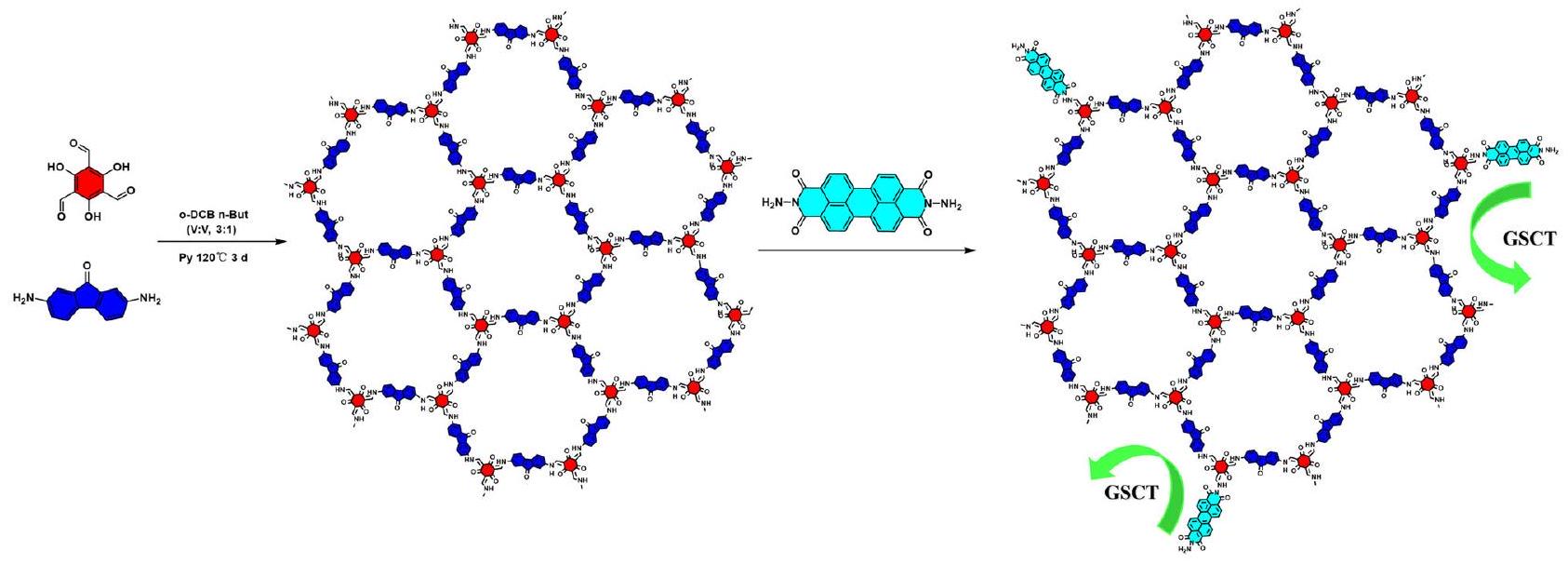
نقل الشحنة في الحالة الأساسية في وصلات الجزيئات الفردية من الأطر العضوية التساهمية لتعزيز تطور الهيدروجين الضوئي. Ground-state charge transfer in single-molecule junctions covalent organic frameworks for boosting photocatalytic hydrogen evolution
يلعب نقل الشحنة في الحالة الأرضية دورًا حيويًا في تحسين أداء التحفيز الضوئي لإطارات الكربون العضوية التساهمية من نوع D-A. ومع ذلك، فإن الدراسات المحدودة قد استكشفت تعديل أداء التحفيز الضوئي في المحفزات القائمة على COFs من خلال نقل الشحنة في الحالة الأرضية. هنا نعرض تشكيل نقل شحنة في الحالة الأرضية شديد الكثافة من خلال نهج فريد من نوعه في الروابط التساهمية. نقوم بتحويل الهياكل الثلاثية الأبعاد المكدسة من نوع COF القائمة على تقاطعات S-scheme (FOOCOF-PDIU) إلى تقاطعات جزيئية فردية متوازية (FOOCOF-PDI). تُظهر هذه البنية الجزيئية الفردية المتوازية نقل شحنة في الحالة الأرضية قوي مقارنة بالتقاطعات المكدسة عشوائيًا التقليدية وCOFs الفردية. يؤدي نقل الشحنة في الحالة الأرضية إلى إعادة توزيع الشحنة وتشكيل لحظة ثنائية القطب، مما يعزز شدة المجال الكهربائي المدمج في التقاطعات الجزيئية الفردية. يعزز هذا المجال الكهربائي المدمج المحسن من تفكك الإثارة وفصل الشحنة، مما يؤدي إلى تحسين كفاءة التحفيز الضوئي. لذلك، تم الحصول على جزيء مزين مستقر من COF مع امتصاص ضوئي واسع، حيث يمكن أن تصل معدل تطور الهيدروجين إلى. يفتح هذا العمل طريقًا لاستغلال الآليات الضوئية الحفازة في COFs استنادًا إلى تأثيرات نقل الشحنة في الحالة الأساسية.
نظرًا لخصائصها الإلكترونية القابلة للتعديل ومساحاتها السطحية العالية، فقد حظيت المحفزات الضوئية القائمة على الإطارات العضوية التساهمية (COFs) باهتمام متزايد مؤخرًا كمرشحين واعدين لتحفيز الطاقة الشمسية.التي ظهرت كمرشحات محتملة للتصوير الضوئي لتقسيم الماء المدفوع بالطاقة الشمسية لإنتاج الهيدروجين. لقد أظهرت هذه المواد إمكانيات كبيرة للتطبيقات الضوئية الحفازة ولكن تواجه التحديات المتعلقة بفصل حاملات الشحنة الضعيف والطاقة العالية لارتباط الإثارة، والتي تحد من كفاءتها في عملية التحفيز الضوئي.
إن بناء الوصلات غير المتجانسة هو نهج فعال لتعزيز كفاءة فصل حاملات الشحنة الناتجة عن الضوء في أشباه الموصلات القائمة على COF.الوصلة غير المتجانسة التي تتكون من تكديس مثالي
تساعد الطبقات المتراكبة على نقل الشحنات خارج المستوى، بينما تعتبر الانحرافات الفعلية في التكديس غير مواتية لفصل الشحنات الناتجة عن الضوء.بينما يمكن أن يؤدي تحسين مستوى المسطحية في أشباه الموصلات إلى تعزيز تفكك الإثارة وفصل حاملات الشحنة في المحفزات الضوئية لأشباه الموصلاتلذلك، فإن تحويل الهياكل المتراكمة إلى وصلات جزيئية متوازية سيساهم بشكل كبير في تعزيز كفاءة نقل الشحنات بين الوصلات غير المتجانسة. ومع ذلك، فإن آلية تعزيز الوصلات الجزيئية قد تم دراستها بشكل أقل حتى الآن، خاصة أن نقل الشحنات في الحالة الأرضية (GSCT) نادراً ما تم الإبلاغ عنه.
تعزز GSCT فصل أزواج الإلكترونات والثقوب الناتجة عن الضوء من خلال آليات مكانية وطاقة. من الناحية المكانية، تسهل GSCT نقل الإلكترونات من جزيئات المانح إلى جزيئات المستقبل، مما يزيد المسافة بين الإلكترونات والثقوب، وبالتالي يقلل من إعادة التركيب. من الناحية الطاقية، تؤدي عملية GSCT إلى بيئة أكثر ملاءمة لنقل الثقوب على المانح بسبب زيادة كثافة الشحنة الموجبة. علاوة على ذلك، فإن محاذاة مستويات الطاقة بين المانح والمستقبل بعد نقل الشحنة تقلل من طاقة الربط الكولومبية، مما يقلل من إعادة التركيب بشكل أكبر.. من المثير للاهتمام أن مثل هذا الانتقال الإلكتروني المولد (GSCT) يُفترض أن يلعب دورًا وسيطًا في فصل الشحنات داخل الوصلات العضوية غير المتجانسة (D-A)، مما يظهر كحالة إلكترونية حقيقية. يُعتقد أن GSCT يعدل حركيات عمليات فصل الشحنات وإعادة التركيب، مما يعزز أداء أجهزة الطاقة الشمسية العضوية.. ومع ذلك، في معظم COFs من نوع D-A، يتم عادةً ملاحظة تكوين حالات مفصولة الشحنة تحت تأثير التحفيز الضوئي. لم يتم التحقيق بشكل موسع في GSCT في COFs من نوع D-A في التحفيز الضوئي.
هنا، يتم التركيب والدراسة لاثنين من المحفزات الضوئية القائمة على FOOCOF، والواجهات غير المتجانسة القائمة على COF ثلاثية الأبعاد، والوصلات الجزيئية الفردية المتجاورة. أظهرت الدراسات أن الوصلات الجزيئية الفردية FOOCOF-PDI، التي تتميز بالروابط التساهمية، تظهر تأثير GSTC أقوى من FOOCOF-PDIU وFOOCOF. تم تأكيد التركيب البلوري والكيميائي لـ FOOCOFPDI الناتج باستخدام حيود الأشعة السينية بالمسحوق (PXRD)، وطيف الأشعة تحت الحمراء بتحويل فورييه (FT-IR)، والرنين المغناطيسي النووي الصلب 13C (13C NMR). تظهر كل من الحسابات التجريبية والنظرية أن ربط جزيئات PDI بنهاية COFs من خلال GSTC يؤدي إلى امتلاك COF-PDI لعزم ثنائي قطب أقوى وحقول كهربائية مدمجة، مما يعزز تفكك الإثارة وهجرة الحاملات. تحت إشعاع الضوء المرئي، مع وجود Pt كعامل مساعد، وصلت نشاط إنتاج الهيدروجين الضوئي إلى.
النتائج والمناقشة
تم تخليق FOOCOF بنفس الطريقة التي استخدمناها في عملنا السابقتم تخليق FOOCOF-PDI في وجود حمض الأسيتيك من خلال تكثف مجموعات الألدهيد في نهاية FOOCOF مع مجموعات الأمين في PDINH في مذيب مختلط من o-dichlorobenzene / n-butanol ) في كما هو موضح في المعلومات الإضافية (الشكل 1). يتم تشكيل الوصلة غير المتجانسة من نوع S-FOOCOF/PDINH (FOO-COF-PDIU) من خلال خلط FOOCOF و PDINH بمساعدة الموجات فوق الصوتية.
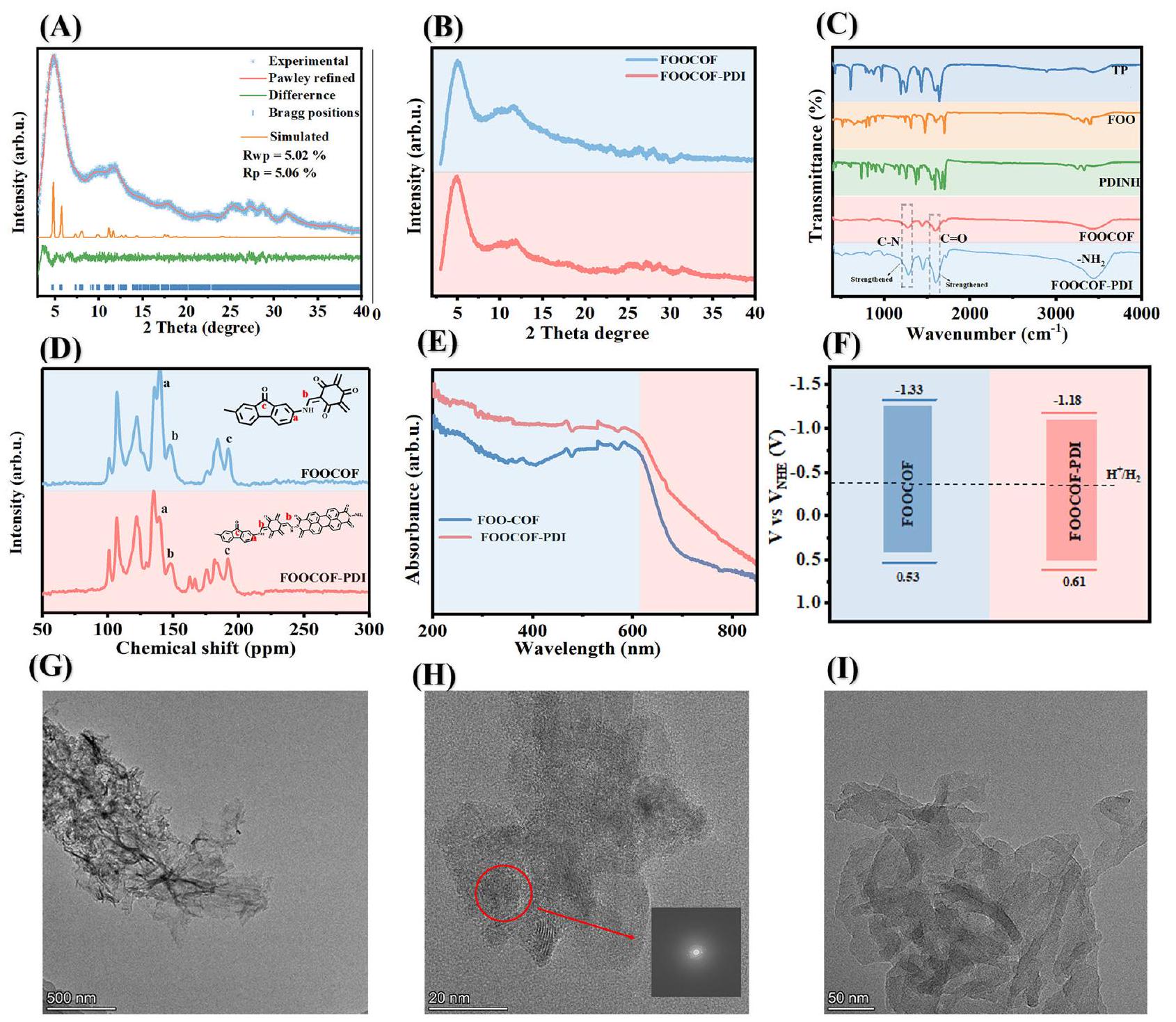
يظهر نمط PXRD لـ FOOCOF في الشكل 2، مع وجود قمم بارزة عند و (الشكل 2A والشكل S1)، الذي يتوافق مع مستويات 001 و 211 و 102 من FOOCOF ( و مع العوامل المتبقية من و ). تظل بنية FOOCOF دون تغيير بعد تعديل PDI، مما يشير إلى أن الاتصال التساهمي لجزيئات PDI له تأثير منخفض على FOOCOF (الشكل 2B). يظهر نمط XRD لـ FOOCOF-PDIU قمم PDINH مميزة (الشكل S2). في طيف FTIR، القمة عند الموافق لـتعزيز الرابطة“، مما يدل على الاتصال التساهمي الناجح لـ PDINH في نهاية FOOCOF (الشكل 2C). في الوقت نفسه، القمة عند 1591 ونُسِبَت إلى و ، على التوالي. كشف طيف الرنين المغناطيسي النووي عن قمة عند 184.5 جزء في المليون، والتي تُنسب إلى مجموعة الكربونيل من وحدة الفلورينون. القمم عند 137 ويتوافق مع روابط C -N و C -NH، مما يؤكد النجاح في تخليق FOOCOF. في طيف NMR لـ FOOCOF-PDI، يظهر الذروة المنسوبة إلى C-N كثافة معززة، مما يؤكد الاتصال التساهمي الناجح لـ PDINH في نهاية FOOCOF (الشكل 2D). كما أكدت تحليل XPS الخصائص الهيكلية لـ FOOCOF-PDI.تم تحليل الطيف إلى ثلاثة قمم عند طاقات ربط تبلغ 284.8 و 286.1 و 288.9 إلكترون فولت، والتي تت correspond إلى، و على التوالي. عرض طيف N 1s قممًا عند 399.6 و 398.8 إلكترون فولت، المنسوبة إلى ذرات النيتروجين من الـ و السندات. في الـالطيف، القمة عند 531.1 إلكترون فولت تت corresponded إلىروابط FOOCOF و PDI؛ القمة عند 532.9 إلكترون فولت نُسبت إلى أنواع الأكسجين الممتصة (الأشكال S3-5). تعرض طيف الانعكاس المنتشر للأشعة فوق البنفسجية والمرئية (UV-DRS) للعينات المحضرة ذيل امتصاص ممتد يصل إلى 750 نانومتر (الشكل 2E والشكل S6)، مما يشير إلى نطاق واسع من الحصاد. لذلك، تم تحديد فجوات الطاقة لـ FOOCOF و FOOCOF-PDI و PDINH على أنهاو 1.89 إلكترون فولت، على التوالي (الشكل S7). وفقًا لـ VB-XPS، تم تحديد نطاقات التكافؤ لـ FOOCOF و FOOCOF-PDI و PDINH على أنهاو 2.37 فولت مقابل RHE، على التوالي.لذلك، تم تحديد نطاقات التكافؤ لـ FOOCOF و FOOCOF-PDI و PDINH على أنها و 1.96 فولت مقابل NHE، على التوالي (الشكل S8-10). وبناءً عليه، كانت نطاقات التوصيل لـ FOOCOF و FOOCOF-PDI و PDINH و 0.07 فولت، على التوالي (الشكل 2F والشكل S11). العينات المحضرة تلبي من الناحية الديناميكية الحرارية المتطلبات اللازمة لتفكيك الماء باستخدام التحفيز الضوئي. في الوقت نفسه، يتم تلبية الشروط الديناميكية الحرارية لتكوين تقاطع غير متجانس من نوع S بين FOOCOF و PDINH.
الشكل 1 | تخليق المحفزات الضوئية COF. مخطط تخليق FOOCOF و FOOCOF-PDI.
الشكل 2 | توصيف المحفزات الضوئية القائمة على COF. A أنماط PXRD التجريبية لـ FOOCOF مع تحسين Pawley المقابل، أنماط التكديس المتداخلة المحاكية (AA)، الفروقات بين التحسين والتجريبي، ومواقع Bragg، (B) أنماط PXRD، (C) طيف FTIR، (D)
CP/MAS الحالة الصلبةطيف NMR، (E) نمط الطيف الانعكاسي التفاضلي UV-vis و(F) مستوى النطاق لـ FOOCOF وFOOCOF-PDI. G صورة TEM لـ FOOCOF، (H) صور TEM عالية الدقة (HRTEM) لـ FOOCOF؛ (I) صور TEM لـ FOOCOF-PDI.
تم استخدام SEM وTEM للتحقيق في التأثيرات الشكلية لربط PDINH بشكل تساهمي مع COFs. تكشف صور SEM (الأشكال S12-14) عن الشكل السلس والطبقي لـ FOOCOF، ولا يؤدي الربط التساهمي لـ PDINH إلى أي تغييرات ملحوظة في شكل العينة. تكشف صور TEM (الشكل 2G والشكل S15) عن هيكل النانو شيت لـ FOOCOF. بالإضافة إلى ذلك، يمكن رؤية الخطوط الشبكية الواضحة في الشكل، مما يدل على البلورية العالية للعينة المحضرة (الشكل 2H). بعد تشكيل وصلات الجزيئات الفردية، لم يتم العثور على أي تغيير واضح في صور TEM (الشكل 2I والشكل S16). في الوقت نفسه، لم يتم العثور على أي جزيء PDINH في FOOCOF-PDI (الشكل 2I والشكل S17). يظهر منحنى امتصاص النيتروجين القابل للعكس لـ FOOCOF خصائص منحنى النوع الرابع النموذجي ويعرض ميزات مسامية متوسطة (الشكل S18). تم حساب مساحة سطح BET لـ FOOCOF لتكونتوزيع حجم المسام هو 1.3 نانومتر، وهو متوافق مع النموذج النظري البالغ 1.3 نانومتر. الربط التساهمي لـ PDI يؤدي إلى انخفاض طفيف في مساحة السطح BET إلى، ومع ذلك، تظل حجم المسام دون تغيير. تم إجراء تحليل الوزن الحراري (TGA) للتحقق من الاستقرار الحراري لـ COFs. كما هو موضح في الشكل S19، FOOCOF يتحلل عندبينما يتم تحسين الاستقرار الحراري بعد الربط التساهمي لـ PDI.
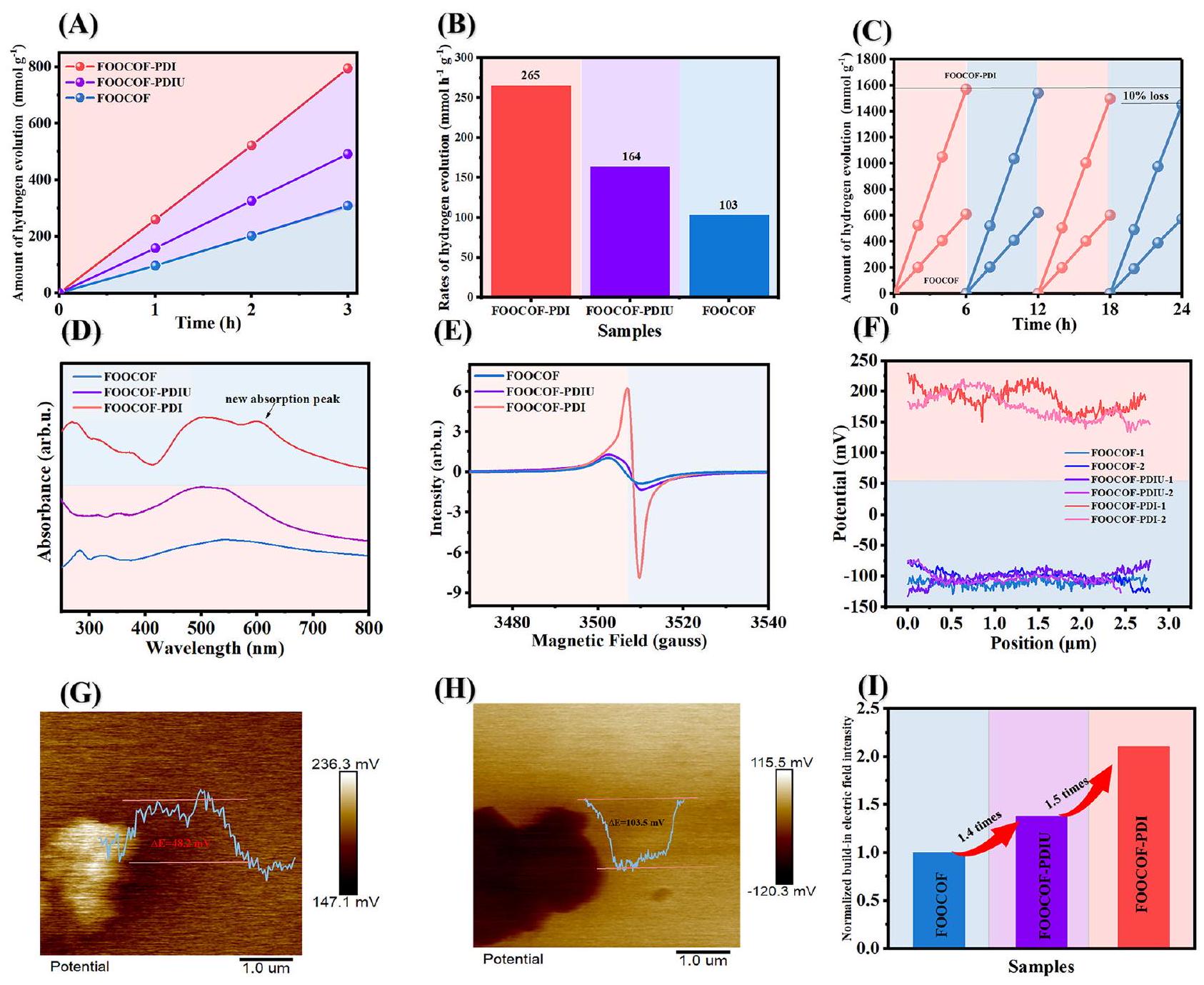
تم استخدام الترسيب الضوئي في الموقع للبلاتين كعامل مساعد وحمض الأسكوربيك بتركيز 0.1 م كعامل تضحية في عملية تحليل الماء الضوئي تحت الضوء المرئي. تم تقييم نشاط تطور الهيدروجين الضوئي لفوكوف وفوكوف-بي دي آي.كعامل مساعد. أظهرت FOOCOF معدل متوسط لتطور الهيدروجين قدرهتحت إشعاع الضوء المرئي لمدة ثلاث ساعات. بعد تشكيل تقاطعات S-scheme، زادت متوسط معدل تطور الهيدروجين لـ FOOCOF-PDIU إلىعند تشكيل الوصلة الجزيئية الفردية، أظهر FOOCOF-PDI زيادة ملحوظة في متوسط معدل تطور الهيدروجين، حيث وصل إلىتحت إشعاع الضوء المرئي لمدة ثلاث ساعات، مما يشير إلى تحسين في النشاط الضوئي التحفيزي للـ COFs عند تشكيل وصلات جزيئية فردية (الشكل 3A، B). في الوقت نفسه، فإن الأداء قابل للمقارنة مع أداء المحفزات الضوئية الأخرى المعتمدة على COF (الجدول S1).عندما يكون محتوى البلاتين أقل مننشاط إنتاج الهيدروجين في العينة أقل منبسبب نقص المواقع النشطة. بسبب تأثير التظليل الضوئي، تتناقص النشاط الضوئي التحفيزي للعناصر تدريجياً
الشكل 3 | الأداء الضوئي المحفز والتوصيف الضوئي الفيزيائي. مسار زمني للتصوير الضوئيتطور العينات المحضرة. ب معدل تطور الهيدروجين المتوسط و (ج) استقرار الدورة للعينات المحضرة. د امتصاص الأشعة فوق البنفسجية، (هـ) طيف ESR في الظلام و (و) فرق الجهد السطحي لـ
FOOCOF و FOOCOF-PDIU و FOOCOF-PDI. إمكانيات السطح عبر KPFM لـ (G) FOOCOF-PDIU و (H) FOOCOF-PDI. I القوة الكهربائية المدمجة العادية لـ FOOCOF و FOOCOF-PDIU و FOOCOF-PDI. انخفض مع زيادة محتوى البلاتين وجرعة المحفز (الأشكال S20 و S21). بالنظر إلى خطأ الوزن عند استخدام 1 ملغ من المحفز، لا يزال نأخذ نشاط إنتاج الهيدروجين عند 2 ملغ كمعيار. يعتمد معدل تطور الهيدروجين لـ FOOCOF-PDI بشكل كبير على الرقم الهيدروجيني لمحلول التفاعل (الشكل S22). نشاط إنتاج الهيدروجين أعلى في الظروف الحمضية في محلول حمض الأسكوربيك مقارنة بالظروف القلوية في محلول TEOA. بالإضافة إلى ذلك، لا يوجد يتم إنتاجه عندما يتم استخدام الإيثانول كعامل تضحية. حافظ FOOCOF-PDI على استقرار بنسبة 90 في المئة في اختبار دورة الـ 24 ساعة (الشكل 3C). كما يتضح من النتائج التجريبية، حافظ العينة على الاستقرار. يمكن أن تُعزى النقاط السوداء على سطح COF إلى المساعدات الحفزية Pt (الأشكال S23 و S24). لتحليل أداء التحفيز الضوئي المعتمد على الطول الموجي لـ FOOCOFPDIs، تم مراقبة الكفاءة الكمية الظاهرة (AQE) كدالة للطول الموجي (الشكل S25). تصل قيمة AQE لـ FOOCOF إلى عند 420 نانومتر.
عندما يشع الضوء فوق البنفسجي على معقد نقل الشحنة، يمكن للإلكترونات أن تخضع لانتقال من الحالة الأساسية إلى مستوى نقل الشحنة، مما يؤدي إلى تشكيل نطاق امتصاص جديد. يُشار إلى هذا النطاق الامتصاصي باسم نطاق امتصاص نقل الشحنة. تم تأكيد تعزيز GSCT لأول مرة بواسطة امتصاص UV-vis (الشكل 3D). ومن الجدير بالذكر أنه بعد تشكيل FOOCOF-PDI، يظهر قمة امتصاص جديدة عند طول موجي أطول ( )، مما يشير إلى تشكيل معقد جديد لنقل الشحنة . ثم، وجدنا أن FOOCOF-PDI أظهر إشارة حالة مظلمة مميزة في الحالة المظلمة من خلال ESR، مما يدل على GSCT (الشكل 3E والشكل S26). علاوة على ذلك، تم التحقيق في تأثير GSCT على قدرة استخراج الحاملات باستخدام مجهر المسح بكلفن (SKPM). كما هو موضح في الشكل 3F والأشكال S27-28، فإن جهد FOOCOF-PDI أعلى بحوالي 0.32 فولت من جهد FOOCOF النقي و FOOCOFPDIU، مما يشير إلى تحول مستوى الفراغ عند الواجهة بسبب نقل الإلكترون.تدعم هذه النتائج نظرية GSCT في FOOCOF-PDI. قمنا بقياس عمر الفلورية للعينات المحضرة، وعمر FOOCOF-PDI أطول من عمر FOOCOF. تم ملاءمة الأعمار المتوسطة لكل من FOOCOF و FOOCOF-PDI باستخدام ديناميات الانحلال الثنائي الأسي. العمر المتوسط المحسوب لـ FOOCOF هو 21.3 نانوثانية. ومع ذلك، عند تشكيل وصلات جزيئية فردية، يزيد العمر المتوسط لـ FOOCOF-PDI بأكثر من 4 مرات (الشكل S29). يشير الزيادة في العمر عند تشكيل المركب بين FOOCOF و PDI إلى تكوين حاملات حرة طويلة العمر.
يمكن أن تؤدي GSCT إلى توليد لحظات ثنائية القطب. بشكل عام، يمكن أن يؤدي لحظة ثنائي القطب لجزيء واحد بشكل فعال إلى نقل الشحنة داخل الجزيء. بمجرد أن تخضع الوحدات الجزيئية لعمليات عالية
الشكل 4 | توصيف فصل الشحنات لمحفزات COF الضوئية. طيف الامتصاص العابر لـ FOOCOF. د الديناميات العابر للامتصاص المنظم لـ FOOCOF. هـ-ز طيف الامتصاص العابر لـ FOOCOF-PDI. ديناميكية الامتصاص العابر الطبيعية لـ FOOCOF-PDI. درجة الحرارة-
طيف الفوتولومينسنس المعتمد على شدة PL المدمجة كدالة لدرجة حرارة (I) FOOCOF و (J) FOOCOF-PDI (الإطار: أطياف PL المعتمدة على درجة الحرارة من 80 إلى 290 كلفن). K استجابة التيار الضوئي و (L) منحنى موت-شوتكي لـ FOOCOF و FOOCOF-PDIU و FOOCOF-PDI.
عند الترتيب المتراص، يتم توليد مجال كهربائي داخلي؛ وإلا، فإن الأقطاب ستلغي بعضها البعض بسبب التجميع غير المنظم.قمنا بتحليل المجال الكهربائي المدمج في العينات المحضرة. بسبب وجود ثنائيات القطب، يتم تعزيز المجال الكهربائي المدمج. بشكل عام، يمكن أن تعمل قوة المجال الكهربائي المدمج كعامل حركي يدفع فصل الشحنات، مما يستدعي تقييمًا كميًا.وفقًا لنظرية زانغ وآخرين، فإن تحديد المجال الكهربائي المدمج في المادة مرتبط بشكل إيجابي أحادي مع كثافة الشحنة السطحية وإمكان الزيتا.باستخدام تقنية قياس القوة باستخدام مسبار كيلفن (KPFM) للكشف عن كثافة الشحنة السطحية للعينة، يتبين أن أعلى جهد لـ FOOCOF-PDI يصل إلى 103.5 مللي فولت (الشكل 3G، H). وبالاقتران مع نتائج جهد زيتا الموضحة في الشكل S30، يؤكد ذلك أن المجال الكهربائي المدمج لـ FOOCOF-PDI أقوى بمقدار 2.1 و 1.5 مرة من ذلك الخاص بـ FOOCOF و FOOCOF-PDIU (الشكل 31).
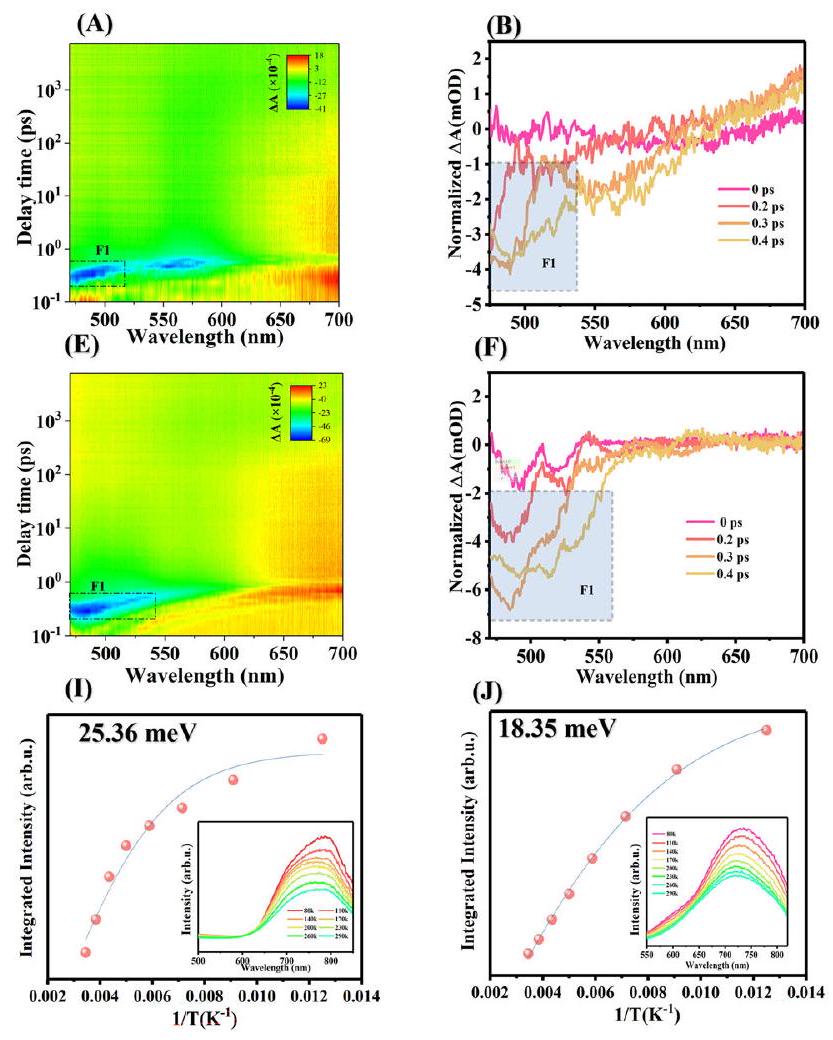
يمكن أن تعزز ثنائيات القطب على الواجهة الناتجة عن نقل الشحنة تفكك الإثارات إلى حاملات حرة، كما تم إثباته في العديد من الدراسات السابقة.تم الكشف المبكر عن الإثارات (في غضون 0.5 بيكوثانية) في COFs المُعدة باستخدام مطيافية الامتصاص العابر بالفيمتوثانية (fs-TAS). أظهر العينة المُعدة قمة سلبية عندوقمة إيجابية عند، والتي تُنسب إلى تبييض الحالة الأساسية (GSB) وامتصاص الحالة المثارة (ESA) على التوالي. تت correspond هذه الإشارات إلى استرخاء الحالة المثارة. لذلك، تساهم أزواج الإلكترونات والثقوب المتولدة ضوئيًا في العينة في كل من إشارات GSB وإشارات ESA الواسعة (الشكل 4A، E، الشكل S31 وS32 A). وقد تم التأكيد على أن الإلكترونات المتولدة ضوئيًا تشارك بشكل رئيسي في النقل الواجهاتي.تظهر الأشكال 4A-H والشكل S32 A-C أن العينة المحضرة أظهرت قمة سلبية واسعة (F1) بالقرب من 500 نانومتر. وبالاقتران مع الفلورية الثابتة، يمكن استنتاج أن F1 هو نطاق الانبعاث المحفز للإثارة الضوئية المتولدة. ومن الجدير بالذكر أن توسيع قمة امتصاص fs-TAS يتوافق مع استرخاء الإثارات المرتبطة وزيادة كثافة الحاملات.. وبالتالي، مقارنةً بـ FOOCOF، فإن اتساع F1 في FOOCOF-PDI يشير إلى ضعف تأثير الإثارة وفعالية أكبر في تفكك الإثارة. للتحليل الكمي الإضافي، تم استخدام دالة مزدوجة أسية لتناسب تدهور F1، مع عرض النتائج في الشكل 4D، H.مرتبط بتفكك الإثارة السريع عند كثافات إثارة عالية، بينمامرتبط بإعادة تركيب الشحنات السريعة والالتقاط. الأصغرقيمة FOOCOF-PDI (0.52 ps) مقارنة بـ FOOCOF (0.62 ps) تؤكد على التفكك الأكثر فعالية للإكسيتونات في FOOCOFPDI، بما يتماشى مع طاقة الربط للإكسيتونات. بعد 0.5 ps، يحدث فصل سريع لزوج الإلكترون-الثقب الناتج عن الضوء، مع وجود بعض انبعاث الفلورسنت، وتخضع الإلكترونات الناتجة عن الضوء المتبقية لعملية التقاط الإلكترون. عندما يقوم الليزر بتحفيز الإلكترونات المثارة ضوئيًا من نطاق التوصيل (CB) للمادة إلى مستويات طاقة أعلى، يؤدي ذلك إلى التقاط الامتصاص الضوئي المخفض أو تقليل النفاذية.. لذلك، تمثل شدة وتدهور الامتصاص الناتج عن الضوء التقاط وتقليل الشحنات. الزيادة فيتشير إلى التأثير المفيد لالتقاط الإلكترونات السطحية طويلة العمر على نقل الإلكترونات النشطة، مما يحقق أداءً عالي الكفاءة في إنتاج الهيدروجين بالتحفيز الضوئي. من خلال المقارنة بين تقاطعات الجزيئات الفردية والتقاطعات غير المتجانسة، وُجد أيضًا أن تشكيل التقاطعات غير المتجانسة يمكن أن يلتقط الإلكترونات طويلة العمر، على الرغم من أن عمر الإلكترون أقصر نسبيًا مقارنة بتقاطعات الجزيئات الفردية (الشكل S32). قمنا بقياس طيف الفلورية الضوئية المعتمد على درجة الحرارة لاختبار طاقة ارتباط الإثارة (الشكل 4I-G). انخفضت شدة الفلورية الضوئية لكل من FOOCOF و FOOCOFPDI مع زيادة درجة الحرارة من 80 إلى 290 كلفن، مما يتوافق مع عملية إعادة التركيب غير الإشعاعي المعتمدة على الحرارة. كانت طاقة ارتباط الإثارة لـ FOOCOF-PDI (18.35 ميلي إلكترون فولت) أقل.
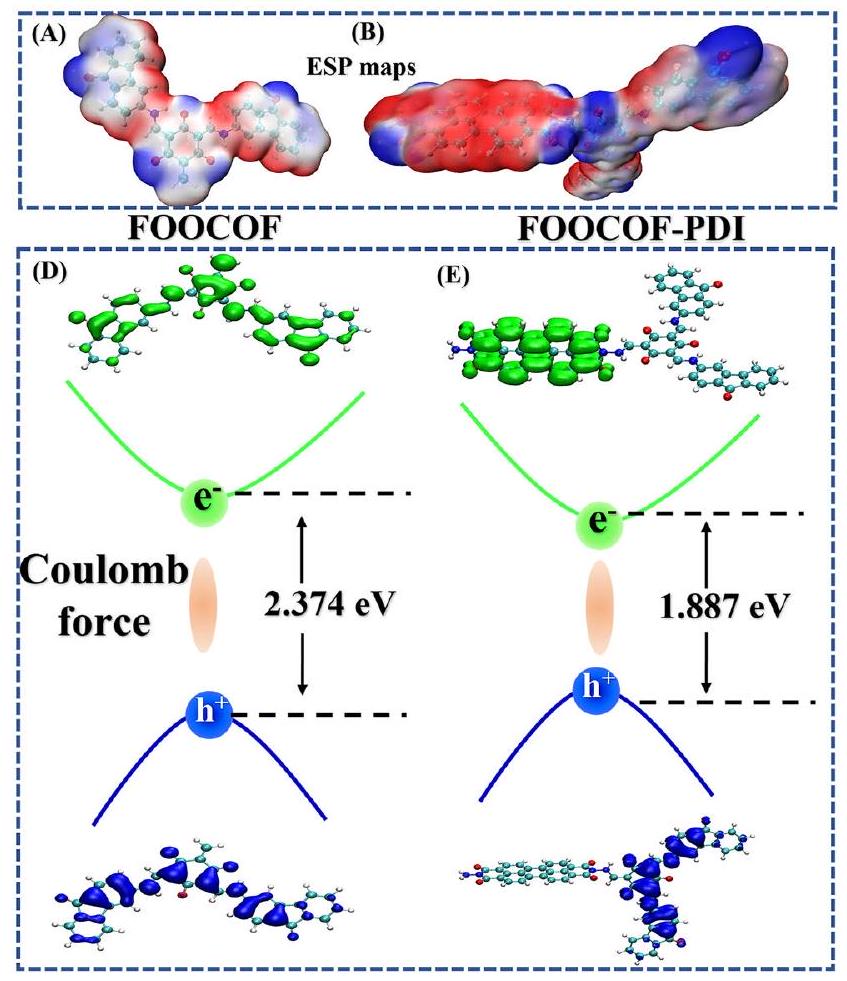
الشكل 5 | الدراسات الحاسوبية لفصل الشحنة. توزيعات الجهد السطحي الكهروستاتيكي (ESP) لـ (A) FOOCOF و (B) FOOCOF-PDI، حيث يمثل الأزرق تراكم الإلكترونات والأحمر نقصها. ج الجهد الكهربائي على طولمحور للطائرة (100). طاقات ارتباط الإثارة والإلكترون-الثقب
توزيعات (D) FOOCOF و (E) FOOCOF-PDI، الأزرق والأخضر يمثلان المراحل الإيجابية والسلبية. الانتقال الإلكتروني المحسوب بواسطة TD-DFT لـ (F) FOOCOF و (G) FOOCOF-PDI، الأزرق والأخضر يمثلان المراحل الإيجابية والسلبية. أقل من تلك الخاصة بـ FOOCOF (25.36 ميلي إلكترون فولت). تقليل طاقة ارتباط الإثارة يعزز الكفاءة الكمية لمواد أشباه الموصلات، والتي تشير إلى كفاءة توليد إلكترون عند امتصاص فوتون. هذه الكفاءة الكمية الأعلى تترجم إلى تحويل أكبر لطاقة الضوء إلى طاقة كيميائية، مما يؤدي إلى تحسين كفاءة إنتاج الهيدروجين. كما أن تقليل طاقة ارتباط الإثارة يطيل من عمر الإلكترونات والفجوات. يسمح العمر الممتد للإلكترونات بمزيد من الوقت للهجرة إلى سطح المحفز والمشاركة في تفاعلات الاختزال، مما يعزز في النهاية الكفاءة الضوئية التحفيزية. تعني شدة التيار الضوئي القوي والانحدار المنخفض زيادة تركيز الشحنة في FOOCOF-PDI نتيجة لتفكك الإثارة الفعال.لقد قمنا بقياس طيف الامتثال الكهروكيميائي (EIS) للكشف عن دور الوصلة الجزيئية الفردية خلال التفاعل الضوئي التحفيزي. في طيف EIS، تظهر العينات المحضرة عادة منطقتين متميزتين، كل منهما تعكس عمليات كهروكيميائية مختلفة. في منطقة التردد العالي، يعرض طيف الامتثال نصف دائرة أصغر مع ميل قريب من 0. تعكس هذه المنطقة بشكل أساسي عمليات نقل الشحنة على سطح القطب، بما في ذلك نقل الإلكترونات وهجرة الأيونات بين مادة القطب والكهارل. يمثل قطر نصف الدائرة مقاومة نقل الشحنة، حيث تشير القيم الأصغر إلى سهولة نقل الشحنة. في منطقة التردد المنخفض، يظهر طيف الامتثال نصف دائرة أكبر مع ميل يقترب من -1. تعكس هذه المنطقة بشكل أساسي انتشار الأيونات داخل مادة القطب، أي معدل انتشار الأيونات داخل هيكل القطب. يشير الميل القريب من -1 إلى مقاومة واربورغ، مما يدل على أن انتشار الأيونات هو العامل الرئيسي الذي يتحكم في معدل التفاعل. تظهر FOOCOF و FOOCOF-PDIU نصف دائرة أكبر مقارنة بـ FOOCOF-PDI، مما يشير إلى أن الوصلة الجزيئية الفردية يمكن أن تقلل من مقاومة نقل الإلكترون في FOOCOF-PDI. (الشكل S33).
لتحقيق تأثير GSCT على تفكك الإثارة، تم حساب الجهد السطحي الكهروستاتيكي (ESP) للهياكل المحسّنة لـ
تم حساب جزيئات FOOCOF و FOOCOF-PDI، كما هو موضح في الشكل 5A و B. كشفت حسابات توسيع لحظة المضاعفات لجزيء ESP عن أوسع توزيع لحظات المضاعفات لجزيء FOOCOF-PDI DFT، مما يدل على وجود مركز واضح لفصل الشحنات. وبالتالي، يضمن الوصل الجزيئي الفردي الوجود المستقر لحقول كهربائية داخلية قوية.، وهو أمر حاسم لتفكك الإثيون، وفصل الحاملات، والهجرة (الشكل S34). بدون وصلة جزيئية واحدة، تميل الإلكترونات المشاركة في التفاعل إلى التراكم عند حواف مادة COF. لا يمكنها التفاعل مع المذيب التفاعلي على الفور. ومع ذلك، بعد تشكيل وصلة جزيئية واحدة، يمكن ملاحظة أن الإلكترونات تميل إلى التراكم في وسط PDI، حيث يكون جزيء PDI بالكامل في اتصال مباشر مع المذيب، مما يعمل كموقع للتفاعل (الشكل 5C). لذلك، فإن تشكيل الوصلة الجزيئية يعزز تفكك الإثيون ويوجه الإلكترونات الحرة إلى مواقع حواف الواجهة، مما يعزز فصل حاملات الشحنة، ويقلل من إعادة تركيب حاملات الشحنة، ويحسن كفاءة التفاعلات الواجهة. تم تحسين الهياكل بالكامل، تلتها حسابات نظرية الكثافة الوظيفية المعتمدة على الزمن (TD-DFT) للحصول على رؤى حول خصائص توليد الإثيون في محفز COF الضوئي. تشير توزيع الإلكترون-الثقب في الحالة المفردة المثارة الأولى (S2) من COFs الاثنين إلى اختلاف في طاقة ربط الإثيون (بسبب الإثارة الموضعية. طاقة ارتباط الإثيون في FOOCOF-PDI هي 1.887 إلكترون فولت، أقل من 2.274 إلكترون فولت في FOOCOF، مما يشير إلى تأثير إثيون أضعف في FOOCOF، وهو ما يتماشى مع نشاطه الضوئي التحفيزي (الشكل 5D، E). بالنظر إلى أن حساب الأجزاء فقط سيتأثر بتأثيرات الحواف والترافق، قمنا بتوسيع النموذج وأجرينا حسابات على FOOCOF و FOOCOF-PDI (الشكل S35). وُجد أنه، تحت الإثارة الضوئية، تتوافق النتائج المحسوبة مع الهياكل المستمرة للأجزاء مع نتائج الأجزاء. وهذا يشير إلى أن تأثير الإثيون قد يكون أحد العوامل المسيطرة في هذه العملية. تمتص FOOCOF و FOOCOF-PDI بشكل رئيسي القمم المكونة من الانتقالات من SO إلى
S2. يمكن وصف حالة S2 من FOOCOF بأنها HOMO-1تظهر الانتقال في S2 خاصية نقل الشحنة الجزيئية الداخلية: تنتقل الإلكترونات من TP إلى موقع الفلورينون. لذلك، فإن الفصل الفعال لزوجات الإلكترونات والثقوب يعزز من عمر الحاملات.بعد تشكيل وصلة الجزيء الفردي، يمكن وصف حالة S2 من FOOCOF بأنها (الشكل 5F، G). تنتقل الإلكترونات من FOOCOF إلى موقع PDI. وهذا يشير إلى أن الإلكترونات تميل إلى التواجد في وسط PDI، وأن جزيء PDI بالكامل هو موقع التفاعل الذي يتصل مباشرة بالمذيب. علاوة على ذلك، قمنا بحساب تغير الطاقة الحرة لجيبس ( لإ adsorptio الهيدروجين عند ذرات الأكسجين في كل من مواقع الفلورينون ومواقع PDI وفقًا لـ FOOCOF و FOOCOFs-PDI. من الملحوظ أن مواقع الفلورينون أكثر ملاءمة من الناحية الطاقية مقارنة بمواقع PDI. علاوة على ذلك، تميل طاقة الارتباط الحر للهيدروجين إلى الانخفاض عند تشكيل وصلات جزيئية فردية (الشكل S36).
باختصار، لقد قمنا بتصميم وتصنيع وصلة الجزيء الواحد FOOCOF-PDI بنجاح. بعد تشكيل هذه الوصلة الجزيئية، تحسنت النشاط الضوئي التحفيزي لـ FOOCOF. في وجود البلاتين كعامل مساعد، تصل نشاط تطور الهيدروجين الضوئي التحفيزي لـ FOOCOF-PDI إلى، وهو أعلى بمقدار 2.57 مرة من FOOCOF. تشير النتائج التجريبية إلى أن تشكيل الوصلات الجزيئية الفردية يحفز GSCT لتعزيز المجال الكهربائي الجوهري. يؤثر تأثير ثنائي القطب بشكل فعال على تعزيز تفكك الإثارة. لهذه الدراسة قيمة نظرية وتطبيقية لدراسة تفكك الإثارة الناتج عن GSCT وفصل الشحنات في المحفزات الضوئية المعتمدة على COF.
طريقة
المواد
كانت جميع المواد الكيميائية والمركبات من مواد ذات درجة تحليلية واستخدمت كما هي دون مزيد من التنقية. كان o-1,2-Dichlorobenzene (1,2-Dichlorobenzene، 99%)، n-BuOH اللامائي (n-Butanol، 99.4%)، Pyrrolidine (Py، 98%)، حمض الأسكوربيك (>99.0%)، 2,7-Dinitro-9-fluorenone (تم شراء (4,9,10-بيريلينتيتراكربوكسيليك دايانهيدريد) من شركة ماكلين للكيماويات، وتم تزويد (2,4,6-ثلاثي هيدروكسي بنزين-1,3,5-ثلاثي الكربالديهيد) من قبل شركة يانشن للتكنولوجيا التابعة للأكاديمية الصينية للعلوم في جيلين.
تركيب PDINH
تم تحضير مونومر PDINH من خلال تفاعل استبدال نووي بين ثنائي أنهدريد 3,4,9,10-بيريلينتتراكربوكسيليك و هيدرازين أحادي الهيدرات. في إجراء نموذجي،تم إذابة الديانهايدريد في 100 مل من الإيثانول داخل دورق دائري سعة 250 مل تحت بيئة نيتروجينية. تم تبريد المزيج إلىاستخدام حمام ثلجي، وتم إدخال هيدرازين أحادي الهيدرات ببطء على مدى فترة 10 دقائق. بعد 30 دقيقة من التحريك في حمام الثلج، تم تسخين التفاعل إلى درجة حرارة الغرفة قبل أن يتم تسخينه تحت الغليان.لمدة ساعة واحدة.
تركيب 2,7-ديامين-9H-فلورين-9-ون
في إجراء نموذجي،تم إذابة 2,7-داينيترو-9-فلورينون في 375 مل من الإيثانول عند درجة حرارة الغرفة. ثم أضيفت إلى هذا المحلول محلول مائي مُعد مسبقًا (650 مل) من كبريتيد الصوديوم غير المائي.وهيدروكسيد الصوديومتمت إعادة تدفق الخليط الناتج لمدة 5 ساعات ثم تم تبريده إلىتم جمع الراسب عن طريق الترشيح وغسله بشكل مكثف بالماء،محلول هيدروكسيد الصوديوم، ماء، جلايكول إيثيلين بارد، إيثر، وهكسان. تجفيف تحت الفراغ، يليه إعادة التبلور من الأسيتون-الإيثانول. )، منح من 2,7-ديامينو-9H-فلورين-9-أون على شكل بلورات إبرية بنفسجية.
تركيب FOOCOF
في أنبوب بايركس، من 2،4،6-ثلاثي هيدروكسي بنزين-1،3،5-ثلاثي الكربالديهيد و 2,7-dيامين-9H-فلورين-9- تم دمجها مع o-1,2-Dichlorobenzene (0.75 مل)، n-BuOH، و بيريدينتم تعريض الخليط للخلط بالموجات فوق الصوتية ثم تم إزالة الغازات منه من خلال ثلاث دورات تجميد-ضخ-ذوبان إلى فراغ قدره 100 مTr أو قبل أن يتم إغلاقه. تم تسخين الأنبوب المغلق إلىوتم الاحتفاظ بها عند هذه الدرجة الحرارة لمدة 72 ساعة. بعد التفاعل، تم فصل المادة الصلبة البنية عن طريق الطرد المركزي وتنقيتها بغسلها مع رباعي هيدروفوران. والأسيتون اللامائي .
تركيب FOOCOF-PDI
في أنبوب بايركس، تم دمج PDINH (1 ملغ) و FOOCOF (10 ملغ) مع o-1,2-ثنائي كلوروبنزين (0.75 مل)، n-BuOH (0.25 مل)، وحمض الأسيتيك.خضعت الخلطة لثلاث دورات من التجميد-الضخ-الذوبان لإزالة الغازات المذابة، محققة ضغط فراغ قدره 100 مللي تور، قبل أن يتم ختمها. تم تسخين الأنبوب المختوم إلىلمدة 36 ساعة. بعد التفاعل، تم ترشيح الصلب البني الناتج، مع استخدام DMSO والإيثانول للمساعدة في الترشيح، وتم تنقيته عن طريق استخراج سوكسهليت المتتابع باستخدام الميثانول والأسيتون والهكسان (كل منها لمدة 24 ساعة).
طريقة وتفاصيل تطور الهيدروجين الضوئي المحفز
قمنا بإجراء تفاعلات التحفيز الضوئي في دورق ثلاثي العنق سعة 100 مل، باستخدام مصباح زينون بقوة 300 واط (PLS-SXE300،لتمثيل ضوء الشمس بشدةاستخدمنا فلتر قطع بطول موجي 420 نانومتر لإزالة الضوء فوق البنفسجي. على وجه التحديد، قمنا بتفريق 2 ملغ من المحفز الضوئي في 40 مل من محلول حمض الأسكوربيك بتركيز 0.1 م. استخدمنا الموجات فوق الصوتية لضمان التشتت المتجانس ثم قمنا بتفريغ الهواء بـلإنشاء بيئة لاهوائية. سمحنا للتفاعل بالاستمرار لمدة ساعة واحدة تحت إضاءة مستمرة. أخيرًا، قمنا باستخراج 0.4 مل من الغاز الناتج بشكل متقطع وحللناه باستخدام جهاز الكروماتوغرافيا الغازية (GC-9560، TCD). قمنا بحساب الكفاءة الكمية الظاهرة (AQE) للعينات على النحو التالي.
أين هو عدد مولات المنتج الناتج (مول) ، هو ثابت أفوجادروهو الكثافة الضوئيةهي منطقة إشعاع الضوء ( ) ، هو وقت إشعاع الضوء (ثانية) ، هو الطول الموجي للضوء أحادي اللون (متر) هو ثابت بلانك ( )، و سرعة الضوء ).
تحليل حيود الأشعة السينية بالمسحوق (PXRD)
تم إجراء تحليل PXRD على جهاز قياس الحيود Rigaku Smartlab باستخدام Cu Kإشعاعتم تشغيل جهاز الحيود في هندسة الانعكاس عند 40 كيلو فولت و40 مللي أمبير.
تحليلات FTIR
تم استخدام مطياف FT-IR نيكوليت أفاتار 6700 (ثيرمو فيشر، الولايات المتحدة الأمريكية) لتسجيل طيف FTIR.
تحليل طيف الامتصاص الانتشاري للأشعة فوق البنفسجية والمرئية في الحالة الصلبة
تم استخدام مطياف الأشعة فوق البنفسجية-المرئية-الأشعة تحت الحمراء القريبة Shimadzu UV-2600 لتحليل طيف الانعكاس المنتشر للأشعة فوق البنفسجية-المرئية للمواد المحفزة.
قياسات الامتصاص الفيزيائي
تمت دراسة الخصائص المسامية لمواد COF من خلال امتصاص النيتروجين عند 77 كلفن باستخدام محلل Autosorb-iQ-MP. قبل القياس، تم إزالة الغازات من العينات تحت الفراغ. ) في درجة حرارة الغرفة ثم تم تسخينه عند لمدة 12 ساعة لضمان الإزالة الكاملة للأنواع الممتصة. الـ تم تحديد المساحة السطحية المحددة بواسطة طريقة BET المطبقة على بيانات إيزوثرم امتصاص النيتروجين في نطاق الضغط النسبي من 0.01 إلى 0.9
طيف الكترونات الأشعة السينية
تم إجراء تحليل العناصر السطحية بواسطة XPS على جهاز VG ESCALAB 250، باستخدامإشعاع ) كمصدر للإثارة. كانت معلمات التحليل كما يلي: حجم البقعةضغط القاعدةجهد التشغيل 12 كيلو فولت، والتيار الخيطي 6 مللي أمبير. تم جمع طيف المسح بطاقة مرور 100 إلكترون فولت وحجم خطوة 1 إلكترون فولت، بينما تم الحصول على الأطياف عالية الدقة بطاقة مرور 50 إلكترون فولت وحجم خطوة 0.1 إلكترون فولت. تم تطبيق تصحيح الشحن على جميع الأطياف من خلال الإشارة إلى قمة C 1s عند 284.80 إلكترون فولت. تم تحسين عدد الدورات لعمليات المسح عالية الدقة لكل عنصر لضمان نسبة إشارة إلى ضوضاء كافية، مع حد أدنى من خمس دورات.
المجهر الإلكتروني الناقل عالي الدقة (HRTEM) والمجهر الإلكتروني الماسح (SEM)
تم إجراء توصيف الميكروهيكل باستخدام مجهر إلكتروني نافذ Thermo Fisher Talos F200S. تم فحص شكل العينات باستخدام مجهر إلكتروني ميداني Zeiss EVO MA 15.
طيف الرنين المغناطيسي للإلكترون (ESR)
جهاز مطياف ميكرو EMX CW من بروكير، يعمل في نطاق X-band ) ومزود بكافتي ER 4119 HS -WI عالية الحساسية، تم استخدامه لإجراء قياسات رنين دوران الإلكترون (ESR) عند 293 كلفن. تم تحفيز العينات بالضوء باستخدام مصباح زينون بقوة 300 واط، مع إزالة الأطوال الموجية التي تقل عن 420 نانومتر بواسطة فلتر قطع (LOT Oriel).
التحليل الكهروكيميائي
تم تصنيع الأقطاب الكهربائية العاملة من خلال إنشاء معلق ضوئي يتكون من 5 ملغ من مسحوق المحفز الضوئي و4 مل من الإيثانول، تلاه تشتت بالموجات فوق الصوتية. إجماليمن المعلق الناتج تم تطبيقه علىركيزة زجاجية من أكسيد القصدير المخلوط بالفلور (FTO) عبر تقنية السكب، تم تكرارها عشر مرات لضمان تغطية متساوية. ثم تم تجفيف الأقطاب في فرن وتم تسخينها. لمدة ساعة واحدة تحت تدفق الغاز. كانت الأقطاب الناتجة ذات طبيعة مسطحة. تم قياس استجابات التيار الضوئي العابر باستخدام جهاز عمل كهربائي كيميائي CHI660E (CHI Shanghai, Inc.) في تكوين قياسي مكون من ثلاثة أقطاب. كانت القطب المصنوع هو القطب العامل، بينما كان سلك البلاتين وتم استخدام أقطاب (KCl المشبع) كأقطاب مضادة ومرجعية، على التوالي. خلية كوارتز بأبعادتم استخدامه للقياسات. إشعاع الضوء المرئيتم توفيره بواسطة مصباح قوس زينون بقدرة 300 واط مزود بفلتر قطع حاد. كانت الجهد المطبق الأولي 0 فولت، وتم تعيين فترة جمع البيانات إلى 0.1 ثانية. تم إجراء قياسات طيف الامتثال الكهروكيميائي (EIS) باستخدام محلل الامتثال IM6e على مدى تردد يتراوح من 0.01 هرتز إلى تحت ظروف مظلمة. مصباح قوس زينون بقدرة 300 واط، بالتزامن مع فلتر قطع ( )، تم استخدامه كمصدر للضوء. كان المحلول الكهربائي يتكون من محلول مائي. تم إنشاء مخططات موت-شوتكي في نفسالالكتروليت، بتردد جهد متناوب قدره 1000 هرتز. تم تحضير محلول الالكتروليت عن طريق إذابة 1.48 جرام منفي الماء المنزوع الأيونات وضبط الحجم النهائي إلى 100 مل، مما ينتج عنه محلول بقيمة pHتم تحضير المحلول الكهربائي حديثًا قبل كل مجموعة من القياسات.
طيف الفلورسنت PL في الحالة المستقرة
تم استخدام مطياف FLS980 من إيدنبرغ إنسترومنتس لقياس طيف الفوتولومينسنس (PL) عند درجات حرارة مختلفة، بالإضافة إلى طيف الفوسفوريسنس. كانت طاقة ارتباط الإثيكون ثم تم تحديده وفقًا للمعادلة:
تحليل AFM-SKPM و KFPM
تم استخدام مجهر القوة الذرية بتقنية قياس الجهد الكهربائي (AFM-KPFM وAFM-SKPM) على مجهر القوة الذرية Bruker Dimension Icon لقياس توزيع الجهد السطحي للعينات.
تحليل جهد زيتا
تم إجراء قياسات جهد زيتا باستخدام جهاز مالفرن زيتاساير نانو ZS ZEN2600.
تحليل طيف الامتصاص العابر
نظام مضخة-استقصاء هيليوس (Ultrafast Systems LLC)، مدفوع بواسطة ليزر فمتوثانية (Coherent، )، تم استخدامه لقياس الامتصاص العابر. نبضات بروب الضوء الأبيض المستمرتم توليدها من خلال تركيز جزءمن الشعاع الأساسي بطول 800 نانومتر إلى بلورة CaF2 بسمك 1 مم. تم اشتقاق نبضات المضخة بطول 365 نانومتر من مضخم بصري بارامترى (TOPAS-800-fs).
تفاصيل الحسابات والمناقشات
وظيفة B3LYP معتم استخدام مجموعة الأساس، كما هو مطبق في Gaussian 09، لتحسين هندسة الجزيئات وتوصيف الهيكل الإلكتروني. تم حساب طاقات الإثارة وقوى الاهتزاز للأنواع العابرة باستخدام TD-DFT عند مستوى TD-B3LYP/6-311G*. تم دمج نماذج الذوبان الضمني وتصحيحات التشتت في الحسابات. تم استخدام Multiwfn (الإصدار 3.8) لتحليل المدارات الإلكترونية، وحسابات الجهد الكهروستاتيكي، وتحليل الثقوب والإلكترونات، بينما تم استخدام VMD (الإصدار 1.9.3) لتصور المدارات. تم بناء نموذج الهيكل الأولي لـ COF داخل Materials Studio (Materials Visualizer) باستخدام مجموعة الفضاء P222 (توبولوجيا sql، FOOCOF) المستمدة من مورد هيكل الكيمياء الشبكية (RCSR). بعد ذلك، تم تقليل الطاقة للهيكل باستخدام مجال القوة الشامل ضمن وحدة Forcite. تم إخضاع أنماط PXRD التجريبية لتعديل Pawley باستخدام وحدة Reflex. تم الحصول على الكثافات المتكاملة باستخدام ملاءمة ملف تعريف pseudo-Voigt. تضمنت المعلمات المعدلة معلمات وحدة الخلية (a، b، c)، ومعلمات FWHM (U، V، W)، ومعلمات الملف الشخصي (NA، NB)، وخطأ النقطة الصفرية. تم استخدام دالة متعددة الحدود من الدرجة العشرين لنمذجة الخلفية. أخيرًا، تم محاكاة أنماط PXRD من الهيكل المحسن باستخدام وحدة Reflex.
تم إجراء حسابات DFT الدورية باستخدام حزمة البرمجيات CP2K. تم وصف طاقة التبادل والتفاعل باستخدام دالة PBE، مع تضمين تصحيح التشتت DFT-D3 الخاص بـ Grimme والذي يتضمن نظام تخفيف BJ. تم استخدام مجموعة أساس MOLOPT، مع إضافة وظائف منتشرة مناسبة، ضمن إطار طريقة الموجات الغاوسية والموجات المستوية. تم استخدام مجموعة أساس DZVP-MOLOPT-GGA-GTH لتحسين الهندسة، وتحديد الطاقة، وتحليل أوضاع الاهتزاز، وحسابات نظرية الكثافة المعتمدة على الزمن. تم تعيين حد الطاقة لمجموعة الموجات المستوية إلى 600 إلكترون فولت، وتم ضمان التقارب ضمنهارتري. تم استخدام الهندسة المحسّنة، التي تتوافق مع هيكل الطاقة الأدنى، في الحسابات اللاحقة. تم تصحيح المعلمات الحرارية الكيميائية باستخدام تقريب المذبذب الهارموني ذو الدوران الصلب (RRHO) ضمن برنامج شيرمو في الظروف القياسية (1.01325 بار و298.15 كلفن). تمثل الجهد الكيميائي لزوج البروتون-الإلكترون (تم معادلته بنصف الغاز في الطور الغازيالجهد الكيميائي عند ظروف القطب الهيدروجيني القياسي (SHE)، وفقًا لنهج القطب الهيدروجيني الحاسوبي (CHE). تغير الطاقة الحرة لكل تم حساب الخطوة الأولية باستخدام المعادلة التالية:
بالنسبة لها، مسار التفاعل هو كما يلي:
توفر البيانات
جميع البيانات متاحة في النص الرئيسي أو المعلومات التكميلية. تم توفير بيانات المصدر مع هذه الورقة.
References
Wang, H. et al. Covalent organic framework photocatalysts: structures and applications. Chem. Soc. Rev. 49, 4135-4165 (2020).
Guan, Q. et al. Metalated covalent organic frameworks: from synthetic strategies to diverse applications. Chem. Soc. Rev. 51, 6307 (2022).
Liang, Z. et al. Covalent organic frameworks: fundamentals, mechanisms, modification, and applications in photocatalysis. Chem. Cata. 2, 2157-2228 (2022).
Lu, M. et al.Rational design of crystalline covalent organic frameworks for efficient photoreduction with . Angew. Chem. Int. Edit. 58, 12392-12397 (2019).
Huang, N. et al. Covalent organic frameworks: a materials platform for structural and functional designs. Nat. Rev. Mater. 1, 16068 (2016).
Diercks, C. S. et al. The atom, the molecule, and the covalent organic framework. Science 355, 1585 (2017).
Chen, Z. et al. Tuning excited state electronic structure and charge transport in covalent organic frameworks for enhanced photocatalytic performance. Nat. Commun. 14, 1106 (2023).
Zhao, C. et al. Recent advances in conjugated polymers for visible-light-driven water splitting. Adv. Mater. 32, 1907746 (2020).
Yang, J. et al. Protonated imine-linked covalent organic frameworks for photocatalytic hydrogen evolution. Angew. Chem. Int. Edit. 60, 19797-19803 (2021).
Ma, S. et al. Photocatalytic hydrogen production on a -carbonlinked covalent organic framework. Angew. Chem. Int. Edit. 61, e202208919 (2022).
Wang, X. et al. Sulfone-containing covalent organic frameworks for photocatalytic hydrogen evolution from water. Nat. Chem. 10, 1180-1189 (2018).
Shen, R. C. et al. Efficient photocatalytic hydrogen evolution by modulating excitonic effects in ni-intercalated covalent organic frameworks. Adv. Energy Mater. 13, 2203695 (2023).
Lu, M. et al. Confining and highly dispersing single polyoxometalate clusters in covalent organic frameworks by covalent linkages for photoreduction. J. Am. Chem. Soc. 144, 1861-1871 (2022).
Zhang, M. et al. Semiconductor/covalent-organic-framework Z-scheme heterojunctions for artificial photosynthesis. Angew. Chem. Int. Edit. 59, 6500-6506 (2020).
Li, Y. et al. In situ photodeposition of platinum clusters on a covalent organic framework for photocatalytic hydrogen production. Nat. Commun. 13, 1355 (2022).
Wang, S. et al. Cobaloxime-integrated covalent organic frameworks for photocatalytic hydrogen evolution coupled with alcohol oxidation. Angew. Chem. Int. Edit. 62, e202311082 (2023).
Wang, Y. et al. Unraveling the photo-induced dynamic behavior of COF-based Z-scheme heterostructure monolithic aerogels. Matter 7, 3145-3162 (2024).
Weng, W. et al. Chiral covalent organic framework films with enhanced photoelectrical performances. J. Am. Chem. Soc. 146, 13201-13209 (2024).
Mukherjee, S. et al. Scalable integration of coplanar heterojunction monolithic devices on two-dimensional . ACS Nano 14, 17543-17553 (2020).
Zhong, H. et al. Fused dithienogermolodithiophene low band gap polymers for high-performance organic solar cells without processing additives. J. Am. Chem. Soc. 135, 2040-2043 (2013).
Lan, Z. A. et al. Reducing the exciton binding energy of donor-acceptor-based conjugated polymers to promote charge-induced reactions. Angew. Chem. Int. Edit. 58, 10236-10240 (2019).
Lan, Z.-A. et al. A fully coplanar donor-acceptor polymeric semiconductor with promoted charge separation kinetics for photochemistry. Angew. Chem. Int. Edit. 60, 16355-16359 (2021).
Xu, K. et al. Ground-state electron transfer in all-polymer donoracceptor heterojunctions. Nat. Mater. 19, 738 (2020).
Park, S. et al. Temperature-dependent electronic ground-state charge transfer in van der waals heterostructures. Adv. Mater. 33, 2008677 (2021).
Sekita, M. et al. Intense ground-state charge-transfer interactions in low-bandgap, panchromatic phthalocyanine-tetracyanobuta-1,3diene conjugates. Angew. Chem. Int. Edit. 55, 5560-5564 (2016).
Deibel, C. et al. Role of the charge transfer state in organic donoracceptor solar cells. Adv. Mater. 22, 4097-4111 (2010).
Akaike, K. et al. Impact of ground-state charge transfer and polarization energy change on energy band offsets at donor/acceptor interface in organic photovoltaics. Adv. Funct. Mater. 20, 715-721 (2010).
Hao, L. et al. Fluorenone-based covalent organic frameworks with efficient exciton dissociation and well-defined active center for remarkable photocatalytic hydrogen evolution. Appl. Catal. B Environ. Energy. 330, 122581 (2023).
Yang, Y. et al. Engineering -ketoamine covalent organic frameworks for photocatalytic overall water splitting. Nat. Commun. 14, 593 (2023).
Weng, W. et al. The effect of enantioselective chiral covalent organic frameworks and cysteine sacrificial donors on photocatalytic hydrogen evolution. Nat. Commun. 13, 5768 (2022).
Bai, J. X. et al. Topology-induced local electric polarization in 2D thiophene-based covalent organic frameworks for boosting photocatalytic evolution. Chinese J. Catal. 59, 225-236 (2024).
Dai, L. et al. Enhancement of visible-light-driven hydrogen evolution activity of 2D π-conjugated bipyridine-based covalent organic frameworks via post-protonation. Angew. Chem. Int. Edit. 62, e202300224 (2023).
Dong, W. B. et al. Isomeric oligo(phenylenevinylene)-based covalent organic frameworks with different orientation of imine bonds and distinct photocatalytic activities. Angew. Chem. Int. Edit. 62, e202216073 (2023).
Li, Z. P. et al. Three-component donor- -acceptor covalent-organic frameworks for boosting photocatalytic hydrogen evolution. J. Am. Chem. Soc. 145, 8364-8374 (2023).
Lin, Z. et al. Boosting photocatalytic hydrogen evolution enabled by -supporting chiral covalent organic frameworks with parallel stacking sequence. Chinese. J. Catal. 64, 87-97 (2024).
Liu, T. et al. Ground-state electron transfer in all-polymer donor:acceptor blends enables aqueous processing of water-insoluble conjugated polymers. Nat. Commun. 14, 8454 (2023).
Kar, S. et al. Unveiling the role of a ground state charge transfer complex in carbon nanoparticles for highly efficient metal-free solar hydrogen production. J. Mater. Chem. A 12, 4712-4726 (2024).
Liao, Q. et al. Tailoring and modifying an organic electron acceptor toward the cathode interlayer for highly efficient organic solar cells. Adv. Mater. 32, 1906557 (2020).
Bao, Q. Y. et al. Trap-assisted recombination via integer charge transfer states in organic bulk heterojunction photovoltaics. Adv. Funct. Mater. 24, 6309-6316 (2014).
Guo, Y. et al. Perylenetetracarboxylic acid nanosheets with internal electric fields and anisotropic charge migration for photocatalytic hydrogen evolution. Nat. Commun. 13, 2067 (2022).
Yang, J. et al. A full-spectrum porphyrin-fullerene D-A supramolecular photocatalyst with giant built-in electric field for efficient hydrogen production. Adv. Mater. 33, 2101026 (2021).
Zhang, Z. J. et al. A highly crystalline perylene imide polymer with the robust built-in electric field for efficient photocatalytic water oxidation. Adv. Mater. 32, 1907746 (2020).
Li, J. et al. Giant enhancement of internal electric field boosting bulk charge separation for photocatalysis. Adv. Mater. 28, 4059-4064 (2016).
Aarnio, H. et al. Spontaneous charge transfer and dipole formation at the interface between P3HT and PCBM. Adv. Energy. Mater. 1, 792-797 (2011).
Bie, C. et al. A bifunctional catalyst enhances photocatalytic evolution and pyruvic acid synthesis. Angew. Chem. Int. Edit. 61, e202212045 (2022).
Yu, J. et al. Excited-state electronic properties in zr-based metalorganic frameworks as a function of a topological network. J. Am. Chem. Soc. 140, 10488-10496 (2018).
Qan, Y. et al. Photocatalytic molecular oxygen activation by regulating excitonic effects in covalent organic frameworks. J. Am. Chem. Soc. 142, 20763-20771 (2020).
Wei, Z. et al. Steering electron-hole migration pathways using oxygen vacancies in tungsten oxides to enhance their photocatalytic oxygen evolution performance. Angew. Chem. Int. Edit. 60, 8236-8242 (2021).
Barman, S. et al. Metal-free catalysis: a redox-active donoracceptor conjugated microporous polymer for selective visible-light-driven reduction to . J. Am. Chem. Soc. 143, 16284-16292 (2021).
Haldar, R. et al. Charge transfer in metal-organic frameworks. Chem. Commun. 59, 1569-1588 (2023).
Liu, D. et al. Rational design of PDI-based linear conjugated polymers for highly effective and long-term photocatalytic oxygen evolution. Adv. Mater. 35, 2300655 (2023).
Shen, R. C. et al. Realizing photocatalytic overall water splitting by modulating the thickness-induced reaction energy barrier of fluorenone-based covalent organic frameworks. Adv. Mater. 35, 2305397 (2023).
شكر وتقدير
يشكر المؤلفون المؤسسة الوطنية للعلوم الطبيعية في الصين (22378148، 52472110، 22308113) ومؤسسة العلوم الطبيعية في مقاطعة قوانغدونغ (2024A1515012433) على دعمهم.
مساهمات المؤلفين
ساهم R.C.S. وC.H. وL.H. بالتساوي في هذا العمل. قام R.C.S. وC.H. بتصميم التجارب. صمم R.C.S. وL.H. وX.L. واصطناعا وميزوا المواد. قام C.H. بإجراء الحسابات النظرية. قام G.J.L. بإجراء تحليل TAS. قام R.C.S. وL.H. بإجراء قياسات TEM. ساعد P.Z. في مناقشة المخطوطة. قام Q.Y. بمراجعة بيانات المخطوطة. تم تفسير البيانات من قبل جميع المؤلفين وتم إعداد المخطوطة بواسطة R.C.S. وX.L. ساهم جميع المؤلفين في هذا العمل، وقرأوا المخطوطة، وجميع البيانات مذكورة في النص الرئيسي والمواد التكميلية.
يجب توجيه المراسلات والطلبات للحصول على المواد إلى غويجي ليانغ، بينغ زانغ، تشيانغ يوي أو شين لي.
معلومات مراجعة الأقران تشكر مجلة Nature Communications يونغفا زو والمراجعين المجهولين الآخرين على مساهمتهم في مراجعة هذا العمل. يتوفر ملف مراجعة الأقران.
ملاحظة الناشر: تظل شركة سبرينجر ناتشر محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
الوصول المفتوح. هذه المقالة مرخصة بموجب رخصة المشاع الإبداعي النسب-غير التجاري-عدم الاشتقاق 4.0 الدولية، التي تسمح بأي استخدام غير تجاري، ومشاركة، وتوزيع، وإعادة إنتاج في أي وسيلة أو صيغة، طالما أنك تعطي الائتمان المناسب للمؤلفين الأصليين والمصدر، وتوفر رابطًا لرخصة المشاع الإبداعي، وتوضح إذا قمت بتعديل المادة المرخصة. ليس لديك إذن بموجب هذه الرخصة لمشاركة المواد المعدلة المشتقة من هذه المقالة أو أجزاء منها. الصور أو المواد الأخرى من طرف ثالث في هذه المقالة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة، ما لم يُشار إلى خلاف ذلك في سطر الائتمان للمادة. إذا لم تكن المادة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة وكان استخدامك المقصود غير مسموح به بموجب اللوائح القانونية أو يتجاوز الاستخدام المسموح به، ستحتاج إلى الحصول على إذن مباشرة من صاحب حقوق الطبع والنشر. لعرض نسخة من هذه الرخصة، قم بزيارة http:// creativecommons.org/licenses/by-nc-nd/4.0/.
(ج) المؤلفون 2025
معهد هندسة الكتلة الحيوية، المختبر الرئيسي لموارد واستخدام نباتات الطاقة، وزارة الزراعة والشؤون الريفية، جامعة جنوب الصين الزراعية، قوانغتشو، الصين. المختبر الرئيسي للمواد والطاقة المستندة إلى البيولوجيا بوزارة التعليم، كلية المواد والطاقة، جامعة جنوب الصين الزراعية، قوانغتشو، الصين. المختبر الرئيسي لمقاطعة قوانغدونغ لاستخدام وحفظ الموارد الغذائية والطبية في المنطقة الشمالية، جامعة شاوغوان، شاوغوان، الصين. المختبر الرئيسي لمواد وأجهزة الإلكترونيات الضوئية منخفضة الأبعاد في هوبى، جامعة هوبى للفنون والعلوم، شيانغيانغ، الصين. المركز الوطني للتعاون الدولي في المواد البيئية منخفضة الكربون المصممة (CDLCEM)، كلية علوم وهندسة المواد، جامعة تشنغتشو، هنان تشنغتشو، الصين. ساهم هؤلاء المؤلفون بالتساوي: رونغتشين شين، كان هوانغ، لي هاو. البريد الإلكتروني: Guijie-liang@hbuas.edu.cn; zhangp@zzu.edu.cn; 2249659235@qq.com; Xinli@scau.edu.cn
Ground-state charge transfer in singlemolecule junctions covalent organic frameworks for boosting photocatalytic hydrogen evolution
Received: 30 June 2024
Accepted: 27 February 2025
Published online: 12 March 2025
Check for updates
Rongchen Shen , Can Huang , Lei Hao , Guijie Liang (1) , Peng Zhang , Qiang Yue & Xin Li (1)
Ground-state charge transfer plays a vital role in improving the photocatalytic performance of D-A type covalent organic frameworks. However, limited studies have explored the modulation of photocatalytic performance in COFsbased photocatalysts through ground-state charge transfer. Here we show the formation of extremely intense ground-state charge transfer via a unique covalent bonding approach. We transform three-dimensional stacked COFbased S-scheme heterojunctions (FOOCOF-PDIU) into co-planar single-molecule junctions (FOOCOF-PDI). This co-planar single-molecule junction structure exhibits strong ground-state charge transfer compared to the traditional randomly stacked heterojunctions and individual COFs. Ground-state charge transfer induces charge redistribution and dipole moment formation, which enhances the built-in electric field intensity in single-molecule junctions. This enhanced built-in electric field promotes exciton dissociation and charge separation, resulting in improved photocatalytic efficiency. Therefore, a stable molecule-decorated COF with broad light absorption has been successfully obtained, whose hydrogen evolution rate can reach . This work opens an avenue for exploiting photocatalytic mechanisms in COFs based on ground-state charge transfer effects.
Due to their tunable electronic properties and high surface areas, covalent organic frameworks (COFs)-based photocatalysts have recently gained increasing attention as promising candidates for solar photocatalysis , which have emerged as potential photocatalysts for solar-driven water splitting to produce hydrogen . These materials have shown great potential for photocatalytic applications but face
challenges related to their poor charge carrier separation and high exciton binding energy, which limit their efficiency in the photocatalytic process .
Constructing heterojunction is an effective approach to enhance the photogenerated charge carrier separation efficiency in COF-based semiconductors . The heterojunction formed by stacking ideal
superimposed layers facilitates the out-of-plane charge transfer, while the actual stacking deviations are unfavorable for photogenerated charge separation . While optimizing the planarity of semiconductors can enhance the exciton dissociation and charge carrier separation in semiconductor photocatalysts . Therefore, converting the stacked heterostructures into co-planar molecular junctions will greatly enhance the interfacial charge carrier transport efficiency between heterojunctions. However, the molecular junction-enhanced mechanism has been less studied so far, especially the ground-state charge transfer (GSCT) has rarely been reported.
GSCT enhances the separation of photogenerated electron-hole pairs through spatial and energetic mechanisms. Spatially, GSCT facilitates electron transfer from donor to acceptor molecules, increasing the distance between electrons and holes, and thus decreasing recombination. Energetically, the GSCT process results in a more favorable environment for hole transport on the donor due to the increased positive charge density. Moreover, the energy level alignment between the donor and acceptor after charge transfer reduces the Coulombic binding energy, further minimizing recombination . Interestingly, such GSCT is posited to play a mediating role in charge separation within D-A organic heterojunctions, manifesting as a real electronic state. The GSCT is thought to modulate the kinetics of charge separation and recombination processes, thereby enhancing the performance of organic photovoltaic devices . However, in most D-A type COFs, the formation of charge-separated states is typically observed under photoexcitation. The GSCT in D-A type COFs has not been extensively investigated in photocatalysis.
Here, the synthesis and study of two FOOCOF-based heterojunction photocatalysts, three-dimensional stacked COF-based S-scheme heterojunctions, and co-planar single-molecule junctions. Studies have revealed that single-molecule junctions FOOCOF-PDI, featuring covalent bonding, exhibit a stronger GSTC effect than FOOCOF-PDIU and FOOCOF. The crystal and chemical structure of the resulting FOOCOFPDI were confirmed using powder X-ray diffraction (PXRD), Fouriertransform infrared (FT-IR) spectroscopy, and solid-state 13 C nuclear magnetic resonance (13 C NMR). Both experimental and theoretical calculations show that bridging PDI molecules to the end of COFs through GSTC results in COF-PDI possessing a stronger dipole moment and built-in electric field, which promotes exciton dissociation and carrier migration. Under visible light irradiation, with Pt as a cocatalyst, its photocatalytic hydrogen production activity reached .
Results and Discussion
FOOCOF was synthesized the same as in our previous work . Under the presence of acetic acid, FOOCOF-PDI was synthesized through
the condensation of aldehyde groups at the end of FOOCOF with the amine groups of PDINH in a mixed solvent of o-dichlorobenzene/ n-butanol ( ) at , as detailed in the additional information (Fig. 1). The FOOCOF/PDINH S-scheme heterojunction (FOO-COF-PDIU) is formed by ultrasound-assisted mixing of FOOCOF and PDINH.
The PXRD pattern of FOOCOF is shown in Fig. 2, featuring prominent peaks at and (Fig. 2A and Figure S1), corresponding to the 001, 211, and 102 planes of FOOCOF ( and with the residual factors of and ). The structure of FOOCOF remains unchanged after PDI modification, indicating the covalent connection of PDI molecules shows a low effect on FOOCOF (Fig. 2B). XRD pattern of FOOCOF-PDIU shows distinct PDINH peaks (Figure S2). In the FTIR spectrum, the peak at corresponding to the bond strengthened , indicating the successful covalent connection of PDINH at the end of FOOCOF (Fig. 2C). Meanwhile, the peak at 1591 and were attributed to and , respectively. The NMR spectrum revealed a peak at 184.5 ppm , which is attributed to the carbonyl group of the fluorenone moiety. Peaks at 137 and corresponded to C -N and C -NH bonds, confirming the successful synthesis of FOOCOF. In the NMR spectrum of FOOCOF-PDI, the peak attributed to C-N exhibits a enhanced intensity, confirming the successful covalent connection of PDINH at the end of FOOCOF (Fig. 2D). XPS analysis further confirmed the structural characteristics of FOOCOF-PDI. The spectrum was resolved into three peaks at binding energies of 284.8, 286.1, and 288.9 eV , corresponding to , and , respectively. The N 1s spectrum displayed peaks at 399.6 and 398.8 eV , attributed to the nitrogen atoms of the and bonds. In the spectrum, the peak at 531.1 eV corresponded to the bonds of FOOCOF and PDI ; the peak at 532.9 eV was attributed to oxygen species adsorbed (Figures S3-5). The prepared samples’ UV-vis diffuse reflectance spectra (UV-DRS) display an extended absorption tail reaching up to 750 nm (Fig. 2E and Figure S6), indicating a broad harvesting range. Therefore, the bandgaps for FOOCOF, FOOCOF-PDI and PDINH are determined to be and 1.89 eV , respectively (Figure S7). According to VB-XPS, the valence bands of FOOCOF, FOOCOF-PDI and PDINH are determined to be and 2.37 V vs RHE, respectively. . Therefore, the valence bands of FOOCOF, FOOCOF-PDI and PDINH are determined to be and 1.96 V vs NHE, respectively (Figure S8-10). Accordingly, the conduction bands of FOOCOF, FOOCOF-PDI and PDINH were and 0.07 V , respectively (Fig. 2F and Figure S11). The prepared samples thermodynamically meet the requirements for photocatalytic water splitting. Meanwhile, the thermodynamic conditions are satisfied for the formation of a S-scheme heterojunction between FOOCOF and PDINH.
Fig. 1 | Synthesis of COF photocatalysts. Scheme of synthesis of the FOOCOF and FOOCOF-PDI.
Fig. 2 | Characterization of COF-based photocatalysts. A Experimental PXRD patterns of FOOCOF with corresponding Pawley refinement, simulated eclipsed (AA) stacking patterns, differences between refinement and experimental, and Bragg positions, (B) PXRD patterns, (C) FTIR spectrum, (D)
Solid-state CP/MAS NMR spectra, (E) UV-vis differential reflectance spectral pattern and (F) band level of FOOCOF and FOOCOF-PDI. G TEM image of FOOCOF, (H) High-resolution TEM (HRTEM) images of FOOCOF; (I) TEM images of FOOCOF-PDI.
SEM and TEM were employed to investigate the morphological impact of PDINH covalently connecting COFs. The SEM images (Figures S12-14) reveal the smooth, layered stacking morphology of FOOCOF, and the covalent attachment of PDINH does not induce any discernible changes in the sample’s morphology. The TEM images (Fig. 2G and Figure S15) further divulge the nanosheet structure of FOOCOF. Additionally, the obvious lattice stripes can be seen in the figure, indicating the high crystallinity of the prepared sample (Fig. 2H). After the formation of single-molecule junctions, no obvious change could be found in the TEM images (Fig. 2I and Figure S16). Meanwhile, no PDINH molecule could be found in the FOOCOF-PDI (Fig. 2I and Figure S17). The reversible nitrogen adsorption isotherm of FOOCOF exhibits the typical type IV isotherm characteristic and displays mesoporous features (Figure S18). The BET surface area of FOOCOF is calculated to be . The pore size distribution is 1.3 nm , which is in agreement with the theoretical model of 1.3 nm . Covalently connecting PDI results in a slight decrease in the BET surface area to , yet the pore size remains unchanged. Thermogravimetric analysis (TGA) was performed to investigate the thermal stability of the COFs. As shown in Figure S19, FOOCOF
decomposes at , while the thermal stability is improved after covalently connecting PDI.
In-situ photodeposition of Pt as a cocatalyst and 0.1 M ascorbic acid as a sacrificial agent were utilized in the photocatalytic water splitting under visible light. The photocatalytic hydrogen evolution activity of FOOCOF and FOOCOF-PDI was evaluated with as the cocatalyst. FOOCOF exhibited an average hydrogen evolution rate of under visible light irradiation for three hours. After formatting S-scheme heterojunctions, the average hydrogen evolution rate of FOOCOF-PDIU increased to . Upon the formation of the single molecular junction, FOOCOF-PDI showed a notable enhancement in the average hydrogen evolution rate, reaching under visible light irradiation for three hours, indicating a improvement in the photocatalytic activity of COFs upon the formation of single molecular junctions (Fig. 3A, B). Meanwhile, the performance is comparable to that of the other COF-based photocatalysts (Table S1) . When the Pt content is less than , the hydrogen production activity of the sample is lower than due to the lack of active sites. Due to the light shielding effect, the photocatalytic activity of the samples gradually
Fig. 3 | Photocatalytic performnace and Photophysical characterization.
A Time course of photocatalytic evolution of as-prepared samples. B Average hydrogen evolution rate and (C) cycling stability for as prepared samples. D UV-vis absorption, (E) ESR spectra in the dark and (F) surface potential difference of
FOOCOF, FOOCOF-PDIU and FOOCOF-PDI. Surface potentials via KPFM for (G) FOOCOF-PDIU and (H) FOOCOF-PDI. I Normalized built-in electric field strength of FOOCOF, FOOCOF-PDIU and FOOCOF-PDI.
decreased with the increase of Pt content and catalysts dose (Figures S20 and S21). Considering the weighing error when using 1 mg of catalyst, we still take the hydrogen production activity at 2 mg as the standard. The hydrogen evolution rate of FOOCOF-PDI is greatly dependent on the pH of the reaction solution (Figure S22). The hydrogen production activity is higher under acidic conditions in ascorbic acid solution than under alkaline conditions in TEOA solution. In addition, no is produced when ethanol is used as a sacrificial agent. FOOCOF-PDI maintained 90 percent stability in the 24 -hour cycle test (Fig. 3C). As evidenced by the experimental results, the sample maintained stability. The black plots on the surface of COF could attributed to the cocatalysts Pt (Figures S23 and S24). To analyze the wavelength-dependent photocatalytic performance of FOOCOFPDIs, the apparent quantum efficiency (AQE) as a function of wavelength was monitored (Figure S25). The AQE value of FOOCOF is up to at 420 nm .
When ultraviolet light irradiates the charge transfer complex, electrons can undergo a transition from the ground state to the charge-transfer level, resulting in the formation of a new absorption band. This absorption band is referred to as the charge transfer absorption band. The enhanced GSCT was first confirmed by UV-vis absorption (Fig. 3D). Notably, after the formation of FOOCOF-PDI, a
new absorption peak arises at a longer wavelength ( ), which indicates the formation of a new charge-transfer complex . Then, we found that FOOCOF-PDI exhibited a distinct dark-state signal in the dark state through ESR, evidencing GSCT (Fig. 3E and Figure S26). Furthermore, the GSCT impact on the carrier extraction capability were investigated using scanning Kelvin probe microscopy (SKPM). As shown in Fig. 3F and Figure S27-28, the potential of FOOCOF-PDI is approximately 0.32 V higher than that of pure FOOCOF and FOOCOFPDIU, indicating interface vacuum level shift due to electron transfer . These results corroborate the GSCT in FOOCOF-PDI. We measured the fluorescence lifetime of the prepared samples, and the lifetime of FOOCOF-PDI is longer than that of FOOCOF. The average lifetimes for both FOOCOF and FOOCOF-PDI were fitted by using biexponential decay kinetics. The calculated average lifetime of FOOCOF is 21.3 ns . However, upon formation single-molecule junctions, the average lifetime of FOOCOF-PDI increases more than 4 times (Figure S29). The enhancement in the lifetime upon complex formation between FOOCOF and PDI suggests the formation of long-lived free carriers.
GSCT can result in the generation of dipole moments . Generally, the dipole moment of a single molecule can effectively drive charge transfer within the molecule. Once the molecular units undergo highly
Fig. 4 | Charge separation characterization of COF photocatalysts.
A-C Transient absorption spectra of FOOCOF. D Normalized transient absorption kinetics for FOOCOF. E-G Transient absorption spectra of FOOCOF-PDI.
H Normalized transient absorption kinetics for FOOCOF-PDI. Temperature-
dependent photoluminescence spectrum of integrated PL intensity as a function of the temperature of (I) FOOCOF and (J) FOOCOF-PDI (inset: temperature-dependent PL spectra from 80 to 290 K). K Photocurrent response and (L) Mott-Schottky curve of FOOCOF, FOOCOF-PDIU and FOOCOF-PDI.
ordered stacking, an intrinsic electric field is generated; otherwise, the dipoles would cancel out due to the disordered assembly . We analyzed the built-in electric field of the prepared samples. Due to the existence of dipoles, the built-in electric field is enhanced. In general, the strength of the built-in electric field can serve as a kinetic factor driving charge separation, thus necessitating a quantitative evaluation .According to the theory of Zhang et al., the determination of the material’s embedded electric field is monotonically positively correlated with its surface charge density and zeta potential . Utilizing KPFM (Kelvin probe force microscopy) to detect the surface charge density of the sample, it is shown that the highest potential of FOOCOF-PDI reaches up to 103.5 mV (Fig. 3G, H). Combined with the zeta potential results shown in Figure S30, it confirms that the built-in electric field of FOOCOF-PDI are 2.1 and 1.5 times stronger than that of FOOCOF and FOOCOF-PDIU (Fig. 31).
The interface dipoles caused by charge transfer can promote the dissociation of excitons into free carriers, as demonstrated in numerous prior studies . The early detection of excitons (within 0.5 ps ) in the prepared COFs was carried out using femtosecond transient absorption spectroscopy (fs-TAS). The prepared sample exhibited a negative peak at and a positive peak at , which are attributed to the ground-state bleach (GSB) and excitedstate absorption (ESA), respectively. These signals correspond to the excited-state relaxation. Therefore, the photogenerated electron and hole pairs in the sample contribute to both the GSB and the broad ESA signals (Fig. 4A, E, Figure S31 and S32 A). It has been confirmed that the photogenerated electrons mainly participate in the interfacial transfer . Figure 4A-H and Figure S32 A-C show that the prepared sample exhibited a broad negative peak (F1) near 500 nm . Combined with steady-state fluorescence, it can be inferred that F1 is the stimulated emission band of photogenerated excitons. Notably, the
broadening of the fs-TAS absorption peak corresponds to the relaxation of bound excitons and the increase in carrier density . Hence, compared to FOOCOF, the broadening of F1 in FOOCOF-PDI indicates a weakening exciton effect and a more efficient exciton dissociation. For further quantitative analysis, a double-exponential function was used to fit the decay of F1, with the results shown in Fig. 4D, H. is related to rapid exciton dissociation at high exciton densities, whereas is associated with rapid charge recombination and capture. The smaller value for FOOCOF-PDI ( 0.52 ps ) compared to FOOCOF ( 0.62 ps ) confirms the more effective exciton dissociation of FOOCOFPDI, consistent with the binding energy of the excitons. After 0.5 ps , rapid separation of photoinduced electron-hole pairs occurs, coexisting with some fluorescence emission, and the remaining photoinduced electrons undergo an electron capture process. When the laser excites the photo-excited electrons from the CB of the material to higher energy levels, it leads to the capture of the reduced photoinduced absorption or the reduction in transmittance . Therefore, the intensity and decay of the photoinduced absorption represent the capture and reduction of charges. The increase in indicates the beneficial effect of capturing long-lived shallow electrons on the transfer of active electrons, achieving high-efficiency photocatalytic hydrogen production performance. Through comparison between single molecule junctions and heterojunctions, it was further found that the formation of heterojunctions can capture long-lived electrons, although the electron lifetime is relatively shorter compared to singlemolecule junctions (Figure S32). We measured the temperaturedependent photoluminescence spectrum to test the exciton binding energy (Fig. 4I-G). The intensity of PL for both FOOCOF and FOOCOFPDI decreased with increasing the temperature from 80 to 290 K , corresponding to the thermally activated nonradiative recombination process. The exciton binding of FOOCOF-PDI ( 18.35 meV ) was lower
Fig. 5 | Computational studies of the chagre separation. Electrostatic surface potential (ESP) distributions of (A) FOOCOF and (B) FOOCOF-PDI, blue and red represented electron accumulation and depletion, respectively. C Electric potential along axile for (100) plane. D Exciton binding energies and electron-hole
distributions of (D) FOOCOF and (E) FOOCOF-PDI, blue and green represent positive and negative phases. TD-DFT calculated electronic transition of (F) FOOCOF and (G) FOOCOF-PDI, blue and green represent positive and negative phases.
than that of FOOCOF ( 25.36 meV ). Reducing the exciton binding energy enhances the quantum efficiency of semiconductor materials, which refers to the efficiency of generating an electron upon absorbing a photon. This higher quantum efficiency translates to greater conversion of light energy into chemical energy, leading to improved hydrogen production efficiency. Decreasing the exciton binding energy also extends the lifetime of electrons and holes. The prolonged lifetime allows electrons more time to migrate to the catalyst surface and participate in reduction reactions, ultimately enhancing photocatalytic efficiency. The vigorous photocurrent intensity and lower slope mean the enhanced charge concentration in FOOCOF-PDI from the efficient exciton dissociation (Fig. ). We have measured electrochemical impedance spectroscopy (EIS) to detect the role of single-molecule junction during the photocatalytic reaction. In the EIS spectrum, the as prepared samples typically exhibits two distinct regions, each reflecting different electrochemical processes. In the high frequency region, the impedance spectrum displays a smaller semicircle with a slope close to 0 . This region primarily reflects charge transfer processes at the electrode surface, including electron transfer and ion migration between the electrode material and the electrolyte. The diameter of the semicircle represents the charge transfer resistance, with smaller values indicating easier charge transfer. In the low frequency region, the impedance spectrum exhibits a larger semicircle with a slope approaching -1 . This region primarily reflects the diffusion of ions within the electrode material, i.e., the diffusion rate of ions within the electrode structure. A slope close to -1 indicates Warburg impedance, signifying that ionic diffusion is the primary factor controlling the reaction rate. FOOCOF and FOOCOF-PDIU exhibit a larger radius compared to FOOCOF-PDI, indicating that the single-molecule junction can reduce electron-transfer impedance in FOOCOF-PDI (Figure S33).
To investigate the influence of GSCT on exciton dissociation, the electrostatic surface potential (ESP) of the optimized structures of
FOOCOF and FOOCOF-PDI molecules was calculated, as depicted in Fig. 5A, B. The calculation of the multipole moment expansion of the molecular ESP revealed the broadest distribution of multipole moments for FOOCOF-PDI DFT, indicating a clear charge separation center. Hence, the single-molecule junction ensures the stable existence of a strong intrinsic electric field , which is crucial for exciton dissociation, carrier separation and migration (Figure S34). Without a single-molecule junction, the electrons involved in the reaction tend to accumulate at the edges of the COF material. They are unable to interact with the reaction solvent immediately. However, after forming a single-molecule junction, it can be observed that the electrons tend to accumulate in the middle of the PDI, as the entire PDI molecule is in direct contact with the solvent, serving as the reaction site (Fig. 5C). Therefore, the formation of the molecular junction promotes the dissociation of excitons and guides free electrons to the interface edge sites, thereby enhancing the separation of charge carriers, reducing the recombination of charge carriers, and improving the efficiency of interfacial reactions. The structures were fully optimized, followed by time-dependent density functional theory (TD-DFT) calculations to gain insights into the exciton generation properties of the COF photocatalyst. The electron-hole distribution in the first excited singlet state (S2) of the two COFs indicates a difference in exciton binding energy ( ) due to localized excitation. The exciton binding energy of FOOCOF-PDI is 1.887 eV , lower than the 2.274 eV of FOOCOF, indicating a weaker exciton effect in FOOCOF, consistent with its photocatalytic activity (Fig. 5D, E). Considering that calculating only fragments would be affected by edge and conjugation effects, we expanded the model and performed calculations on FOOCOF and FOOCOF-PDI (Figure S35). It was found that, under photoexcitation, the calculated results with continuous fragment structures are consistent with the fragment results. This suggests that the exciton effect may be one of the controlling factors in this process. FOOCOF and FOOCOF-PDI mainly absorb peaks composed of transitions from SO to
S2. The S2 state of FOOCOF can be described as HOMO-1 . The transition of S2 exhibits a molecular intra-charge transfer characteristic: electrons transfer from TP to the fluorenone site. Therefore, the effective separation of electron-hole pairs enhances the lifetime of carriers . After the formation of the single-molecule junction, the S2 state of FOOCOF can be described as (Fig. 5F, G). Electrons transfer from FOOCOF to the PDI site. This indicates that electrons tend to reside in the middle of the PDI, and the entire PDI molecule is the reaction site directly in contact with the solvent. Furthermore, we calculated the Gibbs free energy change ( ) for hydrogen adsorption at O atoms in both fluorenone sites and PDI sites accordingly from FOOCOF and FOOCOFs-PDI. It is noticeable that the fluorenone sites are energetically more favorable than the PDI sites. Moreover, the hydrogen adsorption free energy tends to decrease upon the formation of single-molecue junctions (Figure S36).
In summary, we have successfully designed and synthesized the single-molecule junction of FOOCOF-PDI. Following the formation of this molecular junction, the photocatalytic activity of FOOCOF has improved. In the presence of Pt as a cocatalyst, the photocatalytic hydrogen evolution activity of FOOCOF-PDI reaches , which is 2.57 times higher than that of FOOCOF. The experimental results indicate that the formation of the single-molecule junctions induces a GSCT to enhance the intrinsic electric field. The dipole effect effectively enhances exciton dissociation. This work has theoretical and application value for deeply studying the GSCT-inducing exciton dissociation and charge separation in COF-based photocatalysts.
Method
Materials
All chemicals and reagents were of analytical grade materials and used as received without further purification. The o-1,2-Dichlorobenzene (1,2-Dichlorobenzene, 99%), anhydrous n-BuOH (n-Butanol, 99.4%), Pyrrolidine (Py, 98%), Ascorbic acid (>99.0%), 2,7-Dinitro-9-fluorenone ( ) and, 4,9,10-Perylenetetracarboxylic dianhydride were purchased from Macklin Chemicals 2,4,6-trihydroxybenzene-1,3,5-tricarbaldehyde was supplied by Jilin Chinese Academy of SciencesYanshen Technology Co. Ltd.
Synthesis of PDINH
The PDINH monomer was prepared through a nucleophilic substitution reaction between 3,4,9,10-Perylenetetracarboxylic dianhydride and hydrazine monohydrate. In a typical procedure, of the dianhydride was dissolved in 100 mL of ethanol within a 250 mL roundbottom flask under a nitrogen environment. The mixture was cooled to using an ice bath, and of hydrazine monohydrate was then introduced slowly over a 10 -minute period. Following 30 min of agitation in the ice bath, the reaction was warmed to ambient temperature before being heated under reflux at for a duration of 1 h .
Synthesis of 2,7-diamino-9 H-fluoren-9-one
In a typical procedure, of 2,7-dinitro-9-fluorenone was dissolved in 375 mL of ethanol at room temperature. To this solution was added a separately prepared aqueous solution ( 650 mL ) of sodium sulfide nonahydrate and sodium hydroxide . The resulting mixture was refluxed for 5 h and then cooled to . The precipitate was collected by filtration and washed extensively with water, sodium hydroxide solution, water, cold ethylene glycol, ether, and hexane. Vacuum drying, followed by recrystallization from acetone-ethanol ( ), afforded of 2,7-diamino-9H-fluoren-9-one as purple needle-shaped crystals.
Synthesis of FOOCOF
In a Pyrex tube, of 2,4,6-trihydroxybenzene-1,3,5tricarbaldehyde and of 2,7-diamino-9H-fluoren-9-
one were combined with o-1,2-Dichlorobenzene ( 0.75 mL ), n-BuOH , and pyridine . The mixture was subjected to ultrasonic mixing and then degassed by three freeze-pump-thaw cycles to a vacuum of 100 mTr orr before being sealed. The sealed tube was heated to and maintained at this temperature for 72 h . Following the reaction, the brown solid was separated via centrifugation and purified by washing with tetrahydrofuran and anhydrous acetone .
Synthesis of FOOCOF-PDI
In a Pyrex tube, PDINH ( 1 mg ) and FOOCOF ( 10 mg ) were combined with o-1,2-Dichlorobenzene ( 0.75 mL ), n-BuOH ( 0.25 mL ), and acetic acid . The mixture underwent three freeze-pump-thaw cycles to remove dissolved gases, achieving a vacuum pressure of 100 mTorr , before being sealed. The sealed tube was heated to for 36 h . After the reaction, the resulting brown solid was filtered, with DMSO and ethanol aid the filtration, and purified by sequential Soxhlet extraction with methanol, acetone, and hexane (each for 24 h ).
Photocatalytic hydrogen evolution method and parameters
We carried out photocatalytic reactions in a 100 mL three-necked flask, using a 300 W xenon lamp (PLS-SXE300, ) to simulate sunlight at an intensity of . We used a 420 nm cutoff filter to remove UV light. Specifically, we dispersed 2 mg of photocatalyst in 40 mL of 0.1 M ascorbic acid solution. We used ultrasonication to ensure uniform dispersion and then purged with to create an anaerobic environment. We allowed the reaction to proceed for 1 h under continuous illumination. Finally, we extracted 0.4 mL of the generated gas intermittently and analyzed it using a gas chromatograph (GC-9560, TCD). We calculated the apparent quantum efficiency (AQE) of the samples as follows.
where is the mole number of product obtained (mol), is the Avogadro constant is the optical density is the light irradiation area ( ), is the light irradiation time ( s ), is the monochromatic light wavelength ( m ), is Planck’s constant ( ), and is the speed of light ( ).
Powder X-ray diffraction (PXRD) analysis
PXRD analysis was performed on a Rigaku Smartlab diffractometer utilizing Cu K radiation . The diffractometer was operated in reflection geometry at 40 kV and 40 mA .
FTIR analyses
A Nicolet Avatar 6700 FT-IR spectrometer (Thermo Fisher, USA) was utilized to record FTIR spectra.
A Shimadzu UV-2600 UV-vis-NIR spectrophotometer was employed to analyze the UV-Vis diffuse reflection spectra of the photocatalysts.
Physisorption measurements
The porous properties of the COF materials were characterized by nitrogen adsorption at 77 K using an Autosorb-iQ-MP analyzer. Before measurement, samples were outgassed under vacuum ( ) at room temperature and subsequently heated at for 12 h to ensure complete removal of adsorbed species. The
specific surface area was determined by the BET method applied to the nitrogen adsorption isotherm data in the relative pressure range of 0.01-0.9
X-ray photoelectron spectroscopy
Surface elemental analysis was performed by XPS on a VG ESCALAB 250 instrument, utilizing radiation ( ) as the excitation source. The analysis parameters were as follows: spot size , base pressure , operating voltage 12 kV , and filament current 6 mA . Survey spectra were collected with a pass energy of 100 eV and a step size of 1 eV , while high-resolution spectra were obtained with a pass energy of 50 eV and a step size of 0.1 eV . Charge correction was applied to all spectra by referencing the C 1s peak to 284.80 eV . The number of cycles for high-resolution scans was optimized for each element to ensure adequate signal-to-noise ratio, with a minimum of five cycles.
High-resolution transmission electron microscopy (HRTEM) and scanning electron microscopy (SEM)
Microstructural characterization was carried out using a Thermo Fisher Talos F200S TEM. The morphology of the samples was examined using a Zeiss EVO MA 15 field emission SEM.
Electron spin resonance spectroscopy (ESR)
A Bruker EMX CW micro spectrometer, operating at X-band ( ) and equipped with a high-sensitivity ER 4119 HS -WI cavity, was used to perform electron spin resonance (ESR) measurements at 293 K . Samples were photo-excited using a 300 W xenon lamp, with wavelengths below 420 nm removed by a cut-off filter (LOT Oriel).
Electrochemical analysis
The working electrodes were fabricated by first creating a photocatalyst slurry consisting of 5 mg of the photocatalyst powder and 4 mL of ethanol, followed by ultrasonic dispersion. A total of of the resulting slurry was applied to a fluorine-doped tin oxide (FTO) glass substrate via drop-casting, repeated ten times to ensure uniform coverage. The electrodes were then dried in an oven and annealed at for 1 h under a gas flow. The resulting electrodes were planar in nature. Transient photocurrent responses were measured using a CHI660E electrochemical workstation (CHI Shanghai, Inc.) in a standard three-electrode configuration. The fabricated electrode served as the working electrode, while a Pt wire and (saturated KCl ) electrode were used as the counter and reference electrodes, respectively. A quartz cell with dimensions of was used for the measurements. Visible light irradiation was provided by a 300 W Xe arc lamp equipped with a sharp cut-off filter. The initial applied voltage was 0 V , and the data acquisition interval was set to 0.1 s . Electrochemical impedance spectroscopy (EIS) measurements were conducted using an IM6e impedance analyzer over a frequency range from 0.01 Hz to under dark conditions. A 300 W Xe arc lamp, in conjunction with a cut-off filter ( ), was used as the light source. The electrolyte consisted of a aqueous solution. Mott-Schottky plots were generated in the same electrolyte, with an AC potential frequency of 1000 Hz . The electrolyte solution was prepared by dissolving 1.48 g of in deionized water and adjusting the final volume to 100 mL , resulting in a solution with a pH value of . The electrolyte was freshly prepared prior to each set of measurements.
Fluorescence spectrum Steady-state PL
An Edinburgh Instruments FLS980 spectrometer was used to measure photoluminescence (PL) spectra at varying temperatures, as well as the phosphorescence spectrum. The exciton binding energy was
subsequently determined according to the equation::
AFM-SKPM and KFPM analysis
Kelvin probe force microscopy (AFM-KPFM and AFM-SKPM) was employed on a Bruker Dimension Icon atomic force microscope to measure the surface potential distribution of the samples.
Zeta potential analysis
Zeta potential measurements were conducted using a Malvern Zetasizer Nano ZS ZEN2600.
Transient absorption spectroscopy analysis
A Helios pump-probe system (Ultrafast Systems LLC), driven by a femtosecond laser (Coherent, ), was used for transient absorption spectroscopy. White-light continuum probe pulses were generated via focusing a fraction of the 800 nm fundamental beam onto a 1 mm CaF2 crystal. The 365 nm pump pulses were derived from an optical parametric amplifier (TOPAS-800-fs).
Calculations details and discussions
The B3LYP functional with the basis set, as implemented in Gaussian 09, was used to optimize molecular geometries and characterize electronic structure. Excitation energies and oscillator strengths of the transient species were calculated using TD-DFT at the TD-B3LYP/6-311G* level. Implicit solvation models and dispersion corrections were incorporated into the calculations. Multiwfn (version 3.8) was used for electronic orbital analysis, electrostatic potential calculations, and hole-electron analysis, while VMD (version 1.9.3) was used for visualizing the orbitals. The initial COF structural model was constructed within Materials Studio (Materials Visualizer) using the P222 space group (sql topology, FOOCOF) obtained from the Reticular Chemistry Structure Resource (RCSR). Subsequently, the structure was energy-minimized using the universal force field within the Forcite module. Experimental PXRD patterns were subjected to Pawley refinement using the Reflex module. Integrated intensities were obtained using pseudo-Voigt profile fitting. The refined parameters included unit cell parameters (a, b, c), FWHM parameters (U, V, W), profile parameters (NA, NB), and the zero-point error. A 20th-order polynomial function was used to model the background. Finally, PXRD patterns were simulated from the optimized structure using the Reflex module.
Periodic DFT calculations were carried out using the CP2K software package. The exchange-correlation energy was described using the PBE functional, with the inclusion of Grimme’s DFT-D3 dispersion correction incorporating the BJ-damping scheme. A MOLOPT basis set, supplemented with appropriate diffuse functions, was employed within the framework of the Gaussian and plane waves method. The DZVP-MOLOPT-GGA-GTH basis set was used for geometry optimization, energy determination, vibrational mode analysis, and timedependent density functional theory calculations. The energy cutoff for the plane-wave basis was set to 600 eV , and convergence was ensured to within Hartree. The optimized geometry, corresponding to the lowest energy structure, was then used for subsequent calculations. Thermochemical parameters were corrected using the rigidrotor harmonic oscillator (RRHO) approximation within the Shermo program at standard conditions ( 1.01325 bar and 298.15 K ). The chemical potential of the proton-electron pair ( ) was equated to one-half the gas-phase chemical potential at standard hydrogen electrode (SHE) conditions, following the computational hydrogen electrode (CHE) approach. The free energy change for each
elementary step was calculated using the following equation:
For HER, the reaction path is as follows:
Data availability
All data are available in the main text or the supplementary information. Source data are provided with this paper.
References
Wang, H. et al. Covalent organic framework photocatalysts: structures and applications. Chem. Soc. Rev. 49, 4135-4165 (2020).
Guan, Q. et al. Metalated covalent organic frameworks: from synthetic strategies to diverse applications. Chem. Soc. Rev. 51, 6307 (2022).
Liang, Z. et al. Covalent organic frameworks: fundamentals, mechanisms, modification, and applications in photocatalysis. Chem. Cata. 2, 2157-2228 (2022).
Lu, M. et al.Rational design of crystalline covalent organic frameworks for efficient photoreduction with . Angew. Chem. Int. Edit. 58, 12392-12397 (2019).
Huang, N. et al. Covalent organic frameworks: a materials platform for structural and functional designs. Nat. Rev. Mater. 1, 16068 (2016).
Diercks, C. S. et al. The atom, the molecule, and the covalent organic framework. Science 355, 1585 (2017).
Chen, Z. et al. Tuning excited state electronic structure and charge transport in covalent organic frameworks for enhanced photocatalytic performance. Nat. Commun. 14, 1106 (2023).
Zhao, C. et al. Recent advances in conjugated polymers for visible-light-driven water splitting. Adv. Mater. 32, 1907746 (2020).
Yang, J. et al. Protonated imine-linked covalent organic frameworks for photocatalytic hydrogen evolution. Angew. Chem. Int. Edit. 60, 19797-19803 (2021).
Ma, S. et al. Photocatalytic hydrogen production on a -carbonlinked covalent organic framework. Angew. Chem. Int. Edit. 61, e202208919 (2022).
Wang, X. et al. Sulfone-containing covalent organic frameworks for photocatalytic hydrogen evolution from water. Nat. Chem. 10, 1180-1189 (2018).
Shen, R. C. et al. Efficient photocatalytic hydrogen evolution by modulating excitonic effects in ni-intercalated covalent organic frameworks. Adv. Energy Mater. 13, 2203695 (2023).
Lu, M. et al. Confining and highly dispersing single polyoxometalate clusters in covalent organic frameworks by covalent linkages for photoreduction. J. Am. Chem. Soc. 144, 1861-1871 (2022).
Zhang, M. et al. Semiconductor/covalent-organic-framework Z-scheme heterojunctions for artificial photosynthesis. Angew. Chem. Int. Edit. 59, 6500-6506 (2020).
Li, Y. et al. In situ photodeposition of platinum clusters on a covalent organic framework for photocatalytic hydrogen production. Nat. Commun. 13, 1355 (2022).
Wang, S. et al. Cobaloxime-integrated covalent organic frameworks for photocatalytic hydrogen evolution coupled with alcohol oxidation. Angew. Chem. Int. Edit. 62, e202311082 (2023).
Wang, Y. et al. Unraveling the photo-induced dynamic behavior of COF-based Z-scheme heterostructure monolithic aerogels. Matter 7, 3145-3162 (2024).
Weng, W. et al. Chiral covalent organic framework films with enhanced photoelectrical performances. J. Am. Chem. Soc. 146, 13201-13209 (2024).
Mukherjee, S. et al. Scalable integration of coplanar heterojunction monolithic devices on two-dimensional . ACS Nano 14, 17543-17553 (2020).
Zhong, H. et al. Fused dithienogermolodithiophene low band gap polymers for high-performance organic solar cells without processing additives. J. Am. Chem. Soc. 135, 2040-2043 (2013).
Lan, Z. A. et al. Reducing the exciton binding energy of donor-acceptor-based conjugated polymers to promote charge-induced reactions. Angew. Chem. Int. Edit. 58, 10236-10240 (2019).
Lan, Z.-A. et al. A fully coplanar donor-acceptor polymeric semiconductor with promoted charge separation kinetics for photochemistry. Angew. Chem. Int. Edit. 60, 16355-16359 (2021).
Xu, K. et al. Ground-state electron transfer in all-polymer donoracceptor heterojunctions. Nat. Mater. 19, 738 (2020).
Park, S. et al. Temperature-dependent electronic ground-state charge transfer in van der waals heterostructures. Adv. Mater. 33, 2008677 (2021).
Sekita, M. et al. Intense ground-state charge-transfer interactions in low-bandgap, panchromatic phthalocyanine-tetracyanobuta-1,3diene conjugates. Angew. Chem. Int. Edit. 55, 5560-5564 (2016).
Deibel, C. et al. Role of the charge transfer state in organic donoracceptor solar cells. Adv. Mater. 22, 4097-4111 (2010).
Akaike, K. et al. Impact of ground-state charge transfer and polarization energy change on energy band offsets at donor/acceptor interface in organic photovoltaics. Adv. Funct. Mater. 20, 715-721 (2010).
Hao, L. et al. Fluorenone-based covalent organic frameworks with efficient exciton dissociation and well-defined active center for remarkable photocatalytic hydrogen evolution. Appl. Catal. B Environ. Energy. 330, 122581 (2023).
Yang, Y. et al. Engineering -ketoamine covalent organic frameworks for photocatalytic overall water splitting. Nat. Commun. 14, 593 (2023).
Weng, W. et al. The effect of enantioselective chiral covalent organic frameworks and cysteine sacrificial donors on photocatalytic hydrogen evolution. Nat. Commun. 13, 5768 (2022).
Bai, J. X. et al. Topology-induced local electric polarization in 2D thiophene-based covalent organic frameworks for boosting photocatalytic evolution. Chinese J. Catal. 59, 225-236 (2024).
Dai, L. et al. Enhancement of visible-light-driven hydrogen evolution activity of 2D π-conjugated bipyridine-based covalent organic frameworks via post-protonation. Angew. Chem. Int. Edit. 62, e202300224 (2023).
Dong, W. B. et al. Isomeric oligo(phenylenevinylene)-based covalent organic frameworks with different orientation of imine bonds and distinct photocatalytic activities. Angew. Chem. Int. Edit. 62, e202216073 (2023).
Li, Z. P. et al. Three-component donor- -acceptor covalent-organic frameworks for boosting photocatalytic hydrogen evolution. J. Am. Chem. Soc. 145, 8364-8374 (2023).
Lin, Z. et al. Boosting photocatalytic hydrogen evolution enabled by -supporting chiral covalent organic frameworks with parallel stacking sequence. Chinese. J. Catal. 64, 87-97 (2024).
Liu, T. et al. Ground-state electron transfer in all-polymer donor:acceptor blends enables aqueous processing of water-insoluble conjugated polymers. Nat. Commun. 14, 8454 (2023).
Kar, S. et al. Unveiling the role of a ground state charge transfer complex in carbon nanoparticles for highly efficient metal-free solar hydrogen production. J. Mater. Chem. A 12, 4712-4726 (2024).
Liao, Q. et al. Tailoring and modifying an organic electron acceptor toward the cathode interlayer for highly efficient organic solar cells. Adv. Mater. 32, 1906557 (2020).
Bao, Q. Y. et al. Trap-assisted recombination via integer charge transfer states in organic bulk heterojunction photovoltaics. Adv. Funct. Mater. 24, 6309-6316 (2014).
Guo, Y. et al. Perylenetetracarboxylic acid nanosheets with internal electric fields and anisotropic charge migration for photocatalytic hydrogen evolution. Nat. Commun. 13, 2067 (2022).
Yang, J. et al. A full-spectrum porphyrin-fullerene D-A supramolecular photocatalyst with giant built-in electric field for efficient hydrogen production. Adv. Mater. 33, 2101026 (2021).
Zhang, Z. J. et al. A highly crystalline perylene imide polymer with the robust built-in electric field for efficient photocatalytic water oxidation. Adv. Mater. 32, 1907746 (2020).
Li, J. et al. Giant enhancement of internal electric field boosting bulk charge separation for photocatalysis. Adv. Mater. 28, 4059-4064 (2016).
Aarnio, H. et al. Spontaneous charge transfer and dipole formation at the interface between P3HT and PCBM. Adv. Energy. Mater. 1, 792-797 (2011).
Bie, C. et al. A bifunctional catalyst enhances photocatalytic evolution and pyruvic acid synthesis. Angew. Chem. Int. Edit. 61, e202212045 (2022).
Yu, J. et al. Excited-state electronic properties in zr-based metalorganic frameworks as a function of a topological network. J. Am. Chem. Soc. 140, 10488-10496 (2018).
Qan, Y. et al. Photocatalytic molecular oxygen activation by regulating excitonic effects in covalent organic frameworks. J. Am. Chem. Soc. 142, 20763-20771 (2020).
Wei, Z. et al. Steering electron-hole migration pathways using oxygen vacancies in tungsten oxides to enhance their photocatalytic oxygen evolution performance. Angew. Chem. Int. Edit. 60, 8236-8242 (2021).
Barman, S. et al. Metal-free catalysis: a redox-active donoracceptor conjugated microporous polymer for selective visible-light-driven reduction to . J. Am. Chem. Soc. 143, 16284-16292 (2021).
Haldar, R. et al. Charge transfer in metal-organic frameworks. Chem. Commun. 59, 1569-1588 (2023).
Liu, D. et al. Rational design of PDI-based linear conjugated polymers for highly effective and long-term photocatalytic oxygen evolution. Adv. Mater. 35, 2300655 (2023).
Shen, R. C. et al. Realizing photocatalytic overall water splitting by modulating the thickness-induced reaction energy barrier of fluorenone-based covalent organic frameworks. Adv. Mater. 35, 2305397 (2023).
Acknowledgements
The authors thank National Natural Science Foundation of China (22378148, 52472110, 22308113) and Natural Science Fouddation of Guangdong Province (2024A1515012433) for their support.
Author contributions
R.C.S., C.H., and L.H. contributed equally to this work. R.C.S. and C.H., conceived and designed the experiments. R.C.S., L.H., and X.L. designed, synthesized, and characterized the materials. C.H. performed the theoretical calculations. G.J.L. performed the TAS analysis. R.C.S. and L.H. performed the TEM measurements. P.Z. helped with the manuscript discussion. Q.Y. revised the manuscript Data. were interpreted by all authors and the manuscript was prepared by R.C.S. and X.L. All authors contributed to this work, read the manuscript, and all data are reported in the main text and supplemental materials.
Correspondence and requests for materials should be addressed to Guijie Liang, Peng Zhang, Qiang Yue or Xin Li.
Peer review information Nature Communications thanks Yongfa Zhu and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http:// creativecommons.org/licenses/by-nc-nd/4.0/.
(c) The Author(s) 2025
Institute of Biomass Engineering, Key Laboratory of Energy Plants Resource and Utilization, Ministry of Agriculture and Rural Affairs, South China Agricultural University, Guangzhou, China. Key Laboratory for Biobased Materials and Energy of Ministry of Education, College of Materials and Energy, South China Agricultural University, Guangzhou, China. Guangdong Provincial Key Laboratory of Utilization and Conservation of Food and Medicinal Resources in Northern Region, Shaoguan University, Shaoguan, China. Hubei Key Laboratory of Low Dimensional Optoelectronic Materials and Devices, Hubei University of Arts and Science, Xiangyang, China. State Centre for International Cooperation on Designer Low-Carbon & Environmental Materials (CDLCEM), School of Materials Science and Engineering, Zhengzhou University, Henan Zhengzhou, China. These authors contributed equally: Rongchen Shen, Can Huang, Lei Hao. e-mail: Guijie-liang@hbuas.edu.cn; zhangp@zzu.edu.cn; 2249659235@qq.com; Xinli@scau.edu.cn