DOI: https://doi.org/10.1038/s41467-024-45485-8
PMID: https://pubmed.ncbi.nlm.nih.gov/38350981
تاريخ النشر: 2024-02-13
هيدروجيلات مزدوجة الشبكة قوية مع تعزيز ذاتي سريع واهتزاز منخفض استنادًا إلى شبكات متشابكة بشدة
تم القبول: 25 يناير 2024
نُشر على الإنترنت: 13 فبراير 2024
(أ) التحقق من التحديثات
الملخص
تُعزز معظم الهلاميات المائية القاسية من خلال إدخال آليات لتبدد الطاقة، ولكن تحقيق صلابة عالية وفقدان منخفض للطاقة الحركية في الوقت نفسه يمثل تحديًا لأن هيكل تبدد الطاقة لا يمكنه التعافي بسرعة. في هذا العمل، تم تصنيع هلاميات مائية مزدوجة الشبكة شديدة التشابك ذات أداء ميكانيكي عالٍ دون هيكل لتبدد الطاقة، حيث تعمل التشابكات الفيزيائية كروابط فعالة أساسية في الشبكة الأولى. يسمح هيكل التشابك الانزلاقي لشبكة الهلام المائي بتشكيل هيكل موجه موحد للغاية أثناء الشد، مما يؤدي إلى قوة شد عالية.
تظهر آليات التبدد استعادة عالية، لكنها تظهر قوة شد منخفضة ومرونة.
النتائج
تصميم وخصائص الهلاميات مع هيكل شبكة مزدوجة متشابكة بشدة
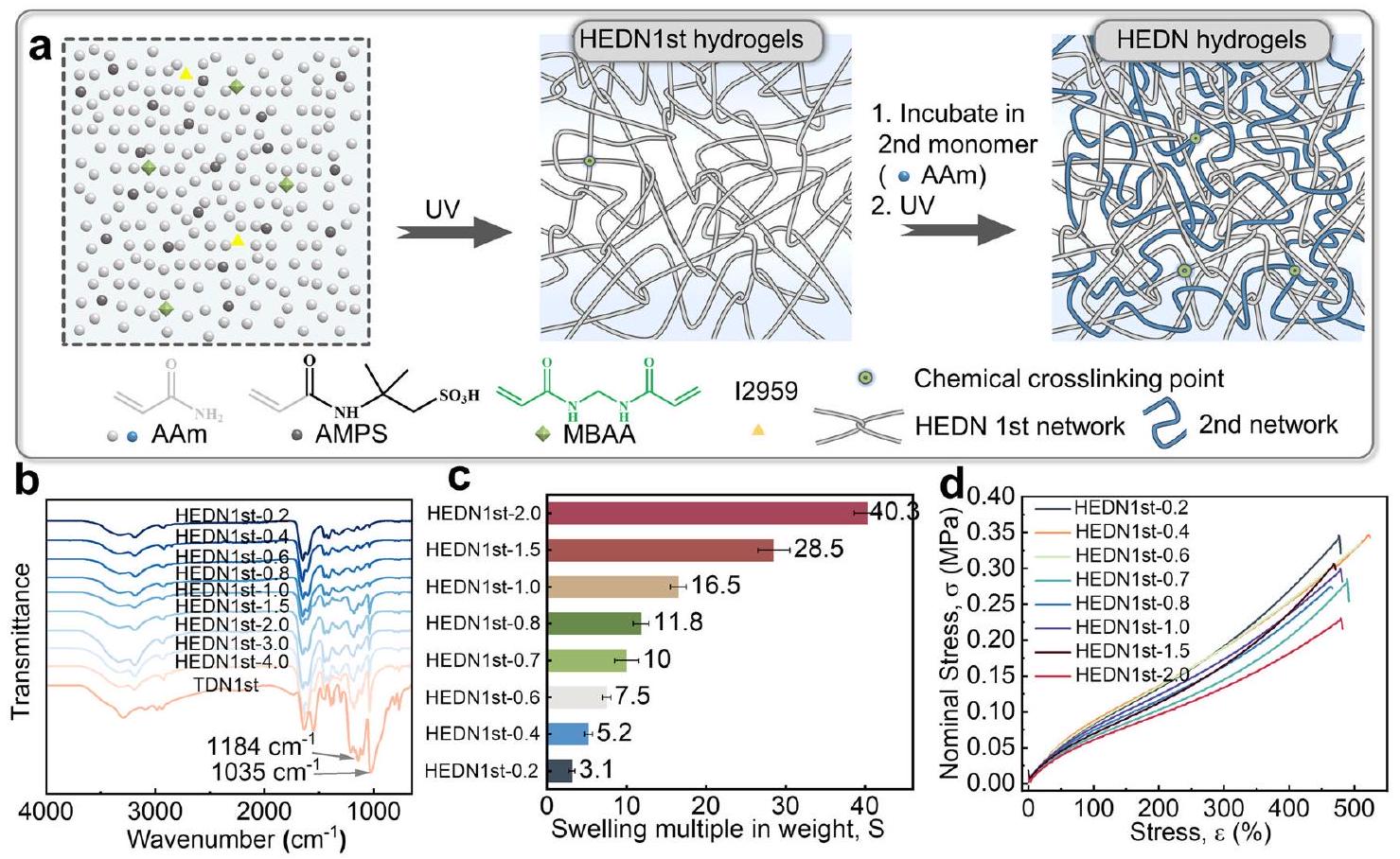
تنخفض قوة الشد لهلامات HEDN1st قليلاً مع زيادة AMPS. نظرًا لأن تركيز محلول البوليمر المسبق لا يتغير، أي أن درجة التشابك هي نفسها، فإن الإطالة عند الكسر والموصلية الأولية تبقى تقريبًا دون تغيير.
المتانة العالية لهلام TDN تنشأ من الكمية الكبيرة من الطاقة الممتصة من خلال كسر الروابط التساهمية في الشبكة الأولى الهشة. ومع ذلك، لا يمكن إصلاح الروابط التساهمية المكسورة، مما يجعل هلام TDN ضعيفًا بعد الشد الأول. تشير الحلقات الهستيرية الكبيرة لهلام TDN في الشكل التكميلي 9 إلى أن العديد من الروابط التساهمية في الشبكة الأولى قد انكسرت لتفريغ الطاقة. منحنى الإجهاد-الانفعال الثاني لهلام TDN مختلف جدًا عن دورة الشد الأولى، وتكون القابلية للعكس
نقاط التشابك أعلى بكثير من تلك الناتجة عن الربط الكيميائي، الذي يلعب دورًا رئيسيًا في تأثير الصلابة. هذا يتماشى مع ما أبلغ عنه سو وآخرون.
تعزيز ذاتي، آلية عالية القابلية للعكس لهياكل الشبكات المزدوجة المتشابكة بشدة

أ. الميكروهيكل لهلام TDN المجفف بالتجميد غير الممدد. يمكن ملاحظة هياكل فراغية أكبر أو أصغر عند علامات الدوائر البرتقالية أو الزرقاء. ب. الميكروهيكل لهلام TDN المجفف بالتجميد الممدد إلى
نقاط التشابك تعمل على توحيد سلاسل البوليمر ذات الأطوال المخفية المختلفة، أي أن جميع سلاسل البوليمر مشدودة بشكل متساوٍ. تظهر هلاميات HEDN تقريبًا عدم وجود كسر في سلاسل البوليمر خلال الشد الأولي، ويحدث فقط توجيه لسلاسل البوليمر. في هذه العملية، يتم تخزين الطاقة على شكل فقدان في الإنتروبيا. عندما يتم إزالة القوة الخارجية، سيتم إطلاق هذه الطاقة على شكل زيادة في الإنتروبيا. عندما يتم شد هلاميات HEDN إلى حد معين، تصبح سلاسل البوليمر موجهة بشكل كبير، ويصبح المادة صلبة جدًا في اتجاه القوة المطبقة، مما يظهر سلوكًا نموذجيًا لزيادة الشد. كما هو موضح في الشكل التكميلي 12b، يستمر معامل هلاميات HEDN في الزيادة مع الشد. تتجاوز قدرة زيادة الشد لـ HEDN-0.8 و HEDN-1.0 الـ 40 (الشكل التكميلي 13)، مما يظهر سلوكًا قويًا من التعزيز الذاتي، وهو ما يساعد في تجنب تلف المادة تحت الشد الكبير.
سلوك التصلب ناتج عن الاتجاه الأعلى لسلاسل البوليمر تحت الضغوط الكبيرة.
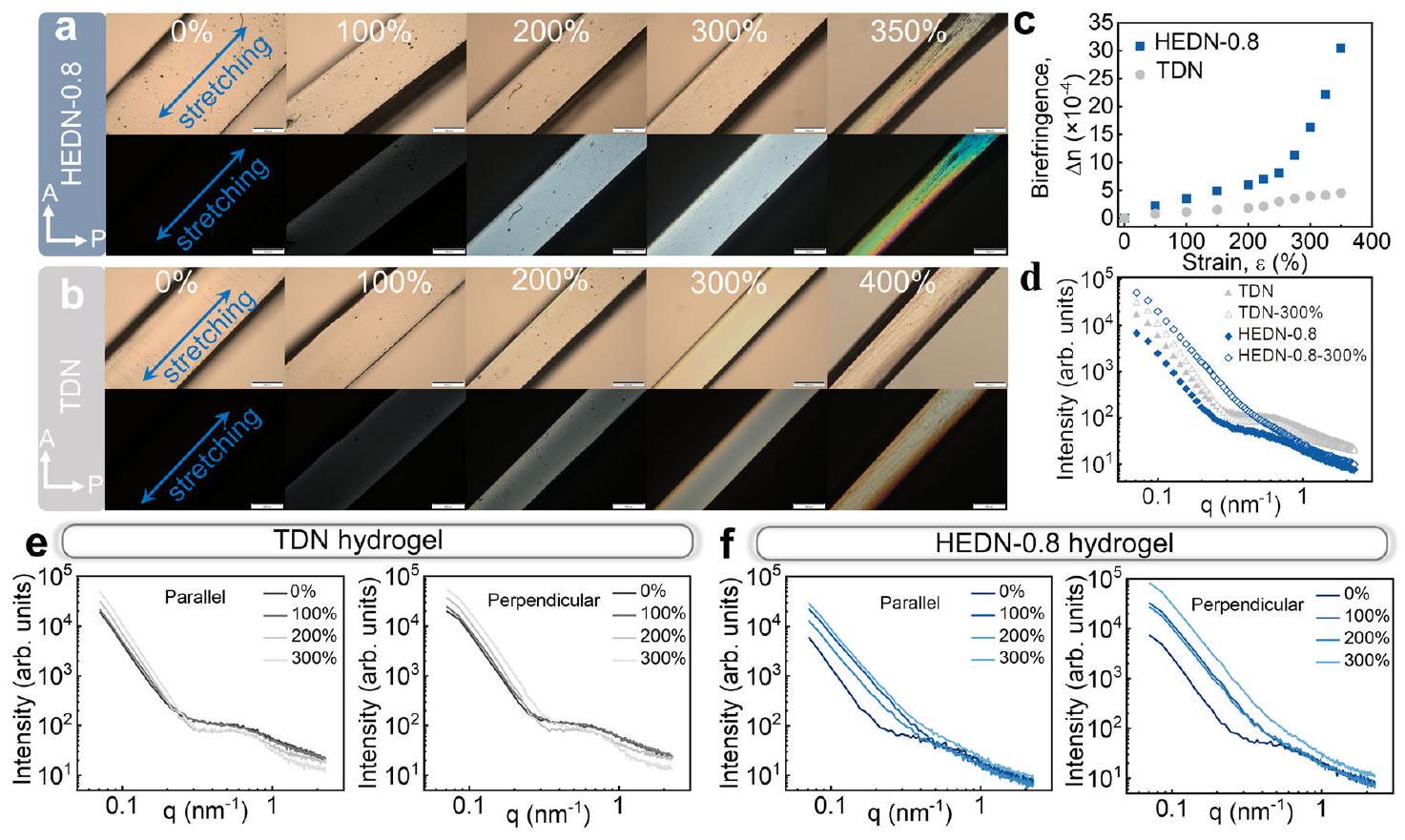
تتحول الكتف في ملف تشتت هيدروجيل TDN قليلاً مع زيادة الإجهاد. يتحول الكتف في الملف الموازي إلى منطقة منخفضة من q، بينما يتحول الكتف في الملف العمودي إلى منطقة عالية من q. بسبب إطالة عينات الهيدروجيل في اتجاهات الشد الموازي، مما يؤدي إلى زيادة في تباعد الميكرو دومينات. في الاتجاه العمودي، يصبح الهيدروجيل أرق، مما يتسبب في اقتراب الميكرو دومينات من بعضها البعض. على النقيض من ذلك، يصبح تشتت هيدروجيل HEDN-0.8 أضعف مع زيادة الإجهاد في كلا الاتجاهين الموازي والعمودي، مما يشير إلى أن الشد يؤدي إلى الاختفاء التدريجي للميكرو دومينات بسبب تأثير انزلاق تشابكات سلاسل البوليمر. كما تشير ملفات وأنماط SAXS إلى أن هيدروجيل HEDN تتمتع بقدرة استرداد عالية (الشكل التكميلي 21). تظهر ملفات SAXS قبل وبعد الشد منحنيات متداخلة تقريبًا تمامًا، مما يشير إلى أن هيدروجيل HEDN تطورت من حالة أولية غير متجانسة إلى هيكل شبكة متجانسة بعد الشد، ثم عادت إلى هيكل غير متجانس بعد التفريغ. هذه الاستعادة من هيكل مشدود منظم للغاية إلى هيكل تشابك غير منظم مدفوعة بالإنتروبيا. علاوة على ذلك، لا يوجد تقريبًا هيكل لتبدد الطاقة في الشبكة. وبالتالي، فإن هيدروجيل HEDN تتمتع بقدرة استرداد عالية.
آلية توزيع الإجهاد عند الشق في هيدروجيل HEDN
و هيدروجيل HEDN مختلف بشكل كبير. طاقة الكسر لهيدروجيل TDN هي فقط
في الوضع المستقطب. اتجاه الشد لعينات الهيدروجيل يشكل
محتويات AMPS المختلفة. منحنيات الشد المتتالية مرتين لهلام HEDN-AAc-3.0 في
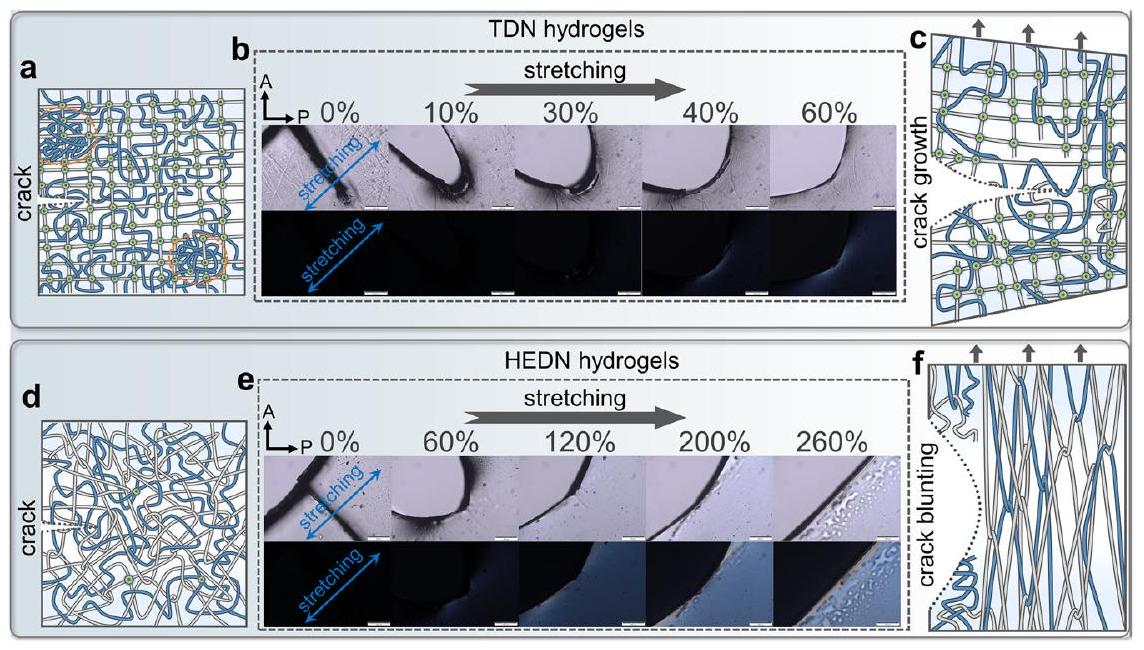
التشابك حول الفتحة يمنح هيدروجيل HEDN طاقة كسر فائقة الارتفاع.
تطبيق استراتيجية الشبكة المزدوجة شديدة التشابك
إلى الطبيعة الضعيفة للإلكتروليت لـ AAc. تضاعف انتفاخ هيدروجيل HEDN1st زاد تدريجياً مع زيادة محتوى AMPS، وكان تضاعف انتفاخ هيدروجيل HEDN1st-4.0 مرتفعاً جداً حيث بلغ 19 بسبب التنافر الكهروستاتيكي القوي بين سلاسل البوليمر الكهربي ذات الكثافة العالية. لا تزال هيدروجيل HEDN-AAc التي تم تحضيرها باستخدام AAc كأحادي الشبكة الثاني تتمتع بخصائص ميكانيكية جيدة. تُظهر الشكل 6b أن قوة الشد لهيدروجيل HEDN-AAc تزداد أولاً ثم تنخفض مع تضاعف الانتفاخ، حيث يتمتع هيدروجيل HEDN-AAc-3.0 بأعلى قوة شد تبلغ 2.1 ميجا باسكال، ويصبح ظاهرة تصلب الشد واضحة تدريجياً. كما أظهرت قوة الضغط لهيدروجيل HEDN-AAc اتجاهًا مشابهًا، حيث أظهر هيدروجيل HEDN-AAc-3.0 قوة ضغط تبلغ 45 ميجا باسكال. تُظهر الشكل 6d أن هيدروجيل HEDN-AAc-3.0
كما يظهر قابلية عكسية جيدة عند
نقاش
طرق
المواد
تحضير هلام HEDN 1
تحضير هلام HEDN (HEDN-AAc)
تم تعريضها تحت مصباح فوق بنفسجي بطول موجي 365 نانومتر لمدة 4 ساعات لتشكيل الشبكة الثانية في الموقع. أخيرًا، تم وضع الهلاميات الناتجة في ماء نقي للحصول على هلاميات HEDN المتورمة المتوازنة (هلاميات HEDN-AAc).
تحضير هيدروجيل TDN
قياس التورم المتعدد في الوزن
قياس محتوى الماء
تحضير الجل بمحتوى مائي محدد
خصائص ATR-FTIR
توصيف النفاذية
اختبار ميكانيكي
درجة الحرارة عند سرعة الشد
اختبار القص النقي
توصيف المجهر الإلكتروني الماسح
اختبار الانكسار المزدوج
تشتت الأشعة السينية بزاوية صغيرة
توصيف المجهر الذري القوي
الميكروسكوب (FastScan Bio، بروكير، الولايات المتحدة الأمريكية). كانت العينات بحاجة إلى أن تُغمر في بركة الماء السائل أثناء القياس.
توفر البيانات
References
- Gong, J. P., Katsuyama, Y., Kurokawa, T. & Osada, Y. Double-network hydrogels with extremely high mechanical strength. Adv. Mater. 15, 1155-1158 (2003).
- Tanaka, Y. et al. Determination of fracture energy of high strength double network hydrogels. J. Phys. Chem. B 109, 11559-11562 (2005).
- Tsukeshiba, H. et al. Effect of polymer entanglement on the toughening of double network hydrogels. J. Phys. Chem. B 109, 16304-16309 (2005).
- Huang, M. et al. Importance of entanglement between first and second components in high-strength double network gels. Macromolecules 40, 6658-6664 (2007).
- Guo, X. et al. Strong and tough fibrous hydrogels reinforced by multiscale hierarchical structures with multimechanisms. Sci. Adv. 9, eadf7075 (2023).
- Li, W. et al. Tough Hydrogels with Isotropic and Unprecedented Crack Propagation Resistance. Adv. Funct. Mater. 32, 220734 (2022).
- Hua, M. et al. Strong tough hydrogels via the synergy of freezecasting and salting out. Nature 590, 594-599 (2021).
- Xu, L., Qiao, Y. & Qiu, D. Coordinatively stiffen and toughen hydrogels with adaptable crystal-domain cross-linking. Adv. Mater. 35, 2209913 (2023).
- Fu, L. et al. Cartilage-like protein hydrogels engineered via entanglement. Nature 618, 740-747 (2023).
- Sun, J. Y. et al. Highly stretchable and tough hydrogels. Nature 489, 133-136 (2012).
- He, G. et al. Strong and reversible covalent double network hydrogel based on force-coupled enzymatic reactions. Angew. Chem. Int. Ed. 134, e202201765 (2022).
- Hua, Y. et al. Ultrafast, tough, and adhesive hydrogel based on hybrid photocrosslinking for articular cartilage repair in water-filled arthroscopy. Sci. Adv. 7, eabg0628 (2021).
- Zhang, M. et al. Toughening double-network hydrogels by polyelectrolytes. Adv. Mater. 35, 2301551 (2023).
- Li, G. et al. Highly conducting and stretchable double-network hydrogel for soft bioelectronics. Adv. Mater. 34, 2200261 (2022).
- Chen, H. et al. Super bulk and interfacial toughness of physically crosslinked double-network hydrogels. Adv. Funct. Mater. 27, 1703086 (2017).
- Liu, S., Bastola, A. K. & Li, L. A 3D printable and mechanically robust hydrogel based on alginate and graphene oxide. ACS Appl. Mater. Interfaces 9, 41473-41481 (2017).
- Gaharwar, A. K., Peppas, N. A. & Khademhosseini, A. Nanocomposite hydrogels for biomedical applications. Biotechnol. Bioeng. 111, 441-453 (2014).
- Liu, D. et al. Tough, transparent, and slippery PVA hydrogel led by syneresis. Small 19, 2206819 (2023).
- Dong, X. et al. Strong and tough conductive organo-hydrogels via freeze-casting assisted solution substitution. Adv. Funct. Mater. 32, 2203610 (2022).
- Kim, J., Zhang, G., Shi, M. & Suo, Z. Fracture, fatigue, and friction of polymers in which entanglements greatly outnumber cross-links. Science 374, 212-216 (2021).
- Xiong, X. et al. Polymerizable rotaxane hydrogels for threedimensional printing fabrication of wearable sensors. Nat. Commun. 14, 1331 (2023).
- Lei, H. et al. Stretchable hydrogels with low hysteresis and antifatigue fracture based on polyprotein cross-linkers. Nat. Commun. 11, 4032 (2020).
- Meng, X. et al. Hysteresis-free nanoparticle-reinforced hydrogels. Adv. Mater. 34, 2108243 (2022).
- Hui, C. Y., Bennison, S. J. & Londono, J. D. Crack blunting and the strength of soft elastic solids. Proc. R. Soc. Lond. A. 459, 1489-1516 (2003).
- Wang, C. et al. Elastic materials with high strength and low hysteresis. Proc. Natl Acad. Sci. USA 116, 5967-5972 (2019).
- Liu, C. et al. Tough hydrogels with rapid self-reinforcement. Science 372, 1078-1081 (2021).
- Wang, J. et al. Low Hysteresis Hydrogel Induced by Spatial Confinement. Adv. Funct. Mater. 33, 2214935 (2023).
- Zhong, M. et al. Quantifying the impact of molecular defects on polymer network elasticity. Science 353, 1264-1268 (2016).
- Yang, C., Yin, T. & Suo, Z. Polyacrylamide hydrogels. I. Network imperfection. J. Mech. Phys. Solids 131, 43-55 (2019).
- Lin, S. & Zhao, X. Fracture of polymer networks with diverse topological defects. Phys. Rev. E 102, 052503 (2020).
- Flory, P. J. Principles of Polymer Chemistry (Cornell University Press, Ithaca, 1953).
- Treloar, L. R. G. The Physics of Rubber Elasticity (Oxford University Press, Oxford, 1975.)
- Li, C. et al. Effects of network structures on the fracture of hydrogel. Extrem. Mech. Lett. 49, 101495 (2021).
- Di Lorenzo, F. & Seiffert, S. Nanostructural heterogeneity in polymer networks and gels. Polym. Chem. 6, 5515-5528 (2015).
- Seiffert, S. Origin of nanostructural inhomogeneity in polymernetwork gels. Polym. Chem. 8, 4472-4487 (2017).
- Seiffert, S. Scattering perspectives on nanostructural inhomogeneity in polymer network gels. Prog. Polym. Sci. 66, 1-21 (2017).
- Fukao, K. et al. Effect of relative strength of two networks on the internal fracture process of double network hydrogels as revealed by in situ small-angle X-ray scattering. Macromolecules 53, 1154-1163 (2020).
- Nakajima, T. et al. A universal molecular stent method to toughen any hydrogels based on double network concept. Adv. Funct. Mater. 22, 4426-4432 (2012).
- Zheng, J. et al. One-pot synthesis of highly mechanical and recoverable double network hydrogels using thermoreversible sol-gel polysaccharide. Adv. Mater. 25, 4171-4176 (2013).
- Chen, Q. et al. A novel design strategy for fully physically linked double network hydrogels with tough, fatigue resistant, and selfhealing properties. Adv. Funct. Mater. 25, 1598-1607 (2015).
- Liu, S. & Lin, L. Ultrastretchable and self-healing double-network hydrogel for 3D printing and strain sensor. ACS Appl. Mater. Interfaces 9, 26429-26437 (2017).
- Zhang, X. et al. Supramolecular nanofibrillar hydrogels as highly stretchable, elastic and sensitive ionic sensors. Mater. Horiz. 6, 326-333 (2019).
- Yang, Y. et al. Highly elastic and ultratough hybrid ionic-covalent hydrogels with tunable structures and mechanics. Adv. Mater. 30, 1707071 (2018).
- Shan, X., Song, S. & Gao, G. Robust and flexible strain sensors based on dual physically cross-linked double network hydrogels for monitoring human-motion. Chem. Eng. J. 354, 817-824 (2018).
- Benitez-Duif, P. A. et al. Ultrastrong Poly (2-Oxazoline)/Poly (Acrylic Acid) double-network hydrogels with cartilage-like mechanical properties. Adv. Funct. Mater. 32, 2204837 (2022).
- Hirsch, M., Alvaro, C. & Esther, A. 3D printing of strong and tough double network granular hydrogels. Adv. Funct. Mater. 31, 2005929 (2021).
- Chong, J. et al. Highly conductive tissue-like hydrogel interface through template-directed assembly. Nat. Commun. 14, 2206 (2023).
- Nian, G., Kim, J., Bao, X. & Suo, Z. Making highly elastic and tough hydrogels from doughs. Adv. Mater. 34, 2206577 (2022).
- Zhang, G. et al. Hydrogels of arrested phase separation simultaneously achieve high strength and low hysteresis. Sci. Adv. 9, eadh7742 (2023).
- Kuhn, W. & Grün, F. Beziehungen zwischen elastischen Konstanten und Dehnungsdoppelbrechung hochelastischer Stoffe. Kolloid-Z. 101, 248-271 (1942).
- Treloar, L. R. G. The photo-elastic properties of rubber. Part I: Theory of the optical properties of strained rubber. Trans. Faraday Soc. 43, 277-284 (1947).
- Sørensen, B. E. A revised Michel-Lévy interference colour chart based on first-principles calculations. Eur. J. Mineral. 25, 5-10 (2013).
- Mai, T. T., Okuno, K., Tsunoda, K. & Urayama, K. Crack-tip strain field in supershear crack of elastomers. ACS Macro Lett. 9, 762-768 (2020).
- Zhang, T., Lin, S., Yuk, H. & Zhao, X. Predicting fracture energies and crack-tip fields of soft tough materials. Extrem. Mech. Lett. 4, 1-8 (2015).
- Long, R., Hui, C. Y., Gong, J. P. & Bouchbinder, E. The fracture of highly deformable soft materials: A tale of two length scales. Annu. Rev. Condens. Matter Phys. 12, 71-94 (2021).
شكر وتقدير
مساهمات المؤلفين
المصالح المتنافسة
معلومات إضافية
المواد التكميلية متاحة على
https://doi.org/10.1038/s41467-024-45485-8.
© المؤلفون 2024
كلية الكيمياء والهندسة الكيميائية، جامعة شنغهاي جياو تونغ، شنغهاي 200240، الصين. المختبر الوطني الرئيسي لمركبات المصفوفة المعدنية، جامعة شنغهاي جياو تونغ، شنغهاي 200240، الصين. البريد الإلكتروني: xlwang@sjtu.edu.cn
DOI: https://doi.org/10.1038/s41467-024-45485-8
PMID: https://pubmed.ncbi.nlm.nih.gov/38350981
Publication Date: 2024-02-13
Tough double network hydrogels with rapid self-reinforcement and low hysteresis based on highly entangled networks
Accepted: 25 January 2024
Published online: 13 February 2024
(A) Check for updates
Abstract
Most tough hydrogels are reinforced by introducing energy dissipation mechanisms, but simultaneously realizing a high toughness and low hysteresis is challenging because the energy dissipation structure cannot recover rapidly. In this work, high mechanical performance highly entangled double network hydrogels without energy dissipation structure are fabricated, in which physical entanglements act as the primary effective crosslinking in the first network. This sliding entanglement structure allows the hydrogel network to form a highly uniform oriented structure during stretching, resulting in a high tensile strength of
dissipation mechanisms exhibit high recovery, their show low tensile strength and toughness
Results
Design and properties of hydrogels with highly entangled double network structure
content. The tensile strength of HEDN1st hydrogels decreases slightly with the increase of AMPS. Since the concentration of the prepolymer solution does not change, i.e., the degree of entanglement is the same, the elongation at break and initial modulus are almost unchanged.
high toughness of the TDN hydrogel originates from the large amount of energy absorbed by covalent bond breakage in the brittle first network. However, the broken covalent bonds cannot be repaired, making the TDN hydrogel weak after the first stretching. The large hysteresis loops of the TDN hydrogel in Supplementary Fig. 9 indicate that many covalent bonds in the first network are broken to dissipate energy. The second stress-strain curve of the TDN hydrogel is very different from the first stretching cycle, and the reversibility
entanglement points is much higher than that of chemical crosslinking, which plays a dominant role in the effect of stiffness. This is consistent with what Suo et al. reported
Self-reinforcement, high reversibility mechanism of highly entangled double network structures
a Microstructure of unstretched freeze-dried TDN hydrogel. Larger or smaller void structures can be observed at the orange or blue circle marks. b Microstructure of freeze-dried TDN hydrogel stretched to
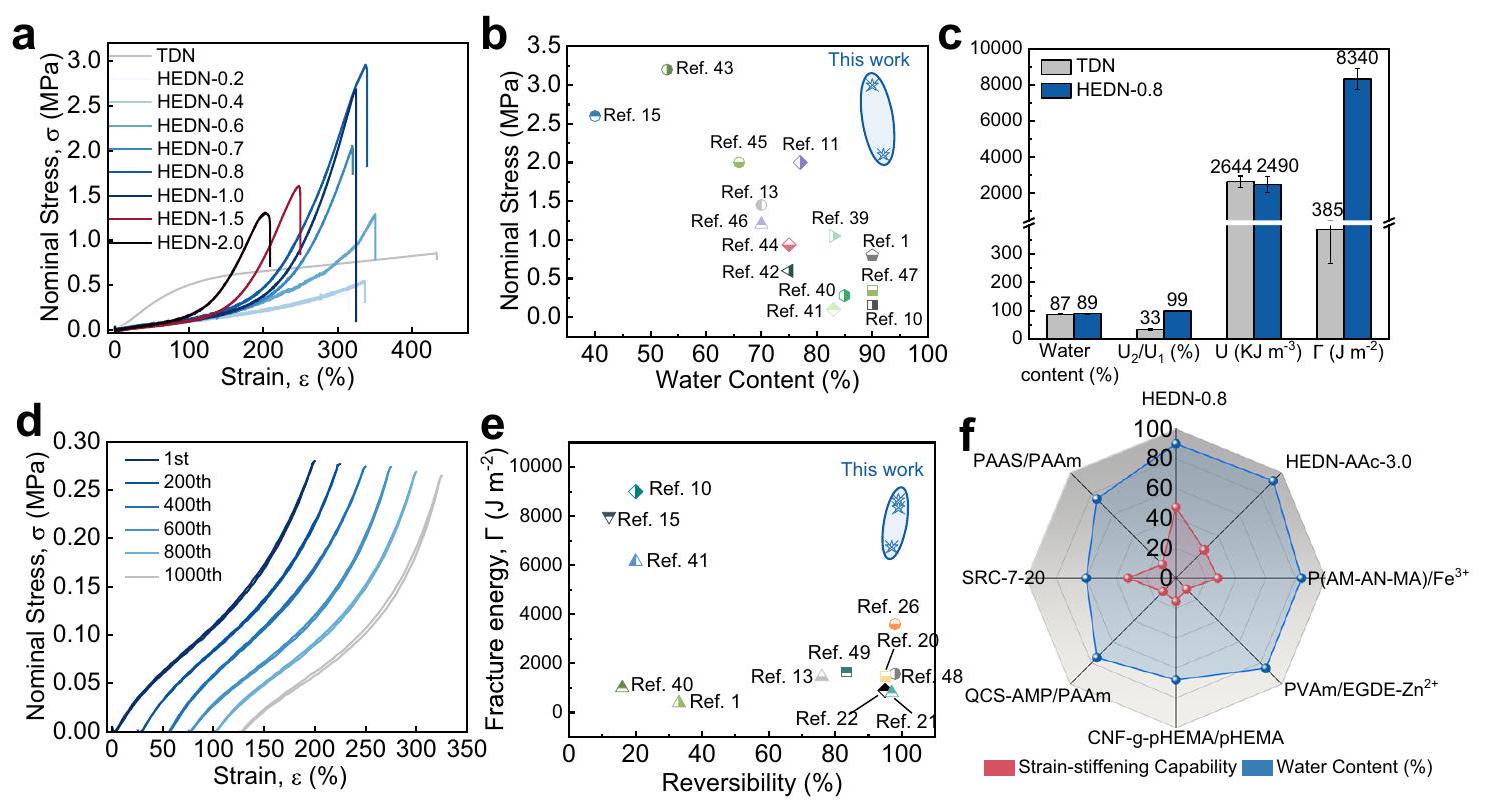
entanglement points homogenizes the polymer chains with different hidden lengths, i.e., all polymer chains are equally stretched. The HEDN hydrogels show almost no polymer chains breakage during the initial stretching, and only polymer chain orientation occurs. In this process, energy is stored as an entropy loss. When the external force is removed, this energy will be released in the form of an entropy increase. When the HEDN hydrogels are stretched to a certain extent, the polymer chains are highly oriented, and the material becomes very rigid in the direction of the applied force, thus exhibiting typical strainstiffening behavior. As shown in Supplementary Fig. 12b, the modulus of HEDN hydrogels continues to increase with strain. The strainstiffening capacity of HEDN-0.8 and HEDN-1.0 exceeds 40 (Supplementary Fig. 13), showing strong self-reinforced behavior, which is conducive to avoiding material damage under large strains.
stiffening behavior is caused by the higher orientation of polymer chains under large strains.
method. The shoulder on the scattering profile of the TDN hydrogel shifts slightly with increasing strain. The shoulder on the parallel profile shifts to the low-q region, while the shoulder on the perpendicular profile shifts to the high-q region. Due to the elongation of the hydrogel samples in the parallel to stretching directions, resulting in an increase in the spacing of the microdomains. In the perpendicular direction, the hydrogels become thinner, causing the microdomains to becomes closer to each other. In contrast, the scattering of HEDN-0.8 hydrogel becomes weaker with increasing strain in both parallel and perpendicular directions, indicating that stretching leads to the gradual disappearance of microdomains due to the sliding effect of polymer chain entanglements. SAXS profiles and patterns also indicate that HEDN hydrogels have high recoverability (Supplementary Fig. 21). The SAXS profiles before and after stretching show almost exactly overlapping curves, indicating that the HEDN hydrogels evolved from a non-uniform initial state to a uniform network structure after stretching, and then returned to a non-uniform structure after unloading. This recovery from a highly ordered stretch structure to a disordered entanglement structure is driven by entropy. Moreover, there is almost no energy dissipation structure in the network. Thus, HEDN hydrogels have high recoverability.
Stress dispersion mechanism at the notch of HEDN hydrogels
and HEDN hydrogels are significantly different. The fracture energy of the TDN hydrogel is only
in polarized mode. The stretching direction of the hydrogel samples forms a
different AMPS contents. d Twice consecutive stretching curves of HEDN-AAc-3.0 hydrogel in
entanglement around the notch gives HEDN hydrogels ultrahigh fracture energy.
Application of the highly entangled double network strategy
to the weak electrolyte nature of AAc. The swelling multiple of HEDN1st hydrogels gradually increased upon further increasing the AMPS content, and the swelling multiple of HEDN1st- 4.0 hydrogel was as high as 19 due to the strong electrostatic repulsion between the high-density polyelectrolyte chains. HEDN-AAc hydrogels prepared with AAc as the second network monomer still have good mechanical properties. Figure 6b shows that the tensile strength of HEDN-AAc hydrogels first increases and then decreases with swelling multiple, HEDN-AAc-3.0 hydrogel has the highest tensile strength of 2.1 MPa , and the strain-stiffening phenomenon is gradually obvious. The compressive strength of HEDN-AAc hydrogel also showed a similar trend, HEDN-AAc-3.0 hydrogel exhibited a compressive strength of 45 MPa . Figure 6d shows that the HEDN-AAc-3.0 hydrogel
also exhibits good reversibility at
Discussion
Methods
Materials
Preparation of HEDN 1st hydrogels
Preparation of HEDN (HEDN-AAc) hydrogels
irradiated under a 365 nm ultraviolet lamp for 4 h to form the second network in situ. Finally, the resulting hydrogels were placed in pure water to obtain balanced swollen HEDN hydrogels (HEDN-AAc hydrogels).
Preparation of TDN hydrogel
Measurement of swelling multiple in weight
Measurement of water content
Preparation of gels with specific water content
ATR-FTIR characterizations
Transmittance characterization
Mechanical Test
temperature at a tensile speed of
Pure shear test
Scanning Electron Microscope characterization
Birefringence test
Small-angle X-ray scattering
Atomic force microscope characterization
microscope (FastScan Bio, Bruker, USA). The samples needed to be immersed in the water-liquid pool during the measurement.
Data availability
References
- Gong, J. P., Katsuyama, Y., Kurokawa, T. & Osada, Y. Double-network hydrogels with extremely high mechanical strength. Adv. Mater. 15, 1155-1158 (2003).
- Tanaka, Y. et al. Determination of fracture energy of high strength double network hydrogels. J. Phys. Chem. B 109, 11559-11562 (2005).
- Tsukeshiba, H. et al. Effect of polymer entanglement on the toughening of double network hydrogels. J. Phys. Chem. B 109, 16304-16309 (2005).
- Huang, M. et al. Importance of entanglement between first and second components in high-strength double network gels. Macromolecules 40, 6658-6664 (2007).
- Guo, X. et al. Strong and tough fibrous hydrogels reinforced by multiscale hierarchical structures with multimechanisms. Sci. Adv. 9, eadf7075 (2023).
- Li, W. et al. Tough Hydrogels with Isotropic and Unprecedented Crack Propagation Resistance. Adv. Funct. Mater. 32, 220734 (2022).
- Hua, M. et al. Strong tough hydrogels via the synergy of freezecasting and salting out. Nature 590, 594-599 (2021).
- Xu, L., Qiao, Y. & Qiu, D. Coordinatively stiffen and toughen hydrogels with adaptable crystal-domain cross-linking. Adv. Mater. 35, 2209913 (2023).
- Fu, L. et al. Cartilage-like protein hydrogels engineered via entanglement. Nature 618, 740-747 (2023).
- Sun, J. Y. et al. Highly stretchable and tough hydrogels. Nature 489, 133-136 (2012).
- He, G. et al. Strong and reversible covalent double network hydrogel based on force-coupled enzymatic reactions. Angew. Chem. Int. Ed. 134, e202201765 (2022).
- Hua, Y. et al. Ultrafast, tough, and adhesive hydrogel based on hybrid photocrosslinking for articular cartilage repair in water-filled arthroscopy. Sci. Adv. 7, eabg0628 (2021).
- Zhang, M. et al. Toughening double-network hydrogels by polyelectrolytes. Adv. Mater. 35, 2301551 (2023).
- Li, G. et al. Highly conducting and stretchable double-network hydrogel for soft bioelectronics. Adv. Mater. 34, 2200261 (2022).
- Chen, H. et al. Super bulk and interfacial toughness of physically crosslinked double-network hydrogels. Adv. Funct. Mater. 27, 1703086 (2017).
- Liu, S., Bastola, A. K. & Li, L. A 3D printable and mechanically robust hydrogel based on alginate and graphene oxide. ACS Appl. Mater. Interfaces 9, 41473-41481 (2017).
- Gaharwar, A. K., Peppas, N. A. & Khademhosseini, A. Nanocomposite hydrogels for biomedical applications. Biotechnol. Bioeng. 111, 441-453 (2014).
- Liu, D. et al. Tough, transparent, and slippery PVA hydrogel led by syneresis. Small 19, 2206819 (2023).
- Dong, X. et al. Strong and tough conductive organo-hydrogels via freeze-casting assisted solution substitution. Adv. Funct. Mater. 32, 2203610 (2022).
- Kim, J., Zhang, G., Shi, M. & Suo, Z. Fracture, fatigue, and friction of polymers in which entanglements greatly outnumber cross-links. Science 374, 212-216 (2021).
- Xiong, X. et al. Polymerizable rotaxane hydrogels for threedimensional printing fabrication of wearable sensors. Nat. Commun. 14, 1331 (2023).
- Lei, H. et al. Stretchable hydrogels with low hysteresis and antifatigue fracture based on polyprotein cross-linkers. Nat. Commun. 11, 4032 (2020).
- Meng, X. et al. Hysteresis-free nanoparticle-reinforced hydrogels. Adv. Mater. 34, 2108243 (2022).
- Hui, C. Y., Bennison, S. J. & Londono, J. D. Crack blunting and the strength of soft elastic solids. Proc. R. Soc. Lond. A. 459, 1489-1516 (2003).
- Wang, C. et al. Elastic materials with high strength and low hysteresis. Proc. Natl Acad. Sci. USA 116, 5967-5972 (2019).
- Liu, C. et al. Tough hydrogels with rapid self-reinforcement. Science 372, 1078-1081 (2021).
- Wang, J. et al. Low Hysteresis Hydrogel Induced by Spatial Confinement. Adv. Funct. Mater. 33, 2214935 (2023).
- Zhong, M. et al. Quantifying the impact of molecular defects on polymer network elasticity. Science 353, 1264-1268 (2016).
- Yang, C., Yin, T. & Suo, Z. Polyacrylamide hydrogels. I. Network imperfection. J. Mech. Phys. Solids 131, 43-55 (2019).
- Lin, S. & Zhao, X. Fracture of polymer networks with diverse topological defects. Phys. Rev. E 102, 052503 (2020).
- Flory, P. J. Principles of Polymer Chemistry (Cornell University Press, Ithaca, 1953).
- Treloar, L. R. G. The Physics of Rubber Elasticity (Oxford University Press, Oxford, 1975.)
- Li, C. et al. Effects of network structures on the fracture of hydrogel. Extrem. Mech. Lett. 49, 101495 (2021).
- Di Lorenzo, F. & Seiffert, S. Nanostructural heterogeneity in polymer networks and gels. Polym. Chem. 6, 5515-5528 (2015).
- Seiffert, S. Origin of nanostructural inhomogeneity in polymernetwork gels. Polym. Chem. 8, 4472-4487 (2017).
- Seiffert, S. Scattering perspectives on nanostructural inhomogeneity in polymer network gels. Prog. Polym. Sci. 66, 1-21 (2017).
- Fukao, K. et al. Effect of relative strength of two networks on the internal fracture process of double network hydrogels as revealed by in situ small-angle X-ray scattering. Macromolecules 53, 1154-1163 (2020).
- Nakajima, T. et al. A universal molecular stent method to toughen any hydrogels based on double network concept. Adv. Funct. Mater. 22, 4426-4432 (2012).
- Zheng, J. et al. One-pot synthesis of highly mechanical and recoverable double network hydrogels using thermoreversible sol-gel polysaccharide. Adv. Mater. 25, 4171-4176 (2013).
- Chen, Q. et al. A novel design strategy for fully physically linked double network hydrogels with tough, fatigue resistant, and selfhealing properties. Adv. Funct. Mater. 25, 1598-1607 (2015).
- Liu, S. & Lin, L. Ultrastretchable and self-healing double-network hydrogel for 3D printing and strain sensor. ACS Appl. Mater. Interfaces 9, 26429-26437 (2017).
- Zhang, X. et al. Supramolecular nanofibrillar hydrogels as highly stretchable, elastic and sensitive ionic sensors. Mater. Horiz. 6, 326-333 (2019).
- Yang, Y. et al. Highly elastic and ultratough hybrid ionic-covalent hydrogels with tunable structures and mechanics. Adv. Mater. 30, 1707071 (2018).
- Shan, X., Song, S. & Gao, G. Robust and flexible strain sensors based on dual physically cross-linked double network hydrogels for monitoring human-motion. Chem. Eng. J. 354, 817-824 (2018).
- Benitez-Duif, P. A. et al. Ultrastrong Poly (2-Oxazoline)/Poly (Acrylic Acid) double-network hydrogels with cartilage-like mechanical properties. Adv. Funct. Mater. 32, 2204837 (2022).
- Hirsch, M., Alvaro, C. & Esther, A. 3D printing of strong and tough double network granular hydrogels. Adv. Funct. Mater. 31, 2005929 (2021).
- Chong, J. et al. Highly conductive tissue-like hydrogel interface through template-directed assembly. Nat. Commun. 14, 2206 (2023).
- Nian, G., Kim, J., Bao, X. & Suo, Z. Making highly elastic and tough hydrogels from doughs. Adv. Mater. 34, 2206577 (2022).
- Zhang, G. et al. Hydrogels of arrested phase separation simultaneously achieve high strength and low hysteresis. Sci. Adv. 9, eadh7742 (2023).
- Kuhn, W. & Grün, F. Beziehungen zwischen elastischen Konstanten und Dehnungsdoppelbrechung hochelastischer Stoffe. Kolloid-Z. 101, 248-271 (1942).
- Treloar, L. R. G. The photo-elastic properties of rubber. Part I: Theory of the optical properties of strained rubber. Trans. Faraday Soc. 43, 277-284 (1947).
- Sørensen, B. E. A revised Michel-Lévy interference colour chart based on first-principles calculations. Eur. J. Mineral. 25, 5-10 (2013).
- Mai, T. T., Okuno, K., Tsunoda, K. & Urayama, K. Crack-tip strain field in supershear crack of elastomers. ACS Macro Lett. 9, 762-768 (2020).
- Zhang, T., Lin, S., Yuk, H. & Zhao, X. Predicting fracture energies and crack-tip fields of soft tough materials. Extrem. Mech. Lett. 4, 1-8 (2015).
- Long, R., Hui, C. Y., Gong, J. P. & Bouchbinder, E. The fracture of highly deformable soft materials: A tale of two length scales. Annu. Rev. Condens. Matter Phys. 12, 71-94 (2021).
Acknowledgements
Author contributions
Competing interests
Additional information
supplementary material available at
https://doi.org/10.1038/s41467-024-45485-8.
© The Author(s) 2024
School of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Shanghai 200240, China. State Key Laboratory of Metal Matrix Composites, Shanghai Jiao Tong University, Shanghai 200240, China. e-mail: xlwang@sjtu.edu.cn