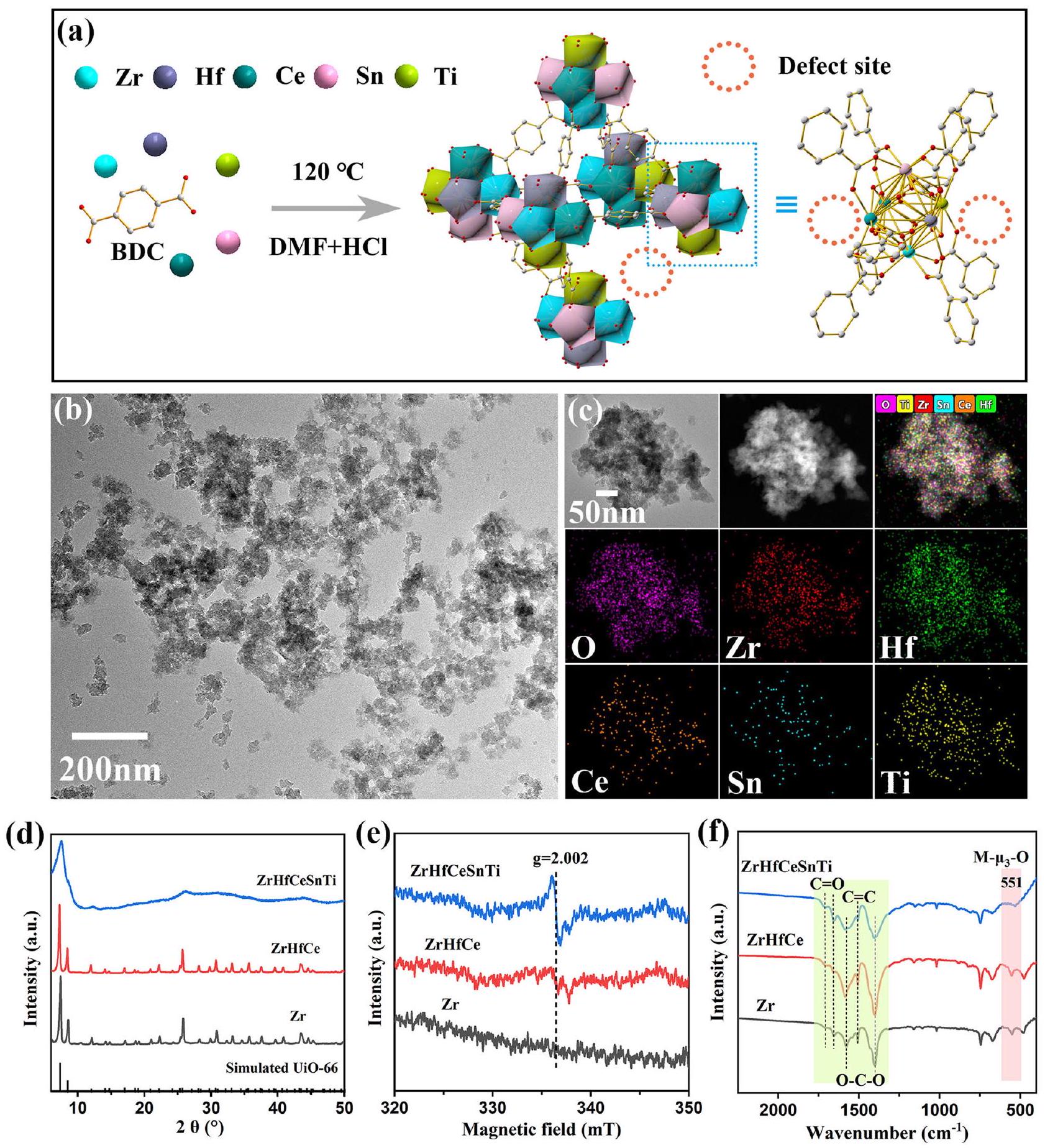
هندسة الانتروبيا لتنشيط UiO-66 لتعزيز الهيدروجين الانتقالي الحفزي Entropy engineering activation of UiO-66 for boosting catalytic transfer hydrogenation
تعد الأطر المعدنية العضوية عالية الإنتروبيا (HE-MOFs) واعدة كمواد متعددة الاستخدامات، ومع ذلك فإن الأمثلة النادرة الحالية محصورة في العناصر ذات التكافؤ المنخفض في الفترة الرابعة، مما يقيد تصميمها وتحسينها لتطبيقات متنوعة. هنا، تم تخليق UiO-66 (ZrHfCeSnTi HE-UiO-66) عالي الإنتروبيا، الغني بالعيوب وصغير الحجم (32 نانومتر) لأول مرة، مستفيدًا من زيادة الإنتروبيا التكوينية لتحقيق تحمل عالٍ للتطعيم بأيونات المعادن المتنوعة. تشوه الشبكة في HE-UiO-66 يؤدي إلى تعرض عالٍ للعقد المعدنية لإنشاء مواقع معدنية غير مشبعة بالتنسيق مع تركيز من , مما يزيد من وفرة مواقع الحمض-القاعدة لويس، وبالتالي تحقيق تحسين كبير في أداء تفاعل الهيدروجين الانتقالي الحفاز (CTH). تظهر التحقيقات المنهجية أن الهيكل الإلكتروني الخاص بـ HE-UiO-66 يعزز التفاعل والترابط مع جزيئات الركيزة ويقلل من حاجز الطاقة لعملية نقل الهيدروجين. تقدم طريقتنا استراتيجية جديدة لبناء مواقع معدنية غير مشبعة بالتنسيق في MOFs.
تتميز المواد عالية الإنتروبيا، كنوع جديد من مواد المحلول الصلب البلوري، بتكوينها الفريد، الذي يتكون من خمسة أنواع أو أكثر من المعادن المتنوعة بنسب متساوية تقريبًا، مع الحفاظ على هيكل بلوري أحادي الطور.. يوفر التكوين الفريد ليس فقط مجموعة واسعة من التركيبات لاكتشاف المواد، ولكن أيضًا بنية ميكروية مميزة لتحسين الخصائص.. يؤدي هذا الجمع بين الأنواع المعدنية المتنوعة وتفاعلاتها إلى خصائص جديدة وغير متوقعة، بما في ذلك تأثيرات الإنتروبيا العالية، وتأثيرات تشوه الشبكة، وتأثيرات الانتشار المتأخر و”تأثيرات الكوكتيل”.. في عام 2004، قدم يي وكانتر مفهوم سبائك الإنتروبيا العالية (HEAs) في وقت واحد.. تم توسيع هذا المفهوم لاحقًا ليشمل الأكسيدات، النيتريدات، الكالكوجينيدات، الفوسفيدات، الفلوريدات والدبوريدات كجزء من هذه العائلة. تختلف هذه المواد عن HEAs ذات الشواغر الأحادية، حيث تسمح الشبكات الكاتيونية والأنونية المستقلة في هذه المواد بمرونة هيكلية أكبر.. يزيد الإنتروبيا التكوينية من تحملها لتشوه الشبكة، مما يسهل دمج واستقرار تركيزات أعلى من الكاتيونات المعدنية غير المتكافئة في الشبكة الأكسيدية.. لتوسيع مكتبة المواد عالية الإنتروبيا، تم مؤخرًا إدخال مفهوم الإنتروبيا العالية في الأطر المعدنية العضوية (MOFs).. ومع ذلك،
يتطلب بناء MOFs عالية الإنتروبيا (HE-MOFs) عملية أكثر تعقيدًا تتطلب تحكمًا دقيقًا في نسبة أيونات المعادن إلى الروابط العضوية، بالإضافة إلى التلاعب في ظروف التفاعل. لذلك، بقدر ما نعلم، لا يزال البحث في تخليق HE-MOFs محدودًا بسبب الاختلاف في القدرة التنسيقية بين أيونات المعادن والروابط، ولم يتم إجراء سوى عدد قليل من الدراسات، تركزت أساسًا على أيونات المعادن ذات التكافؤ المنخفض مثل , و, والتي تعتبر سهلة الاستخدام نسبيًا في تخليق MOF.. لا يزال تخليق HE-MOFs باستخدام أيونات المعادن ذات التكافؤ العالي مثل , و يواجه تحديات ولم يتم الإبلاغ عنه بعد.
لقد حظيت MOFs باهتمام كبير في تفاعلات الهيدروجين الانتقالي الحفاز (CTH) بسبب قدرتها على ضبط الهيكل والخصائص الإلكترونية بدقة.. يوفر إطارها المسامي مساحة سطح عالية وخصائص مسامية محددة جيدًا، ويسمح بضبط دقيق للعقد المعدنية ذات التكافؤ العالي، مما يؤدي إلى خصائص حمضية ممتازة.. تُعتبر Zr/Hf-MOFs ضمن هذه الفئة وتشتهر بخصائصها الاستثنائية كحمض لويس وحمض برونستيد.. من بينها، يُعتبر UiO-66 شائعًا بشكل خاص بسبب إطاره المستقر للغاية، وكثافة الشحنة العالية لعقد ، والحمضية المحتملة لويس، والقدرة على تعديل الخصائص الإلكترونية من خلال التعديلات الهيكلية. يظهر UiO-
66 أنماط هيكلية متناوبة من و، مع اعتقاد أن كثافة الشحنة العالية لمجموعات Zr-O مفيدة لتفاعلات CTH.. حاليًا، يتم استخدام مجموعة متنوعة من UiO-66 وموادها المعدلة بشكل فعال في تفاعلات CTH، مما يظهر أدائها الحفاز الفعال (الجدول S1). علاوة على ذلك، غالبًا ما توجد مواقع Zr/Hf المفتوحة غير المشبعة بالتنسيق داخل إطار UiO-66، سواء كانت تحدث بشكل طبيعي أو يتم تحفيزها عمدًا من خلال هندسة العيوب. تعمل هذه المواقع، الناتجة عن انخفاض الاتصال بين العقد المعدنية والروابط العضوية، كمراكز لويس فعالة وتظهر نشاطًا عاليًا في العمليات الحفازة.. يمكن إنشاء المواقع غير المشبعة داخل إطار UiO-66 عمدًا أثناء التخليق من خلال استخدام المحفزات، الروابط شبه المستقرة أو المواد السطحية لتحفيز تشكيل العيوب، أو من خلال طرق المعالجة بعد التخليق.. ومع ذلك، فإن UiO66 الذي تم تخليقه بهذه الطرق لديه تحمل محدود لمستويات عالية من المواقع غير المشبعة، مما قد يؤدي إلى عدم استقرار أو انهيار الهيكل البلوري.
“هندسة الإنتروبيا” تقدم لنا نهجًا استراتيجيًا لحل هذه المشكلة من خلال رفع الإنتروبيا التكوينية، مما يجعل تشوه الشبكة أكثر تحملًا. وهذا بدوره يسهل دمج واستقرار تركيزات أعلى من الكاتيونات المعدنية المتنوعة وعيوب الشبكة، مما يعزز في النهاية إنشاء مواقع غير مشبعة بالتنسيق. في هذا العمل، تم تصميم وتخليق UiO-66 عالي الإنتروبيا (ZrHfCeSnTi HE-UiO66) الغني بالعيوب لأول مرة من خلال تأثير الإنتروبيا العالية. تستند هذه الطريقة إلى توافق أيونات المعادن و Ce لإنشاء عائلة UiO-66 ، مستفيدة من زيادة الإنتروبيا التكوينية لدمج أيونات المعادن Sn و Ti داخل العقد، ويمكن أن يسهم التشوه الناتج عن الشبكة (المستمد من تأثير تشوه الشبكة، أحد التأثيرات الأربعة للمواد عالية الإنتروبيا) في توليد العديد من المواقع غير المشبعة. مع مثل هذه الاستراتيجية، تعزز العقد المعدنية المعرضة بشكل كبير في HE-UiO-66 زيادة كبيرة في وفرة مواقع الحمض-القاعدة لويس، مما يؤدي إلى اختراق في أداء تفاعل CTH. كشفت حسابات نظرية الكثافة الوظيفية، تتبع النظائر، -TPD، طيف الانعكاس تحت الحمراء بتقنية فورييه وتحليل الانعكاس الكلي تحت الحمراء عن آلية النشاط الحفاز المعزز ومسارات نقل الهيدروجين. يوفر توضيح الهيكل والخصائص لمواقع المعادن غير المشبعة بالتنسيق المعنية في HE-UiO66 إرشادات لتعديل مواقع الحمض-القاعدة لويس في مجموعة واسعة من MOFs المعقدة ذات الوظائف الموسعة.
النتائج
تصميم حفازات HE-UiO-66
في هذا العمل، قمنا بتطوير استراتيجية تُسمى “هندسة الإنتروبيا”، حيث يتم تحفيز تشكيل العيوب في UiO-66 من خلال زيادة الإنتروبيا التكوينية لأيونات المعادن في العقد (الشكل 1a). تعتمد هذه الطريقة على توافق أيونات المعادن و Ce، جميعها قادرة على تشكيل عائلة UiO-66 ، لمنح الإطار (ZrHfCe UiO-66) تحملًا أكبر للتطعيم بأيونات المعادن المتنوعة من خلال رفع الإنتروبيا التكوينية.. تمكّن الخصائص الاستقبالية للنظام من دمج أيونات المعادن Sn و Ti للتطعيم في العقد. في الوقت نفسه، تؤدي الاختلافات في نصف القطر الأيوني، الهيكل الإلكتروني، تأثيرات المجال البلوري، إلخ. بين , و إلى تشوهات كبيرة في الشبكة ، مما يؤدي إلى فقدان جزئي للروابط 12 BDC (حمض البنزينيك 1,4) المرتبطة بالعقد المعدنية.. يؤدي تقليل هذه الروابط BDC إلى تشكيل مواقع معدنية غير مشبعة أو غير متناسقة، أي مواقع عيوب. ونتيجة لذلك، تم تخليق حفاز HE-UiO-66 الغني بالعيوب بنجاح من خلال تأثير الإنتروبيا.
الخصائص الشكلية والهيكلية لحفازات HE-UiO-66
تم مراقبة تأثير الانتروبيا التكوينية على شكل UiO-66 بواسطة المجهر الإلكتروني الماسح (SEM) و
المجهر الإلكتروني الناقل (TEM) (الشكل 1ب، S1 و S2). وُجد أن الزيادة في الانتروبيا التكوينية تسببت في انخفاض حجم الجسيمات من لـ Zr إلى لـ ZrHfCe ثم إلى لـ HE-UiO-66 (الشكل S2). يحدث هذا الظاهرة بسبب إدخال عناصر غير متجانسة مختلفة، مما يقلل من طاقة السطح للجسيمات النانوية وبالتالي يعيق نموها . بالإضافة إلى ذلك، يمكن توضيح ذلك من خلال تأثير الكوكتيل والانتشار المتأخر في المواد ذات الانتروبيا العالية. تشوه الشبكة يزيد من طاقة تنشيط الانتشار للذرات داخل الشبكة، مما يؤدي إلى انخفاض في معدل الانتشار الفعال وبالتالي إبطاء نمو الجسيمات . التراكم غير المنتظم لهذه الجسيمات النانوية الصغيرة يعزز بشكل فعال الهيكل المسامي لـ HE-UiO-66، مما يفيد في تحسين كفاءة نقل الكتلة لجزيئات الركيزة في العملية التحفيزية، وبالتالي تحسين الكفاءة التحفيزية. لتوصيف التوزيع المكاني للعناصر في محفز HE-UiO-66، أظهرت صور المجهر الإلكتروني الناقل ذو الزاوية العالية (HAADF-STEM) وصور رسم الخرائط EDS المقابلة أن , و O كانت موزعة بشكل موحد على مدى الكشف الكامل (الشكل 1ج). علاوة على ذلك، يتوافق الكسر الذري لكل معدن في HE-UiO-66 مع المعايير الخاصة بالمواد ذات الانتروبيا العالية، حيث يقع ضمن نطاق محتوى المعدن ، كما هو موضح بواسطة تحليل البلازما المقترنة بالحث (ICP) (الجدول S2).
تم فحص الهياكل البلورية للعينات بالتفصيل باستخدام حيود الأشعة السينية (XRD) وطيف الرنين المغناطيسي الإلكتروني (EPR) وطيف تحويل فورييه للأشعة تحت الحمراء (FT-IR). جميع العينات، باستثناء العينات غير المتبلورة من Sn و Ti، تظهر مراحل بلورية متبلورة جيدًا تتطابق بشكل جيد مع UiO-66 المحاكى، مما يشير إلى تحضير محفزات HE-UiO-66 (الشكل 1د و S3). بناءً على ترتيب أنصاف الأقطار الأيونية كما يلي: ، تخضع الشبكة لتوسع أولي يتبعه انكماش مع زيادة الانتروبيا، كما يتضح من تحول قمم XRD المتضخمة جزئيًا إلى اليسار أو اليمين في نطاق (الشكل S3). توفر هذه التغييرات دليلًا قويًا على توليد HE-UiO-66 عن طريق إضافة أيونات معدنية بأنصاف أقطار أيونية متغيرة إلى هيكل UiO-66. علاوة على ذلك، تظهر قمم حيود XRD لـ HE-UiO-66 تلاشيًا في الشدة وتوسعًا بسبب الاحتلال العشوائي للأيونات المعدنية داخل الشبكة البلورية. يؤدي الاحتلال العشوائي للأيونات المعدنية في الشبكة البلورية إلى تشوه ملحوظ في الشبكة، مما يؤدي إلى ظهور مستويات ذرية غير متساوية داخل نفس الطبقة وينتج عنه تشتت براغ ملحوظ للأشعة السينية على مستويات بلورية غير منتظمة. وهذا يشير إلى وجود العديد من العيوب (مثل مواقع العيوب المرتبطة المفقودة) أو الاتصالات غير المنتظمة داخل HE-UiO-66 . كما تدعمه طيف EPR (الشكل 1هـ) . يُنسب الذروة عند إلى مراكز معدنية غير مرتبطة تعمل كعيوب في الإطار. مع ارتفاع الانتروبيا التكوينية لـ UiO-66، تخضع الذروة لزيادة كبيرة، مما يشير إلى زيادة متناسبة في كمية العيوب. لا تزال المواقع غير المشبعة الناتجة عن العيوب تتناسق مع مجموعات الهيدروكسيل أو للحفاظ على توازن شحنة السلسلة الرئيسية، كما هو موضح بواسطة تحليل XPS لـ O1s (الشكل S4). الذروة عند 530 eV تتوافق مع الأكسجين الشبكي M-O-M، بينما الذروة عند 532 eV تتوافق مع الأكسجين المرتبط بالمعدن لمجموعة الحمض الكربوكسيلي (M-O-C) أو الأكسجين المرتبط بالمعدن . في وجود العيوب، يحدث انخفاض حتمي في M-O-M و M-O-C، مصحوبًا بزيادة في و ، مما يفسر أيضًا الفرق في الذروتين لـ Zr و ZrHfCe و HE-UiO-66. تم تصوير أطياف FT-IR لجميع العينات في الشكل 1و. تُنسب الذروة الملحوظة عند إلى اهتزاز ، مما يدل على تنسيق الأيونات المعدنية مع الرابطة في حمض التيرفثاليك . من الجدير بالذكر أن شدة ذروة اهتزاز في HE-UiO66 أضعف بكثير من العينات الأخرى، مما يشير إلى أن المزيد من الأيونات المعدنية في HE-UiO-66 غير قادرة على التنسيق مع الروابط وتشكيل مواقع معدنية غير مشبعة. علاوة على ذلك، فإن القمم في منطقة المرتبطة بمجموعات الحمض الكربوكسيلي،
الشكل 1 | التحضير والتوصيف الهيكلي لـ HE-UiO-66. أ رسم تخطيطي لعملية تخليق HE-UiO-66 مع عيوب غنية، (ب) TEM، (ج) HAADF-STEM وصور رسم الخرائط العنصرية المقابلة لـ HE-UiO-66، (د) أنماط XRD، (هـ) أطياف EPR و (و) أطياف FT-IR لسلسلة من عينات UiO-66.
حلقة البنزين ومجموعات الكربونيل ليست متغيرة عبر جميع العينات. وهذا يشير إلى أن زيادة الانتروبيا لا تؤثر على الهيكل، مما يتماشى مع نتائج XRD. باختصار، تؤكد النتائج أعلاه على وجود عيوب وفيرة داخل محفز HE-UiO-66.
تم توصيف توزيع السطح وحجم المسام للعينات التي تم تخليقها حديثًا بواسطة إيزوثرم الامتصاص-الامتزاز. مع زيادة الانتروبيا، تخضع الهيكل المسامي الدقيق لعينات Zr و ZrHfCe لانتقال إلى الهيكل المسامي الدقيق لـ HE-UiO-66، والذي يتميز بتعايش إيزوثرم من النوع الأول والنوع الرابع، متحولًا من إيزوثرم من النوع الأول فقط (الشكل S5أ) . يمكن أيضًا ملاحظة هذا الانتقال بشكل أوضح في توزيع حجم المسام (الشكل S5ب). يتم تفسير هذا الانتقال في هيكل المسام جزئيًا من خلال الانهيار الجزئي للهيكل الناتج عن العيوب، مما يؤدي إلى تحول بعض المسام الدقيقة إلى مسام متوسطة . يؤدي الانخفاض في عدد المسام الدقيقة السائدة إلى انخفاض في المساحة السطحية المحددة، كما هو موضح في الجدول S3. بالإضافة إلى ذلك، تتسبب زيادة الانتروبيا في انكماش الجسيمات إلى وتعتبر التكديس غير المنتظم للجسيمات النانوية عاملًا آخر يساهم في ظهور المسام المتوسطة. باختصار، يوفر الهيكل المسامي الدقيق والمخفض لحجم الجسيمات لـ HE-UiO-66 مزايا في تعزيز كفاءة نقل الكتلة، مما يجعله مناسبًا للتفاعلات التحفيزية ذات الكفاءة العالية.
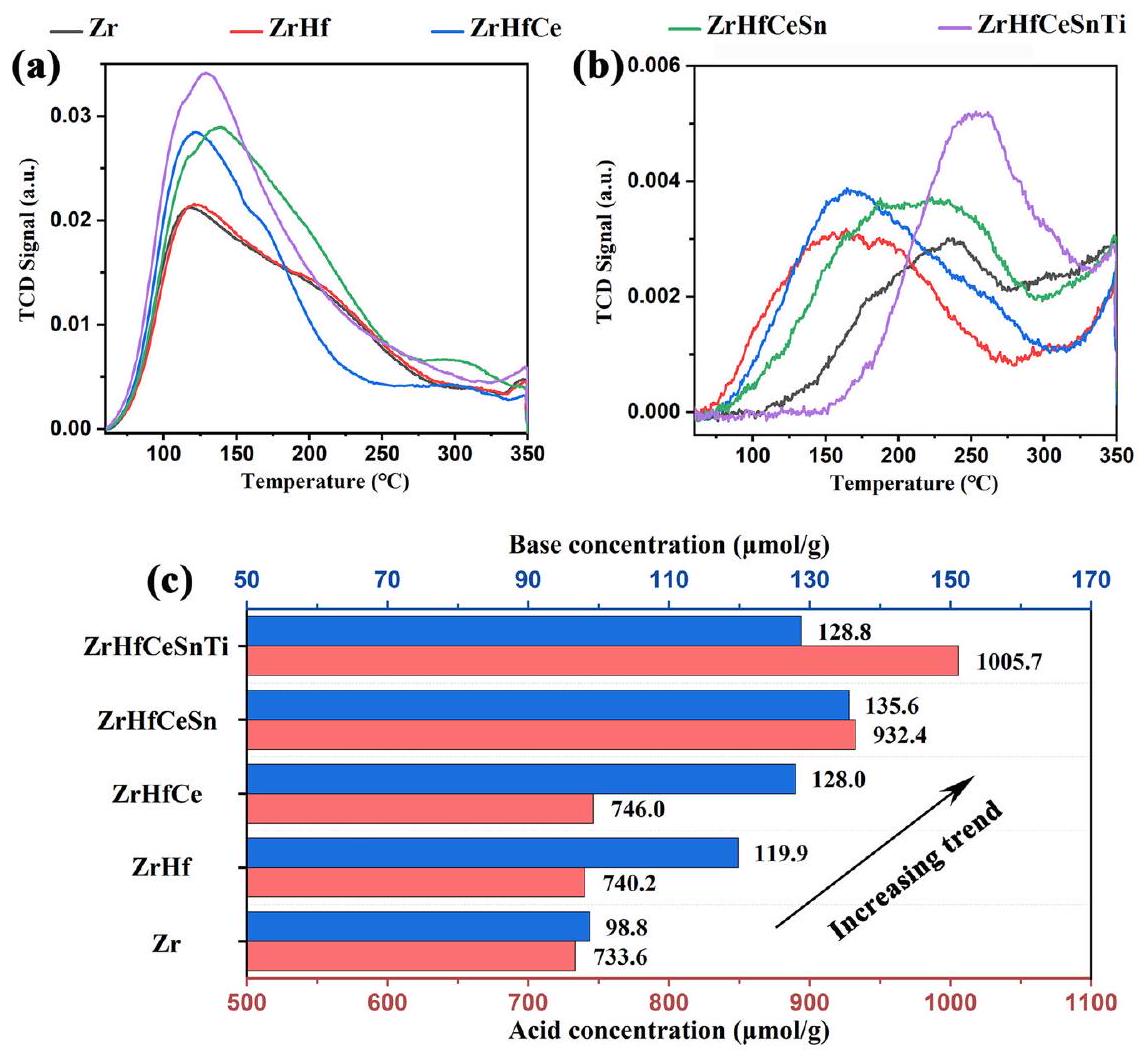
الشكل 2 | التحليل النوعي والكمّي لمواقع الحمض-القاعدة. أ -TPD ملفات تعريف، (ب) -TPD ملفات تعريف، و (ج) مقارنة بين كمية القاعدة والحمض.
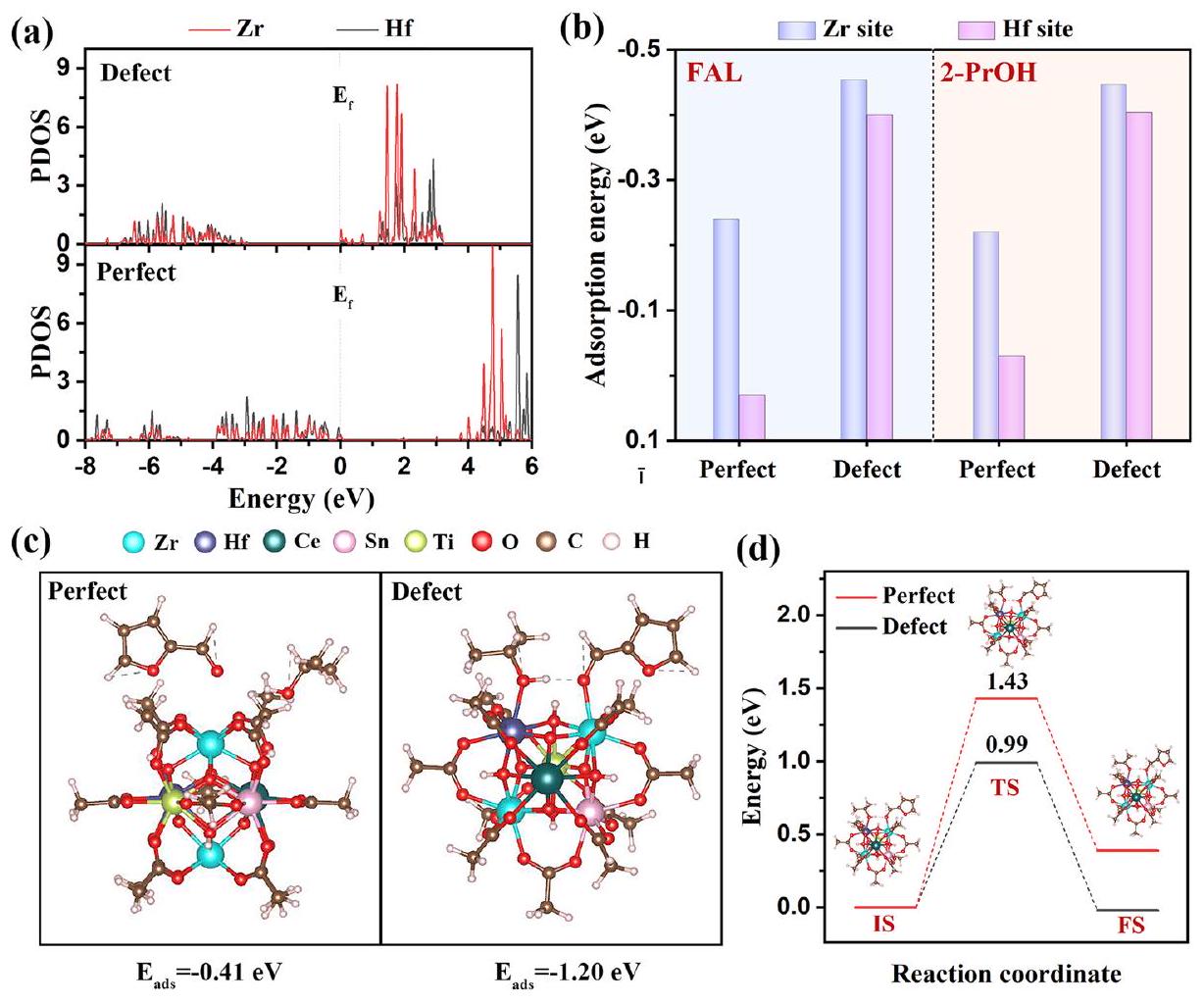
التحليل النوعي والكمّي لمواقع النشاط في HE-UiO-66. تنبع النشاط التحفيزي الاستثنائي لـ و في تفاعل CTH للمركبات الكربونيلية من هيكلها الإلكتروني الفريد وتكوين التنسيق . بشكل عام، كلما كانت كثافة الإلكترون لـ و أقل، كانت المدارات أكثر فراغًا، مما يؤدي إلى طاقة ارتباط أعلى وإنتاج أقوى للأحماض لويس. لم تؤدي التشوهات الشبكية الناتجة عن زيادة الانتروبيا إلى توليد العيوب فحسب، بل أدت أيضًا إلى إنشاء مواقع معدنية غير مشبعة بسبب الروابط المفقودة، مما يسهل جميعه توليد مراكز لويس ذات نشاط تحفيزي عالي. تكشف دراسة تأثير زيادة الانتروبيا على الحالات الإلكترونية لـ و ، التي أجريت عبر أطياف XPS (الشكل S6)، أن طاقات الارتباط لـ Zr و Hf قد زادت بشكل كبير في HE-UiO-66 مقارنةً بالعينات الأخرى. تؤكد هذه الملاحظة زيادة في نقص الإلكترونات في مواقع وتشير إلى تكوين مواقع أحماض لويس أقوى. بالاقتران مع الاستنتاج بأن الروابط المفقودة تعزز توليد مواقع معدنية غير مشبعة، من المتوقع أن تمتلك مادة HE-UiO-66 عددًا أكبر من مواقع أحماض لويس وأقوى. بالإضافة إلى ذلك، فإن وجود العيوب في UiO-66 يؤدي أيضًا إلى امتصاص أو -OH لتثبيت ، مما يؤدي إلى زيادة وفرة في HE-UiO66 (الشكل S4). عادةً، تكون قاعدية أيونات على أعلى من تلك الخاصة بالمواد غير المتبلورة أو البلورية ذات الصلة ، مما يؤدي إلى زيادة وفرة مواقع قاعدة لويس في HE-UiO-66. تم استخدام ملفات امتصاص الحرارة المبرمجة (TPD) للتحليل الكمي للحصول على خصائص الحمض-القاعدة لـ HE-UiO-66 (الشكل 2). تشير النتائج إلى أن تركيز الحمض-القاعدة يميل إلى الزيادة مع زيادة الإنتروبيا التكوينية لـ UiO-66، حيث يظهر HE-UiO-66 أعلى شدة ذروة الامتصاص، مما يشير بشكل أكبر إلى وجود أكبر عدد من مواقع المعادن غير المشبعة المعرضة التي يمكن أن تعمل كأحماض لويس.
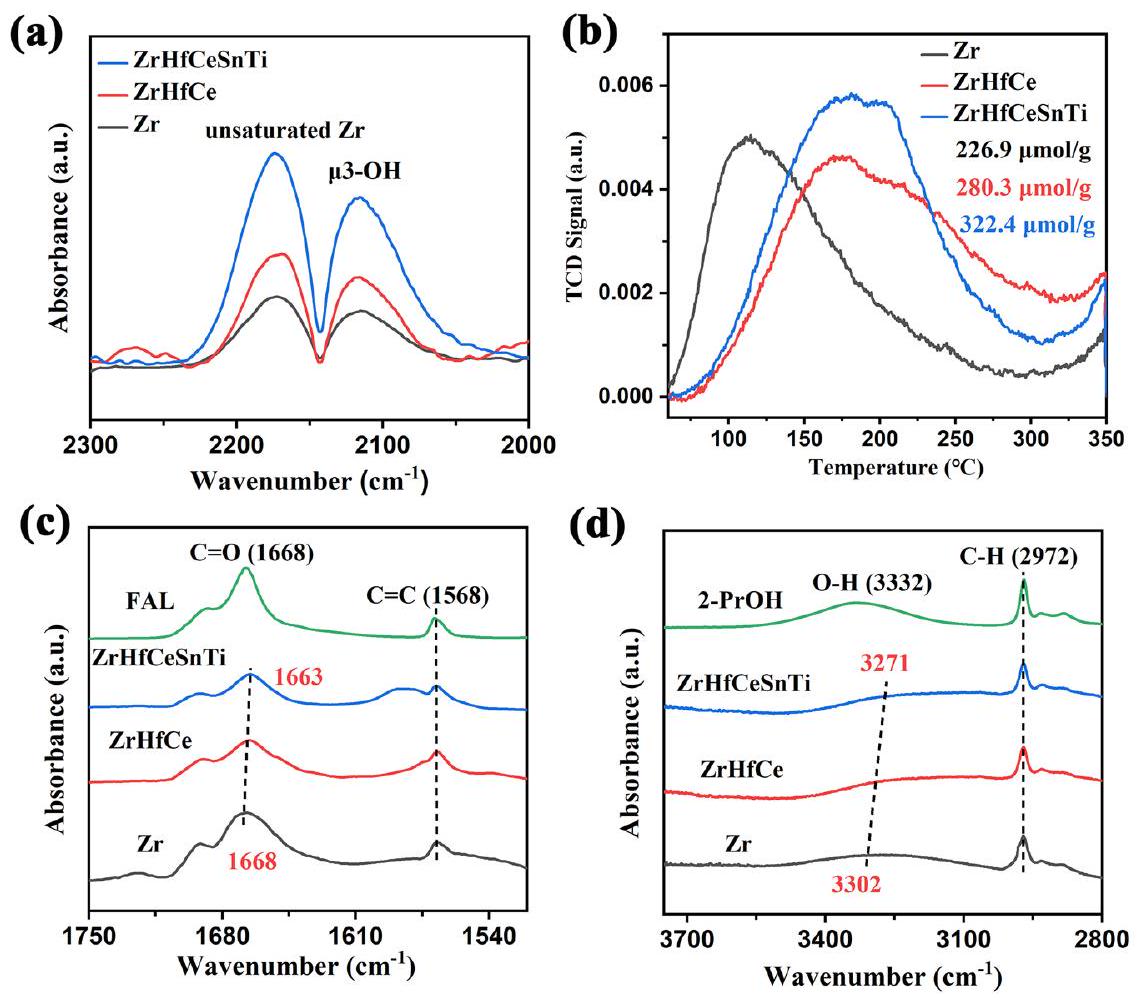
تم استخدام ملفات امتصاص CO-FTIR وملفات CO-TPD لمزيد من تحديد العدد المطلق لمواقع المعادن غير المشبعة في HE-UiO-66 للحصول على مزيد من الفهم لمواقع أحماض لويس. بالنسبة لطيف CO-FTIR (الشكل 3a)، تم تخصيص القمم المرصودة عند 2190 ولـ CO المرتبط بمواقع المعادن غير المشبعة وCO الممتص جسديًا، على التوالي.لوحظ أن HE-UiO-66 لديه أعلى ذروة امتصاص تمثل مواقع المعادن غير المشبعة، مما يشير إلى وجود أكبر عدد من مواقع المعادن غير المشبعة وبالتالي أعلى تركيز لأحماض لويس. تم استخدام ملفات CO-TPD بعد ذلك لتحديد تركيز مواقع المعادن غير المشبعة (الشكل 3b). قبل الاختبار، خضعت العينة لمعالجة مسبقة عند درجة حرارة عالية منلمدة 1 ساعة للقضاء علىمجموعة قادرة على امتصاص CO (مناقشة مفصلة في المعلومات التكميلية، الشكل S8). هذا يضمن أن مواقع المعادن غير المشبعة فقط كانت متاحة لامتصاص CO خلال التحليل. كما هو موضح في الشكل 3b، يظهر HE-UiO-66 أقوى ذروة امتصاص CO، مما يدل على أعلى تركيز لمواقع المعادن غير المشبعة، حيث تصل إلى. لذلك، يثبت هندسة الإنتروبيا أنها استراتيجية فعالة لزيادة مواقع المعادن غير المشبعة المنسقة وتأسيس مواقع أحماض لويس قوية ووفيرة داخل UiO-66.
تقييمات التفاعل تجاه تفاعل CTH للفورفورال
يلعب تفاعل CTH دورًا حاسمًا في إنتاج مواد كيميائية عالية القيمة من الكربونيلات المشتقة من الكتلة الحيوية.هنا، تم اختيار الفورفورال (FAL) كممثل للكربونيلات المشتقة من الكتلة الحيوية للتحقيق في المزايا الحفازة لـ HE-UiO-66 مع مواقع المعادن غير المشبعة المنسقة الوفيرة للتفاعل. نظرًا لأهمية حالة الربط السطحي لـ FAL/الكحول الأيزوبروبيلي (2-PrOH) الممتص على الحفاز، تم التقاط أطياف الانعكاس الكلي المخفف بالأشعة تحت الحمراء (ATR-IR) لفحص التفاعل بين FAL/2-PrOH مع HE-UiO66 وعينات التحكم (الشكل 3c، d). تم ملاحظة الذروة المقابلة لـالرابطة في FAL النقي عند. بالمقارنة مع Zr وZrHfCe، تظهر الذروة في HE-UiO-66 انزياحًا أحمر كبيرًا، مما يدل على تفاعل قوي بين الحفاز والمجموعة. بالإضافة إلى ذلك، لم يتم ملاحظة أي انزياح في موضعالرابطة على
الشكل 3 | التحليل الكمي لمواقع المعادن غير المشبعة المنسقة. أ امتصاص CO-FTIR، (ب) ملفات CO-TPD وأطياف ATR-IR لامتصاص (ج) FAL و(د) 2-PrOH على مجموعة من عينات UiO-66.
حلقة الفوران من الفورفورال (FAL) لم تُلاحظ، مما يشير إلى أن الموقع النشط للحفاز يتفاعل بشكل محدد معالمجموعة من FAL. بالمثل، بالنسبة للذروة المتعلقة برابطة O-H في 2-PrOH، يظهر HE-UiO-66 أيضًا انزياحًا أحمر أكثر وضوحًا، مما يدل على تفاعل قوي بين الحفاز ورابطة O-H في 2-PrOH التي تقلل من طاقة رابطة O-H. تدعم هذه النتائج أن HE-UiO-66، الغني بمواقع المعادن غير المشبعة المنسقة، يحسن الفعالية الحفازة في تفكك 2-PrOH وتنشيط الركيزة FAL من خلال التفاعل الفعال معوالرابطات.
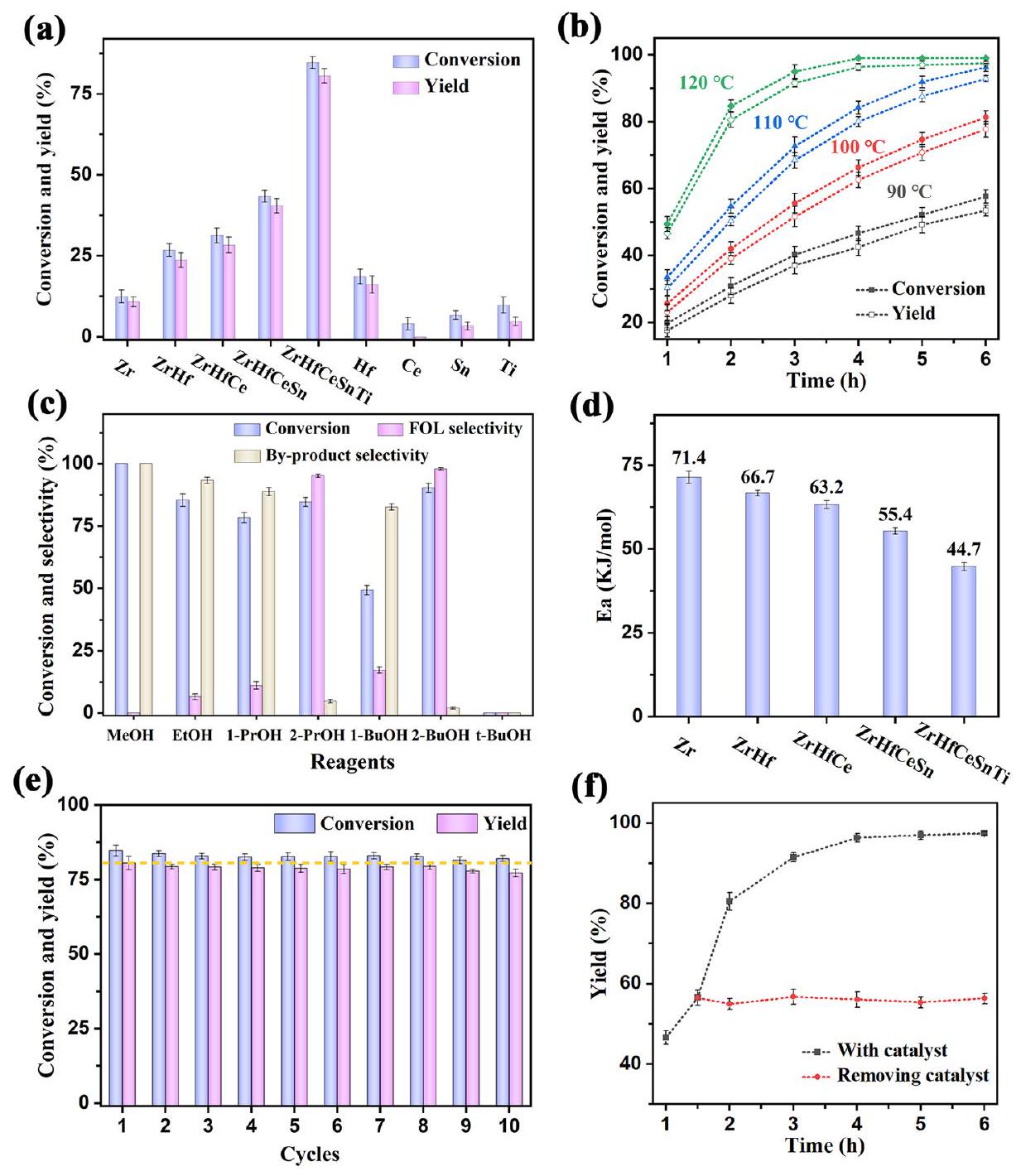
تم إجراء تفاعل CTH لـ FAL إلى كحول الفورفوريل (FOL) باستخدام حفازات مختلفة مع 2-PrOH كمذيب ومانح H عندلمدة ساعتين (الشكل 4a والشكل S9). مع زيادة الإنتروبيا التكوينية لـ UiO-66 (من معدن واحد Zr إلى خمسة معادن ZrHfCeSnTi)، تزداد النشاط الحفاز أيضًا حتى يصل HE-UiO-66 إلى أعلى عائد FOL قدرهوالذي يُعزى إلى توليد مواقع المعادن غير المشبعة المنسقة الوفيرة الناتجة عن هندسة الإنتروبيا. تظهر أرقام التحويل (TONs) اتجاهًا مشابهًا مع زيادة الإنتروبيا التكوينية (الشكل S9). يكشف المزيد من المقارنة بين حفازات UiO-66 المعدنية الفردية أن Zr وHf يظهران نشاطًا حفازًا وانتقائية أفضل، مما يبرز ميلهما الوظيفي المفضل لتفاعل CTH. علاوة على ذلك، تم فحص الأداء الحفاز لبلورات HE-UiO-66 النانوية فيما يتعلق بدرجة حرارة التفاعل والوقت (الشكل 4b)، مما يكشف عن تحسين التحويل والعائد مع زيادة قيم كلا المعاملين. لتوضيح العلاقة بين الحفازات المختلفة ونشاطها الحفاز تحت درجات حرارة وأوقات تفاعل متغيرة، تم إجراء تجارب حركية لتفاعل CTH لـ FAL إلى FOL في 2-PrOH (الشكل 4d والشكل S10). عند كل درجة حرارة تفاعل،كان مرتبطًا خطيًا مع وقت التفاعل، مما يدل على حركية من الدرجة الأولى لتفاعل CTH من FAL إلى FOL مع حفازات مختلفة. تم تحديد طاقة التنشيط الظاهرة (Ea) للتفاعل من معادلة أرهينيوس (). كانت Ea للتفاعل مع HE-UiO-66 كحفازأقل بكثير من تلك الخاصة بـ عينات المرجع، مما يدل على تفاعل CTH أكثر سهولة تم تحفيزه بواسطة HE-UiO-66. يمكن أن يُعزى الانخفاض في Ea إلى زيادة مواقع المعادن غير المشبعة المنسقة كأحماض لويس، والتي قد تكون قد خفضت حاجز طاقة التفاعل وسهلت هدرجة FAL.
للكحوليات كمانحات H تأثير كبير على تفاعل CTH لـ FAL. كما هو متوقع، اختلفت كفاءة التفاعل مع أنواع مختلفة من الكحوليات كما هو موضح في الشكل 4c. تم عزو الكفاءة الأعلى للتفاعل في الكحوليات الثانوية مقارنة بالكحوليات الأولية إلى انخفاض إمكاناتها الاختزالية. من ناحية أخرى، نشأت الأداء الحفاز الضعيف لـ t-BuOH من هيكله الفريد الذي يفتقر إلىمما يجعله غير قادر على المشاركة في التفاعل كمانح H.يُعزز انخفاض الحواجز الاستيريو داخل الكحوليات الأولية مقارنة بالكحوليات الثانوية والثلاثية من قدرتها النووية، مما يعزز مع حموضة HE-UiO-66 الغنية تفاعل الأسيتال في مذيب الكحول الأولي، مما يؤدي إلى توليد منتج ثانوي (مثل أسيتال الفورفورال ثنائي الميثيل). بالإضافة إلى ذلك، فإن إطالة سلسلة الكربون في الكحوليات الأولية تزيد من الحواجز الاستيريو، مما يعيق تفاعل الأسيتال. نتيجة لذلك، يتم الحصول علىالمنتجات الثانوية فقط في المذيبات الميثانولية.باختصار، تلعب الحواجز الاستيريو وإمكانات الاختزال للكحوليات دورًا محوريًا في التأثير على نقل الهيدروجين وتفاعلات الأسيتال. تم اختيار 2-PrOH و2-BuOH كمانحات H تمثيلية لعملية الهدرجة.
استلهمًا من النتائج المرضية في تفاعل CTH لـ FAL، تم توسيع HE-UiO-66 لتفاعل CTH للكربونيلات الأخرى المشتقة من الكتلة الحيوية وكذلك الألدهيدات والكيتونات التمثيلية (الجدول 1). أظهر HE-UiO-66 نشاطًا حفازًا بارزًا لتحويل جميع الكربونيلات إلى كحولياتها المقابلة. أظهرت الألدهيدات تفاعلية أعلى من الكيتونات، وهو ما يُعزى إلى انخفاض الحواجز الاستيريو لمجموعة الألدهيد. بشكل خاص، انخفضت تفاعلية الركائز المستبدلة مقارنة بالركائز غير المستبدلة، مع ملاحظة أقل تفاعلية في الركائز التي تحتوي على مجموعات مانحة للإلكترونات. على سبيل المثال، يظهر FAL نشاطًا حفازًا متفوقًا.
الشكل 4 | أنشطة CTH للحفازات التي تم تحضيرها. أ تفاعل CTH لـ FAL على مختلف الحفازات عندلمدة ساعتين، (ب) تغير أداء HE-UiO-66 مع درجة حرارة التفاعل والوقت، (ج) تفاعل CTH لـ FAL على HE-UiO-66 باستخدام كحوليات مختلفة: ميثانول (MeOH)، إيثانول (EtOH)، بروبانول (1-PrOH)،
2-بروبانول (2-PrOH)، بيوتانول (1-BuOH)، 2-بيوتانول (2-BuOH)، و تيرت-بيوتانول (tBuOH)، (د) Ea لمختلف UiO-66 لتفاعل CTH لـ FAL، (هـ) إعادة الاستخدام و (و) تجربة تسرب HE-UiO-66 لتفاعل CTH لـ FAL. الشروط: FAL (0.17 ممول)، الحفازات (25 ملغ)،ولمدة ساعتين. الخصائص مقارنةً بالفورفورالات المستبدلة الأخرى مثل 5-ميثيلفورفورال و5-هيدروكسي ميثيلفورفورال المشتقة من الكتلة الحيوية (الجدول 1، الإدخالات 1-3). هذه الاتجاهات أكثر وضوحًا في حالة البنزالدهيد/ الأسيتوفينون ومشتقاتهما (الجدول 1، الإدخالات 6-11 و13-16). 4-نيتروبنزالدهيد (الجدول 1، الإدخال 9) و4-كلوروبنزالدهيد (الجدول 1، الإدخال 10) أكثر تفاعلية من غيرها من البنزالدهيدات المستبدلة (الجدول 1، الإدخالات 7، 8). وبالمثل، فإن 4-نيتروبنزوفيكون (الجدول 1، الإدخال 16) أكثر نشاطًا من غيرها من البنزوفيكونات المستبدلة (الجدول 1، الإدخالات 14، 15). تشير هذه النتائج إلى أن التأثير السلبي للمجموعات السالبة الشحنة أقل أهمية من تأثير المجموعات المانحة للإلكترونات. بسبب تأثير الترافق لـالرابطة، يظهر السينامالديهيد (الجدول 1، الإدخال 5) تفاعلية أقل من 3-فينيل بروبانال (الجدول 1، الإدخال 4). علاوة على ذلك، تظهر الكيتونات الأليفاتية (الجدول 1، الإدخال 12) تفاعلية أعلى بكثير من الكيتونات العطرية (الجدول 1، الإدخالات 13-16). في الختام، تتحدد تفاعلية الركائز من خلال كثافة سحابة الإلكترون والازدحام الفراغي لمجموعة الكربونيل. الركائز ذات كثافات سحابة الإلكترون الأعلى تظهر نشاطًا مخفضًا. بالإضافة إلى ذلك، undergo حمض الليفولينيك المشتق من الكتلة الحيوية تحولًا فعالًا من خطوتين إلى الفاليرولاكتون: هدرجة مجموعة الكربونيل تليها الاسترة داخل الجزيئية أو التحويل الاستري (الجدول 1، الإدخال 17). تسلط هذه النتائج الضوء على الإمكانات الكبيرة لـ HE-UiO-66 في تفاعلات CTH في التخليق العضوي وتحويل الكتلة الحيوية. من أجل تقييم إمكانيات تطبيق المحفز في تفاعل CTH لـ FAL، أسفر تجربة زيادة الحجم مع 10 ملليمول من FAL عن عوائد FOL متسقة تفوقأكثر من 8 دورات عندلـ (الشكل S11). يظهر HE-UiO-66 إمكانيات واعدة لإنتاج FOL على نطاق واسع من FAL.
بعد ذلك، تم تقييم إمكانية إعادة استخدام وتنوع HE-UiO-66 من خلال تجارب التسرب وإعادة التدوير. أظهر المحفز قابلية إعادة استخدام ملحوظة، حيث حافظ على أدائه التحفيزي لمدة لا تقل عن 10 دورات (الشكل 4e). بالإضافة إلى ذلك، احتفظت المحفزات المستخدمة بخصائص فيزيائية وكيميائية مستقرة، كما هو موضح في الأشكال S12 و S13، مما يبرز استقرارها الممتاز طوال تفاعل CTH. لتأكيد تنوع المحفز، تم إجراء تجربتين متوازيتين مع HE-UiO-66، حيث تم فصل المحفز في واحدة منها بعد 1.5 ساعة. بعد الترشيح، لم يُلاحظ أي تفاعل إضافي في النظام (الشكل 4f). أظهرت التحليلات عدم الكشف عن، وTi ضمن حد ICP في المحلول المصفى بعد التفاعل، مما يشير إلى عدم تسرب هذه العناصر. هذه النتيجة
الجدول 1 | تفاعل CTH لمركبات الكربونيل المختلفة على HE-UiO-66
دخول
المتفاعل
المنتج الرئيسي
درجة الحرارة
الوقت. (س)
نسبة التحويل (%)
العائد (%)
1
١٢٠
٣
95.0
91.5
2
130
٣
92.6
٨٨.٠
٣
١٣٠
٣
٨٨.٩
٨٣.٧
٤
١٢٠
٣
>99
>99
٥
130
٣
85.4
٨٣.٩
٦
١٢٠
2
98.0
97.4
٧
١٢٠
٣
97.4
96.7
٨
١٢٠
٣
93.7
72.1
9
١٢٠
٣
>99
>99
10
١٢٠
٣
>99
>99
11
١٢٠
٣
90.4
٨٨.٩
12
150
٣
>99
>99
١٣
150
٣
95.7
93.4
14
150
٣
٨٧.٢
٨٦.١
15
150
٣
87.8
٨٦.٠
16
150
٣
94.3
92.8
17
١٦٠
٥
95.4
90.9
الشروط: المحفزركيزة. يعد دليلاً إضافياً على التباين والاستقرار لـ HE-UiO-66.
رؤى ميكانيكية مستندة إلى حسابات حسابية ووضع علامات نظيرية
من المعروف جيدًا أن تفاعلات CTH الفعالة تتطلب تنشيط مجموعة الكربونيل في المتفاعل ومجموعة الهيدروكسيل في الكحول (كمانح للهيدروجين).محفزات قائمة علىتعتبر الأنواع كمواقع حمض لويس دورًا حاسمًا في تنشيط مجموعة الكربونيل، بينما تنشط مواقع قاعدة لويس مجموعة الهيدروكسيل في مصدر الهيدروجين لتسهيل نقل الهيدروجين.المحفز المحسن أداء HE-UiO-66 يُعزى بشكل رئيسي إلى تنظيمتعمل كمواقع حمض لويس وزيادة في تركيز مواقع قاعدة لويس. إن الزيادة في الإنتروبيا التكوينية تسهل توليد مواقع معدنية غير مشبعة بالتنسيق الناتجة عن العيوب بكميات كبيرة، مما يزيد في الوقت نفسه من نقص الإلكترونات في. هذه العوامل تعزز وتزيد من مواقع الأحماض لويس. في الوقت نفسه، تصاحب مواقع القواعد لويس زيادة في مواقع الأحماض لويس. تتوفر مناقشة شاملة لهذه القضايا في التحليلات النوعية والكمية للمواقع النشطة في HE-UiO-66. علاوة على ذلك، أكدت طيف ATR-IR أن HE-UiO-66 حسنت من كفاءة التحفيز لتفكك 2-PrOH و
الشكل 5 | حسابات نظرية الكثافة الوظيفية للنماذج النظرية ذات الصلة. أ كثافة الحالة الجزئية لحزام d لذرات Zr و Hf في HE-UiO-66 المثالي والعيوب. ب طاقات الامتزاز المحسوبة لجزيئات FAL و 2-PrOH من خلال اختيار Zr و Hf كمواقع امتزاز في HE-UiO-66 المثالي والعيوب، على التوالي. ج المحسوبة نموذج الامتزاز وطاقة الامتزاز لـ FAL و 2-PrOH المزدوجين على HE-UiO-66 المثالي والعيوب. د ملفات الطاقة الحرة للهدرجة المحسوبة لـ FAL على سطح HE-UiO-66 المثالي والعيوب.
تنشيط ركيزة FAL من خلال الجمع الفعال مع و الروابط (الشكل 3ج، د). باختصار، يتم التحكم في مواقع حمض-قاعدة لويس بشكل مباشر بواسطة العيوب الناتجة عن تشوه الشبكة في HE-UiO-66، مما يعزز امتصاص وتفعيل جزيئات الركيزة وفي النهاية يحسن الأداء التحفيزي.
في إطار التحقيق في تأثير المواقع غير المشبعة على تفاعل الامتزاز والتفاعل بين جزيئات الركيزة وسطح HE-UiO-66، تم تطبيق نظرية الكثافة الوظيفية (DFT) لتحسين الهندسة، وكثافة الحالة المتوقعة (PDOS)، وطاقة الامتزاز، وحساب ملفات الطاقة الحرة لنقل الهيدروجين. يتم عرض تحسين الهيكل لـ HE-UiO-66 مع وبدون عيوب في الشكل S14. نظرًا لـ هو الموقع النشط الرئيسي لتفاعل CTH، مقارنةً بـ PDOS لفرقة d لذرات Zr و Hf في HE-UiO-66 المثالي والعيوب تكشف عن الأصل الذري وراء النشاط المعزز لتفاعل CTH لمواقع التنسيق غير المشبعة (الشكل 5a). بالنسبة لـ HE-UiO-66 المثالي، تمتلك كل من ذرات Hf و Zr استقرارًا كيميائيًا عاليًا، حيث تكون كثافة الحالات عند مستوى فيرمي أقل من مستوى فيرمي بسبب اكتمال الروابط. بالنسبة لذرات Hf و Zr في HE-UiO-66 المعيب، فإن غياب الروابط يتسبب في تجاوز كثافة الحالات بالقرب من مستوى فيرمي لمستوى فيرمي، مما يستلزم احتجاز جزيئات صغيرة لتعزيز الاستقرار الكيميائي. من حيث الامتزاز، ترتبط المدارات الفارغة في HE-UiO-66 المعيب بإلكترونات الأكسجين في جزيئات صغيرة مثل الكحول الأيزوبروبيلي والفورفورال، مما يحسن أداء الامتزاز. على النقيض من ذلك، فإن HE-UiO-66 المثالي مع ذرات Hf و Zr المربوطة بالكامل لديها قدرة امتزاز ضعيفة. على الرغم من أن هذه الذرات تحتوي على مدارات فارغة فوق مستوى فيرمي، إلا أن مستويات طاقتها الأعلى والمسافة الكبيرة من مستوى فيرمي تجعل المشاركة في التفاعلات تحديًا. يكشف تحليل تكوين الامتزاز أن أطوال الروابط لـ و معيبأقصر من تلك الموجودة في HE-UiO-66 المثالي (الجدول S4). تنشأ هذه الفجوة من الكامل تنسيق زركونيوم (Zr) وهفنيوم (Hf) في الأخير، مما يحد من استيعاب ذرات الأكسجين في جزيئات الركيزة للارتباط بمواقع المعادن. على العكس من ذلك، في HE-UiO-66 المعيب، يمكن لذرات Zr وHf امتصاص جزيئات صغيرة وتحقيق تنسيق كامل. علاوة على ذلك، تم تقديم التكوينات المستقرة حرارياً لامتصاص FAL و2-PrOH الماصة على مواقع Zr وHf في HE-UiO-66 المثالي والمعيب (الأشكال S15-S18). طاقات الامتصاص (تشير النتائج التي تم الحصول عليها إلى أن الأكسجين الكربوني في FAL والأكسجين الهيدروكسي في 2-PrOH لهما ميل أقوى نحولـ FAL و -0.447 إلكترون فولت لـ 2-PrOH)لـ FAL و -0.404 إلكترون فولت لـ 2 مواقع العيوب مقارنة بالمثالي UiO-66 (الشكل 5ب). تشير نتائج الامتزاز إلى تفاعل أقوى ونسبة ارتباط بين جزيئات الركيزة و HE-UiO-66 المعيب مقارنة بـ HE-UiO-66 المثالي، وهو ما يتماشى مع تحليلات PDOS و ATR-IR. وبالمثل، بالنسبة للامتزاز المشترك لـ FAL و 2-PrOH، فإن HE-UiO-66 المعيب أيضًا أقوى (الشكل 5ج). للحصول على رؤى ذرية أعمق حول النشاط التحفيزي المعزز لـ HE-UiO-66 المعيب مع مواقع تنسيق غير مشبعة لتفاعل CTH لـ FAL، تم محاكاة مسار نقل الهيدروجين وعرض توزيع طاقة التفاعل في الشكل 5د، الشكل S19 والجدول S5. بالنسبة لـ HE-UiO-66 المعيب، فإن طاقة نقل الهيدروجين الحرة ( ) لـ FAL (IS ) هي 0.99 eV، وهو أقل من تلك الخاصة بـ HE-UiO-66 المثالي (1.43 eV)، مما يشير إلى حاجز طاقة أصغر لـ HE-UiO-66 المعيب. كما هو متوقع، أكدت نتائج DFT أن وجود مواقع تنسيق غير مشبعة ينظم بشكل فعال الهيكل الإلكتروني لـ HE-UiO-66، مما يعزز التفاعل والارتباط مع جزيئات الركيزة ويخفض حاجز الطاقة لنقل الهيدروجين، وبالتالي يعزز التفاعل.
آلية التحفيز لتفاعل CTH لـ HE-UiO-66، سواء كانت تتضمن نقل هيدروجين مباشر أو غير مباشر، لا تزال غير واضحة . تم إجراء تحقيق في الآلية باستخدام
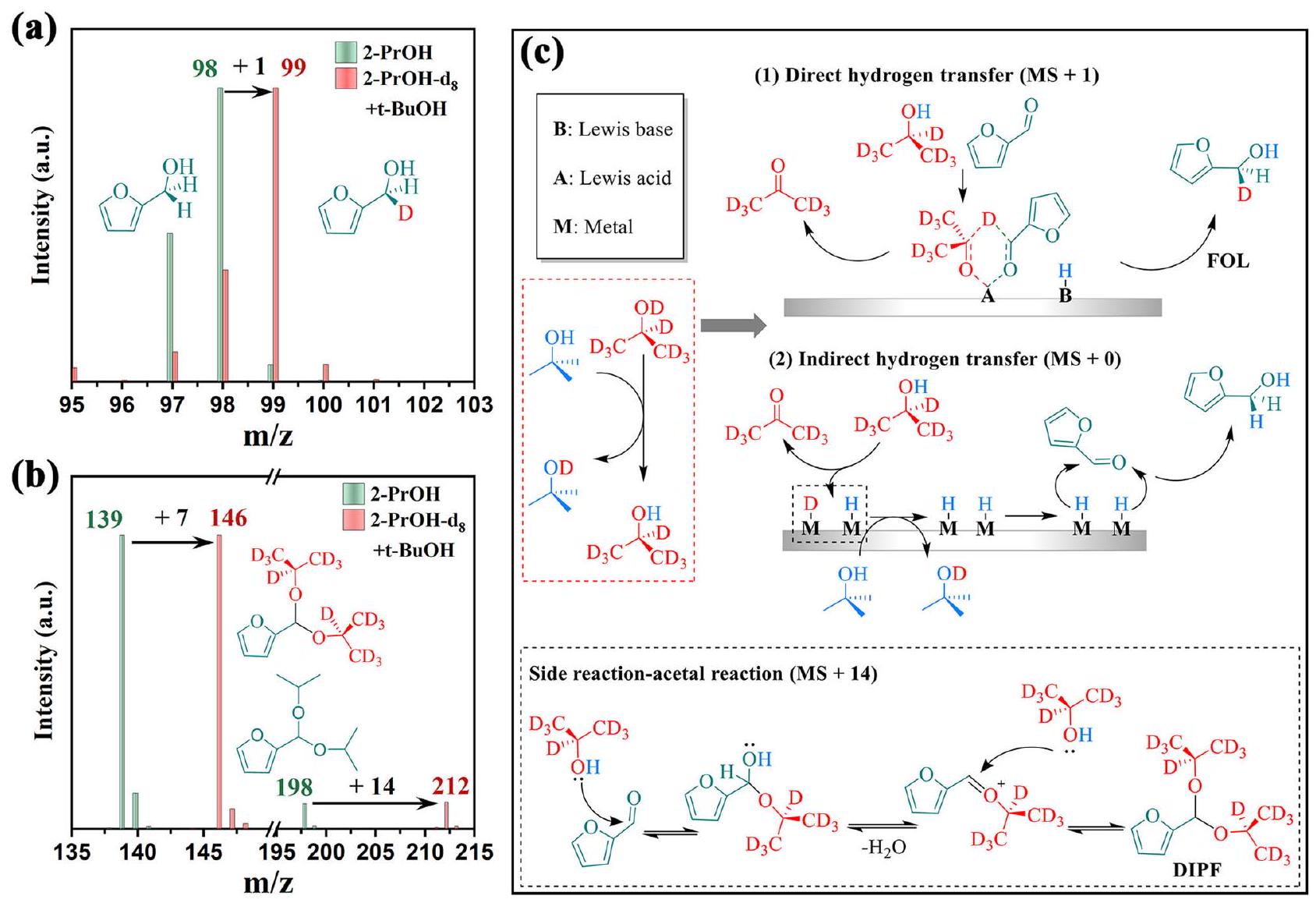
الشكل 6 | تحقيقات الآلية. MS لـ (أ) FOL و (ب) DIPF في مذيبات مختلفة (2-PrOH و 2-PrOH- -بيوتانول)؛ (ج) مخطط لآلية التفاعل عبر نقل هيدروجين مباشر أو غير مباشر وتفاعل جانبي. الشروط: FAL (0.17 مللي مول)، المحفزات (25 ملغ)، المذيبات (5 مل)، و 2 ساعة.
طريقة GC-MS مع 2-PrOH- ومع ذلك، فإن استبدال 2-PrOH- بـ 2-PrOH- لم يوفر وضوحًا حول آلية التفاعل بين نقل الهيدروجين المباشر وغير المباشر (الشكل S20). للتفريق بين عمليات نقل الهيدروجين، تم استخدام خليط من و t – BuOH بنسبة تم استخدامه. على الرغم من أن t -BuOH يفتقر إلى H ولا يساهم كمانح H، إلا أنه يمكنه تبادل ذرات H مع معظم مجموعات 2-PrOH- ورابطة الهيدروجين الديوتيريوم للمعادن (M-D) لتشكيل OH و M-H على التوالي. الأول يشكل 2-PrOH- ، مما يعمل كمانح الهيدروجين الرئيسي لتفاعل CTH لـ FAL (الشكل 6ج). يجب أن تظهر الكحوليات المرغوبة تحولًا في MS بمقدار 1 amu (MS ) عبر نقل هيدروجين مباشر أو عدم تحول (MS ) عبر نقل هيدروجين غير مباشر. كما هو متوقع، كشفت تحليل GC-MS بشكل قاطع عن تحول في الكتلة بمقدار 1 amu، مما يؤكد تحول FAL إلى FOL عبر HE-UiO-66 بعد مسار نقل الهيدروجين المباشر (الشكل 6أ). من المثير للاهتمام، أن المنتج الثانوي 2-(دييسوبروبوكسي ميثيل) فيوران (DIPF) يظهر تحولًا بمقدار 14 amu (MS ) في التفاعل الذي تم إجراؤه في مذيبات 2-PrOH- -بيوتانول (الشكل 6ب). وفقًا لخصائص HE-UiO-66 مع مواقع حمضية وفيرة وتقارير سابقة ، يتم تشكيل DIPF من تفاعل الأسيتال بين FAL و 2-PrOH-d (الشكل 6ج). يمكن التحقق من ذلك بشكل أكبر من خلال التجارب المذكورة أعلاه مع مصادر كحول مختلفة. إن انخفاض الانتقائية ناتج عن قوة تبرع الهيدروجين والازدحام الفراغي لـ 2-PrOH (الشكل 4ج). يمكن شرح عملية التفاعل بإيجاز على النحو التالي: تتفاعل مجموعة الألدهيد، كونها قطبية للغاية، بسهولة مع النيوكليوفيلات بسبب الإيجابية الكهربائية القوية للكربون. يهاجم الأكسجين النيوكليوفيلي في مجموعة الهيدروكسيل في الكحول الكربون في مجموعة الكربونيل بزوج الإلكترونات الوحيد لتشكيل هيمياستال. مجموعة -OH في الهيمياستال غير مستقرة وعرضة للتكثيف الجفاف مع جزيء كحول آخر لتشكيل
أسيتال. لذلك، يظهر تفاعل الأسيتال لـ FAL مع 2-PrOH- تحولًا بمقدار 14 amu، مما يوفر أول نظرة على عملية التفاعل الجانبي من خلال تتبع نظير الديوتيريوم.
نقاش
باختصار، لقد استخدمنا استراتيجية هندسة الإنتروبيا لتخليق HE-UiO-66، وهو مادة MOF عالية الإنتروبيا قادرة على تحفيز تفاعلات CTH بكفاءة تحت ظروف معتدلة. من الجدير بالذكر أن تأثير تشوه الشبكة للمواد عالية الإنتروبيا يمكن استغلاله ليس فقط لتحفيز العيوب، مما يولد تركيزًا كبيرًا من مواقع المعادن غير المشبعة حتى وزيادة العجز الإلكتروني في ، ولكن أيضًا لزيادة طاقات تنشيط الانتشار للذرات في الشبكة ومنع نمو حجم الجسيمات ، مما كان مفيدًا لتحسين كفاءة نقل الكتلة لجزيئات الركيزة. إن الزيادة الناتجة في وفرة وقوة مواقع الحمض والقاعدة لويس حسنت بشكل كبير الأداء التحفيزي لتفاعل CTH. أكدت كل من النتائج التجريبية والحسابات النظرية أن وجود مواقع تنسيق غير مشبعة ينظم بشكل فعال الهيكل الإلكتروني لـ HE-UiO-66، مما يعزز تفاعله وارتباطه مع جزيئات الركيزة ويخفض حاجز الطاقة لنقل الهيدروجين. تم تأكيد مسار نقل الهيدروجين المباشر لتفاعل CTH لـ FAL بواسطة MS المعلم بالنظائر، جنبًا إلى جنب مع مسار التحول للتفاعل الجانبي. إن زيادة الإنتروبيا التوافقية تعزز تحسين الخصائص الفيزيائية والكيميائية لـ HE-UiO-66 (الحجم، الاستقرار، العيوب، مواقع التنسيق غير المشبعة وخصائص الحمض والقاعدة لويس)، مما يوفر منظورًا نحو توسيع وظائف مواد MOF.
طرق
تحضير المحفز
تم تخليق HE-UiO-66 بواسطة الطريقة الحرارية المائية. على وجه التحديد، ، ، حمض البنزين-1،4-ديكربوكسيلي (BDC) (0.665 غرام) و تم إذابته في 15 مل من N,Ndimethylformamide (DMF) لتشكيل محلول متجانس. تم نقل المحلول إلى أنبوب ضغط ACE سعة 48 مل وتفاعل عند لمدة 24 ساعة. بعد التبريد إلى درجة حرارة الغرفة، تم جمع الرواسب بواسطة الطرد المركزي، وغسلها عدة مرات بـ DMF ثم مع الميثانول. تم تجفيف المنتج الناتج تحت الفراغ عند طوال الليل وتم تسميته ZrHfCeSnTi (HE-UiO-66). للمقارنة، تم استخدام طريقة مماثلة لتخليق سلسلة من UiO-66 مع إنتروبيا تكوين مخفضة (إجمالي كمية المولي من أيونات المعادن هو 4 مللي مول)، بما في ذلك أحادي ، ثنائي ZrHf، ثلاثي ZrHfCe، رباعي ZrHfCeSn، من بينها Sn و Ti أحاديان غير متبلورين.
دراسة النشاط التحفيزي
تم إجراء تفاعل CTH لمركب الكربونيل في أنبوب ضغط ACE سعة 15 مل مزود بخلاط مغناطيسي، مغمور في حمام زيت يتحكم في درجة الحرارة. تم خلط 0.17 مللي مول من الركيزة، 5 مل من و 25 ملغ من المحفز في المفاعل. بعد الإغلاق، تم تسخين المفاعل إلى درجة الحرارة المطلوبة ( ) لمدة زمنية محددة ( ) مع التحريك بسرعة 500 دورة في الدقيقة. بعد التفاعل، تم تحليل السائل كميًا باستخدام TRACE ISQ GCMS (عمود TR-WAX-MS ). بدأ برنامج درجة الحرارة عند لمدة دقيقة واحدة، ثم زاد إلى عند لكل دقيقة وتم الاحتفاظ به لمدة دقيقتين. بالنسبة لتجربة CTH الموسعة لـ FAL، تم استخدام أنبوب ضغط ACE سعة 75 مل. في هذه الحالة، تم تحميل 10 مللي مول من FAL، 40 مل من 2-PrOH و 1 غرام من المحفز في أنبوب الضغط، الذي تم إغلاقه وتسخينه إلى لمدة 2 و 4 ساعات. تم تقييم قابلية إعادة استخدام محفز HE-UiO-66 في 10 جولات تفاعل متتالية عند لمدة ساعتين لكل منها. بعد كل جولة، تم فصل المحفز المستخدم بواسطة الطرد المركزي، وغسله بالتتابع مع الإيثانول والأسيتون، وتجفيفه تحت الفراغ عند لمدة ساعتين ثم إعادة استخدامه في الجولة التالية. تم تكرار جميع التجارب المذكورة أعلاه ثلاث مرات للتحقق من قابلية تكرار النتائج التجريبية.
حسابات DFT
تم إجراء حسابات تحسين الهندسة المستندة إلى المبادئ الأولى ضمن نظرية الكثافة الوظيفية (DFT)، باستخدام حزمة محاكاة فيينا Ab Initio (VASP) . تم استخدام طريقة PAW (الموجة المعززة لمشاريع النواة المجمدة) لوصف التفاعل بين النوى الذرية وكثافة الإلكترونات التكافؤية . تم تقريب إمكانات التبادل والتفاعل باستخدام دالة PerdewBurke Ernzerhof (PBE) ضمن تقريب التدرج العام (GGA) . لأخذ تفاعلات Van der Waals (vdW) في الاعتبار، تم تنفيذ مخطط DFT-D3 المصحح للتشتت . تم تعيين طاقة قطع الموجة الطائرة إلى 450 إلكترون فولت. بالنسبة لتحسين الأيونات، تم استخدام خوارزمية التدرج المترافق، مع عتبة تقارب قدرها ذرة للاسترخاء الإلكتروني و لقوة هيلمان-فاينمان على كل ذرة. بالنظر إلى النظام غير الدوري، تم أخذ عينات من منطقة بريلوين في الفضاء العكسي باستخدام مخطط مونكهورست-باك مع -نقاط للشبكات لكل من تحسين الهندسة وحسابات الهيكل الإلكتروني . تم تحديد حاجز طاقة التفاعل باستخدام طريقة الصورة المتسلقة للحزام المرن المدفوع (CI-NEB) . تم حساب طاقة الامتزاز () للمواد الممتزة بواسطة:
حيث ، و تمثل طاقة الأنظمة الكلية بعد الامتزاز، أسطح المحفزات والمواد الممتزة، على التوالي.
توفر البيانات
البيانات التي تدعم الرسوم البيانية في المقالة متاحة ضمن هذه الورقة والمعلومات التكميلية المقابلة. النتائج الأخرى لهذه الدراسة متاحة من المؤلفين المقابلين عند الطلب. يتم توفير بيانات المصدر مع هذه الورقة.
References
Sarkar, A. et al. High-entropy oxides: fundamental aspects and electrochemical properties. Adv. Mater. 31, e1806236 (2019).
Yao, Y. et al. High-entropy nanoparticles: synthesis-structureproperty relationships and data-driven discovery. Science 376, eabn3103 (2022).
Ma, Y. et al. High-entropy energy materials: challenges and new opportunities. Energy Environ. Sci. 14, 2883-2905 (2021).
Pan, Y., Liu, J.-X., Tu, T.-Z., Wang, W. & Zhang, G.-J. High-entropy oxides for catalysis: a diamond in the rough. Chem. Eng. J. 451, 138659 (2023).
Wei, J. et al. Deep eutectic solvent assisted facile synthesis of lowdimensional hierarchical porous high-entropy oxides. Nano Res. 15, 2756-2763 (2021).
Cantor, B., Chang, I. T. H., Knight, P. & Vincent, A. J. B. Microstructural development in equiatomic multicomponent alloys. Mater. Sci. Eng. A 375-3377, 213-218 (2004).
Yeh, B. J.-W. et al. Nanostructured high-entropy alloys with multiple principal elements: novel alloydesign concepts and outcomes. Adv. Eng. Mater. 6, 299-303 (2004).
Feng, D. et al. Holey lamellar high-entropy oxide as an ultra-highactivity heterogeneous catalyst for solvent-free aerobic oxidation of benzyl alcohol. Angew. Chem. Int. Ed. 59, 19503-19509 (2020).
Gu, K. et al. Defect-rich high-entropy oxide nanosheets for efficient 5-hydroxymethylfurfural electrooxidation. Angew. Chem. Int. Ed. 60, 20253-20258 (2021).
Ma, Y. et al. Resolving the role of configurational entropy in improving cycling performance of multicomponent hexacyanoferrate cathodes for sodium-ion batteries. Adv. Funct. Mater. 32, (2022).
Nguyen, T. X., Su, Y. H., Lin, C. C., Ruan, J. & Ting, J. M. A new high entropy glycerate for high performance oxygen evolution reaction. Adv. Sci. 8, 2002446 (2021).
Fereja, S. L. et al. High-entropy oxide derived from metal-organic framework as a bifunctional electrocatalyst for efficient urea oxidation and oxygen evolution reactions. ACS Appl. Mater. Interfaces 14, 38727-38738 (2022).
Gonçalves, J. M. & Ruiz-Montoya, J. G. Emerging high-entropy coordination compounds and their derivatives for energy application. J. Mater. Chem. A 11, 20872-20885 (2023).
Xu, S. et al. High-entropy metal-organic framework arrays boost oxygen evolution electrocatalysis. J. Phys. Chem. C 126, 14094-14102 (2022).
Ma, Y. et al. High-entropy metal-organic frameworks for highly reversible sodium storage. Adv. Mater. 33, e2101342 (2021).
Xu, W. et al. Entropy-driven mechanochemical synthesis of polymetallic zeolitic imidazolate frameworks for fixation. Angew. Chem. Int. Ed. 58, 5018-5022 (2019).
Zhao, X. et al. Ambient fast, large-scale synthesis of entropystabilized metal-organic framework nanosheets for electrocatalytic oxygen evolution. J. Mater. Chem. A 7, 26238-26242 (2019).
Kar, A. K. et al. Unveiling and understanding the remarkable enhancement in the catalytic activity by the defect creation in UIO66 during the catalytic transfer hydrodeoxygenation of vanillin with isopropanol. Appl. Catal. B: Environ. 325, 122385 (2023).
Valekar, A. H. et al. Catalytic transfer hydrogenation of furfural to furfuryl alcohol under mild conditions over Zr-MOFs: exploring the role of metal node coordination and modification. ACS Catal. 10, 3720-3732 (2020).
Feng, X. et al. Engineering a highly defective stable UiO-66 with tunable lewis- bronsted acidity: the role of the hemilabile linker. J. Am. Chem. Soc. 142, 3174-3183 (2020).
Ji, P. et al. Strongly Lewis acidic metal-organic frameworks for continuous flow catalysis. J. Am. Chem. Soc. 141, 14878-14888 (2019).
Masoomi, M. Y., Morsali, A., Dhakshinamoorthy, A. & Garcia, H. Mixed-metal MOFs: unique opportunities in metal-organic framework (MOF) functionality and design. Angew. Chem. Int. Ed. 58, 15188-15205 (2019).
Dai, F., Zhou, S., Qin, X., Liu, D. & Qi, H. Surfactant-assisted synthesis of mesoporous hafnium- imidazoledicarboxylic acid hybrids for highly efficient hydrogen transfer of biomass-derived carboxides. Mol. Catal. 479, 110611 (2019).
SergioRojas-Buzo, García-García, P. & Corma, A. Catalytic transfer hydrogenation of biomass-derived carbonyls over hafnium-based metal-organic frameworks. ChemSusChem 11, 432-438 (2018).
Zhong, L. et al. Hydrogenation of -unsaturated aldehydes over defective UiO-66 with frustrated Lewis pairs: Modulation of densities of defect sites via tailoring ligand-vacancies. Appl. Catal. B: Environ. 342, 123421 (2024).
Qiu, M., Guo, T., Xi, R., Li, D. & Qi, X. Highly efficient catalytic transfer hydrogenation of biomass-derived furfural to furfuryl alcohol using UiO-66 without metal catalysts. Appl. Catal. A: Gen. 602, 117719 (2020).
Chen, X. et al. Tuning node defects as catalytic sites in the metal-organic framework hcp UiO-66. ACS Catal. 10, 2906-2914 (2020).
Tan, K. et al. Defect termination in the UiO-66 family of metalorganic frameworks: the role of water and modulator. J. Am. Chem. Soc. 143, 6328-6332 (2021).
Cho, K. Y. et al. Facile control of defect site density and particle size of UiO-66 for enhanced hydrolysis rates: insights into feasibility of -based metal-organic framework (MOF) catalysts. Appl. Catal. B: Environ. 245, 635-647 (2019).
Feng, Y., Chen, Q., Jiang, M. & Yao, J. Tailoring the properties of UiO66 through defect engineering: a review. Ind. Eng. Chem. Res. 58, 17646-17659 (2019).
Bakuru, V. R., Churipard, S. R., Maradur, S. P. & Kalidindi, S. B. Exploring the Brønsted acidity of UiO-66 (Zr, Ce, Hf) metal-organic frameworks for efficient solketal synthesis from glycerol acetalization. Dalton Trans. 48, 843-847 (2019).
Bakuru, V. R., Churipard, S. R., Maradur, S. P. & Kalidindi, S. B. Exploring the Bronsted acidity of UiO-66 (Zr, Ce, Hf) metal-organic frameworks for efficient solketal synthesis from glycerol acetalization. Dalton Trans. 48, 843-847 (2019).
Wang, W. et al. Chemical bonding of from Zr and Ce single atoms for efficient photocatalytic reduction of under visible light. ACS Catal. 13, 4597-4610 (2023).
Sun, Y. et al. Defect engineering of ceria nanocrystals for enhanced catalysis via a high-entropy oxide strategy. ACS Cent. Sci. 8, 1081-1090 (2022).
Chen, D., Yang, D., Wang, Q. & Jiang, Z. Effects of boron doping on photocatalytic activity and microstructure of titanium dioxide nanoparticles. Ind. Eng. Chem. Res. 45, 4110-4116 (2006).
Feng, N. et al. Boron environments in B-doped and (B, N)-codoped photocatalysts: a combined solid-state NMR and theoretical calculation study. J. Phys. Chem. C. 115, 2709-2719 (2011).
Tomboc, G. M. et al. Stabilization, characterization, and electrochemical applications of high-entropy oxides: critical assessment of crystal phase-properties relationship. Adv. Funct. Mater.
32, (2022).
Zhai, Y., Ren, X., Wang, B. & Liu, S. High-entropy catalyst-a novel platform for electrochemical water splitting. Adv. Funct. Mater. 32 (2022).
Yeh, J.-W., Chang, S.-Y., Hong, Y.-D., Chen, S.-K. & Lin, S.-J. Anomalous decrease in X-ray diffraction intensities of alloy systems with multi-principal elements. Mater. Chem. Phys. 103, 41-46 (2007).
Ma, M. et al. Tandem catalysis of furfural to -valerolactone over polyoxometalate-based metal-organic frameworks: exploring the role of confinement in the catalytic process. Renew. Energy 227, 120474 (2024).
Li, M. et al. Defect-rich hierarchical porous UiO-66(Zr) for tunable phosphate removal. Environ. Sci. Technol. 55, 13209-13218 (2021).
Morterra, C., Giamello, E., Orio, L. & Volante, M. Formation and reactivity of centers at the surface of vacuum-activated monoclinic zirconia. J. Phys. Chem. 94, 3111-3116 (1990).
He, Y., Li, C., Chen, X.-B., Shi, Z. & Feng, S. Visible-light-responsive UiO-66(Zr) with defects efficiently promoting photocatalytic CO2 reduction. ACS Appl. Mater. Interfaces 14, 28977-28984 (2022).
Fan, Y. et al. Low-temperature catalytic degradation of chlorinated aromatic hydrocarbons over bimetallic catalysts. Chem. Eng. J. 414, 128782 (2021).
Kuwahara, Y., Kango, H. & Yamashita, H. Catalytic transfer hydrogenation of biomass-derived levulinic acid and its esters to valerolactone over sulfonic acid-functionalized UiO-66. ACS Sustain. Chem. Eng. 5, 1141-1152 (2016).
Xiang, W. et al. The metal-organic framework UiO-66 with missinglinker defects: a highly active catalyst for carbon dioxide cycloaddition. Appl. Energy 277, 115560 (2020).
Ma, M. et al. Stabilization of unique species in nanocrystals for unprecedented catalytic transfer hydrogenation reaction. Appl. Catal. B: Environ. 350, 123905 (2024).
Zhao, R., Kasipandi, S., Shin, C.-H. & Bae, J. W. Catalytic conversion of biomass-derived levulinic acid to -valerolactone over amphoteric zirconium hydroxide. ACS Catal. 13, 12711-12722 (2023).
Driscoll, D. M. et al. Characterization of undercoordinated Zr defect sites in UiO-66 with vibrational spectroscopy of adsorbed CO. J. Phys. Chem. C 122, 14582-14589 (2018).
Mariscal, R., Maireles-Torres, P., Ojeda, M., Sádaba, I. & López Granados, M. Furfural: a renewable and versatile platform molecule for the synthesis of chemicals and fuels. Energy Environ. Sci. 9, 1144-1189 (2016).
Nie, R. et al. Recent advances in catalytic transfer hydrogenation with formic acid over heterogeneous transition metal catalysts. ACS Catal. 11, 1071-1095 (2021).
Ma, M. et al. Simple basic zirconium carbonate: low temperature catalysis for hydrogen transfer of biomass-derived carboxides. Green. Chem. 21, 5969-5979 (2019).
Ma, M. et al. Humins with efficient electromagnetic wave absorption: a by-product of furfural conversion to isopropyl levulinate via a tandem catalytic reaction in one-pot. Chem. Eur. J. 27, 12659-12666 (2021).
Ma, M. et al. Magnetic nanoparticles as easily separable catalysts for efficient catalytic transfer hydrogenation of biomassderived furfural to furfuryl alcohol. Appl. Catal. A: Gen. 602, 117709 (2020).
Ma, M. et al. Creation of surface frustrated Lewis pairs on highentropy spinel nanocrystals that boosts catalytic transfer hydrogenation reaction. Chem. Eng. J. 470, 144291 (2023).
Fang, W. & Riisager, A. Efficient valorization of biomass-derived furfural to fuel bio-additive over aluminum phosphate. Appl. Catal. B: Environ. 298, 120575 (2021).
An, Z. & Li, J. Recent advances in the catalytic transfer hydrogenation of furfural to furfuryl alcohol over heterogeneous catalysts. Green. Chem. 24, 1780-1808 (2022).
Zhou, S. et al. Sustainable hydrothermal self-assembly of hafnium-lignosulfonate nanohybrids for highly efficient reductive
upgrading of 5-hydroxymethylfurfural. Green. Chem. 21, 1421-1431 (2019).
Zhou, S. et al. Zirconium-lignosulfonate polyphenolic polymer for highly efficient hydrogen transfer of biomass-derived oxygenates under mild conditions. Appl. Catal. B: Environ. 248, 31-43 (2019).
An, Z. et al. Highly active, ultra-low loading single-atom iron catalysts for catalytic transfer hydrogenation. Nat. Commun. 14, 6666 (2023).
Ma, M. et al. One-pot Transfer hydrogenation of methyl levulinate into valerolactone and 1,4-pentanediol over in situ reduced Cu/ in 2-PrOH. ChemistrySelect 5, 1-8 (2020).
Bai, L. et al. Catalyst-free acetalization of biobased furfural into cyclic acetal fuel additives with biogenic ethylene glycol. ACS Sustain. Chem. Eng. 11, 15743-15753 (2023).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169-11186 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758-1775 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3965-3868 (1996).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188-5192 (1976).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901-9904 (2000).
الشكر والتقدير
تم دعم هذا العمل من قبل مؤسسة العلوم الطبيعية في مقاطعة جيلين، الصين (رقم 20240101166JC).
مساهمات المؤلفين
M.M. و G.T. وضعوا الفكرة وصمموا التجارب. قام M.M. و E.C. بتنفيذ معظم التجارب والتوصيفات. كتب M.M. و H.Y. و G.T. المسودة. علق جميع المؤلفين على المسودة.
يجب توجيه المراسلات والطلبات للحصول على المواد إلى جي تيان.
معلومات مراجعة الأقران تشكر مجلة Nature Communications تشونباو (تشارلز) شو والمراجعين الآخرين المجهولين على مساهمتهم في مراجعة الأقران لهذا العمل. يتوفر ملف مراجعة الأقران.
ملاحظة الناشر تظل Springer Nature محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
الوصول المفتوح هذه المقالة مرخصة بموجب رخصة المشاع الإبداعي للاستخدام غير التجاري، والتي تسمح بأي استخدام غير تجاري، ومشاركة، وتوزيع وإعادة إنتاج في أي وسيلة أو صيغة، طالما أنك تعطي الائتمان المناسب للمؤلفين الأصليين والمصدر، وتوفر رابطًا لرخصة المشاع الإبداعي، وتوضح إذا قمت بتعديل المادة المرخصة. ليس لديك إذن بموجب هذه الرخصة لمشاركة المواد المعدلة المشتقة من هذه المقالة أو أجزاء منها. الصور أو المواد الأخرى من طرف ثالث في هذه المقالة مشمولة في رخصة المشاع الإبداعي للمقالة، ما لم يُشار إلى خلاف ذلك في سطر ائتمان للمادة. إذا لم تكن المادة مشمولة في رخصة المشاع الإبداعي للمقالة واستخدامك المقصود غير مسموح به بموجب اللوائح القانونية أو يتجاوز الاستخدام المسموح به، ستحتاج إلى الحصول على إذن مباشرة من صاحب حقوق الطبع والنشر. لعرض نسخة من هذه الرخصة، قم بزيارة http:// creativecommons.org/licenses/by-nc-nd/4.0/.
(ج) المؤلفون 2025
المختبر الوطني الرئيسي للتركيب غير العضوي والكيمياء التحضيرية، كلية الكيمياء، جامعة جيلين، 130012 تشانغتشون، جمهورية الصين الشعبية.
⟶ البريد الإلكتروني: tiange@jlu.edu.cn
Entropy engineering activation of UiO-66 for boosting catalytic transfer hydrogenation
Received: 30 January 2024
Accepted: 27 August 2024
Published online: 03 January 2025
(4) Check for updates
Mingwei Ma , Enpeng Chen , Huijuan Yue , Ge Tian & Shouhua Feng
High-entropy metal-organic frameworks (HE-MOFs) hold promise as versatile materials, yet current rare examples are confined to low-valence elements in the fourth period, constraining their design and optimization for diverse applications. Here, a novel high-entropy, defect-rich and small-sized ( 32 nm ) UiO-66 (ZrHfCeSnTi HE-UiO-66) has been synthesized for the first time, leveraging increased configurational entropy to achieve high tolerance to doping with diverse metal ions. The lattice distortion of HE-UiO-66 induces high exposure of metal nodes to create coordination unsaturated metal sites with a concentration of , which increases the abundance of Lewis acid-base sites, thereby achieving a significant improvement in the performance of the catalytic transfer hydrogenation (CTH) reaction. Systematic investigation manifests that the special electronic structure of HE-UiO-66 enhances the interaction and bonding with substrate molecules and reduces the energy barrier of the hydrogen transfer process. Our approach offers a new strategy for constructing coordination unsaturated metal sites in MOFs.
High-entropy materials, as a new type of crystalline solid solution material, are distinguished by their unique composition, consisting of five or more diverse metal species in nearly equimolar proportions, while maintaining a single-phase crystal structure . The unique composition provides not only a wide range of combinations for materials discovery, but also a distinctive microstructure for property optimization . This combination of diverse metal species and their interactions gives rise to novel and unexpected properties, including high-entropy effects, lattice distortion effects, delayed diffusion effects and “cocktail” effects . In 2004, Yeh and Cantor concurrently introduced the concept of high-entropy alloys (HEAs) . This concept was subsequently extended to include oxides, nitrides, chalcogenides, phosphides, fluorides and diborides as part of this family. Different from HEAs with single-site occupancies, the independent cationic and anionic sublattices in these materials allow for greater structural tunability . The increase in configurational entropy makes them more tolerant to lattice distortion, which facilitates the incorporation and stabilization of higher concentrations of heterovalent metal cations into the oxide lattice . To further expand the library of high entropy materials, the concept of high entropy has recently been introduced into metal-organic frameworks (MOFs) . Nevertheless,
the construction of high-entropy MOFs (HE-MOFs) entails a more intricate process that demands precise control over the ratio of metal ions to organic ligands, as well as manipulation of reaction conditions. Hence, as far as we know, research on HE-MOFs synthesis remains limited due to the difference in coordination ability between metal ions and the ligand, and only a handful of studies have been conducted, primarily centered on low-valent metal ions such as , and , which are relatively straightforward to employ in MOF synthesis . The synthesis of HE-MOFs using high-valent metal ions such as , and still faces challenges and has not yet been reported.
MOFs have garnered considerable attention in catalytic transfer hydrogenation (CTH) reactions due to their ability to fine-tune structure and electronic properties . Their porous framework offers a high surface area and well-defined porosity, and allows precise adjustment of high-valent metal nodes, resulting in excellent acidic properties . Zr/Hf-MOFs fall into this category and are known for their exceptional Lewis and Brønsted acid properties . Among them, UiO-66 is particularly popular due to its highly stable framework, high charge density of nodes, potential Lewis acidity, and ability to modulate electronic properties through structural adjustments. UiO-
66 exhibits alternating structural motifs of and , with the high charge density of its Zr-O clusters thought to be beneficial for CTH reactions . Currently, a variety of UiO-66 and its modified materials are effectively utilized in CTH reactions, showcasing their efficient catalytic performance (Table S1). Moreover, coordination unsaturated open Zr/Hf sites are often found within the UiO-66 framework, either naturally occurring or deliberately induced through defect engineering. These sites, arising from low connectivity between metal nodes and organic linkers, act as efficient Lewis centers and exhibit high activity in catalytic processes . Unsaturated sites within the UiO-66 framework can be intentionally created during synthesis by employing modulators, semi-stable linkers or surfactants to induce defect formation, or through post-synthesis processing methods . However, UiO66 synthesized by these methods has a limited tolerance to high levels of unsaturated sites, potentially resulting in the destabilization or collapse of the crystal structure.
“Entropy engineering” offers us a strategic approach towards solving this issue by elevating configurational entropy, rendering the lattice distortion more tolerant. This in turn facilitates the incorporation and stabilization of higher concentrations of diverse metal cations and lattice defects, ultimately fostering the creation of coordination unsaturated sites. In this work, high-entropy UiO-66 (ZrHfCeSnTi HE-UiO66) rich in defects has been designed and synthesized for the first time through the high-entropy effect. This approach is rooted in the compatibility of and Ce metal ions to create the UiO-66 family , capitalizing on the increased configurational entropy to incorporate Sn and Ti metal ions within the nodes, and the consequent lattice distortion (derived from the lattice distortion effect, one of the four effects for high-entropy materials) can contribute to the generation of numerous unsaturated sites. With such a strategy, the highly exposed metal nodes in HE-UiO-66 promote a significant increase in the abundance of Lewis acid-base sites, leading to a breakthrough in the performance of the CTH reaction. Density functional theory calculations, isotope tracking, -TPD, in situ diffuse reflectance infrared Fourier transform spectra and attenuated total reflectance infrared spectra uncovered the mechanism of the enhanced catalytic activity and hydrogen transfer pathways. Elaborating the structure and properties of the coordination unsaturated metal sites involved in HE-UiO66 provides guidance for tuning the Lewis acid-base sites in a broad range of complex MOFs with expanded functionalities.
Results
Design of HE-UiO-66 catalysts
In this work, we have developed a strategy termed “entropy engineering”, in which defect formation in UiO-66 is induced by increasing the configurational entropy of metal ions at the nodes (Fig. 1a). This approach relies on the compatibility of and Ce metal ions, all of which are capable to form the UiO-66 family , to confer to the framework (ZrHfCe UiO-66) more tolerant to doping with various metal ions by elevating the configurational entropy . The receptive properties of the system enable the incorporation of Sn and Ti metal ions for doping into the nodes. Simultaneously, differences in ionic radius, electronic structure, crystal field effects, etc. among , and metal ions induce significant lattice distortions , resulting in partial loss of the 12 BDC (1,4-benzenedicarboxylic acid) linkers bound to the metal nodes . Reduction of these BDC linkers leads to the formation of undercoordinated or unsaturated metal sites, i.e. defect sites. As a consequence, the defect-rich HE-UiO-66 catalyst was successfully synthesized via the entropic effect.
Morphology and structural characterization of HE-UiO-66 catalysts
The impact of configurational entropy on the morphology of UiO-66 was monitored by scanning electron microscopy (SEM) and
transmission electron microscopy (TEM) (Fig. 1b, S1 and S2). It was found that the increase in configurational entropy caused the particle size to decrease from for Zr to for ZrHfCe and then to for HE-UiO-66 (Fig. S2). This phenomenon occurs due to the introduction of various heteroatoms, which reduces the surface energy of the nanoparticles and consequently impedes their growth . Additionally, this can be elucidated by the cocktail effect and retarded diffusion in high-entropy materials. The lattice distortion raises the diffusion activation energy of atoms within the lattice, leading to a decrease in the effective diffusion rate and subsequently slowing down particle growth . The irregular mutual accumulation of these small nanoparticles effectively enhances the mesoporous structure of the HE-UiO-66 catalyst, which is beneficial for improving the mass transfer efficiency of substrate molecules in the catalytic process, thereby improving the catalytic efficiency. To characterize the spatial distribution of elements in HE-UiO-66 catalyst, high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and the corresponding EDS mapping images evidenced that , and O were uniformly distributed over the whole detection range (Fig. 1c). Furthermore, the atomic fraction of each metal in HE-UiO-66 complies with the criteria for high-entropy materials, falling within the range of metal content , as indicated by inductively coupled plasma (ICP) analysis (Table S2).
Crystal structures of the samples were examined in detail using X-ray diffraction (XRD), electron paramagnetic resonance (EPR) spectra and Fourier transform infrared (FT-IR) spectra. All samples, except the amorphous ones of Sn and Ti , show well-crystallized crystalline phases that match well with simulated UiO-66, implying the preparation of HE-UiO-66 catalysts (Fig. 1d and S3). Based on the ordering of ionic radii as follows: , the lattice undergoes an initial expansion followed by a contraction with increasing entropy, as evidenced by partially enlarged XRD peaks shifting to the left or right in the range (Fig. S3). These alterations provide robust evidence for the generation of HE-UiO-66 by doping metal ions with varying ionic radii into the UiO-66 structure. Moreover, the XRD diffraction peaks of HE-UiO-66 display intensity attenuation and broadening owing to random occupation of metal ions within the crystal lattice. The haphazard occupancy of the metal ions in the crystal lattice induces noticeable lattice distortion, leading to the emergence of uneven atomic planes within the same layer and resulting in pronounced Bragg scattering of X-rays on irregular crystal planes. This suggests that there are numerous defects (e.g. missing linked defect sites) or irregular connections within HE-UiO-66 . It is also further supported by the EPR spectrum (Fig. 1e) . The peak at is attributed to uncoupled metal centers acting as defects in the framework. With the rise in the configuration entropy of UiO-66, the peak undergoes a substantial increase, pointing to a corresponding escalation in the quantity of defects. The defect-induced unsaturated sites still coordinate with hydroxyl groups or to maintain the main chain charge balance, as indicated by the XPS analysis of O1s (Fig. S4). The peak at 530 eV corresponds to the M-O-M lattice oxygen, while the peak at 532 eV corresponds to the metal-bonded oxygen of the carboxylic acid group (M-O-C) or metal-bonded . In the presence of defects, there is an inevitable decrease in M-O-M and M-O-C, coupled with an increase in and , which also explains the difference in the two peaks of Zr, ZrHfCe, and HE-UiO-66. The FT-IR spectra of all samples are depicted in Fig. 1f. The peak observed at is ascribed to the vibration, signifying the coordination of metal ions with the bond in terephthalic acid . It is noteworthy that the intensity of the vibration peak in HE-UiO66 is substantially weaker than in other samples, indicating that more metal ions in HE-UiO-66 are unable to coordinate with bonds and form coordination unsaturated metal sites. Furthermore, peaks in the region associated with carboxylic acid groups,
Fig. 1 | Preparation and structural characterization of HE-UiO-66. a Schematic diagram of the synthesis of HE-UiO-66 with rich defects, (b) TEM, (c) HAADF-STEM and corresponding elemental mapping images of HE-UiO-66, (d) XRD patterns, (e) EPR spectra and (f) FT-IR spectra of a series of UiO-66 samples.
benzene rings and carbonyl groups are not variable across all samples. This indicates that the entropy increase does not affect the structure, aligning with the XRD results. In short, the above results confirm the abundant defects within the HE-UiO-66 catalyst.
The surface and pore size distribution of the as-synthesized samples were characterized by adsorption-desorption isotherm. With the increase in entropy, the microporous structure of Zr and ZrHfCe samples undergoes a transition to the micro-mesoporous structure of HE-UiO-66, characterized by the coexistence of Type I and Type IV isotherms, shifting from a sole Type I isotherm (Fig. S5a) . This transition can also be observed more clearly in the pore size
distribution (Fig. S5b). This pore structure transition is partly explained by the defect-induced partial collapse of the framework structure, leading to the transformation of certain micropores into mesopores . The reduction in the number of dominant micropores results in a decrease in the specific surface area, as shown in Table S3. In addition, the increase in entropy causes the particles to shrink to and the irregular stacking of nanoparticles is another factor contributing to the emergence of mesopores. In summary, the micromesoporous structure and the reduced particle size of HE-UiO-66 provide advantages in enhancing mass transfer efficiency, thereby rendering it auspicious for catalytic reactions of elevated efficacy.
Fig. 2 | Qualitative and quantitative analysis of acid-base sites. a -TPD profiles, (b) -TPD profiles, and (c) a comparison of base and acid amount.
Qualitative and quantitative analysis of active sites in HE-UiO-66 The exceptional catalytic activity of electron-deficient and in the CTH reaction of carbonyl compounds stems from their unique electronic structure and coordination configuration . In general, the lower the electron density of and , the emptier the orbitals, leading to a higher binding energy and stronger Lewis acid production. The lattice distortion induced by the entropy increase not only generated defects, but also led to the creation of coordination unsaturated metal sites due to missing linkers, all of which facilitate the generation of Lewis centers with high catalytic activity. The study of the impact of heightened entropy on the electronic states of and , conducted via XPS spectra (Fig. S6), reveals that the binding energies of Zr and Hf are substantially increased in HE-UiO-66compared to other samples. This observation confirms the increase in electron deficiencies at the sites and indicates the formation of stronger Lewis acid sites. Combined with the conclusion that the missing linkers promote the generation of coordinatively unsaturated metal sites, the HE-UiO-66 material is expected to possess more numerous and stronger Lewis acid sites. Additionally, the presence of defects in UiO-66 also induces the adsorption of or -OH to stabilize the , resulting in a higher abundance of in HE-UiO66 (Fig. S4). Typically, the basicity of ions on is higher than that of the related amorphous or crystalline , leading to a greater abundance of Lewis base sites in HE-UiO-66. The temperature-programmed desorption (TPD) profiles were used for quantitative analysis to obtain the acid-base properties of HE-UiO-66 (Fig. 2). The results indicate that the acid-base concentration tends to increase with increasing configurational entropy of UiO-66, HE-UiO-66 exhibits the highest desorption peak intensity, further suggesting the presence of the maximum exposed unsaturated metal sites that can act as Lewis acids.
CO-FTIR adsorption and CO-TPD profiles were used to further quantify the absolute number of unsaturated metal sites in HE-UiO-66
to gain more insight into its Lewis acid sites. For the CO-FTIR spectra (Fig. 3a), the peaks observed at 2190 and were assigned to CO bound to unsaturated metal sites and physically adsorbed CO, respectively . It was noted that HE-UiO-66 has the highest absorption peak representing unsaturated metal sites, indicating the presence of the most unsaturated metal sites and consequently the highest concentration of Lewis acids. The CO-TPD profiles were subsequently employed to determine the concentration of unsaturated metal sites (Fig. 3b). Prior to testing, the sample underwent a pretreatment at a high temperature of for 1 h to eliminate , a group capable of adsorbing CO (detailed discussion in Supplementary Information, Fig. S8). This ensured that only unsaturated metal sites were available for CO adsorption during the analysis. As depicted in Fig. 3b, HE-UiO-66 displays the strongest CO desorption peak, signifying the highest concentration of unsaturated metal sites, reaching . Therefore, entropy engineering proves to be an effective strategy to increase the coordination unsaturated metal sites and establish strong and abundant Lewis acid sites within UiO-66.
Reaction evaluations toward the CTH reaction of furfural
The CTH reaction plays a crucial role in generating high-value chemicals from biomass-derived carbonyls . Here, furfural (FAL) was chosen as a representative of biomass-derived carbonyls to investigate the catalytic advantages of HE-UiO-66 with abundant coordination unsaturated metal sites for the reaction. Given the importance of the surface bonding state of FAL/isopropyl alcohol (2-PrOH) adsorbed on the catalyst, attenuated total reflectance infrared (ATR-IR) spectra were captured to examine the interaction of FAL/2-PrOH with HE-UiO66 and control samples (Fig. 3c, d). The peak corresponding to the bond in pure FAL is observed at . In comparison with Zr and ZrHfCe , the peak in HE-UiO-66 displays a significant redshift, indicating a robust interaction between the catalyst and the group. Additionally, no shift in the position of the bond on the
Fig. 3 | Quantitative analysis of coordination unsaturated metal sites. a CO-FTIR adsorption, (b) CO-TPD profiles and ATR-IR spectra of the adsorption of (c) FAL and (d) 2-PrOH over a series of UiO-66 samples.
furan ring of furfural (FAL) is observed, suggesting that the active site of the catalyst interacts specifically with the group of FAL. Similarly, for the peak related to the O-H bond in 2-PrOH, HE-UiO-66 also exhibits the most pronounced red-shift, showing a strong interaction between the catalyst and the O-H bond in 2-PrOH that reduces the energy of the O-H bond. These results substantiate that HE-UiO-66, enriched with coordination unsaturated metal sites, improves the catalytic efficacy in the dissociation of 2-PrOH and activation of the substrate FAL by effectively engaging with and bonds.
The CTH reaction of FAL to furfuryl alcohol (FOL) was conducted using different catalysts with 2-PrOH as solvent and H -donor at for 2 h (Fig. 4a and Fig. S9). As the configurational entropy of UiO-66 increases (from single metal Zr to five metal ZrHfCeSnTi), the catalytic activity also increases until HE-UiO-66 achieves the highest FOL yield of , which is attributed to the generation of abundant coordination unsaturated metal sites induced by entropy engineering. Turnover numbers (TONs) show a similar trend with increasing configurational entropy (Fig. S9). Further comparison among the single metal UiO-66 catalysts reveals that Zr and Hf exhibit better catalytic activity and selectivity, highlighting their favorable functional tendency for the CTH reaction. Furthermore, the catalytic performance of HE-UiO-66 nanocrystals was examined with respect to reaction temperature and time (Fig. 4b), revealing improved conversion and yield with increasing values of both parameters. To further clarify the relationship between different catalysts and their catalytic activity under varying reaction temperatures and times, kinetic experiments were carried out for the CTH reaction of FAL to FOL in 2-PrOH (Fig. 4d and Fig. S10). At each reaction temperature, was linearly correlated with the reaction time, indicating the first-order kinetics of the CTH reaction from FAL to FOL with different catalysts. The apparent activation energy (Ea) of the reaction was determined from the Arrhenius equation ( ). The Ea of the reaction with HE-UiO-66 as catalyst was , considerably lower than that of
the reference samples, indicating a more facile CTH reaction catalyzed by HE-UiO-66. The reduction in Ea can be attributed to an increase in the coordination unsaturated metal sites as Lewis acids, which may have lowered the reaction energy barrier and facilitated the hydrogenation of FAL.
Alcohols as H-donors have a significant effect on the CTH reaction of FAL. As anticipated, the reaction efficiency varied with different types of alcohols as depicted in Fig. 4c. The higher reaction efficiency in secondary alcohols compared to primary alcohols was attributed to their lower reduction potentials. In another aspect, the poor catalytic performance of t-BuOH originated from its unique structure lacking , rendering it incapable of participating in the reaction as an H -donor . The lower internal steric hindrance of primary alcohols compared to secondary and tertiary alcohols enhances their nucleophilicity, which in conjunction with the rich acidity of HE-UiO-66 promotes the acetal reaction in the primary alcohol solvent, resulting in the generation of a by-product (e.g. furfural dimethyl acetal). In addition, the elongation of the carbon chain in the primary alcohols increases the steric hindrance, impeding the acetal reaction. As a result, by-products are only obtained in methanol solvents . In summary, steric hindrance and reduction potential of alcohols play a pivotal role in influencing hydrogen transfer and acetal reactions. 2-PrOH and 2-BuOH are selected as representative H -donors for the hydrogenation process.
Inspired by the satisfactory results in the CTH reaction of FAL, HE-UiO-66 was expanded to the CTH reaction of other biomass-derived carbonyls as well as representative aldehydes and ketones (Table 1). HE-UiO-66 demonstrated outstanding catalytic activity for the conversion of all carbonyls to their respective alcohols. Aldehydes exhibited higher reactivity than ketones, which is attributed to the lower steric hindrance of the aldehyde group. In particular, the reactivity of substituted substrates decreased compared to unsubstituted substrates, with the lowest reactivity observed in substrates containing electron-donating groups. For instance, FAL exhibits superior catalytic
Fig. 4 | CTH activities of the as-prepared catalysts. a CTH reaction of FAL over various catalysts at for 2 h , (b) variation of the performance of HE-UiO-66 with reaction temperature and time, (c) CTH reaction of FAL over HE-UiO-66 using various alcohols: Methanol (MeOH), Ethanol (EtOH), n-Propanol (1-PrOH),
2-Propanol (2-PrOH), n-Butanol (1-BuOH), 2-Butanol (2-BuOH), and tert-Butanol (tBuOH ), (d) Ea of different UiO-66 for the CTH reaction of FAL, (e) reusability and (f) leaching experiment of HE-UiO-66 for the CTH reaction of FAL. Conditions: FAL ( 0.17 mmol ), catalysts ( 25 mg ), and 2 h .
properties compared to other substituted furfurals such as biomassderived 5-methylfurfural and 5-hydroxymethylfurfural (Table 1, entries 1-3). This trend is more pronounced in the case of benzaldehyde/ acetophenone and their derivatives (Table 1, entries 6-11 and 13-16). 4-Nitrobenzaldehyde (Table 1, entry 9) and 4-chlorobenzaldehyde (Table 1, entry 10) are more reactive than other substituted benzaldehydes (Table 1, entries 7, 8). Similarly, 4-nitrobenzophenone (Table 1, entry 16) is more active than other substituted benzophenones (Table 1, entries 14, 15). These results indicate that the negative impact of electron-withdrawing groups is less significant than that of electron-donating groups. Due to the conjugation effect of the bond, cinnamaldehyde (Table 1, entry 5) shows lower reactivity than 3-phenylpropanal (Table 1, entry 4). Moreover, aliphatic ketones (Table 1, entry 12) show a much higher reactivity than aromatic ketones (Table 1, entries 13-16). In conclusion, the reactivity of substrates is determined by both the electron cloud density and the steric hindrance of the carbonyl group. Substrates with higher electron cloud densities show reduced activity. Additionally, biomass-derived levulinic acid undergoes an efficient two-step transformation into valerolactone: hydrogenation of the carbonyl group followed by
intramolecular esterification or transesterification (Table 1, entry 17). These results highlight the significant potential of HE-UiO-66 for CTH reactions in organic synthesis and biomass conversion. In order to evaluate the application potential of the catalyst in the CTH reaction of FAL, a scale-up experiment with 10 mmol FAL yielded consistent FOL yields above over 8 cycles at for (Fig. S11). HE-UiO-66 manifests promising potential for large-scale FOL production from FAL.
Subsequently, the reusability and heterogeneity of HE-UiO-66 were assessed through leaching and recycling experiments. The catalyst demonstrated remarkable reusability, maintaining catalytic performance for at least 10 cycles (Fig. 4e). In addition, the catalysts used retained stable physicochemical properties, as illustrated in Figs. S12 and S13, underscoring their excellent stability throughout the CTH reaction. To confirm the heterogeneity of the catalyst, two parallel experiments were conducted with HE-UiO-66, in one of which the catalyst was separated after 1.5 h . After filtration, no further reaction was observed in the system (Fig. 4f). Analysis revealed no detection of , and Ti within the ICP limit in the filtrated solution after the reaction, indicating no leaching of these elements. This result
Table 1 | CTH reaction of different carbonyl compounds over HE-UiO-66
Entry
Reactant
Main product
Temp.
Time. (h)
Conv (%)
Yield (%)
1
120
3
95.0
91.5
2
130
3
92.6
88.0
3
130
3
88.9
83.7
4
120
3
>99
>99
5
130
3
85.4
83.9
6
120
2
98.0
97.4
7
120
3
97.4
96.7
8
120
3
93.7
72.1
9
120
3
>99
>99
10
120
3
>99
>99
11
120
3
90.4
88.9
12
150
3
>99
>99
13
150
3
95.7
93.4
14
150
3
87.2
86.1
15
150
3
87.8
86.0
16
150
3
94.3
92.8
17
160
5
95.4
90.9
Conditions: catalyst , substrate .
serves as additional evidence for heterogeneity and stability of HE-UiO-66.
Mechanistic insights based on computational calculations and isotopically label
It is well-established that efficient CTH reactions require activation of the carbonyl group in the reactant and the hydroxyl group in the alcohol (as the H -donor). In -based catalysts, species as Lewis acid sites play a crucial role in activating the carbonyl group, while Lewis base sites activate the hydroxyl group in the hydrogen source to facilitate hydrogen transfer . The improved catalytic
performance of HE-UiO-66 is mainly attributed to the regulation of acting as Lewis acid sites and the increase in the concentration of Lewis base sites. The rise in configurational entropy facilitates the generation of defect-induced coordination unsaturated metal sites in substantial quantities, concurrently increasing the electron deficiency of . These factors collectively enhance and increase Lewis acid sites. Simultaneously, the Lewis base sites are accompanied by a rise in the Lewis acid sites. A comprehensive discussion of these issues is available in the qualitative and quantitative analyses of active sites in HE-UiO-66. Furthermore, ATR-IR spectra confirmed that HE-UiO-66 improved the catalytic efficiency of 2-PrOH dissociation and
Fig. 5 | Density functional theory calculations of relevant theoretical models. a PDOS of the d-band for the Zr and Hf atoms in perfect and defective HE-UiO-66. b Calculated adsorption energies of FAL and 2-PrOH molecules by selecting Zr and Hf as adsorption sites in perfect and defective HE-UiO-66, respectively.c Calculated
adsorption model and adsorption energy of FAL and 2-PrOH co-adsorbed on perfect and defective HE-UiO-66. d Calculated hydrogenation free energy profiles of FAL on the surface of perfect and defective HE-UiO-66.
FAL substrate activation by effectively combining with and bonds (Fig. 3c, d). In summary, the Lewis acid-base sites are directly governed by the lattice distortion-induced defects of HE-UiO-66, thereby enhancing the adsorption and activation of substrate molecules and ultimately improving the catalytic performance.
In view of investigating the influence of coordination unsaturated sites on the adsorption reaction and the interaction between substrate molecules and the HE-UiO-66 surface, Density Functional Theory (DFT) was applied for geometry optimization, projected density of states (PDOS), adsorption energy and calculated hydrogen transfer free energy profiles. The structural optimization of HE-UiO-66 with and without defects is shown in Fig. S14. Since is the main active site for the CTH reaction, a comparison of the d-band PDOS of Zr and Hf atoms in perfect and defective HE-UiO-66 reveals the atomic origin behind the enhanced CTH activity of coordination unsaturated sites (Fig. 5a). For perfect HE-UiO-66, both Hf and Zr atoms possess high chemical stability, as the density of states at the Fermi level is below the Fermi level owing to ligand completeness. For Hf and Zr atoms in defective HE-UiO-66, the absence of ligands causes the density of states near the Fermi level to exceed the Fermi level, necessitating the trapping of small molecules to enhance chemical stability. In terms of adsorption, the empty orbitals in defective HE-UiO-66 bind to the electrons of the oxygen in small molecules such as isopropyl alcohol and furfural, improving adsorption performance. In contrast, perfect HE-UiO-66 with fully coordinated Hf and Zr atoms have a weak adsorption capacity. Although these atoms do have empty orbitals above the Fermi level, their higher energy levels and considerable distance from the Fermi level make participation in reactions challenging. Analyzing the adsorption configuration reveals that the bond lengths of and in defective are shorter than those in perfect HE-UiO-66 (Table S4). This discrepancy arises from the full
coordination of Zr and Hf in the latter, limiting the accommodation of oxygen atoms in the substrate molecules to bind with metal sites. Conversely, in the defective HE-UiO-66, Zr and Hf atoms can adsorb small molecules and achieve full coordination. Further, the thermodynamically stable adsorption configurations of FAL and 2-PrOH adsorbed on Zr and Hf sites of perfect and defective HE-UiO-66 are presented (Figs. S15-S18). The adsorption energies ( ) obtained indicate that the carbonyl oxygen in FAL and the hydroxyl oxygen in 2-PrOH have a stronger affinity for the for FAL and -0.447 eV for 2-PrOH) and for FAL and -0.404 eV for 2 sites of the defective compared to the perfect UiO-66 (Fig. 5b). The adsorption results suggest a stronger interaction and binding ratio between the substrate molecules and the defective HE-UiO-66 compared to the perfect HE-UiO-66, which is consistent with the PDOS and ATR-IR analyses. Similarly, for the co-adsorption of FAL and 2-PrOH, the defective HE-UiO-66 is also stronger (Fig. 5c). To gain deeper atomic insights into the enhanced catalytic activity of the defective HE-UiO-66 with coordination unsaturated sites for the CTH reaction of FAL, the hydrogen transfer pathway was simulated and the reaction energy distribution is displayed in Fig. 5d, Fig. S19 and Table S5. For the defective HE-UiO-66, the hydrogen transfer free energy ( ) of FAL (IS ) is 0.99 eV , which is lower than that of the perfect HE-UiO-66 ( 1.43 eV ), suggesting a smaller energy barrier for the defective HE-UiO-66. As expected, the DFT results confirmed that the existence of coordination unsaturated sites effectively regulates the electronic structure of HE-UiO-66, enhancing the interaction and binding with substrate molecules and lowering the energy barrier for hydrogen transfer, thereby promoting the reaction.
The catalytic mechanism of the CTH reaction for HE-UiO-66, whether involving direct or indirect hydrogen transfer, remains unclear . An investigation of the mechanism was conducted using an
Fig. 6 | Mechanism investigations. MS of (a) FOL and (b) DIPF in different solvents (2-PrOH and 2-PrOH- -butanol); (c) Schematic of the reaction mechanism via direct or indirect hydrogen transfer and side reaction. Conditions: FAL ( 0.17 mmol ), catalysts ( 25 mg ), solvents ( 5 mL ), and 2 h .
isotope-labeled 2-PrOH- GC-MS method. However, replacing 2-PrOH- with 2-PrOH- did not provide clarity on the reaction mechanism between direct and indirect hydrogen transfer (Fig. S20). To differentiate the hydrogen transfer processes, a mixture of and t – BuOH in a ratio of was employed. Despite t -BuOH lacking H and being non-contributory as an H -donor, it can exchange H atoms with most of the groups of 2-PrOH- and the deuterium hydrogen bonding of metals (M-D) to form OH and M-H respectively. The former forms 2-PrOH- , serving as the primary hydrogen donor for the CTH reaction of FAL (Fig. 6c). The desired alcohols should display a 1 amu MS shift (MS ) via direct hydrogen transfer or no shift (MS ) via indirect hydrogen transfer. As expected, the GC-MS analysis unequivocally revealed a 1 amu mass shift, confirming the transformation of FAL to FOL over HE-UiO-66 following the direct hydrogen transfer route (Fig. 6a). Interestingly, the by-product 2-(diisopropoxymethyl)furan (DIPF) exhibits a 14 amu shift (MS ) in the reaction performed in 2-PrOH- -butanol solvent (Fig. 6b). According to the characteristics of HE-UiO-66 with abundant acid sites and previous reports , DIPF is formed by the acetal reaction of FAL and 2-PrOH-d (Fig. 6c). This can be further verified through the abovementioned experiments with different alcohol sources. The low selectivity is due to the strong hydrogen donation and steric hindrance of 2-PrOH (Fig. 4c). The reaction process can be succinctly explained as follows: the aldehyde group, being highly polar, reacts readily with nucleophiles on account of the strong electropositivity of carbon. The nucleophilic oxygen on the hydroxyl group in the alcohol attacks the carbon of the carbonyl group with its lone pair of electrons to form a hemiacetal. The – OH group in the hemiacetal is unstable and prone to dehydration condensation with another alcohol molecule to form an
acetal. Therefore, the acetal reaction of FAL with 2-PrOH- demonstrates a 14 amu shift, providing the first insight into the side reaction process by deuterium isotope tracing.
Discussion
In summary, we have employed an entropy engineering strategy to synthesize HE-UiO-66, a high-entropy MOF material capable of efficiently catalyzing CTH reactions under mild conditions. Notably, the lattice distortion effect of the high-entropy materials could not only be exploited to induce defects, generating a substantial concentration of coordination unsaturated metal sites up to and increasing the electron deficiencies in , but also to increase the diffusion activation energies of the atoms in the lattice and inhibit the growth of the particle size , which was conducive to improving the mass-transfer efficiency of the substrate molecules. The resulting increase in the abundance and strength of Lewis acid and base sites significantly improved the catalytic performance for the CTH reaction. Both experimental results and theoretical calculations confirmed that the existence of coordination unsaturated sites effectively regulates the electronic structure of HE-UiO-66, enhancing its interaction and binding with substrate molecules and lowering the energy barrier for hydrogen transfer. The direct hydrogen transfer pathway of the CTH reaction of FAL was confirmed by isotope-labeled MS, together with the transformation pathway of the side reaction. The increase in conformational entropy promotes the improvement of the physicochemical properties of HE-UiO-66 (size, stability, defects, coordination unsaturated sites and Lewis acid-base properties), providing a perspective towards the expansion of the functionalization of MOF materials.
Methods
Catalyst preparation
HE-UiO-66 was synthesized by the solvothermal method. Specifically, , , benzene-1,4-dicarboxylic acid (BDC) ( 0.665 g ) and were dissolved in 15 mL of N,Ndimethylformamide (DMF) to form a homogeneous solution. The solution was transferred into a 48 mL ACE pressure tube and reacted at for 24 h . After cooling to room temperature, the precipitate was collected by centrifugation, washed several times with DMF and then with methanol. The resulting product was dried under vacuum at overnight and designated ZrHfCeSnTi (HE-UiO-66). For comparison, a similar method was used to synthesize a series of UiO-66 with reduced configuration entropy (the total molar amount of metal ions is 4 mmol ), including unary , binary ZrHf, ternary ZrHfCe, quaternary ZrHfCeSn, among which unary Sn and Ti are amorphous.
Catalytic activity study
The CTH reaction of the carbonyl compound was carried out in a 15 mL ACE pressure tube equipped with a magnetic stirrer, submerged in a temperature-controlled oil bath. 0.17 mmol of the substrate, 5 mL of and 25 mg of the catalyst were mixed in the reactor. After sealing, the reactor was heated to the desired temperature ( ) for a specified time ( ) with stirring at 500 rpm . Following the reaction, the liquid was quantitatively analyzed using TRACE ISQ GCMS (TR-WAX-MS column ). The temperature programme started at for 1 min , then increased to at per minute and held for 2 min . For the scaled-up CTH experiment of FAL, a 75 mL ACE pressure tube was used. In this case, 10 mmol of FAL, 40 mL of 2-PrOH and 1 g of the catalyst were loaded into the pressure tube, which was sealed and heated to for 2 and 4 h . The reusability of the HE-UiO-66 catalyst was assessed in 10 consecutive reaction runs at for 2 h each. After each run, the used catalyst was separated via centrifugation, washed sequentially with ethanol and acetone, dried under vacuum at for 2 h and then reused in the subsequent run. All of the above experiments were replicated three times to verify the reproducibility of the experimental results.
DFT calculations
First-principle-based geometry optimization calculations were conducted within density-functional theory (DFT), utilizing the Vienna Ab Initio Simulation Package (VASP) code . The frozen-core projector augmented-wave (PAW) method was employed to describe the interaction between atomic cores and valence electron density . The exchange-correlation potential was approximated using the PerdewBurke Ernzerhof (PBE) functional within the generalized gradient approximation (GGA) . To account for Van der Waals (vdW) interactions, the dispersion-corrected DFT-D3 scheme was implemented . The plane-wave cutoff energy was set to 450 eV . For ionic optimization, the conjugate gradient algorithm was utilized, with a convergence threshold of atom for electronic relaxation and for Hellmann-Feynman force on each atom. Considering the nonperiodic system, the Brillouin zone in reciprocal space was sampled using Monkhorst-Pack scheme with -point grids for both geometry optimization and electronic structure calculations . The reaction energy barrier was determined using the climbing image nudged-elastic band (CI-NEB) method . The adsorption energy ( ) of adsorbates was calculated by:
where , and represent the energy of the total systems after adsorption, catalysts surfaces and adsorbates, respectively.
Data availability
The data that support the plots of the article are available within this paper and corresponding supplementary information. Other findings of this study are available from the corresponding authors upon request. Source data are provided with this paper.
References
Sarkar, A. et al. High-entropy oxides: fundamental aspects and electrochemical properties. Adv. Mater. 31, e1806236 (2019).
Yao, Y. et al. High-entropy nanoparticles: synthesis-structureproperty relationships and data-driven discovery. Science 376, eabn3103 (2022).
Ma, Y. et al. High-entropy energy materials: challenges and new opportunities. Energy Environ. Sci. 14, 2883-2905 (2021).
Pan, Y., Liu, J.-X., Tu, T.-Z., Wang, W. & Zhang, G.-J. High-entropy oxides for catalysis: a diamond in the rough. Chem. Eng. J. 451, 138659 (2023).
Wei, J. et al. Deep eutectic solvent assisted facile synthesis of lowdimensional hierarchical porous high-entropy oxides. Nano Res. 15, 2756-2763 (2021).
Cantor, B., Chang, I. T. H., Knight, P. & Vincent, A. J. B. Microstructural development in equiatomic multicomponent alloys. Mater. Sci. Eng. A 375-3377, 213-218 (2004).
Yeh, B. J.-W. et al. Nanostructured high-entropy alloys with multiple principal elements: novel alloydesign concepts and outcomes. Adv. Eng. Mater. 6, 299-303 (2004).
Feng, D. et al. Holey lamellar high-entropy oxide as an ultra-highactivity heterogeneous catalyst for solvent-free aerobic oxidation of benzyl alcohol. Angew. Chem. Int. Ed. 59, 19503-19509 (2020).
Gu, K. et al. Defect-rich high-entropy oxide nanosheets for efficient 5-hydroxymethylfurfural electrooxidation. Angew. Chem. Int. Ed. 60, 20253-20258 (2021).
Ma, Y. et al. Resolving the role of configurational entropy in improving cycling performance of multicomponent hexacyanoferrate cathodes for sodium-ion batteries. Adv. Funct. Mater. 32, (2022).
Nguyen, T. X., Su, Y. H., Lin, C. C., Ruan, J. & Ting, J. M. A new high entropy glycerate for high performance oxygen evolution reaction. Adv. Sci. 8, 2002446 (2021).
Fereja, S. L. et al. High-entropy oxide derived from metal-organic framework as a bifunctional electrocatalyst for efficient urea oxidation and oxygen evolution reactions. ACS Appl. Mater. Interfaces 14, 38727-38738 (2022).
Gonçalves, J. M. & Ruiz-Montoya, J. G. Emerging high-entropy coordination compounds and their derivatives for energy application. J. Mater. Chem. A 11, 20872-20885 (2023).
Xu, S. et al. High-entropy metal-organic framework arrays boost oxygen evolution electrocatalysis. J. Phys. Chem. C 126, 14094-14102 (2022).
Ma, Y. et al. High-entropy metal-organic frameworks for highly reversible sodium storage. Adv. Mater. 33, e2101342 (2021).
Xu, W. et al. Entropy-driven mechanochemical synthesis of polymetallic zeolitic imidazolate frameworks for fixation. Angew. Chem. Int. Ed. 58, 5018-5022 (2019).
Zhao, X. et al. Ambient fast, large-scale synthesis of entropystabilized metal-organic framework nanosheets for electrocatalytic oxygen evolution. J. Mater. Chem. A 7, 26238-26242 (2019).
Kar, A. K. et al. Unveiling and understanding the remarkable enhancement in the catalytic activity by the defect creation in UIO66 during the catalytic transfer hydrodeoxygenation of vanillin with isopropanol. Appl. Catal. B: Environ. 325, 122385 (2023).
Valekar, A. H. et al. Catalytic transfer hydrogenation of furfural to furfuryl alcohol under mild conditions over Zr-MOFs: exploring the role of metal node coordination and modification. ACS Catal. 10, 3720-3732 (2020).
Feng, X. et al. Engineering a highly defective stable UiO-66 with tunable lewis- bronsted acidity: the role of the hemilabile linker. J. Am. Chem. Soc. 142, 3174-3183 (2020).
Ji, P. et al. Strongly Lewis acidic metal-organic frameworks for continuous flow catalysis. J. Am. Chem. Soc. 141, 14878-14888 (2019).
Masoomi, M. Y., Morsali, A., Dhakshinamoorthy, A. & Garcia, H. Mixed-metal MOFs: unique opportunities in metal-organic framework (MOF) functionality and design. Angew. Chem. Int. Ed. 58, 15188-15205 (2019).
Dai, F., Zhou, S., Qin, X., Liu, D. & Qi, H. Surfactant-assisted synthesis of mesoporous hafnium- imidazoledicarboxylic acid hybrids for highly efficient hydrogen transfer of biomass-derived carboxides. Mol. Catal. 479, 110611 (2019).
SergioRojas-Buzo, García-García, P. & Corma, A. Catalytic transfer hydrogenation of biomass-derived carbonyls over hafnium-based metal-organic frameworks. ChemSusChem 11, 432-438 (2018).
Zhong, L. et al. Hydrogenation of -unsaturated aldehydes over defective UiO-66 with frustrated Lewis pairs: Modulation of densities of defect sites via tailoring ligand-vacancies. Appl. Catal. B: Environ. 342, 123421 (2024).
Qiu, M., Guo, T., Xi, R., Li, D. & Qi, X. Highly efficient catalytic transfer hydrogenation of biomass-derived furfural to furfuryl alcohol using UiO-66 without metal catalysts. Appl. Catal. A: Gen. 602, 117719 (2020).
Chen, X. et al. Tuning node defects as catalytic sites in the metal-organic framework hcp UiO-66. ACS Catal. 10, 2906-2914 (2020).
Tan, K. et al. Defect termination in the UiO-66 family of metalorganic frameworks: the role of water and modulator. J. Am. Chem. Soc. 143, 6328-6332 (2021).
Cho, K. Y. et al. Facile control of defect site density and particle size of UiO-66 for enhanced hydrolysis rates: insights into feasibility of -based metal-organic framework (MOF) catalysts. Appl. Catal. B: Environ. 245, 635-647 (2019).
Feng, Y., Chen, Q., Jiang, M. & Yao, J. Tailoring the properties of UiO66 through defect engineering: a review. Ind. Eng. Chem. Res. 58, 17646-17659 (2019).
Bakuru, V. R., Churipard, S. R., Maradur, S. P. & Kalidindi, S. B. Exploring the Brønsted acidity of UiO-66 (Zr, Ce, Hf) metal-organic frameworks for efficient solketal synthesis from glycerol acetalization. Dalton Trans. 48, 843-847 (2019).
Bakuru, V. R., Churipard, S. R., Maradur, S. P. & Kalidindi, S. B. Exploring the Bronsted acidity of UiO-66 (Zr, Ce, Hf) metal-organic frameworks for efficient solketal synthesis from glycerol acetalization. Dalton Trans. 48, 843-847 (2019).
Wang, W. et al. Chemical bonding of from Zr and Ce single atoms for efficient photocatalytic reduction of under visible light. ACS Catal. 13, 4597-4610 (2023).
Sun, Y. et al. Defect engineering of ceria nanocrystals for enhanced catalysis via a high-entropy oxide strategy. ACS Cent. Sci. 8, 1081-1090 (2022).
Chen, D., Yang, D., Wang, Q. & Jiang, Z. Effects of boron doping on photocatalytic activity and microstructure of titanium dioxide nanoparticles. Ind. Eng. Chem. Res. 45, 4110-4116 (2006).
Feng, N. et al. Boron environments in B-doped and (B, N)-codoped photocatalysts: a combined solid-state NMR and theoretical calculation study. J. Phys. Chem. C. 115, 2709-2719 (2011).
Tomboc, G. M. et al. Stabilization, characterization, and electrochemical applications of high-entropy oxides: critical assessment of crystal phase-properties relationship. Adv. Funct. Mater.
32, (2022).
Zhai, Y., Ren, X., Wang, B. & Liu, S. High-entropy catalyst-a novel platform for electrochemical water splitting. Adv. Funct. Mater. 32 (2022).
Yeh, J.-W., Chang, S.-Y., Hong, Y.-D., Chen, S.-K. & Lin, S.-J. Anomalous decrease in X-ray diffraction intensities of alloy systems with multi-principal elements. Mater. Chem. Phys. 103, 41-46 (2007).
Ma, M. et al. Tandem catalysis of furfural to -valerolactone over polyoxometalate-based metal-organic frameworks: exploring the role of confinement in the catalytic process. Renew. Energy 227, 120474 (2024).
Li, M. et al. Defect-rich hierarchical porous UiO-66(Zr) for tunable phosphate removal. Environ. Sci. Technol. 55, 13209-13218 (2021).
Morterra, C., Giamello, E., Orio, L. & Volante, M. Formation and reactivity of centers at the surface of vacuum-activated monoclinic zirconia. J. Phys. Chem. 94, 3111-3116 (1990).
He, Y., Li, C., Chen, X.-B., Shi, Z. & Feng, S. Visible-light-responsive UiO-66(Zr) with defects efficiently promoting photocatalytic CO2 reduction. ACS Appl. Mater. Interfaces 14, 28977-28984 (2022).
Fan, Y. et al. Low-temperature catalytic degradation of chlorinated aromatic hydrocarbons over bimetallic catalysts. Chem. Eng. J. 414, 128782 (2021).
Kuwahara, Y., Kango, H. & Yamashita, H. Catalytic transfer hydrogenation of biomass-derived levulinic acid and its esters to valerolactone over sulfonic acid-functionalized UiO-66. ACS Sustain. Chem. Eng. 5, 1141-1152 (2016).
Xiang, W. et al. The metal-organic framework UiO-66 with missinglinker defects: a highly active catalyst for carbon dioxide cycloaddition. Appl. Energy 277, 115560 (2020).
Ma, M. et al. Stabilization of unique species in nanocrystals for unprecedented catalytic transfer hydrogenation reaction. Appl. Catal. B: Environ. 350, 123905 (2024).
Zhao, R., Kasipandi, S., Shin, C.-H. & Bae, J. W. Catalytic conversion of biomass-derived levulinic acid to -valerolactone over amphoteric zirconium hydroxide. ACS Catal. 13, 12711-12722 (2023).
Driscoll, D. M. et al. Characterization of undercoordinated Zr defect sites in UiO-66 with vibrational spectroscopy of adsorbed CO. J. Phys. Chem. C 122, 14582-14589 (2018).
Mariscal, R., Maireles-Torres, P., Ojeda, M., Sádaba, I. & López Granados, M. Furfural: a renewable and versatile platform molecule for the synthesis of chemicals and fuels. Energy Environ. Sci. 9, 1144-1189 (2016).
Nie, R. et al. Recent advances in catalytic transfer hydrogenation with formic acid over heterogeneous transition metal catalysts. ACS Catal. 11, 1071-1095 (2021).
Ma, M. et al. Simple basic zirconium carbonate: low temperature catalysis for hydrogen transfer of biomass-derived carboxides. Green. Chem. 21, 5969-5979 (2019).
Ma, M. et al. Humins with efficient electromagnetic wave absorption: a by-product of furfural conversion to isopropyl levulinate via a tandem catalytic reaction in one-pot. Chem. Eur. J. 27, 12659-12666 (2021).
Ma, M. et al. Magnetic nanoparticles as easily separable catalysts for efficient catalytic transfer hydrogenation of biomassderived furfural to furfuryl alcohol. Appl. Catal. A: Gen. 602, 117709 (2020).
Ma, M. et al. Creation of surface frustrated Lewis pairs on highentropy spinel nanocrystals that boosts catalytic transfer hydrogenation reaction. Chem. Eng. J. 470, 144291 (2023).
Fang, W. & Riisager, A. Efficient valorization of biomass-derived furfural to fuel bio-additive over aluminum phosphate. Appl. Catal. B: Environ. 298, 120575 (2021).
An, Z. & Li, J. Recent advances in the catalytic transfer hydrogenation of furfural to furfuryl alcohol over heterogeneous catalysts. Green. Chem. 24, 1780-1808 (2022).
Zhou, S. et al. Sustainable hydrothermal self-assembly of hafnium-lignosulfonate nanohybrids for highly efficient reductive
upgrading of 5-hydroxymethylfurfural. Green. Chem. 21, 1421-1431 (2019).
Zhou, S. et al. Zirconium-lignosulfonate polyphenolic polymer for highly efficient hydrogen transfer of biomass-derived oxygenates under mild conditions. Appl. Catal. B: Environ. 248, 31-43 (2019).
An, Z. et al. Highly active, ultra-low loading single-atom iron catalysts for catalytic transfer hydrogenation. Nat. Commun. 14, 6666 (2023).
Ma, M. et al. One-pot Transfer hydrogenation of methyl levulinate into valerolactone and 1,4-pentanediol over in situ reduced Cu/ in 2-PrOH. ChemistrySelect 5, 1-8 (2020).
Bai, L. et al. Catalyst-free acetalization of biobased furfural into cyclic acetal fuel additives with biogenic ethylene glycol. ACS Sustain. Chem. Eng. 11, 15743-15753 (2023).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169-11186 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758-1775 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3965-3868 (1996).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188-5192 (1976).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901-9904 (2000).
Acknowledgements
This work was supported by the Natural Science Foundation of Jilin Province, China (No. 20240101166JC).
Author contributions
M.M. and G.T. conceived the idea and designed the experiments. M.M. and E.C. performed most of the experiments and characterizations. M.M., H.Y. and G.T. cowrite the manuscript. All the authors commented on the manuscript.
Correspondence and requests for materials should be addressed to Ge Tian.
Peer review information Nature Communications thanks Chunbao (Charles) Xu and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http:// creativecommons.org/licenses/by-nc-nd/4.0/.
(c) The Author(s) 2025
State Key Laboratory of Inorganic Synthesis and Preparative Chemistry, College of Chemistry, Jilin University, 130012 Changchun, P. R. China.
⟶ e-mail: tiange@jlu.edu.cn