يؤدي STING إلى تفاقم إصابة نقص تروية القلب الناتجة عن الفيروبتوزيس من خلال استهداف GPX4 للتدهور الذاتي. STING aggravates ferroptosis-dependent myocardial ischemia-reperfusion injury by targeting GPX4 for autophagic degradation
على الرغم من التقدم في تقنيات إعادة التروية التاجية التدخلية بعد احتشاء عضلة القلب، لا يزال جزء ملحوظ من المرضى يعانون من ارتفاع معدلات الوفيات نتيجة إصابة نقص تروية القلب (MI/R). إن الفهم العميق للآليات الكامنة وراء إصابة MI/R أمر حاسم لوضع استراتيجيات لتقليل الضرر القلبي وتعزيز بقاء المرضى. هنا، تم اكتشاف أنه خلال MI/R، يتراكم إشارة DNA مزدوج الشريطة (dsDNA)-حلقة GMP-AMP (cGAS) المحفز لجينات الإنترفيرون (STING)، مصحوبة بمعدلات عالية من الفيروبتوز القلبي. لقد أظهر الحذف المحدد لـ cgas أو Sting في خلايا عضلة القلب، مما أدى إلى تثبيط الإجهاد التأكسدي، أنه يمكن أن يخفف من الفيروبتوز وإصابة I/R. على العكس، فإن تنشيط STING يؤدي إلى تفاقم الفيروبتوز وإصابة I/R. من الناحية الميكانيكية، يستهدف STING مباشرةً إنزيم الجلوتاثيون بيروكسيداز 4 (GPX4) لتسهيل تدهوره من خلال البلعمة الذاتية، من خلال تعزيز اندماج الأوتوفاغوسومات والليزوزومات. تساهم هذه المحور STING-GPX4 في الفيروبتوز القلبي وتشكل دائرة تغذية راجعة إيجابية. إن حجب تفاعل STING-GPX4 من خلال الطفرات في T267 من STING أو N146 من GPX4 يثبت GPX4. من الناحية العلاجية، فإن إدارة GPX4 بواسطة AAV تخفف من الفيروبتوز الناتج عن STING، مما يؤدي إلى تحسين التعافي الوظيفي القلبي من إصابة MI/R. بالإضافة إلى ذلك، فإن تثبيط STING بواسطة H-151 يثبت GPX4 لعكس الفيروبتوز الناتج عن GPX4 وتخفيف إصابة MI/R. بشكل جماعي، تم تحديد آلية جديدة تعتمد على البلعمة الذاتية للفيروبتوز في هذه الدراسة. على وجه التحديد، تؤدي البلعمة الذاتية لـ STING الناتجة عن نقص الأكسجين أو نقص التروية إلى تدهور GPX4، مما يقدم هدفًا علاجيًا واعدًا لأمراض القلب المرتبطة بـ I/R.
تظل أمراض القلب الإقفارية السبب الرئيسي للمراضة والوفيات على مستوى العالم. على الرغم من أن إعادة التروية التاجية التدخلية في الوقت المناسب فعالة في إنقاذ عضلة القلب الإقفارية، إلا أن إصابة نقص تروية القلب (MI/R) لا تزال تسهم بشكل كبير في الوفيات في عدد كبير من المرضى.حتى الآن، على الرغم من الأبحاث المكثفة التي استمرت لعقود، لم يظهر أي عامل مستهدف لإصابة MI/R في المجال السريري.هناك ضرورة طبية ملحة لفهم شامل وعميق للآليات المعقدة التي تحكم موت خلايا عضلة القلب. إن فهم هذه الآليات أمر أساسي لأنه يمكن أن يفتح اكتشاف أهداف علاجية جديدة ومبتكرة، والتي هي في غاية الأهمية للإدارة الفعالة والفعالة للحالة المرضية المرتبطة.
موت الخلايا، وخاصة موت خلايا عضلة القلب (CMs)، هو جانب حاسم ضمن الفيزيولوجيا المرضية لأمراض القلب والأوعية الدموية. من بين أشكال موت الخلايا، يعتبر الفيروبتوز، وهو نمط موت خلايا يعتمد على الحديد يتميز بزيادة أكسدة الدهون، عاملًا رئيسيًا في الفيزيولوجيا المرضية لإصابة MI/R.يتم تنظيم الفيروبتوز بواسطة استقلاب
الحديد، والدهون، والأحماض الأمينية، والجلوتاثيون، وهو مرتبط ارتباطًا وثيقًا بالعديد من حالات القلب. وهذا يجعل استهداف الفيروبتوز نهجًا علاجيًا واعدًا لإصابة MI/R، وبالتالي قد يحدث ثورة في استراتيجيات العلاج لهذه الحالة الشائعة والتهديدة للحياة. لقد أصبح إنزيم الجلوتاثيون بيروكسيداز 4 (GPX4)، الذي يقوم بتحويل بيروكسيدات الدهون إلى كحوليات دهنية أقل ضررًا، عاملًا تنظيميًا محوريًا في تثبيط الفيروبتوز. لقد أظهرت الدراسات السابقة أن الفيروبتوز الناتج عن MI/R يتزامن مع تثبيط GPX4. خلال عملية MI/R، يتزامن انخفاض مستويات GPX4 مع بدء الفيروبتوز. على العكس، فإن زيادة مستويات GPX4 تخفف بشكل فعال من إصابة عضلة القلب وتعزز من وظيفة القلب.ومع ذلك، لا تزال التنظيم الدقيق لمستويات بروتين GPX4 وآلية تدهوره الأساسية غير واضحة. لذلك، قد يمثل اكتشاف العوامل التي تنظم GPX4 هدفًا علاجيًا جذابًا لتعديل الفيروبتوز الذي يحدث خلال عملية MI/R.
يعتبر إنزيم حلقة الجوانين أحادي الفوسفات (GMP)-أدينوسين أحادي الفوسفات (AMP) (cGAMP) (cGAS) ومحفز جينات الإنترفيرون (STING) مكونات مناعية محورية ومحمية بشدة ضمن نظام المناعة الثديي.
إنها جزء لا يتجزأ من آلية الدفاع في الجسم ضد الأحماض النووية المرضية، مع تركيز خاص على DNA مزدوج الشريطة (dsDNA).عند ارتباط DNA غير الطبيعي، ينتج cGAS رسولًا ثانيًا cGAMP. ثم يشارك cGAMP الذي تم تصنيعه حديثًا في تفاعل محدد مع STING، مما يعزز تغييرات في شكله. بمجرد تنشيطه، يرتبط STING بإنزيم TANK-binding kinase 1 (TBK1) وعامل تنظيم الإنترفيرون 3 (IRF3). يؤدي هذا الارتباط إلى سلسلة معقدة من الأحداث الجزيئية، والتي تنتهي في النهاية ببدء التعبير عن الإنترفيرون (IFN).يساهم إنتاج IFN ليس فقط في استجابة الجسم الالتهابية ولكن أيضًا في إحداث تأثيرات مضادة للفيروسات قوية. في السنوات الأخيرة، أبلغ عدد متزايد من الدراسات أن خلل تنظيم الإشارات المرتبطة بـ STING يمكن ملاحظته في مجموعة واسعة من الأمراض البشرية. تشمل هذه الأمراض، ولكن لا تقتصر على، أمراض IFN من النوع الأول،حيث يتم تعطيل مسارات إشارات IFN الطبيعية، وفي السرطان،حيث ينظم STING المناعة المضادة للسرطان بطرق تعتمد على IFN وغير تعتمد على IFN. علاوة على ذلك، كشفت العديد من التقارير أن STING يعمل بشكل مستقل عن المسارات المضادة للفيروسات التقليدية، مثل الشيخوخةوالبلعمة الذاتية.يلعب STING دورًا في دفع شيخوخة الأنسجة وتدهور الأنسجة المرتبط بالشيخوخة المبكرة، وتمثل البلعمة الذاتية وظيفة قديمة متأصلة في مسار cGAS-STING. في الواقع، يحدث جزء كبير من تقدم المرض وموت الخلايا الذي يتم تعديله بواسطة STING بشكل مستقل عن IRF3 وتعبير الجينات المضادة للفيروسات لـ STING. يحدث تطور اعتلال الأوعية المرتبط بـ STING دون الاعتماد على IRF3.يعمل STING البشري، الذي يعمل كقناة بروتون، على فصل قدرته على تحفيز الإنترفيرون عن أدواره الرئيسية في تعزيز ترطيب LC3B وتنشيط inflammasome.في الفاجوسومات، تفاعل STING مباشرة مع Src، وهذا التفاعل أعاق Src من تجنيد Syk وفوسفوريليته.إن استكشاف الوظائف المناعية غير التقليدية لـ STING من المقرر أن يثري بشكل كبير فهمنا لتعقيد المناعة.
في تحقيقنا، استكشفنا كيف ينظم STING الفيروبتوز القلبي عندما تتعرض القلب لإصابة MI/R. تؤدي الأضرار الميتوكوندرية الناتجة عن I/R إلى إطلاق dsDNA، الذي يتم التعرف عليه بعد ذلك بواسطة cGAS، مما يؤدي إلى إنتاج الرسول الثاني cGAMP وتنشيط STING. يمكن أن يؤدي STING مباشرة إلى تحفيز الفيروبتوز القلبي من خلال تفاعله مع GPX4. بمجرد ارتباط STING بـ GPX4، فإنه يحفز زيادة البلعمة الذاتية، مما يؤدي بدوره إلى التدهور اللاحق لـ GPX4، مما يغذي عملية الفيروبتوز القلبي. كما قمنا بتقييم استراتيجيتين علاجيتين فعالتين ضد إصابة MI/R، بما في ذلك استخدام زيادة التعبير عن GPX4 بواسطة AAV وإدارة مضادات STING. في ضوء أبحاثنا، تقدم النتائج التي حصلنا عليها وجهات نظر جديدة حول آلية تطوير الفيروبتوز وتؤسس STING كهدف علاجي محتمل لمنع إصابة MI/R، لتحسين نتائج المرضى وتقليل معدلات الوفيات المرتبطة بأمراض القلب الإقفارية.
النتائج
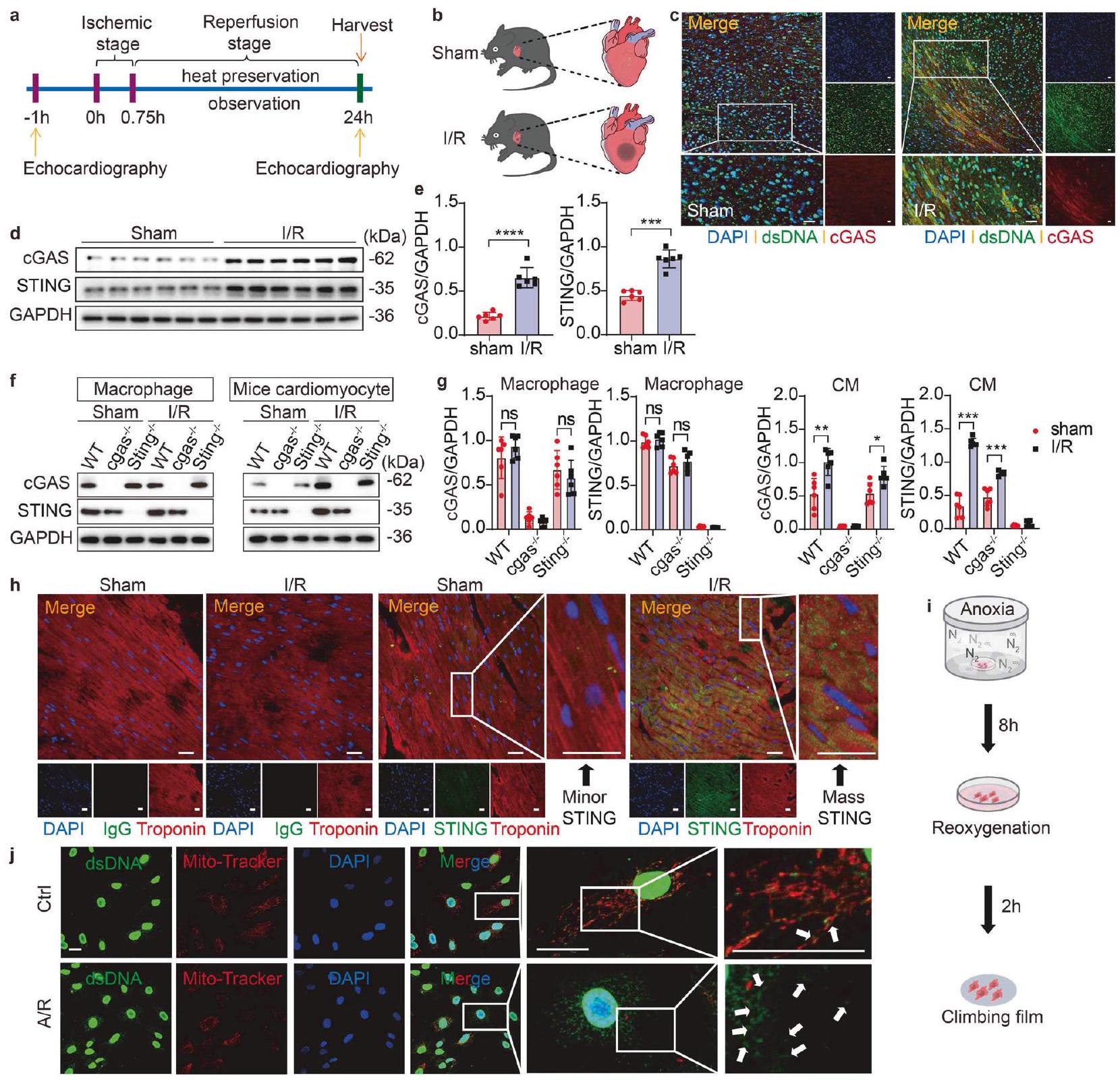
يؤدي I/R إلى زيادة تنظيم cGAS-STING في CMs للبدء في تحديد تأثيرات استشعار الحمض النووي بواسطة cGAS في إصابة نقص التروية/إعادة التروية، قمنا أولاً باختبار التغيرات في مستويات الحمض النووي السيتوزولي. لتوضيح ما إذا كانت إصابة القلب بنقص التروية/إعادة التروية قد تسببت في تلف الحمض النووي للخلايا، قمنا في البداية بإعداد نموذج نقص التروية/إعادة التروية باستخدام فئران ذكرية من سلالة C57BL/6J تبلغ من العمر 8 أسابيع (الشكل 1 أ، ب) وقمنا بتحليل التلوين المناعي المشترك والتعبير عن الحمض النووي مزدوج الشريط (dsDNA) وcGAS في منطقة حدود نقص التروية/إعادة التروية. لوحظت كميات متزايدة بشكل ملحوظ من الحمض النووي السيتوزولي وتنشيط cGAS في خلايا الفئران التي خضعت لنقص التروية/إعادة التروية مقارنةً بالتحكمات التي خضعت لعملية وهمية (الشكل 1 ج)، مما يشير إلى تلف عميق في الحمض النووي وتسرب الحمض النووي إلى السيتوسول في الأنسجة المريضة. بعد التعرف على dsDNA، يخضع cGAS للتنشيط. لمزيد من التحقيق، استخدمنا اختبارات Western blot لتقييم وفرة cGAS وSTING. أظهرت النتائج زيادة ملحوظة في تعبير كل من cGAS وSTING في منطقة الحدود الناتجة عن نقص التروية، والتي حدثت بعد 45 دقيقة من نقص تروية القلب تلتها 24 ساعة من إعادة التروية (الشكل 1d، e). ومع ذلك، لم تظهر مستويات بروتين cGAS وSTING في نواة الاحتشاء تغييرات ملحوظة مقارنة بمجموعة الشام (الشكل التكميلية 1a)، وكانت التغيرات ضئيلة في المنطقة غير الإقفارية، التي لم تتأثر بنقص التروية (الشكل التكميلية 1b). بالتوازي، أظهرت نتائج صبغة المناعية الفلورية لشرائح الأنسجة من كل من مجموعتي نقص التروية والشام أن تعبير STING قد زاد بشكل ملحوظ داخل المنطقة الإقفارية (منطقة الحدود) لمجموعة نقص التروية ولكنه لم يظهر تغييرات ملحوظة في نواة الاحتشاء أو المنطقة غير الإقفارية (الشكل التكميلية 1c).
بالإضافة إلى ذلك، لاكتشاف دليل على تنشيط مسار cGAS-STING بعد نقص التروية/إعادة التروية، قمنا بعزل خلايا العضلة القلبية، والخلايا الليفية، وسكان البلعميات من قلوب الفئران البرية (WT) وcgas.أو ستينغالفئران التي تتبع البروتوكولات المعتمدة، مما أتاح لنا التحقيق في الأدوار المحددة لـ cGAS و STING في أنواع خلايا القلب المختلفة بعد نقص التروية/إعادة التروية. تم تأكيد زيادة cGAS و STING في خلايا القلب من المنطقة الحدودية للقلب بعد نقص التروية/إعادة التروية على مستوى البروتين. ومن الجدير بالذكر أن هذه الزيادات كانت ضئيلة في خلايا القلب المفقودة لـ cgas أو Sting (الشكل 1f، g). ومع ذلك، لم تُلاحظ أي اختلافات واضحة في البلعميات أو الألياف، بغض النظر عما إذا كانت قد خضعت لنقص التروية/إعادة التروية (الشكل 1f، g، الشكل التكميلية 1d). نظرًا لأن النتائج المذكورة أعلاه تشير إلى اعتماد دور cGAS في نقص التروية/إعادة التروية على STING، انتقلنا بعد ذلك إلى التركيز على STING. ثم قمنا بتصميم تجربة تلوين مناعي مشترك لتوضيح نمط التعبير عن STING في خلايا القلب. كشفت صبغة المناعة الفلورية عن زيادة كبيرة في تعبير STING في خلايا القلب الموجودة في المنطقة الحدودية للقلب بعد نقص التروية/إعادة التروية (الشكل 1h)، على عكس التحكم السلبي.
للتحقق من نتائجنا خارج الجسم، أنشأنا نموذج نقص الأكسجين/إعادة الأكسجة (A/R) باستخدام خلايا عضلة القلب الأولية من الفئران (MPCs) المستخرجة من قلوب الفئران (الشكل 1i). تم استخدام تحليل التلوين المشترك لبروتينات الميتوكوندريا (Mito-Tracker) وdsDNA لتحديد الحمض النووي السيتوزولي الذي لم يتواجد في نفس المكان مع النوى أو الميتوكوندريا. ومن المRemarkable، لاحظنا إطلاقًا كبيرًا للـ dsDNA، مصحوبًا بتلف شديد في الميتوكوندريا يظهر على شكل تضخم، تقصير، وتثخين بعد A/R (الشكل 1j)، على عكس MPCs الضابطة.
تشير هذه النتائج معًا إلى أن إشارة dsDNA-cGAS-STING يتم تنشيطها في خلايا عضلة القلب خلال نقص التروية/إعادة التروية. وجهنا تركيزنا نحو فهم الدور المحدد لـ cGAS و STING في خلايا عضلة القلب خلال إصابة نقص التروية.
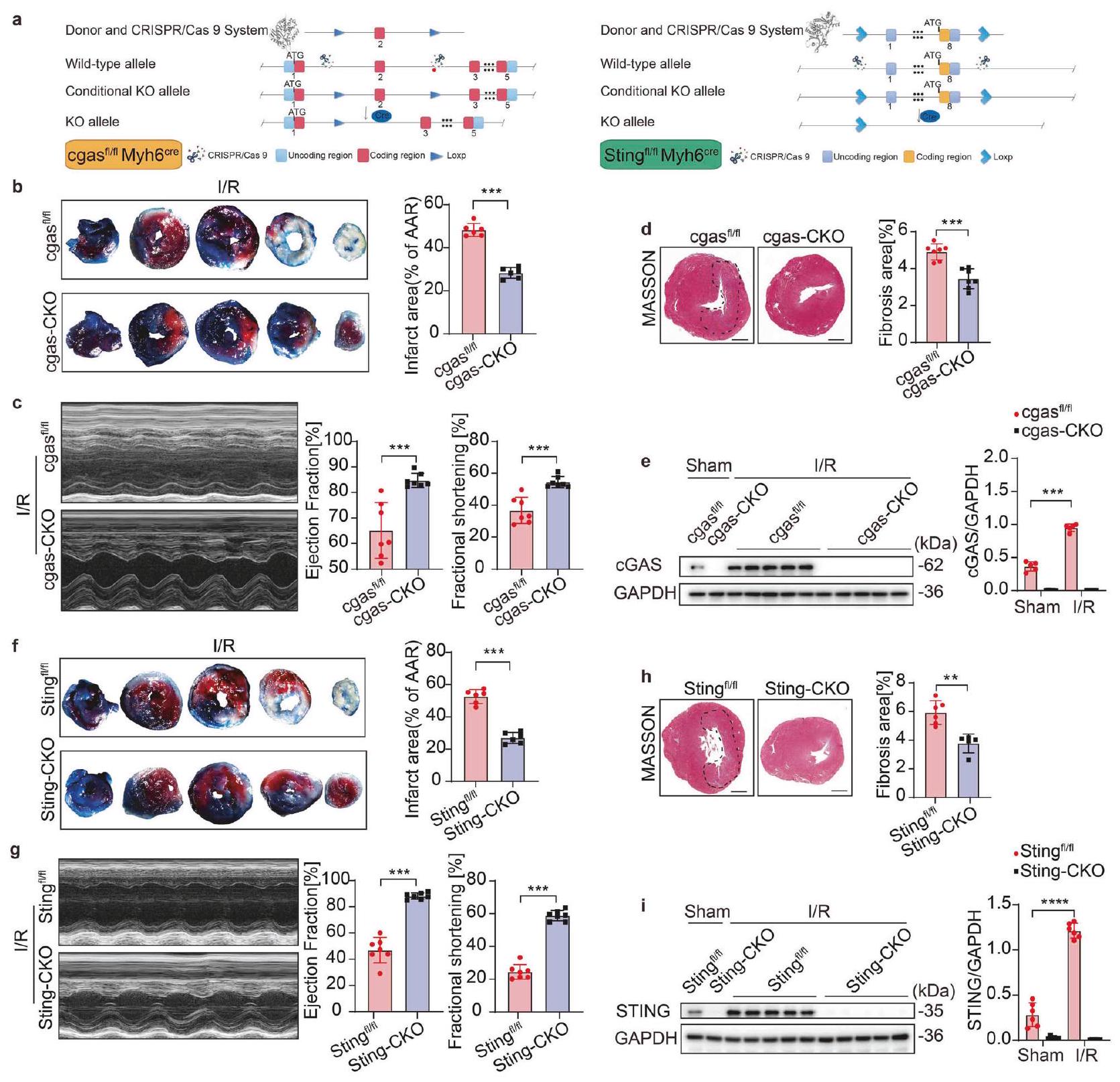
حذف cGAS-STING يحمي من إصابة MI/R دور cGAS أو STING في خلايا القلب غير مفهوم جيدًا. بعد ذلك، قمنا بإنشاء طفرات محددة لخلايا القلب في جين cgas [cgasماي إتش 6 (cgasCKO)] و Sting-knockout [Stingماي إتش 6فئران (Sting-CKO) للتحقيق في دور ووظيفة cGAS و STING المحددين لـ CM في إصابة MI/R (الشكل 2أ). cgas و سيغاس ماي إتش 6، بالإضافة إلى ستينغ وستينغ ماي إتش 6تعرضت للإصابة الناتجة عن نقص التروية. تم تقييم إصابة القلب من خلال قياس مساحة النخر في مقاطع القلب، وتقييم وظيفة القلب، وقياس صبغة ماسون. كانت وظيفة القلب في البداية، الممثلة بنسبة قذف البطين الأيسر ونسبة القصر (LVEF وFS على التوالي)، مشابهة بين الحيوانات المعدلة وراثيًا (KO) وحيوانات التحكم الخاصة بها، دون ملاحظة أي اختلافات ذات دلالة إحصائية (الشكل التوضيحي 1e-h). تؤكد هذه النتائج أن الحيوانات المعدلة وراثيًا لا تظهر أي شذوذات داخلية في وظيفة القلب قبل الإصابة. وفقًا للنتائج، أظهرت فئران cgasCKO مساحة نخر أقل، ووظيفة قلبية أفضل بكثير (مستويات EF وFS) ومنطقة تليف أقل بعد الإصابة الناتجة عن نقص التروية مقارنة بفئران التحكم الخاصة بها (الشكل 2b-d)، مما يشير إلى دور وقائي لقصور cgas المحدد لعضلة القلب ضد إصابة MI/R. أظهرت صبغة ماسون لنسج القلب بعد 7 أيام من إعادة التروية تليفًا أقل في فئران cgas- أو Sting-CKO (الشكل التوضيحي 1i، j).
الشكل 1 تحفز I/R زيادة cGAS-STING في خلايا القلب. تم إخضاع ذكور الفئران (بعمر 8 أسابيع) للعملية وتم euthanized بعد 24 ساعة من I/R أو Sham. أ عرض مخطط سير عملية نموذج I/R للفأر. ب رسم تخطيطي يوضح نموذج إصابة I/R للفأر. ج تحليل مزدوج مناعى للكشف عن dsDNA وcGAS في نفس قسم القلب لمنطقة الحدود WT بعد I/R أو Sham. يتم عرض التفاعلات الإيجابية لشرائح الأنسجة باللون الأخضر (dsDNA) والأحمر (cGAS). يتم عرض التفاعل الإيجابي للتعايش باللون الأصفر. مقياس الرسمم. د، تحليل Western blot وقياس cGAS وSTING في خلايا WT المعزولة من منطقة الحدود بعد I/R أو Sham ). التحليل الغربي وقياس cGAS وSTING في أنواع خلايا مختلفة معزولة من منطقة حدود القلب لـ cgasستينغأو فئران WT بعد الإقفار/إعادة التروية أو الشام. تحليل التألق المناعي المزدوج للكشف عن علامات CMs وSTING في نفس قسم القلب لمنطقة الحدود WT بعد I/R أو Sham. يتم عرض التفاعلات الإيجابية لشرائح الأنسجة باللون الأخضر (STING) والأحمر (تروبونين). يتم عرض التفاعل الإيجابي للتعايش باللون الأصفر. شريط القياس. مخطط تخطيطي يوضح تدفق عملية CM A/R. j تحليل المناعة المزدوجة للكشف عن dsDNA والميتوكوندريا (الموسومة بـ Mito-Tracker) في MPCs بعد A/R. يتم عرض التفاعلات الإيجابية باللون الأخضر (dsDNA) والأحمر (Mito-Tracker). يتم عرض التفاعل الإيجابي للتعايش في اللون الأصفر. تشير الأسهم إلى dsDNA المحرر. شريط القياستُعبر البيانات عن المتوسط ± الانحراف المعياري القياسي. NS غير دالة. و ****P < 0.0001 (اختبار ستودنت ذو الذيلين غير المتزاوج)اختبار). I/R نقص التروية وإعادة التروية، dsDNA الحمض النووي مزدوج الشريط، A/R نقص الأكسجين/إعادة الأكسجة، cGAS سينثاز أحادي فوسفات الجوانوزين-أدينوزين، STING منبه جينات الإنترفيرون، WT النوع البري، cgasفئران knockout لـ cgas، Stingفئران ستينغ knockout، خلايا عضلة القلب CM
لتوضيح دور cGAS في إصابة MI/R، قمنا بعزل خلايا العضلة القلبية من فئران cgas-CKO وفئران التحكم في نموذج I/R داخل الجسم. أظهر تحليل Western blot أن مستوى تعبير cGAS في خلايا العضلة القلبية كان ضئيلاً تحت الظروف الفسيولوجية، بينما أصبح قابلاً للتحفيز بعد I/R (الشكل 2e). من المهم أن نلاحظ أن زيادة cGAS في خلايا العضلة القلبية الناتجة عن I/R كانت غائبة في خلايا العضلة القلبية cgas-CKO.
من المثير للاهتمام أن نتائج مشابهة لوحظت أيضًا في فئران Sting-CKO (الشكل 2f-h). نظرًا لأن STING هو عامل فعال في مجرى cGAS، فإن غياب Sting في فئران Sting-CKO أدى إلى تقليل مساحة النخر، وتحسين وظيفة القلب، وتقليل مساحة التليف مقارنة بفئران التحكم. كما يمكن أن يؤدي نقص التروية وإعادة التروية (I/R) إلى تحفيز تعبير STING في Sting.لم يكن لتأثير نقص التروية/إعادة التروية أي تأثير على مستوى بروتين STING في
الشكل 2 حذف cGAS-STING يحمي من إصابة MI/R. أ رسم تخطيطي لنمط الهيكل لـ cgas أو Sting CM-specific (Myh6-iCre) فئران knockout الشرطية. ب-هـ تأثير cgas-CKO على حجم احتشاء عضلة القلب، FS، EF ومنطقة التليف بعد I/R أو Sham: ب حجم احتشاء عضلة القلب (% من AAR) مع قسم نسيجي تمثيلي.” ); جراحة القلب والأوعية الدموية وقياس نسبة الكسر القذفي (EF%) ونسبة الكسر الانقباضي (FS%) ( ); صبغة ماسون وقياس مساحة التليف شريط القياس; التحليل الغربي وقياس cGAS في خلايا القلب المعزولة من منطقة حدود القلب ( ). تأثير Sting-CKO على حجم احتشاء عضلة القلب، FS، EF ومنطقة التليف بعد I/R: حجم احتشاء عضلة القلب (% من AAR) مع قطع نسيج تمثيلي تخطيط صدى القلب وقياس EF%، وFS% صبغة ماسون وقياس منطقة التليف% , شريط القياس ; التحليل الغربي وقياس STING في خلايا القلب المعزولة من منطقة حدود القلب ( ). المتوسط SEM، , , و. منطقة AAR المعرضة للخطر، IF منطقة الاحتشاء، cgas-CKO cgas Myh , Sting-CKO Sting Myh , FS نسبة الانكماش، EF نسبة الطرد، KO الإزالة
خلايا القلب Sting-CKO. تشير هذه النتائج إلى أن cGAS-STING يلعب دورًا مدمرًا في إصابة القلب بعد I/R، وأن تفاقم إصابة MI/R بواسطة cGAS يعتمد على تنشيط STING.
STING يعزز الفيروبتوسيس القلبي من خلال تعديل إصابة الإجهاد التأكسدي
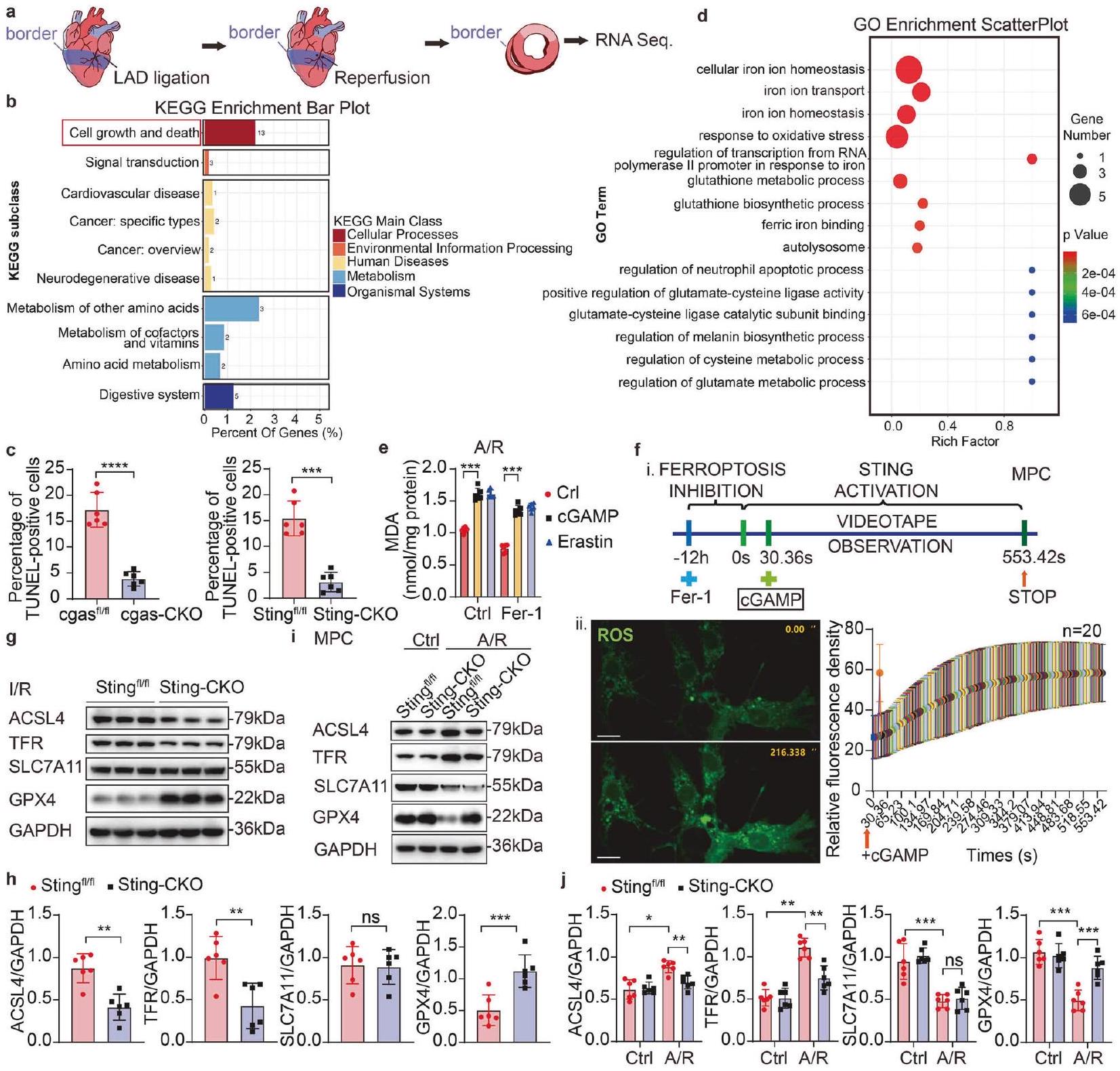
لتوضيح المسار المحدد الذي يساهم من خلاله STING في إصابة MI/R، تم إجراء تسلسل RNA (RNA-seq) على مقاطع القلب من فئران Sting-CKO وفئران التحكم الخاصة بهم بعد I/R (الشكل 3a). أظهر التحليل باستخدام موسوعة كيوتو للجينات والجنوم (KEGG) غنىً كبيرًا للجينات المعنية في مسارات نمو الخلايا والموت بين
الجينات المعبر عنها بشكل مختلف (DEGs) (الشكل 3b). بناءً عليه، قمنا بتقييم مدى موت الخلايا في أنسجة القلب وخلايا القلب المستمدة من فئران cgas-CKO أو Sting-CKO، بالإضافة إلى فئران التحكم الخاصة بهم التي تعرضت لإصابة I/R. بشكل ملحوظ، كان هناك زيادة كبيرة في الخلايا الإيجابية لتونيل في القلوب بعد I/R. على العكس، أظهرت كل من فئران cgas-CKO وSting-CKO مستويات منخفضة بشكل ملحوظ من الخلايا الإيجابية لتونيل مقارنة بمجموعات التحكم الخاصة بهم (الشكل التكميلي 2a، b، الشكل 3c، الشكل التكميلي 2e، f)، مما يشير إلى موت أقل للخلايا. نظرًا لأن تونيل يمكنه أيضًا اكتشاف كسور الحمض النووي الناتجة عن النخر أو عمليات موت الخلايا الأخرى، قمنا بقياس مستويات التعبير لمؤشرات موت الخلايا الرئيسية، مثل Pro-caspase-3، Cleaved-caspase3 و
الشكل 3 STING يعزز الفيروبتوسيس القلبي من خلال تعديل إصابة الإجهاد التأكسدي. أ منطقة القلب لتسلسل RNA بعد I/R. ب رسم بياني لزيادة KEGG لتسلسل RNA. ج رسم بياني إحصائي لصورة المناعية للكشف عن تونيل في مقطع القلب من منطقة الحدود لفئران cgas-CKO أو Sting-CKO وفئران التحكم الخاصة بهم بعد I/R . الصورة المقابلة هي الشكل التكميلي 2a، b. د رسم بياني لتوزيع GO لتسلسل RNA. هـ تحليل MDA للكشف عن أكسدة الدهون . تحليل تصوير المناعية للخلايا الحية للكشف عن ROS في MPC. شريط القياس التحليل الغربي وقياس ACSL4، TFR، SLC7A11 وGPX4 في Sting أو Sting-CKO خلايا القلب بعد I/R التحليل الغربي وقياس ACSL4، TFR، SLC7A11 وGPX4 في Sting أو Sting-CKO MPCs بعد A/R أو النورمكسيا . المتوسط ± SEM، , , و
. ROS أنواع الأكسجين التفاعلية، Fer-1 فيروستاتين-1، cGAMP أحادي فوسفات الغوانوزين الحلقي-أدينوزين أحادي الفوسفات، KEGG موسوعة كيوتو للجينات والجنوم، GO علم الأحياء الجيني، LAD الشريان التاجي الأمامي الأيسر النازل، ACSL4 سينثاز أسيل-CoA من عائلة السلسلة الطويلة 4، TFR مستقبل الترانسفيرين، SLC7A11 ناقل السائل العائلي 7، العضو 11، GPX4 بيروكسيداز الجلوتاثيون 4، MDA مالونديالديهايد
B-cell lymphoma-2 (BCL-2)، باستخدام اختبارات التحليل الغربي. أظهرت النتائج عدم وجود تغيير كبير في وفرة البروتين في فئران Sting-CKO بعد I/R مقارنة بفئران التحكم الخاصة بهم (الشكل التكميلي 2c، d). بشكل جماعي، تؤكد هذه النتائج أن غياب STING يقلل من موت الخلايا في القلب بعد إصابة I/R، ولكن ليس من خلال تعديل الموت المبرمج. نظرًا لوجود العديد من أنماط الموت في الخلايا، وفقًا لتحليل زيادة علم الأحياء الجيني (GO)، أظهرت مقاطع القلب من فئران Sting مساهمة بارزة في مسارات الإشارة المتعلقة بالتحلل الذاتي، والإجهاد التأكسدي والفيروبتوسيس بعد I/R مقارنة بفئران Sting-CKO (الشكل 3d). تم استخدام صبغة تونيل أيضًا كعلامة للفيروبتوسيس. بعد ذلك، بحثنا في تأثير STING على المؤشرات الحيوية للفيروبتوسيس، بما في ذلك أكسدة الدهون، تراكم أنواع الأكسجين التفاعلية (ROS) وتدهور GPX4، بالإضافة إلى التعبير عن بعض البروتينات المتعلقة بالفيروبتوسيس. لقد تم التعرف مؤخرًا على أن أكسدة الدهون تلعب دورًا مباشرًا في تسهيل النخر والفيروبتوسيس. للتحقق مما إذا كان STING يعدل الفيروبتوسيس الناتج عن A/R، قمنا بتحليل إنتاج المالونديالديهايد (MDA)، وهو منتج نهائي لأكسدة الدهون. أظهرت النتائج أنه بعد A/R، كان STING فعالًا للغاية في تعزيز إنتاج MDA، مقارنة بالتحكم الإيجابي Erastin (الشكل 3e)، وفي الوقت نفسه،
أدى تنشيط STING إلى حجب تثبيط MDA في الفيروبتوسيس الناتج عن فيروستاتين-1 (Fer-1، مثبط الفيروبتوسيس). تشير هذه الملاحظات إلى أن STING يعمل في أعلى مستوى من أكسدة الدهون، وبالتالي يلعب دورًا محوريًا في تنظيم الفيروبتوسيس.
نظرًا لأن تلف الميتوكوندريا واستجابة الإجهاد التأكسدي هما حدثان محوريان في I/R، لاختبار العلاقة بين STING والإجهاد التأكسدي في القلب، قمنا أولاً بتأكيد وجود إصابة الإجهاد التأكسدي من خلال الكشف عن إنتاج ROS بعد A/R في خلايا القلب. أدى تعرض خلايا القلب المزروعة لـ A/R إلى زيادة في إنتاج ROS، تم معالجة MPCs مسبقًا بـ Fer1 لمدة 12 ساعة لتقليل مستويات ROS قبل تصوير الخلايا الحية، بعد ذلك، تمت إضافة cGAMP في لتنشيط STING، وتم إنهاء الملاحظات والتصوير بمجرد استقرار مستويات ROS (الشكل 3f). من المثير للاهتمام، أن تثبيط ROS في الفيروبتوسيس بواسطة Fer-1 تم عرقلته بشكل كبير في وجود cGAMP. أشارت النتائج إلى أن تنشيط STING يؤدي إلى زيادة في إنتاج ROS.
بعد تحليل تأثير STING على مؤشرات الفيروبتوسيس، بما في ذلك أكسدة الدهون وتراكم ROS، هدفنا بعد ذلك إلى تأكيد الآلية التي من خلالها يعزز STING الفيروبتوسيس من خلال تقييم أكسدة الدهون ووفرة عوامل الفيروبتوسيس. استخدمنا التحليل الغربي لتقييم وفرة التعبير عن سينثاز أسيل-CoA من عائلة السلسلة الطويلة 4 (ACSL4)، مستقبل الترانسفيرين (TFR)، ناقل السائل العائلي 7، العضو 11 (SLC7A11) وGPX4، والتي تم إثبات أنها أهداف بروتينية للفيروبتوسيس. لاحظنا انخفاضات في التعبير عن ACSL4 وTFR، مع ارتفاعات في GPX4 وعدم وجود تغيير كبير في SLC7A11، في فئران Sting-CKO بعد I/R مقارنة بمجموعاتهم الضابطة (الشكل 3g، h). تم تأكيد زيادة الفيروبتوسيس أيضًا على مستوى البروتين في MPCs بعد A/R (الشكل 3i، j). من المثير للاهتمام، أن نقص Sting منع تدهور GPX4 خلال الفيروبتوسيس الناتج عن A/R، بينما يعتبر تدهور GPX4 ضروريًا لتعزيز أكسدة الدهون في الفيروبتوسيس. بعد ذلك، قمنا بقياس مستويات mRNA لـ gpx4 في الأنسجة القلبية من كل من الفئران Sham والفئران المعالجة بـ I/R باستخدام qPCR. أظهرت نتائجنا أن مستويات mRNA لـ gpx4 ظلت دون تغيير بين مجموعتي Sting وSting-CKO بعد I/R (الشكل التكميلي 2h)، مما يشير إلى أن الانخفاض في بروتين GPX4 ليس بسبب تنظيم التعبير الجيني ولكن بدلاً من ذلك بسبب التنظيم بعد الترجمة. مجتمعة، نستنتج أن STING هو منشط قوي للفيروبتوسيس، يسهل تدهور بروتين GPX4، أكسدة الدهون وتراكم ROS. بعد ذلك، تعمقنا في الأحداث الجزيئية التي كانت فيها STING المرتبطة بـ GPX4 الناتج عن I/R.
استهداف GPX4 بواسطة STING
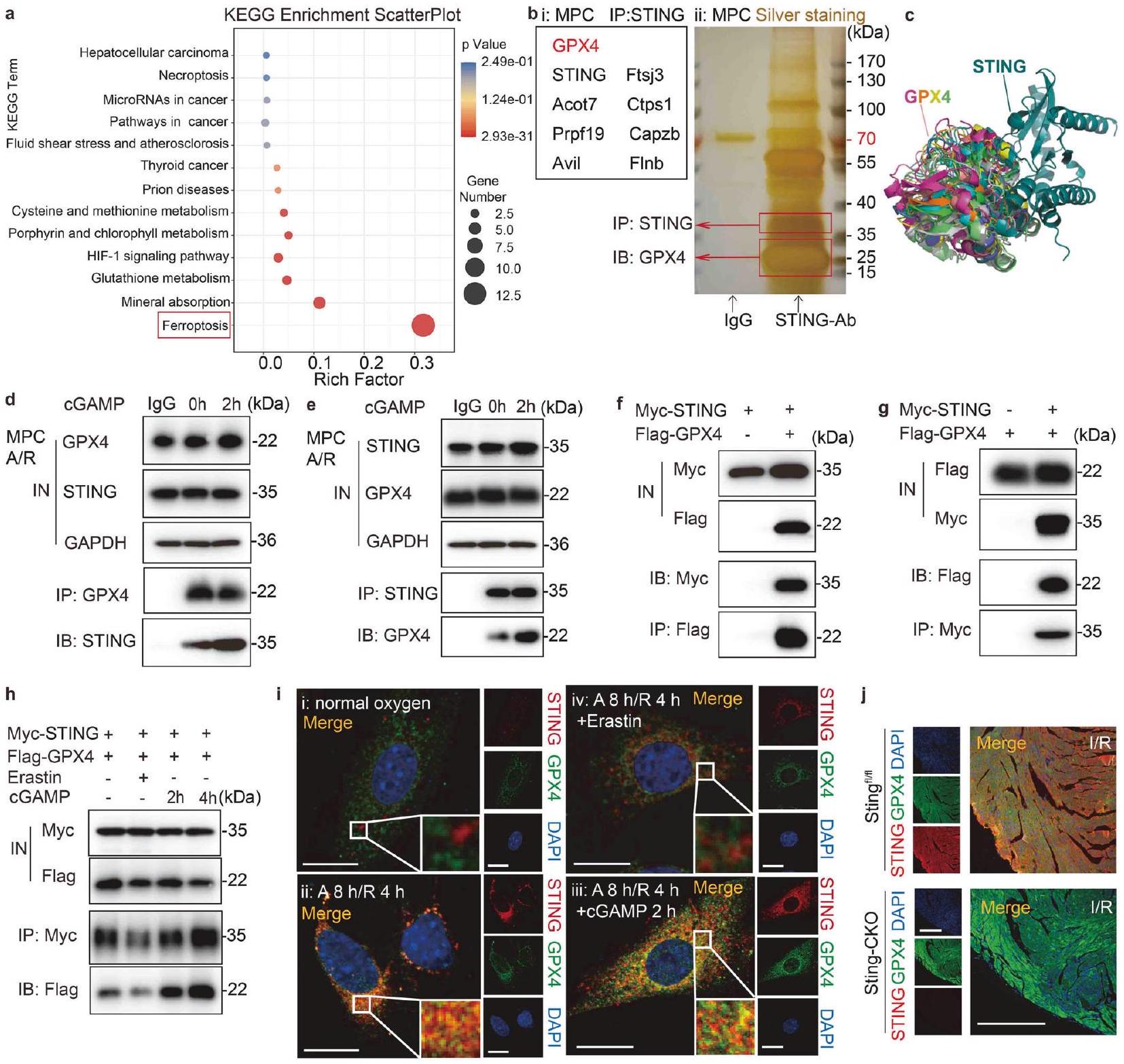
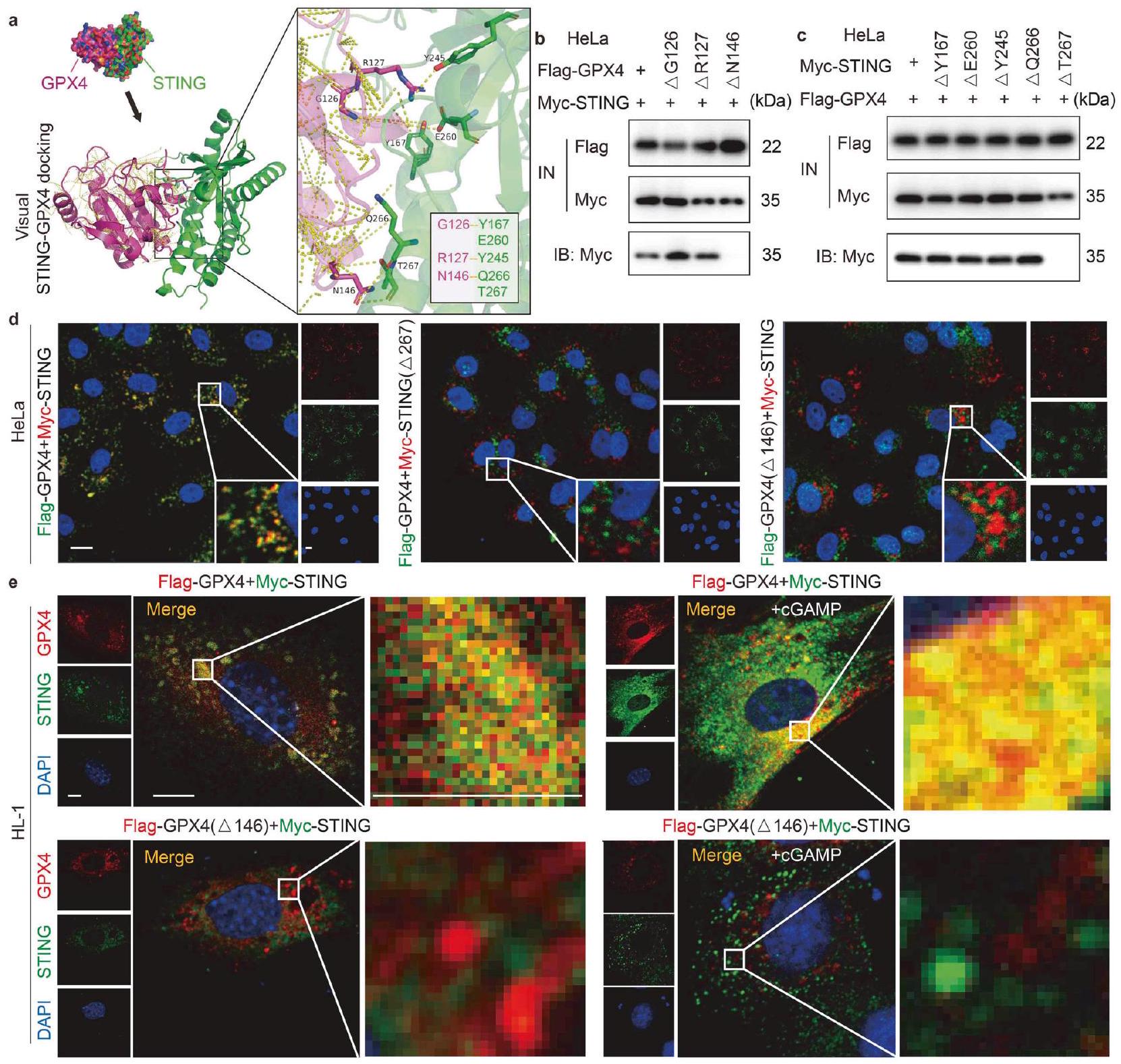
تشير زيادة تنظيم برنامج الفيروبتوزيس بعد تنشيط STING إلى إمكانية أن يؤدي ضرر نقص التروية / إعادة التروية إلى تنشيط غير متوقع لسلسلة نقل الإشارة التي تشمل STING. سعينا لتحديد العوامل الجينية المحتملة واستكشاف جدوى الاستفادة منها لتخفيف تفاقم ضرر نقص التروية / إعادة التروية في الفئران المنشطة بـ STING. أولاً، قمنا بتحليل مسارات KEGG المعبر عنها في الجينات المختلفة المعبر عنها (DEGs) التي تم تحديدها من خلال تسلسل RNA. (الشكل 3أ). من المRemarkably، كانت الجينات المرتبطة بالفيروبتوزيس غنية بشكل ملحوظ بين هذه الجينات المختلفة المعبر عنها (الشكل 4أ). بعد ذلك، تم إجراء تنقية تقارب متسلسلة في MPCs باستخدام أجسام مضادة لارتباط بروتين STING لتحديد الآلية الجزيئية التي ينظم بها STING الفيروبتوزيس. تم استخدام صبغة الفضة لمواد التنقية المناعية (الشكل 4ب i) والتحليل اللاحق باستخدام LC-MS/MS (الشكل 4ب ii) لتحديد بروتين STING الشاب. كشفت هذه الطريقة عن عدة بروتينات، وخاصة عامل الفيروبتوزيس GPX4. استخدمنا PyMOL لعرض النتائج المتوقعة لتحليل الربط الجزيئي بين STING (معرف بنك بيانات البروتين [PDB]: 4F5W) و GPX4 (معرف PDB: 5L71). يوضح الشكل 4ج عشرة أوضاع ربط محتملة، مع
درجة المطابقة-التوافق 8.157، مما يزيد من احتمال أن يتفاعل STING مباشرة مع GPX4.
لتأكيد التفاعل المباشر بين STING و GPX4، استخدمنا جسمًا مضادًا محددًا لـ STING لسحب بروتينه المرافق، ثم جمعنا منتجات تنقية البروتين هذه مع جسم مضاد لـ GPX4 ورصدناها في تجارب الترسيب المناعي (IP). أظهرت تجارب التفاعل المشترك الداخلي أن STING و GPX4 الداخليين شكلا معقدًا في خلايا القلب بعد نقص الأكسجين/العودة (A/R)، مع تعزيز cGAMP لتكوين هذا المعقد (الشكل 4d). وبالمثل، كان من الممكن الكشف عن STING في المنتجات المنقاة داخليًا لـ GPX4 (الشكل 4e)، مما يشير إلى تفاعل مباشر بين هذين البروتينين. بعد ذلك، قمنا بتعبير عن STING المسمى بـ Myc (Myc-STING) و GPX4 المسمى بـ Flag (FlagGPX4) في خلايا HeLa التي تعبر بشكل مستقر عن هذه التركيبات. ثم تم إجراء IP باستخدام Myc أو Flag للتحقق من التفاعل بين Myc-STING و Flag-GPX4. استخدمنا تجارب Western blot لتحليل المدخلات والمستخلص، وأظهرت النتائج أنه يمكن الكشف عن تركيز Myc-STING في المنتج المنقى الذي تم استخراجه بواسطة ببتيد Flag الزائد من Flag-GPX4 (الشكل 4f). وبالمثل، كان من الممكن أيضًا ملاحظة تركيز Flag-GPX4 في المنتج المنقى من Myc-STING (الشكل 4g). علاوة على ذلك، وجدنا أن التفاعل بين GPX4 و STING قد زاد بطريقة تعتمد على الوقت في خلايا HeLa المعالجة بـ cGAMP (الشكل 4h)، بينما قلل Erastin من قوة التفاعل.
لتأكيد التفاعل بين STING و GPX4، تم إجراء تلوين مناعي لتحليل التوضع المشترك لـ STING و GPX4 في MPCs. تحت ظروف نقص الأكسجين، أظهرت MPCs تعبيرًا منخفضًا عن STING وعدم وجود توضع مشترك مع GPX4 (الشكل 4ii). بعد حدوث نقص الأكسجين الحاد، تم تنشيط STING وتكوين نقاط متجمعة مع GPX4 (الشكل 4i ii)، بينما كان هناك تجميع هائل وتوضع مشترك مع تحفيز cGAMP (الشكل 4i iii). من المثير للاهتمام، عند استخدام Erastin، كان هناك القليل من التوضع المشترك بين GPX4 و STING (الشكل 4 i iv). تتماشى هذه النتائج بشكل وثيق مع النتائج التجريبية التي تم الحصول عليها من دراسة Co-IP. معًا، تشير هذه الأدلة بقوة إلى أن STING و GPX4 يتفاعلان مباشرة في CMs بعد نقص الأكسجين/العودة. علاوة على ذلك، مع زيادة تنشيط STING بواسطة cGAMP، لوحظ توزيع أكثر وضوحًا للتجمع بين STING و GPX4، مما يشير إلى تأثير ارتباط معزز بين البروتينين.
نظرًا للتواجد المشترك المعروف والتفاعل بين STING و GPX4 في خلايا الفئران والبشر، بالإضافة إلى الدور المذكور سابقًا لـ STING في تنظيم الفيروبتوز، افترضنا أن GPX4 قد يكون له دور في تعديل إصابة نقص التروية/إعادة التروية التي يساهم فيها STING. تم تحليل التلوين المناعي لشرائح القلب من Stingوأظهرت فئران Sting-CKO زيادة تعبير STING الناجم عن نقص التروية/إعادة التروية وتوضعه المشترك مع GPX4 في منطقة حدود نقص التروية/إعادة التروية لـ Sting.القلب (الشكل 4j). على العكس من ذلك، أصبح GPX4 أكثر سطوعًا ولم يكن هناك تداخل مع STING عندما تم حذف Sting بشكل محدد. هذه النتائج أكدت بشكل أكبر التأثير الذي تم اكتشافه سابقًا لـ STING في تحفيز الفيروبتوزيس والارتباط بين STING و GPX4 بعد نقص التروية/إعادة التروية. من الجدير بالذكر أن هذا الارتباط لديه القدرة على إثارة انخفاض في تعبير GPX4، مما يتطلب مزيدًا من الجهود البحثية.
باختصار، يحدث تفاعل مباشر بين STING و GPX4، ووسيط المنشط STING cGAMP يسهل التواصل بين GPX4 و STING. هذا التأثير محفوظ في خطوط خلايا الفئران والبشر. حيث أن غياب Sting أدى فعليًا إلى زيادة التعبير عن GPX4 في شرائح القلب بعد نقص التروية/إعادة التروية، مما يتماشى مع النتائج السابقة التي تشير إلى أن STING يمكن أن يحفز الفيروبتوز. استنادًا إلى هذه الملاحظات، نفترض أن STING قد يرتبط مباشرة بـ GPX4، مما يؤدي إلى تثبيط نشاطه وفي النهاية يؤدي إلى الفيروبتوز.
يتفاعل STING و GPX4 مباشرة عند بقايا الأحماض الأمينية N146 من GPX4 و T267 من STING لتوضيح موقع الربط حيث يرتبط STING بـ GPX4، قمنا بإجراء فحص دقيق لرسوم PDB.
الشكل 4 استهداف STING بواسطة GPX4. أ تم عرض مسار الفيروبتوز بواسطة KEGG. ب تحليل التلوين الفضي و LC-MS/MS للتنقية بالت affinity المزدوج باستخدام أجسام مضادة لستينغ في MPCs بعد A/R. ج صور تظهر عشرة أوضاع محتملة للتواصل وفقًا لنتائج الربط الجزيئي بين STING (معرف PDB: 4F5W) و GPX4 (معرف PDB: 5L71). د اختبار Co-IP لـ MPCs لفحص ما إذا كان GPX4 الداخلي يتفاعل مع STING وما إذا كانت هذه التركيبة تتأثر بتحفيز نقطة قصيرة لـ cGAMP. IB: STING، IP: GPX4. هـ اختبار Co-IP لـ MPCs لفحص ما إذا كان STING الداخلي يتفاعل مع GPX4 وما إذا كانت هذه التركيبة تتأثر بتحفيز نقطة قصيرة لـ cGAMP. IB: GPX4، IP: STING. و اختبار Co-IP لخلايا HeLa التي تم نقلها معًا بـ Flag-GPX4 و Myc-STING لفحص ما إذا كان STING يتفاعل مع GPX4. IB: Myc، IP: Flag.اختبار Co-IP لخلايا HeLa التي تم نقلها معًا بـ Flag-GPX4 و Myc-STING لفحص ما إذا كان GPX4 يتفاعل مع STING. IB: Flag، IP: Myc. اختبار Co-IP لخلايا HeLa التي تم نقلها معًا بـ Flag-GPX4 و Myc-STING تحت تأثير Erastin أو cGAMP لفحص ما إذا كان تفاعل GPX4 و STING يتأثر بمدة cGAMP أو Erastin. IB: Flag، IP: Myc. تحليل مزدوج للتألق المناعي للكشف عن STING و GPX4 وتحديد تواجدهم المشترك في خلايا HeLa التي تم نقلها معًا بـ Myc-STING و Flag-GPX4 تحت إضافة cGAMP أو Erastin. يتم عرض التفاعل الإيجابي لعلامة Myc باللون الأحمر، والتفاعل الإيجابي لعلامة Flag باللون الأخضر. يتم عرض التفاعل الإيجابي للتواجد المشترك باللون الأصفر. شريط القياستحليل المناعة المزدوجة باستخدام الفلورسنت للكشف عن GPX4 و STING في نفس مقطع القلب لـ Stingأو منطقة الحدود Sting-CKO. يتم عرض التفاعلات الإيجابية لشرائح الأنسجة باللون الأخضر (GPX4) والأحمر (STING). يتم عرض التفاعل الإيجابي للتعايش باللون الأصفر. شريط القياس. كروماتوغرافيا السائل LC، مطيافية الكتلة MS، بنك بيانات البروتين PDB، الترسيب المناعي المشترك Co-IP، المدخل IN، التحليل المناعي IB، الترسيب المناعي IP، بلازميد GPX4 المعلم بعلم العلم Flag-GPX4، بلازميد STING المعلم بعلم Myc Myc-STING
والبيانات المرتبطة. تم اختيار أوليغومر Apo STING (غير المرتبط بالليغاند) (معرف PDB: 4F5W) و GPX4 (معرف PDB: 5L71) لتوقع تفاعل البروتين. كشفت نتائج التوقع أن STING قد يشارك في ربط البروتين مع GPX4 عند مواقع الأحماض الأمينية Y167 و E260 و Y245 و Q266 أو T267. وبالمثل، يمكن أن يشارك GPX4 في ربط البروتين مع STING عند مواضع الأحماض الأمينية G126 و R127 أو N146 (الشكل 5a). استنادًا إلى التوقعات المذكورة أعلاه، تم إجراء تجارب IP على خلايا HeLa التي تعبر عن طفرات Flag-GPX4 وطفرات Myc-STING لتحديد منطقة التفاعل بين STING و GPX4. في البداية، تم استخدام ثلاثة مقاطع ببتيد GPX4، و تم تقييمها. مع تحليل البقعة الغربية
الشكل 5 يتفاعل STING و GPX4 مباشرة عند بقايا الأحماض الأمينية N146 من GPX4 و T267 من STING. أ صور تمثيلية لنتائج ربط الجزيئات لإظهار مواقع الاتصال المحتملة بين GPX4 و STING. ب اختبار Co-IP لخلايا HeLa التي تم نقلها بشكل مشترك مع Flag-GPX4 أو طفرة نقطة Flag-GPX4 ( G126]، علم-GPX4[ ] أو علم-GPX4[ ” ]) و Myc-STING، لفحص موقع الاتصال المحتمل بين GPX4 و STING. IB: Myc، IP: Flag. ج تجربة Co-IP لخلايا HeLa التي تم نقلها معًا بـ Flag-GPX4 و Myc-STING أو طفرة نقطة Myc-STING (Myc-STING[ ], Myc-STING[ مايك-ستينغ ], Myc-STING[ ] أو Myc-STING[ ]) لفحص موقع الاتصال المحتمل بين GPX4 و STING. IB: Myc، IP: Flag. d تحليل المناعة المزدوجة بالفلور للكشف عن STING و GPX4 وتوطينهما المشترك في خلايا HeLa التي تم نقلها معًا بـ Myc-STING و Flag-GPX4، Flag-GPX4 و Myc-STING[ T267] أو Myc-STING و FlagGPX4[ ]. التفاعل الإيجابي لعلامة Myc موضح باللون الأحمر، والتفاعل الإيجابي لعلامة Flag موضح باللون الأخضر. التفاعل الإيجابي للتوضع المشترك معروض باللون الأصفر. شريط القياستحليل المناعة المزدوجة باستخدام الفلورسنت للكشف عن STING و GPX4 وتحديد تواجدهم المشترك في خلايا HL-1 التي تم نقلها معًا بـ Myc-STING و Flag-GPX4 أو Myc-STING و Flag-GPX4. ] تحت إضافة cGAMP أو عدمها. يتم عرض التفاعل الإيجابي لعلامة Myc باللون الأخضر، والتفاعل الإيجابي لعلامة Flag باللون الأحمر. يتم عرض التفاعل الإيجابي للتعايش في اللون الأصفر. شريط القياس مقياس الصورة المكبرة. جلايسين (Gly)، أرجينين (Arg)، أسباراجين (Asn)، تيروزين (Tyr)، حمض الجلوتاميك (Glu)، جلوتامين (Gln)، ثريونين (Thr)
النتائج، وجدنا أن فقط Flag-GPX4-المتحور فقد قدرته على التفاعل مع STING، مما يشير إلى دور حاسم لهذا الحمض الأميني في التفاعل بين البروتينين (الشكل 5ب). من ناحية أخرى، فإن المتحورات المميزة بـ Myc-STING مع طفرات حمض أميني محددة، وهي Myc-STING-مايك-ستينغمايك-ستينغ-مايك-ستينغ-و Myc-STINGتم بناء النتائج. أظهرت النتائج أن غياب الحمض الأميني T267 في STING ألغى التفاعل بين STING و GPX4 (الشكل 5c).
علاوة على ذلك، في خلايا HeLa التي تعبر بشكل مستقر عن Myc-STING وFlag-GPX4، لاحظنا أن GPX4 فقد تزامنه مع STING عندما كان موقع T267 من STING غائبًا (الشكل 5d)، حتى لو تم التعبير عن الجينوم الكامل لـ GPX4. وبالمثل، اختفى التفاعل بين STING وGPX4 عندما فقد GPX4 موقعه N146 (الشكل 5d). بشكل جماعي، تثبت هذه البيانات بشكل قاطع أن STING يتفاعل جسديًا مع GPX4-N146 من خلال الحمض الأميني T267 من STING.
باستخدام أجهزة المجهر الضوئي عالي الدقة (SIM) لتعزيز وضوح صور التلوين المناعي، لاحظنا ارتباطًا قويًا وتداخلًا بين STING و GPX4 في خلايا HL-1 التي تم نقلها بجينات STING الموسومة بـ Myc و GPX4 الموسومة بـ Flag. من خلال استخدام هذا النهج، تمكنا من تحقيق فهم أكثر دقة لطريقة التفاعل بين هذين البروتينين في السياق الخلوي (الشكل 5e). كما هو موضح في الشكل 4h، فإن توصيل cGAMP قد سرع أيضًا من تشكيل مركب STING-GPX4. ومن الجدير بالذكر أنه عندما تم نقل Flag-GPX4-ΔN146 و Myc-STING في نفس الوقت، فقد فقد GPX4 تداخله مع STING. في هذه المرحلة، حتى عند توصيل cGAMP، لم يكن بالإمكان استعادة التداخل بين البروتينين، مما يبرز الدور الحاسم لهذا الحمض الأميني في تسهيل التفاعل بين هذين البروتينين.
يُعزز STING الموت الخلوي الناتج عن الحديد من خلال التحلل الذي يتوسطه الالتهام الذاتي-الليزوزوم لـ GPX4
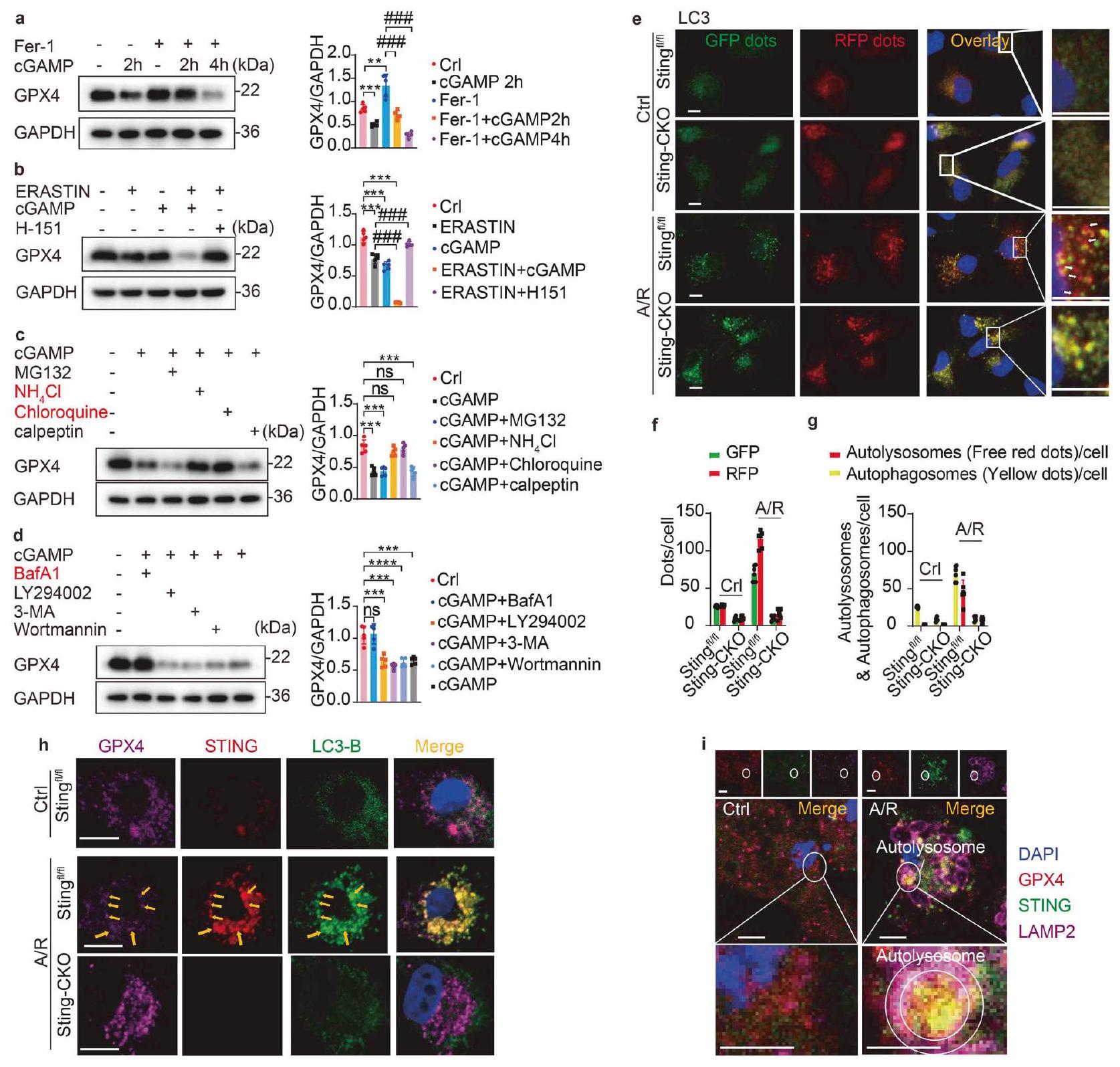
تحلل بروتين GPX4 هو حدث محوري في الفيروبتوز، وتوليد الجذور الحرة، وأكسدة الدهون غير القابلة للعكس، مما يؤدي في النهاية إلى موت الخلايا.في الشكل 4j، نلاحظ أيضًا التأثير المحتمل لتدهور STING على GPX4 داخل الأنسجة القلبية. بعد ذلك، تحققنا من تأثير STING على تدهور GPX4 خلال الفيروبتوز. نظرًا لملاحظتنا أن نقص Sting منع تدهور GPX4 خلال الفيروبتوز الناتج عن I/R أو A/R، استكشفنا المزيد من التأثير المنبه لم Activator STING، cGAMP، على تدهور GPX4. من الجدير بالذكر أننا لاحظنا أن cGAMP أدى إلى انخفاض في مستويات بروتين GPX4، على عكس التحكم السلبي Fer-1 (الشكل 6a). أدى علاج MPCs بمثبط الفيروبتوز Fer-1 إلى ارتفاع مستويات بروتين GPX4، لكن هذا الارتفاع تم منعه في وجود cGAMP. بالإضافة إلى ذلك، يمكن قمع نشاط GPX4 بواسطة Erastin، الذي يشترك في تأثيرات مشابهة لـ cGAMP كما ذُكر سابقًا، مما يؤدي إلى تراكم بيروكسيدات الدهون داخل الخلايا. لتقييم ما إذا كان cGAMP يعزز تدهور GPX4، استخدمنا Erastin في تجاربنا. إن إعطاء cGAMP بدأ بالفعل تدهور GPX4 (الشكل 6b). علاوة على ذلك، كان إضافة H-151 (مثبط STING) قادرًا أيضًا على حماية الفيروبتوز ومنع تدهور GPX4 الناتج عن Erastin. مجتمعة، يمكن أن يؤدي تنشيط STING إلى تحفيز تدهور GPX4.
بعد ذلك، تعمقنا في الآلية الأساسية التي تحكم تنظيم تحلل GPX4 بواسطة STING. يحدث تحلل البروتين بعد النسخ بشكل أساسي من خلال آليتين: التحلل البروتيني المعتمد على اليوبكويتين والتحلل الذاتي المعتمد على الليسوزوم.تظهر النتائج المعروضة في الشكل 3d أيضًا إثراء المسارات الأوتوليزوزومية في Stingالفئران خلال إصابة نقص التروية/إعادة التروية، مما يشير إلى أن STING قد يمارس تأثيرًا تنظيميًا على تحلل GPX4 عبر المسار الذاتي. لذلك، استخدمنا عدة مثبطات لتحلل البروتين، بما في ذلك مثبطات البروتيازوم (MG-132 وكالببتين) ومثبطات الليزوزوم (كلوروكين (CQ) و ) لحظر هذه المسارات الانحلالية بشكل محدد. ومن المثير للاهتمام أن تطبيق مثبطات الليزوزوم CQ أو يمكن أن تمنع بشكل فعال تحلل GPX4 الذي يسببه cGAMP وساهمت في وفرة GPX4 (الشكل 6c)، مما يشير إلى مشاركتها في عملية تعديل توازن GPX4 خلال التعرض لـ A/R. يعمل بافيلوميسين A1 (Baf A-1) كمثبط في المرحلة المتأخرة من الالتهام الذاتي، مما يمنع بشكل فعال نضوج الحويصلات الالتهامية من خلال تعطيل عملية الاندماج بين الأوتوفاجوسومات والليزوزومات. تعمل LY294002 و3-MA وWortmannin كمثبطات تستهدف المرحلة الأولية من الالتهام الذاتي، مما يمنع بشكل فعال تشكيل الأوتوفاجوسومات. من خلال تحليل اختبارات Western blot، اكتشفنا أن Baf A-1 فقط كان قادرًا على منع تحلل GPX4 الذي يسببه cGAMP (الشكل 6d)، مما يشير إلى أن STING سهل التحلل الذاتي لـ GPX4 من خلال تعزيز اندماج الأوتوفاجوسومات والليزوزومات.
الالتهام الذاتي، وهو عملية خلوية محورية مسؤولة عن تحلل البروتينات،برز كإمكانية كبيرة عامل يتوسط في تقليل GPX4، الذي تم تحفيزه بواسطة STING بعد نقص التروية/إعادة التروية. كما هو موضح في الشكل التوضيحي 2g، يمكن أن يؤدي حذف STING إلى كبح عملية الالتهام الذاتي التي تم تحفيزها بواسطة نقص التروية/إعادة التروية. من أجل تأكيد تدخل STING في التحلل الذاتي لـ GPX4 من خلال تعديل ارتباط الحويصلات الذاتية مع الليزوزومات، استخدمنا نظام مؤشر الالتهام الذاتي ثنائي الفلورية RFP-GFPLC3 لتوسيم وتتبع التغيرات في البروتينات المرتبطة بالأنابيب الدقيقة بدقة.سلسلة الضوء 3 ب (LC3B) وتدفق الالتهام الذاتي. كشفت الملاحظات الأولية عن تنشيط قوي لعملية الالتهام الذاتي تحت ظروف نقص الأكسجين / إعادة التروية (الشكل 6e-g). ربطت بروتين LC3 الفلوري الهجين نفسها بقوة بغشاء الأوتوفاجوسوم واندماجت مع الليسوسوم لتشكيل الأوتوليسوسومات. ومن الجدير بالذكر أنه خلال هذه العملية، تلاشى توهج GFP، مما يدل على الانتقال السلس من الأوتوفاجوسومات إلى الأوتوليسوسومات. ومع ذلك، في خلايا MPCs المفقودة لجين Sting، لوحظ زيادة ملحوظة في النقاط الفلورية الصفراء، مع وجود نقاط حمراء أقل. تدعم هذه الظاهرة بقوة نتائجنا من اختبار Western blot، مما يشير إلى وجود عائق في اندماج الأوتوفاجوسومات والليسوسومات، فضلاً عن حدوث اضطراب في عملية نضوج الأوتوفاجوسومات. في جوهر الأمر، يمنع غياب Sting القضاء على GPX4 من خلال عرقلة اتحاد الأوتوفاجوسومات والليسوسومات.
يُعترف بـ STING لقدرتها على بدء عملية الالتهام الذاتي من خلال دهن LC3B والتكوين اللاحق للأوتوفاغوسومات، بشكل مستقل عن تنشيط TBK1 وتحفيز الإنترفيرون.بعد ذلك، قمنا بدراسة تأثير STING و LC3B و GPX4 في الفيروبتوز. باستخدام تحليل المناعة الفلورية ثلاثية الألوان، لاحظنا أن ظروف نقص الأكسجين/الإعادة (A/R) نشطت STING، مما أدى إلى تكوين نقاط LC3 في STING.الخلايا MPCs ولكن ليس الخلايا الناقصة في Sting (الشكل 6h). تتفق هذه النتيجة مع نتائج الأدبيات المبلغ عنها سابقًا.تمت ملاحظة التوضع الواضح لنقاط LC3 مع كل من STING و GPX4 في Sting MPCs ولكن ليس في الخلايا التي تفتقر إلى STING، مما يشير إلى أن GPX4 تم استهدافه بواسطة البلعمة الذاتية المستحثة بواسطة STING بعد A/R. لتحديد موقع تفاعل STING و GPX4، قمنا بتلوين مشترك لـ STING و GPX4 وعلامات حجرة التفاعل بين الشبكة الإندوبلازمية وجهاز جولجي (ERGIC) وبروتين الغلاف I (COP-I) و LC3 في MPCs. أظهرت نتائجنا أن مركب STING-GPX4 يتوضع في البداية في ERGIC. يتم بعد ذلك نقل مركب STING-GPX4 نحو ERGIC، وحويصلات COP-I، والبلعميات الذاتية لـ LC3 (الشكل التكميلي 3).
بالإضافة إلى ذلك، فإن بروتين الغشاء المرتبط بالليزوزوم 2B (LAMP2B) يتوسط آلية اندماج الأوتوفاغوسوم مع الليزوزوم.تم تصور تشكيل الأوتوفاغوسوم المحفز بواسطة A/R بواسطة LAMP2B. كشفت الصور أن النقاط GPX4STING التي تم تحفيزها بواسطة A/R كانت محاطة بحلقة غشاء الأوتوليسوزوم المسمى بـ LAMP2B (الشكل 6i). وهذا يشير إلى أن التفاعل بين STING و GPX4 يحدث في ERGIC وأن STING يسهل تجنيد GPX4 إلى الأوتوفاغوسومات من أجل التحلل الأوتوفاغي اللاحق. تشير هذه النتائج إلى أن الأوتوفاغي المحفز بواسطة STING سهل القضاء على GPX4 في CMs بعد A/R.
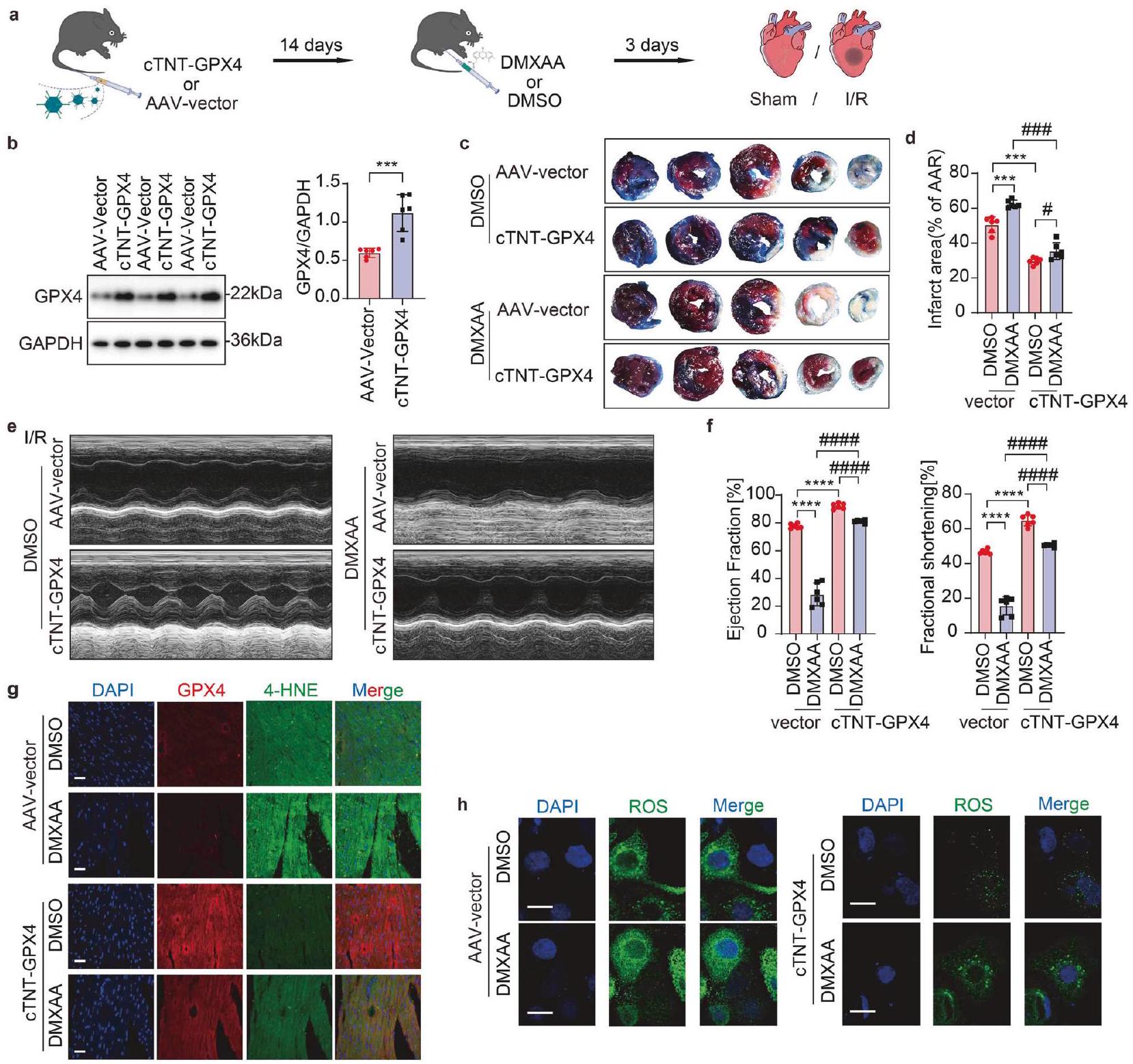
علاج GPX4 المعتمد على AAV يحمي وظيفة القلب من الإصابة الشديدة الناتجة عن نقص التروية/إعادة التروية الناتجة عن تنشيط STING استنادًا إلى نتائجنا، تحققنا بعد ذلك من أن GPX4 يعمل كعامل تابع لـ STING، وتعمقنا أكثر في إمكانية GPX4 كهدف علاجي لإصابة I/R المرتبطة بـ STING. تم إنتاج فيروس مرتبط بالأدينوفيروس (AAV) يستهدف GPX4 (AAV-GPX4) مدفوعًا بمروج cTNT في خلايا القلب وموضحًا بـ GFP (المشار إليه بـ cTNT-GPX4) لفحص ما إذا كان الإفراط في التعبير عن GPX4 يمكن أن يخفف من التدهور التدريجي لوظيفة القلب في الفئران المنشطة بـ STING تحت ظروف داخل الجسم. تم إعطاء AAV-GPX4-GFP من خلال حقن في الوريد الذيل قبل 14 يومًا من تحفيز I/R. بعد ذلك، تم إجراء حقن داخل البطن لـ DMXAA (منشط STING) أو DMSO كل يوم لمدة ثلاثة أيام قبل عملية I/R (الشكل 7a). قبل إنشاء النموذج،
الشكل 6 STING يعزز الفيروبتوز عبر التحلل بواسطة الالتهام الذاتي-الليزوزومي لـ GPX4. أ التحليل الغربي وقياس تعبير GPX4 في MPCs المتأثرة بـ Fer-1 أو cGAMP ( ). ب. التحليل الغربي وقياس تحلل GPX4 في MPCs الناتج عن إيراستين، cGAMP أو H-151 . تحليل Western blot وقياس تحلل GPX4 في MPCs الناتج عن cGAMP وحجبه باستخدام MG-132، الكلوروكين والكالببتين ). تم إجراء تحليل Western blot وقياس تحلل GPX4 في خلايا MPCs الناتج عن cGAMP ووقفه باستخدام Baf A-1 وLY294002 و3-MA وWortmannin . تحليل المناعة الفلورية لتدفق الالتهام الذاتي المقدم بواسطة mRFP-GFP-LC3 في Stingأو Sting-CKO MPCs بعد A/R أو لا. ردود الفعل الإيجابية للاوتوفاجي تظهر في النقاط الحمراء. شريط القياس. تحليل التألق المناعي الثلاثي للكشف عن GPX4 و STING و LC3B في Sting أو Sting-CKO MPCs. يتم عرض التفاعلات الإيجابية للتواجد المشترك باللون الأصفر (تواجد LC3B وSTING المشترك) أو الوردي (التواجد المشترك الثلاثي). شريط القياس تحليل ثلاثي المناعة الفلورية للكشف عن GPX4 و STING و LAMP2B في MPCs بعد A/R أو عدمه. يتم عرض التفاعلات الإيجابية للتعايش في اللون الأصفر (تعايش GPX4 و STING) أو الوردي (تعايش الثلاثة). شريط القياسالمتوسط ± الخطأ المعياري، NS غير دال، **P < 0.01،, **** و ### . باف A-1 بافيلوميسين A1، بروتين الغشاء المرتبط بالليزوزوم 2B LAMP2B
تم تأكيد كفاءة التعبير المفرط عن GPX4 في خلايا القلب المعزولة من ثلاثة أو ستة فئران باستخدام تقنية Western blot وصبغة المناعة الفلورية (الشكل 7ب، الشكل التوضيحي 4أ). يمكن أن يؤدي توصيل DMXAA إلى زيادة حجم الاحتشاء، بينما قلل التعبير المفرط عن GPX4 من حجم الاحتشاء في الفئران المنشطة بواسطة STING (الشكل 7ج، د)، مما يشير إلى دور وقائي للتعبير المفرط عن GPX4 ضد إصابة I/R الناتجة عن تنشيط STING. أظهر التحليل بالموجات فوق الصوتية للقلب أن تنشيط STING قد زاد بالفعل من سوء وظيفة القلب بعد I/R، ومع ذلك، فإن التعبير المفرط عن GPX4 قد حسن بشكل كبير من التدهور. وظيفة القلب في الفئران المنشّطة بواسطة STING بعد إعطاء DMXAA، كما يتضح من زيادة EF وFS بعد نقص التروية/إعادة التروية (الشكل 7e، f).
4-هيدروكسي-2-نونال (4-HNE)، الذي يُعتبر منتجًا ثانويًا معروفًا ناتجًا عن أكسدة الدهون، يعمل كمؤشر حاسم للفيروبتوز.في نتائجنا، أدى تنشيط STING إلى ارتفاع كبير في مستويات 4-HNE، مما يشير إلى زيادة في أكسدة الدهون (الشكل 7g)، بينما كان التعبير المفرط عن GPX4 قادرًا على عكس هذه الزيادة في أكسدة الدهون التي تسببها DMXAA، مما يشير إلى تقليل عملية الفيروبتوز. الكشف في المختبر
الشكل 7 العلاج بواسطة AAV المعتمد على GPX4 يحمي وظيفة القلب ضد الإصابة الشديدة الناتجة عن نقص التروية/إعادة التروية الناتجة عن تنشيط STING. تأثير AAV-cTNTGPX4 على علاج خلل القلب في فئران C57BL/6J مع إضافة DMXAA أو DMSO بعد نقص التروية/إعادة التروية: أ رسم تخطيطي يوضح مسار الزمن لخلل القلب الناتج عن نقص التروية/إعادة التروية الذي يتلقى AAV أو DMXAA. ب تحليل الغربي للتحقق من التعبير الزائد الناجح عن GPX4 في خلايا القلب العضلية. ). ، حجم احتشاء عضلة القلب (% من منطقة نقص التروية) مع قطع نسيجية تمثيلية ). تخطيط صدى القلب وقياس نسبة الكسر القذفي (EF%) ونسبة الكسر الانقباضي (FS%)تحليل المناعة المزدوجة باستخدام الفلورسنت لـ GPX4 و 4-HNE في نفس مقطع القلب من منطقة الحدود. مقياس الرسمتحليل تصوير المناعة الفلورية للكشف عن ROS في خلايا القلب للفئران المصابة بـ AAV-cTNT-GPX4 أو AAV-Control تحت إضافة DMSO أو DMXAA بعد A/R. مقياس الرسمالمتوسط ± الخطأ المعياري، غير دال،و ###P < 0.001. فيروس الأدينو المرتبط AAV، 4-HNE 4-هيدروكسي نونينال، cTNT-GPX4 AAV-Mus-GPX4-cTNT-C-GFP، DMXAA فاديميزان
مستويات تراكم ROS في MPCs دعمت هذه الملاحظة بشكل قوي (الشكل 7h). للتعمق أكثر في تأثير محور cGAS-STING-GPX4 على الميتوكوندريا، أجرينا عدة تجارب. شملت هذه التجارب قياسات لمستوى ATP، وتقييم للجهد الكهربائي للغشاء الميتوكوندري.وتحديد معدل استهلاك الأكسجين (OCR) لتقييم صحة الميتوكوندريا، ومحتوى ATP، والجهد الغشائي، والقدرة التنفسية تحت ظروف A/R. أظهرت النتائج أن حجب إشارة cGAS/STING أو التعبير المفرط عن GPX4 أعاد جزئيًا مستويات ATP،التنفس الميتوكوندري القاعدي، إنتاج ATP وكلا من السعة التنفسية القصوى والاحتياطية (الشكل التكميلي 4b-g، الشكل التكميلي 5a-c)، مما يدل على التجديد الجزئي لوظيفة الميتوكوندريا الناتج عن حذف cGAS/STING أو فرط التعبير عن GPX4.
قبل كل شيء، يمكن الاستنتاج أنه على الرغم من تعزيز ضرر نقص التروية/إعادة التروية في الفئران التي تعبر عن STING بشكل مفرط، فإن زيادة تعبير GPX4 تحتفظ بقدرة ملحوظة على منع تأكسد الدهون والموت الخلوي الناتج عن الحديد، مما يحمي وظيفة القلب. في الختام، فإن حذف cGAS/STING أو زيادة تعبير GPX4 يحمي من تدهور وظيفة القلب والضرر الميتوكوندري بعد نقص التروية/إعادة التروية في الفئران التي تم تنشيط STING فيها، مما يشير إلى استراتيجية علاجية محتملة لتخفيف الآثار السلبية لزيادة تعبير STING على وظيفة القلب.
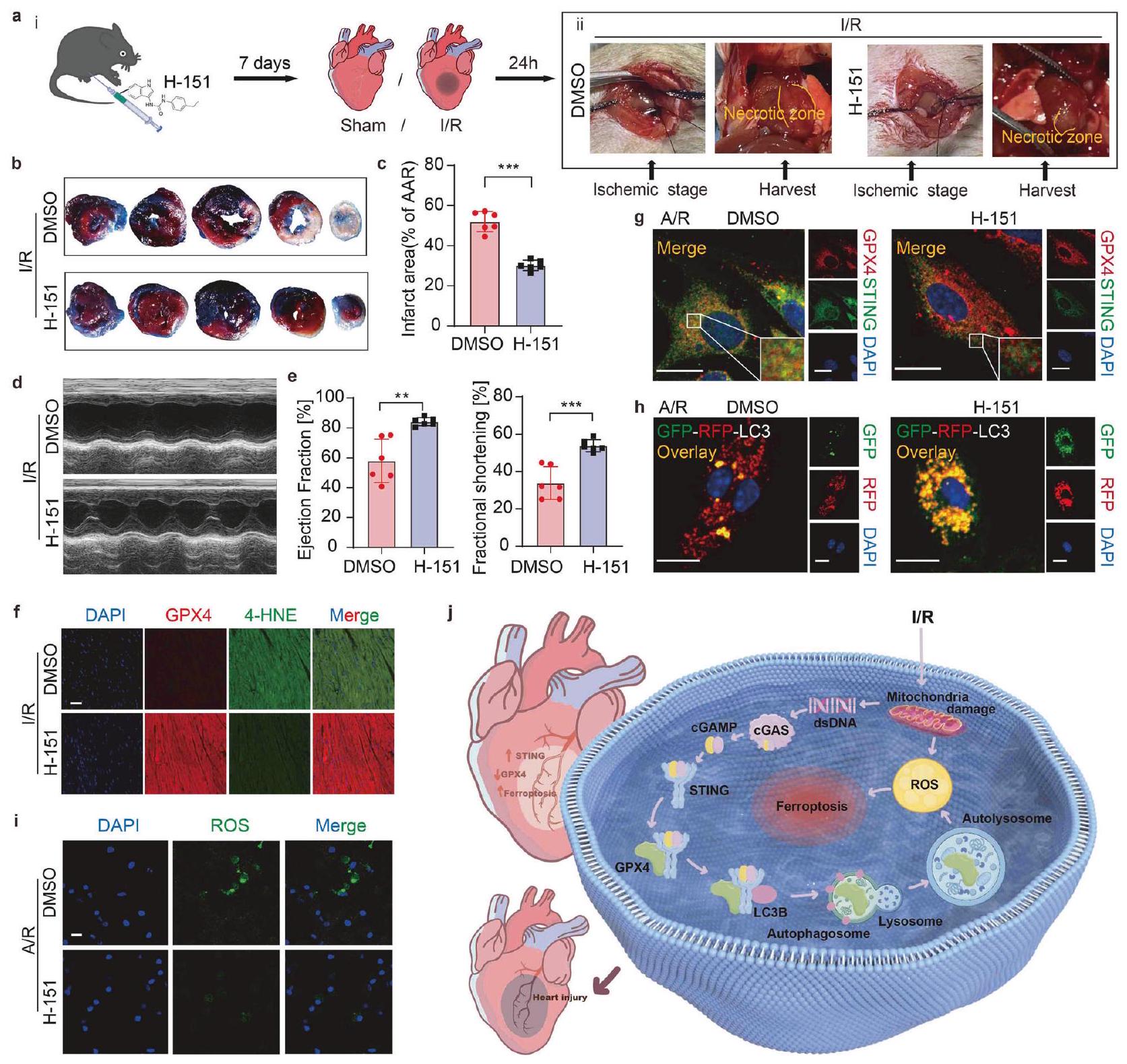
STING هو هدف علاجي محتمل لتثبيط خلل القلب بعد نقص التروية/إعادة التروية. للتعمق أكثر في استراتيجيات علاج I/R التي تستفيد من مسار الإشارة STING-GPX4، اخترنا H-151 لتقييمه
الشكل 8 STING هو هدف علاجي محتمل لتخفيف خلل القلب بعد نقص التروية/إعادة التروية. تأثير H-151 على خلل القلب في فئران C57BL/6J بعد نقص التروية/إعادة التروية: أ. i. مخطط بياني يوضح مسار الزمن لخلل القلب الناتج عن نقص التروية/إعادة التروية مع إضافة H-151 أو DMSO. ii. صور فوتوغرافية تمثيلية لقلوب مع إضافة DMSO أو H-151 بعد نقص التروية/إعادة التروية. ب، ج. حجم احتشاء عضلة القلب (% من AAR) مع قطع نسيج تمثيلية.. د، هـ تخطيط صدى القلب وقياس نسبة الكسر القذفي ونسبة الكسر القلبيتحليل المناعة المزدوجة باستخدام الفلورسنت لـ GPX4 و 4-HNE في نفس مقطع القلب من منطقة الحدود. مقياس الرسمتحليل المناعة الفلورية لـ GPX4 و STING في خلايا القلب للفئران بعد إضافة DMSO أو H-151 بعد نقص الأكسجين وإعادة التروية. مقياس الرسمتحليل المناعة الفلورية لتدفق الالتهام الذاتي المقدم بواسطة mRFP-GFP-LC3 في خلايا القلب للفئران بعد إضافة DMSO أو H-151 بعد نقص الأكسجين وإعادة التروية. تظهر التفاعلات الإيجابية للالتهام الذاتي كنقاط حمراء. مقياس الرسمتحليل تصوير المناعة الفلورية للكشف عن ROS في خلايا القلب من الفئران بعد إضافة DMSO أو H-151 بعد نقص الأكسجين وإعادة التروية. مقياس الرسمرسم تخطيطي يوضح آلية إصابة MI/R المعززة بواسطة STING. تم رسم جزء من الصورة بواسطة Figdraw. المتوسط ± الانحراف المعياري، NS غير ذي دلالة، **، و ****
الإمكانات في التخفيف من الآثار الضارة لـعلى وظيفة القلب ولتحديد العوامل العلاجية المحتملة لعلاج إصابة نقص التروية. قبل جراحة نقص التروية، تم إعطاء الفئران H-151 عن طريق الحقن داخل الصفاق كل يومين لمدة إجمالية قدرها 7 أيام (الشكل 8a i). في الوقت نفسه، كان من الواضح درجة نخر عضلة القلب في الصور القلبية الحية (الشكل 8a ii)، مما يظهر السيطرة الفعالة التي يمارسها H-151 على مدى النخر داخل منطقة الوظيفة القلبية. ومن الجدير بالذكر أنه في وجود H-151، لوحظ تقليل في حجم الاحتشاء (الشكل 8b، c) واستعادة وظيفة القلب (الشكل 8d، e). علاوة على ذلك، كانت إضافة H-151 قادرة على تخفيف الزيادة في
4-HNE الناتج عن نقص التروية/إعادة التروية، مما يشير إلى تقليل أكسدة الدهون والفيروبتوز (الشكل 8f).
بعد ذلك، قمنا بالتعمق في الآلية الدوائية التي تكمن وراء التأثيرات المحسنة لـ H-151 على إصابة عضلة القلب بعد نقص التروية/إعادة التروية في المختبر. في نتائجنا، أوقف H-151 التفاعل بين STING و GPX4 (الشكل 8g)، وحجب تدفق الالتهام الذاتي (الشكل 8h)، وبالتالي قلل من تراكم ROS (الشكل 8i) للتخفيف من الفيروبتوزيس القلبي.
تُبرز تأثيرات H-151 إمكانيته في تقليل منطقة إصابة نقص التروية/الإعادة (I/R) والحفاظ على وظيفة القلب. معًا، يظهر STING كهدف علاجي واعد لتخفيف تدهور القلب الذي يحدث بعد نقص التروية/إعادة التروية، مع تأثيراته الضارة التي يتم تعديلها من خلال تنظيم GPX4، مما يشير إلى طريق واعد للتدخل في إعادة تشكيل القلب.
لإظهار الأهمية السريرية لهذه الدراسة وللتحقيق في وجود محور الإشارة STING-GPX4 لدى المرضى الذين يعانون من مرض القلب الإقفاري، قمنا بإجراء قياسات لبروتينات المصل لدى المرضى قبل وبعد التدخل التاجي عن طريق الجلد (PCI). الخصائص الأساسية للمرضى المشاركين في هذه الدراسة موضحة في الجدول التكميلي S3. كشفت نتائجنا أنه بعد PCI، كان هناك ارتفاع في مستوى تعبير بروتين STING في مصل المرضى، مصحوبًا بانخفاض في مستوى تعبير GPX4 (الشكل التكميلي 5d). تشير هذه النتيجة إلى أن محور STING-GPX4 قد يلعب دورًا حاسمًا لدى المرضى الذين يعانون من نقص التروية/إعادة التروية (I/R). بالإضافة إلى ذلك، لاحظنا زيادة في تعبير بروتين STING وانخفاض في محتوى بروتين GPX4 في مستخلص خلايا القلب (CMs) المشتقة من خلايا جذعية جنينية بشرية، والتي تم تحفيزها بواسطة ظروف نقص التروية/إعادة التروية (الشكل التكميلي 5e). تؤكد هذه النتائج مرة أخرى على الدور البيولوجي المحتمل المهم لمحور STING-GPX4 في قلب الإنسان.
نقاش
يحدث الفيروبتوزيس خلال إصابة نقص التروية وإعادة التروية القلبية (MI/R) ويصاحبه تدهور GPX4. هنا، نقدم عدة أدلة على أن STING يلعب دورًا ابتدائيًا في الفيروبتوزيس القلبي الناتج عن MI/R من خلال الارتباط المباشر بـ GPX4 وتحفيز تدهور GPX4 عبر آلية الالتهام الذاتي-الليزوزومي. يؤدي نقص التروية وإعادة التروية إلى ارتفاع الحمض النووي السيتوبلازمي، مما ينشط إشارة dsDNA-cGASSTING في خلايا القلب (CMs). يؤدي حذف cGAS أو STING في CMs إلى تقليل الإجهاد التأكسدي، والفيروبتوزيس، وإصابة MI/R، بينما يؤدي تنشيط STING إلى تفاقمها. من الناحية الآلية، يتفاعل STING مع GPX4 عند بقايا محددة (N146 من GPX4 وT267 من STING) لبدء الالتهام الذاتي والتدهور الليزوزومي. يشكل STING حلقة تغذية راجعة إيجابية عبر محور dsDNA-cGAS-STING-GPX4، مما يؤدي إلى تفاقم الفيروبتوزيس. تظهر استراتيجيتان للعلاج لإصابة MI/R – زيادة التعبير عن GPX4 بواسطة AAV ومضادات STING – وعدًا في منع وتقليل إصابة I/R، مما يبرز الأهداف السريرية المحتملة. بشكل جماعي، كشفت نتائجنا أن تراكم STING خلال MI/R يحفز الفيروبتوزيس من خلال التدخل في GPX4.
أحد الأساليب الفعالة لحماية القلب في اضطرابات عضلة القلب هو منع موت خلايا القلب.أظهرت الدراسات الحديثة أن مسار الإشارة المناعية cGAS-STING يلعب دورًا في مجموعة متنوعة من الأمراض الإقفارية، بما في ذلك حالات الإقفار الدماغي والكلوي والمعوي والكبدي.علاوة على ذلك، تم الإشارة إلى أن cGAS له دور في الاستجابة الالتهابية التي لوحظت في احتشاء عضلة القلب (MI).يُعرف IRF3، وهو جزيء إشارة تابع لمسار cGAS-STING، بدوره في العملية المرضية التي تلي احتشاء العضلة القلبية.حتى الآن، كانت الجهود البحثية الرئيسية المتعلقة بمسار cGAS-STING بعد احتشاء العضلة القلبية تركزت على السيتوكينات الفردية، مثل الإنترفيرون (IFN)،أو مناعة البلعميات.لا يزال هناك فجوة في فهم الأهمية الوظيفية المباشرة لـ cGAS-STING في إصابة القلب الناتجة عن نقص التروية وإعادة التروية (MI/R) وتقدم الضرر الناتج عن الفيروبتوز. في البحث الحالي، أكدنا أن STING يحفز الفيروبتوز في خلايا العضلة القلبية (CMs) خلال إصابة MI/R.
تتميز عملية نقص التروية وإعادة التروية بمرحلة أولية من نقص التروية ونقص الأكسجة، تليها مرحلة إعادة التروية التي تتميز بإعادة استعادة تدفق الدم. ومن الجدير بالذكر أن هذه المرحلة من إعادة التروية تحمل أكبر وأخطر الأضرار المرتبطة بنقص التروية وإعادة التروية، والتي تشمل إعادة برمجة التمثيل الغذائي، وإصابة الحمض النووي، وزيادة التعبير عن الجينات المؤيدة للالتهاب، وخلل في الميتوكوندريا. إن فهم الآليات المعقدة التي تكمن وراء هذه العمليات المرضية أمر بالغ الأهمية لتطوير استراتيجيات علاجية فعالة للتخفيف من الآثار الضارة لنقص التروية وإعادة التروية.أفادت الدراسات أن مستويات الحمض النووي المتداولة كانت مرتفعة بشكل ملحوظ بعد نقص تروية القلب.نحن نواصل أكدت أنه خلالعند الإصابة، يحدث درجة عميقة من تلف الحمض النووي، مما يؤدي إلى تسرب الحمض النووي إلى السيتوسول، مما يحفز بعد ذلك زيادة تنظيم مسار الإشارة cGAS-STING في خلايا القلب بدلاً من الخلايا الليفية أو البلعميات خلال إصابة نقص التروية الحادة. بالإضافة إلى ذلك، وجدت دراستنا أعلى تنشيط لمسار إشارة cGAS-STING في المنطقة الحدودية، مقارنةً بالندبة أو المنطقة غير الإقفارية، مما يتماشى مع حقيقة أنها المنطقة الأكثر ديناميكية مع إجهاد أكسيدي نشط ومسارات موت الخلايا، بما في ذلك الفيروبتوز. بالإضافة إلى ذلك، يبدو أن STING هو عامل أساسي لا غنى عنه لتمكين cGAS من تنفيذ دوره الوظيفي في سياق نقص التروية.
نظرًا لأن دور cGAS و STING في خلايا القلب غير مفهوم جيدًا، قمنا بإنشاء نموذج حذف جيني محدد لخلايا القلب لـ cgas.ماي إتش 6 [cgasCKO])، حذف STING (Stingماي 6فئران [Sting-CKO]) وفئران التحكم الخاصة بهم للتحقيق في الأهمية الوظيفية لـ cGAS و STING المحددين لعضلة القلب في إصابة نقص التروية/إعادة التروية. لتجنب التأثيرات الخلوية والجزيئية المرتبطة بالشيخوخة على عضلة القلب وآلياتها الواقية القلبية ضد إصابة نقص التروية/إعادة التروية، استخدمنا فئران بعمر 8 أسابيع للنمذجة.بالنظر إلى التأثير الضئيل للجنس على الـ I/نموذج، اخترنا الفئران الذكور.لكن لا يزال لدينا قصور في إنشاء النماذج. بينما تظل الفئران نموذجًا مقبولًا على نطاق واسع لدراسة الآليات الجزيئية، فإننا ندرك الحاجة إلى التحقق من النتائج الرئيسية في نماذج حيوانية أكبر أو أنظمة مستمدة من البشر في الدراسات المستقبلية لتحسين الصلة الانتقالية.
لأن تركيزنا على إصابة نقص التروية الحادة والتعبير عن الوسائط الجزيئية للالتهاب والتسلل الخلوي كان بحاجة إلى التحقيق خلال المرحلة الأولىاخترنا النقطة الزمنية الحرجة بعد 24 ساعة من إعادة تدفق الدم للتجارب على الحيوانات. ومع ذلك، كانت عملية شفاء الاحتشاء غير مكتملة في هذا الوقت.لتقييم آثار نقص cGAS وSTING على تليف القلب الناتج عن إصابة نقص التروية/إعادة التروية، قمنا بتمديد فترة إعادة التروية إلى 7 أيام. تشير هذه النتائج إلى أن حذف cGAS وSTING يوفر حماية قلبية تتجاوز المرحلة الحادة، مما يقلل من التليف ويعزز الشفاء، مما يبرز الدور الحاسم لـ cGAS-STING في إصابة ما بعد نقص التروية/إعادة التروية وإصلاحها.
نظرًا لأن الخلايا الجذعية القلبية تفتقر إلى القدرات المناعية، أوضح ز. ج. تشين وعلماء آخرون أن STING ينظم البلعمة الذاتية، التي تعتبر وظيفة خلوية أساسية تعمل بشكل مستقل عن مسار الإشارة المناعية التقليدي. وبالتالي، فإن تركيزنا الأساسي في سياق الخلايا الجذعية القلبية هو دراسة المسار غير المناعي الذي يتوسطه STING. إصابة نقص التروية/إعادة التروية هي عملية مرضية معقدة تتميز بأشكال متعددة من موت الخلايا، مثل النخر، والبرم، والنخر المبرمج، والفيروبتوز، والبلعمة الذاتية، وتتطلب مساهمات كل آلية مزيدًا من المناقشة.في دراستنا، ركزنا على الفيروبتوز كعامل مهم في تقليل حجم الاحتشاء بناءً على دوره المعروف في الإصابة الإقفارية، وخاصة من خلال آليات أكسدة الدهون والأضرار التأكسدية المعتمدة على الحديد. بالتأكيد، تتداخل آليات موت الخلايا خلال إصابة الإقفار/إعادة التروية. ركز بحثنا فقط على التأثير التنظيمي لـ STING على الفيروبتوز، والذي له قيود، خاصة نقص تحليل العوامل المتعلقة بالنخر الخلوي المبرمج والفيروبتوز. يتطلب الأمر استكشافًا إضافيًا لآليات موت الخلايا الأخرى خلالمضمون في المستقبل.
على الرغم من أن الخلايا الإيجابية لتونيل تمثل مزيجًا من مسارات موت الخلايا المختلفة، كانت النتائج من تسلسل RNA لدينا (RNA-seq)، استنادًا إلى قلوب Sting-CKO وقلوب التحكم بعد نقص التروية/إعادة التروية، ملحوظة. كشفت هذه النتائج عن جانب جديد من الفيزيولوجيا المرضية لنقص التروية/إعادة التروية، يتميز بتنظيم الأوتوليزوزومات جنبًا إلى جنب مع الفيروبتوز. عند التعرض لإصابة نقص التروية/إعادة التروية، أصبح من الواضح بشكل كبير وجود إثراء كبير لمسارات تنظيم الفيروبتوز في Sting.الفئران مقارنة بفئران Sting-CKO. وهذا يقدم دليلاً قوياً يدعم الفكرة القائلة بأن الفيروبتوز، وهو شكل من أشكال موت الخلايا المعتمد على الحديد، يلعب دوراً محورياً في إصابة MI/R التي تنظمها STING. إن تحلل بروتين GPX4 هو حدث محوري في الفيروبتوز، مما يؤدي بدوره إلى تلف الميتوكوندريا وتوليد أنواع الأكسجين التفاعلية (ROS).يؤدي إلى أكسدة الدهون غير القابلة للعكس، وبالتالي، موت الخلايا.قمنا بعد ذلك بتأكيد تأثير STING على علامات الفيروبتوز، بما في ذلك تراكم ROS، وأكسدة الدهون، وتدهور GPX4، بالإضافة إلى التعبير عن بعض البروتينات المرتبطة بالفيروبتوز (عضو 4 من عائلة سينثيتاز أسيل-CoA طويلة السلسلة، مستقبل الترانسفيرين، وعائلة ناقلات المواد 7، العضو 11). استنادًا إلى الأدلة، أوضحنا أن التأثير التنظيمي لـ STING على إصابة I/R لا يتم من خلال موت الخلايا المبرمج أو آليات موت الخلايا الأخرى، بل من خلال تحفيز الفيروبتوز الخلوي. باختصار، تشير نتائج بحثنا إلى أن STING يعمل كمحفز قوي للفيروبتوز، مما يعزز بشكل فعال تدهور GPX4، وأكسدة الدهون، وتراكم ROS. يقوم STING بتعزيز الفيروبتوز القلبي من خلال تعديل إصابة الإجهاد التأكسدي.
تشير الدراسات الحديثة إلى أن GPX4 يعزز تنشيط STING من خلال الحفاظ على توازن الأكسدة والاختزال للدهون.ومع ذلك، فإن التأثير المباشر لـ STING على GPX4 وآلياته الأساسية في الفيروبتوزيس الناتج عن نقص التروية/إعادة التروية لا يزال غامضًا وغير مستكشف. تقدم نتائجنا رؤى حول الآليات التنظيمية المعقدة التي تحكم وظيفة GPX4 و STING خلال أحداث نقص التروية/إعادة التروية. وبالتالي، تساهم اكتشافاتنا في تعزيز الإطار المعرفي القائم بشأن تنظيم الفيروبتوزيس على مستوى الأنسجة خلال نقص التروية/إعادة التروية.
الاستخدام الواسع للمنشطات والمثبطات يعزز فهمنا لتأثير التحلل المباشر لـ STING على GPX4. حيث يحدث تحلل البروتين بعد النسخ بشكل أساسي من خلال آليتين: التحلل البروتيني المعتمد على اليوبكويتين والتحلل اللينوسومي المعتمد على الالتهام الذاتي.قمنا باستخدام مجموعة متنوعة من مثبطات تحلل البروتين (MG-132، كالببتين، كلوروكين،بايفيلوميسين A1، LY294002، 3-MA وورتمانين) لتعطيل تحلل GPX4 الذي يسببه cGAMP. يُعرف STING بقدرته على بدء عملية الالتهام الذاتي من خلال دهن البروتينات المرتبطة بالأنابيب الدقيقة.سلسلة الضوء 3B (LC3B) والتكوين اللاحق للأوتوفاغوسومات، بشكل مستقل عن تنشيط TBK1 وتحفيز الإنترفيرون.قمنا بإجراء تحليل التوطين المشترك باستخدام التألق المناعي لـ GPX4 و STING و compartment الوسيط بين الشبكة الإندوبلازمية وجهاز جولجي (ERGIC) وبروتين الغلاف I (COP-1) و LC3B وبروتين الغشاء المرتبط بالليزوزوم 2، بالإضافة إلى تتبع تدفق الالتهام الذاتي باستخدام GFP-RFP-LC3 في CMs من نوع Sting-CKO والمجموعة الضابطة. اكتشفنا أن cGAMP يحفز نقل STING إلى ERGIC عند حدوث نقص التروية/إعادة التروية. عند ERGIC، يرتبط STING بـ GPX4 لتشكيل معقد STING-GPX4، الذي يتم نقله بعد ذلك إلى حويصلات COP-I و الأوتوفاجوسومات LC3. ثم حفز STING تراكم نقاط LC3B. بعد ذلك، اندمج معقد GPX4-STING مع الليزوزومات وتم احتواؤه بواسطة غشاء الأوتوليزوزوم المميز ببروتين الغشاء المرتبط بالليزوزوم 2B.في النهاية يؤدي ذلك إلى تحلل GPX4. تقدم هذه الاكتشافات رؤى حول الآليات المعقدة التي تحكم تحلل GPX4 من خلال البلعمة الذاتية، التي تتوسطها التفاعل بين STING و LC3B. في غياب STING، يتم عرقلة تدفق البلعمة الذاتية، مما يؤدي إلى عدم قدرة GPX4 على التواجد مع الأوتوفاغوسومات. ونتيجة لذلك، تعيق هذه الاضطرابات تحلل GPX4، مما يقلل بدوره من الإجهاد التأكسدي الخلوي والفيروبتوز. إن هذا الفهم المعزز للآليات الجزيئية التي تكمن وراء تفاعلات GPX4-STING-البلعمة الذاتية يحمل وعدًا لوضع استراتيجيات علاجية مستهدفة قابلة للتطبيق على سيناريوهات الأمراض ذات الصلة.
نظرًا لأن الآليات التي تحكم الفيروبتوزيس معقدة ولا تزال غير مفهومة تمامًا، فإن بحثنا يكشف عن آلية تنظيمية جديدة للفيروبتوزيس. في هذه الآلية، يتم تحديد STING كبروتين شريك يعزز تحلل GPX4 خلال إصابة نقص التروية/إعادة التروية. على عكس الت ubiquitin المباشر، فإن دور STING في تحلل GPX4 من المحتمل أن ينطوي على قدرته على تنظيم الالتهام الذاتي.على الرغم من أن STING ليس إنزيم E3، إلا أنه ينظم البلعمة الذاتية من خلال الانتقال من الشبكة الإندوبلازمية نحو ERGIC ومن ثم إلى جهاز جولجي، مما يعزز ترسيب الدهون LC3 وتكوين الأوتوفاجوسوم. لدينا تظهر النتائج أن STING يتفاعل مع GPX4، مما يسهل استقطابه إلى الجسيمات الذاتية للتفكك الليزوزومي من خلال الآلية الذاتية بدلاً من ubiquitination المباشر. أوضح دراستنا الآلية وسلطت الضوء على الآثار الأوسع للاوتوفاجي التي يتم تحفيزها بواسطة STING في مسارات موت الخلايا. ستركز التحقيقات المستقبلية على تحديد الليغازات E3 المحتملة أو المتكيفات الذاتية المشاركة في هذه العملية لتوضيح الآليات الجزيئية بشكل أكبر.
دراستنا لا تفسر فقط الأهمية الوظيفية لـ STING في تعزيز الفيروبتوزيس القلبي خلال نقص التروية/إعادة التروية، بل تتناول أيضًا تأثيراته الوقائية على وظيفة القلب بعد إصابة نقص التروية/إعادة التروية. ومن الجدير بالذكر أن الأبحاث التي استخدمت علاج GPX4 المعتمد على AAV كشفت أيضًا أن GPX4 يعمل كعامل تابع لـ STING. مثبط STINGيمتلك القدرة على عرقلة التوطن المشترك لـ STING و GPX4، مما يعطل تدفق الالتهام الذاتي، وبالتالي يقلل من تراكم ROS، وفي النهاية يخفف من شدة إصابة نقص التروية/إعادة التروية. معًا، تظهر خيارات العلاج الفعالة هاتين إمكانيات علاجية واعدة في إدارة إصابة نقص التروية/إعادة التروية، مما يبرز الدور الكبير لـ STING في تنظيم تحلل GPX4 والنتيجة المترتبة على ذلك من الفيروبتوزيس القلبي أثناء إصابة MI/R.
الميتوكوندريا هي محددات حاسمة لبقاء أو موت خلايا عضلة القلب خلال نقص التروية / إعادة التروية.تظهر نتائجنا بشكل خاص حول وظيفة الميتوكوندريا والصحة وحالة ATP أن حجب إشارة cGAS/STING أو تنشيط GPX4 يمكن أن يحد من تقدم المرض ويعيد جزئيًا وظيفة الميتوكوندريا. ومع ذلك، لا تزال هذه الأساليب تؤدي إلى ميتوكوندريا معيبة وإنتاج ATP معطل، مما يبرز الحاجة إلى استراتيجيات علاجية إضافية تستهدف صحة الميتوكوندريا بشكل خاص لتحقيق إصلاح قلبي كامل.
قبل كل شيء، من خلال تحديد STING كهدف علاجي محتمل في سياق إصابة MI/R، نقدم اتجاهًا جديدًا للتحقيقات المستقبلية في العلاجات لإصابة I/R. إن صياغة نهج علاجي يهدف إلى تعزيز وظيفة أو تعبير GPX4 لديه القدرة على التخفيف من الآثار الضارة الناتجة عن تنشيط STING خلال إصابة I/R على وظيفة القلب. يبقى تحويل الحماية القلبية من الأدلة التجريبية القوية إلى فوائد سريرية للمرضى الذين يعانون من احتشاء عضلة القلب الحاد أو الذين يخضعون لجراحة القلب والأوعية الدموية أولوية ملحة. للتحقق من القابلية السريرية لنتائجنا الحيوانية، أجرينا دراسة على مجموعة من المرضى الذين يخضعون لعلاج PCI وأكدنا الصلة السريرية لأبحاثنا، مما يظهر تنشيط محور إشارة STING-GPX4 في المرضى الذين يعانون من مرض القلب الإقفاري، مما يشير إلى أنه قد يكون له تأثير كبير على إصابة I/R. بالطبع، يكمن الاختلاف الحاسم بين الدراسات التجريبية والسريرية في وجود العديد من الأمراض المصاحبة واستخدام الأدوية المتعددة في المرضى، والتي غالبًا ما يتم نمذجتها بشكل غير كافٍ في الدراسات الحيوانية. على سبيل المثال، قد تحد مثبطات الصفائح الدموية من التأثيرات الوقائية للعلاجات التجريبية لاحتشاء عضلة القلب، بينما يمكن أن تلغي تخدير البروبوفول فوائد التكييف الإقفاري. تسلط هذه العوامل الضوء على أهمية تطوير نماذج قبل السريرية ذات صلة سريرية لتتوافق بشكل أفضل مع تعقيدات الأمراض البشرية.
في الختام، كشفت دراستنا للمرة الأولى عن الآليات التي من خلالها يبدأ STING عملية الفيروبتوز في خلايا القلب ويزيد من إصابة نقص التروية/إعادة التروية. قدمنا نموذجًا حيث يحفز نقص التروية/إعادة التروية تراكم الحمض النووي المزدوج الشريطة في السيتوبلازم، مما ينشط إشارة cGAS-cGAMP-STING. ثم يرتبط STING المنشط مباشرة بـ GPX4 ويحفز تدهور GPX4 عبر الالتهام الذاتي، مما يؤدي إلى إجهاد أكسيدي وفي النهاية يحفز الفيروبتوز في خلايا القلب. قد يستهدف استهداف STING بشكل محدد تخفيف الفيروبتوز الناتج عن نقص التروية/إعادة التروية والأضرار القلبية. تشير هذه النتائج إلى أن محور STING-GPX4 يلعب دورًا حاسمًا في الفيروبتوز القلبي. لا يكشف هذا فقط عن الآليات الجزيئية الجديدة التي تدعم موت الخلايا المرتبط بـ GPX4، ولكن أيضًا يحدد STING كهدف علاجي واعد لإدارة إصابة نقص التروية/إعادة التروية.
المواد والأساليب
بيانات الأخلاقيات
تم إجراء جميع تجارب الحيوانات وفقًا صارمًا لإرشادات المعاهد الوطنية للصحة (NIH) لرعاية واستخدام الحيوانات المختبرية. وقد منحت لجنة الحيوانات المختبرية في مستشفى تشينغلو بجامعة شاندونغ (رقم: DWLL-2021-206) موافقتها الشاملة، مما يضمن تنفيذًا أخلاقيًا وقابلًا للمسائلة خلال عملية البحث بأكملها. تم إجراء الدراسة السريرية وفقًا للمبادئ المنصوص عليها في إعلان هلسنكي وحصلت على موافقة لجنة الأخلاقيات في مستشفى تشينغلو، جامعة شاندونغ (رقم الموافقة: KYLL-2022(ZM)-1344). تم الحصول على موافقة مستنيرة من جميع المرضى الذين تم تسجيلهم في الدراسة.
نموذج حيواني للاحتشاء/إعادة التروية (I/R)
لإقامة نموذج حيواني للإصابة/الشفاء، تم اختيار ذكور الفئران من سلالة C57BL/6J بعمر 8 أسابيع. تم تثبيت هذه الحيوانات بشكل آمن على لوحة الفئران التي تم الحفاظ على درجة حرارتها ثابتة.قبل العملية، تم تعقيم منطقة الصدر بشكل كامل باستخدام اليودوفور. تم تخدير الفئران باستخدام التخدير بالاستنشاق معإيزوفلوران بمعدلباستخدام مقص معقم، تم إجراء شق جراحي بطول حوالي 2 سم على الصدر في النقطة التي كان فيها نبض القلب الأكثر وضوحًا.تم وضع غرزة على شكل حبل، وتم تشريح الصدر فوق القلب من خلال الفضاء بين الضلوع الرابع باستخدام ملقط منحني. تم الضغط برفق على الصدر لتسهيل إخراج القلب من تجويف الصدر ووضعه داخل الفتحة المخيطة، مع التأكد من بقاء الحد الأدنى من الهواء في تجويف الصدر. ثم تم استخدام غرزة جراحية 6-0 لربط عقدة جراحية حول الشريان التاجي الأمامي الأيسر النازل، مع بروز أحد طرفي الغرزة خارج الصدر. على الفور، تم إعادة القلب إلى تجويف الصدر، وتم طرد أي هواء زائد، وتم خياطة الشق الجلدي. بعد العملية، تم نقل الفئران إلى غرفة التعافي من التخدير. بعد 45 دقيقة، تم تحرير العقدة لإعادة تدفق الدم إلى القلب، وتمت إعادة الفئران إلى أقفاص التربية الخاصة بها لمتابعة الرعاية.
بعد ذلك، تم استئصال القلوب وصبغها لتحديد مدى نخر عضلة القلب بدقة، معبرًا عنه كنسبة من منطقة الإقفار المعرضة للخطر (AAR) التي لم يتم ترويتها. تم صبغ المنطقة الإقفارية التي تحتوي على أنسجة قابلة للحياة باللون الأحمر باستخدام 2،3،5-ثلاثي فينيل تيترازوليوم، بينما تم تمييز المنطقة غير الإقفارية بوضوح باللون الأزرق باستخدام صبغة إيفان. ثم تم تجميد القلوب عندلمدة 10 دقائق وقطع إلى شرائح (تم قياس 6 شرائح/قلب). تم قياس منطقة الاحتشاء، ومنطقة AAR، ومنطقة LV باستخدام برنامج ImageJ من NIH. تم حساب حجم الاحتشاء كنسبة منطقة الاحتشاء إلى منطقة AAR. تم تصوير هذه الشرائح باستخدام ميكروسكوب تصويري كبير (Leica M205 FA)، وتم تحليل الصور لاحقًا بمساعدة برنامج NIH Image.
تخطيط صدى القلب
بعدفي مرحلة النمذجة، تم تقييم الهيكل والوظيفة القلبية للفئران باستخدام تخطيط صدى القلب عبر الصدر. تم استخدام نظام التصوير VisualSonic VeVo 2100، الذي نشأ من تورونتو، كندا، لهذا التقييم. قبل بدء الإجراء، تم تخدير الفئران عن طريق الاستنشاقإيزوفلوران. بعد ذلك، تم وضعهم على منصة مدفأة تم الحفاظ على درجة حرارتها عندوكانت متصلة بشكل آمن بجهاز تخطيط القلب الكهربائي (ECG) للمراقبة المستمرة. مع تطبيق تخطيط صدى القلب بطريقة M-mode، تم توثيق قطر البطين الأيسر في مرحلة الانبساط (LVIDd) وقطر البطين الأيسر في مرحلة الانقباض (LVIDs) بدقة على المحور الطولي الجانبي. في النهاية، تم حساب نسبة قذف البطين الأيسر ونسبة القصر تلقائيًا لتمكين تقييم دقيق.
عزل مجموعات خلايا قلبية متميزة
تم عزل مجموعات خلايا قلبية متميزة من قلوب الفئران المصابة بالاحتشاء بعد 24 ساعة من نقص تروية القلب- إعادة التروية (MI/R). تم ذلك وفقًا للبروتوكولات الموجودة مسبقًا.تم استخدام تقنيات موحدة لعزل خلايا القلب العضلية (CMs).للعزل الخلايا الليفية القلبية (CFs) والبلعميات، تم استخدام مجموعة تفكيك العضلات الهيكلية (Miltenyi Biotech، شنغهاي، الصين). لفصل البلعميات عن CFs، تم استخدام كريات مغناطيسية مغطاة بمضاد F4/80 (رقم الكاتالوج 130-110-443؛ Miltenyi Biotech) وفقًا للتعليمات الصارمة للجهة المصنعة. بعد ذلك، تم جمع الخلايا المنقاة من خلال الطرد المركزي عند لمدة 5 دقائق عند ، استعدادًا للاستخراج اللاحق للبروتينات.
تم تحديد منطقة الاحتشاء على أنها الجزء الواقع بين الخياطة وقمة القلب. تم وصف منطقة حدود الاحتشاء بعناية على أنها المنطقة الهامشية التي تميز الأنسجة المحتشية عن عضلة القلب غير المحتشية المجاورة في المقطع العرضي القصير. باستخدام الميكروتوم (RM2235؛ لايكا للأنظمة الدقيقة، مانهايم، ألمانيا)، تم قطع أنسجة القلب الموضوعة 1 مم أسفل موقع الربط بدقة إلىشرائح عرضية سميكة، مرتبة على طول المحور الأفقي الطويل. تم تطبيق القطع التسلسلي لكل من عمليات صبغ MT، مما سهل تقييم تليف عضلة القلب.
لصبغة المناعية الفلورية لـ dsDNA و cGAS و STING و GPX4 و 4-HNE، تم أولاً معالجة الشرائح لإزالة الشمع. ثم تم إجراء استرجاع المستضد باستخدام مجموعة مخصصة (C1034؛ سولار بيو، بكين، الصين). بعد ذلك، تم غمر الشرائح في 0.1% تريتون X-100 (GC204003؛ سيرفيس بيو، ووهان، الصين) في محلول ملحي مخفف بالفوسفات (PBS) لمدة 10 دقائق لزيادة نفاذيتها. بعد ذلك، تم تحضينها معمصل الماعز العادي (G1208؛ سيرفيس بيو، ووهان، الصين) في PBS لمدة 60 دقيقة في درجة حرارة الغرفة ) لحجب مواقع الربط غير المحددة. ثم تم تطبيق الأجسام المضادة المحددة لـ dsDNA و cGAS و STING و GPX4 أو 4-HNE، وتمت معالجة الشرائح في الليل عند. بعد ذلك، تم غسل الأقسام ثلاث مرات بمحلول PBS لإزالة الأجسام المضادة غير المرتبطة. بعد ذلك، تم إجراء حضانة مع الأجسام المضادة الثانوية أليكسا فلور 594 (ab150120؛ أبكام، كامبريدج، ماساتشوستس، الولايات المتحدة الأمريكية) وأليكسا فلور 488 (ab150081؛ أبكام، الولايات المتحدة الأمريكية) (مخففة بنسبة 1:200) لمدة ساعة واحدة في الظلام عند لتصوير الارتباط المحدد للأجسام المضادة الأولية. وأخيرًا، تم تلوين النوى بلون مضاد.-دياميدينو-2-فينيل إندول (DAPI، ab104139؛ أبكام، المملكة المتحدة) لتمكين التعرف الواضح على الموقع الخلوي.
زراعة الخلايا
تم الحصول على خلايا HEK293T وHeLa وHL-1 من شركة KeyGene BioTech في الصين. تم عزل خلايا القلب الأولية من الفئران (MPCs) من cgas.ستينغ، cgas-CKO، Sting-CKO، بالإضافة إلى أقرانهم من مجموعة التحكم أو فئران C57BL/6J من النوع البري (WT). تم زراعة هذه الخلايا في وسط ديوبيكو المعدل من إيجل (DMEM) يحتوي على 10% من مصل الجنين البقري (FBS؛ 9014-81-7، سيغما ألدريش، ألمانيا). تم الحفاظ على زراعة الخلايا عند درجة حرارة .
تلوين خلايا IF والتحليل بالليزر المجهري
في عملية صبغ المناعية الخلوية، تم زراعة خلايا هيلا، خلايا HL-1، أو خلايا القلب عند كثافةخلايا لكل مليلتر على شريحة زجاجية بقطر 14 مليمتر (WHB، الصين). بعد المعالجة بالليغاندات، تم تثبيت الخلايا على الفور باستخدامبارافورمالدهيد (بيوتايم، الصين) لمدة 5 دقائق. بعد ذلك، تم زيادة نفاذية غشاء الخلية عن طريق معالجة الخلايا بـتم استخدام Triton X-100 (Beyotime، الصين) لمدة 5 دقائق إضافية. بعد ذلك، تم حجب مواقع الربط غير المحددة عن طريق حضن الخلايا في محلول يحتوي على 10% من مصل الحمار (Solarbio Science & Technology، الصين) في درجة حرارة الغرفة لمدة ساعة واحدة. ثم، تم حضن العينات مع الأجسام المضادة الأولية المحددة طوال الليل في. بعد ذلك، تم شطف العينات ثلاث مرات تمت معالجة العينات مع PBS (علوم وتكنولوجيا سولاربيو، الصين). ثم تم حضانة العينات مع الأجسام المضادة الثانوية الفلورية المعينة لمدة ساعة واحدة في درجة حرارة الغرفة، تلتها ثلاث جولات أخرى من الشطف مع PBS. في النهاية، تم معالجة العينات بالتلوين المضاد مع DAPI (ab104139؛ أبكام، المملكة المتحدة) لتحديد النوى.
تحليل الإحصائيات
تم تقديم البيانات كمتوسط ± الخطأ المعياري للمتوسط (SEM). تم إجراء جميع التحليلات الإحصائية بعناية باستخدام برنامج GraphPad Prism 9 (GraphPad، سان دييغو، كاليفورنيا، الولايات المتحدة الأمريكية). تم الإشارة إلى SEM بواسطة أشرطة الخطأ. في البداية، تم فحص التوزيع الطبيعي، وبعد ذلك تم تطبيق اختبار شابيرو-ويلك لتقييم تجانس التباين. أظهرت البيانات توزيعًا طبيعيًا تقريبيًا. )، مما يشير إلى ملاءمة الطرق الإحصائية المعلمية. لتقييم الفروق ذات الدلالة الإحصائية بين مجموعتين لبيانات موزعة بشكل طبيعي، يتم استخدام اختبار ت الطالب غير المتزاوج ذو الطرفين تم استخدام اختبار. بالنسبة للبيانات غير الموزعة بشكل طبيعي، تم استخدام طريقة إحصائية غير معلمية. على وجه التحديد، تم استخدام اختبار كروسكال-واليس، وتبعه اختبار دان بعد الاختبار لإجراء مقارنات متعددة. اعتُبرت الفروقات ذات دلالة إحصائية بناءً على معايير صارمة، على وجه التحديد،للدلالة الهامشية،لأهمية معتدلة،لأهمية قوية، و ****للفروق ذات الدلالة العالية، أو كغير دالة. تم تكرار ثلاثة تجارب مستقلة على الأقل لإجراء التحليل الإحصائي. كل نقطة بيانات تضمنت على الأقل ثلاث تكرارات بيولوجية.
توفر البيانات
يجب أن تكون جميع البيانات والمواد متاحة ضمن المواد المقدمة أو في مستودع عام. بيانات تسلسل RNA التي تم إنتاجها في هذه الدراسة متاحة للجمهور في مجموعة بيانات Gene Expression Omnibus (GEO) GSE291453.
شكر وتقدير
تم دعم هذا العمل من خلال منح من المؤسسة الوطنية للعلوم الطبيعية في الصين (رقم 82270487، 82241203، 82200502، 84270277، 82200507)؛ البرنامج الوطني الرئيسي للبحث والتطوير في الصين (2024YFA1307002، 2021YFF0501403)؛ مؤسسة شاندونغ الإقليمية للعلوم الطبيعية (ZR2023JQ030، 2021ZDSYS05، 2024CXPT080، ZR2024ZD09، ZR2002QH089)؛ برنامج استقطاب المواهب في الجامعات (BP0719033)؛ صندوق المعهد المركزي للبحوث غير الربحية من الأكاديمية الصينية للعلوم الطبية (2023-PT320-06)؛ برنامج علماء تايشان في مقاطعة شاندونغ (تشانغ م، تشانغ ج، وتشانغ ج)؛ وصندوق البحث الأساسي للجامعات المركزية (رقم 2023QNTD003 إلى م. ز.). كما نتقدم بالشكر لبايجيه ليو (شركة بكين يونغ شين كانغ تاي لتطوير التكنولوجيا المحدودة) على إرشاداتها الفنية القيمة.
مساهمات المؤلفين
قام C.Z. و M.Z. بتصميم الدراسة وتوجيهها والإشراف على البحث. قامت Xiaohong W. بأداء معظم التجارب، وجمعت جميع البيانات، وشاركت في تحليل النتائج، وعرضت البيانات في أشكال قابلة للنشر، وكتبت المقال. ساهم T.C. في تجارب الحيوانات. ساهم N.L. وXiao W. في الكشف بالليزر عن المناعية الفلورية. ساهم Z.M. في تصوير تخطيط صدى القلب. شارك Y.Z. في مناقشات المشروع. شارك S.C. وJ.Z. وL.Cai. وC.L. وYu.Z. وL.Cao وQ.L. وW.Q. وR.R. وH.Z. وC.G. وQ.D. وW.S. وY.H. في مناقشات المشروع وتحليل البيانات. جميع المؤلفين قرأوا ووافقوا على المقال.
المصالح المتنافسة: يعلن المؤلفون عدم وجود مصالح متنافسة. ملاحظة الناشر: تظل شركة سبرينغر ناتشر محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
REFERENCES
Heusch, G. Myocardial ischaemia-reperfusion injury and cardioprotection in perspective. Nat. Rev. Cardiol. 17, 773-789 (2020).
Heusch, G. Myocardial ischemia/reperfusion: Translational pathophysiology of ischemic heart disease. Medicine 5, 10-31 (2024).
Wu, X., Li, Y., Zhang, S. & Zhou, X. Ferroptosis as a novel therapeutic target for cardiovascular disease. Theranostics 11, 3052-3059 (2021).
Xu, S. et al. Naringenin alleviates myocardial ischemia/reperfusion injury by regulating the nuclear factor-erythroid factor 2-related factor 2 (Nrf2) /System xc-/ glutathione peroxidase 4 (GPX4) axis to inhibit ferroptosis. Bioengineered 12, 10924-10934 (2021).
Lu, H. et al. Britanin relieves ferroptosis-mediated myocardial ischaemia/reperfusion damage by upregulating GPX4 through activation of AMPK/GSK3β/ Nrf2 signalling. Pharm. Biol. 60, 38-45 (2022).
Dvorkin, S., Cambier, S., Volkman, H. E. & Stetson, D. B. New frontiers in the cGASSTING intracellular DNA-sensing pathway. Immunity 57, 718-730 (2024).
Kemmoku, H. et al. Single-molecule localization microscopy reveals STING clustering at the trans-Golgi network through palmitoylation-dependent accumulation of cholesterol. Nat. Commun. 15, 220 (2024).
Liu, S. et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347, aaa2630 (2015).
Konno, H., Konno, K. & Barber, G. N. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell 155, 688-698 (2013).
Crow, Y. J. & Casanova, J. L. STING-associated vasculopathy with onset in infancya new interferonopathy. N. Engl. J. Med. 371, 568-571 (2014).
Zhu, Y. et al. STING: a master regulator in the cancer-immunity cycle. Mol. Cancer 18, 152 (2019).
Sladitschek-Martens, H. L. et al. YAP/TAZ activity in stromal cells prevents ageing by controlling cGAS-STING. Nature 607, 790-798 (2022).
Gui, X. et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 567, 262-266 (2019).
Warner, J. D. et al. STING-associated vasculopathy develops independently of IRF3 in mice. J. Exp. Med. 214, 3279-3292 (2017).
Liu, B. et al. Human STING is a proton channel. Science 381, 508-514 (2023).
Chen, T. et al. The nucleotide receptor STING translocates to the phagosomes to negatively regulate anti-fungal immunity. Immunity 56, 1727-1742.e1726 (2023).
Heusch, G. et al. Health position paper and redox perspectives on reactive oxygen species as signals and targets of cardioprotection. Redox Biol. 67, 102894 (2023).
Yang, W. S. & Stockwell, B. R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 26, 165-176 (2016).
Korge, P., Ping, P. & Weiss, J. N. Reactive oxygen species production in energized cardiac mitochondria during hypoxia/reoxygenation: modulation by nitric oxide. Circ. Res. 103, 873-880 (2008).
Yang, W. S. et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317-331 (2014).
Zhu, S. et al. HSPA5 Regulates Ferroptotic Cell Death in Cancer Cells. Cancer Res. 77, 2064-2077 (2017).
Pohl, C. & Dikic, I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 366, 818-822 (2019).
Kaushal, G. P. & Shah, S. V. Autophagy in acute kidney injury. Kidney Int. 89, 779-791 (2016).
Qiao, L. et al. LAMP2A, LAMP2B and LAMP2C: similar structures, divergent roles. Autophagy 19, 2837-2852 (2023).
Dalleau, S., Baradat, M., Guéraud, F. & Huc, L. Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance. Cell Death Differ. 20, 1615-1630 (2013).
Li, Q. et al. Inhibition of double-strand DNA-sensing cGAS ameliorates brain injury after ischemic stroke. EMBO Mol. Med. 12, e11002 (2020).
Cao, D. J. et al. Cytosolic DNA Sensing Promotes Macrophage Transformation and Governs Myocardial Ischemic Injury. Circulation 137, 2613-2634 (2018).
Hu, Q. et al. Released Mitochondrial DNA Following Intestinal Ischemia Reperfusion Induces the Inflammatory Response and Gut Barrier Dysfunction. Sci. Rep. 8, 7350 (2018).
Bindi, E. et al. Mitochondrial DNA: A Biomarker of Disease Severity in Necrotizing Enterocolitis. Eur. J. Pediatr. Surg. 30, 85-89 (2020).
Rech, L. et al. Small molecule STING inhibition improves myocardial infarction remodeling. Life Sci. 291, 120263 (2022).
King, K. R. et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat. Med. 23, 1481-1487 (2017).
Visan, I. Myocardial infarct inflammation. Nat. Immunol. 19, 99 (2018).
Zhong, W. et al. Aging aggravated liver ischemia and reperfusion injury by promoting STING-mediated NLRP3 activation in macrophages. Aging Cell 19, e13186 (2020).
Maekawa, H. et al. Mitochondrial Damage Causes Inflammation via cGAS-STING Signaling in Acute Kidney Injury. Cell Rep. 29, 1261-1273.e1266 (2019).
Ter Horst, E. N. et al. Elevated monocyte-specific type I interferon signalling correlates positively with cardiac healing in myocardial infarct patients but interferon alpha application deteriorates myocardial healing in rats. Basic Res. Cardiol. 114, 1 (2018).
Wang, L. et al. Plasma nuclear and mitochondrial DNA levels in acute myocardial infarction patients. Coron. Artery Dis. 26, 296-300 (2015).
Boengler, K., Schulz, R. & Heusch, G. Loss of cardioprotection with ageing. Cardiovasc Res. 83, 247-261 (2009).
Kleinbongard, P., Lieder, H., Skyschally, A. & Heusch, G. No sex-related differences in infarct size, no-reflow, and protection by ischaemic pre-conditioning in Göttingen minipigs. Cardiovasc Res. 119, 561-570 (2023).
Guo, Y. et al. Genetic background, gender, age, body temperature, and arterial blood pH have a major impact on myocardial infarct size in the mouse and need to be carefully measured and/or taken into account: results of a comprehensive analysis of determinants of infarct size in 1,074 mice. Basic Res. Cardiol. 107, 288 (2012).
Ferdinandy, P. et al. Interaction of Cardiovascular Nonmodifiable Risk Factors, Comorbidities and Comedications With Ischemia/Reperfusion Injury and Cardioprotection by Pharmacological Treatments and Ischemic Conditioning. Pharm. Rev. 75, 159-216 (2023).
Christia, P. et al. Systematic characterization of myocardial inflammation, repair, and remodeling in a mouse model of reperfused myocardial infarction. J. Histochem Cytochem 61, 555-570 (2013).
Jia, M. et al. Redox homeostasis maintained by GPX4 facilitates STING activation. Nat. Immunol. 21, 727-735 (2020).
Haag, S. M. et al. Targeting STING with covalent small-molecule inhibitors. Nature 559, 269-273 (2018).
Heusch, G. Mitochondria at the heart of cardiovascular protection: p66shc-friend or foe? Eur. Heart J. 36, 469-471 (2015).
Kleinbongard, P. et al. Co-morbidities and co-medications as confounders of cardioprotection-Does it matter in the clinical setting? Br. J. Pharm. 177, 5252-5269 (2020).
Heusch, G. Critical Issues for the Translation of Cardioprotection. Circ. Res. 120, 1477-1486 (2017).
Cai, W. et al. Alox15/15-HpETE Aggravates Myocardial Ischemia-Reperfusion Injury by Promoting Cardiomyocyte Ferroptosis. Circulation 147, 1444-1460 (2023).
Ma, H. et al. Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/ reperfusion injury: role of autophagy paradox and toxic aldehyde. Eur. Heart J. 32, 1025-1038 (2011).
Ackers-Johnson, M. et al. A Simplified, Langendorff-Free Method for Concomitant Isolation of Viable Cardiac Myocytes and Nonmyocytes From the Adult Mouse Heart. Circ. Res. 119, 909-920 (2016).
Vukicevic, S. et al. Bone morphogenetic protein 1.3 inhibition decreases scar formation and supports cardiomyocyte survival after myocardial infarction. Nat. Commun. 13, 81 (2022).
State Key Laboratory for Innovation and Transformation of Luobing Theory; Key Laboratory of Cardiovascular Remodeling and Function Research of MOE, NHC, CAMS and Shandong Province; Department of Cardiology, Qilu Hospital of Shandong University, Jinan 250012, China
Correspondence: Meng Zhang (zhangmeng@sdu.edu.cn) or Cheng Zhang (zhangc@sdu.edu.cn)
STING aggravates ferroptosis-dependent myocardial ischemia-reperfusion injury by targeting GPX4 for autophagic degradation
Xiaohong Wang , Tao Chen , Sizhe Chen , Jie Zhang® , Liangyu Cai , Changhao Liu , Yujie Zhang , Xiao Wu , Na Li , Zhiyong Ma , Lei Cao , Qian Li , Chenghu Guo , Qiming Deng , Wenqian Qi , Yonghao Hou , Ruiqing Ren , Wenhai Sui , Haonan Zheng , Yun Zhang , Meng Zhang and Cheng Zhang
Abstract
Despite advancements in interventional coronary reperfusion technologies following myocardial infarction, a notable portion of patients continue to experience elevated mortality rates as a result of myocardial ischemia-reperfusion (MI/R) injury. An in-depth understanding of the mechanisms underlying MI/R injury is crucial for devising strategies to minimize myocardial damage and enhance patient survival. Here, it is discovered that during MI/R, double-stranded DNA (dsDNA)-cyclic GMP-AMP synthase (cGAS)stimulator of interferon genes (STING) signal accumulates, accompanied by high rates of myocardial ferroptosis. The specific deletion of cgas or Sting in cardiomyocytes, resulting in the inhibition of oxidative stress, has been shown to mitigate ferroptosis and I/R injury. Conversely, activation of STING exacerbates ferroptosis and I/R injury. Mechanistically, STING directly targets glutathione peroxidase 4 (GPX4) to facilitate its degradation through autophagy, by promoting the fusion of autophagosomes and lysosomes. This STING-GPX4 axis contributes to cardiomyocyte ferroptosis and forms a positive feedback circuit. Blocking the STING-GPX4 interaction through mutations in T267 of STING or N146 of GPX4 stabilizes GPX4. Therapeutically, AAV-mediated GPX4 administration alleviates ferroptosis induced by STING, resulting in enhanced cardiac functional recovery from MI/R injury. Additionally, the inhibition of STING by H-151 stabilizes GPX4 to reverse GPX4-induced ferroptosis and alleviate MI/R injury. Collectively, a novel autophagy-dependent ferroptosis mechanism is identified in this study. Specifically, STING autophagy induced by anoxia or ischemia-reperfusion leads to GPX4 degradation, thereby presenting a promising therapeutic target for heart diseases associated with I/R.
Ischemic heart disease remains the predominant cause of morbidity and mortality globally. Despite timely interventional coronary reperfusion being effective in salvaging ischemic myocardium, myocardial ischemia-reperfusion (MI/R) injury remains a significant contributor to mortality in a substantial patient population. To date, despite intense decades-long research, no specific MI/R injury targeting agent has arrived at the clinical area. There exists an urgent medical imperative to comprehensively and deeply understand the intricate mechanisms that govern cardiomyocyte death. Grasping these mechanisms is fundamental as it can unlock the discovery of novel and innovative therapeutic targets, which are of utmost importance for the efficient and effective management of the associated pathological state.
Cell death, especially the death of cardiomyocytes (CMs), is a crucial aspect within the pathophysiology of cardiovascular diseases. Among the forms of cell death, ferroptosis, an irondependent cell death modality marked by excessive lipid peroxidation, has emerged as a key factor in MI/R injury pathophysiology. Ferroptosis is regulated by the metabolism of
iron, lipids, amino acids, and glutathione, and is closely associated with multiple heart conditions. This makes targeting ferroptosis a promising therapeutic approach for MI/R injury, and thus potentially revolutionizing treatment strategies for this common and life-threatening condition. Glutathione peroxidase 4 (GPX4), having the function of converting lipid peroxides into lipid alcohols with less harmfulness, has become a pivotal regulatory factor in inhibiting ferroptosis. Previous studies have demonstrated that ferroptosis triggered by MI/R is concomitant with the suppression of GPX4. During the MI/R process, a reduction in GPX4 levels coincides with the initiation of ferroptosis. In contrast, elevating GPX4 levels effectively mitigates myocardial injury and enhances cardiac function. However, the precise regulation of GPX4 protein levels and its underlying degradation mechanism remain elusive. Therefore, the discovery of factors that regulate GPX4 might present an attractive therapeutic target for the modulation of ferroptosis occurring during the MI/R process.
The cyclic guanosine monophosphate (GMP)-adenosine monophosphate (AMP) (cGAMP) synthase (cGAS) and stimulator of interferon genes (STING) stand as pivotal and highly conserved immune components within the mammalian immune system.
They are integral in the body’s defense mechanism against pathological nucleic acids, with a particular focus on doublestranded DNA (dsDNA). Upon abnormal DNA binding, cGAS produces a second messenger cGAMP. This newly synthesized cGAMP then engages in a specific interaction with STING, promoting its conformational changes. Once activated, STING binds to TANK-binding kinase 1 (TBK1) and interferon regulatory factor 3 (IRF3). This binding occurrence sets off a complex cascade of molecular events, which ultimately culminate in the initiation of interferon (IFN) expression. The production of IFN not only contributes to the body’s inflammatory response but also elicits powerful antiviral effects. In recent years, an increasing number of studies have reported that STING-associated signaling dysregulation can be observed in a wide array of human pathologies. These include, but are not limited to, type I IFN diseases, where the normal IFN signaling pathways are disrupted, and in cancer, where STING regulates anticancer immunity in both IFNdependent and IFN-independent manners. Moreover, many reports revealed that STING functions independently of classical antiviral pathways, such as in aging and autophagy. STING plays a role in driving tissue senescence and premature agingrelated tissue degeneration, and autophagy represents an ancestral function intrinsic to the cGAS-STING pathway. Indeed, a large part of disease progression and cell death modulated by STING occurs independently of IRF3 and STING’s antiviral gene expression. The development of STING-associated vasculopathy occurs without relying on IRF3. Human STING, which functions as a proton channel, has its interferon-inducing ability separated from its key roles in promoting LC3B lipidation and inflammasome activation. In phagosomes, STING directly interacted with Src, and this interaction hindered Src from recruiting Syk and phosphorylating it. The exploration of STING’s non-traditional immune functions is poised to substantially enrich our comprehension of immune complexity.
In our investigation, we explored how STING modulates myocardial ferroptosis when the heart undergoes MI/R injury. I/R-induced mitochondrial damage leads to the release of dsDNA, which is subsequently recognized by cGAS, resulting in the production of the second messenger cGAMP and activation of STING. STING can directly trigger myocardial ferroptosis via its interaction with GPX4. Once STING binds to GPX4, it prompts augmented autophagy, which in turn causes the subsequent degradation of GPX4, fueling the process of myocardial ferroptosis. We also evaluated two effective therapeutic strategies against MI/R injury, including the utilization of AAV-mediated GPX4 overexpression and the administration of STING antagonists. In the light of our research, the results we’ve obtained offer fresh perspectives on the development mechanism of ferroptosis and establish STING as a potential therapeutic target to prevent MI/R injury, to improve patient outcomes and to reduce mortality rates associated with ischemic heart disease.
RESULTS
I/R triggers cGAS-STING upregulation in CMs
To begin to define effects of cGAS-mediated DNA sensing in I/R injury, we first tested for changes in cytosolic DNA levels. To elucidate whether heart I/R caused cell DNA damage, we initially set up an I/R model using male C57BL/6J mice that were 8 weeks old (Fig. 1a, b) and analyzed the coimmunostaining and expression of double-stranded DNA (dsDNA) and cGAS in the I/R border region. Markedly increased amounts of cytosolic DNA and cGAS activation were observed in cells from mice undergoing I/R compared to Sham-operated controls (Fig. 1c), suggesting profound DNA damage and the leakage of DNA into the cytosol in diseased tissues. Following the recognition of dsDNA, cGAS undergoes activation. To further investigate, we employed Western blot assays to assess the abundance of cGAS and STING
proteins. The results revealed a significant upregulation of both cGAS and STING expression in the I/R border region, occurring after 45 min of heart ischemia followed by 24 h of reperfusion (Fig. 1d, e). However, cGAS and STING protein levels in the infarct core did not show significant changes compared to the Sham group (Supplementary Fig. 1a), and the variations were negligible in the non-ischemic region, which remains unaffected by I/R (Supplementary Fig. 1b). In parallel, the immunofluorescence staining results for tissue sections from both the I/R and Sham groups indicated that the expression of STING was markedly enhanced within the ischemic region (border region) of the I/R group but showed no significant changes in the infarct core or non-ischemic region (supplementary Fig. 1c).
In addition, to detect evidence of cGAS-STING pathway activation post-I/R, we isolated CMs, fibroblasts and macrophages populations from the hearts of WT, cgas or Sting mice following established protocols, which allowed us to investigate the specific roles of cGAS and STING in different cardiac cell types post-I/R. The upregulation of cGAS and STING in CMs from the border region of the I/R heart was confirmed at the protein level. Notably, these increases were negligible in cgas- or Sting-null CMs (Fig. 1f, g). However, no apparent differences were observed in the macrophages or fibroblasts, regardless of whether they underwent I/R (Fig. 1f, g, Supplementary Fig. 1d). Since the results mentioned above implied a dependency of cGAS’s role in I/R on STING, we next shifted our focus to STING. Then we devised an immunofluorescence co-localization experiment to further elucidate the expression pattern of STING in CMs. The immunofluorescence staining revealed a significant upregulation of STING expression in CMs located in the border region of the I/R heart (Fig. 1h), in contrast to the negative control.
To verify our results ex vivo, we established an anoxia/ reoxygenation (A/R) model with mouse primary CMs (MPCs) obtained from mouse hearts (Fig. 1i). The coimmunostaining analysis of live-cell mitochondrial probes (Mito-Tracker) and dsDNA was used to determine the cytosolic DNA that did not colocalize with either the nuclei or mitochondria. Remarkably, we observed a significant release of dsDNA, accompanied by severe mitochondrial damage manifesting as enlargement, shortening, and thickening post A/R (Fig. 1j), in contrast to the control MPCs.
Together, these findings point out that dsDNA-cGAS-STING signaling is activated in the heart CMs during I/R. We directed our focus towards understanding the specific role of cGAS and STING in CMs during I/R injury.
Deletion of cGAS-STING protects against MI/R injury
The role of cGAS or STING in CMs is poorly understood. Next, we generated CM-specific cgas-knockout [cgas Myh6 (cgasCKO)] and Sting-knockout [Sting Myh6 (Sting-CKO)] mice for the investigation of the role and functionality of CM-specific cGAS and STING in MI/R injury (Fig. 2a). cgas and cgas Myh6 , as well as Sting and Sting Myh6 were subjected to I/R. Heart injury was assessed by quantifying the area of necrosis in heart sections, assessing cardiac function, and measuring Masson staining. Cardiac function at baseline, represented by left ventricular ejection fraction and fractional shortening (LVEF and FS respectively), was comparable between the KO and their control animals, with no significant differences observed (Supplementary Fig. 1e-h). These findings confirm that the knockout animals do not exhibit intrinsic abnormalities in cardiac function prior to injury. According to the results, cgasCKO mice exhibited a reduced necrosis area, much better cardiac function (EF and FS levels) and decreased fibrosis area following I/R compared to their control mice (Fig. 2b-d), indicating a protective role of CM-specific cgas knockout against MI/R injury. Masson staining of 7-day-reperfusion heart tissue showed reduced fibrosis in cgas- or Sting-CKO mice (Supplementary Fig. 1i, j).
Fig. 1 I/R triggers cGAS-STING upregulation in CMs. Male mice (aged 8 weeks) were subjected to the operation and euthanized at 24 h post-I/ R or Sham. a Display the mouse I/R model operation flowchart. b Schematic diagram showing the mouse I/R injury model. c Double immunofluorescence analysis for detecting dsDNA and cGAS in the same heart section of the WT border region post I/R or Sham. The positive reactions of tissue sections are displayed in green (dsDNA) and red (cGAS). The positive reaction for co-localisation is displayed in yellow. Scale bar m. d, e Western blot and quantification of cGAS and STING in WT cells isolated from border region post I/R or Sham ( ). Western blot and quantification of cGAS and STING in various cell types isolated from the heart border region of cgas , Sting , or WT mice post I/R or Sham . Double immunofluorescence analysis for detecting markers of CMs and STING in the same heart section of the WT border region post I/R or Sham. The positive reactions of tissue sections are displayed in green (STING) and red (Troponin). The positive reaction for co-localisation is displayed in yellow. Scale bar . i Schematic diagram showing the CM A/R operation flowchart. j Double immunofluorescence analysis for detecting dsDNA and mitochondria (labeled with Mito-Tracker) in MPCs post-A/R. The positive reactions are displayed in green (dsDNA) and red (Mito-Tracker). The positive reaction for co-localisation is displayed in yellow. Arrowheads indicate released dsDNA. Scale bar . Data are expressed as the mean ± SEM. NS non-significant, and ****P < 0.0001 (unpaired twotailed Student’s test). I/R Ischemia reperfusion, dsDNA double-stranded DNA, A/R anoxia/ reoxygenation, cGAS cyclic guanosine monophosphate-adenosine monophosphate synthase, STING stimulator of interferon genes, WT wild type, cgas cgas knockout mice, Sting , Sting knockout mice, CM cardiomyocyte
To further delineate the role of cGAS in MI/R injury, we isolated CMs from cgas-CKO mice and their control mice of the in vivo I/R model. Western blot analysis revealed that the expression level of cGAS in CMs was minimal under physiological conditions, whereas it became inducible following I/R (Fig. 2e). Importantly, this cGAS upregulation in CMs triggered by I/R was absent in cgas-CKO CMs.
Interestingly, similar findings were also observed in Sting-CKO mice (Fig. 2f-h). Since STING is a downstream effector of cGAS, the absence of Sting in Sting-CKO mice led to reduced necrosis area, better cardiac function, and smaller fibrosis area compared to their control mice. I/R could also induce STING expression in Sting CMs (Fig. 2i). Notably, I/R had no effect on STING protein level in
Fig. 2 Deletion of cGAS-STING protects against MI/R injury. a Structural pattern diagram of cgas or Sting CM-specific (Myh6-iCre) conditional knockout mice. b-e Effect of cgas-CKO on myocardial infarct size, FS, EF and fibrosis area following I/R or Sham: b Myocardial infarct size (% of AAR) with representative tissue sectioning ( ); c Echocardiography and measured EF%, and FS% ( ); d Masson staining and measured fibrosis area , scale bar ; e Western blot and quantification of cGAS in CMs isolated from the heart border region ( ). Effect of Sting-CKO on myocardial infarct size, FS, EF and fibrosis area following I/R: Myocardial infarct size (% of AAR) with representative tissue sectioning Echocardiography and measured EF%, and FS% Masson staining and measured fibrosis area% , scale bar ; Western blot and quantification of STING in CMs isolated from the heart border region ( ). Mean SEM, , , and . AAR area at risk, IF infarct area, cgas-CKO cgas Myh , Sting-CKO Sting Myh , FS fraction shortening, EF ejection fraction, KO knockout
Sting-CKO CMs. These findings indicated that cGAS-STING plays a destructive role in cardiac injury post-I/R, and the exacerbation of MI/R injury by cGAS is dependent on the activation of STING.
STING amplifies myocardial ferroptosis through modulation of oxidative stress injury
To elucidate the specific pathway through which STING contributes to MI/R injury, RNA sequencing (RNA-seq) was conducted on heart sections from Sting-CKO mice and their control mice following I/R (Fig. 3a). Analysis with Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways revealed a significant enrichment of genes involved in cell growth and death pathways among the
differentially expressed genes (DEGs) (Fig. 3b). Accordingly, we evaluated the extent of cell death in heart tissues and CMs derived from cgas-CKO or Sting-CKO mice, as well as their control mice subjected to I/R injury. Remarkably, a significant increase in Tunelpositive cells was discernible in the hearts subsequent to I/R. Conversely, both cgas-CKO and Sting-CKO mice exhibited notably reduced levels of Tunel-positive cells when compared to their respective control groups (Supplementary Fig. 2a, b, Fig. 3c, Supplementary Fig. 2e, f), suggesting less cell death. Considering that Tunel can also detect DNA breaks caused by necrosis or other cell death processes, we quantified the expression levels of key cell death markers, such as Pro-caspase-3, Cleaved-caspase3 and
Fig. 3 STING amplifies myocardial ferroptosis through modulation of oxidative stress injury. a Heart region for RNA-seq post I/R. b KEGG Enrichment Bar Plot of RNA-seq. c Statistical chart of immunofluorescence image for detecting Tunel in the heart section of the border region of cgas-CKO or Sting-CKO mice and their control mice post-I/R . The corresponding image is supplementary Fig. 2a, b. d GO Enrichment Scatter Plot of RNA-seq. e MDA analysis for detecting lipid peroxidation . Live cell immunofluorescence imaging analysis for detecting ROS in MPC. Scale bar Western blot and quantification of ACSL4, TFR, SLC7A11 and GPX4 in Sting or Sting-CKO CMs post-I/R Western blot and quantification of ACSL4, TFR, SLC7A11 and GPX4 in Sting or Sting-CKO MPCs post-A/R or normoxia . Mean ± SEM, , , and . ROS reactive oxygen species, Fer-1 ferrostatin-1, cGAMP cyclic guanosine monophosphate-adenosine monophosphate, KEGG Kyoto Encyclopedia of Genes and Genomes, GO Gene Ontology, LAD left anterior descending coronary artery, ACSL4 acyl-CoA synthetase long-chain family member 4, TFR transferrin receptor, SLC7A11 solute carrier family 7, member 11, GPX4 glutathione peroxidase 4, MDA malondialdehyde
B-cell lymphoma-2 (BCL-2), using Western blot assays. The results showed no significant change of protein abundance in Sting-CKO mice post-I/R compared with their control mice (Supplementary Fig. 2c, d). Collectively, these results confirm that the absence of STING attenuates cell death in the heart following I/R injury, but not through the modulation of apoptosis.
Since there are many modes of death in cells, according to Gene Ontology (GO) enrichment analysis, the heart sections from Sting mice exhibited a prominent contribution to autolysosomerelated, oxidative stress and ferroptosis signaling pathways following I/R compared to Sting-CKO mice (Fig. 3d). Tunel staining
is also used as a marker for ferroptosis. We next investigated the effect of STING on the biomarkers of ferroptosis, including lipid peroxidation, reactive oxygen species (ROS) accumulation and GPX4 degradation, as well as the expression of some ferroptosisrelated proteins. Lipid peroxidation has, of late, been recognized as playing a direct role in facilitating necrosis and ferroptosis. To investigate whether STING modulates ferroptosis triggered by A/R, we assayed the generation of malondialdehyde (MDA), an end product of lipid peroxidation. The results showed that after A/R, STING was highly effective in promoting the production of MDA, comparable to the positive control Erastin (Fig. 3e), meanwhile,
STING activation blocked the inhibition of MDA in ferroptosis induced by ferrostatin-1 (Fer-1, a ferroptosis inhibitor). These observations suggest that STING functions upstream of lipid peroxidation, thereby playing a pivotal role in ferroptosis regulation.
Given that mitochondrial damage and oxidative stress response are pivotal events in I/R, to further test the relationship of STING and oxidative stress in heart, we first confirmed the presence of oxidative stress injury by detecting the generation of ROS following A/R in CMs. Exposure of cultured CMs to A/R triggered an elevation in ROS production, MPCs were pretreated with Fer1 for 12 h to attenuate ROS levels before live cell imaging, subsequently, cGAMP was added at to activate STING, and observations and imaging were terminated once ROS levels stabilized (Fig. 3f). Interestingly, the inhibition of ROS in ferroptosis by Fer-1 was significantly impeded in the presence of cGAMP. The results indicated that STING activation leads to an increase in ROS generation.
Having analyzed the impact of STING on ferroptosis biomarkers, encompassing lipid peroxidation and ROS accumulation, we next aimed to confirm the mechanism underlying STING’s promotion of ferroptosis by assessing lipid peroxidation and the abundance of ferroptosis factors. We used Western blot to evaluate the abundance of expression for acyl-CoA synthetase long-chain family member 4 (ACSL4), transferrin receptor (TFR), solute carrier family 7, member 11(SLC7A11) and GPX4, all of which are established to be protein targets of ferroptosis. We observed decreases in the expression of ACSL4 and TFR, alongside elevations in GPX4 and no significant change in SLC7A11, in Sting-CKO mice following I/R compared to their controls (Fig. 3g, h). Up-regulation of ferroptosis was further confirmed at the protein level in MPCs post-A/R (Fig. 3i, j). Interestingly, Sting deficiency blocked the degradation of GPX4 during ferroptosis triggered by A/R, while GPX4 degradation is essential for promoting lipid peroxidation in ferroptosis. Next, we measured the gpx4 mRNA levels in cardiac tissue from both Sham and I/R-treated mice using qPCR. Our results showed that gpx4 mRNA levels remained unchanged between the Sting and Sting-CKO groups post-I/R (Supplementary Fig. 2h), suggesting that the reduction in GPX4 protein is not due to transcriptional downregulation but rather to post-translational regulation. Taken together, we conclude that STING is a potent activator of ferroptosis, facilitating GPX4 protein degradation, lipid peroxidation and ROS accumulation. Subsequently, we delved into the molecular occurrences in which I/R-mediated STING was associated with the reduction of GPX4.
Targeting of GPX4 by STING
The upregulation of the ferroptosis program following STING activation hinted at the potential for MI/R injury to promote unanticipated activation of a signal-transduction chain cascade involving STING. We sought to identify potential gene factors and explore the feasibility of leveraging them to mitigate the exacerbation of MI/R injury in STING-activated mice. Firstly, we analyzed the KEGG pathways expressed in DEGs identified through RNA-seq. (Fig. 3a). Remarkably, ferroptosis-related genes were significantly enriched among these DEGs (Fig. 4a). Next, tandem affinity purification was performed in MPCs using STING protein affinity antibodies to determine the molecular mechanism by which STING regulates ferroptosis. Silver staining of the immune purification materials (Fig. 4b i) and subsequent analysis using LC-MS/MS (Fig. 4bii) were employed to identify the STING protein chaperone. This method revealed several proteins, particularly the ferroptosis factor GPX4. We used PyMOL to display the predicted results of molecular docking analysis between STING (Protein Data Bank [PDB] ID: 4F5W) and GPX4 (PDB ID: 5L71). Figure 4c depicts ten potential binding modes, with a
Match-Align score of 8.157, increasing the likelihood that STING may directly interact with GPX4.
To further corroborate the direct interaction between STING and GPX4, we employed a STING-specific antibody to pull down its protein chaperone, then combined this protein purification products with GPX4 antibody and monitored them in immunoprecipitation (IP) assays. The endogenous co-IP experiments revealed that endogenous STING and GPX4 formed a complex in CMs post-A/R, with cGAMP further enhancing the formation of this complex (Fig. 4d). Correspondingly, STING was detectable in the endogenous purified products of GPX4 (Fig. 4e), indicating a direct interaction between these two proteins. Next, we expressed Myc-tagged STING (Myc-STING) and Flag-tagged GPX4 (FlagGPX4) plasmids in HeLa cells that stably overexpressed these constructs. Myc or Flag IP was then performed to investigate the interaction between Myc-STING and Flag-GPX4. We used Western blot assays to analyze the input and eluate, and the results showed that Myc-STING enrichment could be detected in the purified product eluted by excessive Flag peptide of Flag-GPX4 (Fig. 4f). Correspondingly, Flag-GPX4 enrichment was also observable in the purified product of Myc-STING (Fig. 4g). Furthermore, we found that the interaction between GPX4 and STING was augmented in a time-dependent manner in HeLa cells treated with cGAMP (Fig. 4h), while Erastin diminished the strength of the interaction.
To confirm the interaction between STING and GPX4, immunostaining was performed to analyze the co-localization of STING and GPX4 in MPCs. Under normoxic conditions, MPCs exhibited low expression of STING and no co-localization with GPX4 (Fig. 4ii). Following acute A/R, STING was activated and formed aggregate dots with GPX4 (Fig. 4i ii), while the two had massive aggregation and co-localization with cGAMP stimulation (Fig. 4i iii). Interestingly, upon Erastin, GPX4 had few co-localization with STING (Fig. 4 i iv ). These findings align closely with the experimental results obtained from the Co-IP study. Together, these evidence strongly indicates that STING and GPX4 interact directly in CMs post-A/R. Furthermore, with increasing STING activation by cGAMP, a more pronounced cluster distribution of STING and GPX4 is observed, indicating an enhanced binding effect between the two proteins.
Given the known co-localization and interaction between STING and GPX4 in both mouse and human cells, coupled with the previously mentioned role of STING in regulating ferroptosis, we surmised that GPX4 might be involved in modulating I/R injury which STING contribute to. Immunostaining analysis of heart slices from Sting and Sting-CKO mice highlighted the I/R-induced enhancement of STING expression and its co-localization with GPX4 in I/R border region of Sting hearts (Fig. 4j). Conversely, GPX4 became much brighter and had no co-localization with STING when Sting was specifically deleted. These results further confirmed the previously discovered effect of STING on inducing ferroptosis and the binding between STING and GPX4 post-I/R. It is noteworthy to mention that this binding has the potential to elicit a downregulation in GPX4 expression, necessitating further investigative efforts.
In summary, a direct interaction occurs between STING and GPX4 and the STING activator cGAMP mediates the crosstalk between GPX4 and STING. This effect is conserved in mouse and human cell lines. As the absence of Sting actually led to an increased expression in GPX4 in heart slices following I/R, which aligns with previous findings indicating that STING can induce ferroptosis. Based on these observations, we speculate that STING may directly bind to GPX4, thereby inhibiting its activity and ultimately leading to ferroptosis.
STING and GPX4 directly interact at the amino acid residues N146 of GPX4 and T267 of STING
To further elucidate the binding site where STING associates with GPX4, we conducted a meticulous examination of PDB graphics
Fig. 4 Targeting of STING by GPX4. a KEGG displayed ferroptosis pathway enrichment. b Silver staining and LC-MS/MS analysis for tandem affinity purification using STING protein affinity antibodies in MPCs post-A/R. c Images showing ten potential contact modes according to molecular-docking results between STING (PDB ID: 4F5W) and GPX4 (PDB ID: 5L71). d Co-IP assay of MPCs to examine whether endogenous GPX4 interacts with STING and this combination is influenced by short point-stimulation of cGAMP. IB: STING, IP: GPX4. e Co-IP assay of MPCs to examine whether endogenous STING interacts with GPX4 and this combination is influenced by short point-stimulation of cGAMP. IB: GPX4, IP: STING. f Co-IP assay of HeLa cells co transfected with Flag-GPX4 and Myc-STING to examine whether STING interacts with GPX4. IB: Myc, IP: Flag. Co-IP assay of HeLa cells co transfected with Flag-GPX4 and Myc-STING to examine whether GPX4 interacts with STING. IB: Flag, IP: Myc. h Co-IP assay of HeLa cells co transfected with Flag-GPX4 and Myc-STING under Erastin or cGAMP delivering to examine whether GPX4 and STING interaction is influenced by cGAMP time-grant or Erastin. IB: Flag, IP: Myc. i Double immunofluorescence analysis for detecting STING and GPX4 and their co-localisation in HeLa cells co transfected with Myc-STING and Flag-GPX4 under cGAMP or Erastin addition. The positive reaction for the Myc-label is shown in red, and that for the Flag-label is shown in green. The positive reaction for co-localisation is displayed in yellow. Scale bar . j Double immunofluorescence analysis for detecting GPX4 and STING in the same heart section of the Sting or Sting-CKO border region. The positive reactions of tissue sections are displayed in green (GPX4) and red (STING). The positive reaction for colocalisation is displayed in yellow. Scale bar . LC liquid chromatography, MS mass spectrometry, PDB Protein Data Bank, Co-IP coimmunoprecipitation, IN input, IB immunoblotting, IP immunoprecipitation, Flag-GPX4 Flag-labeled GPX4 plasmid, Myc-STING Myc-labeled STING plasmid
and associated data. Apo STING (non-ligand bound) oligomer (PDB ID: 4F5W) and GPX4 (PDB ID: 5L71) were selected for protein interaction prediction. The prediction results revealed that STING might engage in protein docking with GPX4 at the amino acid positions of Y167, E260, Y245, Q266, or T267. Correspondingly, GPX4 could potentially engage in protein docking with STING at
the amino acid positions of G126, R127, or N146 (Fig. 5a). Based on the aforementioned predictions, IP experiments were conducted on HeLa cells expressing Flag-GPX4 mutants and Myc-STING mutants to determine the interaction domain between STING and GPX4. Initially, three GPX4 peptide segments, and , were evaluated. With the analysis of the Western blot
Fig. 5 STING and GPX4 directly interact at the amino acid residues N146 of GPX4 and T267 of STING. a Representative images of molecular docking results to show the potential contact sites between GPX4 and STING. b Co-IP assay of HeLa cells co-transfected with Flag-GPX4 or Flag-GPX4 point mutation (Flag-GPX4[ G126], Flag-GPX4[ ] or Flag-GPX4[ ]) and Myc-STING, to examine the potential contact site between GPX4 and STING. IB: Myc, IP: Flag. c Co-IP assay of HeLa cells co-transfected with Flag-GPX4 and Myc-STING or Myc-STING point mutation (Myc-STING[ ], Myc-STING[ ], Myc-STING[ ], Myc-STING[ ] or Myc-STING[ ]) to examine the potential contact site between GPX4 and STING. IB: Myc, IP: Flag. d Double immunofluorescence analysis for detecting STING and GPX4 and their colocalisation in HeLa cells co-transfected with Myc-STING and Flag-GPX4, Flag-GPX4 and Myc-STING[ T267] or Myc-STING and FlagGPX4[ ]. The positive reaction for the Myc-label is shown in red, and that for the Flag-label is shown in green. The positive reaction for co-localisation is displayed in yellow. Scale bar . e Double immunofluorescence analysis for detecting STING and GPX4 and their colocalisation in HL-1 cells co-transfected with Myc-STING and Flag-GPX4 or Myc-STING and Flag-GPX4 [ ] under cGAMP addition or not. The positive reaction for the Myc-label is shown in green, and that for the Flag-label is shown in red. The positive reaction for co-localisation is displayed in yellow. Scale bar . Scale bar of the enlarged image . G Gly, R Arg, N Asn, Y Tyr, E Glu, Q Gln, T Thr
results, we found that only Flag-GPX4- mutant lost its capacity to interact with STING, indicating a critical role of this amino acid in the interaction between the two proteins (Fig. 5b). On the other hand, Myc-tagged STING mutants with specific amino acid mutations, namely Myc-STING- , Myc-STING, Myc-STING- , Myc-STING- and Myc-STING, were constructed. The results demonstrated that the absence of the T267 amino acid in STING abolished the interaction between STING and GPX4 (Fig. 5c).
Furthermore, in HeLa cells stably overexpressing Myc-STING and Flag-GPX4, we observed that GPX4 lost its co-localization with STING when the T267 site of STING was absent (Fig. 5d), even if the entire genome of GPX4 was overexpressed. Similarly, the interaction between STING and GPX4 disappeared when GPX4 lost its N146 site (Fig. 5d). Collectively, these data conclusively demonstrate that STING physically interacts with GPX4-N146 through the T267 amino acid of STING.
Utilizing super-resolution imaging microscopy (SIM) devices to enhance the clarity of immunostaining images, we observed robust binding and co-localization between STING and GPX4 in HL-1 cells transfected with Myc-tagged STING and Flag-tagged GPX4 plasmids. Employing this approach, we were able to achieve a more precise comprehension regarding the mode of interaction between these two proteins within the cellular context (Fig. 5e). As depicted in Fig. 4h, cGAMP delivery further accelerated STINGGPX4 complex formation. Notably, when Flag-GPX4-ΔN146 and Myc-STING were simultaneously transfected, GPX4 lost its colocalization with STING. At this point, even upon the delivery of cGAMP, the co-localization between the two proteins could not be restored, thus highlighting the pivotal role of this amino acid in facilitating the interaction between these two proteins.
STING promotes ferroptosis via autophagy-lysosome-mediated degradation of GPX4
The degradation of GPX4 protein is a pivotal event in ferroptosis, ROS generation and irreversible lipid peroxidation, ultimately resulting in cell death. In Fig. 4j, we also observe the potential degradation effect of STING on GPX4 within the myocardial tissue. Next, we verified the impact of STING on GPX4 degradation during ferroptosis. Given our observation that Sting deficiency blocked the degradation of GPX4 during ferroptosis induced by I/R or A/R, we further explored the stimulatory effect of the STING activator, cGAMP, on GPX4 degradation. Notably, we observed that cGAMP resulted in a decrease in the levels of GPX4 protein, contrary to the negative control Fer-1 (Fig. 6a). Treating MPCs with the ferroptosis inhibitor Fer-1 led to an elevation of GPX4 protein levels, but this elevation was blocked in the presence of cGAMP. Additionally, GPX4 activity can be suppressed by Erastin, which shares similar effects to cGAMP as previously mentioned, leading to an accumulation of intracellular lipid peroxides. To evaluate whether cGAMP enhances the degradation of GPX4, we utilized Erastin in our experiments. Administration of cGAMP indeed initiated the degradation of GPX4 (Fig. 6b). Furthermore, the addition of H-151 (a STING inhibitor) was also able to protect ferroptosis and inhibit the degradation of GPX4 induced by Erastin. Taken together, STING activation could elicit the degradation of GPX4.
Subsequently, we delved into the underlying mechanism governing the regulation of GPX4 degradation by STING. Protein degradation post-transcriptionally occurs primarily through two mechanisms: ubiquitin-mediated proteasomal degradation and autophagy-mediated lysosomal degradation. The findings presented in Fig. 3d also demonstrate the enrichment of autolysosomal pathways in Sting mice during I/R injury, suggesting that STING may exert regulatory influence on the degradation of GPX4 via the autophagic pathway. Therefore, we employed several protein degradation inhibitors, including proteasomal inhibitors (MG-132 and calpeptin) and lysosome inhibitors (chloroquine (CQ) and ) to specifically block these degradation pathways. Interestingly, only the application of the lysosome inhibitors CQ or could effectively block GPX4 degradation induced by cGAMP and contributed to GPX4 abundance (Fig. 6c), indicating their participation in the modulation process of GPX4 homeostasis during A/R exposure. Bafilomycin A1(Baf A-1) serves as a late-stage inhibitor of autophagy, effectively preventing the maturation of autophagic vesicles by disrupting the fusion process between autophagosomes and lysosomes. LY294002, 3-MA and Wortmannin act as inhibitors targeting the initial stage of autophagy, effectively blocking the formation of autophagosomes. Through the analysis of Western blot assays, we discovered that only Baf A-1 was capable of blocking GPX4 degradation induced by cGAMP (Fig. 6d), indicating that STING facilitated the autophagic degradation of GPX4 by promoting the fusion of autophagosomes and lysosomes.
Autophagy, a pivotal cellular process responsible for the degradation of proteins, emerged as a significant potential
factor mediating the reduction of GPX4, which was induced by STING subsequent to I/R. As shown in supplementary Fig. 2g, STING deletion could suppress autophagy that was triggered by I/ R. In order to further corroborate the intervention of STING in the autophagic degradation of GPX4 through its modulation of autophagosome-lysosome binding, we employed the RFP-GFPLC3 dual fluorescence autophagy indicator system for meticulously labeling and tracking alterations in microtubule-associated proteins light chain 3 B (LC3B) and autophagic flux. Initial observations revealed a robust activation of autophagy under A/R conditions (Fig. 6e-g). The chimeric fluorescent protein-LC3 firmly anchored itself to the autophagosome membrane and merged with the lysosome to form autolysosomes. Notably, during this process, GFP fluorescence waned, signifying the seamless transition from autophagosomes to autolysosomes. However, in StingCKO MPCs, a remarkable increase in yellow fluorescent dots was observed, with less red dots. This phenomenon robustly corroborates our Western blot findings, indicating a hindrance in the fusion of autophagosomes and lysosomes, as well as a disruption in the maturation process of autophagosomes. In essence, the absence of Sting prevents the elimination of GPX4 by impeding the union of autophagosomes and lysosomes.
STING is recognized for its ability to initiate autophagy through the lipidation of LC3B and the subsequent formation of autophagosomes, independently of TBK1 activation and interferon induction. We next examined the effect of STING, LC3B and GPX4 in ferroptosis. Utilizing tricolor immunofluorescence analysis, we observed that A/R conditions activated STING, resulting in the formation of LC3 puncta in Sting MPCs but not Sting-deficient cells (Fig. 6h). This finding concurs with previously reported literature results. The clear co-localization of LC3 puncta with both STING and GPX4 was observed in Sting MPCs but not in cells that lack STING, suggesting GPX4 was targeted by STINGinduced autophagy post-A/R. To localize the interaction of STING and GPX4, we co-stained for STING, GPX4, and markers of endoplasmic reticulum-Golgi intermediate compartment (ERGIC), coat protein I (COP-I), and LC3 in MPCs. Our results demonstrated that the STING-GPX4 complex initially localizes to the ERGIC. The STING-GPX4 complex is subsequently trafficked towards the ERGIC, vesicles of COP-I, and autophagosomes of LC3 (Supplementary Fig. 3).
Additionally, lysosomal-associated membrane protein 2B (LAMP2B) mediates the mechanism of autophagosomelysosomal fusion. A/R-stimulated autophagosome formation was visualized by LAMP2B. Imaging revealed that the GPX4STING puncta induced by A/R was enclosed by the LAMP2Blabeled autolysosome membrane ring (Fig. 6i). This suggests that the interaction between STING and GPX4 occurs in the ERGIC and that STING facilitates the recruitment of GPX4 into autophagosomes for subsequent autophagic degradation. These findings suggest that autophagy induced by STING facilitated the elimination of GPX4 in CMs post-A/R.
AAV-mediated GPX4 therapy shields cardiac function against the severe I/R injury triggered by STING activation
Given our findings, we subsequently verified that GPX4 functions as a downstream factor of STING, and further delved into the potential of GPX4 as a therapeutic target for STINGassociated I/R injury. An adeno-associated virus (AAV) targeting GPX4 (AAV-GPX4) driven by the cTNT promoter in CMs and labeled with GFP (referred to cTNT-GPX4) was generated to examine whether the overexpression of GPX4 can alleviate the progressive decline of cardiac function in STING-activated mice under in-vivo conditions. AAV-GPX4-GFP was administered through tail-vein injection 14 days before the induction of I/R. Subsequently, intraperitoneal injection of DMXAA (a STING activator) or DMSO was carried out every day for three days before I/R operation (Fig. 7a). Prior to model establishment, the
Fig. 6 STING promotes ferroptosis via autophagy-lysosome-mediated degradation of GPX4. a Western blot and quantification of GPX4 expression in MPCs influenced by Fer-1 or cGAMP ( ). b Western blot and quantification of GPX4 degradation in MPCs induced by Erastin, cGAMP or H-151 . c Western blot and quantification of GPX4 degradation in MPCs induced by cGAMP and its blocking using MG-132, , chloroquine and calpeptin ( ). d Western blot and quantification of GPX4 degradation in MPCs induced by cGAMP and its blocking using Baf A-1, LY294002, 3-MA and Wortmannin . Immunofluorescence analysis for autophagic flow presented by mRFP-GFP-LC3 in Sting or Sting-CKO MPCs post-A/R or not. The positive reactions for autophagy are displayed in red dots. Scale bar . Triple immunofluorescence analysis for detecting GPX4, STING, and LC3B in Sting or Sting-CKO MPCs. The positive reactions for co-localisation are displayed in yellow (LC3B and STING co-localisation) or pink (co-localisation of the three). Scale bar . i Triple immunofluorescence analysis for detecting GPX4, STING, and LAMP2B in MPCs post A/R or not. The positive reactions for co-localisation are displayed in yellow (GPX4 and STING co-localisation) or pink (co-localisation of the three). Scale bar . Mean ± SEM, NS non-significant, **P < 0.01, , **** and ### . Baf A-1 Bafilomycin A1, LAMP2B lysosomal-associated membrane protein 2B
overexpression efficiency of GPX4 in CMs isolated from three or six mice was confirmed using Western blot and immunofluorescence staining (Fig. 7b, Supplementary Fig. 4a). Delivering DMXAA could increase the infarct size, while GPX4 overexpression minimized the infarct size in STING-activated mice (Fig. 7c, d), suggesting a protective role of GPX4 overexpression against I/R injury induced by STING activation. Echocardiographic analysis revealed that STING activation indeed exacerbated cardiac dysfunction following I/R, however, GPX4 overexpression significantly ameliorated the deteriorating
cardiac function in STING-activated mice upon DMXAA administration, as evidenced by increased EF and FS post-I/R (Fig. 7e, f).
4-Hydroxy-2-nonenal (4-HNE), which is a widely acknowledged byproduct emerging from lipid peroxidation, functions as a crucial indicator of ferroptosis. In our results, STING activation led to a significant elevation in 4-HNE levels, indicating increased lipid peroxidation (Fig. 7g), while GPX4 overexpression was able to reverse this increase in lipid peroxidation caused by DMXAA, suggesting a downregulation of ferroptosis. The in vitro detection
Fig. 7 AAV-mediated GPX4 therapy shields cardiac function against the severe I/R injury triggered by STING activation. Effect of AAV-cTNTGPX4 on heart dysfunction therapy in C57BL/6J mice with DMXAA or DMSO addition post-I/R: a Schematic diagram depicting the time course of I/R-induced cardiac dysfunction receiving AAV or DMXAA. b Western blot of verifying successful overexpression of GPX4 in CMs ( ). , Myocardial infarct size (% of AAR) with representative tissue sectioning ( ). Echocardiography and measured EF% and FS% ( ). g Double immunofluorescence analysis for GPX4 and 4-HNE in the same heart section of the border region. Scale bar . h Immunofluorescence imaging analysis for detecting ROS in AAV-cTNT-GPX4 or AAV-Control infection mice CMs under DMSO or DMXAA addition following A/R. Scale bar . Mean ± SEM, NS, non-significant, and ###P < 0.001 . AAV adeno-associated virus, 4-HNE 4-Hydroxynonenal, cTNT-GPX4 AAV-Mus-GPX4-cTNT-C-GFP, DMXAA Vadimezan
of ROS accumulation levels in MPCs robustly corroborated this observation (Fig. 7h). To delve deeper into the impact of the cGAS-STING-GPX4 axis on mitochondria, we conducted several experiments. These experiments encompassed measurements of the ATP level, assessment of the mitochondrial membrane potential , and determination of the oxygen consumption rate (OCR) to assess mitochondrial health, ATP content, membrane potential, and respiratory capacity under A/R conditions. The results showed that, blocking cGAS/STING signaling or overexpressing GPX4 partially restored ATP levels, , basal mitochondrial respiration, ATP production and both maximal and spare respiratory capacities (Supplementary Fig. 4b-g, Supplementary Fig. 5a-c), signifying the partial rejuvenation of mitochondrial function induced by cGAS/STING deletion or GPX4 overexpression.
Above all, it can be concluded that, despite the potentiation of I/R damage in STING-overexpressing mice, the upregulation of GPX4 retains a remarkable capacity to prevent lipid peroxidation and ferroptosis, thereby safeguarding cardiac function. In conclusion, cGAS/STING deletion or GPX4 overexpression protects against deteriorating cardiac dysfunction and mitochondrial damage following I/R in STING-activated mice, pointing towards a potential therapeutic strategy to alleviate the adverse effects of STING overexpression on cardiac function.
STING is a potential therapeutic target to inhibit cardiac dysfunction post-I/R
To delve deeper into I/R treatment strategies that harness the STING-GPX4 signaling pathway, we selected H-151 to evaluate its
Fig. 8 STING is a potential therapeutic target to alleviate cardiac dysfunction post-I/R. Effect of H-151 on heart dysfunction in C57BL/6J mice post-I/R: a i. Schematic diagram depicting the time course of I/R-induced cardiac dysfunction receiving H-151 or DMSO. ii. Representative photographic images of hearts with DMSO or H-151 addition post-I/R. b, c Myocardial infarct size (% of AAR) with representative tissue sectioning . d, e Echocardiography and measured EF% and FS% . f Double immunofluorescence analysis for GPX4 and 4-HNE in the same heart section of the border region. Scale bar Immunofluorescence analysis for GPX4 and STING in DMSO or H-151 addition mice CMs post A/R. Scale bar . h Immunofluorescence analysis for autophagic flux presented by mRFP-GFP-LC3 in DMSO or H-151 addition mice CMs post A/R. The positive reactions for autophagy are displayed in red dots. Scale bar . i Immunofluorescence imaging analysis for detecting ROS in DMSO or H-151 addition mice CMs post A/R. Scale bar . j Schematic diagram showing the mechanism of STING-promoted MI/R injury. Part of the image was drawn by Figdraw. Mean ± SEM, NS non-significant, ** , and ****
potential in alleviating the harmful impacts of on cardiac function and to identify potential therapeutic agents for the treatment of I/R injury. Prior to I/R surgery, mice were intraperitoneally administered with H-151 every two days for a total duration of 7 days (Fig. 8a i). Concurrently, the degree of myocardial necrosis was clearly discernible in the in vivo cardiac images (Fig. 8a ii), demonstrating the effective control exerted by H-151 on the extent of necrosis within the cardiac functional area. Notably, in the presence of H-151, a reduction in infarct size (Fig. 8b, c) and restoration of heart function (Fig. 8d, e) were observed. Furthermore, H -151 addition was able to alleviate the increase of
4-HNE caused by I/R, suggesting a downregulation of lipid peroxidation and ferroptosis (Fig. 8f).
Subsequently, we delved into the pharmacological mechanism underlying the ameliorative effects of H-151 on myocardial injury following I/R in vitro. In our results, H -151 impeded the interaction between STING and GPX4 (Fig. 8g), blocked the autophagic flux (Fig. 8h), thus minimized ROS accumulation (Fig. 8i) to mitigate myocardial ferroptosis.
The effect of H-151 highlights its potential in reducing the area of I/R injury and preserving cardiac function. Taken together, STING emerges as a promising therapeutic target to attenuate the
cardiac impairment that occurs post-I/R, with its deleterious effects being modulated through the regulation of GPX4, suggesting a promising avenue for the intervention of cardiac remodeling.
To demonstrate the clinical significance of this study and to investigate the presence of the STING-GPX4 signaling axis in patients suffering from ischemic heart disease, we conducted measurements of serum proteins in patients both before and after percutaneous coronary intervention (PCI). The baseline characteristics of the patients involved in this study are outlined in Supplementary Table S3. Our results revealed that following PCI, there was an elevation in the expression level of STING protein in the patients’ serum, accompanied by a decrease in the expression level of GPX4 (Supplementary Fig. 5d). This finding suggests that the STING-GPX4 axis might play a crucial role in patients experiencing I/R. Additionally, we observed an increased expression in STING protein and a reduction in GPX4 protein content in the lysate of CMs that were derived from human embryonic stem cells, which were induced by A/R conditions (Supplementary Fig. 5e). These results further underscore the potential significant biological role of the STING-GPX4 axis in the human heart.
DISCUSSION
Ferroptosis occurs during myocardial ischemia-reperfusion (MI/R) injury and is accompanied by the degradation of GPX4. Here, we provide multiple lines of evidence that STING plays an initiating role in MI/R-induced myocardial ferroptosis by directly binding to GPX4 and triggering autophagy-lysosome-mediated GPX4 degradation. I/R elevates cytoplasmic DNA, activating dsDNA-cGASSTING signaling in cardiomyocytes (CMs). Deletion of cGAS or STING in CMs reduces oxidative stress, ferroptosis, and MI/R injury, while STING activation exacerbates them. Mechanistically, STING interacts with GPX4 at specific residues (N146 of GPX4 and T267 of STING) to initiate autophagy and lysosomal degradation. STING forms a positive feedback loop via the dsDNA-cGAS-STING-GPX4 axis, exacerbating ferroptosis. Two treatment strategies for MI/R injury-AAV-mediated GPX4 overexpression and STING antagonistsshow promise for preventing and reducing I/R injury, highlighting potential clinical targets. Collectively, our findings revealed that the accumulation of STING during MI/R induces ferroptosis by interfering with GPX4.
One effective cardioprotective approach in myocardial disorders is to prevent the death of cardiac cells. Recent studies have shown that cGAS-STING immuno-signaling pathway plays a role in various ischemic diseases, encompassing cerebral, renal, enteral, and hepatic I/R scenarios. Furthermore, cGAS has been implicated as a player in the inflammatory response observed in myocardial infarction (MI). IRF3, a downstream signaling molecule of the cGAS-STING pathway, is recognized for its role in the pathological process following MI. To date, major research efforts regarding the cGAS-STING pathway post-MI have focused on individual cytokines, such as interferon (IFN), or macrophage immunity. There remains a gap in understanding the direct functional relevance of cGAS-STING in MI/R injury of the heart and the progression of ferroptotic damage. In the present research, we confirmed that STING induced ferroptosis in CMs during MI/R injury.
The I/R process is characterized by an initial phase of ischemia and hypoxia, followed by a reperfusion stage characterized by the re-establishment of blood flow. Notably, this reperfusion phase has the most substantial and harmful damage associated with I/R, encompassing metabolic reprogramming, DNA impairment, the enhanced expression of pro-inflammatory genes, and mitochondrial malfunction. Understanding the intricate mechanisms underlying these pathological processes is crucial for developing effective therapeutic strategies to mitigate the harmful effects of I/R. Studies have reported that circulating levels of DNA were significantly elevated following cardiac ischemia. We further
confirmed that during injury, there occurs a profound degree of DNA damage, resulting in the leakage of DNA into the cytosol, subsequently triggering the upregulation of the cGAS-STING signaling pathway in CMs rather than fibroblasts or macrophages during acute I/R injury. Besides, our study found the highest cGASSTING signaling activation in the border region, compared to scar or non-ischemic region, aligning with the fact that it is the most dynamic area with active oxidative stress and cell death pathways, including ferroptosis. Additionally, STING appears to be an indispensable downstream factor for cGAS to execute its functional role in the context of I/R.
Since the role of cGAS and STING in CMs is poorly understood, we generated CM-specific cgas-knockout (cgas Myh6 [cgasCKO]), STING-knockout (Sting Myh6 [Sting-CKO]) mice and their control mice to investigate the functional importance of CMspecific cGAS and STING in I/R injury. To avoid aging-related subcellular and molecular impacts on CMs and their cardioprotective mechanisms against I/R injury, we used 8 -week-old mice for modeling. Considering the negligible gender impact on the I/ model, we opted for male mice. But we still have shortcomings in model establishment. While mice remain a widely accepted model for studying molecular mechanisms, we recognize the need to validate key findings in larger animal models or human-derived systems in future studies to improve translational relevance.
Because our focus on acute I/R injury and the expression of molecular mediators of inflammation and cellular infiltration needed to be investigated during the first we selected the critical time point of 24 h after reperfusion for animal experiments. However, infarction healing was incomplete at this time. To assess the effects of cGAS and STING deficiency on I/R injury-induced cardiac fibrosis, we extended reperfusion to 7 days. These findings suggest that cGAS and STING deletion offer cardioprotection beyond the acute phase, mitigating fibrosis and enhancing healing, highlighting the crucial role of the cGAS-STING in post-I/R injury and repair.
As CMs are devoid of immune capabilities, ZJ Chen and other scientists elucidated that STING regulates autophagy, which serves as a fundamental cellular function that operates independently of its canonical immune signaling pathway. Consequently, our primary focus in the context of CM is to investigate the nonimmune pathway mediated by STING. I/R injury is a complex pathological process characterized by multiple forms of cell death, such as necrosis, apoptosis, necroptosis, ferroptosis, and autophagy, and the contributions of each mechanism require further discussion. In our study, we focused on ferroptosis as a significant contributor to infarct size reduction based on its well-established role in ischemic injury, particularly through mechanisms of lipid peroxidation and iron-dependent oxidative damage. Certainly, the cell death mechanisms during I/R injury intertwine. Our research focused solely on the regulatory effect of STING on ferroptosis, which has limitations, especially the lack of analysis of factors related to programmed cell necrosis and pyroptosis. Further exploration of other cell death mechanisms during is warranted in the future.
Although Tunel-positive cells represent a combination of different cell death pathways, the findings from our RNA sequencing (RNA-seq), based on Sting-CKO and their control hearts post-I/R, were noteworthy. These results revealed a novel aspect of MI/R pathophysiology, characterized by the regulation of autolysosomes alongside ferroptosis. Upon I/R insult, a significant enrichment of pathways regulating ferroptosis became highly evident in Sting mice compared to Sting-CKO mice. This offers strong evidence supporting the concept that ferroptosis, a form of cell death reliant on iron, plays a pivotal role in MI/R injury regulated by STING. The degradation of GPX4 protein is a pivotal event in ferroptosis, which in turn prompts mitochondrial damage and the generation of reactive oxygen species (ROS), leads to
irreversible lipid peroxidation, and, consequently, cell death. We next confirmed the effect of STING on the biomarkers of ferroptosis, including ROS accumulation, lipid peroxidation and GPX4 degradation, as well as the expression of some ferroptosisrelated proteins (acyl-CoA synthetase long-chain family member 4, transferrin receptor and solute carrier family 7, member 11). Based on the evidence, we have elucidated that the regulatory impact of STING on I/R injury is not mediated through cell apoptosis or other programmed cell death mechanisms, but rather through the induction of cell ferroptosis. In summary, our research outcomes indicate that STING serves as a potent activator of ferroptosis, effectively promoting the degradation of GPX4, lipid peroxidation and ROS accumulation. STING amplifies myocardial ferroptosis through modulation of oxidative stress injury.
Recent studies report that GPX4 promotes the activation of STING through the maintenance of lipid redox homeostasis. However, the direct influence of STING on GPX4 and its underlying mechanisms in ferroptosis triggered by MI/R have remained elusive and unexplored. Our findings provide insights into the complex regulatory mechanisms governing the function of GPX4 and STING in the course of I/R events. Thus, our discoveries contribute to augmenting the existing knowledge framework regarding the tissue-level regulation of ferroptosis during MI/R.
Widespread use of agonists and inhibitors enhances our understanding of STING’s direct degradation effect on GPX4. As protein degradation post-transcriptionally occurs primarily through two mechanisms: ubiquitin-mediated proteasomal degradation and autophagy-dependent lysosomal degradation, we employed various protein degradation inhibitors (MG-132, calpeptin, chloroquine, , Bafilomycin A1, LY294002, 3-MA and Wortmannin) to disturb GPX4 degradation induced by cGAMP. STING is recognized for its ability to initiate autophagy through the lipidation of microtubule-associated proteins light chain 3B (LC3B) and the subsequent formation of autophagosomes, independently of TBK1 activation and interferon induction. We performed immunofluorescence co-localization analysis of GPX4, STING, endoplasmic reticulum-Golgi intermediate compartment (ERGIC), coat protein I (COP-1), LC3B, and lysosomal-associated membrane protein 2, along with tracking the autophagic flux using GFP-RFP-LC3 in Sting-CKO and control CMs. We discovered that cGAMP induces STING trafficking to the ERGIC upon I/R. At the ERGIC, STING binds to GPX4 to form the STING-GPX4 complex, which is subsequently trafficked to COP-I vesicles and LC3 autophagosomes. Then STING triggered the accumulation of LC3B puncta. The GPX4-STING complex subsequently fused with lysosomes and was encapsulated by autolysosome membrane marked by lysosomal-associated membrane protein 2B, ultimately leading to GPX4 degradation. These discoveries offer perspectives on the intricate mechanisms governing the degradation of GPX4 through autophagy, mediated by the interaction between STING and LC3B. In the absence of STING, the autophagic flux is impeded, resulting in the inability of GPX4 to colocalize with autophagosomes. Consequently, this disruption impedes the degradation of GPX4, which in turn mitigates cellular oxidative stress and ferroptosis. This enhanced understanding of the molecular mechanisms underlying GPX4-STING-autophagy interactions holds promise for the formulation of targeted treatment strategies applicable to related disease scenarios.
Given that the mechanisms governing ferroptosis are complex and remain incompletely comprehended, in an effort to decipher these intricacies, our research discloses a novel ferroptosisregulatory mechanism. In this mechanism, STING is pinpointed as a partner protein that promotes the degradation of GPX4 during I/R injury. Unlike direct ubiquitination, STING’s role in GPX4 degradation likely involves its ability to regulate autophagy. Although STING is not an E3 ligase, it modulates autophagy by trafficking from the ER towards the ERGIC and further to the Golgi, promoting LC3 lipidation and autophagosome biogenesis. Our
findings demonstrate that STING interacts with GPX4, facilitating its recruitment to autophagosomes for lysosomal degradation through the autophagic machinery rather than direct ubiquitination. Our study clarified the mechanism and highlighted STINGmediated autophagy’s broader implications in cell death pathways. Future investigations will focus on identifying potential E3 ligases or autophagy adaptors involved in this process to further elucidate the molecular mechanisms.
Our study not only deciphers the functional significance of STING in promoting myocardial ferroptosis during MI/R, but also delves into its protective effects on cardiac function subsequent to I/R injury. Notably, the investigations employing AAV-mediated GPX4 therapy further revealed that GPX4 functions as a downstream factor of STING. STING inhibitor possesses the capacity to hinder the co-localization of STING and GPX4, disrupting autophagic flux, thereby diminishing ROS accumulation, and ultimately mitigating the severity of I/R injury. Together, the two effective treatment options demonstrate promising therapeutic potential in managing I/R injury, highlighting the substantial role of STING in regulating the degradation of GPX4 and the consequent myocardial ferroptosis during MI/R injury.
Mitochondria are pivotal determinants of myocardial cell survival or death during I/R. Specifically, our findings on mitochondrial function, health, and ATP status collectively demonstrate that blocking cGAS/STING signaling or activating GPX4 can limit disease progression and partially restore mitochondrial function. However, these approaches still result in defective mitochondria and impaired ATP production, highlighting the need for additional therapeutic strategies specifically targeting mitochondrial health to achieve complete cardiac repair.
Above all, by identifying STING as a prospective therapeutic target in the context of MI/R injury, we present a novel direction for future investigations into treatments for I/R. The formulation of a therapeutic approach aimed at augmenting GPX4 function or expression has the potential to mitigate the harmful impacts induced by STING activation during I/R injury on heart function. Translating cardiac protection from robust experimental evidence to clinical benefits for patients with acute myocardial infarction or undergoing cardiovascular surgery remains an urgent priority. To verify the clinical applicability of our animal findings, we conducted a study on a cohort of patients undergoing PCI treatment and confirmed the clinical relevance of our research, demonstrating the activation of the STINGGPX4 signaling axis in patients with ischemic heart disease, suggesting that it could potentially have a significant impact on I/R injury. Of course, a critical disparity between experimental and clinical studies lies in the presence of multiple comorbidities and polypharmacy in patients, which are often inadequately modeled in animal studies. For instance, platelet inhibitors may limit the protective effects of experimental therapies for myocardial infarction, while propofol anesthesia can negate the benefits of ischemic conditioning. These factors highlight the importance of developing clinically relevant preclinical models to better align experimental therapies with the complexities of human disease.
In conclusion, for the first time, our study unveiled the mechanisms through which STING initiates ferroptosis in CMs and exacerbates MI/R injury. We put forward a model in which MI/ R stimulates the accumulation of cytoplasmic dsDNA, thereby activating cGAS-cGAMP-STING signaling. The activated STING then directly binds to GPX4 and induces autophagic degradation of GPX4, induces oxidative stress and eventually triggers ferroptosis in CMs. Specifically targeting STING might alleviate I/R-induced ferroptosis and cardiac damage. These results indicate that the STING-GPX4 axis plays a crucial role in myocardial ferroptosis. This not only discloses the novel molecular mechanisms underpinning GPX4-related cell death, but also designates STING as a promising therapeutic target for the management of MI/R injury.
MATERIALS AND METHODS
Ethics statements
All animal experiments were carried out in strict compliance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. The Laboratory Animal Committee of Shandong University Qilu Hospital (No. DWLL-2021-206) gave its comprehensive approval, guaranteeing ethical and accountable execution during the entire research process. The clinical study was performed in line with the principles set forth in the Declaration of Helsinki and obtained approval from the Ethics Committee of Qilu Hospital, Shandong University (Approval Number: KYLL-2022(ZM)-1344). Informed consent was obtained from all patients who were enrolled in the study.
Animal model of ischemia/ reperfusion (I/R)
For establishing the I/R animal model, male C57BL/6J mice aged 8 weeks were chosen. These animals were securely fixed on a mouse plate maintained at a constant temperature of . Prior to the operation, the precordial area was thoroughly sterilized using iodophor. The mice were anesthetized using inhalation anesthesia with isoflurane at a rate of . Using sterile scissors, a surgical incision of approximately 2 cm was made on the chest at the point where the heartbeat was most prominent. A purse-string suture was placed, and the thorax above the heart was dissected through the fourth intercostal space using curved forceps. The chest was gently pressed to ease the heart out of the chest cavity and position it within the sutured opening, ensuring minimal air remained in the chest cavity. A 6-0 surgical suture was then used to tie a surgical slipknot around the left anterior descending coronary artery, with one end of the suture protruding outside the chest. Promptly, the heart was returned to the chest cavity, any excess air was expelled, and the epidermal incision was sutured closed. Post-operatively, the mice were transferred to the anesthesia recovery room. 45 min later, the slipknot for cardiac reperfusion was released, and the mice were returned to their rearing cages for continued care.
Subsequently, the hearts were excised and stained to accurately determine the extent of myocardial necrosis, expressed as a percentage of the ischemic area at risk (AAR) that was not perfused. The ischemic region containing viable tissue was distinctly stained red using 2,3,5-triphenyltetrazolium, while the non-ischemic region was clearly marked blue with Evan’s blue. The hearts were then frozen at for 10 min and cut into slices ( 6 slices/heart). Infarct, AAR, and LV areas were measured using ImageJ software from NIH. Infarct size was calculated as the ratio of infarct area to AAR area. These slices were photographed using a gross imaging microscope (Leica M205 FA), and the images were subsequently analyzed with the aid of NIH Image software.
Echocardiogram
Following a modeling phase, the cardiac structure and function of mice were evaluated using a Transthoracic Echocardiogram. The VisualSonic VeVo 2100 Imaging System, which originated from Toronto, Canada, was utilized for this assessment. Before commencing the procedure, the mice were anesthetized by inhaling isoflurane. Subsequently, they were positioned on a heated platform kept at a temperature of and were securely attached to an electrocardiogram (ECG) for continuous monitoring. With the application of M-mode echocardiography, the left ventricular diastolic diameter (LVIDd) and left ventricular systolic diameter (LVIDs) on the parasternal long axis were carefully documented. Ultimately, the left ventricular ejection fraction and shortening fraction were computed automatically to enable a precise evaluation.
Distinct cardiac cell populations isolation
The isolation of distinct cardiac cell populations from the infarcted hearts of mice was performed 24 h following myocardial ischemia-
reperfusion (MI/R). This was done in accordance with pre-existing protocols. Standardized techniques were employed to isolate cardiomyocytes (CMs). For the isolation of cardiac fibroblasts (CFs) and macrophages, the Skeletal Muscle Dissociation Kit (Miltenyi Biotech, Shanghai, China) was utilized. To separate macrophages from CFs, Anti-F4/80-coated magnetic beads (Cat. No. 130-110-443; Miltenyi Biotech) were employed in strict accordance with the manufacturer’s instructions. Subsequently, the purified cells were gathered through centrifugation at for 5 min at , in preparation for the subsequent extraction of proteins.
Immunofluorescence, Masson’s trichrome (MT), and 4-HNE staining
The infarct area was demarcated as the portion situated between the suture and the heart’s apex. The infarct border area was carefully characterized as the marginal zone that differentiates the infarcted tissue from the adjacent non-infarcted myocardium in the short-axis cross-section. Employing a microtome (RM2235; Leica Microsystems, Inc., Mannheim, Germany), heart tissues positioned 1 mm below the ligation site were accurately cut into thick transverse slices, arranged along the horizontal long axis. Serial sectioning was applied for both MT staining processes, facilitating the assessment of myocardial fibrosis.
For the immunofluorescence staining of dsDNA, cGAS, STING, GPX4, and 4-HNE, the sections were first subjected to dewaxing. Antigen retrieval was then carried out using a dedicated kit (C1034; Solarbio, Beijing, China). Subsequently, the sections were immersed in 0.1% Triton X-100 (GC204003; Servicebio, Wuhan, China) in phosphate-buffered saline (PBS) for 10 min to increase their permeability. Next, they were incubated with normal goat serum (G1208; Servicebio, Wuhan, China) in PBS for 60 min at room temperature ( ) to block non-specific binding sites. Antibodies specific to dsDNA, cGAS, STING, GPX4, or 4-HNE were then applied, and the sections were incubated overnight at . After that, the sections were washed three times with PBS to eliminate unbound antibodies. Subsequently, incubation with Alexa Fluor 594 (ab150120; Abcam, Cambridge, MA, USA) and Alexa Fluor 488 (ab150081; Abcam, USA) secondary antibodies (diluted 1:200) was performed for 1 h in the dark at to visualize the specific binding of the primary antibodies. Finally, the nuclei were counterstained with -diamidino-2-phenylindole (DAPI, ab104139; Abcam, UK) to enable clear identification of cellular localization.
Cell culture
HEK293T, HeLa, and HL-1 cells were procured from KeyGene BioTech in China. Mouse primary cardiomyocytes (MPCs) were isolated from cgas , Sting , cgas-CKO, Sting-CKO, as well as their control littermates or C57BL/6J wild-type (WT) mice. These cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS; 9014-81-7, Sigma Aldrich, Germany). The cell cultures were maintained at a temperature of .
Cell IF staining and laser confocal analysis
In the process of immunocytochemistry staining, HeLa cells, HL-1 cells, or CMs were plated at a density of cells per milliliter on a 14 -millimeter coverslip (WHB, China). Following treatment with ligands, the cells were immediately fixed using paraformaldehyde (Beyotime, China) for 5 min . Next, cell membrane permeability was increased by treating the cells with Triton X-100 (Beyotime, China) for an additional 5 min. Subsequently, non-specific binding sites were blocked by incubating the cells in a buffer containing 10% Donkey Serum (Solarbio Science & Technology, China) at room temperature for 1 h . Then, the samples were incubated with the specific primary antibodies overnight at . After that, the samples were rinsed three times
with PBS (Solarbio Science & Technology, China). The samples were then incubated with the assigned fluorescent secondary antibody for 1 h at room temperature, followed by another three rounds of rinsing with PBS. Ultimately, the samples were subjected to counterstaining with DAPI (ab104139; Abcam, UK) to mark the nuclei.
Statistics analysis
Data were presented as the mean ± the standard error of the mean (SEM). All statistical analyses were carefully carried out with the use of GraphPad Prism 9 (GraphPad, San Diego, CA, USA). The SEM was denoted by the error bars. Initially, the normal distribution was examined, and subsequently, the Shapiro-Wilk test was applied for the assessment of variance homogeneity. The data exhibited approximate normal distribution ( ), indicating the suitability of parametric statistical methods. To assess statistically significant differences between two groups for normally distributed data, an unpaired two-tailed Student’s test was employed. For non-normally distributed data, a nonparametric statistical method was employed. Specifically, the Kruskal-Wallis test was utilized, and this was followed by Dunn’s post-hoc test for conducting multiple comparisons. Differences were deemed statistically significant based on stringent criteria, specifically, for marginal significance, for moderate significance, for strong significance, and **** for highly significant differences, or as not significant. At least three independent experiments was repeated to perform statistical analysis. Each data point included at least three biological repeats.
DATA AVAILABILITY
All data and materials be made available within the submitted material or in a public repository. The RNA-sequencing data generated in this study are publicly available in Gene Expression Omnibus (GEO) dataset GSE291453.
ACKNOWLEDGEMENTS
This work was supported by grants of the National Natural Science Foundation of China (No. 82270487, 82241203, 82200502, 84270277, 82200507); the National Key Research and Development Program of China (2024YFA1307002, 2021YFF0501403); the Shandong Provincial Natural Science Foundation (ZR2023JQ030, 2021ZDSYS05, 2024CXPT080, ZR2024ZD09, ZR2002QH089); the Program of Introducing Talents of Discipline to Universities (BP0719033), Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2023-PT320-06); the Taishan Scholars Program of Shandong Province (Zhang M, Zhang C and Zhang J), and the Fundamental Research Funds for the Central Universities (No. 2023QNTD003 to M.Z.). We also extend our gratitude to Baijie Liu (Beijing YongXinKangTai Technology Development Co., Ltd.) for her invaluable technical guidance.
AUTHOR CONTRIBUTIONS
C.Z. and M.Z. conceived the study, directed, and supervised the research. Xiaohong W. performed most experiments, collected all the data, participated in the analysis of results, presented the data in publishable figures, and wrote the paper. T.C. contributed to animal experiments. N.L. and Xiao W. contributed to laser confocal detection of immunofluorescence. Z.M. contributed to echocardiography imaging. Y.Z. participated in project discussions. S.C., J.Z., L.Cai., C.L., Yu.Z., L.Cao, Q.L., W.Q., R.R., H.Z., C.G., Q.D., W.S. and Y.H. participated in project discussions and data analysis. All authors have read and approved the article.
Competing interests: The authors declare no competing interests.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
REFERENCES
Heusch, G. Myocardial ischaemia-reperfusion injury and cardioprotection in perspective. Nat. Rev. Cardiol. 17, 773-789 (2020).
Heusch, G. Myocardial ischemia/reperfusion: Translational pathophysiology of ischemic heart disease. Medicine 5, 10-31 (2024).
Wu, X., Li, Y., Zhang, S. & Zhou, X. Ferroptosis as a novel therapeutic target for cardiovascular disease. Theranostics 11, 3052-3059 (2021).
Xu, S. et al. Naringenin alleviates myocardial ischemia/reperfusion injury by regulating the nuclear factor-erythroid factor 2-related factor 2 (Nrf2) /System xc-/ glutathione peroxidase 4 (GPX4) axis to inhibit ferroptosis. Bioengineered 12, 10924-10934 (2021).
Lu, H. et al. Britanin relieves ferroptosis-mediated myocardial ischaemia/reperfusion damage by upregulating GPX4 through activation of AMPK/GSK3β/ Nrf2 signalling. Pharm. Biol. 60, 38-45 (2022).
Dvorkin, S., Cambier, S., Volkman, H. E. & Stetson, D. B. New frontiers in the cGASSTING intracellular DNA-sensing pathway. Immunity 57, 718-730 (2024).
Kemmoku, H. et al. Single-molecule localization microscopy reveals STING clustering at the trans-Golgi network through palmitoylation-dependent accumulation of cholesterol. Nat. Commun. 15, 220 (2024).
Liu, S. et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347, aaa2630 (2015).
Konno, H., Konno, K. & Barber, G. N. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell 155, 688-698 (2013).
Crow, Y. J. & Casanova, J. L. STING-associated vasculopathy with onset in infancya new interferonopathy. N. Engl. J. Med. 371, 568-571 (2014).
Zhu, Y. et al. STING: a master regulator in the cancer-immunity cycle. Mol. Cancer 18, 152 (2019).
Sladitschek-Martens, H. L. et al. YAP/TAZ activity in stromal cells prevents ageing by controlling cGAS-STING. Nature 607, 790-798 (2022).
Gui, X. et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 567, 262-266 (2019).
Warner, J. D. et al. STING-associated vasculopathy develops independently of IRF3 in mice. J. Exp. Med. 214, 3279-3292 (2017).
Liu, B. et al. Human STING is a proton channel. Science 381, 508-514 (2023).
Chen, T. et al. The nucleotide receptor STING translocates to the phagosomes to negatively regulate anti-fungal immunity. Immunity 56, 1727-1742.e1726 (2023).
Heusch, G. et al. Health position paper and redox perspectives on reactive oxygen species as signals and targets of cardioprotection. Redox Biol. 67, 102894 (2023).
Yang, W. S. & Stockwell, B. R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 26, 165-176 (2016).
Korge, P., Ping, P. & Weiss, J. N. Reactive oxygen species production in energized cardiac mitochondria during hypoxia/reoxygenation: modulation by nitric oxide. Circ. Res. 103, 873-880 (2008).
Yang, W. S. et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317-331 (2014).
Zhu, S. et al. HSPA5 Regulates Ferroptotic Cell Death in Cancer Cells. Cancer Res. 77, 2064-2077 (2017).
Pohl, C. & Dikic, I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 366, 818-822 (2019).
Kaushal, G. P. & Shah, S. V. Autophagy in acute kidney injury. Kidney Int. 89, 779-791 (2016).
Qiao, L. et al. LAMP2A, LAMP2B and LAMP2C: similar structures, divergent roles. Autophagy 19, 2837-2852 (2023).
Dalleau, S., Baradat, M., Guéraud, F. & Huc, L. Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance. Cell Death Differ. 20, 1615-1630 (2013).
Li, Q. et al. Inhibition of double-strand DNA-sensing cGAS ameliorates brain injury after ischemic stroke. EMBO Mol. Med. 12, e11002 (2020).
Cao, D. J. et al. Cytosolic DNA Sensing Promotes Macrophage Transformation and Governs Myocardial Ischemic Injury. Circulation 137, 2613-2634 (2018).
Hu, Q. et al. Released Mitochondrial DNA Following Intestinal Ischemia Reperfusion Induces the Inflammatory Response and Gut Barrier Dysfunction. Sci. Rep. 8, 7350 (2018).
Bindi, E. et al. Mitochondrial DNA: A Biomarker of Disease Severity in Necrotizing Enterocolitis. Eur. J. Pediatr. Surg. 30, 85-89 (2020).
Rech, L. et al. Small molecule STING inhibition improves myocardial infarction remodeling. Life Sci. 291, 120263 (2022).
King, K. R. et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat. Med. 23, 1481-1487 (2017).
Visan, I. Myocardial infarct inflammation. Nat. Immunol. 19, 99 (2018).
Zhong, W. et al. Aging aggravated liver ischemia and reperfusion injury by promoting STING-mediated NLRP3 activation in macrophages. Aging Cell 19, e13186 (2020).
Maekawa, H. et al. Mitochondrial Damage Causes Inflammation via cGAS-STING Signaling in Acute Kidney Injury. Cell Rep. 29, 1261-1273.e1266 (2019).
Ter Horst, E. N. et al. Elevated monocyte-specific type I interferon signalling correlates positively with cardiac healing in myocardial infarct patients but interferon alpha application deteriorates myocardial healing in rats. Basic Res. Cardiol. 114, 1 (2018).
Wang, L. et al. Plasma nuclear and mitochondrial DNA levels in acute myocardial infarction patients. Coron. Artery Dis. 26, 296-300 (2015).
Boengler, K., Schulz, R. & Heusch, G. Loss of cardioprotection with ageing. Cardiovasc Res. 83, 247-261 (2009).
Kleinbongard, P., Lieder, H., Skyschally, A. & Heusch, G. No sex-related differences in infarct size, no-reflow, and protection by ischaemic pre-conditioning in Göttingen minipigs. Cardiovasc Res. 119, 561-570 (2023).
Guo, Y. et al. Genetic background, gender, age, body temperature, and arterial blood pH have a major impact on myocardial infarct size in the mouse and need to be carefully measured and/or taken into account: results of a comprehensive analysis of determinants of infarct size in 1,074 mice. Basic Res. Cardiol. 107, 288 (2012).
Ferdinandy, P. et al. Interaction of Cardiovascular Nonmodifiable Risk Factors, Comorbidities and Comedications With Ischemia/Reperfusion Injury and Cardioprotection by Pharmacological Treatments and Ischemic Conditioning. Pharm. Rev. 75, 159-216 (2023).
Christia, P. et al. Systematic characterization of myocardial inflammation, repair, and remodeling in a mouse model of reperfused myocardial infarction. J. Histochem Cytochem 61, 555-570 (2013).
Jia, M. et al. Redox homeostasis maintained by GPX4 facilitates STING activation. Nat. Immunol. 21, 727-735 (2020).
Haag, S. M. et al. Targeting STING with covalent small-molecule inhibitors. Nature 559, 269-273 (2018).
Heusch, G. Mitochondria at the heart of cardiovascular protection: p66shc-friend or foe? Eur. Heart J. 36, 469-471 (2015).
Kleinbongard, P. et al. Co-morbidities and co-medications as confounders of cardioprotection-Does it matter in the clinical setting? Br. J. Pharm. 177, 5252-5269 (2020).
Heusch, G. Critical Issues for the Translation of Cardioprotection. Circ. Res. 120, 1477-1486 (2017).
Cai, W. et al. Alox15/15-HpETE Aggravates Myocardial Ischemia-Reperfusion Injury by Promoting Cardiomyocyte Ferroptosis. Circulation 147, 1444-1460 (2023).
Ma, H. et al. Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/ reperfusion injury: role of autophagy paradox and toxic aldehyde. Eur. Heart J. 32, 1025-1038 (2011).
Ackers-Johnson, M. et al. A Simplified, Langendorff-Free Method for Concomitant Isolation of Viable Cardiac Myocytes and Nonmyocytes From the Adult Mouse Heart. Circ. Res. 119, 909-920 (2016).
Vukicevic, S. et al. Bone morphogenetic protein 1.3 inhibition decreases scar formation and supports cardiomyocyte survival after myocardial infarction. Nat. Commun. 13, 81 (2022).
State Key Laboratory for Innovation and Transformation of Luobing Theory; Key Laboratory of Cardiovascular Remodeling and Function Research of MOE, NHC, CAMS and Shandong Province; Department of Cardiology, Qilu Hospital of Shandong University, Jinan 250012, China
Correspondence: Meng Zhang (zhangmeng@sdu.edu.cn) or Cheng Zhang (zhangc@sdu.edu.cn)