نقص الأكسجين يحفز تعديل بروتين الميتوكوندريا باللاكتيل للحد من الفسفرة التأكسدية Hypoxia induces mitochondrial protein lactylation to limit oxidative phosphorylation
تستهلك الفسفرة التأكسدية (OXPHOS) الأكسجين لإنتاج ATP. ومع ذلك، لا يزال الآلية التي توازن بين نشاط OXPHOS وتوافر الأكسجين داخل الخلايا غامضة. هنا، نبلغ أن بروتين الميتوكوندريا الذي يتم تعديل اللاكتيل له يتم تحفيزه بواسطة نقص الأكسجين داخل الخلايا للحد من OXPHOS. نوضح أن إنزيم الألانين-تRNA سينثيتاز (AARS2) هو إنزيم نقل اللاكتيل للبروتينات، حيث يتم تعزيز تحلل بروتينه بواسطة هيدروكسيل البرولين 377 الذي يحفزه إنزيم الهيدروكسيل PHD2 الذي يستشعر الأكسجين. يؤدي نقص الأكسجين إلى تراكم AARS2 لتعديل اللاكتيل لليسين 336 في مركب ديهيدروجيناز البيروفات (PDHA1) والليسين 457/8 في إنزيم كارنيتين بالميتويل ترانسفيراز 2 (CPT2)، مما يؤدي إلى تعطيل كلا الإنزيمين وتثبيط OXPHOS عن طريق الحد من تدفق الأسيتيل-CoA من البيروفات وأكسدة الأحماض الدهنية، على التوالي. يمكن عكس تعديل اللاكتيل لـ PDHA1 وCPT2 بواسطة SIRT3 لتنشيط OXPHOS. في خلايا عضلات الفئران، يتم تحفيز تعديل اللاكتيل بواسطة نقص الأكسجين داخل الخلايا الناتج عن أكسدة اللاكتات أثناء التمرين للحد من وقت استنفاد التحمل العالي الكثافة، والذي يمكن زيادته أو تقليله عن طريق تقليل أو زيادة مستويات تعديل اللاكتيل، على التوالي. تكشف نتائجنا أن تعديل بروتين الميتوكوندريا باللاكتيل يدمج إشارات نقص الأكسجين داخل الخلايا واللاكتات لتنظيم OXPHOS.
الفوسفوريلATION التأكسدية (OXPHOS) هي العملية الرئيسية لإنتاج ATP التي تتطلب الأكسجين. تقوم خلايا العضلات بزيادة تنظيم OXPHOS لزيادة إمدادات الطاقة أثناء التمرين.يتم تحقيق ذلك جزئيًا من خلال التحول إلى أكسدة اللاكتات عبر OXPHOS.على الرغم من أن هذه العمليات تتماشى مع زيادة إمداد ATP الموزع عبر الشبكة الميتوكوندرية لعملية انقباض العضلات أثناء التمرين،يثير هذا سؤالاً حول كيفية الحفاظ على توازن بين استهلاك الأكسجين من خلال عملية الفسفرة التأكسدية (OXPHOS) التي تولد ATP والظروف الناجمة عن نقص الأكسجين التي تخلقها التمارين. وذلك لأنه، على الرغم من أن إمدادات الأكسجين داخل العضلات تزداد من خلال تحسين قدرة الانتشار الرئوي، وإنتاج القلب، وقدرة الدم على حمل الأكسجين، واستخراج الأكسجين من العضلات الهيكلية أثناء التمرين،نقص الأكسجين، أي انخفاض الضغط الجزئي للأكسجين ) من تور، في حالة الراحة، إلى 3-4 تور تحت التمرين عند حوالي 65% من استهلاك الأكسجين الأقصىيتم تحفيزه في العضلات. علاوة على ذلك، فإن الآلية التي تستخدمها خلايا العضلات لمنع الإفراط في إنتاج أنواع الأكسجين التفاعلية (ROS)، والتي تحدث تحت نقص الأكسجين المرتبط بالتمارين الهوائية.ويؤدي إلى الإجهاد التأكسدي في الخلايا والأنسجة، لا يزال غير واضح.
تتكيف الفسيولوجيا الخلوية مع توفر الأكسجين من خلال استشعار توفر الأكسجين عبر الهيدروكسيلاز البروتين (PHDs) – فون هيبل-لينداو (VHL).-عامل نقص الأكسجين القابل للتحفيز (HIFa)محور. تستخدم الدكتوراه الأكسجين و-كيتوجلوتارات كركائز لهيكله هيدروكسيلي لذرات البرولين في البروتينات التي تحتوي على تسلسلات معترف بها من قبل PHD.تُعترف البروتينات الهيدروكسيليّة، مثل HIF1a، وتُحلل لاحقًا بواسطة VHL، وهو إنزيم E3.تعتبر عوامل النسخ HIFas عوامل تنظيمية تتحكم في مجموعة من العمليات، مثل امتصاص الجلوكوز، وتحلل السكر،وتكوين الأوعية الدمويةالتي توفر الركائز لعملية الفسفرة التأكسدية. ومع ذلك، فإن عوامل الاستجابة لنقص الأكسجين لا تنظم مباشرة عملية الفسفرة التأكسدية في خلايا العضلات، بل تمارس فقط تنظيمًا نسخيًا، والذي يستغرق ساعات ليكون فعالًا، وبالتالي تتأخر عن التغيرات الناتجة عن التمرين في عملية الفسفرة التأكسدية في العضلات التي تستغرق دقائق فقط لتظهر بعد بدء التمرين.
يتم تحفيز اللاكتات بسرعة بواسطة التمرين أو نبض القلب، بسبب زيادة التحلل الجليكولي الهوائي وتحلل الجليكوجين الذي يرفع مستوى البيروفات.الذي يتم تحويله بعد ذلك بشكل حراري ديناميكي ملائم إلى لاكتات بواسطة إنزيم لاكتات ديهيدروجيناز A (LDHA).بجانب LDHA، يتم تحديد توازن اللاكتات أيضًا بواسطة مركب ديهيدروجيناز البيروفات (PDC) الذي يوجه البيروفات إلى الأسيتيلCoA. ) كمادة أساسية في نظام أكسدة الفوسفور. في خلايا العضلات، فإن PDC،
الذي يتم تفعيله أثناء التمرين بسبب التغيرات التي تحدث في NADH/NADنسبة وأيونات المعادن،يمكن أن يتم تعطيله عن طريق الأسيتيل.ومن خلال ارتباط PHD3 بـ PDC E1 بيتا (PDHE1B)،كلاهما يؤديان إلى زيادة في فسفرة السيرينات 232 و293 و300 من PDHA1،الوحدة الحفازة من PDC. الحد من AcCoA، الذي ينشأ من الجلوكوز، والجليكوجين، واللاكتاتالتحلل عبرأو من أكسدة الأحماض الدهنية (FAO)، تثبط OXPHOS وتقلل من استهلاك الأكسجين. ومع ذلك، فإن اللاكتات، بينما تعمل كمصدر وقود مفضل لعضلات القلب والخلايا عبر OXPHOS، تثبط تحلل الدهون.ونقل الأحماض الدهنية إلى الميتوكوندريا بواسطة الكارنيتين بالميتويل ترانسفيراز 1 (CPT1)،يظهر أن اللاكتات تمارس تنظيمًا إيجابيًا وسلبيًا لعملية الفسفرة التأكسدية.
تظل الآليات الكامنة وراء استشعار اللاكتات وكذلك توليد إشارات اللاكتات لتنظيم التنفس الخلوي المؤكسد غير واضحة. يتم تسليط الضوء على إشارات اللاكتات من خلال تعديل الليسين في البروتينات بواسطة اللاكتات.على الرغم من أن إنزيم اللاكتيل ترانسفيراز الحقيقي لا يزال بعيد المنال. الطفرات في إنزيم الألانين-تRNA سينثيتاز 2 (AARS2)،الذي يحفز تشكيل الألانيل-tRNA في تخليق البروتينات الميتوكوندريةويحفز الألانيلات من الليسين في البروتينات،تسبب في ضعف OXPHOS، واضطراب حمض اللاكتيك وفقدان وظيفة العضلات، مع آليات غير محددة.لقد أثار اهتمامنا احتمال أن يعمل AARS2 كإنزيم نقل اللاكتيل المحتمل الذي ينظم عملية الفسفرة التأكسدية في العضلات. في هذه الدراسة، هدفنا إلى تحديد ما إذا كان AARS2 يعمل كإنزيم نقل اللاكتيل الميتوكوندري الذي يدمج إشارات نقص الأكسجين داخل الخلايا وإشارات اللاكتات لتنظيم عملية الفسفرة التأكسدية في خلايا العضلات.
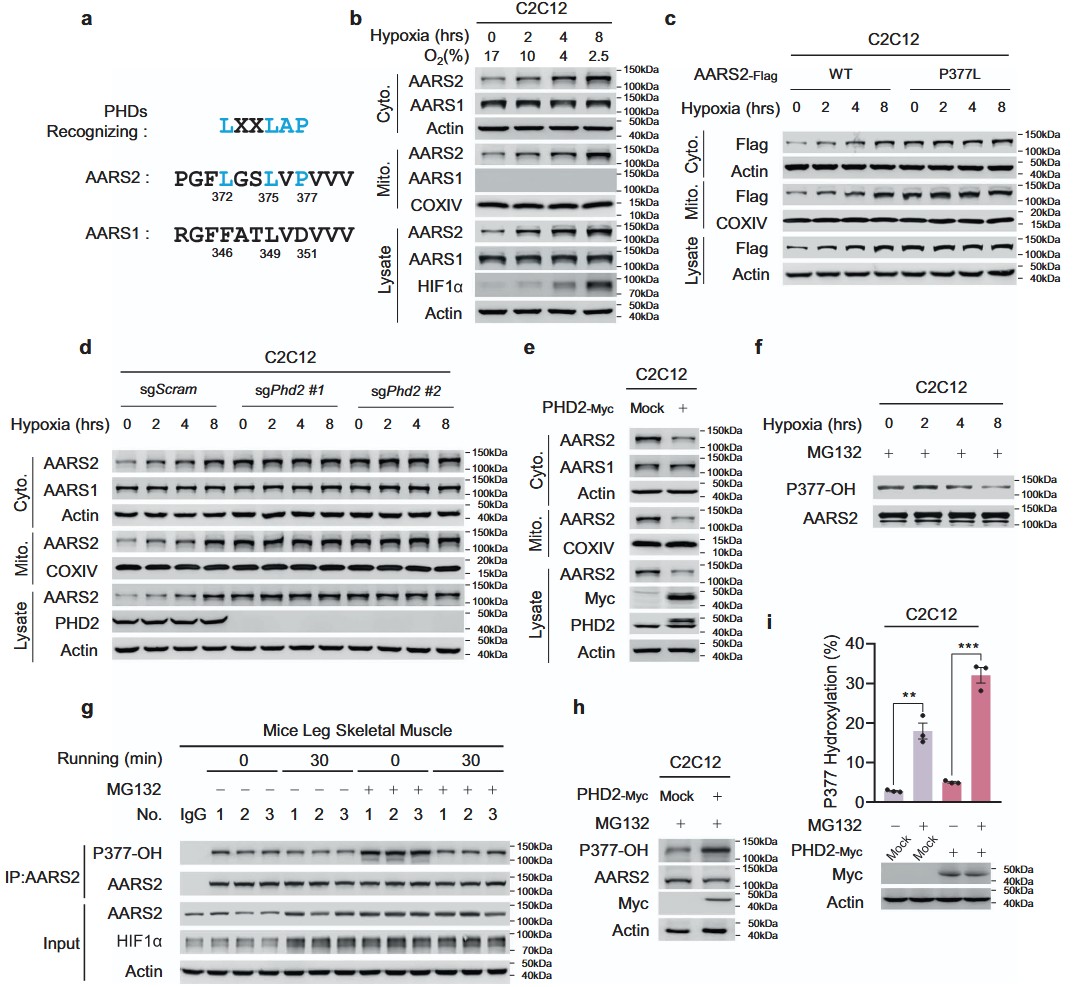
النتائج AARS2 يحتوي على تسلسل يتعرف عليه PHD ويتم هيدروكسيله بواسطة PHD2
البروتين AARS2 البرولين 377 (P377) يقع في تسلسل يتعرف على PHD،الذي لا يوجد في التسلسل المقابل لإنزيم الألانين-تRNA سينثيتاز 1 (AARS1) (الشكل 1أ)، مما يشير إلى أن P377 قد يتم هيدروكسيله، مما يسمح بمستويات AARS2 مشابهة لـ HIF1a،لتنظيمه خلال نقص الأكسجين. وقد تم تأكيد ذلك من خلال الملاحظة أنه، على الرغم من أن نقص الأكسجين أدى بشكل زمني إلى زيادة تعبير AARS2 في كل من الميتوكوندريا والسيتوبلازم لخلايا الميوبلasts المتكاثرة C2C12، وخلايا القلب HL-1 للفئران، وخلايا الكبد الأولية للفئران، إلا أن AARS1 تم التعبير عنه فقط في السيتوسول ولم يتم تحفيزه بواسطة نقص الأكسجين (الشكل 1ب؛ المعلومات التكميلية، الشكل S1أ، ب). كما أدى نقص الأكسجين إلى زيادة مستويات بروتين AARS2 في خلايا الميوبلasts الأولية للفئران مع مرور الوقت (المعلومات التكميلية، الشكل S1ج). علاوة على ذلك، لم تتغير مستويات بروتين AARS2 مع نقص الأكسجين عندما تم تحويل P377 إلى ليوسين (الشكل 1ج)، مما دعم الملاحظة أن زيادة مستويات بروتين AARS2 التي يسببها نقص الأكسجين تتطلب P377. أدى إسكاة أو حذف Phd2، ولكن ليس غيره من هيدروكسيلاز البرولين الحساس للأكسجين، مثل Phd1 و Phd3، في خلايا C2C12 إلى رفع مستويات AARS2 الذاتية (المعلومات التكميلية، الشكل S1د) ومنع تراكم AARS2 الذاتي الناتج عن نقص الأكسجين (الشكل 1د). هذه النتائج، جنبًا إلى جنب مع الملاحظة أن زيادة تعبير PHD2 قللت من مستويات AARS2 في خلايا C2C12 (الشكل 1هـ)، في حين أن تثبيط PHDs باستخدام روكسايدوستات زاد منها (المعلومات التكميلية، الشكل S1هـ)، أكدت أن PHD2 يعمل كـ هيدروكسيلاز برولين ومستشعر للأكسجين لـ AARS2.
بحثنا عن ببتيدات تحتوي على P377 (P377OH) هيدروكسيلية في 120 طيف كتلة مزدوجة (MS/MS) لمكتبة ببتيدات إنزيم غلوتاميك-سي (Glu-C) من خلايا HEK293T التي تعبر عن AARS2 بشكل مفرط (معلومات إضافية، الجدول S1)، ووجدنا 3 أطياف تتطابق مع الطيف المتوقع لـ MS/MS لببتيد AARS2 المحتوي على P377OH (معلومات إضافية، الشكل S1f). نظرًا لأن ببتيد AARS2 المحتوي على P377OH الذي تم تصنيعه أنتج طيفًا مطابقًا لذلك الطيف المحدد (معلومات إضافية، الشكل S1f)، استنتجنا أن P377 تم هيدروكسيله في الجسم الحي. لذلك، لاكتشاف P377OH في الخلايا، قمنا بإنشاء جسم مضاد محدد لموقع P377OH يتعرف بشكل خاص علىفي الببتيد الخاص بـ AARS2 السليم (معلومات إضافية، الشكل S1g، h). تم تقليل مستويات P377OH المعبر عنها بشكل غير طبيعي لـ AARS2 في خلايا C2C12 وخلايا العضلات الأولية من الفئران بشكل يعتمد على الوقت نتيجة التعرض لنقص الأكسجين في وجود MG132، وهو مثبط للبروتيازوم (الشكل 1f؛ معلومات إضافية، الشكل S1i). انخفضت مستويات P377OH في عضلات أرجل الفئران التي تم فيها تثبيط تحلل البروتين بواسطة MG132 (الشكل 1g) بعد أن تم تحفيز نقص الأكسجين عن طريق الجري (معلومات إضافية، الشكل S1j)، مما يؤكد أن P377OH يتم تنظيمه بواسطة توفر الأكسجين بشكل فسيولوجي. بالإضافة إلى ذلك، زاد التعبير المفرط لـ PHD2 من مستويات P377OH في خلايا C2C12، وعلاوة على ذلك، عزز كل من MG132 و PHD2 مستويات P377OH الخلوية بشكل تآزري (الشكل 1h، i). أكدت هذه النتيجة أن P377OH، الذي يعمل على الأرجح كمنظم سلبي لـ AARS2، يتم تعديله بدوره بواسطة PHD2.
AARS2 يعمل كركيزة لآلية البروتيازوم PHD2-VHL
تمت التعبير عن VHL بشكل غير طبيعي وترافق مع AARS2 المفرط التعبير وAARS2 الداخلي في خلايا C2C12 (الشكل 2أ، ب)، مما يتماشى مع تفاعل AARS2 الداخلي مع PHD2 وVHL الداخلي في خلايا العضلات الأولية للفئران (معلومات إضافية، الشكل S1ك)، مما يشير إلى أن AARS2 هو ركيزة لـ VHL. عند التعبير المشترك في خلايا C2C12، كان التفاعل بين AARS2 البري وVHL أكبر من ذلك بين AARS2 الذي يحمل P377L وVHL (الشكل 2ج)؛ علاوة على ذلك، تم تقليل تفاعل AARS2-VHL وزيادته بواسطة نقص الأكسجة (الشكل 2د) وزيادة التعبير عن PHD2 (الشكل 2هـ)، على التوالي، مما يشير إلى أن P377OH زاد من تفاعل VHL-AARS2. زاد التعبير المفرط عن VHL من مستويات يوبكويتين AARS2 المعبر عنها في خلايا HEK293T (الشكل 2و)، بينما قلل من مستويات بروتين AARS2 الداخلي في الميتوكوندريا والسيتوسول في خلايا C2C12 (الشكل 2ز). على العكس، زاد حذف Vhl (KO) في خلايا C2C12 من مستويات AARS2 الداخلية وألغى الزيادة في مستويات AARS2 الناتجة عن نقص الأكسجة (الشكل 2ح). أكدت هذه النتائج مجتمعة أن VHL يقوم بتفكيك AARS2.
AARS2 يثبط إنتاج Ac-CoA و OXPHOS
أدى الإفراط في التعبير عن AARS2 إلى تقليل معدل استهلاك الأكسجين (OCR) في خلايا C2C12 (الشكل 3أ). تم حذف Aars2 (Aars2زادت OCR وانخفضت استجابة OCR لنقص الأكسجين في خلايا C2C12، وتم إنقاذ هذه العواقب من خلال إعادة إدخال AARS2 البري، ولكن ليس الطفرة AARS2 P377L إلى Aars2.الخلايا (الشكل 3ب، ج)، مما يشير إلى أن AARS2 استجاب لنقص الأكسجين عن طريق تثبيط التنفس الخلوي. بالإضافة إلى ذلك، أدى الإفراط في التعبير عن AARS2 إلى انخفاض مستويات Ac-CoA ولكنه زاد من مستويات البيروفات واللاكتات في خلايا C2C12 (الشكل 3د)، مما يوحي بأن AARS2 ينظم سلبًا PDC، وهو معقد الإنزيم الذي يحول البيروفات إلى. وقد تم دعم ذلك بشكل أكبر من خلال الملاحظة التي تفيد بأن التعبير المفرط عن AARS2 بشكل غير طبيعي لزيادة مستويات AARS2 في الخلايا أدى إلى تقليل النشاط النوعي لـ PDHA1 (الشكل 3e)، في حين أن حذف Aars2 زاد من هذا النشاط (الشكل 3f). زادت أنشطة PDHA1 بينما تم إلغاء استجابتها لنقص الأكسجين في خلايا Aars2 KO C2C12؛ ومع ذلك، تم استعادة الاستجابات لنقص الأكسجين من خلال إعادة إدخال AARS2 من النوع البري، ولكن ليس الطفرة P377L (معلومات إضافية، الشكل S2a). علاوة على ذلك، لم يؤثر التعبير المفرط عن AARS2 على مستويات الفسفرة للسرينات 232 و293 و300 من PDHA1 (معلومات إضافية، الشكل S2b)، والتي من المعروف أنها تعطل PDHA1، مما يعني أن AARS2 يعطل PDHA1 من خلال آلية جديدة تحت سيطرة نقص الأكسجين وAARS2.
نظرًا لأن تثبيط CPT1/2/FAO قد يساهم أيضًا في تثبيط استهلاك الأكسجين، من خلال تقليل إمداد Ac-CoA إلى OXPHOS،تم استخدام -بالميتات للتحقيق فيما إذا كان CPT1/2/FAO مثبطًا بواسطة AARS2 (معلومات إضافية، الشكل S2c). أدى الإفراط في التعبير عن AARS2 إلى تقليل التسمية المزدوجة (M2)-إنتاج أسيتيل-CoA من-بالميتات (الشكل 3g)، مما يشير إلى أن AARS2 قام بتثبيط CPT1/2/FAO. فقط الكارنيتين بالميتويل ترانسفيراز 2 (CPT2) الداخلي، وليس CPT1 أو إنزيمات FAO، تفاعلت مع AARS2 المعبر عنها بشكل خارجي (معلومات إضافية، الشكل S2d). علاوة على ذلك، أدى الإفراط في التعبير عن AARS2 إلى تقليل نشاط CPT2 (الشكل 3h)، في حين أن KO Aars2 زاد منه (الشكل 3i)، مما يؤكد أن AARS2 يقوم بتعطيل CPT2 ربما عبر
الشكل 1 الأكسجين ينظم مستويات بروتين AARS2 عبر PHD2. أ يحمل AARS2 تسلسلًا يتعرف عليه PHD. تم محاذاة تسلسل الأحماض الأمينية (aa) من 369 إلى 380 من AARS2 وتسلسله المقابل AARS1 (aa 343-354) مع تسلسل التعرف على PHD. تم تمييز بقايا PHD المتوافقة باللون الأزرق. ب يتم تنظيم مستويات بروتين AARS2 بواسطة نقص الأكسجين. AARS1 و AARS2 و HIF1 في الميتوكوندريا والسيتوسول.تم الكشف عن مستويات في خلايا C2C12 المزروعة في غرفة نقص الأكسجين لفترات زمنية محددة. تم استخدام COXIV والأكتين لإظهار العزل الناجح للميتوكوندريا والسيتوبلازم، على التوالي (تم استخدام نفس الطريقة لتجزئة الخلايا الفرعية من الآن فصاعدًا). تم مراقبة مستويات الأكسجين في الغرفة. c P377 مطلوب لتنظيم مستويات بروتين AARS2 بواسطة نقص الأكسجين. تم التعبير عن AARS2 بشكل مستقر. و AARS2 تم قياس مستويات بروتين (P377L) في خلايا C2C12 بعد زراعة الخلايا في غرفة نقص الأكسجين لمدة الزمن المحددة. د، هـ ينظم PHD2 مستويات بروتين AARS2. تم قياس مستويات AARS1 و AARS2 الميتوكوندرية والسيتوسولية في خلايا C2C12 أو خلايا Phd2 KO C2C12 المزروعة في غرفة نقص الأكسجين لمدة الزمن المحددة (د) وفي خلايا C2C12 التي تعبر عن PHD2 بشكل مفرط (هـ). ف-ي ينظم نقص الأكسجين و PHD2 هيدروكسلة P377. تم تحديد مستويات P377OH عبر اختبار الويسترن بلوت في خلايا C2C12 المزروعة في غرفة نقص الأكسجين لمدة الزمن المحددة في وجود MG132 في وسط الزراعة لمنع التحلل البروتيني (ف)؛ في عضلات الساق الهيكلية للفئران المستريحة والتي تم حقنها وريدياً مع أو بدونMG132 مرتين في الأسبوع لمدة 4 أسابيع متتالية ) ( ); في خلايا C2C12 التي تعبر عن PHD2 بشكل مفرط والمعالجة بـ MG132 ( تم قياس مستويات P377OH في خلايا C2C12 التي تعبر عن PHD2 بشكل مفرط في وجود وغياب MG132 بواسطة مطيافية الكتلة (i) (جميع البيانات مُبلغ عنها كمتوسطSEM لثلاث تجارب مستقلة. تم تقييم الدلالة الإحصائية بواسطة تحليل التباين الثنائي.؛.
تعديل تساهمي. علاوة على ذلك، أدى نقص Aars2 إلى تنشيط CPT2 وتقليل استجابة نشاط CPT2 لنقص الأكسجين، والتي تم إنقاذها من خلال إعادة إدخال AARS2 البري، ولكن ليس الطفرة AARS2 P377L (معلومات إضافية، الشكل S2e). أدى نقص Cpt2 في خلايا C2C12 إلى جعلإنتاج غير مستجيب لزيادة التعبير عن AARS2 (الشكل 3g). أخيرًا، لم يكن هناك نشاط محدد لـ CPT1 (المكمل) لم تتأثر المعلومات (الشكل S2f) أو مستوى البروتين (المعلومات التكميلية، الشكل S2g) بزيادة تعبير AARS2. تشير هذه البيانات مجتمعة إلى أن AARS2 يقوم بتعطيل CPT2 لمنع أكسدة الأحماض الدهنية.
استخدمنا-المُوسومة بالجلوتامين لتتبع– كيتوجلوتارات (-KG) تدفق، وهو النقطة التي تتغذى فيها الأحماض الأمينية في دورة TCA (معلومات إضافية، الشكل S2h). الـالمستويات لم تتأثر بزيادة تعبير AARS2 (المعلومات التكميلية، الشكل S2i)، مما يشير إلى أن استقلاب الأحماض الأمينية لم يكن منظمًا بواسطة AARS2. هذه النتائج مجتمعة تشير إلى أن AARS2 يثبط التنفس عن طريق تقليل إمداد Ac-CoA من التحلل السكري واستقلاب الأحماض الدهنية من خلال تعطيل PDHA1 وCPT2، وليس عبر استقلاب الأحماض الأمينية (المعلومات التكميلية، الشكل S2j).
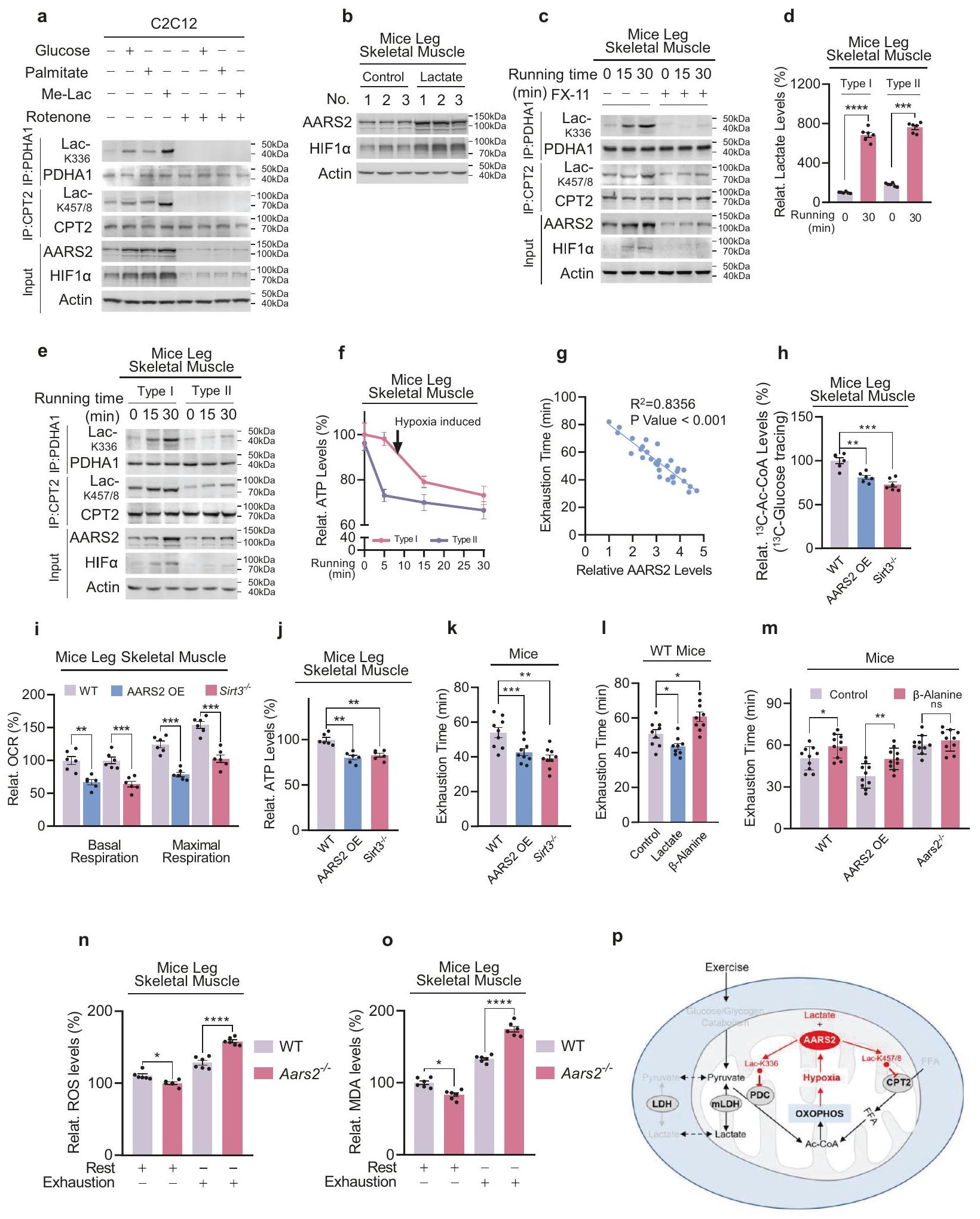
بعد ذلك، تحققنا مما إذا كانت نقص الأكسجين يتوسط تنظيم PDHA1 و CPT2 عبر AARS2 في الجسم الحي. كانت مستويات بروتين HIFas و AARS2 تم تحفيز ذلك في عضلات الساق الهيكلية للفئران بطريقة تعتمد على الوقت بينما كانت الفئران تجري على جهاز المشي (الشكل 3j). علاوة على ذلك، كان مستوى Ac-CoA في العضلات قد انخفض بشكل معتدل بينما استمرت الأنشطة المحددة لـ PDC- (الشكل 3k) و CPT2- (الشكل 3l) في الانخفاض، واستمرت نقص الأكسجة في عضلات الساق (معلومات إضافية، الشكل S3a) بالإضافة إلى مستويات اللاكتات والبيروفات في الزيادة، بعد بدء الجري (الشكل 3m). وكان ذلك متزامناً مع زيادة قصيرة في النشاط الكلي لـ PDC في عضلات الساق للفئران.
الشكل 2 AARS2 يعمل كركيزة لآلية البروتيازوم PHD2-VHL.يتفاعل VHL مع AARS2. تم الكشف عن AARS2 المترافق مع المناعة، والمتعبّر عنه بشكل مشترك (أ) وAARS2 الداخلي (ب) في خلايا C2C12 التي تعبر عن VHL بشكل غير طبيعي. يظهر الطفرة P377L affinity ضعيفة لـ VHL. تم الكشف عن التفاعلات بين VHL وAARS2، وبين VHL وP377L عبر الترسيب المناعي المشترك عندما تم التعبير عنهما بشكل مشترك في خلايا C2C12. تم توفير تحليل كثافة الصور. د، هـ تفاعل VHL-AARS2 يتم تنظيمه بواسطة نقص الأكسجة وPHD2. تم تحديد كمية AARS2 المترافقة مع VHL تحت الظروف الطبيعية ونقص الأكسجة لمدة 8 ساعات (د)، وكذلك مع أو بدون التعبير المفرط عن PHD2 (هـ) في خلايا C2C12. تم توفير تحليل كثافة الصور.تتوسط VHL تحلل البروتينات بواسطة البروتيازوم. تم اختبار AARS2 المعبر عنه بشكل غير طبيعي في خلايا HEK293T من حيث الت ubiquitination عندما تم التعبير عنه بمفرده وعندما تم التعبير عنه مع اليوبكويتين أو VHL، أو كليهما، في وجود وغياب MG132. g تقوم VHL بتقليل مستويات بروتين AARS2. تم قياس مستويات AARS2 الميتوكوندري والسايتوزولي في خلايا C2C12 مع أو بدون فرط التعبير عن VHL.تسبب نقص الأكسجين في تراكم AARS2 بطريقة تعتمد على VHL. تم قياس مستويات AARS2 الميتوكوندري والسايتوسولي في خلايا C2C12 وخلايا C2C12 المعدلة وراثيًا VhI، التي تم زراعتها في غرفة نقص الأكسجين لمدة الأوقات المحددة. جميع البيانات مُبلغ عنها كمتوسط.SEM لثلاث تجارب مستقلة. تم تقييم الدلالة الإحصائية بواسطة اختبار الطالب غير المتزاوج ذو الطرفين.-اختبار: . العضلات، التي تلاها بعد ذلك انخفاض إلى مستويات أقل من مستويات الراحة (الشكل 3ن). كان الارتفاع الأولي في نشاط PDC الكلي متسقًا مع انخفاض أولي في فسفرة PDHA1 (معلومات إضافية، الشكل S3ب) وعلاج الفوسفاتاز لتقليل تأثيرات الفسفرة (المعلومات التكميلية، الشكل S3c) ألغى الزيادة الأولية (الشكل 3n). ومع ذلك، لم يتأثر الانخفاض في نشاط PDC مع وقت الجري حتى بعدعلاج الفوسفاتاز (الشكل 3ن). هذه، جنبًا إلى جنب مع الملاحظة بأن مستويات البروتين لمكونات PDC وCPT2 (المعلومات التكميلية، الشكل S3د) ولا مستويات البروتين وأنشطة LDHs (المعلومات التكميلية، الشكل S3د، هـ) تأثرت طوال 30 دقيقة من الجري، مما يشير إلى أن AARS2 قد يتوسط آلية تعديل تساهمي تعطل PDC دون أن تتضمن فسفرة PDHA1.
اللاكتات يعطل PDHA1 و CPT2 اعتمادًا على AARS2
انخفضت أنشطة PDHA1 وCPT2 الخاصة عند إضافة اللاكتات (الشكل 4أ). تم استخدام ميثيل-لاكتات القابل لاختراق غشاء الخلية (Me-Lac) لتحليل تأثيرات اللاكتات. لوحظت زيادة في معدلات استهلاك الأكسجين (OCRs) بالإضافة إلى انخفاض أنشطة PDHA1 وCPT2 الخاصة في خلايا C2C12 المعالجة بـ 2 مللي مول من Me-Lac، مما زاد من مستويات حمض اللاكتيك الميتوكوندري إلى 0.6 مللي مول إلى 1.0 مللي مول؛ ومع ذلك، فإن Me-Lac بتركيز 10 مللي مول، الذي زاد من حمض اللاكتيك الميتوكوندري إلى 3.2 مللي مول، أدى إلى انخفاض في معدلات استهلاك الأكسجين، بالإضافة إلى الأنشطة الخاصة لـ PDHA1 وCPT2 (الشكل 4ب-د؛ معلومات إضافية، الشكل S4أ). كانت نتائج معالجة 2 مللي مول من Me-Lac متوافقة مع الملاحظة التي تفيد بأن اللاكتات تغذي خلايا العضلات عبر دورة TCA.ومع ذلك، فإن معالجة 10 مللي مول من Me-Lac تؤدي إلى مستويات داخل الخلايا من اللاكتات تشبه تلك الموجودة في العضلات أثناء التمرين، مما يشير إلى أن اللاكتات تلعب أيضًا أدوارًا مثبطة في OXPHOS، على الأرجح من خلال تعديل تساهمي لـ PDHA1 وCPT2، وهو مفهوم تم تأكيده من خلال انخفاض نشاط PDHA1 وCPT2 في خلايا C2C12، ولكن ليس في خلايا Aars2 KO C2C12 (معلومات إضافية، الشكل S4b، c). تم تأكيد هذا المفهوم بشكل أكبر من خلال فحص إنتاج Ac-CoA في خلايا C2C12 وHL-1 التي تقوم بأكسدة اللاكتات، والتي كشفت أن 10 مللي مول من Me-Lac قد خفضتالإنتاج من-جلوكوز (الشكل 4e، f) و-بالميتات (الشكل 4g، h) بطريقة تعتمد على AARS2. ومن المثير للاهتمام أن فقدان AARS2 زاد منالإنتاج من-جلوكوز و-بالميتات (الشكل 4e-h)، مما يشير إلى أن اللاكتات أوقفت PDHA1 وCPT2 عبر AARS2، وأن اللاكتات لا توقف PDHA1 وCPT2 وOXPHOS عبر حموضتها. علاوة على ذلك، فإن 10 مللي مول من Me-Lac أوقف OCR في الخلايا العضلية الأولية بطريقة تعتمد على AARS2 (معلومات إضافية، الشكل S4d). علاوة على ذلك، كان لتأثير Me-Lac ضئيل على-أنتاج كجم من-جلوتامين (معلومات إضافية، الشكل S4e)، والذي كان متسقًا مع أن تحلل الأحماض الأمينية لم يكن منظمًا بواسطة حمض اللبنيك.
AARS2 يقوم بتعطيل PDHA1 و CPT2 عن طريق إضافة مجموعة لاكتيل إلى هذه البروتينات
حمض اللبنيك مشابه هيكليًا للألانين (الشكل 5أ). تم نمذجة ارتباط حمض اللبنيك بـ AARS2 باستخدام تحليل ربط الجزيئات (المعلومات التكميلية، الشكل S4f) وتم تأكيده من خلال قياس الحرارة التلقائية (ITC) (المعلومات التكميلية، الشكل S4g).
علاوة على ذلك، تفاعلت AARS2 المفرطة التعبير مع كل من PDHA1 الداخلي (المعلومات التكميلية، الشكل S4h) وCPT2 المعبر عنه بشكل خارجي (المعلومات التكميلية، الشكل S4i). تشير هذه النتائج مجتمعة إلى أن AARS2 قد تعمل كـ lactyltransferase لـ PDHA1 وCPT2. لتأكيد هذه الفرضية، بحثنا أولاً عن بقايا الليسين الملبدة باللاكتيل في PDHA1 وCPT2. باستخدام مكتبة ببتيد تريبتيك من PDHA1 المنقى ومكتبة ببتيد Glu-C من CPT2 المنقى، وجدنا أن الليسين 336 في PDHA1 (المعلومات التكميلية، الشكل S5a) والليسين 457 و458 في CPT2 (المعلومات التكميلية، الشكل S5b) قد تكون ملبدة باللاكتيل (Lac-K336، Lac-K457/8). تم تأكيد وجود Lac-K336، بالإضافة إلى Lac-K457/8، من خلال مطابقة طيف MS/MS للببتيدات الاصطناعية التي تحتوي على Lac-K336 وLac-K457/8 مع تلك الموجودة في مكتبة الببتيد (المعلومات التكميلية، الشكل S5a، b).
قمنا بتقييم قدرة AARS2 على إضافة مجموعة لاكتيل إلى ببتيدات PDHA1 K336 وCPT2 K457/8 الاصطناعية والبروتينات السليمة PDHA1 وCPT2 في المختبر. على عكس AARS2المتحور، الذي لديه تبديل الأسباراجين 104 إلى التيروزين، يعادل تبديل الأسباراجين 71 إلى التيروزين في AARS1 (AARS1الصيغة غير النشطة من AARS1 ) (معلومات إضافية، الشكل S6a)، قام AARS2 بإضافة اللاكتيل إلى الببتيد الاصطناعي PDHA1 الذي يحتوي على K336 بالإضافة إلى ببتيد CPT2 الذي يحتوي على K457/8 (الشكل 5b؛ معلومات إضافية، الشكل S6b) من خلال آليات تعتمد على اللاكتات وATP وقابلة للتثبيط بواسطة البيروفوسفات. ومع ذلك، لم يقم AARS2 بإضافة اللاكتيل إلى تلك الببتيدات التي تم تغيير بقايا الليسين فيها إلى ألانين (معلومات إضافية، الشكل S6c). هذه النتائج اقترحت أن الآلية التي يستخدمها AARS2 لإضافة اللاكتات إلى الليسين هي نفسها المستخدمة لإضافة الألانين إلى الليسين. (معلومات إضافية، الشكل S6d). كان تأثير AARS2 على ببتيد PDHA1 الاصطناعي المحتوي على K336 ضمن مستويات اللاكتات الفسيولوجية البالغة 5.21 مللي مول، في حين أن رقم الدوران (كان AARS2 لهذا الببتيد 1.19 (الشكل 5c)، مما يشير إلى أن AARS2 كان ناقل لاكتيل فعال.
قمنا بتوليد أجسام مضادة متعددة النسائل محددة الموقع التي تعرفت على Lac-K336 (المعلومات التكميلية، الشكل S6e) وLacK457/8 (المعلومات التكميلية، الشكل S6f). أدى العلاج بـ 10 مللي مول من Me-Lac إلى زيادة مستويات Lac-K336 وLac-K457/8 من PDHA1 المعبر عنه بشكل خارجي (الشكل 5d) وCPT2 (الشكل 5e) في خلايا C2C12 وHL-1 (المعلومات التكميلية، الشكل S6g). ومع ذلك، لم يتم ملاحظة اللاتيل عندما تم حذف Aars2 (الشكل 5d، e). أيضًا، زادت مستويات Lac-K336 وLac-K457/8 في الخلايا العضلية الأولية التي تعبر عن AARS2 بشكل مفرط وانخفضت بعد حذف Aars2 (المعلومات التكميلية، الشكل S6h). تشير هذه النتائج إلى أن AARS2 يعمل كـ lactyltransferase لـ PDHA1 K336 وCPT2 K457/8. علاوة على ذلك، يتماشى مع الملاحظة بأن اللاكتات يمكن أن تُنقل إلى الميتوكوندريا بواسطة الناقل أحادي الكربوكسيلات 1 (MCT1)،زيادة مكملات Me-Lac في وسائط الثقافة، بينما تم تثبيط MCT1 باستخدام حمض الأسيانو-4-هيدروكسي سيناميك. ) انخفضت مستويات اللاكتات الميتوكوندريا (الشكل 5f)، وكانت هذه النتيجة متوافقة مع علاج a-CHCA الذي قلل من مستويات Lac-K336 وLac-K457/458 في خلايا C2C12 (الشكلهذه النتائج، جنبًا إلى جنب مع الملاحظة بأن مستويات اللاكتات الميتوكوندريالية زادت بشكل متناسب مع مستويات اللاكتات في المستخلص، كلاهما في نطاق AARS2.نحو اللاكتات، في عضلات الساق الهيكلية للفئران بعد الجري (الشكل 5i)، مما يشير إلى أن اللاكتات الناتجة عن التمرين قد تنظم اللاكتيل في الميتوكوندريا.
تشغيل مستويات Lac-K336 وLac-K457/8 المعززة في خلايا عضلات الفئران (الشكل 5j)، بما يتماشى مع الاكتشاف الذي أظهر أن نقص الأكسجين زاد من كل من اللاكتات في السائل الخلوي والميتوكوندريا (المكملات المعلومات، الشكل S6i) ومستويات Lac-K336 وLac-K457/8 في خلايا C2C12 (الشكل 5k، I). زاد تثبيط PHD عبر الروكسايدوستات من Lac-K336 وLac-K457/8 (المعلومات التكميلية، الشكل S6j، k) في خلايا C2C12، مما يبرز الأهمية الفسيولوجية لـ Lac-K336 وLac-K457/8. أظهرت التقديرات عبر MS أن
الشكل 3 AARS2 يثبط إنتاج Ac-CoA و OXOPHOS. أ، ب AARS2 ينظم OCR. تأثيرات التعبير المفرط عن AARS2 (يسار، قياسات OROBOROS Oxygraph-2K؛ يمين، الكمية) (أ)، و Aars2 KO باستخدام sgRNAs مستقلة (ب) على OCR.تدفق لكل حجم من الخلايا،تم تحديدهاتقوم AARS2 بتنظيم OCR استجابةً لمستويات الأكسجين. تأثيرات إعادة إدخال AARS2 من النوع البري أو P377L إلى Aarsتم تحديد خلايا C2C12 في خلايا تحت ظروف طبيعية من الأكسجين وخلايا تعرضت لنقص الأكسجين لمدة 8 ساعات. ). AARS2 ينظم مستويات المستقلبات. المستويات النسبية (بالنسبة لمستويات خلايا C2C12) من اللاكتات، البيروفات، و Ac-CoA في خلايا C2C12 وخلايا C2C12 التي تعبر عن AARS2 بشكل مفرط.تم تقييمها.تنظم AARS2 نشاط PDHA1 المحدد. تم تحديد الأنشطة المحددة النسبية (بالنسبة لتلك المستخلصة من خلايا C2C12) لـ PDHA1 المنقى من خلايا C2C12 التي تعبر عن AARS2 بشكل مفرط (e) وخلايا C2C12 التي تم حذف Aars2 منها (f). ). تنظم AARS2 إنتاج Ac-CoA بواسطة CPT2. النسب المئوية للمواد غير المعلمة ( )، مُعَلَّمَة واحدة ( ) ، وذو تسميات مزدوجة ( ) أسيتيل-CoA من -بالميتات في خلايا C2C12 وخلايا C2C12 التي تم فيها حذف Cpt2 باستخدام sgRNAs مستقلة، تم تحديدها مع أو بدون فرط التعبير عن AARS2؛تم إجراء مطاردة بالبالميتات لـتمت مقارنة النسب M+2 في خلايا C2C12 مع وبدون فرط التعبير عن AARS2 من حيث الدلالة.تنظم AARS2 نشاط CPT2 المحدد. تم تحديد النشاط النسبي المحدد (بالنسبة لتلك المستمدة من خلايا C2C12) لـ CPT2، المعزول من خلايا C2C12 التي تعبر عن AARS2 بشكل مفرط (h) وخلايا Aars2 KO C2C12 (i).الجري يحفز HIFتعبير s و AARS2 في عضلات الساق الهيكلية للفئران. HIF1، HIF2، HIF3تم تحديد مستويات AARS1 و AARS2 في عضلات الساق الهيكلية للفئران قبل وبعد الجري لمدة الزمن المحددة. ). ك، إن الجري ينشط PDC و CPT2 في الجسم الحي. تم تحديد الأنشطة النوعية النسبية (بالنسبة لتلك الموجودة في الفئران المستريحة) لـ PDC (ك) و CPT2 (ل) في عضلات الساق الهيكلية للفئران بعد أن بدأت الفئران الجري لفترات زمنية محددة ( ). تراكم اللاكتات الناتج عن الجري التحمل وتقليل أسيتيل-CoA. تم تحديد مستويات اللاكتات والبيوفيرات وأسيتيل-CoA في عضلات الساق الهيكلية للفئران بعد السماح للفئران بالجري لمدة الفترات المحددة. ). يُعطّل AARS2 PDC من خلال آلية غير الفسفرة. تُظهر الأنشطة النسبية لـ PDC (بالنسبة لكمية البروتين) في عضلات الساق الهيكلية للفئران البرية و Aars2-/- التي تم أخذ عينات منها في نقاط زمنية مختلفة بعد بدء الجري مع أو بدون إزالة الفسفرة التي تم تحفيزها بواسطةتم تحديد الفوسفاتاز، ). تم تحديد الأنشطة في الوقت 0 على أنها جميع البيانات مُبلغ عنها كمتوسطSEM لثلاث تجارب مستقلة. تم تقييم الدلالة الإحصائية بواسطة اختبار الطالب غير المتزاوج ذو الطرفين.-اختبار t وتحليل التباين ثنائي الاتجاه:؛؛؛; لا أهمية.
كانت مستويات Lac-K336 و Lac-K457/8 في عضلات الساق الهيكلية للفئران مرتفعة كما هو الحال فيتحت التعبير المفرط المتزامن لـ AARS2 وعلاج Me-Lac (الشكل 5 م)، وبلغتبعد جري الفئران لمدة 30 دقيقة (الشكل 5ن) أو عندما تتعرض عضلات الفئران لنقص الأكسجين (الشكل 50)، مما يشير إلى أن جزءًا كبيرًا من PDHA1 وCPT2 يتم لاكتيله تحت نقص الأكسجين داخل خلايا العضلات وارتفاع مستوى اللاكتات في الجسم، وهي حالة تحاكي تمارين التحمل.
تحويل K336 من PDHA1 و K457/8 من CPT2 إلى K336Q ومن المقلد اللاكتيل، الجلوتامين (Q)، قلل من أنشطة كلا الإنزيمين بينما تم تغيير K336 و K457/8 إلى K336R وإن المقلد غير اللاكتيل، الأرجينين (R)، أثر بشكل ضئيل على أنشطتهم المحددة (الشكل 5p، q)، مما يشير إلى أن اللاكتيل قد عطل PDHA1 وCPT2. على الرغم من أن زيادة التعبير عن AARS2 (الشكل 5p، q) وإضافة Me-Lac (الشكل 5r، s) زادت من Lac-K336 وLac-K457/8 وعطلت PDHA1 وCPT2، إلا أنها لم تغير من لاكتيل اللاكتيل لموقع اللاكتيل الخالي من الطفرات في PDHA1 وCPT2، وهو ما يتماشى مع الملاحظة التي تفيد بأن Aarsأظهرت عضلة الساق في الفأر زيادة في نشاط PDC وCPT2 (المعلومات التكميلية، الشكل S6I)، مما يؤكد أن اللاكتيلation هي التي تقف وراء تعطيل PDHA1 وCPT2 بواسطة AARS2.
أخيرًا، فشل مستوى الألانين الفسيولوجي في تثبيط اللاكتيلation K336 في المختبر (معلومات إضافية، الشكل S6m)؛ كان لتكملة الميثيل-ألانين تأثير ضئيل على Lac-K336 وLac-K457/8 وعلى الأنشطة المحددة لـ PDHA1 وCPT2 (معلومات إضافية، الشكل S6n، o)، مما يلغي إمكانية تأثير الألانين على اللاكتيلation، وأن الألانيلation البروتيني، الناتج عن زيادة تنظيم AARS2، ينظم أنشطة PDHA1 وCPT2.
SIRT3 يعكس تعديل اللاكتيل لـ PDHA1 و CPT2
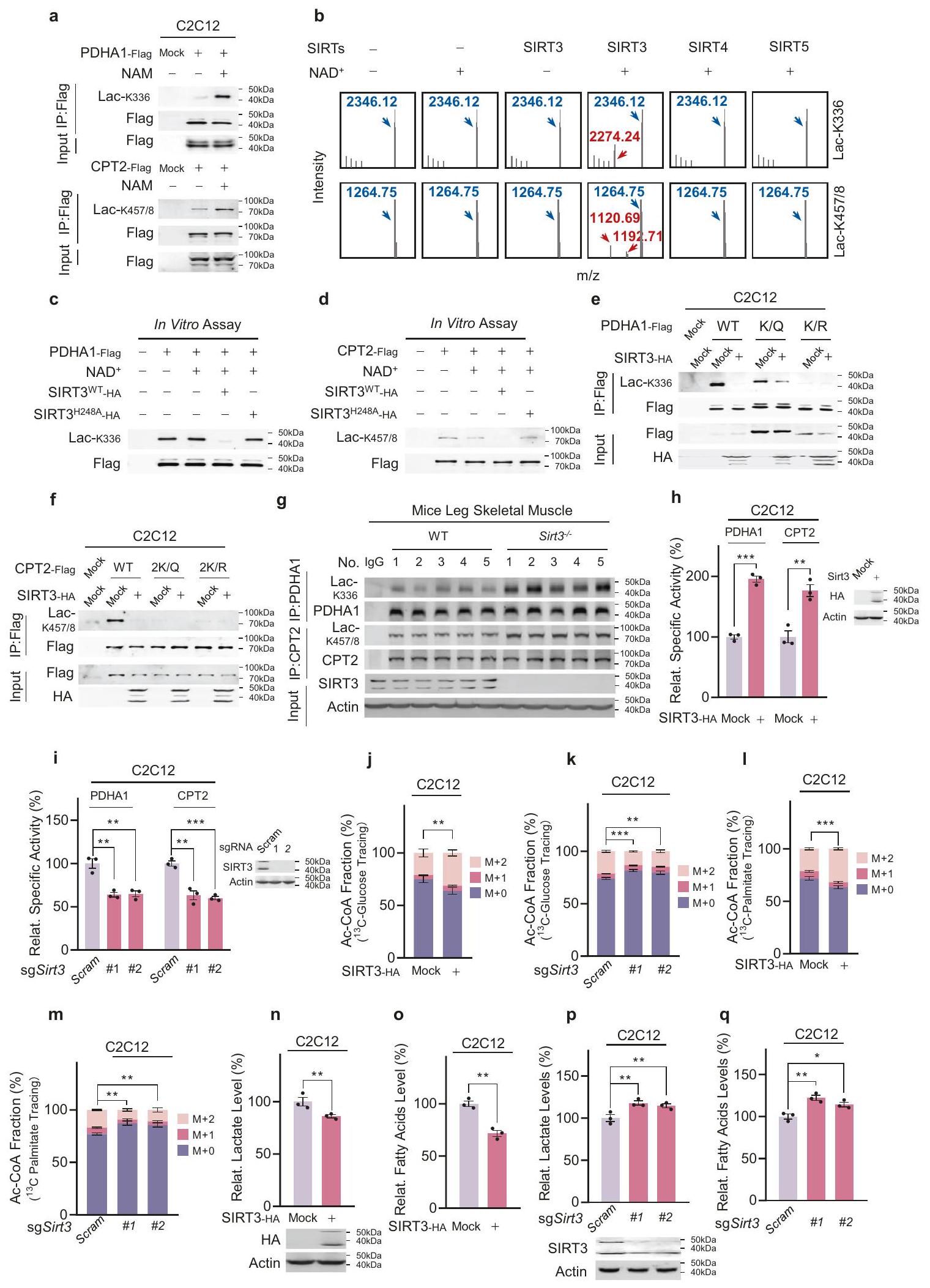
للتحقيق فيما إذا كانت مستويات Lac-K336 وLac-K457/8 منظمة ديناميكيًا، قمنا بتقييم استجابتهما للنيكوتيناميد (NAM)، وهو مثبط عام للسيرتوينات والديميدازات التي تزيل تعديلات البروتين المرتبطة بالاميد المتعددة.علاج NAM زاد من مستويات Lac-K336 وLac-K457/8 للبروتينات المعبر عنها بشكل غير طبيعي PDHA1 وCPT2 في خلايا C2C12، على التوالي (الشكل 6a). نظرًا لأن Lac-K336 وLac-K457/8 هما تعديلات بروتينية ميتوكوندرية، قمنا بتقييم أنشطة الديلاكتيلز لإنزيمات السيتوينات الميتوكوندرية، وهي SIRT3 وSIRT4 وSIRT5. قامت SIRT3 بإزالة اللاكتيل من ببتيدات Lac-K336 وLac-K457/8 الاصطناعية عبر NAD.مسار معتمد، في حين أن المجال التحفيزي لـ SIRT4 أو SIRT5 لم يفعل (الشكل 6ب)، مما يشير إلى أن SIRT3 هو ديلكتيلز لـ Lac-K336 وLac-K457/8. وقد تم تأكيد ذلك بشكل أكبر من خلال الملاحظة أن كل من PDHA1 وCPT2 تفاعلا مع SIRT3. (معلومات إضافية، الشكل S7a، b)، وأن النوع البري من SIRT3 قلل من مستويات Lac-K336 وLac-K457/8 من PDHA1 النقي السليم (الشكل 6c) وCPT2 (الشكل 6d)، في حين أن SIRT3 المعطل بواسطة الأميدازطافرةلم يحدث. علاوة على ذلك، أدى الإفراط في التعبير عن SIRT3 إلى تقليل مستويات Lac-K336 وLac-K457/8 في خلايا C2C12 من النوع البري، ولكن ليس في تلك التي تحمل طفرات PDHA1 وCPT2 الخالية من مواقع اللاكتيل (الشكل 6e، f). علاوة على ذلك، تم تنقية PDHA1 وCPT2 من عضلات الساق الهيكلية لفئران Sirt3 KO (Sirt3أظهرت الفئران مستويات أعلى من Lac-K336 وLac-K457/8، على التوالي، مقارنة بتلك الموجودة في عضلات الساق لدى الفئران من النوع البري (الشكل 6g).
كانت الأنشطة المحددة لـ PDHA1 و CPT2 المعبر عنها في خلايا C2C12 التي تعبر عن SIRT3 بشكل مفرط وخلايا Sirt3 KO أعلى وأقل، على التوالي، من تلك المعبر عنها في خلايا C2C12 (الشكل 6h، i). ومع ذلك، لم يؤثر لا التعبير المفرط عن SIRT3 ولا Sirt3 KO على الأنشطة المحددة لطفرات PDHA1 و CPT2 التي لا تحتوي على مواقع اللاكتيل (معلومات إضافية، الشكل S7c-f). علاوة على ذلك، زاد التعبير المفرط عن SIRT3 وقلل Sirt3 KOالإنتاج من-جلوكوز (الشكل 6j، k) و-بالميتات (الشكل 6ل، م)، على التوالي، في خلايا C2C12.
علاوة على ذلك، أدى الإفراط في التعبير عن SIRT3 إلى انخفاض مستويات اللاكتات (الشكل 6ن) والأحماض الدهنية الحرة (الشكل 60) في خلايا C2C12، في حين أن نقص Sirt3 زاد من هذه المستويات (الشكل 6ص، ق). كانت هذه النتائج متسقة مع أن SIRT3 قام بإزالة اللاكتيل وعكس تأثيرات اللاكتيل على PDHA1 وCPT2. أخيرًا، قد يؤدي الإفراط في التعبير عن SIRT3 إلى إزالة اللاكتيل وكذلك إزالة الأسيتيل من PDHA1.نتج عن ذلك تنشيط PDHA1 بشكل أكثر وضوحًا في خلايا C2C12 مقارنةً بخلايا Aars2 KO C2C12، مما يشير إلى أن اللاكتيل هو آلية رئيسية لتعطيل PDHA1 (معلومات إضافية، الشكل S7g).
يدفع OXPHOS عملية اللاكتيل إلى تقييد OXPHOS وقدرة التحمل في الجري
تعرض أنسجة أرجل الفئران في ظروف نقص الأكسجين في المختبر أدى إلى تحفيز Lac-K336 و Lac-K457/8 في النوع البري، ولكن ليس في Aars2.أنسجة أرجل الفئران، مما يشير إلى أن نقص الأكسجين يحفز AARS2 بالإضافة إلى Lac-K336 وLac-K457/8 (معلومات إضافية، الشكل S8a). لتأكيد أن نقص الأكسجين داخل العضلات الناتج عن التمارين قد يكون مدفوعًا بتفعيل عملية التنفس الخلوي في العضلات التي تعمل كمصرف للأكسجين،إن إضافة وسائط الثقافة إما بالجلوكوز أو بالبالميتات أو باللاكتات المنخفضة لتعزيز OXPHOS (المعلومات التكميلية، الشكل S8b) أدت إلى تحفيز HIF1a جنبًا إلى جنب مع AARS2 و Lac-K336 و Lac-K457/8 في خلايا C2C12، بينما أدى إلغاء OXPHOS الخلوي باستخدام الروتينون إلى تقليل هذه التأثيرات (الشكل 7a). هذه النتائج تدعم تلك الدراسات التي أظهرت أن تعزيز OXPHOS يحفز HIF1a في أنواع خلايا أخرى.وأكدت أن تعزيز OXPHOS يؤدي إلى نقص الأكسجين داخل العضلات و
الشكل 4: اللاكتات تعطل PDHA1 وCPT2 اعتمادًا على AARS2. أ. تعطل علاجات اللاكتات PDHA1 وCPT2 الخلوية. تم معالجة خلايا C2C12 بـ 10 مللي مول من اللاكتات. تم تحديد الأنشطة النوعية النسبية (مقارنة بتلك من خلايا C2C12 غير المعالجة) لـ PDHA1 وCPT2.تقلل علاجات Me-Lac من معدلات استهلاك الأكسجين الخلوي. معدلات استهلاك الأكسجين الخلوي لخلايا C2C12 التي لم تُعالج أو التي عولجت بـ 2 مللي مول أو 10 مللي مول من Me-Lac لـكانوا مصممين.تعمل علاجات Me-Lac على تعطيل PDHA1 وCPT2 الخلوية. الأنشطة النوعية النسبية (بالنسبة لتلك الخلايا C2C12 غير المعالجة) لـ PDHA1 (ج) وCPT2 (د) في خلايا C2C12 التي لم تُعالج أو التي عولجت بـ 2 مللي مول أو 10 مللي مول من Me-Lac.تم تحديدها.تقلل علاجات اللاكتات من تدفق Ac-CoA من التحلل السكري وأكسدة الأحماض الدهنية.المستويات في-جلوكوز ( ) و -بالميتات ( ) (إلى خلايا C2C12 غير المعالجة) و ( خلايا C2C12 المعالجة فقط، خلايا C2C12 KO Aars2 ) وخلايا HL-1، Aars خلايا (تم الكشف عنها قبل وبعد علاجها بـ. وقت المطاردة لـوكان زمن -بالميتات 1 ساعة و 12 ساعة، على التوالي. جميع البيانات مُبلغ عنها كمتوسط.SEM لثلاث تجارب مستقلة. تم تقييم الدلالة الإحصائية بواسطة اختبار الطالب غير المتزاوج ذو الطرفين.-اختبار t وتحليل التباين ثنائي الاتجاه:; ؛; **** ; لا أهمية.
اللاكتيلation. علاوة على ذلك، فإن إعادة الأكسجة للخلايا C2C12 المعالجة بنقص الأكسجين لم تقلل فقط من مستويات Lac-K336 وLac-K457/8، بل أيضًا فعلت PDHA1 وCPT2 وزادت من إنتاج Ac-CoA (معلومات إضافية، الشكل S8c، d)، مما يتماشى مع أن نقص الأكسجين الناتج عن OXPHOS هو المحرك لتثبيط OXPHOS. ومع ذلك، فإن إعادة الأكسجة أثرت بشكل ضئيل على نشاط SIRT3 (معلومات إضافية، الشكل S8e)، مما يشير إلى أن OXPHOS يستجيب بسرعة للاكتيلation التي تتوسطها AARS2 ولكن ليس للديلاكتيلation التي تتوسطها SIRT3.
حقنة منأدى حامض اللبنيك إلى تحفيز HIF1a وAARS2 في عضلة الفخذ الخلفية للفأر (الشكل 7ب)، وLac-K336/Lac-K457/8 (المعلومات التكميلية، الشكل S8f)، بينما تم تثبيط LDHA باستخدامقلل FX-11 من إنتاج اللاكتات في عضلات الفئران أثناء الجري (المعلومات التكميلية، الشكل S8g) وألغى إلى حد كبير التحفيز المرتبط بالجري لـ HIF1a و AARS2 و Lac-K336 و Lac-K457/8 في أفخاذ الساق الخلفية للفئران (الشكل 7c)، مما يعكس أن مثبط LDHA الأوكزامات يثبط Lac-K336 و Lac-K457/8 في خلايا C2C12 (المعلومات التكميلية، الشكل S8h). تبرز هذه البيانات حقيقة أن إنتاج اللاكتات والأكسدة يلعبان دورًا رئيسيًا في تحفيز نقص الأكسجة في خلايا العضلات واللاكتيل أثناء التمرين. ومن الجدير بالذكر أن الجري لمدة 30 دقيقة، الذي أدى إلى تراكم كبير للاكتات في كل من عضلات الفئران الهيكلية من النوع الأول الغني بالميتوكوندريا وقدرة التحمل والنوع الثاني الفقير بالميتوكوندريا وقدرة السرعة (الشكل 7d)، أدى إلى المزيد من تم نطق HIF1a و AARS2 و Lac-K336 و Lac-K457/8 في العضلات من النوع الأول (الشكل 7e)، مما يشير إلى أن اللاكتيل يضبط أكسدة الفوسفور في العضلات الهيكلية من النوع الأول وقدرة التحمل في التمارين أكثر مما يضبط أكسدة الفوسفور في العضلات الهيكلية من النوع الثاني وقدرة العدو. تدعم الملاحظة بأن انخفاض ATP في العضلات من النوع الأول حدث بالتزامن مع تحفيز Lac-K336 و Lac-K457/8، على عكس انخفاض ATP في العضلات من النوع الثاني الذي حدث قبل تحفيز Lac-K336 و Lac-K457/8 (الأشكال 7f، 5j)، هذه الفرضية. علاوة على ذلك، Aars2 من النوع البري، منخفض اللاكتيلو Sirt3 المفرط في اللاكتيلكانت الفئران جميعها تتمتع بقدرات جري مشابهة (معلومات إضافية، الشكل S8i)، لأن حقن حمض اللبنيك في فخذ الساق الخلفية للفأر أثر فقط بشكل ضئيل على قدراتهم في الجري (معلومات إضافية، الشكل S8j). بالمقابل، كانت أوقات نفاد الطاقة أثناء الجري عالي الكثافة لدى الفئران مرتبطة بشكل إيجابي معتدل بمستويات SIRT3 في عضلات الساق الأساسية (معلومات إضافية، الشكل S8k) ومرتبطة بشكل عكسي قوي بمستويات AARS2 في عضلات الساق الأساسية (الشكل 7g؛ معلومات إضافية، الشكل S8I). علاوة على ذلك، فإن زيادة التعبير عن AARS2 بشكل محدد في عضلات الهيكل العظمي للفئران من خلال عبور فئران تعبير hAARS2 مع فئران CKMM-cre (معلومات إضافية، الشكل S8m) وإسقاط Sirt3، زادت كل من Lac-K336 وLac-K457/8 (الشكل 6g؛ معلومات إضافية، الشكل S8n)، لكنها قللت من تدفق Ac-CoA (الشكل 7h)، وOCR (الشكل 7i)، ومستويات ATP (الشكل 7j) في عضلات الساق الهيكلية وأدت إلى تقصير نفاد الطاقة أثناء الجري.
للكاتيون
PDHA1
K336
nالوقت (الشكل 7k). علاوة على ذلك، على عكس حقن اللاكتات الذي قلل من وقت نفاد جهد الفئران،حقن الألانين في عضلة الفخذ في الساق الخلفية قلل من Lac-K336 وLac-K457/8 (معلومات إضافية، الشكل S8f)، مما يتماشى مع ذلك.الألانين كان نظيرًا للألانين (الشكل 5أ) الذي يرتبط بالألانين- جيب الربط لـ AARS2 (معلومات إضافية، الشكل S4f، g)، مما يقلل من مستويات Lac-K336 و Lac-K457/8 في المختبر (معلومات إضافية، الشكل S9a، b)، وينشط PDHA1 و CPT2 (معلومات إضافية، الشكل S9c، d)، ويزيد من مستويات Ac-CoA (معلومات إضافية، الشكل S9e)، OCR (معلومات إضافية
الشكل 5 يقوم AARS2 بتعطيل PDHA1 وCPT2 عن طريق إضافة مجموعة لاكتيل إلى هذه البروتينات. أ حمض اللبنيك،-ألانين، والألانين هما نظيران. محاذاة الهياكل الكيميائية للألانين، وحمض اللبنيك، و-ألانين. يقوم AARS2 بإضافة اللاكتيل إلى ببتيدات PDHA1 K336 وCPT2 K457/8 في المختبر. الكشف عن ببتيدات PDHA1 K336 وCPT2 K457/8 الاصطناعية المضافة باللاكتيل بواسطة AARS2 وAARS2 باستخدام تقنية قياس الطيف الكتلي.ملخصة (لا يوجد منتج مكتشف،: تم اكتشاف المنتج : تم الكشف عن منتج أقل؛ انظر المعلومات التكميلية، الشكل S6b لنتائج MS). ج AARS2 هو ناقل لاكتيل فعال.وتم تحديد نشاط AARS2 المؤتلف تجاه اللاكتات عندما تم الاحتفاظ بالببتيد PDHA1 المحتوي على K336 الاصطناعي في. د، هـ KO يلغي التحفيز المرتبط باللاكتات لـ Lac-K336 و Lac-K457/8. تم الكشف عن مستويات Lac-K336 (د) ومستويات Lac-K457/8 (هـ) لـ PDHA1 و CPT2 المنقاة من خلايا C2C12 وخلايا C2C12 التي تم فيها حذف Aars2 باستخدام sgRNAs مستقلة، مع أو بدون 10 مللي مول من Me-Lac في وسط الثقافة.إن تثبيط استيراد اللاكتات إلى الميتوكوندريا يقلل من مستويات Lac-K336 وLac-K457/8. خلايا C2C12 التي لم تُعالج أو التي تم علاجها بـ-CHCA لمدة ساعتين، أو مع 10 مللي مول من اللاكتات لمدة 4 ساعات. لاكتات الميتوكوندريا (ف) (تم الكشف عن Lac-K336 (g) وLac-K457/8 (h). يزيد الجري بشكل متناسب من مستويات اللاكتات في الميتوكوندريا. تم الكشف عن مستويات اللاكتات في الميتوكوندريا والمستخلصات في عضلات الساق الهيكلية للفئران قبل وبعد 30 دقيقة من الجري.تم تأكيد نجاح استخراج وعزل الميتوكوندريا من خلال صبغ كل من علامة الميتوكوندريا COXIV وعلامة السيتوسول GAPDH (كما هو موضح لاحقًا). الجري يحفز Lac-K336 وLac-K457/8. تم الكشف عن مستويات Lac-K336 وLac-K457/8 في PDHA1 وCPT2 المنقاة من عضلات الساق الهيكلية للفأر بعد الجري لفترات زمنية محددة.تم الكشف عن مستويات PDHA1 وCPT2 في خلايا C2C12 قبل وبعد تعرضها لنقص الأكسجين. زادت العلاجات المشتركة لـ AARS2 واللاكتات من مستويات Lac-K336 و Lac-K457/8. تم تقدير مستويات Lac-K336 و Lac-K457/8 في عضلات الساق الهيكلية للفئران المستريحة والفئران التي تعبر عن AARS2 بشكل مفرط والتي تم إعطاؤها حقن اللاكتات في عضلة الساق باستخدام تقنية الطيف الكتلي (MS). ). ، زاد الجري ونقص الأكسجين من مستويات Lac-K336 وLac-K457/8. مستويات Lac-K336 وLac-K457/8 في عضلات الساق الهيكلية للفأر قبل وبعد 30 دقيقة من الجري (n) وتم ضخها بمحلول كريبس رينجر المؤكسج أو بدون.في نظام اختبار العضلات المعزولة السليمة 1200 A لمدة 30 دقيقة (o) تم تقديرها باستخدام MS ( ). يعمل AARS2 على تعطيل PDHA1 وCPT2 من خلال تحفيز اللاكتيل. تأثيرات زيادة تعبير AARS2 على مستويات اللاكتيل والأنشطة المحددة ) من PDHA1 ( ) و CPT2 ( ) وتم الكشف عن طفرات مواقع اللاكتيل الخاصة بهم.يؤدي اللاكتات إلى تعطيل PDHA1 وCPT2 من خلال تحفيز اللاكتيل. تأثيرات 10 مللي مول من Me-Lac على Lac-K336 والأنشطة المحددة لـ PDHA1 وطفرات موقع اللاكتيل الخاصة به. ) ، وتم الكشف عن تأثيرات Me-Lac على Lac-K457/8 والنشاطات المحددة لـ CPT2 وطفرات موقع اللاكتيل (s) الخاصة به ( جميع البيانات مُبلغ عنها كمتوسطSEM لثلاث تجارب مستقلة. تم تقييم الدلالة الإحصائية بواسطة اختبار الطالب غير المتزاوج ذو الطرفين.-اختبار t وتحليل التباين ثنائي الاتجاه:؛؛; لا أهمية. المعلومات، الشكل S9f)، ومستويات ATP (المعلومات التكميلية، الشكل S9g) في خلايا C2C12.أدى تناول -ألانين إلى زيادة في وقت نفاد الجري في فئران C57BL/6 (الشكل 7I) وكذلك في الفئران التي تحتوي على AARS2، ولكن ليس في الفئران التي تفتقر إلى AARS2 (الشكل 7m). وأخيرًا، تم دعم الفكرة القائلة بأن Lac-K336 وLac-K457/8 تثبط OXPHOS وتقيّد الجري التحمل من خلال الاكتشاف بأن كل من التنفس الأساسي والحد الأقصى لعضلات الساق الهيكلية للفئران قد زاد بسبب نقص Aars2، ولكنه انخفض بسبب فرط تعبير AARS2 أثناء التمرين، وكانت استجابات التنفس مرتبطة بمستويات AARS2 (معلومات إضافية، الشكل S9h).
يمكن تسليط الضوء على الأدوار الفسيولوجية المحتملة التي تلعبها Lac-K336 وLac-K457/8 في التمرين من خلال فحص Aars2 الخالي من Lac-K336 وLac-K457/8.الفئران. بينما Aars2تحملت الفئران جريًا مكثفًا وأظهرت الفئران التي تعبر عن AARS2 بشكل مفرط ضعفًا في الجري المكثف (الشكل 7م)، Aars2تراكمت الفئران مستويات أعلى من أنواع الأكسجين التفاعلية (ROS) ومؤشر مالونديالديهايد (MDA) الناتج عن ROS، وعلامة تلف الأكسدة الناتجة عن تأكسد الدهون.في عضلات الساق الهيكلية لديهم أكثر من تلك الموجودة في عضلات الساق الهيكلية للفئران من النوع البري، في وقت الإرهاق (الشكل 7ن، 7ع). وهذا يشير إلى أن خلايا العضلات قد تستخدم بروتين الميتوكوندريا اللاكتيل لحماية نفسها من الأضرار التأكسدية المحتملة أثناء تمارين التحمل. وبالتالي، بينما يتطلب تعزيز OXPHOS تحويل الأسيتيل- CoA الناتج عن اللاكتات والجلوكوز والأحماض الدهنية إلى ATP أثناء تمارين التحمل، فإن تدفق Ac-CoA إلى OXPHOS يتم تثبيطه بالتغذية الراجعة بواسطة نقص الأكسجين داخل الخلايا الناتج عن OXPHOS (الشكل 7ص).
نقاش
لقد أظهرنا أن نظام الفسفرة المؤكسدة (OXPHOS) يخضع لتنظيم سلبي بواسطة اللاكتيل من البروتينات الميتوكوندرية، والذي يتم تحفيزه بواسطة نقص الأكسجين داخل الخلايا الناتج عن OXPHOS. لذلك، يمثل اللاكتيل من البروتينات الميتوكوندرية آلية تغذية راجعة للحد من OXPHOS. بالإضافة إلى ارتباط حمض اللبنيك بمستقبلات الأحماض الهيدروكربوكسيلية.وتفعيل ميتالوبروتيناز المصفوفة-2 لتنشيط TGF-إشارةتمثل عملية اللاكتيلation التي تتوسطها AARS2 وسيلة أخرى للإشارة باللاكتات. نظرًا لأن اللاكتيلation البروتينية الميتوكوندرية تستجيب لنقص الأكسجين داخل الخلايا، يجب أن تشمل أهميتها على الأقل منع عيوب نقص الأكسجين، مثل الإفراط في إنتاج الجذور الحرة الضارة (ROS) والأضرار الناتجة عنها كما لوحظ في الدراسة الحالية. من المتوقع أن تكون هناك أهمية فسيولوجية أو مرضية أكبر للاكتيلation البروتينية الميتوكوندرية لأن المستويات العالية تحدث استجابات حمض اللبنيك ونقص الأكسجة في عمليات فسيولوجية مثل التمرين وتطور الأجنة، وفي عمليات مرضية مثل السرطانات.
تدمج عملية لاكتيل بروتين الميتوكوندريا بين الأيض ونقص الأكسجين، وهما مترابطان، للحد من عملية الفسفرة التأكسدية. إن ارتفاع حمض اللاكتيك في تطور الأجنة والسرطان يعود إلى زيادة الحاجة إلى المواد الأيضية بما في ذلك سلف حمض اللاكتيك البيروفات، وارتفاع حمض اللاكتيك أثناء التمرين يعود إلى تعزيز التحلل السكري وتحلل الجليكوجين الذي يزيد من مستوى البيروفات. علاوة على ذلك، يمكن أكسدة حمض اللاكتيك فقط عبر عملية الفسفرة التأكسدية، التي تستهلك الأكسجين داخل الخلايا. هذه العوامل، بالإضافة إلى أن مستويات حمض اللاكتيك تختلف بشكل كبير في هذه العمليات، تجعل من لاكتيل بروتين الميتوكوندريا آلية فعالة للحد من عملية الفسفرة التأكسدية. ومن الجدير بالذكر أنه على الرغم من أن اللاكتات يمكن أن تكون ركيزة لعملية الفسفرة التأكسدية عندما لا يكون الأكسجين داخل الخلايا مستنفدًا، إلا أن لاكتيل بروتين الميتوكوندريا يؤثر على تثبيط عملية الفسفرة التأكسدية عندما يتم تحفيز نقص الأكسجين داخل الخلايا، كما هو الحال خلال المرحلة المرهقة من تمارين التحمل. إنها تختلف عن آليات تنظيم عملية الفسفرة التأكسدية الأخرى، مثل الأسيتيل، والتنظيم الألستيري لـ PDHA1، التي تدمج المغذيات (أسيتيل-CoA) والإشارات الهرمونية.لتسهيل إمداد ATP بعد بدء التمرين، حيث لا يتم تحفيز نقص الأكسجة داخل الخلايا وتكون الحالة الحرجةلأقصى تنفس ميتوكوندري يكون منخفضًا كماتُورلضمان وظيفة كاملة لنظام أكسدة الفسفور المؤكسد. لا نستبعد إمكانية أن تنظيم بروتين الميتوكوندريا عن طريق اللاكتيل قد يؤثر أيضًا على القدرة على ممارسة الرياضة من خلال التأثير على خلايا أخرى غنية بالميتوكندريا وتستخدم اللاكتات، بما في ذلك خلايا القلب، كما لاحظنا في خلايا القلب المزروعة، ونظرًا لأن طفرات AARS2 قد ارتبطت باعتلال عضلة القلب.
يُعرف كل من AARS2 و SIRT3 كمنظّمين لعملية الفسفرة التأكسدية (OXPHOS) واللاكتات ووظيفة العضلات. تؤدي طفرة AARS2 إلى ضعف OXPHOS، واعتلال عضلة القلب الميتوكوندري، وفقدان وظيفة العضلات، وتراكم اللاكتات في الأنسجة.فقدان SIRT3 يسبب ضعف في OXPHOS وتراكم اللاكتات.علاوة على ذلك، يتم تحفيز SIRT3 خلال التدريب،مما يشير إلى أن SIRT3 قد يلعب دورًا في التكيف مع التدريب. يمتلك SIRT3 أدوارًا متعددة في تنظيم OXPHOS. في ظل الظروف الهوائية، يقلل SIRT3 من أسيتيل PDHA1 لتعزيز إنتاج اللاكتات وأكسدة الأحماض الدهنية في عضلات الفئران.بينما تحت ظروف نقص الأكسجين، يقلل من لاكتيلation PDHA1 لتعزيز OXPHOS. من خلال أي من العمليتين، يعزز SIRT3 إنتاج ATP في خلايا العضلات أثناء التمرين. علاوة على ذلك، قد يؤدي تقليل لاكتيلation البروتين الميتوكوندري من خلال استهداف كل من AARS2 و SIRT3 إلى تعزيز مؤقت لوظيفة أكسدة الفسفور المؤكسد في خلايا العضلات وقدرة التحمل أثناء التمارين كما لاحظنا في الفئران. ومع ذلك، لا ينبغي تشجيع هذه الأساليب التي تعزز قدرة التحمل أثناء التمارين، حيث إن فقدان حماية بروتين الميتوكوندريا من اللاكتيل قد يؤدي إلى تلف الجذور الحرة.
المواد والأساليب
المواد الكيميائية والأجسام المضادة
المواد الكيميائية والمواد المتفاعلة: ATP (رقم الكاتالوج A6559)، لاكتات (رقم الكاتالوج L6661)، ميثيل لاكتات (رقم الكاتالوج 230340)،-ألانين (رقم الكاتالوج 146064)، L-ألانين (رقم الكاتالوج A7627)، DAPI (رقم الكاتالوج D9542)، MG132 (رقم الكاتالوج M8699)، PPi (رقم الكاتالوج 433314)، NAM (رقم الكاتالوج
الشكل 6 SIRT3 يعكس Lac-K336 وLac-K457/8. أ. تزيد علاجات NAM من Lac-K336 وLac-K457/8. تم الكشف عن مستويات Lac-K336 وLac-K457/8 من PDHA1 وCPT2 المنقاة من خلايا C2C12 وخلايا C2C12 المعالجة بـ NAM. تم فحص الخلايا بعد 3 ساعات من علاج 5 مليمول NAM. ب. يقوم SIRT3 بإزالة اللاكتيل من Lac-K336 وLac-K457/8. تم تحليل قدرة SIRT3، وSIRT4 المجال التحفيزي، وSIRT5 على إزالة اللاكتيل من الببتيدات الاصطناعية التي تحتوي على Lac-K336 وLac-K457/8 عبر MS للكشف عن تكوين الأنواع اللاكتيلية. ج-و. يقلل SIRT3 من مستويات Lac-K336 وLac-K457/8. تم الكشف عن مستويات Lac-K336 وLac-K457/8 في PDHA1 المنقى (ج) وCPT2 (د) قبل وبعد إزالة اللاكتيل باستخدام SIRT3 أو SIRT3 غير الفعال تحفيزياً.تم الكشف عن تأثيرات تعبير SIRT3 على النوع البري، وPDHA1 الخالي من اللاكتيل (e)، وCPT2 (f) في خلايا C2C12.إزالة Sirt3 تزيد من مستويات Lac-K336 وLac-K457/8 في الجسم الحي. مستويات Lac-K336 وLac-K457/8 من النوع البري وSirt3تم الكشف عن عضلات الهيكل العظمي لساق الفأر. h، i SIRT3 ينظم أنشطة PDHA1 و CPT2. الأنشطة النسبية لـ PDHA1 و CPT2 التي تم تنقيتها من خلايا C2C12 والتي تعبر عن SIRT3 بشكل مفرط ( ) أو خلايا C2C12 KO Sirt3 (i) تم مقارنتها ( ). ينظم SIRT3 إنتاج Ac-CoA من التحلل السكري وأكسدة الأحماض الدهنية.، و “نسب من Ac-CoA من-جلوكوز ( ) و -بالميتات ( ) تم اكتشافها في خلايا C2C12 التي تعبر عن SIRT3 بشكل مفرط ( ) أو التي تم فيها حذف Sirt3 باستخدام sgRNAs مستقلة ( ) ( ). وقت المطاردة لـ -جلوكوز وكان زمن الـ -بالميتات 1 ساعة و 12 ساعة، على التوالي.SIRT3 ينظم مستويات اللاكتات والأحماض الدهنية الحرة. اللاكتات ( ) ومستويات الأحماض الدهنية الحرة ( ) في خلايا C2C12 التي تعبر عن SIRT3 بشكل مفرط ( ) أو خلايا C2C12 التي تم فيها حذف Sirt3 عبر sgRNAs مستقلة ( ) تم اكتشافها ( جميع البيانات مُبلغ عنها كمتوسطSEM لثلاث تجارب مستقلة. تم تقييم الدلالة الإحصائية بواسطة اختبار Student ذو الطرفين غير المتزاوج.-اختبار t وتحليل التباين ثنائي الاتجاه:؛؛. 72340)-بالميتات (رقم الكاتالوج 605573) ومجموعة عد الخلايا-8 (رقم الكاتالوج 96992) تم شراؤها من سيغما-ألدريش. أوليغوميسين A (رقم الكاتالوج S1478)، FCCP (رقم الكاتالوج S8276)، α-CHCA (رقم الكاتالوج S8612) وروكسايدوستات (رقم الكاتالوج S1007) تم شراؤها من سيلك. FX-11 (رقم الكاتالوج HY-16214) تم شراؤه من ميد كيم إكسبريس. تم شراء فوسفاتاز البروتين لامبدا (رقم الكاتالوج P0753) من نيو إنجلاند بايو لابز.تم شراء C-glucose (رقم الكاتالوج CLM1396) من مختبرات كامبريدج لل isotopes. تم شراء كريات سيباروز بروتين-A (رقم الكاتالوج 16-156) من ميرك ميليبور. تم شراء بنسلين-ستربتوميسين (رقم الكاتالوج 15070063) من إنفيتروجين. CellTiter-Gloتم شراء مجموعة الكواشف (رقم الكات G9241) من شركة بروميجا. تم شراء مجموعة اختبار نشاط إنزيم اللاكتات ديهيدروجيناز (رقم الكات K726) ومجموعة قياس الأحماض الدهنية الحرة (رقم الكات K612) من شركة بيوفيجن. تم شراء مجموعة اختبار نشاط إنزيم بيروفات ديهيدروجيناز (PDH) في صفيحة ميكرو (رقم الكات ab109902) ومجموعة اختبار نشاط SIRT3 (رقم الكات ab156067) من شركة أبكام. تم شراء الببتيدات الاصطناعية من شركة توب ببتيد. تم شراء الأوليغونيوكليوتيدات الاصطناعية من شركة جينفارما.
الأجسام المضادة: تم شراء anti-AARS1 (رقم الكاتالوج sc165990) من سانتا كروز. تم شراء anti-VHL (رقم الكاتالوج ab140989)، anti-HIF2a (رقم الكاتالوج ab109616)، anti-HIF3a (رقم الكاتالوج ab2165)، anti-CPT2 (رقم الكاتالوج ab110293)، anti-PDHA1-pS293 (رقم الكاتالوج ab177461)، anti-COXIV (رقم الكاتالوج ab202554)، anti-GAPDH (رقم الكاتالوج ab1439) و anti-Actin (رقم الكاتالوج ab1440) من أبكام. تم شراء anti-AARS2 (رقم الكاتالوج 22696-1-AP)، anti-PDHA1 (رقم الكاتالوج 18068-1-AP)، anti-DLD (رقم الكاتالوج 16431-1-AP)، anti-DLAT (رقم الكاتالوج 13426-1-AP)، anti-PDHB (رقم الكاتالوج 14744-1-AP)، anti-PHD1 (رقم الكاتالوج 12984-1-AP)، anti-PHD2 (رقم الكاتالوج 19886-1-AP)، anti-PHD3 (رقم الكاتالوج 18325-1-AP)، anti-CPT1A (رقم الكاتالوج 15184-1-AP)، anti-EHHADH (رقم الكاتالوج 26570-1-AP)، anti-ACADS (رقم الكاتالوج 16623-1-AP)، anti-ACADM (رقم الكاتالوج 55210-1-AP) و anti-ACADL (رقم الكاتالوج 17526-1-AP) من بروتين تك. تم شراء anti-HIF1a (رقم الكاتالوج 36169S) من تكنولوجيا الإشارات الخلوية. تم شراء anti-PDHA1-pS232 (رقم الكاتالوج LS-C265964/64724) من لايف سبان بيوساينس. تم شراء anti-PDHA1-pS300 (رقم الكاتالوج ABS194) من شركة EMD Millipore. تم شراء anti-Laminin (رقم الكاتالوج L9393) من سيغما-ألدريش. تم شراء anti-Flag (رقم الكاتالوج M20008)، anti-HA (رقم الكاتالوج M20002)، anti-Myc (رقم الكاتالوج M20003) من أب مارت. تم شراء Goat anti-rabbit IgG (رقم الكاتالوج 111-035-003) و Goat anti-mouse IgG (رقم الكاتالوج 115-035-003) من جاكسون. تم تصنيع anti-LacK336، anti-LacK457/8، anti-P377OH و anti-ECHS1 محليًا في هذه الدراسة.
الحيوانات
تم الحصول على ذكور فئران C57BL/6J من شركة شنغهاي SLAC للحيوانات المخبرية المحدودة (شنغهاي، الصين). تم وضع الفئران في أقفاص تحت دورة ضوء/ظلام مدتها 12 ساعة في غرفة تتحكم في درجة الحرارة والرطوبة، وتم إطعامها طعام مختبر قياسي وماء حسب الحاجة. ثم تم تقسيمها عشوائيًا إلى مجموعات ضابطة وتجريبية. تم إنتاج فئران ترانسجينية hAARS2 مشروطة في خلفية جينية C57BL/6 عن طريق إدخال CAG-LSL-hAARS2-WPRE-PA في جين Rosa26 باستخدام نظام تحرير الجينوم المعتمد على CRISPR-Cas9 من قبل مركز شنغهاي لنماذج الكائنات الحية، Inc. (شنغهاي، الصين). تم إنتاج فئران hAARS2 ترانسجينية محددة عن طريق تزاوج فئران hAARS2 ترانسجينية مشروطة مع فئران CKMM-cre (رقم مخزون جاكسون #029100). Aars2تم الحصول على فئران تحتوي على موقعين loxP يحيطان بالإكسون 1-5 من شركة جيم فارما تك المحدودة (شنغهاي، الصين). تم إنتاج فئران ترانسجينية محددة للعضلات عن طريق تزاوج فئران Aars2 KO الشرطية مع فئران CKMMcre. Sirt3تم الحصول على الفئران من مركز شنغهاي لنماذج الكائنات الحية، وتم تأكيد أنماطها الجينية عبر تفاعل البوليميراز المتسلسل (PCR). تم إجراء جميع إجراءات الحيوانات وفقًا للجنة رعاية الحيوانات في جامعة فودان.
زراعة الخلايا
تم شراء خطوط خلايا العضلات الهيكلية للفئران C2C12، وخلايا عضلة القلب للفئران HL-1، وخلايا الكلى الجنينية البشرية HEK293T من مجموعة الثقافة الأمريكية (الولايات المتحدة الأمريكية). تم زراعة جميع خطوط الخلايا المذكورة أعلاه في وسط دالبكو المعدل من إيجل (DMEM) (هايكلاون، الولايات المتحدة الأمريكية) مع إضافةمصل جنيني من الأبقار (جيبكو، الولايات المتحدة الأمريكية) فيتحتفي حاضنة مرطبة. تم اختبار جميع خطوط الخلايا وتوثيقها من خلال تقييم شكلها ومعدل نموها؛ جميعها خالية من الميكوبلازما.
تم عزل الخلايا الكبدية الأولية من الفئران باستخدام طريقة التروية ذات الخطوتين. تم حقن الفئران داخل الصفاق بـ 1% نيمبوتال، وتم قسطرة الأوردة البابية. بعد ذلك، تم استخدام 0.5 مللي مول من EDTA وتم تسخين الكولاجيناز IV (سيغما ألدريتش، الولايات المتحدة الأمريكية) مسبقًا، وتم ضخه بشكل متتابع في الكبد حتى تفتت هيكل الكبد وتشتت الخلايا. وتم تصفية التعليق الخلوي الناتج من خلالتم تصفية (NEST، ووكسي، الصين)، وغسلها بمحلول ويليام E المتوسط (جيبكو، الولايات المتحدة الأمريكية)، وتنقيتها باستخدام محلول بيركول (GE، الولايات المتحدة الأمريكية). تم زراعة خلايا الكبد بكثافةخلايا/بئر في وسط ويليام الخالي من المصل. تم التحقق من خلايا الكبد الأولية للفئران من خلال تقييم شكل الخلايا وتعبير الألبومين. تم عزل خلايا العضلات الأولية للفئران وتم تعديل ظروف الزراعة من البروتوكولات المنشورة.تم عزل عضلات الأطراف الخلفية من الفئران البالغة ثم تم هضمها في وسط DMEM يحتوي علىمل من الكولاجيناز II (سيغما ألدريتش، الولايات المتحدة الأمريكية) وهيبز. تم تصفية المعلقات من خلالثممرشحات. تم تعليق الخلايا في وسط F-10 يحتوي على 20% من مصل العجل الجنينى.عامل نمو الألياف الأساسية (bFGF) فيطبق مغطى مسبقًا بمادة ماتريجل، محفوظ فيلمدة 72 ساعة. ثم تم نقل الخلايا إلى طبق غير مغطى بمادة ماتريجيل لمدة 45 دقيقة لإزالة الخلايا الليفية التي التصقت في هذه الخطوة. تم نقل السائل الخلوي إلى طبق جديد مغطى مسبقًا بمادة ماتريجيل وزرعه.
الأجسام المضادة المحددة للموقع
تم إنتاج أجسام مضادة متعددة النسائل محددة لموقع اللاكتيل في أرنب نيوزيلندي ذكر عمره 3 أشهر باستخدام ببتيد PDHA1 المحتوي على Lac-K336 أو ببتيد CPT2 المحتوي على Lac-K457/8. تم ربط هذه الببتيدات من خلال سيستين في الطرف C إلى هيموسيانين قوقع المفتاح (KLH). تم تحصين الأرانب بـتم حقن المستحضر في معزز فريد الكامل. تم إجراء حقن معززة كل 4 أسابيع. تم استئصال دم الأرانب بعد 10-14 يومًا من الحقنة النهائية. تم جمع 50 مل من الدم من كل أرنب وتم طرده مركزيًا. تم تنقية الطور العلوي (المصل) باستخدام ببتيدات المستضد وفقًا للبروتوكولات المنشورة. تم إنتاج أجسام مضادة متعددة النسائل محددة بموقع الهيدروكسيل باستخدام ببتيد AARS2 المحتوي على OH-P377 كما هو موضح أعلاه. المستضد المستخدم:
ببتيد PDHA1 المحتوي على Lac-K336: ASVEEL(K-Lac)EIDVEV-C ببتيد CPT2 المحتوي على Lac-K457/8: EFLK(K-Lac)QKLS-C ببتيد AARS2 المحتوي على OH-P377: C-GSLV(P-OH)VVVET
البلازميد
تم استنساخ AARS2 و VHL في pcDNA3.1(b+)-Flag و pcDNA3.1(b+)Myc، على التوالي، بين مواقع Xhol و EcoRI. تم استنساخ PDHA1 و CPT2 في pcDNA3.1(b+)-Flag بين مواقع Xhol و EcoRI. تم استنساخ PDH2 في pcDNA3.1(b+)-Myc بين مواقع Xhol و EcoRI، وتم استنساخ SIRT3 في pcDNA3.1(b+)-HA بين مواقع Xhol و EcoRI.
تحويل الخلايا
تم إجراء نقل البلازميد باستخدام بولي إيثيلين إيمين (PEI، بوليساينس، الولايات المتحدة الأمريكية) أو ليبوفكتامين 8000 (بيوتايم، شنغهاي، الصين).
لإجراء نقل الجينات باستخدام PEI، تم إضافة البلازميد و PEI إلى DMEM خالي من المصل مع التحريك بقوة، وتم احتضان المزيج لمدة 15 دقيقة قبل إضافته إلى وسط زراعة الخلايا. أما بالنسبة لنقل الجينات باستخدام Lipofectamine 8000، فقد تم إضافة البلازميد و Lipofectamine 8000 إلى DMEM خالي من المصل، وتم خلطه برفق باستخدام ماصة، ثم أضيف مباشرة إلى وسط زراعة الخلايا. تم استبدال وسط الزراعة كلبعد التحويل. تم جمع الخلايا بعد 36 ساعة.
علاجات الخلايا
مي-لاكتات، ألانين،-ألانين وNAM. تم زراعة الخلايا في وسط RMPI 1640 القياسي (US Biological، الولايات المتحدة الأمريكية)، وتم نقلها إلى RMPI 1640 خالي من الأحماض الأمينية لمدة 30 دقيقة قبل المعالجات. التركيزات المحددة منلاكتات، ألانين، وتم إضافة -ألانين إلى وسط الثقافة عندما وصلت كثافة الخلايا إلىالتقاء عندقبل الحصاد. تمثل معالجة NAM في إضافة 5 مللي مول من NAM إلى وسط DMEM خالي من المصلقبل الحصاد.
الشكل 7 يدفع OXPHOS إلى اللاتيل لتحديد حدود OXPHOS وقدرة التحمل في الجري. أ. يحفز OXPHOS نقص الأكسجة داخل الخلايا. HIFومستويات AARS2 في خلايا C2C12 في قاعدة DMEM والجلوكوز-بالميتات-وميلك-تم اختبار وسائط DMEM المدعمة في وجود أو غيابروتينون الذي يثبط OXPHOS.حقن اللاكتات يسبب نقص الأكسجين في عضلات الفئران. HIF1تم الكشف عن مستويات AARS2 في أفخاذ الساقين الخلفيتين للفأر قبل وبعدتم حقن اللاكتات في عضلة الساق الخلفية (إنتاج اللاكتات ضروري لتحفيز نقص الأكسجة واللاكتيل في عضلات أرجل الفئران. HIF1تم تقييم مستويات AARS2 واللاكتيل في أفخاذ أرجل الفئران الخلفية غير المعالجة والفئران التي تم معالجتها مسبقًا بـ FX-11 عندما بدأت في الجري. يؤدي الجري إلى تراكم اللاكتات في عضلات أرجل الفئران الهيكلية. تم تحديد مستويات اللاكتات النسبية في عضلات الفئران الهيكلية من النوع الأول والنوع الثاني قبل وبعد 30 دقيقة من الجري.تسبب الجري في زيادة ملحوظة في اللاكتيل في العضلات من النوع الأول. HIF1تم تحديد مستويات AARS2، بالإضافة إلى Lac-K336 وLac-K457/8 في عضلات الهيكل العظمي للفئران من النوع الأول والنوع الثاني عند بدء الجري.انخفاض ATP في العضلات الهيكلية من النوع الأول مرتبط بزيادة في Lac-K336 وLac-K457/8. تم مراقبة مستويات ATP في عضلات الساق الهيكلية من النوعين الأول والثاني في الفئران بعد الجري لفترات زمنية محددة.تظهر التغيرات في Lac-K336 و Lac-K457/8 (الشكل 5j). g مستويات AARS2 الأساسية في عضلات الساق الهيكلية للفئران مرتبطة عكسياً مع وقت نفاد جهدها أثناء الجري. كان هناك ارتباط بين وقت نفاد الجهد أثناء الجري عالي الكثافة ومستويات AARS2 في عضلات الساق أثناء الراحة. ). يعيق Lac-K336 و Lac-K457/8 تدفق Ac-CoA في عضلات الهيكل العظمي للفئران.-مستويات Ac-CoA في النوع البري، وذوات التعبير المفرط لـ AARS2 وSirt3تم الكشف عن عضلات الهيكل العظمي لساق الفأر بعد ساعة واحدة من تلقي كل فأر “حقن الجلوكوز عبر الوريد الذيلتقلل اللاكتيلation من معدلات استهلاك الأكسجين في العضلات الهيكلية للفئران وإنتاج ATP. مستويات استهلاك الأكسجين (i) و ATP (j) في الميتوكوندريا في عضلات الساق الهيكلية في حالة الراحة، وذوات التعبير المفرط عن AARS2، و Sirt3.تم تحليل الفئرانيعيق Lac-K336 وLac-K457/8 وقت نفاد جري الفئران. تم قياس وقت نفاد جري الفئران عالي الكثافة في النوع البري، والفئران التي تعبر عن AARS2 في العضلات، وSirt3.فئرانأنا أرضع و-ألانين ينظم بشكل عكسي وقت نفاد جري الفئران. أوقات نفاد الجري عالية الكثافة للفئران غير المعالجة أو المعالجة باللاكتات أوتم قياس -ألانين عن طريق حقن عضلة الساق، على التوالي. ). -ألانين يطيل وقت إرهاق الفئران بطريقة تعتمد على AARS2. أوقات إرهاق الجري عالي الكثافة من النوع البري، وذوي التعبير المفرط عن AARS2، وAars2فئران مع أو بدون عضلة الساقحقن الألانينتم قياس (.فقدان اللاتيل يؤدي إلى زيادة تلف الجذور الحرة. مستويات الجذور الحرة ( ) ومستويات المالونديالديهايد المحفزة ( النوع البري و Aarsالفئران في وقت نفاد الطاقة أثناء الجري ) تم مقارنتها. يوضح الرسم البياني أن التمارين تعزز أكسدة اللاكتات من خلال OXPHOS، مما يعزز نقص الأكسجة داخل الخلايا الذي يحفز AARS2 و Lac-K336 و Lac-K457/8 لتثبيط تدفقات Ac-CoA من اللاكتات/تحلل السكر و FAO و OXPHOS. الآليات التي تم الكشف عنها في هذه الدراسة موضحة بأسهم حمراء. جميع البيانات مُبلغ عنها كمتوسط.SEM لثلاث تجارب مستقلة. تم تقييم الدلالة الإحصائية بواسطة اختبار الطالب غير المتزاوج ذو الطرفين.-اختبار t وتحليل التباين ثنائي الاتجاه:؛؛؛; لا دلالة.
روتينون. تم إضافة الروتينون إلى وسائط الثقافة للحصول على محلول قبل 20 دقيقة من جمع الخلايا. تم إضافة مثبط ناقل أحادي الكربوكسيلات a-CHCA (5 مللي مول، التركيز النهائي) إلى وسط الثقافة قبل ساعتين من الحصاد.
روكسادستات. روكسادستات (تم إضافة (التركيز النهائي) إلى وسط الثقافة قبل الحصاد بـ 6 ساعات.
نقص الأكسجين. تم زراعة الخلايا تحت ظروف النورموكسي قبل نقلها إلى حاضنة نقص الأكسجين (ثيرمو فيشر ساينتيفيك، الولايات المتحدة الأمريكية) مع خليط غازي يحتوي علىوتم التوازن مع النيتروجين. تم جمع الخلايا في النقاط الزمنية المحددة التي لم تتجاوز 8 ساعات بعد التعرض لنقص الأكسجين. تم مراقبة مستويات الأكسجين في الحجرة في أوقات أخذ العينات.
عزل الميتوكوندريا الخلوية
محلول عازل مفرط التخفيف يحتوي على 20 مللي مول من HEPES (رقم الهيدروجين 7.5)EDTA، وتم إضافة خليط من مثبطات البروتياز والفوسفاتاز إلى الخلايا بعد غسلها بمحلول PBS. ثم تم كشط الخلايا وكسرها باستخدام حقنة. وتم طرد المستحلب في جهاز الطرد المركزي عندلمدة 10 دقائق لفصل النواة وبقية الحطام. ثم تم طرد السائل الطافي في جهاز الطرد المركزي عندولمدة 30 دقيقة. كانت السائل الناتج يتكون من البلازما. بعد ذلك، تم غسل الراسب ثلاث مرات بمحلول مفرط التوتر يحتوي على 20 مللي مول من HEPES (رقم الهيدروجيني 7.5)،EDTA، في PBS. كان الراسب النهائي يحتوي على الميتوكوندريا. تم التحقق من نقاء البلازما والميتوكوندريا باستخدام علامات البلازما والميتوكوندريا، الأكتين، وCOXIV، من خلال اختبارات الوسترن بلوت.
حقن الفئران
حقن عضلة الساق. لاكتات، ألانين، وتم تحضير -ألانين فيمحلول ملحي للحصول على تركيز نهائي قدره 500 مللي مول، تم ضبط الرقم الهيدروجيني له على 7.4.تم حقن جرعة من كل دواء في عضلة الساق الخلفية للفئران عن طريق الحقن العضلي. تم حقن حجم مماثل منتم حقن محلول ملحي في نفس العضلات كتحكم.
حقن MG132 عن طريق الوريد. تم تحضير MG132 فيمحلول ملحي للحصول على تركيز نهائي قدره 500 مللي مولار، وتم ضبط الرقم الهيدروجيني على 7.4. تم حقن الفئران عن طريق الوريد بـتم استخدام MG132 مرتين في الأسبوع لمدة 4 أسابيع قبل استخدامه للتجارب وتم أخذ عينات من العضلات للكشف.
حقن FX-11. تم حقن الفئران عن طريق البطن بـدي إم إس أو كتحكم أوتم إعطاء مثبط LDHA FX-11 المذاب في DMSO يوميًا لمدة أسبوعين، وبعد ذلك تم إجراء اختبارات الجري على الفئران.
اختبار تعب الفأر أثناء الجري والتمارين عالية الكثافة
تم تعديل اختبار أداء العدو للفئران من البروتوكولات المنشورة.أولاً، تم وضع الفئران التي تبلغ من العمر 20 أسبوعًا على جهاز المشي ZH-PT/5S المزود بشبكة تحفيز كهربائية خلفية توفر صدمة كهربائية بقوة 0.2 مللي أمبير. تم تسخين الفئران أولاً على جهاز المشي لمدة 3 دقائق عندتم زيادة السرعة إلىلمدة 30 ثانية. بعد ذلك، قمنا بزيادة السرعة منإلىمع زيادة قدرها، مع استمرار كل زيادة لمدة 15 ثانية (إجماليتم تكرار النمط بسرعة متزايدة حتى لم يعد بإمكان الفئران مواكبة السرعة واستسلمت عند الشبكة.
تم تعديل اختبار إرهاق تمارين الشدة العالية للفئران من البروتوكولات المنشورة.باختصار، تم تكييف الفئران التي تبلغ من العمر 20 أسبوعًا مع جهاز المشي قبل يومين من التجارب عن طريق الجري على لمدة 5 دقائق و دقيقة واحدة لمدة 5 دقائق تليهالمدة دقيقة واحدة كل يوم. بالنسبة لتجارب التمرين، تم زيادة السرعة.كل 5 دقائق حتىتم الوصول إلى النقطة التي تم فيها زيادة الميل بمقدار 5 درجات كل 5 دقائق حتى الإرهاق.
تحضير عضلات الساق السريعة والبطيئة للفأر
تم تعديل إعداد العضلات من البروتوكولات المنشورة.باختصار، تم euthanize الفئران وتم تشريح عضلات السوليوس (SOL) التي تتكون أساسًا من العضلات البطيئة (النوع الأول)، وعضلات الباسطة للأصابع الطويلة (EDL) التي تتكون أساسًا من العضلات السريعة (النوع الثاني)، وتم إخضاعها للاختبارات التالية.
تروية نقص الأكسجين في العضلات
تم euthanizing الفئران وتم تشريح عضلات الساق الهيكلية الخاصة بها ووضعها على الفور في محلول كريبس رينجر.د-جلوكوز, HEPES) في حوض مائي معزول من نظام اختبار العضلات السليمة 1200 A (أورورا ساينتيفيك، كندا)، ومن ثم تم تهويته مع أو بدونوفيلمدة 30 دقيقة.
عزل الميتوكوندريا من نسيج العضلات
تم تشريح عضلات أرجل الفأر وغسلها في محلول عزل الميتوكوندريا البارد الذي يحتوي على 70 مللي مول من السكروز، 210 مللي مول من المانيتول، 5 مللي مول من MOPS،إي جي تي إيه، وألبومين مصل البقر خالي من الأحماض الدهنية (pH 7.2). ثم تم تجانس الأنسجة باستخدام مطحنة الأنسجة (شنغهاي جينغ شين، شنغهاي، الصين) عندتمت عملية الطرد المركزي للمستحلب عندولمدة 10 دقائق لإزالة النوى وفضلات الخلايا. ثم تم طرد السائل الطافي الغني بالميتوكندريا.وتم استخدام الراسب الناتج عن الميتوكوندريا لمدة 10 دقائق في الاختبارات.
تحليل
تم جمع الدم الشرياني على الفور في أنابيب مضادة للتخثر من الشريان الأبهر البطني للفئران التي جرت لفترات زمنية محددة.تم تحديده باستخدام جهاز تحليل الإلكتروليتات وغاز الدم IDEXX VetStat (IDEXX B.V.، هولندا).
اختبار ROS
تم إجراء اختبارات ROS لعضلات الساق في الفئران باستخدام مجموعة اختبار ROS (رقم الكاتالوج E-BC-K138-F، شركة Elabscience Biotechnology Inc.، هيوستن، تكساس، الولايات المتحدة الأمريكية) وفقًا لتعليمات الشركة المصنعة. تم هضم الأنسجة إلى تعليق خلوى مفرد عبر هضم الكولاجيناز. ثم،تم حضانة الخلايا معتم استخدام كاشف DCFH-DA لمدة 30 دقيقة. بعد الطرد المركزي بسرعة 1000 دورة في الدقيقة لمدة 5 دقائق، تم غسل كريات الخلايا مرتين وإعادة تعليقها في الكاشف 3 قبل تحليلها في جهاز قياس التدفق FACS Calibur.
اختبارات MDA
تم تقييم أكسدة الدهون باستخدام مجموعة اختبار MDA (ab233471، Discovery Drive، Cambridge Biomedical Campus، Cambridge، CB2 0AX، المملكة المتحدة) وفقًا لتعليمات الشركة المصنعة. لتحضير مستخلص الأنسجة، تم تجانس 50 ملغ من العضلات الهيكلية الطازجة بسرعة على الثلج فيمحلول فوسفات (pH 3-3.2) معترايتون X100 يليه الطرد المركزي عندلمدة 15 دقيقة عندتمت إزالة المواد غير القابلة للذوبان، وتم تخزين السائل العلوي على الثلج لاستخدامه لاحقًا. قبل الاستخدام، تم توازن جميع المواد والمركبات المحضرة إلى درجة حرارة الغرفة مع التحريك بلطف. بعد ذلك،تم إضافة عينات الاختبار، وعيّنة تحكم فارغة (محلول التخفيف)، ومعايير MDA المخففة بالتسلسل إلى لوحة ميكروية ذات قاع شفاف مكونة من 96 بئرًا. تلا ذلك حضانة خليط التفاعل في درجة حرارة الغرفة لمدة. ثم، تم إضافة محلول التفاعل لزيادة حجم العينة الإجمالي إلىحسنًا. تم تحضين خليط التفاعل النهائي في درجة حرارة الغرفة لمدةتم قياس الامتصاص عند نقطة النهاية باستخدام جهاز قراءة الامتصاص مع تصحيح مسار الضوء عند.
تألق المناعي لنسج العضلات
تم إعداد مقاطع بارافين لعضلات الساق الهيكلية للفئران من النوع البري، والفئران التي تعبر عن AARS2 بشكل محدد في العضلات، والفئران التي جرت. تم إزالة البارافين من المقاطع، وإعادة ترطيبها، واسترجاع المستضد في محلول استرجاع المستضد EDTA (pH 8.0)، وتم حجبها بـتمت معالجة العينة بمحلول BSA لمدة 30 دقيقة. تم صبغ النوى بصبغة DAPI. تم صبغ HIF1a وpS293-PDHA1 وAARS2 وCOXIV باستخدام الأجسام المضادة الأولية والثانوية الخاصة بها. تم التقاط صور للشرائح المحضرة تحت ميكروسكوب الفلورسنت.
نسيج العضلات-علاج الفوسفاتاز
تم التضحية بالفئران وتم تجانس 50 ملغ من العضلات الهيكلية الطازجة بسرعة على الثلج فيمحلول التحليل الذي يحتوي علىتريس- HCl (pH 7.5) و150 مللي مولار NaCl، وتم الطرد المركزي عندلمدة 15 دقيقة عند، وبعد ذلك تم إزالة المواد غير القابلة للذوبان. ثم تم جمع السائل العلوي على الثلج للاختبار التالي. بعد ذلك، تم استخدام 400 وحدة من -فوسفاتاز (نيو إنجلاند بيو لاب) وتمت إضافتها إلى السائل الفائق وتم تحضينها عندلمدة 30 دقيقة، وتم استخدام السائل الطافي لاختبار نشاط PDHA1.
تداخل RNA صغير
تم تصنيع الأوليغونيوكليوتيدات المستخدمة في كتم التعبير عن VHL و PHD1 و PHD2 و PHD3 و AARS2 و SIRT3 بواسطة GenePharma (شنغهاي، الصين). تم إدخال الأوليغونيوكليوتيدات إلى الخلايا.قبل الحصاد. تم التحقق من كفاءة التثبيط باستخدام اختبارات الوسترن بلوت. تسلسلات siRNA المستخدمة لاستهداف الجينات هي كما يلي:
Phd1 siRNA: 5′-أكوتشاويوغويوتشويوأغغغ-3′ Phd2 siRNA: 5′-UGGAUUUGUACCAUUCUUCUG-3′ siRNA Phd3:-يو سي جي إيه إيه إيه سي يو سي تي يو جي إيه إيه جي إيه إيه جي جي-3′
إزالة الجين بواسطة CRISPR/Cas9
لإسقاط Phd2 و Vhl و Aars2 و Cpt2 و Sirt3 في خلايا C2C12، تم استخدام بروتوكولات CRISPR/Cas9. تم نقل RNA الدليل الذي يحتوي على تسلسل الهدف إلى خلايا C2C12، والتي تم اختيارها بعد 36 ساعة باستخدامبرومايسين (أمريسكو، الولايات المتحدة الأمريكية) لمدة 3 أيام. تم جمع الخلايا التي نجت في النهاية وزرعها في لوحة 96 بئر بكثافة
1 خلية/بئر. تم التحقق من نجاح إلغاء كل جين من خلال اختبارات الوسترن بلوت. تسلسل الدلائل كما يلي: nsgPhd2 #1: ف:-CACCGACACCGGCAAGTTCACGGAC-3′ R: 5′-AAACGTCCGTGAACTTGCCGGTGTC-3′ nsgPhd2 #2: 5′-CACCGCAAGTTCACGGACGGGCAGC-3′ ر:-AAACGCTGCCCGTCCGTGAACTTGC-3′ nsgAars2 #1: ف:-CACCGCTGCTAAGTACCGGGGCCGT-3′ R: 5′-AAACACGGCCCCGGTACTTAGCAGC-3′ nsgAars2 #2: ف:-CACCGCGAGCGGCTGCGCTTTGACG-3′ ر:-AAACCGTCAAAGCGCAGCCGCTCGC-3′ nsgCpt2 #1: F: 5′-CACCGATCGTCCCGGGCGGCAATGG-3′ R: 5′-AAACCCATTGCCGCCCGGGACGATC-3′ nsgCpt2 #2: ف:-CACCGAGGCGCGGCATCATCGTCCC-3′ ر: -AAACGGGACGATGATGCCGCGCCTC-3′ nsgVhl #1: ف:-CACCGCGTTCCAATAATGCCCCGGA- ر:-AAACTCCGGGGCATTATTGGAACGC-3′ nsgVhl #2: F: 5′-CACCGCCGATCTTACCACCGGGCAC-3′ ر: -AAACGTGCCCGGTGGTAAGATCGGC-3′ nsgSirt3 #1: ف:-CACCGCCAGTACAGACAGGGCAGCGGGG-3′ ر: -AAACCCCCGCTGCCCTGTCTGTACTGGC- nsgSirt3 #2: F: 5′-CACCGCTTTCAACAAACCTCCAGGGAGG-3′ R: 5′-AAACCCTCCCTGGAGGTTTGTTGAAAGC-3′
الترسيب المناعي
تم تحلل الخلايا باستخدام محلول التحلل الذي يحتوي علىمحلول عازل NP-40 (50 ملليمول من تريس-هيدروكلوريد، pH 7.5)نونيديت بي-أبروتينينمل من لوبيبتينتم تجانس الأنسجة في محلول التحلل قبل عملية التحلل. تمت إضافة كريات الأجسام المضادة المرتبطة إلى المستخلص لزيادة تركيز البروتينات المستهدفة. تم غسل البروتينات الغنية على الكريات بمحلول 0.5% NP-40 ثلاث مرات قبل خلطها بمحلول تحميل SDS-PAGE لتحرير البروتينات للتحليل.
اختبار الويسترن بلوت
تم تحطيم الخلايا المزروعة وغليها مع محلول تحميل SDS-PAGE. تم تجانس أنسجة الفئران باستخدام محلول التحلل الذي يحتوي على 0.5% NP-40، و50 مليمول من تريس- HCl (pH 7.5).Nonidet P-40، ومزيج من مثبطات البروتياز (سيغما-ألدريتش)، قبل أن يتم غليه معتحميل المخزن. بعد الطرد المركزي عندولمدة 15 دقيقة، تم جمع السائل الطافي من المستخلصات لاختبار الوسترن بلوت وفقًا للإجراءات القياسية. تم الكشف عن إشارات الوسترن بلوت باستخدام جهاز Typhoon FLA 9500 (GE Healthcare، ليتل تشالفونت، المملكة المتحدة).
اختبار بقعة النقطة
تم تخفيف الببتيدات بـبالتخفيفات المحددة. تم نقل العينات إلى أغشية نيتروسليلوز، وتركها لتجف في الهواء عند درجة حرارة الغرفة. تم حجب الغشاء بـالحليب، ثم تم حضنه مع جسم مضاد أولي للتعرف على الببتيد وجسم مضاد ثانوي للكشف. تم الكشف عن إشارات التلطيخ باستخدام جهاز Typhoon FLA 9500 (GE، الولايات المتحدة الأمريكية).
اختبار اليوبكويتين
لتحليل اليوبكويتين، تم نقل البلازميدات المسمّاة بـ HA-tagged ubiquitin والهدف معًا إلى الخلايا. بعد 36 ساعة من النقل، تم إضافة MG132 إلى الوسط من أجلقبل الحصاد. تم جمع الخلايا، وتحليلها، وغليها في محلول 1% SDS (تريس-هيدروكلوريك، الرقم الهيدروجينيتم إجراء الترسيب المناعي في مستخلصات مخففة بعشرة أضعاف مع EDTA و 1 مM DTT لمدة 10 دقائق.تم تحليل عازلة NP-40، وتم تحليل ubiquitination وفقًا لبروتوكولات الوسترن بلوت القياسية.
اختبار نشاط PDHA1
تم التعبير عن PDHA1-Flag و PDHB-Myc بشكل مشترك في الخلايا المحددة. تم تحلل الخلايا على الثلج باستخداممحلول NP-40 المدعوم بالبروتياز المثبطات. تم تنقية PDHA1 باستخدام كرات العلم (سيغما ألدريتش) ثم تم غسلها ثلاث مرات بـعازل NP-40. تم قياس PDHB الذي تم تنقيته مع PDHA1 عبر تقنية الويسترن بلوت باستخدام الأجسام المضادة لمركب Myc، واستخدم لتطبيع نشاط PDHA1. تم إجراء اختبار نشاط PDHA1 كما هو موصوف سابقًا.باختصار، يحتوي محلول التفاعل على (درجة الحموضة 7.0)، تم إضافة البيروفات الصوديوم، 0.2 مللي مول من ثيامين ثنائي الفوسفات، و0.1 مللي مول من 2,6-ثنائي كلوروفينول إندوفينول (2,6-DCPIP) إلى مركب PDHA1/PDHB المنقى لبدء التفاعل. تم الحفاظ على التفاعلات عندتمت مراقبة التفاعل من خلال قياس انخفاض 2,6-DCPIP عند 600 نانومتر باستخدام مطياف ضوئي من شركة روش (بازل، سويسرا).
اختبار نشاط PDC
تم قياس نشاط إنزيم مركب PDC باستخدام مجموعة اختبار نشاط إنزيم PDH (Abcam، الولايات المتحدة الأمريكية) وفقًا لتعليمات الشركة المصنعة. تم جمع الخلايا وأنسجة الفئران لاستخراج البروتينات. تم تحميل العينات على اللوحة وتمت الحضانة في درجة حرارة الغرفة لمدة 3 ساعات. تمت إضافة محلول الاختبار إلى كل بئر، وتم تحديد نشاط PDH من خلال متابعة تقليلإلى NADH، المرتبط بتقليل صبغة المراسل لإنتاج منتج تفاعل ملون (أصفر)، تم مراقبة تركيزه من خلال قياس الامتصاص عند 450 نانومتر.
اختبار نشاط CPT2
تم قياس نشاط CPT2 من خلال قياس الانخفاض في مستويات CoA. تم إجراء التفاعلات في “خليط التفاعل يحتوي على 50 مللي مول من تريس- HCl (رقم الهيدروجيني 7.5)EDTA، 2 مللي مول CoA، و2 مللي مول بالميتيل-ل-كارنيتين. بعد ذلك، تم ترسيب CPT2 من خلايا HEK293T. تم بدء الاختبار في حاضنة اهتزاز حرارية عند 1000 دورة في الدقيقة و، وتم جمع الخليط في، و 30 دقيقة. تم إنهاء التفاعل بإضافةمن 10 مللي مولار DTNB. تم مراقبة السائل العلوي باستخدام قارئ الميكرو بلايت عند 410 نانومتر.
اختبار نشاط LDH
تم استخدام مجموعة اختبار لقياس نشاط LDH بطريقة الكروماتوغرافيا اللونية (بيوفيجن، الولايات المتحدة الأمريكية). باختصار،تم تجانس الخلايا مع محلول الفحص البارد، وتم حضنها على الثلج لمدة 10 دقائق، ثم تم الطرد المركزي عندلمدة 15 دقيقة. بعد ذلك، تم نقل السائل العلوي إلى أنبوب جديد، ومن خليط التفاعل (عينةمحلول العينة، وتم نقل مزيج التفاعل إلى لوحة 96 بئر. تم قياس الامتصاص على الفور عند 450 نانومتر في وضع الحركة فيلـتم قياس نشاط LDH بناءً على التغير في الامتصاص خلال فترة زمنية محددة.
اختبار نشاط CPT1A
تم قياس نشاط CPT1A في مستخلصات الخلايا طيفيًا من خلال متابعة إطلاق CoA-SH من بالميتويل-CoA باستخدام الكاشف العام للثيول DTNB. تم تحضين خلطات التفاعل التي تحتوي على محلول DTNB ومستخلص الخلايا في درجة حرارة الغرفة لمدة 20 دقيقة لإزالة جميع مجموعات الثيول التفاعلية. بعد التحضين، تم قياس الامتصاص عند 412 نانومتر. لبدء التفاعل، تم استخدام بالميتويل-CoA.تم إضافة محلول L-carnitine (1 مللي مول، التركيز النهائي في 1 م مول من Tris، pH 8.0) إلى خلطات التفاعل. تم حضن خلطات التفاعل لمدة 10 دقائق عندبعد الحضانة، تم قياس الامتصاص عند 412 نانومتر. الفرق بين الامتصاص مع وبدون الركائز يقيس إطلاق CoA-SH. تم تعريف النشاط على أنه نانومول من CoA-SH المحرر.تم تحديد محتوى البروتين في مستخلصات الخلايا وفقًا لطريقة برادفورد.
اختبار نشاط SIRT3
تم استخدام مجموعة اختبار نشاط SIRT3 (Abcam، الولايات المتحدة الأمريكية) وفقًا لتعليمات الشركة المصنعة. يحتوي خليط التفاعل على محلول الاختبار، ببتيد الفلورو-ركيزة، NAD، المطور، والعينات. اقرأ شدة الفلورية لمدة 30 دقيقة إلى 60 دقيقة بفواصل زمنية تتراوح بين 1 دقيقة إلى 2 دقيقة باستخدام مقياس فلورية لوحات الميكروتيتر مع تحفيز عند والانبعاث في قم بقياس وحساب معدل التفاعل بينما تظل سرعة التفاعل ثابتة.
حمض اللبنيك في الدم
تم قياس تركيز اللاكتات في دم الوريد الذيل باستخدام جهاز Lactate Scout4 (SensLab GmbH، ألمانيا). تم قطع ذيل كل فأر باستخدام شفرة، وتم مسح القطره الأولى من الدم. تم الضغط برفق على الذيل، وكانت القطره الثانية من الدم كافية لملء الـ غرفة القياس لمستشعر اللاكتات التي تتيح تحديد التركيز.
اختبارات OCR للخلايا والأنسجة
تم إجراء اختبارات OCR باستخدام وحدة OROBOROS Oxygraph-2K (Oroboros Instruments GmbH، النمسا). تم تطبيع قيم استهلاك الأكسجين وفقًا لعدد الخلايا أو محتوى الميتوكوندريا.
بالنسبة للخلايا المزروعة، حواليتم هضم الخلايا وإعادة تعليقها في PBS. تم تحديد معدل التنفس الأساسي باستخدام الركيزة (5 مللي مول من البيروفات و0.1 مللي مول من الماليك). تم تحديد قدرة إنتاج ATP والتنفس الأقصى بشكل متتابع باستخدام علاجات أوليغوميسين A وFCCP (سيلك، الصين) على التوالي.
تم عزل الميتوكوندريا من عينات عضلات الفئران وفقًا لبروتوكول منشور، وتم إعادة تعليق عينات الميتوكوندريا المعزولة في محلول التنفس (0.5 مللي مول EDTA، 3 مللي مول MgCl، 60 مللي مول حمض اللاكتوبيونيك، 20 مللي مول التورين،هيبز، 110 مللي مولار د-سكروز وبروتين ألبومين مصل البقر خالي من الأحماض الدهنية، pH 7.2).
اختبار كيناتي AARS2
تم تحويل المتجه pGEX-6p-1-AARS2 إلى سلالة الإشريكية القولونية BL21 (DE3). ثم تم تحفيز الخلايا المحولة باستخدام 0.2 مللي مول من IPTG عندماكان. بعد زراعتها في الليل في تم جمع البكتيريا، وتجانسها في محلول عازل يحتوي على 50 ملليمول من TrisHCl و150 ملليمول من NaCl، ثم تم الطرد المركزي عندولمدة 30 دقيقة. تم تحميل السائل العلوي على جهاز AKTA Purifier (GE، الولايات المتحدة) مع عمود His-Ni. تم قياس تركيز البروتين الذي تم الحصول عليه باستخدام مجموعة اختبار بروتين BSA.
تم تحديد المعلمات الحركية لـ AARS2 من خلال قياس لاكتيلation الركائز الببتيدية (PDHA1: MVNSNLASVEELKEIDVEVR؛ CPT2: EFLKKQKLS).وتم تقدير مستويات اللاكتات من خلال تغيير مستويات اللاكتات منبينما كانت الركائز الببتيدية ثابتة عندتم إجراء تفاعلات اللاكتيل فيخليط التفاعل يحتوي على 50 ملليمول من HEPES عند pH 7.5تم تحديد الببتيد المشتق من اللاكتيل باستخدام مطياف الكتلة (MS) مع تركيزات معروفة من الببتيدات الاصطناعية (PDHA1: MVNSNLASVEELK) بوجود 4 مللي مول من ATP و10 نانومول من AARS2.عيد الفطر; CPT2: EFLK ) كضوابط داخلية. وتم استنتاجها باستخدام معادلة ميكاليز-مينتن.
ITC
تم تحديد تفاعل البروتين مع الجزيئات الصغيرة باستخدام جهاز MicroCal PEAQ-ITC (مالفرن باناليتيكال، المملكة المتحدة) فياللاكتات، الألانين، وتم إذابة “-ألانين” في 50 مللي مول من تريس-هيدروكلوريد و150 مللي مول من كلوريد الصوديوم بتركيزتم تخفيف AARS2 المؤتلف إلى، ثم أضيفت إلى خلية العينة. تم إسقاط الجزيئات في خلية العينة كل دقيقتين لمدة 20 مرة.
تفاعلات اللاكتيل في المختبر
تم إجراء تفاعلات اللاكتيل في المختبر في “خليط التفاعل يحتوي على 50 مللي مول من HEPES (رقم هيدروجيني 7.5)لاكتات، 4 مللي مول ATP، 10 نانومول AARS2، والببتيد الركيزة الاصطناعية. تم ضبط الرقم الهيدروجيني النهائي لمزيج التفاعل على 7.5 قبل إضافة AARS2. تم السماح للتفاعل بالاستمرار لمدة 3 ساعات عندتم إزالة الأملاح من الببتيد باستخدام C18 ZipTip (Millipore، الولايات المتحدة الأمريكية)، وتم إخضاعه للتحليل باستخدام مطياف الكتلة MALDI-TOF/TOF (SCIEX-5800، AB Sciex، فريمينغهام، ماساتشوستس، الولايات المتحدة الأمريكية). تسلسلات الببتيدات الاصطناعية هي كما يلي:
تم إجراء تفاعلات إزالة اللاكتيل في المختبر فيخليط التفاعل يحتوي على 50 مللي مول من HEPES (رقم هيدروجيني 7.5)دي تي تي، الببتيد اللاكتيل الاصطناعي أو بروتين PDHA1 و CPT2 المؤتلف، SIRT3 و 1 مللي مول من PMSF. تم السماح للتفاعل بالاستمرار لمدة 4 ساعات فيتم إزالة الأملاح من الببتيد عن طريق تمريره عبر C18 ZipTip قبل إخضاعه للتحليل باستخدام مطياف الكتلة MALDI-TOF/TOF (SCIEX-5800). تم إخضاع PDHA1 و CPT2 الناتجين لتحليل Western blot واختبار نشاط الإنزيم. تسلسلات الببتيدات الاصطناعية هي كما يلي:
تحديد AARS2 P377OH و PDHA1Lac-K336 و CPT2 Lac-K457/8 باستخدام تقنية MS/MS
تم تنقية بروتينات AARS2 أو CPT2 التي تم التعبير عنها بشكل مفرط في الخلايا باستخدام التنقية بال affinity وتم هضمها باستخدام إنزيم البروتيناز Glu-C. تم تنقية PDHA1 المفرط التعبير من خلايا C2C12 باستخدام التنقية بال affinity وتم هضمه باستخدام التربسين. تم تحليل الببتيدات الهيدروكسيلية أو اللاكتيلية عبر تسلسل MS/MS.
تمت معالجة قمم الكروماتوغرافيا السائلة التي تم فصلها في نفس الوقت مع تلك الخاصة بالببتيدات الاصطناعية القياسية بواسطة تحليل الطيف الكتلي باستخدام مطياف الكتلة Orbitrap Fusion (ThermoFisher Scientific، الولايات المتحدة الأمريكية). تم مقارنة عدد الببتيدات و/أو مناطق العينات بتلك الخاصة بالببتيدات الاصطناعية القياسية. تم ضبط جهاز الطيف الكتلي في وضع يعتمد على البيانات للتبديل تلقائيًا بين اكتساب MS و MS/MS. تم الحصول على طيف MS الكامل (m/z 350-1600) في جهاز Orbitrap بدقة كتلة تبلغ 60,000 عندتم تحديد هدف AGC عند 300,000 ، وكانت أقصى مدة حقن 50 مللي ثانية. تم إجراء اكتساب MS/MS في Orbitrap بدورة زمنية مدتها 3 ثوانٍ، بدقة 15,000.. كانت عتبة الكثافة 50,000، وكان الحد الأقصى لوقت الحقن 200 مللي ثانية. تم تعيين هدف AGC على 200,000، وكانت نافذة العزلتم تكسير الأيونات بحالات شحن 2+ و3+ و4+ بشكل متتابع عبر تفكك تصادمي عالي الطاقة مع طاقة تصادم موحدة.، وتم تعيين الكتلة الثابتة الأولى على 120. في جميع الحالات، تم تسجيل مسح ميكروي واحد يستمر لمدة 30 ثانية باستخدام الاستبعاد الديناميكي. تم تحقيق تقدير الببتيد المستهدف المخلّق عبر قياس الكتلة. باختصار، تم حساب نسبة إشارة الببتيد المخلّق (إجمالي عدد الأيونات (TIC) للصيغة المخلّقة) إلى إجمالي إشارة الببتيد (TIC للصيغة المخلّقة + TIC للصيغة غير المخلّقة) وفقًا للمعادلة التالية: TICK-Lac/(TICK-Lac + TICnon-K-Lac) = نسبة K-Lac (RK-Lac). تسلسلات الببتيدات الاصطناعية هي كما يلي:
تحديد الكمية باستخدام LC-MS/MS لـ AARS2 P377OH و PDHA1Lac-K336 و CPT2 lac-K457/8
تم التعبير عن AARS2 و PDHA1 و CPT2 بشكل غير طبيعي في خلايا C2C12 المزروعة التي تعبر عن AARS2 بشكل مفرط أو التي تم معالجتها باللاكتات. تم جمع الخلايا وتحليلها في محلول 0.1% NP-40 (50 مليمول من Tris-HCl، pHتم استخدام كريات مغناطيسية مضادة لـ FLAG M2 (Sigma-Aldrich) لترسيب PHD2 و PDHA1 و CPT2 لمدة 3 ساعات فيثم تم غسل الخرز مرتين بـمحلول NP-40، مرتين مع، وثلاث مرات مع ، الذي تلاه هضم التربسين على الحبة طوال الليل في. تم جمع الببتيدات الناتجة في السائل الفائق عن طريق الطرد المركزي وتجفيفها في مكنة تفريغ السرعة (Eppendorf، هامبورغ، ألمانيا). تم تخزين الببتيدات المستخلصة فيلتحليل LC-MS/MS. طريقة تم نشرها سابقًاتم استخدامه لتحديد كمية الببتيدات المستهدفة الميثيلية. باختصار، تم حساب نسبة إشارة الببتيد الميثيلي (إجمالي عدد الأيونات (TIC) للشكل الميثيلي) إلى إجمالي إشارة الببتيد (TIC للشكل الميثيلي + TIC للشكل غير الميثيلي) وفقًا للمعادلة التالية: TICK-Lac/(TICK-Lac + TICnon-K-Lac) = نسبة K-Lac (RK-Lac).
تحليل المستقلبات باستخدام LC-MS/MS
تقريباًتم معالجة الخلايا بمحلول ميثانول مائي باردلإيقاف عملية الأيض الخلوي بسرعة. تم كشط الخلايا من الأنابيب، ووضعها فيبين عشية وضحاها. بالنسبة لعينات عضلات الفئران، تم تجانس حوالي 80 ملغ من عضلة ساق الفأر في محلول مائي بارد من الميثانول. ) في تمت عملية الطرد المركزي للعينات فيولمدة 15 دقيقة، وتم جمع السائل الطافي. ثم تم تجفيف السائل الطافي بالتجميد وإعادة حله فيميثانول/ ماءتم الحصول على المستقلبات المنفصلة باستخدام الكروماتوغرافيا السائلة عالية الأداء مع مضخة LC-20AB (شيمادزو، كيوتو، اليابان) وLuna العمود (P/N 00B-4378-B0؛ ، ; فينومينيكس، تورانس، كاليفورنيا، الولايات المتحدة الأمريكية). كانت المرحلة المتنقلة تتكون من المحلول A (، و حمض الأسيتيك المذاب في 500 مل من الماء) والمذيب ب. كان برنامج الإيلاشن كما يلي:و, معدل تدفق المضخة كان، وكان جهاز مطياف الكتلة المستخدم هو نظام 4000 QTRAP (AB Sciex، فريمينغهام، ماساتشوستس، الولايات المتحدة الأمريكية) الذي يعمل في أوضاع مراقبة التفاعلات المتعددة. تم مراقبة أيونات المستقلبات عند: PYR 87-43؛ LAC 89-43؛ Ac-CoA 810-60؛ ATP 509-79؛ و AKG 145-101. تم الحصول على كل قياس على الأقل في ثلاث نسخ.
اختبار تدفق الأيض
تم إجراء تجارب تدفق الأيض عندما وصلت الخلايا إلىالتقاء. الوسيلة لـتجارب وسم الجلوكوز احتوت على 10 مللي مول (U-الجلوكوز (مختبرات كامبريدج لل isotopes، ماساتشوستس، الولايات المتحدة الأمريكية) و 2 مللي مول من الجلوتامين غير المعلم (سيغما-ألدريتش، الولايات المتحدة الأمريكية). الوسط لـتجارب وسم الجلوتامين احتوت على 10 مللي مول من الجلوكوز غير الموسوم و2 مللي مول من (U- ) الجلوتامين (مختبرات كامبريدج لل isotopes). وسط لـ تجارب وسم بالبالميتات احتوت على 0.1 مللي مولار (U-بالميتات (سيغما-ألدريتش). بعد المعالجة، تم غسل الخلايا ثلاث مرات بمحلول PBS ثم تم معالجتها بالميثانول البارد مسبقًا.في الوقت نفسه، تم استخدام أطباق موازية لعد أعداد الخلايا. ثم تم تحليل استخلاصات المستقلبات باستخدام LC-MS مع عمود C18. تم تحديد وفرة المستقلبات النسبية من خلال تطبيع وفرة كل مستقلب إلى المعيار الداخلي وعدد الخلايا. تم دمجتم الإشارة إلى الذرات على أنها، حيث كانت n هي عدد ذرات.
النمذجة الهيكلية
نظرًا لعدم ملاحظة أي هياكل مرتبطة بالركيزة في AARS2 البشري حتى الآن، قمنا بنمذجة هيكل منطقة ارتباط الألانين باستخدام Phyre2 (مجموعة المعلوماتية الحيوية الهيكلية، كلية إمبريال، لندن). أشارت توقعات الهيكل إلى أنه كان متجانسًا هيكليًا عاليًا لـ AARS2 في Aquifex aeolicus، وهو إنزيم الألانيل-tRNA (اسم مستعار: alaS، رمز PDB: 1YFS) المعقد مع الألانين. وبالتالي، قمنا بمزيد من التراصالألانين واللاكتات في الهيكل المعقد وطبقت نموذج AARS2 على نموذج alaS من Aquifex aeolicus.
طرق إحصائية
تم إجراء التحليل الإحصائي باستخدام برنامج Prism 8.0 (GraphPad Software, Inc.، سان دييغو، كاليفورنيا، الولايات المتحدة الأمريكية) وExcel (Microsoft Corp.، ريدموند، كاليفورنيا، الولايات المتحدة الأمريكية). تم التعبير عن النتائج المجمعة كمتوسط.تمت المقارنات بين المجموعات من خلال اختبار ت student’s غير المقترن ذو الطرفين.-اختبار وANOVA ثنائي الاتجاه. تم تحديد الدلالة الإحصائية عند; لا دلالة لـ ns؛ ; ؛؛.
REFERENCES
Brooks, G. A. The science and translation of lactate shuttle theory. Cell Metab. 27, 757-785 (2018).
Bendahan, D., Chatel, B. & Jue, T. Comparative NMR and NIRS analysis of oxygendependent metabolism in exercising finger flexor muscles. Am. J. Physiol. Reg. Intgr. Comp. Physiol. 313, R740-R753 (2017).
Glancy, B. et al. Mitochondrial reticulum for cellular energy distribution in muscle. Nature 523, 617-620 (2015).
Bassett, D. R. Jr. & Howley, E. T. Limiting factors for maximum oxygen uptake and determinants of endurance performance. Med. Sci. Sports Exerc. 32, 70-84 (2000).
Vargas-Mendoza, N. et al. Oxidative stress, mitochondrial function and adaptation to exercise: new perspectives in nutrition. Life 11, 1269 (2021).
Liu, W. Y., He, W. & Li, H. Exhaustive training increases uncoupling protein 2 expression and decreases Bcl-2/Bax ratio in rat skeletal muscle. Oxid. Med. Cell. Longev. 2013, 1-7 (2013).
Maxwell, P. H. et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399, 271-275 (1999).
Wang, G. L. & Semenza, G. L. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc. Natl. Acad. Sci. USA 90, 4304-4308 (1993).
Epstein, A. C. et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107, 43-54 (2001).
Jaakkola, P. et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O-2-regulated prolyl hydroxylation. Science 292, 468-472 (2001).
Denko, N. C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 8, 705-713 (2008).
Pugh, C. W. & Ratcliffe, P. J. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat. Med. 9, 677-684 (2003).
Henderson, G. C., Horning, M. A., Wallis, G. A. & Brooks, G. A. Pyruvate metabolism in working human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 292, E366 (2007).
Peters, S. J. Regulation of PDH activity and isoform expression: diet and exercise. Biochem. Soc. Trans. 31, 1274-1280 (2003).
Mourtzakis, M., Saltin, B., Graham, T. & Pilegaard, H. Carbohydrate metabolism during prolonged exercise and recovery: interactions between pyruvate dehydrogenase, fatty acids, and amino acids. J. Appl. Physiol. 100, 1822-1830 (2006).
Watt, M. J. et al. Rapid upregulation of pyruvate dehydrogenase kinase activity in human skeletal muscle during prolonged exercise. J. Appl. Physiol. 97, 1261-1267 (2004).
Gudiksen, A. et al. Lack of skeletal muscle IL-6 affects pyruvate dehydrogenase activity at rest and during prolonged exercise. PLoS One 11, e0156460 (2016).
Fan, J. et al. Tyr phosphorylation of PDP1 toggles recruitment between ACAT1 and SIRT3 to regulate the pyruvate dehydrogenase complex. Mol. Cell 53, 534-548 (2014).
Kikuchi, D., Minamishima, Y. A. & Nakayama, K. Prolyl-hydroxylase PHD3 interacts with pyruvate dehydrogenase (PDH)-E1 beta and regulates the cellular PDH activity. Biochem. Bioph. Res. Commun. 451, 288-294 (2014).
Spriet, L. L. et al. Pyruvate dehydrogenase activation and kinase expression in human skeletal muscle during fasting. J. Appl. Physiol. 96, 2082-2087 (2004).
Bowker-Kinley, M. M., Davis, W. I., Wu, P. F., Harris, R. A. & Popov, K. M. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem. J. 329, 191-196 (1998).
Bricker, D. K. et al. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, drosophila, and humans. Science 337, 96-100 (2012).
Herzig, S. et al. Identification and functional expression of the mitochondrial pyruvate carrier. Science 337, 93-96 (2012).
Bergersen, L. H. Lactate transport and signaling in the brain: potential therapeutic targets and roles in body-brain interaction. J. Cereb. Blood Flow Metab. 35, 176-185 (2015).
Hoque, R., Farooq, A., Ghani, A., Gorelick, F. & Mehal, W. Z. Lactate reduces liver and pancreatic injury in Toll-like receptor- and inflammasome-mediated inflammation via GPR81-mediated suppression of innate immunity. Gastroenterology 146, 1763-1774 (2014).
Saddik, M., Gamble, J., Witters, L. A. & Lopaschuk, G. D. Acetyl-CoA carboxylase regulation of fatty acid oxidation in the heart. J. Biol. Chem. 268, 25836-25845 (1993).
He, X. D. et al. Sensing and transmitting intracellular amino acid signals through reversible lysine aminoacylations. Cell Metab. 27, 151-166 (2018).
Zhang, D. et al. Metabolic regulation of gene expression by histone lactylation. Nature 574, 575-580 (2019).
Gotz, A. et al. Exome sequencing identifies mitochondrial alanyl-tRNA synthetase mutations in infantile mitochondrial cardiomyopathy. Am. J. Hum. Genet. 88, 635-642 (2011).
Euro, L. et al. Structural modeling of tissue-specific mitochondrial alanyl-tRNA synthetase (AARS2) defects predicts differential effects on aminoacylation. Front. Genet. 6, 21 (2015).
Filosto, M. et al. A novel mitochondrial tRNA(Ala) gene variant causes chronic progressive external ophthalmoplegia in a patient with Huntington disease. Mol. Genet. Metab. Rep. 6, 70-73 (2016).
Konovalova, S. & Tyynismaa, H. Mitochondrial aminoacyl-tRNA synthetases in human disease. Mol. Genet. Metab. 108, 206-211 (2013).
Tuppen, H. A. L., Blakely, E. L., Turnbull, D. M. & Taylor, R. W. Mitochondrial DNA mutations and human disease. Biochim. Biophys. Acta 1797, 113-128 (2010).
Vo, M.-N. et al. ANKRD16 prevents neuron loss caused by an editing-defective tRNA synthetase. Nature 557, 510-515 (2018).
Hussien, T. H. R. Lactate as a metabolic signal in gene expression: genechip analysis in L6 myocytes. Med. Sci. Sport Exer. 39, S289 (2007).
Taglia, I. et al. AARS2-related ovarioleukodystrophy: clinical and neuroimaging features of three new cases. Acta Neurol. Scand. 138, 278-283 (2018).
Ivan, M. et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for sensing. Science 292, 464-468 (2001).
Yao, P. & Fox, P. L. Aminoacyl-tRNA synthetases in medicine and disease. EMBO Mol. Med. 5, 332-343 (2013).
Chen, Y. J. et al. Lactate metabolism is associated with mammalian mitochondria. Nat. Chem. Biol. 12, 937-943 (2016).
Lee, D. C. et al. A lactate-induced response to hypoxia. Cell 161, 595-609 (2015).
Isoe, T. et al. High glucose activates HIF-1-mediated signal transduction in glomerular mesangial cells through a carbohydrate response element binding protein. Kidney Int. 78, 48-59 (2010).
Seo, J. et al. Fatty-acid-induced FABP5/HIF-1 reprograms lipid metabolism and enhances the proliferation of liver cancer cells. Commun. Biol. 3, 638 (2020).
Sonveaux, P. et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS One 7, e33418 (2012).
Morales, M. & Munne-Bosch, S. Malondialdehyde: facts and artifacts. Plant Physiol. 180, 1246-1250 (2019).
Abrantes, H. D. et al. The lactate receptor HCAR1 modulates neuronal network activity through the activation of G (alpha) and G (beta gamma) subunits. J. Neurosci. 39, 4422-4433 (2019).
Baumann, F. et al. Lactate promotes glioma migration by TGF-beta 2-dependent regulation of matrix metalloproteinase-2. Neuro. Oncol. 11, 368-380 (2009).
Zhao, S. et al. Regulation of cellular metabolism by protein lysine acetylation. Science 327, 1000-1004 (2010).
Richardson, R. S., Leigh, J. S., Wagner, P. D. & Noyszewski, E. A. Cellular Po-2 as a determinant of maximal mitochondrial consumption in trained human skeletal muscle. J. Appl. Physiol. 87, 325-331 (1999).
Fan, Y., Han, J., Yang, Y. & Chen, T. Novel mitochondrial alanyl-tRNA synthetase 2 (AARS2) heterozygous mutations in a Chinese patient with adult-onset leukoencephalopathy. BMC Neurol. 22, 214 (2022).
Palacios, O. M. et al. Diet and exercise signals regulate SIRT3 and activate AMPK and PGC-1a in skeletal muscle. Aging 1, 771-783 (2009).
Jing, E. et al. Sirt3 regulates metabolic flexibility of skeletal muscle through reversible enzymatic deacetylation. Diabetes 62, 3404-3417 (2013).
Imamura, M. et al. MondoA deficiency enhances sprint performance in mice. Biochem. J. 464, 35-48 (2014).
Liu, F. et al. Effects of hereditary moderate high fat diet on metabolic performance and physical endurance capacity in C57BL/6 offspring. Mol. Med. Rep. 17, 4672-4680 (2018).
Carrell, E. M., Coppola, A. R., McBride, H. J. & Dirksen, R. T. Orai1 enhances muscle endurance by promoting fatigue-resistant type I fiber content but not through acute store-operated entry. FASEB J. 30, 4109-4119 (2016).
Kammoun, M. et al. Impact of TIEG1 on the structural properties of fast- and slowtwitch skeletal muscle. Muscle Nerve 55, 410-416 (2017).
Nemeria, N. et al. Inhibition of the Escherichia coli pyruvate dehydrogenase complex E1 subunit and its tyrosine 177 variants by thiamin 2-thiazolone and thiamin 2-thiothiazolone diphosphates. Evidence for reversible tight-binding inhibition. J. Biol. Chem. 276, 45969-45978 (2001).
Schulze, W. X. & Usadel, B. Quantitation in mass-spectrometry-based proteomics. Annu. Rev. Plant Biol. 61, 491-516 (2010).
شكر وتقدير
تم دعم هذا العمل من خلال منح من البرامج الرئيسية للتنمية في الصين (2018YFA0800300 إلى S.Z.; 2018 YFA0801300 إلى W.X.)، ومؤسسة العلوم الطبيعية الوطنية في الصين (92253305، 31821002، 32230054، 31930062، 91857000 و92157001 إلى S.Z.; 32171298 إلى W.X.; 81971449 و82171672 إلى Y.Y.)، وبرنامج قادة البحث الأكاديمي في شنغهاي (21XD1423000 إلى W.X.).
مساهمات المؤلفين
قام S.Z. و W.X. بتصميم المشروع وكتابة المخطوطة. قام Y.M. و J.Z. و Q.Z. و Y.W. و X.H. و K.Z. و H.Y. و H.Z. و Y.Z. و P.L. بإجراء التجارب الخلوية البيوكيميائية. قام Q.Z. و Y.W. و Z.Z. بإجراء اختبارات الجري على الفئران. قام B.W. بإجراء تجارب المحاكاة الحاسوبية. أشرف Y.L. و Y.Y. و J.Z. على جميع التجارب. قرأ جميع المؤلفين وناقشوا المخطوطة.
الوصول المفتوح، هذه المقالة مرخصة بموجب رخصة المشاع الإبداعي ترخيص النسبة 4.0 الدولي، الذي يسمح بالاستخدام والمشاركة والتكيف والتوزيع والاستنساخ بأي وسيلة أو صيغة، طالما أنك تعطي الائتمان المناسب للمؤلفين الأصليين والمصدر، وتوفر رابطًا لترخيص المشاع الإبداعي، وتوضح ما إذا تم إجراء تغييرات. الصور أو المواد الأخرى من طرف ثالث في هذه المقالة مشمولة في ترخيص المشاع الإبداعي الخاص بالمقال، ما لم يُشار إلى خلاف ذلك في سطر الائتمان للمواد. إذا لم تكن المادة مشمولة في ترخيص المشاع الإبداعي الخاص بالمقال وكان استخدامك المقصود غير مسموح به بموجب اللوائح القانونية أو يتجاوز الاستخدام المسموح به، فستحتاج إلى الحصول على إذن مباشرة من صاحب حقوق الطبع والنشر. لعرض نسخة من هذا الترخيص، قم بزيارة http://creativecommons.org/licenses/by/4.0/.
مستشفى التوليد وأمراض النساء بجامعة فودان، مختبر شنغهاي الرئيسي لإعادة تشكيل الأيض والصحة، المختبر الرئيسي للدولة للهندسة الوراثية، كلية علوم الحياة، مستشفى الأطفال بجامعة فودان، ومعاهد العلوم الطبية الحيوية، جامعة فودان، شنغهاي، الصين.قسم بيولوجيا السرطان، معهد دانا-فاربر للسرطان، بوسطن، ماساتشوستس، الولايات المتحدة الأمريكية.قسم الكيمياء الحيوية والدوائية الجزيئية، كلية الطب بجامعة هارفارد، بوسطن، ماساتشوستس، الولايات المتحدة الأمريكية.المختبر الرئيسي لتنظيم التكاثر (معهد شنغهاي للبحوث في تنظيم الأسرة)، شنغهاي، الصين.مستشفى الشعب الخامس في جامعة فودان، جامعة فودان، شنغهاي، الصين.المختبر الرئيسي لعلم الكيمياء النباتية لهضبة التبت في مقاطعة تشينغهاي، كلية الصيدلة، جامعة القوميات في تشينغهاي، شيونغ، تشينغهاي، الصين.مختبر قوانغدونغ الإقليمي الرئيسي لعلم الوراثة الوبائي للأورام الخبيثة وتنظيم الجينات، المختبر المشترك بين قوانغدونغ وهونغ كونغ لطب RNA، معهد RNA الطبي الحيوي، مركز الأبحاث الطبية، مستشفى صن يات سن التذكاري، جامعة صن يات سن، قوانغتشو، قوانغدونغ، الصين.ساهم هؤلاء المؤلفون بالتساوي: يونزي ماو، جياوجياو تشانغ، تشيان تشو، شيايدي هي.البريد الإلكتروني:xuwei_0706@fudan.edu.cn؛zhaosm@fudan.edu.cn
Oxidative phosphorylation (OXPHOS) consumes oxygen to produce ATP. However, the mechanism that balances OXPHOS activity and intracellular oxygen availability remains elusive. Here, we report that mitochondrial protein lactylation is induced by intracellular hypoxia to constrain OXPHOS. We show that mitochondrial alanyl-tRNA synthetase (AARS2) is a protein lysine lactyltransferase, whose proteasomal degradation is enhanced by proline 377 hydroxylation catalyzed by the oxygen-sensing hydroxylase PHD2. Hypoxia induces AARS2 accumulation to lactylate PDHA1 lysine 336 in the pyruvate dehydrogenase complex and carnitine palmitoyltransferase 2 (CPT2) lysine 457/8, inactivating both enzymes and inhibiting OXPHOS by limiting acetyl-CoA influx from pyruvate and fatty acid oxidation, respectively. PDHA1 and CPT2 lactylation can be reversed by SIRT3 to activate OXPHOS. In mouse muscle cells, lactylation is induced by lactate oxidation-induced intracellular hypoxia during exercise to constrain high-intensity endurance running exhaustion time, which can be increased or decreased by decreasing or increasing lactylation levels, respectively. Our results reveal that mitochondrial protein lactylation integrates intracellular hypoxia and lactate signals to regulate OXPHOS.
Oxidative phosphorylation (OXPHOS) is the major ATP-producing process that requires oxygen. Muscle cells upregulate OXPHOS to increase energy supply during exercise. This is achieved partly by switching to lactate oxidation via OXPHOS. Although these processes are consistent with the increase in ATP supply distributed via the mitochondrial reticulum for muscle contraction during exercise, it raises a question regarding how muscle cells maintain a balance between oxygen-consuming OXPHOS that generates ATP and the hypoxic conditions created by exercise. This is because, although the intramuscular oxygen supply is increased by enhanced pulmonary diffusion capacity, cardiac output, the oxygen-carrying capacity of the blood, and skeletal muscle oxygen extraction during excercise, the hypoxia, i.e., the decrease of partial pressure of oxygen ( ) from Torr, at rest, to 3-4 Torr under exercise at about 65% of maximal oxygen consumption is induced in muscles. Moreover, the mechanism utilized by muscle cells to prevent the overproduction of reactive oxygen species (ROS), which occurs under endurance exercise-associated hypoxia and leads to oxidative stress in cells and tissues, remains unclear.
Cellular physiology adapts to oxygen availability by sensing oxygen availability via the prolyl hydroxylases (PHDs)-von HippelLindau (VHL) -hypoxia-inducible factor a (HIFa) axis. PHDs use oxygen and -ketoglutarate as substrates to hydroxylate proline residues in proteins containing PHD-recognizing sequences. Hydroxylated proteins, such as HIF1a, are subsequently recognized and degraded by VHL, an E3 ligase. HIFas are transcription factors that regulate an array of processes, such as glucose uptake, glycolysis, and vascularization that provide substrates for OXPHOS. However, HIFas do not directly regulate muscle cell OXPHOS, but only exert transcriptional regulation, which takes hours to be effective, and thus lag behind exercise-induced changes in OXPHOS in muscles that take only minutes to manifest after starting to exercise.
Lactate is quickly induced by exercise or heartbeat, due to enhanced aerobic glycolysis and glycogen catabolism that elevates pyruvate, which is then thermodynamic favorably converted to lactate by lactate dehydrogenase A (LDHA). Besides LDHA, lactate homeostasis is also determined by the pyruvate dehydrogenase complex (PDC) that channels pyruvate to acetylCoA ( ) as an OXPHOS substrate. In muscle cells, the PDC,
which is activated during exercise due to changes taking place in the NADH/NAD ratio and metal ions, can be inactivated by acetylation, and by PHD3 binding to PDC E1 beta (PDHE1B), both leading to an increase in phosphorylation of serines 232, 293, and 300 of PDHA1, the catalytic subunit of PDC. Limiting AcCoA , originating from glucose, glycogen, and lactate catabolism via or from fatty acid oxidation (FAO), inhibits OXPHOS and reduces oxygen consumption. However, lactate, while serving as a preferred fuel source for muscle and heart cells via OXPHOS, inhibits lipolysis and carnitine palmitoyltransferase 1 (CPT1)mediated fatty acid import into mitochondria, showing that lactate exerts both positive and negative regulation of OXPHOS.
Mechanisms underlying the sensing of lactate as well as the generation of lactate signals to regulate OXPHOS remain unclear. Lactate signaling is highlighted by the modification of lysine in proteins by lactate, although a bona fide lactyltransferase remains elusive. Mutations in mitochondrial alanyl-tRNA synthetase 2 (AARS2), which catalyzes alanyl-tRNA formation in mitochondrial protein synthesis and induces lysine alanylation in proteins, cause impaired OXPHOS, lactic acid deregulation and muscle function loss, with underlying mechanisms undefined. We were intrigued by the possibility that AARS2 might act as a potential lactyltransferase that regulates muscle OXPHOS. In this study, we aimed to determine whether AARS2 acts as a mitochondrial lactyltransferase that integrates intracellular hypoxia and lactate signals to regulate muscle cell OXPHOS.
RESULTS
AARS2 contains PHD-recognizing sequence and is hydroxylated by PHD2
AARS2 proline 377 (P377) lies in a PHD-recognizing sequence, which is not present in the corresponding sequence of cytosolic alanyl-tRNA synthetase 1 (AARS1) (Fig. 1a), indicating that P377 may be hydroxylated, allowing AARS2 levels similar to HIF1a, to be regulated during hypoxia. This was confirmed by the observation that, although hypoxia time-dependently induced AARS2 expression in both mitochondria and the cytoplasm of mouse proliferating myoblasts C2C12 cells, mouse HL-1 cardiomyocytes, and primary mouse hepatocytes, AARS1 was expressed only in the cytosol and was not induced by hypoxia (Fig. 1b; Supplementary information, Fig. S1a, b). Hypoxia also induced AARS2 protein levels in mouse primary myoblasts over time (Supplementary information, Fig. S1c). Moreover, AARS2 protein levels did not change with hypoxia when P377 was switched to leucine (Fig. 1c), which substantiated the observation that hypoxia-mediated upregulation of AARS2 protein levels requires P377. Silencing or knocking out Phd2, but not other oxygen-sensing proline hydroxylases, such as Phd1 and Phd3, in C2C12 cells elevated the levels of endogenous AARS2 (Supplementary information, Fig. S1d) and prevented hypoxia-mediated accumulation of endogenous AARS2 (Fig. 1d). These results, together with the observation that PHD2 overexpression decreased AARS2 levels in C2C12 cells (Fig. 1e), whereas inhibiting PHDs with roxadustat increased them (Supplementary information, Fig. S1e), confirmed that PHD2 acts as a proline hydroxylase and oxygen sensor for AARS2.
We searched for hydroxylated P377 (P377OH)-containing peptides in the 120 tandem mass (MS/MS) spectra of a Glu-C protease peptide library (Supplementary information, Table S1) of AARS2 overexpressing HEK293T cells, and found 3 spectra matching the projected MS/MS spectrum of a P377OHcontaining AARS2 peptide (Supplementary information, Fig. S1f). As a synthetic P377OH-containing Glu-C AARS2 peptide generated a spectrum identical to that of the identified MS/MS spectrum (Supplementary information, Fig. S1f), we concluded that P377 was hydroxylated in vivo. Therefore, to detect P377OH in cells, we generated a P377OH site-specific antibody that specifically recognizes in the peptide of intact AARS2
(Supplementary information, Fig. S1g, h). The P377OH levels of ectopically expressed AARS2 in C2C12 cells and mouse primary myoblasts were time-dependently decreased by hypoxia exposure in the presence of MG132, a proteasome inhibitor (Fig. 1f; Supplementary information, Fig. S1i). P377OH levels decreased in mouse leg muscles in which protein degradation was inhibited by MG132 (Fig. 1g) after hypoxia was induced by running (Supplementary information, Fig. S1j), confirming that P377OH is regulated by oxygen availability physiologically. Additionally, PHD2 overexpression increased P377OH levels in C2C12 cells, further to which, both MG132 and PHD2 enhanced the cellular P377OH levels synergistically (Fig. 1h, i). This result confirmed that P377OH, which possibly acts as a negative regulator of AARS2, is in turn modulated by PHD2.
AARS2 acts as a substrate of the PHD2-VHL proteasomal machinery
Ectopically expressed VHL co-precipitated with overexpressed and endogenous AARS2 in C2C12 cells (Fig. 2a, b), consistent with endogenous AARS2 interacting with endogenous PHD2 and VHL in mouse primary myoblasts (Supplementary information, Fig. S1k), suggesting that AARS2 is a substrate of VHL. When co-expressed in C2C12 cells, the interaction between wild-type AARS2 and VHL was greater than that between AARS2 harboring P377L and VHL (Fig. 2c); moreover, the AARS2-VHL interaction was decreased and increased by hypoxia (Fig. 2d) and PHD2 overexpression (Fig. 2e), respectively, indicating that P377OH increased the VHL-AARS2 interaction. VHL overexpression increased the ubiquitination levels of AARS2 expressed in HEK293T cells (Fig. 2f), whereas it reduced endogenous mitochondrial and cytosolic AARS2 protein levels in C2C12 cells (Fig. 2g). Conversely, Vhl knockout (KO) in C2C12 cells increased endogenous AARS2 levels and abrogated the hypoxiamediated increase in AARS2 levels (Fig. 2h). These results collectively confirmed that VHL degrades AARS2.
AARS2 inhibits Ac-CoA production and OXPHOS
AARS2 overexpression decreased the oxygen consumption rate (OCR) in C2C12 cells (Fig. 3a). Aars2 KO (Aars2 ) increased OCR and diminished OCR response to hypoxia in C2C12 cells, and these consequences were rescued by reintroducing wild-type AARS2, but not the AARS2 P377L mutant into Aars2 cells (Fig. 3b, c), indicating that AARS2 responded to hypoxia by inhibiting cellular respiration. Additionally, AARS2 overexpression decreased Ac-CoA levels but increased pyruvate and lactate levels in C2C12 cells (Fig. 3d), suggesting that AARS2 negatively regulated PDC, the enzyme complex that converts pyruvate to . This was further supported by the observation that ectopically overexpressing AARS2 to double cellular AARS2 levels decreased the specific activity of PDHA1 (Fig. 3e), whereas Aars2 KO increased it (Fig. 3f). PDHA1 activities increased while their responses to hypoxia were abrogated in Aars2 KO C2C12 cells; the responses to hypoxia were however, restored by reintroducing wild-type, but not P377L mutant AARS2 (Supplementary information, Fig. S2a). Moreover, AARS2 overexpression did not alter the phosphorylation levels of serines 232, 293, and 300 of PDHA1 (Supplementary information, Fig. S2b), which are known to inactivate PDHA1, implying that AARS2 inactivates PDHA1 via a novel mechanism under the control of hypoxia and AARS2.
As CPT1/2/FAO inhibition may also contribute to OCR inhibition, by decreasing Ac-CoA supply to OXPHOS, -palmitate was used to investigate whether CPT1/2/FAO was inhibited by AARS2 (Supplementary information, Fig. S2c). AARS2 overexpression decreased double-labeled (M2) -Ac-CoA production from -palmitate (Fig. 3g), indicating that AARS2 inhibited CPT1/2/FAO. Only endogenous carnitine palmitoyltransferase 2 (CPT2), and not CPT1 or FAO enzymes, interacted with ectopically expressed AARS2 (Supplementary information, Fig. S2d). Moreover, AARS2 overexpression decreased CPT2 activity (Fig. 3h), whereas Aars2 KO increased it (Fig. 3i), confirming that AARS2 inactivates CPT2 possibly via a
Fig. 1 Oxygen regulates AARS2 protein levels via PHD2. a AARS2 carries a PHD-recognizing sequence. The amino acid (aa) sequence from 369 to 380 of AARS2 and its corresponding AARS1 sequence (aa 343-354) were aligned to the PHD-recognizing sequence. Consensus PHDrecognizing residues are marked in blue. b AARS2 protein levels are regulated by hypoxia. Mitochondrial and cytosolic AARS1, AARS2 and HIF1 levels were detected in C2C12 cells cultured in a hypoxia chamber for the indicated time durations. COXIV and actin were used to demonstrate the successful isolation of mitochondria and cytoplasm, respectively (the same method was employed for subcellular fractionation henceforth). Chamber oxygen levels were monitored. c P377 is required for the regulation of AARS2 protein levels by hypoxia. Stably expressed AARS2 and AARS2 protein (P377L) levels in C2C12 cells were measured after the cells were cultured in a hypoxia chamber for the indicated time durations. d, e PHD2 regulates AARS2 protein levels. Mitochondrial and cytosolic AARS1 and AARS2 levels were measured in C2C12 cells or Phd2 KO C2C12 cells cultured in a hypoxia chamber for the indicated time durations (d) and in C2C12 cells overexpressing PHD2 (e). f-i Hypoxia and PHD2 regulate P377 hydroxylation. P377OH levels were determined via western blot assay in C2C12 cells cultured in a hypoxia chamber for indicated time durations in the presence of MG132 in the culture media to prevent proteasomal degradation (f); in the resting and 30 min running mouse leg skeletal muscles that were intravenously injected with or without MG132 twice a week for 4 consecutive weeks ( ) ( ); in the PHD2 overexpressing C2C12 cells treated with MG132 ( ). P377OH levels in PHD2 overexpressing C2C12 cells in the presence and absence of MG132 were quantified by mass spectrometry (i) ( ). All data are reported as mean SEM of three independent experiments. Statistical significance was assessed by two-way ANOVA: ; .
covalent modification. Furthermore, Aars2 KO activated CPT2 and diminished responses of CPT2 activity to hypoxia, which were rescued by reintroducing wild-type AARS2, but not AARS2 P377L mutant (Supplementary information, Fig. S2e). Cpt2 KO in C2C12 cells rendered production unresponsive to AARS2 overexpression (Fig. 3g). Finally, neither CPT1 specific activity (Supplementary
information, Fig. S2f) nor protein level (Supplementary information, Fig. S2g) was affected by AARS2 overexpression. These data collectively suggested that AARS2 inactivates CPT2 to inhibit FAO.
We used -labeled glutamine to trace -ketoglutarate ( -KG) influx, which is the point at which amino acids feed into the TCA cycle (Supplementary information, Fig. S2h). The levels
were not affected by AARS2 overexpression (Supplementary information, Fig. S2i), suggesting that amino acid catabolism was not regulated by AARS2. These results collectively suggested that AARS2 inhibits respiration by decreasing Ac-CoA supply from glycolysis and FAO by inactivating PDHA1 and CPT2, and not via amino acid catabolism (Supplementary information, Fig. S2j).
Next, we verified whether hypoxia mediates PDHA1 and CPT2 regulation via AARS2 in vivo. HIFas and AARS2 protein levels were
induced in mouse leg skeletal muscles in a time-dependent manner while mice were running on a treadmill (Fig. 3j). Moreover, muscle Ac-CoA level, which was moderately decreased while muscle PDC- (Fig. 3k) and CPT2- (Fig. 3l) specific activities kept decreasing, and hypoxia in leg muscle (Supplementary information, Fig. S3a) as well as lactate and pyruvate levels kept increasing, after running started (Fig. 3m). This was coincident with a brief increase in the total PDC activity in mouse leg skeletal
Fig. 2 AARS2 acts as a substrate of the PHD2-VHL proteasomal machinery. VHL interacts with AARS2. Co-immunoprecipitated, coexpressed (a) and endogenous (b) AARS2 was detected in C2C12 cells ectopically expressing VHL. c P377L mutant shows a weakened affinity for VHL. Interactions between VHL and AARS2, and between VHL and P377L were detected via co-immunoprecipitation when they were coexpressed in C2C12 cells. Densitometric analysis for images was provided. d, e VHL-AARS2 interaction is regulated by hypoxia and PHD2. The amount of AARS2 co-purified VHL was determined under normoxia and 8 h hypoxia (d), as well as with or without PHD2 overexpressing (e) in C2C12 cells. Densitometric analysis for images was provided. VHL mediates proteasomal degradation. AARS2 ectopically expressed in HEK293T cells was assayed for ubiquitination when expressed alone and when co-expressed with ubiquitin or VHL, or both, under the presence and absence of MG132. g VHL downregulates AARS2 protein levels. Mitochondrial and cytosolic AARS2 levels were measured in C2C12 cells with or without VHL overexpression. Hypoxia induces AARS2 accumulation in a VHL-dependent manner. Mitochondrial and cytosolic AARS2 levels were measured in C2C12 cells and VhI KO C2C12 cells both cultured in a hypoxia chamber for the indicated time durations. All data are reported as mean SEM of three independent experiments. Statistical significance was assessed by unpaired two-tailed Student’s -test: .
muscles, which was then followed by a decrease to levels below the resting levels (Fig. 3n). The initial increase in total PDC activity was consistent with an initial decrease in PDHA1 phosphorylation (Supplementary information, Fig. S3b) and phosphatase treatment to diminish phosphorylation effects (Supplementary information, Fig. S3c) abrogated initial increase (Fig. 3n). However, the decrease in PDC activity with running time was unaffected even after phosphatase treatment (Fig. 3n). These, together with the observation that neither the protein levels of PDC components and CPT2 (Supplementary information, Fig. S3d) nor the protein levels and activities of LDHs (Supplementary information, Fig. S3d, e) were affected throughout 30 min running, suggesting that AARS2 may mediate a covalent modification mechanism which inactivates PDC without involving PDHA1 phosphorylation.
Lactate inactivates PDHA1 and CPT2 dependent on AARS2
Lactate decreased PDHA1 and CPT2-specific activities (Fig. 4a). Cell membrane permeable methyl-lactate (Me-Lac) was employed to analyze lactate effects. Increased OCRs, as well as decreased PDHA1 and CPT2-specific activities, were observed in C2C12 cells treated with 2 mM Me-Lac, which increased the mitochondrial lactic acid levels from 0.6 mM to 1.0 mM ; however, Me-Lac at 10 mM , which increased mitochondrial lactic acid to 3.2 mM , decreased OCRs, as well as the specific activities of PDHA1 and CPT2 (Fig. 4b-d; Supplementary information, Fig. S4a). The 2 mM Me-Lac treatment results were consistent with the observation that lactate fuels muscle cells via the TCA cycle. However, the 10 mM Me-Lac treatment, results in intracellular lactate levels mimicking those in muscle during exercise, suggesting that lactate also plays inhibitory roles in OXPHOS, likely via a covalent modification to PDHA1 and CPT2, a notion confirmed by Me-lac decreasing PDHA1 and CPT2 activity in C2C12 cells, but not in Aars2 KO C2C12 cells (Supplementary information, Fig. S4b, c). The above notion was further confirmed by examining Ac-CoA production in lactate-oxidizing C2C12 and HL-1 cells, which revealed that 10 mM Me-Lac decreased production from -glucose (Fig. 4e, f) and -palmitate (Fig. 4g, h) in an AARS2-dependent manner. Intriguingly, loss of AARS2 increased production from -glucose and -palmitate (Fig. 4e-h), indicating that lactate inactivated PDHA1 and CPT2 via AARS2, and that lactate does not inactivate PDHA1 and CPT2 and OXPHOS via its acidity. Moreover, 10 mM Me-Lac inhibited OCR in primary myoblasts in an AARS2-dependent manner (Supplementary information, Fig. S4d). Further, Me-Lac negligibly affected -a-KG production from -glutamine (Supplementary information, Fig. S4e), which was consistent with that amino acid catabolism was not regulated by lactic acid.
AARS2 inactivates PDHA1 and CPT2 by lactylating these proteins
Lactic acid is structurally analogous to alanine (Fig. 5a). The binding of lactic acid to AARS2 was modeled using molecule docking analysis (Supplementary information, Fig. S4f) and confirmed via isothermal titration calorimetry (ITC) (Supplementary information, Fig. S4g).
Moreover, overexpressed AARS2 interacted with both endogenous PDHA1 (Supplementary information, Fig. S4h) and ectopically expressed CPT2 (Supplementary information, Fig. S4i). These results collectively suggested that AARS2 may function as a lactyltransferase for PDHA1 and CPT2. To confirm this hypothesis, we first searched for lactylated lysine residues in PDHA1 and CPT2. Using a tryptic peptide library of purified PDHA1 and a Glu-C peptide library of purified CPT2, we found that PDHA1 lysine 336 (Supplementary information, Fig. S5a) and CPT2 lysine 457 and 458 (Supplementary information, Fig. S5b) were possibly lactylated (Lac-K336, Lac-K457/8). The presence of Lac-K336, as well as Lac-K457/8, was confirmed by matching the MS/MS spectra of synthetic Lac-K336- and Lac-K457/8containing PDHA1 and CPT2 peptides to those from the peptide library (Supplementary information, Fig. S5a, b).
We assessed the ability of AARS2 to lactylate synthetic PDHA1 K336, CPT2 K457/8 peptides and intact PDHA1 and CPT2 proteins in vitro. As opposed to the AARS2 mutant, which has the asparagine 104 to tyrosine switch, equivalent to asparagine 71 to tyrosine switch in the AARS1 (AARS1 , the inactivated form of AARS1 ) (Supplementary information, Fig. S6a), AARS2 lactylated the synthetic K336-containing PDHA1 peptide as well as the K457/8-containing CPT2 peptide (Fig. 5b; Supplementary information, Fig. S6b) via lactate- and ATP-dependent and pyrophosphate-inhibitable mechanisms. However, AARS2 did not lactylate those peptides in which lysine residues had been changed to alanine (Supplementary information, Fig. S6c). These results suggested that the mechanism via which AARS2 attaches lactate to lysine is identical to that is used to attach alanine to lysine (Supplementary information, Fig. S6d). The of AARS2 acting on a synthetic K336containing PDHA1 peptide was within the physiological lactate levels of 5.21 mM , while the turnover number ( ) of AARS2 for this peptide was 1.19 (Fig. 5c), indicating that AARS2 was an efficient lactyltransferase.
We generated polyclonal site-specific antibodies that recognized Lac-K336 (Supplementary information, Fig. S6e) and LacK457/8 (Supplementary information, Fig. S6f). Treatment with 10 mM Me-Lac increased the Lac-K336 and Lac-K457/8 levels of ectopically expressed PDHA1 (Fig. 5d) and CPT2 (Fig. 5e) in C2C12 and HL-1 cells (Supplementary information, Fig. S6g). However, the lactylation was not observed when Aars2 was knocked out (Fig. 5d, e). Also, Lac-K336 and Lac-K457/8 increased in AARS2overexpressing primary myoblasts and decreased after Aars2 KO (Supplementary information, Fig. S6h). These results suggest that AARS2 acts as the lactyltransferase of PDHA1 K336 and CPT2 K457/ 8. Moreover, consistent with the observation that lactate can be transported into mitochondria by monocarboxylate transporter 1 (MCT1), Me-Lac supplementations in the culture media increased, whereas inhibition of MCT1 with a-cyano-4hydroxycinnamic acid ( ) decreased mitochondrial lactate levels (Fig. 5f), and this result was consistent with a-CHCA treatment which reduced Lac-K336 and Lac-K457/458 levels in C2C12 cells (Fig. ). These results, together with the observation that mitochondrial lactate levels increased proportionally with lysate lactate levels, both are in the range of AARS2 toward lactate, in mouse leg skeletal muscle after running
(Fig. 5i), indicating that exercise-induced lactate may regulate mitochondrial lactylation.
Running enhanced Lac-K336 and Lac-K457/8 levels in mouse muscle cells (Fig. 5j), consistent with the finding that hypoxia increased both lysate and mitochondria lactate (Supplementary
information, Fig. S6i) and Lac-K336 and Lac-K457/8 levels in C2C12 cells (Fig. 5k, I). PHD inhibition via roxadustat increased Lac-K336 and Lac-K457/8 (Supplementary information, Fig. S6j, k) in C2C12 cells, thereby highlighting the physiological significance of Lac-K336 and Lac-K457/8. Estimation via MS showed that
Fig. 3 AARS2 inhibits Ac-CoA production and OXOPHOS. a, b AARS2 regulates OCR. The effects of overexpressing AARS2 (left, OROBOROS Oxygraph-2K measurements; right, quantitation) (a), and Aars2 KO using independent sgRNAs (b) on OCR ( influx per volume cells, ) were determined ( ). c AARS2 regulates OCR in response to oxygen levels. The effects of re-introducing wide-type AARS2 or P377L into Aars C2C12 cells were determined in cells under normoxic conditions and cells which were exposed to hypoxia for 8 h ( ). AARS2 regulates metabolite levels. Relative levels (to those of C2C12 cells) of lactate, pyruvate, and Ac-CoA in C2C12 cells and C2C12 cells overexpressing AARS2 ( ) were evaluated. e, AARS2 regulates PDHA1-specific activity. Relative specific activities (to those from C2C12 cells) of PDHA1 purified from C2C12 cells overexpressing AARS2 (e) and Aars2 KO C2C12 cells (f) were determined ( ). AARS2 regulates CPT2-mediated Ac-CoA production. The percentages of unlabeled ( ), single labeled ( ), and double-labeled ( ) Ac-CoA from -palmitate in C2C12 cells and C2C12 cells in which Cpt2 had been knocked out using independent sgRNAs, were determined with or without AARS2 overexpression; -palmitate chasing was performed for . The M+2 percentages in C2C12 cells with and without AARS2 overexpression were compared for significance. , i AARS2 regulates CPT2-specific activity. The relative specific activity (to those from C2C12 cells) of CPT2, isolated from both AARS2-overexpressing (h) and Aars2 KO C2C12 cells (i) was determined ( ). j Running induces HIF s and AARS2 expression in mouse leg skeletal muscles. HIF1 , HIF2 , HIF3 , AARS1, and AARS2 levels in mouse leg skeletal muscles were determined before and after running for the indicated durations ( ). k, I Running inactivates PDC and CPT2 in vivo. The relative (to those of resting mice) specific activities of PDC (k) and CPT2 (l) in mouse leg skeletal muscles were determined after mice started running for indicated durations ( ). Endurance running-induced lactate accumulation and Ac-CoA reduction. The lactate, pyruvate, and Ac-CoA levels in mouse leg skeletal muscles were determined after mice were allowed to run for the indicated durations ( ). AARS2 inactivates PDC via a mechanism other than phosphorylation. The relative PDC activities (to protein amount) of wild-type and Aars2-/- mouse leg skeletal muscles sampled at different time points after starting to run with or without dephosphorylation induced by phosphatase, were determined ( ). The activities at time 0 were set as . All data are reported as mean SEM of three independent experiments. Statistical significance was assessed by unpaired two-tailed Student’s -test and two-way ANOVA: ; ; ; ; ns no significance.
Lac-K336 and Lac-K457/8 levels in mouse leg skeletal muscles were as high as under simultaneous AARS2 overexpression and Me-Lac treatment (Fig. 5 m ), and reached after mice running for 30 min (Fig. 5n) or when mice muscle exposed to hypoxia (Fig. 50), indicating that a substantial portion of PDHA1 and CPT2 is lactylated under intracellular muscle cell hypoxia and high lactate in vivo, a condition that mimics endurance exercise.
Converting the K336 of PDHA1 and K457/8 of CPT2 to K336Q and of the lactylation mimetic, glutamine (Q), reduced the activities of both enzymes while changing K336 and K457/8 to K336R and of the non-lactylation mimetic, arginine (R), negligibly affected their specific activities (Fig. 5p, q), indicating that lactylation inactivated PDHA1 and CPT2. Although AARS2 overexpression (Fig. 5p, q) and Me-Lac supplementation (Fig. 5r, s) increased Lac-K336 and Lac-K457/8 and inactivated PDHA1 and CPT2, they did not alter lactylation of lactylation site-null PDHA1 and CPT2 mutants, which was consistent with the observation that Aars mouse leg muscle showed elevated PDC and CPT2 activities (Supplementary information, Fig. S6I), confirming that lactylation underlies AARS2-mediated PDHA1 and CPT2 inactivation.
Finally, physiologic level of alanine failed to inhibit K336 lactylation in vitro (Supplementary information, Fig. S6m); methyl-alanine supplementation negligibly affected Lac-K336 and Lac-K457/8 and the specific activities of PDHA1 and CPT2 (Supplementary information, Fig. S6n, o), eliminating the possibility that alanine affects lactylation, and protein alanylation, induced by AARS2 upregulation, regulates PDHA1 and CPT2 activities.
SIRT3 reverses PDHA1 and CPT2 lactylation
To investigate whether Lac-K336 and Lac-K457/8 levels are dynamically regulated, we evaluated their responses to nicotinamide (NAM), a general inhibitor of sirtuins and deamidases that remove multiple amide-bonded protein modifications. NAM treatment increased Lac-K336 and Lac-K457/8 levels of ectopically expressed PDHA1 and CPT2 in C2C12 cells, respectively (Fig. 6a). Because Lac-K336 and Lac-K457/8 are mitochondrial protein modifications, we assessed the delactylase activities of mitochondrial sirtuins, namely SIRT3, SIRT4, and SIRT5. SIRT3 delactylated synthetic Lac-K336 and Lac-K457/8 peptides via an NAD dependent pathway, whereas the catalytic domain of SIRT4 or SIRT5 did not (Fig. 6b), indicating that SIRT3 is a delactylase for Lac-K336 and Lac-K457/8. This was further confirmed by the observation that both PDHA1 and CPT2 interacted with SIRT3
(Supplementary information, Fig. S7a, b), and that wild-type SIRT3 decreased Lac-K336 and Lac-K457/8 levels of purified intact PDHA1 (Fig. 6c) and CPT2 (Fig. 6d), whereas amidase-inactivated SIRT3 mutant did not. Moreover, SIRT3 overexpression decreased Lac-K336 and Lac-K457/8 levels in wild-type C2C12 cells, but not in those carrying lactylation site-null PDHA1 and CPT2 mutants (Fig. 6e, f). Furthermore, PDHA1 and CPT2 purified from the leg skeletal muscles of Sirt3 KO mice (Sirt3 mice) showed higher Lac-K336 and Lac-K457/8 levels, respectively, than those from the leg muscles in wild-type mice (Fig. 6g).
The specific activities of PDHA1 and CPT2 expressed in SIRT3overexpressing and Sirt3 KO C2C12 cells were higher and lower, respectively, than those expressed in C2C12 cells (Fig. 6h, i). However, neither SIRT3 overexpression nor Sirt3 KO affected the specific activities of lactylation site-null PDHA1 and CPT2 mutants (Supplementary information, Fig. S7c-f). Moreover, SIRT3 overexpression and Sirt3 KO increased and decreased production from -glucose (Fig. 6j, k) and -palmitate (Fig. 6l, m), respectively, in C2C12 cells.
Furthermore, SIRT3 overexpression decreased lactate (Fig. 6n) and free fatty acids levels (Fig. 60) in C2C12 cells, whereas Sirt3 KO increased these levels (Fig. 6p, q). These results were consistent with that SIRT3 delactylated and reversed the effects of lactylation on PDHA1 and CPT2. Lastly, SIRT3 overexpression, which may delactylate as well as deacetylate PDHA1, resulted in much more pronounced PDHA1 activation in C2C12 cells than in Aars2 KO C2C12 cells, suggesting that lactylation is a major PDHA1 inactivating mechanism (Supplementary information, Fig. S7g).
OXPHOS drives lactylation to feedback limit OXPHOS and endurance running ability
In vitro hypoxia exposure of mouse leg tissues induced Lac-K336 and Lac-K457/8 in wild-type, but not in Aars2 mouse leg tissues, indicating that hypoxia induces AARS2 as well as Lac-K336 and Lac-K457/8 (Supplementary information, Fig. S8a). To confirm that exercise-induced intramuscular hypoxia may be driven by activated muscle cell OXPHOS that acts as an oxygen sink, supplementation of culture media with either glucose, palmitate, or low lactate to enhance OXPHOS (Supplementary information, Fig. S8b) induced HIF1a alongside AARS2, Lac-K336 and Lac-K457/ 8 in C2C12 cells, whereas abrogating cell OXPHOS with rotenone diminished these effects (Fig. 7a). These results substantiated those of studies which demonstrated that enhancement of OXPHOS induced HIF1a in other cell types and confirmed that enhanced OXPHOS drives intramuscular hypoxia and
Fig. 4 Lactate inactivates PDHA1 and CPT2 dependent on AARS2. a Lactate treatments inactivate cellular PDHA1 and CPT2. C2C12 cells were treated with 10 mM lactate. The relative (to those from untreated C2C12 cells) specific activities of PDHA1 and CPT2 were determined . b Me-Lac treatments decrease cellular OCRs. OCRs of C2C12 cells that were untreated or treated with 2 mM or 10 mM Me-Lac for were determined. Me-Lac treatments inactivate cellular PDHA1 and CPT2. Relative specific activities (to those of untreated C2C12 cells) of PDHA1 (c) and CPT2 (d) in C2C12 cells that were untreated or treated with 2 mM or 10 mM Me-Lac ( ) were determined. Lactate treatments decrease Ac-CoA influx from glycolysis and FAO. Relative levels in -glucose ( ) and -palmitate ( ) (to untreated C2C12 cells) and ( only)-treated C2C12 cells, Aars2 KO C2C12 cells ( ) and HL-1 cells, Aars cells ( ) were detected before and after being treated with . The chasing time for and -palmitate was 1 h and 12 h , respectively. All data are reported as mean SEM of three independent experiments. Statistical significance was assessed by unpaired two-tailed Student’s -test and two-way ANOVA: ; ; ; **** ; ns no significance.
lactylation. Moreover, re-oxygenating hypoxia-treated C2C12 cells not only decreased Lac-K336 and Lac-K457/8 levels, but also activated PDHA1 and CPT2 and increased Ac-CoA production (Supplementary information, Fig. S8c, d), consistent with that OXPHOS-induced intramuscular hypoxia is the driver of OXPHOS inhibition. However, re-oxygenation negligibly affected SIRT3 activity (Supplementary information, Fig. S8e), suggesting that OXPHOS promptly responds to AARS2-mediated lactylation but not SIRT3-mediated delactylation.
An injection of lactate to mouse hind leg thigh muscle induced HIF1a, AARS2 (Fig. 7b), and Lac-K336/Lac-K457/8 (Supplementary information, Fig. S8f), while inhibition of LDHA using FX-11 decreased mouse muscle lactate production during running (Supplementary information, Fig. S8g) and largely abrogated running-linked induction of HIF1a, AARS2 and Lac-K336 and Lac-K457/8 in mouse hind leg thighs (Fig. 7c), echoing that LDHA inhibitor oxamate inhibits Lac-K336 and Lac-K457/8 in C2C12 cells (Supplementary information, Fig. S8h). These data highlight the fact that lactate production and oxidation play major roles in inducing muscle cell hypoxia and lactylation during exercise. Notably, 30 min running, which resulted in a substantial accumulation of lactate in both mitochondria-rich endurance ability-relative type I and mitochondria-less sprint ability-relative type Il mouse skeletal muscles (Fig. 7d), induced more
pronounced HIF1a, AARS2 and Lac-K336 and Lac-K457/8 in type I muscles (Fig. 7e), suggesting that lactylation regulates type I skeletal muscle OXPHOS and endurance exercise ability more than it regulates type II skeletal muscle OXPHOS and sprint ability. The observation that the ATP drop in type I muscle occurred coincidentally with Lac-K336 and Lac-K457/8 induction, as opposed to the ATP drop in type II muscle which occurred prior to Lac-K336 and Lac-K457/8 induction (Figs. 7f, 5j), supports this hypothesis. Moreover, wild-type, hypo-lactylated Aars2 , and hyper-lactylated Sirt3 mice all had comparable sprint abilities (Supplementary information, Fig. S8i), because lactate injection in mouse hind leg thigh only exerted a negligible effect on their sprinting abilities (Supplementary information, Fig. S8j). By contrast, mouse high-intensity running exhaustion times were moderately positively correlated with their basal leg muscle SIRT3 levels (Supplementary information, Fig. S8k) and strongly inversely related to their basal leg muscle AARS2 levels (Fig. 7g; Supplementary information, Fig. S8I). Moreover, AARS2 overexpressing specifically in mouse skeletal muscle by crossing hAARS2 expression mice with CKMM-cre mice (Supplementary information, Fig. S8m) and knocking out Sirt3, both increased Lac-K336 and Lac-K457/8 (Fig. 6g; Supplementary information, Fig. S8n), but decreased Ac-CoA influx (Fig. 7h), OCR (Fig. 7i), and ATP levels (Fig. 7j) in leg skeletal muscles and shortened running exhaustion
for lactate
PDHA1
K336
time (Fig. 7k). Furthermore, in contrast to lactate injection which shortened mouse running exhaustion time, -alanine injection to hind leg thigh muscle decreased Lac-K336 and Lac-K457/8 (Supplementary information, Fig. S8f), consistent with that alanine was an alanine analog (Fig. 5a) that binds to the alanine-
binding pocket of AARS2 (Supplementary information, Fig. S4f, g), thus decreasing Lac-K336 and Lac-K457/8 levels in vitro (Supplementary information, Fig. S9a, b), activating PDHA1 and CPT2 (Supplementary information, Fig. S9c, d), and increasing Ac-CoA levels (Supplementary information, Fig. S9e), OCR (Supplementary
Fig. 5 AARS2 inactivates PDHA1 and CPT2 by lactylating these proteins. a Lactic acid, -alanine, and alanine are analogs. Alignment of chemical structures of alanine, lactic acid, and -alanine. b AARS2 lactylates PDHA1 K336 and CPT2 K457/8 peptides in vitro. MS-based detection of lactylated synthetic PDHA1 K336 and CPT2 K457/8 peptides catalyzed by AARS2 and AARS2 are summarized ( : no product detected, : product detected, : less product detected; see Supplementary information, Fig. S6b for MS results). c AARS2 is an efficient lactyltransferase. The and of recombinant AARS2 toward lactate were determined when the synthetic K336-containing PDHA1 peptide was kept at . d, e KO abolishes lactate-linked induction of Lac-K336 and Lac-K457/8. Lac-K336 levels (d) and Lac-K457/ 8 levels (e) were detected for PDHA1 and CPT2 purified from C2C12 cells and C2C12 cells in which Aars2 had been knocked out using independent sgRNAs, with or without 10 mM Me-Lac in the culture media. Inhibiting the import of lactate into mitochondria decreases Lac-K336 and Lac-K457/8 levels. C2C12 cells that were either untreated, treated with -CHCA for 2 h , or with 10 mM lactate for 4 h . Mitochondrial lactate (f) ( ), Lac-K336 (g) and Lac-K457/8 (h) were detected. i Running proportionally increases lactate levels in mitochondria. Mitochondrial and lysate lactate levels in mouse leg skeletal muscles were detected before and after 30 min of running ( ). Success mitochondria extraction isolation was confirmed by staining both mitochondria marker COXIV and cytosolic marker GAPDH (here in after). j Running induces Lac-K336 and Lac-K457/8. Lac-K336 and Lac-K457/8 levels in purified PDHA1 and CPT2 of mouse leg skeletal muscles were detected after running for indicated time durations ( ). k, I Hypoxia-induced Lac-K336 and Lac-K457/8. Lac-K336 (k) and Lac-K457/8 (I) levels of PDHA1 and CPT2 in C2C12 cells were detected before and after they were exposed to hypoxia for Combined AARS2 and lactate treatments increased Lac-K336 and Lac-K457/8 levels. Lac-K336 and Lac-K457/8 levels in the leg skeletal muscles of resting mice and AARS2-overexpressing mice given lactate gastrocnemius muscle injections were estimated using MS ( ). , Running and hypoxia increased Lac-K336 and Lac-K457/8 levels. Lac-K336 and Lac-K457/8 levels in the mouse leg skeletal muscles before and after 30 min of running (n) and perfused with Krebs Ringer solution aerated with or without in a 1200 A Isolated Intact Muscle Test System for 30 min (o) were estimated using MS ( ). AARS2 inactivates PDHA1 and CPT2 by inducing lactylation. The effects of AARS2 overexpression on lactylation levels and specific activities ( ) of PDHA1 ( ) and CPT2 ( ) and their respective lactylation site mutants were detected. Lactate inactivates PDHA1 and CPT2 by inducing lactylation. The effects of 10 mM Me-Lac on Lac-K336 and the specific activities of PDHA1 and its lactylation site mutants ( ), and the effects of Me-Lac on Lac-K457/8 and the specific activities of CPT2 and its lactylation site mutants (s) were detected ( ). All data are reported as mean SEM of three independent experiments. Statistical significance was assessed by unpaired two-tailed Student’s -test and two-way ANOVA: ; ; ; ns no significance.
information, Fig. S9f), and ATP levels (Supplementary information, Fig. S 9 g ) in C2C12 cells. -alanine resulted in an increase in running exhaustion time in C57BL/6 mice (Fig. 7I) as well as in mice with intact AARS2, but not in mice lacking AARS2 (Fig. 7m). Lastly, the notion that Lac-K336 and Lac-K457/8 inhibit OXPHOS and constrain endurance running was further supported by the finding that both basal and maximum respiration of mouse leg skeletal muscles, were increased by Aars2 KO, but decreased by AARS2 overexpression during exercise, and the respiration responses were correlated to AARS2 levels (Supplementary information, Fig. S9h).
Possible physiologic roles played by Lac-K336 and Lac-K457/8 in exercise may be highlighted by examining Lac-K336- and Lac-K457/8-null Aars2 mice. While Aars2 mice endured extensive running and AARS2-overexpressing mice displayed impaired extensive running (Fig. 7m), Aars2 mice accumulated higher levels of ROS and ROS-induced Malondialdehyde (MDA), lipid peroxidation and oxidative damage marker in their leg skeletal muscles than those in the leg skeletal muscles of wild-type mice, at the time of exhaustion (Fig. 7n, o). This suggested that muscle cells may use mitochondrial protein lactylation to protect against possible oxidative damage during endurance exercise. Thus, while enhanced OXPHOS is required to convert lactate-, glucose- and fatty acids-generated Ac-CoA to ATP during endurance exercise, Ac-CoA influx to OXPHOS is feedback inhibited by OXPHOSinduced intracellular hypoxia (Fig. 7p).
DISCUSSION
We demonstrated that OXPHOS is subject to negative regulation by mitochondrial protein lactylation, which is induced by OXPHOS-induced intracellular hypoxia. Therefore, mitochondrial protein lactylation represents a feedback mechanism to constrain OXPHOS. In addition to lactic acid binding to hydroxycarboxylic acid receptor and activating matrix metalloproteinase-2 to activate TGF- signaling, AARS2-mediated lactylation represents another means of lactate signaling. Given that mitochondrial protein lactylation responds to intracellular hypoxia, its significances should at least include preventing drawbacks of hypoxia, such as ROS overproduction and ROS damage as observed in the current study. More physiologic or pathologic significances of mitochondrial protein lactylation are anticipated because high
lactic acid and hypoxia responses occur in such physiologic processes as exercise and embryonic development, and in such pathologic processes as cancers.
Mitochondrial protein lactylation integrates metabolism and hypoxia, which are interconnected, to constrain OXPHOS. High lactic acid in embryonic and cancer development is due to an increased requirement for anabolites including lactic acid precursor pyruvate, and high lactic acid in exercise is due to enhanced glycolysis and glycogen catabolism-hiked pyruvate. Moreover, lactic acid can only be oxidized via OXPHOS, which consumes intracellular oxygen. These, together with that lactic acid levels vary substantially in these processes, make mitochondrial protein lactylation an efficient mechanism to constrain OXPHOS. Notably, although lactate can be a substrate of OXPHOS when intracellular oxygen is not depleted, mitochondrial protein lactylation takes effects to inhibit OXPHOS when intracellular hypoxia is induced, such as during the exhausting stage of endurance exercise. It is distinct from other OXPHOS-regulating mechanisms, such as acetylation and allosteric regulation of PDHA1, which integrate nutrients (acetyl-CoA) and hormonal signals to facilitate ATP supply after the start of exercise, during which intracellular hypoxia is not induced and the critical for maximal mitochondrial respiration is as low as Torr to ensure a fully functional OXPHOS. We don’t exclude the possibility that mitochondrial protein lactylation may also regulate exercise ability by affecting other mitochondrion-rich and lactateutilizing cells including heart cells, as we observed in cultured heart cells and given that AARS2 mutations have been linked to cardiomyopathy.
AARS2 and SIRT3 are both known regulators of OXPHOS, lactate and muscle function. AARS2 mutation causes impaired OXPHOS, mitochondrial cardiomyopathy, muscle function loss and tissue lactate accumulation. SIRT3 loss causes impaired OXPHOS and lactate accumulation. Moreover, SIRT3 is induced during training, suggesting that SIRT3 may play a role in training adaption. SIRT3 has multiple roles in regulating OXPHOS. Under aerobic conditions, SIRT3 decreases PDHA1 acetylation to promote lactate production and FAO in mouse muscle, while under hypoxic conditions, it decreases PDHA1 lactylation to promote OXPHOS. Via either process, SIRT3 promotes muscle cell ATP production during exercise. Furthermore, decreasing mitochondrial protein lactylation by targeting both AARS2 and SIRT3 may
temporarily boost muscle cell OXPHOS and exercise endurance as we observed in mice. However, these endurance exercise-boosting approaches should not be encouraged, as loss of mitochondrial protein lactylation protection may lead to ROS damage.
MATERIALS AND METHODS
Reagents and antibodies
Chemical and reagents: ATP (Cat# A6559), lactate (Cat# L6661), methylactate (Cat# 230340), -alanine (Cat# 146064), L-alanine (Cat# A7627), DAPI (Cat# D9542), MG132 (Cat# M8699), PPi (Cat# 433314), NAM (Cat#
Fig. 6 SIRT3 reverses Lac-K336 and Lac-K457/8. a NAM treatments increase Lac-K336 and Lac-K457/8. Lac-K336 and Lac-K457/8 levels of PDHA1 and CPT2 purified from C2C12 cells and NAM-treated C2C12 cells were detected. Cells were examined 3 h after 5 mM NAM treatment. b SIRT3 delactylates Lac-K336 and Lac-K457/8. The ability of SIRT3, SIRT4 catalytic domain and SIRT5 to delactylate synthetic Lac-K336- and Lac-K457/8-containing peptides was analyzed via MS to detect the formation of lactylated species. c-f SIRT3 decreases Lac-K336 and LacK457/8 levels. The Lac-K336 and Lac-K457/8 levels in purified PDHA1 (c) and CPT2 (d) were detected before and after being delactylated with SIRT3 or catalytic dead SIRT3 , and the SIRT3 expression effects on wild-type, lactylation-null PDHA1 (e), and CPT2 (f) were detected in C2C12 cells. Ablation of Sirt3 increases Lac-K336 and Lac-K457/8 levels in vivo. Lac-K336 and Lac-K457/8 levels of wild-type and Sirt3 mouse leg skeletal muscles were detected . h, i SIRT3 regulates PDHA1 and CPT2 activities. Relative activities of PDHA1 and CPT2 that were purified from C2C12 cells and SIRT3-overexpressing ( ) or Sirt3 KO (i) C2C12 cells were compared ( ). j-m SIRT3 regulates Ac-CoA production from glycolysis and FAO. The , and fractions of Ac-CoA from -glucose ( ) and -palmitate ( ) were detected in C2C12 cells either overexpressing SIRT3 ( ) or in which Sirt3 had been knocked out using independent sgRNAs ( ) ( ). The chasing time for -glucose and -palmitate were 1 h and 12 h , respectively. SIRT3 regulates lactate and free fatty acids levels. Lactate ( ) and free fatty acid levels ( ) in C2C12 cells overexpressing SIRT3 ( ) or C2C12 cells in which Sirt3 had been knocked out via independent sgRNAs ( ) were detected ( ). All data are reported as mean SEM of three independent experiments. Statistical significance was assessed by unpaired two-tailed Student’s -test and two-way ANOVA: ; ; .
72340), -palmitate (Cat# 605573) and cell counting kit-8 (Cat# 96992) were purchased from Sigma-Aldrich. Oligomycin A (Cat# S1478), FCCP (Cat# S8276), a-CHCA (Cat# S8612) and roxadustat (Cat# S1007) were purchased from Selleck. FX-11 (Cat# HY-16214) was purchased from MedChemExpress. Lambda protein phosphatase (Cat# P0753) was purchased from New England BioLabs. C-glucose (Cat# CLM1396) was purchased from Cambridge Isotope Labs. Protein-A sepharose beads (Cat# 16-156) was purchased from Merck Millipore. Penicillin-Streptomycin (Cat# 15070063) was purchased from Invitrogen. CellTiter-Glo reagent kit (Cat# G9241) was purchased from Promega. Lactate dehydrogenase activity colorimetric assay kit (Cat# K726) and free fatty acid quantification kit (Cat# K612) were purchased from Biovision. Pyruvate dehydrogenase (PDH) enzyme activity microplate assay kit (Cat# ab109902) and SIRT3 activity assay kit (Cat# ab156067) were purchased from Abcam. Synthetic peptides were purchased from Toppeptide. Synthetic oligonucleotides were purchased from Genepharma.
Antibodies: anti-AARS1 (Cat# sc165990) was purchased from Santa Cruz. Anti-VHL (Cat# ab140989), anti-HIF2a (Cat# ab109616), anti-HIF3a (Cat# ab2165), anti-CPT2 (Cat# ab110293), anti-PDHA1-pS293 (Cat# ab177461), anti-COXIV (Cat# ab202554), anti-GAPDH (Cat# ab1439) and anti-Actin (Cat# ab1440) were purchased from Abcam. Anti-AARS2 (Cat# 22696-1-AP), anti-PDHA1 (Cat# 18068-1-AP), anti-DLD (Cat# 16431-1-AP), anti-DLAT (Cat# 13426-1-AP), anti-PDHB (Cat# 14744-1-AP), anti-PHD1 (Cat# 12984-1-AP), anti-PHD2 (Cat# 19886-1-AP), anti-PHD3 (Cat# 18325-1-AP), anti-CPT1A (Cat# 15184-1-AP), anti-EHHADH (Cat# 26570-1-AP), anti-ACADS (Cat# 16623-1-AP), anti-ACADM (Cat# 55210-1-AP) and antiACADL (Cat# 17526-1-AP) were purchased from Proteintech. Anti-HIF1a (Cat# 36169S) was purchased from Cell Signaling Technology. Anti-PDHA1-pS232 (Cat# LS-C265964/64724) was purchased from LifeSpan Biosciences. Anti-PDHA1-pS300 (Cat# ABS194) was purchased from EMD Millipore Corporation. Anti-Laminin (Cat# L9393) was purchased from Sigma-Aldrich. Anti-Flag (Cat# M20008), anti-HA (Cat# M20002), anti-Myc (Cat# M20003) were purchased from Abmart. Goat anti-rabbit IgG (Cat# 111-035-003) and Goat anti-mouse IgG (Cat# 115-035-003) were purchased from Jackson. Anti-LacK336, anti-LacK457/8, anti-P377OH, and anti-ECHS1 were home-made in this study.
Animals
Male C57BL/6J mice were obtained from Shanghai SLAC Laboratory Animal Co., Ltd. (Shanghai, China). Mice were housed in cages under a 12 h light/dark cycle in a temperature- and humidity-controlled room, and fed standard laboratory chow and water ad libitum. They were then randomly divided into control and experimental groups. Conditional hAARS2 transgenic mice were generated in a C57BL/6 genetic background by inserting CAG-LSL-hAARS2-WPRE-PA in the Rosa26 gene using a CRISPR-Cas9-mediated genome editing system by Shanghai Model Organisms Center, Inc. (Shanghai, China). Specific hAARS2 transgenic mice were generated by crossing conditional hAARS2 transgenic mice with CKMM-cre mice (Jackson Stock Number #029100). Aars2 mice containing two loxP sites flanking exon1-5 were obtained from GemPharmatech Co., Ltd. (Shanghai, China). Muscle-specific transgenic mice were generated by crossing Aars2 conditional KO mice with CKMMcre mice. Sirt3 mice were obtained from the Shanghai Model Organisms Center, Inc., and their genotypes were confirmed via PCR. All animal procedures were conducted in accordance with the animal care committee of Fudan University.
Cell culture
Mouse myoblast C2C12, mouse cardiomyocytes HL-1 and human embryonic kidney HEK293T cell lines were purchased from the American Type Culture Collection (USA). All cell lines mentioned above were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Hyclone, USA) supplemented with fetal bovine serum (Gibco, USA) at under in a humidified incubator. All cell lines were tested and authenticated by evaluating their morphology and growth rate; all were mycoplasma-free.
Mouse primary hepatocytes were isolated using a two-step perfusion method. Mice were intraperitoneally injected with 1% Nembutal, and their portal veins were catheterized. Subsequently, 0.5 mM EDTA and collagenase IV (Sigma Aldrich, USA) were preheated, and successively perfused into the liver until the liver structure disintegrated and cells dispersed. The cell suspensions, so obtained, were filtered through a filter (NEST, Wuxi, China), washed with William’s E Medium (Gibco, USA), and purified using Percoll solution (GE, USA). Hepatocytes were seeded at a density of cells/well in serum-free William’s E Medium. Mouse primary hepatocytes were verified by evaluating their cell morphology and albumin expression. Mouse primary myoblasts were isolated and the culturing condition was modified from published protocols. The hind limb muscles were isolated from adult mice and then digested in DMEM containing mL collagenase II (Sigma Aldrich, USA) and HEPES. The suspensions were filtered through and then filters. Cells were suspended in F-10 medium containing 20% FBS and basic fibroblast growth factor (bFGF) in the Matrigel pre-coated dish, kept at for 72 h . Cells were then transfered to non-Matrigel coated dish for 45 min to remove fibroblasts which adhered in this step. The cell supernatant was transfered to a new Matrigel pre-coated dish and cultured.
Site-specific antibodies
Lactylation site-specific polyclonal antibodies were generated in 3-monthold male New Zealand rabbits using synthetic Lac-K336-containing PDHA1 peptide or Lac-K457/8-containing CPT2 peptide. These peptides were conjugated through a C-terminal cysteine to keyhole limpet hemocyanin (KLH). Rabbits were immunized with of the conjugate in Freunds complete adjuvant. Booster injections were performed every 4 weeks. The rabbits were exsanguinated 10-14 days after the final boost. 50 mL blood collected from each rabbit was centrifuged. The supernatant (serum) was purified with antigen peptides according to published protocols. Hydroxylation site-specific polyclonal antibodies were generated with OH-P377containing AARS2 peptide as described above. Antigen used:
AARS2 and VHL were cloned into pcDNA3.1(b+)-Flag and pcDNA3.1(b+)Myc, respectively, between Xhol and EcoRI sites. PDHA1 and CPT2 were cloned into pcDNA3.1(b+)-Flag between Xhol and EcoRI sites. PDH2 was cloned into pcDNA3.1(b+)-Myc between Xhol and EcoRI sites, and SIRT3 was cloned into pcDNA3.1(b+)-HA between Xhol and EcoRI sites.
Cell transfection
Plasmid transfections were performed using polyethylenimine (PEI, Polyscience, USA) or Lipofectamine 8000 (Beyotime, Shanghai, China).
For PEI transfection, the plasmid and PEI were added into serum-free DMEM with vigorous shaking, and the mixture was incubated for 15 min before being added into the cell culture medium. For Lipofectamine 8000 transfection, the plasmid and Lipofectamine 8000 were added into serum-free DMEM, mixed gently with a pipette, and then immediately added to the cell culture medium. The culture medium was replaced every after transfection. Cells were harvested after 36 h .
Cell treatments
Me-lactate, alanine, -alanine, and NAM. Cells were cultured in a standard RMPI 1640 medium (US Biological, USA), and transferred to amino acid-free RMPI 1640 for 30 min before treatments. Indicated concentrations of lactate, alanine, and -alanine were added into the culture media when cell density reached confluence at before harvest. NAM treatment consisted of adding 5 mM NAM to serum-free DMEM medium before harvest.
Fig. 7 OXPHOS drives lactylation to feedback limit OXPHOS and endurance running ability. a OXPHOS induces intracellular hypoxia. HIF and AARS2 levels of C2C12 cells in DMEM base and glucose- , palmitate- and Me-lac- supplemented DMEM media were tested in the presence or absence of rotenone which inhibits OXPHOS. Lactate injection induces mouse muscle hypoxia. HIF1 and AARS2 levels of mouse hind leg thighs were detected before and after lactate was injected into gastrocnemius muscle ( ). c Lactate production is essential for inducing hypoxia and lactylation in mouse leg muscles. The HIF1 , AARS2, and lactylation levels of mouse hind leg thighs of untreated mice and mice pre-treated with FX-11 were assessed when they started to run. d Running leads to the accumulation of lactate in mouse leg skeletal muscles. Relative lactate levels in type I and type II mouse skeletal muscles were determined before and after 30 min of running ( ). e Running induces more pronounced lactylation in type I muscle. HIF1 and AARS2, as well as Lac-K336 and Lac-K457/8 levels in type I and type II mice skeletal muscles, were determined when started to run. Drop of ATP in type I skeletal muscle is correlated with an increase in Lac-K336 and Lac-K457/8. ATP levels in type I and II mouse leg skeletal muscles were monitored after running for indicated times ( ). Changes in Lac-K336 and Lac-K457/8 are shown (Fig. 5j). g Basal AARS2 levels in mouse leg skeletal muscle are inversely correlated with their running exhaustion time. High-intensity running exhaustion time and resting leg muscle AARS2 levels were correlated ( ). Lac-K336 and Lac-K457/8 inhibit the influx of mouse skeletal muscle Ac-CoA. The -Ac-CoA levels in wild-type, AARS2 overexpressing and Sirt3 mouse leg skeletal muscle were detected 1 h after each mouse received a -glucose injection via the tail vein ( ). i, j Lactylation reduces mouse skeletal muscle OCRs and ATP production. OCR (i) and ATP levels (j) in mitochondria in the leg skeletal muscles of resting, AARS2 overexpressing, and Sirt3 mice, were analyzed Lac-K336 and Lac-K457/8 inhibit mouse running exhaustion time. Mouse high-intensity running exhaustion time was measured in wild-type, muscle AARS2-overexpressing, and Sirt3 mice ( ). I Lactate and -alanine inversely regulate mouse running exhaustion time. High-intensity running exhaustion times of untreated mice or treated with lactate or -alanine via gastrocnemius muscle injection were measured, respectively ( ). -alanine prolongs mice exhaustion time in an AARS2-dependent manner. High-intensity running exhaustion times of wild-type, AARS2 overexpressing, and Aars2 mice with or without gastrocnemius muscle -alanine injection ( ) were measured. , o Loss of lactylation results in higher ROS damage. ROS levels ( ) and induced malondialdehyde levels ( ) of wild-type and Aars mice at the time of running exhaustion ( ) were compared. Diagram showing that exercise promotes lactate oxidation through OXPHOS, which promotes intracellular hypoxia that induces AARS2 and Lac-K336 and Lac-K457/8 to feedback inhibit Ac-CoA influxes from lactate/glycolysis and FAO and OXPHOS. The mechanisms revealed in this study are marked by red arrows. All data are reported as mean SEM of three independent experiments. Statistical significance was assessed by unpaired two-tailed Student’s -test and two-way ANOVA: ; ; ; ; ns no significance.
Rotenone. Rotenone was added to culture media to obtain a solution 20 min before cell harvesting.
a-CHCA. Monocarboxylate transporter inhibitor a-CHCA ( 5 mM , final concentration) was added to the culture medium 2 h before harvesting.
Roxadustat. Roxadustat ( , final concentration) was added to the culture medium 6 h before harvesting.
Hypoxia. Cells were cultured under normoxia before being transferred to a hypoxia incubator (ThermoFisher Scientific, USA) with a gas mixture containing and balanced with nitrogen. Cells were harvested at the indicated time points that were not longer than 8 h after hypoxic exposure. Chamber oxygen levels were monitored at sampling times.
Isolation of cell mitochondria
A hypotonic buffer containing 20 mM HEPES ( pH 7.5 ), EDTA, and with a protease and phosphatase inhibitor cocktail was added to cells after washing with PBS. The cells were then scraped and broken with a syringe. The homogenate was centrifuged at for 10 min to separate the nucleus and other debris. The supernatant was then centrifuged at and for 30 min . The obtained supernatant consisted of plasma. Following this, the precipitate was washed thrice with a hypertonic buffer containing 20 mM HEPES ( pH 7.5 ), EDTA, , in PBS. The final precipitate contained mitochondria. The purity of plasma and mitochondria was verified using plasma and mitochondria markers, Actin, and COXIV, via western blot assays.
Mouse injections
Gastrocnemius muscle injection. Lactate, alanine, and -alanine were prepared in saline to obtain a final concentration of 500 mM , the pH of which was adjusted to 7.4 . A dose of each drug was intramuscularly injected to the mice gastrocnemius muscle. A similar volume of saline was injected into the same muscles as a control.
Intravenous MG132 injection. MG132 was prepared in saline to obtain a final concentration of 500 mM , the pH of which was adjusted to 7.4. Mice were injected intravenously with MG132 twice a week for 4 weeks before being used for experimentation and muscles were sampled for detection.
FX-11 injection. Mice were intraperitoneally injected with DMSO as a control or DMSO-dissolved LDHA inhibitor FX-11 daily for 2 weeks, following which the mouse running tests were carried out.
Mouse sprint and high-intensity exercise fatigue test
Mouse sprint performance test was modified from published protocols : First, 20 -week-old mice were placed on a ZH-PT/5S treadmill equipped with a rear electrical stimulus grid set that delivered a 0.2 mA electric shock. Mice were first warmed up on the treadmill for 3 min at . Speed was increased to for 30 s . Afterward, we increased the speed from to with an increment of , with each increment lasting 15 s (total ). The pattern was repeated with increasing speed until the mice could no longer keep up with the speed and gave up at the grid.
Mouse high-intensity exercise fatigue test was modified from published protocols. Briefly, 20 -week-old mice were acclimated to the treadmill 2 days prior to the experiments by running at for 5 min and min for 5 min followed by for 1 min every day. For the exercise experiments, speed was increased every 5 min until was reached, at which point the incline was increased 5 inclines every 5 min until exhaustion.
Mouse leg fast and slow muscle preparation
Muscle preparation was modified from published protocols. Briefly, mice were euthanized and the soleus (SOL) muscles, mainly composed of slow muscle (type I), and the extensor digitorum longus (EDL), mainly composed of fast muscle (type II), were dissected and subjected to the following tests.
Muscle hypoxia perfusion
Mice were euthanized and their leg skeletal muscles were dissected and immediately placed in Krebs Ringer solution D-glucose, , HEPES) in a water-jacketed bath of the 1200 A Isolated Intact Muscle Test System (Aurora Scientific, Canada), and subsequently aerated with or without and at for 30 min .
Muscle tissue mitochondria isolation
Mouse leg muscles were dissected and rinsed in ice-cold mitochondrial isolation buffer containing 70 mM sucrose, 210 mM mannitol, 5 mM MOPS, EGTA, and fatty acid-free bovine serum albumin (pH 7.2). The tissues were then homogenized with a tissue grinder (Shanghai Jing Xin, Shanghai, China) at . The homogenate was centrifuged at and for 10 min to remove nuclei and cell debris. The supernatant enriched in mitochondria was then centrifuged at and for 10 min . The mitochondria precipitate so obtained was used for assays.
assay
Arterial blood was immediately collected in anticoagulation tubes from aorta abdominalis of mice that had run for indicated times. was determined using IDEXX VetStat (IDEXX B.V., The Netherlands) Electrolyte and a Blood Gas Analyzer.
ROS assay
Mouse leg skeletal muscle ROS assays were conducted using a ROS Assay Kit (Cat# E-BC-K138-F, Elabscience Biotechnology Inc., Houston, TX, USA) according to the manufacturer’s instructions. The tissues were digested into a single-cell suspension via collagenase digestion. Then, cells were incubated with DCFH-DA reagent for 30 min . Following centrifugation at 1000 rpm for 5 min , the cell pellets were washed twice and resuspended in Reagent 3 before being analyzed in a FACS Calibur flow cytometer.
MDA assays
Lipid peroxidation was assessed using an MDA assay Kit (ab233471, Discovery Drive, Cambridge Biomedical Campus, Cambridge, CB2 0AX, UK) according to the manufacturer’s instructions. For tissue extract preparation, 50 mg fresh skeletal muscle was rapidly homogenized on ice in Na Phosphate buffer (pH 3-3.2) with TritonX100 followed by centrifugation at for 15 min at . Insoluble material was removed, and the supernatant was stored on ice for later assays. Prior to being used, all materials and prepared reagents were equilibrated to room temperature with gentle agitation. Next, test samples, a blank control (dilution buffer), and serially diluted MDA standards were added to a 96-well clear bottom microplate. This was followed by incubating the reaction mixture at room temperature for . Then, of Reaction Solution was added to bring the total assay volume up to well . The final reaction mixture was incubated at room temperature for . End-point absorbance was measured using an absorbance plate reader with path-check correction at .
Muscle tissue immunofluorescence
Paraffin sections for the leg skeletal muscles of wild-type, muscle-specific AARS2 overexpressing mice, and mice that had run were prepared. Sections were deparaffinized, rehydrated, antigen retrieved in EDTA antigen retrieval buffer ( pH 8.0 ), and blocked with BSA for 30 min . Nuclei were stained with DAPI. HIF1a, pS293-PDHA1, AARS2, and COXIV were stained with their primary and secondary antibodies, respectively. Images of the prepared sections were captured under a fluorescence microscope.
Muscle tissue -phosphatase treatment
The mice were sacrificed and 50 mg fresh skeletal muscle was rapidly homogenized on ice in lysis buffer containing Tris- HCl ( pH 7.5 ), and 150 mM NaCl , and centrifuged at for 15 min at , following which insoluble material was removed. The supernatant was then collected on ice for the subsequent assay. Next, 400 units of -phosphatase (New England Biolab) and were added to the supernatant and incubated at for 30 min , and the supernatant was used for PDHA1 activity assay.
Small RNA interference
Oligonucleotides used for small interfering RNA (siRNA)-mediated silencing of VHL, PHD1, PHD2, PHD3, AARS2, and SIRT3 were synthesized by GenePharma (Shanghai, China). Oligonucleotides were transfected into cells before being harvested. Knockdown efficiency was verified using western blott assays. The siRNA sequences used for targeting genes are as follows:
To knock out Phd2, Vhl, Aars2, Cpt2, and Sirt3 in C2C12 cells, CRISPR/Cas9 protocols were employed. Guide RNA containing the target sequence was transfected into C2C12 cells, which were then selected after 36 h using puromycin (Amresco, USA) for 3 days. The cells that eventually survived were harvested and seeded into a 96-well plate at a density of
1 cell/well. Successful KO of each gene was verified via western blot assays.
The guide sequences are as follows:
sgPhd2 #1:
F: -CACCGACACCGGCAAGTTCACGGAC-3′
R: 5′-AAACGTCCGTGAACTTGCCGGTGTC-3′
sgPhd2 #2:
F: 5′-CACCGCAAGTTCACGGACGGGCAGC-3′
R: -AAACGCTGCCCGTCCGTGAACTTGC-3′
sgAars2 #1:
F: -CACCGCTGCTAAGTACCGGGGCCGT-3′
R: 5′-AAACACGGCCCCGGTACTTAGCAGC-3′
sgAars2 #2:
F: -CACCGCGAGCGGCTGCGCTTTGACG-3′
R: -AAACCGTCAAAGCGCAGCCGCTCGC-3′
sgCpt2 #1:
F: 5′-CACCGATCGTCCCGGGCGGCAATGG-3′
R: 5′-AAACCCATTGCCGCCCGGGACGATC-3′
sgCpt2 #2:
F: -CACCGAGGCGCGGCATCATCGTCCC-3′
R: -AAACGGGACGATGATGCCGCGCCTC-3′
sgVhl #1:
F: -CACCGCGTTCCAATAATGCCCCGGA-
R: -AAACTCCGGGGCATTATTGGAACGC-3′
sgVhl #2:
F: 5′-CACCGCCGATCTTACCACCGGGCAC-3′
R: -AAACGTGCCCGGTGGTAAGATCGGC-3′
sgSirt3 #1:
F: -CACCGCCAGTACAGACAGGGCAGCGGGG-3′
R: -AAACCCCCGCTGCCCTGTCTGTACTGGC-
sgSirt3 #2:
F: 5′-CACCGCTTTCAACAAACCTCCAGGGAGG-3′
R: 5′-AAACCCTCCCTGGAGGTTTGTTGAAAGC-3′
Immunoprecipitation
Cells were lysed using lysis buffer containing NP- 40 buffer ( 50 mM Tris-HCl, pH 7.5), Nonidet P- aprotinin, mL leupeptin, pepstatin, and 1 mM (PMSF). Tissues were homogenized in the lysis buffer before lysis. Affinity antibody beads were added to the lysate to enrich target proteins. The enriched proteins on beads were washed with 0.5% NP-40 buffer thrice before being mixed with SDS-PAGE loading buffer to release proteins for analysis.
Western blot assay
Cultured cells were lysed and boiled with SDS-PAGE loading buffer. Mouse tissues were homogenized using lysis buffer containing 0.5% NP-40 buffer, 50 mM Tris- HCl (pH 7.5), Nonidet P-40, and a mixture of protease inhibitors (Sigma-Aldrich), before being boiled with loading buffer. After centrifugation at and for 15 min , the supernatant of the lysates was collected for western blot assay according to standard procedures. Western blot signals were detected using the Typhoon FLA 9500 (GE Healthcare, Little Chalfont, UK).
Dot blot assay
Peptides were diluted with at the indicated dilutions. Samples were blotted onto nitrocellulose membranes, and allowed to air dry at room temperature. The membrane was blocked with milk, and then incubated with a primary antibody for peptide recognition and a secondary antibody for detection. Blotting signals were detected using a Typhoon FLA 9500 (GE, USA).
Ubiquitination assay
For ubiquitination analysis, HA-tagged ubiquitin and target plasmids were co-transfected into cells. After 36 h of transfection, MG132 was added to the medium for before harvesting. Cells were collected, lysed, and boiled in 1% SDS buffer (Tris-HCl, pH EDTA, 1 mM DTT) for 10 min . Immunoprecipitation was performed in 10-fold diluted lysates with NP-40 buffer, and ubiquitination was analyzed following standard western blot protocols.
PDHA1 activity assay
PDHA1-Flag and PDHB-Myc were co-expressed in indicated cells. Cells were lysed on ice using NP-40 buffer supplemented with protease
inhibitors. PDHA1 was immuno-purified using flag beads (Sigma Aldrich) and then washed thrice with NP-40 buffer. PDHB co-purified with PDHA1 was quantified via Myc-western blot and used to normalize the activity of PDHA1. The PDHA1 activity assay was performed as previously described. Briefly, a reaction buffer containing (pH 7.0), sodium pyruvate, 0.2 mM thiamin diphosphate, and 0.1 mM 2,6-dichlorophenolindophenol (2,6-DCPIP) was added to the purified PDHA1/PDHB complex to initiate the reaction. The reactions were maintained at . The reaction was monitored by measuring the reduction of 2,6 -DCPIP at 600 nm on a Roche spectrophotometer (Basel, Switzerland).
PDC activity assay
The enzyme activity of the PDC complex was measured using a PDH Enzyme Activity Microplate Assay Kit (Abcam, USA) according to the manufacturer’s instructions. Cells and mouse tissues were harvested to extract proteins. Samples were loaded on the plate and incubated at RT for 3 h . The assay solution was added to each well, and PDH activity was determined by following the reduction of to NADH, coupled with the reduction of a reporter dye to yield a colored (yellow) reaction product, the concentration of which was monitored by measuring absorbance at 450 nm .
CPT2 activity assay
CPT2 activity was assayed by measuring the decrease in CoA levels. The reactions were conducted in a reaction mix containing 50 mM Tris- HCl (pH 7.5), EDTA, 2 mM CoA , and 2 mM Palmity-L-Carnitine. Subsequently, CPT2 was immunoprecipitated from HEK293T cells. The assay was initiated in a thermostatic oscillation incubator at 1000 rpm and , and the mixture was collected at , and 30 min . The reaction was terminated by adding of 10 mM DTNB. The supernatant was monitored using a microplate reader at 410 nm .
LDH activity assay
An LDH Activity Colorimetric Assay Kit (Biovision, USA) was used. Briefly, cells were homogenized with ice-cold assay buffer, incubated on ice for 10 min , and centrifuged at for 15 min . Next, the supernatant was transferred to a fresh tube, and of the reaction mixture ( sample, assay buffer, and reaction mix) was transferred to a 96-well plate. Absorbance was immediately measured at 450 nm in kinetic mode at for . LDH activity was measured based on the change in absorption during a specific time frame.
CPT1A activity assay
CPT1A activity in cell homogenates was assayed spectrophotometrically by following the release of CoA-SH from palmitoyl-CoA using the general thiol reagent DTNB. Reaction mixtures containing DTNB buffer and cell homogenate were incubated at room temperature for 20 min to eliminate all the reactive thiol groups. After incubation, the absorbance was measured at 412 nm . To start the reaction, palmitoyl-CoA ( , final concentration) and L-carnitine solution ( 1 mM , final concentration in 1 M Tris, pH 8.0 ) were added to the reaction mixtures. Reaction mixtures were incubated for 10 min at . After incubation, the absorbance was measured at 412 nm . The difference between absorbance with and without substrates measures the release of CoA-SH. Activity was defined as nmole CoA-SH released protein. The protein content of the cell homogenates was determined according to the method of Bradford.
SIRT3 activity assay
SIRT3 Activity Assay Kit (Abcam, USA) was used according to the manufacturer’s instructions. The reaction mixture contains assay buffer, Fluoro-Substrate peptide, NAD, developer, and samples. Read fluorescence intensity for 30 min to 60 min at 1 min to 2 min intervals using microtiter plate fluorometer with excitation at and emission at . Measure and calculate the rate of reaction while the reaction velocity remains constant.
Blood lactate
The blood lactate concentration in tail vein blood was measured using a Lactate Scout4 (SensLab GmbH, Germany). The tail of each mouse was cut using a blade, and the first droplet of blood was wiped away. The tail was gently pressed, and the second droplet of blood was sufficient to fill the
measurement chamber of the lactate sensor enabling the concentration to be determined.
Cell and tissue OCR assays
OCR assays were performed using the OROBOROS Oxygraph-2K module (Oroboros Instruments GmbH, Austria). Oxygen consumption values were normalized to the cell number or mitochondrial content.
For cultured cells, approximately cells were digested and resuspended in PBS. The basal respiratory rate was determined with the substrate ( 5 mM pyruvate and 0.1 mM malate). The ATP production capacity and maximal respiration were successively determined using Oligomycin A and FCCP (Selleck, China) treatments, respectively.
For mouse muscle samples, mitochondria were isolated according to a published protocol and the isolated mitochondria samples were resuspended in respiration buffer ( 0.5 mM EDTA, 3 mM MgCl , 60 mM lactobionic acid, 20 mM taurine, HEPES, 110 mM D-sucrose and fatty acid-free BSA, pH 7.2 ).
AARS2 kinetic assay
The pGEX-6p-1-AARS2 vector was transformed into the Escherichia coli strain, BL21 (DE3). The transformed cells were then induced with 0.2 mM IPTG when was . After cultured overnight at , the bacteria were collected, homogenized in a buffer containing 50 mM TrisHCl and 150 mM NaCl , and centrifuged at and for 30 min . The supernatant was loaded onto an AKTA Purifier (GE, USA) with a His-Ni column. The concentration of protein so obtained was measured using the BSA protein assay kit.
The kinetic parameters of AARS2 were determined by measuring the lactylation of peptide substrates (PDHA1: MVNSNLASVEELKEIDVEVR; CPT2: EFLKKQKLS). and of lactate were estimated by varying lactate levels from , while peptide substrates were fixed at . Lactylation reactions were carried out in a reaction mix containing 50 mM pH 7.5 HEPES, lactate, 4 mM ATP, and 10 nM AARS2. Lactylated peptide produced was determined using MS with known concentrations of synthetic peptides (PDHA1: MVNSNLASVEELK EIDVEVR; CPT2: EFLK ) as internal controls. and were deduced using the Michaelis-Menten equation.
ITC
Protein-small molecule interaction was determined using MicroCal PEAQ-ITC (Malvern Panalytical, UK) at . Lactate, alanine, and -alanine were dissolved in 50 mM Tris- HCl and 150 mM NaCl at a concentration of . Recombinant AARS2 was diluted to , and then added to the sample cell. Molecules were dropped into the sample cell every 2 min for 20 times.
In vitro lactylation reactions
In vitro lactylation reactions were performed in a reaction mix containing 50 mM HEPES ( pH 7.5 ), lactate, 4 mM ATP, 10 nM AARS2, and synthetic substrate peptide. The final pH of the reaction mixture was adjusted to 7.5 before AARS2 was added. The reaction was allowed to continue for 3 h at . The peptide was desalted using C18 ZipTip (Millipore, USA), and subjected to analysis using a MALDI-TOF/TOF mass spectrometer (SCIEX-5800, AB Sciex, Framingham, MA, USA). Synthetic peptides sequences are as follows:
In vitro de-lactylation reactions were conducted in a reaction mix containing 50 mM HEPES (pH 7.5), DTT, , synthetic lactylated peptide or recombinant PDHA1 and CPT2 protein, SIRT3, and 1 mM PMSF. The reaction was allowed to continue for 4 h at . The peptide was desalted by passing it through a C18 ZipTip before subjecting it to analysis using a MALDI-TOF/TOF mass spectrometer (SCIEX-5800). The resulting PDHA1 and CPT2 were subjected to western blot analysis and enzyme activity assay. Synthetic peptides sequences are as follows:
MS/MS identification of AARS2 P377OH, PDHA1Lac-K336, and CPT2 Lac-K457/8
AARS2 or CPT2 proteins that were overexpressed in cells were affinity purified and digested with endoproteinase Glu-C. Overexpressed PDHA1 from C2C12 cells were affinity-purified and digested with trypsin. Hydroxylated or lactylated peptides were analyzed via MS/MS sequencing.
LC peaks eluted at the same time as those of standard synthetic peptides were subjected to MS analysis with an Orbitrap Fusion mass spectrometer (ThermoFisher Scientific, USA). Peptide counting and/or sample areas were compared to those of standard synthetic peptides. The MS machine was set in a data-dependent mode to switch automatically between MS and MS/MS acquisition. Survey full-scan MS spectra (m/z 350-1600) were acquired in the Orbitrap with a mass resolution of 60,000 at . The AGC target was set to 300,000 , and the maximum injection time was 50 ms . MS/MS acquisition was performed in Orbitrap with a cycle duration of 3 s , at a resolution of 15,000 at . The intensity threshold was 50,000 , and the maximum injection time was 200 ms . The AGC target was set to 200,000, and the isolation window was . lons with charge states of 2+, 3+, and 4+ were sequentially fragmented via a high-energy collisional dissociation with a normalized collision energy of , and the fixed first mass was set as 120. In all cases, one micro scan lasting 30 s was recorded using dynamic exclusion. Quantification of the targeted lactylated peptide was achieved via MS quantitation. Briefly, a ratio of lactylated peptide signal (the total ion counts (TIC) of lactylated form) to the total peptide signal (TIC of lactylated form + TIC of non-lactylated form) was calculated according to the following equation: TICK-Lac/(TICKLac + TICnon-K-Lac) = Ratio of K-Lac (RK-Lac). Synthetic peptides sequences are as follows:
LC-MS/MS quantitation of AARS2 P377OH, PDHA1Lac-K336, and CPT2 lac-K457/8
AARS2, PDHA1, and CPT2 were ectopically expressed in cultured C2C12 cells overexpressing AARS2 or treated with lactate. Cells were harvested and lysed in 0.1% NP-40 buffer ( 50 mM Tris-HCl, pH Nonidet P-40), and anti-FLAG M2 magnetic beads (Sigma-Aldrich) were used to precipitate PHD2, PDHA1, and CPT2 for 3 h at . The beads were then washed twice with NP-40 buffer, twice with , and thrice with , which was followed by on-bead trypsin digestion overnight at . The resulting peptides in supernatants were collected via centrifugation and dried in a speed vacuum (Eppendorf, Hamburg, Germany). The obtained peptides were stored at for LC-MS/MS analysis. A previously published method was used to quantify targeted lactylated peptides. Briefly, a ratio of lactylated peptide signal (the total ion counts (TIC) of lactylated form) to the total peptide signal (TIC of lactylated form + TIC of non-lactylated form) was calculated according to the following equation: TICK-Lac/(TICK-Lac + TICnon-K-Lac) = Ratio of K-Lac (RK-Lac).
Metabolite assay with LC-MS/MS
Approximately cells were treated with cold aqueous methanol solution ( ) to quickly stop cell metabolism. Cells were scraped off from tubes, and placed at overnight. For mouse muscle samples, approximately 80 mg of mouse leg muscle was homogenized in a cold aqueous methanol solution ( ) at . Samples were centrifuged at and for 15 min , and supernatants were collected. The supernatants were then lyophilized and re-dissolved in methanol/ water ( ). The separated metabolites were obtained using highperformance liquid chromatography with an LC-20AB pump (Shimadzu, Kyoto, Japan) and the Luna column (P/N 00B-4378-B0; , ; Phenomenex, Torrance, CA, USA). The mobile phase comprised eluent A ( , and acetic acid dissolved in 500 mL water) and eluent B . The elution program was as follows: and , . The flow rate of the pump was , and the mass spectrometer used was the 4000 QTRAP system (AB Sciex, Framingham, MA, USA) operated in multiple reaction monitoring modes. The metabolite ions were monitored at: PYR 87-43; LAC 89-43; Ac-CoA 810-60; ATP 509-79; and AKG 145-101. Each measurement was obtained at least in triplicate.
Metabolic flux assay
Metabolic flux experiments were performed when cells reached confluence. The medium for glucose labeling experiments contained 10 mM (U- ) glucose (Cambridge Isotope Labs, MA, USA) and 2 mM unlabeled glutamine (Sigma-Aldrich, USA). Medium for glutamine labeling experiments contained 10 mM unlabeled glucose and 2 mM (U- ) glutamine (Cambridge Isotope Labs). Medium for palmitate labelling experiments contained 0.1 mM (U- ) palmitate (Sigma-Aldrich). After treatment, cells were washed thrice with PBS and subsequently treated with pre-cold methanol ( ). At the same time, parallel dishes were used to count cell numbers. Metabolite extractions were then analyzed using LC-MS with a C18 column. Relative metabolite abundances were determined by normalizing the abundances of each metabolite to the internal standard and cell number. The incorporation of atoms was denoted as , where n was the number of atoms.
Structural modeling
Since no substrate-binding structures in human AARS2 have been observed to date, we modeled the structure of the alanine binding region with Phyre2 (Structural Bioinformatics Group, Imperial College, London). Prediction of the structure indicated that it was a high structural homolog of AARS2 in Aquifex aeolicus, which is an alanyl-tRNA synthetase (alias: alaS, PDB code: 1YFS) complexed with alanine. Thus, we further docked alanine and lactate into the complex structure and superimposed the modeled AARS2 to that of alaS from Aquifex aeolicus.
Statistical methods
Statistical analysis was performed using Prism 8.0 (GraphPad Software, Inc., San Diego, CA, USA) and Excel (Microsoft Corp., Redmond, CA, USA). Pooled results were expressed as the mean SEM. Comparisons between groups were made via unpaired two-tailed Student’s -test and two-way ANOVA. Statistical significance was set at ; ns no significance; ; ; ; .
REFERENCES
Brooks, G. A. The science and translation of lactate shuttle theory. Cell Metab. 27, 757-785 (2018).
Bendahan, D., Chatel, B. & Jue, T. Comparative NMR and NIRS analysis of oxygendependent metabolism in exercising finger flexor muscles. Am. J. Physiol. Reg. Intgr. Comp. Physiol. 313, R740-R753 (2017).
Glancy, B. et al. Mitochondrial reticulum for cellular energy distribution in muscle. Nature 523, 617-620 (2015).
Bassett, D. R. Jr. & Howley, E. T. Limiting factors for maximum oxygen uptake and determinants of endurance performance. Med. Sci. Sports Exerc. 32, 70-84 (2000).
Vargas-Mendoza, N. et al. Oxidative stress, mitochondrial function and adaptation to exercise: new perspectives in nutrition. Life 11, 1269 (2021).
Liu, W. Y., He, W. & Li, H. Exhaustive training increases uncoupling protein 2 expression and decreases Bcl-2/Bax ratio in rat skeletal muscle. Oxid. Med. Cell. Longev. 2013, 1-7 (2013).
Maxwell, P. H. et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399, 271-275 (1999).
Wang, G. L. & Semenza, G. L. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc. Natl. Acad. Sci. USA 90, 4304-4308 (1993).
Epstein, A. C. et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107, 43-54 (2001).
Jaakkola, P. et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O-2-regulated prolyl hydroxylation. Science 292, 468-472 (2001).
Denko, N. C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 8, 705-713 (2008).
Pugh, C. W. & Ratcliffe, P. J. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat. Med. 9, 677-684 (2003).
Henderson, G. C., Horning, M. A., Wallis, G. A. & Brooks, G. A. Pyruvate metabolism in working human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 292, E366 (2007).
Peters, S. J. Regulation of PDH activity and isoform expression: diet and exercise. Biochem. Soc. Trans. 31, 1274-1280 (2003).
Mourtzakis, M., Saltin, B., Graham, T. & Pilegaard, H. Carbohydrate metabolism during prolonged exercise and recovery: interactions between pyruvate dehydrogenase, fatty acids, and amino acids. J. Appl. Physiol. 100, 1822-1830 (2006).
Watt, M. J. et al. Rapid upregulation of pyruvate dehydrogenase kinase activity in human skeletal muscle during prolonged exercise. J. Appl. Physiol. 97, 1261-1267 (2004).
Gudiksen, A. et al. Lack of skeletal muscle IL-6 affects pyruvate dehydrogenase activity at rest and during prolonged exercise. PLoS One 11, e0156460 (2016).
Fan, J. et al. Tyr phosphorylation of PDP1 toggles recruitment between ACAT1 and SIRT3 to regulate the pyruvate dehydrogenase complex. Mol. Cell 53, 534-548 (2014).
Kikuchi, D., Minamishima, Y. A. & Nakayama, K. Prolyl-hydroxylase PHD3 interacts with pyruvate dehydrogenase (PDH)-E1 beta and regulates the cellular PDH activity. Biochem. Bioph. Res. Commun. 451, 288-294 (2014).
Spriet, L. L. et al. Pyruvate dehydrogenase activation and kinase expression in human skeletal muscle during fasting. J. Appl. Physiol. 96, 2082-2087 (2004).
Bowker-Kinley, M. M., Davis, W. I., Wu, P. F., Harris, R. A. & Popov, K. M. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem. J. 329, 191-196 (1998).
Bricker, D. K. et al. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, drosophila, and humans. Science 337, 96-100 (2012).
Herzig, S. et al. Identification and functional expression of the mitochondrial pyruvate carrier. Science 337, 93-96 (2012).
Bergersen, L. H. Lactate transport and signaling in the brain: potential therapeutic targets and roles in body-brain interaction. J. Cereb. Blood Flow Metab. 35, 176-185 (2015).
Hoque, R., Farooq, A., Ghani, A., Gorelick, F. & Mehal, W. Z. Lactate reduces liver and pancreatic injury in Toll-like receptor- and inflammasome-mediated inflammation via GPR81-mediated suppression of innate immunity. Gastroenterology 146, 1763-1774 (2014).
Saddik, M., Gamble, J., Witters, L. A. & Lopaschuk, G. D. Acetyl-CoA carboxylase regulation of fatty acid oxidation in the heart. J. Biol. Chem. 268, 25836-25845 (1993).
He, X. D. et al. Sensing and transmitting intracellular amino acid signals through reversible lysine aminoacylations. Cell Metab. 27, 151-166 (2018).
Zhang, D. et al. Metabolic regulation of gene expression by histone lactylation. Nature 574, 575-580 (2019).
Gotz, A. et al. Exome sequencing identifies mitochondrial alanyl-tRNA synthetase mutations in infantile mitochondrial cardiomyopathy. Am. J. Hum. Genet. 88, 635-642 (2011).
Euro, L. et al. Structural modeling of tissue-specific mitochondrial alanyl-tRNA synthetase (AARS2) defects predicts differential effects on aminoacylation. Front. Genet. 6, 21 (2015).
Filosto, M. et al. A novel mitochondrial tRNA(Ala) gene variant causes chronic progressive external ophthalmoplegia in a patient with Huntington disease. Mol. Genet. Metab. Rep. 6, 70-73 (2016).
Konovalova, S. & Tyynismaa, H. Mitochondrial aminoacyl-tRNA synthetases in human disease. Mol. Genet. Metab. 108, 206-211 (2013).
Tuppen, H. A. L., Blakely, E. L., Turnbull, D. M. & Taylor, R. W. Mitochondrial DNA mutations and human disease. Biochim. Biophys. Acta 1797, 113-128 (2010).
Vo, M.-N. et al. ANKRD16 prevents neuron loss caused by an editing-defective tRNA synthetase. Nature 557, 510-515 (2018).
Hussien, T. H. R. Lactate as a metabolic signal in gene expression: genechip analysis in L6 myocytes. Med. Sci. Sport Exer. 39, S289 (2007).
Taglia, I. et al. AARS2-related ovarioleukodystrophy: clinical and neuroimaging features of three new cases. Acta Neurol. Scand. 138, 278-283 (2018).
Ivan, M. et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for sensing. Science 292, 464-468 (2001).
Yao, P. & Fox, P. L. Aminoacyl-tRNA synthetases in medicine and disease. EMBO Mol. Med. 5, 332-343 (2013).
Chen, Y. J. et al. Lactate metabolism is associated with mammalian mitochondria. Nat. Chem. Biol. 12, 937-943 (2016).
Lee, D. C. et al. A lactate-induced response to hypoxia. Cell 161, 595-609 (2015).
Isoe, T. et al. High glucose activates HIF-1-mediated signal transduction in glomerular mesangial cells through a carbohydrate response element binding protein. Kidney Int. 78, 48-59 (2010).
Seo, J. et al. Fatty-acid-induced FABP5/HIF-1 reprograms lipid metabolism and enhances the proliferation of liver cancer cells. Commun. Biol. 3, 638 (2020).
Sonveaux, P. et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS One 7, e33418 (2012).
Morales, M. & Munne-Bosch, S. Malondialdehyde: facts and artifacts. Plant Physiol. 180, 1246-1250 (2019).
Abrantes, H. D. et al. The lactate receptor HCAR1 modulates neuronal network activity through the activation of G (alpha) and G (beta gamma) subunits. J. Neurosci. 39, 4422-4433 (2019).
Baumann, F. et al. Lactate promotes glioma migration by TGF-beta 2-dependent regulation of matrix metalloproteinase-2. Neuro. Oncol. 11, 368-380 (2009).
Zhao, S. et al. Regulation of cellular metabolism by protein lysine acetylation. Science 327, 1000-1004 (2010).
Richardson, R. S., Leigh, J. S., Wagner, P. D. & Noyszewski, E. A. Cellular Po-2 as a determinant of maximal mitochondrial consumption in trained human skeletal muscle. J. Appl. Physiol. 87, 325-331 (1999).
Fan, Y., Han, J., Yang, Y. & Chen, T. Novel mitochondrial alanyl-tRNA synthetase 2 (AARS2) heterozygous mutations in a Chinese patient with adult-onset leukoencephalopathy. BMC Neurol. 22, 214 (2022).
Palacios, O. M. et al. Diet and exercise signals regulate SIRT3 and activate AMPK and PGC-1a in skeletal muscle. Aging 1, 771-783 (2009).
Jing, E. et al. Sirt3 regulates metabolic flexibility of skeletal muscle through reversible enzymatic deacetylation. Diabetes 62, 3404-3417 (2013).
Imamura, M. et al. MondoA deficiency enhances sprint performance in mice. Biochem. J. 464, 35-48 (2014).
Liu, F. et al. Effects of hereditary moderate high fat diet on metabolic performance and physical endurance capacity in C57BL/6 offspring. Mol. Med. Rep. 17, 4672-4680 (2018).
Carrell, E. M., Coppola, A. R., McBride, H. J. & Dirksen, R. T. Orai1 enhances muscle endurance by promoting fatigue-resistant type I fiber content but not through acute store-operated entry. FASEB J. 30, 4109-4119 (2016).
Kammoun, M. et al. Impact of TIEG1 on the structural properties of fast- and slowtwitch skeletal muscle. Muscle Nerve 55, 410-416 (2017).
Nemeria, N. et al. Inhibition of the Escherichia coli pyruvate dehydrogenase complex E1 subunit and its tyrosine 177 variants by thiamin 2-thiazolone and thiamin 2-thiothiazolone diphosphates. Evidence for reversible tight-binding inhibition. J. Biol. Chem. 276, 45969-45978 (2001).
Schulze, W. X. & Usadel, B. Quantitation in mass-spectrometry-based proteomics. Annu. Rev. Plant Biol. 61, 491-516 (2010).
ACKNOWLEDGEMENTS
This work was supported by Grants from the State Key Development Programs of China (2018YFA0800300 to S.Z.; 2018 YFA0801300 to W.X.), the National Natural Science Foundation of China (92253305, 31821002, 32230054, 31930062, 91857000 and 92157001 to S.Z.; 32171298 to W.X.; 81971449 and 82171672 to Y.Y.), and Program of Shanghai Academic Research Leader (21XD1423000 to W.X.).
AUTHOR CONTRIBUTIONS
S.Z. and W.X. conceived the project and wrote the manuscript. Y.M., J.Z., Q.Z., Y.W., X.H., K.Z., H.Y., H.Z., Y.Z. and P.L. performed the biochemical cell experiments. Q.Z., Y.W. and Z.Z. performed the mice running tests. B.W. performed computer simulation assays. Y.L., Y.Y. and J.Z. supervised all experiments. All authors read and discussed the manuscript.
Open Access This article is licensed under a Creative Commons
Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http:// creativecommons.org/licenses/by/4.0/.
The Obstetrics & Gynecology Hospital of Fudan University, Shanghai Key Laboratory of Metabolic Remodeling and Health, State Key Laboratory of Genetic Engineering, School of Life Sciences, Children’s Hospital of Fudan University, and Institutes of Biomedical Sciences, Fudan University, Shanghai, China. Department of Cancer Biology, Dana-Farber Cancer Institute, Boston, MA, USA. Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, MA, USA. NHC Key Lab of Reproduction Regulation (Shanghai Institute of Planned Parenthood Research), Shanghai, China. Shanghai Fifth People’s Hospital of Fudan University, Fudan University, Shanghai, China. Key Laboratory for Tibet Plateau Phytochemistry of Qinghai Province, College of Pharmacy, Qinghai University for Nationalities, Xining, Qinghai, China. Guangdong Provincial Key Laboratory of Malignant Tumor Epigenetics and Gene Regulation, Guangdong-Hong Kong Joint Laboratory for RNA Medicine, RNA Biomedical Institute, Medical Research Center, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou, Guangdong, China. These authors contributed equally: Yunzi Mao, Jiaojiao Zhang, Qian Zhou, Xiadi He. email: xuwei_0706@fudan.edu.cn; zhaosm@fudan.edu.cn