مواقع ذرات مفردة قوية Fe-N4-C6O2 لتنشيط PMS بكفاءة وزيادة تفاعل FeIV = O Robust Fe-N4-C6O2 single atom sites for efficient PMS activation and enhanced FeIV = O reactivity
تنظيم الميكروبيئةتحفيز الذرات الفردية (SACs) يتحكم بشكل حاسم في تنشيط بيروكسيمونوكبريتات (PMS). على الرغم من أن استبدال الذرات غير المتجانسة التقليدي في التنسيق الأولي يعزز النشاط، إلا أنه يعطل الحديد-التناظر والتسويات تعزز الاستقرار. هنا، نقترح إضافة الأكسجين في الغلاف التنسيقي الثانوي لبناء، مما يعزز المجال الكهربائي المحلي مع الحفاظ على تنسيق التناظر الأصلي، وبالتالي يوازن بين نشاطه واستقراره. هذه الطريقة تقلل من تشوه بنية رابطة الحديد-نيتروجين (تم تقليل سعة الرابطة من إلى ) أثناء تفعيل PMS عن طريق خفض كثافة الإلكترونات في مركز الحديد لتعزيز رابطة الحديد-النيتروجين، مما يحقق متانة تحفيزية ممتدة ( ). في الوقت نفسه، يقلل مجال التنسيق الضعيف من طاقة المدارات، تعزز النوكليوفيلية-هجوم الحديد الأكسو عالي التكافؤ نحو البيسفينول أ، وزيادة معدل تحلله بمقدار 41.6 مرة. تُظهر هذه الدراسة هندسة التنسيق الثانوي كاستراتيجية قابلة للتطبيق لحل التوازن بين النشاط والاستقرار في تصميم المحفزات ذات النشاط الانتقائي، مما يوفر آفاقًا واعدة لتطوير المحفزات البيئية.
لقد جذبت عمليات الأكسدة المتقدمة (AOPs) القائمة على بيروكسيمونوكبريتات (PMS) اهتمامًا كبيرًا في التحكم في الملوثات وإعادة تأهيل البيئة نظرًا لكفاءتها العالية في نطاق واسع من درجة الحموضة.مؤخراً، تم استخدام المحفزات ذات الذرة الواحدة (SACs) مع المعادن- (م- تُستخدم المواقع، حيث يتم تنسيق مراكز المعادن مع أربعة ذرات نيتروجين، على نطاق واسع لتفعيل PMS بسبب نشاطها التحفيزي المتميز، والانتقائية الممتازة، وكفاءة استخدام المعادن العالية بشكل استثنائي.القدرة على تبرع الإلكترون من هذهيمكن أن تؤدي المواقع إلى تفعيل متلازمة ما قبل الحيض لإنتاج الجذور الكبريتيةالجذور الهيدروكسيلية، والمعادن عالية القيمة – أوكسو ( )، إلخ، مما يسهل بشكل فعال تحلل المواد العضوية المختلفة الملوثات. ومع ذلك، فإن طاقات تفكك الروابط العالية لـوربما تشكل الروابط O-O في PMS تحديًا كبيرًا لتفكيكها، مما يعيق تفعيل PMS عند M-مواقعلذلك، فإن تطوير المحفزات عالية الأداء لتعزيز تفعيل PMS أمر حاسم للأكسدة الفعالة للملوثات.
مؤخراً، تم اقتراح العديد من الاستراتيجيات لتنشيط نظام إدارة المشاريع بكفاءة من خلال زيادة كثافة المراكز المعدنية أو تنظيم بيئتها المحلية المحيطة في الأنظمة الحفزية المعتمدة على المعادن (M-SACs).من بين هذه الاستراتيجيات، تعتبر الاستراتيجية الأكثر شيوعًا هي الاستبدال الجزئي للنيتروجين في الغلاف التنسيقي الأول بعناصر أجنبية، مما يخلق مجالًا كهربائيًا محليًا معززًا لتسهيل التفعيل.
من و السندات فيلسوء الحظ، هذه الاستراتيجية تدمر الهيكل المتماثل لـالمواقع، مما قد يقلل من استقرارها على المدى الطويل من خلال إزالة المعادن المتسارعة. على سبيل المثال، وجد داي وآخرون أن النشاط التحفيزي لـتقلص بشكل ملحوظ بعد دورتين من التفاعل داخلأظهرت مجموعة بينغ أنيمكن أن يزيل كل الباراسيتامول خلال 40 دقيقة، لكن لم يتم تحقيق التحلل الكامل للباراسيتامول حتى بعد تمديد وقت التفاعل إلى 180 دقيقة في دورة إعادة الاستخدام الثانية.. لذلك، لا يزال من التحديات الكبيرة تحقيق التوازن بين النشاط العالي وطول مدة صلاحية SACs لتفعيل PMS.
مختلف عن تشويب الغلاف التنسيقي الأول، فإن تشويب الذرات غير المتجانسة في الغلاف التنسيقي الثاني لـيمكن أن تحسن المواقع الأداء التحفيزي لـالمواقع من خلال تعديل الهيكل الإلكتروني لمركز المعدن من خلال التفاعلات طويلة المدى بين d-p، وفي الوقت نفسه الحفاظ علىهيكل التنسيقعلى سبيل المثال، تم إثبات أن تأثير سحب الإلكترون الناتج عن إضافة الكبريت في الغلاف التنسيقي الثاني كان مفيدًا لتثبيت ذرات الحديد الفردية، مما يعزز بشكل كبير نشاط اختزال النترات واستقراره.المواقع مقارنةً مع لوحة الحديد. ومع ذلك، فإن تأثير موقع قشرة التخصيب بالكبريت على الاستقرار التحفيزي لـلم يتم التحقيق في المواقعبالمقارنة مع اختزال النترات، تفرض تفعيل PMS متطلبات أكثر صرامة على الاستقرار التحفيزي.المواقع بسبب بيئتها المؤكسدة القوية. فيما يتعلق بأن الكهربية السالبة لـأعلى من ذلك لـنتوقع أن يؤدي تشبع الأكسجين إلى تقليل كثافة الإلكترونات في المركز المعدني بشكل أكثر وضوحًا من خلال التفاعل بعيد المدى بين d-p وتأسيس تدرج كهربائي أقوى حول M-المواقع، مما يسهل امتصاص وتفعيل PMS السالب الشحنة (). في هذه الأثناء، قد يؤدي تشبع الأكسجين في الغلاف التنسيقي الثاني إلى تعزيز تكوين فراغات كربونية حول المركز المعدني وتغيير قدرة الربط بين المعدن والذرات التنسيقية المحيطة، مما يؤدي إلى قمع إزالة المعدن.
في هذا البحث، نطور استراتيجية ما قبل التنسيق لتصميم تشبع الذرات غير المتجانسة من خلال التحكم بدقة في استبدال ذرات النيتروجين المجاورة في القشرة التنسيقية الأولى وذرات الكربون البعيدة في القشرة التنسيقية الثانية لمركز الحديد، بهدف توضيح تأثيرات تشبع الأكسجين في القشرة التنسيقية الأولى أو الثانية على التفاعل والاستقرار.المواقع من خلال التوصيف المنهجي، تجارب التحلل، حسابات نظرية الكثافة الوظيفية (DFT) ومحاكاة الديناميكا الجزيئية (MD). تسلط هذه الدراسة الضوء على تصميم SACs مع أداء معزز في تنشيط PMS من خلال معالجة التوازن بين النشاط والاستقرار المستدام.
النتائج
التركيب والتوصيف
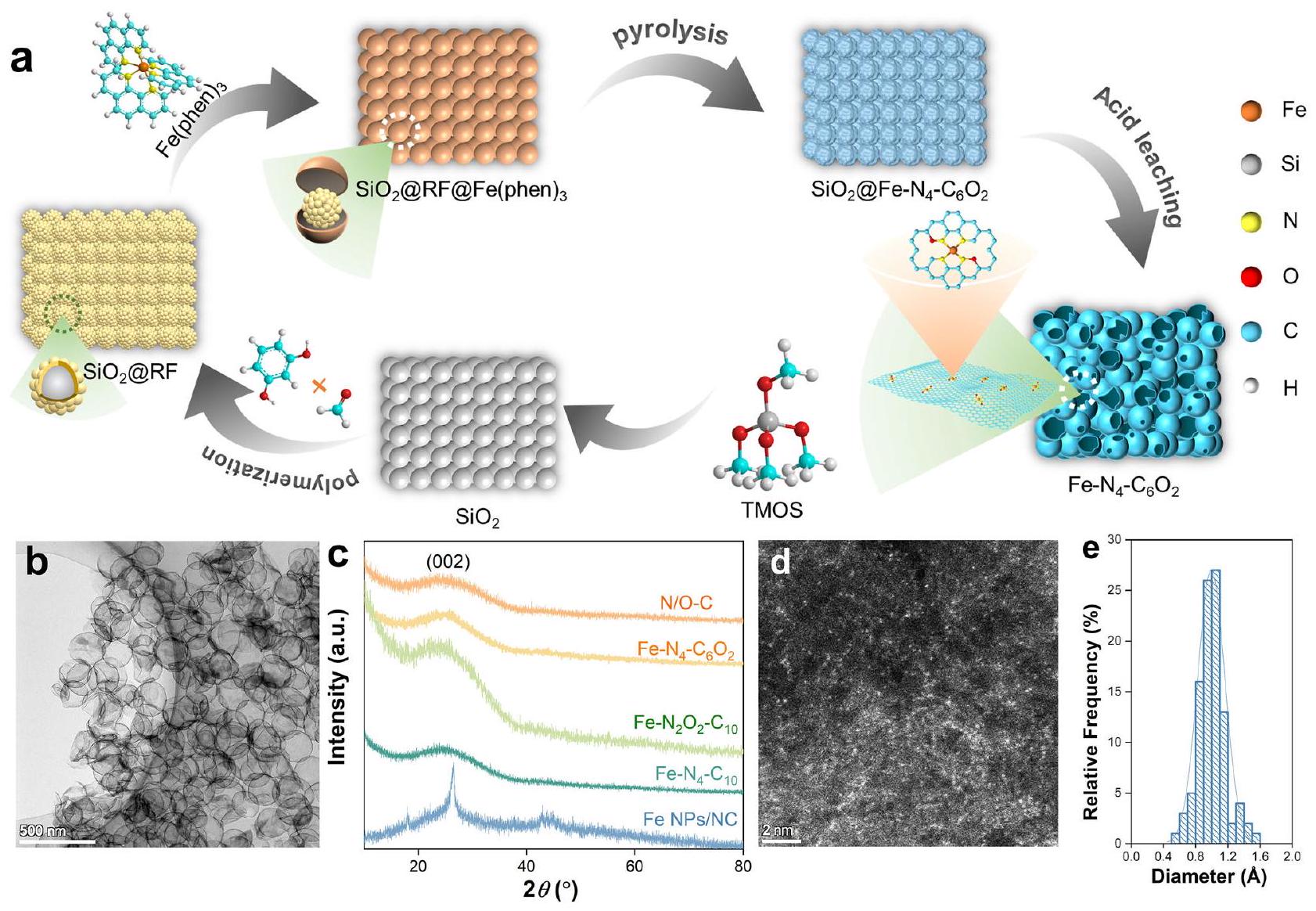
في-SACs مع الأكسجين المخلوط في الأول أو القشرة الثانية ( ) تم تصنيعها بواسطة استراتيجية ما قبل التنسيق (الشكل 1أ). أولاً، قمنا بتصنيع حاملين ذوي هيكل قشرة-نواة باستخدام السيليكا ( ) كطبقة أساسية. كانت إحدى طبقات القشرة مكونة من كربون مخدر بالنيتروجين (N-C) بدون أي مجموعات وظيفية تحتوي على الأكسجين ( ) بينما كانت الأخرى تتكون من ريسورسينولفورمالديهايد (RF) المعزز بمجموعات وظيفية تحتوي على الأكسجين ( ) (الشكل التوضيحي التكميلي 1a-c). في هذه الأثناء، تم تحضير معقدات تآزر الحديد-النيتروجين/الأكسجين (I) وتآزر الحديد-النيتروجين (II) باستخدام (S, S)-(+)-N, N’-bis(3,5-di-tert-butylsalicylidene)-1, 2-cyclohexanediamin كمقدمة للنيتروجين/الأكسجين و1,10-phenanthroline كمقدمة للنيتروجين، على التوالي. بعد ذلك، تم ترسيب المعقدات (I) و(II) على و باستخدام تقنية التبخر الدوار، والتي تم إخضاعها بعد ذلك لعمليات التحلل الحراري عند درجات حرارة عالية وإزالة القالب للحصول على الحديد و ، على التوالي. Fe-SAC بدون إضافة الأكسجين (Fe- ) تم تخليقه أيضًا بنفس الإجراء باستثناء ترسيب المركب (I) على . للمقارنة، تم تشويب N/O تم تصنيع جزيئات الكربون (N/O-C)، N-C، وجزيئات الحديد المدعومة على الكربون المخدر بالنيتروجين (جزيئات الحديد/NC) أيضًا (الجدول التكميلي 1).
كانت جميع الثلاثة Fe-SACs ذات هيكل مجوف رقيق للغاية وموحد وذو مسام هرمية (الشكل 1b والأشكال التكميلية 1-3)، مما يسهل التعرض الكامل لمواقع النشاط وانتقال الكتلة أثناء تنشيط PMS. كانت أنماط حيود الأشعة السينية للمواد المسحوقة (PXRD) تحتوي فقط على قمة عريضة واحدة عندالمطابقة للطائرة (002) لحامل الكربون، دون أي قمم ملحوظة ناتجة عن الحديد البلوري أو أكاسيد الحديد (الشكل 1c). في هذه الأثناء، تم استبعاد وجود جزيئات الحديد من خلال صور المجهر الإلكتروني الناقل عالي الدقة (HR-TEM) وأنماط حيّز الإلكترون المختار (SAED) (الشكل التكميلي 1). صور المجهر الإلكتروني الماسح بتصحيح الانحرافات في مجال الظلام الحلقي العالي الزاوية (ACHAADF STEM) لـأشارت بوضوح إلى بقع ساطعة بأحجام تتوافق مع ذرات الحديد الفرديةتم توزيعها بشكل موحد في الخلفية المظلمة O-N/C (الشكل 1d، e)، مؤكداً التشتت الذري للحديد.
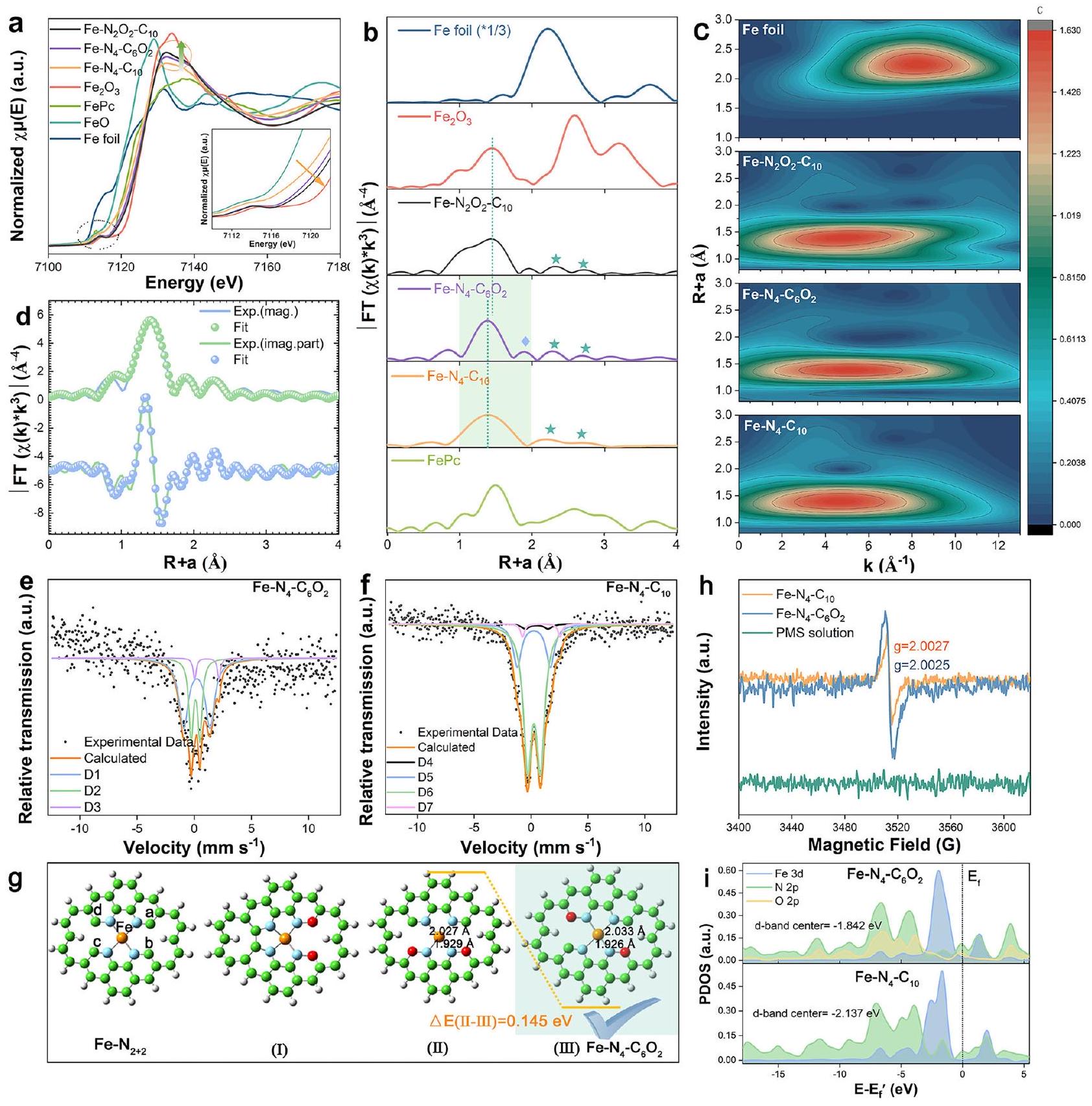
ثم قمنا بفحص التركيب الإلكتروني وبيئة التنسيق لذرات الحديد في ثلاثة من المحفزات أحادية الذرة للحديد باستخدام هيكل الامتصاص بالأشعة السينية، وطيف الإلكترون الضوئي بالأشعة السينية (XPS)، وطيف مطيافية ميسباور. كما هو موضح في طيف امتصاص الأشعة السينية بالقرب من حافة الامتصاص (XANES) (الشكل 2أ)، فإن قمة ما قبل الحافة وحافة الامتصاص لـ و انتقلت نحو طاقات أعلى، مصحوبة بزيادة شدة قمة الخط الأبيض، مما يشير إلى أن تشويب الأكسجين غير بيئة التنسيق لمراكز الحديد وقلل من كثافاتها الإلكترونية.. من الواضح أن هذه التغييرات كانت أكثر وضوحًا في طيف XANES لـ، مما يدل على مسافتها الأقصر وتفاعلها الأقوى بين الأكسجين والحديد. تحويل فورييه-طيف الامتصاص الدقيق للأشعة السينية الممتد الموزون (FT-EXAFS) لـكشف عن قمة واسعة وغير متناسقة مع وجود الحد الأقصى للقمة عندقريب منالارتداد الخلفي، مما يدل على وجود و التنسيق المزدوج في (الشكل 2ب). تم العثور على نتائج مماثلة في التقارير الحديثة حول ، و . بشكل مختلف، القمة الرئيسية لـ و كانت متناظرة وتم إزاحتها إلى اليسار إلىينتمي إلى مسار تشتت الحديد-نيتروجين الفردي. ومع ذلك، هناك قمة جديدة فينطاق R -space ظهر في طيف، مما يشير إلى أن البيئات التنسيقية المختلفة لـ و . هذه النتائج أكدت أن الأكسجين تم إضافته إلى الأغلفة التنسيقية الخارجية لـفي. علاوة على ذلك، تلك القمم مع (علامات الخماسي) لم يكن بالإمكان تعيينها لمسار تشتت الحديد-حديد، لأن تحويلات الموجات (WT) لخطوط EXAFS لثلاث عينات من Fe-SACs كانت تحتوي فقط على حد أقصى واحد من الشدة يتوافق مع تنسيق الحديد -N/O عند قيمة لـفي نطاق R من (الشكل 2ج). بعد ذلك، كشفت عملية التناسب الكمي باستخدام طريقة المربعات الصغرى لمنحنيات FT-EXAFS أن الأعداد التنسيقية المثلى للحديد في جميع الثلاثة Fe-SACs كانت تقريبًا 4 (الشكل 2د، الشكل التكميلي 4 والجدول 2). كانت الفروق تكمن في أن شمل كلا من و مسارات الانعكاس الخلفي، مما يؤكد البناء الناجح لـبيئة تنسيق مزدوجةبينما أظهرت SACs الحديدية الأخرى مسارات تشتت خلفي من نوع Fe-N فقط. كما هو متوقع، كان هناك طولان متميزان ومتساويان من روابط Fe-N و تمت ملاحظتها في“، مما يشير إلى أن ذرتي الأكسجين تم إدخالهما في الغلاف التنسيقي الثاني، بما يتماشى مع النتائج المبلغ عنها لـمع وجود الكبريت المخدر في الغلاف التنسيقي الثاني. على العكس، SAC بدون إضافة الأكسجين أظهرت فقط مجموعة واحدة من طول روابط الحديد والنيتروجين عند.
قمنا بمزيد من التوصيف و استخدامطيف موصبر لتوضيح بيئات التنسيق للحديد. تم تحليلها بشكل مفصلعرضت طيفيات ميسباور حصريًا ثنائيات، دون أي أحادي أو سداسي مرتبط بها.-حديد،، أو (الشكل 2e، f). وفقًا لتحول الإيزومر ( ) وانقسام الرباعي ( ) القيم، طيف ميسباور لـيمكن أن تكون ملائمة جيدًا مع ثلاثة دبلتات (D1-D3)، تتوافق
الشكل 1 | تخليق وتوصيف Fe-SACs. أ رسم تخطيطي لـ Fe-التركيب. صور TEM لـ. نمط PXRD لمحفزات مختلفة. d صور HADDF-STEM لـحجم جزيئات الحديد فيتُقدم بيانات المصدر كملف بيانات مصدر.
إلى دوران متوسط (MS) (D1)، دوران متوسط (MS) (D2) والدوران العالي (HS) (D3) ، على التوالي (الجدول التكميلي 3) حيثيمثل العيبالموقع في هيكل طبقة الجرافيت غير السليمة (الشكل التكميلي 5). بالمقابل، يمكن تحديد أربعة مزدوجات مختلفة (D4-D7) فيالتي تم تعيينها إلى الدوران المنخفض (LS) (D4)، LS حديد (D5)، MS (D6) و HS Fe (D7) على التوالي نسبة شدة D-band إلى G-band الأعلى نسبيًا ) في من ذلكأثبتت أن تشبع الأكسجين خلق المزيد من العيوب (الشكل التكميلي 6). في الوقت نفسه، تم تأكيد وجود روابط Fe-N و N-O أيضًا من خلال طيف HR-XPS للحديد.، و في (الأشكال التكميلية 7-8). لذلك، استنتجنا أنه تم إضافة ذرتين من الأكسجين في الغلاف التنسيقي الثاني لـ.
بالنظر إلى وجود ذرات الأكسجين المخدرة في الغلاف التنسيقي الثاني لـقمنا ببناء ثلاثة احتمالاتالنماذج باستخدام DFT (الشكل 2g). على وجه التحديد، يمكن أن تكون ذرتا الأكسجين في أي من المواضع المتقابلة a و b (التكوين I)، أو المتقابلة و المواقع (التكوين II)، أو العكس و المواقع (التكوين III) ضمن الغلاف التنسيقي الثاني، على التوالي. ومع ذلك، أدت تحسينات التكوين I إلى ارتباط ذرات الكربون في المواقع c و d، مما يتعارض مع الحديد. الهيكل وبالتالي أدى إلى استبعاده. كانت استقرار التكوين II أقل بكثير من استقرار التكوين III، على الرغم من توافقه مع المتطلبات لـالهيكل. لذلك، تم اختيار التكوين III كـالنموذج في هذه الدراسة. يمكن أن يُعزى تشكيل التكوين III إلى إعادة ترتيب الذرات الحرارية أثناء التحلل الحراري (الشكل التوضيحي 9). منع سلف الحديد -N المرتبط بشكل مستقر دخول ذرات الأكسجين إلى القشرة التنسيقية الأولى، مما يوفر المزيد من الفرص لكي يستبدلوهمالذرات في الغلاف التنسيقي الثاني لـ. بعد ذلك، أدت قاعدة الثمانية إلكترونات والكهروسلبية العالية لذرات الأكسجين إلى فقدان ارتباط الذرات وانحراف في طول رابطة الحديد-النيتروجين. في النهاية، تصادم ذرات مع الجزيئات في حركة حرارية غير منتظمة ستؤدي إلى إطلاقها من سطح المحفز لتشكيل فراغات كربونية (CVs)، مصاحبة لتحول في التكوين منإلى. مختلف عن امتلكت إشارة منحنى لورينز المحسنة بشكل كبير، مما يدل على وجود المزيد من الإلكترونات غير المتزاوجة في (الشكل 2h)، الذي ساهم في تحسين كفاءة نقل الإلكترون خلال التفاعل الحفزي. يمكن أن يُعزى الزيادة في الإلكترونات غير المتزاوجة إلى إدخال الأكسجين وتكوين الـ CVs. يمكن أن يُضعف إدخال الأكسجين بشكل فعال مجال الربط، مما يزيد من حالة الدوران وعدد الإلكترونات غير المتزاوجة في مركز الحديد (الجدول التكميلي 3). إن تكوين الـ CVs من خلال فقدان ذرات الكربون سيترك إلكترونات إضافية تعيد توزيعها بين الجيران.ذرات لتكوين المزيد من الإلكترونات غير المتزاوجةعلاوة على ذلك، كشفت تحليل الشحنة الطبيعية للسكان (NPA) وتحليل الكثافة الجزئية للحالة (PDOS) أن إضافة الأكسجين في الغلاف التنسيقي الثاني قللت من كثافة الإلكترون لذرة الحديد وضاقت عرض نطاق d الخاص بها من خلال تغيير التركيب الإلكتروني لرباط النيتروجين (الشكل 2i والشكل التكميلي 10).، وبالتالي تغيير مركز نطاق d للحديد في (-1.842 إلكترون فولت مقابل -2.137 إلكترون فولت لـ ) أقرب إلى مستوى فيرمي ( ) (الشكل 2i)، وزيادة التفاعل الذاتي لـ Fe-SAC تجاه تنشيط PMS.
تفعيل PMS وتحلل الملوثات
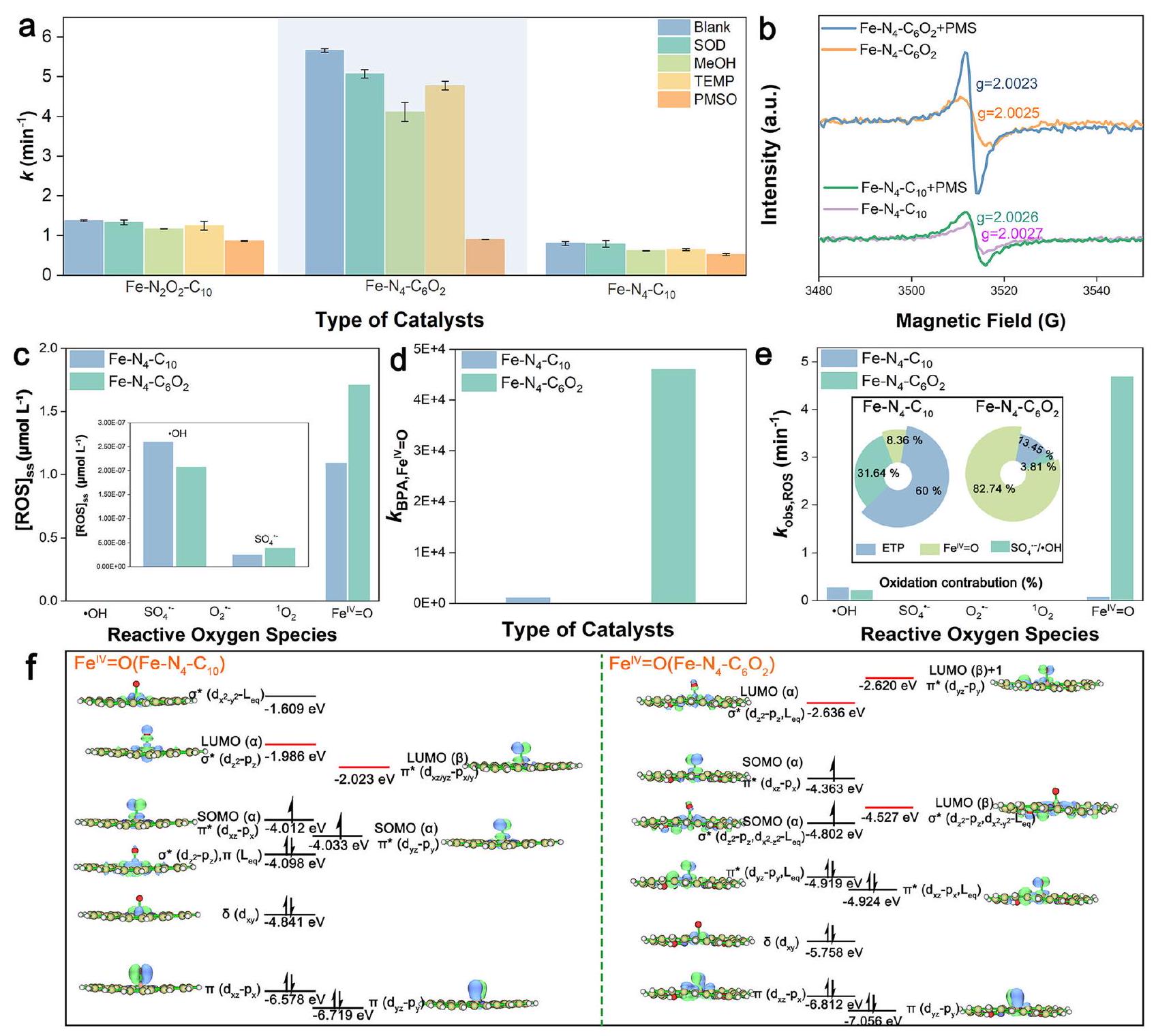
استخدمنا حسابات DFT للتحقيق في تأثير تشويب الأكسجين على تنشيط PMS وتكوين أنواع الأكسجين التفاعلية (ROS)، واستخدمنا نجمة (*) للدلالة على الامتصاص السطحي.
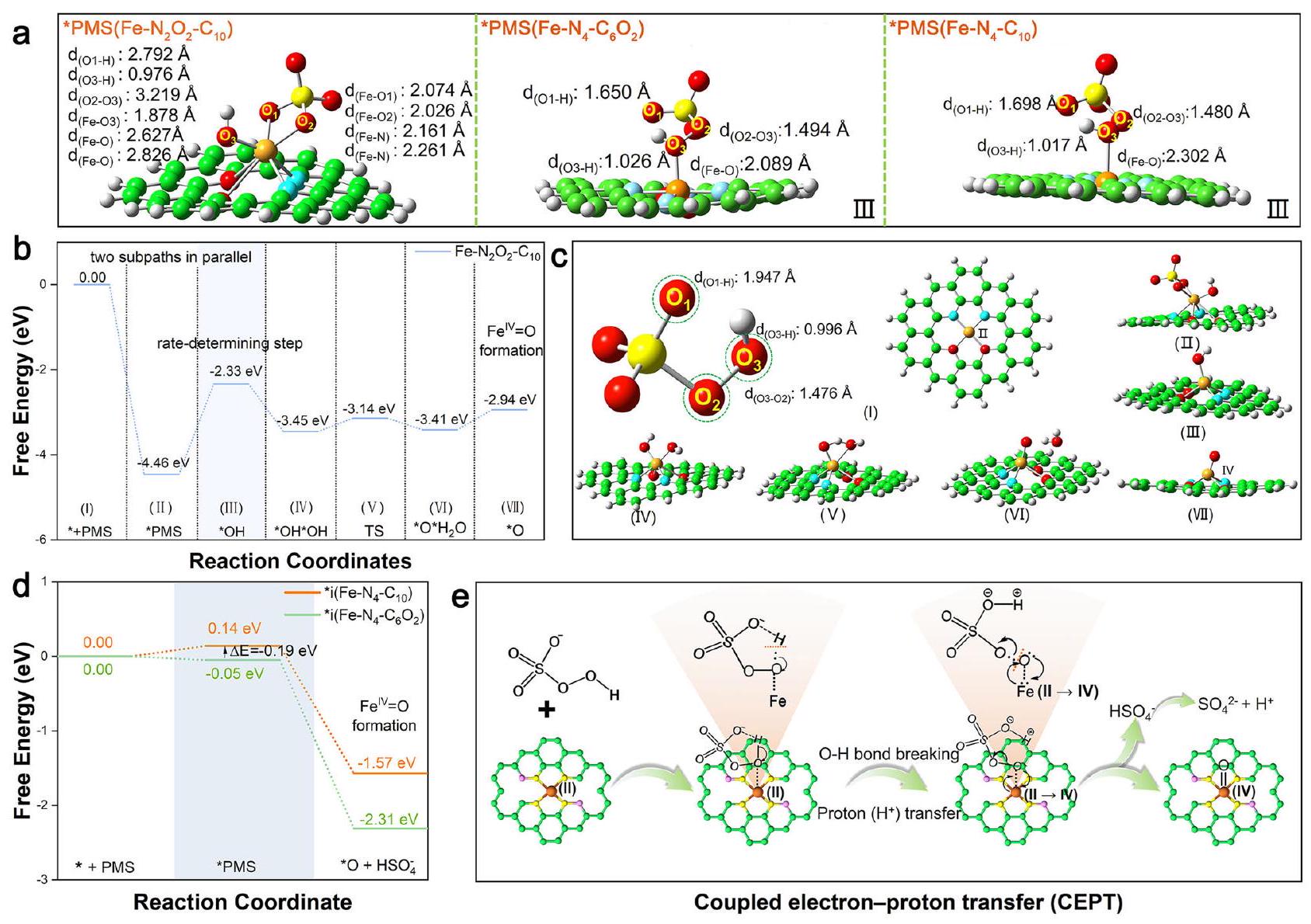
الشكل 2 | التوصيف الطيفي والحساب الهيكلي للعوامل المساعدة. أ XANES (الإدراج: عرض مكبر لـ XANES)، ب طيف FT-EXAFS (-الموزونة)، و c طيف WT-EXAFS لأوراق الحديد المختلفة و Fe-SACs. d منحنى التناسب في فضاء R لـ EXAFS المقابل لـطيف مويسباور لـ وفي-. الهندسيّات المحسّنة لثلاثة أنواع ممكنةهيكل الموقع النشط. طيف EPR وPDOS لـ Fe-SACsتُقدم بيانات المصدر كملف بيانات مصدر. الأنواع ولتمثيل الأنواع الثلاثة المختلفة من ذرات الأكسجين في PMS (الأشكال التكميلية 11-12). كما هو متوقع، فإن الشحنة الإيجابية الأعلى لمركز الحديد وحقله الكهربائي المحلي القوي المحيط به فيعززت امتصاص PMS وأثارت الانقسام السريع للبروكسيد ) الرابطة ( الشكل 3أ)، بينما ساعدت الكثافة المنخفضة للإلكترونات في مركز الحديد على تعزيز تفاعله القوي مع إزاحة ذرة الحديد من مستوى الحامل وإطالة وروابط الحديد والأكسجين إلى قد يؤدي إلى إزالة المعادن. ومع ذلك، فإن تقليل الشحنة الموجبة على مراكز الحديد وحقله الكهربائي المحلي المحيط بها في و فشل في تلبية متطلبات الكسر المباشر لـالرابطة الناتجة عن امتصاصهم لـ PMS، وبالتالي قد تشكل معقدات (I) و (II) و (III) مع PMS عبر، و على التوالي (الشكل 3 أ والمكمل
الشكل 11)، بينما تنشيط الـالرابطة حدثت فقط في المركب (III). لذلك، تم اعتماد المركب (III) للحسابات التالية.
بعد ذلك، يتم تشكيل الحديد الأكسو عالي التكافؤ ) والراديكالات في أنظمة Fe-SACs/PMS الثلاثة تم استكشافها نظريًا. كما هو موضح في الشكل 3b، c، كان من الممكن أيضًا من الناحية الديناميكية الحرارية توليد فيالنظام من خلال المسار التقليدي لـ(أنا) →متلازمة ما قبل الحيض (II) * OH (III) (الرابع) حالة الانتقال (TS، V). بشكل مختلف، المسافة المناسبة بين و H من *PMS( ) و *متلازمة ما قبل الحيض( ) سهلت تشكيل الروابط الهيدروجينية داخل الجزيء، والتفاعل بين الحديد وضعف و الروابط، مما يفضل انقسامها لإنتاج من خلال الاقتران غير الكلاسيكي
الشكل 3 | الآلية الجزيئية لتنشيط PMS على Fe-SACs. أ تكوين الامتزاز لـ PMS على Fe-SACs.الطاقة الحرة لـالجيل فينظام PMS. ج هياكل الوسائط خلال معلومات فينظام PMS. د الطاقات الحرة لـالجيل فيمتلازمة ما قبل الحيضأنظمة PMS. الرسم التخطيطي لـ CEPT. يتم توفير بيانات المصدر كملف بيانات مصدر. مسار نقل الإلكترون-بروتون (CEPT) (الشكل 3d، e والشكل التوضيحي 12). في هذا المسار، تم نقل البروتونات بسهولة منإلىوالإلكترونان المتبقيان فيالمدار سيكون مرتبطًا بالإلكترونين من الحديد لتشكيلالرابطة. من الجدير بالذكر أن الشوائب البعيدة من الأكسجين وCVs عززت امتصاص PMS من خلال خفض طاقة الامتصاص من -0.21 إلكترون فولت لـإلى -0.46 إلكترون فولت لـ (الشكل التوضيحي 12)، وأثر على هيكل *PMS من خلال توفير مجال كهربائي محلي معزز بشكل معتدل، بما في ذلك ممدود ( لـ مقابل لـ ) و ( لـ مقابل لـالسندات، فضلاً عن تقليل المسافة بين و لـ مقابل لـ ). هذه التغييرات حسنت بشكل فعال الديناميكا الحرارية ( -2.31 إلكترون فولت لـ و -1.57 إلكترون فولت لـ ) وإمكانيات الحركة لـ نظام إدارة الممتلكات لـتوليد عبر مسار CETP. أخيرًا، تم حساب الجدوى الديناميكية الحرارية لتكوين الجذور في ثلاثة أنظمة Fe-SACs/PMS من خلال مسار نقل الإلكترون الفردي، متبعًا اتجاه الحديد.متلازمة ما قبل الحيض مع طاقة غيبس الحرة ) من متلازمة ما قبل الحيض معمنمعمن -0.41 إلكترون فولت (الشكل التكميلي 13). ومع ذلك، فإن القيمة المنخفضة بشكل ملحوظ المرتبطة بـ تشكيللـلـلـ ) اقترح أن كانت عملية التكوين أسهل بكثير من تلك الخاصة بالمتطرفين في ثلاثة أنظمة Fe-SACs/PMS.
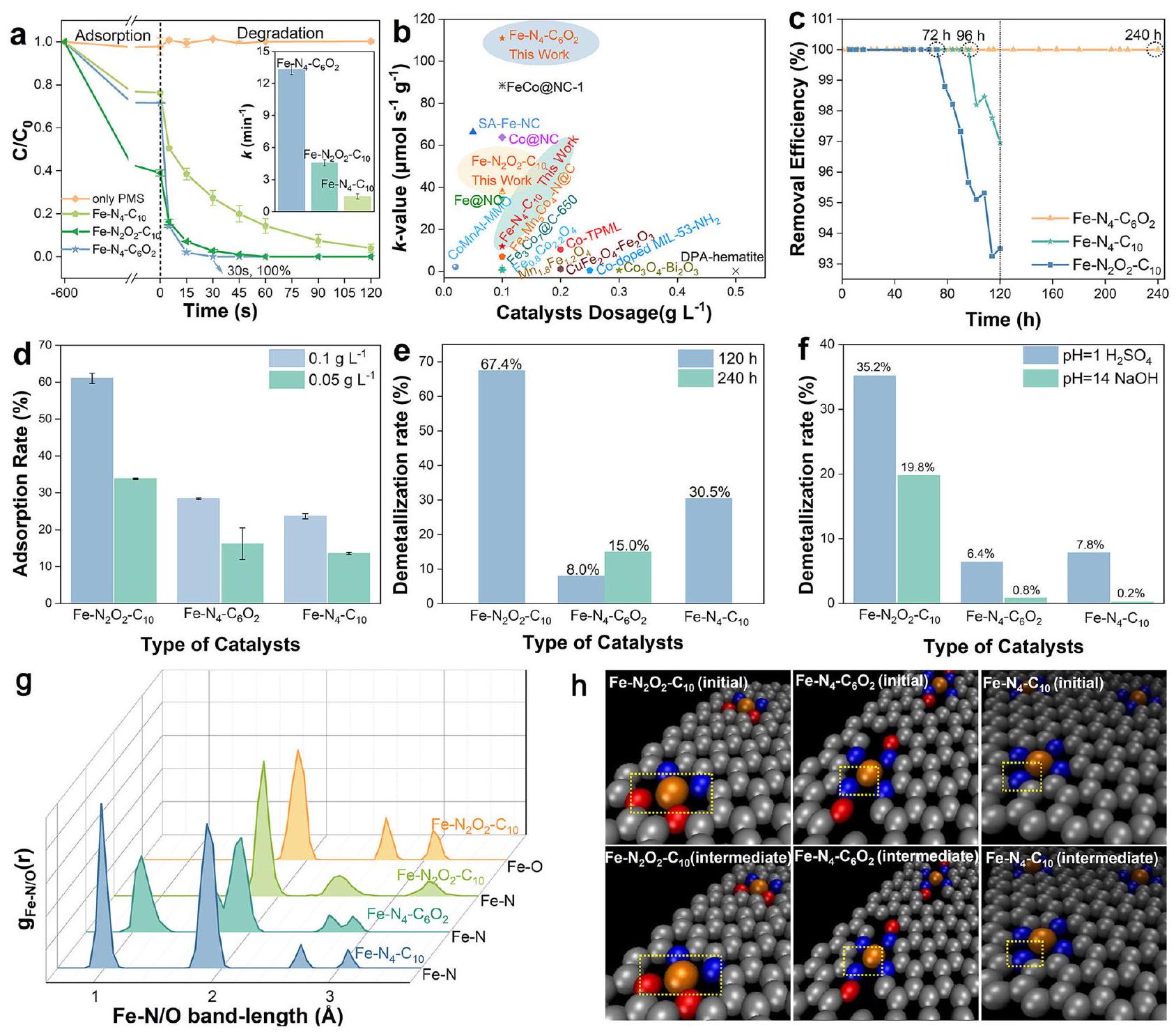
ثم قمنا بتقييم أداء تنشيط PMS لمركبات Fe-SACs من خلال إزالة بيسفينول A (BPA)، ووجدنا أن إضافة الأكسجين زادت بشكل كبير من النشاط. و N/O-C، بينما أظهرت أفضل نشاط تحفيزي بين ثلاثة عينات من Fe-SACs (الشكل 4a والشكل التكميلي 14). يمكن أن يتم تكسير BPA بالكامل تمت إزالته في 30 ثانية بثابت معدل زائف من الدرجة الأولى ( ) بقدر ما فيمن و من PMS (الشكل 4 أ والشكل التكميلي 15). من المثير للإعجاب، أداء إزالة BPA من حتى تجاوزت تلك المتجانسةتجاريوجزيئات نانو الحديد/الكربون، ونانو الحديد صفر القيمة/الكاولين، وغيرها من المواد المركبة المعروفة (الشكل 4ب والشكل التكميلي 16). والأهم من ذلك،أظهرت توافقًا واسع النطاق مع الرقم الهيدروجيني وحافظت علىكفاءة إزالة الملوثات تحت تأثير أيونات غير عضوية بتركيز عالٍ ومصفوفات مائية شائعة (الشكل التوضيحي 17). علاوة على ذلك، فإن الحديد-يمكن لنظام /PMS إزالة ملوثات متنوعة بشكل فعال (الجدول التكميلي 4)، لكن معدلات تحللها تأثرت بهياكلها. على سبيل المثال، تم تحلل الملوثات الفينولية الغنية بالإلكترونات مثل 2،4-ثنائي كلوروفينول (2، 4-DCP)، وبارا كلوروفينول (4CP)، والفينول (PE) بالكامل في غضون 1.5 دقيقة، مع ثابت معدل التفاعل من الدرجة الزائفة الأولى ( ) تتراوح من 5.522 إلى (الشكل التوضيحي 18). ومع ذلك، كانت الملوثات التي تحتوي على مجموعات سحب الإلكترون، مثل p-nitrophenol (PNP) وfipronil (FP) وp-nitrobenzyl alcohol (PNBA) وp-nitrobenzaldehyde (PNBD)، من الصعب أن يتم تحللها بالكامل خلال 20 دقيقة حتى عند تركيز عالٍ من PMS يبلغ 3 مللي مول.كانت القيم منخفضة كماتشير الانتقائية العالية تجاه الملوثات الغنية بالإلكترونات إلى أن الأكسدة التفاعلية غير الجذرية قد تكون السائدة في هذا النظام..
من الجدير بالذكر أن أنشطة N/O-C وN-C كانت أقل بكثير من أنشطة الثلاثة Fe-SACs (الشكل التكميلي 14)، مما يؤكد أن مواقع الحديد الموزعة على المستوى الذري تلعب دورًا لا غنى عنه في تفعيل PMS بشكل فعال. لذلك، تم التحقيق بشكل أكبر في النشاط المحدد لموقع الحديد الفردي، وتردد دوران BPA.
الشكل 4 | تقييم النشاط والاستقرار لـ Fe-SACs. أ تدهور BPA في أنظمة PMS المنشطة بواسطة محفزات مختلفة (الإدراج: ثابت معدل الإزالة الزائف من الدرجة الأولى المقابل لـ BPA). تمثل أشرطة الخطأ الانحراف المعياري المستمد من تجربتين مكررتين. الظروف التجريبية: [المحفزات] =. ب تركيز الملوثات المحسوب على أساس جرعة المحفز ثابت معدل التفاعل الديناميكي -قيمة) لإزالة BPA في أنظمة المحفزات/PMS المختلفة. ج اختبار التشغيل المستمر لتحلل BPA في نظام التدفق المستمر. متوسط معدل التدفق: جرعة المحفز:
20 ملغ. معدل الامتزاز لـ Fe-SACs على BPA عند جرعات مختلفة. معدلات إزالة المعادن من مختلف Fe-SACs بعد (e) الإزالة المستمرة لـ BPA لمدة 120 ساعة و240 ساعة و(f) 36 ساعة من التآكل بواسطة الأحماض/القلويات القوية. g ملفات توزيع الجذور الحرة Fe-N وFe-O لـ Fe-SACs عند. لقطات من Fe-SACs تم الحصول عليها من محاكاة الديناميكا الجزيئية في تم توفير التكوين الأولي وحالة وسيطة لإظهار التمدد لـالسند كما هو موضح في المربع الأصفر. تم توفير بيانات المصدر كملف بيانات المصدر. كان (TOF) لمواقع الحديد بترتيب، مما يشير إلى أن تشبع الأكسجين في الغلاف التنسيقي الأول يمكن أن يعزز نشاط مواقع الحديد بشكل أكثر فعالية من تشبع الأكسجين في الغلاف التنسيقي الثاني (الشكل التكميلي 19). ويعزى ذلك إلى الأكسجين المشبع فيقلل بشكل كبير من كثافة الإلكترون في مركز الحديد وخلق مجال كهربائي محلي أقوى حوله من خلال التفاعلات المباشرة وتعطيل الحديد.هيكل تنسيق متماثل، مما يعزز امتصاص وتفعيل PMS السالب الشحنة، بما يتماشى مع الحسابات النظرية. على الرغم من أن معدل الدوران (TOF) لمواقع الحديد فيكان أعلى بمقدار 10.6 مرات من ذلك فيمحتوى الحديد في ( ) كان فقط من ذلك في ( الجدول التكميلي 5)، مما أدى إلى انخفاض النشاط الظاهر للأول (الشكل 4أ). بعد ذلك، حاولنا زيادة محتوى الحديد فيمن خلال زيادة نسبة سوائل الحديد، مما أدى بدلاً من ذلك إلى تكتل المعادن بشكل كبير وانخفاض النشاط التحفيزي. (الشكل التوضيحي الإضافي 20)، مما يشير إلى أن تشويب الأكسجين في الغلاف التنسيقي الأول قد أضعف استقرار ذرات الحديد المفردة ذات التحميل العالي. بعد ذلك، تم إجراء تجارب مستمرة لتدهور BPA التحفيزي لفحص استقرار Fe-SACs أثناء التفاعل (الشكل 4c)، والتي كشفت عن أقوى استقرار لـبين الثلاثة Fe-SACs، على الرغم من أن سعة امتصاصه لبسطة BPA لم تكن جيدة مثل تلك الخاصة بـ (الشكل 4د). بعد 120 ساعة من العلاج المستمر، كانت نسبة تسرب الحديد من كان مجردأقل بكثير من تلك الخاصة بـ و (الشكل 4e). من اللافت للنظر، أن تفاعل لم تنخفض بعد 240 ساعة من التفاعل وظل هيكلها تقريبًا دون تغيير حتى بعد 500 ساعة من التشغيل (الأشكال التكميلية 21-24 والجدول 6). نظرًا لتعقيد درجة الحموضة في المياه المختلفة، قمنا أيضًا بمراقبة تسرب الحديد من ثلاثة من مواد الكربون المنشط المدعمة بالحديد تحت ظروف حمضية وقاعدية شديدة (الشكل 4f)، ووجدنا أن معدل تسرب الحديد منكان أقل حتى من ذلكعند درجة حموضة حمضية تقريبًا نفس ما هو عليهفي محلول قلوي. بالإضافة إلى ذلك، فإن معدل إزالة المعادن منكان أعلى بكثير منمع عامل 4.5 أو 99 في ظل ظروف حمضية أو قلوية. لذلك، فإن تشبع الأكسجين في الغلاف التنسيقي الأول عزز بشكل كبير نشاط مواقع المعادن ذات الذرة الواحدة على حساب استقرارها، ولكن يمكن تحقيق علاقة متوازنة بين النشاط واستقرار مواقع المعادن ذات الذرة الواحدة من خلال تصميم تكوين التنسيق لـ Fe-SAC بشكل عقلاني مع تشبع الأكسجين في الغلاف التنسيقي الثاني.
بالنسبة إلى أن إزالة المعادن من Fe-SACs تبدأ من الإطالة والانكسار لـ الروابط، تم إجراء محاكاة الديناميكا الجزيئية لاستكشاف استقرار SACs الحديدية المعتمد على الهيكل من حيث تقلبات طول روابط Fe-N/O. يشير طول رابطة Fe-N/O الأقصر نسبيًا ونطاق التقلبات الأضيق إلى الروابط المستقرة لـ Fe-N/O في المحفز مع ميل أقل لإزالة المعادن أثناء التفاعلات. الاهتزازات الحرارية لـ رابطة الـ، ، و يمكن تصورها في الأفلام التكميلية 1-3، على التوالي. كما هو موضح في الشكل.دالة التوزيع الشعاعي (RDF) لـ Fe-N/O، المشار إليها بـتم الحصول عليه من خلال حساب وعدّ تكرار حدوث ذرات النيتروجين/الأكسجين على مسافةمن ذرة الحديد. عند درجة حرارة الغرفة،مناحتوى SAC على أربعة قمم مميزة في النطاق من. ومع ذلك، فإن تقلباتو Oفي كانت SAC أبعد عن مركز الحديد، مما أشار إلى أن تدمير هيكل التنسيق المتناظر له أضعف و الروابط، مما يزيد من ميل إزالة المعادن. ومن المثير للاهتمام أن سعة الحركة الحرارية لـ السندات فيانخفض إلى، كاشفًا عن قوته الأكبر بكثيررابطة لمقاومة الاضطرابات الخارجية. هذه النتائج توضح أكثر أنيمكن أن يعزز التلاعب في الغلاف التنسيقي الثاني التفاعل بين ذرات الحديد الفردية وذرات النيتروجين المنسقة من خلال تقليل كثافة الإلكترونات في الحديد، مما يمنع بشكل فعال تسرب ذرات الحديد أثناء تنشيط PMS.
تحقيق في الآلية
قمنا بعد ذلك بالتحقيق في الأنواع التفاعلية الناتجة في أنظمة Fe-SACs/PMS الثلاثة. مساهمة الجذور الحرة السوبر أكسيد () تم استبعاد تأثيره على تحلل BPA أولاً من خلال تجارب إخماد سوبر أكسيد ديسموتاز (SOD) وقياسات الرنين المغناطيسي الإلكتروني (EPR) (الشكل 5a والشكل التكميلي 25a). وجود الميثانول (MeOH) أوقف فقط بشكل طفيف تحلل BPA في أنظمة Fe-SACs/PMS الثلاثة، مما يدل على مساهمة ضعيفة لـوتحلل BPA (الشكل 5 أ والجدول التكميلي 7). عند استخدام 5،5-ثنائي ميثيل-1-بيريدون-N-أكسيد (DMPO) كعامل حبس، لوحظ فقط إشارة لأكسيدات DMPO (DMPOX) في أنظمة Fe-SAC/PMS الثلاثة، والتي قد تكون ناتجة عن أكسدة DMPO بواسطةأو أنواع غير راديكالية أخرى، دون ظهور • OH والإشارات (الشكل التوضيحي 25ب-د). كشفت النتائج أعلاه أن مسارات الأكسدة غير الجذرية مثل عملية نقل الإلكترون (ETP)،أو الأكسجين الأحادي (حدثت الأكسدة بشكل رئيسي في هذه الأنظمة، بينما قد لا يُعزى التأثير المثبط لـ MeOH فقط إلى إخماد الجذور، ولكن إلى امتصاصه التنافسي على Fe-SACs مع BPA و/أو تفاعله التنافسي مع أنواع الأكسجين التفاعلية الأخرى، مثلتأثيرات التثبيط الضعيفة نسبيًا لمثيل فينيل سلفوكسيد (PMSO) على و الأنظمة اقترحت الدور السائد لـ ETP أو عملية الأكسدة، بدلاً منالأكسدة، في هذين النظامين. من المدهش أن تم العثور على PMSO لتقليلتحلل BPA فينظام PMS، مما يدل على أن إضافة الأكسجين في الغلاف التنسيقي الثاني يمكن أن تحول مسار تحلل BPA من ETP أوعملية الأكسدة إلىنظير. علاوة على ذلك، أكدت تجارب إخماد الميثانول وPMSO بشكل أكبر أنكانت الأنواع السائدة في تحلل 2، 4-DCP، 4-CP، PE، PNP،
FP و PNBD و PNBA (الأشكال التكميلية 26-28). ومن الجدير بالذكر أن معدلات التحلل الأعلى للملوثات الفينولية الغنية بالإلكترونات في هذا النظام كانت تُعزى إلى بيئتها ذات الكثافة الإلكترونية العالية، مما سهل الهجوم الكهروفيلي بواسطة. نظرًا لتفاعلات PMSO مع • OH، وستشكل منتجات مختلفة (المعادلات S15-S17)، حيث يمكن أكسدة PMSO إلى ميثيل فينيل سلفون. ) من خلال مسار نقل ذرة الأكسجين (OAT) بواسطة استخدمنا أكسدة PMSO للتحقق بشكل أكبر من توليد الجذور وتم ملاحظة أن استهلاك PMSO في الـكان نظام إدارة الممتلكات أقل بكثير من ذلك فيمتلازمة ما قبل الحيضأنظمة PMS، مما يشير إلى أن *PMS كانت النوع التفاعلي الأساسي فينظام إدارة الأداء، بدلاً من المتطرفين و (الشكل التوضيحي التكميلي 29). في هذه الأثناء، تحويل في و الأنظمة كانت قريبة منيؤكد أنتم توليدها بشكل أساسي في هذين النظامين بدلاً من الجذور. علاوة على ذلك، إشارة متساوية الاتجاه عندنُسب إلىالمدارات ذات الانقسام الكبير في الحقل الصفري والإلكترونات غير المتزاوجة، تم اكتشافها في و الأنظمة. تم تعزيز إشارة EPR هذه وتم تحويلها أيضًا إلى قيمة أقل بواسطة تنشيط PMS، مما يؤكد مرة أخرى وجود (الشكل 5ب) .
بعد ذلك، قمنا بالتحقيق في ETP وعمليات الأكسدة في أنظمة Fe-SACs/PMS الثلاثة. لم يثبط إضافة 2، 2، 6، 6-تترا ميثيل بيبيريدين (TEMP) بشكل كبير تحلل BPA (الشكل 5a)، واستبدالمعلم تعزز إشارة 2، 2، 6، 6-تترا ميثيل بيبيريدين-N-أوكسي (TEMPO) (الشكل التكميلي 30)، مما يشير إلى المساهمة الضئيلة لـ. بعد استبعاد المتطرفين والتداخل، كانت معدلات التحلل المعتمدة على الرقم الهيدروجيني لبسفينول أ (BPA) و2، 4-DCP في نظام FeSACs/PMS مختلفة تمامًا عن تلك فينظام الأكسدة (الشكل التكميلي 31). استبعدت هذه النتائججيل. بعد ذلك، تم إجراء اختبارات مطيافية رامان في الموقع واختبارات الجهد الزمني للتحقق بشكل أكبر من التفاعل بين مواقع الحديد وPMS، بالإضافة إلى عمليات نقل الإلكترون (الشكل التكميلي 32). لاحظنا ثلاثة قمم مميزة عند 880 و980 وفي حل PMS، بما يتوافق مع اهتزازات الشد لـ، و على التوالي. الـ و انخفضت القمم بشكل كبير، بينمازادت القمم بشكل ملحوظ عند إضافة Fe-SACs، مما يدل على تنشيط PMS السريع. في الوقت نفسه، ظهرت قمتان جديدتان عند و ، جنبًا إلى جنب مع انزياح أحمر لـذروة مميزة، تشير إلى تشكيل معقدات *PMS وبالإضافة إلى تبرع الإلكترونات من مواقع الحديد إلى، والتي تم تأكيدها بشكل أكبر من خلال الاتجاه الأول المتزايد والاتجاه اللاحق المتناقص لجهد الدائرة المفتوحة لقطب Fe-SAC عند إضافة PMS و. وبالتالي، نستنتج أن مسار ETP شارك في تحلل BPA في أنظمة Fe-SACs/PMS الثلاثة، حيث تقوم FeSACs بوساطة نقل الإلكترون من BPA إلى PMS. ومن الجدير بالذكر، أن Fe و امتلكت تقلبات محتملة أقوى منبعد إضافة PMS و BPA، مما يشير إلى أن تشويب الأكسجين في أي من القشرة التنسيقية الأولى أو الثانية يمكن أن يعزز ETP. وذلك لأن الأكسجين المشوب يمكن أن يقلل من طاقة أدنى مدار جزيئي غير مشغول (LUMO) في مركبات *PMS من خلال تعديل مستويات طاقة المدار الجزيئي وتوزيعات الإلكترونات في Fe-SACs، مما ضيق الفجوة الطاقية مع أعلى مدار جزيئي مشغول (HOMO) لـ BPA، وبالتالي يسهل بشكل فعال نقل الإلكترونات من BPA إلى PMS (الشكل التكميلي 33).
ثم قمنا بتحديد تركيزات الحالة المستقرة لمختلف أنواع الجذور الحرة للأكسجين من خلال قياس حركيات المنافسة بين الجذور الحرة للأكسجين ومركبات الاستكشاف للتحقيق بعمق في تأثير تشبع الأكسجين في الغلاف التنسيقي الثاني على العملية التحفيزية والتفاعل (الشكل 5ج-هـ، الشكل التوضيحي 34، الجداول 8-9). كما هو موضح في الشكل 5ج، فإن التركيزات الثابتة لـتم إنتاجه في الحديد-متلازمة ما قبل الحيضنظام إدارة الممتلكاتو
الشكل 5 | التحقيق الميكانيكي في نظام Fe-SACs/PMS: تحديد الأنواع التفاعلية ومحاكاةالبنية الإلكترونية. أ تأثير المواد الماصة على تحلل BPA في نظام Fe-SACs/PMS. تمثل أشرطة الخطأ الانحراف المعياري المستمد من تجربتين مكررتين. الظروف التجريبية:العوامل المساعدة، ، [TEMP] . طيف EPR لـ Fe-SACs و Fe-SACs/PMS. ج التركيزات الثابتة لمختلف أنواع ROS في نظام Fe-SACs/PMS (الإطار: الثابت- تركيزات الدولة من و ). د معدل التفاعل من الدرجة الثانية بين BPA وفي نظام Fe-SACs/PMS. e الثوابت المعدلة لمعدل التفاعل الملاحظة لـ ROS و BPA في نظام Fe-SACs/PMS (الإطار: مساهمات الأكسدة لمختلف ROS في تحلل BPA). f التركيب المداري الجزيئي ومستوى الطاقة لـ. توضح الصورة أعلاه فقط المدارات في محيط مدار LUMO. محور N-Fe-N فيحيث يتم تعريف N المتصل مباشرة بـ O على أنه محور x. يتم توفير بيانات المصدر كملف بيانات مصدر. على التوالي، والتي كانتأوامر من حيث الحجم أعلى من ذلك لـ و ، مما يؤكد بقوة أن / متلازمة ما قبل الحيض وكان من المرجح أن تنتج متلازمة ما قبل الحيض الأنواع بدلاً من الجذور، بما يتماشى مع التوقعات النظرية. ومع ذلك، فإن ثابت معدل التفاعل من الدرجة الثانية لـ الأنواع المنتجة فيالنظام مع BPA كان فقطأقل بكثير من ذلك لدى المتطرفين، مما يجعل مساهمة لأكسدة BPA أن تكون فقط، أقل بكثير من تلك الخاصة بـ ETP (60.0%) والأكسدة الجذرية (31.6%) (الشكل 5d و e والجدول التكميلي 8-9). هذه الحقائق تشير إلى أنالأنواع المنتجة فيكان نظام PMS يمتلك تفاعلية منخفضة تجاه BPA. من المدهش أن ثابت سرعة التفاعل من الدرجة الثانية لـالأنواع المنتجة فيتم زيادة نظام إدارة المشاريع (PMS) مع BPA إلى-ضعف ذلك من /PMS (الشكل 5d)، مما يشير إلى أن تشويب الأكسجين في الثاني يمكن أن يزيد غلاف التنسيق بشكل كبير من تفاعلنحو BPA. وبالتالي، فإن مساهمةإلى إزالة BPA زادت بشكل حاد إلىفينظام إدارة الممتلكات.
نظريًا، إما أو مدارات مضادة للرابطة عندشظية فييمكن أن تتلقى إلكترونات أجنبية لتعطيلالرابطة، مما يؤدي إلى نقل الأكسجين النهائي وأكسدة الركيزة (الشكل التوضيحي 35أ). أشارت مطيافية موسباور إلى أن تشبع الأكسجين وتوليد الفولتامات يمكن أن يزيد بشكل كبير من حالة الدوران (الشكل التوضيحي 35ب). وفقًا لنظرية حقل الربط، فإن المركز المعدني عادة ما يمتلك حالة دوران عالية عندما يكون مرتبطًا برابطة ضعيفة، والعكس صحيح. لذلك، فإن حالة الدوران المعززة تشير إلى أن تشويب الأكسجين وتوليد الفولتامات يمكن أن يضعف بشكل فعال قوة حقل التنسيق، مما يؤدي إلى انخفاض في طاقة مدارات الحديد 3d لتقليل الطاقة المدارية بشكل أكبر.
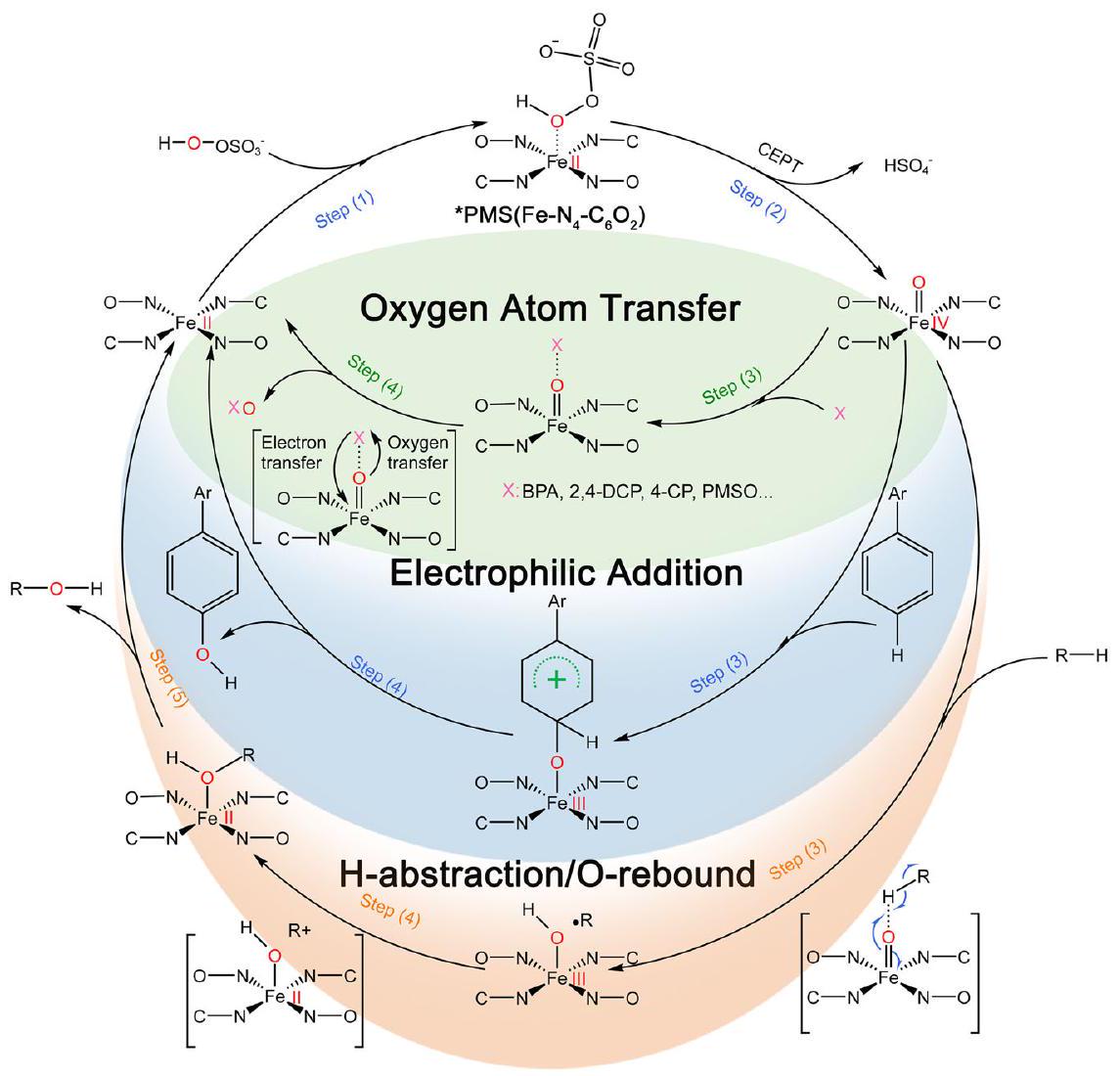
شكل.رسم تخطيطي لآليات التفاعلمع الملوثات. الـتقوم الأنواع بأكسدة الملوثات من خلال تفاعل OAT، والإضافة الكهربية، وآلية سحب الهيدروجين/ارتداد الأكسجين.
شظية في، وتحسين التفاعل التأكسدي لـ. بعد ذلك، استخدمنا حسابات DFT لتوضيح زيادة التفاعل الجوهري لـتم إنتاجه فينظام إدارة PMS. كما هو موضح في الشكل 5f،تم إنتاجه فينظام إدارة PMS يمتلك – و -مسارات الهجوم لأكسدة الملوثات. الحديد/الأكسجين (مدارات ( -2.023 إلكترون فولت ) و ( ) مداراتيمكن أن تقبل الإلكترونات من الركائز الملوثة، مما يقلل من ترتيب الروابط لـوتفضيل نقل O النهائي. في -مسار الهجوم، يجب أن تتداخل الملوثات مع المدارات من اتجاه عمودي علىرابطة، والتي أدت إلى تصادم مكاني بين الملوثات والليغاندات الاستوائية لـتم إنتاجه فينظام إدارة الممتلكات.-مسار الهجوم،كان المدار ذو طاقة عالية (-1.986 إلكترون فولت)، مما يتطلب حاجز طاقة كبير للتغلب عليه لبدء-الهجوم. بشكل عام، مساران التفاعل الاثنين لـتم إنتاجه فيأظهر نظام PMS تفاعلاً معتدلاً تجاه الملوثات.تم إنتاجه فيالنظام، أصبحت المكونات المدارية أكثر تعقيدًا وانخفضت مستويات الطاقة المدارية بشكل ملحوظ بسبب إضافة ذرات الأكسجين وانخفاض شدة حقل الربط. من بينها، كانت طاقات ( ليق) و , ليق) تم تقليل المدارات إلى -4.527 إلكترون فولت و -2.636 إلكترون فولت، وهو أقل بكثير من ذلك لـ ) مدار ( -2.620 eV ). نتيجة لذلك، يمكن لإلكترونات الملوث ملء المستويات المنخفضةمداراتتم إنتاجه فيالنظام على طول محور Z دون عوائق ستيركية، مما سهل تشكيل حالة الانتقال المتعامدة بين الملوث-O-Fe، وبالتالي تسريع التفاعل بين الملوث مع.
أخيرًا، حددنا الوسائط الرئيسية في عملية التحلل باستخدام تقنيات الكروماتوغرافيا السائلة-مطياف الكتلة (LCMS) وكروماتوغرافيا الغاز-مطياف الكتلة (GC-MS) (الأشكال التكميلية 36-39، الجداول 10-11)، واقترحنا اثنين مسارات التحلل المحتملة لبسطة BPA فينظام PMS (الشكل التكميلي 40، يمكن العثور على التفاصيل في المعلومات الداعمة). في هذين المسارين، تم أكسدة BPA بواسطةمن خلال تفاعل OAT، والإضافة الكهربية، وآليات سحب الهيدروجين / إعادة ارتداد الأكسجين (الشكل 6). كان جوهر هذه الآليات الثلاثة للأكسدة هو ملء إلكترونات الملوثات في المستويات المنخفضة.مداراتلتشكيل وسائط ملوثة-O-Fe، وبالتالي تحقيق أكسدة الملوث وتقليلمن الجدير بالذكر أن انخفاض الكربون العضوي الكلي (TOC) في محلول BPA وصل إلى مستوى عالٍ يصل إلىفي دقيقتين عندما تكون تركيزكانوكان قريبًا منفي دقيقتين حتى عندما تم تقليل تركيز المحفز إلى النصف (الشكل التكميلي 41)، مما أشار إلى أنأظهرت SAC أداءً جيدًا في عمليات الأكسدة المتقدمة، حيث كانت قادرة على تحلل المركبات العضوية إلى مركبات غير ضارة تمامًا وتقليل الأثر البيئي.
نقاش
باختصار، أبلغنا عن تخليق SAC عن طريق استبدال C في الغلاف التنسيقي الثاني لـالمواقع التي تحتوي على O وأظهرتيمكن أن تقوم المواقع بتنشيط PMS بشكل أكثر كفاءة واستقرارًا من خلال توفير مجال كهربائي محلي معزز دون تدمير هيكل التنسيق المتناظر في الغلاف التنسيقي الأول. يمكن أن يعزز تشبع الأكسجين في الغلاف التنسيقي الثاني قوة رابطة Fe-N من خلال تقليل كثافة الإلكترونات في مركز الحديد وضعف السعة لـالرابطة خلال تفعيل PMS، وبالتالي منع إزالة المعادن بشكل فعالالمواقع. والأهم من ذلك، أن هذا التلاعب بالأكسجين خفض أيضًا الطاقة لـالمدارات عن طريق إضعاف مجال التنسيق لتعزيز النواة الكهربية-هجوم علىنحو الملوثات الغنية بالإلكترونات. يسلط هذا العمل الضوء على أهمية إضافة عناصر في الغلاف التنسيقي الثاني في تعزيز استقرار SAC ويقدم تصميمًا استراتيجية لمعالجة التوازن بين النشاط العالي والثبات الدائم في تحضير SAC.
طرق
المواد والكيماويات
أوكزونالأكسجين النشطتم تزويد (AR) و BPA من قبل شركة شنغهاي ألايدين للتكنولوجيا الحيوية المحدودة (شنغهاي، الصين) وشركة سينوفارم للمواد الكيميائية المحدودة (شنغهاي، الصين) على التوالي. تفاصيل المواد الكيميائية الأخرى موضحة في الطريقة التكميلية 1. تم استخدام جميع المواد الكيميائية كما هي وتم إذابتها في الماء النقي..
تحضير المحفز
لتحضيرو 10 فيناثرولين مونوهيدراتتم تفريقها بالموجات فوق الصوتية في 60 مل من الإيثانول المطلق للحصول على مركب تنسيق الحديد-النيتروجين (I). ثم،تمت إضافة إلى المحلول وتم التحريك تحت الغليان عندلمدة 4 ساعات. بعد ذلك، تم ترسيب المركب المُعد مسبقًا (I) علىعن طريق تبخر تحت ضغط منخفض فيثم تم تسخين المادة الصلبة المجففة إلى (محروق لمدة ساعتين) بمعدل فيالجو. بعد ذلك، تم توزيع المادة الصلبة السوداء الناتجة فيوتم التحريك في درجة حرارة الغرفة لمدة 4 ساعات لإزالة. تم تكرار هذه العملية ثلاث مرات. بعد ذلك، تم غسل المادة الصلبة عدة مرات بكمية كبيرة من الماء النقي حتى وصل السائل المصفى إلى درجة الحموضة المحايدة. أخيرًا، تم تسجيل المسحوق الأسود الجاف على أنه لتركيب، ظلت الخطوات متسقة مع تلك الخاصة بـباستثناء 10 فيناثرولين مونوهيدرات تم استبدالها بـ ( )( + )- -بيس(3,5-دي-تيرت-بيوتيل ساليسيلدين)-1,2-سيكلوهكسانديامين، على التوالي. من أجل تخليق ، ظلت الخطوات متسقة مع تلك الخاصة بـباستثناء ترسيب المركب (I) على.
تركيب الحوامل (، و ) وغيرها من المحفزات (Fe-NPs/NC، N/O-C، وN-C) تم تضمينها في الطريقة التكميلية 2. الكميات المحددة لكل مادة كيميائية مستخدمة في عملية تخليق المحفزات موضحة في الجدول التكميلية 1.
توصيف المحفزات
طرق التوصيف بما في ذلك مجهر المسح الإلكتروني بالانبعاث الميداني (FESEM)، مجهر الإلكترون الناقل عالي الدقة (HR-TEM)، حيود الأشعة السينية (PXRD)، مطياف الأشعة السينية للأشعة السطحية (XPS)، مطياف رامان، مطياف الانبعاث الضوئي البلازمي المقترن بالحث (ICP-OES)، طريقة بروناوير-إيميت-تيلر (BET)، مجهر STEM عالي الدقة مع تباين كثافة الكتلة (AC-HAADF)، طيف امتصاص الأشعة السينية (XAS)، مطياف الرنين المغناطيسي الإلكتروني (EPR)، وتم وصف مطيافية ميسباور في الطريقة التكميلية 3.
إجراءات التجربة
تجارب تحلل BPA، بالإضافة إلى تأثيرات جرعات PMSجرعات المحفز، الأيونات المتعايشةوتم وصف المثبطات على تحلل BPA في الطريقة التكميلية 4.
طرق تحليلية
تم تفصيل الطرق التحليلية للمركبات المستهدفة، بما في ذلك الكروماتوغرافيا السائلة (LC) وكروماتوغرافيا السائلة-مطياف الكتلة (LCMS) وكروماتوغرافيا الغاز-مطياف الكتلة (GC-MS)، في الطريقة التكميلية 5.
تحليل EPR لـ
تم خلط 20 ملغ من Fe SAC معمن الماء النقي أو محلول PMS المشبع، ثم تم نقله إلى أنبوب كوارتز لتحليل EPR. تم إجراء جميع اختبارات EPR عند 77 ك.
صيغة الحساب
صيغ الحساب للثوابت السريعة الظاهرةTOFالقيمة، وTOC مقدمة في الطريقة التكميلية 6.
تحضير الأقطاب الكهربائية
طريقة تحضير إلكترود Fe-SAC موضحة بالتفصيل في الطريقة التكميلية 7.
تركيز الحالة المستقرة ومساهمة الجذور الحرة للأكسجين
تم تفصيل الطرق التجريبية ومشتقات المعادلات لتقييم التركيز الثابت للأنواع التفاعلية للأكسجين (ROS) ومساهمتها في تحلل BPA في الطريقة التكميلية 8.
اختبار الاستقرار على المدى الطويل
تم ترسيب تعليق Fe-SAC على غشاء الإستر السليلوزي المختلط (حجم المسام، D. 25 مم) من خلال الترشيح ثم وضعت في فلتر (i.D. 25 مم). المساحة الفعالة للغشاء الميكروفيلتري المركب Fe-SAC/MCE هيوسعة تحميل المحفز هي. بعد ذلك، يتم استخدام فلتر Fe-SAC/MCE ومضخة بيرستالتية لتشكيل مفاعل تدفق مستمر لتقييم الاستقرار على المدى الطويل لـ Fe-SAC.
نظري
تم إجراء الحسابات النظرية باستخدام برنامج Gaussian 16وبرنامج Multiwfn 3.8استنادًا إلى نظرية الكثافة. تم إجراء المحاكاة الديناميكية التي توضح السلوكيات الديناميكية للمادة من خلال تطبيق محاكاة الديناميكا الجزيئية بورن-أوبنهايمر (BOMD)، كما تم تنفيذها ضمن حزمة CP2K/Quickstep المتاحة للجمهور (الطريقة التكميلية 9).
توفر البيانات
جميع بيانات الدراسة مدرجة في المقال والمعلومات التكميلية. تم توفير بيانات المصدر مع هذه الورقة.
References
Yao, Y. et al. Rational regulation of Co-N-C coordination for highefficiency generation of toward nearly selective degradation of organic pollutants. Environ. Sci. Technol. 56, 8833-8843 (2022).
Zhang, L. S. et al. Carbon nitride supported high-loading Fe singleatom catalyst for activation of peroxymonosulfate to generate with 100 % selectivity. Angew. Chem. Int. Ed. 60, 21751-21755 (2021).
Wang, J. et al. Facile synthesis of atomic Fe-N-C materials and dual roles investigation of sites in Fenton-like reactions. Adv. Sci. 8, 2101824 (2021).
Xiong, Y. et al. Single-atom Fe catalysts for Fenton-like reactions: roles of different N species. Adv. Mater. 34, e2110653 (2022).
Huang, B. et al. Coupled surface-confinement effect and pore engineering in a single-Fe-atom catalyst for ultrafast Fenton-like reaction with High-Valent Iron-Oxo complex oxidation. Environ. Sci. Technol. 57, 15667-15679 (2023).
Gao, Y. et al. Unraveling the high-activity origin of single-atom iron catalysts for organic pollutant oxidation via peroxymonosulfate activation. Environ. Sci. Technol. 55, 8318-8328 (2021).
Chen, Q. et al. Atomically dispersed dual active sites singleatom nanozymes for cascade catalysis and peroxymonosulfate activation to degrade dyes. J. Hazard. Mater. 422, 126929 (2022).
. et al. single-atom catalyst for efficient peroxymonosulfate activation and selective generation. Angew. Chem. Int. Ed. 62, e202303267 (2023).
Cui, J. et al. Regulating the metal-support interaction: double jump to reach the efficiency apex of the -catalyzed Fenton-like reaction. ACS Catal. 12, 14954-14963 (2022).
Chen, Y., Zhang, G., Liu, H. & Qu, J. Confining free radicals in close vicinity to contaminants enables ultrafast fenton-like processes in the interspacing of membranes. Angew. Chem. Int. Ed. 58, 8134-8138 (2019).
Kuang, J., Guo, H., Si, Q., Guo, W. & Ma, F. Nitrogen vacancies regulated the local electron density of iron sites in to boost the generation of high-valent iron-oxo species in a peracetic acidbased Fenton-like process. Appl. Catal. B Environ. 337, 122990 (2023).
Yang, T., Fan, S., Li, Y. & Zhou, Q. Fe-N/C single-atom catalysts with high density of sites toward peroxymonosulfate activation for high-efficient oxidation of bisphenol A: electron-transfer mechanism. Chem. Eng. J. 419, 129590 (2021).
Zhang, B., Li, X., Akiyama, K., Bingham, P. A. & Kubuki, S. Elucidating the mechanistic origin of a spin state-dependent catalyst toward organic contaminant oxidation via peroxymonosulfate activation. Environ. Sci. Technol. 56, 1321-1330 (2022).
Chu, C. et al. Cobalt single atoms on tetrapyridomacrocyclic support for efficient peroxymonosulfate activation. Environ. Sci. Technol. 55, 1242-1250 (2021).
Zhou, X. et al. Identification of Fenton-like active Cu sites by heteroatom modulation of electronic density. Proc. Natl. Acad. Sci. USA 119, e2119492119 (2022).
Cheng, C. et al. Generation of and its contribution to Fenton-like reactions on a single-atom iron-N-C Catalyst. Angew. Chem. Int. Ed. 62, e202218510 (2023).
Pan, Y. et al. Regulating the coordination structure of single-atom Fe- catalytic sites for benzene oxidation. Nat. Commun. 10, 4290 (2019).
Wang, B. et al. A site distance effect induced by reactant molecule matchup in single-atom catalysts for Fenton-like reactions. Angew. Chem. Int. Ed. 61, e202207268 (2022).
Miao, J. et al. Single-Atom catalytic sites enable efficient peroxymonosulfate activation by forming highly reactive Oxo Species. Environ. Sci. Technol. 57, 4266-4275 (2023).
Zhou, Z. et al. Fe-based single-atom catalysis for oxidizing contaminants of emerging concern by activating peroxides. J. Hazard. Mater. 418, 126294 (2021).
Song, J. et al. Asymmetrically coordinated moieties for selective generation of high-valence Co-Oxo species via coupled Electron-Proton Transfer in Fenton-like reactions. Adv. Mater. 35, e2209552 (2023).
Dai, H. et al. Regulating electronic structure of Fe single-atom site by S/N dual-coordination for efficient Fenton-like catalysis. J. Hazard. Mater. 465, 133399 (2024).
Peng, L., Duan, X., Shang, Y., Gao, B. & Xu, X. Engineered carbon supported single iron atom sites and iron clusters from Fe-rich Enteromorpha for Fenton-like reactions via nonradical pathways. Appl. Catal. B Environ. 287, 119963 (2021).
Li, J. et al. Atomically dispersed Fe atoms anchored on S and N -codoped carbon for efficient electrochemical denitrification. Proc. Natl. Acad. Sci. USA 118, e2105628118 (2021).
Chen, S. et al. Unveiling the proton-feeding effect in sulfur-doped Fe-N-C single-atom catalyst for enhanced electroreduction. Angew. Chem. Int. Ed. 61, e202206233 (2022).
Li, Q. et al. Fe isolated single atoms on S, N codoped carbon by copolymer pyrolysis strategy for highly efficient oxygen reduction reaction. Adv. Mater. 30, e1800588 (2018).
Wang, J. et al. Suppressing local charge symmetry of iron single atoms for efficient electrocatalytic nitrate reduction to ammonia ORR. Adv. Funct. Mater. 33, 2304277-2304285 (2023).
Xu, H. et al. Improving PMS oxidation of organic pollutants by single cobalt atom catalyst through hybrid radical and non-radical pathways. Appl. Catal. B Environ. 263, 118350 (2020).
Wang, J. et al. Suppressing thermal migration by fine-tuned metalsupport interaction of iron single-atom catalyst for efficient ORR. Adv. Funct. Mater. 33, 2304277-2304285 (2023).
Miao, Q. et al. sites in carbon nanosheets by templatepyrolysis of COFs for RR. Chem. Eng. J. 450, 138427 (2022).
Wang, Z. et al. Cobalt single atoms anchored on oxygen-doped tubular carbon nitride for efficient Peroxymonosulfate activation: simultaneous coordination structure and morphology modulation. Angew. Chem. Int. Ed. 61, e202202338 (2022).
Tan, Z. et al. Highly efficient electrocatalysis of oxygen to hydroxyl radical by single-atom catalyst for refractory organic pollutant removal. Appl. Catal. B-Environ. Energy 355, 124170 (2024).
Kramm, U. I. et al. Structure of the catalytic sites in Fe/N/C-catalysts for -reduction in PEM fuel cells. Phys. Chem. Chem. Phys. 14, 11673-11688 (2012).
Wang, Z. L. et al. Gelatin-derived sustainable carbon-based functional materials for energy conversion and storage with controllability of structure and component. Sci. Adv. 1, e1400035 (2015).
Kramm, U. I., Lefevre, M., Larouche, N., Schmeisser, D. & Dodelet, J. P. Correlations between mass activity and physicochemical properties of Fe/N/C catalysts for the ORR in PEM fuel cell via 57Fe Mossbauer spectroscopy and other techniques. J. Am. Chem. Soc. 136, 978-985 (2014).
Kramm, U.I. et al. Influence of the electron-density of FeN4-centers towards the catalytic activity of pyrolyzed FeTMPPCl-based ORRelectrocatalysts. J. Electrochem. Soc. 158, B69-B78 (2011).
Shi, L. et al. Photoassisted construction of holey defective photocatalysts for efficient visible-light-driven production. Small 14, 1703142 (2018).
Zhang, X. et al. Unraveling the dual defect sites in graphite carbon nitride for ultra-high photocatalytic evolution. Energ. Environ. Sci. 15, 830-842 (2022).
Hammer, B. & Norskov, J. K. Why gold is the noblest of all the metals. Nature 376, 238-240 (1995).
Liu, J., Guo, Y., Fu, X., Luo, J. & Zhi, C. Strengthening absorption ability of Co-N-C as efficient bifunctional oxygen catalyst by modulating the d band center using MoC. Green Energy Environ. 8, 459-469 (2023).
Wu, Q. Y., Yang, Z. W., Wang, Z. W. & Wang, W. L. Oxygen doping of cobalt-single-atom coordination enhances peroxymonosulfate activation and high-valent cobalt-oxo species formation. Proc. Natl. Acad. Sci. USA 120, e2219923120 (2023).
Zhao, J., Sun, X., Huang, X. & Li, J. Reaction mechanisms of methanol oxidation by biomimetic complex. Int. J. Quantum Chem. 116, 692-701 (2016).
Yang, M. et al. Unveiling the origins of selective oxidation in singleatom catalysis via intensified radical and nonradical pathways. Environ. Sci. Technol. 56, 11635-11645 (2022).
Chen, Y. et al. Facile fabrication of rGO/PPy/nZVI catalytic microreactor for ultrafast removal of p-nitrophenol from water. Appl. Catal. B Environ. 324, 122270 (2023).
Pestovsky, O. & Bakac, A. Aqueous Ferryl(IV) Ion: kinetics of oxygen atom transfer to substrates and oxo exchange with solvent water. Inorg. Chem. 45, 814-820 (2006).
Ji, J. et al. Defects on : tuning redox reactions for sustainable degradation of organic pollutants. Angew. Chem. Int. Ed. 60, 2903-2908 (2021).
Gorman, A. & Rodgers, M. Singlet molecular oxygen. Chem. Soc. Rev. 10, 205-231 (1981).
Mo, F. et al. The optimized Fenton-like activity of Fe single-atom sites by Fe atomic clusters-mediated electronic configuration modulation. Proc. Natl. Acad. Sci. USA 120, e2300281120 (2023).
Miao, J. et al. Spin-state-dependent peroxymonosulfate activation of single-atom M-N Moieties via a radical-free pathway. ACS Catal. 11, 9569-9577 (2021).
Yun, E. T., Lee, J. H., Kim, J., Park, H. D. & Lee, J. Identifying the nonradical mechanism in the peroxymonosulfate activation process: singlet oxygenation versus mediated electron transfer. Environ. Sci. Technol. 52, 7032-7042 (2018).
Sanchez, P. M., Ocampo-Pérez, R., Rivera-Utrilla, J. & Mota, A. J. Comparative study of the photodegradation of bisphenol A by HO , and radicals in aqueous phase. Sci. Total Environ. 463, 423-431 (2013).
Ding, J. et al. Electrochemical activation of persulfate on BDD and DSA anodes: electrolyte influence, kinetics and mechanisms in the degradation of bisphenol A. J. Hazard. Mater. 388, 121789 (2020).
Ren, W. et al. Origins of electron-transfer regime in persulfatebased nonradical oxidation processes. Environ. Sci. Technol. 56, 78-97 (2021).
Frisch, M. J. et al. Gaussian 16, Revision A.03. Gaussian, Inc., Wallingford CT (2016).
Lu, T. & Chen, F. W. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580-592 (2012).
Lu, T. A comprehensive electron wavefunction analysis toolbox for chemists, Multiwfn. J. Chem. Phys. 161, 082503 (2024).
Lu, T. & Chen, F. W. Calculation of molecular orbital composition. Acta Chim. Sin. 69, 2393 (2011).
شكر وتقدير
تم دعم هذا العمل ماليًا من قبل البرنامج الوطني الرئيسي للبحث والتطوير في الصين (2023YFC3708002)، ومؤسسة العلوم الطبيعية الوطنية في الصين (U22A20402، 22076059، 22376076، 22076061، 21936003)، ومركز البحث الدولي المشترك لتكنولوجيا الاستشعار الحيوي الذكي والصحة، وبرنامج العلوم والتكنولوجيا في شنتشن (JCYJ20220818095601002)، وصناديق البحث الأساسية للجامعات المركزية (CCNU22JC014، CCNU24JL011).
مساهمات المؤلفين
قام H.X. و L.Z. بالإشراف على المشروع وتقديم الدعم المالي. قام T.C. بتصميم التجارب. قام G.Z. بإجراء حسابات DFT وتحليلها. كتب T.C. و H.X. و H.S. و G.Z. و L.Z. الورقة. قدم Y.H. و S.Y. و D.Z. و H.D. و Y.X. و S.H. المساعدة التجريبية.
ملاحظة الناشر: تظل شركة سبرينغر ناتشر محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
الوصول المفتوح هذه المقالة مرخصة بموجب رخصة المشاع الإبداعي النسب-غير التجارية-عدم الاشتقاق 4.0 الدولية، التي تسمح بأي استخدام غير تجاري، ومشاركة، وتوزيع، واستنساخ في أي وسيلة أو صيغة، طالما أنك تعطي الائتمان المناسب للمؤلفين الأصليين والمصدر، وتوفر رابطًا لرخصة المشاع الإبداعي، وتوضح إذا قمت بتعديل المادة المرخصة. ليس لديك إذن بموجب هذه الرخصة لمشاركة المواد المعدلة المشتقة من هذه المقالة أو أجزاء منها. الصور أو المواد الأخرى من طرف ثالث في هذه المقالة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة، ما لم يُشار إلى خلاف ذلك في سطر الائتمان للمادة. إذا لم تكن المادة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة وكان استخدامك المقصود غير مسموح به بموجب اللوائح القانونية أو يتجاوز الاستخدام المسموح به، فستحتاج إلى الحصول على إذن مباشرة من صاحب حقوق الطبع والنشر. لعرض نسخة من هذه الرخصة، قم بزيارة http:// creativecommons.org/licenses/by-nc-nd/4.0/. (ج) المؤلف(ون) 2025
المختبر الوطني الرئيسي لمبيدات الآفات الخضراء؛ مركز أبحاث الهندسة لاستخدام الطاقة الضوئية في التحكم في التلوث وتقليل الكربون، وزارة التعليم؛ كلية الكيمياء، جامعة وسط الصين العادية، ووهان، جمهورية الصين الشعبية.مركز الابتكار التعاوني للمواد الكيميائية العضوية المتقدمة الذي تم إنشاؤه بالتعاون بين المحافظة والوزارة؛ المختبر الرئيسي لوزارة التعليم لتخليق وتطبيق الجزيئات العضوية الوظيفية؛ كلية الكيمياء والهندسة الكيميائية، جامعة هوبى، ووهان، جمهورية الصين الشعبية.مدرسة علوم البيئة والهندسة، محطة المراقبة والبحث الوطنية لنظام إيكولوجي بحيرة إرهائي في يونان، معهد دالي للبحوث في يونان، جامعة جياو تونغ في شنغهاي، شنغهاي، جمهورية الصين الشعبية. البريد الإلكتروني: gbzhang@hubu.edu.cn; huixu@ccnu.edu.cn; zhanglizhi@sjtu.edu.cn
Robust single atom sites for efficient PMS activation and enhanced reactivity
Received: 28 June 2024
Accepted: 26 February 2025
Published online: 10 March 2025
Check for updates
Tiantian Chen¹, Ganbing Zhangoº , Hongwei Sun ¹, Yetong Hua , Shu Yang , Dandan Zhou , Haoxin Di , Yiling Xiong , Shenghuai Hou , Hui Xu® & Lizhi Zhang
The microenvironment regulation of single atom catalysts (SACs) critically governs peroxymonosulfate (PMS) activation. Although conventional heteroatom substitution in primary coordination enhances activity, it disrupts Fe- symmetry and compromises stability. Herein, we propose oxygen doping in the secondary coordination shell to construct , which amplifies the localized electric field while preserving the pristine coordination symmetry, thus trading off its activity and stability. This approach suppresses Fe-N bond structural deformation (bond amplitude reduced from to ) during PMS activation by lowering Fe center electron density to strengthen Fe-N bond, achieving extended catalytic durability ( ). Simultaneously, the weakened coordination field lowers the orbital energy, promoting electrophilic -attack of high-valent ironoxo towards bisphenol A , and increasing its degradation rate by 41.6 -fold. This work demonstrates secondary coordination engineering as a viable strategy to resolve the activity-stability trade-off in SAC design, offering promising perspectives for developing environmental catalysts.
Advanced oxidation processes (AOPs) based on peroxymonosulfate (PMS) have attracted great attention for pollutant control and environmental remediation due to their high efficiency in a wide pH range . Recently, single-atom catalysts (SACs) with metal- (M- ) sites, where the metal centers are coordinated with four nitrogen atoms, are widely used for the PMS activation because of their outstanding catalytic activity, excellent selectivity, and exceptionally high metal utilization efficiency . The electron donating potential of these sites can trigger the PMS activation to produce sulfate radicals ( ), hydroxyl radicals , and high-valence metal-oxo ( ), etc., thus effectively facilitating the degradation of various organic
pollutants . However, the high bond dissociation energies of and O-O bonds in PMS pose a significant challenge to their cleavage, thereby disfavoring the PMS activation at M- sites . Therefore, the development of high-performance catalysts to promote the PMS activation is crucial for the effective oxidation of contaminants.
Recently, many strategies have been proposed for the efficient PMS activation via increasing the density of metal centers or regulating their local environment surrounding in metal-SACs (M-SACs) . Among these strategies, the most common one is the partial replacement of N in the first coordination shell with foreign elements, thereby creating an enhanced localized electric field to facilitate the activation
of and bonds in . Unfortunately, this strategy destroys the symmetric structure of sites, potentially decreasing their long-term stability via the accelerated demetallation. For instance, Dai et al. found that the catalytic activity of significantly diminished after two cycles of reaction within . Peng’s group demonstrated that could remove all paracetamol within 40 min , but complete degradation of paracetamol was not realized even after extending the reaction time to 180 min at the second reuse cycle . Therefore, it is still a great challenge to balance the high activity and the long durability of SACs for the PMS activation.
Different from the first coordination shell doping, the heteroatom doping in the second coordination shell of sites could improve the catalytic performance of sites by modulating the electronic structure of the metal center through d-p long-range interactions, and simultaneously maintain the coordination structure . For instance, the electron-withdrawing effect of S doping in the second coordination shell was demonstrated to be beneficial for anchoring single Fe atoms, significantly enhancing the nitrate electroreduction activity and stability of sites in comparison with Fe-plate. However, the influence of S doping shell location on the catalytic stability of sites was not investigated . In comparison with nitrate electroreduction, the PMS activation imposes much stricter requirements on the catalytic stability of sites owing to its strong oxidative environment. Regarding that the electronegativity of is higher than that of , we anticipate that the O doping can reduce the electron density in the metal center more significantly through d-p long-range interaction and establish a stronger electric field gradient around M- sites, facilitating the adsorption and activation of negatively charged PMS ( ). Meanwhile, the O doping in the second coordination shell might favor the formation of carbon vacancies around the metal center and alter the binding ability between the metal and surrounding coordination atoms, thus suppressing demetallation.
Herein, we develop a pre-coordination strategy to engineer heteroatom doping by precisely controlling the substitution of neighboring first coordination shell N atoms and distanced second coordination shell C atoms of Fe center, aiming to clarify the effects of O doping in the first or second coordination shell on the reactivity and stability of sites through systematical characterization, degradation experiments, density functional theory (DFT) calculations and molecular dynamics (MD) simulations. This study sheds light on the design of SACs with enhanced PMS activation performance by addressing the trade-off between activity and sustainable stability.
Results
Synthesis and characterization
Fe-SACs with oxygen doped in the first or the second shell ( ) were synthesized by a pre-coordination strategy (Fig. 1a). First, we synthesized two core-shell structured carriers with utilizing silica ( ) as the core layer. One of shell layers was composed of nitrogen-doped carbon (N-C) without any oxygen functional groups ( ), while the other consisted of resorcinolformaldehyde (RF) enriched with oxygen functional groups ( ) (Supplementary Fig. 1a-c). Meanwhile, Fe-N/O cocoordination (I) and Fe-N coordination (II) complexes were prepared using (S, S)-(+)-N, N’-bis(3,5-di-tert-butylsalicylidene)-1, 2-cyclohexanediamin as the N/O precursors and 1,10-phenanthroline as the N precursor, respectively. Subsequently, complexes (I) and (II) were deposited on and using the rotary evaporation technique, which were then subjected to the processes of high-temperature pyrolysis and template removal to obtain Fe and , respectively. Fe-SAC without O -doping (Fe- ) was also synthesized with the same procedure except for the deposition of complex (I) on . For comparison, N/O co-doped
carbon (N/O-C), N-C, and Fe nanoparticles supported on N-doped C (Fe NPs/NC) were also fabricated (Supplementary Table 1).
All the three Fe-SACs were of ultrathin, uniform, hierarchically porous hollow structure (Fig. 1b and Supplementary Figs. 1-3), facilitating the full exposure of active sites and the mass transfer during the PMS activation. Their powder X-ray diffraction (PXRD) patterns only contained one broad peak at corresponding to the (002) plane of the carbon carrier, without any discernible peaks arisen from crystalline iron or iron oxides (Fig. 1c) . Meanwhile, the existence of iron particles was ruled out by their high-resolution transmission electron microscope (HR-TEM) images and the selected area electron diffraction (SAED) patterns (Supplementary Fig. 1). Aberration-corrected high-angle annular dark field scanning electron microscopy (ACHAADF STEM) images of clearly indicated that bright spots of sizes, consistent with single Fe atoms , were uniformly dispersed in the dark O-N/C background (Fig. 1d, e) , confirming the atomically dispersion of Fe.
We then checked the electronic structure and coordination environment of Fe atoms in three Fe -SACs by X-ray absorption fine structure, X-ray photoelectron spectroscopy (XPS), and Mössbauer spectroscopy. As shown in X-ray absorption near-edge structure (XANES) spectra (Fig. 2a), the pre-edge peak and absorption edge of and shifted towards higher energies, accompanied by the intensity increase of the white-line peak, indicating that the O doping altered the coordination environment of Fe centers and reduced their electron densities . Obviously, these changes were more pronounced in the XANES spectrum of , indicative of its shorter distance and stronger interaction between O and Fe . The Fourier-transformed -weighted extended X-ray absorption fine structure (FT-EXAFS) spectrum of revealed a broad and unsymmetric peak with the peak maximum located at , close to the backscattering, indicating the presence of and dual coordination in (Fig. 2b). Similar results were found in the recent reports on , and . Differently, the main peak of and were symmetric and left-shifted to , belonging to the single Fe-N scattering path. However, a new peak in the range of R -space appeared in the spectrum of , indicating that the different coordination environments of and . These results confirmed that O was doped into outer coordination shells of in . Moreover, those peaks with (pentagram markers) could not be assigned to Fe-Fe scattering path, because the wavelet transforms (WT) EXAFS contrours of three Fe-SACs samples had only one intensity maximum corresponding to the Fe -N/O coordination at value of in the R range of (Fig. 2c). Subsequently, quantitative leastsquares fitting of the FT-EXAFS curves revealed that the optimal coordination numbers of Fe in all three Fe-SACs were approximately 4 (Fig. 2d, Supplementary Fig. 4 and Table 2). The distinction lied in the fact that encompassed both and backscattering paths, thereby confirming the successful construction of a dual coordination environment , while the other two Fe-SACs solely exhibited Fe-N backscattering paths. As expected, two distinct and equally proportioned Fe-N bond lengths of and were observed in , indicating that the two O atoms were doped into the second coordination shell, consistent with the reported results of with S doped in the second coordination shell . Contrarily, SAC without O doping showed only one set of FeN bonding length at .
We further characterized and using Mössbauer spectroscopy to elucidate their coordination environments of Fe. Their deconvoluted Mössbauer spectra exclusively exhibited doublets, without any singlet or sextet associated with -Fe, , or (Fig. 2e, f). According to the isomer shift ( ) and quadrupole splitting ( ) values, Mössbauer spectrum of could be well fitted with three doublets (D1-D3), corresponding
Fig. 1 | Synthesis and characterization of Fe-SACs. a Schematic diagram of the Fe- synthesis. b TEM images of . c PXRD pattern of different catalysts. d HADDF-STEM images of . e The particle size of Fe in . Source data are provided as a Source Data file.
to medium-spin (MS) (D1), medium-spin (MS) (D2), and high-spin (HS) (D3), respectively (Supplementary Table 3) , wherein represents the defective site in the non-intact graphite layer structure (Supplementary Fig. 5). In contrast, four different doublets (D4-D7) could be identified in , which were assigned to low-spin (LS) (D4), LS Fe (D5), MS (D6), and HS Fe (D7), respectively . The relatively higher intensity ratio of D-band to G-band ( ) in than that of evidenced that the O doping created more defects (Supplementary Fig. 6). Meanwhile, the presence of Fe-N and N-O bonds were also confirmed by HR-XPS spectra of Fe , and in (Supplementary Figs. 7-8). Therefore, we concluded that two O atoms were doped in the second coordination shell of .
Considering the presence of doped O atoms in the second coordination shell of , we constructed three possible models using DFT (Fig. 2g). Specifically, the two O atoms could be situated at either ipsilateral a and b positions (configuration I), or ipsilateral and positions (configuration II), or opposite and positions (configuration III) within the second coordination shell, respectively. However, the optimization of configuration I led to the connection of C atoms at positions c and d , which contradicted the Fe structure and consequently resulted in its exclusion. The stability of configuration II was significantly lower than that of configuration III, despite its conformity to the requirements for the structure. Therefore, configuration III was selected as the model in this study. The formation of configuration III could be attributed to the thermal rearrangement of atoms during pyrolysis (Supplementary Fig. 9). The stably coordinated Fe -N precursor prevented O atoms entering the first coordination shell, and providing more opportunity
for them to replace atoms in the second coordination shell of . Subsequently, the eight-electron rule and high electronegativity of the O atoms led to the loss of a linkage of the atoms and a deviation in Fe-N bond length . Eventually, the collision of atoms with molecules in irregular thermal motion would lead to their release from the catalyst surface to form carbon vacancies (CVs), accompanying with a configuration transformation from to . Different from possessed much enhanced Lorenz curve signal, indicative of more unpaired electrons in (Fig. 2h), which contributed to improving the electron transfer efficiency during the catalytic reaction. The increase in unpaired electron could be attributed to the O introduction and the CVs formation. The O introduction could effectively weaken the ligand field, increasing the spin state and the number of unpaired electrons in the Fe center (Supplementary Table 3). The CVs formation via the loss of C atoms would leave extra electrons redistribute among neighboring atoms to form more unpaired electrons . Furthermore, the natural population analysis (NPA) charge and partial density of state (PDOS) analysis revealed that the O doping in the second coordination shell reduced the electron density of Fe atom and narrowed its d-band width via changing the electronic structure of N ligand (Fig. 2i and Supplementary Fig. 10) , thus shifting the d-band center of Fe in ( -1.842 eV vs -2.137 eV for ) closer to the Fermi level ( ) (Fig. 2i), and enhancing the intrinsic reactivity of Fe-SAC towards the PMS activation.
PMS activation and pollutant degradation
We utilized DFT calculations to investigate the influence of O doping on the PMS activation and the reactive oxygen species (ROS) formation, and employed an asterisk (*) to denote the surface adsorbed
Fig. 2 | Spectroscopic characterization and structural calculation of catalysts. a XANES (inset: magnified view of XANES), b FT-EXAFS spectra ( -weighted), and c WT-EXAFS spectra of different Fe foil and Fe-SACs. d Corresponding EXAFS R-space fitting curve of Mössbauer spectroscopy of
and Fe- . Optimized geometries of three types possible active site structure. h EPR spectra and i PDOS of Fe-SACs . Source data are provided as a Source Data file.
species and to represent the three different types of oxygen atoms in PMS (Supplementary Figs. 11-12). As expected, the higher positive charge of Fe center and its surrounding strong local electric field in promoted the PMS adsorption and triggered the rapid cleavage of peroxide ( ) bond (Fig. 3a), while the low electron density of Fe center facilitated its robust interaction with , delocalizing Fe atom from the carrier plane and elongating the and Fe-O bonds to to potentially result in demetallation. However, the reduction of positive charge on Fe centers and their surrounding local electric field in and failed to satisfy the requirements for direct breakage of bond induced by their PMS adsorption, and thus might form complexes (I), (II), and (III) with PMS via , and , respectively (Fig. 3a and Supplementary
Fig. 11), while the activation of the bond only occurred in complex (III). Therefore, complex (III) was adopted for the following calculations.
Subsequently, the formation of high-valent iron-oxo ( ) and radicals in the three Fe-SACs/PMS systems was explored theoretically. As shown in Fig. 3b, c, it was also thermodynamically feasible for the generation of in the system via the traditional pathway of (I) →PMS (II) * OH (III) (IV) transition state (TS, V) . Differently, the appropriate distance between and H of *PMS( ) and *PMS( ) facilitated the formation of intramolecular hydrogen bonds, and the interaction between Fe and weakened the and bonds, thus favoring their cleavage to produce through the non-classical coupled
Fig. 3 | Molecular mechanism of PMS activation on Fe-SACs. a The adsorption configuration of PMS on Fe-SACs. The free energy for the generation in the PMS system. c The structures of intermediates during
formation in the PMS system. d The free energies for the generation in the PMS and PMS systems. e The schematic diagram of the CEPT. Source data are provided as a Source Data file.
electron-proton transfer (CEPT) pathway (Fig. 3d, e and Supplementary Fig. 12) . In this pathway, protons were easily transferred from to , and the two remaining electrons in the orbital would be coupled with the two electrons of Fe to form the bond. Notably, the distantly doped O and CVs promoted the PMS adsorption by lowering the adsorption energy from -0.21 eV for to -0.46 eV for (Supplementary Fig. 12), and affected the *PMS structure by providing a moderately enhanced local electric field, including elongated ( for vs for ) and ( for vs for ) bonds, as well as a reduced distance between and for vs for ). These changes effectively improved the thermodynamic ( -2.31 eV for and -1.57 eV for ) and kinetic possibilities of the PMS system for the generation via CETP pathway. Finally, the thermodynamic feasibility of radical formation in three Fe-SACs/PMS systems through the single electron transfer pathway was calculated, obeying the trend of Fe PMS with Gibbs free energy ( ) of PMS with of with of -0.41 eV (Supplementary Fig. 13). However, the significantly lower associated with the formation for for for ) suggested that the formation was much easier than that of radicals in three Fe-SACs/PMS systems.
We then evaluated the PMS activation performance of Fe-SACs by the bisphenol A (BPA) removal, and found that the O doping significantly enhanced the activity of and N/O-C, while exhibited the best catalytic activity among the three Fe-SACs samples (Fig. 4a and Supplementary Fig. 14). BPA could be completely
removed in 30 s with a pseudo-first-order rate constant ( ) as high as at of and of PMS (Fig. 4 a and Supplementary Fig. 15). Impressively, the BPA removal performance of even surpassed those of homogeneous , commercial and Fe NPs/NC, nZVI/kaolinite, and other reported M-SACs (Fig. 4b and Supplementary Fig. 16). More importantly, displayed broad pH compatibility and maintained contaminant removal efficiency under high-concentration inorganic ions interference and common water matrices (Supplementary Fig. 17). Moreover, the Fe- /PMS system could effectively remove various pollutants (Supplementary Table 4), but their degradation rates were affected by their structures. For example, electron-rich phenolic pollutants such as 2,4 -dichlorophenol (2, 4-DCP), p-chlorophenol (4CP), and phenol (PE) were completely degraded within 1.5 min, with the pseudo-first-order rate constant ( ) ranging from 5.522 to (Supplementary Fig. 18). Nevertheless, contaminants with electron-withdrawing groups, such as p-nitrophenol (PNP), fipronil (FP), p-nitrobenzyl alcohol (PNBA), and p-nitrobenzaldehyde (PNBD), were difficult to be completely degraded within 20 min even at a high PMS concentration of 3 mM , and the values were as low as . The high selectivity towards electron-rich pollutants indicated that non-radical reactive oxidation may be dominant in this system .
Notably, the activities of N/O-C and N-C were significantly lower than those of the three Fe-SACs (Supplementary Fig. 14), thus confirming that atomically dispersed Fe sites play an indispensable role in the effective PMS activation. Therefore, the specific activity of individual Fe site was further investigated, and the BPA turnover frequency
Fig. 4 | Activity and stability evaluation of Fe-SACs. a The degradation of BPA in different catalysts-activated PMS systems (inset: corresponding pseudo-first-order rate constant of BPA removal). The error bars represent the standard deviation derived from two repeated experiments. Experimental conditions: [Catalysts] = . b The catalyst dosepollutant concentration normalized kinetic rate constant ( -value) of BPA removal in different catalysts/PMS systems. c Continuous operation test of BPA degradation in the continuous flow system. Average flow rate: , catalyst dosage:
20 mg . d The adsorption rate of Fe-SACs on BPA at different dosages. The demetallation rates of different Fe-SACs after (e) continuous removal of BPA for 120 h and 240 h and (f) 36 h of corrosion by strong acids/bases. g Fe-N and Fe-O radical distribution function profiles of Fe-SACs at . h Snapshots of Fe-SACs obtained from MD simulations at . The initial configuration and an intermediate state are provided to show the elongation of the bond as marked by the yellow box. Source data are provided as a Source Data file.
(TOF) of Fe sites was in the order of , which suggested that the O doping in the first coordination shell could enhance the activity of Fe sites more effectively than the O doping in the second coordination shell (Supplementary Fig. 19). This is attributed to the doped oxygen in significantly reduced the electron density of the Fe center and created a stronger local electric field around it via direct interactions and disruption of the Fe symmetric coordination structure, thus enhancing the adsorption and activation of negatively charged PMS, consistent with theoretical calculations. Although the TOF of iron sites in was 10.6 times higher than that in , the iron content in ( ) was only of that in ( , Supplementary Table 5), resulting in the lower apparent activity of the former (Fig. 4a). Next, we attempted to increase the Fe content of by increasing the proportion of Fe precursors, which instead resulted in its significant metal agglomeration and catalytic reactivity decrease
(Supplementary Fig. 20), suggesting that the O doping in the first coordination shell disfavored the stability of high-loading Fe single atoms. Subsequently, continuous catalytic BPA degradation experiments were carried out to examine the stability of Fe-SACs during the reaction (Fig. 4c), which revealed the most robust stability of among the three Fe-SACs, even though its BPA adsorption capacity was not as good as that of (Fig. 4d). After 120 h of continuous treatment, the iron leaching ratio of was merely , much lower than those of and (Fig. 4e). Strikingly, the reactivity of did not decline after 240 h of reaction and its structure kept almost unchanged even after 500 h of operation (Supplementary Figs. 21-24 and Table 6). Considering the complexity of pH in different waters, we also monitored the iron leaching of three Fe-SACs under extreme acidic and basic conditions (Fig. 4f), and found that the Fe leaching rate of was even less than that of at acidic pH and almost the
same as that of in alkaline solution. Additionally, the demetallation rate of was much higher than that of , with a factor of 4.5 or 99 under acidic or alkaline conditions. Therefore, the O doping in the first coordination shell significantly enhanced the activity of single-atom metal sites at the expense of their stability, but a well-balanced relationship between activity and stability of singleatom metal sites could be achieved by rationally designing the Fe-SAC coordination configuration with the O doping in the second coordination shell.
Regarding that the demetallation of Fe-SACs initiates from the elongation and breakage of bonds, MD simulations were conducted to explore the structure-dependent stability of Fe-SACs in terms of Fe-N/O bond length fluctuations. A relatively shorter Fe-N/O bond length and a narrower range of fluctuations indicate the stable Fe-N/O bonds of the catalyst with less demetallation tendency during reactions. The thermal vibrations of bond of the , , and could be visualized in Supplementary Movies 1-3, respectively. As shown in Fig. , the radial distribution function (RDF) of Fe-N/O, denoted as , was obtained by calculating and counting the frequency of the occurrence of N/O atoms at a distance from Fe atom. At room temperature, the of SAC contained four distinct peaks in the range of . However, the fluctuations of and O in SAC were farther away from the Fe center, which indicated that the destruction of its symmetric coordination structure weakened the and bonds, thereby increasing the demetallation tendency. Fascinatingly, the thermal motion amplitude of bonds in decreased to , revealing its much stronger bond to resist external perturbations. These results further illustrated that the doping in the second coordination shell could reinforce the interaction between Fe single atoms and coordinating N atoms by reducing the electron density of Fe, effectively inhibiting the leaching of Fe atoms during the PMS activation.
Mechanism investigation
We subsequently investigated the reactive species generated in the three Fe-SACs/PMS systems. The contribution of superoxide radicals ( ) to the BPA degradation was first excluded through the superoxide dismutase (SOD) quenching experiments and electron paramagnetic resonance (EPR) measurements (Fig. 5a and Supplementary Fig. 25a). The presence of methanol (MeOH) only slightly inhibited the BPA degradation in the three Fe-SACs/PMS systems, indicative of weak contribution of and to the BPA degradation (Fig. 5a and Supplementary Table 7). When using 5,5-dimethyl-1-pyridone-N-oxide (DMPO) as a trapping agent, only a signal of DMPO oxides (DMPOX) was observed in the three Fe-SAC/PMS systems, which might be originated from oxidation of DMPO by or other non-radical species, without the appearance of • OH and signals (Supplementary Fig. 25b-d). The above results revealed that non-radical oxidation pathways such as electron transfer process (ETP), or singlet oxygen ( ) oxidation mainly took place in these systems, while the inhibitory effect of MeOH may not be solely attributed to the radicals quenching, but to its competitive adsorption on Fe-SACs with BPA and/or its competitive reaction with other ROS, such as . The relatively weak inhibition effects of methyl phenyl sulfoxide (PMSO) on and systems suggested the dominant role of ETP or oxidation process, rather than the oxidation, in these two systems. Surprisingly, PMSO was found to suppress of BPA degradation in the PMS system, demonstrating that the doping of O in the second coordination shell could shift the BPA degradation pathway from the ETP or oxidation process to the counterpart. Furthermore, the MeOH and PMSO quenching experiments further confirmed that was the dominant species for the degradation of 2, 4-DCP, 4-CP, PE, PNP,
FP, PNBD, and PNBA (Supplementary Figs.26-28). Notably, the higher degradation rates of electron-rich phenolic pollutants in this system were attributed to their high electron density environment, which facilitated the electrophilic attack by . Given that the reactions of PMSO with • OH, and would form different products (Eqs. S15-S17), where PMSO can be oxidized to methyl phenyl sulfone ( ) through the oxygen atom transfer (OAT) pathway by , we utilized the PMSO oxidation to further check the generation of radicals and . It was observed that the consumption of PMSO in the PMS system was significantly lower than that in the PMS and PMS systems, suggesting that *PMS was the primary reactive species in the PMS system, rather than radicals and (Supplementary Fig. 29). Meanwhile, the conversion of in the and systems was close to , confirming that was predominantly generated in these two systems rather than radicals. Furthermore, an isotropic signal at , attributed to the orbitals with large zero-field splitting and unpaired electrons, was detected in the and systems. This EPR signal was enhanced and also shifted to a lower value by the PMS activation, reconfirming the presence of (Fig. 5b) .
Subsequently, we investigated the ETP and the oxidation processes in the three Fe-SACs/PMS systems. The addition of 2, 2, 6, 6-tetramethylpiperidine (TEMP) did not significantly inhibit the BPA degradation (Fig. 5a), and the replacement of with did not enhance the 2, 2, 6, 6-tetramethylpiperidine-N-oxyl (TEMPO) signal (Supplementary Fig. 30), indicating the negligible contribution of . After excluding radicals and interference, the pH dependent degradation rate changes of BPA and 2, 4-DCP in the FeSACs/PMS system were completely different from those in the oxidation system (Supplementary Fig. 31). These results ruled out the generation . Subsequently, in-situ Raman spectroscopy and chronopotentiometry tests were conducted to further check the interaction between Fe sites and PMS, as well as the electron transfer processes (Supplementary Fig. 32). We observed three distinct peaks at 880,980 , and in the PMS solution, corresponding to the stretching vibrations of , and , respectively . The and peaks decreased drastically, while peaks increased significantly upon addition of Fe-SACs, indicating the rapid PMS activation. Meanwhile, two new peaks emerged at and , along with a red-shift of the characteristic peak, suggesting the formation of *PMS complexes and , as well as electron donation from Fe sites to , which were further confirmed by the first increased and subsequent decreased trend of open-circuit potential of Fe-SAC electrode upon the addition of PMS and . Consequently, we conclude that the ETP pathway engaged in the BPA degradation in the three Fe-SACs/PMS systems, where FeSACs mediate the electron transfer from BPA to PMS. Notably, Fe and possessed the stronger potential fluctuations than after the addition of PMS and BPA, suggesting that the O doping in either the first or second coordination shell could promote the ETP. This is because the doped O could reduce the energy of the lowest unoccupied molecular orbital (LUMO) in the *PMS complexes by modulating the molecular orbital energy levels and electron distributions of the Fe-SACs, which narrowed the energy gap with the highest occupied molecular orbital (HOMO) of BPA, thus effectively facilitating the transfer of electrons from BPA to PMS (Supplementary Fig. 33).
We then quantified the steady-state concentrations of different ROS through measuring the competition kinetics between ROS and probe compounds to deeply investigate the effect of O doping in the second coordination shell on the catalytic process and the reactivity (Fig. 5c-e, Supplementary Fig. 34, Tables 8-9). As illustrated in Fig. 5c, the steady-state concentrations of produced in the Fe- PMS and PMS system were and
Fig. 5 | Mechanistic interrogation of Fe-SACs/PMS system: identification of reactive species and simulation of electronic structure. a Effect of scavengers on BPA degradation in the Fe-SACs/PMS system. The error bars represent the standard deviation derived from two repeated experiments. Experimental conditions: , [catalysts] , , [TEMP] . b EPR spectra of Fe-SACs and Fe-SACs/PMS. c The steady-state concentrations of different ROS in the Fe-SACs/PMS system (inset: the steady-
state concentrations of and ). d The second-order reaction rate between BPA and in the Fe-SACs/PMS system. e The observed reaction rate constants of ROS and BPA in the Fe-SACs/PMS system (inset: the oxidation contributions of different ROS to BPA degradation). f The molecular orbital composition and energy level of . The above illustration displays only the orbitals in the vicinity of the LUMO orbital. The N-Fe-N axis in where N is directly connected to O is defined as the x -axis. Source data are provided as a Source Data file. , respectively, which were orders of magnitude higher than that of and , strongly validating that / PMS and PMS were more likely to produce species rather than radicals, consistent with theoretical predictions. However, the second-order reaction rate constant of species produced in the system with BPA was only , much lower than that of the radicals , rendering the contribution of to the BPA oxidation to be only , much lower than those of ETP (60.0%) and radical oxidation (31.6%) (Fig. 5d, e and Supplementary Tables 8-9). These facts suggested that the species produced in the PMS system possessed low reactivity towards BPA. Surprisingly, the second-order reaction rate constant of species produced in the PMS system with BPA increased to -fold that of /PMS (Fig. 5d), suggesting that the O doping in the second
coordination shell could significantly increase the reactivity of towards BPA. Consequently, the contribution of to the BPA removal sharply increased to in the PMS system.
Theoretically, either or antibonding orbitals at the fragment in could receive foreign electrons to disrupt the bond, triggering the terminal O transfer and the substrate oxidation (Supplementary Fig. 35a). The Mössbauer spectroscopy indicated the O doping and the CVs generation could significantly increase the spin state of (Supplementary Fig. 35b). According to the ligand field theory, the metal center typically possesses a high spin state when it is coordinated with a weakfield ligand and vice versa. Therefore, the enhanced spin state indicated that the O doping and the CVs generation could effectively weaken the strength of coordination field, resulting in a decrease in the energy of the Fe 3d orbitals to further reduce the orbital energy of
Fig. Reaction mechanisms diagram of with pollutants. The species oxidizes pollutants through the OAT reaction, electrophilic addition, and H-abstraction/O-rebound mechanism.
fragment in , and improving the oxidative reactivity of . Subsequently, we employed DFT calculations to clarify the increased intrinsic reactivity of produced in the PMS system. As shown in Fig. 5f, the produced in the PMS system possessed the – and -attack pathways for the pollutants oxidation. Its Fe/O ( orbitals ( -2.023 eV ) and ( ) orbitals could accept electrons from pollutant substrates, thereby reducing the bond order of and favoring the terminal O transfer. In the -attack pathway, the pollutants needed to overlap orbitals from a direction perpendicular to the bond, which resulted in a spatial collision between the pollutants and the equatorial ligands of produced in the PMS system. In the -attack pathway, its orbital had a high energy ( -1.986 eV ), requiring a considerable energy barrier to be overcome to initiate -attack. Overall, the two reaction pathways of produced in the PMS system exhibited moderate reactivity towards pollutant. For the produced in the system, the orbital components became more complicate and the orbital energy levels were significantly lowered due to the doping of oxygen atoms and the decrease of ligand field intensity. Among them, the energies of its ( Leq) and , Leq) orbitals were respectively reduced to -4.527 eV and -2.636 eV , significantly lower than that of its ( ) orbital ( -2.620 eV ). As a result, the electrons of the pollutant could fill the low-lying orbitals of produced in the system along the Z-axis without steric hindrance, which facilitated the formation of the pollutant-O-Fe collinear transition state, thereby accelerating the reaction between the pollutant with .
Finally, we determined the main intermediates in the degradation process by liquid chromatography-mass spectrometry (LCMS) and gas chromatography-mass spectrometry (GC-MS) techniques (Supplementary Figs. 36-39, Tables 10-11), and proposed two
potential BPA degradation pathways in the PMS system (Supplementary Fig. 40, details can be found in the Supporting Information). In these two pathways, BPA was oxidized by through the OAT reaction, electrophilic addition, and H -abstrac-tion/O-rebound mechanisms (Fig. 6). The core of these three oxidation mechanisms was the filling of pollutant electrons into the low-lying orbitals of to form pollutant-O-Fe intermediates, thus realizing the oxidation of pollutant and the reduction of . Notably, the decrease of total organic carbon (TOC) in BPA solution reached as high as in 2 min when the concentration of was , and it was close to in 2 min even when the catalyst concentration was reduced by half (Supplementary Fig. 41), which indicated that the SAC exhibited a good performance in AOPs, which was able to cause the organic compounds to be degraded into harmless ones completely and reduce the environment impact.
Discussion
In summary, we reported the synthesis of SAC by substituting C in the second coordination shell of sites with O and demonstrated sites could activate PMS more efficiently and stably by providing an enhanced localized electric field without destroying their symmetric coordination structure in the first coordination shell. The O doping in the second coordination shell could enhance the strength of the Fe-N bond by reducing the electron density of Fe center and weaken the amplitude of bond during the PMS activation, therefore effectively preventing the demetallation of sites. More importantly, this O doping also lowered the energy of orbitals by weakening the coordination field to promote the electrophilic -attack of towards electron-rich contaminants. This work sheds light on the importance of second coordination shell doping in enhancing the stability of SAC and offers a design
strategy to address the trade-off between high activity and durable stability in SAC preparation.
Methods
Materials and chemicals
Oxone ( , active oxygen ) and BPA (AR) were supplied by Shanghai Aladdin Biochemical Technology Co., Ltd (Shanghai, China) and Sinopharm Chemical Reagent Co., Ltd (Shanghai, China), respectively. Details of the other chemicals are presented in Supplementary Method 1. All chemicals were used as received and dissolved by ultrapure water .
Catalyst preparation
For the synthesis of and 10phenanthroline monohydrate were ultrasonically dispersed in 60 mL absolute ethanol to obtain Fe-N coordination complex (I). Then, was added to the solution and stirred under reflux at for 4 h . Next, the pre-prepared complex (I) was deposited on by reduced pressure evaporation at . The dried solid was then heated to (calcined for 2 h ) at a rate of in an atmosphere. Subsequently, the resulting black solid was dispersed into and stirred at room temperature for 4 h to remove . This procedure was repeated three times. Afterward, the solid was washed repeatedly with a large amount of ultrapure water until the filtrate reached neutral pH . Finally, the dried black powder was recorded as . For the synthesis of , the steps remained consistent with those of except that 10phenanthroline monohydrate and were replaced by ( )( + )- -bis(3,5-di-tert-butylsalicylidene)-1,2-cyclohexanediamin and , respectively. For the synthesis of , the steps remained consistent with those of except for the deposition of complex (I) on .
The synthesis of carriers ( , and ) and other catalysts (Fe-NPs/NC, N/O-C, and N-C) were included in Supplementary Method 2. The specific amounts of each reagent used in the catalyst synthesis process are presented in Supplementary Table 1.
Characterization of the catalysts
The characterization methods including Field emission scanning electron microscope (FESEM), HR-TEM, PXRD, XPS, Raman, inductively coupled plasma optical emission spectrometry (ICP-OES), Brunauer-Emmett-Teller (BET), AC-HAADF STEM, X-ray absorption spectra (XAS), EPR, and Mössbauer spectroscopy are described in Supplementary Method 3.
Experiment procedures
The degradation experiments of BPA, as well as the effects of PMS dosages , catalyst dosages , coexisting ions , and quenchers on BPA degradation are described in Supplementary Method 4.
Analytical methods
The analytical methods for the target compounds, including liquid chromatography (LC), liquid chromatography-mass spectrometry (LCMS), and gas chromatography-mass spectrometry (GC-MS), are detailed in Supplementary Method 5.
EPR analysis for
20 mg of Fe SAC was mixed with of pure water or saturated PMS solution, and subsequently transferred to a quartz tube for EPR analysis. All EPR tests were performed at 77 k .
Calculation formula
The calculation formulas for apparent rate constants , TOF, -value, and TOC are presented in Supplementary Method 6.
Electrode preparation
The preparation method for the Fe-SAC electrode is detailed in Supplementary Method 7.
Steady-state concentration and contribution of ROS
The experimental methods and formula derivation for the quantitative assessment of ROS steady-state concentration and their contribution to BPA degradation are detailed in Supplementary Method 8.
Long-term stability test
The Fe-SAC suspension was deposited on a Mixed Cellulose Esters (MCE) membrane (pore size , D. 25 mm ) through filtration and then placed in a filter (i.D. 25 mm ). The effective membrane area of the Fe-SAC/MCE composite microfiltration membrane is and the loading capacity of the catalyst is . Next, the Fe-SAC/ MCE filter and peristaltic pump are used to form a continuous flow reactor to evaluate the long-term stability of Fe-SAC.
Theoretical
The theoretical calculations were performed using Gaussian 16 software and Multiwfn 3.8 program on the basis of DFT. The dynamic simulations elucidating the material’s dynamic behaviors were conducted through the application of Born-Oppenheimer molecular dynamics (BOMD) simulations, as implemented within the openly accessible CP2K/Quickstep package (Supplementary Method 9).
Data availability
All study data are included in the article and Supplementary Information. Source data are provided with this paper.
References
Yao, Y. et al. Rational regulation of Co-N-C coordination for highefficiency generation of toward nearly selective degradation of organic pollutants. Environ. Sci. Technol. 56, 8833-8843 (2022).
Zhang, L. S. et al. Carbon nitride supported high-loading Fe singleatom catalyst for activation of peroxymonosulfate to generate with 100 % selectivity. Angew. Chem. Int. Ed. 60, 21751-21755 (2021).
Wang, J. et al. Facile synthesis of atomic Fe-N-C materials and dual roles investigation of sites in Fenton-like reactions. Adv. Sci. 8, 2101824 (2021).
Xiong, Y. et al. Single-atom Fe catalysts for Fenton-like reactions: roles of different N species. Adv. Mater. 34, e2110653 (2022).
Huang, B. et al. Coupled surface-confinement effect and pore engineering in a single-Fe-atom catalyst for ultrafast Fenton-like reaction with High-Valent Iron-Oxo complex oxidation. Environ. Sci. Technol. 57, 15667-15679 (2023).
Gao, Y. et al. Unraveling the high-activity origin of single-atom iron catalysts for organic pollutant oxidation via peroxymonosulfate activation. Environ. Sci. Technol. 55, 8318-8328 (2021).
Chen, Q. et al. Atomically dispersed dual active sites singleatom nanozymes for cascade catalysis and peroxymonosulfate activation to degrade dyes. J. Hazard. Mater. 422, 126929 (2022).
. et al. single-atom catalyst for efficient peroxymonosulfate activation and selective generation. Angew. Chem. Int. Ed. 62, e202303267 (2023).
Cui, J. et al. Regulating the metal-support interaction: double jump to reach the efficiency apex of the -catalyzed Fenton-like reaction. ACS Catal. 12, 14954-14963 (2022).
Chen, Y., Zhang, G., Liu, H. & Qu, J. Confining free radicals in close vicinity to contaminants enables ultrafast fenton-like processes in the interspacing of membranes. Angew. Chem. Int. Ed. 58, 8134-8138 (2019).
Kuang, J., Guo, H., Si, Q., Guo, W. & Ma, F. Nitrogen vacancies regulated the local electron density of iron sites in to boost the generation of high-valent iron-oxo species in a peracetic acidbased Fenton-like process. Appl. Catal. B Environ. 337, 122990 (2023).
Yang, T., Fan, S., Li, Y. & Zhou, Q. Fe-N/C single-atom catalysts with high density of sites toward peroxymonosulfate activation for high-efficient oxidation of bisphenol A: electron-transfer mechanism. Chem. Eng. J. 419, 129590 (2021).
Zhang, B., Li, X., Akiyama, K., Bingham, P. A. & Kubuki, S. Elucidating the mechanistic origin of a spin state-dependent catalyst toward organic contaminant oxidation via peroxymonosulfate activation. Environ. Sci. Technol. 56, 1321-1330 (2022).
Chu, C. et al. Cobalt single atoms on tetrapyridomacrocyclic support for efficient peroxymonosulfate activation. Environ. Sci. Technol. 55, 1242-1250 (2021).
Zhou, X. et al. Identification of Fenton-like active Cu sites by heteroatom modulation of electronic density. Proc. Natl. Acad. Sci. USA 119, e2119492119 (2022).
Cheng, C. et al. Generation of and its contribution to Fenton-like reactions on a single-atom iron-N-C Catalyst. Angew. Chem. Int. Ed. 62, e202218510 (2023).
Pan, Y. et al. Regulating the coordination structure of single-atom Fe- catalytic sites for benzene oxidation. Nat. Commun. 10, 4290 (2019).
Wang, B. et al. A site distance effect induced by reactant molecule matchup in single-atom catalysts for Fenton-like reactions. Angew. Chem. Int. Ed. 61, e202207268 (2022).
Miao, J. et al. Single-Atom catalytic sites enable efficient peroxymonosulfate activation by forming highly reactive Oxo Species. Environ. Sci. Technol. 57, 4266-4275 (2023).
Zhou, Z. et al. Fe-based single-atom catalysis for oxidizing contaminants of emerging concern by activating peroxides. J. Hazard. Mater. 418, 126294 (2021).
Song, J. et al. Asymmetrically coordinated moieties for selective generation of high-valence Co-Oxo species via coupled Electron-Proton Transfer in Fenton-like reactions. Adv. Mater. 35, e2209552 (2023).
Dai, H. et al. Regulating electronic structure of Fe single-atom site by S/N dual-coordination for efficient Fenton-like catalysis. J. Hazard. Mater. 465, 133399 (2024).
Peng, L., Duan, X., Shang, Y., Gao, B. & Xu, X. Engineered carbon supported single iron atom sites and iron clusters from Fe-rich Enteromorpha for Fenton-like reactions via nonradical pathways. Appl. Catal. B Environ. 287, 119963 (2021).
Li, J. et al. Atomically dispersed Fe atoms anchored on S and N -codoped carbon for efficient electrochemical denitrification. Proc. Natl. Acad. Sci. USA 118, e2105628118 (2021).
Chen, S. et al. Unveiling the proton-feeding effect in sulfur-doped Fe-N-C single-atom catalyst for enhanced electroreduction. Angew. Chem. Int. Ed. 61, e202206233 (2022).
Li, Q. et al. Fe isolated single atoms on S, N codoped carbon by copolymer pyrolysis strategy for highly efficient oxygen reduction reaction. Adv. Mater. 30, e1800588 (2018).
Wang, J. et al. Suppressing local charge symmetry of iron single atoms for efficient electrocatalytic nitrate reduction to ammonia ORR. Adv. Funct. Mater. 33, 2304277-2304285 (2023).
Xu, H. et al. Improving PMS oxidation of organic pollutants by single cobalt atom catalyst through hybrid radical and non-radical pathways. Appl. Catal. B Environ. 263, 118350 (2020).
Wang, J. et al. Suppressing thermal migration by fine-tuned metalsupport interaction of iron single-atom catalyst for efficient ORR. Adv. Funct. Mater. 33, 2304277-2304285 (2023).
Miao, Q. et al. sites in carbon nanosheets by templatepyrolysis of COFs for RR. Chem. Eng. J. 450, 138427 (2022).
Wang, Z. et al. Cobalt single atoms anchored on oxygen-doped tubular carbon nitride for efficient Peroxymonosulfate activation: simultaneous coordination structure and morphology modulation. Angew. Chem. Int. Ed. 61, e202202338 (2022).
Tan, Z. et al. Highly efficient electrocatalysis of oxygen to hydroxyl radical by single-atom catalyst for refractory organic pollutant removal. Appl. Catal. B-Environ. Energy 355, 124170 (2024).
Kramm, U. I. et al. Structure of the catalytic sites in Fe/N/C-catalysts for -reduction in PEM fuel cells. Phys. Chem. Chem. Phys. 14, 11673-11688 (2012).
Wang, Z. L. et al. Gelatin-derived sustainable carbon-based functional materials for energy conversion and storage with controllability of structure and component. Sci. Adv. 1, e1400035 (2015).
Kramm, U. I., Lefevre, M., Larouche, N., Schmeisser, D. & Dodelet, J. P. Correlations between mass activity and physicochemical properties of Fe/N/C catalysts for the ORR in PEM fuel cell via 57Fe Mossbauer spectroscopy and other techniques. J. Am. Chem. Soc. 136, 978-985 (2014).
Kramm, U.I. et al. Influence of the electron-density of FeN4-centers towards the catalytic activity of pyrolyzed FeTMPPCl-based ORRelectrocatalysts. J. Electrochem. Soc. 158, B69-B78 (2011).
Shi, L. et al. Photoassisted construction of holey defective photocatalysts for efficient visible-light-driven production. Small 14, 1703142 (2018).
Zhang, X. et al. Unraveling the dual defect sites in graphite carbon nitride for ultra-high photocatalytic evolution. Energ. Environ. Sci. 15, 830-842 (2022).
Hammer, B. & Norskov, J. K. Why gold is the noblest of all the metals. Nature 376, 238-240 (1995).
Liu, J., Guo, Y., Fu, X., Luo, J. & Zhi, C. Strengthening absorption ability of Co-N-C as efficient bifunctional oxygen catalyst by modulating the d band center using MoC. Green Energy Environ. 8, 459-469 (2023).
Wu, Q. Y., Yang, Z. W., Wang, Z. W. & Wang, W. L. Oxygen doping of cobalt-single-atom coordination enhances peroxymonosulfate activation and high-valent cobalt-oxo species formation. Proc. Natl. Acad. Sci. USA 120, e2219923120 (2023).
Zhao, J., Sun, X., Huang, X. & Li, J. Reaction mechanisms of methanol oxidation by biomimetic complex. Int. J. Quantum Chem. 116, 692-701 (2016).
Yang, M. et al. Unveiling the origins of selective oxidation in singleatom catalysis via intensified radical and nonradical pathways. Environ. Sci. Technol. 56, 11635-11645 (2022).
Chen, Y. et al. Facile fabrication of rGO/PPy/nZVI catalytic microreactor for ultrafast removal of p-nitrophenol from water. Appl. Catal. B Environ. 324, 122270 (2023).
Pestovsky, O. & Bakac, A. Aqueous Ferryl(IV) Ion: kinetics of oxygen atom transfer to substrates and oxo exchange with solvent water. Inorg. Chem. 45, 814-820 (2006).
Ji, J. et al. Defects on : tuning redox reactions for sustainable degradation of organic pollutants. Angew. Chem. Int. Ed. 60, 2903-2908 (2021).
Gorman, A. & Rodgers, M. Singlet molecular oxygen. Chem. Soc. Rev. 10, 205-231 (1981).
Mo, F. et al. The optimized Fenton-like activity of Fe single-atom sites by Fe atomic clusters-mediated electronic configuration modulation. Proc. Natl. Acad. Sci. USA 120, e2300281120 (2023).
Miao, J. et al. Spin-state-dependent peroxymonosulfate activation of single-atom M-N Moieties via a radical-free pathway. ACS Catal. 11, 9569-9577 (2021).
Yun, E. T., Lee, J. H., Kim, J., Park, H. D. & Lee, J. Identifying the nonradical mechanism in the peroxymonosulfate activation process: singlet oxygenation versus mediated electron transfer. Environ. Sci. Technol. 52, 7032-7042 (2018).
Sanchez, P. M., Ocampo-Pérez, R., Rivera-Utrilla, J. & Mota, A. J. Comparative study of the photodegradation of bisphenol A by HO , and radicals in aqueous phase. Sci. Total Environ. 463, 423-431 (2013).
Ding, J. et al. Electrochemical activation of persulfate on BDD and DSA anodes: electrolyte influence, kinetics and mechanisms in the degradation of bisphenol A. J. Hazard. Mater. 388, 121789 (2020).
Ren, W. et al. Origins of electron-transfer regime in persulfatebased nonradical oxidation processes. Environ. Sci. Technol. 56, 78-97 (2021).
Frisch, M. J. et al. Gaussian 16, Revision A.03. Gaussian, Inc., Wallingford CT (2016).
Lu, T. & Chen, F. W. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580-592 (2012).
Lu, T. A comprehensive electron wavefunction analysis toolbox for chemists, Multiwfn. J. Chem. Phys. 161, 082503 (2024).
Lu, T. & Chen, F. W. Calculation of molecular orbital composition. Acta Chim. Sin. 69, 2393 (2011).
Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (2023YFC3708002), the National Natural Science Foundation of China (U22A20402, 22076059, 22376076, 22076061, 21936003), International Joint Research Center for Intelligent Biosensing Technology and Health, Shenzhen Science and Technology Program (JCYJ20220818095601002), and the Fundamental Research Funds for the Central Universities (CCNU22JC014, CCNU24JL011).
Author contributions
H.X. and L.Z. supervised the project and provided financial support. T.C. conceived and designed the experiments. G.Z. performed DFT calculations and its analysis. T.C., H.X., H.S., G.Z., and L.Z. wrote the paper. Y.H., S.Y., D.Z., H.D., Y.X., and S.H. provided experimental assistance.
Correspondence and requests for materials should be addressed to Ganbing Zhang, Hui Xu or Lizhi Zhang.
Peer review information Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http:// creativecommons.org/licenses/by-nc-nd/4.0/.
(c) The Author(s) 2025
State Key Laboratory of Green Pesticide; Engineering Research Center of Photoenergy Utilization for Pollution Control and Carbon Reduction, Ministry of Education; College of Chemistry, Central China Normal University, Wuhan, PR China. Collaborative Innovation Center for Advanced Organic Chemical Materials Co-constructed by the Province and Ministry; Ministry-of-Education Key Laboratory for the Synthesis and Applications of Organic Functional Molecules; College of Chemistry and Chemical Engineering, Hubei University, Wuhan, PR China. School of Environmental Science and Engineering, National observation and Research Station of Erhai Lake Ecosystem in Yunnan, Yunnan Dali Research Institute, Shanghai Jiao Tong University, Shanghai, PR China.
-e-mail: gbzhang@hubu.edu.cn; huixu@ccnu.edu.cn; zhanglizhi@sjtu.edu.cn