DOI: https://doi.org/10.1038/s12276-024-01180-8
PMID: https://pubmed.ncbi.nlm.nih.gov/38424190
تاريخ النشر: 2024-03-01
نقص الأكسجين، الإجهاد التأكسدي، وتفاعل إشارات HIFs وNRF2 في السرطان
الملخص
الأكسجين ضروري للحياة ويعمل كمتقبل نهائي للإلكترونات في إنتاج الطاقة الميتوكوندري. تتكيف الخلايا مع مستويات الأكسجين المتغيرة من خلال أنظمة استجابة معقدة. تقوم عوامل الاستجابة لنقص الأكسجين (HIFs)، بما في ذلك HIF-1a وHIF-2a، بتنظيم الاستجابة الخلوية لنقص الأكسجين، من خلال تنشيط الجينات لزيادة إمدادات الأكسجين وتقليل الاستهلاك. في ظل ظروف وجود فائض من الأكسجين وما ينتج عنه من إجهاد أكسيدي، يقوم عامل النواة المرتبط بالحديد 2 (NRF2) بتنشيط المئات من الجينات لإزالة المؤكسدات والبقاء الخلوي التكيفي. يُعتبر نقص الأكسجين والإجهاد الأكسيدي من السمات الأساسية للأورام الصلبة، وتلعب HIFs وNRF2 أدوارًا محورية في نمو الأورام وتقدمها. إن التفاعل المعقد بين نقص الأكسجين والإجهاد الأكسيدي داخل بيئة الورم يضيف طبقة أخرى من التعقيد إلى أنظمة الإشارات HIF وNRF2. تهدف هذه المراجعة إلى توضيح التغيرات الديناميكية ووظائف مسارات الإشارات HIF وNRF2 استجابةً لظروف نقص الأكسجين والإجهاد الأكسيدي، مع التأكيد على تداعياتها داخل بيئة الورم. بالإضافة إلى ذلك، استكشفت هذه المراجعة التفاعل المعقد بين HIFs وNRF2، مقدمة رؤى حول أهمية هذه التفاعلات لتطوير استراتيجيات جديدة لعلاج السرطان.
مقدمة
خلل في الجزيئات الكبيرة، وفي النهاية، موت الخلايا. استجابةً لذلك، تزداد مستويات التعبير عن الجينات المعنية بإزالة الجذور الحرة الناتجة عن فرط الأكسجين وإصلاح الخلايا التالفة. يعتبر عامل النسخ الرئيسي الذي يتوسط هذه الاستجابة هو عامل النسخ المرتبط بالعامل 2 (NRF2) الذي يعزز التكيف وبقاء الخلايا في ظل ظروف الإجهاد التأكسدي.
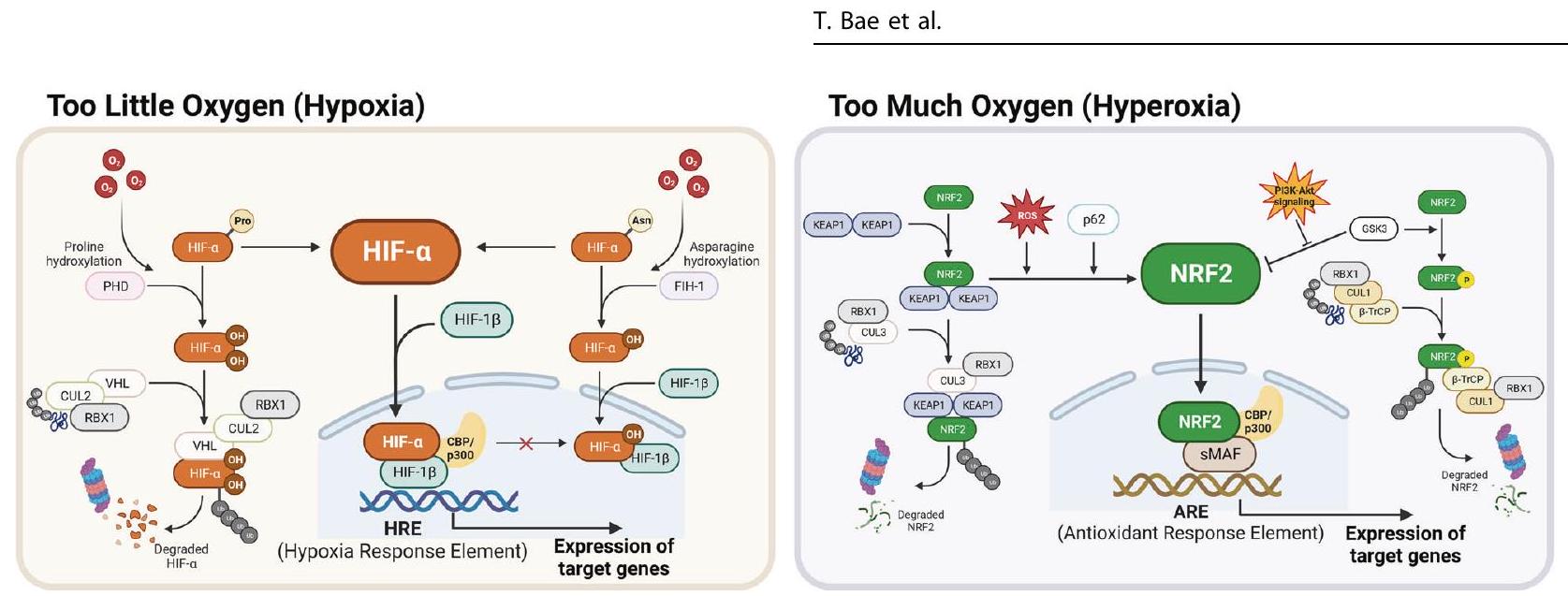
أنظمة الاستجابة التكيفية للمتغيرات
مرتبط بتحول الأيض الجليكولي واحتجاز دورة الخلية. على النقيض من ذلك، يصبح HIF-2a نشطًا تدريجيًا تحت ظروف نقص الأكسجين المستمرة، مما يزيد من مستويات التعبير للجينات المشاركة في تكوين كريات الدم الحمراء وخصائص الخلايا الجذعية الورمية.
استجابة متقاطعة: نقص الأكسجين – NRF2 والإجهاد التأكسدي – HIFs
بعد نقص الأكسجين الحاد في خلايا الإنسان
يساهم في زيادة تنظيم HIF
التفاعل بين HIFs و NRF2
| سرطان | آلية | الظواهر | مرجع |
| سرطان الثدي | حذف N-glycan المفرد (N418Q) لبروتين ErbB3 (HER3) يقلل من تأثير هناجولين-
|
نمو الخلايا، الهجرة | ١٠١ |
| خفض NRF2 يقلل من HIF-1
|
انتشار | 98 | |
| يرتبط NRF2 بـ ARE على بعد 30 كيلوبايتًا upstream من موقع HIF1A. | ٥٧ | ||
| زيادة التعبير عن NRF2 تعزز تعبير الإنزيمات الرئيسية في مسار الفوسفات البنتوز، بما في ذلك G6PD وTKT، وHIF اللاحق.
|
انتشار، هجرة | 99 | |
| خفض NRF2 يزيد من مستويات miR-181c لتثبيط HIF-1
|
إعادة برمجة الأيض، الالتهام الذاتي | 65 | |
| سرطان القولون والمستقيم | تقليل NRF2 يقلل من الخلوية والميتوكوندريا
|
نمو الورم، تكوين الأوعية الدموية | ٤٥ |
| NRF2 و HIF و NF-кВ تتواجد بكثرة في قلب الكتل المتنامية. | CSC، مقاومة الضغط | 165 | |
| خفض NRF2 يزيد من مستويات miR-181a-2-3p ويثبط HIF-
|
نمو الورم، خصائص خلايا السرطان الجذعية | 66 | |
| سرطان بطانة الرحم | miR-148b يقلل من NRF2 و HIF-1
|
انتشار | 192 |
| سرطان المريء الحرشفي | حجب الشRNA لـ NRF2 HIF-1
|
الهجرة، الغزو | 53 |
| سرطان المعدة | تزيد نقص الأكسجين أو VEGFA من تعبير VEGFR2 لتسهيل الانتقال النووي لـ NRF2، بينما أدى تقليل NRF2 إلى تثبيط HIF-1.
|
البقاء، الغزو | 100 |
| تثبيط TRPM2 يزعزع استقرار NRF2 و HIF-1
|
فيروبتوسيس | 184 | |
| ورم دبقي متعدد الأشكال | تثبيط NRF2 يثبط الميتوكوندريا
|
التكاثر، نمو الورم، تكوين الأوعية الدموية | 52 |
| تنشيط NRF2 الناتج عن الإجهاد التأكسدي ينشط NIX ويزيد من مستوى HIF-
|
خلايا الساق، البقاء، نمو الورم | 63 | |
| نقص الأكسجين (12 ساعة) يحفز HIF-1
|
التكاثر، خلايا السرطان الجذعية، التحول الظهاري، مقاومة الأدوية، نمو الورم، تكوين الأوعية الدموية | 164 | |
| سرطان الخلايا الكبدية | تنظم NRF2 مستوى النسخ لـ HIF1A عبر توافق ARE على محفزه تحت ظروف نقص الأكسجين الخفيف
|
مقاومة السيسبلاتين | ٥٦ |
| تثبيط NRF2 يقلل من HIF-1
|
مقاومة 5-FU، بقاء الخلايا، الهجرة، الغزو، نمو الورم | ١١٢ | |
| يرتبط NRF2 بنطاق التحلل المعتمد على الأكسجين (ODD) لـ HIF-1
|
نمو الورم | ٥٥ | |
| سرطان الكلى | تتراكم خلايا نقص FH الفومارات لاستقرار NRF2 المستقل عن ROS و HIF المعتمد على ROS
|
انتشار | 67 |
| سرطان الغدة الرئوية | نقص الأكسجين المتقطع يعزز إنتاج ROS بواسطة NOX1 لتنشيط NRF2 من خلال تثبيت البروتين و Trx1 اللاحق، مما يزيد من HIF-1.
|
البقاء | 60 |
| تكون السرطان الرئوي | iAs يحفز HIF-1 المعتمد على NRF2
|
CSC، إعادة برمجة الأيض | 51 |
| سرطان المبيض | زيادة في أنواع الأكسجين التفاعلية (ROS) بواسطة هرمون تحفيز الجريب (FSH) تنشط NRF2، مما يعزز HIF-1 بشكل أكبر.
|
تكوين الأوعية الدموية | ٥٤ |
| سرطان البنكرياس والرئة | نقص NRF2 يعيق HIF-1
|
انتشار | 193 |
تفاعل إيجابي
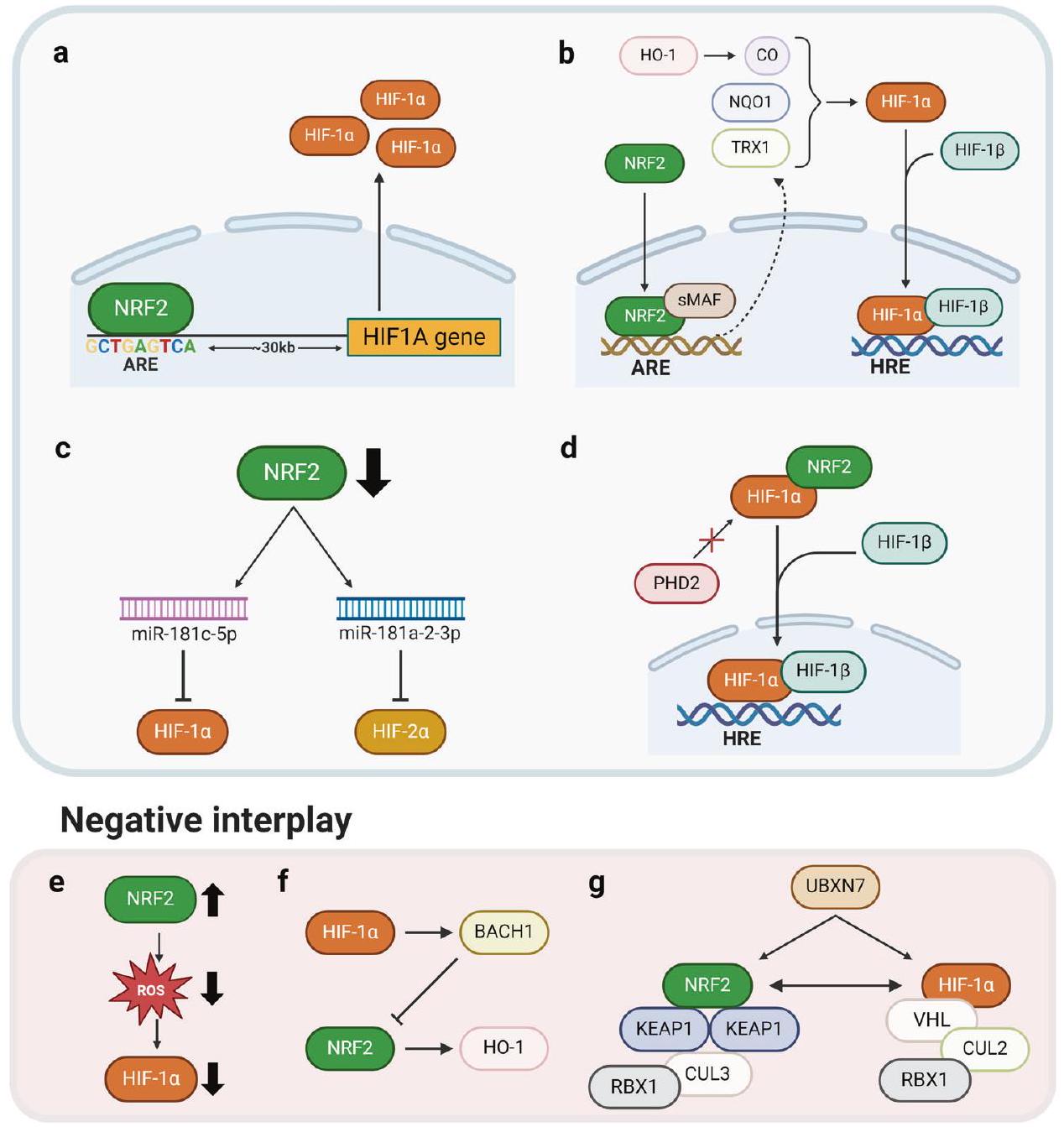
الجينات المستهدفة (الجدول 2). تتماشى هذه النتائج مع وجود ارتباط متبادل بين مستويات تعبير HIF-1a وNRF2، حيث ترتبط المستويات العالية من هذه العوامل النسخية بنتائج سيئة لدى مرضى الورم الدبقي.
تكاثر السرطان والبقاء
| أنماط سرطان | جينات الهدف NRF2 | جينات أهداف HIFs |
| التكاثر/البقاء/التحول الأيضي | NOTCH1
|
NOTCH1
|
| مقاومة العلاج | BCL-2، ABCC1، ABCG2، ABCC2، ABCC3
|
BCL-2
|
| تكوين الأوعية الدموية | VEGF
|
VEGF، SDF1، SCF، PGF، ANGPT2
|
| EMT/نقائل | NOTCH1
|
NOTCH1
|
| خلايا السرطان الجذعية | OCT4، NANOG
|
OCT4
|
| فيروبتوزيس | SLC7A11، GCLM، HMOX1، GCLC، FPN، FTL، FTH1، TXNRD1، GPX4، NQO1
|
SLC7A11، GCLM
|
ديهيدروجيناز A و PDK1
كانت مستويات التعبير العالية لـ NRF2 مرتبطة إيجابيًا بتعبير HIF-1a و HO-1
مقاومة العلاج
تكوين الأوعية الدموية للأورام
المستويات عند التعرض لمحفزات نقص الأكسجين
أدمغة الفئران المصابة بارتفاع ضغط الدم الوريدي، وقد أدى نقص Nrf2 إلى تثبيط تشكيل الأنابيب الوعائية في خلايا بطانة الأوعية الدقيقة الدماغية الأولية
EMT وانتشار السرطان
خصائص خلايا السرطان الجذعية
يُعتقد أنها تسبب فشل علاج السرطان، حيث تعزز الخصائص الخبيثة، بما في ذلك النقائل، والغزو، ومقاومة العلاج، والهروب المناعي.
مقاومة للفيروبتوز
كمنظم سلبي للفيروبتوز
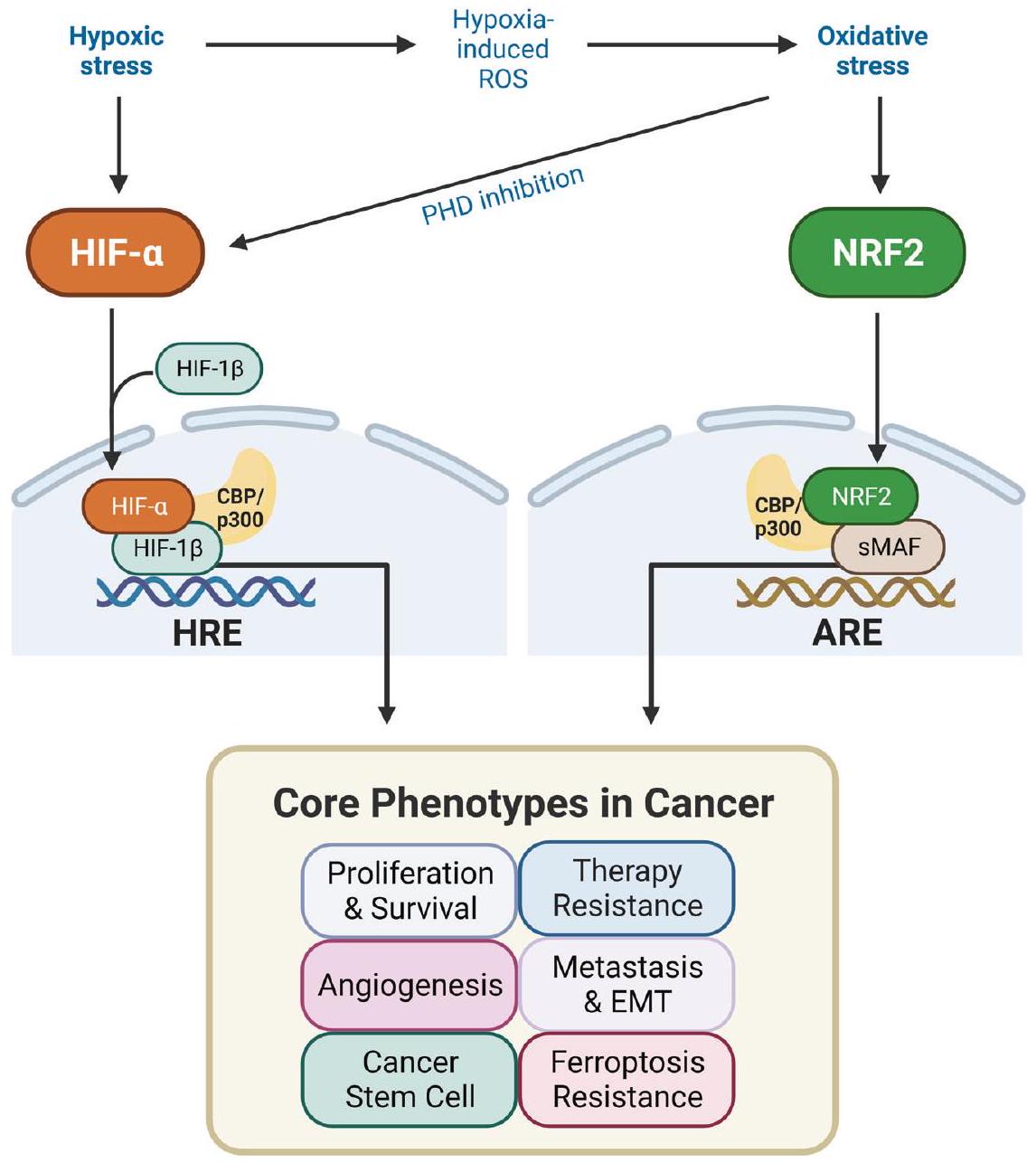
الملاحظات الختامية
الإجهاد الناجم عن نقص الأكسجين والإجهاد التأكسدي، والذي من المحتمل أن يكون حتميًا في ظل ظروف نقص الأكسجين. على النقيض من ذلك، تتغير مستويات NRF2 تحت ظروف نقص الأكسجين بطريقة تعتمد على السياق، مما يشير إلى أن الإجهاد التأكسدي المرتبط بنقص الأكسجين لا يصاحب بالضرورة تنشيط NRF2. تشير هذه العلاقة إلى أن تثبيط NRF2 في الأورام التي تشهد زيادة في كل من HIF وNRF2 قد يكون استراتيجية أكثر كفاءة بالنظر إلى استجابة HIFs لنقص الأكسجين والإجهاد التأكسدي. على سبيل المثال، يمنع العلاج باستخدام البروساتول، وهو مثبط لتخليق بروتين NRF2، تراكم HIF-1a الناتج عن نقص الأكسجين ويقلل من استهلاك الجلوكوز في خلايا سرطان القولون.
REFERENCES
- Raymond, J. & Segrè, D. The effect of oxygen on biochemical networks and the evolution of complex life. Science 311, 1764-1767 (2006).
- Wicks, E. E. & Semenza, G. L. Hypoxia-inducible factors: cancer progression and clinical translation. J. Clin. Investig. 132, e159839 (2022).
- Halliwell, B. Biochemistry of oxidative stress. Biochem Soc. Trans. 35, 1147-1150 (2007).
- Semenza, G. L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 29, 625-634 (2010).
- Yamamoto, M., Kensler, T. W. & Motohashi, H. The KEAP1-NRF2 system: a thiolbased sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 98, 1169-1203 (2018).
- O’Malley, J., Kumar, R., Inigo, J., Yadava, N. & Chandra, D. Mitochondrial stress response and cancer. Trends Cancer 6, 688-701 (2020).
- Vaupel, P., Höckel, M. & Mayer, A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid. Redox Signal 9, 1221-1235 (2007).
- Nakamura, H. & Takada, K. Reactive oxygen species in cancer: current findings and future directions. Cancer Sci. 112, 3945-3952 (2021).
- Rojo de la Vega, M., Chapman, E. & Zhang, D. D. NRF2 and the hallmarks of cancer. Cancer Cell 34, 21-43 (2018).
- Hermes-Lima, M. et al. Preparation for oxidative stress under hypoxia and metabolic depression: revisiting the proposal two decades later. Free Radic. Biol. Med. 89, 1122-1143 (2015).
- Lee, G. et al. Oxidative dimerization of PHD2 is responsible for its inactivation and contributes to metabolic reprogramming via HIF-1a activation. Sci. Rep. 6, 18928 (2016).
- Fukuda, R. et al. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 129, 111-122 (2007).
- Semenza, G. L. The genomics and genetics of oxygen homeostasis. Annu. Rev. Genomics Hum. Genet. 21, 183-204 (2020).
- Wang, G. L., Jiang, B. H., Rue, E. A. & Semenza, G. L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O 2 tension. Proc. Natl Acad. Sci. USA 92, 5510-5514 (1995).
- Schofield, C. J. & Ratcliffe, P. J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 5, 343-354 (2004).
- Bruick, R. K. & McKnight, S. L. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 294, 1337-1340 (2001).
- Appelhoff, R. J. et al. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor*. J. Biol. Chem. 279, 38458-38465 (2004).
- Mahon, P. C., Hirota, K. & Semenza, G. L. FIH-1: a novel protein that interacts with HIF-1a and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 15, 2675-2686 (2001).
- Cowman, S. J. & Koh, M. Y. Revisiting the HIF switch in the tumor and its immune microenvironment. Trends Cancer 8, 28-42 (2022).
- Holmquist-Mengelbier, L. et al. Recruitment of HIF-1a and HIF-2a to common target genes is differentially regulated in neuroblastoma: HIF-2a promotes an aggressive phenotype. Cancer Cell 10, 413-423 (2006).
- Koh, M. Y. & Powis, G. Passing the baton: the HIF switch. Trends Biochem. Sci. 37, 364-372 (2012).
- Manalo, D. J. et al. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 105, 659-669 (2005).
- Semenza, G. L. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharm. Sci. 33, 207-214 (2012).
- Covello, K. L. et al. HIF-2a regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 20, 557-570 (2006).
- Sowter, H. M., Ratcliffe, P. J., Watson, P., Greenberg, A. H. & Harris, A. L. HIF-1dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 61, 6669-6673 (2001).
- Dodson, M. et al. Modulating NRF2 in disease: timing is everything. Annu. Rev. Pharm. Toxicol. 59, 555-575 (2019).
- Torrente, L. & DeNicola, G. M. Targeting NRF2 and its downstream processes: opportunities and challenges. Annu. Rev. Pharm. Toxicol. 62, 279-300 (2022).
- Ho, Y. S., Dey, M. S. & Crapo, J. D. Antioxidant enzyme expression in rat lungs during hyperoxia. Am. J. Physiol. 270, L810-L818 (1996).
- Cho, H. Y., Reddy, S. P., Debiase, A., Yamamoto, M. & Kleeberger, S. R. Gene expression profiling of NRF2-mediated protection against oxidative injury. Free Radic. Biol. Med. 38, 325-343 (2005).
- Papaiahgari, S., Zhang, Q., Kleeberger, S. R., Cho, H. Y. & Reddy, S. P. Hyperoxia stimulates an Nrf2-ARE transcriptional response via ROS-EGFR-PI3K-Akt/ERK MAP kinase signaling in pulmonary epithelial cells. Antioxid. Redox Signal. 8, 43-52 (2006).
- Taguchi, K. & Yamamoto, M. The KEAP1-NRF2 system in cancer. Front. Oncol. 7, 85 (2017).
- Rada, P. et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase
-TrCP axis. Mol. Cell Biol. 32, 3486-3499 (2012). - Suzuki, T., Takahashi, J. & Yamamoto, M. Molecular basis of the KEAP1-NRF2 signaling pathway. Mol. Cells 46, 133-141 (2023).
- Iso, T., Suzuki, T., Baird, L. & Yamamoto, M. Absolute amounts and status of the Nrf2-Keap1-Cul3 complex within cells. Mol. Cell Biol. 36, 3100-3112 (2016).
- Baird, L. & Yamamoto, M. The molecular mechanisms regulating the KEAP1NRF2 pathway. Mol. Cell Biol. 40, e00099-20 (2020).
- Horie, Y. et al. Molecular basis for the disruption of Keap1-Nrf2 interaction via Hinge & Latch mechanism. Commun. Biol. 4, 576 (2021).
- Hayes, J. D. & Dinkova-Kostova, A. T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 39, 199-218 (2014).
- Hernansanz-Agustín, P. et al. Acute hypoxia produces a superoxide burst in cells. Free Radic. Biol. Med. 71, 146-156 (2014).
- Chandel, N. S. et al. Mitochondrial reactive oxygen species trigger hypoxiainduced transcription. Proc. Natl Acad. Sci. USA 95, 11715-11720 (1998).
- Guzy, R. D. et al. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 1, 401-408 (2005).
- Toth, R. K. & Warfel, N. A. Strange bedfellows: nuclear factor, erythroid 2-like 2 (Nrf2) and hypoxia-inducible factor 1 (HIF-1) in tumor hypoxia. Antioxidants 6, 27 (2017).
- Tello, D. et al. Induction of the mitochondrial NDUFA4L2 protein by HIF-1a decreases oxygen consumption by inhibiting Complex I activity. Cell Metab. 14, 768-779 (2011).
- Xin, X., Li, Y. & Liu, H. Hesperidin ameliorates hypobaric hypoxia-induced retinal impairment through activation of Nrf2/HO-1 pathway and inhibition of apoptosis. Sci. Rep. 10, 19426 (2020).
- Potteti, H. R. et al. Nrf2-AKT interactions regulate heme oxygenase 1 expression in kidney epithelia during hypoxia and hypoxia-reoxygenation. Am. J. Physiol. Ren. Physiol. 311, F1025-f1034 (2016).
- Kim, T.-H. et al. NRF2 blockade suppresses colon tumor angiogenesis by inhibiting hypoxia-induced activation of HIF-1a. Cancer Res. 71, 2260-2275 (2011).
- Bondi, C. D. et al. Suppression of NRF2 Activity by HIF-1
promotes fibrosis after ischemic acute kidney injury. Antioxidants 11, 1810 (2022). - Loboda, A. et al. HIF-1 induction attenuates Nrf2-dependent IL-8 expression in human endothelial cells. Antioxid. Redox Signal. 11, 1501-1517 (2009).
- Pan, Y. et al. Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol. Cell Biol. 27, 912-925 (2007).
- Watanabe, Y. et al. Glutathione adducts induced by ischemia and deletion of glutaredoxin-1 stabilize HIF-1a and improve limb revascularization. Proc. Natl Acad. Sci. USA 113, 6011-6016 (2016).
- Hamanaka, R. B., Weinberg, S. E., Reczek, C. R. & Chandel, N. S. The mitochondrial respiratory chain is required for organismal adaptation to hypoxia. Cell Rep. 15, 451-459 (2016).
- Bi, Z. et al. Nrf2 and HIF1a converge to arsenic-induced metabolic reprogramming and the formation of the cancer stem-like cells. Theranostics 10, 4134-4149 (2020).
- Ji, X. et al. Knockdown of Nrf2 suppresses glioblastoma angiogenesis by inhibiting hypoxia-induced activation of HIF-1a. Int. J. Cancer 135, 574-584 (2014).
- Shen, H. et al. Blockage of Nrf2 suppresses the migration and invasion of esophageal squamous cell carcinoma cells in hypoxic microenvironment. Dis. Esophagus 27, 685-692 (2014).
- Zhang, Z. et al. Reactive oxygen species regulate FSH-induced expression of vascular endothelial growth factor via Nrf2 and HIF1a signaling in human epithelial ovarian cancer. Oncol. Rep. 29, 1429-1434 (2013).
- Zheng, J. et al. Overactivated NRF2 induces pseudohypoxia in hepatocellular carcinoma by stabilizing HIF-1alpha. Free Radic. Biol. Med. 194, 347-356 (2023).
- Jin, X., Gong, L., Peng, Y., Li, L. & Liu, G. Enhancer-bound Nrf2 licenses HIF-1alpha transcription under hypoxia to promote cisplatin resistance in hepatocellular carcinoma cells. Aging (Albany NY) 13, 364-375 (2020).
- Lacher, S. E., Levings, D. C., Freeman, S. & Slattery, M. Identification of a functional antioxidant response element at the HIF1A locus. Redox. Biology 19, 401-411 (2018).
- Oh, E. T. et al. NQO1 inhibits proteasome-mediated degradation of HIF-1a. Nat. Commun. 7, 13593 (2016).
- Csiki, I. et al. Thioredoxin-1 modulates transcription of cyclooxygenase-2 via hypoxia-inducible factor-1alpha in non-small cell lung cancer. Cancer Res. 66, 143-150 (2006).
- Malec, V. et al. HIF-1 alpha signaling is augmented during intermittent hypoxia by induction of the Nrf2 pathway in NOX1-expressing adenocarcinoma A549 cells. Free Radic. Biol. Med. 48, 1626-1635 (2010).
- Chin, B. Y. et al. Hypoxia-inducible factor 1alpha stabilization by carbon monoxide results in cytoprotective preconditioning. Proc. Natl Acad. Sci. USA 104, 5109-5114 (2007).
- Choi, Y. K. et al. Carbon monoxide promotes VEGF expression by increasing HIF1alpha protein level via two distinct mechanisms, translational activation and stabilization of HIF-1alpha protein. J. Biol. Chem. 285, 32116-32125 (2010).
- Jung, J. et al. Mitochondrial NIX promotes tumor survival in the hypoxic niche of glioblastoma. Cancer Res. 79, 5218-5232 (2019).
- Jung, K. A., Lee, S. & Kwak, M. K. NFE2L2/NRF2 activity is linked to mitochondria and AMP-activated protein kinase signaling in cancers through miR-181c/ mitochondria-encoded cytochrome c oxidase regulation. Antioxid. Redox Signal 27, 945-961 (2017).
- Lee, S., Hallis, S. P., Jung, K.-A., Ryu, D. & Kwak, M.-K. Impairment of HIF-1amediated metabolic adaption by NRF2-silencing in breast cancer cells. Redox Biol. 24, 101210 (2019).
- Hallis, S. P., Kim, S. K., Lee, J.-H. & Kwak, M.-K. Association of NRF2 with HIF-2ainduced cancer stem cell phenotypes in chronic hypoxic condition. Redox Biol. 60, 102632 (2023).
- Sullivan, L. B. et al. The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol. Cell 51, 236-248 (2013).
- Early, J. O. et al. Circadian clock protein BMAL1 regulates IL-1
in macrophages via NRF2. Proc. Natl Acad. Sci. USA 115, E8460-E8468 (2018). - Di Gregorio, J. et al. UBXN7 cofactor of CRL3KEAP1 and CRL2VHL ubiquitin ligase complexes mediates reciprocal regulation of NRF2 and HIF-1a proteins. Biochim. Biophys. Acta 1868, 118963 (2021).
- Lu, C. et al. Nrf2 activation is required for ligustrazine to inhibit hepatic steatosis in alcohol-preferring mice and hepatocytes. Toxicol. Sci. 155, 432-443 (2017).
- Li, Y. et al. Inhibitor of apoptosis-stimulating protein of p53 inhibits ferroptosis and alleviates intestinal ischemia/reperfusion-induced acute lung injury. Cell Death Differ. 27, 2635-2650 (2020).
- Lin, H. C. et al. Andrographolide inhibits hypoxia-induced HIF-1a-driven endothelin 1 secretion by activating Nrf2/HO-1 and promoting the expression of prolyl hydroxylases
in human endothelial cells. Environ. Toxicol. 32, 918-930 (2017). - Talks, K. L. et al. The expression and distribution of the hypoxia-inducible factors HIF-1a and HIF-2a in normal human tissues, cancers, and tumor-associated macrophages. Am. J. Pathol. 157, 411-421 (2000).
- Schito, L. & Semenza, G. L. Hypoxia-inducible factors: master regulators of cancer progression. Trends Cancer 2, 758-770 (2016).
- Krieg, M. et al. Up-regulation of hypoxia-inducible factors HIF-1alpha and HIF2alpha under normoxic conditions in renal carcinoma cells by von HippelLindau tumor suppressor gene loss of function. Oncogene 19, 5435-5443 (2000).
- Ravi, R. et al. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1a. Genes Dev. 14, 34-44 (2000).
- Zhong, H. et al. Modulation of hypoxia-inducible factor 1a expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics1. Cancer Res. 60, 1541-1545 (2000).
- Isaacs, J. S. et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: Novel role of fumarate in regulation of HIF stability. Cancer Cell 8, 143-153 (2005).
- Greco, S., Gaetano, C. & Martelli, F. HypoxamiR regulation and function in ischemic cardiovascular diseases. Antioxid. Redox Signal. 21, 1202-1219 (2014).
- Menegon, S., Columbano, A. & Giordano, S. The dual roles of NRF2 in cancer. Trends Mol. Med. 22, 578-593 (2016).
- Robertson, H., Dinkova-Kostova, A. T. & Hayes, J. D. NRF2 and the Ambiguous Consequences of Its Activation during Initiation and the Subsequent Stages of Tumourigenesis. Cancers (Basel) 12, 3609 (2020).
- Kitamura, H. & Motohashi, H. NRF2 addiction in cancer cells. Cancer Sci. 109, 900-911 (2018).
- Guichard, C. et al. Integrated analysis of somatic mutations and focal copynumber changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 44, 694-698 (2012).
- Network, C.G.A.R. Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543-550 (2014).
- Shibata, T. et al. Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology 135, 1358-1368 (2008).
- Muscarella, L. A. et al. Frequent epigenetics inactivation of KEAP1 gene in nonsmall cell lung cancer. Epigenetics 6, 710-719 (2011).
- DeNicola, G. M. et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106-109 (2011).
- Komatsu, M. et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 12, 213-223 (2010).
- Adam, J. et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 20, 524-537 (2011).
- Ji, X. et al. Correlation of Nrf2 and HIF-1alpha in glioblastoma and their relationships to clinicopathologic features and survival. Neurol. Res. 35, 1044-1050 (2013).
- Papandreou, I., Cairns, R. A., Fontana, L., Lim, A. L. & Denko, N. C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 3, 187-197 (2006).
- Semenza, G. L., Roth, P. H., Fang, H. M. & Wang, G. L. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 269, 23757-23763 (1994).
- Sakagami, H. et al. Loss of HIF-1a impairs GLUT4 translocation and glucose uptake by the skeletal muscle cells. Am. J. Physiol. Endocrinol. Metab. 306, E1065-E1076 (2014).
- Bellot, G. et al. Hypoxia-induced autophagy is mediated through hypoxiainducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell Biol. 29, 2570-2581 (2009).
- Chee, N. T., Lohse, I. & Brothers, S. P. mRNA-to-protein translation in hypoxia. Mol. Cancer 18, 49 (2019).
- Singh, A., Bodas, M., Wakabayashi, N., Bunz, F. & Biswal, S. Gain of Nrf2 function in non-small-cell lung cancer cells confers radioresistance. Antioxid. Redox Signal 13, 1627-1637 (2010).
- Malhotra, D. et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 38, 5718-5734 (2010).
- Zhang, H. S. et al. NRF2 facilitates breast cancer cell growth via HIF1a-mediated metabolic reprogramming. Int. J. Biochem. Cell Biol. 95, 85-92 (2018).
- Zhang, H. S. et al. Nrf2 promotes breast cancer cell migration via up-regulation of G6PD/HIF-1alpha/Notch1 axis. J. Cell Mol. Med. 23, 3451-3463 (2019).
- Yang, Y. et al. Abnormal phenotype of Nrf2 is associated with poor prognosis through hypoxic/VEGF-A-Rap1b/VEGFR2 pathway in gastric cancer. Aging (Albany NY) 14, 3293-3312 (2022).
- Takamiya, R. et al. The single N-glycan deletion mutant of soluble ErbB3 protein attenuates heregulin beta1-induced tumor progression by blocking of the HIF-1 and Nrf2 pathway. Biochem. Biophys. Res. Commun. 454, 364-368 (2014).
- Kazi, A. A. et al. Nonhypoxic regulation and role of hypoxia-inducible factor 1 in aromatase inhibitor resistant breast cancer. Breast Cancer Res. 16, R15 (2014).
- Liu, L. et al. Hypoxia-inducible factor-1 alpha contributes to hypoxia-induced chemoresistance in gastric cancer. Cancer Sci. 99, 121-128 (2008).
- Chen, N. et al. BCL-xL is a target gene regulated by hypoxia-inducible factor1alpha. J. Biol. Chem. 284, 10004-10012 (2009).
- Erler, J. T. et al. Hypoxia-mediated down-regulation of Bid and Bax in tumors occurs via hypoxia-inducible factor 1-dependent and -independent mechanisms and contributes to drug resistance. Mol. Cell Biol. 24, 2875-2889 (2004).
- Yang, T., Xu, F., Sheng, Y., Zhang, W. & Chen, Y. A targeted proteomics approach to the quantitative analysis of ERK/Bcl-2-mediated anti-apoptosis and multidrug resistance in breast cancer. Anal. Bioanal. Chem. 408, 7491-7503 (2016).
- Choi, B.-h & Kwak, M.-K. Shadows of NRF2 in cancer: resistance to chemotherapy. Curr. Opin. Toxicol. 1, 20-28 (2016).
- Homma, S. et al. Nrf2 enhances cell proliferation and resistance to anticancer drugs in human lung cancer. Clin. Cancer Res. 15, 3423-3432 (2009).
- Kurz, E. U., Cole, S. P. C. & Deeley, R. G. Identification of DNA-protein interactions in the
Flanking and Untranslated Regions of the Human Multidrug Resistance Protein (MRP1) gene: evaluation of a putative antioxidant response ele-ment/AP-1 binding site. Biochem. Biophys. Res. Commun. 285, 981-990 (2001). - Singh, A. et al. Expression of ABCG2 (BCRP) is regulated by Nrf2 in cancer cells that confers side population and chemoresistance phenotype. Mol. Cancer Ther. 9, 2365-2376 (2010).
- Niture, S. K. & Jaiswal, A. K. Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. J. Biol. Chem. 287, 9873-9886 (2012).
- Duan, X. et al. Nrf2-siRNA enhanced the anti-tumor effects of
in 5-fluorouracil-resistant hepatocellular carcinoma by inhibiting HIF-1alpha/ HSP70 signaling. J. Hepatocell. Carcinoma 9, 1341-1352 (2022). - Hanahan, D. & Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 86, 353-364 (1996).
- Forsythe, J. A. et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 16, 4604-4613 (1996).
- Ceradini, D. J. et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 10, 858-864 (2004).
- Gao, C. et al. SCF, regulated by HIF-1a, promotes pancreatic ductal adenocarcinoma cell progression. PLoS ONE 10, e0121338 (2015).
- Simon, M.-P., Tournaire, R. & Pouyssegur, J. The angiopoietin-2 gene of endothelial cells is up-regulated in hypoxia by a HIF binding site located in its first intron and by the central factors GATA-2 and Ets-1. J. Cell. Physiol. 217, 809-818 (2008).
- Florczyk, U. et al. Nrf2 regulates angiogenesis: effect on endothelial cells, bone marrow-derived proangiogenic cells and hind limb ischemia. Antioxid. Redox Signal. 20, 1693-1708 (2013).
- Ichihara, S. et al. Ablation of the transcription factor Nrf2 promotes ischemiainduced neovascularization by enhancing the inflammatory response. Arterioscler. Thromb. Vasc. Biol. 30, 1553-1561 (2010).
- Wei, Y. et al. Nrf2 acts cell-autonomously in endothelium to regulate tip cell formation and vascular branching. Proc. Natl Acad. Sci. USA 110, E3910-E3918 (2013).
- Li, L. et al. Interplay between VEGF and Nrf2 regulates angiogenesis due to intracranial venous hypertension. Sci. Rep. 6, 37338 (2016).
- Feng, R. et al. Nrf2 activation drive macrophages polarization and cancer cell epithelial-mesenchymal transition during interaction. Cell Commun. Signal 16, 54 (2018).
- Shao, S. et al. Curcumin suppresses hepatic stellate cell-induced hepatocarcinoma angiogenesis and invasion through downregulating CTGF. Oxid. Med. Cell. Longev. 2019, 8148510 (2019).
- Hapke, R. Y. & Haake, S. M. Hypoxia-induced epithelial to mesenchymal transition in cancer. Cancer Lett. 487, 10-20 (2020).
- Zhong, H. et al. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 59, 5830-5835 (1999).
- Xu, X. et al. Snail is a direct target of hypoxia-inducible factor 1a (HIF1a) in hypoxia-induced endothelial to mesenchymal transition of human coronary endothelial cells*. J. Biol. Chem. 290, 16653-16664 (2015).
- Yang, M.-H. et al. Direct regulation of TWIST by HIF-1 a promotes metastasis. Nat. Cell Biol. 10, 295-305 (2008).
- Zhang, W. et al. HIF-1a promotes epithelial-mesenchymal transition and metastasis through direct regulation of ZEB1 in colorectal cancer. PLoS ONE 10, e0129603 (2015).
- Wang, R. et al. Hypoxia-inducible factor-dependent ADAM12 expression mediates breast cancer invasion and metastasis. Proc. Natl Acad. Sci. USA 118, e2020490118 (2021).
- Wang, H. et al. NRF2 activation by antioxidant antidiabetic agents accelerates tumor metastasis. Sci. Transl. Med. 8, 334ra351 (2016).
- Zhang, C. et al. NRF2 promotes breast cancer cell proliferation and metastasis by increasing RhoA/ROCK pathway signal transduction. Oncotarget 7, 73593-73606 (2016).
- Jeong, Y. et al. Role of KEAP1/NRF2 and TP53 mutations in lung squamous cell carcinoma development and radiation resistance. Cancer Discov. 7, 86-101 (2017).
- Yazaki, K. et al. ROS-Nrf2 pathway mediates the development of TGF-
induced epithelial-mesenchymal transition through the activation of Notch signaling. Eur. J. Cell Biol. 100, 151181 (2021). - Vilchez Mercedes, S. A. et al. Nrf2 modulates the hybrid epithelial/mesenchymal phenotype and notch signaling during collective cancer migration. Front. Mol. Biosci. 9, 807324 (2022).
- Rachakonda, G. et al. Increased cell migration and plasticity in Nrf2-deficient cancer cell lines. Oncogene 29, 3703-3714 (2010).
- Ryu, D., Lee, J.-H. & Kwak, M.-K. NRF2 level is negatively correlated with TGF-
induced lung cancer motility and migration via NOX4-ROS signaling. Arch. Pharmacal Res. 43, 1297-1310 (2020). - Batlle, E. & Clevers, H. Cancer stem cells revisited. Nat. Med. 23, 1124-1134 (2017).
- Yang, L. et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 5, 8 (2020).
- Wright, M. H. et al. Brca1 breast tumors contain distinct CD44+/CD24- and CD133+cells with cancer stem cell characteristics. Breast Cancer Res. 10, R10 (2008).
- Dalerba, P. et al. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl Acad. Sci. USA 104, 10158-10163 (2007).
- Heddleston, J. M. et al. Hypoxia inducible factors in cancer stem cells. Br. J. Cancer 102, 789-795 (2010).
- Samanta, D., Gilkes, D. M., Chaturvedi, P., Xiang, L. & Semenza, G. L. Hypoxiainducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc. Natl Acad. Sci. USA 111, E5429-E5438 (2014).
- Conley, S. J. et al. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc. Natl Acad. Sci. USA 109, 2784-2789 (2012).
- Keith, B. & Simon, M. C. Hypoxia-inducible factors, stem cells, and cancer. Cell 129, 465-472 (2007).
- Covello, K. L. et al. HIF-2alpha regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 20, 557-570 (2006).
- Lu, H. et al. Chemotherapy-induced S100A10 recruits KDM6A to facilitate OCT4mediated breast cancer stemness. J. Clin. Investig. 130, 4607-4623 (2020).
- Qin, J. et al. Hypoxia-inducible factor 1 alpha promotes cancer stem cells-like properties in human ovarian cancer cells by upregulating SIRT1 expression. Sci. Rep. 7, 10592 (2017).
- Yan, Y. et al. HIF-2a promotes conversion to a stem cell phenotype and induces chemoresistance in breast cancer cells by activating Wnt and Notch pathways. J. Exp. Clin. Cancer Res. 37, 256 (2018).
- Yan, Y. et al. A novel HIF-2a targeted inhibitor suppresses hypoxia-induced breast cancer stemness via SOD2-mtROS-PDI/GPR78-UPR(ER) axis. Cell Death Differ. 29, 1769-1789 (2022).
- Micucci, C., Matacchione, G., Valli, D., Orciari, S. & Catalano, A. HIF2a is involved in the expansion of CXCR4-positive cancer stem-like cells in renal cell carcinoma. Br. J. Cancer 113, 1178-1185 (2015).
- Diehn, M. et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 458, 780-783 (2009).
- Hallis, S. P., Kim, J. M. & Kwak, M.-K. Emerging Role of NRF2 Signaling in Cancer Stem Cell Phenotype. Mol. Cells 46, 153-164 (2023).
- Chang, C.-W. et al. ROS-independent ER stress-mediated NRF2 activation promotes warburg effect to maintain stemness-associated properties of cancerinitiating cells. Cell Death Dis. 9, 194 (2018).
- Chang, C. W. et al. Distinct subpopulations of head and neck cancer cells with different levels of intracellular reactive oxygen species exhibit diverse stemness, proliferation, and chemosensitivity. Cancer Res. 74, 6291-6305 (2014).
- Gao, L. et al. Nrf2 signaling promotes cancer stemness, migration, and expression of ABC transporter genes in sorafenib-resistant hepatocellular carcinoma cells. PLoS ONE 16, e0256755 (2021).
- Zhu, J. et al. Nrf2 is required to maintain the self-renewal of glioma stem cells. BMC Cancer 13, 380 (2013).
- Ryoo, I.-g, Choi, B.-h, Ku, S.-K. & Kwak, M.-K. High CD44 expression mediates p62associated NFE2L2/NRF2 activation in breast cancer stem cell-like cells: implications for cancer stem cell resistance. Redox Biol. 17, 246-258 (2018).
- Park, J., Kim, S. K., Hallis, S. P., Choi, B.-H. & Kwak, M.-K. Role of CD133/NRF2 axis in the development of colon cancer stem cell-like properties. Front. Oncol. 11, 808300 (2022).
- Kim, D., Choi, B. H., Ryoo, I. G. & Kwak, M. K. High NRF2 level mediates cancer stem cell-like properties of aldehyde dehydrogenase (ALDH)-high ovarian cancer cells: inhibitory role of all-trans retinoic acid in ALDH/NRF2 signaling. Cell Death Dis. 9, 896 (2018).
- Noman, A. S. M. et al. Chemotherapeutic resistance of head and neck squamous cell carcinoma is mediated by EpCAM induction driven by IL-6/p62 associated Nrf2-antioxidant pathway activation. Cell Death Dis. 11, 663 (2020).
- Okazaki, K. et al. Enhancer remodeling promotes tumor-initiating activity in NRF2-activated non-small cell lung cancers. Nat. Commun. 11, 5911 (2020).
- Kim, D. H. et al. Nuclear factor erythroid-derived 2 -like 2 -induced reductive stress favors self-renewal of breast cancer stem-like cells via the FoxO3a-Bmi-1 axis. Antioxid. Redox Signal. 32, 1313-1329 (2020).
- Fragoulis, A. et al. Nrf2 induces malignant transformation of hepatic progenitor cells by inducing
-catenin expression. Redox Biol. 57, 102453 (2022). - Zhang, G., Tao, X., Ji, B. & Gong, J. Hypoxia-driven M2-polarized macrophages facilitate cancer aggressiveness and temozolomide resistance in glioblastoma. Oxid. Med. Cell. Longev. 2022, 1614336 (2022).
- Kipp, A. P., Deubel, S., Arnér, E. S. J. & Johansson, K. Time- and cell-resolved dynamics of redox-sensitive Nrf2, HIF and NF-кB activities in 3D spheroids enriched for cancer stem cells. Redox Biol. 12, 403-409 (2017).
- Dixon, ScottJ. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060-1072 (2012).
- Li, J. et al. Ferroptosis: past, present and future. Cell Death Dis. 11, 88 (2020).
- Yang, W. S. & Stockwell, B. R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 15, 234-245 (2008).
- Friedmann Angeli, J. P. et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 16, 1180-1191 (2014).
- Viswanathan, V. S. et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453-457 (2017).
- Fuhrmann, D. C., Mondorf, A., Beifuß, J., Jung, M. & Brüne, B. Hypoxia inhibits ferritinophagy, increases mitochondrial ferritin, and protects from ferroptosis. Redox Biol. 36, 101670 (2020).
- Miess, H. et al. The glutathione redox system is essential to prevent ferroptosis caused by impaired lipid metabolism in clear cell renal cell carcinoma. Oncogene 37, 5435-5450 (2018).
- Green, Y. S. et al. ISCA2 inhibition decreases HIF and induces ferroptosis in clear cell renal carcinoma. Oncogene 41, 4709-4723 (2022).
- Yang, M. et al. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci. Adv. 5, eaaw2238 (2019).
- Singhal, R. et al. HIF-2a activation potentiates oxidative cell death in colorectal cancers by increasing cellular iron. J. Clin. Invest. 131, e143691 (2021).
- Su, X. et al. HIF-a activation by the prolyl hydroxylase inhibitor roxadustat suppresses chemoresistant glioblastoma growth by inducing ferroptosis. Cell Death Dis. 13, 861 (2022).
- Dodson, M., Castro-Portuguez, R. & Zhang, D. D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 23, 101107 (2019).
- Fan, Z. et al. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis 6, e371-e371 (2017).
- Shin, D., Kim, E. H., Lee, J. & Roh, J.-L. Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radic. Biol. Med. 129, 454-462 (2018).
- Yang, J. et al. Cetuximab promotes RSL3-induced ferroptosis by suppressing the Nrf2/HO-1 signalling pathway in KRAS mutant colorectal cancer. Cell Death Dis. 12, 1079 (2021).
- Koppula, P. et al. A targetable CoQ-FSP1 axis drives ferroptosis- and radiationresistance in KEAP1 inactive lung cancers. Nat. Commun. 13, 2206 (2022).
- Anandhan, A. et al. NRF2 controls iron homeostasis and ferroptosis through HERC2 and VAMP8. Sci. Adv. 9, eade9585 (2023).
- Chen, L. D. et al. Nrf2 plays protective role during intermittent hypoxia-induced ferroptosis in rat liver (BRL-3A) cells. Sleep. Breath. 27, 2069-2076 (2023).
- Li, D. et al. Silencing TRPM2 enhanced erastin- and RSL3-induced ferroptosis in gastric cancer cells through destabilizing HIF-1a and Nrf2 proteins. Cytotechnology 74, 559-577 (2022).
- Kikuchi, H., Pino, M. S., Zeng, M., Shirasawa, S. & Chung, D. C. Oncogenic KRAS and BRAF differentially regulate hypoxia-inducible factor-1alpha and -2alpha in colon cancer. Cancer Res. 69, 8499-8506 (2009).
- Cahuzac, K. M. et al. AKT activation because of PTEN loss upregulates xCT via GSK3
NRF2, leading to inhibition of ferroptosis in PTEN-mutant tumor cells. Cell Rep. 42, 112536 (2023). - Kang, H. J. et al. HER2 confers drug resistance of human breast cancer cells through activation of NRF2 by direct interaction. Sci. Rep. 4, 7201 (2014).
- Lu, Y. et al. Brusatol inhibits HIF-1 signaling pathway and suppresses glucose uptake under hypoxic conditions in HCT116 cells. Sci. Rep. 6, 39123 (2016).
- Chen, F. et al. Triptolide, a Chinese herbal extract, enhances drug sensitivity of resistant myeloid leukemia cell lines through downregulation of HIF-1alpha and Nrf2. Pharmacogenomics 14, 1305-1317 (2013).
- Liu, Y. et al. Low-dose triptolide in combination with idarubicin induces apoptosis in AML leukemic stem-like KG1a cell line by modulation of the intrinsic and extrinsic factors. Cell Death Dis. 4, e948 (2013).
- Jin, J. et al. Cardamonin inhibits breast cancer growth by repressing HIF-1alphadependent metabolic reprogramming. J. Exp. Clin. Cancer Res. 38, 377 (2019).
- Qu, J., Zhang, L., Li, L. & Su, Y. miR-148b functions as a tumor suppressor by targeting endoplasmic reticulum metallo protease 1 in human endometrial cancer cells. Oncol. Res. 27, 81-88 (2018).
- Kuper, A. et al. Overcoming hypoxia-induced resistance of pancreatic and lung tumor cells by disrupting the PERK-NRF2-HIF-axis. Cell Death Dis. 12, 82 (2021).
- Wakabayashi, N. et al. Regulation of Notch1 signaling by Nrf2: implications for tissue regeneration. Sci. Signal. 3, ra52-ra52 (2010).
- He, F., Ru, X. & Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 21, 4777 (2020).
- Gustafsson, M. V. et al. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Develop. Cell 9, 617-628 (2005).
- Lv, Y. et al. Hypoxia-inducible factor-1alpha induces multidrug resistance protein in colon cancer. Onco Targets Ther. 8, 1941-1948 (2015).
- Lee, P. J. et al. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia*. J. Biol. Chem. 272, 5375-5381 (1997).
- Jang, J. et al. Primary cilium-autophagy-Nrf2 (PAN) axis activation commits human embryonic stem cells to a neuroectoderm fate. Cell 165, 410-420 (2016).
- Petruzzelli, R., Christensen, D. R., Parry, K. L., Sanchez-Elsner, T. & Houghton, F. D. HIF2a regulates NANOG expression in human embryonic stem cells following hypoxia and reoxygenation through the interaction with an Oct-Sox cis regulatory element. PLoS ONE 9, e108309 (2014).
الشكر والتقدير
مساهمات المؤلفين
المصالح المتنافسة
معلومات إضافية
معلومات إعادة الطبع والإذن متاحة على http://www.nature.com/reprints
ملاحظة الناشر تظل Springer Nature محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
© المؤلفون 2024
المعهد البحثي المتكامل لعلوم الأدوية، الجامعة الكاثوليكية في كوريا، بوشون، جيونغجي-دو 14662، جمهورية كوريا. قسم الصيدلة، كلية الدراسات العليا في الجامعة الكاثوليكية في كوريا، بوشون، جيونغجي-دو 14662، جمهورية كوريا. كلية الصيدلة، الجامعة الكاثوليكية في كوريا، بوشون، جيونغجي-دو 14662، جمهورية كوريا. ساهم هؤلاء المؤلفون بالتساوي: تايغيون باي، ستيفانوس برانوتو هاليس. البريد الإلكتروني: mkwak@catholic.ac.kr
DOI: https://doi.org/10.1038/s12276-024-01180-8
PMID: https://pubmed.ncbi.nlm.nih.gov/38424190
Publication Date: 2024-03-01
Hypoxia, oxidative stress, and the interplay of HIFs and NRF2 signaling in cancer
Abstract
Oxygen is crucial for life and acts as the final electron acceptor in mitochondrial energy production. Cells adapt to varying oxygen levels through intricate response systems. Hypoxia-inducible factors (HIFs), including HIF-1a and HIF-2a, orchestrate the cellular hypoxic response, activating genes to increase the oxygen supply and reduce expenditure. Under conditions of excess oxygen and resulting oxidative stress, nuclear factor erythroid 2-related factor 2 (NRF2) activates hundreds of genes for oxidant removal and adaptive cell survival. Hypoxia and oxidative stress are core hallmarks of solid tumors and activated HIFs and NRF2 play pivotal roles in tumor growth and progression. The complex interplay between hypoxia and oxidative stress within the tumor microenvironment adds another layer of intricacy to the HIF and NRF2 signaling systems. This review aimed to elucidate the dynamic changes and functions of the HIF and NRF2 signaling pathways in response to conditions of hypoxia and oxidative stress, emphasizing their implications within the tumor milieu. Additionally, this review explored the elaborate interplay between HIFs and NRF2, providing insights into the significance of these interactions for the development of novel cancer treatment strategies.
INTRODUCTION
macromolecule dysfunction and, ultimately, cell death. In response, the expression levels of genes involved in hyperoxiaderived ROS removal and damaged cell repair increase. Nuclear factor erythroid 2-related factor 2 (NRF2) is the primary transcription factor mediating this response and promotes the adaptation and survival of cells under oxidative stress conditions
ADAPTIVE RESPONSE SYSTEMS TO VARIABLE
associated with glycolytic metabolic shift and cell cycle arrest. In contrast, HIF-2a gradually becomes activated under persistent hypoxic conditions, elevating the expression levels of genes involved in erythropoiesis and tumor stemness
Cross response: hypoxia-NRF2 and oxidative stress-HIFs
following acute hypoxia in human cells
contributes to HIF upregulation
The interplay between HIFs and NRF2
| Cancer | Mechanism | Phenotypes | Ref |
| Breast cancer | Single N-glycan deletion (N418Q) of ErbB3 protein (HER3) attenuates heregulin-
|
Cell growth, migration | 101 |
| NRF2 knockdown decreases HIF-1
|
Proliferation | 98 | |
| NRF2 binds to the ARE at 30 kilobases upstream of HIF1A locus. | 57 | ||
| Overexpression of NRF2 promotes the expression of key enzymes in PPP, including G6PD and TKT, and subsequent HIF
|
Proliferation, migration | 99 | |
| NRF2 knockdown increases miR-181c levels to inhibit HIF-1
|
Metabolic reprogramming, autophagy | 65 | |
| Colorectal cancer | NRF2 knockdown reduces cellular and mitochondrial
|
Tumor growth, angiogenesis | 45 |
| NRF2, HIF, and NF-кВ are enriched in the core of growing spheroids. | CSC, stress resistance | 165 | |
| NRF2 knockdown increases miR-181a-2-3p levels and inhibits HIF-
|
Tumor growth, CSC properties | 66 | |
| Endometrial cancer | miR-148b decreases NRF2 and HIF-1
|
Proliferation | 192 |
| Esophageal squamous carcinoma | NRF2 shRNA blockade HIF-1
|
Migration, invasion | 53 |
| Gastric cancer | Hypoxia or VEGFA increases the expression of VEGFR2 to facilitate NRF2 nuclear translocation, while knockdown of NRF2 inhibited HIF-1
|
Survival, invasion | 100 |
| TRPM2 silencing destabilizes NRF2 and HIF-1
|
Ferroptosis | 184 | |
| Glioblastoma | NRF2 inhibition suppresses mitochondrial
|
Proliferation, tumor growth, angiogenesis | 52 |
| Oxidative stress-induced NRF2 transactivation activates NIX and increases the level of HIF-
|
CSC, survival, tumor growth | 63 | |
| Hypoxia ( 12 h ) induces HIF-1
|
Proliferation, CSC, EMT, drug resistance, tumor growth, angiogenesis | 164 | |
| Hepatocellular carcinoma | NRF2 regulates the transcript level of HIF1A via ARE consensus on its promoter under mild hypoxic (
|
Cisplatin resistance | 56 |
| NRF2 inhibition reduces HIF-1
|
5-FU resistance, cell viability, migration, invasion, tumor growth | 112 | |
| NRF2 binds to the oxygen-dependent degradation (ODD) domain of HIF-1
|
Tumor growth | 55 | |
| Renal carcinoma | FH-deficient cells accumulate fumarate to stabilize ROSindependent NRF2 and ROS-dependent HIF-
|
Proliferation | 67 |
| Lung adenocarcinoma | Intermittent hypoxia enhances NOX1-mediated ROS production to activate NRF2 by protein stabilization and subsequent Trx1, which increases HIF-1
|
Survival | 60 |
| Lung carcinogenesis | iAs induces NRF2-dependent HIF-1
|
CSC, metabolic reprogramming | 51 |
| Ovarian cancer | Increases in ROS by follicle-stimulating hormone (FSH) activate NRF2, which further enhances HIF-1
|
Angiogenesis | 54 |
| Pancreatic and lung cancer | Depletion of NRF2 impairs HIF-1
|
Proliferation | 193 |
Positive interplay
target genes (Table 2). Consistent with these findings, HIF-1a and NRF2 expression levels are mutually associated, and high levels of these transcription factors correlate with poor outcomes in glioblastoma patients
Cancer proliferation and survival
| Cancer phenotypes | NRF2 target genes | HIFs target genes |
| Proliferation/Survival/Metabolic shift | NOTCH1
|
NOTCH1
|
| Therapy resistance | BCL-2, ABCC1, ABCG2, ABCC2, ABCC3
|
BCL-2
|
| Angiogenesis | VEGF
|
VEGF, SDF1, SCF, PGF, ANGPT2
|
| EMT/Metastases | NOTCH1
|
NOTCH1
|
| Cancer stem cells | OCT4, NANOG
|
OCT4
|
| Ferroptosis | SLC7A11, GCLM, HMOX1, GCLC, FPN, FTL, FTH1, TXNRD1, GPX4, NQO1
|
SLC7A11, GCLM
|
dehydrogenase A and PDK1
high NRF2 expression levels were positively correlated with HIF-1a and HO-1 expression
Therapeutic resistance
Tumor angiogenesis
levels upon exposure to hypoxic stimuli
brains of venous hypertensive rats, and Nrf2 knockout suppressed vascular tube formation in primary brain microvascular endothelial cells
EMT and cancer metastasis
Cancer stem cell traits
thought to cause cancer treatment failure, as they drive malignant properties, including metastasis, invasion, therapeutic resistance, and immune escape
Resistance to ferroptosis
as a negative regulator of ferroptosis
Concluding remarks
hypoxic and oxidative stress, which is likely inevitable under lowoxygen conditions. In contrast, changes in NRF2 under hypoxic conditions vary in a context-dependent manner, indicating that hypoxia-associated oxidative stress does not necessarily accompany NRF2 activation. This relationship suggests that inhibiting NRF2 in tumors with both HIF and NRF2 upregulation could be a more efficient strategy considering the response of HIFs to hypoxia and oxidative stress. For instance, treatment with brusatol, an inhibitor of NRF2 protein synthesis, prevents hypoxia-induced HIF-1a accumulation and reduces glucose consumption in colorectal cancer cells
REFERENCES
- Raymond, J. & Segrè, D. The effect of oxygen on biochemical networks and the evolution of complex life. Science 311, 1764-1767 (2006).
- Wicks, E. E. & Semenza, G. L. Hypoxia-inducible factors: cancer progression and clinical translation. J. Clin. Investig. 132, e159839 (2022).
- Halliwell, B. Biochemistry of oxidative stress. Biochem Soc. Trans. 35, 1147-1150 (2007).
- Semenza, G. L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 29, 625-634 (2010).
- Yamamoto, M., Kensler, T. W. & Motohashi, H. The KEAP1-NRF2 system: a thiolbased sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 98, 1169-1203 (2018).
- O’Malley, J., Kumar, R., Inigo, J., Yadava, N. & Chandra, D. Mitochondrial stress response and cancer. Trends Cancer 6, 688-701 (2020).
- Vaupel, P., Höckel, M. & Mayer, A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid. Redox Signal 9, 1221-1235 (2007).
- Nakamura, H. & Takada, K. Reactive oxygen species in cancer: current findings and future directions. Cancer Sci. 112, 3945-3952 (2021).
- Rojo de la Vega, M., Chapman, E. & Zhang, D. D. NRF2 and the hallmarks of cancer. Cancer Cell 34, 21-43 (2018).
- Hermes-Lima, M. et al. Preparation for oxidative stress under hypoxia and metabolic depression: revisiting the proposal two decades later. Free Radic. Biol. Med. 89, 1122-1143 (2015).
- Lee, G. et al. Oxidative dimerization of PHD2 is responsible for its inactivation and contributes to metabolic reprogramming via HIF-1a activation. Sci. Rep. 6, 18928 (2016).
- Fukuda, R. et al. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 129, 111-122 (2007).
- Semenza, G. L. The genomics and genetics of oxygen homeostasis. Annu. Rev. Genomics Hum. Genet. 21, 183-204 (2020).
- Wang, G. L., Jiang, B. H., Rue, E. A. & Semenza, G. L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O 2 tension. Proc. Natl Acad. Sci. USA 92, 5510-5514 (1995).
- Schofield, C. J. & Ratcliffe, P. J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 5, 343-354 (2004).
- Bruick, R. K. & McKnight, S. L. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 294, 1337-1340 (2001).
- Appelhoff, R. J. et al. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor*. J. Biol. Chem. 279, 38458-38465 (2004).
- Mahon, P. C., Hirota, K. & Semenza, G. L. FIH-1: a novel protein that interacts with HIF-1a and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 15, 2675-2686 (2001).
- Cowman, S. J. & Koh, M. Y. Revisiting the HIF switch in the tumor and its immune microenvironment. Trends Cancer 8, 28-42 (2022).
- Holmquist-Mengelbier, L. et al. Recruitment of HIF-1a and HIF-2a to common target genes is differentially regulated in neuroblastoma: HIF-2a promotes an aggressive phenotype. Cancer Cell 10, 413-423 (2006).
- Koh, M. Y. & Powis, G. Passing the baton: the HIF switch. Trends Biochem. Sci. 37, 364-372 (2012).
- Manalo, D. J. et al. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 105, 659-669 (2005).
- Semenza, G. L. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharm. Sci. 33, 207-214 (2012).
- Covello, K. L. et al. HIF-2a regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 20, 557-570 (2006).
- Sowter, H. M., Ratcliffe, P. J., Watson, P., Greenberg, A. H. & Harris, A. L. HIF-1dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 61, 6669-6673 (2001).
- Dodson, M. et al. Modulating NRF2 in disease: timing is everything. Annu. Rev. Pharm. Toxicol. 59, 555-575 (2019).
- Torrente, L. & DeNicola, G. M. Targeting NRF2 and its downstream processes: opportunities and challenges. Annu. Rev. Pharm. Toxicol. 62, 279-300 (2022).
- Ho, Y. S., Dey, M. S. & Crapo, J. D. Antioxidant enzyme expression in rat lungs during hyperoxia. Am. J. Physiol. 270, L810-L818 (1996).
- Cho, H. Y., Reddy, S. P., Debiase, A., Yamamoto, M. & Kleeberger, S. R. Gene expression profiling of NRF2-mediated protection against oxidative injury. Free Radic. Biol. Med. 38, 325-343 (2005).
- Papaiahgari, S., Zhang, Q., Kleeberger, S. R., Cho, H. Y. & Reddy, S. P. Hyperoxia stimulates an Nrf2-ARE transcriptional response via ROS-EGFR-PI3K-Akt/ERK MAP kinase signaling in pulmonary epithelial cells. Antioxid. Redox Signal. 8, 43-52 (2006).
- Taguchi, K. & Yamamoto, M. The KEAP1-NRF2 system in cancer. Front. Oncol. 7, 85 (2017).
- Rada, P. et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase
-TrCP axis. Mol. Cell Biol. 32, 3486-3499 (2012). - Suzuki, T., Takahashi, J. & Yamamoto, M. Molecular basis of the KEAP1-NRF2 signaling pathway. Mol. Cells 46, 133-141 (2023).
- Iso, T., Suzuki, T., Baird, L. & Yamamoto, M. Absolute amounts and status of the Nrf2-Keap1-Cul3 complex within cells. Mol. Cell Biol. 36, 3100-3112 (2016).
- Baird, L. & Yamamoto, M. The molecular mechanisms regulating the KEAP1NRF2 pathway. Mol. Cell Biol. 40, e00099-20 (2020).
- Horie, Y. et al. Molecular basis for the disruption of Keap1-Nrf2 interaction via Hinge & Latch mechanism. Commun. Biol. 4, 576 (2021).
- Hayes, J. D. & Dinkova-Kostova, A. T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 39, 199-218 (2014).
- Hernansanz-Agustín, P. et al. Acute hypoxia produces a superoxide burst in cells. Free Radic. Biol. Med. 71, 146-156 (2014).
- Chandel, N. S. et al. Mitochondrial reactive oxygen species trigger hypoxiainduced transcription. Proc. Natl Acad. Sci. USA 95, 11715-11720 (1998).
- Guzy, R. D. et al. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 1, 401-408 (2005).
- Toth, R. K. & Warfel, N. A. Strange bedfellows: nuclear factor, erythroid 2-like 2 (Nrf2) and hypoxia-inducible factor 1 (HIF-1) in tumor hypoxia. Antioxidants 6, 27 (2017).
- Tello, D. et al. Induction of the mitochondrial NDUFA4L2 protein by HIF-1a decreases oxygen consumption by inhibiting Complex I activity. Cell Metab. 14, 768-779 (2011).
- Xin, X., Li, Y. & Liu, H. Hesperidin ameliorates hypobaric hypoxia-induced retinal impairment through activation of Nrf2/HO-1 pathway and inhibition of apoptosis. Sci. Rep. 10, 19426 (2020).
- Potteti, H. R. et al. Nrf2-AKT interactions regulate heme oxygenase 1 expression in kidney epithelia during hypoxia and hypoxia-reoxygenation. Am. J. Physiol. Ren. Physiol. 311, F1025-f1034 (2016).
- Kim, T.-H. et al. NRF2 blockade suppresses colon tumor angiogenesis by inhibiting hypoxia-induced activation of HIF-1a. Cancer Res. 71, 2260-2275 (2011).
- Bondi, C. D. et al. Suppression of NRF2 Activity by HIF-1
promotes fibrosis after ischemic acute kidney injury. Antioxidants 11, 1810 (2022). - Loboda, A. et al. HIF-1 induction attenuates Nrf2-dependent IL-8 expression in human endothelial cells. Antioxid. Redox Signal. 11, 1501-1517 (2009).
- Pan, Y. et al. Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol. Cell Biol. 27, 912-925 (2007).
- Watanabe, Y. et al. Glutathione adducts induced by ischemia and deletion of glutaredoxin-1 stabilize HIF-1a and improve limb revascularization. Proc. Natl Acad. Sci. USA 113, 6011-6016 (2016).
- Hamanaka, R. B., Weinberg, S. E., Reczek, C. R. & Chandel, N. S. The mitochondrial respiratory chain is required for organismal adaptation to hypoxia. Cell Rep. 15, 451-459 (2016).
- Bi, Z. et al. Nrf2 and HIF1a converge to arsenic-induced metabolic reprogramming and the formation of the cancer stem-like cells. Theranostics 10, 4134-4149 (2020).
- Ji, X. et al. Knockdown of Nrf2 suppresses glioblastoma angiogenesis by inhibiting hypoxia-induced activation of HIF-1a. Int. J. Cancer 135, 574-584 (2014).
- Shen, H. et al. Blockage of Nrf2 suppresses the migration and invasion of esophageal squamous cell carcinoma cells in hypoxic microenvironment. Dis. Esophagus 27, 685-692 (2014).
- Zhang, Z. et al. Reactive oxygen species regulate FSH-induced expression of vascular endothelial growth factor via Nrf2 and HIF1a signaling in human epithelial ovarian cancer. Oncol. Rep. 29, 1429-1434 (2013).
- Zheng, J. et al. Overactivated NRF2 induces pseudohypoxia in hepatocellular carcinoma by stabilizing HIF-1alpha. Free Radic. Biol. Med. 194, 347-356 (2023).
- Jin, X., Gong, L., Peng, Y., Li, L. & Liu, G. Enhancer-bound Nrf2 licenses HIF-1alpha transcription under hypoxia to promote cisplatin resistance in hepatocellular carcinoma cells. Aging (Albany NY) 13, 364-375 (2020).
- Lacher, S. E., Levings, D. C., Freeman, S. & Slattery, M. Identification of a functional antioxidant response element at the HIF1A locus. Redox. Biology 19, 401-411 (2018).
- Oh, E. T. et al. NQO1 inhibits proteasome-mediated degradation of HIF-1a. Nat. Commun. 7, 13593 (2016).
- Csiki, I. et al. Thioredoxin-1 modulates transcription of cyclooxygenase-2 via hypoxia-inducible factor-1alpha in non-small cell lung cancer. Cancer Res. 66, 143-150 (2006).
- Malec, V. et al. HIF-1 alpha signaling is augmented during intermittent hypoxia by induction of the Nrf2 pathway in NOX1-expressing adenocarcinoma A549 cells. Free Radic. Biol. Med. 48, 1626-1635 (2010).
- Chin, B. Y. et al. Hypoxia-inducible factor 1alpha stabilization by carbon monoxide results in cytoprotective preconditioning. Proc. Natl Acad. Sci. USA 104, 5109-5114 (2007).
- Choi, Y. K. et al. Carbon monoxide promotes VEGF expression by increasing HIF1alpha protein level via two distinct mechanisms, translational activation and stabilization of HIF-1alpha protein. J. Biol. Chem. 285, 32116-32125 (2010).
- Jung, J. et al. Mitochondrial NIX promotes tumor survival in the hypoxic niche of glioblastoma. Cancer Res. 79, 5218-5232 (2019).
- Jung, K. A., Lee, S. & Kwak, M. K. NFE2L2/NRF2 activity is linked to mitochondria and AMP-activated protein kinase signaling in cancers through miR-181c/ mitochondria-encoded cytochrome c oxidase regulation. Antioxid. Redox Signal 27, 945-961 (2017).
- Lee, S., Hallis, S. P., Jung, K.-A., Ryu, D. & Kwak, M.-K. Impairment of HIF-1amediated metabolic adaption by NRF2-silencing in breast cancer cells. Redox Biol. 24, 101210 (2019).
- Hallis, S. P., Kim, S. K., Lee, J.-H. & Kwak, M.-K. Association of NRF2 with HIF-2ainduced cancer stem cell phenotypes in chronic hypoxic condition. Redox Biol. 60, 102632 (2023).
- Sullivan, L. B. et al. The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol. Cell 51, 236-248 (2013).
- Early, J. O. et al. Circadian clock protein BMAL1 regulates IL-1
in macrophages via NRF2. Proc. Natl Acad. Sci. USA 115, E8460-E8468 (2018). - Di Gregorio, J. et al. UBXN7 cofactor of CRL3KEAP1 and CRL2VHL ubiquitin ligase complexes mediates reciprocal regulation of NRF2 and HIF-1a proteins. Biochim. Biophys. Acta 1868, 118963 (2021).
- Lu, C. et al. Nrf2 activation is required for ligustrazine to inhibit hepatic steatosis in alcohol-preferring mice and hepatocytes. Toxicol. Sci. 155, 432-443 (2017).
- Li, Y. et al. Inhibitor of apoptosis-stimulating protein of p53 inhibits ferroptosis and alleviates intestinal ischemia/reperfusion-induced acute lung injury. Cell Death Differ. 27, 2635-2650 (2020).
- Lin, H. C. et al. Andrographolide inhibits hypoxia-induced HIF-1a-driven endothelin 1 secretion by activating Nrf2/HO-1 and promoting the expression of prolyl hydroxylases
in human endothelial cells. Environ. Toxicol. 32, 918-930 (2017). - Talks, K. L. et al. The expression and distribution of the hypoxia-inducible factors HIF-1a and HIF-2a in normal human tissues, cancers, and tumor-associated macrophages. Am. J. Pathol. 157, 411-421 (2000).
- Schito, L. & Semenza, G. L. Hypoxia-inducible factors: master regulators of cancer progression. Trends Cancer 2, 758-770 (2016).
- Krieg, M. et al. Up-regulation of hypoxia-inducible factors HIF-1alpha and HIF2alpha under normoxic conditions in renal carcinoma cells by von HippelLindau tumor suppressor gene loss of function. Oncogene 19, 5435-5443 (2000).
- Ravi, R. et al. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1a. Genes Dev. 14, 34-44 (2000).
- Zhong, H. et al. Modulation of hypoxia-inducible factor 1a expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics1. Cancer Res. 60, 1541-1545 (2000).
- Isaacs, J. S. et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: Novel role of fumarate in regulation of HIF stability. Cancer Cell 8, 143-153 (2005).
- Greco, S., Gaetano, C. & Martelli, F. HypoxamiR regulation and function in ischemic cardiovascular diseases. Antioxid. Redox Signal. 21, 1202-1219 (2014).
- Menegon, S., Columbano, A. & Giordano, S. The dual roles of NRF2 in cancer. Trends Mol. Med. 22, 578-593 (2016).
- Robertson, H., Dinkova-Kostova, A. T. & Hayes, J. D. NRF2 and the Ambiguous Consequences of Its Activation during Initiation and the Subsequent Stages of Tumourigenesis. Cancers (Basel) 12, 3609 (2020).
- Kitamura, H. & Motohashi, H. NRF2 addiction in cancer cells. Cancer Sci. 109, 900-911 (2018).
- Guichard, C. et al. Integrated analysis of somatic mutations and focal copynumber changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 44, 694-698 (2012).
- Network, C.G.A.R. Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543-550 (2014).
- Shibata, T. et al. Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology 135, 1358-1368 (2008).
- Muscarella, L. A. et al. Frequent epigenetics inactivation of KEAP1 gene in nonsmall cell lung cancer. Epigenetics 6, 710-719 (2011).
- DeNicola, G. M. et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106-109 (2011).
- Komatsu, M. et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 12, 213-223 (2010).
- Adam, J. et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 20, 524-537 (2011).
- Ji, X. et al. Correlation of Nrf2 and HIF-1alpha in glioblastoma and their relationships to clinicopathologic features and survival. Neurol. Res. 35, 1044-1050 (2013).
- Papandreou, I., Cairns, R. A., Fontana, L., Lim, A. L. & Denko, N. C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 3, 187-197 (2006).
- Semenza, G. L., Roth, P. H., Fang, H. M. & Wang, G. L. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 269, 23757-23763 (1994).
- Sakagami, H. et al. Loss of HIF-1a impairs GLUT4 translocation and glucose uptake by the skeletal muscle cells. Am. J. Physiol. Endocrinol. Metab. 306, E1065-E1076 (2014).
- Bellot, G. et al. Hypoxia-induced autophagy is mediated through hypoxiainducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell Biol. 29, 2570-2581 (2009).
- Chee, N. T., Lohse, I. & Brothers, S. P. mRNA-to-protein translation in hypoxia. Mol. Cancer 18, 49 (2019).
- Singh, A., Bodas, M., Wakabayashi, N., Bunz, F. & Biswal, S. Gain of Nrf2 function in non-small-cell lung cancer cells confers radioresistance. Antioxid. Redox Signal 13, 1627-1637 (2010).
- Malhotra, D. et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 38, 5718-5734 (2010).
- Zhang, H. S. et al. NRF2 facilitates breast cancer cell growth via HIF1a-mediated metabolic reprogramming. Int. J. Biochem. Cell Biol. 95, 85-92 (2018).
- Zhang, H. S. et al. Nrf2 promotes breast cancer cell migration via up-regulation of G6PD/HIF-1alpha/Notch1 axis. J. Cell Mol. Med. 23, 3451-3463 (2019).
- Yang, Y. et al. Abnormal phenotype of Nrf2 is associated with poor prognosis through hypoxic/VEGF-A-Rap1b/VEGFR2 pathway in gastric cancer. Aging (Albany NY) 14, 3293-3312 (2022).
- Takamiya, R. et al. The single N-glycan deletion mutant of soluble ErbB3 protein attenuates heregulin beta1-induced tumor progression by blocking of the HIF-1 and Nrf2 pathway. Biochem. Biophys. Res. Commun. 454, 364-368 (2014).
- Kazi, A. A. et al. Nonhypoxic regulation and role of hypoxia-inducible factor 1 in aromatase inhibitor resistant breast cancer. Breast Cancer Res. 16, R15 (2014).
- Liu, L. et al. Hypoxia-inducible factor-1 alpha contributes to hypoxia-induced chemoresistance in gastric cancer. Cancer Sci. 99, 121-128 (2008).
- Chen, N. et al. BCL-xL is a target gene regulated by hypoxia-inducible factor1alpha. J. Biol. Chem. 284, 10004-10012 (2009).
- Erler, J. T. et al. Hypoxia-mediated down-regulation of Bid and Bax in tumors occurs via hypoxia-inducible factor 1-dependent and -independent mechanisms and contributes to drug resistance. Mol. Cell Biol. 24, 2875-2889 (2004).
- Yang, T., Xu, F., Sheng, Y., Zhang, W. & Chen, Y. A targeted proteomics approach to the quantitative analysis of ERK/Bcl-2-mediated anti-apoptosis and multidrug resistance in breast cancer. Anal. Bioanal. Chem. 408, 7491-7503 (2016).
- Choi, B.-h & Kwak, M.-K. Shadows of NRF2 in cancer: resistance to chemotherapy. Curr. Opin. Toxicol. 1, 20-28 (2016).
- Homma, S. et al. Nrf2 enhances cell proliferation and resistance to anticancer drugs in human lung cancer. Clin. Cancer Res. 15, 3423-3432 (2009).
- Kurz, E. U., Cole, S. P. C. & Deeley, R. G. Identification of DNA-protein interactions in the
Flanking and Untranslated Regions of the Human Multidrug Resistance Protein (MRP1) gene: evaluation of a putative antioxidant response ele-ment/AP-1 binding site. Biochem. Biophys. Res. Commun. 285, 981-990 (2001). - Singh, A. et al. Expression of ABCG2 (BCRP) is regulated by Nrf2 in cancer cells that confers side population and chemoresistance phenotype. Mol. Cancer Ther. 9, 2365-2376 (2010).
- Niture, S. K. & Jaiswal, A. K. Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. J. Biol. Chem. 287, 9873-9886 (2012).
- Duan, X. et al. Nrf2-siRNA enhanced the anti-tumor effects of
in 5-fluorouracil-resistant hepatocellular carcinoma by inhibiting HIF-1alpha/ HSP70 signaling. J. Hepatocell. Carcinoma 9, 1341-1352 (2022). - Hanahan, D. & Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 86, 353-364 (1996).
- Forsythe, J. A. et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 16, 4604-4613 (1996).
- Ceradini, D. J. et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 10, 858-864 (2004).
- Gao, C. et al. SCF, regulated by HIF-1a, promotes pancreatic ductal adenocarcinoma cell progression. PLoS ONE 10, e0121338 (2015).
- Simon, M.-P., Tournaire, R. & Pouyssegur, J. The angiopoietin-2 gene of endothelial cells is up-regulated in hypoxia by a HIF binding site located in its first intron and by the central factors GATA-2 and Ets-1. J. Cell. Physiol. 217, 809-818 (2008).
- Florczyk, U. et al. Nrf2 regulates angiogenesis: effect on endothelial cells, bone marrow-derived proangiogenic cells and hind limb ischemia. Antioxid. Redox Signal. 20, 1693-1708 (2013).
- Ichihara, S. et al. Ablation of the transcription factor Nrf2 promotes ischemiainduced neovascularization by enhancing the inflammatory response. Arterioscler. Thromb. Vasc. Biol. 30, 1553-1561 (2010).
- Wei, Y. et al. Nrf2 acts cell-autonomously in endothelium to regulate tip cell formation and vascular branching. Proc. Natl Acad. Sci. USA 110, E3910-E3918 (2013).
- Li, L. et al. Interplay between VEGF and Nrf2 regulates angiogenesis due to intracranial venous hypertension. Sci. Rep. 6, 37338 (2016).
- Feng, R. et al. Nrf2 activation drive macrophages polarization and cancer cell epithelial-mesenchymal transition during interaction. Cell Commun. Signal 16, 54 (2018).
- Shao, S. et al. Curcumin suppresses hepatic stellate cell-induced hepatocarcinoma angiogenesis and invasion through downregulating CTGF. Oxid. Med. Cell. Longev. 2019, 8148510 (2019).
- Hapke, R. Y. & Haake, S. M. Hypoxia-induced epithelial to mesenchymal transition in cancer. Cancer Lett. 487, 10-20 (2020).
- Zhong, H. et al. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 59, 5830-5835 (1999).
- Xu, X. et al. Snail is a direct target of hypoxia-inducible factor 1a (HIF1a) in hypoxia-induced endothelial to mesenchymal transition of human coronary endothelial cells*. J. Biol. Chem. 290, 16653-16664 (2015).
- Yang, M.-H. et al. Direct regulation of TWIST by HIF-1 a promotes metastasis. Nat. Cell Biol. 10, 295-305 (2008).
- Zhang, W. et al. HIF-1a promotes epithelial-mesenchymal transition and metastasis through direct regulation of ZEB1 in colorectal cancer. PLoS ONE 10, e0129603 (2015).
- Wang, R. et al. Hypoxia-inducible factor-dependent ADAM12 expression mediates breast cancer invasion and metastasis. Proc. Natl Acad. Sci. USA 118, e2020490118 (2021).
- Wang, H. et al. NRF2 activation by antioxidant antidiabetic agents accelerates tumor metastasis. Sci. Transl. Med. 8, 334ra351 (2016).
- Zhang, C. et al. NRF2 promotes breast cancer cell proliferation and metastasis by increasing RhoA/ROCK pathway signal transduction. Oncotarget 7, 73593-73606 (2016).
- Jeong, Y. et al. Role of KEAP1/NRF2 and TP53 mutations in lung squamous cell carcinoma development and radiation resistance. Cancer Discov. 7, 86-101 (2017).
- Yazaki, K. et al. ROS-Nrf2 pathway mediates the development of TGF-
induced epithelial-mesenchymal transition through the activation of Notch signaling. Eur. J. Cell Biol. 100, 151181 (2021). - Vilchez Mercedes, S. A. et al. Nrf2 modulates the hybrid epithelial/mesenchymal phenotype and notch signaling during collective cancer migration. Front. Mol. Biosci. 9, 807324 (2022).
- Rachakonda, G. et al. Increased cell migration and plasticity in Nrf2-deficient cancer cell lines. Oncogene 29, 3703-3714 (2010).
- Ryu, D., Lee, J.-H. & Kwak, M.-K. NRF2 level is negatively correlated with TGF-
induced lung cancer motility and migration via NOX4-ROS signaling. Arch. Pharmacal Res. 43, 1297-1310 (2020). - Batlle, E. & Clevers, H. Cancer stem cells revisited. Nat. Med. 23, 1124-1134 (2017).
- Yang, L. et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 5, 8 (2020).
- Wright, M. H. et al. Brca1 breast tumors contain distinct CD44+/CD24- and CD133+cells with cancer stem cell characteristics. Breast Cancer Res. 10, R10 (2008).
- Dalerba, P. et al. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl Acad. Sci. USA 104, 10158-10163 (2007).
- Heddleston, J. M. et al. Hypoxia inducible factors in cancer stem cells. Br. J. Cancer 102, 789-795 (2010).
- Samanta, D., Gilkes, D. M., Chaturvedi, P., Xiang, L. & Semenza, G. L. Hypoxiainducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc. Natl Acad. Sci. USA 111, E5429-E5438 (2014).
- Conley, S. J. et al. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc. Natl Acad. Sci. USA 109, 2784-2789 (2012).
- Keith, B. & Simon, M. C. Hypoxia-inducible factors, stem cells, and cancer. Cell 129, 465-472 (2007).
- Covello, K. L. et al. HIF-2alpha regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 20, 557-570 (2006).
- Lu, H. et al. Chemotherapy-induced S100A10 recruits KDM6A to facilitate OCT4mediated breast cancer stemness. J. Clin. Investig. 130, 4607-4623 (2020).
- Qin, J. et al. Hypoxia-inducible factor 1 alpha promotes cancer stem cells-like properties in human ovarian cancer cells by upregulating SIRT1 expression. Sci. Rep. 7, 10592 (2017).
- Yan, Y. et al. HIF-2a promotes conversion to a stem cell phenotype and induces chemoresistance in breast cancer cells by activating Wnt and Notch pathways. J. Exp. Clin. Cancer Res. 37, 256 (2018).
- Yan, Y. et al. A novel HIF-2a targeted inhibitor suppresses hypoxia-induced breast cancer stemness via SOD2-mtROS-PDI/GPR78-UPR(ER) axis. Cell Death Differ. 29, 1769-1789 (2022).
- Micucci, C., Matacchione, G., Valli, D., Orciari, S. & Catalano, A. HIF2a is involved in the expansion of CXCR4-positive cancer stem-like cells in renal cell carcinoma. Br. J. Cancer 113, 1178-1185 (2015).
- Diehn, M. et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 458, 780-783 (2009).
- Hallis, S. P., Kim, J. M. & Kwak, M.-K. Emerging Role of NRF2 Signaling in Cancer Stem Cell Phenotype. Mol. Cells 46, 153-164 (2023).
- Chang, C.-W. et al. ROS-independent ER stress-mediated NRF2 activation promotes warburg effect to maintain stemness-associated properties of cancerinitiating cells. Cell Death Dis. 9, 194 (2018).
- Chang, C. W. et al. Distinct subpopulations of head and neck cancer cells with different levels of intracellular reactive oxygen species exhibit diverse stemness, proliferation, and chemosensitivity. Cancer Res. 74, 6291-6305 (2014).
- Gao, L. et al. Nrf2 signaling promotes cancer stemness, migration, and expression of ABC transporter genes in sorafenib-resistant hepatocellular carcinoma cells. PLoS ONE 16, e0256755 (2021).
- Zhu, J. et al. Nrf2 is required to maintain the self-renewal of glioma stem cells. BMC Cancer 13, 380 (2013).
- Ryoo, I.-g, Choi, B.-h, Ku, S.-K. & Kwak, M.-K. High CD44 expression mediates p62associated NFE2L2/NRF2 activation in breast cancer stem cell-like cells: implications for cancer stem cell resistance. Redox Biol. 17, 246-258 (2018).
- Park, J., Kim, S. K., Hallis, S. P., Choi, B.-H. & Kwak, M.-K. Role of CD133/NRF2 axis in the development of colon cancer stem cell-like properties. Front. Oncol. 11, 808300 (2022).
- Kim, D., Choi, B. H., Ryoo, I. G. & Kwak, M. K. High NRF2 level mediates cancer stem cell-like properties of aldehyde dehydrogenase (ALDH)-high ovarian cancer cells: inhibitory role of all-trans retinoic acid in ALDH/NRF2 signaling. Cell Death Dis. 9, 896 (2018).
- Noman, A. S. M. et al. Chemotherapeutic resistance of head and neck squamous cell carcinoma is mediated by EpCAM induction driven by IL-6/p62 associated Nrf2-antioxidant pathway activation. Cell Death Dis. 11, 663 (2020).
- Okazaki, K. et al. Enhancer remodeling promotes tumor-initiating activity in NRF2-activated non-small cell lung cancers. Nat. Commun. 11, 5911 (2020).
- Kim, D. H. et al. Nuclear factor erythroid-derived 2 -like 2 -induced reductive stress favors self-renewal of breast cancer stem-like cells via the FoxO3a-Bmi-1 axis. Antioxid. Redox Signal. 32, 1313-1329 (2020).
- Fragoulis, A. et al. Nrf2 induces malignant transformation of hepatic progenitor cells by inducing
-catenin expression. Redox Biol. 57, 102453 (2022). - Zhang, G., Tao, X., Ji, B. & Gong, J. Hypoxia-driven M2-polarized macrophages facilitate cancer aggressiveness and temozolomide resistance in glioblastoma. Oxid. Med. Cell. Longev. 2022, 1614336 (2022).
- Kipp, A. P., Deubel, S., Arnér, E. S. J. & Johansson, K. Time- and cell-resolved dynamics of redox-sensitive Nrf2, HIF and NF-кB activities in 3D spheroids enriched for cancer stem cells. Redox Biol. 12, 403-409 (2017).
- Dixon, ScottJ. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060-1072 (2012).
- Li, J. et al. Ferroptosis: past, present and future. Cell Death Dis. 11, 88 (2020).
- Yang, W. S. & Stockwell, B. R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 15, 234-245 (2008).
- Friedmann Angeli, J. P. et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 16, 1180-1191 (2014).
- Viswanathan, V. S. et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453-457 (2017).
- Fuhrmann, D. C., Mondorf, A., Beifuß, J., Jung, M. & Brüne, B. Hypoxia inhibits ferritinophagy, increases mitochondrial ferritin, and protects from ferroptosis. Redox Biol. 36, 101670 (2020).
- Miess, H. et al. The glutathione redox system is essential to prevent ferroptosis caused by impaired lipid metabolism in clear cell renal cell carcinoma. Oncogene 37, 5435-5450 (2018).
- Green, Y. S. et al. ISCA2 inhibition decreases HIF and induces ferroptosis in clear cell renal carcinoma. Oncogene 41, 4709-4723 (2022).
- Yang, M. et al. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci. Adv. 5, eaaw2238 (2019).
- Singhal, R. et al. HIF-2a activation potentiates oxidative cell death in colorectal cancers by increasing cellular iron. J. Clin. Invest. 131, e143691 (2021).
- Su, X. et al. HIF-a activation by the prolyl hydroxylase inhibitor roxadustat suppresses chemoresistant glioblastoma growth by inducing ferroptosis. Cell Death Dis. 13, 861 (2022).
- Dodson, M., Castro-Portuguez, R. & Zhang, D. D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 23, 101107 (2019).
- Fan, Z. et al. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis 6, e371-e371 (2017).
- Shin, D., Kim, E. H., Lee, J. & Roh, J.-L. Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radic. Biol. Med. 129, 454-462 (2018).
- Yang, J. et al. Cetuximab promotes RSL3-induced ferroptosis by suppressing the Nrf2/HO-1 signalling pathway in KRAS mutant colorectal cancer. Cell Death Dis. 12, 1079 (2021).
- Koppula, P. et al. A targetable CoQ-FSP1 axis drives ferroptosis- and radiationresistance in KEAP1 inactive lung cancers. Nat. Commun. 13, 2206 (2022).
- Anandhan, A. et al. NRF2 controls iron homeostasis and ferroptosis through HERC2 and VAMP8. Sci. Adv. 9, eade9585 (2023).
- Chen, L. D. et al. Nrf2 plays protective role during intermittent hypoxia-induced ferroptosis in rat liver (BRL-3A) cells. Sleep. Breath. 27, 2069-2076 (2023).
- Li, D. et al. Silencing TRPM2 enhanced erastin- and RSL3-induced ferroptosis in gastric cancer cells through destabilizing HIF-1a and Nrf2 proteins. Cytotechnology 74, 559-577 (2022).
- Kikuchi, H., Pino, M. S., Zeng, M., Shirasawa, S. & Chung, D. C. Oncogenic KRAS and BRAF differentially regulate hypoxia-inducible factor-1alpha and -2alpha in colon cancer. Cancer Res. 69, 8499-8506 (2009).
- Cahuzac, K. M. et al. AKT activation because of PTEN loss upregulates xCT via GSK3
NRF2, leading to inhibition of ferroptosis in PTEN-mutant tumor cells. Cell Rep. 42, 112536 (2023). - Kang, H. J. et al. HER2 confers drug resistance of human breast cancer cells through activation of NRF2 by direct interaction. Sci. Rep. 4, 7201 (2014).
- Lu, Y. et al. Brusatol inhibits HIF-1 signaling pathway and suppresses glucose uptake under hypoxic conditions in HCT116 cells. Sci. Rep. 6, 39123 (2016).
- Chen, F. et al. Triptolide, a Chinese herbal extract, enhances drug sensitivity of resistant myeloid leukemia cell lines through downregulation of HIF-1alpha and Nrf2. Pharmacogenomics 14, 1305-1317 (2013).
- Liu, Y. et al. Low-dose triptolide in combination with idarubicin induces apoptosis in AML leukemic stem-like KG1a cell line by modulation of the intrinsic and extrinsic factors. Cell Death Dis. 4, e948 (2013).
- Jin, J. et al. Cardamonin inhibits breast cancer growth by repressing HIF-1alphadependent metabolic reprogramming. J. Exp. Clin. Cancer Res. 38, 377 (2019).
- Qu, J., Zhang, L., Li, L. & Su, Y. miR-148b functions as a tumor suppressor by targeting endoplasmic reticulum metallo protease 1 in human endometrial cancer cells. Oncol. Res. 27, 81-88 (2018).
- Kuper, A. et al. Overcoming hypoxia-induced resistance of pancreatic and lung tumor cells by disrupting the PERK-NRF2-HIF-axis. Cell Death Dis. 12, 82 (2021).
- Wakabayashi, N. et al. Regulation of Notch1 signaling by Nrf2: implications for tissue regeneration. Sci. Signal. 3, ra52-ra52 (2010).
- He, F., Ru, X. & Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 21, 4777 (2020).
- Gustafsson, M. V. et al. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Develop. Cell 9, 617-628 (2005).
- Lv, Y. et al. Hypoxia-inducible factor-1alpha induces multidrug resistance protein in colon cancer. Onco Targets Ther. 8, 1941-1948 (2015).
- Lee, P. J. et al. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia*. J. Biol. Chem. 272, 5375-5381 (1997).
- Jang, J. et al. Primary cilium-autophagy-Nrf2 (PAN) axis activation commits human embryonic stem cells to a neuroectoderm fate. Cell 165, 410-420 (2016).
- Petruzzelli, R., Christensen, D. R., Parry, K. L., Sanchez-Elsner, T. & Houghton, F. D. HIF2a regulates NANOG expression in human embryonic stem cells following hypoxia and reoxygenation through the interaction with an Oct-Sox cis regulatory element. PLoS ONE 9, e108309 (2014).
ACKNOWLEDGEMENTS
AUTHOR CONTRIBUTIONS
COMPETING INTERESTS
ADDITIONAL INFORMATION
Reprints and permission information is available at http://www.nature.com/reprints
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
© The Author(s) 2024
Integrated Research Institute for Pharmaceutical Sciences, The Catholic University of Korea, Bucheon, Gyeonggi-do 14662, Republic of Korea. Department of Pharmacy, Graduate School of The Catholic University of Korea, Bucheon, Gyeonggi-do 14662, Republic of Korea. College of Pharmacy, The Catholic University of Korea, Bucheon, Gyeonggi-do 14662, Republic of Korea. These authors contributed equally: Taegeun Bae, Steffanus Pranoto Hallis. email: mkwak@catholic.ac.kr