نهج شامل قائم على المحولات لتوقعات امتصاص الغاز بدقة عالية في الإطارات العضوية المعدنية A comprehensive transformer-based approach for high-accuracy gas adsorption predictions in metal-organic frameworks
فصل الغاز أمر حاسم للإنتاج الصناعي وحماية البيئة، حيث توفر الهياكل العضوية المعدنية (MOFs) حلاً واعدًا بفضل خصائصها الهيكلية القابلة للتعديل وتركيباتها الكيميائية. تعتبر الطرق التقليدية للمحاكاة، مثل الديناميكا الجزيئية، معقدة وتتطلب حسابات مكثفة. على الرغم من أن طرق التعلم الآلي المعتمدة على هندسة الميزات تؤدي بشكل أفضل، إلا أنها عرضة للتكيف الزائد بسبب نقص البيانات المعلّمة. علاوة على ذلك، تم تصميم هذه الطرق عادةً لمهام فردية، مثل التنبؤ بسعة امتصاص الغاز تحت ظروف معينة، مما يقيّد استخدام مجموعات البيانات الشاملة التي تشمل جميع سعات الامتصاص. لمعالجة هذه التحديات، نقترح Uni-MOF، وهو إطار مبتكر لتعلم تمثيل MOF ثلاثي الأبعاد على نطاق واسع، مصمم للتنبؤ بالغازات لأغراض متعددة. على وجه التحديد، يعمل Uni-MOF كمنظم سعة امتصاص الغاز متعدد الاستخدامات لمواد MOF، مستخدمًا تمثيلات ثلاثية الأبعاد نقية تم تعلمها من أكثر من 631,000 هيكل MOF وCOF تم جمعها. تظهر نتائجنا التجريبية أن Uni-MOF يمكنه استخراج التمثيلات الهيكلية تلقائيًا والتنبؤ بسعات الامتصاص تحت ظروف تشغيل مختلفة باستخدام نموذج واحد. بالنسبة للبيانات المحاكاة، يظهر Uni-MOF دقة تنبؤية عالية بشكل ملحوظ عبر جميع مجموعات البيانات. بالإضافة إلى ذلك، تتوافق القيم التي يتنبأ بها Uni-MOF مع نتائج تجارب الامتصاص. علاوة على ذلك، يظهر Uni-MOF إمكانات كبيرة للتطبيق الواسع في التنبؤ بمجموعة واسعة من الخصائص الأخرى.
فصل الغاز هو تحدٍ صناعي كبير يتطلب اهتمامًا فوريًا، نظرًا لدوره الحاسم في تطبيقات متنوعة. على سبيل المثال، فصل منضروري للحصول على غاز طبيعي عالي الجودة وتحقيق احتجاز الكربون بشكل فعال، الاستخدام والتخزين لأسباب بيئيةفصل الغاز له أيضًا تداعيات على مجالات أخرى، مثل إنتاج الأكسجين عالي النقاء.والنيتروجينلأغراض صناعية وتنقية الغازات النبيلة للتشخيص الطبي، والليزر. نظرًا لـ
تعتبر الأبحاث في هذا المجال ضرورية لتقدم التكنولوجيا وتلبية متطلبات الصناعة.
أطر المعادن العضوية (MOFs) ظهرت كنوع من المواد الواعدة في مجال فصل الغازات بسبب خصائصها الفريدة.تتكون MOFs من أيونات معدنية ومواد عضوية، مما يمنحها هياكل مسامية مرتبة للغاية وأحجام فتحات قابلة للتعديل. تجعل هذه الخصائص منها مثالية لمجموعة متنوعة من تطبيقات فصل الغاز.تتيح القدرة على التحكم في حجم المسام والتركيب الكيميائي لمركبات الإطار المعدني العضوي (MOFs) الامتصاص الانتقائي والفصل بين الغازات المختلفة. تظهر مركبات الإطار المعدني العضوي ذات أحجام المسام المختلفة سعات متفاوتة لامتصاص الغاز.وتعديل التركيب الكيميائييمكن أن تؤثر على تفضيل الغازات الممتصة. إن القدرة على الامتصاص الانتقائي وفصل الغازات المختلفة تجعل من MOFs مادة واعدة لمجموعة متنوعة من التطبيقات الصناعية والبيئية..
بينما يعدّ إمكانات المواد الإطار الجزيئي (MOFs) لامتصاص الغاز واعدًا، فإن التنبؤ بدقة بسعة امتصاصها لا يزال يمثل تحديًا. الديناميات الجزيئيةمونت كارلو (MC)وطرق المحاكاة/الحساب الأخرىتم تطبيقها لتوفير قيم مرجعية، ولكن هذه الأساليب مكلفة حسابياً ومعقدة للتنفيذ، مما يحد من تطبيقها في الحسابات واسعة النطاق والمتعددة الغازات وعالية الإنتاجية. علاوة على ذلك، فإن النطاق الواسع من ظروف التشغيل لامتصاص الغاز يزيد من تعقيد التنبؤات.
أظهرت تقنيات تعلم الآلة إمكانيات كبيرة في التنبؤ بدقة بخصائص المواد البلورية.، مما يقلل من تكلفة التجارب التقليدية القائمة على التجربة والخطأ، ويقضي على الحاجة إلى المحاكاة المكلفة. ومع ذلك، غالبًا ما تعتمد هذه الطرق على هندسة الميزات بناءً على معرفة الخبراء في المجال، مما يؤدي إلى الإفراط في التكيف والأداء المنحاز عند استخدام كمية محدودة من البيانات المصنفة. مع ظهور التعلم العميق، والشبكات العصبية البيانية،وTransformersلقد أثبتت نجاحها في التنبؤ بخصائص MOF. تتضمن هذه النماذج معلومات هيكلية مباشرة مثل الروابط الكيميائية، والذرات، والإحداثيات المكانية كمدخلات، وتتعلم تلقائيًا الميزات الهيكلية من خلال أساليب مدفوعة بالبيانات. بفضل القدرة التمثيلية القوية لهذه الأطر القائمة على التعلم العميق، يمكن للميزات المتعلمة أن تقضي بفعالية على التحيزات التي أدخلتها هندسة الميزات المذكورة سابقًا.
على الرغم من أدائها العالي وقدراتها التنبؤية القوية، فإن النماذج الحالية لتوقع خصائص الامتزاز عادة ما تكون مصممة لمهام فردية، وتحديداً لتوقع امتصاص غاز معين تحت ظروف معينة. ومع ذلك، فإن مجموعات البيانات المتاحة لهذه التوقعات ذات المهام الفردية غالباً ما تكون محدودة، مما يعيق قابلية تعميم النماذج والاستخدام الكامل لقدراتها. من ناحية أخرى، يمكن أن يؤدي دمج البيانات المعلّمة من غازات الامتزاز المختلفة عبر بيئات درجات حرارة وضغط مختلفة إلى إنشاء مجموعة بيانات كبيرة مناسبة للتدريب عبر جميع ظروف العمل. قد يؤدي زيادة حجم البيانات أيضاً إلى تعزيز قدرة النموذج على التعميم وتحسين استخدامه العملي في الصناعة. لذلك، فإن إطار عمل موحد للامتزاز ضروري لتقدم هذه النماذج. بالإضافة إلى ذلك، قد يؤدي دمج تعلم التمثيل (أو التدريب المسبق) لهياكل MOF غير المعلّمة على نطاق واسع إلى تحسين أداء النموذج وكذلك قدرة التمثيل. لقد تم تنفيذ خدعة التدريب المسبق على نطاق واسع بالاشتراك مع نماذج كبيرة لاكتشاف الأدوية.حيث تم استخدام هياكل جزيئية ثلاثية الأبعاد نقية لتدريب هذه النماذج مسبقًا. كما أظهرت النتائج التجريبية أن النماذج المدربة مسبقًا تتفوق على الطرق السابقة، لا سيما في توقع الخصائص.مقترحًا تحسينًا ملحوظًا من خلال التدريب المسبق.
استلهمًا من ذلك، نقترح إطار عمل Uni-MOF كحل متعدد الأغراض للتنبؤ بامتصاص الغاز لمركبات الإطار المعدني العضوي (MOFs) تحت ظروف مختلفة باستخدام تعلم تمثيل الهيكل. بالمقارنة مع نماذج أخرى قائمة على Transformer مثل MOFormer و MOFTransformer نظام Uni-MOF لدينا، كإطار عمل قائم على المحولات، ليس فقط أن التدريب المسبق يمكنه التعرف على واستعادة الهيكل ثلاثي الأبعاد للمواد المسامية النانوية وبالتالي تحسين متانة النموذج بشكل كبير، ولكن مهمة الضبط الدقيق تأخذ أيضًا في الاعتبار ظروف التشغيل مثل درجة الحرارة والضغط وجزيئات الغاز المختلفة، مما يجعل Uni-MOF مناسبًا لكل من البحث العلمي والتطبيقات العملية. يتطلب UniMOF، كأداة شاملة لتقدير امتصاص الغاز لمواد MOF، فقط ملف معلومات البلورة (CIF) للـ MOF، جنبًا إلى جنب مع الغاز المرتبط، ودرجة الحرارة، ومعلمات الضغط، للتنبؤ بخصائص امتصاص الغاز للمواد المسامية النانوية عبر مجموعة واسعة من ظروف التشغيل. إطار عملنا سهل الاستخدام ويسمح باختيار الوحدات. بالإضافة إلى ذلك، فإنه يعالج بفعالية مشكلة الإفراط في التكيف من خلال دمج بيانات امتصاص معلمة من أنظمة مختلفة مع التعلم التمثيلي من كميات هائلة من البيانات الهيكلية غير المعلمة. هذا يعوض عن نقص البيانات عالية الجودة وغير الكافية، مما يؤدي في النهاية إلى دقة أعلى في توقعات امتصاص الغاز. استخدمت دراستنا نهج التعلم الذاتي المراقب على قاعدة بيانات تحتوي على أكثر من 631,000 هيكل من MOF وCOF. كانت النتائج ملحوظة، حيث أظهرت دقة توقع عالية. كشفت تجارب الضبط الدقيق أن إطار عمل UniMOF قوي في قواعد البيانات التي تحتوي على بيانات وفيرة. عند تطبيقه على قواعد بيانات تحتوي على عينات كافية من ظروف العمل، كان إطار عمل UniMOF قادرًا على تصفية المواد الممتصة عالية الأداء تحت ضغط عالٍ بدقة من خلال تغذية البيانات المعلمة التي تم الحصول عليها عند ضغط منخفض من خلال محاكاة محلية. يجب أن نؤكد أن Uni-MOF يوفر نهجًا مريحًا لسعات امتصاص الغاز تحت ضغط عالٍ، والتي تكون عمومًا أكثر تطلبًا حسابيًا بالنسبة للمحاكاة التقليدية. النتائج متسقة مع نتائج الفحص التجريبية. علاوة على ذلك، فإن أداء Uni-MOF على مجموعات البيانات عبر الأنظمة تجاوز ذلك في المهام ذات النظام الواحد. من خلال الاستفادة من الدعم من بيانات امتصاص الغاز الأخرى، توقع Uni-MOF بدقة خصائص امتصاص الغازات غير المعروفة.
إطار Uni-MOF يحقق دقة التعرف على المواد على المستوى الذري، بينما يجعل النموذج المتكامل Uni-MOF أكثر قابلية للتطبيق على المشكلات الهندسية. لا شك أن تحقيق نماذج موحدة حقًا هو الاتجاه المستقبلي في مجال المواد، بدلاً من التركيز فقط على المجالات المتخصصة. Uni-MOF هو ممارسة رائدة في تعلم الآلة في امتصاص الغاز.
النتائج والمناقشة
نظرة عامة على سير العمل
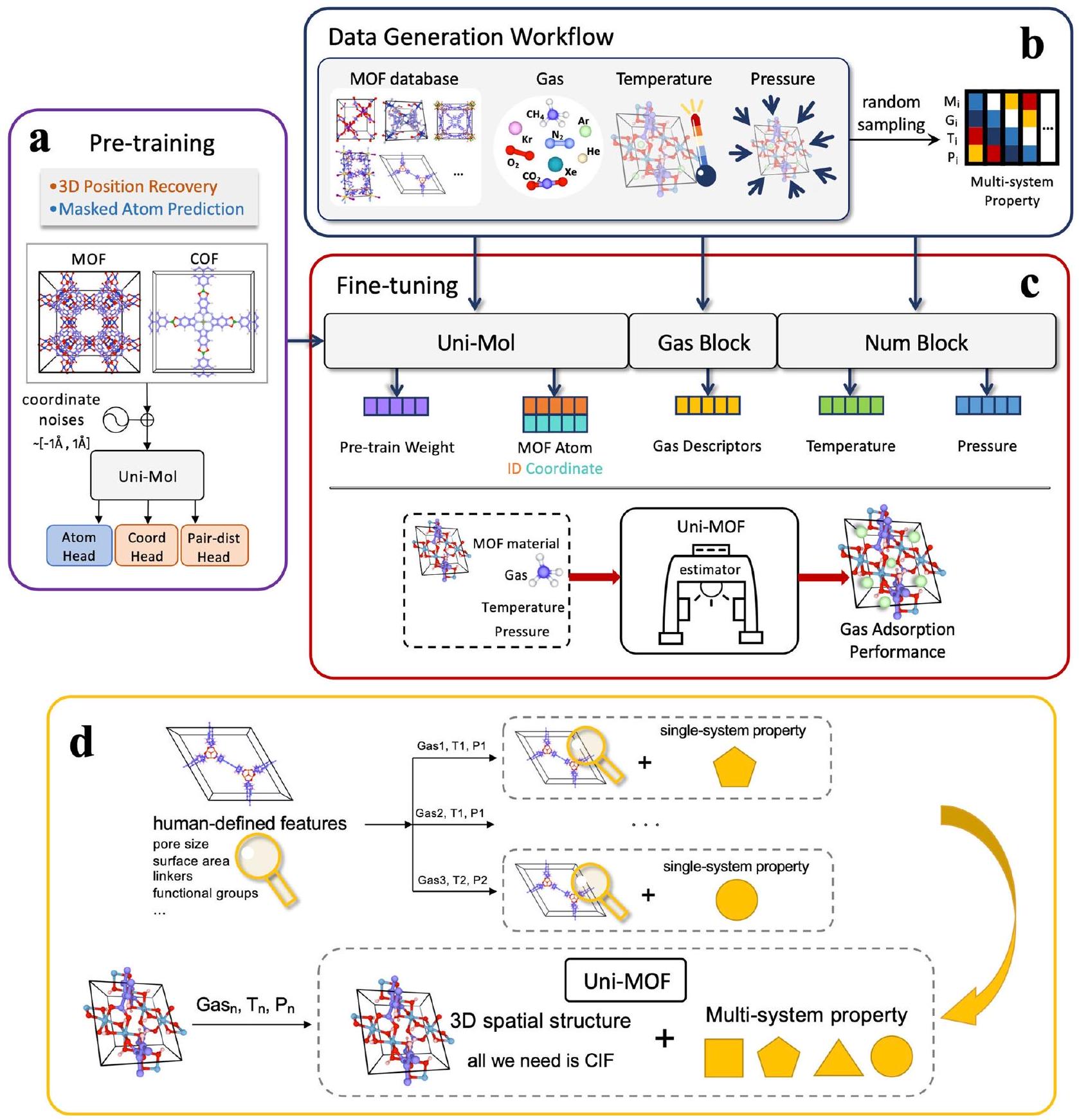
يتكون إطار عمل Uni-MOF من التدريب المسبق على البلورات النانوية ثلاثية الأبعاد والتعديل الدقيق للتنبؤ متعدد المهام في التطبيقات اللاحقة. توفر الشكل 1 تمثيلاً تخطيطياً لإطار عمل Uni-MOF. يعزز التدريب المسبق على مواد البلورات ثلاثية الأبعاد بشكل كبير أداء التنبؤ للمهام اللاحقة، خاصةً بالنسبة للبيانات غير المعلّمة على نطاق واسع. لمعالجة مشكلة مجموعات البيانات التدريبية غير المراقبة بشكل كافٍ، جمعنا مجموعة بيانات واسعة من هياكل MOF وأنشأنا أكثر من 300,000 MOF باستخدام ToBaCCo.3.0.إن البناء عالي الإنتاجية لمواد الإطار العضوي (COFs) استنادًا إلى استراتيجية علم المواد الجينومي مع خوارزميات التجميع شبه التفاعلية (QReaxAA) ممكن، مما يؤدي إلى مكتبة شاملة منمن خلال التكوين المكاني للمواد، يتمكن Uni-MOF من تعلم الخصائص الهيكلية للمواد، وأهمها معلومات الروابط الكيميائية، بشكل جيد جداً. من أجل تمكين Uni-MOF من تعلم مواد أكثر تنوعاً وبالتالي تحسين القدرة على التعميم لمجموعة أوسع من المواد، قمنا بتقديم MOFs و COFs بشكل افتراضي وتجريبي خلال عملية التدريب المسبق. مشابهة لمهمة وضع العلامات القابلة للتغطية في BERT.و يونيتستخدم Uni-MOF مهمة توقع للذرات المmasked، مما يعزز النماذج المدربة مسبقًا لاكتساب فهم عميق للهياكل المكانية للمواد. لتعزيز قوة التدريب المسبق وتعميم التمثيل المتعلم، قمنا بإدخال ضوضاء على الإحداثيات الأصلية للـ MOFs، كما هو موضح في الشكل 1a. في مرحلة التدريب المسبق، وضعنا مهمتين. 1) إعادة بناء
الشكل 1 | نظرة عامة تخطيطية لإطار Uni-MOF. سير عمل ما قبل التدريب. في مرحلة ما قبل التدريب، بالإضافة إلى توقع أنواع الذرات المmasked، تم استخدام مهمة إزالة الضوضاء من الموضع ثلاثي الأبعاد لتعلم التمثيل المكاني ثلاثي الأبعاد. ضوضاء موحدة منيتم إضافته إلىلإحداثيات ذرية بشكل عشوائي، ثم يتم حساب ترميز الموقع المكاني بناءً على الإحداثيات التالفة.سير عمل توليد البيانات. يمكن جمع أو توليد مجموعات بيانات الأداء عبر الأنظمة من خلال أخذ عينات عشوائية من عمليات تشغيل مختلفة. الشروط. ج سير العمل لتعديل Uni-MOF. تم بناء نموذج موحد لتوقع امتصاص الغاز Uni-MOF من خلال تضمين الأوزان المدربة مسبقًا، ومواد MOF، والغاز، ودرجة الحرارة، والضغط. د سير العمل العام لـ Uni-MOF. بالنسبة لإطار عمل Uni-MOF متعدد الأغراض، لا تتطلب أي حسابات تحليلية إضافية للمواد، ويمكن توقع الخصائص تحت ظروف العمل المتنوعة استنادًا فقط إلى ملف المعلومات البلورية (CIF) لمواد MOF. MOF تعني الإطار المعدني العضوي. المواقع الثلاثية الأبعاد النقية من البيانات الم noisy، و 2) التنبؤ بالذرات المخفية. يمكن أن تعزز هذه المهام من قوة النموذج وتحسن أداء التنبؤ في المراحل التالية.
بالإضافة إلى التكوينات المكانية المتنوعة، فإن مجموعة شاملة من نقاط بيانات خصائص المواد ضرورية أيضًا لتدريب النموذج. لإثراء مجموعة البيانات، أنشأنا عملية توليد بيانات مخصصة (كما هو موضح في الشكل 1b). على سبيل المثال، استخدمنا قاعدة بيانات CoRE MOF التي تتضمن MOFs التي تم تخليقها بنجاح، والغازات التي لها أهمية في مجال الفصل، بالإضافة إلى درجة الحرارة الشائعة. ونطاق الضغط التشغيلي تحت النظام المقابل. من خلال أخذ عينات عشوائية من هذه المواد المختلفة، والغازات، ودرجات الحرارة، ومجموعات الضغط، يمكن توليد حجم كبير من بيانات امتصاص المواد لتحسين Uni-MOF. تعمل عملية توليد هذه البيانات على تحسين كفاءة توليد البيانات ويمكن أن تشكل مجموعة بيانات تم أخذ عينات منها على نطاق واسع. تسرد الجدول 1 جميع قواعد البيانات المستخدمة في هذه الدراسة. تعتبر قاعدة البيانات المستمدة من المحاكاة والمتنوعة مفيدة لتحسين النموذج وتحسينه، مما يحقق في النهاية هدف الفحص الافتراضي لأداء المواد.
الجدول 1 | الهيكل وموارد البيانات
أنواع البيانات
المصادر
التوفر
برمجيات
حجم
هيكل
مجموعة
هيموف
–
137,000+
هيكل
مجموعة
توباكو
–
10,000+
هيكل
مجموعة
موف CoRE
–
12,000+
هيكل
مجموعة
CCDC
–
12,000+
هيكل
مجموعة
كوَر كوف
–
600+
هيكل
مجموعة
مؤشرات التغير المناخي العالمية
–
160,000+
هيكل
جيل
هذا العمل
توباكو.3.0
300,000+
بيانات امتصاص الإعلانات
مجموعة
hMOF_ MOFX_DB
–
2,400,000+
بيانات امتصاص الإعلانات
مجموعة
قاعدة بيانات CoRE_MOFX
–
460,000+
بيانات امتصاص الإعلانات
جيل
هذا العمل
راسبا
99,000+
بيانات الملكية الأخرى
جيل
هذا العمل
زيو++
149,000+
تستند عملية ضبط Uni-MOF الموضحة في الشكل 1c إلى استخراج التمثيلات المكتسبة من خلال التدريب المسبق، بالإضافة إلى توليد وجمع مجموعات بيانات واسعة باستخدام سير العمل الخاص بنا. خلال عملية الضبط، قمنا بتدريب النموذج باستخدام حوالي 3,000,000 نقطة بيانات مصنفة عبر ظروف امتصاص مختلفة لمركبات MOFs وCOFs، مما يتيح التنبؤ الدقيق بسعات الامتصاص. مع قاعدة البيانات المتنوعة من البيانات المستهدفة عبر الأنظمة، يمكن لـ Uni-MOF المضبوط التنبؤ بخصائص الامتصاص المتعددة الأنظمة لمركبات MOFs تحت حالات عشوائية، بما في ذلك الغازات المختلفة، ودرجات الحرارة، والضغوط. نتيجة لذلك، يعد Uni-MOF إطارًا موحدًا ومتاحة بسهولة للتنبؤ بأداء امتصاص مواد MOF.
فوق كل شيء، يزيل Uni-MOF الحاجة إلى عمل إضافي في تحديد الميزات الهيكلية المعرفة من قبل الإنسان. بدلاً من ذلك، يكفي CIF للـ MOFs، جنبًا إلى جنب مع معلمات الغاز ودرجة الحرارة والضغط ذات الصلة. تضمن استراتيجية التعلم الذاتي والإشراف وقواعد البيانات الوفيرة أن Uni-MOF يمكنه التنبؤ بخصائص امتصاص الغاز للمواد النانوية المسامية في مجموعة واسعة من معلمات التشغيل، مما يجعله مقدرًا كفؤًا لامتصاص الغاز لمواد MOF.
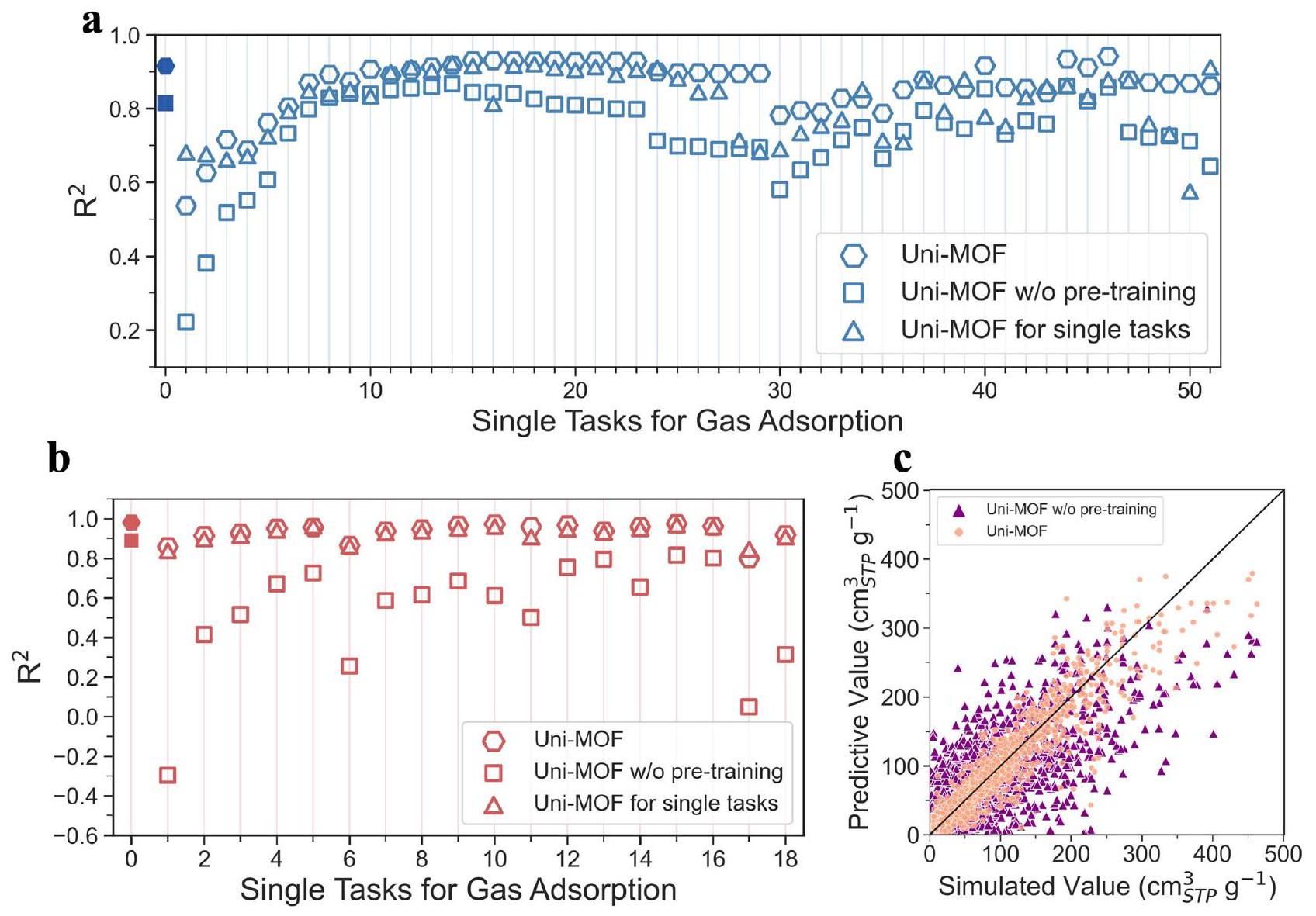
الأداء العام في قواعد البيانات الكبيرة
لتقييم القدرة التنبؤية لـ Uni-MOF كإطار شامل لتوقع أداء الامتصاص، تم تجميع قاعدتي بيانات مختلطتين لامتصاص الغاز، وهما hMOF_MOFX-DB و CoRE_MOFX-DB، مع غازات الامتصاص المكونة من [ ] و [ ]، على التوالي. بالإضافة إلى ذلك، تم إنشاء قاعدة بيانات CoRE_MAP_DB من خلال سير العمل لمحاكاة MC الخاصة بنا لامتصاص سبعة غازات ( , ).
نظرًا لاختلاف مصادر البيانات في قواعد البيانات الثلاث، قمنا بإجراء تدريب النموذج لكل قاعدة بيانات بشكل منفصل لضمان اتساق مجموعات البيانات. تفاصيل هذه القواعد الثلاث، بما في ذلك نطاقات درجة الحرارة والضغط، مدرجة في الجدول التكميلي 1. لمنع تحيز البيانات وضمان بقاء مجموعة الاختبار غير مرئية للنموذج، قمنا بتقسيم مجموعة البيانات إلى ثلاث مجموعات بيانات مختلفة (تدريب، تحقق، واختبار) بنسبة 8:1:1 وفقًا لهيكل MOF بدلاً من التقسيم العشوائي، أي أنه لا يوجد مادة متطابقة بين مجموعات البيانات الثلاث. يضمن هذا التقسيم أن يحقق النموذج توقع المواد الجديدة في مجموعة التحقق ومجموعة الاختبار، بدلاً من تلك المواد التي تم رؤيتها بالفعل. يتم تحسين معلمات النموذج خلال عملية التدريب وتنعكس في نتائج مجموعة التحقق. يتم حفظ النموذج الأمثل المقابل لمجموعة التحقق، وتمثل نتائج التنبؤ في مجموعة الاختبار التي لم تُرَ من قبل الأداء النهائي ( الموضح هنا) للنموذج، مما يتجنب بشكل معقول الإفراط في التكيف.
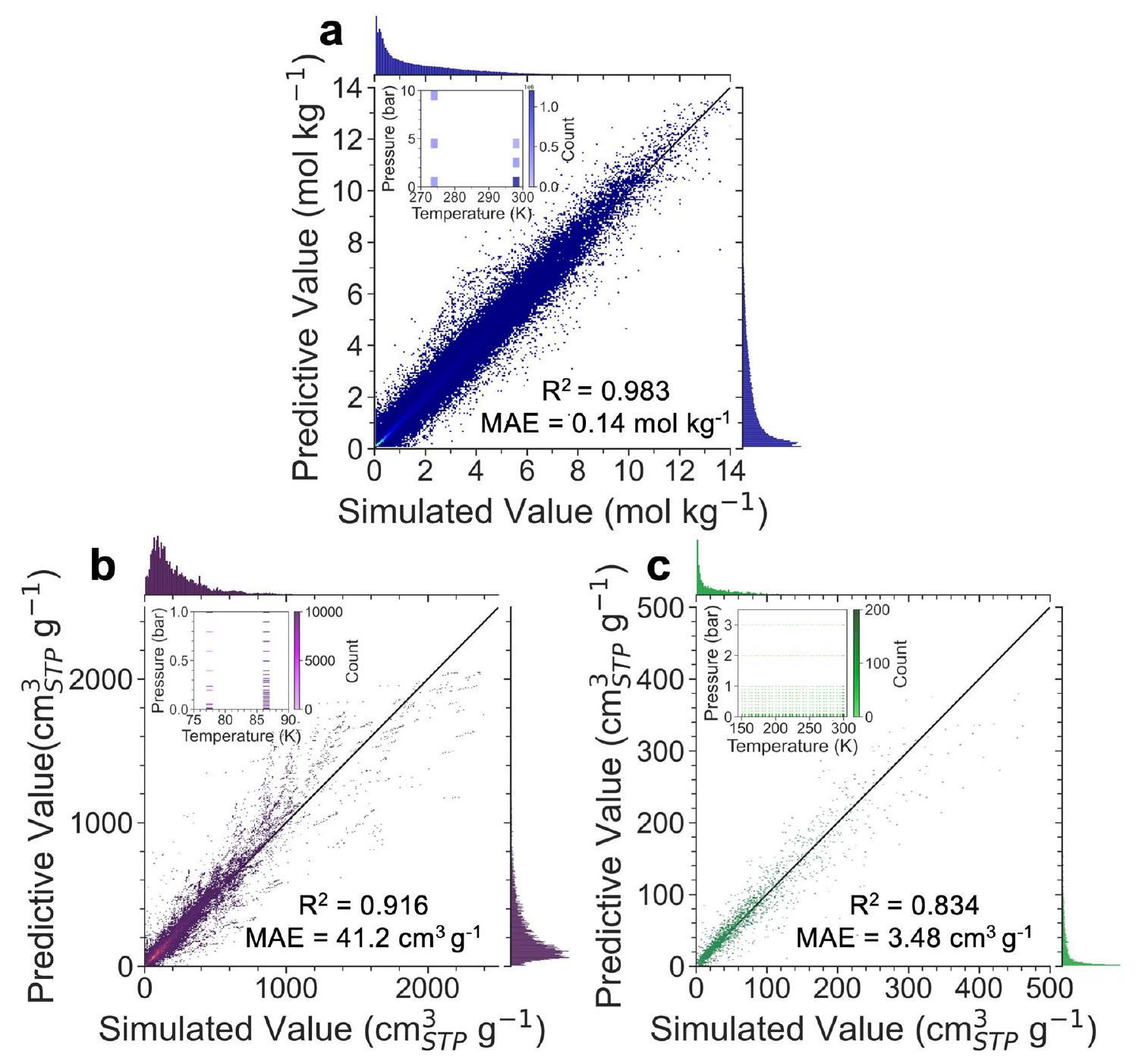
تظهر مجموعات البيانات المجمعة، hMOF_MOFX_DB و CoRE_MOFX_DB، توزيعات درجة الحرارة والضغط مركزة نسبيًا، كما هو موضح في الشكل الفرعي من الشكل 2a، b. من الجدير بالذكر أن كلا قاعدتي البيانات تقدمان بيانات كافية، مع أكثر من 2,000,000 و 400,000 نقطة بيانات، على التوالي. تظهر نتائج التنبؤ أن Uni-MOF قوي بشكل ملحوظ عند تطبيقه على قواعد البيانات التي تمتلك بيانات كافية مع حالات تشغيل مركزة نسبيًا، مثل hMOF_MOFX_DB و CoRE_MOFX_DB، مع قيم تبلغ 0.98 و 0.92، على التوالي. بالمقابل، تحتوي قاعدة بيانات CoRE_MAP_DB الخاصة بنا، التي تعلمنا منها من الجدول التكميلي 1، على أقل بقليل من 100,000 نقطة بيانات. تشمل عينة واسعة من امتصاص سبعة غازات ممتصة في أكثر من 10,000 MOFs، تغطي نطاق درجة حرارة من ونطاق ضغط من ، كما هو موضح في الشكل الفرعي من الشكل 2c. بالنسبة لقاعدة بيانات موزعة على نطاق واسع، لا يزال بإمكان Uni-MOF تحقيق دقة تنبؤ ممتازة مع قيمة تبلغ 0.83، مما يظهر عموميتها الجيدة.
يتضمن التحليل أيضًا مقياسين آخرين للخطأ، وهما متوسط الخطأ المطلق (MAE) وجذر متوسط مربع الخطأ (RMSE). ومع ذلك، تظهر مجموعة بيانات CoRE_MOFX أخطاء أكبر، خاصة في RMSE. تحتوي مجموعة بيانات CoRE_MOFX على كميات امتصاص Ar و عند 77 كلفن و 87 كلفن، وهي درجات حرارة أقل بكثير مقارنة بقواعد البيانات الأخرى. وبالتالي، فإن قيم الامتصاص في CoRE_MOFX أكبر عمومًا. يتم تصوير منحنيات الامتصاص ذات أكبر الأخطاء في الشكل التكميلي 1. عادةً ما تحتوي مواد MOF ذات أكبر خطأ على Cu (مع أقل معلمة طاقة تبلغ 2.52 كلفن) كموقع معدني. وهذا يعني أن مواد MOF ذات مواقع معدنية ذات معلمات طاقة منخفضة أو أحجام مسام كبيرة هي العوامل المحددة لتوقع بيانات درجات الحرارة المنخفضة. من المثير للاهتمام، حتى مع أكبر الأخطاء، يمكن لـ Uni-MOF إعادة إنتاج اتجاه الامتصاص بدقة من الضغط المنخفض إلى العالي. بالإضافة إلى ذلك، تحتوي قاعدة بيانات CoRE_MOFX_DB، كقاعدة بيانات هيكلية تجريبية مجمعة، على أكثر من 1800 مادة غير مرتبة من أكثر من 12,000 MOFs. وُجد أن MOFs غير المرتبة تؤثر سلبًا على أداء النموذج، حيث تؤدي الهياكل الداخلية غير الواقعية والعينات المحدودة من المواد إلى صعوبة توقع امتصاص الغاز، كما يتضح من القيم الشاذة ja5111317_ja5111317_si_003_clean و LELDOX_clean في الجدول التكميلي 3. لذلك، تم استبعاد المواد غير المرتبة من قاعدة بيانات CoRE_MAP_DB المولدة، مما يؤدي إلى تقليل الأخطاء ويكون أكثر اقتراحًا للاستخدام في توقع امتصاص الغاز في المواد النانوية المسامية. لا تأخذ القوة المستخدمة في هذا العمل في الاعتبار تأثير المواقع المعدنية المفتوحة. بالنسبة لأعلى عشرة قيم شاذة في قاعدة بيانات CoRE_MOFX_DB (الموضحة في الجدول التكميلي 4)، فإن 80% منها تحتوي على مواقع معدنية مفتوحة. وهذا يشير إلى أن الانحرافات الكبيرة بين المحاكاة وتوقعات Uni-MOF قد تكون بسبب التفاعل المفقود بين المواقع المعدنية المفتوحة والغاز الممتص الذي تم اعتباره في المحاكاة.
بالإضافة إلى ذلك، اكتشفنا أن دقة التنبؤ في مرحلة ما قبل التدريب يمكن أن تصل إلى 0.98 قبل وبعد دمج مواد COF. وهذا يشير إلى أن Uni-MOF قادر على تعلم الهيكل المكاني ثلاثي الأبعاد بفعالية للعديد من المواد النانوية المسامية. أما بالنسبة للمهام اللاحقة، فقد تجاوز الأداء التنبؤي ( , RMSE، و MAE) لـ Uni-MOF لجميع قواعد البيانات الثلاث أداء Uni-MOF مع تدريب MOF فقط، كما هو موضح في الجداول التكملية 5، 6. وهذا يشير إلى أن دمج COF لا يحافظ فقط على قوة النموذج ولكن أيضًا يعززها، مما يوضح تفوق إطار عمل Uni-MOF لدينا.
توقع امتصاص الغاز التجريبي
على الرغم من الأداء التنبؤي الممتاز لـ Uni-MOF على النتائج المحاكاة، نتساءل كيف سيتصرف الإطار مقارنةً بالنتائج التجريبية المجمعة الحقيقية. في هذه الدراسة، اخترنا مواد مختبرية شائعة الاستخدام، مثل MOF-5 و MOF-177 , وقارننا سعة امتصاص الغاز المتوقعة مع البيانات التجريبية الموجودة. نظرًا لأن قاعدة بيانات CoRE_MAP_DB تحتوي على مجموعة متنوعة من ظروف التشغيل و
الشكل 2 | الأداء العام لـ Uni-MOF في قواعد البيانات الكبيرة. العلاقة بين القيمة المتوقعة والمحاكاة لكمية امتصاص الغاز لـ (أ) قاعدة بيانات hMOF_MOFX_DB ( )، (ب) قاعدة بيانات CoRE_MOFX_DB ( ) و (ج) قاعدة بيانات CoRE_MAP_DB . الشكل الفرعي هو
الهياكل MOF التي تم التحقق منها تجريبيًا، اخترنا الأوزان المدربة باستخدام هذه القاعدة البيانات لتوقع أداء الامتصاص للمواد التجريبية. يعرض الشكل 3 النتائج المتوقعة تحت ظروف مختلفة، والتفاصيل مدرجة في الجداول التكملية 8-10.
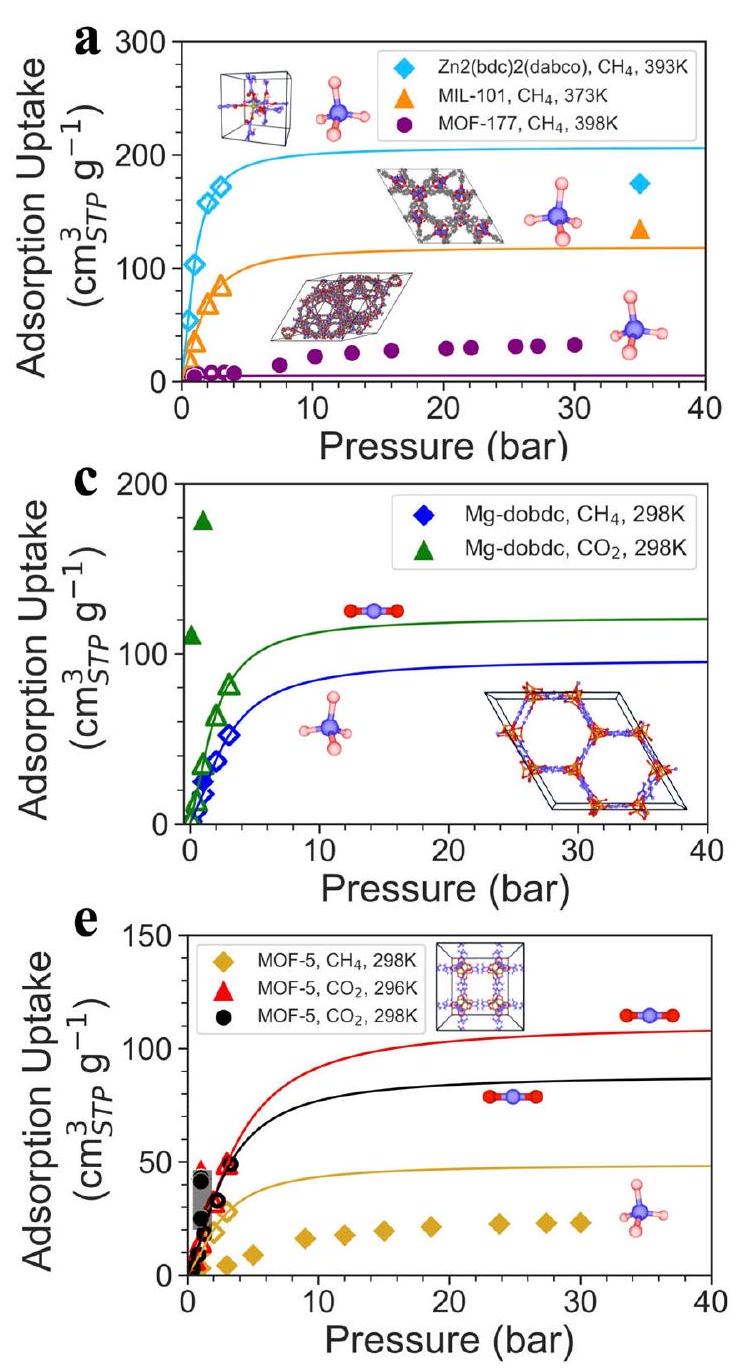
يعرض الشكل 3a منحنى امتصاص لانغموير الذي تم الحصول عليه من خلال ملاءمة سعة امتصاص الميثان المتوقعة تحت ضغط منخفض (أقل من 5 بار). يظهر أن سعة الامتصاص عند الضغط العالي التي تظهرها منحنيات امتصاص لانغموير تتوافق مع القيم التجريبية. على وجه التحديد، سعة الامتصاص عند الضغط العالي لـ (Zn2(bdc)2(dabco)، الميثان، 393 كلفن) (MIL-101، الميثان، 373 كلفن) > (MOF-177، الميثان، 398 كلفن). وهذا يشير إلى أن إطار عمل Uni-MOF قادر على تصفية الممتزات عالية الأداء بدقة تحت الضغط العالي بناءً فقط على سعة الامتصاص المتوقعة تحت الضغط المنخفض. ومع ذلك، لا نزال نلاحظ انحرافات كبيرة بين العديد من القيم المتوقعة والتجريبية تحت الضغط المنخفض، خاصة في حالات Mg -dobdc و MOF-5. قد لا تمثل المحاكاة القيم التجريبية بدقة لـ MOFs ذات المواقع المعدنية المفتوحة، مثل Mg-dobdc . ومع ذلك، فإن MOF-5 يحتوي على مواقع معدنية قريبة، وقد أظهرت الدراسات السابقة أن المحاكاة يمكن أن تصف بشكل فعال قيم امتصاص الغاز التجريبية . على الرغم من هذه النتائج، لاحظنا أن أداء امتصاص الغاز لـ MOFs يختلف حتى تحت نفس ظروف التشغيل (راجع
الجداول التكميلية 8-10). وهذا يشير إلى أن هناك طرقًا مختلفة وأخطاء موضوعية كبيرة موجودة في القيم التجريبية. لذلك، لم نقم باختيار البيانات عمدًا، بل هدفنا إلى تقديم تمثيل شامل للبيانات المجمعة قدر الإمكان. على سبيل المثال، تمثل التقاطعات السوداء في الشكل 3e القيم التجريبية من مصادر أدبية متنوعة تحت ظروف تشغيل متطابقة. تظهر النتائج أنه، على الرغم من التباينات الكبيرة في القيم التجريبية، فإن إطار عمل Uni-MOF يحافظ على مستوى عالٍ من الدقة في تصنيف المواد، مما يجعله مناسبًا لمعالجة التحديات الهندسية.
علاوة على ذلك، يتم إدخال القيم التجريبية لتصحيح منحنيات الامتزاز. على سبيل المثال، توضح الشكل 3ب منحنى الامتزاز لانغموير الذي تم الحصول عليه من خلال ملاءمة كل من بيانات الامتزاز المتوقعة والتجريبية. بينما نستخدم مجموعات بيانات محاكاة لمعالجة ندرة البيانات، يمكننا أيضًا إدخال القيم التجريبية بشكل صحيح لتصحيح منحنيات الامتزاز، مما يساعد على توقع أكثر دقة لأداء الامتزاز عند الضغط العالي حيث تصبح تفاعلات الغاز-الغاز أكثر أهمية. في الشكل 3ب، يمكن ملاحظة أن منحنيات الامتزاز المصححة لها ارتباط قوي مع سعة الامتزاز التجريبية إلى حد ما. تظهر النتائج أن Uni-MOF لا يمتلك فقط القدرة على تصفية أداء الامتزاز لنفس الغاز في مواد مختلفة ولكن أيضًا
الشكل 3 | منحنيات الامتزاز بناءً على توقعات الضغط المنخفض والقيم التجريبية للضغط العالي، كل منحنى يمثل ملاءمة لانغموير. أ) منحنيات الامتزاز المتوقعة لـ Uni-MOF و (ب) منحنيات الامتزاز المصححة تجريبيًا لامتزاز الميثان في Zn2(bdc)2(dabco) و MIL-101 و MOF-177 عند، و398 ك، على التوالي. ج تم التنبؤ بامتصاص Uni-MOF و (د) تم تصحيح تجريبي لثوابت الامتصاص للميثان وثاني أكسيد الكربون في Mg-dobdc عند 298 ك. هـ Uni-
تنبؤات MOF وتصحيحات تجريبية لثوابت الامتصاص للميثان وثاني أكسيد الكربون في MOF-5 عند 298 كلفن و296 كلفن. كل مجموعة من الرسوم البيانية الأفقية (أي، و و و ) يمثل نفس النظام، أي نفس MOF وجزيء الغاز. العلامة الفارغة تمثل القيمة المتوقعة من UniMOF، والعلامة المملوءة تمثل البيانات التجريبية المستندة إلى الأدبيات السابقة.
يمكنه بدقة فحص أداء الامتزاز لغازات مختلفة في نفس المادة (الشكل 3c، d) أو عند درجات حرارة مختلفة (الشكل 3e، f).
في المستقبل القريب، سيتطلب تقاطع الذكاء الاصطناعي (AI) وعلوم المواد حل القضايا العملية والعلمية. ومع ذلك، فإن تحقيق تنفيذ العمليات بواسطة الذكاء الاصطناعي في مجال تقنيات التعلم الآلي التي تتطلب كميات هائلة من البيانات لا يزال يمثل تحديًا كبيرًا، نظرًا لنقص البيانات التجريبية وتنوع تقنيات التصنيع وظروف التوصيف المعنية. لقد حقق بحثنا تقدمًا كبيرًا في علوم المواد من خلال دمج ظروف التشغيل في إطار Uni-MOF لضمان كفاية البيانات وتمكين وظائف الفحص التي تتماشى مع النتائج التجريبية.
التنبؤ عبر الأنظمة
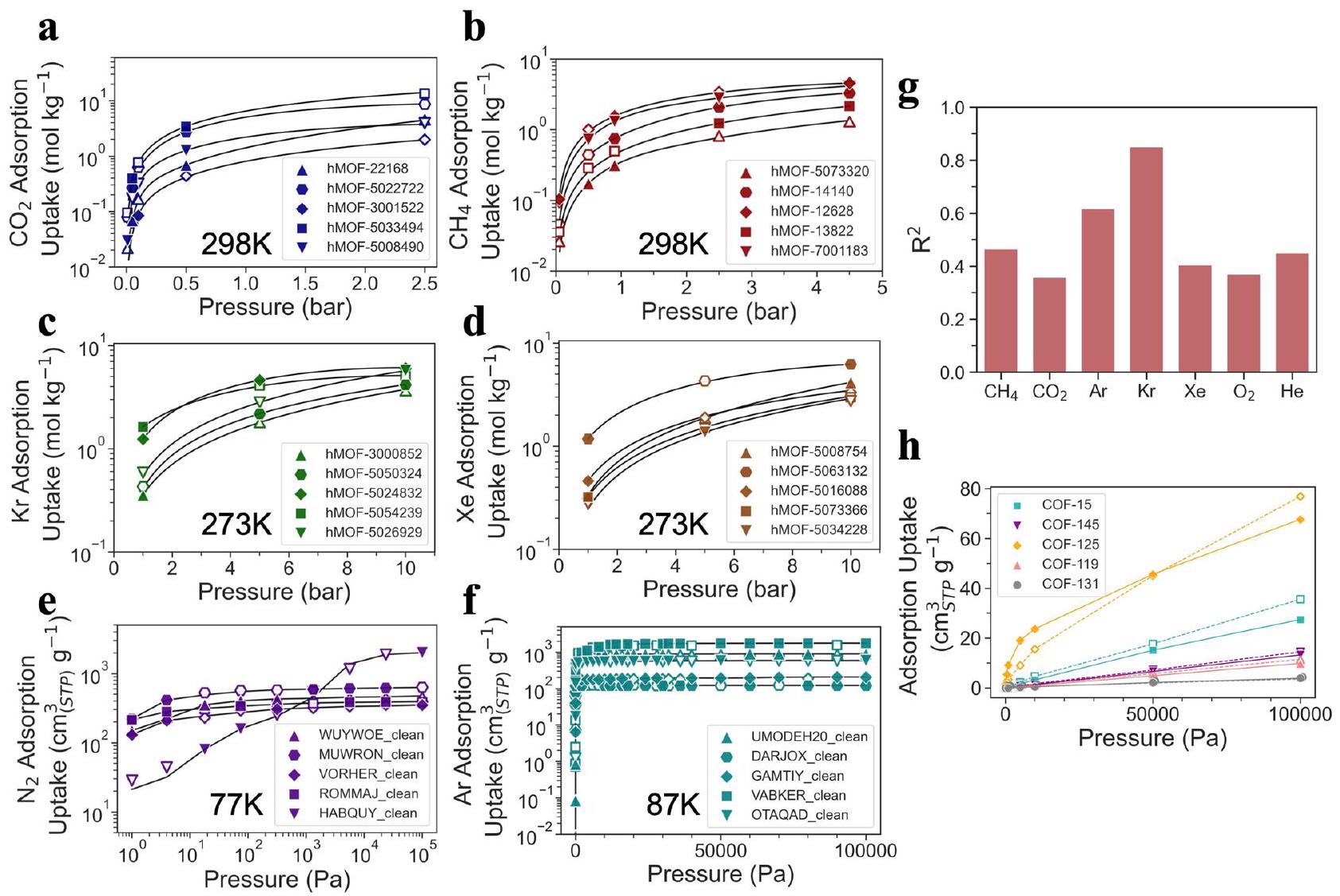
من أجل عرض القدرات التنبؤية لـ Uni-MOF فيما يتعلق بالخصائص عبر الأنظمة، تم اختيار خمسة مواد بشكل عشوائي من كل من الأنظمة الستة (ثاني أكسيد الكربون عند 298 كلفن، الميثان عند 298 كلفن، الكريبتون عند 273 كلفن، الزينون عند 273 كلفن، النيتروجين عند 77 كلفن والأرجون عند 87 كلفن) الموجودة في قواعد البيانات hMOF_MOFX_DB و
تم أخذ عينات شاملة من CoRE_MOFX_DB من حيث درجة الحرارة والضغط. ثم تم مقارنة القيم المتوقعة والمحاكاة لامتصاص الغاز عند ضغوط متغيرة، مع تقديم النتائج في الشكل 4a-f. ستؤدي ملاءمة منحنيات الامتصاص من كل من توقعات Uni-MOF والقيم المحاكاة إلى تقليل الأخطاء البصرية بشكل مصطنع. من أجل القضاء على تحيز البيانات، تم الحصول على منحنيات الامتصاص في جميع الحالات فقط من القيم المحاكاة. من الواضح أنه، نظرًا لأن منحنيات الامتصاص تم الحصول عليها بشكل كامل من خلال القيم المحاكاة، فإن القيم المتوقعة لامتصاص الغاز التي تم إنشاؤها بواسطة Uni-MOF لقاعدة بيانات hMOF_MOFX_DB و CoRE_MOFX_DB تتماشى عن كثب مع القيم المحاكاة عبر جميع الحالات. تدعم هذه النتيجة أيضًا الدقة العالية للتوقعات التي تم توضيحها في الشكل 2a، b.
نظرًا لقدرة Uni-MOF على التنبؤ بالخصائص عبر الأنظمة، كنا مهتمين بإمكاناته في التنبؤ بسعة الامتصاص للغازات غير المعروفة. تحتوي قاعدة بيانات CoRE_MAP_DB على مجموعة متنوعة من نقاط بيانات الامتصاص لغازات مختلفة، مثل الميثان وثاني أكسيد الكربون والأرجون والكراتون والزينون والأكسجين والهليوم. لتقييم القدرة التنبؤية لـ Uni-MOF، قمنا بتقسيم نقاط بيانات CoRE_MAP_DB بشكل عشوائي حسب الغاز الممتص إلى ثلاث مجموعات بيانات (تدريب،
الشكل 4 | حالات التنبؤ عبر النظام Uni-MOF. القيم المتوقعة والمحاكاة لكمية امتصاص الغاز مقابل الضغط في قاعدة بيانات hMOF_MOFX_DB لـ (أ) ثاني أكسيد الكربون عند 298 كلفن، (ب) الميثان عند 298 كلفن، (ج) الكريبتون عند 273 كلفن و (د) الزينون عند 273 كلفن، وفي قاعدة بيانات CoRE_MOFX_DB لـ (هـ) النيتروجين عند 77 كلفن و (و) الأرجون عند 87 كلفن. تشير العلامات المختلفة في كل شكل إلى مواد امتصاص MOF مختلفة. تمثل العلامة الفارغة الامتصاص المتوقع لـ Uni-MOF، وتمثل العلامة المملوءة الامتصاص المحاكى في قاعدة البيانات. يتم الحصول على منحنيات الامتصاص فقط من القيم المحاكية لإزالة التحيز في البيانات. تعلم نقل الغاز تحت ظروف متعددة الأنظمة، مع التقسيم العشوائي إلى ثلاثة مجموعات بيانات (مجموعة التدريب، مجموعة التحقق، ومجموعة الاختبار بنسبة ) وفقًا لغازات الامتصاص. متوقع ومحاكىإيزوثرمات الامتزاز عند 300 كلفن في مواد COF، تمثل أنواع العلامات المختلفة COFs مختلفة. العلامة الفارغة تعني القيمة المتوقعة، بينما تمثل العلامة المملوءة القيمة المحاكاة.يمثل معامل التحديد. يتم توفير بيانات المصدر كملف بيانات المصدر. صحيح، ومجموعة بيانات الاختبار بنسبة” )، ثم تم التنبؤ بسعة الامتزاز لكل غاز على حدة. تم تصوير التنبؤات الناتجة في الشكل 4g وتلخيصها في الجدول التكميلي 11. من المRemarkably، أظهر Uni-MOF قوة في التنبؤ بسعة الامتزاز للغازات غير المعروفة، محققًا دقة تنبؤ عالية (0.85 للكراتون ودقة توقع تزيد عن 0.35 لجميع الغازات غير المعروفة.
يمكن ملاحظة أن أداء التنبؤ يختلف بين الغازات. على سبيل المثال،و Xe جميعها غازات نبيلة حيث يظهر Kr أفضل أداء بينها مع0.85، في حين أن أداء التنبؤ لـ Xe أقل من ذلك ( ). توزيع مشابه لخصائص مادة MOF عبر ، وتمت ملاحظة قواعد بيانات Xe، كما هو موضح في الشكل الفرعي المقابل في الشكل التوضيحي 2، مما يشير إلى أن اختلافات أخذ عينات مواد MOF لها تأثير ضئيل في هذه الحالة. على الرغم من أن قطر جزيء Xe أكبر، مما يجعله أقل ملاءمة للامتزاز في المسام الصغيرة، إلا أنه يظهر حد امتصاص أعلى (موضح في الشكل التوضيحي 3) مقارنة بالأرجون والكرپتون. قد يكون هذا نتيجة للمعلمات الطاقية الأكبر لـعند التفاعل مع ذرات مادة MOF. في الشكل الفرعي من الشكل التكميلية 2c، المواد ذات سعة امتصاص Xe العالية (تتجاوز ) تم تمييزها باللون الأصفر. هذه المواد عادة ما تظهر أحجام مسام أكبر ونسب فراغات، حيث تمتلك أكبر قطر تجويف (LCD) أكبر من كسر فراغ يتجاوز 0.68، وPLD لا يقل عن (قريب من القطر الحركي للزينون مع على الرغم من أنه من الصعب لإقامة ارتباط شامل بين توقعات النموذج والخصائص الفيزيائية الكيميائية للغازات، يتبين أن الغازات التي تمتلك معلمات طاقة أكبر عادة ما تظهر أداءً تنبؤياً أقل، مثل، وXe. يمكن أن يُعزى هذا الظاهرة إلى الأهمية المتزايدة لتفاعلات جزيئات الغاز مع زيادة الضغط، جنبًا إلى جنب مع التعقيد الذي تسببه المعلمات عالية الطاقة، مما يؤدي في النهاية إلى تعقيد تعلم النقل.
على الرغم من التحديات، يمكن ملاحظة نتائج مشجعة، حيث يتنبأ النموذج بدقة بامتصاص الكريبتون المعتدل، استنادًا إلى سلوك الامتصاص للأرجون والزينون ضمن نفس فئة الغازات الخاملة.
في هذه الدراسة، نعرض الأداء العام لـ Uni-MOF في تعلم نقل الغاز، باستخدام قاعدة بيانات تتكون من أقل من 100,000 نقطة بيانات. وهذا يشير إلى أنه حتى في ظل عدم اختيار ظروف التنبؤ المثلى بشكل محدد، فإن Uni-MOF قادر في مجالات الهندسة. للتخفيف من الإفراط في التكيف، يتم عادةً تقسيم البيانات إلى ثلاث مجموعات. ومع ذلك، فإن تعلم النقل عبر الغازات المتنوعة يمثل تحديًا كبيرًا، خاصة عند مواجهة عدد محدود من أنواع الغاز ونقاط البيانات. نتيجة لذلك، من أجل تحسين استخدام البيانات النادرة، يتم أيضًا عرض النتائج التي تم الحصول عليها من تقسيم البيانات إلى مجموعتين بناءً على أنواع الغاز في الشكل التوضيحي 5. كشفت النتائج عن تحسين في دقة تعلم النقل لكل غاز إلى حد ما. على عكس التنبؤ بالامتصاص بين المواد المختلفة، فإن التنبؤ بـ
الشكل 5 | مقارنة بين Uni-MOF و Uni-MOF بدون تدريب مسبق. مقارنة الأداء بين Uni-MOF و Uni-MOF بدون تدريب مسبق في (أ) CoRE_MOFX_DB و (ب) hMOF_MOFX_DB. يمثل الرمز المملوء أداء التنبؤ لقاعدة البيانات بالكامل. يمثل الرمز الفارغ أداء التنبؤ لـ مجموعة البيانات الفرعية (غاز معين، درجة حرارة وضغط). يمكن العثور على التفاصيل في الجدول التكميلي 15-17. ج مقارنة الارتباط بين القيمة المتوقعة والقيمة المحاكاة لكمية امتصاص الغاز عبر Uni-MOF و Uni-MOF بدون تدريب مسبق لقاعدة بيانات CoRE_MAP_DB.يعني معامل التحديد. سلوك الامتزاز للغازات غير المعروفة هو تحدٍ كبير. تؤثر الاختلافات في حجم الجزيئات، وطاقة الامتزاز السطحية، والقوى بين الجزيئات بين أنواع الغاز المختلفة بشكل كبير على آلية وسلوك الامتزاز. على الرغم من هذه التعقيدات، فإن Uni-MOF يظهر عمومية استثنائية، كما يتضح من قدرته على التنبؤ بدقة بخصائص الامتزاز للغازات غير المعروفة بدعم فقط من بيانات الامتزاز لغازات أخرى.
علاوة على ذلك، قمنا بالتحقيق في قدرة Uni-MOF على التنبؤ بسلوك امتصاص الغاز في COFs. حوالي 500 نقطة بيانات منتمت محاكاة امتصاص الإعلانات في COFs CoRE عند 300 كلفن. على الرغم من الحجم المحدود لقاعدة بيانات امتصاص COF، فإن Uni-MOF يحافظ على أداء تنبؤي عالي، محققًا 0.76. بالإضافة إلى ذلك، كما هو موضح في الشكل 4h، يظهر Uni-MOF قدرة تصنيف عالية لامتصاص مواد متنوعة تحت ضغوط مختلفة.
إطار عمل يطبق التدريب المسبق
من أجل التحقق من أن استراتيجية التعلم الذاتي المراقب للتدريب المسبق يمكن أن تحسن بشكل فعال من قوة التحمل وأداء التنبؤ في Uni-MOF، قمنا بإنشاء ومقارنة Uni-MOF بدون تدريب مسبق مع Uni-MOF على ثلاثة قواعد بيانات، وهي CoRE_MOFX_DB و hMOF_MOFX_DB و CoRE_MAP_DB. تظهر قواعد بيانات CoRE_MOFX_DB و hMOF_MOFX_DB درجة عالية من تركيز البيانات، مما يتيح تقسيمها إلى قواعد بيانات أصغر تحتوي على بيانات الامتصاص لمواد مختلفة تحت ظروف عمل متطابقة (أي نفس الغاز، ودرجة الحرارة، والضغط). قمنا بتدريب كل من قواعد بيانات CoRE_MOFX_DB و hMOF_MOFX_DB باستخدام Uni-MOF و Uni-MOF بدون تدريب مسبق، ومن ثم تم حساب معاملات التحديد لكل مجموعة بيانات صغيرة. بالإضافة إلى ذلك، يتم تدريب كل مجموعة بيانات صغيرة بشكل منفصل باستخدام إطار عمل Uni-MOF لاشتقاق معامل التحديد المقابل.
كما هو موضح في الشكل 5a، تمثل العلامات المملوءة الأداء التنبؤي لقاعدة بيانات CoRE_MOFX_DB بالكامل. يمكن ملاحظة أن Uni-MOF يتفوق على Uni-MOF بدون التدريب المسبق. تتيح استراتيجية التعلم الذاتي المسبق للنموذج تعلم التكوين ثلاثي الأبعاد للمواد المسامية النانوية بعمق، مما يحسن دقة ضبط النموذج. في هذا الجزء، يتم تقسيم مجموعات البيانات بطريقة تتضمن كل مجموعة بيانات النظام الفرعي ذات الصلة. لذلك، تمثل جميع العلامات السداسية في الشكل 5a الأداء التنبؤي العام لـ Uni-MOF. في الواقع، يظهر Uni-MOF أداءً عاليًا متساويًا على مجموعة البيانات الكاملة كما هو الحال على مجموعة بيانات فردية، حتى عندما يعتمد أداء مجموعة بيانات واحدة على بيانات أكثر اتساعًا من مهمة التنبؤ المحددة لها. بالنسبة لضبط المهام ذات النظام الفردي، أي التدريب بشكل فردي لكل مجموعة بيانات صغيرة، فإن الأداء التنبؤي بالكاد تجاوز أداء Uni-MOF. وبالتالي، نعلم أن أخذ عينات بيانات واسعة النطاق يمكن أن يعزز قدرة التنبؤ لنموذج التعلم. يمكن تلخيص نفس الاستنتاجات بالنسبة لـ hMOF_MOFX_DB من الشكل 5b.
ومع ذلك، بسبب العينة المتناثرة من قاعدة بيانات CoRE_MAP_DB، لا يمكن تقسيم مجموعات البيانات الصغيرة المحدودة وفقًا لنفس الإجراء كما في قاعدتي البيانات السابقتين. لذلك، يتم مقارنة العلاقات بين القيم المتوقعة والمحاكاة من Uni-MOF وUni-MOF بدون تدريب مسبق، كما هو موضح في الشكل 5c. وبالمثل، مقارنةً بـ Uni-MOF بدون تدريب مسبق، كانت أداء Uni-MOF
الشكل 6 | توقع وتحليل الميزات الهيكلية. العلاقة بين القيمة المتوقعة والقيمة الحاسوبية لـ (أ) قطر الحد من المسام (PLD) )، (ب) أكبر قطر تجويف (LCD) ( )، (ج) نسبة الفراغ، و (د) حجم المسام ( ) من MOFs في قواعد بيانات hMOF (الشكل الرئيسي) و CoRE_MOF (الشكل الفرعي). مقارنة تقدير كثافة النواة (KDE) لميزات هيكلية مختلفة [PLD ( ) ، LCD ( )، كسر الفراغ، مساحة السطح ( )، حجم ( )] بين CoRE_MOF مع جميع قيم امتصاص الأرجون و CoRE_MOF مع الأعلى أداء امتصاص الأرجون عند 87 كلفن و هو معامل التحديد، MOF تعني الإطار المعدني العضوي. يتم توفير بيانات المصدر كملف بيانات المصدر. تحسنت من 0.70 إلى 0.83، مما أثبت بشكل أكبر أهمية استراتيجية التدريب المسبق لـ Uni-MOF.
تنبؤ الميزات الهيكلية والفحص عالي الإنتاجية
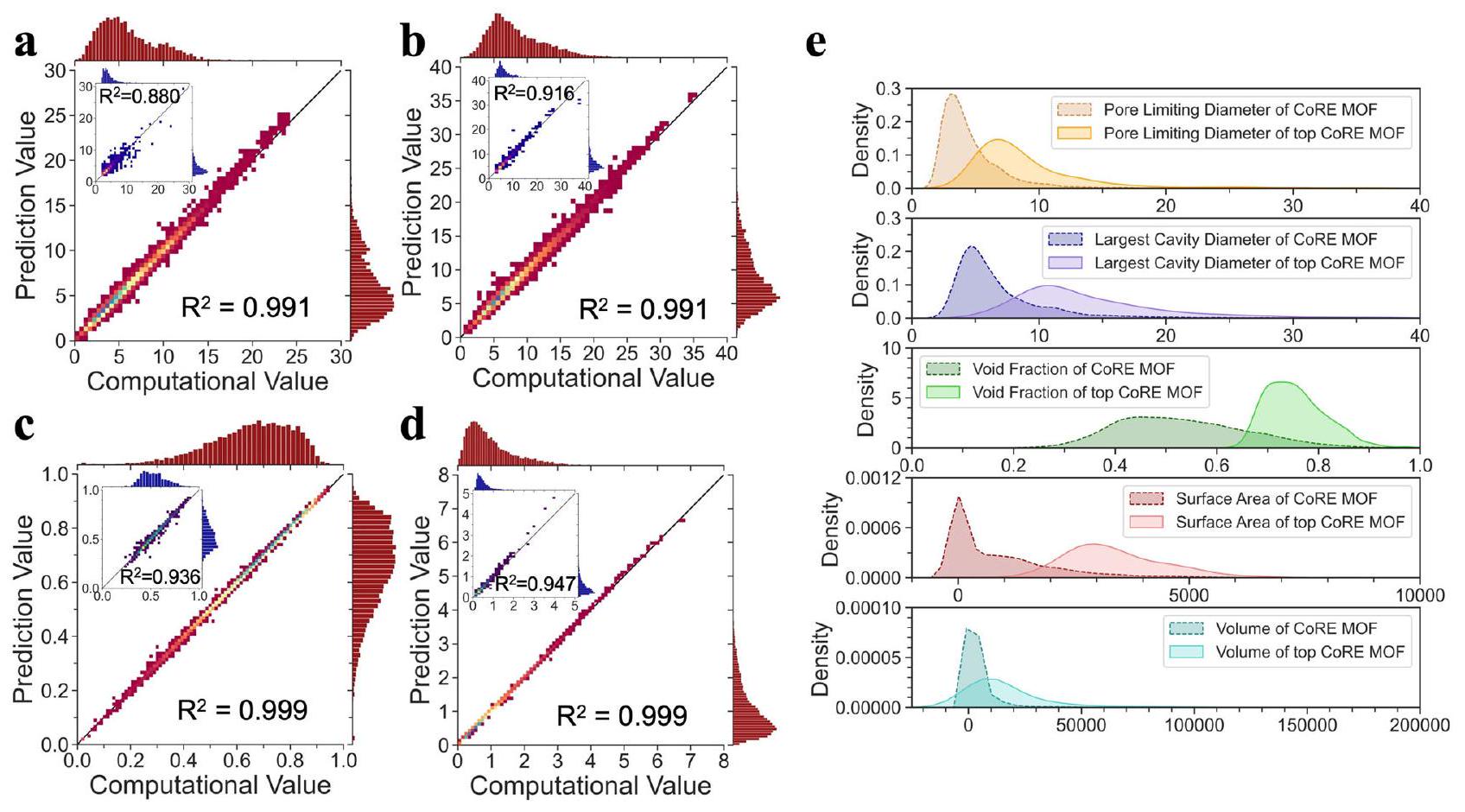
بينما يتمتع إطار Uni-MOF بقدرة على تمييز الترتيب المكاني للمواد النانوية المسامية، نهدف إلى استكشاف إمكانياته في التنبؤ بالخصائص الهيكلية. تُعرض نتائج التنبؤ بالخصائص الهيكلية لـ Uni-MOF لمكتبات مواد hMOF و CoRE_MOF في الشكل 6a-d.
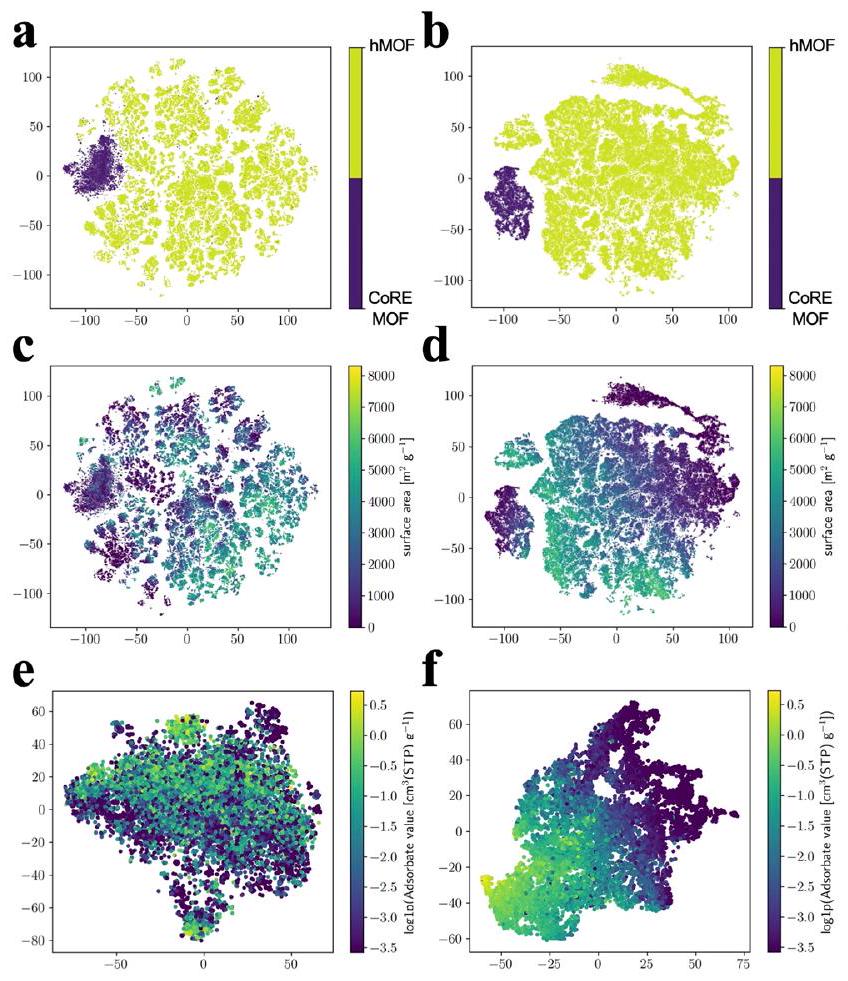
يمكن ملاحظة أن أداء التنبؤ لبارامترات الهيكلية لـ hMOF يتفوق عمومًا على أداء CoRE_MOF. من خلال استخدام مجموعة من الكتل البنائية الهيكلية (عقد معدنية، روابط، ومجموعات وظيفية)، تم استكشاف مساحة التصميم لمجموعة بيانات MOF الافتراضية (hMOFs) بالكامل، وتوزعت الميزات الهيكلية بشكل أكثر توازنًا مقارنة بمجموعة بيانات CoRE_MOF. تميل المواد المركبة تجريبيًا من MOF (CoRE_MOF) إلى تفضيلات هيكلية لعقد معدنية (مثل الزنك، والنحاس، والكادميوم) والروابط. أظهرت الأبحاث السابقة أن MOF التجريبي (CoRE-2019) يتركز بشكل أساسي في منطقة المسام الصغيرة، بينما يتم توزيع hMOF (التبغ) بشكل أكثر توازنًا مع تحول طفيف نحو منطقة المسام الكبيرة.يمكن العثور على نفس الملاحظة في الشكل التوضيحي التكميلي 6، حيث يظهر الشكل التوضيحي التكميلي 6a والشكل التوضيحي التكميلي 6b الفروق بين هياكل CoRE_MOF و hMOF باستخدام طريقة تضمين الجوار العشوائي الموزع t (t-SNE). من خلال الشكل التوضيحي التكميلي 6c-f، يمكن ملاحظة أن الفتحات في CoRE_MOF تتركز بوضوح في منطقة الفتحات الصغيرة، خاصةً PLD. من ناحية أخرى، لا يزال يمكن ملاحظة تدرج الفتحات في hMOF بسبب غنى عيناته الهيكلية ونقاط البيانات، على الرغم من أن الفتحات بشكل عام صغيرة. وبالتالي، بالنسبة لأنواع مختلفة من قواعد البيانات، فإن توقع الميزات الهيكلية يؤدي بشكل مختلف، خاصةً بالنسبة لـ PLD. ومن الجدير بالذكر أنه في مواد مثل hMOFs، تصل قدرته التنبؤية إلى معامل تحديد أكبر من 0.99، مما يدل على مستوى عالٍ من
الدقة والموثوقية. لقد تم التعرف على أن Uni-MOF يظهر قدرة قوية في التنبؤ بالخصائص الهيكلية للمواد، وذلك بفضل استخدام التدريب المسبق على عدد كبير من الهياكل ثلاثية الأبعاد. وبالتالي، لا يتنبأ Uni-MOF بدقة بالأداء المطلوب لمواد MOF فحسب، بل يتنبأ أيضًا بدقة بخصائصها الهيكلية، وهو ما يحمل أهمية كبيرة لأبحاث وتطبيقات المواد.
يمكن إنشاء عدد لا يحصى من المواد النانوية المسامية مع وحدات بناء ثانوية متنوعة، مما يؤدي إلى هياكل MOF متنوعة بشكل استثنائي، مما يجعل تحليل هيكل وخصائص MOF مبادرة استراتيجية للغاية. في هذا العمل، تم جمع وتوليد امتصاصات الغاز المتعددة تحت ظروف تشغيل مختلفة، حيث تعتبر امتصاصات الأرجون عند 87 كلفن و1 بار تمثيلية وقابلة للتحليل. يمثل الأرجون الغالبية العظمى من الغازات النبيلة في الغلاف الجوي (9340 جزء في المليون في الظروف المحيطة)، وقد تم استخدامه على نطاق واسع للعزل والإضاءة بقيمة تجارية تبلغ 3.1 دولار أمريكي.. الشكل 6e يظهر مقارنة لتقدير كثافة النواة (KDE) لخصائص هيكلية مختلفة بين CoRE_MOF بالكامل مع قيم امتصاص الأرجون وMOFs في CoRE_MOF مع الأداء الأعلى لامتصاص الأرجون. تصور KDE توزيع البيانات باستخدام منحنى كثافة الاحتمال المستمر بطريقة أقل ازدحامًا.
نظرًا لأن توزيع بعض الخصائص الهيكلية محدود، فقد يؤدي ذلك إلى تشوهات (مثل القيمة السلبية لمساحة السطح والحجم). ومع ذلك، لا يزال يقدم اتجاهات قابلة للتفسير ودقيقة في هيكل أفضل مواد امتصاص MOF. المعلمات النموذجية التي تصف أحجام المسام هي كما يلي. 1) قطر المسام المحدود (PLD) هو أكبر كرة حرة؛ 2) LCD هو أكبر كرة مدرجة على طول مسار الكرة الحرة. وبالتالي، فإن LCD أكبر بطبيعتها من PLD في نفس المادة. بالمقارنة مع قاعدة بيانات MOF بالكامل، فإن أفضل من MOFs لديها قيم توزيع أكبر من 5-10 و لـ PLD وLCD، على التوالي. تتوزع الكسر الفارغ لقاعدة البيانات بالكامل بشكل معتدل حول 0.5، بينما تلك الخاصة بأفضل MOFs أكبر بكثير، وتتوزع بشكل رئيسي حول 0.75. نظرًا لأن معظم الامتصاص
الشكل 7 | تصور التمثيلات الهيكلية لـ MOFs في مجموعات بيانات hMOF وCoRE_MOF، يتم حساب التضمينات ذات الأبعاد المنخفضة بواسطة طريقة t-SNE (تضمين الجوار العشوائي الموزع). التمثيلات المسترجعة بعد (أ)، (ج)، و(هـ) التدريب المسبق و(ب)، (د)، و(و) الضبط الدقيق مقابل الخصائص الأخرى. التضمينات ذات الأبعاد المنخفضة مقابل تسميات مجموعة البيانات، حيث يمثل اللون الداكن CoRE_MOF واللون الفاتح يمثل hMOF. توضح التمثيلات مقابل مساحة السطح في لـ hMOF و
CoRE_MOF مجتمعة. هـ، تظهر العلاقة بين التمثيلات بالنسبة لقيم الامتصاص لـ Ar عند لمجموعة بيانات CoRE_MOF فقط. الذكاء الاصطناعي القابل للتفسير (XAI). ج توضيح هيكل hMOF-5004238 و(ح)، (ط) خريطة حرارية لتفاعلات الذرات التي تتعلم من خوارزمية الانتباه متعدد الرؤوس في الرأس 10 والرأس 18. تعني MOF إطار العمل المعدني العضوي. يتم توفير بيانات المصدر كملف بيانات مصدر.
يحدث على سطح المواد النانوية المسامية، تصبح مساحة السطح واحدة من أهم المحددات، وتظهر النتائج أن أفضل 10% من MOFs تمتلك مساحة سطح كبيرة جدًا تبلغ حوالي، وهو ما يتماشى مع الحس السليم. تحدد مساحة السطح عملية الامتصاص السطحي، بينما تحدد LCD عملية الامتصاص الداخلي. لذلك، تم تصنيف أفضل 10% من MOFs إلى ثلاث فئات وفقًا لأداء امتصاصها على الأرجون، وتم استكشاف التوزيع العددي لهذين العاملين الرئيسيين (مساحة السطح/LCD) (موضح في الشكل التكميلي 8). يمكن للمرء أن يرى أنه عند 87 كلفن و1 بار، فإن أفضل مواد امتصاص MOF للأرجون تمتلك بشكل رئيسي LCD تبلغ حوالي ومساحة سطح تبلغ حوالي.
نمذجة تمثيل الهيكل المادي
للتحقق من أن هياكل MOF تم تعلمها بشكل جيد في كل من مراحل التدريب المسبق والضبط الدقيق، نقوم بتصور الخصائص الهيكلية، والتي هي متجهات ذات أبعاد 512، باستخدام طريقة t-SNE. تظهر النتائج في الشكل 7. كما هو موضح في الشكل 7أ، ب، فإن الميزات المتعلمة قادرة على تصنيف MOFs إما من مجموعات بيانات CoRE_MOF أو hMOF. قد يلاحظ المرء أيضًا أن الحدود بين CoRE_MOF وhMOF في الميزات المضبوطة بدقة أكثر وضوحًا بكثير، مما يشير إلى تحسين كبير لهذه الميزات بعد الضبط الدقيق. بشكل ملحوظ، يتم توضيح ذلك بوضوح عندما نرسم التضمينات مقابل مساحات السطح لـ MOFs، كما هو موضح في الشكل 7د، حيث تقع الهياكل ذات مساحات السطح الصغيرة في الزوايا العليا اليمنى والسفلى اليسرى، وتكون الهياكل ذات مساحات السطح الكبيرة في المركز. بالمقارنة مع ميزات التدريب المسبق، الشكل 7ج، يمكن أن يحسن الضبط الدقيق بشكل كبير جودة التمثيل.
من ناحية أخرى، نحن أكثر فضولًا بشأن ما إذا كانت التمثيلات الهيكلية المتعلمة مرتبطة ارتباطًا وثيقًا بـ
سلوكيات الامتصاص. لمعالجة هذه المسألة، نقوم بتصور التضمينات الهيكلية مقابل قيم الامتصاص لكل من Ar و، ونظهر نتيجة Ar هنا كمثال، انظر الشكل. كما يمكن ملاحظته، فإن التمثيلات المتعلمة في مرحلة التدريب المسبق غير قادرة على تصنيف الهياكل ذات سعات الامتصاص المختلفة بينما يتم تجميع الهياكل ذات قيم الامتصاص المتنوعة معًا، انظر الشكل 7هـ. ومع ذلك، بعد الضبط الدقيق، يتم فصل الهياكل ذات سلوكيات الامتصاص المختلفة بشكل جيد، مما يظهر علاقة جيدة بين التمثيلات المتعلمة وهدف قيم الامتصاص، انظر الشكل 7و. هذا يفسر جيدًا وظائف مرحلة الضبط الدقيق في التأثير بشكل أكبر على التمثيلات الهيكلية وكذلك معلمات النموذج الأخرى.
علاوة على ذلك، يمكن لآلية الانتباه متعدد الرؤوس في Transformer تعلم التفاعلات داخل الهيكل المادي. باستخدام خوارزمية الانتباه ذات 64 رأسًا، يتم تمثيل مناظر التفاعل الذري لـ hmof-5004238 في رأسين مختلفين في الشكل 7ح، ط. كما هو موضح في الشكل 7ح، يمكن ملاحظة تفاعلات قوية بين مواقع المعادن (Zn)، وزنك وذرات الأكسجين، وكذلك ذرات الأكسجين. الشكل 7ط يصور أيضًا التفاعل بين سلاسل الكربون الخطية (C7-C15). بالإضافة إلى ذلك، هناك ارتباط ملحوظ بين ذرة O 0 وذرات Zn (). تؤكد التوضيح الهيكلي في الشكل 7 ج هذه النتيجة، حيث ترتبط الذرات الأربعة كيميائيًا. بخلاف ذلك، يتم تصوير المناظر الكيميائية المختلفة لـ hmof-5004238 في رؤوس مختلفة في الشكل التكميلي 9. بهذه الطريقة، يحدد Uni-MOF مواد هائلة ويقدم توقعات موثوقة لخصائص متنوعة.
الاستنتاج
في هذه الدراسة، قدمنا Uni-MOF، إطار عمل متعدد الأغراض يمكنه التنبؤ بدقة بامتصاص الغاز في مواد MOF. نحن أيضًا
تم إنشاء وجمع وتنظيم قواعد بيانات ذات صلة بالمواد النانوية المسامية ومجموعات بيانات امتصاص الغاز. تم إجراء التعلم الذاتي المشرف لقاعدة بيانات تحتوي على أكثر من 631,000 من MOFs و COFs، مما أسفر عن دقة توقع عالية تبلغ 0.98. وهذا يشير إلى أن إطار تعلم التمثيل القائم على التدريب المسبق ثلاثي الأبعاد يتعلم بفعالية المعلومات الهيكلية المعقدة لـ MOFs مع تجنب الإفراط في التكيف. قمنا بتطبيق Uni-MOF للتنبؤ بأداء امتصاص الغاز لثلاث قواعد بيانات رئيسية وحققنا دقة توقع عالية تصل إلى 0.98 في قاعدة بيانات تحتوي على بيانات كافية. في حالة مجموعة بيانات تم أخذ عينات منها بشكل كافٍ، لا يحافظ Uni-MOF على دقة توقع تزيد عن 0.83 فحسب، بل يختار أيضًا بدقة المواد الممتصة عالية الأداء تحت ضغط عالٍ من خلال التنبؤ فقط بالامتصاص تحت ضغط منخفض، بما يتماشى مع نتائج الفحص التجريبي. وبالتالي، يمثل Uni-MOF اختراقًا كبيرًا في مجال علوم المواد فيما يتعلق بتطبيق تقنيات التعلم الآلي. علاوة على ذلك، يظهر إطار عمل Uni-MOF أداءً متفوقًا على مجموعات بيانات الأنظمة المتقاطعة مقارنةً بالمهام ذات النظام الواحد ويمكنه التنبؤ بدقة بخصائص امتصاص الغازات غير المعروفة بدقة توقع عالية تصل إلى 0.85، مما يوضح قدرته التنبؤية القوية وعموميتها. من خلال التدريب المسبق المكثف للهياكل ثلاثية الأبعاد، يتعلم Uni-MOF بفعالية الميزات الهيكلية لـ MOFs، محققًا معامل تحديد عالي يبلغ 0.99 لـ hMOFs. بالإضافة إلى ذلك، تؤكد تحليل t-SNE أن مرحلة الضبط الدقيق يمكن أن تتعلم المزيد من الميزات الهيكلية، ويتم التعرف على الهياكل ذات سلوكيات الامتصاص المختلفة بشكل جيد، مما يشير إلى وجود ارتباط قوي بين التمثيلات المتعلمة وأهداف امتصاص الغاز.
تغطي قاعدة بياناتنا نطاق ضغط قدره بار ونطاقات درجات حرارة قدرها و ، مما يجعلها مناسبة لمعظم قضايا امتصاص الغاز. لقد أظهر Uni-MOF نتائج توقع دقيقة لمجموعة شاملة من هياكل MOF ومواد COF المتميزة. حتى في ظل بعض الظروف القاسية، تظل الاتجاهات المتوقعة موثوقة للغاية. مع Uni-MOF، يمكن دعم التحديث المستمر لقواعد البيانات وحتى سيناريوهات الاستخدام، مما يبرز عالميته بشكل أكبر. باختصار، يعمل إطار عمل Uni-MOF كمنصة تنبؤية متعددة الاستخدامات لمواد MOF، حيث يعمل كمنظم لامتصاص الغاز لـ MOFs، حيث يظهر دقة عالية في التنبؤ بامتصاص الغاز تحت ظروف تشغيل متنوعة وله تطبيقات واسعة في مجال علوم المواد.
طرق
جمع المواد والبيانات وتوليدها
تم استخدام هياكل MOF/COF للتدريب المسبق إما من قاعدة البيانات المتاحة حاليًا أو تم إنشاؤها باستخدام البرنامج المقابل. هناك ثروة من قواعد بيانات MOF/COF الموجودة، بما في ذلك قواعد البيانات التي تم تصنيعها بواسطة الكمبيوتر لـ hMOFs ، وToBaCCo (البناء البلوري القائم على الطوبولوجيا) لـ MOFs، وقواعد البيانات على مستوى التجربة لـ CoRE (التجريبية الجاهزة للحساب) لـ MOFs ، وCoRE COFs وCCDC (مركز كامبريدج للبيانات البلورية)، إلخ. واحدة من قواعد البيانات المتكاملة عبر الإنترنت هي MOFXDB، حيث يتوفر أكثر من هيكل COF. بعيدًا عن استكشاف المواد النانوية المسامية في مكتبة المواد، استخدمنا برنامج ToBaCCo.3.0 لإنشاء أكثر من 306,773 هيكل MOF. يأخذ برنامج ToBaCCo كمدخلات مخطط طوبولوجي، ويبحث عن كتل بناء متوافقة من مجموعة محددة، ويقوم بإنشاء جميع الهياكل الممكنة باستخدام الطوبولوجيا بالاشتراك مع كتل بناء العقد المختلفة وكتل بناء الحواف. على وجه الخصوص، يقوم البرنامج بإنشاء هياكل مع ثلاثة مجلدات كمتغيرات إدخال، أي “قوالب”، “عقد”، “حواف”. لتوليد أكبر عدد ممكن من هياكل MOF، نستخدم جميع الحواف كما هو موضح في “edges_database”، وجميع العقد كما هو موضح في “nodes_database” وجميع القوالب كما هو موضح في “template_database”. بالنسبة للمهمة اللاحقة، أي امتصاص الغاز بواسطة MOFs، جمعنا البيانات من مصادر عبر الإنترنت مثل MOFXDB، مكونين مجموعات بيانات لأكثر من 2,400,000 امتصاص لـ hMOFs على
خمسة غازات () عند و وأكثر من 460,000 امتصاص لـ CoRE MOFs على غازين () عند و . في هذا العمل، يتم اعتبار بيانات امتصاص الغاز لمكون واحد فقط. بالإضافة إلى ذلك، أجرينا محاكاة Grand Canonical Monte Carlo (GCMC) باستخدام برنامج RASPA لإنتاج مجموعة بيانات أخرى لأكثر من 99,000 امتصاص للغاز، مع 50,000 دورة تهيئة و50,000 دورة إضافية مستخدمة لعينات سعة الامتصاص. تم الحصول على الامتصاصات المجمعة ضمن و بار، مع الأخذ في الاعتبار سبعة أنواع من جزيئات الغاز (). تم وصف التفاعلات بين جزيئات الغاز والذرات في مواد الامتصاص من حيث إمكانات Lennard-Jones (12-6)، وتم تعيين نصف القطر القطعي إلى مع تصحيح الذيل. تم وصف معلمات حقل القوة لذرات الإطار بواسطة UFF (حقل القوة العالمي) . تم تقدير معلمات حقل القوة لجزيئات الغاز النبيلة (أي الأرجون، والكراتون، والزينون) من مبدأ الحالات المقابلة لمعاملات الفيريل الثانية (المذكورة في الجدول التكميلي 19). تم استخدام نموذج الجزيء Transferable Potentials for Phase Equilibria أيضًا. في عملنا، لا يتم اعتبار الشحنة الجزئية لذرات الإطار بسبب تكلفة الحساب والانحرافات الكبيرة الملحوظة في نتائج الامتصاص باستخدام طرق تعيين شحنات جزئية مختلفة .
تحليل المواد
المفتاح لعلوم المواد الحاسوبية عالية الإنتاجية هو أدوات البرمجيات القوية. هنا، نستخدم Python Materials Genomics (pymatgen)، مكتبة بايثون مفتوحة المصدر وقوية، لاشتقاق خصائص المواد المفيدة من بيانات الهيكل البلوري الخام وإجراء تحليل شامل للمواد. يتم استخراج خصائص المواد، بما في ذلك متجهات الشبكة، وزوايا الشبكة، وحجم وحدة الخلية، والذرات، والإحداثيات، باستخدام pymatgen. سيتم استخدام أنواع الذرات والإحداثيات في مرحلة التدريب المسبق لاستراتيجية التعلم الذاتي المشرف. تم استخدام حزمة بايثون OpenMetalDetector ، لتحليل مجموعات من الإطارات العضوية المعدنية لمواقع المعادن المفتوحة.
إطار عمل Uni-MOF
التدريب المسبق. استخدمنا Uni-Mol كإطار عمل للتدريب المسبق، وهو إطار عمل مخصص للتدريب المسبق ثلاثي الأبعاد مصمم للجزيئات. لقد أظهر Uni-Mol أداءً عاليًا في مهام مختلفة لاحقة في مجال اكتشاف الأدوية. ومع ذلك، نظرًا للاختلاف التام في الهيكل والتوزيع المكاني ثلاثي الأبعاد لمواد MOF مقارنة بالأدوية الصغيرة، بالإضافة إلى ظروف الحدود الدورية (PBC) للهياكل المسامية وعدد الذرات الثقيلة الأكبر بكثير في البلورات، قمنا بإجراء تعديلات ضرورية على Uni-Mol بناءً على هذه الحقائق.
نستفيد من رأس إضافي لمصفوفة الشبكة للحفاظ على معلومات الهندسة الخلوية. يتم اعتبار وجود PBC في تعلم تمثيل MOF حيث أن PBC طبيعي لمواد MOF. بالإضافة إلى توقع الذرات المmasked واستعادة الإحداثيات في التدريب المسبق لـ Uni-Mol، يتم استخدام رأس انحدار لتوقع مصفوفة لتعلم معلومات PBC.
حيث تمثيل مؤشرات Uni-Mol لرمز التصنيف، يشير رمز التصنيف (CLS) إلى الرمز الخاص في تسلسل إدخال الذرات، والذي يستخدم لتمثيل الجزيء بالكامل في مخرجات Uni-Mol. هي مصفوفة الشبكة، نستخدم شبكة عصبية أمامية (FFN) من للتنبؤ بمصفوفة الشبكة مع تحسين خسارة MSE مباشرة. في التدريب المسبق لـ Uni-MOF هو
مجموع و الأصلية. في ، يعتبر توقع الذرات المmasked واستعادة الإحداثيات عنصرين رئيسيين من عناصر الخسارة. لمزيد من التفاصيل، يُرحب بالقراء بالرجوع إلى الورقة الأصلية لـ Uni-Mol. 2) نقترح نواة محكومة بحواف لتشفير المواقع الهندسية المكانية. تشترك MOFs في ترتيب مختلف تمامًا للذرات مقارنة بجزيئات الأدوية في الفضاء ثلاثي الأبعاد، مع هياكل مسامية ومتوسط يزيد عن 1000 ذرة ثقيلة في خلية واحدة. تستخدم UniMol نواة غاوسية لتشفير المعلومات المكانية، بينما تعاني MOF في مرحلة ما قبل التدريب من عدم استقرار التدريب مع نواة غاوسية ذات مسافة أزواج وعدد ذرات أكبر بكثير. لمعالجة ذلك، نقترح نواة مسافة محكومة بحواف.
أينهو المسافة الإقليدية لزوج الذرات، و هو نوع الحافة من زوج الذراتيرجى ملاحظة أن الحافة هنا ليست الرابطة الكيميائية، ونوع الحافة يتحدد بواسطة أنواع الذرات في الزوج ij.هو التحويل الأفيني مع المعلمات و ، فإنه يقرّبالمتوافق مع نوع حافته.يؤشر إلى نواة المسافة المحجوزة بواسطة الحواف، والتي هي مجموع إسقاط المسافة والتضمين المحجوز بواسطة الحواف. FFN هو الشبكة العصبية الأمامية المتعلقة بالتحويل غير الخطي للمسافة.، و LN هو عملية LayerNorm. يتم استخدام بوابة سيغمويد كتحويل أفيني لـلوزن نوع تضمين زوج الحواف.
التعديل الدقيق. توقع امتصاص الغاز المتعدد تحت ظروف تشغيل مختلفة، يجب أن يتم تزويد نموذج التعديل الدقيق ليس فقط بالهيكل المكاني ثلاثي الأبعاد ولكن أيضًا بالغاز وظروف التشغيل (أي، درجة الحرارة والضغط). لذلك، تم اقتراح كتل الغاز وكتل درجة الحرارة/الضغط في Uni-MOF لتشكيل وحدة توقع أداء عبر النظام.
كتلة الغاز تمثل الغاز كمجموعة من معرف الغاز ووصف الخصائص الجوهرية للغاز.
أينفهرس معرف الغازيمثل الغازالمؤشرات (المذكورة في الجدول التكميلي 2). تمثيل الغازهو تجميع لتمثيل معرف الغاز ورسم طبقة الشبكة العصبية التغذوية لوصف الغاز.
نستخدم التقطيع على مسافات متساوية (EDD) والتقطيع اللوغاريتمي (LD) لترميز درجة الحرارة والضغط، على التوالي. يقوم EDD أولاً بتعيين القيمة العددية إلى الدلو المقابل بعرض متساوٍ، ثم يطبق خريطة التضمين. يستخدم LD التحويل اللوغاريتمي مع EDD لاستيعاب الميزات المحتملة اللوغاريتمية.
حيث يتم الإشارة إلى عرض الفاصل كـ تشير إلى الميزات العددية، و هي القيم الأصلية لدرجة الحرارة والضغط للبيئة المعنية. بالنسبة لخاصية درجة الحرارة، نستخدم و التضمينات، وللضغط الميزة، نختار استخدام و مع الأخذ في الاعتبار التحويل اللوغاريتمي المحتمل في الضغط.
تحليل عالي الإنتاجية لقاعدة بيانات MOF الكبيرة
للمزيد من التحقيق في تأثير هيكل المادة على امتصاص الغاز، زيوتم استخدام حزمة البرمجيات، لتحليل المواد المسامية البلورية، لإجراء تحليل للهيكل والطوبولوجيا لجيومترية المادة. تشمل الميزات الهيكلية LCD وPLD ونسبة الفراغ وحجم الفراغ والمساحة السطحية المحددة. يمكن أن يؤثر حجم الفراغ الداخلي بشكل كبير على أداء المواد النانوية المسامية. توجد تعريفات دقيقة للحجم من طرق مختلفة، خاصة حجم المسام القابل للوصول وغير القابل للوصول (Ac-PC، NAc-PC) وحجم المسام القابل للاحتلال وغير القابل للاحتلال (Ac-PO، NAc-PO).. يعتمد مفهوم الوصول على حجم المجس، ومجس ذو نصف قطر يتم استخدامه في عملنا من أجل ربط أحجام المسام بشكل مباشر مع النيتروجين المقاس تجريبيًا (قطر كيناتيكي لـالخطوط المتساوية الحرارة. الحجم القابل للوصول المحسوب هنا يُعرف أيضًا بأنه الحجم المتاح لمركز المجس الكروي، والذي يتوافق مع حجم المسام القابل للوصول لمركز المجس.
تصوير البيانات الإحصائية
في هذا العمل، مكتبات تصور البيانات في بايثون مثل سيبورنومكتبة ماتplotlibتم استخدامه لإنشاء الرسوم البيانية الإحصائية المعلوماتية. يتم رسم الهياكل ثلاثية الأبعاد باستخدام خدمة الويب Bohrium.فيhttps://bohrium.dp.tech.
بيئة الحوسبة
تم إجراء التدريب المسبق والتدريب الدقيق لنموذج Uni-MOF على وحدات معالجة الرسوميات V100/A100، وتم إجراء محاكاة مونت كارلو على مجموعة معالجات مركزية من Bohrium..
ملخص التقرير
معلومات إضافية حول تصميم البحث متاحة في ملخص تقارير مجموعة نيتشر المرتبط بهذه المقالة.
توفر البيانات
البيانات المقدمة في هذه الدراسة متاحة في ملف المخطوطة، وملف المعلومات التكميلية وملفات بيانات المصدر. تم إيداع بيانات المصدر في هذه الدراسة في قاعدة بيانات figshare.تحت رمز الوصول لـhttps://doi.org/10.6084/m9.figshare.24996317.
توفر الشيفرة
الكود لتشغيل نموذج Uni-MOF متاح على GitHub (https://github. com/dptech-corp/Uni-MOF) .
Sholl, D. S. & Lively, R. P. Seven chemical separations to change the world. Nature 532, 435-437 (2016).
Kohl, A. L. & Nielsen, R. Gas Purification (Elsevier, 1997).
Yang, R. T. Gas Separation by Adsorption Processes, Vol. 1 (World Scientific, 1997).
Boyd, P. G. et al. Data-driven design of metal-organic frameworks for wet flue gas capture. Nature 576, 253-256 (2019).
Koytsoumpa, E. I., Bergins, C. & Kakaras, E. The economy: review of capture and reuse technologies. J. Supercrit. Fluids 132, 3-16 (2018).
Kather, A. & Scheffknecht, G. The oxycoal process with cryogenic oxygen supply. Naturwissenschaften 96, 993-1010 (2009).
Jee, J.-G., Kim, M.-B. & Lee, C.-H. Pressure swing adsorption processes to purify oxygen using a carbon molecular sieve. Chem. Eng. Sci. 60, 869-882 (2005).
Van Groenestijn, J. & Kraakman, N. Recent developments in biological waste gas purification in europe. Chem. Eng. J. 113, 85-91 (2005).
Zhuang, L.-L., Yang, T., Zhang, J. & Li, X. The configuration, purification effect and mechanism of intensified constructed wetland for wastewater treatment from the aspect of nitrogen removal: a review. Bioresour. Technol. 293, 122086 (2019).
Akerib, D. et al. Chromatographic separation of radioactive noble gases from xenon. Astropart. Phys. 97, 80-87 (2018).
Lu, Z.-T. et al. Tracer applications of noble gas radionuclides in the geosciences. Earth Sci. Rev. 138, 196-214 (2014).
Furukawa, H., Cordova, K. E., O’Keeffe, M. & Yaghi, O. M. The chemistry and applications of metal-organic frameworks. Science 341, 1230444 (2013).
Ding, M., Cai, X. & Jiang, H.-L. Improving mof stability: approaches and applications. Chem. Sci. 10, 10209-10230 (2019).
Wang, J., Zhou, M., Lu, D., Fei, W. & Wu, J. Virtual screening of nanoporous materials for noble gas separation. ACS Appl. Nano Mater. 5, 3701-3711 (2022).
Yang, Q., Liu, D., Zhong, C. & Li, J.-R. Development of computational methodologies for metal-organic frameworks and their application in gas separations. Chem. Rev. 113, 8261-8323 (2013).
Li, J.-R., Kuppler, R. J. & Zhou, H.-C. Selective gas adsorption and separation in metal-organic frameworks. Chem. Soc. Rev. 38, 1477-1504 (2009).
Knebel, A. & Caro, J. Metal-organic frameworks and covalent organic frameworks as disruptive membrane materials for energyefficient gas separation. Nat. Nanotechnol. 17, 911-923 (2022).
Zhou, M. & Wu, J. Inverse design of metal-organic frameworks for c2h4/c2h6 separation. npj Comput. Mater. 8, 256 (2022).
Wang, J., Zhou, M., Lu, D., Fei, W. & Wu, J. Computational screening and design of nanoporous membranes for efficient carbon isotope separation. Green. Energy Environ. 5, 364-373 (2020).
Lin, R.-B., Xiang, S., Zhou, W. & Chen, B. Microporous metal-organic framework materials for gas separation. Chem 6, 337-363 (2020).
Banerjee, D. et al. Metal-organic framework with optimally selective xenon adsorption and separation. Nat. Commun. 7, ncomms11831 (2016).
Iftimie, R., Minary, P. & Tuckerman, M. E. Ab initio molecular dynamics: Concepts, recent developments, and future trends. Proc. Natl Acad. Sci. 102, 6654-6659 (2005).
Hollingsworth, S. A. & Dror, R. O. Molecular dynamics simulation for all. Neuron 99, 1129-1143 (2018).
Rubinstein, R. Y. & Kroese, D. P. Simulation and the Monte Carlo Method (John Wiley & Sons, 2016).
Weinan, E., Ren, W. & Vanden-Eijnden, E. String method for the study of rare events. Phys. Rev. B 66, 052301 (2002).
Zhou, M. & Wu, J. A gpu implementation of classical density functional theory for rapid prediction of gas adsorption in nanoporous materials. J. Chem. Phys. 153, 074101 (2020).
Altintas, C., Altundal, O. F., Keskin, S. & Yildirim, R. Machine learning meets with metal organic frameworks for gas storage and separation. J. Chem. Inf. Model. 61, 2131-2146 (2021).
Abdi, J. & Mazloom, G. Machine learning approaches for predicting arsenic adsorption from water using porous metal-organic frameworks. Sci. Rep. 12, 16458 (2022).
Nandy, A. et al. Mofsimplify, machine learning models with extracted stability data of three thousand metal-organic frameworks. Sci. Data 9, 74 (2022).
Scarselli, F., Gori, M., Tsoi, A. C., Hagenbuchner, M. & Monfardini, G. The graph neural network model. IEEE Trans. Neural Netw. 20, 61-80 (2008).
Xie, T. & Grossman, J. C. Crystal graph convolutional neural networks for an accurate and interpretable prediction of material properties. Phys. Rev. Lett. 120, 145301 (2018).
Vaswani, A. et al. Attention is all you need. Advances in Neural Information Processing Systems (eds Guyon, I. et al.) Vol. 30 (Curran Associates, Inc., 2017).
Kang, Y., Park, H., Smit, B. & Kim, J. A multi-modal pre-training transformer for universal transfer learning in metal-organic frameworks. Nat. Mach. Intell. 5, 309-318 (2023).
Cao, Z., Magar, R., Wang, Y. & Barati Farimani, A. Moformer: self-supervised transformer model for metal-organic framework property prediction. J. Am. Chem. Soc. 145, 2958-2967 (2023).
Zhou, G. et al. Uni-mol: a universal 3d molecular representation learning framework. In Proc. International Conference on Learning Representations (2023).
Colón, Y. J., Gómez-Gualdrón, D. A. & Snurr, R. Q. Topologically guided, automated construction of metal-organic frameworks and their evaluation for energy-related applications. Cryst. Growth Des. 17, 5801-5810 (2017).
Gómez-Gualdrón, D. A. et al. Evaluating topologically diverse metal-organic frameworks for cryo-adsorbed hydrogen storage. Energy Environ. Sci. 9, 3279-3289 (2016).
Lan, Y. et al. Materials genomics methods for high-throughput construction of cofs and targeted synthesis. Nat. Commun. 9, 5274 (2018).
Kenton, J. D. M.-W. C. & Toutanova, L. K. Bert: Pre-training of deep bidirectional transformers for language understanding. In Proc. of naacL-HLT, vol. 1, 2 (2019).
Nath, K., Ahmed, A., Siegel, D. J. & Matzger, A. J. Microscale determination of binary gas adsorption isotherms in mofs. J. Am. Chem. Soc. 144, 20939-20946 (2022).
Zhao, Z., Li, Z. & Lin, Y. Adsorption and diffusion of carbon dioxide on metal- organic framework (mof-5). Ind. Eng. Chem. Res. 48, 10015-10020 (2009).
Walton, K. S. et al. Understanding inflections and steps in carbon dioxide adsorption isotherms in metal-organic frameworks. J. Am. Chem. Soc. 130, 406-407 (2008).
Nugent, P. et al. Porous materials with optimal adsorption thermodynamics and kinetics for co2 separation. Nature 495, 80-84 (2013).
Duff, D. G., Ross, S. M. & Vaughan, D. H. Adsorption from solution: an experiment to illustrate the langmuir adsorption isotherm. J. Chem. Educ. 65, 815 (1988).
Krishna, R. & van Baten, J. M. In silico screening of metal-organic frameworks in separation applications. Phys. Chem. Chem. Phys. 13, 10593-10616 (2011).
Pillai, R. S., Pinto, M. L., Pires, J., Jorge, M. & Gomes, J. R. Understanding gas adsorption selectivity in irmof-8 using molecular simulation. ACS Appl. Mater. Interfaces 7, 624-637 (2015).
Moosavi, S. M. et al. Understanding the diversity of the metalorganic framework ecosystem. Nat. Commun. 11, 1-10 (2020).
Kalmutzki, M. J., Hanikel, N. & Yaghi, O. M. Secondary building units as the turning point in the development of the reticular chemistry of mofs. Sci. Adv. 4, eaat9180 (2018).
van der Maaten, L. & Hinton, G. Visualizing data using t-sne. J. Mach. Learn. Res. 9, 2579-2605 (2008).
Chung, Y. G. et al. In silico discovery of metal-organic frameworks for precombustion co2 capture using a genetic algorithm. Sci. Adv. 2, e1600909 (2016).
Chung, Y. G. et al. Advances, updates, and analytics for the com-putation-ready, experimental metal-organic framework database: core mof 2019. J. Chem. Eng. Data 64, 5985-5998 (2019).
Tong, M., Lan, Y., Yang, Q. & Zhong, C. Exploring the structureproperty relationships of covalent organic frameworks for noble gas separations. Chem. Eng. Sci. 168, 456-464 (2017).
Hammersley, J. Monte Carlo Methods (Springer Science & Business Media, 2013).
Dubbeldam, D., Calero, S., Ellis, D. E. & Snurr, R. Q. Raspa: molecular simulation software for adsorption and diffusion in flexible nanoporous materials. Mol. Simul. 42, 81-101 (2016).
Rappé, A. K., Casewit, C. J., Colwell, K., Goddard III, W. A. & Skiff, W. M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 114, 10024-10035 (1992).
Boato, G. & Casanova, G. A self-consistent set of molecular parameters for neon, argon, krypton and xenon. Physica 27, 571-589 (1961).
Martin, M. G. & Siepmann, J. I. Transferable potentials for phase equilibria. 1. united-atom description of n-alkanes. J. Phys. Chem. B 102, 2569-2577 (1998).
Altintas, C. & Keskin, S. Role of partial charge assignment methods in high-throughput screening of mof adsorbents and membranes for co 2/ch 4 separation. Mol. Syst. Des. Eng. 5, 532-543 (2020).
Ong, S. P. et al. Python materials genomics (pymatgen): a robust, open-source python library for materials analysis. Comput. Mater. Sci. 68, 314-319 (2013).
Willems, T. F., Rycroft, C. H., Kazi, M., Meza, J. C. & Haranczyk, M. Algorithms and tools for high-throughput geometry-based analysis of crystalline porous materials. Microporous Mesoporous Mater. 149, 134-141 (2012).
Ongari, D. et al. Accurate characterization of the pore volume in microporous crystalline materials. Langmuir 33, 14529-14538 (2017).
Waskom, M. L. seaborn: statistical data visualization. J. Open Source Softw. 6, 3021 (2021).
Hunter, J. D. Matplotlib: a 2d graphics environment. Comput. Sci. Eng. 9, 90-95 (2007).
تم دعم D.L. من قبل المؤسسة الوطنية للعلوم الطبيعية في الصين، رقم U1862204 و 21878175. تم دعم هذا البحث جزئيًا من قبل جامعة تسينغhua وتقنية DP.
مساهمات المؤلفين
ج.و. (وانغ) و ج.ل. ساهموا بالتساوي في هذا العمل. د.ل.، ز.ج. و ج.و. (وو) صمموا وأشرفوا على المشروع. ج.و. (وانغ)، ج.ل. و ز.ج. قاموا بجمع البيانات، وتطوير النموذج الرائد، وصياغة الورقة. قدم H.W. وM.Z. وG.K. وL.Z. نصائح مهمة وقاموا بمراجعة الورقة. ساهم جميع المؤلفين في مناقشة النتائج وعلقوا على الورقة.
يجب توجيه المراسلات والطلبات للحصول على المواد إلى جيانجونغ وو، وزيفينغ قاو أو ديانان لو.
معلومات مراجعة الأقران تشكر مجلة Nature Communications إيلليا كيفليشفيلي والمراجعين الآخرين المجهولين على مساهمتهم في مراجعة هذا العمل. يتوفر ملف مراجعة الأقران.
ملاحظة الناشر: تظل شركة سبرينجر ناتشر محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
الوصول المفتوح هذه المقالة مرخصة بموجب رخصة المشاع الإبداعي النسب 4.0 الدولية، التي تسمح بالاستخدام والمشاركة والتكيف والتوزيع وإعادة الإنتاج بأي وسيلة أو صيغة، طالما أنك تعطي الائتمان المناسب للمؤلفين الأصليين والمصدر، وتوفر رابطًا لرخصة المشاع الإبداعي، وتوضح إذا ما تم إجراء تغييرات. الصور أو المواد الأخرى من طرف ثالث في هذه المقالة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة، ما لم يُشار إلى خلاف ذلك في سطر الائتمان للمواد. إذا لم تكن المادة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة وكان استخدامك المقصود غير مسموح به بموجب اللوائح القانونية أو يتجاوز الاستخدام المسموح به، فستحتاج إلى الحصول على إذن مباشرة من صاحب حقوق الطبع والنشر. لعرض نسخة من هذه الرخصة، قم بزيارةhttp://creativecommons.org/رخصة/بواسطة/4.0/. (ج) المؤلف(ون) 2024
قسم الهندسة الكيميائية، جامعة تسينغhua، بكين 100084، الصين.دي بي تكنولوجي، بكين 100089، الصين.مدرسة الطاقة المتقدمة، جامعة صن يات سن، شنتشن 518107، الصين.معهد الذكاء الاصطناعي للعلوم، بكين 100190، الصين.مختبر جيانغسو الرئيسي للأجهزة الوظيفية والمواد القائمة على الكربون، معهد المواد اللينة الوظيفية والنانوية (FUNSOM)، جامعة سوتشو، سوتشو 215123، الصين.قسم الهندسة الكيميائية والبيئية، جامعة كاليفورنيا، ريفرسايد، كاليفورنيا 92521، الولايات المتحدة الأمريكية.ساهم هؤلاء المؤلفون بالتساوي: جينغكي وانغ، جيابينغ ليو.البريد الإلكتروني: jwu@engr.ucr.edu; gaozf@dp.tech; ludiannan@mail.tsinghua.edu.cn
A comprehensive transformer-based approach for high-accuracy gas adsorption predictions in metal-organic frameworks
Received: 23 May 2023
Accepted: 20 February 2024
Published online: 01 March 2024
(A) Check for updates
Jingqi Wang , Jiapeng Liu , Hongshuai Wang , Musen Zhou , Guolin ® , Linfeng Zhang , Jianzhong Wu (B) , Zhifeng Gao (B) & Diannan Lu®
Gas separation is crucial for industrial production and environmental protection, with metal-organic frameworks (MOFs) offering a promising solution due to their tunable structural properties and chemical compositions. Traditional simulation approaches, such as molecular dynamics, are complex and computationally demanding. Although feature engineering-based machine learning methods perform better, they are susceptible to overfitting because of limited labeled data. Furthermore, these methods are typically designed for single tasks, such as predicting gas adsorption capacity under specific conditions, which restricts the utilization of comprehensive datasets including all adsorption capacities. To address these challenges, we propose Uni-MOF, an innovative framework for large-scale, three-dimensional MOF representation learning, designed for multi-purpose gas prediction. Specifically, Uni-MOF serves as a versatile gas adsorption estimator for MOF materials, employing pure three-dimensional representations learned from over 631,000 collected MOF and COF structures. Our experimental results show that Uni-MOF can automatically extract structural representations and predict adsorption capacities under various operating conditions using a single model. For simulated data, Uni-MOF exhibits remarkably high predictive accuracy across all datasets. Additionally, the values predicted by Uni-MOF correspond with the outcomes of adsorption experiments. Furthermore, Uni-MOF demonstrates considerable potential for broad applicability in predicting a wide array of other properties.
Gas separation is a significant industrial challenge that requires immediate attention, given its critical role in various applications. For example, separating from is essential for obtaining highquality natural gas and effectively achieving the carbon capture,
utilization and storage for environmental reasons . Gas separation also has implications for other fields, such as the production of highpurity oxygen and nitrogen for industrial purposes and the purification of noble gases for medical diagnosis, and lasers . Given its
importance, research in this area is crucial for advancing technology and meeting industry demands.
Metal-organic frameworks (MOFs) have emerged as a kind of promising material in the field of gas separation due to their unique properties . MOFs are composed of metal ions and organic ligands, which provide them with highly ordered pore structures and adjustable aperture sizes. These properties make them ideal for various gas separation applications . The ability to control the pore size and chemical composition of MOFs allows for selective adsorption and separation of different gases. MOFs with different pore sizes exhibit varied capacities for gas adsorption , and tuning the chemical composition can affect the preference for adsorbate gases. The ability to selectively adsorb and separate different gases makes MOFs a promising material for various industrial and environmental applications .
While the potential of MOFs for gas adsorption is promising, accurately predicting their adsorption capacity remains a challenge. Molecular dynamics , Monte Carlo (MC) and other simulation/ calculation methods have been applied to provide reference values, but these approaches are computationally expensive and complicated for implementation, limiting their application to large-scale, multi-gas, and high-throughput calculations. Moreover, the vast range of operating conditions for gas adsorption further complicates the predictions.
Machine learning techniques have demonstrated significant potential in accurately predicting properties of crystalline materials , reducing the cost of traditional trial and error experiments, and eliminating the need for expensive simulations. However, these methods often rely on feature engineering based on expert domain knowledge, leading to overfitting and biased performance when using a limited amount of labeled data. With the emergence of deep learning, graph neural networks, and Transformers have proven successfully in predicting MOF properties. These models directly incorporate structural information such as chemical bonds, atoms, and spatial coordinates as inputs, and automatically learn structural features through data-driven approaches. Thanks to the powerful representation capacity of these deep-learning frameworks, learned features can effectively eliminate biases introduced by feature engineering mentioned earlier.
Despite their high performance and powerful predictive capabilities, existing models for predicting adsorption properties are typically designed for single tasks, specifically predicting the adsorption uptake of a particular gas under certain conditions. However, the available datasets for these single task predictions are often limited, thereby hindering the models generalizability and full utilization of their capabilities. On the other hand, the combination of labeled data from various adsorbate gases across different temperature and pressure environments can create a substantial dataset suitable for training across the entire working conditions. The increased data size may also enhance the model ability to generalize and improve their practical industrial use. Therefore, a unified adsorption framework is necessary for advancing these models. Additionally, integrating representation learning (or pre-training) for large-scale unlabeled MOF structures may further improve the model performance as well as representation ability. The pre-training trick has been widely implemented in combination with large-scale models to discover drugs , where pure threedimensional molecular structures were used to pre-train these models. Experimental results have also demonstrated that pre-trained models outperform previous methods, particularly in property prediction , suggesting remarkable improvement through pre-training.
Inspired by this, we propose the Uni-MOF framework as a multipurpose solution for predicting gas adsorption of MOFs under different conditions using structural representation learning. Compared with other Transformer-based models such as MOFormer and MOFTransformer , our Uni-MOF, as a Transformer-based framework,
not only can the pre-training recognize and recover the threedimensional structure of nanoporous materials and thus greatly improve the robustness of the model, but the fine-tuning task also further takes into account the operating conditions such as temperature, pressure, and different gas molecules, which makes Uni-MOF suitable for both scientific research and practical applications. UniMOF, as a comprehensive gas adsorption estimator for MOF materials, requires only the crystallographic information file (CIF) of the MOF, along with the associated gas, temperature, and pressure parameters, to predict the gas adsorption properties of nanoporous materials over a wide range of operating conditions. Our framework is easy to use and allows for module selection. Additionally, it effectively addresses the issue of overfitting by integrating various cross-system absorption labeled data with representation learning from massive amounts of unlabeled structural data. This compensates for the lack of highquality and insufficient data, ultimately leading to higher accuracy in gas adsorption predictions. Our study utilized a self supervised learning approach on a database containing over 631,000 MOF and COF structures. The results were remarkable, demonstrating a high prediction accuracy. Fine-tuning experiments revealed that the UniMOF framework is robust in databases with ample data. When applied to databases with sufficient sampling of working conditions, our UniMOF framework is able to screen high performance adsorbents under high pressure accurately by feeding only the labeled data obtained at low pressure by home-brew simulations. We must stress that Uni-MOF provides a convenient approach for high pressure adsorption capacities, which are generally more computationally demanding for traditional simulations. The results are consistent with experimental screening outcomes. Furthermore, the performance of Uni-MOF on cross-system datasets exceeded that on single-system tasks. By leveraging support from other gas adsorption data, Uni-MOF accurately predicted the adsorption properties of unknown gases.
Uni-MOF framework achieves material recognition accuracy at the atomic level, while the integrated model makes Uni-MOF more applicable to engineering problems. Undoubtedly, accomplishing truly unified models is the future direction of the materials field, rather than just focusing on specialized areas. Uni-MOF is a pioneering practice of Machine Learning in gas adsorption.
Results and discussion
Overview of workflow
The Uni-MOF framework comprises pre-training on three-dimensional nanoporous crystals and fine-tuning for multitask prediction in downstream applications. Figure 1 provides a schematic representation of the Uni-MOF framework. The pre-training of three-dimensional crystal materials significantly enhances the prediction performance of downstream tasks, particularly for large-scale unlabeled data. To address the issue of inadequately supervised training datasets, we collected an extensive dataset of MOF structures and generated over 300,000 MOFs using ToBaCCo.3.0 . High-throughput construction of COFs based on materials genomics strategy with quasi-reactive assembly algorithms (QReaxAA) is feasible, leading to a comprehensive library of . Through the spatial configuration of materials, Uni-MOF is capable of learning the material structural properties, most importantly the chemical bonding information, very well. In order to enable Uni-MOF to learn more diverse materials and thus improve the generalization ability to a broader range of materials, we introduced MOFs and COFs both virtually and experimentally during the pretraining process. Similar to the masking tagging task in BERT and Uni, Uni-MOF employs a prediction task for masked atoms, thereby promoting the pre-trained models to acquire an in-depth understanding of the material spatial structures. To enhance the robustness of pre-training and generalize the learned representation, we introduced noises to the original coordinates of MOFs, as depicted in Fig. 1a. In the pre-training stage, we devised two tasks. 1) reconstructing the
Fig. 1 | Schematic overview of Uni-MOF framework. a pre-training workflow. In the pre-training stage, in addition to predicting the types of masked atoms, a threedimensional position denoising task was used to learn the three-dimensional spatial representation. Uniform noise of is added to the of atomic coordinates randomly, and then the spatial position encoding is calculated based on the corrupted coordinates. data generation workflow. Cross-system performance datasets can be collected or generated by random sampling of different operating
conditions. c workflow of Uni-MOF fine-tuning. A unified gas adsorption prediction model Uni-MOF is built by the embedding of pre-trained weight, MOF material, gas, temperature, and pressure. d overall workflow of Uni-MOF. For the multi-purpose Uni-MOF framework, no additional analytical calculations for materials are required, and the properties under varied working conditions can be predicted based solely on the crystallographic information file (CIF) of MOF materials. MOF means metal-organic framework.
pristine three-dimensional positions from the noisy data, and 2) predicting the masked atoms. These tasks can augment the model robustness and improve downstream prediction performance.
In addition to diverse spatial configurations, a comprehensive set of material property data points is also crucial for model training. To enrich the dataset, we established a custom data generation process (as illustrated in Fig. 1b). For example, we utilized the CoRE MOF database that comprises successfully synthesized MOFs, gases that are significant in the separation field, as well as the common temperature
and pressure operating range under the corresponding system. By randomly sampling from these various materials, gases, temperatures, and pressure pools, a significant volume of adsorption uptake data can be generated for Uni-MOF fine-tuning. This data generation process improves the efficiency of data generation and can form a widely sampled dataset, Table 1 lists all the databases applied in this study. The simulation-derived database with diversity is beneficial for model fine-tuning and optimization, ultimately accomplishing the objective of virtual screening for material performance.
Table 1 | Structure & Data resources
Data Types
Sources
Availability
Software
Size
Structure
collection
hMOF
–
137,000+
Structure
collection
ToBaCCo
–
10,000+
Structure
collection
CoRE MOF
–
12,000+
Structure
collection
CCDC
–
12,000+
Structure
collection
CoRE COF
–
600+
Structure
collection
GCOFs
–
160,000+
Structure
generation
this work
ToBaCCo.3.0
300,000+
Adsorption Uptake Data
collection
hMOF_ MOFX_DB
–
2,400,000+
Adsorption Uptake Data
collection
CoRE_ MOFX_DB
–
460,000+
Adsorption Uptake Data
generation
this work
RASPA
99,000+
Other Property Data
generation
this work
Zeo++
149,000+
The fine-tuning of Uni-MOF depicted in Fig. 1c is based on the extraction of representations acquired through pre-training, as well as the generation and collection of extensive datasets using our homebrew workflows. During the fine-tuning process, we trained the model using around 3,000,000 labeled data points across various adsorption conditions of MOFs and COFs, enabling accurate prediction of adsorption capacities. With the diverse database of cross-system targeted data, the fine-tuned Uni-MOF can predict the multi-system adsorption property of MOFs under arbitrary states, including different gases, temperatures, and pressures. As a result, Uni-MOF is a unified and readily available framework for predicting the adsorption performance of MOF adsorbents.
Above all, Uni-MOF obviates the requirement for additional labor in identifying human-defined structural features. Instead, the CIF of MOFs, along with pertinent gas, temperature, and pressure parameters, suffices. The self-supervised learning strategy and abundant databases ensure that Uni-MOF can foretell gas adsorption properties of nanoporous material in a wide range of operating parameters, thereby rendering it a proficient gas adsorption estimator for MOF materials.
Overall performance in large-scale databases
In order to evaluate the predictive capability of Uni-MOF as a comprehensive framework for adsorption performance prediction, two mixed-state databases for gas adsorption, namely hMOF_MOFX-DB and CoRE_MOFX-DB, were compiled with adsorbate gases consisting of [ ] and [ ], respectively. In addition, the CoRE_MAP_DB database was generated via our home-brew MC simulation workflow for the adsorption uptake of seven gases ( , ).
Since the data sources of the three databases are different, we conducted model training for each database separately in order to ensure the consistency of data sets. Details of these three databases, including temperature and pressure ranges, are listed in Supplementary Table 1. To prevent data bias and ensure that the test set remained unseen by the model, we divided the data set into three different data sets (train, valid, and test) with the ratio of 8:1:1 according to the MOF structure instead of randomly splitting, that is, there is no identical material between the three datasets. The splitting ensures that the model accomplishes the prediction of new materials in validation and test set, rather than those materials that have already been seen. The model parameters are optimized during the training process and reflected in the results of the validation set. The optimal model corresponding to the validation set is saved, and the prediction results in the never-before-seen test set represent the final performance ( shown here) of the model, thus reasonably avoiding over-fitting.
The collected datasets, hMOF_MOFX_DB and CoRE_MOFX_DB, exhibit relatively concentrated temperature and pressure distributions, as depicted in the sub-figure of Fig. 2a, b. Notably, both databases offer adequate data, with over 2,000,000 and 400,000 data points, respectively. The prediction results demonstrate that Uni-MOF is remarkably robust when applied to databases that possess sufficient data with relatively concentrated operating states, such as hMOF_MOFX_DB and CoRE_MOFX_DB, with values of 0.98 and 0.92 , respectively. In contrast, our CoRE_MAP_DB database, which we have learned from Supplementary Table 1, contains slightly less than 100,000 data points. It encompasses an extensive sampling of the adsorption of seven adsorbed gases in over 10,000 MOFs, covering a temperature range of and a pressure range of , as depicted in the sub-figure of Fig. 2c. For such a widely distributed database, Uni-MOF can still achieve excellent prediction accuracy with an value of 0.83 , demonstrating its good generalizability.
The analysis also incorporates two other error metrics, i.e., Mean Absolute Error (MAE) and Root Mean Square Error (RMSE). However, the CoRE_MOFX dataset shows larger errors, particularly in RMSE. CoRE_MOFX dataset contains Ar and adsorption amounts at 77 K and 87 K , which are significantly lower temperatures compared to the other two databases. Consequently, the adsorption values in CoRE_MOFX are generally larger. The adsorption isotherms with the most significant errors are depicted in Supplementary Fig. 1. MOF materials with the largest error typically have Cu (with the lowest energy parameter of 2.52 K ) as the metal site. This implies that MOF materials with low energy parameter metal sites or large pore sizes are the limiting factors for low-temperature data prediction. Intriguingly, even with the highest errors, Uni-MOF can accurately reproduce the adsorption trend from low to high pressure. Additionally, CoRE_MOFX_DB, as the collected experimental structural database, contains 1800+ disordered materials out of 12,000+ MOFs. Disordered MOFs were found to adversely affect the model performance, as the unrealistic internal structures and limited material samples lead to the challenging prediction of gas adsorption, as demonstrated by the outlier ja5111317_ja5111317_si_003_clean and LELDOX_clean in Supplementary Table 3. Therefore, disordered materials were excluded from the generated CoRE_MAP_DB database, which results in reduced errors and is more suggested to use for prediction of gas adsorption in nanoporous materials. The force field used in this work does not account the effect of open-metal sites. For the top ten outliers in the CoRE_MOFX_DB database (shown in Supplementary Table 4), 80% of them have open-metal sites. This suggests that the significant deviations between simulations and Uni-MOF predictions could be due to the missing interaction between open-metal sites and adsorbate considered in the simulation.
Additionally, we discovered that the prediction accuracy in the pre-training stage could reach 0.98 before and after incorporating COF materials. This indicates that Uni-MOF is capable of effectively learning the three-dimensional spatial structure of multiple nanoporous materials. As for the downstream tasks, the predictive performance ( , RMSE, and MAE) of Uni-MOF for all three databases surpassed that of Uni-MOF with only MOF pre-training, as illustrated in Supplementary Tables 5, 6. This indicates that incorporating COF not only maintains but also enhances the model robustness, further demonstrating the superiority of our Uni-MOF framework.
Experimental adsorption uptake prediction
Despite the excellent prediction performance of Uni-MOF on simulated results, we are wondering how the framework would behave in comparison to the real experimentally collected results. In this study, we have chosen commonly used laboratory materials, such as MOF-5 and MOF-177 , and compared the predicted gas adsorption capacity with existing experimental data. Considering that the CoRE_MAP_DB database contains a diverse range of operating conditions and
Fig. 2 | Overall performance of Uni-MOF in large-scale databases. The correlation between predicted and simulated value of gas adsorption amount for (a) Database of hMOF_MOFX_DB ( ), (b) Database of CoRE_MOFX_DB ( ) and (c) Database of CoRE_MAP_DB . Sub-figure is the
experimentally validated MOF structures, we chose the weights trained using this database to predict the adsorption performance of experimental materials. Figure 3 displays the predicted results under different conditions, details are listed in Supplementary Tables 8-10.
Figure 3a presents the Langmuir adsorption isotherm obtained by fitting the predicted methane adsorption capacity under low pressure (less than 5 bar). It shows that the high pressure adsorption capacity displayed by the Langmuir adsorption isotherm is consistent with the experimental values. Specifically, the high pressure adsorption capacity of (Zn2(bdc)2(dabco), methane, 393 K ) (MIL-101, methane, 373 K ) > (MOF-177, methane, 398 K ). This suggests that the Uni-MOF framework is capable of accurately screening high performance adsorbents under high pressure based solely on prediction adsorption capacity under low pressure. Nevertheless, we still notice significant deviations between many predicted and experimental values under low pressure, especially in the cases of Mg -dobdc and MOF-5. Simulations may not precisely represent experimental values for MOFs with open metal sites, such as Mg-dobdc . However, MOF-5 has close metal sites, and previous studies have shown that simulations can effectively depict experimental gas adsorption values . Despite these findings, we observed that the gas adsorption performance of MOFs varies even under the same operating conditions (refer to
Supplementary Tables 8-10). This suggests that different methods and significant objective errors exist in the experimental values. Therefore, we did not purposely select data but instead aimed to provide as comprehensive a representation of the collected data as possible. For example, the black intersections in Fig. 3e represent experimental values from various literature sources under identical operating conditions. The results demonstrate that, despite significant variations in experimental values, the Uni-MOF framework maintains a high level of accuracy in material ranking, rendering it suitable for addressing engineering challenges.
Furthermore, the experimental values are introduced to correct the adsorption isotherms. For example, Fig. 3b shows the Langmuir adsorption isotherm obtained by fitting both the predicted and experimental adsorption data. While we use simulated datasets to address data scarcity, we can also properly introduce experimental values to correct adsorption isotherms, which helps a more quantitative prediction of adsorption performance at high-pressure where the gas-gas interaction becomes more significant. In Fig. 3b, one can observe that the corrected adsorption isotherms have a strong correlation with experimental adsorption capacity to some extent. The results exhibit that Uni-MOF not only has the ability to screen the adsorption performance of the same gas in different materials but also
Fig. 3 | Adsorption isotherms based on low-pressure predictions and highpressure experimental values, each curve represents Langmuir fit. a Uni-MOF predicted and (b) experimentally corrected adsorption isotherms of methane adsorption in Zn2(bdc)2(dabco), MIL-101 and MOF-177 at , and 398 K , respectively. c Uni-MOF predicted and (d) experimentally corrected adsorption isotherms of methane and carbon-dioxide adsorption in Mg-dobdc at 298 K . e Uni-
MOF predicted and (f) experimentally corrected adsorption isotherms of methane and carbon-dioxide adsorption in MOF-5 at 298 K and 296 K . Each set of horizontal plots (i.e., and and and ) represents the same system, that is, the same MOF and gas molecule. The hollow marker represents the predicted value of UniMOF, and the filled marker represents the experimental data referenced from previous literature.
can accurately screen the adsorption performance of different gases in the same material (Fig. 3c, d) or at different temperatures (Fig. 3e, f).
In the foreseeable future, the intersection of Artificial Intelligence (AI) and materials science will necessitate the resolution of practical and scientific issues. Nonetheless, the attainment of process implementation by AI in the realm of machine learning techniques that entail copious amounts of data remains a formidable challenge, given the dearth of experimental data and the diverse array of synthetic technology and characterization conditions implicated. Our research has made a significant stride in materials science by incorporating operating conditions into the Uni-MOF framework to ensure data adequacy and enable screening functions that are consistent with experimental findings.
Cross-system forecasting
In order to showcase the predictive capabilities of Uni-MOF with regard to cross-system properties, five materials were randomly selected from each of the six systems (carbon-dioxide at 298 K , methane at 298 K , krypton at 273 K , xenon at 273 K , nitrogen at 77 K and argon at 87 K ) contained in databases hMOF_MOFX_DB and
CoRE_MOFX_DB, which have been thoroughly sampled in terms of temperature and pressure. The predicted and simulated values of gas adsorption uptake at varying pressures were then compared, with the results presented in Fig. 4a-f. Adsorption isotherms fitting from both Uni-MOF predictions and simulated values would artificially reduce visual errors. In order to eliminate data bias, adsorption isotherms in all cases were obtained only by simulated values. It is evident that, due to the fact that the adsorption isotherms were obtained purely through simulated values, the predicted values of adsorption uptake generated by Uni-MOF for the hMOF_MOFX_DB and CoRE_MOFX_DB databases align closely with the simulated values across all cases. This finding is further supported by the high prediction accuracy demonstrated in Fig. 2a, b.
Given the ability of Uni-MOF to predict properties across systems, we were intrigued by its potential to forecast the adsorption capacity of unknown gases. The CoRE_MAP_DB database contains a diverse array of adsorption data points for various gases, such as methane, carbon dioxide, argon, krypton, xenon, oxygen, and helium. To evaluate the predictive capability of Uni-MOF, we randomly divided the CoRE_MAP_DB data points by adsorbate gas into three datasets (train,
Fig. 4 | Uni-MOF cross-system prediction cases. The predicted and simulated values of gas adsorption amount versus pressure in hMOF_MOFX_DB database for (a) Carbon-dioxide at 298 K , (b) Methane at 298 K , (c) Krypton at 273 K and (d) Xenon at 273 K , and in CoRE_MOFX_DB database for (e) Nitrogen at 77 K and (f) Argon at 87 K . Different markers in each figure denote different MOF adsorbents. The hollow marker represents the predicted adsorption uptake of Uni-MOF, and the filled marker represents the simulated adsorption uptake in the database. The adsorption isotherms are obtained only by simulated values to eliminate data bias.
g Gas transfer learning under multi-system conditions, with the random division into three datasets (train, validation, and test dataset with a ratio of ) according to adsorbate gases. Predicted and simulated adsorption isotherms at 300 K in COF materials, different marker types represent different COFs. The hollow marker means predicted value, the filled marker represents simulated value. represents the coefficient of determination. Source data are provided as a Source Data file.
valid, and test dataset with a ratio of ), then predicted the adsorption capacity for each gas separately. The resulting predictions are depicted in Fig. 4g and summarized in Supplementary Table 11. Remarkably, Uni-MOF demonstrated robustness in predicting the adsorption capacity of unknown gases, achieving a high prediction accuracy ( ) of 0.85 for krypton and a prediction accuracy above 0.35 for all unknown gases.
One can observe that prediction performance varies among gases. For instance, , and Xe are all noble gases with Kr demonstrating the best performance among them with of 0.85 , while the prediction performance of Xe is inferior ( ). A similar distribution of MOF material features across , and Xe databases is observed, as illustrated in the corresponding sub-figure of Supplementary Fig. 2, indicating that MOF material sampling differences have minimal impact in this case. Despite the larger molecular diameter of Xe , making it less favorable for adsorption in small pores, it shows a higher adsorption limit (depicted in Supplementary Fig. 3) compared to argon and krypton. This may be a result of the greater energy parameters of when interacting with MOF material atoms. In the sub-figure of Supplementary Fig. 2c, materials with high Xe adsorption capacity (exceeding ) are highlighted in yellow. These materials generally exhibit larger pore sizes and void fractions, possessing an largest cavity diameter (LCD) greater than a void fraction exceeding 0.68 , and a PLD of at least (close to the kinetic diameter of xenon with ). Although it is challenging to
establish a comprehensive connection between model predictions and the physicochemical properties of gases, it is discernible that gases possessing larger energy parameters typically demonstrate inferior predictive performance, such as , and Xe . This phenomenon can be attributed to the increased significance of gas molecule interactions with escalating pressure, coupled with the complexity introduced by high energy parameters, ultimately complicating transfer learning.
Notwithstanding the challenges, encouraging outcomes can still be observed, as the model precisely predicts the adsorption of moderate krypton, premised on the adsorption behavior of argon and xenon within the same inert gas category.
In this study, we showcase the general performance of Uni-MOF for gas transfer learning, employing a database comprising fewer than 100,000 data points. This suggests that even the optimal prediction conditions were not specifically selected, Uni-MOF is capable in engineering domains. To mitigate over-fitting, the data is typically divided into three sets. However, transfer learning across diverse gases presents a considerable challenge, especially when confronted with a restricted number of gas varieties and data points. As a result, in order to optimize the utilization of the scarce data, the results obtained by dividing the data into two datasets based on the gas types are also shown in Supplementary Fig. 5. Results revealed an improvement in transfer learning accuracy for each gas to some extent. Unlike the prediction of adsorption between different materials, the prediction of
Fig. 5 | Uni-MOF and Uni-MOF w/o pre-training comparison. The performance comparison of Uni-MOF and Uni-MOF w/o pre-training in (a) CoRE_MOFX_DB and (b) hMOF_MOFX_DB. The filled marker represents the prediction performance of the entire database. The hollow marker represents the prediction performance of
the sub-dataset (certain gas, temperature and pressure). Details can be found in Supplementary Table 15-17. c The comparison of correlation between predicted and simulated value of gas adsorption amount via Uni-MOF and Uni-MOF w/o pretraining for CoRE_MAP_DB database. means the coefficient of determination.
adsorption behavior for unknown gases is a formidable challenge. Variations in molecular size, surface adsorption energy, and intermolecular forces among different gas types have a significant impact on the adsorption mechanism and behavior. Despite this complexity, Uni-MOF exhibits exceptional generalizability, as evidenced by its ability to accurately predict the adsorption properties of unknown gases with only the support of adsorption data from other gases.
Furthermore, we investigated the capability of Uni-MOF to predict the gas adsorption behavior in COFs. Around 500 data points of adsorption uptake in CoRE COFs at 300 K were simulated. Despite the limited size of the COF adsorption database, Uni-MOF maintains high predictive performance, achieving an of 0.76 . Additionally, as shown in Fig. 4h, Uni-MOF exhibits high ranking capability for the adsorption of diverse materials under various pressures.
Framework applying pre-training
In order to verify that the self-supervised learning strategy of pretraining can effectively improve the robustness and downstream prediction performance of the Uni-MOF, we established and compared Uni-MOF w/o pre-training against Uni-MOF on three databases, namely CoRE_MOFX_DB, hMOF_MOFX_DB, and CoRE_MAP_DB. The CoRE_MOFX_DB and hMOF_MOFX_DB databases exhibit a high degree of data concentration, thereby enabling their partitioning into smaller databases containing adsorption data for various materials under identical working conditions (i.e., same gas, temperature, and pressure). We trained both CoRE_MOFX_DB and hMOF_MOFX_DB databases using Uni-MOF and Uni-MOF w/o pre-training, and subsequently
calculated the coefficients of determination for each small dataset. In addition, each small dataset is trained separately using the Uni-MOF framework to derive the corresponding coefficient of determination.
As shown in Fig. 5a, the filled markers represent the predictive performance of the whole CoRE_MOFX_DB database. It can be seen that the Uni-MOF performs better than Uni-MOF w/o pre-training. The self-supervised learning strategy of pre-training allows the model to learn the three-dimensional configuration of nanoporous materials in depth, thus improving the accuracy of model fine-tuning. In this part, the data sets are divided in a manner where each set comprises the relevant subsystem data. Therefore, all the hexagonal markers in Fig. 5a represent the overall predicted performance of Uni-MOF. Indeed, Uni-MOF demonstrates equally high performance on the complete data set as it does on an individual data set, even when the performance of a single data set relies on more extensive data than its specific prediction task. For fine-tuning of single-system tasks, that is, training individually for each small data set, the predicted performance hardly exceeded the performance of Uni-MOF. Thus, we know that far-ranging data sampling can further promote the prediction capacity of the learning model. The same conclusions can be summarized for hMOF_MOFX_DB from Fig. 5b.
However, due to the scattered sampling of the CoRE_MAP_DB database, limited small data sets cannot be divided following the same procedure as the previous two databases. Therefore, the correlations between predicted and simulated values from Uni-MOF and Uni-MOF w/o pre-training are compared, shown in Fig. 5c. Similarly, compared with Uni-MOF w/o pre-training, the performance of Uni-MOF was
Fig. 6 | Structural features prediction and analysis. The correlation between predicted and computational value of (a) Pore Limiting Diameter (PLD) ( ), (b) Largest Cavity Diameter (LCD) ( ), (c) void fraction, and (d) pore volume ( ) of MOFs in hMOF (main figure) and CoRE_MOF (sub-figure) databases.
e Comparison of the kernel density estimate (KDE) for different structural features
[PLD ( ), LCD ( ), void fraction, surface area ( ), volume ( )] between the CoRE_MOF with all argon adsorption values and the CoRE_MOF with the top performance of argon adsorption at 87 K and is the coefficient of determination, MOF means metal-organic framework. Source data are provided as a Source Data file.
improved from 0.70 to 0.83 , which further proved the significance of the pre-training strategy for Uni-MOF.
Structural features prediction and high-throughput screening
While the Uni-MOF framework is adept at discerning the spatial arrangement of nanoporous materials, we aim to further explore its potential in forecasting structural attributes. The structural feature prediction results of Uni-MOF for the hMOF and CoRE_MOF materials libraries are presented in Fig. 6a-d.
It can be observed that the prediction performance of hMOF structural parameters is generally superior to that of CoRE_MOF. By employing a combination of structural building blocks (metal nodes, ligands, and functional groups), the design space of the hypothetical MOF (hMOFs) dataset was fully explored, and structural features are more evenly distributed compared to the CoRE_MOF dataset. Experimentally synthesized MOF materials (CoRE_MOF) tend to have structural preferences for metal nodes (e.g., with zinc, copper, and cadmium) and linkers. Previous research has demonstrated that the experimental MOF (CoRE-2019) is primarily concentrated in the small pore region, while the hMOF (Tobacco) is more evenly distributed with a slight shift of focus towards the large pore region . The same observation can be found in Supplementary Fig. 6, where Supplementary Fig. 6a and Supplementary Fig. 6b show the differences between CoRE_MOF and hMOF structures using the t-distributed stochastic neighbor embedding (t-SNE) method. Through Supplementary Fig. 6c-f, it can be seen that the apertures of CoRE_MOF are obviously concentrated in the small aperture region, especially the PLD. hMOF, on the other hand, can still observe the aperture gradient due to its rich structural sampling and data points, even though the apertures are generally small. Thus, for different types of databases, the prediction of structural features performs differently, especially for PLD. Notably, in materials such as hMOFs, its predictive capability attains a coefficient of determination greater than 0.99 , signifying a high level of
precision and dependability. One has come to recognize that Uni-MOF demonstrates a strong capability in predicting the structural features of materials, owing to the utilization of pre-training on a substantial number of three-dimensional structures. Thus, Uni-MOF not only accurately predicts the desired performance of MOF materials but also precisely predicts their structural features, which holds significant importance for material research and application.
Innumerable nanoporous materials can be created with varied secondary building units , leading to exceptionally diverse MOF structures, making MOF structure-property analysis a very strategic initiative. In this work, multi-gas adsorption uptakes under various operating conditions were collected and generated, where the argon uptakes at 87 K and 1 bar is representative and analyzable. Argon accounts for the vast majority of noble gases in the atmosphere ( 9340 ppm at ambient conditions), and has been widely used for insulation and illumination with a commercial value of 3.1 USD . Figure 6e shows a comparison of the kernel density estimate (KDE) for different structural features between the entire CoRE_MOF with argon adsorption values and MOFs in CoRE_MOF with the top performance of argon adsorption. KDE visualizes the distribution of data using a continuous probability density curve in a less cluttered way.
As the distribution of some structural features is bounded, it may lead to distortions (such as the minus value of surface area and volume). However, it still presents interpretable and accurate trends in the structure of top MOF adsorbents. The typical parameters describing pore sizes are as follows. 1) pore limiting diameter (PLD) is the largest free sphere; 2) LCD is the largest included sphere along the free sphere path. Thus, LCD is intrinsically larger than PLD in the same material. Compared with the entire MOF database, the top of MOFs have larger distribution values of 5-10 and for PLD and LCD, respectively. The void fraction of the whole database is moderately distributed around 0.5 , while that of the top MOFs is much larger, mainly distributed around 0.75 . Since most of the adsorption
Fig. 7 | Visualization of structural representations of MOFs in the hMOF and CoRE_MOF datasets, the low-dimensional embeddings are computed by t-SNE (t-distributed stochastic neighbor embedding) method. The representations retrieved after (a), (c), and (e) pre-training and (b), (d), and (f) fine-tuning versus the other properties. The low-dimensional embeddings versus dataset labels, where darker color represents CoRE_MOF and lighter color represents hMOF. illustrate the representations versus surface area in for hMOF and
CoRE_MOF combined. e, show the relationship of representations with respect to the adsorbate values of Ar at for the CoRE_MOF dataset only. Explainable Artificial Intelligence (XAI). g Illustration of hMOF-5004238 structure and (h), (i) heat map of atomic interactions learning from multi-head attention algorithm in head 10 and head 18. MOF means metal-organic framework. Source data are provided as a Source Data file.
occurs on the surface of nanoporous materials, the surface area becomes one of the most critical determinants, and the results show that the top 10% of MOFs possess a very large surface area of about , which is consistent with common sense. The surface area determines the surface adsorption process, while the LCD determines the internal absorption process. Therefore, the top 10% MOFs were classified into three tiers according to their adsorption performance on argon, and the numerical distribution of these two key factors (surface area/LCD) was explored (shown in Supplementary Fig. 8). One can see that at 87 K and 1 bar , the most promising MOF adsorbents for argon mainly possess an LCD of about and a surface area of about .
Modeling of material structural representation
To validate that the MOF structures are well learned in both the pretraining and fine-tuning stages, we visualize the structural features, which are 512 -dimensional vectors, using the t -SNE method . The results are shown in Fig. 7. As illustrated in Fig. 7a, b, the learned features are capable of classifying MOFs either from CoRE_MOF or hMOF datasets. One may also notice that the boundary between CoRE_MOF and hMOF in the fine-tuned features is much more obvious, suggesting a significant improvement of these features after the finetuning. Remarkably, this is clearly demonstrated when we draw the embeddings versus the surface areas of MOFs, as shown in Fig. 7d, where the structures with small surface areas are located in the upperright and lower-left corners, and the structures with large surface areas are in the center. In comparison to the pre-training features, Fig. 7c, the fine-tuning can significantly improve the representation quality.
On the other hand, we are more curious about whether the learned structural representations are closely correlated with the
adsorption behaviors. To address this issue, we visualize the structural embeddings versus the adsorbate values of both Ar and , and show the result of Ar here as an example, see Fig. . As one can observe, the representations learned in the pre-training stage are not able to classify the structures with different adsorbate capacities while structures with various adsorbate values are grouped together, see Fig. 7e. However, after the fine-tuning, the structures with different adsorbate behaviors are well separated, demonstrating a good relationship between the learned representations and the target of adsorbate values, see Fig. 7f. This well explained the functions of the fine-tuning stage in further affecting the structural representations as well as other model parameters.
Furthermore, the multi-head attention mechanism of the Transformer can learn the interactions within the material structure. With the 64-Heads attention algorithm, the atomic interaction landscapes of hmof-5004238 in two different heads are represented in Fig. 7h, i. As shown in Fig. 7h, strong interactions can be observed between the metal sites ( Zn ), Zn and O atom, and also O atoms. Figure 7i also depicts the interaction between the linear carbon chains (C7-C15). In addition, there is a noticeable correlation between the O 0 atom and the Zn atoms ( ). The structural illustration in Fig. 7 g further confirms this result, as the four atoms are chemically linked. Beyond this, the various chemical landscapes of hmof-5004238 in different heads are depicted in Supplementary Fig. 9. In this manner, Uni-MOF identifies tremendous materials and provides reliable predictions for diverse properties.
Conclusion
In this study, we introduced Uni-MOF, a multi-purpose framework that can accurately predict gas adsorption in MOF materials. We also
generated, collected, and organized relevant databases of nanoporous materials and gas adsorption datasets. The self-supervised learning of a database containing over 631,000 MOFs and COFs was performed, resulting in a high prediction accuracy of 0.98 . This indicates that the representation learning framework based on three-dimensional pretraining effectively learns the complex structural information of MOFs while avoiding over-fitting. We applied Uni-MOF to predict the gas adsorption performance of three major databases and achieved a high prediction accuracy of up to 0.98 in database with sufficient data. In the case of a sufficiently sampled dataset, Uni-MOF not only maintains a predictive accuracy above 0.83 , but also accurately selects highperformance adsorbents under high pressure by solely predicting adsorption under low pressure, consistent with experimental screening results. Thus, Uni-MOF represents a significant breakthrough in the field of material science with regards to the application of machine learning techniques. Furthermore, our Uni-MOF framework shows superior performance on cross-system datasets compared to singlesystem tasks and can accurately predict the adsorption properties of unknown gases with a high prediction accuracy of up to 0.85 , demonstrating its strong predictive ability and generality. Through extensive pre-training of three-dimensional structures, Uni-MOF effectively learns the structural features of MOFs, achieving a high coefficient of determination of 0.99 for hMOFs. Additionally, t-SNE analysis confirms that the fine-tuning stage can further learn structural features, and structures with different adsorbate behaviors are well identified, indicating a strong correlation between learned representations and gas adsorption targets.
Our database encompasses a pressure range of bar and temperature ranges of and , making it suitable for the majority of gas adsorption issues. Uni-MOF has demonstrated precise prediction outcomes for a fairly comprehensive collection of MOF structures and distinct COF materials. Even under some extreme conditions, the predicted trend remains highly dependable. With UniMOF, the continuous updating of databases and even usage scenarios are supportable, which further emphasizes its universality. In summary, Uni-MOF framework serves as a versatile predictive platform for MOF materials, functioning as a gas adsorption estimator for MOFs, as it exhibits high precision in predicting gas adsorption under diverse operating conditions and has broad applications in the field of material science.
Methods
Materials and data collection and generation
The MOF/COF structures used for pre-training are either collected from the currently available database or generated using the corresponding program. There is a wealth of existing MOF/COF databases, including computer-synthesized databases of hMOFs , ToBaCCo (Topologically Based Crystal Constructor) MOFs, and experimentallevel databases of CoRE (Computation-Ready Experimental) MOFs , CoRE COFs and CCDC (The Cambridge Crystallographic Data Centre), etc. One integrated database online is MOFXDB, where more than COF structures are available. Apart from exploring nanoporous materials in the materials library, we employed the ToBaCCo.3.0 program to generate over 306,773 MOF structures. The ToBaCCo program takes as input a topological blueprint, searches compatible node building blocks from a defined set, and constructs all possible structures using the topology in combination with different node building blocks and edge building blocks. In particular, the program generates structures with three folders as input variables, i.e., “templates”, “nodes”, “edges”. To generate as many MOF structures as possible, we use all edges as provided in “edges_database”, all nodes as provided in “nodes_database” and all templates as provided in “template_database”. For the downstream task, i.e., gas adsorption uptake by MOFs, we collected data from online sources such as MOFXDB, composing datasets of more than 2,400,000 sorptions of hMOFs on
five gases ( ) at and and over 460,000 sorptions of CoRE MOFs on two gases ( ) at and . In this work, only single component gas adsorption data are considered. In addition, we conducted Grand Canonical Monte Carlo (GCMC) simulations using the RASPA software to produce another 99,000+ gas adsorption uptake dataset, with 50,000 initialization cycles and an additional 50,000 cycles employed for adsorption capacity samples. The collected sorptions were obtained within and bar, considering seven types of gas molecules ( ). Interactions between gas molecules and atoms in adsorbent materials were described in terms of LennardJones (12-6) potential, and the cutoff radius is set to with the tailcorrection. The force field parameters of framework atoms were described by UFF (Universal Force Field) . The force field parameters of noble gas molecules (i.e., argon, krypton, and xenon) were estimated from the principle of corresponding states for the second virial coefficients (listed in Supplementary Table 19). The molecular model Transferable Potentials for Phase Equilibria was also used. In our work, the partial charge of the framework atoms is not considered because of computational cost and significant deviations observed in adsorption results by using different partial charges assignment methods .
Material analysis
The key to high-throughput computational materials science is robust software tools. Here, we use the Python Materials Genomics (pymatgen), a robust and open-source Python library, to derive useful material properties from raw crystallographic structural data and conduct comprehensive materials analysis. Materials properties, including lattice vectors, lattice angles, unit cell volume, atoms, and coordinates, are extracted using the pymatgen. The atom types and coordinates will be used in the pre-training stage for the selfsupervised learning strategy. The Python package OpenMetalDetector , was used to analyze collections of Metal Organic Frameworks for open metal sites.
Uni-MOF framework
Pre-training. We employed Uni-Mol as the pre-training framework, which is a dedicated pure three-dimensional pre-training framework designed for molecules. Uni-Mol has shown high performance in various downstream tasks in the field of drug discovery. However, due to the completely different structure and three-dimensional spatial distribution of MOF materials compared to small molecule drugs, as well as the periodic boundary conditions (PBC) of porous structures and the much larger number of heavy atoms in crystals, we made necessary modifications to Uni-Mol based on these facts.
We leverage an extra head of the lattice matrix to preserve cell geometric information. The presence of PBC is considered in MOF representation learning as PBC is natural for MOF materials. Besides masked atoms prediction and coordinates recovery in Uni-Mol pretraining, a regression head to prediction lattice matrix is used to learn the PBC information.
where indices representation of Uni-Mol for classification token, classification token (CLS) refers to the special token in the input sequence of atoms, which is used to represent the entire molecule in the output of Uni-Mol. is the lattice matrix, we use Feedforward Neural Network (FFN) of to predict lattice matrix with optimizing MSE loss directly. in Uni-MOF pre-training is a
summation of and original . In , the atommasked prediction and coordinates recovery are two major components of loss items. For more details, the readers are welcome to refer to original paper of Uni-Mol.
2) We propose an edge gated kernel for geometric spatial positional encoding. MOFs share totally different arrangement of atoms compared to drug molecules in three-dimensional space, with porous structures and an average above 1000 heavy atoms in a single cell. UniMol uses a Gaussian kernel to encode spatial positional information, while in MOF pre-training suffers from training instability with a Gaussian kernel of much larger pair distance and atom counts. To address this, we propose an edge gated distance kernel.
where is the Euclidean distance of atom pair , and is the edge type of atom pair . Please note the edge here is not the chemical bond, and edge type is determined by the atom types of pair ij. is the affine transformation with parameters and , it affines corresponding to its edge type. indices the edge gated distance kernel, which is a summation of distance projection and edge gated embedding. FFN is the Feedforward Neural Network about the non-linear transformation of distance , and LN is the LayerNorm operation. A sigmoid gated is used as the affine transformation of to weight edge pair type embedding.
Fine-tuning. Prediction of multi-gas adsorption under different operating conditions, the fine-tuning model should be fed with not only the three-dimensional spatial structure but also the gas and operating conditions (i.e., temperature and pressure). Therefore, gas block and temperature/pressure blocks are proposed in Uni-MOF to form a cross-system performance prediction module.
Gas block The gas representation is a combination of gas id and gas intrinsic property-related descriptors.
where indices the gas id of gas represents the gas descriptors (listed in Supplementary Table 2). The gas representation is a concatenation of gas id embedding and FFN layer mapping of gas descriptors.
Num block We use Equal Distance Discretization (EDD) and Logarithm Discretization (LD) for temperature and pressure encoding, respectively. EDD first maps the numerical value into the corresponding bucket with equal width, then applies embedding mapping. LD utilizes the logarithm transform with EDD to accommodate logarithmic likely features.
where the interval width is noted as denotes numerical features, and are the original temperature and pressure values of the corresponding environment. For the temperature feature, we use and embeddings, and for the pressure
feature, we choose to use and with consideration of logarithmic likely transformation in pressure.
High-throughput analysis of large MOF database
To further investigate the effect of material structure on gas adsorption, Zeo , a software package for crystalline porous materials analysis, was used to perform an analysis of the structure and topology of the material geometry. Structural features include the LCD, PLD, void fraction, void volume, and specific surface area. The internal void volume can largely influence the performance of nanoporous materials. Precise definitions exist for volume from different methods, especially the accessible and nonaccessible probe center pore volume (Ac-PC, NAc-PC), accessible and nonaccessible probe-occupiable pore volume (Ac-PO, NAc-PO) . The concept of accessibility relies on the probe size, and a probe of the radius of is used in our work in order to directly relate the pore volumes to experimentally measured Nitrogen (kinetic diameter of ) isotherms. Accessible volume calculated here is also defined as the volume available to the center of the spherical probe, corresponding to the accessible probe center pore volume.
Statistical data visualization
In this work, Python data visualization libraries such as seaborn and matplotlib were used for informative statistical graphics. Threedimensional structures are drawn using the web service Bohrium at https://bohrium.dp.tech.
Computing environment
Uni-MOF pre-training and fine-tuning are performed on V100/A100 GPUs, and Monte Carlo simulations are performed on CPU cluster of Bohrium .
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data presented in this study are available in the manuscript file, the Supplementary Information file and Source Data files. Source Data in this study have been deposited in the figshare database under accession code of https://doi.org/10.6084/m9.figshare.24996317.
Code availability
Code to run the Uni-MOF model is available in GitHub (https://github. com/dptech-corp/Uni-MOF) .
The notebook demo can be found at https://bohrium.dp.tech/ notebook/cca98b584a624753981dfd5f8bb79674.
References
Sholl, D. S. & Lively, R. P. Seven chemical separations to change the world. Nature 532, 435-437 (2016).
Kohl, A. L. & Nielsen, R. Gas Purification (Elsevier, 1997).
Yang, R. T. Gas Separation by Adsorption Processes, Vol. 1 (World Scientific, 1997).
Boyd, P. G. et al. Data-driven design of metal-organic frameworks for wet flue gas capture. Nature 576, 253-256 (2019).
Koytsoumpa, E. I., Bergins, C. & Kakaras, E. The economy: review of capture and reuse technologies. J. Supercrit. Fluids 132, 3-16 (2018).
Kather, A. & Scheffknecht, G. The oxycoal process with cryogenic oxygen supply. Naturwissenschaften 96, 993-1010 (2009).
Jee, J.-G., Kim, M.-B. & Lee, C.-H. Pressure swing adsorption processes to purify oxygen using a carbon molecular sieve. Chem. Eng. Sci. 60, 869-882 (2005).
Van Groenestijn, J. & Kraakman, N. Recent developments in biological waste gas purification in europe. Chem. Eng. J. 113, 85-91 (2005).
Zhuang, L.-L., Yang, T., Zhang, J. & Li, X. The configuration, purification effect and mechanism of intensified constructed wetland for wastewater treatment from the aspect of nitrogen removal: a review. Bioresour. Technol. 293, 122086 (2019).
Akerib, D. et al. Chromatographic separation of radioactive noble gases from xenon. Astropart. Phys. 97, 80-87 (2018).
Lu, Z.-T. et al. Tracer applications of noble gas radionuclides in the geosciences. Earth Sci. Rev. 138, 196-214 (2014).
Furukawa, H., Cordova, K. E., O’Keeffe, M. & Yaghi, O. M. The chemistry and applications of metal-organic frameworks. Science 341, 1230444 (2013).
Ding, M., Cai, X. & Jiang, H.-L. Improving mof stability: approaches and applications. Chem. Sci. 10, 10209-10230 (2019).
Wang, J., Zhou, M., Lu, D., Fei, W. & Wu, J. Virtual screening of nanoporous materials for noble gas separation. ACS Appl. Nano Mater. 5, 3701-3711 (2022).
Yang, Q., Liu, D., Zhong, C. & Li, J.-R. Development of computational methodologies for metal-organic frameworks and their application in gas separations. Chem. Rev. 113, 8261-8323 (2013).
Li, J.-R., Kuppler, R. J. & Zhou, H.-C. Selective gas adsorption and separation in metal-organic frameworks. Chem. Soc. Rev. 38, 1477-1504 (2009).
Knebel, A. & Caro, J. Metal-organic frameworks and covalent organic frameworks as disruptive membrane materials for energyefficient gas separation. Nat. Nanotechnol. 17, 911-923 (2022).
Zhou, M. & Wu, J. Inverse design of metal-organic frameworks for c2h4/c2h6 separation. npj Comput. Mater. 8, 256 (2022).
Wang, J., Zhou, M., Lu, D., Fei, W. & Wu, J. Computational screening and design of nanoporous membranes for efficient carbon isotope separation. Green. Energy Environ. 5, 364-373 (2020).
Lin, R.-B., Xiang, S., Zhou, W. & Chen, B. Microporous metal-organic framework materials for gas separation. Chem 6, 337-363 (2020).
Banerjee, D. et al. Metal-organic framework with optimally selective xenon adsorption and separation. Nat. Commun. 7, ncomms11831 (2016).
Iftimie, R., Minary, P. & Tuckerman, M. E. Ab initio molecular dynamics: Concepts, recent developments, and future trends. Proc. Natl Acad. Sci. 102, 6654-6659 (2005).
Hollingsworth, S. A. & Dror, R. O. Molecular dynamics simulation for all. Neuron 99, 1129-1143 (2018).
Rubinstein, R. Y. & Kroese, D. P. Simulation and the Monte Carlo Method (John Wiley & Sons, 2016).
Weinan, E., Ren, W. & Vanden-Eijnden, E. String method for the study of rare events. Phys. Rev. B 66, 052301 (2002).
Zhou, M. & Wu, J. A gpu implementation of classical density functional theory for rapid prediction of gas adsorption in nanoporous materials. J. Chem. Phys. 153, 074101 (2020).
Altintas, C., Altundal, O. F., Keskin, S. & Yildirim, R. Machine learning meets with metal organic frameworks for gas storage and separation. J. Chem. Inf. Model. 61, 2131-2146 (2021).
Abdi, J. & Mazloom, G. Machine learning approaches for predicting arsenic adsorption from water using porous metal-organic frameworks. Sci. Rep. 12, 16458 (2022).
Nandy, A. et al. Mofsimplify, machine learning models with extracted stability data of three thousand metal-organic frameworks. Sci. Data 9, 74 (2022).
Scarselli, F., Gori, M., Tsoi, A. C., Hagenbuchner, M. & Monfardini, G. The graph neural network model. IEEE Trans. Neural Netw. 20, 61-80 (2008).
Xie, T. & Grossman, J. C. Crystal graph convolutional neural networks for an accurate and interpretable prediction of material properties. Phys. Rev. Lett. 120, 145301 (2018).
Vaswani, A. et al. Attention is all you need. Advances in Neural Information Processing Systems (eds Guyon, I. et al.) Vol. 30 (Curran Associates, Inc., 2017).
Kang, Y., Park, H., Smit, B. & Kim, J. A multi-modal pre-training transformer for universal transfer learning in metal-organic frameworks. Nat. Mach. Intell. 5, 309-318 (2023).
Cao, Z., Magar, R., Wang, Y. & Barati Farimani, A. Moformer: self-supervised transformer model for metal-organic framework property prediction. J. Am. Chem. Soc. 145, 2958-2967 (2023).
Zhou, G. et al. Uni-mol: a universal 3d molecular representation learning framework. In Proc. International Conference on Learning Representations (2023).
Colón, Y. J., Gómez-Gualdrón, D. A. & Snurr, R. Q. Topologically guided, automated construction of metal-organic frameworks and their evaluation for energy-related applications. Cryst. Growth Des. 17, 5801-5810 (2017).
Gómez-Gualdrón, D. A. et al. Evaluating topologically diverse metal-organic frameworks for cryo-adsorbed hydrogen storage. Energy Environ. Sci. 9, 3279-3289 (2016).
Lan, Y. et al. Materials genomics methods for high-throughput construction of cofs and targeted synthesis. Nat. Commun. 9, 5274 (2018).
Kenton, J. D. M.-W. C. & Toutanova, L. K. Bert: Pre-training of deep bidirectional transformers for language understanding. In Proc. of naacL-HLT, vol. 1, 2 (2019).
Nath, K., Ahmed, A., Siegel, D. J. & Matzger, A. J. Microscale determination of binary gas adsorption isotherms in mofs. J. Am. Chem. Soc. 144, 20939-20946 (2022).
Zhao, Z., Li, Z. & Lin, Y. Adsorption and diffusion of carbon dioxide on metal- organic framework (mof-5). Ind. Eng. Chem. Res. 48, 10015-10020 (2009).
Walton, K. S. et al. Understanding inflections and steps in carbon dioxide adsorption isotherms in metal-organic frameworks. J. Am. Chem. Soc. 130, 406-407 (2008).
Nugent, P. et al. Porous materials with optimal adsorption thermodynamics and kinetics for co2 separation. Nature 495, 80-84 (2013).
Duff, D. G., Ross, S. M. & Vaughan, D. H. Adsorption from solution: an experiment to illustrate the langmuir adsorption isotherm. J. Chem. Educ. 65, 815 (1988).
Krishna, R. & van Baten, J. M. In silico screening of metal-organic frameworks in separation applications. Phys. Chem. Chem. Phys. 13, 10593-10616 (2011).
Pillai, R. S., Pinto, M. L., Pires, J., Jorge, M. & Gomes, J. R. Understanding gas adsorption selectivity in irmof-8 using molecular simulation. ACS Appl. Mater. Interfaces 7, 624-637 (2015).
Moosavi, S. M. et al. Understanding the diversity of the metalorganic framework ecosystem. Nat. Commun. 11, 1-10 (2020).
Kalmutzki, M. J., Hanikel, N. & Yaghi, O. M. Secondary building units as the turning point in the development of the reticular chemistry of mofs. Sci. Adv. 4, eaat9180 (2018).
van der Maaten, L. & Hinton, G. Visualizing data using t-sne. J. Mach. Learn. Res. 9, 2579-2605 (2008).
Chung, Y. G. et al. In silico discovery of metal-organic frameworks for precombustion co2 capture using a genetic algorithm. Sci. Adv. 2, e1600909 (2016).
Chung, Y. G. et al. Advances, updates, and analytics for the com-putation-ready, experimental metal-organic framework database: core mof 2019. J. Chem. Eng. Data 64, 5985-5998 (2019).
Tong, M., Lan, Y., Yang, Q. & Zhong, C. Exploring the structureproperty relationships of covalent organic frameworks for noble gas separations. Chem. Eng. Sci. 168, 456-464 (2017).
Hammersley, J. Monte Carlo Methods (Springer Science & Business Media, 2013).
Dubbeldam, D., Calero, S., Ellis, D. E. & Snurr, R. Q. Raspa: molecular simulation software for adsorption and diffusion in flexible nanoporous materials. Mol. Simul. 42, 81-101 (2016).
Rappé, A. K., Casewit, C. J., Colwell, K., Goddard III, W. A. & Skiff, W. M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 114, 10024-10035 (1992).
Boato, G. & Casanova, G. A self-consistent set of molecular parameters for neon, argon, krypton and xenon. Physica 27, 571-589 (1961).
Martin, M. G. & Siepmann, J. I. Transferable potentials for phase equilibria. 1. united-atom description of n-alkanes. J. Phys. Chem. B 102, 2569-2577 (1998).
Altintas, C. & Keskin, S. Role of partial charge assignment methods in high-throughput screening of mof adsorbents and membranes for co 2/ch 4 separation. Mol. Syst. Des. Eng. 5, 532-543 (2020).
Ong, S. P. et al. Python materials genomics (pymatgen): a robust, open-source python library for materials analysis. Comput. Mater. Sci. 68, 314-319 (2013).
Willems, T. F., Rycroft, C. H., Kazi, M., Meza, J. C. & Haranczyk, M. Algorithms and tools for high-throughput geometry-based analysis of crystalline porous materials. Microporous Mesoporous Mater. 149, 134-141 (2012).
Ongari, D. et al. Accurate characterization of the pore volume in microporous crystalline materials. Langmuir 33, 14529-14538 (2017).
Waskom, M. L. seaborn: statistical data visualization. J. Open Source Softw. 6, 3021 (2021).
Hunter, J. D. Matplotlib: a 2d graphics environment. Comput. Sci. Eng. 9, 90-95 (2007).
D.L. was supported by National Natural Science foundation of China, No. U1862204 and 21878175. This research was supported in part by the Tsinghua University and the DP Technology.
Author contributions
J.W.(Wang) and J.L. contributed equally to this work. D.L., Z.G. and J.W.(Wu) designed and guided the project. J.W.(Wang), J.L. and Z.G.
performed the data collection, developed the breakthrough model and drafted the paper. H.W., M.Z., G.K. and L.Z. provided important advice and revised the paper. All authors contributed to the discussion of results and commented on the paper.
Correspondence and requests for materials should be addressed to Jianzhong Wu, Zhifeng Gao or Diannan Lu.
Peer review information Nature Communications thanks Ilia Kevlishvili, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/ licenses/by/4.0/.
(c) The Author(s) 2024
Department of Chemical Engineering, Tsinghua University, Beijing 100084, China. DP Technology, Beijing 100089, China. School of Advanced Energy, Sun Yat-Sen University, Shenzhen 518107, China. AI for Science Institute, Beijing 100190, China. Jiangsu Key Laboratory for Carbon-Based Functional & Materials Devices, Institute of Functional & Nano Soft Materials (FUNSOM), Soochow University, Suzhou 215123, China. Department of Chemical and Environmental Engineering, University of California, Riverside, CA 92521, USA. These authors contributed equally: Jingqi Wang, Jiapeng Liu. e-mail: jwu@engr.ucr.edu; gaozf@dp.tech; ludiannan@mail.tsinghua.edu.cn