نقل الإلكترون متعدد القنوات المحفز بواسطة البوليفانادات في الإطار المعدني العضوي لتنشيط بيروكسيمونوكبريتات المعزز Multi-channel electron transfer induced by polyvanadate in metal-organic framework for boosted peroxymonosulfate activation
عمليات تنشيط بيروكسيمونوكبريتات (PMS) الحفزية لا تعتمد فقط على نقل الإلكترونات من المراكز المعدنية السائدة بسبب التركيب المعقد وبيئة الواجهة للحفازات. هنا يتم تصنيع إطار معدني عضوي قائم على الكوبالت يحتوي على بوليفانادات.عنقود bpy -بيبيريدينيتم تقديمه. يُظهر المحفز نشاط تحلل متفوق تجاه مجموعة متنوعة من الملوثات الدقيقة، مع أعلى مستوى مشغول من المدارات الجزيئية (HOMO) أعلى، من خلال هجوم غير جذري. تُظهر طيفية الامتصاص بالأشعة السينية وحسابات نظرية الكثافة الوظيفية (DFT) أن مواقع الكوبالت تعمل ككل من مصيدة PMS ومانح للإلكترونات. تكشف التوصيفات الطيفية في الموقع وحسابات DFT أن ذرات الأكسجين الطرفية في الـيمكن أن يتفاعل الإسفنجة الإلكترونية مع ذرات الهيدروجين الطرفية في PMS لتشكيل روابط هيدروجينية، مما يعزز توليدالوسيط من خلال كل من سحب الديناميكية وعملية نقل الإلكترون المباشر. علاوة على ذلك،يظهر قدرة طويلة الأمد على تنقية المياه، تصل إلى 40 ساعة، تجاه المياه العادمة الفعلية المنبعثة من مصنع إنتاج الأوفلوكساسين. لا يقدم هذا العمل فقط محفزًا فعالًا مع إسفنجة إلكترونية لمعالجة البيئة المائية عبر مسار غير جذري، ولكنه يوفر أيضًا رؤى أساسية حول آلية تفاعل شبيهة بفنتون.
تم التحقق من أن التفاعلات الشبيهة بفنتون التي يتم تحفيزها بواسطة بيروكسيديسلفات (PDS) أو بيروكسيمونوسلفات (PMS) هي تقنية واعدة لتنقية المياه، مستفيدة من إنتاج الجذور و/أو غير الجذور من خلال التفاعل بين البيرسلفات والمراكز النشطة.حتى الآن، تم تكريس العديد من الجهود لضبط آلية الأكسدة بشكل اتجاهي أو السعي لتحقيق كفاءة أعلى في نقل الإلكترونات خلال تفاعلات شبيهة بفنتون. ومع ذلك، فإن عمليات تنشيط PDS/PMS قد تُحفز من خلال نقل الإلكترون بواسطة مكونات أخرى، مما يتطلب تنظيمًا دقيقًا للعوامل المساعدة من مستوى الجزيء أو حتى الذرة.
إطارات معدنية عضوية (MOFs)، كمواد هجينة غير عضوية-عضوية متعددة الاستخدامات، يمكن ضبطها بدقة من مستوى الجزيئات.بجانب
يمكن استخدام بعض تجمعات البوليوكسوميتالات (POM) كروابط غير عضوية لبناء هياكل MOFs خاصة بخصائص فريدة.. مؤخرًا، تم استخدام بعض الهياكل الإطارية المعدنية العضوية (MOFs) التي تحتوي على مركبات الأكسيد المتعدد (POM) في مجالات تفاعل الاختزالتفاعل تطور الهيدروجينوخلية شمسية، والاستفادة من تلك المادة POM كمستودع أو إسفنجة للإلكترونات يمكن أن تلتقط، وتخزن، وتنقل الإلكترونات لإنشاء مواقع امتصاص ومراكز تحفيز متنوعة. ومع ذلك، فإن جزيئات PMS تمتلك هيكلاً كيميائياً أكثر تعقيداً من تلك الخاصة بـ و لذلك، كان من المتوقع أن يكون البيئة الحفازة بالقرب من واجهة MOF القائم على POM معقدة بسبب وجود الأكسجينات المنسقة غير المشبعة، مما أدى إلى أن عملية تنشيط PMS قد تعتمد على تفاعلات أخرى مثل قناة الإلكترون المتعدد والقوى بين الجزيئات بدلاً من الاعتماد فقط على نقل الإلكترون لمراكز المعادن الانتقالية. لذلك، كان من المثير استكشاف آلية تنشيط PMS الحفازة من أجل التنظيم المباشر لهيكل الحفاز والتحقيق في علاقة الهيكل بالنشاط.
في هذا العمل، تم استخدام مادة قائمة على بوليوكسي فاندات.تم تصنيعها لتحقيق تفعيل PMS لتحلل الملوثات العضوية، مما قد يسهل التحقيق في العلاقة بين بيئة التنسيق السطحية المعقدة للحفاز وتفاعل شبيه فنتون. أكدت هذه الدراسة أن نقل الشحنة بين PMS ومواقع المعادن في الحفازات المعتمدة على المعادن لم يكن المسار الوحيد للتفعيل. أظهرت حسابات نظرية الكثافة الوظيفية (DFT) أنه كان من الصعب تشكيل المركبات ذات الدوران العالي بين PMS ضعيف المجال ومراكز الكوبالت كمواقع الامتصاص المثلى، مما يعني أن مصدر نقل الشحنة لم يكن فقط المركز المعدني. أوضحت تقنيات رامان في الموقع وتحويل فورييه للأشعة تحت الحمراء (FTIR) التغير الديناميكي لـرابطة من PMS والمعادن-سندات ( و ) بالإضافة إلى نقل الإلكترون بين و PMS خلال تفاعل شبيه فنتون، الذي عزز تشكيل لإنتاج الأكسجين الأحادي بشكل أكبر ) و جذور الكبريتات ( ) لأكسدة أوفلوكساسين (OFX). ساعدت آلية الأكسدة التي تهيمن عليها غير الجذور (bpy) لإظهار قدرة علاجية طويلة الأمد متفوقة تجاه المياه العادمة الفعلية وقدرة سمية مرضية من ردود الفعل التجريبية البيولوجية. فتحت هذه الدراسة بابًا لتصميم وتخليق المحفزات مثل مجموعة POM التي تعمل كإسفنجة إلكترونية لتحقيق مسارات غير جذرية لتنقية المياه.
النتائج
توصيفات
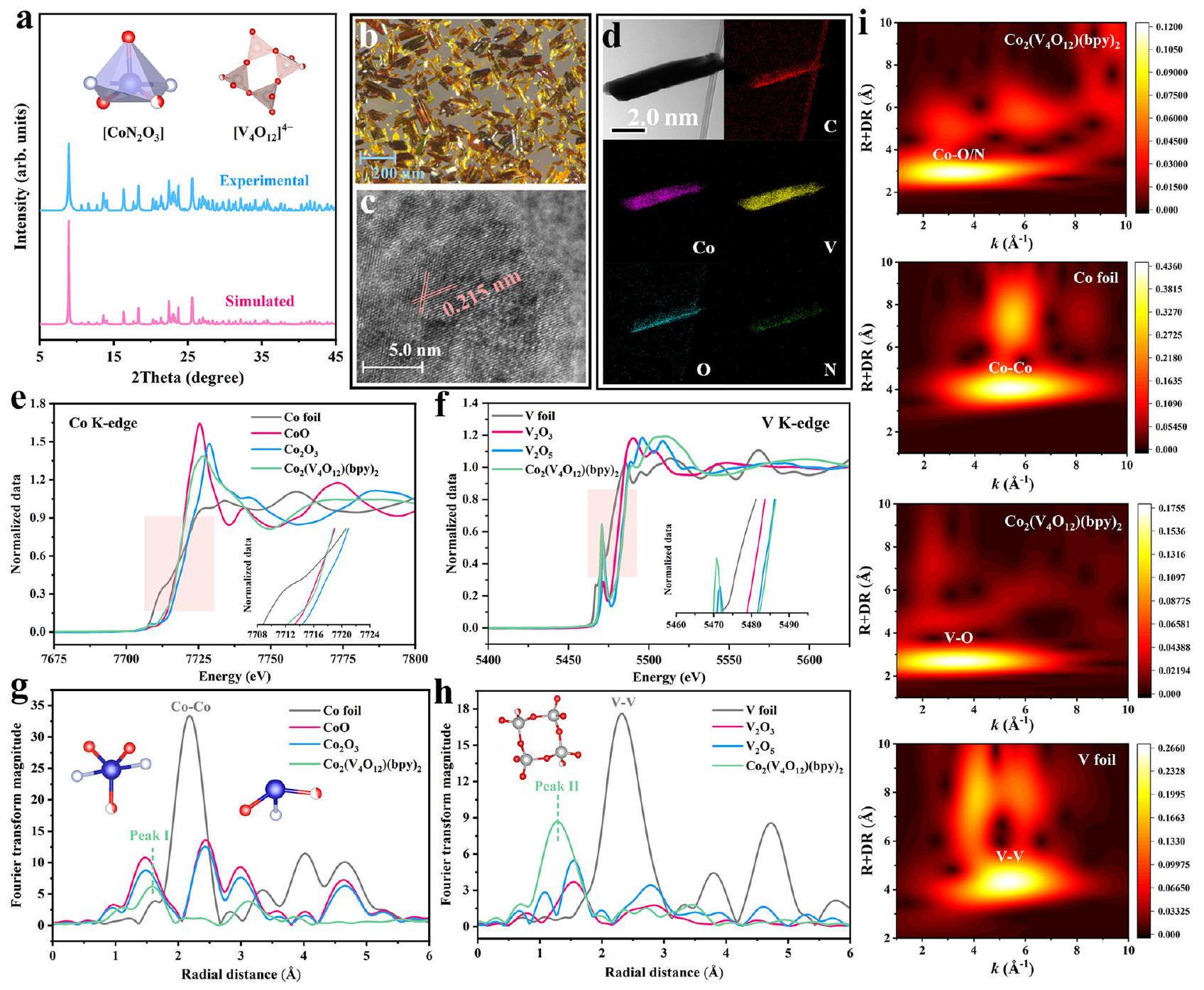
تم تحليل بنية البلورة المفردة (CCDC 2323958) لتأكيد البنية المتطابقة للمنتج المحضر.إلى السابقينفي الذييتم تنسيقها بخمسة بواسطة ذرتين نيتروجين من bpy وثلاث ذرات أكسجين من ثلاثة مختلفةالعناقيد. لوحظ أن هناك ستة ذرات أكسجين طرفية فيمنسقة إلى ستة مختلفين الذرات عبر الروابط، وذرتا الأكسجين الطرفيتان الأخريان غير منسقتين (الشكل التوضيحي 1 والجدول التوضيحي 1). أنماط حيود الأشعة السينية للبودرة (PXRD) (الشكل 1a) للعينات المحضرة حديثًا تطابقت بشكل جيد مع تلك المحاكية من CCDC 2323958 و 152985، مما يشير إلى نقاء عالي للمحفز المستخدم في الاختبارات والتحديدات المتعاقبة. أظهرت صورة المجهر الضوئي (الشكل 1ب) أنعرض بلورة كتلة صفراء بمقياس الميكرون، وهو ما تم تأكيده من خلال صورة المجهر الإلكتروني الماسح (SEM) (الشكل التكميلي 2). وأكدت المجهر الإلكتروني الناقل عالي الدقة (HR-TEM) الواجهة المكشوفة (332) لـمن (bpy) (الشكل 1c والشكل التكميلي 3). عرضت خريطة العناصر المقابلة (الشكل 1d) توزيعًا موحدًا للعناصر المميزة مثل ، و O .
تمت دراسة الحالة الكيميائية وبيئة التنسيق لكل من الكوبالت والفاناديوم باستخدام طيف الامتصاص بالأشعة السينية بالقرب من حافة الطاقة (XANES) وهيكل الامتصاص الدقيق بالأشعة السينية الممتد (EXAFS). تم دمج طيف XANES لحافة العنصر K لكل من الكوبالت (الشكل 1e) والفاناديوم (الشكل 1f) مع فضاء k. وتمت البيانات الملائمة (الشكل التكميلي 4) توضيح أن حالات التكافؤ لـ Co و V كانت قريبة من +2 و +5 على التوالي. أشارت طيفيات EXAFS (الشكل 1g) إلى أن ذرات Co منتم تنسيقها مع ذرات الأكسجين والنيتروجين بطول رابطة (الذروة الأولى). بالإضافة إلى ذلك، لم يتم تقديم أي تجمعات من الكوبالت أو جزيئات بلورية فيهيكل بسبب عدم وجود قمة واضحة لـرابطة عندتمت ملاحظتها، الذي تطابق جيدًا مع نتائج تحليلات بنية البلورات. أظهرت طيف EXAFS لعنصر الفاناديوم (الشكل 1h) أن رابطة V-O (الذروة II) بطولكان مختلفًا عن تلك الخاصة بـ و ، مما يوضح أن التفاعلات بين ذرات الفاناديوم والأكسجين فيكانوا أقوى من أولئك في و تحويل المويجات (WT) لـ (ببي) تم إجراء ذلك لتوضيح القمم المذكورة أعلاه بشكل أكبر. كما هو موضح في الشكل 1i والشكل التكميلي 5، القمم عند و منيمكن أن يُنسب العينة إلى وروابط V-O، على التوالي. أظهرت مخططات WT لرقائق الكوبالت، ورقائق الفاناديوم ونتائج تحديد بنية البلورة المفردة أنه لا يوجد و يمكن ملاحظة الرابطة في.
النشاط الحفزي وآلية الأكسدة المعنية
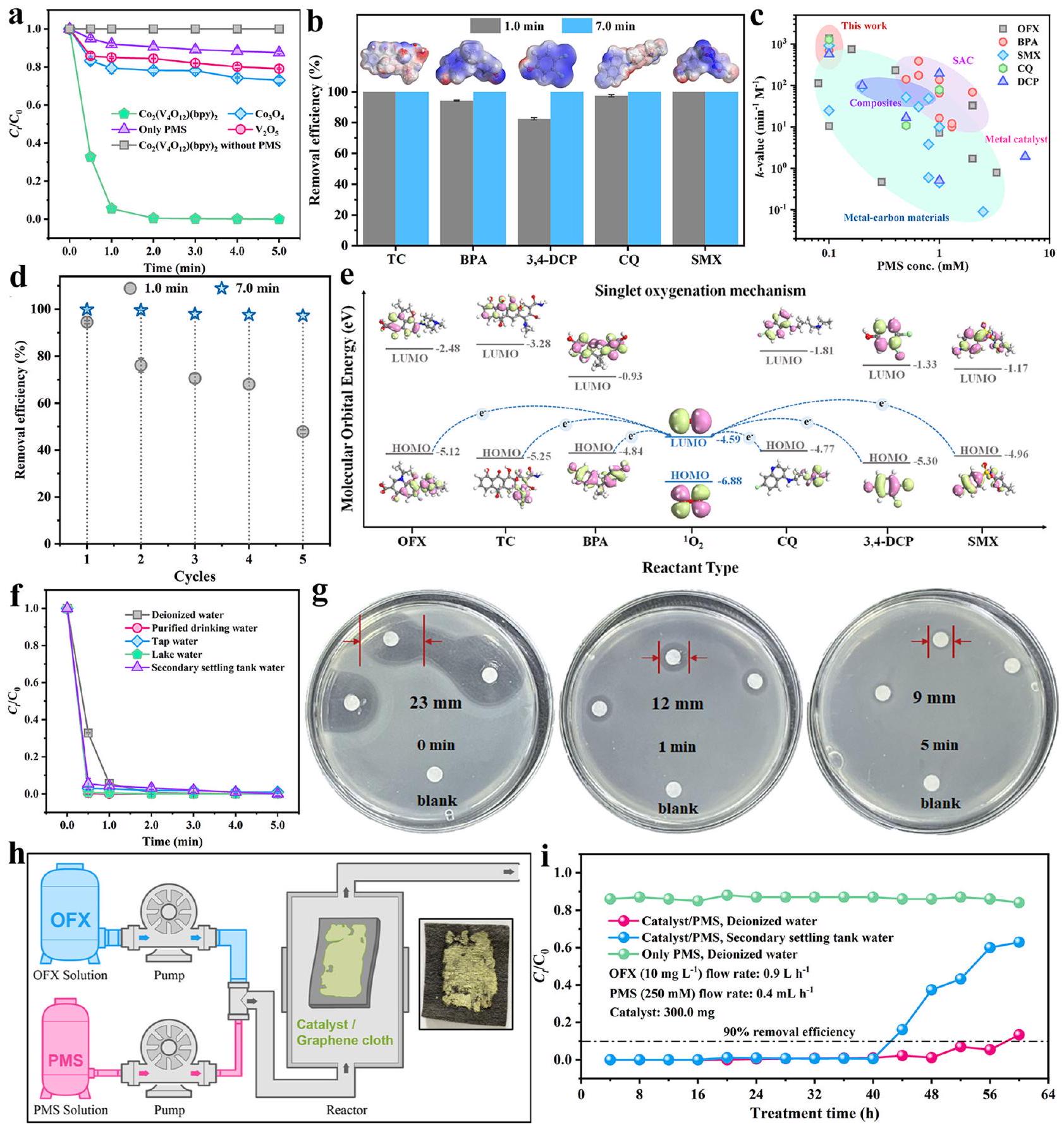
تم اختيار OFX كملوث دقيق مستهدف للتحقيق في أداء تنشيط PMSتمت دراسة قدرات تنشيط نظام PMS المختلفة دون تعديل الرقم الهيدروجيني. (الشكل 2أ والأشكال التكميلية 6، 7). الـ (bpy) عرضت أفضل تفعيل لنظام إدارة المشاريع لتحلل OFX مع أكبر ثابت معدل تفاعل ظاهر ) من (الشكل التوضيحي التكميلي 8أ)، الذي تم تأكيده بشكل أكبر من خلال تجارب استهلاك PMS (الشكل التوضيحي التكميلي 8ب). بالإضافة إلى ذلك، حوالي يمكن تحقيق إزالة إجمالي الكربون العضوي (TOC) في غضون 5.0 دقائق فينظام PMS (الشكل التوضيحي 8c)، مما يؤكد القدرة الأفضل على التمعدنمن و . الـ (bpy) يمكن أن تحقق أيضًا كفاءات إزالة فائقة الارتفاع تجاه ملوثات عضوية دقيقة أخرى مثل التتراسيكلين (TC) ، والبيسفينول أ (BPA) ، والديكلوروفينول 3,4 (3,4-DCP) ، والفوسفات الكلوروكين (CQ) ، والسلفاميثوكسازول (SMX) (الشكل 2b والشكل التكميلي 8d) ، حيث أن المعنية-قيمة ( تجاوزت تقريبًا جميع المحفزات المبلغ عنها لتنشيط PMS (الشكل 2c والجدول التكميلي 3). تم التأكيد على أنأظهرت إعادة الاستخدام الجيدة (الشكل 2d). لم يُلاحظ سوى تراجع طفيف في النشاط التحفيزي بعد خمس عمليات إعادة تدوير، ربما بسبب أن بعض المواقع النشطة مشغولة بال intermediates المتكونة (الشكل التوضيحي 9).. شركة التسرب ( ) و V ( كانت ( ) أقل بكثير من معايير جودة البيئة للمياه السطحية (GB 3838-2002)، مما يدل على الاستقرار الكبير للمياه في المحفز. كما أكدت ملاحظات أنماط PXRD، وطيف FTIR، وصورة SEM، وصورة HR-TEM على الاستقرار الجيد لـ (ببي) (الشكل التوضيحي التكميلي 10a-d).
لتحديد الأنواع الرئيسية من الجذور الحرة للأكسجين (ROSs) أثناء تنشيط PMS، تم إجراء تجارب إخماد كيميائية (الملاحظة التكميلية 5)، حيث تم اختيار الميثانول (MeOH) و التيرت-بيوتانول (TBA) والبينزوكوينون (BQ) والكحول الفورفوريلي (FFA) كمواد موقفة.ومبرد، على التواليإدخال الميثانول والأحماض الدهنية الحرة إلىيمكن أن يمنع نظام PMS كفاءات تحلل OFX من 100% إلى 68.2%“، على التوالي (الشكل التكميلي 11a). بينما لم يُلاحظ تأثير يذكر على كفاءات تحلل OFX الحفزي عند إضافة TBA و BQ. لوحظ أن PMS يمكن أن يتم استهلاكه مباشرة بواسطة FFA (الشكل التكميلي 11b)، وهو ما يتماشى مع الدراسات السابقة.. لذلك، L-هيستيدين، كآخرالمُطفئ، تم اعتماده لإطفاءلتحديد مساهمته بشكل أكبر. أظهرت النتائج (الشكل التكميلي 11c، d) أن كفاءة معدل تحلل OFX ومعدل التحلل قد تم تثبيطها بشكل خطير. من المهم أن معدل التحفيز لـ OFX فيكان أسرع من ذلك في الماء المنزوع الأيونات (الشكل التكميلي 11c، d)، مما يؤكد المساهمة الكبيرة لـفي (bpy) نظام إدارة المشاريع. أظهرت النتائج أعلاه أن
الشكل 1 | التوصيفات الكيميائية والهيكلية لـ. أنماط PXRD للمنتج المصنوع حديثًامحفزصورة المجهر الضوئي وصورة HR-TEM لـصور رسم الخرائط العنصرية للعناصر المميزة منطيف XANES لـ (e) الكوبالت و (f) الفاناديوم
حد K في (أدخل: حافة الامتصاص لـ Co و V، على التوالي). طيف EXAFS لـ (g) Co و (h) V K-edge في . رسومات WT لحواف Co و V K في EXAFS للرقائق Co و V و Co2. -أ و قد تلعب مساهمة رئيسية خلال عملية التحلل. كما أظهرت نتائج تحليلات الرنين المغناطيسي الإلكتروني (EPR) (الشكل التكميلي 12) أن DMPO-SO4 القويو TEMP-يمكن ملاحظة الإشارات، مما يعنيوتم إنتاجها فينظام إدارة الممتلكات. بالإضافة إلى ذلك،تم إجراء تحليلات كمية عبر تقنية EPR (الشكل التكميلي 12b) لتجنب المبالغة في تقدير مساهمته، مما كشف أن المنتج كما هوفينظام (bpy)/PMS يمكن أن يستمر لفترة أطول فيمن. وشدة TEMP- انخفض الإشارة بوضوح عند إضافة OFX فيمتلازمة ما قبل الحيضالنظام، مؤكدًا الدور الهام لـخلال عملية تحلل OFX.جوالجو (الشكل التكميلي 13) وتجارب إخماد الأنيلين (الشكل التكميلي 11a) أكدت أن توليدقد يُعزى إلىالتحلل بدلاً من الجذور الحرة السوبر أكسيدتحول. في هذه الأثناء، نتائج أكدت طريقة الكشف بالفلوريسcence (الشكل التكميلي 14) أنلم يكن بالإمكان إنتاجه بكثرة فينظام إدارة الممتلكاتاستبعدت تجارب خلط PMS والتقاط المعادن عالية التكافؤ (الشكل التكميلي 15) مساهمة مسارات غير جذرية أخرى مثل عملية نقل الإلكترون والوسطية عالية التكافؤ من الكوبالت، على التوالي.تجارب المجسات الكيميائية (الملاحظات التكميلية 6، 7)أكد أن المنتج و في يمكن لنظام PMS كنوع نشط رئيسي أن يحقق الإزالة السريعة تجاه OFX (الشكل التكميلي 16). ومع ذلك، فإن إزالة TOC تعتمد بشكل رئيسي على عملية الأكسدة للجذور المرتبطة بالسطح والامتزاز بين الوسائط والمحفز (الشكل التكميلي 17 والملاحظة التكملية 8).لقد تم الإبلاغ عن أنه يمكن أكسدة الملوثات الغنية بالإلكترونات بشكل انتقائي في نظام غير جذري.تم حساب الجهود الكهروستاتيكية (ESP) لبعض المركبات العضوية المختارة، حيث أظهرت أن TC و BPA و 3،4-DCP و CQ و SMX، التي تحتوي على مجموعات وظيفية لتجميع الإلكترونات، عرضت ESP أعلى من حمض البنزويك (BA) وحمض النيتروبنزويك (NBA) (الشكل 2b والشكل التكميلي 18). الفرق فيتمت دراسة قدرة الأكسدة تجاه مجموعة مختارة من المواد العضوية من وجهة نظر مسار نقل الشحنة (الشكل 2e). يمكن ملاحظة بوضوح أن أعلى مدار جزيئي مشغول (HOMO) للملوثات الغنية بالإلكترونات مثل TC (-5.25 eV) و BPA (-4.84 eV) ، و SMX كانت أقل قليلاً من أدنى مدار جزيئي غير مشغول (LUMO) لـ، مما يشير إلى أن يمكن أن تسحب بسهولة الإلكترونات من أعلى مستوى مملوء بالإلكترونات (HOMO) لتلك الملوثات الغنية بالإلكترونات لتحقيق المزيد من تحلل المواد العضوية.. بينما الـ LUMO لـكان أعلى بكثير من HOMO لـ BA (-6.14 eV) و NBA (-6.70 eV)، مما يدل على أنه كان من الصعب إجراء تفاعل الأكسدة (الشكل التكميلي 19).
الشكل 2 | النشاط التحفيزي، آلية الأكسدة والتطبيقات البيئية فياختبار نشاط تفاعل شبيه فنتون على محفزات مختلفة.إزالة العديد من الملوثات الدقيقة بواسطة (الإدراج: ESP المقابل للملوثات العضوية). ج مقارنة كفاءة إزالة عدة ملوثات دقيقة بواسطة المحفزات المتطورة في نظام تنشيط PMS (تم تقديم المعلومات ذات الصلة في الجدول التكميلي 3). د اختبار القابلية لإعادة التدوير لـآلية الأكسدة لـلعدة ملوثات دقيقة (الإطار: HOMO المقابل) و LUMO للملوثات العضوية).تأثيرات مياه الصرف الصحي المحاكاة المختلفة على كفاءات إزالة OFX.تثبيط OFX ووسائط تحلله على نمو E. coli. h توضيح تخطيطي للريكتور التجريبي. i تطور تركيز OFX في أنظمة مختلفة في الريكتور. الظروف التجريبية: [المحفز] (في، د، ف)، (في ب). تمثل أشرطة الخطأ في الأشكال الانحرافات المعيارية من الاختبارات الثلاثية.
يمكن أن تتفاعل الأنيونات والمواد العضوية المتواجدة في الماء الفعلي مع أنواع الأكسجين التفاعلية (ROS) لتقليل كفاءات التحفيز.. أظهرت النتائج أن المواد المتواجدة معًا كان لها تأثير ضئيل على تحلل OFX في نظام إدارة الممتلكات (الشكل التوضيحي 20)، بسبب ذلكامتلكت القدرة على الأكسدة المنخفضة والقدرة الأفضل على الأكسدة الانتقائية مقارنة بالجذور.يمكن أن ينشط PMS بكفاءة لتحقيق OFX التحلل في نطاق pH الواسع من 3.12 إلى 8.04 (الأشكال التكميلية 21، 22)، مع الأخذ في الاعتبار أنأظهر تحملاً قوياً للحل. أيضًا، (bpy) يمكن لنظام PMS تحقيق إزالة كاملة تجاه OFX في عينات المياه المحاكية من حلول المياه الفعلية (مياه الأنهار، البحيرات، مياه الصنبور، مياه الشرب المنقاة ومياه خزان الترسيب الثانوي) خلال 5.0 دقائق (الشكل 2f والشكل التوضيحي 23). كشفت هذه النتائج أننظام إدارة PMS أظهر تفوقًا قدرة مقاومة التداخل، تعكس تفوقفي التطبيقات العملية لتحلل OFX.
بالإضافة إلى تحديد منتجات UPLC-MS، تم حساب مؤشر فوكوي لـ OFX (الأشكال التكميلية 24-26) لتحديد المواقع التفاعلية.، الذي ساعد في اقتراح خمسة مسارات محتملة لإزالة OFX. أظهرت التغيرات السمية لل intermediates المتكونة (الشكل التوضيحي 27) أن عملية تنشيط PMS علىيمكن أن يحلل OFX إلى وسائط سامة منخفضة، بل يمكن أن يقوم بتعدين OFX بالكامل إلى و تم تحديد تأثير تثبيط النمو لـ OFX ووسائطه على الإشريكية القولونية (E. coli) (الملاحظة التكميلية 9)، مما يؤكد انخفاض سمية الوسائط. كما هو موضح في الشكل 2g، فإن أقطار مناطق التثبيط لـ OFX ومنتجات التحلل في 1.0 دقيقة و 5.0 دقائق ضد. القولونية كانت 23.0 و 12.0 و 9.0 مم (تجربة قريبة من الفراغ بدون OFX)، مما يدل على تحلل OFX على مدىأدى إلى تقليل السمية تجاه E. coli. أظهر تجربة نمو النبات (الشكل التكميلي 28) أن معدل الإنبات وحالة نمو براعم الفاصوليا المزروعة بالمياه المعالجة كانت مشابهة لبراعم الفاصوليا المزروعة بالمياه المنزوعة الأيونات. تشير النتائج أعلاه إلى أنأظهر نظام PMS قدرة مرضية على التمعدن وقدرة على إزالة السموم.
لتقييم آفاق التطبيق العملي لـ (ببي) كمُنشط لمتلازمة ما قبل الحيض،تم تثبيت المسحوق على قماش الجرافين (الشكل 2h) لإجراء العلاج طويل الأمد لكل من مياه الصرف الصحي المحاكية التي تحتوي على OFX المكونة من الماء النقي ومياه خزان الترسيب الثانوي الفعلية (الشكل التكميلية 29). أظهر الشكل 2i أن كفاءة إزالة OFX في كل من الماء النقي ومياه خزان الترسيب الثانوي الفعلية يمكن أن تحافظ علىحتى 56 ساعة و 40 ساعة فينظام PMS، بينما كانت كفاءة إزالة OFX لنظام PMS الفردي أقل من. بالإضافة إلى ذلك، فإن المعزولضمنت استقرار المحفز خلال عملية التفاعل (الشكل التكميلي 30أ، ب)، حيث تم تسرب الكوبالت ( ) و كانت التركيزات أقل بكثير من المستوى الأقصى المستهدف للملوثات المحدد من قبل المعايير الوطنية الصينية (Co: GB 3838-2002؛ V: GB 26452-2011). نمط PXRD (الشكل التكميلي 31) وصورة SEM (الشكل التكميلي 32) للمواد المستخدمةبعد أن تطابقت التفاعل بشكل جيد مع العينة الجديدة، مما يدل على المرحلة المستقرة، والتركيب، والشكل الخارجي. في هذه الأثناء، يمكن أن تصل كفاءة إزالة TOC إلى حوالي خلال 48 ساعة فيمتلازمة ما قبل الحيضنظام المياه النقية (الشكل التوضيحي 30c)، مما يعني أن OFX يمكن أن يتعرض جزئيًا للتعادل المعدني إلى و . هذه النتائج أكدت أن كان المحفز يمتلك إمكانيات كبيرة للتطبيق العملي.
تحديد مواقع التحفيز وآلية الإثارة
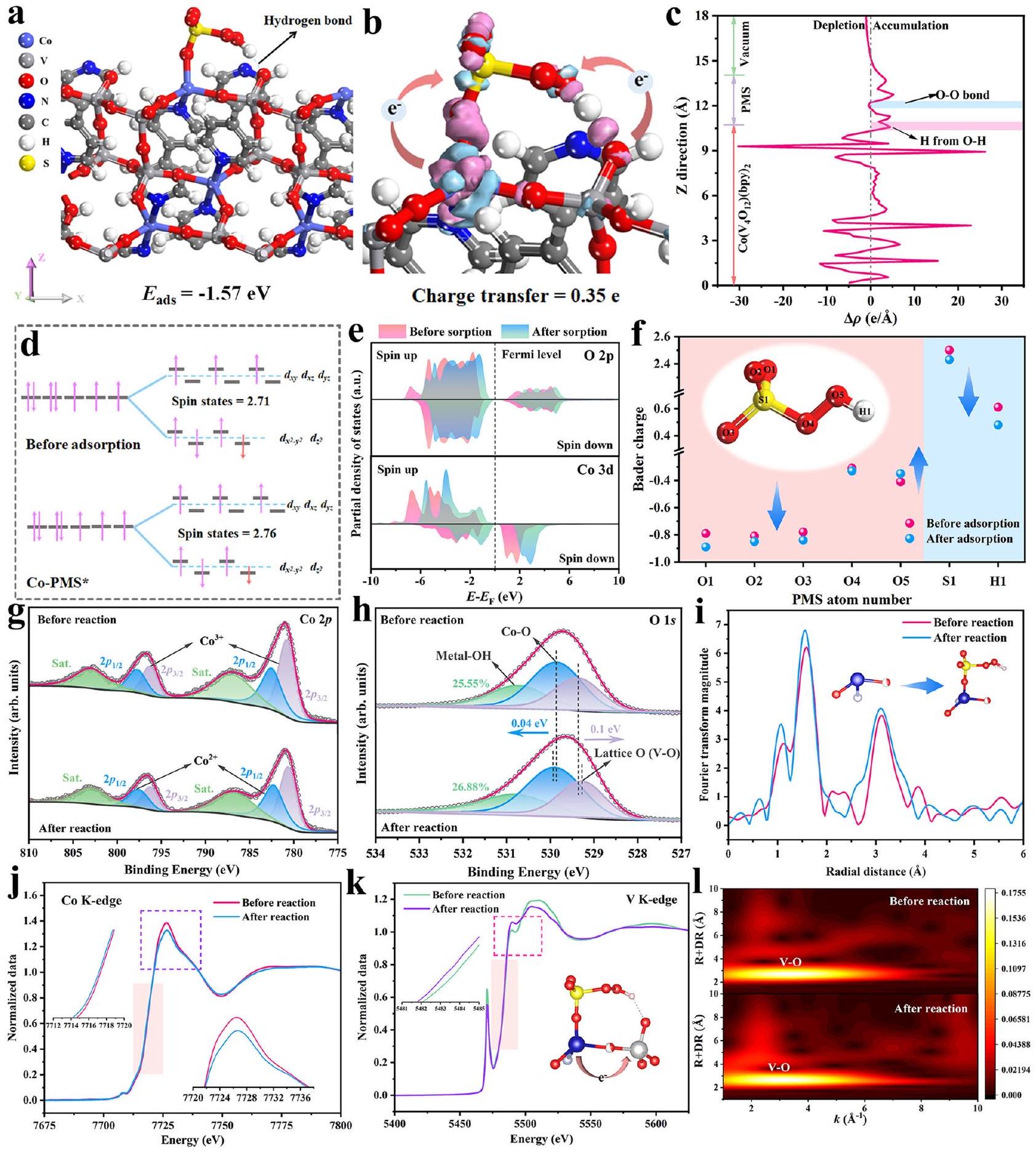
تم استخدام حسابات DFT لبناءنموذج الكتلة مع دوران متوازي، والذي كان أكثر استقرارًا من دوران مضاد (الشكل التكميلي 33).تم بناء نموذج السطح (الشكل التوضيحي 34a) على طول وجه البلورة (001)، حيث كانت ذرات الكوبالت المجاورة لطبقة الفراغ مواقع تنسيق غير مشبعة. أظهر التحليل الطيفي الإلكتروني (EPR) استجابة واضحة عند (الشكل التوضيحي 34ب)، يؤكد وجود الفراغات. بشكل عام، قد يكون من المرجح أن تعمل منطقة تراكم الإلكترونات كمركز تحفيزيأظهرت نتائج كثافة الحالات وكثافة الحالات الجزئية (PDOS) أن المدار الذي يشغله الإلكترون قريب من مستوى فيرمي. ) كانت تُنسب بشكل رئيسي إلى مساهمة Co و Oالمدارات (الشكل التوضيحي 35)، التي تكون فيها الحالة الإلكترونية للكوبيالت أظهر انقسامًا واضحًا بسبب تأثير جان-تيلر (الشكل التكميلي 35b). تم بناء ستة نماذج محتملة لامتصاص PMS (الأشكال التكملية 36، 37) لحساب طاقة الامتزاز ( )، مع الأخذ في الاعتبار أن كل من Co و V يمكن أن يُنظر إليهما كمواقع امتصاص محتملة للتفاعل مع PMS. كما هو موضح في الشكل 3a، مواقع Co من (bpy) السطح مرتبط بـ O 1 من PMS، وكان الرابطة الهيدروجينية تكونت بين الهيدروجين النهائي لـ PMS والأكسجين النهائي لـ. الـتم حسابه ليكون -1.57 إلكترون فولت، وهو أكثر سلبية بكثير من تلك النماذج الأخرى، مما يؤكد أنكان نموذج الامتصاص الأمثل.
استنادًا إلى النموذج الأمثل، تم حساب فرق كثافة الإلكترون المقابل. أظهرت الشكل 3b أن PMS يمكن أن تسحب الإلكترون (0.35 e) من سطح المحفز، حيث كانت منطقة تراكم الإلكترونات تركزت بشكل رئيسي على الرابطة الهيدروجينية بدلاً من رابطة الأكسجين-الأكسجين (Fig. 3c). للتحقيق بشكل أعمق في مصدر الإلكترون لتنشيط PMS، تم استخدام Coتم حساب توزيع الإلكترونات المدارية بناءً على قاعدة هوند ومبدأ استبعاد باولي (الشكل 3d). على الرغم من اعتبار مواقع الكوبالت كمواقع امتصاص مثالية تجاه PMS، فإن الزيادة الضعيفة في حالات الدوران بعد امتصاص PMS تشير إلى أن المركبات ذات الدوران العالي مع إمكانات أكسدة قوية لم تتشكل بسهولة. لذلك، قد لا تعتبر مواقع الكوبالت هي المراكز الوحيدة لنقل الإلكترونات، وهو ما يتماشى أيضًا مع نتائج تحليل PDOS. كما هو موضح في الشكل 3e والشكل التكميلي 38، جميع الكوبالتيا و V الأوربيتالز أظهرت انزياحًا أزرق مميزًا، مما يشير إلى أن PMS يمكن أن تسحب الإلكترونات من ذرات Co و O و V. نتائج توزيع شحنة مولكين (الشكل 3f) أشارت إلى أن ذرة H في نموذج PMS بعد الامتصاص يمكن أن تحصل على المزيد من الإلكترونات مقارنةً بتلك الموجودة في الذرات الأخرى في نموذج PMS الأصلي. كان طول رابطة O-H في PMS بعد الامتصاص أطول من طولها في الحالة النقية (الشكل التكميلي 39)، بينما ظل طول رابطة O-O في البيروكسو تقريبًا ثابتًا. النتائج المذكورة أعلاه تشير إلى أنكان من الأسهل كسر رابطة PMS مقارنة برابطة البيروكسو O-O خلال عملية التنشيط، بسبب تأثير رابطة الهيدروجين ونقل الشحنة، وهو ما تم تأكيده بشكل أكبر من خلال مخططات الطاقة الحرة لجيبس (الشكل التكميلي 40). للتحقيق في الدور المهم لرابطة الهيدروجين، تم بناء نموذج امتصاص PMS بدون رابطة هيدروجين وحسابه. وقد وُجد من الشكل التكميلي 41 أنكان نموذج امتصاص PMS مع الرابطة الهيدروجينية أعلى من نموذج امتصاص PMS بدون الرابطة الهيدروجينية (-1.15 eV)، مما يوضح أن نمط امتصاص PMS مع الرابطة الهيدروجينية كان أسهل في التكوين من منظور الديناميكا الحرارية. لتأكيد دور تفاعل الرابطة الهيدروجينية بشكل أكبر،مع أكبر كهرسلبية تم إدخاله في نظام التفاعل للتنافس مع الأكسجين منلتشكيل رابطة هيدروجينية. إدخالأدى إلى التثبيط الملحوظ لكفاءة ومعدل تحلل OFX (الشكل التوضيحي 42)، بسبب ذلكقد يفضل التفاعل معمن PMS لتشكيل روابط هيدروجينية قوية لتقليل تكوين الروابط الهيدروجينية بين PMS والعامل المساعد. وقد اعتُبر أن أيونات الكوبالت كمواقع امتصاص PMS لعبت أيضًا دورًا رئيسيًا خلال تفاعل شبيه فنتون. كما هو موضح في الشكل التكميلي 43، تم اختيار حمض الإيثيلين ديامين رباعي الأسيتيك والفوسفات لحماية مواقع الكوبالت لتقليل عملية امتصاص PMS بشكل أكبر. كما هو متوقع، انخفضت كل من أداء إزالة OFX واستهلاك PMS، مما يؤكد المساهمة الكبيرة لمواقع الكوبالت. بالإضافة إلى ذلك، قدرة تنشيط PMS لمواقع V بناءً علىتمت دراسة النموذج أيضًا (الشكل التوضيحي التكميلي 37e). كما هو موضح في الشكل التوضيحي التكميلي 44a، يمكن نقل 0.13 e من سطحإلى PMS. أظهرت الشكل التوضيحي الإضافي 44b أن منطقة تراكم الإلكترونات كانت تركز بشكل رئيسي على الروابط الهيدروجينية بدلاً من رابطة الأكسجين-الأكسجين في PMS، وهو ما كان مشابهًا لنموذج Co-O1. علاوة على ذلك، تم بناء نموذج امتصاص PMS بموقعين مزدوجين (الشكل التوضيحي الإضافي 45). كانت المدارات المضادة للرابطة التي تشكلت بين موقع الامتصاص وPMS مليئة بمزيد من الإلكترونات بسبب الانزياح الأزرق لـ-مركز النطاق، مما أدى إلى تدهور قدرة امتصاص PMS (الشكل التوضيحي التكميلي 46a، b). كما هو موضح في الشكل التوضيحي التكميلي 47a، كان نموذج امتصاص PMS في المواقع المزدوجة أقل من ذلك في المواقع الفردية لـ Co أو V. كان من المثير للاهتمام أن تفعيل PMS لم يعتمد فقط على نقل الإلكترون من مواقع المعادن Co فينظام PMS، بسبب ذلك لم يتغير عدد نقل الإلكترون (0.37 e) من المحفز إلى PMS (الشكل التكميلي 47b). انخفاضدلت على الأنواع النشطة المتكونة
الشكل 3 | تحديد مواقع التحفيز واستكشاف آلية الإثارة. أ نموذج الامتصاص الأمثل لـ PMS علىوطاقة الامتزاز المقابلة.فرق كثافة الإلكترون لنموذج الامتصاص الأمثل لـ PMS. ج فرق كثافة الإلكترون المتوسطة على المستوىحالة ملء إلكترونات مدارات d لعنصر الكوبالت وحالة الدوران قبل وبعد امتصاص PMS (e) الكثافة الإلكترونية لحالة O 2p و Co قبل وبعد امتصاص PMS.تغيير شحنة مولكين لنموذج PMS. طيف XPS لـ (g) Coقبل وبعد التفاعل. طيف EXAFS لحافة كوبالت K، (j) طيف حافة كوبالت K و (k) طيف حافة فانياديوم K XANES بالإضافة إلى (l) مخططات WT لـقبل وبعد التفاعل (أدخل: حافة الامتصاص والخط الأبيض لـ Co و V، على التوالي). كانوا أكثر عرضة للإزالة، مما كان أكثر ملاءمة لتحقيق تنشيط PMS.
طيف التحليل الطيفي للأشعة السينية (XPS)، طيف XANES وطيف EXAFS لـتم إجراء تفاعلات قبل وبعد مع حسابات DFT لتحديد مسار نقل الإلكترون أثناء التفاعل الحفزي. أظهرت الشكل 3 ج أن النسبة المئوية لـزاد منإلى بعد التفاعل الحفزي، مما يدل على أنقد يتم تحويله إلىبسبب انتقال الإلكترون من مواقع الكوبالت إلى. تم نقل بعض الإلكترونات من مواقع الكوبالت إلى PMS لتفاعل التنشيط، وتم التقاط إلكترونات أخرى وتخزينها فيخزان الإلكترونات. في الوقت نفسه، انتقلت طاقة الربط لـ Co-N (الشكل التكميلي 48a) و Co-O (الشكل 3h) جميعها إلى مستوى طاقة أعلى من ذلك الخاص بالكاتاليزور النقي، مما يؤكد عملية نقل الإلكترون من
المواقع المشتركة. بالإضافة إلى ذلك، أظهرت الشكل التكميلية 48b أن الانزياح الأحمر بمقدار 0.08 إلكترون فولت حدث لطاقة الربط لـمن المحفز النقي وبعد التفاعل، مما يشير إلى أنحيث يمكن لمستقبل الشحنة تخزين ونقل الإلكترونكما عرض الشكل 3 ح أيضًا أن طاقة الربط لـقبل وبعد التفاعل، انتقل من 529.65 إلكترون فولت إلى 529.73 إلكترون فولت، مما يوضح أن ذرات الأكسجين يمكن أن تعمل كمانحات للإلكترونات لنقل الإلكترونات إلى PMS. أشارت طيف EXAFS عند حافة كوبالت (الشكل 3i) إلى أن شدة القمة زادت بعد تفاعل تنشيط PMS، والذي يمكن أن يُعزى إلى تكوينالرابطة. إن تغير الخط الأبيض (الشكل 3j) للكوبيوم بعد التفاعل يشير إلى إعادة توزيع كثافة الإلكترون، حيث أن ضعف شدة الخط الأبيض يدل على فقدان الإلكترونات في مواقع الكوبيوم. في الوقت نفسه، فإن زيادة شدة الخط الأبيض للمعادن الفاناديوم بعد التفاعل (الشكل 3k) توضح أن الفاناديوم يسحب الإلكترونات مما يؤدي إلى زيادة كثافة الشحنة.، والتي تطابقت جيدًا مع تحليلات XPS. علاوة على ذلك، أظهرت مخططات WT لـ Co (الشكل التكميلي 49) و V (الشكل 31) أن رابطة Co-N/O ورابطة V-O قد انتقلت من و إلى و بعد التفاعل، على التوالي، مما يشير إلى أن كلارابطة وقدم الرابطة تباينًا ديناميكيًا خلال تفاعل تنشيط PMS.
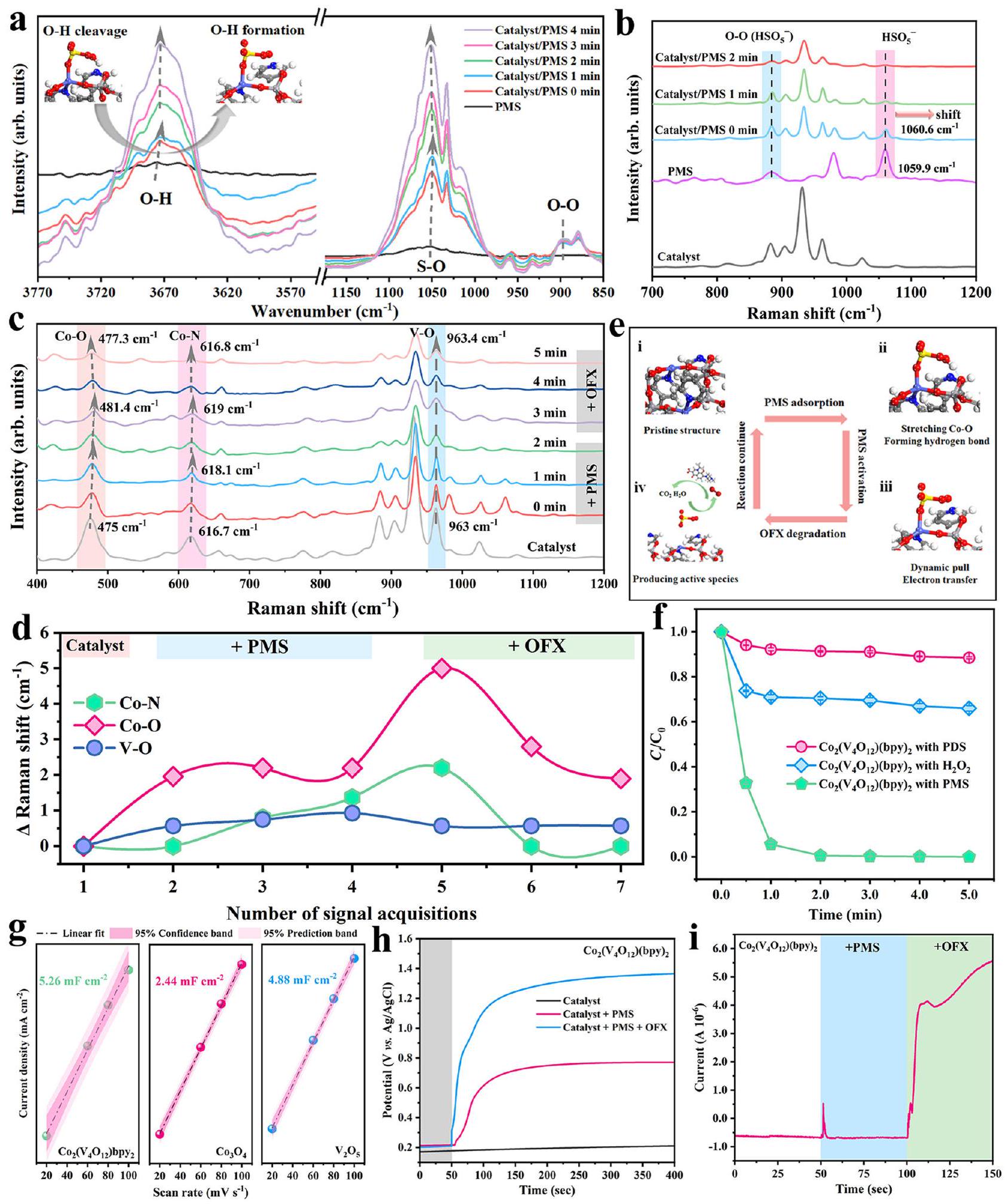
في الموقع FTIR تحتتم إجراء قياسات الطيف Raman في الموقع (الشكل التكميلي 50ج، د) لاستكشاف التفاعل بين الجزيئات بين PMS و (bpy) خلال تفعيل نظام إدارة المشاريعأظهرت طيف FTIR لمركب PMS الفردي (الشكل التكميلي 51) أن القمم عند و تم تعيينهم إلىرابطة و رابطة S-O، على التواليعندما تم إضافة PMS بتركيز معين إلى النظام، أظهر ذروة رابطة O-H (الشكل 4a) انزياحًا أزرق سريعًا منإلى. ثم، تم الاحتفاظ بقمة رابطة O-H عند بعد 1.0 دقيقة من التفاعل، والذي قد يُعزى إلى الاستهلاك السريع لـ PMS فينظام إدارة المشاريع. التغير الديناميكي لـيمكن أن يُعزى الذروة إلى: (ط)الرابطة من PMS أظهرت تفاعلًا قويًا مع سطح المحفز؛ و (ii)تم كسر الرابطة في نظام إدارة الممتلكات بسبب التفاعل، مما أدى إلى تشكيل الجديد والثابترابطة بين H من PMS و O النهائية منعلى سطح المحفز. كما أشارت تحليلات XPS (الشكل 3h) إلى أن محتوى مجموعة الهيدروكسيل على المحفز زاد بعد تفاعل شبيه فنتون، والذي قد يُعزى إلى التفاعل بين الأكسجين الطرفي من المحفز والهيدروجين الطرفي من PMS. في الوقت نفسه، يمكن أن يُعزى الانزياح الطفيف لقمة S-O (الشكل 4a) إلى تأثير الامتزاز لمركز المعدن تجاه PMS، والذي كان قابلاً للمقارنة مع حسابات DFT. والأهم من ذلك، تمتد خاصية O-O عند (الشكل 4أ) ظهر تحت ظروف الغلاف الجوي وغياب الأكسجين المذاب، مما يؤكد بشكل أكبر على توليد فينظام إدارة PMS بسبب تدخلعلىتم القضاء على الرابطة. بالإضافة إلى ذلك، تم اعتماد تقنية رامان في الموقع مع تحفيز الليزر عند 532 نانومتر لتوصيف التغير في PMS خلال التفاعل. الاهتزاز المميز لـانتقلت تدريجياً إلى أعداد موجية أعلى (الشكل 4ب)، مما يؤكد عملية نقل الإلكترون من المحفز إلى. أيضًا، القمة الجديدة عند حوالي اختفى بعد (bpy) تم إضافته إلى حل إدارة الممتلكات، مما يشير إلى أن المركبات ذات الدوران العالي (المحفز-PMS*) ذات القدرة على الأكسدة القوية لم تتشكل. علاوة على ذلك، تم إجراء تجارب رامان في الموقع لتسهيل ملاحظة التغير الديناميكي لمراكز المعادن، حيث كانت قمم رامان عند و يمكن أن يُنسب إلىوترددات تمدد V-O (الشكل 4c)، على التوالي. بالمقارنة مع الهيكل الديناميكي لـ و الرابطة، يمكن تجاهل التغير الديناميكي الطفيف في رابطة V-O (الشكل 4d) بسبب الخصائص الهيكلية المستقرة لـعنقود، يتطابق بشكل جيد مع صورة HR-TEM. كما هو موضح في الشكل التكميلية 52، الشبكة البلورية لـالمستخدمكان شبه مطابق لذلك في الجديد. على العكس، كان هناك اهتزاز ملحوظ في التمدد لـ و تشير الروابط إلى الألفة المتبادلة بين مواقع الكوبالت وPMS لأن المراكز المعدنية ستؤدي إلى استجابة هيكلية لـ امتصاص PMS. من المثير للاهتمام أن الاستنتاجات المذكورة تتوافق جيدًا مع نتائج تحليل حسابات DFT. الزاوية المضمنة لـتغير منإلىعندما تم امتصاص PMS على مواقع الكوبالت، بينما كانت الزاوية المضمنة لـفقط انتقل منإلىبعد امتصاص PMS (الشكل التكميلي 53). في الختام، بجانب الإلكترونات المنقولة من مواقع Co إلى PMS، أشارت النتائج الطيفية في الموقع المجمعة مع حسابات DFT إلى أن O-H في PMS تفضل أن تنكسر بسبب جذب الروابط الهيدروجينية وانتقال الإلكترونات بينو متلازمة ما قبل الحيض، مما يعني تشكيلقد ينتج المزيد و لتدهور OFX (الشكل 4e).
استنادًا إلى المناقشات أعلاه، تم اعتماد ثلاثة أنواع من POM-MOFs وثلاثة أنواع من MOFs القائمة على الكوبالت للتحقيق في عمومية الآلية المقترحة (الشكل التكميلي 54). أشارت نتائج تجارب التبريد وتجارب المجس الكيميائي (الأشكال التكملية 55-57) إلى أنساهمت بشكل كبير في تحلل OFX في نظام POM-MOFs/PMS، والذي يمكن أن يُعزى إلى تأثير نقل الإلكترونات الاتجاهي للبوليفانادات. بينما كانت مواقع الكوبالت تُعتبر مانحة للإلكترونات في MOFs المعتمدة على الكوبالت لتفعيل PMS، مما أدى إلى مشاركة الجذور وغير الجذور في التفاعل الأكسدي لإزالة OFX. بالإضافة إلى ذلك، تم تفعيل PDS وبيروكسيد الهيدروجين (تمت أيضًا إجراء ( ) لاستكشاف الدور الهام لتفاعلات المضيف-الضيف على تفاعلات فنتون الشبيهة الأخرى. كما هو متوقع، أشارت النتائج (الشكل 4f) إلى أن النشاط التحفيزي لـكان التنشيط أفضل من تنشيط PDS، حيث كانت معدل تحلل المحفزكانت التفعيل أعلى بحوالي 3.58 مرة من تفعيل PDS (الشكل التوضيحي 58a).نموذج الامتزاز (الشكل التكميلي 58b) ونموذج امتزاز PDS (الشكل التكميلي 58c) أظهروا أن ذرة الهيدروجين الطرفية منيمكن أن تولد أيضًا تفاعلات مع ذرة الأكسجين الطرفية الكتلة، بينما كان بإمكان امتصاص PDS الاعتماد فقط على مواقع الكوبالت. أظهرت الطاقة المحسوبة للامتصاص أن نموذج امتصاص PMS عرض أعلى طاقة امتصاص ( )، تليها نموذج الامتزاز ) ونموذج امتصاص PDS ( ). لذلك، فإن نقل الإلكترون مباشرة من يمكن أن تحقق السرعةتفعيل لإنتاج أنواع نشطة لتفكيك OFX. على النقيض من ذلك، كانت عملية تفعيل PDS تعتمد فقط على نقل الإلكترون من مواقع Co، والتي أظهرت أداءً تحفيزيًا ضعيفًا في تفكيك OFX. كما أكدت التوصيفات الكهروكيميائية أننظام PMS يمتلك قدرة أفضل على نقل الإلكترونات من تلك الخاصة بـنظام و (bpy) نظام PMS (الشكل التوضيحي التكميلي 58d، e). بالإضافة إلى ذلك، تم إجراء تجارب كيميائية كهربائية لاختبار المساحة السطحية الحقيقية لمختلف المحفزات. أظهر الشكل التوضيحي التكميلي 59 أن منحنيات الفولتمترية الدورية (CV) لـ و بعد 20 دورة تنشيط كيميائي كهربائي، والتي يمكن أن تحسب السعة الثنائية الطبقةمن و استنادًا إلى الكثافة الحالية ومعدلات المسح المختلفةالشكل 4 ج أظهر أنمنكان أعلى من تلك الخاصة بـ و . الأعلىتشير القيمة إلى مساحة السطح النشطة الكهروكيميائية الأقوىجهد الدائرة المفتوحة لـارتفعت عندما تم إضافة PMS إلى الإلكتروليت (الشكل 4 ح والشكل التكميلي 60 أ، ب)، وارتفع الجهد تدريجياً مع إضافة PMS و OFX. الأمبيرومتريةأظهرت المنحنيات (الشكل 4i والشكل التكميلي 60c، d) أن إضافة PMS و OFX يمكن أن تعزز شدة التيار، مما يشير إلى أن OFX كملوث غني بالإلكترونات يمكن أن يعزز عملية نقل الإلكترونات بين الجزيئات المضيفة والضيوف. كانت هذه النتيجة متوافقة أيضًا مع تجارب الفولتمترية المسحية الخطية (LSV) واستهلاك PMS. أظهرت منحنيات LSV (الشكل التكميلي 61a) أن التيار كان صغيرًا جدًا وقريبًا من الإهمال في الحالة الفردية.النظام. مقارنةً بمنحنيات LSV فينظام إدارة الممتلكات، زاد بشكل كبير بعد إضافة OFX، مما يشير إلى تدهور. بالإضافة إلى ذلك، نتائج تجارب استهلاك PMS
الشكل 4 | التحقيق في عملية تنشيط PMS والاختبارات الكهروكيميائية في الموقع. أ طيف FTIR في الموقع لـنظام إدارة الأداء تحتجوطيف رامان في الموقع لـ، PMS، وتفاعلها. ج تحول القمم المختلفة في طيف رامان في الموقع. د تحول القمم المميزة لطيف رامان في الموقع خلال أوقات التفاعل المختلفة. هـ تحليلات خطوة بخطوة لآلية تنشيط PMS خلال تفاعل شبيه فنتون. و تأثيرات منبهات جزيئات الضيف المختلفة على كفاءات تحلل OFX.
الظروف التجريبية: [المحفز]، [PDS] = [ ] = 3.5 مللي مول , [OFX] = (في ف). قياسات ECSA بناءً على نتائج منحنيات CV لـ و . منحنيات الجهد المفتوح الدائرة و (i) منحنى التيار الزمني الأمبيرومتري في أنظمة مختلفة باستخدامكقطب عمل. تمثل أشرطة الخطأ في الأشكال الانحرافات المعيارية من الاختبارات الثلاثية. (الشكل التوضيحي 61ب) أشار إلى أن إدخال OFX يمكن أن يسرع من تحلل PMS.
نقاش
بشكل عام،تم تصنيع MOF يحتوي على بوليفانادات ليتم استخدامه كمحفز لـ PMS للمرة الأولى لتحقيق إزالة فعالة للملوثات الدقيقة ذات HOMO عالي عبر مسار غير جذري. التأثير التآزري بين مواقع Co وأدى إلى زيادة معدل التحفيز لـنحو OFX، حوالي 25.16 و 39.37 مرة أعلى من تلك الخاصة بـ و آلية الأكسدة التي تهيمن عليها المسار غير الجذري منحت (bpy) تحمل بيئي جيد، حيث أن الأيونات المختلفة المتواجدة معًا ودرجة الحموضة (3.12-8.04) لم تؤثر على كفاءات تنشيط PMS. بالإضافة إلى ذلك،أظهر نظام PMS معالجة مرضية على المدى الطويل لمدة 40.0 ساعة تجاه مياه الصرف الصحي الفعلية. أظهرت أقطار مناطق التثبيط في التجربة ضد E. coli قدرة جيدة على إزالة السموم.كمحفز لـ PMS. بشكل ملحوظ، تم إجراء تجارب مختلفة، وتوصيفات، وحسابات DFT للتحقق من التفاعلات المتعددة بين المضيف والضيف على واجهة المحفز المعقدة. أكدت حسابات DFT المدمجة مع XAS أن مواقع Co لعبت دورًا رئيسيًا في احتجاز PMS. ومع ذلك، فإن الواجهةمع خصائص إسفنجة الإلكترون والصلابة أدت إلى توليد تفاعل قوي بين الأكسجين الطرفي منوالمحطة H من PMS، التي تم تأكيدها بواسطة حسابات DFT، وتقنيات FTIR وRaman في الموقع. قدمت روابط الهيدروجين قناة نقل إلكتروني اتجاهية للمحفز، وعملية السحب الديناميكية عززت توليد المفتاحالمتوسط، الذي كان مفيدًا للإنتاجلتدهور الملوثات الدقيقة. علاوة على ذلك، كانت هذه الآلية للتفعيل مناسبة أيضًا لأنظمة POM-MOFs/PMS الأخرى. قدم هذا العمل حالة جيدة لتصنيع محفز/مفعل فعال يحتوي على بوليوكسوميتالات لغرض تنقية المياه. في المستقبل، سيتم بذل المزيد من الجهود لتوضيح الأداءات الحفازة والآليات لمزيد من MOFs مع مجموعات POM مختلفة، واستكشاف إمكانية تطبيقها في معالجة البيئة المائية على نطاق واسع.
طرق
المواد الكيميائية
تم سرد جميع المواد الكيميائية (الملاحظة التكميلية 1) التي تم استخدامها في تخليق المحفزات (الملاحظة التكميلية 2) وتقييم التجارب في هذا العمل في المعلومات التكميلية.
تركيب
المصفوفة لـ bpy وتم نقل 13.0 مل من الماء المقطر إلى أوتوكلاف مبطن بتفلون سعة 25.0 مل، والذي تم تسخينه عندلمدة 72.0 ساعة. البني الأرجوانيتم جمع البلورات بعد أن تم تبريد الأوتوكلاف إلى درجة حرارة الغرفة، والتي تم تجفيفها في فرن عندلمدة 12.0 ساعة بعد غسلها بالماء المقطر والإيثانولتم سرد تقنيات التوصيف التفصيلية في الملاحظات التكميلية 3 و 11.
اختبار النشاط التحفيزي
تم تقييم أداء المحفزات المختلفة لتفعيل PMS وتحلل الملوثات بواسطة OFX كملوث نموذج مقاوم. تم إجراء جميع التجارب في مفاعل سعة 60.0 مل مع تحريك ثابت بسرعة 200.0 دورة في الدقيقة تحت درجة حرارة الغرفة، حيثتم إضافة المحفز و0.1 مللي مول من PMS إلى 50.0 مل من المحلول المائي الذي يحتوي على الملوث بتركيز ابتدائي منتم توفير طرق تركيزات المتبقيات في الملاحظة التكميلية 4. تم ضبط قيم pH التي تتراوح من 2.0 إلى 11.0 للمحلول المستهدف باستخدامأو محاليل NaOH بتركيزات مناسبة. تم إجراء جميع التجارب ثلاث مرات على الأقل.
تفاصيل حساب DFT
في هذه الدراسة، تم إجراء حسابات من المبادئ الأولى في إطار نظرية الكثافة (DFT) باستخدام برنامج ماتييرالز ستوديو 2020 (وحدة كاستيب).تم وصف طاقة التبادل-الارتباط بواسطة شكل بيردو-بورك-إرنزرهوف (PBE) من تقريب التدرج العام (GGA) لوظيفة طاقة التبادل-الارتباط.تم تحديد حد الطاقة عند 500 إلكترون فولت وتم تعيين تباعد النقاط ليكون أصغر من-نقطة كانتحالة الدوران وDFTتم اعتبارها بسبب وجود الكوبالت والفاناديوم، حيث تم تحديد قيمة U للكوبالت والفاناديوم على أنها 3.2 و 2.5، على التوالي.كانت معايير التقارب لتحسين هندسة البلورات مثل تحمل الطاقة، وتحمل القوة القصوى، وتحمل الإجهاد الأقصى، وتحمل الإزاحة القصوى أقل منذرة و تم تقديم عناصر الحساب المحددة في الملاحظة التكميلية 10.
ملخص التقرير
معلومات إضافية حول تصميم البحث متاحة في ملخص تقارير مجموعة نيتشر المرتبط بهذه المقالة.
. et al. A green edge-hosted zinc single-site heterogeneous catalyst for superior Fenton-like activity. Proc. Natl Acad. Sci. 120, e2221228120 (2023).
Hodges, B. C., Cates, E. L. & Kim, J. H. Challenges and prospects of advanced oxidation water treatment processes using catalytic nanomaterials. Nat. Nanotechnol. 13, 642-650 (2018).
Ren, W. et al. Origins of electron-transfer regime in persulfatebased nonradical oxidation processes. Environ. Sci. Technol. 56, 78-97 (2022).
Anipsitakis, G. P. & Dionysiou, D. D. Degradation of organic contaminants in water with sulfate radicals generated by the conjunction of peroxymonosulfate with cobalt. Environ. Sci. Technol. 37, 4790-4797 (2003).
Guo, J. et al. Fenton-like activity and pathway modulation via singleatom sites and pollutants comediates the electron transfer process. Proc. Natl Acad. Sci. 121, e2313387121 (2024).
Zhang, L. S. et al. Carbon nitride supported high-loading Fe singleatom catalyst for activation of peroxymonosulfate to generate with 100 % selectivity. Angew. Chem. Int. Ed. Engl. 60, 21751-21755 (2021).
Wu, X. L. et al. Directional and ultrafast charge transfer in oxygen-vacancy-rich ZnO@single-atom cobalt core-shell junction for photo-Fenton-like reaction. Angew. Chem. Int. Ed. Engl. 62, e202305639 (2023).
Yao, Y. et al. Rational regulation of coordination for highefficiency generation of toward nearly selective degradation of organic pollutants. Environ. Sci. Technol. 56, 8833-8843 (2022).
Howarth, A. J. et al. Chemical, thermal and mechanical stabilities of metal-organic frameworks. Nat. Rev. Mater. 1, 15018 (2016).
Islamoglu, T. et al. Postsynthetic tuning of metal-organic frameworks for targeted applications. Acc. Chem. Res. 50, 805-813 (2017).
Horn, M. R. et al. Polyoxometalates (POMs): from electroactive clusters to energy materials. Energy Environ. Sci. 14, 1652-1700 (2021).
Liu, Y. et al. Fabricating polyoxometalates-stabilized single-atom site catalysts in confined space with enhanced activity for alkynes diboration. Nat. Commun. 12, 4205 (2021).
Zhang, H. et al. Three mechanisms in one material: uranium capture by a polyoxometalate-organic framework through combined complexation, chemical reduction, and photocatalytic reduction. Angew. Chem. Int. Ed. Engl. 58, 16110-16114 (2019).
Du, D.-Y. et al. Recent advances in porous polyoxometalate-based metal-organic framework materials. Chem. Soc. Rev. 43, 4615-4632 (2014).
Wang, Y.-R. et al. Oriented electron transmission in polyoxometalate-metalloporphyrin organic framework for highly selective electroreduction of . Nat. Commun. 9, 4466 (2018).
Kong, X. J., Lin, Z., Zhang, Z. M., Zhang, T. & Lin, W. Hierarchical integration of photosensitizing metal-organic frameworks and nickel-containing polyoxometalates for efficient visible-light-driven hydrogen evolution. Angew. Chem. Int. Ed. Engl. 55, 6411-6416 (2016).
Dong, Y. et al. Regulating crystallization and lead leakage of perovskite solar cell via novel polyoxometalate-based metal-organic framework. Small 19, e2301824 (2023).
Li, Z.-H., Yin, C., Wang, R.-J., Wang, P. & Guo, H.-Y. Synthesis and crystal structure of an inorganic-organic hybrid compound bpy) (bpy = 4, 4′-bipyridine). Acta Phys. Chim. Sin. 19, 1133-1137 (2003).
Chen, Z. et al. Single-atom Mo-Co catalyst with low biotoxicity for sustainable degradation of high-ionization-potential organic pollutants. Proc. Natl Acad. Sci. 120, e2305933120 (2023).
Wang, B. et al. A site distance effect induced by reactant molecule matchup in single-atom catalysts for Fenton-like reactions. Angew. Chem. Int. Ed. Engl. 134, e202207268 (2022).
Guo, Z. Y. et al. Mn-O covalency governs the intrinsic activity of CoMn spinel oxides for boosted peroxymonosulfate activation. Angew. Chem. Int. Ed. Engl. 60, 274-280 (2021).
Liu, F. et al. Covalent organic frameworks for direct photosynthesis of hydrogen peroxide from water, air and sunlight. Nat. Commun. 14, 4344 (2023).
Ren, W. et al. Activation of peroxydisulfate on carbon nanotubes: electron-transfer mechanism. Environ. Sci. Technol. 53, 14595-14603 (2019).
Li, M., You, S., Duan, X. & Liu, Y. Selective formation of reactive oxygen species in peroxymonosulfate activation by metal-organic framework-derived membranes: a defect engineering-dependent study. Appl. Catal. B 312, 121419 (2022).
Li, Y.-H. et al. Nearly zero peroxydisulfate consumption for persistent aqueous organic pollutants degradation via nonradical processes supported by in-situ sulfate radical regeneration in defective MIL-88B (Fe). Appl. Catal. B 331, 122699 (2023).
Yao, Y. et al. Rational regulation of Co-N-C coordination for highefficiency generation of toward nearly selective degradation of organic pollutants. Environ. Sci. Technol. 56, 8833-8843 (2022).
Mi, X. et al. Almost peroxymonosulfate conversion to singlet oxygen on single-atom sites. Angew. Chem. Int. Ed. Engl. 60, 4588-4593 (2021).
Wei, Z. et al. Artificial photosynthesis of through reversible photoredox transformation between catechol and o-benzoquinone on polydopamine-coated CdS. ACS Catal. 12, 11436-11443 (2022).
Yang, M. et al. Unveiling the origins of selective oxidation in singleatom catalysis via intensified radical and nonradical pathways. Environ. Sci. Technol. 56, 11635-11645 (2022).
Wang, L. et al. A polymer tethering strategy to achieve high metal loading on catalysts for Fenton reactions. Nat. Commun. 14, 7841 (2023).
Zhang, Y.-J. et al. Simultaneous nanocatalytic surface activation of pollutants and oxidants for highly efficient water decontamination. Nat. Commun. 13, 3005 (2022).
Yao, Y. et al. Synthesis of “sea urchin”-like carbon nanotubes/porous carbon superstructures derived from waste biomass for treatment of various contaminants. Appl. Catal. B 219, 563-571 (2017).
Yu, H. et al. Singlet oxygen synergistic surface-adsorbed hydroxyl radicals for phenol degradation in CoP catalytic photo-Fenton. Chin. J. Catal. 43, 2678-2689 (2022).
Ren, W. et al. The intrinsic nature of persulfate activation and N-doping in carbocatalysis. Environ. Sci. Technol. 54, 6438-6447 (2020).
Shao, P. et al. Revisiting the graphitized nanodiamond-mediated activation of peroxymonosulfate: singlet oxygenation versus electron transfer. Environ. Sci. Technol. 55, 16078-16087 (2021).
Yan, Y. et al. Merits and limitations of radical vs. nonradical pathways in persulfate-based advanced oxidation processes. Environ. Sci. Technol. 57, 12153-12179 (2023).
Fang, Q. et al. Generation and identification of in catalysts/ peroxymonosulfate systems for water purification. Water Res. 245, 120614 (2023).
Stuyver, T., Porft, F., Geerlings, P. & Shaik, S. How do local reactivity descriptors shape the potential energy surface associated with chemical reactions? The valence bond delocalization perspective. J. Am. Chem. Soc. 142, 10102-10113 (2020).
Li, Y.-H. et al. Seignette salt induced defects in Zr-MOFs for boosted adsorption: universal strategy and mechanism insight. Chem. Eng. J. 442, 136276 (2022).
Zhou, X. et al. Identification of Fenton-like active Cu sites by heteroatom modulation of electronic density. Proc. Natl Acad. Sci. 119, e2119492119 (2022).
Wang, Z. et al. Elevating the d-band center of six-coordinated octahedrons in through Fe-Incorporated topochemical deintercalation. Adv. Energy Mater. 11, 2003023 (2020).
Sun, C., Zhao, P., Yang, Y., Li, Z. & Sheng, W. Lattice oxygen-induced d-band shifting for enhanced hydrogen oxidation reaction on nickel. ACS Catal. 12, 11830-11837 (2022).
Ran, M. et al. Selective production of CO from organic pollutants by coupling piezocatalysis and advanced oxidation processes. Angew. Chem. Int. Ed. Engl. 62, e202303728 (2023).
Zhang, N. et al. Lewis acid Fe-V pairs promote nitrate electroreduction to ammonia. Adv. Funct. Mater. 33, 2211537 (2023).
Zhang, D. et al. Dynamic active-site induced by host-guest interactions boost the Fenton-like reaction for organic wastewater treatment. Nat. Commun. 14, 3538 (2023).
Lin, W. & Frei, H. Photochemical and FT-IR probing of the active site of hydrogen peroxide in Ti silicalite sieve. J. Am. Chem. Soc. 124, 9292-9298 (2002).
Qin, C. et al. Dual donor-acceptor covalent organic frameworks for hydrogen peroxide photosynthesis. Nat. Commun. 14, 5238 (2023).
Wang, X. et al. Understanding the deposition and reaction mechanism of ammonium bisulfate on a vanadia SCR catalyst: a combined DFT and experimental study. Appl. Catal. B 260, 118168 (2020).
Hu, J. et al. Oxygen vacant in situ embedded on carbon spheres: cooperatively tuning electron transfer for boosted peroxymonosulfate activation. J. Mater. Chem. A 9, 16489-16499 (2021).
López, X., Carbó, J. J., Bo, C. & Poblet, J. M. Structure, properties and reactivity of polyoxometalates: a theoretical perspective. Chem. Soc. Rev. 41, 7537-7571 (2012).
McCrory, C. C., Jung, S., Peters, J. C. & Jaramillo, T. F. Benchmarking heterogeneous electrocatalysts for the oxygen evolution reaction. J. Am. Chem. Soc. 135, 16977-16987 (2013).
Tong, Y. et al. Vibronic superexchange in double perovskite electrocatalyst for efficient electrocatalytic oxygen evolution. J. Am. Chem. Soc. 140, 11165-11169 (2018).
Wang, F. et al. High-efficient peroxymonosulfate activation for rapid atrazine degradation by derived from MIL-88A(Fe). J. Hazard. Mater. 440, 129723 (2022).
Monteiro, F. F. et al. On the mechanical, electronic, and optical properties of the boron nitride analog for the recently synthesized biphenylene network: a DFT study. J. Mol. Model. 29, 215 (2023).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758-1775 (1999).
Jain, A. et al. Formation enthalpies by mixing GGA and GGA+U calculations. Phys. Rev. B 84, 045115 (2011).
شكر وتقدير
تم دعم هذا العمل من قبل المؤسسة الوطنية للعلوم الطبيعية في الصين (22176012، 52370025، 52100069، 22325602، 12175263، 22176060)، وصندوق مشروع تنمية جامعة بكين للهندسة المدنية والمعمارية (X23034) وبرنامج العلوم والتكنولوجيا في شنتشن (JCYJ2O220531093205013)، وبرنامج قادة البحث الأكاديمي/التقني في شنغهاي (23XD1421000).
مساهمات المؤلفين
صمم Y.-H.L. و C.-C.W. التجارب. قام M.-Y.L. و Y.-H.L. و L.M. و X.-J.L. بتنفيذ التجارب. طور Y.-H.L. و M.-Y.L. و S.G. و J.C. و Y.M. و H.J. و C.-C.W. نموذج التفاعل وحللوه. أشرف C.-C.W. و H.J. و M.X. على الدراسة والتجارب. قام Y.-H.L. و C.-C.W. و H.J. و M.X. بإجراء تحليلات البيانات وإعداد هذه المخطوطة. قدم C.-C.W. و M.X. اقتراحات بناءة لمراجعة المخطوطة.
يجب توجيه المراسلات والطلبات للحصول على المواد إلى تشونغ-تشين وانغ، هاودونغ جي أو مينغيانغ شينغ.
معلومات مراجعة الأقران تشكر مجلة Nature Communications بوفان زانغ والمراجعين الآخرين المجهولين على مساهمتهم في مراجعة هذا العمل. يتوفر ملف مراجعة الأقران.
مختبر بكين الرئيسي للمواد الوظيفية لهياكل البناء وإعادة تأهيل البيئة، كلية الهندسة البيئية والطاقة، جامعة بكين للهندسة المدنية والمعمارية، بكين، جمهورية الصين الشعبية.مختبر أبحاث الكفاءة البيئية والموارد، كلية البيئة والطاقة، جامعة بكين، مدرسة الدراسات العليا في شنتشن، شنتشن، قوانغدونغ، جمهورية الصين الشعبية.المركز الوطني للبحوث الهندسية لمعالجة مياه الصرف الصناعي واستعادة الموارد، كلية الموارد والهندسة البيئية، جامعة شرق الصين للعلوم والتكنولوجيا، شنغهاي، جمهورية الصين الشعبية.المختبر الرئيسي لتأثيرات المواد النانوية على الصحة والسلامة، معهد الفيزياء عالية الطاقة، الأكاديمية الصينية للعلوم، بكين، جمهورية الصين الشعبية.ساهم هؤلاء المؤلفون بالتساوي: مينغ يان لان، يو هانغ لي.البريد الإلكتروني:wangchongchen@bucea.edu.cn;jihaodong@pku.edu.cn; mingyangxing@ecust.edu.cn
Multi-channel electron transfer induced by polyvanadate in metal-organic framework for boosted peroxymonosulfate activation
Received: 11 March 2024
Accepted: 9 August 2024
Published online: 22 August 2024
Ming-Yan Lan , Yu-Hang Li , Chong-Chen Wang , Xin-Jie Li , Jiazhen Cao , Linghui Meng , Shuai Gao , Yuhui Ma , Haodong Ji Mingyang Xing
Catalytic peroxymonosulfate (PMS) activation processes don’t solely rely on electron transfer from dominant metal centers due to the complicated composition and interface environment of catalysts. Herein the synthesis of a cobalt based metal-organic framework containing polyvanadate [ cluster, bpy -bipyridine , is presented. The catalyst demonstrates superior degradation activity toward various micropollutants, with higher highest occupied molecular orbital (HOMO), via nonradical attack. The X-ray absorption spectroscopy and density functional theory (DFT) calculations demonstrate that Co sites act as both PMS trapper and electron donor. In situ spectral characterizations and DFT calculations reveal that the terminal oxygen atoms in the electron sponge could interact with the terminal hydrogen atoms in PMS to form hydrogen bonds, promoting the generation of intermediate via both dynamic pull and direct electron transfer process. Further, exhibits long-term water purification ability, up to 40 h , towards actual wastewater discharged from an ofloxacin production factory. This work not only presents an efficient catalyst with an electron sponge for water environmental remediation via nonradical pathway, but also provides fundamental insights into the Fenton-like reaction mechanism.
Fenton-like reactions triggered by peroxydisulfate (PDS) or peroxymonosulfate (PMS) activation were verified to be promising technology for water purification, profiting from the production of radicals and/or non-radicals via interacting between persulfates and active centers . Up to now, many efforts were devoted to directionally adjusting the oxidation mechanism or pursuing higher electron transfer efficiency during
the Fenton-like reactions . However, beyond the reciprocating from the transition metal centers, the PDS/PMS activation processes might be induced by electron transfer by other components, which required fine regulation of catalysts from the molecule or even atom level.
Metal-organic frameworks (MOFs), as versatile inorganic-organic hybrid materials, could be finely tuned from molecule level . Besides
functional organic linkers, some polyoxometalate (POM) clusters could be used as inorganic linkers to construct special MOFs with unique properties . Recently, a few MOFs containing POM were used in the fields of reduction reaction , hydrogen evolution reaction and solar cell , benefiting from that POM as electron reservoir or sponge could presumably capture, reserve and transfer electrons to create various sorption sites and catalytic centers. However, PMS molecules owned more complicated chemical structure than those of and . Therefore, it was expected that the catalytic environment near the interface of POM-based MOF was complicated due to the presence of unsaturated coordination oxygens, leading to that the PMS activation process might rely on other interactions like multielectron channel and inter-molecular forces rather than solely on electron transfer of transition metal centers. Therefore, it was interesting to explore the catalytic PMS activation mechanism for the subsequent direct regulation of catalyst structure and the investigation of structure-activity relationship.
In this work, a polyoxovanadate-based was fabricated to achieve PMS activation for organic pollutants degradation, which could facilitate to investigate the relationship between the complex surface coordination environment of catalyst and Fenton-like reaction. This study affirmed that charge transfer between PMS and metal sites in metal-based catalysts was not the only activation pathway. The density functional theory (DFT) calculations exhibited that it was difficult to form the high-spin complexes between weak-field PMS and Co centers as the optimal sorption sites, implying that source of charge transfer was not just the metal center. In situ Raman and Fourier transform infrared (FTIR) illustrated the dynamic variation of bond from PMS and metal- bonds ( and ) as well as electron transfer between and PMS during the Fenton-like reaction, which promoted the formation of to further produce singlet oxygen ( ) and sulfate radicals ( ) for ofloxacin ( OFX ) oxidation. Nonradical dominated oxidation mechanism helped (bpy) to exhibit superior long-term treatment ability toward actual wastewater and satisfactory detoxification ability feedback from biological experiments. This work opened a door for designing and synthesizing catalysts with electron sponge like POM cluster to accomplish nonradical pathways for water purification.
Results
Characterizations of
The single crystal structure (CCDC 2323958) was analyzed to affirm the identical structure of as-prepared to the previous ones , in which is five-coordinated by two nitrogen atoms from bpy and three oxygen atoms from three different clusters. It was observed that six terminal oxygen atoms in are coordinated to six different atoms via bonds, and the other two terminal oxygen atoms are uncoordinated (Supplementary Fig. 1 and Supplementary Table 1). The powder X-ray diffraction (PXRD) patterns (Fig. 1a) of as-prepared matched well with the simulated ones from CCDC 2323958 and 152985, implying high purity of the catalyst used in the successive tests and determinations. The optical microscope image (Fig. 1b) showed that displayed yellow clump crystal with the micron scale, which was affirmed by scanning electron microscopy (SEM) image (Supplementary Fig. 2). High-resolution transmission electron microscopy (HR-TEM) affirmed the exposed facet (332) of of (bpy) (Fig. 1c and Supplementary Fig. 3). The corresponding elemental mapping (Fig. 1d) displayed uniform distribution of characteristic elements like , and O .
The chemical state and coordination environment of Co and V were investigated using X-ray adsorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectra. The Co (Fig. 1e) and V (Fig. 1f) element K-edge XANES combined with k-space
and fitted data (Supplementary Fig. 4) illustrated that the valence states of Co and V were near +2 and +5 , respectively. The EXAFS spectra (Fig. 1g) indicated that Co atoms from were coordinated to O and N atoms with the bond length of (Peak I). In addition, no Co clusters or crystalline particles was presented in structure due to that no obvious peak of the bond at were observed , which matched well with the results of crystal structure analyses. The EXAFS spectra of V element (Fig. 1h) demonstrated that V-O bond (Peak II) with the length of was different from those of and , illustrating that the interactions between V and O atoms in were stronger than those in and . The wavelet transform (WT) of (bpy) was performed to further clarify the above-mentioned peaks. As illustrated in Fig. 1i and Supplementary Fig. 5, the peaks at and of sample could be ascribed to and V-O bonds, respectively. The WT plots of Co foil, V foil and the single crystal structure determination results revealed that no and bond could be observed in .
Catalytic activity and involved oxidation mechanism
OFX was selected as target micropollutant to investigate the PMS activation performance of . The PMS activation abilities of different systems were investigated without adjusting pH (Fig. 2a and Supplementary Figs. 6, 7). The (bpy) displayed the best PMS activation for OFX degradation with the biggest reaction apparent rate constant ( ) of (Supplementary Fig. 8a), which was further affirmed by PMS consumption experiments (Supplementary Fig. 8b). Additionally, ca. of total organic carbon (TOC) removal could be achieved within 5.0 min in PMS system (Supplementary Fig. 8c), verifying the better mineralizing capacity of than and . The (bpy) could also accomplish ultra-high elimination efficiencies toward other organic micropollutants like tetracycline (TC), bisphenol A (BPA), 3,4-dichlorophenol (3,4-DCP), chloroquine phosphate (CQ), and sulfamethoxazole (SMX) (Fig. 2b and Supplementary Fig. 8d), in which the corresponding -value ( ) surpassed almost all reported catalysts for PMS activation (Fig. 2c and Supplementary Table 3). It was affirmed that displayed good reusability (Fig. 2d). Only slight decline of catalytic activity could be observed after five recycles’ operation, possibly due to that some active sites are occupied by the formed intermediates (Supplementary Fig. 9) . The leaching Co ( ) and V ( ) were far below the environmental quality standards for surface water (GB 3838-2002), indicating the great water stability of catalyst. Also, the observations of PXRD patterns, FTIR spectrum, SEM image and HR-TEM image affirmed the decent stability of (bpy) (Supplementary Fig. 10a-d).
To identify the primary reactive oxygen species (ROSs) during PMS activation, chemical quenching experiments were conducted (Supplementary Note 5), in which methanol (MeOH), tert-butanol (TBA), benzoquinone (BQ), and furfuryl alcohol (FFA) were selected as quenchers of and quencher, respectively . The introduction of MeOH and FFA into PMS system could inhibit the OFX degradation efficiencies from 100% to 68.2% and , respectively (Supplementary Fig. 11a). While negligible influence on catalytic OFX degradation efficiencies could be observed when TBA and BQ were added. It was observed that PMS could be directly consumed by FFA (Supplementary Fig. 11b), comparable to the previous studies . Therefore, L -histidine, as another quencher, was adopted to quench to further identify its contribution. The results (Supplementary Fig. 11c, d) indicated that both the OFX degradation efficiency and rate were seriously inhibited. Importantly, the OFX catalytic rate in was faster than that in deionized water (Supplementary Fig. 11c, d), further confirming the significant contribution of in (bpy) PMS system. The above results demonstrated that
Fig. 1 | Chemical and structural characterizations of . a PXRD patterns of as-fabricated catalyst. Optical microscope image and (c) HR-TEM image of . d Elemental mapping images of characteristic elements from . XANES spectra of the (e) Co and (f) V
K-edge in (insert: the absorption edge of Co and V , respectively). EXAFS spectra of the (g) Co and (h) V K-edge in . i WT plots of the Co and V K-edge EXAFS for the Co foil, V foil and Co2 . -a and might play primary contribution during the degradation process. Also, electron paramagnetic resonance (EPR) analyses results (Supplementary Fig. 12) showed that strong DMPO-SO4 and TEMP- signals could be observed, implying and were yielded in PMS system. Besides, quantitative analyses via EPR technology (Supplementary Fig. 12b) were performed to avoid overestimating its contribution, revealing that the as-produced in (bpy)/PMS system could survive longer in than in . And the intensity of TEMP- signal decrease evidently when OFX was added in PMS system, confirming the significant role of during the OFX degradation process. atmosphere, atmosphere (Supplementary Fig. 13) and aniline quenching experiments (Supplementary Fig. 11a) verified that the generation of might be attributed to decomposition rather than superoxide radicals ( ) transformation . Meanwhile, the results of detection by fluorescence method (Supplementary Fig. 14) confirmed that could not be abundantly generated in PMS system . PMS-premixing and high-valent metal capture experiments (Supplementary Fig. 15) excluded the contribution of other nonradical pathways like electron transfer process and high-valent Co-intermediate, respectively . Chemical probe experiments (Supplementary Notes 6,7 affirmed that the produced and in PMS system as the major active species could realize the rapid removal toward OFX (Supplementary Fig. 16). However, the TOC removal mainly relied on the oxidation process of surface-bound radicals and the adsorption between intermediates and catalyst (Supplementary Fig. 17 and Supplementary Note 8) . It had been reported that electron-rich contaminants could be selectively oxidized in non-radical system . The electrostatic potentials (ESP) of some selected organics were calculated, exhibiting that TC, BPA, 3,4-DCP, CQ and SMX with functional groups of electron aggregation displayed higher ESP than benzoic acid (BA) and nitrobenzoic acid (NBA) (Fig. 2b and Supplementary Fig. 18). The difference of oxidation ability toward different selected organics was investigated from the view of charge transfer pathway (Fig. 2e). It could be clearly observed that the highest occupied molecular orbital (HOMO) of electron-rich contaminants like TC ( -5.25 eV ), BPA ( -4.84 eV ), and SMX were slightly lower than the lowest unoccupied molecular orbital (LUMO) of , indicating that could readily withdraw electrons from HOMO of those electronrich contaminants to further achieve organics degradation . While the LUMO of was much higher than the HOMO of BA ( -6.14 eV ) and NBA ( -6.70 eV ), demonstrating that it was difficult to perform the oxidation reaction (Supplementary Fig. 19).
Fig. 2 | Catalytic activity, oxidation mechanism and environmental applicant in system. a The Fenton-like reaction activity test over different catalysts. Removal of multiple micropollutants by (inset: the corresponding ESP of the organic pollutants). c Comparison of the kinetics of multiple micropollutants elimination efficiency by the state-of-the-art catalysts on PMS activation system (The related information was presented in Supplementary Table 3). d Recyclability test of . e The oxidation mechanism of for multiple micropollutants (inset: the corresponding HOMO
and LUMO of the organic pollutants). Influences of different simulated wastewater on OFX elimination efficiencies. Inhibitions of OFX and its degradation intermediates on E. coli growth. h Schematic illustration of pilot reactor. i OFX concentration evolution of different systems in the reactor. Experimental conditions: [Catalyst] (in a, d, f), (in b). The error bars in the figures represented the standard deviations from triplicate tests.
The co-existing anions and organic matters in the actual water could react with ROSs to inhibit the catalytic efficiencies . The results demonstrated that co-existing substances exerted negligible impact on OFX degradation in PMS system (Supplementary Fig. 20), due to that possessed the lower oxidation potential and better selective oxidation ability than radicals. Moreover, the could efficiently activate PMS to accomplish the OFX
degradation in the wide pH range from 3.12 to 8.04 (Supplementary Figs. 21, 22), considering that exhibited strong tolerance to solution . Also, (bpy) PMS system could achieve complete removal toward OFX in the water samples simulated from actual water solutions (river, lake, tap, purified drinking and secondary settling tank water) within 5.0 min (Fig. 2f and Supplementary Fig. 23). These findings revealed that PMS system displayed superior
anti-interference ability, reflecting the superiority of in practical applications for the OFX degradation.
Besides UPLC-MS products identification, the Fukui index of OFX was calculated (Supplementary Figs. 24-26) to identify the reactive sites , which assisted to propose five possible OFX elimination pathways. The toxicity changes of the formed intermediates (Supplementary Fig. 27) showed that PMS activation process over could degrade OFX into lowly toxic intermediates, even completely mineralize OFX into and . The growth inhibition influence of OFX and its intermediates on Escherichia coli (E. coli) was determined (Supplementary Note 9), confirming the low toxicity of intermediates. As displayed in Fig. 2g, the diameters of inhibition zones for OFX, the degradation products in 1.0 min and 5.0 min against . coli were 23.0 , 12.0 , and 9.0 mm (near blank experiment without OFX ), demonstrating that the OFX degradation over resulted in decreasing toxicity toward E. coli. The plant growth experiment (Supplementary Fig. 28) exhibited that the germination rate and growth status of bean sprouts cultivated with the treated wastewater were comparable to the bean sprouts cultivated with deionized water. The above results indicated that PMS system displayed satisfactory mineralization ability and detoxification ability.
To evaluate the practical application prospects of (bpy) as PMS activator, powder was fixed onto the graphene cloth (Fig. 2h) to perform the long term treatment to both simulated wastewater containing OFX formulated from pure water and actual secondary sedimentation tank water were performed (Supplementary Fig. 29). Figure 2i exhibited that the OFX elimination efficiency in both pure water and actual secondary sedimentation tank water could maintain up to 56 h and 40 h in PMS system, while the OFX removal efficiency of the individual PMS system was less than . Additionally, the immobilized ensured the catalyst stability during the reaction process (Supplementary Fig. 30a, b), in which the leached Co ( ) and concentrations were far below the maximum pollutant level target set by the Chinese National Standard (Co: GB 3838-2002; V: GB 26452-2011). The PXRD pattern (Supplementary Fig. 31) and SEM image (Supplementary Fig. 32) of the used after the reaction matched well with the fresh sample, demonstrating the stable phase, composition and morphology of . Meanwhile, the TOC removal efficiency could reach about within 48 h in PMS pure water system (Supplementary Fig. 30c), implying that OFX could be partly mineralized into and . These results confirmed that the catalyst possessed great potential for practical application.
Catalytic sites identification and excitation mechanism
DFT calculations were utilized to construct bulk model with spin parallel, which was more stable than spin antiparallel (Supplementary Fig. 33). The surface model (Supplementary Fig. 34a) was constructed along the ( 001 ) crystal facet, in which Co atoms adjacent the vacuum layer were unsaturated coordination sites. The EPR exhibited an obvious response at (Supplementary Fig. 34b), affirming the existence of vacancies . Generally, the electron accumulation area might be more likely to act as the catalytic center . The results of density of states and partial density of states (PDOS) indicated that electron occupied orbital near Fermi level ( ) were mainly attributed to the contribution of Co and O orbitals (Supplementary Fig. 35), in which electronic state of Co displayed obvious split due to the Jahn-Teller effect (Supplementary Fig. 35b). Six possible PMS sorption models (Supplementary Figs. 36, 37) were built to calculate adsorption energy ( ), considering that both Co and V could be seen as the potential sorption sites to interact with PMS. As shown in Fig. 3a, Co sites from (bpy) surface bonded with O 1 from PMS, and the hydrogen bond was
formed between the terminal hydrogen of PMS and the terminal oxygen of . The was calculated to be -1.57 eV , much more negative than those of other models, confirming that was the optimal sorption model.
Based on optimal model, the corresponding electron density difference was calculated. Figure 3b indicated that PMS could draw electron ( 0.35 e) from the surface of catalyst, in which the electron accumulation area mainly focused on hydrogen bond rather than peroxo O-O bond (Fig. 3c). To further investigate the source of electron for PMS activation, the Co orbital electron distribution was calculated based on Hund’s rule and Pauli exclusion principle (Fig. 3d). Although Co sites were regarded as the optimal sorption sites toward PMS, weak increase of spin states after PMS adsorption indicated that high-spin complexes with strong oxidation potential were not facilely formed. Therefore, Co sites might not be considered as the only electron transport centers, which was also consistent with the analyses results of PDOS. As shown in Fig. 3e and Supplementary Fig. 38, all the Co , O and V orbitals presented distinct blue shift, indicating that PMS could draw electrons from Co, O, V atoms. Mulliken charge population results (Fig. 3f) indicated that H atom in PMS model after adsorption could obtain more electrons than those of other atoms in original PMS model. The bond length of O-H bond of PMS after adsorption was longer than that of pristine one (Supplementary Fig. 39), while the bond length of peroxo O-O bond nearly remain constant. The above-mentioned results indicated that bond of PMS was easier to be broken than peroxo O-O bond during activation process, due to the pull of hydrogen bond and charge transfer, which was further confirmed by the Gibbs free energy diagrams (Supplementary Fig. 40). To investigate the significant role of hydrogen bond, PMS adsorption model without hydrogen bond was built and calculated. It was found from Supplementary Fig. 41 that the of PMS adsorption model with hydrogen bond was higher than that of PMS adsorption model without hydrogen bond ( -1.15 eV ), illustrating that PMS adsorption pattern with hydrogen bond was easier to be formed from thermodynamic perspective. To further affirm the role of hydrogen bond interaction, with biggest electronegativity was introduced into the reaction system to compete with O from for forming hydrogen bond. The introduction of led to the noticeable inhibition of OFX degradation efficiency and rate (Supplementary Fig. 42), due to that could prefer to interact with from PMS to form strong hydrogen bond to decrease the formation of hydrogen bond between PMS and catalyst. It was deemed that Co cations as the PMS sorption sites also played the key role during the Fenton-like reaction. As shown in Supplementary Fig. 43, ethylene diamine tetraacetic acid and phosphate were selected to shield the Co sites to further inhibit the PMS sorption process. As expected, both the OFX elimination performances and PMS consumption decreased, confirming the significant contribution of Co sites. In addition, the PMS activation ability of V sites based on the model was also investigated (Supplementary Fig. 37e). As shown in Supplementary Fig. 44a, 0.13 e could be transferred from the surface of to PMS. Supplementary Fig. 44b indicated that the electron accumulation area mainly focused on hydrogen bond rather than peroxo O-O bond in PMS, which was similar to Co-O1 model. Moreover, the dual sites PMS adsorption model was also constructed (Supplementary Fig. 45). The antibonding orbital formed between the adsorption site and PMS was filled with more electrons due to the blue shift of the -band center , which further led to the deterioration of the PMS adsorption ability (Supplementary Fig. 46a, b). As shown in Supplementary Fig. 47a, the of dual sites PMS adsorption model was lower than that of individual Co or V sites. It was interesting that PMS activation didn’t merely rely on electron transfer from Co metal sites in PMS system, due to that the electron transfer number ( 0.37 e ) from catalyst to PMS was not altered (Supplementary Fig. 47b). The decrease of signified that the formed active species
Fig. 3 | Catalytic sites identification and excitation mechanism exploration. a Optimal PMS adsorption model on and corresponding adsorption energy. Electron density difference of optimal PMS adsorption model. c Planar-averaged electron density difference . d Electron filled situation of Co d orbital and spin state before and after PMS adsorption (e) PDOS of O 2p and Co before and after PMS adsorption. Change of Mulliken charge of PMS model. XPS spectra of (g) Co before and after reaction. i Co K-edge EXAFS spectra, (j) Co K-edge and (k) V K-edge XANES spectra as well as (l) WT plots of before and after reaction (insert: the absorption edge and white line of Co and V , respectively).
were more susceptible to desorption, which was more conducive to achieve PMS activation.
X-ray photoelectron spectroscopy (XPS), XANES spectra and EXAFS spectra of before and after reaction combined with DFT calculations were performed to ascertain the electron transfer pathway during the catalytic reaction. Figure 3 g exhibited that the percentage of increased from to
after the catalytic reaction, indicating that might be converted to due to electron transfer from Co sites to . Some electrons were delivered from Co sites to PMS for activation reaction, and other electrons were captured and stored in electron reservoir. Meanwhile, the binding energy of Co-N (Supplementary Fig. 48a) and Co-O (Fig. 3h) all shifted to higher energy level than that of pristine catalyst, affirming the electron transfer process from
Co sites. Additionally, Supplementary Fig. 48b showed that the red shift of 0.08 eV happened to the binding energy of of pristine catalyst and after reaction, indicating that as the charge receptor could store and transfer electron . Figure 3 h also displayed that the binding energy of before and after reaction shifted from 529.65 eV to 529.73 eV , illustrating that O atoms could act as electron donors to transfer electrons to PMS. Co K-edge EXAFS spectra (Fig. 3i) indicated that the peak intensity increased after PMS activation reaction, which could be attributed to the formation of bond. The white line variation (Fig. 3j) of Co after reaction implied the redistribution of electron density, in which the weak intensity of white line indicated the electronic loss of Co sites. Meanwhile, the enhanced white line intensity of V metal after reaction (Fig. 3k) demonstrated that V draw electrons to cause the increasing charge density , which matched well with XPS analyses. Furthermore, the WT plots of Co (Supplementary Fig. 49) and V (Fig. 31) displayed that Co-N/O bond and V-O bond shifted from and to and after reaction, respectively, indicating that both bond and bond presented dynamic variation during the PMS activation reaction.
In situ FTIR under atmosphere (Supplementary Fig. 50a, b) and In situ Raman spectroscopy (Supplementary Fig. 50c, d) were performed to further investigate the intermolecular interaction between PMS and (bpy) during the PMS activation . The FTIR spectra of individual PMS (Supplementary Fig. 51) showed that the peaks at and were assigned to the bond and S-O bond, respectively . When PMS with a certain concentration was added into the system, the peak of O-H bond (Fig. 4a) exhibited a rapid blue shift from to . Then, the peak of O-H bond was kept at after 1.0 min reaction, which might be ascribed to the rapid PMS consumption in PMS system. The dynamic variation of peak could be attributed to: (i) bond from PMS presented strong interaction with catalyst surface; and (ii) bond in PMS was broken due to the interaction, which led to the formation of the new and steady bond between H from PMS and terminal O from on the catalyst surface. XPS analyses also indicated (Fig. 3h) that the content of hydroxyl group on the catalyst increased after Fenton-like reaction, which might be attributed to the interaction between terminal O from catalyst and terminal H from PMS. Meanwhile, the slight shift of S-O peak (Fig. 4a) could be attributed to the adsorption effect of metal center toward PMS, which was comparable to DFT calculations. More significantly, the characteristic stretching of O-O at (Fig. 4a) appeared under the conditions of atmosphere and the absence of dissolved oxygen , further verifying the generation of in PMS system due to that the interference of on bond was eliminated. Additionally, In situ Raman with laser excitation at 532 nm was adopted to characterize the change of PMS during the reaction. The characteristic vibration of gradually shifted to higher wavenumbers (Fig. 4b), affirming the electron transfer process from catalyst to . Also, the new peak at about disappeared after (bpy) was added into the PMS solution , indicating that the high-spin complexes (catalyst-PMS*) with strong oxidation ability was not formed. Moreover, In situ Raman experiments were performed to facilitate to observe the dynamic variation of metal centers, in which the Raman peaks at and could be ascribed to the and V-O stretching frequencies (Fig. 4c), respectively . Compared with the dynamic structure of and bond, the slight dynamic change of V-O bond could be neglected (Fig. 4d) due to the stable structural characteristics of cluster , matching well with HR-TEM image. As shown in Supplementary Fig. 52, the crystal lattice of of the used was almost identical to that in the fresh one. On the contrary, noticeable stretching vibration of and bonds indicated the mutual affinity between Co sites and PMS because the metal centers will induce structural response to
adsorption of PMS. Interestingly, above conclusions matched well with the analysis results of DFT calculations. The included angle of changed from to when PMS was adsorbed on the Co sites, while the included angle of only shifted from to after PMS adsorption (Supplementary Fig. 53). In conclusion, beside the electrons transferred from Co sites to PMS, In situ spectral results combined with DFT calculations jointly indicated that the O-H of PMS preferred to be broken due to the pull of hydrogen bond and electron transfer between and PMS, implying the formation of might further produce and for OFX degradation (Fig. 4e).
Based on the above discussions, three kinds of POM-MOFs and three kinds of Co based MOFs were adopted to investigate the universality of the proposed mechanism (Supplementary Fig. 54). The results of quenching and chemical probe experiments (Supplementary Fig. 55-57) indicated that played the major contribution for OFX degradation in POM-MOFs/PMS system, which could be attributed to the electron directional transfer effect of polyvanadates. While the Co sites were regarded as the electron donators in Co based MOFs for PMS activation, leading to that the radicals and nonradicals jointly participated the oxidation reaction for OFX elimination. Additionally, the activations of PDS and hydrogen peroxide ( ) were also performed to explore the significant role of host-guest interactions on other Fenton-like reactions. As expected, the results (Fig. 4f) indicated that the catalytic activity of activation was better than that of PDS activation, in which the catalytic degradation rate of activation was about 3.58 times higher than that of PDS activation (Supplementary Fig. 58a). adsorption model (Supplementary Fig. 58b) and PDS adsorption model (Supplementary Fig. 58c) displayed that the terminal H atom of could also generate interactions with terminal O atom of cluster, while PDS adsorption could only rely on Co sites. The calculated adsorption energy indicated that PMS adsorption model displayed highest sorption energy ( ), followed by adsorption model ( ) and PDS adsorption model ( ). Therefore, the directly electron transfer from could achieve the rapid activation to produce active species for OFX degradation. In contrast, PDS activation process could only rely on the electron transfer from Co sites, which displayed poor catalytic OFX degradation performances. Electrochemical characterizations also confirmed that PMS system possessed better electron transfer ability than those of system and (bpy) PMS system (Supplementary Fig. 58d, e). In addition, electrochemical experiments were performed to test the real surface area of various catalysts. Supplementary Fig. 59 showed that cyclic voltammetry (CV) curves of and after 20 electrochemical activation cycles, which could calculate the double-layer capacitance of and based on the current density and different scan rates . Figure 4 g displayed that the of was higher than those of and . The higher value indicated the stronger electrochemical active surface area . The open-circuit potential of rose when PMS was added into the electrolyte (Fig. 4 h and Supplementary Fig. 60a, b), and the potential gradually elevated with the addition of PMS and OFX. Amperometric curves (Fig. 4i and Supplementary Fig. 60c, d) displayed that the addition of PMS and OFX could promote current intensity, indicating that OFX as the electron-rich pollutant could boost the electron transfer process between host and guest molecules. This result was also in agreement with the linear sweep voltammetry (LSV) and PMS consumption experiments. The LSV curves (Supplementary Fig. 61a) showed that the current was very small and almost negligible in the individual system. Compared to the LSV curves in PMS system, the current increased significantly after adding OFX, indicating the degradation of . Additionally, the results of PMS consumption experiments
Fig. 4 | In situ investigation of PMS activation process and electrochemical tests. a In situ FTIR spectra of PMS system under atmosphere. In situ Raman spectra of , PMS, and their reaction. c The shift of different peaks in the In situ Raman spectrum. d The shift of the characteristic peaks of the In situ Raman spectra during the different reaction time. e Stepwise analyses of PMS activation mechanism during Fenton-like reaction. f Influences of different guest molecule activators on OFX degradation efficiencies.
Experimental conditions: [Catalyst] ,
[PDS] = [ ] = 3.5 mM , [OFX] = (in f). g ECSA measurements based on the results of CV curves of and . Open-circuit potential curves and (i) Amperometric i-t curve in different system using as working electrode. The error bars in the figures represented the standard deviations from triplicate tests.
(Supplementary Fig. 61b) indicated that the introduction of OFX could accelerate PMS decomposition.
Discussion
In all, , a MOF containing polyvanadate was fabricated to be used as PMS activator for the first time to accomplish efficient elimination of micropollutants with high HOMO via nonradical pathway. The synergistic effect between Co sites and led to enhanced catalytic rate of toward OFX, about 25.16 and 39.37 times higher than those of and . The oxidation mechanism dominated by nonradical pathway endowed (bpy) good environmental tolerance, in which different co-existing ions and pH (3.12-8.04) could not produce influence on PMS activation efficiencies. In addition, PMS system exhibited satisfactory long-time treatment of 40.0 h toward actual wastewater. The diameters of inhibition zones experiment against E. coli indicated good detoxification ability of as PMS activator. Significantly, various experiments, characterizations and DFT calculations were performed to verify the multiple host-guest interactions on complicated interface of catalyst. DFT calculations integrated with XAS affirmed that Co sites played key role for PMS trap. However, interfacial with the properties of electron sponge and robustness led to the generation of strong interaction between terminal O from and terminal H from PMS, which was confirmed by DFT calculations, In situ FTIR and In situ Raman technologies. Formed hydrogen bond provided directional electron transfer channel for catalyst, and the dynamic pull process promoted the generation of key intermediate, which was beneficial to produce for micropollutant degradation. Furthermore, this activation mechanism was also suitable to other POM-MOFs/PMS systems. This work provided a good case to fabricate efficient catalyst/activator containing polyoxometalate for the purpose of water purification. In future, more efforts will be made to clarify the catalytic performances and mechanisms of more MOFs with different POM clusters, and to tap the possibility of their application for large-scale water environment remediation.
Methods
Chemicals
All the chemical reagents (Supplementary Note 1) which were used for catalyst syntheses (Supplementary Note 2) and experiments evaluation in this work were listed in Supplementary information.
Synthesis of
The matrix of bpy and 13.0 mL distilled water was transferred into a 25.0 mL Teflon lined autoclave, which was heated at for 72.0 h . The purple-brown crystals were harvested after the autoclave was cooled to room temperature, which were dried in an oven at for 12.0 h after being washed by distilled water and ethanol . The detail characterization technologies were listed in Supplementary Notes 3 and 11.
Catalytic activity test
The performances of the various catalysts for PMS activation and pollutant degradation were evaluated by OFX as the model recalcitrant pollutant. All experiments were conducted in a 60.0 mL reactor with a constant stirring of 200.0 rpm under room temperature, in which catalyst and 0.1 mM PMS were added into 50.0 mL of aqueous solution containing pollutant with initial concentration of . The residual concentrations methods were provided in Supplementary Note 4. The pH values ranging from 2.0 to 11.0 of the targeted solution were adjusted with or NaOH solutions with suitable concentrations. All the experiments were carried out at least three times.
DFT calculation details
In this study, the first-principles calculations under frame of DFT were performed using Materials Studio 2020 (CASTEP module) . The exchange-correlation energy was described by the Perdew-Burke-Ernzerhof (PBE) form of generalized-gradient approximation (GGA) exchange-correlation energy functional . The energy cutoff was set as 500 eV and the -point spacing was set to be smaller than -point was . The spin state and DFT were considered due to the existence of Co and V , in which the U value of Co and V was set as 3.2 and 2.5 , respectively . The convergence standards for optimizing crystal geometry like energy tolerance, maximum force tolerance, maximum stress tolerance and maximum displacement tolerance were less than atom and . The specific calculation items were presented in Supplementary Note 10.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings of the study are included in the main text and supplementary information files. Source data are provided with this paper. The link of CCDC 152985: https://www. ccdc.cam.ac.uk/structures/Search?Ccdcid=152985; the link of CCDC 2323958: https://www.ccdc.cam.ac.uk/structures/Search?Ccdcid= 2323958&DatabaseToSearch=Published. The DFT calculations model after structure optimization including the atomic coordinates is provided in Supplementary Data 1. Source data are provided with this paper.
References
. et al. A green edge-hosted zinc single-site heterogeneous catalyst for superior Fenton-like activity. Proc. Natl Acad. Sci. 120, e2221228120 (2023).
Hodges, B. C., Cates, E. L. & Kim, J. H. Challenges and prospects of advanced oxidation water treatment processes using catalytic nanomaterials. Nat. Nanotechnol. 13, 642-650 (2018).
Ren, W. et al. Origins of electron-transfer regime in persulfatebased nonradical oxidation processes. Environ. Sci. Technol. 56, 78-97 (2022).
Anipsitakis, G. P. & Dionysiou, D. D. Degradation of organic contaminants in water with sulfate radicals generated by the conjunction of peroxymonosulfate with cobalt. Environ. Sci. Technol. 37, 4790-4797 (2003).
Guo, J. et al. Fenton-like activity and pathway modulation via singleatom sites and pollutants comediates the electron transfer process. Proc. Natl Acad. Sci. 121, e2313387121 (2024).
Zhang, L. S. et al. Carbon nitride supported high-loading Fe singleatom catalyst for activation of peroxymonosulfate to generate with 100 % selectivity. Angew. Chem. Int. Ed. Engl. 60, 21751-21755 (2021).
Wu, X. L. et al. Directional and ultrafast charge transfer in oxygen-vacancy-rich ZnO@single-atom cobalt core-shell junction for photo-Fenton-like reaction. Angew. Chem. Int. Ed. Engl. 62, e202305639 (2023).
Yao, Y. et al. Rational regulation of coordination for highefficiency generation of toward nearly selective degradation of organic pollutants. Environ. Sci. Technol. 56, 8833-8843 (2022).
Howarth, A. J. et al. Chemical, thermal and mechanical stabilities of metal-organic frameworks. Nat. Rev. Mater. 1, 15018 (2016).
Islamoglu, T. et al. Postsynthetic tuning of metal-organic frameworks for targeted applications. Acc. Chem. Res. 50, 805-813 (2017).
Horn, M. R. et al. Polyoxometalates (POMs): from electroactive clusters to energy materials. Energy Environ. Sci. 14, 1652-1700 (2021).
Liu, Y. et al. Fabricating polyoxometalates-stabilized single-atom site catalysts in confined space with enhanced activity for alkynes diboration. Nat. Commun. 12, 4205 (2021).
Zhang, H. et al. Three mechanisms in one material: uranium capture by a polyoxometalate-organic framework through combined complexation, chemical reduction, and photocatalytic reduction. Angew. Chem. Int. Ed. Engl. 58, 16110-16114 (2019).
Du, D.-Y. et al. Recent advances in porous polyoxometalate-based metal-organic framework materials. Chem. Soc. Rev. 43, 4615-4632 (2014).
Wang, Y.-R. et al. Oriented electron transmission in polyoxometalate-metalloporphyrin organic framework for highly selective electroreduction of . Nat. Commun. 9, 4466 (2018).
Kong, X. J., Lin, Z., Zhang, Z. M., Zhang, T. & Lin, W. Hierarchical integration of photosensitizing metal-organic frameworks and nickel-containing polyoxometalates for efficient visible-light-driven hydrogen evolution. Angew. Chem. Int. Ed. Engl. 55, 6411-6416 (2016).
Dong, Y. et al. Regulating crystallization and lead leakage of perovskite solar cell via novel polyoxometalate-based metal-organic framework. Small 19, e2301824 (2023).
Li, Z.-H., Yin, C., Wang, R.-J., Wang, P. & Guo, H.-Y. Synthesis and crystal structure of an inorganic-organic hybrid compound bpy) (bpy = 4, 4′-bipyridine). Acta Phys. Chim. Sin. 19, 1133-1137 (2003).
Chen, Z. et al. Single-atom Mo-Co catalyst with low biotoxicity for sustainable degradation of high-ionization-potential organic pollutants. Proc. Natl Acad. Sci. 120, e2305933120 (2023).
Wang, B. et al. A site distance effect induced by reactant molecule matchup in single-atom catalysts for Fenton-like reactions. Angew. Chem. Int. Ed. Engl. 134, e202207268 (2022).
Guo, Z. Y. et al. Mn-O covalency governs the intrinsic activity of CoMn spinel oxides for boosted peroxymonosulfate activation. Angew. Chem. Int. Ed. Engl. 60, 274-280 (2021).
Liu, F. et al. Covalent organic frameworks for direct photosynthesis of hydrogen peroxide from water, air and sunlight. Nat. Commun. 14, 4344 (2023).
Ren, W. et al. Activation of peroxydisulfate on carbon nanotubes: electron-transfer mechanism. Environ. Sci. Technol. 53, 14595-14603 (2019).
Li, M., You, S., Duan, X. & Liu, Y. Selective formation of reactive oxygen species in peroxymonosulfate activation by metal-organic framework-derived membranes: a defect engineering-dependent study. Appl. Catal. B 312, 121419 (2022).
Li, Y.-H. et al. Nearly zero peroxydisulfate consumption for persistent aqueous organic pollutants degradation via nonradical processes supported by in-situ sulfate radical regeneration in defective MIL-88B (Fe). Appl. Catal. B 331, 122699 (2023).
Yao, Y. et al. Rational regulation of Co-N-C coordination for highefficiency generation of toward nearly selective degradation of organic pollutants. Environ. Sci. Technol. 56, 8833-8843 (2022).
Mi, X. et al. Almost peroxymonosulfate conversion to singlet oxygen on single-atom sites. Angew. Chem. Int. Ed. Engl. 60, 4588-4593 (2021).
Wei, Z. et al. Artificial photosynthesis of through reversible photoredox transformation between catechol and o-benzoquinone on polydopamine-coated CdS. ACS Catal. 12, 11436-11443 (2022).
Yang, M. et al. Unveiling the origins of selective oxidation in singleatom catalysis via intensified radical and nonradical pathways. Environ. Sci. Technol. 56, 11635-11645 (2022).
Wang, L. et al. A polymer tethering strategy to achieve high metal loading on catalysts for Fenton reactions. Nat. Commun. 14, 7841 (2023).
Zhang, Y.-J. et al. Simultaneous nanocatalytic surface activation of pollutants and oxidants for highly efficient water decontamination. Nat. Commun. 13, 3005 (2022).
Yao, Y. et al. Synthesis of “sea urchin”-like carbon nanotubes/porous carbon superstructures derived from waste biomass for treatment of various contaminants. Appl. Catal. B 219, 563-571 (2017).
Yu, H. et al. Singlet oxygen synergistic surface-adsorbed hydroxyl radicals for phenol degradation in CoP catalytic photo-Fenton. Chin. J. Catal. 43, 2678-2689 (2022).
Ren, W. et al. The intrinsic nature of persulfate activation and N-doping in carbocatalysis. Environ. Sci. Technol. 54, 6438-6447 (2020).
Shao, P. et al. Revisiting the graphitized nanodiamond-mediated activation of peroxymonosulfate: singlet oxygenation versus electron transfer. Environ. Sci. Technol. 55, 16078-16087 (2021).
Yan, Y. et al. Merits and limitations of radical vs. nonradical pathways in persulfate-based advanced oxidation processes. Environ. Sci. Technol. 57, 12153-12179 (2023).
Fang, Q. et al. Generation and identification of in catalysts/ peroxymonosulfate systems for water purification. Water Res. 245, 120614 (2023).
Stuyver, T., Porft, F., Geerlings, P. & Shaik, S. How do local reactivity descriptors shape the potential energy surface associated with chemical reactions? The valence bond delocalization perspective. J. Am. Chem. Soc. 142, 10102-10113 (2020).
Li, Y.-H. et al. Seignette salt induced defects in Zr-MOFs for boosted adsorption: universal strategy and mechanism insight. Chem. Eng. J. 442, 136276 (2022).
Zhou, X. et al. Identification of Fenton-like active Cu sites by heteroatom modulation of electronic density. Proc. Natl Acad. Sci. 119, e2119492119 (2022).
Wang, Z. et al. Elevating the d-band center of six-coordinated octahedrons in through Fe-Incorporated topochemical deintercalation. Adv. Energy Mater. 11, 2003023 (2020).
Sun, C., Zhao, P., Yang, Y., Li, Z. & Sheng, W. Lattice oxygen-induced d-band shifting for enhanced hydrogen oxidation reaction on nickel. ACS Catal. 12, 11830-11837 (2022).
Ran, M. et al. Selective production of CO from organic pollutants by coupling piezocatalysis and advanced oxidation processes. Angew. Chem. Int. Ed. Engl. 62, e202303728 (2023).
Zhang, N. et al. Lewis acid Fe-V pairs promote nitrate electroreduction to ammonia. Adv. Funct. Mater. 33, 2211537 (2023).
Zhang, D. et al. Dynamic active-site induced by host-guest interactions boost the Fenton-like reaction for organic wastewater treatment. Nat. Commun. 14, 3538 (2023).
Lin, W. & Frei, H. Photochemical and FT-IR probing of the active site of hydrogen peroxide in Ti silicalite sieve. J. Am. Chem. Soc. 124, 9292-9298 (2002).
Qin, C. et al. Dual donor-acceptor covalent organic frameworks for hydrogen peroxide photosynthesis. Nat. Commun. 14, 5238 (2023).
Wang, X. et al. Understanding the deposition and reaction mechanism of ammonium bisulfate on a vanadia SCR catalyst: a combined DFT and experimental study. Appl. Catal. B 260, 118168 (2020).
Hu, J. et al. Oxygen vacant in situ embedded on carbon spheres: cooperatively tuning electron transfer for boosted peroxymonosulfate activation. J. Mater. Chem. A 9, 16489-16499 (2021).
López, X., Carbó, J. J., Bo, C. & Poblet, J. M. Structure, properties and reactivity of polyoxometalates: a theoretical perspective. Chem. Soc. Rev. 41, 7537-7571 (2012).
McCrory, C. C., Jung, S., Peters, J. C. & Jaramillo, T. F. Benchmarking heterogeneous electrocatalysts for the oxygen evolution reaction. J. Am. Chem. Soc. 135, 16977-16987 (2013).
Tong, Y. et al. Vibronic superexchange in double perovskite electrocatalyst for efficient electrocatalytic oxygen evolution. J. Am. Chem. Soc. 140, 11165-11169 (2018).
Wang, F. et al. High-efficient peroxymonosulfate activation for rapid atrazine degradation by derived from MIL-88A(Fe). J. Hazard. Mater. 440, 129723 (2022).
Monteiro, F. F. et al. On the mechanical, electronic, and optical properties of the boron nitride analog for the recently synthesized biphenylene network: a DFT study. J. Mol. Model. 29, 215 (2023).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758-1775 (1999).
Jain, A. et al. Formation enthalpies by mixing GGA and GGA+U calculations. Phys. Rev. B 84, 045115 (2011).
Acknowledgements
This work was supported by National Natural Science Foundation of China (22176012, 52370025, 52100069, 22325602, 12175263, 22176060), the Cultivation project Funds for Beijing University of Civil Engineering and Architecture (X23034) and the Shenzhen Science and Technology Program (JCYJ2O220531093205013), and the Program of Shanghai Academic/Technology Research Leader (23XD1421000).
Author contributions
Y.-H.L. and C.-C.W. designed the experiments. M.-Y.L., Y.-H.L., L.M. and X.-J.L. performed the experiments. Y.-H.L., M.-Y.L., S.G., J.C., Y.M., H.J. and C.-C.W. developed and analyzed the reaction model. C.-C.W., H.J. and M.X. supervised the study and experiments. Y.-H.L, C.-C.W., H.J. and M.X. performed the data analyses and prepared this manuscript. C.-C.W. and M.X. provided constructive suggestions for the manuscript revision.
Correspondence and requests for materials should be addressed to Chong-Chen Wang, Haodong Ji or Mingyang Xing.
Peer review information Nature Communications thanks Bofan Zhang and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Beijing Key Laboratory of Functional Materials for Building Structure and Environment Remediation, School of Environment and Energy Engineering, Beijing University of Civil Engineering and Architecture, Beijing, PR China. Eco-environment and Resource Efficiency Research Laboratory, School of Environment and Energy, Peking University Shenzhen Graduate School, Shenzhen, Guangdong, PR China. National Engineering Research Center of Industrial Wastewater Detoxication and Resource Recovery, School of Resources and Environmental Engineering, East China University of Science and Technology, Shanghai, PR China. Key Laboratory for Biomedical Effects of Nanomaterials and Nanosafety, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing, PR China. These authors contributed equally: Ming-Yan Lan, Yu-Hang Li. e-mail: wangchongchen@bucea.edu.cn; jihaodong@pku.edu.cn; mingyangxing@ecust.edu.cn