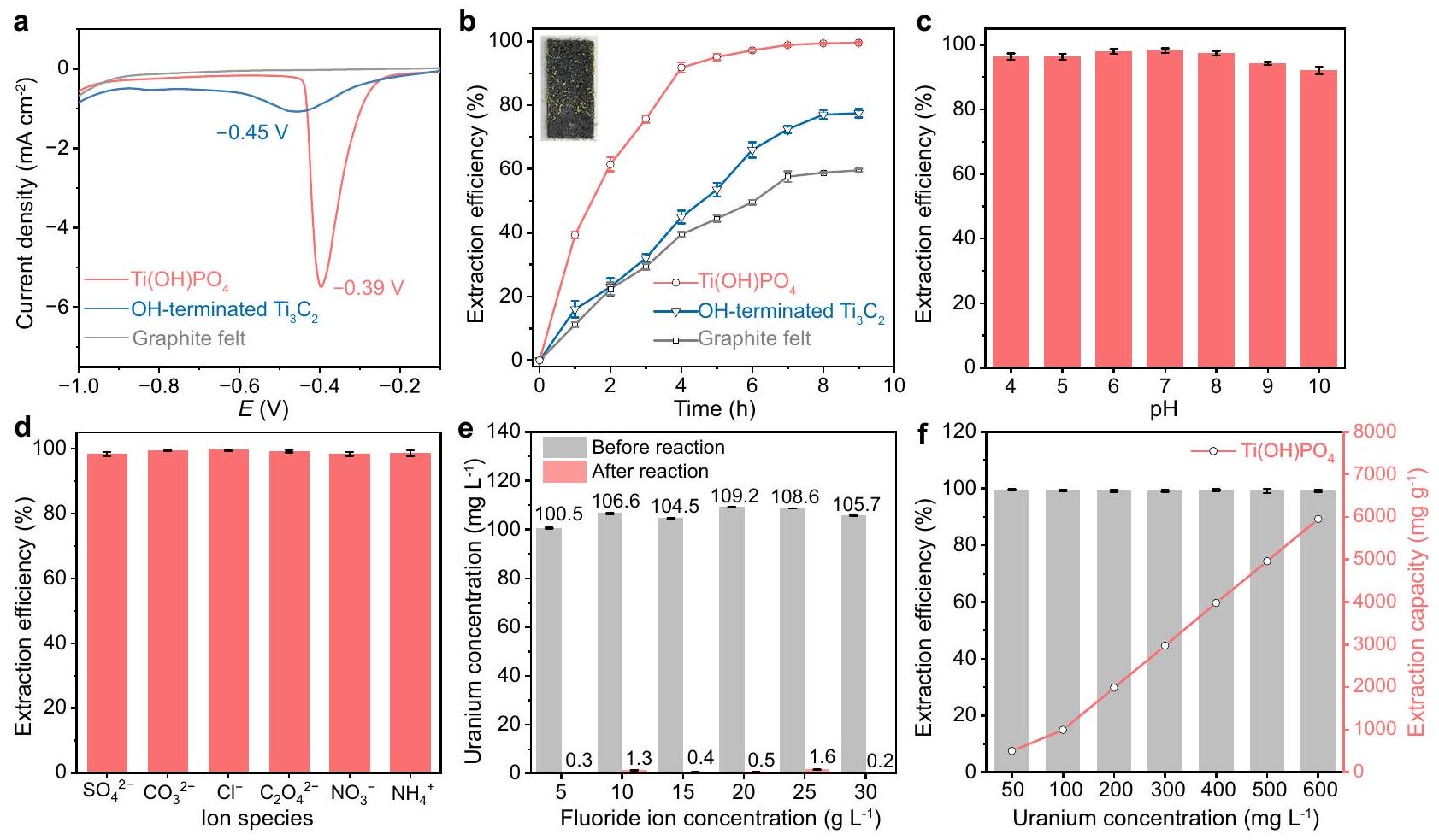
التم توزيع الأنابيب النانوية بشكل متساوٍ على شعيرات الجرافيت لتقييم تأثير مواقع أزواج الأيونات على استخراج اليورانيوم الكهروكيميائي. محلول مائي يحتوي علىمن و منتم اعتمادها كإلكتروليت لمحاكاة مياه الصرف الناتجة عن الإنتاج النووي. لقد أظهرنا ذروة الانخفاض لـفي هذا المحلول الكهربائي بواسطة اختبار الفولتمترية ذات المسح الخطي (LSV) (الشكل 3أ).أظهرت القضبان النانوية قمة انخفاض حادة عند -0.39 فولت مقابل.الذي يتوافق مع تقليصإلى اليورانيوم الخماسي التكافؤ. بالمقارنة، فإن هذه القمم البكرمُنهىوكانت الألياف الجرافيتية العارية تقع في مواقع أكثر سلبية مع شدة ضعيفة إلى حد ما. للتحقق من أصل قمة الانخفاض، قمنا بإجراء اختبارات الفولتمترية الدورية (CV) في وجود أو غيابفيمن(الشكل التوضيحي 6). الكثافة الحالية الإجمالية لمنحنى CV زاد بشكل كبير بعد إضافة، مما يشير إلى أن شارك في تركيب السعة ذات الطبقتين. بالإضافة إلى ذلك، اختفى ذروة الاختزال في غياب ، مما يدل بشكل أكبر على نسبة ذروة تقليل اليورانيوم. أشارت هذه النتائج إلى كانت القضبان النانوية تمتلك نشاطًا خاصًا لاستخراج اليورانيوم في وجود.
كما هو موضح في الشكل 3ب، كفاءة الاستخراج الكهروكيميائي لـعلىكانفي غضون 5 ساعات، وهو ما كان متفوقًا بشكل ملحوظ على ذلك الذي تم إنهاؤه بـ OH.وذلك من شعيرات الجرافيت العارية ). مزيد من المناقشة حول مجموعة الإنهاء لـ أشار إلى أن مجموعة -OH كانت موقع الاستخراج، بدلاً من -F (الشكل التوضيحي 7 والجدول التوضيحي 2). ومن الجدير بالذكر أن كفاءة الاستخراج النهائية لـمنحقق، مما أتاح المراقبة المباشرة لشرائط الترسيب الكبيرة من اليورانيوم البلوري (الإطار في الشكل 3ب). بالإضافة إلى ذلك، مقارنةً بطريقة الامتصاص غير الكهربائية، فإن كفاءة الاستخراج الكهروكيميائي لـعلىأظهرت القضبان النانوية تعزيزًا بمقدار 2 ضعف (الشكل التوضيحي 8). لجمع اليورانيوم من القطب، تم أخذ 0.1 مول من محلول HCl كعامل إزالة. بعد 10 ثوانٍ من وقت الغسل، تلاشت البلورات الشبيهة بالشريط الأصفر تدريجيًا في محلول الإزالة (الشكل التوضيحي 9). ونتيجة لذلك، كانت الطريقة الكهروكيميائية معكأقطاب تمثل بروتوكولًا فعالًا لاستخراج اليورانيوم في وجود تركيز عالٍ من.
استقرار الاستخراج الكهروكيميائي لـعلىتم تقييم النانورودات بشكل إضافي. بعد 5 دورات من استخراج اليورانيوم، كانت كفاءة الاستخراج لـعلىاحتفظ بقيمة عالية من (الشكل التوضيحي 10). علاوة على ذلك، بغض النظر عن التلاعب الواسع في الرقم الهيدروجيني من 4 إلى 10، فإن كفاءة الاستخراج لـعلىتجاوز (الشكل 3ج). علاوة على ذلك، قدرة مقاومة التداخلتم إثباته من خلال كفاءة الاستخراج في وجود الأيونات المتزامنة، خاصةً، و “، التي كانت موجودة بشكل شائع في مياه الصرف الصحي الناتجة عن الإنتاج النووي (الشكل 3د). كما اختبرنا التغيرات في درجة الحموضة وتركيزات الأيونات قبل وبعد تفاعل استخراج اليورانيوم الكهربائي (الشكل التكميلي 11). زادت جميع قيم درجة الحموضة للمحلول الكهربائي بعد التفاعل الكهربائي بسبب تطور الهيدروجين في العملية الكهربائية. ظلت كفاءة استخراج U(VI) عند مستوى عالٍ منمما يشير إلى أن أيًا من أيونات التداخل لم تظهر تأثيرًا واضحًا علىالاستخراج. علاوة على ذلك، أظهرت تركيزات الأنيونات في الإلكتروليت انخفاضًا طفيفًا بعد التفاعل الكهروكيميائي، والذي تم نسبه إلى الامتصاص الطفيف للأيونات بواسطة القطب. كما قمنا بتقييم تأثيرتغير التركيز في استعادة اليورانيوم (الشكل 3e). بغض النظر عن التركيز الأولي لـ، الـتم تقليل التركيز من حواليإلىبعد التفاعل بمستوى عالٍ من، مما يشير إلى أن لم تظهر التركيزات تأثيرًا واضحًا علىالاستخراج لـالقطب الكهربائي (الشكل التوضيحي 12أ). وقد تم نسب هذه الظاهرة إلى الارتباط الانتقائي لـعن طريق مواقع أزواج الأيونات، بدلاً من. علاوة على ذلك، فإن الانخفاضات الطفيفة في تركيز تمت ملاحظتها بعد التفاعل الكهروكيميائي، والذي تم نسبه إلى الامتزاز بواسطةموقع الكاتيون والتبلور المشترك الإضافي بواسطة منتج اليورانيوم (الشكل التكميلي 12b). علاوة على ذلك، مع التركيز الأولي لـتفاوتت منإلىكفاءة الاستخراج منبواسطةظل باستمرار فوق، مما يشير إلى قدرة الاستخراج بـدون ظاهرة تشبع الاستخراج (الشكل 3f). تشير هذه النتائج إلى الاستخدام المحتمل لـمع مواقع أزواج الأيونات تجاه استخراج اليورانيوم الكهروكيميائي في مياه الصرف الناتجة عن الإنتاج النووي.
تطور أنواع اليورانيوم
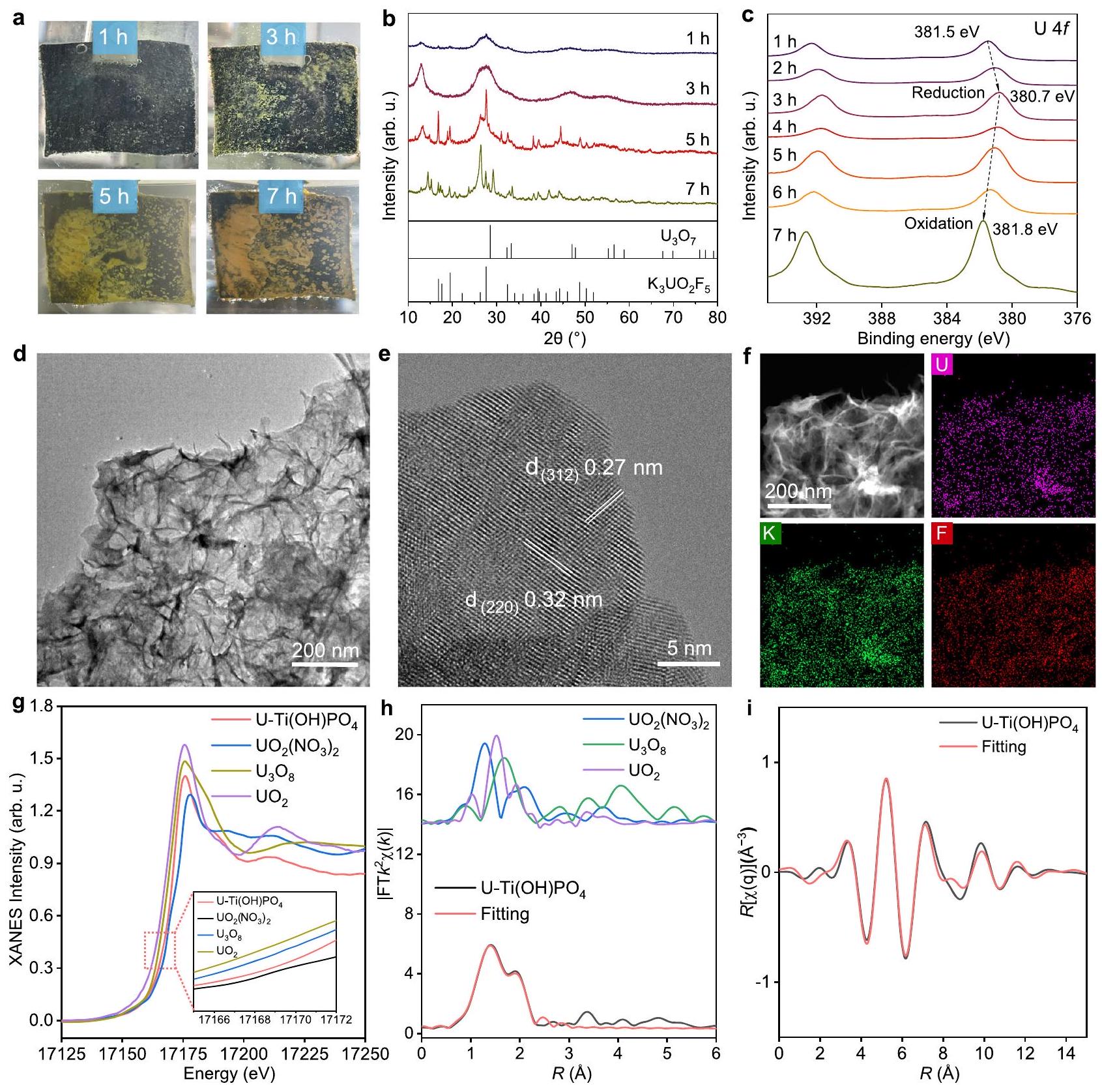
الاستخراج الكهربائي الكيميائي الفعال لليورانيومأنابيب نانوية تحتوي على مواقع أزواج أيونية في وجود تركيزات عالية منألهمنا لاستكشاف عملية ترسيب اليورانيوم على القطب. تجربة الاستخراج المعتمدة على الزمن حولتم إجراء دراسة على القضبان النانوية في إلكتروليت مائي يحتوي علىمن و من(الشكل 4أ). في نقطة الزمن 1 ساعة، ظهرت الأنواع المترسبة وظهرت بلون رمادي. مع تقدم التفاعل إلى 3 ساعات و5 ساعات، تم تحويل الرواسب الرمادية تدريجياً إلى المنتج الأصفر، مع تجمع الرواسب الصلبة على القطب. أخيراً، تحول لون الرواسب إلى حالة مستقرة من الأصفر الداكن عند 7 ساعات. للتحقق من الأنواع خلال استخراج اليورانيوم، جمعنا الرواسب الصلبة في نقاط زمنية مختلفة. كما تشير أنماط XRD، فإن الرواسب الرمادية عند 1 ساعة و3 ساعات كانت تُنسب إلى (JCPDS #42-1215)، والتي كانت نوعًا خاصًا من يحتوي على واليورانيوم رباعي التكافؤ (U(IV)) (الشكل 4ب). يمكن تفسير وجود U(IV) من خلال التحلل الذاتي لـبالمقارنة، كانت نتائج حيود الأشعة السينية (XRD) للترسبات الصفراء عند 5 ساعات و 7 ساعات متوافقة بشكل جيد مع (JCPDS #38-0023) مع أنواع المجال من ، مما يشير إلى أكسدة اليورانيوم منخفض التكافؤ في الرواسب خلال التفاعل. الـكانت الأنواع أيضًا مسؤولة عن الانخفاضات الطفيفة في تركيزفي تجربة الشكل التوضيحي الإضافي 12. البوتاسيوم في المنتج النهائي لاستخراج اليورانيومنشأ من KF في الإلكتروليت، والذي يمكن استبداله بـ Na فيعند استخدام NaF كإلكتروليت (الشكل التكميلي 13).
تم التحقق من نتيجة تطور اليورانيوم من خلال طيف XPS U4f في نقاط زمنية مختلفة (الشكل 4c). مع تقدم التفاعل إلى 3 ساعات، كانت قممتم تحويلها سلبًا من 381.5 إلكترون فولت إلى 380.7 إلكترون فولت، مما يدل على الانخفاض المستمر لليورانيوم خلال التفاعل. مع تقدم التفاعل إلى 4 ساعات و 7 ساعات، تم تحويل الرواسب الرمادية تدريجيًا إلى المنتج الأصفر، مع ظهور قمم لـانتقل بشكل إيجابي إلى 381.8 إلكترون فولت، مما يدل على الأكسدة وجود اليورانيوم منخفض التكافؤ في الرواسب أثناء التفاعل. من أجل تفسير دقيق، كانت الرواسب الرمادية تحتوي على المزيد من المحتوى من و من تلك الرواسب الصفراء (الشكل التكميلي 14). الوسيط لـوالمنتج النهائي لـتمت ملاحظتها أيضًا من خلال ترسبات دورات متعددة من اختبار CV، مما تحقق من عملية تطور أنواع اليورانيوم (الشكل التكميلي 15). كانت تبديل أنواع اليورانيوم في الترسب مصحوبة بتطور الشكل. عند زمن تفاعل قدره ساعة واحدة، عرضت الترسبات الرمادية الأولية لليورانيوم شكل إبرة نانوية مع بلورة منخفضة (الشكل التكميلي 16). مع مرور الوقت، تطورت الإبر النانوية تدريجيًا إلى صفائح نانوية، مع وجود حواف شبكية بلورية مرئية. أظهر الشكل 4d صورة TEM نموذجية للترسبات الصفراء النهائية، والتي أظهرت شكل الصفيحة النانوية المرنة. علاوة على ذلك، كانت الحواف الشبكية لهذه الصفيحة النانوية تمتلك تباعدًا بين الطبقات قدره 0.32 نانومتر، والذي ينتمي إلى وجه (220). (الشكل 4e). في قياس EDS، كانت عناصر اليورانيوم والبوتاسيوم والفلور موزعة بشكل موحد في الأوراق النانوية، مما يوضح المزيد من التركيب لـفي الرواسب الصفراء النهائية (الشكل 4f). مصحوبة بتطور شكل رواسب اليورانيوم، شكلتحولت من نانوصفائح متراصة نقية إلى نانوصفائح مفككة من خلال توسيع المسافة بين الطبقات (الشكل التكميلي 17). أظهر هذا النتيجة إدخال اليورانيوم في المسافات بين الطبقات منخلال التفاعل الكهروكيميائي، الذي أكد ميزة المسافة الأكبر بين الطبقاتمن قطر.
للتحقق من تطور الأنواع المعنية في استخراج اليورانيوم، قمنا بالتحقيق مباشرة في حالة التكافؤ وبيئة التنسيق لليورانيوم في القطب الكهربائي بعد 3 ساعات خلال التفاعل الكهروكيميائي. ومن الجدير بالذكر أن الرواسب الوسيطة الرئيسية لم تفصل عن مادة الاستخراج في هذه الحالة. كما هو موضح بواسطة-edge XANES الطيف (الشكل 4 ج)، تم تحديد القمم المميزة عند 17175.7 eV في الطيف على أنها قمة الخط الأبيض، والتي تتوافق مع انتقال الإلكترونات من الحالة المملوكةمدار إلى غير المشغول. بشكل ملحوظ، كانت موضع حافة الامتصاص للمنتجات الكهروكيميائية يقع بين و ، مع انحراف طفيف نحو الجانب المنخفض من E. هذه الملاحظة تشير إلى أن حالات التكافؤ لـكانت الكاتيونات تتراوح بين +4 و +6، مما يشير إلى أن استخراج اليورانيوم خضع لعملية الاختزال الكهروكيميائي. توضح الشكل 4h تحويل فورييه (FT)-طيف الامتصاص الدقيق للأشعة السينية الممتد (EXAFS) الموزون للمنتج الكهروكيميائي. كان منحنى تحويل فورييه للمنتج الكهروكيميائي يحتوي على قمتين رئيسيتين عند 1.4 والتي نُسبت إلى تنسيق U-O للقشرة الأولى والثانية. تم الإشارة إلى القشرة الأولى والثانية على أنها و ، والتي تشير على التوالي إلى الأكسجين المحوري في أنواع اليورانيوم والأكسجين في موقع الامتزاز لمواقع الأيونات الزوجية. من خلال ملاءمة EXAFS، كانت أعداد التنسيق لـ و كانت 2.2 و3.4، على التوالي (الشكل 4i والجدول 1). تشير هذه النتيجة إلى أن عملية الامتزاز كانت مدعومة بثلاث ذرات أكسجين في مواقع أزواج الأيونات على السطحالنانو القضبان. بشكل خاص، كان رابط U-F موجودًا مع عدد تنسيق يبلغ 1.1، مما يدل على أن المادة المستهدفة التي شاركت في استخراج اليورانيوم كانت هي الإجماليبدلاً من، الذي نتج عن تفاعل كولوم بين مواقع أزواج الأيونات و.
آلية التفاعل
الامتصاص غير العادي بشكل عاموتحفزنا تطورات الأنواع المقابلة لاستكشاف آلية التفاعل الجوهرية. استنادًا إلى تطور الأنواع، قمنا بإنشاء مخطط لمحاكاة عملية التفاعل (الشكل 5أ).تم امتصاصه على مواقع أزواج الأيوناتالنانو القضبان دون فصل رابطة اليورانيوم-فلور. بعد ذلك، تم تقليل اليورانيوم إلى أنواع منخفضة التكافؤ، مما أضعف القوة الكولومبية لليورانيوم و، مما يؤدي إلى تشكيل وسيط المنتج. ثم تم أكسدة اليورانيوم منخفض التكافؤ غير المستقر، تلا ذلك التبلور معوتشكيل النهائي.
الشكل 4 | تطور أنواع اليورانيوم. أ صور لتطور أنواع اليورانيوم خلال استخراج اليورانيوم.نمط XRD للترسبات الصلبة المجمعة في نقاط زمنية مختلفة.طيف XPS لـأقطاب كهربائية في نقاط زمنية مختلفة لاستخراج الكهروكيميائية. د صورة TEM للترسبات الصفراء النهائية ذات شكل النانو. هـ قياس HRTEM و وقياس EDS لـ تركيب الـفي الرواسب الصفراء النهائية. الـ -طيف XANES الحدي للترسب الوسيط. في الزاوية: منطقة XANES ما قبل الحافة مكبرة. فاينانشيال تايمز -طيف EXAFS الموزون للمنتج الكهروكيميائي. i المعادلتوافق الفضاء للمنحنيات الناتجة عن المنتج الكهروكيميائي. يتم توفير بيانات المصدر كملف بيانات مصدر.
بالنظر إلى امتصاص الكليكان العامل الرئيسي الذي أثر على كفاءة استخراج اليورانيوم، قمنا بمحاكاة نظرية لامتصاصعلى موقع زوج الأيوناتالأنابيب النانوية (الشكل 5ب، الجدول التكميلي 3، والبيانات التكميلية 1).
الجدول 1 | المعلمات الهيكلية لـفي الـ U-الحافة المستخرجة من ملاءمات منحنى EXAFS الكمية
مسار
-عامل
2.2
1.7
٤.١
-11.1
0.004
3.4
2.3
٤.٠
6.8
0.004
1.1
2.1
٥.٠
-11.0
0.004
أخذكمثال، قمنا بمقارنة تكوين الامتزاز المحسن على السطح المنتهي بـ OH- و مععلى السطح. المجموعة المنتهية بـ OHامتلكت مواقع -OH العادية للسطح العاريالامتزاز، الذي يمتلك طاقة امتزاز قدرها -2.6 إلكترون فولت لـدون تفاعلو. من أجل نموذج، تنسيق السطحكان غير مشبع، مما أدى إلى التفاعل مع. على هذا النحو، الـاستقر علىمواقع أزواج الأيونات من خلال كلا و التفاعلات. في هذا النموذج، ثلاثة ذرات أكسجين فيشارك في الامتزاز لـ، متسقة مع نتيجة EXAFS التجريبية. علاوة على ذلك، فإن طاقة الامتزاز لـعلى مواقع أزواج الأيوناتكان -4.5 إلكترون فولت، وهو ما كان أكثر سلبية بكثير من ذلك على مواقع -OH في السطح المنتهي بـ OHتمكنت طاقة الامتزاز من أن تكون
الشكل 5 | آلية تفاعل مواقع أزواج الأيونات. أ الرسم التخطيطي لآلية التفاعل المعقولةمواقع أزواج الأيونات.تكوينات الامتزاز المحسّنةممتص على (منتهية بـ OH) و الأكسجين المحوري في أنواع اليورانيومذرة الأكسجين في موقع الامتزاز،طاقة الامتزاز لـتُقدم بيانات المصدر كملف بيانات مصدر. تم تقليلها أكثر من خلال إدخال فراغات إضافية من الأكسجين (الشكل التوضيحي التكميلي 18). علاوة على ذلك، فإن زاوية الرابطة لـمنالنموذج كانالذي كان أقل من ذلكمن (منتهية بـ OH). نظرًا لأن تقليل يتطلب تقليل زاوية الرابطة، مواقع أزواج الأيونات لـيمكن أن يسهل تقليلفي. ونتيجة لذلك، قامت مواقع أزواج الأيونات بتثبيت الامتزاز لـوروجت لتقليلالذي كان مسؤولاً عن استخراج اليورانيوم بتركيزات عالية من.
تجربة مياه الصرف النووي الحقيقية
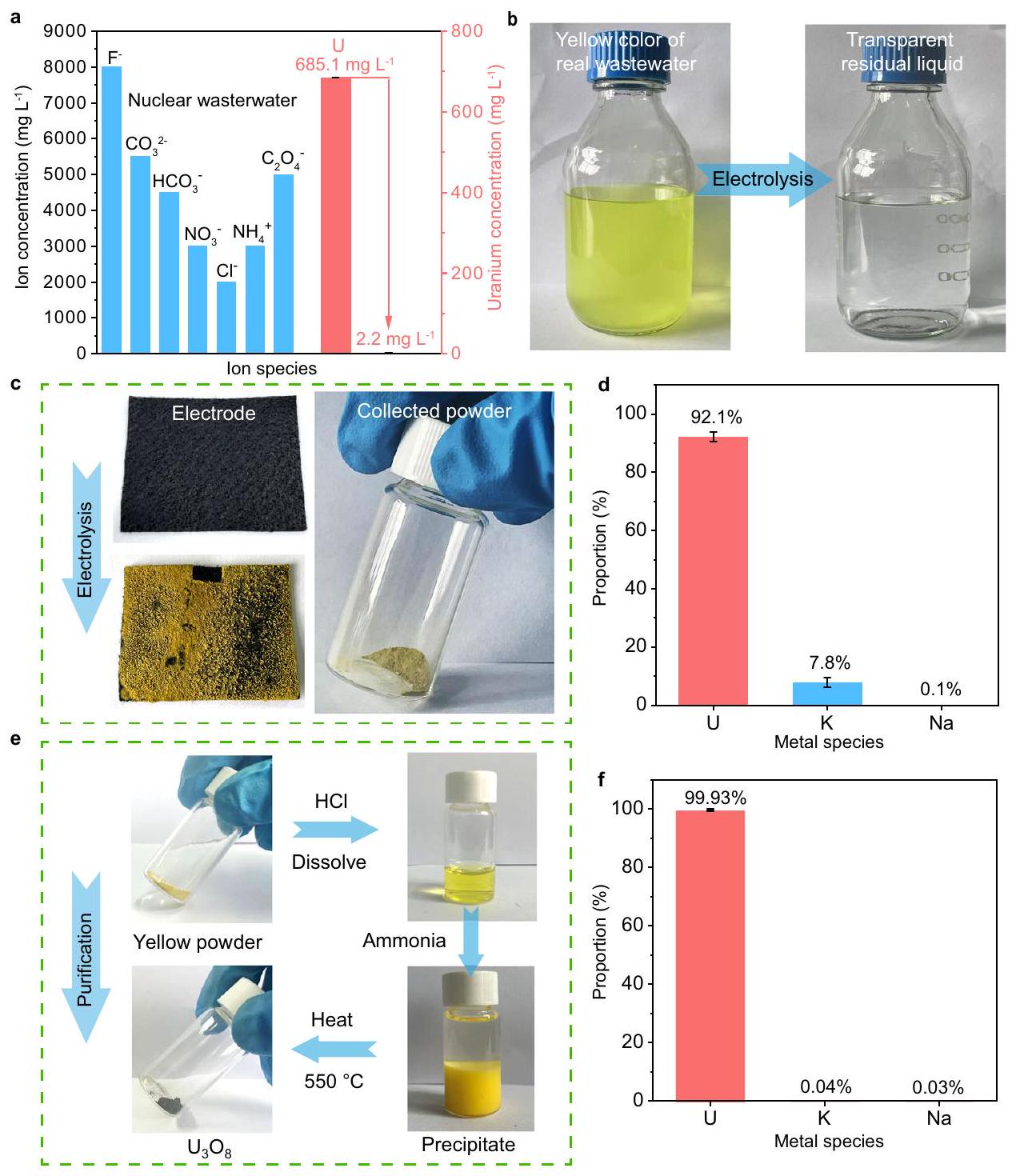
مدفوعًا بالتأثير المميز لمواقع أزواج الأيونات على استخراج اليورانيوم في تركيزات عالية مناستكشفنا استخراج اليورانيوم من المياه النووية الملوثة الحقيقية على. كانت هذه المياه العادمة تحتوي على مستويات عالية من ، و “، الذي تم إنتاجه من غسل الحاوية في منشأة إنتاج الوقود النووي. بعد 7 ساعات من التحليل الكهربائي عند كثافة تيار ثابتة منتركيزتم تقليصه من قيمة أولية قدرهاإلىمعكفاءة الاستخراج وقدرة الاستخراج (الشكل 6أ). كانت قيمة كفاءة الاستخراج هذه أكبر بمقدار 1.7 مرة مقارنة بتلك الخاصة بـ OH -المُنهية، مما يدل على فعالية مع مواقع أزواج الأيونات (الشكل التكميلي 19). مع تقدم عملية التحليل الكهربائي، بدأ اللون الأصفر لمياه الصرف الحقيقية يتلاشى تدريجياً، مما أدى في النهاية إلى سائل متبقي عديم اللون وشفاف (الشكل 6ب). تم عرض تركيز جميع أنواع المعادن في السائل المتبقي الشفاف في الجدول التكميلي 4. بالمقارنة مع مياه الصرف الحقيقية الصفراء مياه الصرف الصحي،وانخفضت على التوالي بنسبة ( انخفاض) ( انخفاض)، والذي تم نسبته بشكل رئيسي إلى امتصاص القطب، بدلاً من الاستخراج الكهروكيميائي (الشكل التكميلي 20). الغالبية العظمى من الممتصويمكن إزالته من القطب الكهربائي من خلال عملية الغسيل. بالإضافة إلى ذلك، فإن التركيزات الأولية المنخفضة (أقل منكانت عناصر المعادن النادرة غير ذات أهمية جوهرية لعملية استخراج اليورانيوم ونقاء رواسب اليورانيوم. لذلك، كان من الممكن استخراج اليورانيوم بكفاءة من القطب.
بعد التفاعل الكهروكيميائي، تم ترسيب اليورانيوم بكثافة على القطب، والذي يمكن إزالته ببساطة عن طريق ترسيب KOH، وجمعه كمسحوق صلب (الشكل 6c). قمنا بتحليل نسبة الأنواع المعدنية في هذا المسحوق عن طريق قياس الطيف الضوئي الانبعاثي البلازمي المقترن بالحث (ICP-OES) (الشكل 6d). وصلت نسبة اليورانيوم إلى 92.1% بين العناصر المعدنية في المسحوق، مما كان مريحًا لعملية الفصل وإعادة استخدام اليورانيوم لاحقًا. بالإضافة إلى ذلك، استخدمنا عملية ترسيب-تكلس نموذجية لتنقية منتجات اليورانيوم بشكل أكبر (الشكل 6e). في البداية، قمنا بجمع المنتج المستخرج من اليورانيوم وذوبانه في HCl. بعد ذلك، تم إضافة كمية زائدة من الأمونيا إلى المحلول أعلاه لتشكيل راسب من ثنائي يورانات الأمونيوم. أخيرًا، تم جمع ثنائي يورانات الأمونيوم عن طريق الطرد المركزي وتسخينه عندلمدة 5 ساعات لتشكيل منتج يورانيوم أسود-أخضر. وفقًا لاختبارات XRD (الشكل التوضيحي 21) واختبارات ICP-OES (الشكل 6f)، تم تحديد منتج اليورانيوم الأسود-الأخضر المنقى على أنه (JCPDS #31-1425) بمحتوى يورانيوم من بين العناصر المعدنية، التي يمكن استخدامها مباشرة في إنتاج اليورانيوم.
الشكل 6 | تجربة المياه النووية الحقيقية لمواقع أزواج الأيونات. أ كفاءة الاستخراج الكهروكيميائي لـعلىفي مياه الصرف النووي الحقيقية. تغير لون المياه النووية الحقيقية قبل وبعد التحليل الكهربائي. تم ترسيب اليورانيوم على القطب والمسحوق المجموع المقابل بعد التحليل الكهربائي. نسبة اليورانيوم بين الأنواع المعدنية في المسحوق. المخطط التخطيطي لعملية تنقية اليورانيوم بالتساقط والتكلس.نسبة اليورانيوم بين الأنواع المعدنية في منتج التنقية. تمثل أشرطة الخطأ الانحراف المعياري لثلاث قياسات. تم توفير بيانات المصدر كملف بيانات مصدر.
وبالتالي، فإن استخراج اليورانيوم الكهربائي الكيميائي بواسطةقدمت استراتيجية قابلة للتطبيق لاستعادة الموارد من مياه الصرف الصحي الحقيقية الناتجة عن الإنتاج النووي. كما قمنا بتقييم التكلفة التشغيلية التقريبية. كانت الكهرباء المستهلكة 31.5 واط ساعة، مما يعني أن استخراج كل كيلوغرام من اليورانيوم يتطلب كهرباء قدرها، مما يتوافق مع تكلفة الكهرباء الدولارات الأمريكية (وفقًا لسعر الصين). لتكلفةالقطب الكهربائي، كان السعر حوالي 0.2 دولار أمريكي عند اعتبار الشراء لـالمادة السابقة، المادة الاصطناعية، ودعم الجرافيت. بالنظر إلى استخدام إعادة تدوير القطب، كانت تكلفة مادة القطب22 دولار أمريكي لاستخراج 1 كجم من اليورانيوم. لذلك، كانت التكلفة الإجمالية حوالي 22 دولار أمريكي لاستخراج 1 كجم من اليورانيوم مع الأخذ في الاعتبار استهلاك الطاقة وتكاليف المواد.
نقاش
باختصار،تم إنشاء مواقع أزواج الأيونات فيكمادة لاستخراج اليورانيوم من المياه النووية الملوثة التي تحتوي على تركيزات عالية منمن إنتاج الوقود. في الإلكتروليتات المحاكاة مع حتىمنيمتلك كفاءة استخراج قدرهافي 5 ساعات. تكشف الدراسة الميكانيكية أنيمكن أن يكون مرتبطًا بقوة في شكل وروابط Ti-F. مع تقدم التفاعل، فإن المادة الممتصة يتحول من الحالة المخفضةإلى الحالة البلورية. في تجربة حقيقية لمياه الصرف النووي، معارضكفاءة استخراج اليورانيوم خلال 7 ساعات بغض النظر عن التداخلمن، بقدرة استخراج تبلغدون تشبع. بعد الجمع البسيط، يتم الحصول على منتج مسحوق يحتوي على نقاء عالٍ من اليورانيوم، مما يظهر نجاح إعادة تدوير اليورانيوم في مياه الصرف الصحي الحقيقية. لا يقدم عملنا فقط مادة فعالة لاستخراج اليورانيوم لمقاومة التركيزات العالية منولكنه يوفر أيضًا استراتيجية فعالة لاستعادة اليورانيوم في مياه الصرف النووي الحقيقية والمعقدة.
طرق
المواد الكيميائية والمواد
تم شراء المسحوق (400 شبكة) من شركة 11 للتكنولوجيا المحدودة. حمض الهيدروكلوريك (النقاء) وحمض الفيتيك (PA، 70% تم تزويد فلوريد الليثيوم (LiF، نقاء 99%) ونترات اليورانيوم من مصنع تشنغدو كيلونغ الكيميائي.تم الحصول على النقاء من شركة ألاaddin للكيماويات في شنغهاي. شعيرات الجرافيت (محتوى الكربون ) والإيثانول (AR) تم شراؤهما من شركة CeTech المحدودة. محلول غشاء نافيون (NR50، تم الحصول على ) من شركة شانغهاي ماكلين للكيماويات الحيوية المحدودة. تم جمع المياه النووية الحقيقية من منشأة إنتاج الوقود النووي. تم استخدام جميع المواد الكيميائية والمذيبات دون تنقية.
توصيفات
تم عرض صور المجهر الإلكتروني الماسح (SEM) على جهاز JSMمجهر إلكتروني مسح ضوئي. تم عرض صور المجهر الإلكتروني الناقل (TEM) والمجهر الإلكتروني الناقل عالي الدقة (HRTEM) وطيف الأشعة السينية المشتتة للطاقة (EDS) على مجهر إلكتروني ناقل JEM 2100 F يعمل بجهد تسريع قدره 200 كيلو فولت. تم الحصول على صور المجهر الضوئي الذري (AFM) على جهاز Bruker Dimension ICON في وضع ScanAsyt. تم إجراء تحليلات طيف الانبعاث الضوئي للبلازما المقترنة بالحث (ICPOES) على جهاز PerkinElmer ICP 2100 وPerkinElmer Optima 5300 DV. تم جمع بيانات حيود الأشعة السينية (XRD) على جهاز Smartlab SE المجهز بـ المصدر. تم الحصول على أطياف رامان (Raman) باستخدام مطياف رامان بالليزر Thermo Fischer DXR الذي يعمل عند طول موجي 532 نانومتر. تم تحديد مطيافية الأشعة تحت الحمراء بتحويل فورييه (FT-IR) من مطياف Nicolet iS 10. تم تحديد العيوب وخصائص السطح للمواد بواسطة مطيافية الرنين المغناطيسي الإلكتروني (ESR) باستخدام Bruker EMXplus. تم إجراء تحليل مطيافية الأشعة السينية للألكترونات (XPS) من خلال Thermo Kalpha، المجهز بألمنيوم K أحادي اللون.مصدر الأشعة السينية. تم تحديد تركيز الأنيونات في الإلكتروليت بواسطة جهاز كروماتوغرافيا الأيونات (IC). تم استخدام توصيف حالات الأكسدة للعينات بواسطة هيكل الامتصاص الدقيق للأشعة السينية (XAFS) من خلال وضع النقل لـالحافة و U-حافة الامتصاص في مختبر الإشعاع السنكروتروني الوطني (NSNR). تم معايرة مواقع حواف الامتصاص خلال قياسات XAFS. تم جمع بيانات هيكل الامتصاص الدقيق للأشعة السينية الممتد (EXAFS) من وحدة ATHENA، المدمجة ضمن مجموعة برامج IFEFIT. تم عرض بيانات المسح الخطي للجهد (LSV) والجهد الدوري (CV) على محطة العمل الكيميائية الكهربائية CHI 660E.
الحسابات النظرية
لقد قمنا بإجراء حسابات DFT في نظامنا باستخدام حزمة المحاكاة من فيينا (VASP، الإصدار: VASP 5.4.4)تم استخدام دالة PBE لوصف التبادل الإلكترونيتم اعتماد طريقة الموجات المعززة بالمش projector لوصف التفاعلات بين نوى الأيونات والإلكترونات التكافؤية.. كانت الطاقة القصوى لمجموعة الأساس الموجي الطائرة 500 إلكترون فولت. عينة مخطط بريلوان مخطط مونكهورست-باك باستخدام شبكة نقطية. خلال تحسين الهندسة وحساب البنية الإلكترونية، تم تحسين مواقع الذرات حتى أصبحت الطاقة والقوة القصوى ذرة و تم اعتبار تشتت فان der Waals من خلال استخدام طريقة DFT-D3 لـ Grimme في جميع الحسابات..
بناءمع مواقع أزواج الأيونات
الطبقات المقشرة المنتهية بـ OH-تم تخليق النانوصفائح وفقًا لطريقة MILD من خلال الحفر الانتقائي في الموقع باستخدام HF لطبقة الألمنيوممقدمة. تحديداً،تم حله معمحلول في كوب تيفلون، يتبعه الإضافة البطيئة لـفي حمام ثلجي تحت التحريك المغناطيسي. تم إجراء التفاعل عندتحت ظروف تهوية غير كافية لمدة 24 ساعة لتجنب المخاطر الأمنية الناتجة عن استخدام HF. الكومة على شكل أكورديون -F- المنتهيةتم جمعه وطرده مركزيًا عندعدة مرات بالماء المقطر لإزالة أي حمض متبقي حتى وصل السائل العلوي إلى درجة حموضة 6. بعد ذلك، كانت المعجونة أعلاه مغمور في الماء منزوع الأيونات (100 مل) لتكوين تعليق متجانس عن طريق الموجات فوق الصوتية لمدة ساعتين تحت الحماية من. بعد ذلك، تم تقشير السطح المنتهي بـ OHتم جمع السائل المائي العلوي في مكعب الطرد المركزي بعد الطرد المركزي مرة أخرى عندلمدة 5 دقائق. أخيرًا، تم تحضير التعليق للتجارب عن طريق تخفيف OH -المُنتهي المُستخرج.تعليق على. الـتم تخليق مواقع أزواج الأيونات من خلال طريقة كيميائية رطبة سهلة. عادةً، تم تقشيرها مع إنهاء OH.تم خلط التعليق (2 مل) مع الماء المنزوع الأيونات (40 مل) وتم تحريكه لمدة 30 دقيقة في درجة حرارة الغرفة للحصول على محلول موزع بشكل موحد. ثم، تم إضافة حمض الفيتيك (1.6 مل) إلى المحلول الموزع أعلاه بالتنقيط واستمر التحريك لمدة ساعة أخرى. بعد ذلك، تم وضع المحلول المتجانس في أوتوكلاف مبطن بتفلون (50 مل) وتم تسخينه عند لمدة 12 ساعة. أخيرًا، الـتم جمع مواقع أزواج الأيونات بعد غسلها بالماء المقطر والإيثانول عدة مرات، وتم تجفيفها بالتجميد لمدة 24 ساعة.
تجارب استخراج اليورانيوم الكهروكيميائي في مياه الصرف النووي المحاكاة
تم قياس تجارب استخراج اليورانيوم الكهروكيميائي في محاكاة مياه الصرف النووي باستخدام نظام ثلاثي الأقطاب. تم تصنيع الأقطاب العاملة على النحو التالي:مواقع أزواج الأيوناتالكربون الأسود (2.00 ملغ) ومحلول غشاء نافيون ( ) تم خلطها مع الإيثانول ( 2 مل ) وتمت الموجات فوق الصوتية حتى أصبحت حبرًا متجانسًا. تم طلاء الحبر المحضر مباشرة على شعيرات الجرافيت ( ) وجفف في سلك البلاتين (Pt) وتم استخدام الأقطاب الكهربائية كقطب مضاد وقطب مرجعي، على التوالي. تم إجراء تجارب استخراج اليورانيوم تحت تيار ثابت منتم اختبار أداء استخراج اليورانيوم الكهروكيميائي في محلول مائي (50 مل) يحتوي علىمن و من(تم الحصول عليها من محلول KF الكهربائي). تم اختبار تجارب الامتصاص غير الجهدية على نظام ثلاثي الأقطاب غير مزود بالطاقة في 50 مل من الماء النووي المحاكي. تم إعدادها كما هوإلكترود، سلك بلاتيني، وتم استخدام الأقطاب الكهربائية أيضًا كقطب عمل وقطب مضاد وقطب مرجعي على التوالي. كانت حالة اختبار الامتزاز مشابهة لتلك الخاصة بالاستخراج الكهروكيميائي باستثناء استبعاد التيار أو الجهد الخارجي. كانت تأثيرات التركيز الابتدائي لليورانيوم (من 50 إلىدرجة الحموضة للمحلول (من 4 إلى 10)، الأيونات المتداخلة (بما في ذلك)، وتأثير (من 5 إلى تم قياس أداء استخراج اليورانيوم الكهروكيميائي بالتفصيل. تم تحديد تركيزات اليورانيوم والكاتيونات الأخرى في المحلول الكهربائي قبل وبعد التحليل الكهربائي بواسطة ICP-OES. تم تحديد تركيزات الأنيونات في المحلول الكهربائي قبل وبعد التحليل الكهربائي بواسطة IC. في عملية الإزالة، تم نقل القطب العامل الذي يحتوي على اليورانيوم المستخرج إلى محلول مائي من HCl (0.1 مول). تم إجراء تجارب الاستخراج ثلاث مرات وتم استخدام أشرطة الخطأ في المنحنيات. تم إجراء اختبارات LSV و CV في مياه الصرف النووي المحاكية بمعدل مسح.
تجارب استخراج اليورانيوم الكهروكيميائي في مياه الصرف النووي الحقيقية
تم قياس تجارب استخراج اليورانيوم الكهروكيميائي في مياه الصرف النووي الحقيقية باستخدام نظام قطبين نموذجي. تم قطع شعيرات الجرافيت إلىأشكال كركائز للأقطاب الكهربائية.مواقع أزواج الأيونات (40.0 ملغ)، الكربون الأسود (8.0 ملغ)، وحل غشاء نافيون ( ) تم خلطها مع الإيثانول ( 8 مل ) وتمت الموجات فوق الصوتية لمدة ساعة واحدة لتكوين حبر متجانس. ثم تم طلاء الحبر المحضر بشكل موحد على شعيرات الجرافيت لتحضير القطب العامل، وتم استخدام سلك البلاتين كقطب مضاد. تم إجراء استخراج اليورانيوم تحت تيار ثابت من في 400 مل من مياه الصرف النووي الحقيقية. تم غمر الإلكترود المودع في الإيثانول وتم تنقيته من خلال تفاعل فوق صوتي، و تم جمع المسحوق عن طريق التجفيف. تم قياس وتحليل نسب الأنواع المعدنية في هذا المسحوق بواسطة ICP-OES. تم تحديد تغييرات التركيز لليورانيوم والكاتيونات الأخرى في مياه الصرف النووي الحقيقية قبل وبعد التحليل الكهربائي بواسطة ICP-OES.
تنقية منتجات اليورانيوم
تمت تنقية اليورانيوم من خلال عملية ترسيب-تحميص نموذجية. في البداية، تم جمع منتج اليورانيوم من الاستخراج الكهروكيميائي وذوبانه في حمض الهيدروكلوريك. بعد ذلك، تم إضافة كمية زائدة من الأمونيا إلى المحلول أعلاه لتكوين راسب من ثنائي يورانات الأمونيوم. أخيرًا، تم جمع ثنائي يورانات الأمونيوم عن طريق الطرد المركزي وتسخينه فيلمدة 5 ساعات لتشكيل منتج يورانيوم أسود-أخضر.
أين و تمثل تركيز اليورانيوم الابتدائي والنهائي، على التوالي.
أين و تمثل تركيز اليورانيوم الابتدائي والنهائي، على التوالي؛ و تشير إلى كتلة المحفز وحجم الإلكتروليت، على التوالي.
أين، و هي الطاقات الكلية للمواد الممتصة-الركيزة (AS)، والركيزة (S)، والمواد الممتصة (A)، على التوالي.
توفر البيانات
البيانات التي تدعم نتائج الدراسة مدرجة في النص الرئيسي وملفات المعلومات التكميلية. تم توفير بيانات المصدر مع هذه الورقة. تتوفر ملفات بيانات المصدر في Figshare تحت رمز الوصولhttps://doi.org/10.6084/m9.figshare.25603905تم توفير بيانات المصدر مع هذه الورقة.
References
Chen, T. et al. Advanced photocatalysts for uranium extraction: elaborate design and future perspectives. Coord. Chem. Rev. 467, 214615 (2022).
Zhang, S., Li, H. & Wang, S. Construction of an ion pathway boosts uranium extraction from seawater. Chem 6, 1504-1505 (2020).
Tsouris, C. Uranium extraction: fuel from seawater. Nat. Energy 2, 17022 (2017).
Mei, D., Liu, L. & Yan, B. Adsorption of uranium (VI) by metal-organic frameworks and covalent-organic frameworks from water. Coord. Chem. Rev. 475, 214917 (2023).
Hu, Y. et al. Photochemically triggered self-extraction of uranium from aqueous solution under ambient conditions. Appl. Catal. B: Environ. 322, 122092 (2023).
Zhou, Y. et al. Amidoxime-functionalized MXene beads for the effective capture of uranium from wastewater with high fluoride concentrations. Chem. Eng. J. 471, 144647 (2023).
Ohashi, Y., Murashita, S. & Nomura, M. Extraction of uranium from solid waste containing uranium and fluorine. Miner. Eng. 61, 32-39 (2014).
Li, Z. et al. Exciton dissociation and transfer behavior and surface reaction mechanism in Donor-Acceptor organic semiconductor photocatalytic separation of uranium. Appl. Catal. B: Environ. 332, 122751 (2023).
Busquim e Silva, R., Kazimi, M. S. & Hejzlar, P. Nuclear fuel recycling: national and regional options for the US nuclear energy system. Energy Environ. Sci. 3, 996-1010 (2010).
Wang, C. et al. Uranium in situ electrolytic deposition with a reusable functional graphene-foam electrode. Adv. Mater. 33, 2102633 (2021).
Song, P. S., Min, B. Y., Choi, W. K., Jung, C. H. & Oh, W. Z. Effects of a slag former on the absorption of cerium and uranium oxide within a slag during a melting of stainless steel contaminated with uranium. Sep. Purif. Technol. 60, 136-141 (2008).
Xing, S., Luo, M., Wu, Y., Liu, D. & Dai, X. Rapid determination of uranium isotopes in calcium fluoride sludge by tandem quadrupole ICP-MS/MS. J. Anal. Spectrom. 34, 2027-2034 (2019).
Liu, C. et al. A half-wave rectified alternating current electrochemical method for uranium extraction from seawater. Nat. Energy 2, 17007 (2017).
Wang, Y. et al. Electrochemical-mediated regenerable Fell active sites for efficient uranium extraction at ultra-low cell voltage. Angew. Chem. Int. Ed. 62, e202217601 (2023).
Riedhammer, J., Halter, D. P. & Meyer, K. Nonaqueous electrochemistry of uranium complexes: A guide to structure-reactivity tuning. Chem. Rev. 123, 7761-7781 (2023).
Keener, M. et al. Redox-switchable carboranes for uranium capture and release. Nature 577, 652-655 (2020).
Liu, T. et al. Removal and recovery of uranium from groundwater using direct electrochemical reduction method: performance and implications. Environ. Sci. Technol. 53, 14612-14619 (2019).
Yang, H. et al. Functionalized iron-nitrogen-carbon electrocatalyst provides a reversible electron transfer platform for efficient uranium extraction from seawater. Adv. Mater. 33, 2106621 (2021).
Das, N., Das, A., Sarma, K. P. & Kumar, M. Provenance, prevalence and health perspective of co-occurrences of arsenic, fluoride and uranium in the aquifers of the Brahmaputra River floodplain. Chemosphere 194, 755-772 (2018).
Shen, J. & Schäfer, A. Removal of fluoride and uranium by nanofiltration and reverse osmosis: a review. Chemosphere 117, 679-691 (2014).
Miskowiec, A., Niedziela, J. L., Kirkegaard, M. C. & Shields, A. E. Analysis of water coupling in inelastic neutron spectra of uranyl fluoride. Sci. Rep. 9, 10476 (2019).
Li, X. et al. MXene chemistry, electrochemistry and energy storage applications. Nat. Rev. Chem. 6, 389-404 (2022).
Rosenkranz, A., Righi, M. C., Sumant, A. V., Anasori, B. & Mochalin, V. N. Perspectives of 2D MXene tribology. Adv. Mater. 35, 2207757 (2023).
Vahidmohammadi, A., Rosen, J. & Gogotsi, Y. The world of twodimensional carbides and nitrides (MXenes). Science 372, eabf1581 (2021).
Robertson, D. D. & Tolbert, S. H. A direct and clean route to MXenes. Science 379, 1189-1190 (2023).
Zhang, X. et al. MXene aerogel scaffolds for high-rate lithium metal anodes. Angew. Chem. Int. Ed. 57, 15028-15033 (2018).
Sun, B. et al. Redox-active metaphosphate-like terminals enable high-capacity MXene anodes for ultrafast Na-ion storage. Adv. Mater. 34, 2108682 (2022).
Lei, J. et al. Enhanced photoreduction of on nanosheets by oxygen defect engineering. Chem. Eng. J. 416, 129164 (2021).
Zeng, Y. et al. Oxygen-vacancy and surface modulation of ultrathin nickel cobaltite nanosheets as a high-energy cathode for advanced Zn-ion batteries. Adv. Mater. 30, 1802396 (2018).
Li, R. et al. Flexible and high-performance electrochromic devices enabled by self-assembled 2D TiO MXene heterostructures. Nat. Commun. 12, 1587 (2021).
Zhang, H. et al. MXene-derived quantum dots distributed on porous carbon nanosheets for stable and long-life Li-S batteries: enhanced polysulfide mediation via defect engineering. Adv. Mater. 33, 2008447 (2021).
Li, Y. et al. Synergetic effect of defects rich and MXene as cocatalysts for enhanced photocatalytic production activity of . Chem. Eng. J. 383, 123178 (2020).
Al-Temimy, A. et al. Impact of cation intercalation on the electronic structure of MXenes in sulfuric acid. ACS Appl. Mater. Interfaces 12, 15087-15094 (2020).
Wang, P.-F. et al. Elucidation of the Jahn-Teller effect in a pair of sodium isomer. Nano Energy 77, 105167 (2020).
Wang, Z. et al. Single-atom catalysts with ultrahigh catalase-like activity through electron filling and orbital energy regulation. Adv. Funct. Mater. 33, 2209560 (2023).
Huang, J., Liu, Z., Huang, D., Jin, T. & Qian, Y. Efficient removal of uranium (VI) with a phytic acid-doped polypyrrole/carbon felt electrode using double potential step technique. J. Hazard. Mater. 433, 128775 (2022).
Lv, S.-Y. et al. A non-polluting method for rapidly purifying uraniumcontaining wastewater and efficiently recovering uranium through electrochemical mineralization and oxidative roasting. J. Hazard. Mater. 416, 125885 (2021).
Ye, Y. et al. Electrochemical removal and recovery of uranium: effects of operation conditions, mechanisms, and implications. J. Hazard. Mater. 432, 128723 (2022).
Chen, T. et al. Ternary boron carbon nitrides hollow nanotubes with tunable p-n homojunction for photo-assisted uranium extraction: a combined batch, EXAFS and DFT calculations. Appl. Catal. B: Environ. 318, 121815 (2022).
Chen, T. et al. Actinide-uranium single-atom catalysis for electrochemical nitrogen fixation. Sci. Bull. 67, 2001-2012 (2022).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15-50 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169-11186 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865-3868 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953-17979 (1994).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758-1775 (1999).
Chadi, D. J. Special points for Brillouin-zone integrations. Phys. Rev. B 16, 1746-1747 (1977).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
شكر وتقدير
تم دعم هذا العمل من قبل NSFC (رقم U23A2O105، R.H.; 22303002، Y.L.; 22106126، T.C.; و U2267224، W.Z.)، وصندوق مفتوح لمختبر CNNC الرئيسي لاستخراج اليورانيوم من مياه البحر (KLUES2O22O1، W.Z.)، ومشروع المختبر الوطني الرئيسي لمواد الطاقة الصديقة للبيئة في SWUST (رقم 20fksy19، W.Z.). تم إجراء الحسابات العددية في مركز هيفي للحوسبة المتقدمة. يمد المؤلفون شكرهم لمختبر Shiyanjia.www.shiyanjia.comللمساعدة القيمة التي قدمتها في تحليل TEM و XPS.
مساهمات المؤلفين
قدم T.L. أهم المساهمات، على الرغم من أن جميع المؤلفين ساهموا في العمل. صمم T.L. و R.H. و W.Z. الدراسات وكتبوا الورقة. قام T.L. و H.J. بتنفيذ معظم التجارب. قدم T.C. و H.Z. و K.H. المشورة والمواد. قام C.J. و Y.L. بإجراء حسابات DFT. أشرف R.H. و W.Z. على البحث. ساهم جميع المؤلفين في تحليل البيانات وعلقوا على المخطوطة.
يجب توجيه المراسلات والطلبات للحصول على المواد إلى يان ليو، وينكون زو أو رونغ هي.
معلومات مراجعة الأقران تشكر مجلة Nature Communications تشانغهيون رو والمراجع الآخر المجهول على مساهمتهما في مراجعة هذا العمل. يتوفر ملف مراجعة الأقران.
ملاحظة الناشر: تظل شركة سبرينجر ناتشر محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
الوصول المفتوح هذه المقالة مرخصة بموجب رخصة المشاع الإبداعي النسب 4.0 الدولية، التي تسمح بالاستخدام والمشاركة والتكيف والتوزيع وإعادة الإنتاج بأي وسيلة أو صيغة، طالما أنك تعطي الائتمان المناسب للمؤلفين الأصليين والمصدر، وتوفر رابطًا لرخصة المشاع الإبداعي، وتوضح إذا ما تم إجراء تغييرات. الصور أو المواد الأخرى من طرف ثالث في هذه المقالة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة، ما لم يُشار إلى خلاف ذلك في سطر الائتمان للمواد. إذا لم تكن المادة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة وكان استخدامك المقصود غير مسموح به بموجب اللوائح القانونية أو يتجاوز الاستخدام المسموح به، فسيتعين عليك الحصول على إذن مباشرة من صاحب حقوق الطبع والنشر. لعرض نسخة من هذه الرخصة، قم بزيارةhttp://creativecommons.org/رخصة/بواسطة/4.0/. (ج) المؤلف(ون) 2024
المختبر الوطني الرئيسي لمواد الطاقة الصديقة للبيئة، كلية الدفاع الوطني، كلية علوم الحياة والهندسة، كلية المواد والكيمياء، المركز الوطني للتعاون في ابتكار التخلص من النفايات النووية والسلامة البيئية، معهد التكامل المدني والعسكري في سيتشوان، جامعة جنوب غرب العلوم والتكنولوجيا، مينيانغ، جمهورية الصين الشعبية.كلية الكيمياء وعلوم المواد، جامعة آنهوي نورمال، ووهو، جمهورية الصين الشعبية. البريد الإلكتروني: ly0201@ahnu.edu.cn; zhuwenkun@swust.edu.cn; her@swust.edu.cn
Ion pair sites for efficient electrochemical extraction of uranium in real nuclear wastewater
Received: 11 December 2023
Accepted: 6 May 2024
Published online: 16 May 2024
(A) Check for updates
Tao Lin , Tao Chen , Chi Jiao , Haoyu Zhang , Kai Hou , Hongxiang Jin , Yan Liu , Wenkun Zhu & Rong He ¹
Abstract
Electrochemical uranium extraction from nuclear wastewater represents an emerging strategy for recycling uranium resources. However, in nuclear fuel production which generates the majority of uranium-containing nuclear wastewater, fluoride ion ( ) co-exists with uranyl ( ), resulting in the complex species of and thus decreasing extraction efficiency. Herein, we construct ion pair extraction sites in for efficient electrochemical uranium extraction in wastewater from nuclear fuel production. These sites selectively bind with through the combined Ti-F and multiple bonds. In the uranium extraction, the uranium species undergo a crystalline transition from to . In real nuclear wastewater, the uranium is electrochemically extracted with a high efficiency of and finally purified as uranium oxide powder, corresponding to an extraction capacity of without saturation. This work paves an efficient way for electrochemical uranium recycling in real wastewater of nuclear production.
Uranium is the key fundamental resource in the nuclear industry . With the development of the nuclear industry, uranium resources in terrestrial ore will be depleted within less than a century, accompanied by the massive generation of uranium-containing nuclear wastewater . In practical situations, the majority of uraniumcontaining nuclear wastewater is produced by the procedures of fuel production, such as uranium enrichment, uranium conversion, and fuel element fabrication, which requires the wide usage of uranium fluoride . After the hydrolysis of uranium fluoride, the uranium in nuclear wastewater commonly exists in the form of uranyl ( ), together with the co-existing high concentration of fluoride ion . As such, extracting uranium under the interference of high concentrations of is an important issue for environmental protection and the recycling of uranium resources . For this nuclear wastewater, the traditional adsorption or ion exchange method commonly requires the pre-precipitation of by , resulting in the formation of uranium-containing as radioactive solid waste . Accordingly,
searching for other technologies for uranium extraction is highly desired under the interference of high concentrations of .
As an emerging technology, electrochemical uranium extraction has attracted ever-increasing attention due to the fast kinetics, increased extraction capacity, and resistance to the interference of anions . At present, electrochemical uranium extraction has been applied in the aqueous systems without . For example, the single atoms with functional amidoxime groups were reported to achieve an extraction capacity of from real seawater over 24 h . However, despite the significant progress, the electrochemical uranium extraction in real nuclear wastewater with the existence of is still challenging, because of the rather complex species of uranium due to the coordination effect . Specifically, the dominant hexavalent uranium ( ) species generally lies on a series of uranyl fluoride , such as , and , instead of bare . In this case, the conventional uranium extraction sites suffer from the competing coordination of uranium and , thus resulting in the poor extraction efficiency
of uranium under the interference of high concentrations of . Therefore, the construction of specific sites for the binding of is an essential prerequisite for achieving efficient electrochemical uranium extraction in real nuclear wastewater with high concentrations of .
Herein, we develop the with ion pair extraction sites for selective binding of and efficient electrochemical uranium extraction under the interference of high concentration of . Both synchrotron X-ray absorption fine structure (XAFS) and theoretical simulation demonstrate that the ion pair extraction sites are strongly bound with through the combined interactions of and . During the uranium extraction, the is reduced to as a gray deposit followed by further oxidation and crystallization as . In 400 mL of real nuclear wastewater produced by fuel production, the achieves an electrochemical extraction efficiency of for uranium within 7 h , with an extraction capacity of without saturation. The uranium-containing powder is successfully collected from the real nuclear wastewater, with of uranium proportion among the metal elements in the powder.
Results
Construction of with ion pair sites
The construction of ion pair sites originated from the reconstruction of lamellar OH -terminated , as illustrated in Fig. 1a. Initially, the Al
layers of commercial bulk were etched to form the accordionlike stack of F-terminated nanosheets. Followed by the exfoliation process under protection in aqueous solution, the stacked F-terminated nanosheets were transformed into lamellar OH terminated nanosheets (Supplementary Fig. 1). The flexible exfoliated lamellar OH-terminated nanosheets were confirmed by the high-resolution transmission electron microscope (HRTEM) images, and the X-ray diffraction (XRD) pattern (Supplementary Fig. 2). As shown by Fourier transform infrared spectroscopy (FT-IR) measurement, the exfoliated lamellar OH -terminated nanosheets lacked the peak of C-F stretching at relative to accordion-like stacked nanosheets, demonstrating the replacement of -F terminations by -OH terminations during the exfoliation process (Supplementary Fig. 3). After that, a wet chemical treatment was applied to transform the exfoliated lamellar OH-terminated into , which contained an ion pair of and . As shown by the transmission electron microscopy (TEM) images, the Ti(OH)PO4 exhibited a nanorod-like morphology (Fig. 1b). The atomic force microscopy (AFM) analysis and the corresponding three-dimensional graph of nanorods indicated the narrow height distribution due to the regular stacking (Supplementary Fig. 4). Figure 1c showed the HRTEM image of a free-standing nanocrystal. The nanorod exhibited a periodic layer-by-layer structure with a
Fig. 1 | Construction of ion pair sites. a The schematic illustration for the preparation of the with ion pair sites. TEM image of nanorods. c HRTEM image of nanorods. Inset: HRTEM image of the boundary of nanorods. d EDS analysis of nanorods. e XRD pattern, f Raman spectra, and static contact angle of nanorods and lamellar
OH-terminated nanosheets. The highlighted areas in Fig. If show the peak of OH -terminated and the P-O-P and P-O peaks of , respectively. Credits: a (schematic models) copyright Hangzhou SPHERE Technology Co., Ltd. Source data are provided as a Source Data file.
Fig. 2 | Bonding and electronic structure of ion pair sites. a FT-IR spectrum, b ESR spectrum, c Ti XPS spectrum, and XANES spectrum of nanorods and lamellar OH -terminated nanosheets. e Energy diagram of the
splitting orbits of peak. and represent two different sub-energy levels in the shell of the Ti atom, respectively. and represent the energy level splitting of orbitals in the coordination field. Source data are provided as a Source Data file.
spacing of 0.725 nm , which was larger than the diameter of uranyl or uranyl fluoride ( ). The inset was the edge region of the rod structure, which displayed the lattice fringe with interlayer spacing of 0.34 nm and indicated the (111) facets of . Determined by energy dispersive X-ray spectroscopy (EDS), the Ti, O, and P elements filled the outline of the selected region, indicating the uniform distribution of the elemental components in the nanorod (Fig. 1d).
The physical phase of nanorods was further validated. Two sets of diffraction peaks appeared in the XRD pattern of the nanorods (Fig. 1e). The two sets of characteristic peaks were identified as the standard monoclinal phase (JCPDS #360697) and (JCPDS #52-1529). Notably, the was the dimerization of after a dehydration process, indicating the transformable hydroxyl in . Figure 1f shows the Raman spectra of and lamellar OH terminated . After the reconstruction of OH -terminated into , the peak of pristine OH-terminated disappeared, accompanied by the characteristic peaks of bonds and bonds . Moreover, the exhibited a water static contact angle of , which was lower than that of pristine lamellar terminated (Fig. 1g). Such phenomenon resulted from the incorporation of hydroxyl into lattice with the enhanced hydrogen bonds in , further demonstrating the existence form of .
The ion pair sites in were investigated by the analysis of the bonding and electronic structure of . Determined by FT-IR measurement (Fig. 2a), the stretching vibrations of , and P-O-H in group were observed in the spectrum of , which differed from that of OH -terminated . In the electron spin
resonance (ESR) spectra (Fig. 2b), the exhibited a sharp resonance signal at the g-factor of 2.004, indicating the massive oxygen vacancies in . Moreover, the X-ray photoelectron spectroscopy (XPS) analysis was performed to check the chemical bonds of , which displayed the , and O signals in the survey spectra (Supplementary Fig. 5a). The oxidative state of P in the group was confirmed by the spectrum of (Supplementary Fig. 5b). In the spectrum, the peak of the O in the vicinity of oxygen vacancy was observed at 531.2 eV , further demonstrating the defective nature of (Supplementary Fig. 5c). Meanwhile, possessed the dominant signal of Ti-C bond, together with the slight signal of and bonds, which was attributed to the OH termination (Fig. 2c). By comparison, the Ti spectrum of showed three sets of peaks, which were assigned as , and . The massive low valent ( ) indicated the defective nature of , which was constructed by the oxygen vacancies during the transformation of terminated into .
To further demonstrate the electronic structure of Ti in nanorods, we tested the -edge X-ray absorption near-edge structure (XANES) spectra of hydroxyl-terminated and . As shown in Fig. 2d, both the edge and edge of nanorods exhibited a slight shift of to transition and a dramatic shift of to transition relative to pristine OH -terminated . This result was illustrated by the energy diagram of the splitting orbits of the Ti peak (Fig. 2e). Typically, Ti possessed five orbits, which could be divided into two orbits and three orbits . The binding energy of and orbits was associated with two factors, including the coordination environment and the electron filling of Ti. For the coordination environment, the transformation of in
Fig. 3 | Electrochemical uranium extraction of ion pair sites. a LSV tests on -terminated , and bare graphite felt electrodes. represents potential. b The electrochemical extraction efficiency of on terminated , and bare graphite felt electrodes. Inset: macroscopic deposition stripes of crystalline uranium. The stability of electrochemical extraction of U(VI) on electrode under different pH conditions, with different co-
existing ionic species. The change in uranium concentration after electrochemical uranium extraction under electrolytes with different concentrations (ranging from to ). The electrochemical extraction efficiency of on as the initial concentration of varied from to . Error bars represent standard deviation of three measurements. Source data are provided as a Source Data file.
OH-terminated into Ti-O in induced the positive shift of both and orbits in Ti -edge XANES, owing to the higher electronegativity of O than that of C . Moreover, for the factor of electron filling, possessed empty and orbits, whereas defective possessed abundant and , which respectively had one and two electrons in orbits. Due to the lower energy of orbits than orbits, the additional electrons in were located at orbits, resulting in the dramatic energy decrease of orbits. As a comprehensive result of the coordination environment and the electron filling of Ti , the peak in showed an obvious shift, whereas peak displayed a negligible shift relative to OH -terminated in -edge XANES (Supplementary Table 1). The and the group in constructed the ion pair sites, which were the key species for the uranium extraction under the interference of high concentration of .
Electrochemical uranium extraction
The nanorods were uniformly spread on graphite felt to evaluate the effect of ion pair sites on the electrochemical uranium extraction. An aqueous solution containing of and of was adopted as the electrolyte to simulate the wastewater of nuclear production. We demonstrated the reduction peak of in this electrolyte by linear sweep voltammetry (LSV) test (Fig. 3a). The nanorods exhibited a sharp reduction peak at -0.39 V vs. , which corresponded to the reduction of into pentavalent uranium . By comparison, these peaks of pristine terminated and the bare graphite felt were located at more negative positions with rather weak intensity. To double-check the origin of the reduction peak, we performed Cyclic Voltammetry (CV) tests in the presence or absence of in of (Supplementary Fig. 6). The overall current density of the CV curve
dramatically increased after the addition of , indicating that the participated in the composition of double-layer capacitance . Besides, the reduction peak disappeared in the absence of , further demonstrating the ascription of the uranium reduction peak. These results indicated the nanorods possessed a special activity for uranium extraction in the presence of .
As shown in Fig. 3b, the electrochemical extraction efficiency of on was within 5 h , which was significantly superior to that of OH -terminated and that of bare graphite felt ( ). Further discussion on the termination group of indicated the -OH group was the extraction site, instead of -F (Supplementary Fig. 7 and Supplementary Table 2). Notably, the ultimate extraction efficiency of from attained , which enabled the direct observation of macroscopic deposition stripes of crystalline uranium (inset of Fig. 3b). In addition, compared with the non-voltage adsorption method, the electrochemical extraction efficiency of on nanorods exhibited a 2 -fold enhancement (Supplementary Fig. 8). To collect the uranium from the electrode, 0.1 mol of HCl solution was taken as the desorbing agent. After 10 seconds of washing time, the yellow stripe-like crystallization on the surface of the electrode gradually dispersed in the desorbing solution (Supplementary Fig. 9). As a result, the electrochemical method with as an electrode represented an efficient protocol for uranium extraction in the presence of a high concentration of .
The stability of electrochemical extraction of on the nanorods was further evaluated. After 5 cycles of uranium extraction, the extraction efficiency of on retained a high value of (Supplementary Fig. 10). Moreover, regardless of the wide manipulation of pH from 4 to 10 , the extraction efficiency of on exceeded (Fig. 3c). Furthermore, the
anti-interference capability of was demonstrated by extraction efficiency in the presence of co-existing ions, especially , and , which commonly existed in the wastewater of nuclear production (Fig. 3d). We also tested the changes in pH and ion concentrations before and after the electrochemical uranium extraction reaction (Supplementary Fig. 11). All the pH values of the electrolyte increased after the electrochemical reaction because of the hydrogen evolution in the electrochemical process. The extraction efficiency of U(VI) remained at a high level of , indicating that none of the interference ions exhibited obvious influence on the extraction. Moreover, the concentrations of anions in the electrolyte displayed a negligible decrease after the electrochemical reaction, which was attributed to the slight adsorption of ions by the electrode. We also evaluated the influence of concentration change on uranium recovery (Fig. 3e). Regardless of the initial concentration of , the concentration was reduced from approximately to after the reaction with a high level of , indicating that the concentrations did not exhibit an obvious influence on the extraction for the electrode (Supplementary Fig. 12a). Such a phenomenon was attributed to the selective binding of by ion pair sites, instead of . Furthermore, slight decreases in the concentration of were observed after the electrochemical reaction, which was ascribed to the adsorption by the cation site and further co-crystallization by the uranium product (Supplementary Fig. 12b). Moreover, as the initial concentration of varied from to , the extraction efficiency of by remained consistently above , indicating an extraction capacity of without the phenomenon of extraction saturation (Fig. 3f). These results suggested the potential use of with ion pair sites towards the electrochemical uranium extraction in the wastewater of nuclear production.
Evolution of uranium species
The effective electrochemical uranium extraction of nanorods with ion pair sites in the presence of high concentrations of inspired us to explore the process of uranium deposition on the electrode. The time-dependent extraction experiment on nanorods was conducted in an aqueous electrolyte containing of and of (Fig. 4a). At the time point of 1 h , the deposited species appeared and exhibited a gray color. With the reaction proceeding to 3 h and 5 h , the gray deposit was gradually transformed into the yellow product, together with the aggregation of the solid deposits on the electrode. The color of the deposit finally transformed to a stable state of deep yellow at 7 h . To validate the species during the uranium extraction, we collected the solid deposits at different time points. As indicated by XRD patterns, the gray deposits at 1 h and 3 h were ascribed to (JCPDS #42-1215), which was a special species of containing and tetravalent uranium (U(IV)) (Fig. 4b). The existence of U(IV) can be explained by the dismutation of . By comparison, the XRD of yellow deposits at 5 h and 7 h well fitted by (JCPDS #38-0023) with domain species of , indicating the oxidation of low valent uranium in the deposits during the reaction. The species was also responsible for the slight decreases in the concentration of in the experiment of Supplementary Fig. 12. The K in the final uranium extraction product originated from the KF in the electrolyte, which could be replaced by Na in when using NaF as electrolyte (Supplementary Fig. 13).
The result of uranium evolution was verified by the U4f XPS spectra at different time points (Fig. 4c). With the reaction proceeding to 3 h , the peaks of were negatively shifted from 381.5 eV to 380.7 eV , indicating the continuous reduction of uranium during the reaction. With the reaction proceeding to 4 h and 7 h , the gray deposit was gradually transformed into the yellow product, together with the peaks of positively shifted to 381.8 eV , indicating the oxidation
of low valent uranium in the deposits during the reaction . For a precise interpretation, the gray deposits possessed more content of and than that of the yellow deposits (Supplementary Fig. 14). The intermediate of and final product of were also observed through the deposits of multiple cycles of CV test, which verified the evolution process of uranium species (Supplementary Fig. 15). The alternation of the uranium species in the deposit was accompanied by the shape evolution. At a reaction time of 1 h , the initial gray uranium deposits displayed a nanoneedle morphology with low crystallization (Supplementary Fig. 16). As time proceeded, the nanoneedles gradually evolved into nanosheets, with visible crystalline lattice fringes. Figure 4d showed a typical TEM image of the final yellow deposits, which showed the flexible nanosheet morphology. Moreover, the lattice fringes of this nanosheet possessed the interplanar spacing of 0.32 nm , belonging to the (220) facet of (Fig. 4e). In the EDS measurement, the U, K, and F elements were uniformly distributed in the nanosheets, further demonstrating the composition of the in the final yellow deposits (Fig. 4f). Accompanied by the shape evolution of the uranium deposit, the morphology of transformed from pristine stacked nanosheets to exfoliated nanosheets through the expansion of interlayer spacing (Supplementary Fig. 17). This result demonstrated the uranyl intercalation into the interlayer spaces of during the electrochemical reaction, which verified the advantage of larger interlayer spacing of than the diameter of .
To validate the species evolution involved in the uranium extraction, we directly investigated the valence state and coordination environment of uranium in the electrode at 3 h during the electrochemical reaction. Notably, the key intermediate deposit was not separated from the extraction material in this case. As shown by the -edge XANES spectra (Fig. 4 g ), the characteristic peaks at 17175.7 eV in the spectrum were identified as the white line peak, corresponding to the transition of electrons from the occupied orbital to the unoccupied . Significantly, the absorption edge position of electrochemical products was situated between and , with a slight shift towards the low-E side. This observation implied that the valence states of cations encompassed a range between +4 and +6 , indicating the uranium extraction underwent the electrochemical reduction process. Figure 4h illustrated the Fourier transform (FT) -weighted extended X-ray absorption fine structure (EXAFS) spectra of the electrochemical product. The FT curve of the electrochemical product had two main peaks at 1.4 and , which were attributed to the U-O coordination of the first and second shells . The first and second shells were denoted as and , which respectively referred to the axial O of uranyl species and the O atom at the adsorption site of ion pair sites. Through the EXAFS fitting, the coordination numbers of and were 2.2 and 3.4, respectively (Fig. 4i and Table 1). This result suggested that the adsorption process was assisted by three O atoms in the surface ion pair sites of nanorods. Specially, the U-F bond co-existed with a coordination number of 1.1, demonstrating the target adsorbate involved in the uranium extraction was the overall instead of , which resulted from the Coulomb interaction between ion pair sites and .
Reaction mechanism
The unusual adsorption of the overall and the corresponding species evolution motivate us to explore the intrinsic reaction mechanism. Based on the species evolution, we constructed a scheme to simulate the reaction process (Fig. 5a). The was adsorbed on the ion pair sites of nanorods without the separation of the U-F bond. After that, the uranyl was reduced to low-valent species, which weakened the Coulombian force of uranyl and , thus resulting in the formation of an intermediate product. The metastable low-valent uranium was then oxidized, followed by crystallizing with and forming the final .
Fig. 4 | Evolution of uranium species. a Photographs of the evolution of uranium species during the uranium extraction. XRD pattern of the collected solid deposits at different time points. XPS spectrum of electrodes at different electrochemical extraction time points. d TEM image of the final yellow deposits with nanosheet morphology. e HRTEM and f EDS measurement of the
composition of the in the final yellow deposits. The -edge XANES spectra of intermediate deposit. Inset: magnified pre-edge XANES region. The FT -weighted EXAFS spectra of the electrochemical product. i The corresponding space fitting curves of the electrochemical product. Source data are provided as a Source Data file.
Considering the adsorption of the overall was the key factor that influenced the extraction efficiency of uranium, we theoretically simulated the adsorption of on the ion pair site of nanorods (Fig. 5b, Supplementary Table 3, and Supplementary Data 1).
Table. 1 | Structural parameters of at the U -edge extracted from quantitative EXAFS curve-fittings
Path
-factor
2.2
1.7
4.1
-11.1
0.004
3.4
2.3
4.0
6.8
0.004
1.1
2.1
5.0
-11.0
0.004
Taking as an example, we compared the optimized adsorption configuration on OH-terminated and with on the surface. The OH -terminated possessed the normal -OH sites for bare adsorption, which possessed adsorption energy of -2.6 eV for without the interaction of and . For the model, the coordination of surface was unsaturated, giving rise to the interaction with . As such, the has stabilized on the ion pair sites through both the and interactions. In this model, three O atoms in the participated in the adsorption of , consistent with the experimental EXAFS result. Moreover, the adsorption energy of on the ion pair sites of was -4.5 eV , which was much more negative than that on the -OH sites of OH -terminated . The adsorption energy was able to be
Fig. 5 | Reaction mechanism of ion pair sites. a The schematic diagram of the rational reaction mechanism of ion pair sites. Optimized adsorption configurations of adsorbed on the (OH-terminated) and the axial O of uranyl species, the O atom at the adsorption site, the adsorption energy of . Source data are provided as a Source Data file.
further decreased by introducing additional O vacancies (Supplementary Fig. 18). Furthermore, the bond angle of of model was , which was lower than that of (OH-terminated). Given that the reduction of requires the decrease of bond angle, the ion pair sites of can facilitate the reduction of in . As a result, the ion pair sites stabilized the adsorption of and promoted the reduction of , which was responsible for the uranium extraction in high concentrations of .
Experiment of real nuclear wastewater
Motivated by the distinctive effect of ion pair sites on the uranium extraction in high concentrations of , we explored the uranium extraction from real nuclear wastewater on . This wastewater consisted of high levels of , and , which was produced by the washing of container in a nuclear fuel production facility. After 7-h electrolysis at a constant current density of , the concentration of was reduced from an initial value of to , with of extraction efficiency and of extraction capacity (Fig. 6a). Such value of extraction efficiency was 1.7-time larger relative to that of OH -terminated , indicating the effectiveness of with ion pair sites (Supplementary Fig. 19). With the proceeding of the electrolysis, the yellow color of real wastewater gradually faded, finally resulting in a colorless and transparent residual liquid (Fig. 6b). The concentration of all metal species in the transparent residual liquid was displayed in the Supplementary Table 4. Compared with the yellow real
wastewater, the and respectively decreased by ( decrease) and ( decrease), which was mainly ascribed to the adsorption of the electrode, instead of the electrochemical extraction (Supplementary Fig. 20). The majority of adsorbed and could be removed from the electrode by washing process. Additionally, the low initial concentrations (below ) of trace metal elements were essentially inconsequential to the uranium extraction process and purity of uranium deposits. Therefore, the uranium was able to be efficiently extracted from the electrode.
After the electrochemical reaction, the uranium was densely deposited on the electrode, which can be desorbed, simply precipitated by KOH , and collected as a solid powder (Fig. 6c). We analyzed the proportion of metal species in this powder by inductively coupled plasma optical emission spectrometry (ICP-OES) measurement (Fig. 6d). The proportion of uranium reached 92.1% among the metal elements in the powder, which was convenient for the following separation and reuse of uranium. In addition, we employed a typical precipitation-calcination process for the further purification of uranium products (Fig. 6e). Initially, we collected and dissolved uranium extraction product in HCl . Subsequently, excess ammonia was added to the above solution to form a precipitate of ammonium diuranate. Finally, ammonium diuranate was collected by centrifugation and heated at for 5 h to form a black-green uranium product. According to XRD (Supplementary Fig. 21) and ICP-OES tests (Fig. 6f), the purified black-green uranium product was identified as (JCPDS #31-1425) with a uranium content of among the metal elements, which can be directly used in the uranium production.
Fig. 6 | Experiment of real nuclear wastewater of ion pair sites. a The electrochemical extraction efficiency of on in real nuclear wastewater.
b The color change of real nuclear wastewater before and after electrolysis. c The deposited uranium on the electrode and the corresponding collected powder after electrolysis. d The proportion of uranium among the metal species in the powder.
e The schematic diagram of the precipitation-calcination purification process for uranium. The proportion of uranium among the metal species in the purification product. Error bars represent standard deviation of three measurements. Source data are provided as a Source Data file.
Consequently, the electrochemical uranium extraction by provided a feasible strategy for resource recovery in real wastewater of nuclear production. We also evaluated the approximate operating cost. The consumed electricity was 31.5 Wh , which means the extraction per kg of U requires electricity of , corresponding to an electricity cost of US dollars (according to the price of China). For the cost of electrode, the price was approximately 0.2 US dollars considering the purchase of precursor, synthetic material, and graphite support. Considering the recycling use of the electrode, the cost of electrode material was US dollars for extracting 1 kg of uranium. Therefore, the overall cost was approximately 22 US dollars for the extraction of 1 kg U considering both energy consumption and material costs.
Discussion
In summary, the ion pair sites are constructed in as uranium extraction material for nuclear wastewater containing high concentrations of from fuel production. In simulated electrolytes with
up to of possesses an extraction efficiency of in 5 h . The mechanistic study reveals that can be strongly bound in the form of and Ti-F bonds. With the reaction proceeding, the adsorbed is transformed from the reduced state to the crystalline state . In a real nuclear wastewater experiment, the exhibits extraction efficiency of uranium within 7 h regardless of the interferential of , with an extraction capacity of without saturation. After the simple collection, the powder product containing high-purity of uranium is obtained, demonstrating the successful recycling of uranium in real wastewater. Our work not only presents an efficient uranium extraction material for resisting high concentrations of , but also provides an efficient strategy for uranium recovery in real and complex nuclear wastewater.
Methods
Chemicals and materials
powder ( 400 mesh) was purchased from 11 Technology Co., Ltd. Hydrochloric acid ( purity) and Phytic acid (PA, 70%
purity) were supplied by Chengdu Kelong Chemical Factory. Lithium fluoride (LiF, 99% purity) and Uranium nitrate ( purity) were obtained from Aladdin Shanghai Chemical. Graphite felt (Carbon content ) and ethanol (AR) were purchased from CeTech Co., Ltd. Nafion membrane solution (NR50, ) was acquired from Shanghai Macklin Biochemical Co., Ltd. The real nuclear wastewater was collected from a nuclear fuel production facility. All reagents and solvents were used without purification.
Characterizations
Scanning electron microscopy (SEM) images were displayed on a JSM Scanning Electron Microscope. Transmission electron microscopy (TEM), high-resolution transition electron microscopy (HRTEM), and energy dispersive X-ray spectroscopy (EDS) images were displayed on a JEM 2100 F transmission electron microscope operating at an accelerating voltage of 200 kV . Atomic force microscopy (AFM) images were obtained on a Bruker Dimension ICON testing at ScanAsyt mode. Inductively coupled plasma optical emission spectrometry (ICPOES) analyses were performed on PerkinElmer ICP 2100 and PerkinElmer Optima 5300 DV. X-ray diffraction (XRD) data were collected on a Smartlab SE diffractometer equipped with a source. Raman spectra (Raman) were obtained on a Thermo Fischer DXR Laser Raman spectrometer testing at a wavelength of 532 nm . Fourier transform infrared (FT-IR) spectroscopy was determined from Nicolet iS 10 Spectrometer. The defects and surface properties of materials were identified by electron spin resonance (ESR) spectroscopy using a Bruker EMXplus. X-ray photoelectron spectroscopy (XPS) analysis was performed through Thermo Kalpha, equipped with a monochromatic Al K X-ray source. The concentration of anions in the electrolyte was determined by an ion chromatograph (IC). The oxidation states characterization of the samples was utilized by the X-ray absorption fine structure (XAFS) through the transmission mode for edge and U -edge in the National Synchrotron Radiation Laboratory (NSNR). The absorption edge positions were calibrated during the XAFS measurements. Extended X-ray absorption fine structure (EXAFS) data were collected from the ATHENA module, integrated within the IFEFIT software suite. Linear sweep voltammetry (LSV) and Cyclic Voltammetry (CV) data were displayed on the CHI 660E electrochemistry workstation.
Theoretical calculations
We have performed the DFT calculations in our system by using the Vienna ab initio simulation package (VASP, Version: VASP 5.4.4) . The PBE function was used for describing electronic exchange . The projector augmented wave method was adopted to describe the interactions between the ion cores and valence electrons . The energy cutoff for the plane-wave basis set was 500 eV . The Brillouin scheme sampled the Monkhorst-Pack scheme using a -point grid . During the geometry optimization and electronic structure calculation, the atomic positions were optimized until the energy and the maximum force were atom and , respectively. The van der Waals dispersion by employing the DFT-D3 method of Grimme was considered for all the calculations .
Construction of with ion pair sites
The exfoliated lamellar OH-terminated nanosheets were synthesized following the MILD method by in situ HF selective etching of the Al layer of precursor. Specifically, was dissolved with solution in a Teflon beaker, followed by the slow addition of in an ice bath under magnetic stirring. The reaction was conducted at under ventilated conditions for 24 h to avoid safety hazards caused by HF use. The accordion-like stack -F-terminated was collected and centrifuged at several times with deionized water to remove any vestigial acid until the supernatant reached a pH of 6 . Subsequently, the slurry above was
immersed in deionized water ( 100 mL ) to form a homogeneous suspension via sonication for 2 h under the protection of . After that, the exfoliated OH -terminated aqueous supernatant in a centrifuge cube was collected after centrifugation once again at for 5 mins. Finally, the suspension for the experiments was prepared by diluting the exfoliated OH -terminated suspension to . The ion pair sites were synthesized via a facile wet chemical method. Typically, the exfoliated OH -terminated suspension ( 2 mL ) was mixed with deionized water ( 40 mL ) and stirred for 30 mins at room temperature to obtain a uniformly dispersed solution. Then, phytic acid ( 1.6 mL ) was added to the above-dispersed solution dropwise and kept stirring for another 1 h . Afterward, the homogeneous solution was placed into a Teflon-lined autoclave ( 50 mL ) and heated at for 12 h . Finally, the ion pair sites were collected after washing with deionized water and ethanol several times, and freeze-dried for 24 h .
Electrochemical uranium extraction experiments in simulated nuclear wastewater
The electrochemical uranium extraction experiments in simulating nuclear wastewater were measured with a three-electrode system. The working electrodes were fabricated as follows: ion pair sites , carbon black ( 2.00 mg ), and Nafion Membrane solution ( ) were mixed with ethanol ( 2 mL ) and sonicated until they became a homogeneous ink. The as-prepared ink was directly brushcoated on graphite felt ( ) and dried at . The platinum (Pt) wire and electrode were used as the counter electrode and reference electrode, respectively. The uranium extraction experiments were conducted under a constant current of . The electrochemical uranium extraction performances were tested in an aqueous solution ( 50 mL ) containing of and of ( were sourced from KF electrolyte). The non-voltage adsorption experiments were tested on an unpowered three-electrode system in 50 mL of simulating nuclear water. The as-prepared electrode, Pt wire, and electrode were also used as the working electrode, counter electrode, and reference electrode respectively. The condition of the adsorption test was similar to that of electrochemical extraction except for excluding external current or voltage. The effects of the initial concentration of uranium (from 50 to ), pH of the solution (from 4 to 10), interfering ions (including ), and the influence of (from 5 to ) on the electrochemical uranium extraction performance were measured in detail. The concentrations of the uranium and other cations in the electrolyte before and after electrolysis were determined by ICP-OES. The concentrations of the anions in the electrolyte before and after electrolysis were determined by IC. In the desorption process, the working electrode with extracted uranium was transferred into HCl aqueous solution ( 0.1 mol ). The extraction experiments were conducted three times and the error bars were used in the curves. LSV and CV tests were performed in simulated nuclear wastewater at a scan rate of .
Electrochemical uranium extraction experiments in real nuclear wastewater
The electrochemical uranium extraction experiments in real nuclear wastewater were measured with a typical two-electrode system. Graphite felt was cut into shapes as electrode substrates. ion pair sites ( 40.0 mg ), carbon black ( 8.0 mg ), and Nafion Membrane solution ( ) were mixed with ethanol ( 8 mL ) and sonicated for 1 h to form a homogeneous ink. The as-prepared ink was then uniformly brush-coated on graphite felt to prepare the working electrode, and Pt wire was used as the counter electrode. The uranium extraction was operated under a constant current of in 400 mL of real nuclear wastewater. The deposited electrode was immersed in ethanol and purified through an ultrasonic reaction, and
the powder was collected by drying. The proportions of metallic species in this powder were measured and analyzed by ICP-OES. The concentration changes of the uranium and other cations in real nuclear wastewater before and after electrolysis were determined by ICP-OES.
Uranium product purification
The purification of uranium was performed through a typical precipitation-calcination process. Initially, the uranium product from the electrochemical extraction was collected and dissolved in HCl . Subsequently, excess ammonia was added to the above solution to form a precipitate of ammonium diuranate. Finally, ammonium diuranate was collected by centrifugation and heated at for 5 h to form a black-green uranium product.
Calculations
For quantifying the extraction efficiency ( ), the uranium extraction capacity ( ), and adsorption energy ( ) were calculated according to the following equations:
Where and represent the initial and final uranium concentration, respectively.
Where and represent the initial and final uranium concentration, respectively; and denote the mass of the catalyst and volume of the electrolyte, respectively.
Where , and are the total energies of the adsorbate-substrate (AS), substrate (S), and adsorbate (A), respectively.
Data availability
The data that supports the findings of the study are included in the main text and supplementary information files. Source data are provided with this paper. Source Data files are available in Figshare under accession code https://doi.org/10.6084/m9.figshare.25603905. Source data are provided with this paper.
References
Chen, T. et al. Advanced photocatalysts for uranium extraction: elaborate design and future perspectives. Coord. Chem. Rev. 467, 214615 (2022).
Zhang, S., Li, H. & Wang, S. Construction of an ion pathway boosts uranium extraction from seawater. Chem 6, 1504-1505 (2020).
Tsouris, C. Uranium extraction: fuel from seawater. Nat. Energy 2, 17022 (2017).
Mei, D., Liu, L. & Yan, B. Adsorption of uranium (VI) by metal-organic frameworks and covalent-organic frameworks from water. Coord. Chem. Rev. 475, 214917 (2023).
Hu, Y. et al. Photochemically triggered self-extraction of uranium from aqueous solution under ambient conditions. Appl. Catal. B: Environ. 322, 122092 (2023).
Zhou, Y. et al. Amidoxime-functionalized MXene beads for the effective capture of uranium from wastewater with high fluoride concentrations. Chem. Eng. J. 471, 144647 (2023).
Ohashi, Y., Murashita, S. & Nomura, M. Extraction of uranium from solid waste containing uranium and fluorine. Miner. Eng. 61, 32-39 (2014).
Li, Z. et al. Exciton dissociation and transfer behavior and surface reaction mechanism in Donor-Acceptor organic semiconductor photocatalytic separation of uranium. Appl. Catal. B: Environ. 332, 122751 (2023).
Busquim e Silva, R., Kazimi, M. S. & Hejzlar, P. Nuclear fuel recycling: national and regional options for the US nuclear energy system. Energy Environ. Sci. 3, 996-1010 (2010).
Wang, C. et al. Uranium in situ electrolytic deposition with a reusable functional graphene-foam electrode. Adv. Mater. 33, 2102633 (2021).
Song, P. S., Min, B. Y., Choi, W. K., Jung, C. H. & Oh, W. Z. Effects of a slag former on the absorption of cerium and uranium oxide within a slag during a melting of stainless steel contaminated with uranium. Sep. Purif. Technol. 60, 136-141 (2008).
Xing, S., Luo, M., Wu, Y., Liu, D. & Dai, X. Rapid determination of uranium isotopes in calcium fluoride sludge by tandem quadrupole ICP-MS/MS. J. Anal. Spectrom. 34, 2027-2034 (2019).
Liu, C. et al. A half-wave rectified alternating current electrochemical method for uranium extraction from seawater. Nat. Energy 2, 17007 (2017).
Wang, Y. et al. Electrochemical-mediated regenerable Fell active sites for efficient uranium extraction at ultra-low cell voltage. Angew. Chem. Int. Ed. 62, e202217601 (2023).
Riedhammer, J., Halter, D. P. & Meyer, K. Nonaqueous electrochemistry of uranium complexes: A guide to structure-reactivity tuning. Chem. Rev. 123, 7761-7781 (2023).
Keener, M. et al. Redox-switchable carboranes for uranium capture and release. Nature 577, 652-655 (2020).
Liu, T. et al. Removal and recovery of uranium from groundwater using direct electrochemical reduction method: performance and implications. Environ. Sci. Technol. 53, 14612-14619 (2019).
Yang, H. et al. Functionalized iron-nitrogen-carbon electrocatalyst provides a reversible electron transfer platform for efficient uranium extraction from seawater. Adv. Mater. 33, 2106621 (2021).
Das, N., Das, A., Sarma, K. P. & Kumar, M. Provenance, prevalence and health perspective of co-occurrences of arsenic, fluoride and uranium in the aquifers of the Brahmaputra River floodplain. Chemosphere 194, 755-772 (2018).
Shen, J. & Schäfer, A. Removal of fluoride and uranium by nanofiltration and reverse osmosis: a review. Chemosphere 117, 679-691 (2014).
Miskowiec, A., Niedziela, J. L., Kirkegaard, M. C. & Shields, A. E. Analysis of water coupling in inelastic neutron spectra of uranyl fluoride. Sci. Rep. 9, 10476 (2019).
Li, X. et al. MXene chemistry, electrochemistry and energy storage applications. Nat. Rev. Chem. 6, 389-404 (2022).
Rosenkranz, A., Righi, M. C., Sumant, A. V., Anasori, B. & Mochalin, V. N. Perspectives of 2D MXene tribology. Adv. Mater. 35, 2207757 (2023).
Vahidmohammadi, A., Rosen, J. & Gogotsi, Y. The world of twodimensional carbides and nitrides (MXenes). Science 372, eabf1581 (2021).
Robertson, D. D. & Tolbert, S. H. A direct and clean route to MXenes. Science 379, 1189-1190 (2023).
Zhang, X. et al. MXene aerogel scaffolds for high-rate lithium metal anodes. Angew. Chem. Int. Ed. 57, 15028-15033 (2018).
Sun, B. et al. Redox-active metaphosphate-like terminals enable high-capacity MXene anodes for ultrafast Na-ion storage. Adv. Mater. 34, 2108682 (2022).
Lei, J. et al. Enhanced photoreduction of on nanosheets by oxygen defect engineering. Chem. Eng. J. 416, 129164 (2021).
Zeng, Y. et al. Oxygen-vacancy and surface modulation of ultrathin nickel cobaltite nanosheets as a high-energy cathode for advanced Zn-ion batteries. Adv. Mater. 30, 1802396 (2018).
Li, R. et al. Flexible and high-performance electrochromic devices enabled by self-assembled 2D TiO MXene heterostructures. Nat. Commun. 12, 1587 (2021).
Zhang, H. et al. MXene-derived quantum dots distributed on porous carbon nanosheets for stable and long-life Li-S batteries: enhanced polysulfide mediation via defect engineering. Adv. Mater. 33, 2008447 (2021).
Li, Y. et al. Synergetic effect of defects rich and MXene as cocatalysts for enhanced photocatalytic production activity of . Chem. Eng. J. 383, 123178 (2020).
Al-Temimy, A. et al. Impact of cation intercalation on the electronic structure of MXenes in sulfuric acid. ACS Appl. Mater. Interfaces 12, 15087-15094 (2020).
Wang, P.-F. et al. Elucidation of the Jahn-Teller effect in a pair of sodium isomer. Nano Energy 77, 105167 (2020).
Wang, Z. et al. Single-atom catalysts with ultrahigh catalase-like activity through electron filling and orbital energy regulation. Adv. Funct. Mater. 33, 2209560 (2023).
Huang, J., Liu, Z., Huang, D., Jin, T. & Qian, Y. Efficient removal of uranium (VI) with a phytic acid-doped polypyrrole/carbon felt electrode using double potential step technique. J. Hazard. Mater. 433, 128775 (2022).
Lv, S.-Y. et al. A non-polluting method for rapidly purifying uraniumcontaining wastewater and efficiently recovering uranium through electrochemical mineralization and oxidative roasting. J. Hazard. Mater. 416, 125885 (2021).
Ye, Y. et al. Electrochemical removal and recovery of uranium: effects of operation conditions, mechanisms, and implications. J. Hazard. Mater. 432, 128723 (2022).
Chen, T. et al. Ternary boron carbon nitrides hollow nanotubes with tunable p-n homojunction for photo-assisted uranium extraction: a combined batch, EXAFS and DFT calculations. Appl. Catal. B: Environ. 318, 121815 (2022).
Chen, T. et al. Actinide-uranium single-atom catalysis for electrochemical nitrogen fixation. Sci. Bull. 67, 2001-2012 (2022).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15-50 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169-11186 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865-3868 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953-17979 (1994).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758-1775 (1999).
Chadi, D. J. Special points for Brillouin-zone integrations. Phys. Rev. B 16, 1746-1747 (1977).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Acknowledgements
This work was supported by NSFC (No. U23A2O105, R.H.; 22303002, Y.L.; 22106126, T.C.; and U2267224, W.Z.), the Open Fund of CNNC Key Laboratory for Uranium Extraction from Seawater (KLUES2O22O1, W.Z.), the Project of State Key Laboratory of Environment-Friendly Energy Materials in SWUST (No. 20fksy19, W.Z.). Numerical computations were performed at Hefei Advanced Computing Center. The authors extend their gratitude to Shiyanjia Lab (www.shiyanjia.com) for providing invaluable assistance with the TEM and XPS analysis.
Author contributions
T.L. made the most important contributions, although all authors made contributions to the work. T.L., R.H., and W.Z. designed the studies and wrote the paper. T.L. and H.J. performed most of the experiments. T.C., H.Z., and K.H. provided advice and reagents. C.J. and Y.L. performed DFT calculations. R.H. and W.Z. supervised the research. All authors contributed to data analysis and commented on the manuscript.
Correspondence and requests for materials should be addressed to Yan Liu, Wenkun Zhu or Rong He.
Peer review information Nature Communications thanks Changhyun Roh and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/ licenses/by/4.0/.
(c) The Author(s) 2024
State Key Laboratory of Environment-friendly Energy Materials, School of National Defense, School of Life Science & Engineering, School of Materials & Chemistry, National Co-innovation Center for Nuclear Waste Disposal & Environmental Safety, Sichuan Civil-military Integration Institute, Southwest University of Science & Technology, Mianyang, P. R. China. School of Chemistry and Materials Science, Anhui Normal University, Wuhu, P. R. China.
-e-mail: ly0201@ahnu.edu.cn; zhuwenkun@swust.edu.cn; her@swust.edu.cn