يحفز ثنائي Pd1–Au1 المدعوم من MOF تفاعل الهيدروجين الجزئي للأسيتيلين في الإيثيلين بطاقة تنشيط شبه خالية من الحواجز A MOF-supported Pd1–Au1 dimer catalyses the semihydrogenation reaction of acetylene in ethylene with a nearly barrierless activation energy
إزالة الأسيتيلين من تيارات الإيثيلين أمر أساسي في الصناعة لتصنيع البولي إيثيلين. هنا نوضح أن هناك تعريفًا جيدًاثنائي، مثبت على جدران إطار معدني عضوي (MOF)، يحفز الهيدروجين الجزئي الانتقائي للأسيتيلين إلى الإيثيلين معتحويل ( من الأسيتيلين) و الانتقائية في تدفقات الإيثيلين الغنية للغايةأسيتيلينإيثيلين، ظروف تفاعل أمامية صناعية محاكاة). تسير التفاعل بطاقة تنشيط ظاهرة قدرهايعمل حتى في، ومع نوافذ التشغيل ( ) ووزن سرعات الفضاء بالساعة ( ضمن المواصفات الصناعية. تُظهر دراسة ميكانيكية تجريبية وحاسوبية مشتركة التعاون بين كلا الذرتين، وبين الذرات والدعم، لتمكين الهيدروجين الجزئي بدون حواجز للأسيتيلين.
البولي إيثيلين هو واحد من أكثر المواد الكيميائية طلبًا في جميع أنحاء العالم، مع تقدير للإنتاج السنويمليون طن. وبالتالي، يجب إنتاج مليون طن من الإيثيلين كل عام، بشكل رئيسي من خلال التكسير الحفزي، الذي ينتج أيضًا كميات متغيرة من الأسيتيلين (>1%) ويحتاج هذا التيار من الإيثيلين إلى التنقية قبل البلمرة، وبالتالي فإن تفاعل الهدرجة الانتقائية للأسيتيلين في تدفقات الإيثيلين هو واحد من أعلى العمليات حجمًا في البتروكيماويات. على الرغم من أن هذا التفاعل الهيدروجيني مفضل من الناحية الديناميكية الحرارية ( )، مطلوب محفز، ليس فقط لتجاوز الحواجز الحركية ولكن أيضًا للتحكم في الانتقائية لـ العملية، لأن تفاعلات غير مرغوب فيها أخرى، مثل هدرجة الإيثيلين، والهدرجة المباشرة للأسيتيلين إلى الإيثان، والتكثيف، هي أيضًا مواتية من الناحية الديناميكية الحرارية. و على التوالي). يتكون المحفز الصلب المستخدم حاليًا في الصناعة من أنواع من البالاديوم والفضة، مع إضافات أخرى (صوديوم، كالسيوم، إلخ)، ويجب أن تحتوي تيارات الخروج الناتجة على من الأسيتيلين و الإيثان ليكون مقبولاً صناعيًادرجة حرارة التفاعلوسرعة الفضاء بالوزن الساعي (WHSV)مطلوب تجنب تفاعلات الهدرجة الزائدة والتبلمر غير المرغوب فيه (تكوين الأخضر
الزيت، الكوك)، والذي من شأنه تعطيل المحفز الصلب. بالإضافة إلى ذلك، فإن نطاق درجة الحرارة التشغيلية عادةً ما يكونمطلوب للسيطرة على الهروب المحتمل خلال العملية ذات الحجم الكبير. كل هذه المتطلبات الصارمة، جنبًا إلى جنب مع تعقيد المحفز الصلب المستخدم حاليًا، قد دفعت المجتمع العلمي للبحث عن المحفزات والآليات الجزيئية القابلة للتنبؤ. وبالتالي، فإن الزيادة في المنشورات حول هذا الموضوع خلال السنوات العشر الماضية ليست مفاجئة (انظر أدناه).
في هذا السياق، لا يزال من الصعب العثور على محفز قادر على تحقيق المتطلبات الصناعية المذكورة أعلاه لتفاعل الهيدروجين الجزئي للأسيتيلين، خاصة تحت ظروف البداية (الأسيتيلين وظروف التفاعل الغنية بالمواد. تظهر مراجعة للأدبيات المفتوحة وجود العديد من المحفزات الصلبة بتراكيب معدنية مختلفة، وحوامل وظروف تشغيل متنوعة (تركيب التغذية، درجة الحرارة، معدل تدفق الوزن الساعي، إلخ)، ولكن لا يوجد أي منها يتطابق بالكامل مع المتطلبات الموضحة أعلاه. إحدى المراجعات الحديثةونشر حديثتلخيص البيانات الأكثر صلة (تركيب المتفاعلات، درجة حرارة التفاعل، تحويل الأسيتيلين، الانتقائية وإنتاجية المحفز) لـأنظمة التحفيز. كما نقوم هنا بتلخيص بعض الأمثلة الأخرى ذات الصلة والحديثة جداً (انظر أدناه). إدراك المعلومات العلمية الهائلة وإمكانات هذه الأنظمة الحفازة بعد المزيد من التحسين، فإن النتائج المقدمة، في كثير من الحالات، بعيدة جداً عن أي استخدام صناعي. على سبيل المثال، يتم عادةً تقديم تحويلات الأسيتيلين >99%، ولكن تحديد من الأسيتيلين في التيار النهائي ( التحويل) نادرًا ما يُرى. علاوة على ذلك، تستخدم العديد من التقارير بشكل متكرر غازًا خاملًا مخففًا أو تزيل الإيثيلين من التيار (باستخدام الأسيتيلين فقط) من أجل تحسين التحكم في الانتقائية، وهو بالطبع ليس نهجًا واقعيًا. ظروف التفاعل في النهاية (باستخدام فائض طفيف منتُستخدم أيضًا لتقليل هدرجة الإيثيلين، ومع ذلك، على حساب تعزيز البوليمرات غير المرغوب فيها. علاوة على ذلك، فإن درجات حرارة التفاعل عادةً ما تكونلزيادة الإنتاجية، ويُزعم بالفعل أن التفاعلات عند درجات حرارة منخفضة تحدث عند العمل تحتعندما يتم تشغيل العملية الصناعية عند فقط.
تحليل الأدبيات يظهر أيضًا أن تصميم المحفز لتفاعل الهيدروجين الجزئي للأسيتيلين يسيطر عليه بقوة مفهومين: مواقع تحفيزية فردية معزولة.وتعديل المحفزات الإلكترونية عن طريق السبائككلا المفهومين يتبعان نفس الاتجاه الميكانيكي، الذي يتضمن إعداد مواقع معدنية تحفيزية حيث يحدث انبعاث سريع لمنتج الإيثيلين، دون المساس بعملية الامتصاص السريعة الطبيعية للأسيتيلين.الانفصال على المعدن. في الواقع، يبدو أن المحفز الصلب المستخدم حاليًا يجمع بين المفهومين من خلال تخفيف مواقع البالاديوم على تجمعات الفضة، مع إضافات إضافية لتنظيم الانتقائية.تم وصف سبائك البالاديوم المختلفة لتحفيز التفاعل بنشاط معزز مقارنةً بنظيراتها غير السبائكية، وبشكل خاص،لقد أظهرت الذرات الفردية المدعومة على مجموعات المعادن المختلفة والجسيمات النانوية نشاطًا تحفيزيًا رائعًا (انظر أدناه). يُقبل في الأدبيات أن يمكن أن ينفصل (المراجع 11، 12، 20، 23، 28)، وبالتالي لم تعد مجموعات البالاديوم مطلوبة. ونتيجة لذلك، فإن الحدود النهائية لدمج عزل مواقع التحفيز مع السبائك ستكون ثنائي البالاديوم غير المتجانس، أي، المعدن)، حيث يقوم M بضبط النشاط التحفيزي للبلاديوم. في الواقع، دراسة حديثة باستخدام التعلم الآليلقد أظهر أن ثنائيات المعادن، مثليجب أن تكون محفزات مثالية لتفاعل الهيدروجين الجزئي للأسيتيلين، ودراسة تجريبية حديثة جدًا تستخدم التحفيزثنائيات على دعائم الجرافين من النانوماس.
الإطارات العضوية المعدنية (MOFs) هي مواد بلورية مسامية ذات مساحات سطحية عالية للغاية.كما أنهم يظهرون كيمياء مثيرة للاهتمام بين المضيف والضيف.-نتيجة مباشرة لمساحتهم الوظيفية الفارغة التي تسمح باستخدامهم كنانردات كيميائية لإنتاج/تثبيت أنواع كيميائية معدنية غير مسبوقة. بناءً على هذا النهج، أفادت مجموعاتنا مؤخرًا بتكوين بلورات محددة جيدًا من البالاديوموالبلاتينمحفزات ذات ذرة واحدة مدعومة على هياكل الإطار العضوي المعدنية، وأيضًا التحفيز (مرجع 35) و (مرجع 36) ثنائيات، مع تشتت عالٍ للغاية وتحميلات معدنية (حتىلذا، بدا لنا من المعقول محاولة تخليق مركب هيترو معدنيديمر لاختبار الهيدروجين الجزئي الحفزي للأسيتيلين. هنا نعرض تخليق مادة محددة جيدًاديمر، معتحميلات، على إطار معدني عضوي مشتق من الحمض الأميني-ميثيل-L-سيستين، الذي يتم تثبيته من خلال روابط ثيوإيثر داخل ضيقة ( ) مسام بقطر. هذا المادة الصلبة تحفز الهيدروجين الجزئي للأسيتيلين تحت ظروف صناعية محاكاة في المرحلة الأمامية، من 35 إلى ما يصل إلى ، مع تحويل والانتقائية تحت ظروف التفاعل المحسّنة، مما يعطي الإيثان كمنتج ثانوي وحيد. يتقدم التفاعل بطاقة تنشيط ظاهرة ( ) من ، وبالتالي فهي تقريبًا بلا حواجز، وتدعم الدراسات التجريبية والحاسوبية أن هو الموقع الرئيسي لامتصاص خلال الآلية بأكملها، وأنوتمثل ذرات الكبريت في MOF معدلات إلكترونية لـ، المساعدة خلال المفتاحخطوة الانفصال.
النتائج
تركيب وتوصيف
MOF قوي للغاية وبلوري مع الصيغةميسيماكس (ميسي موكسبيس [S-ميثيل سيستين] أوكسالييل دايميدتم استخدامه كنانوراكتر كيميائي لتشكيل ثنائيات، التي يتم الاحتفاظ بها وتثبيتها بواسطة مجموعات الثيوالكيل التي تزين قنوات MOF (الشكل 1)، بهدف إنتاج محفز فعال وتنافسي لتفاعل نصف هدرجة الأسيتيلين (انظر الشكل التكميلي 1، الجدول التكميلي 1 والنقاش التكميلي لمزيد من السياق). ونتيجة لذلك، تم الحصول على تجميع جديد من المضيف والضيف، مع الصيغة@ (الشكل 1ب، هـ)، تم الحصول عليه أخيرًا. تعتمد الاستراتيجية الاصطناعية على عملية ما بعد التركيب ذات الخطوتين، تتكون أولاً من الإدخال المتتالي لـ و الأملاح داخل قنوات MOF والتقليل المتزامن في الموقع معلتشكيلالأبعاد داخل MOF. كما سمح التوزيع المتجانس للمجموعات المحتوية على الكبريت داخل جدران القنوات بتوزيع متجانس للمعادن غير المتجانسة.الأبعاد (الأشكال التكميلية 2-5). كانت تقنية حيود الأشعة السينية أحادية البلورة (SCXRD) قادرة على كشف الهيكل الدقيق للمحفز المعدني المدعوم في المستوى الذري (الشكل 1 والجدول التكميلية 2) بسبب البلورية العالية والمتانة.من هذا النوع من MOF. هو متشابه مع MOF 1 النقي، يتبلور في الشكل الحلزوني مجموعة الفضاء، وتظهر شبكة ثلاثية الأبعاد من ستراتنيوم(II)-نحاس(II) ذات خصائص كيرالية، تتميز بقنوات سداسية بحجم تقريبي يبلغ 0.6 نانومتر (الشكل التكميلي 4). يتم تمثيل الشبكة الأساسية بواسطة نمط متصل سداسي الأحادية. ) ومبني من وحدات ثنائي النحاس (II) المربوطة بواسطة ترانز-أوكزاميداتو {Cu [( سلاسل ثيوإيثر ثنائي الميثيل تضمن الربط واستقرارالدايمرات، التي تتوزع بالتالي بشكل موحد داخل قنوات MOF (الشكل 1ب-د والأشكال التكميلية 2-4).
تتصل ذرات البالاديوم بمواقع ربط الكبريت مع مسافة رابطة Pd-S تبلغ 2.41 (2) (الشكل 1e والشكل التكميلي 5)، أطول من ما تم ملاحظته سابقًامسافات الروابط. يتم ترتيب سلاسل الثيو إيثر من MOF في أكثر تكوين مستقر لها حيث يتم تثبيت أحد الجزئين المختلفين بلورياً في تكوين منحنٍ وبالتالي يحمل البالاديوم. من خلال ذلك، يتم توجيه المجموعات الميثيلية الطرفية نحو الفراغات البينية الأصغر، التي تتشكل على طول محور (الشكل 1ب والشكل التكميلي 3). السلسلة الأخرى من ثيوإيثر ثنائي الميثيل مستقرة في شكل ممدود أكثر، موجهة نحو مركز المسام (الشكل التكميلي 3). هذه المسام السداسية تستوعب ذرات الذهب الموجودة على بعد 3.21 Å من ذرات البالاديوم، وهو مسافة بين الذرات أقصر من مجموع أنصاف أقطار فان der Waals، مما يبرر التصنيف كـديمر، ويتفق مع الميل لجذب البالاديوم والذهب بسبب نقل الشحنة الصافية من البالاديوم إلى الذهب.
الشكل 1|بنية البلورة بالأشعة السينية لـ @1.a,b, تشكيل ثنائيات تظهر هياكل و محدد بواسطة SCXRD.قناة واحدة منفي و الطائرة. تمثل ذرات النحاس والسترونتيوم من الشبكة بواسطة بوليهدرا باللونين السماوي والبنفسجي، على التوالي، فيبينما يتم تمثيلها بعصي رمادية في (ج، د). يتم تصوير الرابطة العضوية كعصي رمادية ومجموعات الثيوإيثر كأصفر.
تلتصق في جميع الحالات. تمثل الكرات البرتقالية والزرقاء ذرات الذهب والبلاديوم، على التوالي، فيثنائيات. تمثل الخطوط المنقطة الزرقاء والبرتقالية والحمراء و التفاعلات، على التوالي. e، تفاصيل هيكل البلورة بالأشعة السينية توضح التفاعلات الرئيسية بين المضيف والضيف والمعلمات الهيكلية ذات الصلة لـثنائيات.
ت stabilized ذرات الذهب من خلال التفاعلات التي تشمل ذرات الأكسجين من وحدات الأوكزامات، التي تنتمي إلى نواة MOF، وتحدد جدران MOF على مسافة من. من الجدير التأكيد على مزيد من مسافةذلك، على الرغم من أنه أطول منيمكن اعتبار طول الرابطة تفاعلًا stabilizing فعالًا جدًا (الشكل 1e والشكل التكميلي 5) كما تدعمه قياسات الامتصاص الانتقائي للأشعة السينية (EXAFS) (انظر أدناه). يتم تثبيت ذرات الذهب من خلال تفاعلات تشمل ذرات الأكسجين من وحدات الأوكزامات، التي تنتمي إلى نواة MOF وتحدد جدران MOF والارتباط الضعيف بالكبريت على مسافات تبلغ 2.91 و Å، على التوالي (الشكل 1e والشكل التكميلية 5). لا توجد أمثلة على دقة بلورية يبدو أن الديمر قد تم الإبلاغ عنه سابقًا. ومع ذلك، فإن كلا من و المسافات قابلة للمقارنة بتلك التي تم الإبلاغ عنها سابقًا لمجموعات النانو من البلاتين والذهبتفسير الـ SCXRD يقدم وصفًا لبيئة البالاديوم-ذهب حيث تكون ذرات البالاديوم رباعية التنسيق بواسطةوتوقع (لم يتم اكتشافه بواسطة خرائط كمتوقعة لجزيئات المذيب في البلورات المسامية، ولكن تم تأكيدها من خلال تجارب EXAFS) في بيئة مشوهة للغاية، ومع الذهب مرتبط فقط بـ Pd ومع عدد تنسيق يبلغ 2، في هندسة خطية محتملة، مشوهة بشكل كبير، ويرجع ذلك أساسًا إلى فعالية التفاعلات الضعيفة بين الذهب والكبريت المذكورة أعلاه. تساهم القيود الهندسية التي تفرضها طوبولوجيا الإطار العضوي المعدني ومرونة وحدات الميثيل سيستين، التي تم متوسط جميع التشكيلات الممكنة لها في وحدة خلوية واحدة بواسطة البلورة، في تعزيز التشوه عن الخطية.
حسابات نظرية الكثافة الوظيفية (DFT) (انظر أدناه) تدعم بيانات SCXRD. التركيب البلوري التجريبي لـ @1 (الشكل 1e) يظهر أن ذرة البالاديوم مرتبطة مباشرة بذرة كبريت واحدة من سلسلة الثيوإيثر عند ويقيم على مسافة أطول، من، من ذرة كبريت ثانية، وأن ذرة الذهب حققت الاستقرار عند طول slightly أطول مسافةوأطول بكثيرفصل. على النقيض من ذلك، تشير الحسابات النظرية إلى أن النظام الأكثر استقرارًا هو حيث يكون ذرة البالاديوم مرتبطة مباشرةً باثنتين من ذرات الكبريت على مسافات متشابهة من ومجموعة ثيو إيثر واحدة عند مسافة أطول من Au-S (يستقر ذرة الذهب. ومع ذلك، فإن الانحراف الظاهر بين النتائج التجريبية (SCXRD) والنتائج النظرية (حسابات DFT) يوفر، في الواقع، الإجابة الأكثر واقعية على الهيكل المقترح لـ @1. المرونة الجوهرية والاستثنائية لسلاسل الثيوإيثر المحصورة في المسام، التي تم اكتشافها من خلال الاضطراب الحراري والديناميكي، ومن المتوقع في البلورات المسامية، يعمل بشكل تآزري لتمكين اقتراب الثاني
الشكل 2 | التجارب الحفزية لتفاعل الهيدروجين الجزئي. أ، ب، تأثيرنسبة الأستيلين (أ) ودرجة الحرارة (ب) في الهدرجة الانتقائية للأستيلين مع @1. ج، الاستقرار على مر الزمن لـ @1 تحت ظروف التفاعل المثلى: نسبة الأسيتيلين،منالأسيتيلين في الإيثيلين، وقت تفاعل 24 ساعة ودرجة حرارة التفاعل.
ذرة الكبريت نحو ذرة البالاديوم. فيما يتعلق بالذهب، من المعقول اعتبار آلية مشابهة حيث تضمن مرونة وحدات الميثيل سيستين اقتراب ذرات الذهب من ذرات الكبريت، متحركة من مسافة 3.66 إلى. هذه الحركة مدعومة بانكسار متزامن لـالتفاعل، الذي يعمل أولاً على استقرار أيونات الذهب المحملة، والتي يتم تقليلها بعد ذلك إلىوأخيرًا تم استقراره بواسطة سلاسل ثيوإيثر ديناميكية عالية (الشكل التكميلي 5).
تؤكد قياسات EXAFS ارتباط ذرات المعدن بسلاسل الثيوإيثر الكبريت (الشكل التكميلي 6). البالاديومإشارات EXAFS (الشكل التوضيحي 6a) والتحويل فورييه النسبي المقابل (الشكل التوضيحي 6b) لـ PdAu-MOF، قبل وبعد عملية اختزال جزئي (تشمل فقط اثنين منالإضافات)، تظهر تشكيل رابطة جديدة متوافقة مع و ، يمكن تحديده بسهولة من خلال المقارنة مع مراجع Pd و PdO. تكون تشكيل الروابط الجديدة أكثر وضوحًا في الجزء السفليالنطاق والنتائج تؤدي إلى تغيير طفيف في القمة الرئيسية في تحويل فورييه، وهو ما يُرى أيضًا في النموذج البلوري لملاءمة القشرة الأولى للبلاديوم (الشكل التكميلية 7). تظهر النتائج الرقمية للملاءمة (الجدول التكميلية 3) أن التنسيق الرباعي النموذجي الموجود في معقدات البلاديوم أحادي المعدن موجود أيضًا في @1، مشوهة إلى حد ما، مع الكبريت (وربما الأكسجين) كذرات منسقة في القشرة الأولى. لم تكن قياسات EXAFS للذهب واضحة جدًا فيما يتعلق بتكوين السندات لأن الأخيرة أقل أهمية في @1 وكميات صغيرة من جزيئات الذهب النانوية التي تتشكل بشكل لا مفر منه غطت النتائج (الشكل التوضيحي 8). ومع ذلك، يمكن أن يُرى أن@1 يحتوي على كميات كبيرة من الإشارات المتوافقة مع روابط Au-S، على عكس رقائق الذهب.
لكشف حالات الأكسدة للبلاديوم والذهب في الإطار العضوي المعدني (MOF)، تم استخدام هيكل الامتصاص القريب من حافة الأشعة السينية (XANES)، وطيف الإلكترون الضوئي للأشعة السينية (XPS)، وطيف الانعكاس المنتشر بالأشعة تحت الحمراء بتقنية تحويل فورييه مع مجس CO (DRIFTS) والرنين المغناطيسي النووي بتدوير الزاوية السحرية للحالة الصلبة معاستقصاءتم إجراء تحليلات (ss-MAS-NMR).
تحليل XANES للبلاديوم @1 بعد عملية تقليل جزئي (تشمل فقط اثنين الإضافات؛ الأشكال التكميلية 9 و 10 والجدول التكميلية 4) تظهر تشكيل معدن أحادي مؤكسد الموقع. لم يتم تضمين تحليل XANES للذهب المقابل هنا لأن تشكيل بعض جزيئات الذهب النانوية يخفي النتائج. طيف XPS لـقبل وبعد التخفيض الكامل مع (الشكل التكميلي 11أ، ب، على التوالي) يظهر أن السطر قبل التخفيض هو ثنائي مع طاقات ربط لـ و من 337.5 و 342.8 إلكترون فولت، على التوالي، وهي نموذجية لـالكاتيونات ومماثلة للقيم المبلغ عنها الأخرى. من ناحية أخرى، فإن فك التداخل للذهبالمنطقة تظهر ثلاثة أزواج مزدوجة لـ و الانتقالات عند 86.4 و90.5 إلكترون فولت (الثنائية الأولى)، 85.3 و89.5 إلكترون فولت (الثنائية الثانية)، و84.1 و87.7 إلكترون فولت (الثنائية الثالثة)، والتي تُنسب إلىومعدنيعلى التوالي، ويقترح أن تم تقليل الكاتيونات جزئيًا بالفعل قبل معالجة MOF بـ. بعد الاختزال، تتغير الوضعية بشكل جذري بالنسبة للبلاديوم والثنائي الملحوظ في و 340.8 إلكترون فولت ( يمكن أن يُعزى إلى إيجابية طفيفةنوع. بالمثل، الـالمنطقة تظهر زوجًا عندو 87.1 إلكترون فولت (ذهب ) الذي يدل على الذهب المخفض بالكامل تتوافق هذه الطاقات الربط تمامًا مع تلك التي لوحظت في سبائك AuPd الثنائية المعدنية الأخرى المبلغ عنها.علاوة على ذلك، تم تعيين طاقات الربط لـ وأو فيتم تقليل طاقة الديمر (83.5 و 87.1 إلكترون فولت، على التوالي) قليلاً مقارنةً بتلك الملاحظة، لنفس المناطق، قبل الاختزال (84.1 و 87.7 إلكترون فولت، على التوالي). تشير هذه الحقيقة إلى أن ذرات الذهب تكتسب كثافة إلكترونية من ذرات البالاديوم في عملية تشكيل الديمر.. الـ و طاقات الربط، وطاقات الربط من و المناطق، تم جمعها في الجدول التكميلي 5.
طيف مجس CO الخاص بـ DRIFTS @1 (الشكل التوضيحي الإضافي 12، الأعلى) يظهر إشارتين رئيسيتين عند 1,975 ووالتي يمكن أن تُخصص لتقليل كبير و الأنواع، على التوالي. إشارة صغيرة عند يمكن أيضًا ملاحظته، والذي يمكن أن يُنسب إلى جزء صغير من الفرد الذرات المتبقية في المادة، بالمقارنة مع @1 (الشكل التوضيحي الإضافي 12، الأسفل). أتحليل ss-MAS-NMR @1، MOF من AuPd غير المخفض وتم تنفيذ @1 في جو منلمدة 8 ساعات في درجة حرارة الغرفة. تتيح هذه التقنية التمييز النوعي بين المواقع الكاتيونية المخفضة جزئيًا (حمض لويس) ومواقع المعادن المخفضة لأن شدة CO المرتبطة بالأولى أعلى بمقدار 1 مرة.. علاوة على ذلك، في حالة معادن البالاديوم والذهب، تركنا عمدًا كمية صغيرة من الهواء أثناء القياس للحصول علىإذا كانت مواقع المعادن الذهبية موجودة. تظهر النتائج (الشكل التكميلي 13) أن كلا من@1 و MOF AuPd غير المخفض يحتويان على كميات كبيرة من المواقع الكاتيونية (إشارات بين 150 و 180 جزء في المليون) ويكاد لا ينتجان (إشارة عند 122 جزء في المليون) ولكن، على النقيض، @1 يحتوي على ما يقرب من نصف كمية المواقع الكاتيونية ويشكل كميات كبيرة من بعبارة أخرى، تم تقليل نصف محتوى المعدن تمامًا إلى حالة الفلز.@1، ربما ذهب، بينما يحتفظ النصف الآخر ببعض الشحنة الكاتيونية. تتطابق هذه النتائج معًا بشكل جيد مع الهيكل المقترح لـ @1.
بالإضافة إلى التوصيف الهيكلي، الطبيعة الكيميائية لـتم تأسيس @1 أيضًا بواسطة تقنيات توصيف مختلفة. التحليلات العنصرية ( ) وقياس الطيف الكتلي باستخدام بلازما مقترنة بالحث (الجدول التكميلي 6) أكد التركيب الكيميائي المقترح. مسحوق الأشعة السينية التجريبية
الشكل 3 | دليل ميكانيكي تجريبي لـ @1 و @1.a، طاقة التنشيط الظاهرة تفاعل هدرجة الأسيتيلين بشكل عام. ب، قياس KIE ( ) في تفاعل هدرجة الأسيتيلين. الرموز المملوءة، ; رموز فارغة، . طاقة تنشيط انقسام الهيدروجين المستخرجة من معدلات تكوين HD. د، طاقة تنشيط عملية انقسام الهيدروجين.
نمط الحيود لـ @1 (الشكل التوضيحي التكميلي 14) يتطابق مع النظري، مما يؤكد كل من النقاء والتجانس لعينة الكتلة. تؤكد التحليل الحراري الوزني محتوى المذيب من @1 (الشكل التوضيحي الإضافي 15) ويسمح بإقامة الصيغة الكيميائية المقترحة. إجمالي الفراغات المحتملة القابلة للوصول في @1 المبلغ هو 1,274.0 ، محاسبة عنحجم وحدة الخليةيقل القطر الافتراضي للقنوات منفي MOF 1 النقي إلى @1 (الشكل التوضيحي التكميلي 3)، بسبب وجود ثنائيات داخل القنوات، وتؤكد منحنيات الامتزاز عند 77 كلفن المسامية الدائمة لـ (الشكل التوضيحي التكميلي 16، الأعلى). من الجدير بالذكر هنا أن حجم المسام الصغير هذا مناسب لأداء الهيدروجين الجزئي للأسيتيلين دون تفاعلات اقتران تنافسية، حيث يسمح حجم المسام 0.56 نانومتر بدخول وانتشار الجزيئات ولكن تعيق تشكيل وانتشار الجزيئات الأكبر (نتائج التحفيز). تجارب الامتصاص التنافسية للأسيتيلين والإيثيلين فيأظهر أن فقطللأسيتيلين في الإيثيلين النقي ينتج عنه انخفاض كبير في امتصاص الإيثيلين (الشكل التكميلي 16، الوسط)، مما يشير إلى وجود ألفة أقوى لمواقع MOF للأسيتيلين. طيف تحويل فورييه للأشعة تحت الحمراء للمعالجةتظهر العينة @1 الإشارات المرتبطة بامتصاص الأسيتيلين (الشكل التكميلي 16، الأسفل).
تم استخدام مجهر الإلكترون الناقل ذو المجال المظلم الزاوي العالي لتحليل @1 المادة (الشكل التوضيحي التكميلي 17). تظهر النتائج أن كل من البالاديوم والذهب موزعين بشكل متجانس عبر MOF وفقًا لقياسات الأشعة السينية المشتتة بالطاقة، جنبًا إلى جنب مع العناصر الأخرى في MOF (أي الكبريت والسترونتيوم). تتوافق النسب الوزنية الملحوظة للبالاديوم والذهب مع الصيغة الجزيئية المحسوبة لـ @1 (الشكل التوضيحي التكميلي 18).
نتائج تحفيزية
تم خلطه معمسحوق (300 ملغ) وتم تحميله في مفاعل أنبوبي ثابت السرير؛ تركيبات مختلفة من معدلات التدفق/خليط الهيدروكربونات ( ) وتم فحص درجات الحرارة للعثور على الظروف التشغيلية المثلى لتفاعل نصف هدرجة الأسيتيلين (الشكل 2أ، ب؛ انظر الجدول التكميلي 7 للقيم العددية). لا تؤثر درجة حرارة التفاعل بشكل كبير على تحويل الأسيتيلين، على الأقل من 35 إلى، مما يشير إلى طاقة تنشيط منخفضة جداً للتفاعل على @1 المحفز (الشكل 3). لم تُلاحظ قمم التفاعل الثنائي حتى ، مما يدل على نقص في البلمرة. تؤكد التحليلات العنصرية والاستخراجات من المحفز المستخدم مع المذيبات العضوية (الطرق) عدم حدوث تكوين المنتجات، الزيوت الخضراء أو الكوك. كشفت التغيرات في معدل التدفق أن النسبة المثلى لـكان نسبة الأسيتيلين 10:1، وهو ما يتوافق مع ظروف هدرجة الأسيتيلين النموذجية في المقدمة.كان الإنتاج المحقق تحت هذه الظروف، من حيث WHSV، هو، والأسيتيلين المتبقي كانحد الكشف لجهاز الكروماتوغرافيا الغازية المستخدم، والذي يتوافق معالعائد، عادةً معالانتقائية. لتقييم التركيز (أجزاء لكل ) من الأسيتيلين المتبقي، تم تركيب حلقة أكبر بعشر مرات في جهاز الكروماتوغرافيا الغازية؛ ومع ذلك، لم نتمكن من تقييم كمية الأسيتيلين بدقة تتجاوز القيم ، بسبب تشبع إشارة تدهور التحريض الحر وتداخل القمم. في أي حال، فإن قيم التحويل والانتقائية والإنتاجية التي تم الحصول عليها لـ تتوافق المحفزات مع المواصفات الصناعية. الاستقرار على مر الزمن لـ @1 يظهر ثابتاً تحويل الأسيتيلين والانتقائية بعد فترة من الاستقرار (الشكل 2c).
محتوى معدن البلاديوم والذهب في @1 هو 2.9 و ، على التوالي (الجدول التكميلي 8). توفر هذه المحتويات العالية جداً من المعادن في المادة الصلبة ميزة واضحة لمرور التفاعل في التدفق من حيث الإنتاجية لكل وحدة كتلة من المحفز. لأغراض المقارنة، تحتوي MOFs على نوع واحد فقط من ذرات المعادن، هنا يسمى @1 (مرجع 33) و @1، تم إعدادها واختبارها أيضًا كعوامل حفازة (الجدول التكميلي 7)، ونظرًا لأن محتوى المعادن النبيلة يبقى مشابهًا لهذه المواد العضوية المعدنية (الجدول التكميلي 8)، يمكن اعتبار التجارب قابلة للمقارنة. ثم تم إجراء تفاعل هدرجة الأسيتيلين مع و/أومن Au@1 للحفاظ على كمية البالاديوم والذهب ثابتة، على التوالي.@1 لم يحفز هدرجة الأسيتيلين عند معدلات تدفق عالية، كـ يفعل، ويتطلب وقت إقامة ونسبة الأستيلين 3 أضعاف أعلى (الجدول التكميلي 9). تحت هذه الظروف المحسّنة للتفاعل،حقق @1 تحويل أسيتيلين بنسبة 99.6% (مع بقاء 40 جزء في المليون) ومعدل تدفق الوزن الساكن (WHSV) من (سرعة الغاز في الفضاء الساعي، ; الشكل التوضيحي الإضافي 19)، وهي قيم تختلف كثيرًا عن النشاط التحفيزي لـلم تُظهر أي نشاط تحفيزي تحت جميع ظروف التفاعل المختبرة (الجدول التكميلي 9) ولم تُغير النشاط التحفيزي لـ @1 عند مزجه جسديًا معه. استقرار/قابلية إعادة التدوير لـتم تأكيد @1 بواسطة XPS وحيود الأشعة السينية البودرة: أظهرت إجراء هذه التحليلات بعد التجارب الحفزية عدم وجود فرق كبير مقارنة بالتحليلات التي أجريت قبل التحفيز (الأشكال التكميلية 20 و21).
قمنا بالتحقيق في كيفية سلوك التحفيز للميكروبوروس @1 المواد متنوعة بحجم المسام. نظرًا لأن تحضير MOF مشابه بقنوات بأحجام مختلفة ليس بالأمر السهل (فإن الكتل البنائية المعتمدة على الأحماض الأمينية المستخدمة تؤدي إلى تشكيل هذا MOF المحدد وليس غيره)، قمنا بتحضير واختبار زيوت ميكروية ذات بنية مشابهة مع تركيبة كيميائية مماثلة ولكن بأحجام مسام مختلفة قليلاً. على وجه التحديد، قمنا بتحضير مواد Pd-Au-MS مختلفة (MS، المناخل الجزيئية) بأحجام مسام 3 و5 و. الثلاثة وتتوافق مواد حجم المسام مع عوامل التجفيف النموذجية، التي تحتوي على و كردود فعل، وحجم المسام يتوافق مع الزيوليتتم تجنب الزيوليت لتفادي أي عملية قد تكون محفزة بواسطة حمض برونستيد. تم إدخال معادن البالاديوم والذهب بواسطة طريقة النقع الرطب (الطرق)، بهذا الترتيب المعدني، وتم تأكيد السلامة الهيكلية لإطار الزيوليت بواسطة حيود الأشعة السينية (الشكل التوضيحي 22). يجب الإشارة هنا إلى أنه يمكن ضبط محتوى المعادن في الزيوليت (من 1 إلى ) لتجنب تجمع المعادن، وأن هذه الكمية من المعدن يمكن أن تكون أقل بكثير من في @1 (7.1 wt% بلاديوم). ومع ذلك، كانت الكمية الإجمالية للمعادن في التفاعل متساوية مع تلك الموجودة في MOF من خلال إضافة المزيد من الزيوليت.
أظهرت الزيوليت المحتوية على البالاديوم والذهب نشاطًا تحفيزيًا مختلفًا اعتمادًا على حجم المسام. تم ضبط ظروف التفاعل لتحقيق تحويل بنسبة 90% من الأسيتيلين لـ @1 عامل مساعد لمقارنة النشاط التحفيزي للمواد المختلفة بشكل أفضل. و المواد حققت تحويل كامل للأسيتيلين، بينما أظهرت Pd-Au-MS 3 Å تحويلاً قدره (الشكل التوضيحي 23). تؤدي أبعاد مادة الدعم الأصغر MS إلى قيود في تحقيق تحويل كامل للأسيتيلين لأن القنوات التي تكون صغيرة جدًا في القطر يمكن أن تعيق انتشار ونقل المتفاعلات والمنتجات داخل هيكل المحفز. ونتيجة لذلك، تصبح جزيئات الأسيتيلين أقل وصولاً إلى المواقع النشطة التحفيزية، مما يؤدي إلى تحويلات غير مكتملة.
كما زادت نسبة الإيثان/الإيثيلين مع زيادة حجم المسام من 3 إلى، مع استقرار النسبة عند حجم مسام 7 Å. لم يتم العثور على الزيوت الخضراء تحت أي ظروف. مادة ( القناة) تظهر سلوكًا تحفيزيًا وسيطًا، مشابهًا لـ MSالزيوليت. يجب أن يُلاحظ هنا أن المقارنة الحفزية المباشرة بين الزيوليتات وMOF من خلال اعتبار حجم المسام فقط ليست صحيحة تمامًا لأن الزيوليتات تحتوي على قنوات ثلاثية الأبعاد مترابطة مع تجاويف. يجب أيضًا أن يُلاحظ هنا أن النشاط الحفزي لـ @1 أعلى بكثير من أي زيلوت عندما تم دفع ظروف التفاعل لتحقيق أقصى معدلات الإنتاج، حتى بعد تحسين ظروف التفاعل، محتوى المعدن، ترتيب الإدماج (الذهب قبل البالاديوم) أونسبة التغذية في الزيوليت (تم إجراء الدراسة على مادة MS 5 Å؛ الأشكال التكميلية 24 وتدعم هذه النتائج الفرضية القائلة بأن حجم المسام الصغيرة للـ MOF مع تكوين عدد كثيف منالنوع الحفاز هو وراء نشاطه الحفاز العالي للغاية.
الدراسات الميكانيكية
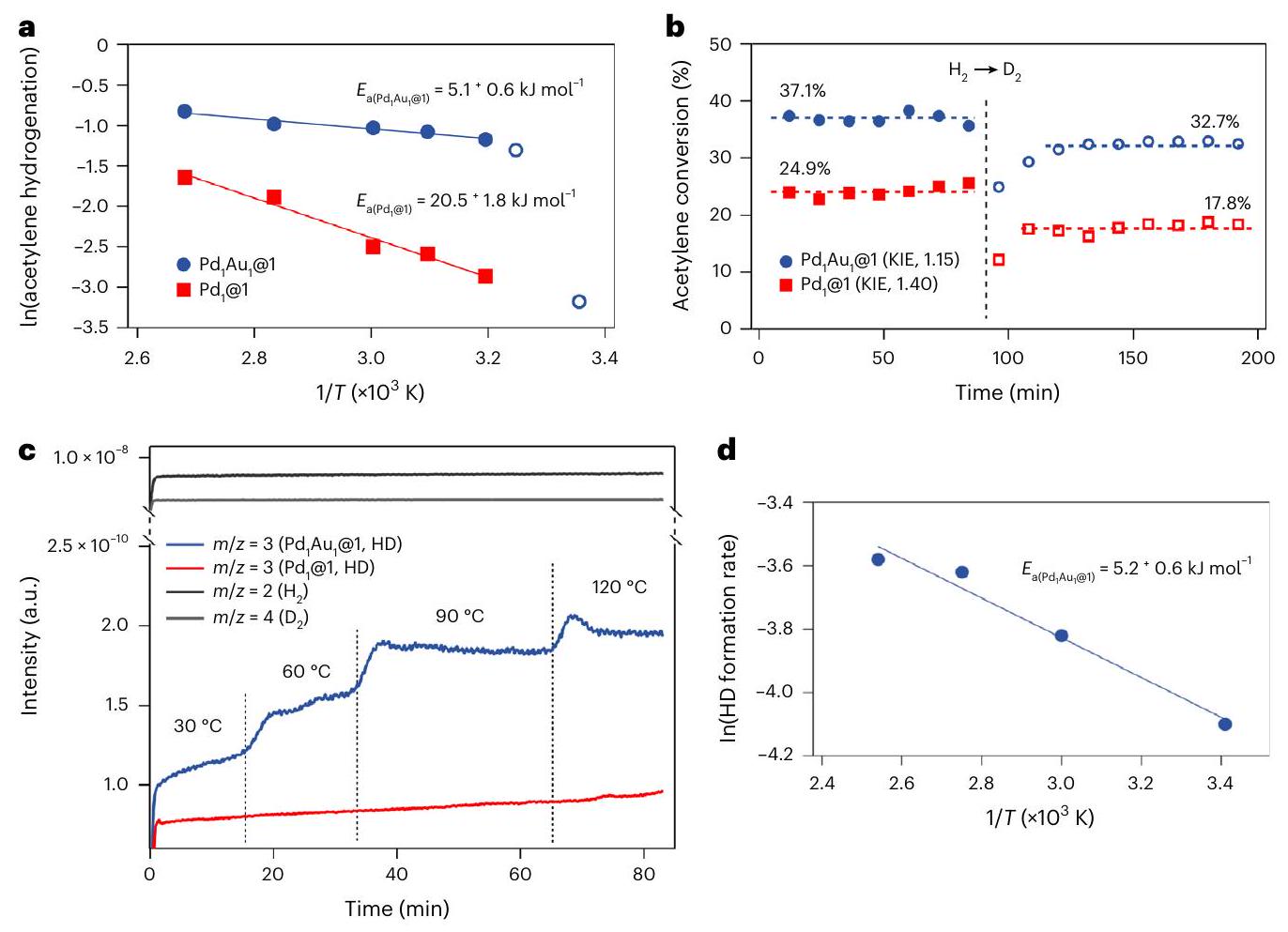
تم إجراء تجارب حركية ونظيرية. توضح الشكل 3 أن الطاقة التنشيطية المقاسة لعملية هدرجة الأسيتيلين معكعامل مساعد منخفض بشكل ملحوظ،أدنى قيمة يمكننا العثور عليها في الأدبيات لأي محفز في هذه التفاعل هي (مرجع 8) لـ مادة سبيكة. طاقة التنشيط لذرات البالاديوم المفردة المضافة إليها الفضة، والتي تشبه بشكل أكبر المحفز الصناعي، هي (المرجع 12). لأغراض المقارنة، تم العثور على ذرات البالاديوم المفردة المسبوكة بالفضة أو الذهب أو النحاس أن لها طاقات تنشيط تتراوح من 37.3 إلى (المرجع 5). وقد تم الإبلاغ عن أن إضافات الثيوإيثر تزيد من الانتقائية للأسيتيلين.، ومن ثم البيئة التنسيقية لـ يجب أن تلعب السبيكة دورًا.
تم قياس تأثيرات النظائر الحركية (KIEs) أيضًا (الشكل 3ب). تظهر النتائج أن التغيير منإلىلا ينتج أي تغيير كبير في معدل التفاعل. تأثير نظير كيميائي أولي خفيف جداً ( ) تم تسجيله عندما @1 تم استخدامه كعامل مساعد، مما قد يعني أن عملية الانقسام ليست الخطوة المحددة، على الرغم من أنه لا يمكن التأكيد على ذلك بشكل قاطع بسبب انخفاض الطاقة التنشيطية الظاهرة الملحوظة. الحقيقة أنيحدث الانفصال بسهولة و@1 لا يحفز تفاعل النقاط إلى البالاديوم كذرة المعدن النشطة خلال التفاعل لأن الذهب غالبًا ما يتطلب التكتل.أو روابط قويةفصل. عندما تم استخدام @1، وهو أولي خفيفتم قياسه، مما يشير إلى أن وجود الذهب يسهلالانفصال، الذي يتوافق بشكل جيد مع الطاقة التنشيطية الأكبر الملحوظة (الشكل 3أ). تم إجراء تجارب تبادل لدراسة الطبيعة المحدودة بدقة لـعملية الانفصال (الشكل 3ج). كشفت تجارب تبادل النظائر أن طاقة التنشيط لعملية انقسام الهيدروجين هيلـ (الشكل 3د)، متساوي ضمن الخطأ مع الطاقة التنشيطية الظاهرة العالمية، وبالتالي يشير تجريبيًا إلى أن خطوة الانفصال تحد من تفاعل الهدرجة: عملية شبه تلقائية. على الرغم من استخدام ثلاث مرات أكثر من العينة مقارنة بـ @1 خلال الـ قياس التبادللم يتم الحصول على معلومات الانفصال مع @1 بسبب تشكيل HD المنخفض جداً، الذي نربطه بارتفاع النسبي التدفقات المطلوبة لهدرجة الأسيتيلين على @1 المحفز (الجدول التكميلي 9).
الحسابات النظرية
حسابات DFT الدورية لـالديمر داخل MOF، في ستة تكوينات مختلفة، يدعم التوصيف البلوري لـ@1 وجعل من الممكن اختيار نموذج واقعي لموقع النشاط لاستخدامه في الدراسة الميكانيكية (الشكل التكميلي 26 والجدول التكميلي 10).تظل الديمرات مستقرة في معظم المواقع المدروسة، مع تحسينأطوال الروابط لـ، ومع تحسين و مسافاتتكون ذرات البالاديوم دائمًا مشحونة بشكل إيجابي قليلاً بينما تظل ذرات الذهب شبه محايدة. يُظهر النظام الأكثر استقرارًا الذي تم الحصول عليه، والمُعَلم بـ F، أن ذرة البالاديوم تتفاعل مباشرة مع ذرتين من الكبريت، وأن ذرة الذهب مدعومة بواسطة ذرة واحدة من الكبريت.
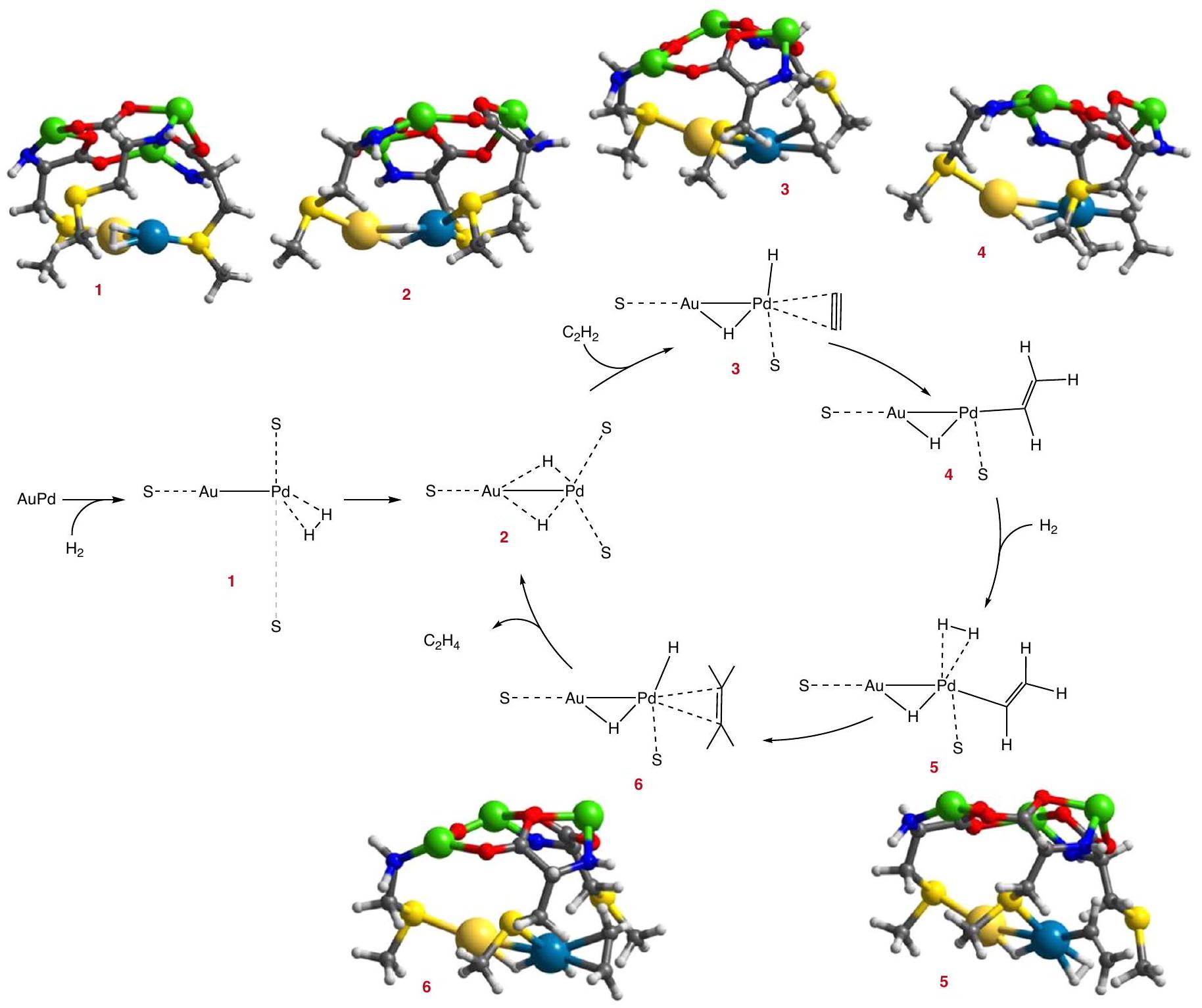
بمجرد تحسين موقع المعدن الحفاز، تم دراسة آلية تفاعل نصف هدرجة الأسيتيلين باستخدام جزء تمثيلي من النظام.. الخطوة الأولى التي تم اعتبارها هي تفكك الجزيئات، الذي يحدث على ذرة البالاديوم. الموقع الأكثر ملاءمة للارتباط بـيكون عكس ذرة الذهب، مكونًا الهيكل 1-A (الشكل التوضيحي 27). ومع ذلك، فإن الطاقة التنشيطية المحسوبة لـكسر الروابط في هذا التكوين مرتفع للغاية،، مما يتعارض مع النتائج التجريبية. بدلاً من ذلك، يمكن أيضًا ربط البالاديوم عن طريق إزاحة أحد
الشكل 4 | الحسابات النظرية. الوسائط المحسوبة الأكثر احتمالاً والآلية المقترحة لهدرجة الأسيتيلين معالعامل المساعد. يتم تصوير ذرات الذهب والبلاديوم والنحاس والكبريت والأكسجين والنيتروجين والكربون والهيدروجين ككرات ذهبية وزرقاء وخضراء وصفراء وحمراء وزرقاء ورمادية وبيضاء، على التوالي.
مجموعات الثيو إيثر لتشكيل الهيكل 1-B. على الرغم من أن معقد الامتزاز أقل استقرارًا، فإن طاقة التنشيط اللازمة للتفككاتباع هذا المسار أقل بكثير، (انظر ملف الطاقة في الشكل التكميلية 28a)، ويمكن تفضيل هذا المسار في وجود إضافيالجزيئات (المسار C في الأشكال التكميلية 27 و 28).
في جميع الحالات، يكون ناتج الخطوة الأولية الأساسية هو هيكل، يُسمى 2، مع وجود أحد ذرات الهيدروجين مرتبطًا بين البالاديوم والذهب، والآخر إما مرتبطًا بشكل أحادي بالبالاديوم كما في الهياكل 2-A و2-C، أو مرتبطًا بين البالاديوم والذهب كما في الهيكل 2-B (الشكل التكميلي 27). يعمل ذرة الذهب كعامل تعديل لذرة البالاديوم، لزيادة النشاط التحفيزي والانتقائية.، ويساعد أيضًا خلال الـحدث انفصام.
يتطلب امتصاص الأسيتيلين على الهيكل 2-A فصل أحد مجموعات الثيوإيثر عن البالاديوم (الشكل التكميلي 29) ونقل الهيدروجين المرتبط بالبالاديوم إلى أحد ذرات الكربون من خلال حالة الانتقال TS34-A، لإنتاج الوسط.مع طاقة تنشيط قدرهايتطلب المسار مشاركة طرف ثانٍجزيء (الهيكل 5-A في
الشكل التوضيحي التكميلي 29)، الذي ينفصل على البالاديوم من خلال TS5-6، مكونًا إيثيلين ممتص مع ذرة هيدروجين على البالاديوم (الهيكل 6-A). تم الحصول على طاقة التنشيط لهذه الخطوة،، مرة أخرى مرتفعة جداً مقارنة بالبيانات التجريبية. على النقيض من ذلك، فإن مساراً مشابهاً يبدأ من الهيكل 2-B مفضل طاقياً، دون وجود وسائط تفاعل مستقرة جداً، وطاقات تنشيط تبلغ فقط 17 وللخطوتين الابتدائيتين اللتين تم النظر فيهما (انظر الهندسات المحسّنة في الشكل التكميلية 30 وملف الطاقة في الشكل التكميلية 28b، الخط البرتقالي). السبب هو أن الانفصال الأولي لأحد مجموعات الثيوإيثر عن البالاديوم يسهل امتصاص والأسيتيلين على طول المسار. ومن المثير للاهتمام أن إزالة الإيثيلين من الهيكل 6-B يعيد توليد الوسيط النشط 2-B مع ذرتين من الهيدروجين الممتصتين، ومن ثم فإن الانفصال الأولي والمكلف من الناحية الطاقية لـعلى البالاديوم لم يعد ضروريًا في الدورة الحفازة الأكثر احتمالًا (الشكل 4).
تمت دراسة الهدرجة التنافسية للإيثيلين لإنتاج الإيثان أيضًا بشكل حسابي. بدءًا من الهيكل 6-B، تتضمن أول عملية نقل للهيدروجين لإنتاج الوسيط 7-B تفعيلًا منخفضًا. طاقة فقط (الأشكال التكميلية 31 و 32). ومع ذلك، فإن الأنواع الوسيطة التي تم الحصول عليها ليست مستقرة للغاية، حيث أن التفاعل المشترك الضروري لامتصاص ثانٍالجزيء ماص للحرارة، والطاقة التنشيطية الإضافية اللازمة لتفكيكهوتوليد الإيثان في نفس الوقت هومقارنة بين ملفات الطاقة لتفاعلات هدرجة الأسيتيلين (باللون البرتقالي) والإيثيلين (باللون الأرجواني) على نفس الموقع النشط (الشكل التكميلي 28c) تُظهر الفرق الطاقي بين التفاعلين، حيث أن تكوين الإيثان يبدو غير مفضل بوضوح.
يمكن القول هنا إن نموذج الكتلة المستخدم في هذه المحاكاة الحاسوبية لا يمكنه محاكاة الهيكل الدوري لمادة MOF بدقة، حيث أن العيب الرئيسي هو إغفال تفاعلات التشتت. ومع ذلك، تم استخدام نهج الكتلة على نطاق واسع لنمذجة العمليات الحفازة في MOFs حيث تكون المواقع النشطة معزولة ومحددة جيدًا نظرًا لتكلفتها الحاسوبية المنخفضة. لذلك، قمنا أيضًا بإجراء الحسابات باستخدام كتلة تمثل بدقة الميزات الرئيسية للموقع النشط لمقارنة آليات التفاعل المختلفة بتكلفة حاسوبية معقولة. تؤكد النتائج دقة نموذج الكتلة المستخدم بعد إعادة حساب تفككمع نموذج دوري لـ MOF ورمز حزمة المحاكاة الأولية في فيينا لأن كلا الطريقتين تؤديان إلى نفس المسارين المحتملين A و B، اللذين يختلفان في طريقةالارتباط بالثنائي PdAu (الشكل التكميلي 33). في المسار A،يرتبط بذرة البالاديوم على الجانب المقابل من الذهب، مكونًا مركبًا أكثر استقرارًا 1-A يتطلب طاقة تنشيط أعلى لتفكك الـرابطة (الخطوط الزرقاء في الشكل التوضيحي 33)، بينما في المسار B، يرتبطيؤدي استبدال البالاديوم بأحد مجموعات الثيوإيثر إلى نظام أقل استقرارًا 1-B الذي يسهل بوضوحفصل (الخطوط البرتقالية في الشكل التكميلي 33). مقارنة بين ملفات الطاقة الدورية (الخطوط الكاملة) وملفات الطاقة العنقودية (الخطوط المتقطعة) تظهر أن جميع الوسائط وحالات الانتقال تكون أكثر استقرارًا في النموذج الدوري الأكثر واقعية، ولكن نظرًا لأن هذا التأثير ثابت إلى حد كبير، فإن الطاقات النسبية لا تختلف بشكل كبير. تؤدي الطريقتان الحاسبيتان إلى الاستنتاج أنيتطلب الانفصال بعد المسار A التغلب على حاجز طاقة تنشيط محظور منلنموذج الدوري ولنهج الكتلة)، بينما الإزاحة الأولية لمجموعة الثيو إيثر في المسار B تسهل بوضوح كسر الروابط الهيدروجينية، مع طاقات تنشيط محسوبة تبلغ 5 ولأساليب الدوريات والعناقيد، على التوالي. تؤكد تشابه الهندسة والخطوات الأساسية والطاقة أن نموذج العنقود المستخرج من الهيكل الدوري مناسب لإجراء الدراسة الميكانيكية، وأن تشبع ذرات النيتروجين بالهيدروجين بدلاً منلا يغير بشكل كبير النتائج التي تم الحصول عليها.
تتوافق الظروف التجريبية المثلى للهيدروجين الجزئي للأسيتيلين مع بيئة غنية بالهيدروجين، معنسبة الأسيتيلين من. لذلك، فإن احتمال العثور على اثنين من الجزيئات القريبة منالعنقود كما هو موصوف في المسار C لـالانفصال مرتفع. تحت هذه الظروف التفاعلية، هناك احتمال أن تكون ذرات الهيدروجين الموجودة فييجب معالجة مسألة إمكانية مشاركة مواقع الجسر في تفاعل الهدرجة بشكل أكبر. لهذا، تم إجراء نماذج حسابية جديدة (الشكل التوضيحي 34). أظهرت النماذج أنه بعد تفكك الأولالجزيء، على الأقل واحد من ذرتي الهيدروجين يشغل موقعًا مستقرًا فيمواقع الجسور (انظر الهياكل 2-A و 2-B). ثم، يتم امتصاص الأسيتيلين بشكل ثنائي التنسيق إلى ذرة البالاديوم (الهياكل 3-A و 3-B) وتحدث الخطوة الأولى لنقل الهيدروجين، مما يؤدي إلى تكوين الأنواع المرتبطة بشكل أحادي بالبلاتين، بينما يبقى ذرة الهيدروجين الأخرى في مواقع الجسور (الهياكل 4-A و4-B). في هذه المرحلة، كانت جميع جهودنا للعثور على حالة انتقالية لهجوم ذرة الهيدروجين المربوطة على الـفشلت الأنواع، سواء باستخدام نهج التجميع أو النماذج الدورية. على النقيض من ذلك، فإن التفاعل المشترك لامتصاص ثانٍالجزيء القريب من أو على ذرة البالاديوم (الهياكل 5-A و 5-B) وتفككه اللاحق الذي يشكل الإيثيلين الم adsorb مباشرة (الهياكل 6-A و 6-B) هو بسيط وذو طاقة. ممكن. بعد إزالة الإيثيلين، يتم تجديد الهياكل غير المتفاعلة 2-A أو 2-B مع ذرة الهيدروجين المربوطة، جاهزة للتفاعل مع جزيء أسيتيلين آخر وبدء دورة تحفيزية جديدة.
بالإضافة إلى ذلك، للنظر أيضًا في تأثير درجة الحرارة على حاجز الطاقة للتفاعل، قمنا بإنشاء ملفات تعريف للطاقة المستمرة وطاقة غيبس الحرة تحت ظروف الحالة القياسية (1 ضغط جوي و298 كلفن) لتفكك الهيدروجين والهيدروجين الجزئي للأسيتيلين. لهذا، نتبع المسارات A وB (الشكل التكميلي 35). كما هو متوقع، فإن طاقات غيبس الحرة (الخطوط المتقطعة) أعلى من الطاقات الإلكترونية (الخطوط الكاملة) وتتبع نفس الاتجاهات عند مقارنة المسارين المقترحين. تتضمن المسار A حد أدنى أكثر استقرارًا وحواجز تنشيط أعلى، في حين أن المسارأكثر سلاسة، مما يسهل الدورة الحفزية. تؤكد طاقات التنشيط المطلقة وطاقة غيبس الحرة جدوى المسار B (الجدول التكميلي 11).
الفرق الكبير في طاقات التنشيط المحسوبة لـالانفصال بعد المسارات و يعود ذلك إلى الطريقة المختلفة للتفاعل معمع البالاديوم، مما يؤدي إلى تمزق أسهل لـالرابطة في المسار B. الترتيب ثلاثي الأبعاد لموقع التفاعل في MOF يجعل من الصعب ملاحظة اختلافات الهندسة في الصور في الشكل 4 أو الشكل التكميلي 34، وبالتالي، لتسهيل هذا التحليل، يتم رسم المتفاعلات (1-A و 1-B)، وحالات الانتقال (TS12-A و TS12-B) والمنتجات (2-A و 2-B) في الشكل التكميلي 36، ويتم تلخيص أهم المسافات المحسنة في الجدول التكميلي 12. يجب ملاحظة أنه يتم الحصول على معلمات هندسية مشابهة باستخدام نماذج العنقود والنماذج الدورية. بالنسبة للمتفاعليتفاعل مع ذرة البالاديوم على الجانب المقابل لذرة الذهب، بحيث تهاجر واحدة من ذرتي الهيدروجين مسافة طويلة لتصل إلى موقع الجسر بين الذهب والبالاديوم في المنتج.. في حالة الانتقال TS12-A، يكون ذرة الهيدروجين المهاجرة مرتبطة عمودياً بذرة البالاديوم رباعية التنسيق، بعيداً عن ذرة الهيدروجين الأخرى ( ) ومن الذهب (2.7-2.8 ). يتم تفسير الاستقرار المنخفض لهذه الحالة الانتقالية من خلال حقيقة أن ذرة الهيدروجين المهاجرة مدعومة فقط برابطة واحدة مع البالاديوم. بالمقابل، في المتفاعل 1-B، يتفاعل مع البالاديوم في جوار الـرابطة، مما يؤدي إلى تحسين تفعيلرابطة (مسافة الرابطة المحسّنة) في 1-A و في 1-ب). بالإضافة إلى ذلك،يتم تسهيل الانفصال من خلال حالة الانتقال TS12-B من خلال تفاعلات أقرب بين ذرة الهيدروجين المهاجرة وذرة الهيدروجين الأخرى (المحسّنة و في النماذج العنقودية والدورية) ومع الذهب (1.55 وفي النماذج العنقودية والدورية). تجعل هذه التفاعلات الإضافية TS12-B أكثر استقرارًا بوضوح من TS12-A. تظهر المشاهد المعززة الإضافية لـ 2-A و3-A و2-B و3-B الموضحة في الشكل التكميلية 37 أن الاتجاه النسبي لذرة الهيدروجين على البالاديوم، وذرتي الكبريت، والأسيتيلين الممتص في 3-A يأتي مباشرة من 2-A، بينما يؤدي امتصاص الأسيتيلين في 2-B إلى إزاحة واحدة من ذرات الهيدروجين المربوطة إلى موقعها في 3-B.
فيما يتعلق بامتصاص الأسيتيلين والإيثيلين علىالمواقع، لأنه يحدث في بيئة غنية بالأوليفينات، يجب أخذ إزالة الهيدروجين أو الهيدروجنة المفضلة للإيثيلين بعين الاعتبار. لتوضيح هذه المسألة، قمنا بإجراء حسابات باستخدام النهج الدوري، بما في ذلك الهيكل الكامل لمادة الإطار العضوي المسامي (MOF) وجميع التفاعلات ذات الصلة الموجودة في بيئة الموقع النشط. مع الأخذ في الاعتبار الآلية المقترحة، تم اعتبار نموذجين مختلفين للموقع النشط: ثنائي البلاتين والذهب النظيف الموجود فقط في بداية التفاعل؛ والهيكل 2-B مع ذرتين من الهيدروجين الممتصتين، أي الحالة الأولية والنهائية للدورة الحفازة الحقيقية. نظرًا لأن القيمة المطلقة للتفاعلات المحسوبة من نوع فان دير فالس تعتمد على العدد الإجمالي للذرات في النظام، فإن جميع طاقات التفاعل المحسوبة تكون أكبر بشكل منهجي للإيثيلين مقارنةً بالأسيتيلين؛ وبالتالي، لإجراء مقارنة موثوقة، أخذنا الحالة الأولية للأسيتيلين أو الإيثيلين الموضوعة في المنطقة البينية لمادة الإطار العضوي المسامي كمرجع، بالقرب من الموقع النشط ولكن لم تتفاعل معه بعد (الشكل التكميلي 38). أدت المحاولات لامتصاص الأسيتيلين أو الإيثيلين على ذرة الذهب في الموقع النشط إلى نفس الحالة المرجعية الأولية، مما يؤكد أنها مستقرة. ويمكن أن تكون مناسبة كحالة مرجعية. أدت عملية امتصاص الأسيتيلين والإيثيلين على ذرة البالاديوم إلى هياكل مستقرة حيث تكون الجزيئات دائمًا-مرتبط بذرة البالاديوم (الشكل التكميلي 38c، d). إن طاقات التفاعل المحسوبة بالنسبة للحالة المرجعية أكبر على السطح النظيفأكبر من الموقع 2-B، وفي كلا الحالتين أكبر قليلاً بالنسبة للأسيتيلين مقارنة بالإيثيلين (الجدول التكميلي 13)، مما يؤكد تفضيل امتصاص الأسيتيلين على المواقع النشطة.
إن نقل الإلكترونات من البالاديوم إلى الذهب مبرر أيضًا من خلال تحليل شحنات بادر الذرية على البالاديوم والذهب المستمدة من حسابات DFT الدورية لنماذج المحفزات المختلفة (الجدول التكميلي 10). الشحنات الذرية الصافية على البالاديوم والذهب في الذرة الواحدة @1 و نموذج المحفز @1، مع ذرات البالاديوم والذهب الموضوعة في موضعين مختلفين داخل الإطار العضوي المعدني (في القنوات وفي المناطق البينية)، تظهر شحنة إيجابية طفيفة،للبلاديوم وللذهب، في حين أن الشحنات المحسوبة لستة مواقع مختلفة من ثنائي البلاتين والذهب (الموسوم A-F) تعطي شحنة إيجابية صافية على البلاتين، تزداد بين و بينما تصبح ذرات الذهب قريبة من الحياد أو حتى مشحونة سلبًا قليلاً في بعض الحالات. الاستثناء الوحيد هو الهيكل، حيث يتم كسر الديمر ( ) وتتصرف الذرتان المعدنيتان كأنهما نوعان فرديان، كلاهما مشحون إيجابياً.
الاستنتاجات
تفاعل نصف هدرجة الأسيتيلين في تيارات الإيثيلين تحت ظروف صناعية محاكاة (الواجهة الأمامية) يتم بشكلعائد (من الأسيتيلين المتبقي) وحتىالانتقائية مع محفز محدد جيدًا يتكون من جزيءتم تحضير الديمر ودعمه، في الموقع، على إطار معدني عضوي مفعل بالثيو إيثر. أداء هذه المادة ملحوظ مقارنةً بالعوامل الحفازة الموجودة، حيث تعمل تحت طاقة تنشيط شبه خالية من الحواجز. )، عند درجات حرارة تتراوح بين 35 إلى ، مع الإيثان كمنتج ثانوي وحيد. تكشف دراسة ميكانيكية تجريبية وحاسوبية مجمعة أن ذرة البالاديوم تعمل كموقع تحفيزي رئيسي بينما تساعد ذرة الذهب وموحدات الثيوإيثر في الـ MOF خلال الـ خطوة الانفصال. توفر هذه الدراسة رؤى حول آلية هذه التفاعل الصناعي وتجمع بين مفاهيم المواقع الحفازة المعزولة الفردية والسبائك على محفز هيترو معدني على المستوى الذري.
طرق
المواد
كانت جميع المواد الكيميائية من جودة درجة الكاشف. تم شراؤها من مصادر تجارية واستخدمت كما هي. بلورات/عينات متعددة البلورات منتم إعدادها كما تم الإبلاغ عنه سابقًا (طرق إضافية).
أهرامات خضراء متقنة الشكل من، المناسبة لتقنية حيود الأشعة السينية، تم الحصول عليها من خلال اتباع عملية ما بعد التركيب ذات الخطوتين. أولاً، تم الحصول على بلورات من تم تعليقها، بالتناوب، في و محاليل (0.2 مليمول من الملح المقابل من الذهب أو البالاديوم لكل مليمول من ) لـ تم تكرار العملية خمس مرات، ثم تم تعليق المادة الصلبة الناتجة في خطوة ما بعد التركيب الثانية في 5 مل من مادة أخرى.حل يتجاوزمقسمة إلى 12 جزءًا (كل جزء يتكون من 1 مول من لكل مول من )، تم إضافته تدريجياً على مدى 72 ساعة. بعد كل إضافة، تم السماح للمزيج بالتفاعل لمدة 1.5 ساعة. بعد هذه الفترة، تم غسل العينات بلطف باستخدام الحل وتم تصفيته على الورق. التحليل: تم حساب (%) لـ : C، 19.48؛ H، 3.49؛ S، 10.40؛ N، 4.54. تم العثور على: ، 2,961 و.
تم تنفيذ إجراء على مقياس جرام، للتجارب الحفزية، باستخدام نفس الإجراء الاصطناعي ولكن مع زيادة كميات من عينة مسحوقومبالغ معادلة من و مع نفس النتائج الناجحة وعائد مرتفع جداً ( ). التحليل: محسوب (%) لـ 4.54. تم العثور على: كربون، 19.29؛ هيدروجين، 3.29؛ كبريت، 10.40؛ نيتروجين، 4.57. الأشعة تحت الحمراء (KBr):، 2,954 و.
يمكن العثور على مزيد من التفاصيل حول إعداد المواد في الطرق التكميلية.
SCXRD
بيانات البلورة لـسداسي، مجموعة الفضاء (2) ك، (11) (9) ، يمكن العثور على مزيد من التفاصيل في المعلومات التكميلية.
الامتصاص التنافسي للأسيتيلين/الإيثيلين
تم قياس الإيزوثرمات عالية الدقة باستخدام جهاز ميكروميريتكس ASAP 2010 الحجمي. حوالي 50 ملغ منتم استخدام الشكل البودري لهذه التجارب. تم وضع العينة داخل حامل زجاجي للعينة وغمرها في حمام حراري سائل دائري لضمان التحكم الدقيق في درجة الحرارة. قبل تجارب الامتزاز، خضعت العينة لعملية تفريغ طوال الليل عند 373 كلفن تحت فراغ عالي. تم قياس الإيزوثرمات باستخدام الإيثيلين النقي من جهة، بينما تم استخدام خليط غازي يتكون من 1% أسيتيلين في الإيثيلين من جهة أخرى. ثم تم الحصول على إيزوثرم الامتزاز عند 298 كلفن مع الحفاظ على العينة عند درجة حرارة ثابتة طوال التجربة. يمكن العثور على مزيد من التفاصيل في الطرق التكميلية.
قياسات المجهر
تم إجراء قياسات مجهر الإلكترون الناقل بتقنية الحقل المظلم الحلقي بزاوية عالية على مجهر FEI Titan3 Themis 60-300 المصحح للانحرافات المزدوجة والمونكروماتور، والذي يعمل عند 300 كيلو فولت. يمكن العثور على مزيد من التفاصيل في الطرق التكميلية.
قياسات XPS
تم إعداد العينات عن طريق لصق MOF، دون غربلة، على لوحة من الموليبدينوم باستخدام شريط السيلوفان، تلاها تجفيف بالهواء. تم إجراء القياسات على نظام K-Alpha XPS باستخدام AI K أحادي اللون. مصدر ( يمكن العثور على مزيد من التفاصيل في الطرق التكميلية.
ss-MAS-NMR مع استكشاف
تم تسجيل طيف C ss-MAS-NMR على جهاز مطياف Bruker AVIII HD 400 WB باستخدام مسبار بقطر 3.2 مم يدور العينة بسرعة 15 كيلوهرتز، باستخدامنبضات لـ، مع فصل أثناء الاستحواذ و5 ثوانٍ كفترة تأخير لإعادة التدوير. يمكن العثور على مزيد من التفاصيل في الطرق التكميلية.
تقنيات طيف الامتصاص بالأشعة السينية
تم إجراء قياسات XANES و EXAFS على خط شعاع NOTOS في مصدر الضوء السنكروتروني ALBA، برشلونة، إسبانيا. يمكن العثور على مزيد من التفاصيل في الطرق التكميلية.
تجارب التحفيز
تفاعلات الهدرجة. تم دراسة الهدرجة في الطور الغازي للأسيتيلين (1%) في الإيثيلين (99%). تم إجراء التفاعلات في مفاعل سرير محشو معمن محفز MOF، بمعدل تدفق إجمالي قدره، دون أي غاز ناقل خامل. تم تغذية الهيدروجين بنسب تتراوح من 80:1 إلى 1:30 بالنسبة لإجمالي مولات الإيثيلين المدخلة إلى المفاعل، ولكن عادة ما كانت تتم عند نسب تتراوح من 1:10 إلى 1:30. تم متابعة التفاعلات بواسطة كروماتوغرافيا الغاز عبر الإنترنت، التي أجريت في عمود مناسب لفصل الهيدروكربونات من C1 إلى C6. تم تطهير المفاعل لمدة 30 دقيقة بـبعد تحميل المحفز، ثم تم تسخينه إلى درجة حرارة التفاعل.
درجات حرارة التفاعل المدروسة تراوحت منإلىقبل بدء التفاعلات، تم تمرير الكاشف عبر تجاوز المفاعل وتم معايرة المناطق على جهاز الكروماتوغرافيا الغازية. تم وضع المحفز في مركز أنبوب المفاعل، محاطًا بالصوف الزجاجي.تم استخدامه كمواد حشو ومقاومة للحرارة في المفاعل الأنبوب؛ تظهر التجارب الفارغة أن أي تحويل يحدث في وجوده.
تنظيف @1 بواسطة استخراج Soxhlet. بعد استخدام المادة لتحفيز تفاعل الهيدروجين الجزئي للأسيتيلين، تم استعادة السرير المعبأ من المفاعل ونخلها لإزالة الصوف الزجاجي. تم استعادة المحفز ( ) تم وضعه على ورق الترشيح وأدخل في إعداد سوكسهليت لاستخراجه باستخدام ثنائي كلورو الميثان، الذي وُضع في قارورة سعة 250 مل فوق لوحة تحريك مبرمجة على درجة حرارة تم ترك الاستخراج يعمل لمدة ساعة واحدة (حوالي عشرة دورات تعبئة وغسل في إعداد سوكسليت). تم تحليل محتويات الاستخراج بواسطة الكروماتوغرافيا الغازية-مطياف الكتلة. تبادل النظائر. تم تحميل مفاعل السرير الثابت بـ 2 ملغ من @1. بعد الحفاظ على معدل تدفق الأرجون لـ و تمت إضافتها إلى تدفق الأرجون (معدل التدفق الكلي، ). هذا المزيج من و تم تدفقه لمدة 15 دقيقة عبر تجاوز المفاعل لمعايرة الإشارة، ثم لمدة تقارب 15 دقيقة عبر السرير الثابت للحصول على القياس. تم الحفاظ على الضغط ثابتًا عند 1 بار، وتم الحفاظ على درجة الحرارة عندخلال التجربة. تم تكرار نفس الإجراء التجريبي مع عينة وزنها 6 ملغ من. المركبات عند مخرج المفاعل ( ) تم قياسها بواسطة مطيافية الكتلة عبر الإنترنت ( و 3 ، على التوالي).
توفر البيانات
جميع البيانات التي تدعم نتائج هذا العمل متاحة من المؤلفين المقابلين عند الطلب. تم إيداع بيانات البلورات للهيكل المبلغ عنه في هذه الورقة في قاعدة بيانات كامبريدج للتركيبات برقم CCDC 2249968.
References
Martín, A. J., Mondelli, C., Jaydev, S. D. & Pérez-Ramírez, J. Catalytic processing of plastic waste on the rise. Chem 7, 1487-1533 (2021).
Shittu, T. D. & Ayodele, O. B. Catalysis of semihydrogenation of acetylene to ethylene: current trends, challenges, and outlook. Front. Chem. Sci. Eng. 16, 1031-1059 (2022).
Szesni, N. et al. Catalyst composition for selective hydrogenation with improved characteristics. US patent 13/276,403 (2013).
Zou, S. et al. Grafting nanometer metal/oxide interface towards enhanced low-temperature acetylene semihydrogenation. Nat. Commun. 12, 5770 (2021).
Pei, G. X. et al. Performance of Cu-alloyed Pd single-atom catalyst for semihydrogenation of acetylene under simulated front-end conditions. ACS Catal. 7, 1491-1500 (2017).
Pei, G. X. et al. Promotional effect of Pd single atoms on Au nanoparticles supported on silica for the selective hydrogenation of acetylene in excess ethylene. New J. Chem. 38, 2043-2051 (2014).
Pei, G. X. et al. Ag alloyed Pd single-atom catalysts for efficient selective hydrogenation of acetylene to ethylene in excess ethylene. ACS Catal. 5, 3717-3725 (2015).
Li, R. et al. Selective hydrogenation of acetylene over Pd-Sn catalyst: identification of intermetallic alloy and crystal plane-dependent performance. Appl. Catal. B 279, 119348 (2020).
Pachulski, A., Schödel, R. & Claus, P. Performance and regeneration studies of catalysts for the selective hydrogenation of acetylene. Appl. Catal. A 400, 14-24 (2011).
Hu, M. et al. MOF-confined sub- 2 nm atomically ordered intermetallic PdZn nanoparticles as high-performance catalysts for selective hydrogenation of acetylene. Adv. Mater. 30, 201801878 (2018).
Huang, F. et al. Atomically dispersed Pd on nanodiamond/ graphene hybrid for selective hydrogenation of acetylene. J. Am. Chem. Soc. 140, 13142-13146 (2018).
Qin, C., Guo, Q., Guo, J. & Chen, P. Atomically dispersed Pd atoms on a simple MgO support with an ultralow loading for selective hydrogenation of acetylene to ethylene. Chem. Asian J. 16, 1225-1228 (2021).
Tejeda-Serrano, M. et al. Isolated sites catalyze the hydrogenation of acetylene in ethylene flows under front-end industrial conditions. J. Am. Chem. Soc. 140, 8827-8832 (2018).
Wang, J. et al. Au@Pt nanotubes within CoZn-based metal-organic framework for highly efficient semihydrogenation of acetylene. iScience 23, 101233 (2020).
Bu, J. et al. Selective electrocatalytic semihydrogenation of acetylene impurities for the production of polymer-grade ethylene. Nat. Catal. 4, 557-564 (2021).
Guo, Y. et al. Photo-thermo semihydrogenation of acetylene on Pd/TiO single-atom catalyst. Nat. Commun. 13, 2648 (2022).
Yang, Z. et al. microrods-supported Pd catalysts for semihydrogenation of acetylene: acidic properties tuned reaction kinetics behaviors. Chem. Eng. J. 445, 136681 (2022).
Zhang, W. et al. Bismuth-modulated surface structural evolution of intermetallic alloy catalysts for selective propane dehydrogenation and acetylene semihydrogenation. ACS Catal. 12, 10531-10545 (2022).
. et al. Anchoring Pd species over defective alumina to achieve high atomic utilization and tunable electronic structure for semihydrogenation of acetylene. Appl. Catal. A 642, 118690 (2022).
Ru, W. Control of local electronic structure of Pd single atom catalyst by adsorbate induction. Small 18, 2103852 (2022).
Luo, Q. et al. Alloyed PdCu nanoparticles within siliceous zeolite crystals for catalytic semihydrogenation. ACS Mater. Au 2, 313-320 (2022).
Delgado, J. A. et al. Controlled one-pot synthesis of PdAg nanoparticles and their application in the semihydrogenation of acetylene in ethylene-rich mixtures. ChemNanoMat 8, e202200058 (2022).
Liu, Y. et al. Polyoxometalate-based metal-organic framework as molecular sieve for highly selective semihydrogenation of acetylene on isolated single Pd atom sites. Angew. Chem. Int. Ed. 60, 22522-22528 (2021).
Song, Y. et al. Understanding the role of coordinatively unsaturated Al sites on nanoshaped Al for creating uniform Ni-Cu alloys for selective hydrogenation of acetylene. ACS Catal. 13, 1952-1963 (2023).
Wang, J., Xu, H., Che, C., Zhu, J. & Cheng, D. Rational design of PdAg catalysts for acetylene selective hydrogenation via structural descriptor-based screening strategy. ACS Catal. 13, 433-444 (2023).
Kley, K. S., De Bellis, J. & Schueth, F. Selective hydrogenation of highly concentrated acetylene streams over mechanochemically synthesized PdAg supported catalysts. Catal. Sci. Technol 13, 119-131 (2023).
Huang, F. et al. Low-temperature acetylene semihydrogenation over the Pd-Cu dual-atom catalyst. J. Am. Chem. Soc. 144, 18485-18493 (2022).
Jian, M., Liu, J.-X. & Li, W.-X. Hydroxyl improving the activity, selectivity and stability of supported Ni single atoms for selective semihydrogenation. Chem. Sci 12, 10290-10298 (2021).
Feng, H. et al. Machine-learning-assisted catalytic performance predictions of single-atom alloys for acetylene semihydrogenation. ACS Appl. Mater. Interfaces 14, 25288-25296 (2022).
Furukawa, H., Cordova, K. E., O’Keeffe, M. & Yaghi, O. M. The chemistry and applications of metal-organic frameworks. Science 341, 974 (2013).
Kitagawa, S. & Matsuda, R. Chemistry of coordination space of porous coordination polymers. Coord. Chem. Rev. 251, 2490-2509 (2007).
Escamilla, P. et al. Metal-organic frameworks as chemical nanoreactors for the preparation of catalytically active metal compounds. Chem. Commun. 59, 836-851 (2023).
Tiburcio, E. et al. Soluble/MOF-supported palladium single atoms catalyze the ligand-, additive-, and solvent-free aerobic oxidation of benzyl alcohols to benzoic acids. J. Am. Chem. Soc. 143, 2581-2592 (2021).
Rivero-Crespo, M. A. et al. Confined water clusters in a MOF catalyze the low-temperature water-gas shift reaction with both oxygen atoms coming from water. Angew. Chem. Int. Ed. 57, 17094-17099 (2018).
Mon, M. et al. Synthesis of densely packaged, ultrasmall clusters within a thioether-functionalized MOF: catalytic activity in industrial reactions at low temperature. Angew. Chem. Int. Ed. 57, 6186-6191 (2018).
Tiburcio, E. et al. Highly efficient MOF-driven silver subnanometer clusters for the catalytic Büchner ring expansion reaction. Inorg. Chem. 61, 11796-11802 (2022).
Fortea-Pérez, F. R. et al. The MOF-driven synthesis of supported palladium clusters with catalytic activity for carbene-mediated chemistry. Nat. Mater. 16, 760-766 (2017).
Reina, M., Wallace, W. T., Wyrwas, R. B., Whetten, R. L. & Martínez, A. Binding of multiple molecules to small gold cluster anions ( ). Int. J. Quantum Chem. 119, e25987 (2019).
Dobrzyńska, J., Dąbrowska, M., Olchowski, R. & Dobrowolski, R. An ion-imprinted thiocyanato-functionalized mesoporous silica for preconcentration of gold(III) prior to its quantitation by slurry sampling graphite furnace AAS. Microchim. Acta 185, 564 (2018).
Oliver-Meseguer, J. et al. Generation and reactivity of electron-rich carbenes on the surface of catalytic gold nanoparticles. J. Am. Chem. Soc. 140, 3215-3218 (2018).
Silva, T. A. G. et al. Restructuring of gold-palladium alloyed nanoparticles: a step towards more active catalysts for oxidation of alcohols. ChemCatChem 11, 4021-4027 (2019).
Nishimura, S., Ikeda, N. & Ebitani, K. Selective hydrogenation of biomass-derived 5-hydroxymethylfurfural (HMF) to 2,5-dimethylfuran (DMF) under atmospheric hydrogen pressure over carbon supported PdAu bimetallic catalyst. Catal. Today 232, 89-98 (2014).
Cerezo-Navarrete, C. et al. Ruthenium nanoparticles canopied by heptagon-containing saddle-shaped nanographenes as efficient aromatic hydrogenation catalysts. Chem. Sci. 13, 13046-13059 (2022).
Hyun, K. et al. Tailoring a dynamic metal-polymer interaction to improve catalyst selectivity and longevity in hydrogenation. Angew. Chem. Int. Ed. 60, 12482-12489 (2021).
Dong, J., Robinson, J. R., Gao, Z.-H. & Wang, L.-S. Selective semihydrogenation of polarized alkynes by a gold hydride nanocluster. J. Am. Chem. Soc. 144, 12501-12509 (2022).
Campos, J. Dihydrogen and acetylene activation by a gold(I)/ platinum(0) transition metal-only frustrated Lewis pair. J. Am. Chem. Soc. 139, 2944-2947 (2017).
Dasgupta, A. et al. Atomic control of active-site ensembles in ordered alloys to enhance hydrogenation selectivity. Nat. Chem. 14, 523-529 (2022).
Gao, R. et al. with electronic coupling single-site pair sites for low-temperature semihydrogenation of alkynes. J. Am. Chem. Soc. 144, 573-581 (2022).
شكر وتقدير
يتم الاعتراف بالدعم المالي من المشاريع PID2020-115100GB-I00، PID2020112590 GB-C21، PID2019-104778GB-IO0، CEX2021-001230-S وCEX2019-000919-M (الممولة من وزارة التعليم والابتكار الإسبانية MCIINN). كما نُعبر عن شكرنا لحكومة فالنسيا (SEJI/2020/034 وPROMETEO/2021/054)، ووزارة التعليم والجامعة والبحث الإيطالية (رمز المشروع PE0000021، ‘شبكة الانتقال المستدام للطاقة – NEST’) ومجلس البحث الأوروبي (814804، MOF-reactors). يشكر J.F.-S. برنامج رامون واي كاجال (RYC2019-027940-I). يشكر N.M. وM.M. برنامج خوان دي لا سيرفا على عقد (MCIINN، FJC2018-035455-I وFJC2019-040523-I، على التوالي). يشكر M.B. معهد التكنولوجيا الكيميائية على منح عقد. تم إجراء الميكروسكوب الإلكتروني الناقل عالي الدقة في DME-UCA، جامعة قادس بدعم مالي من FEDER/MINECO (PID2019-110018GA-I00 وPID2019-107578GA-IO0). نشكر أيضًا Diamond Light Source على منح وقت الأشعة (خط الأشعة I19) وتوفير مرافق الإشعاع السنكروتروني (اقتراح CY28808-1). كما نُعبر عن شكرنا على منحة دراسية (ID 100010434) LCF/BQ/DI19/11730029 (J.B.-S) من مؤسسة ‘لا كايسا’. تم إجراء الحسابات على مجموعة Tirant III من خدمة المعلوماتية بجامعة فالنسيا. تشكل هذه الدراسة جزءًا من برنامج المواد المتقدمة (MFA/2022/048) وقد تم دعمها من قبل MCIINN بتمويل من الاتحاد الأوروبي NextGenerationEU (PRTR-C17.11) ومن حكومة فالنسيا. نحن ممتنون لـ A. Vidal وM. Palomino على إجراء تجارب NMR وامتصاص الأسيتيلين، على التوالي.
مساهمات المؤلفين
قام ج.ب.-س. بتنفيذ وتفسير تفاعلات التدفق والتجارب الميكانيكية التجريبية. قام ن.م. و إ.ت. بتنفيذ تخليق مواد MOF. قام م.ب. بتنفيذ تفاعلات التدفق وإعداد الزيوليتات. قام م.م. بتنفيذ بعض من عمليات توصيف المواد. قام ج.س.هـ.-ج. بتنفيذ وتفسير قياسات المجهر. قام ج.م. بتنفيذ تجارب مطيافية امتصاص الأشعة السينية. قام م.ب. بتنفيذ والإشراف على الدراسات الحاسوبية. قام ج.ف.-س. بتصميم تخليق MOF وتفسير النتائج. قام د.أ. بتنفيذ وحل وتفسير تجارب SCXRD. أشرف إ.ب. على تخليق وتوصيف المادة وكتب الورقة، وأشرف أ.ل.-ب. على الجزء التحفيزي وكتب الورقة. ساهم جميع المؤلفين في كتابة الورقة.
المصالح المتنافسة
تم تقديم طلب براءة اختراع في إسبانيا، برقم الطلب P202330601؛ ويظهر J.B.-S. وM.B. وJ.F.-S. وE.P. وA.L.-P. كمخترعين. يعلن المؤلفون الآخرون عدم وجود مصالح متنافسة.
معهد التكنولوجيا الكيميائية، جامعة بوليتكنيك فالنسيا – المجلس الأعلى للبحوث العلمية، فالنسيا، إسبانيا. قسم الكيمياء غير العضوية، معهد العلوم الجزيئية، جامعة فالنسيا، باتيرنا، إسبانيا. قسم علوم المواد والهندسة المعدنية والكيمياء غير العضوية، كلية العلوم، جامعة قادس، الحرم الجامعي بورتو ريال، بورتو ريال، إسبانيا. خلايا-ألبا سينكروترون، برشلونة، إسبانيا. قسم الكيمياء والتقنيات الكيميائية، جامعة كالابريا، ريندي، كوزنزا، إيطاليا. ساهم هؤلاء المؤلفون بالتساوي: جوردي باليستيروس-سوبراناس، نوريا مارتين، ماتي باكيتش. البريد الإلكتروني: donatella.armentano@unical.it; emilio.pardo@uv.es; anleyva@itq.upv.es
The removal of acetylene from ethylene streams is key in industry for manufacturing polyethylene. Here we show that a well-defined dimer, anchored to the walls of a metal-organic framework (MOF), catalyses the selective semihydrogenation of acetylene to ethylene with conversion ( of acetylene) and selectivity in extremely rich ethylene streams ( acetylene, ethylene, , simulated industrial front-end reaction conditions). The reaction proceeds with an apparent activation energy of , working even at , and with operational windows ( ) and weight hourly space velocities ( ) within industrial specifications. A combined experimental and computational mechanistic study shows the cooperativity between both atoms, and between atoms and support, to enable the barrierless semihydrogenation of acetylene.
Polyethylene is one of the most demanded chemicals worldwide, with an estimated annual production million tonnes . Thus, million tonnes of ethylene must be produced every year, mainly by catalytic cracking, which also produces variable amounts of acetylene (>1%) and . This ethylene stream needs to be purified before polymerization, and thus the selective semihydrogenation reaction of acetylene in ethylene flows is one of the highest volume processes in petrochemistry. Although this hydrogenation reaction is thermodynamically favoured ( ), a catalyst is required, not only to overcome kinetic barriers but also to control the selectivity of
the process, because other undesired reactions, such as ethylene hydrogenation, direct hydrogenation of acetylene to ethane and dimerization, are also thermodynamically favourable ( and , respectively). The solid catalyst currently used in industry is composed of palladium and silver species, together with other additives (sodium, calcium, etc.), and the resulting exit stream must contain of acetylene and of ethane to be industrially acceptable . Both a reaction temperature and a weight hourly space velocity (WHSV) are required to avoid overhydrogenation reactions and undesired polymerizations (formation of green
oil, coke), which would deactivate the solid catalyst. In addition, an operational temperature window of typically is required to control potential runaways during the high-volume process. All these strict requirements. together with the complexity of the solid catalyst currently in use. have spurred the scientific community to look for catalysts and predictable molecular mechanisms. Thus, the surge in publications on the topic during the last ten years is not surprising (see below).
In this context, it is still difficult to find a catalyst able to accomplish the above-mentioned industrial requisites for the acetylene semihydrogenation reaction, particularly under front-end (acetylene- and -rich) reaction conditions. A survey of the open literature shows a plethora of solid catalysts with different metal compositions, carriers and diverse operating conditions (feed composition, temperature, WHSV, etc.), but none of them matches, in full, the requisites outlined above. One recent review and a recent publication summarize the most relevant data (reactant composition, reaction temperature, acetylene conversion, selectivity and catalyst productivity) for catalytic systems. We also summarize here (see below) some other relevant and very recent examples . Recognizing the tremendous scientific information and the potential of these catalytic systems after further optimization, the provided results are, in many cases, very far from any industrial use. For instance, acetylene conversions >99% are generally provided, but the specification of of acetylene in the final stream ( conversion) is rarely seen. Furthermore, many reports repeatedly employ a diluting inert gas or remove ethylene from the stream (just using acetylene) for a better selectivity control, which is of course not a realistic approach. Tail-end reaction conditions (usingjust a slight excess of ) are also employed to minimize ethylene hydrogenation, however, at the expense of boosting undesired polymerizations. Moreover, reaction temperatures are commonly to increase productivity, and indeed low-temperature reactions are claimed when working below , when the industrial process is operated at just .
Analysis of the literature also shows that catalyst design for the acetylene semihydrogenation reaction is strongly dominated by two concepts: isolated single catalytic sites and electronic catalyst modulation by alloying . Both concepts follow the same mechanistic direction, which involves the preparation of catalytic metal sites at which a rapid desorption of the ethylene product occurs, without compromising the natural fast acetylene adsorption and dissociation on the metal. Indeed, the currently used solid catalyst apparently joins both concepts by diluting palladium sites on silver aggregates, with additional modifiers to regulate selectivity . Different palladium alloys have been described to catalyse the reaction with enhanced activity in comparison to the non-alloyed counterparts, and, in particular, single atoms supported on different metal clusters and nanoparticles have shown superb catalytic activity (see below) . It is accepted in the literature that can dissociate (refs. 11,12,20,23,28), and thus palladium ensembles are no longer required. As a consequence, the ultimate frontier of joining catalytic site isolation and alloying would be a palladium heterodimer, that is, metal), where M tunes the catalytic activity of palladium. Indeed, a recent machine-learning study has shown that metal dimers, such as , should be optimal catalysts for the acetylene semihydrogenation reaction, and a very recent experimental study employs catalytic dimers on nanodiamond graphene supports .
Metal-organic frameworks (MOFs) are porous crystalline materials with extremely high surface areas . They also exhibit a very interesting host-guest chemistry -a direct consequence of their functional empty space-that permits their use as chemical nanoreactors to produce/stabilize unprecedented metal chemical species . Following this approach, our groups have recently reported the formation of well-defined palladium and platinum single-atom catalysts supported on MOFs, and also catalytic (ref. 35) and (ref. 36)
dimers, with extremely high dispersion and metal loadings (up to ). Thus, it seemed plausible to us to attempt the synthesis of an heterometallic dimer to test the catalytic semihydrogenation of acetylene. Here we show the synthesis of a well-defined dimer, with loadings, on a MOF derived from the amino acid -methyl-L-cysteine, which is stabilized through thioether linkages inside narrow ( ) diameter pores. This solid material catalyses the semihydrogenation of acetylene under simulated front-end industrial conditions, from 35 to up to , with conversion and selectivity under optimized reaction conditions, giving ethane as the only by-product. The reaction proceeds with an apparent activation energy ( ) of , and is thus nearly barrierless, and experimental and computational studies support that is the main adsorption site during the whole mechanism, and that and MOF sulfur atoms act as electronic modifiers for , assisting during the key dissociation step.
Results
Synthesis and characterization of
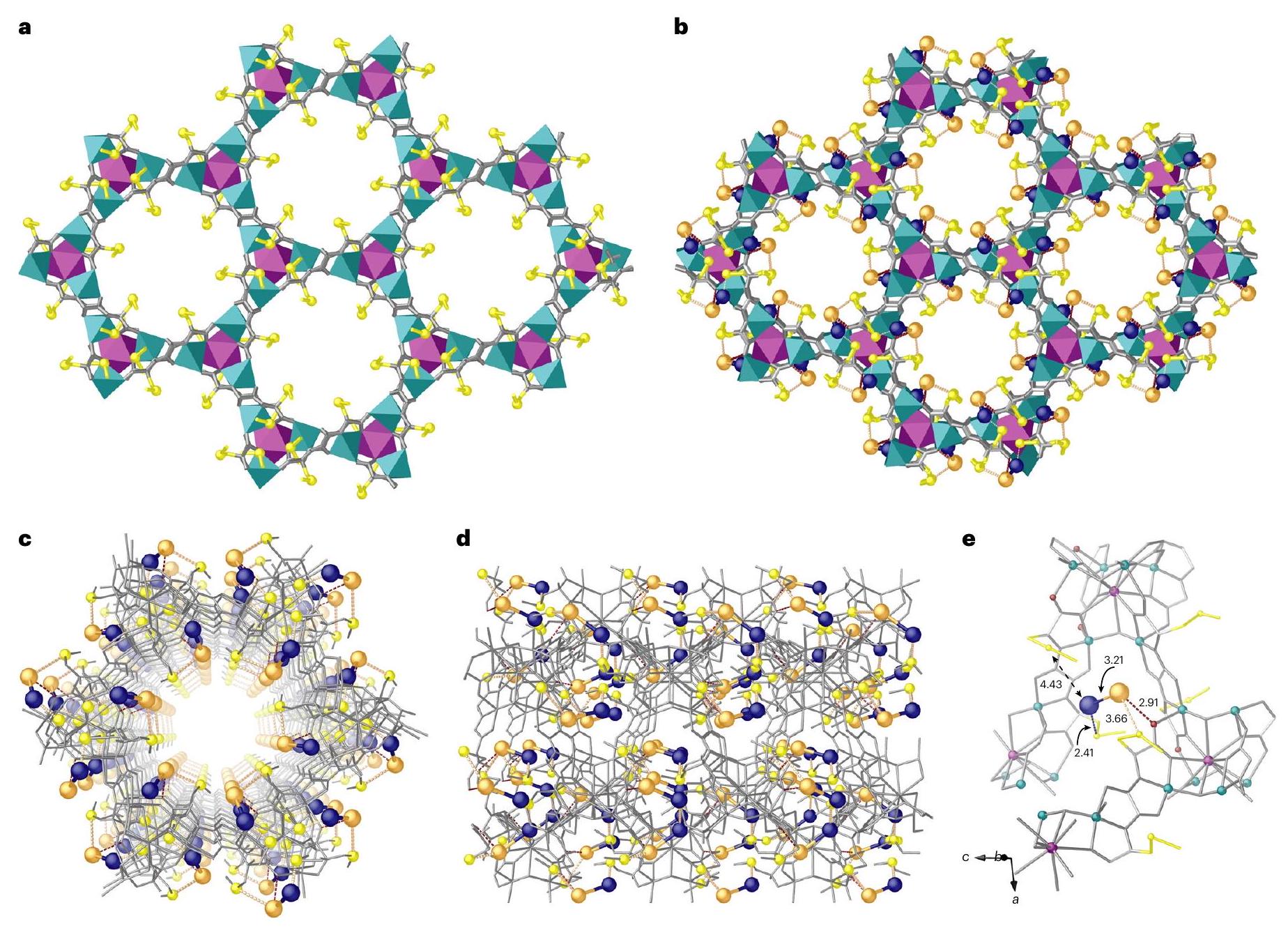
A highly robust and crystalline MOF with the formula mecysmox (mecysmox bis[S-methylcysteine] oxalyl diamide) was used as a chemical nanoreactor for the in situ sequential formation of the dimers, which are retained and stabilized by the thioalkyl groups decorating MOF channels (Fig. 1), with the aim of producing an effective and competitive catalyst for the acetylene semihydrogenation reaction (see Supplementary Fig. 1, Supplementary Table 1 and Supplementary Discussion for more context). As a consequence, a new host-guest aggregate, with the formula @ (Fig. 1b,e), was finally obtained. The synthetic strategy relies on a dual-step postsynthetic process consisting first of the successive insertion of and salts within the MOF’s channels and concomitant in situ reduction with , to form dimers inside the MOF. The homogeneous distribution of sulfur-containing groups within the walls of the channels also allowed a homogeneous distribution of heterometallic dimers (Supplementary Figs. 2-5). Single-crystal X-ray diffraction (SCXRD) was able to reveal the exact structure of the solid supported metal catalyst at the atomic level (Fig. 1 and Supplementary Table 2) due to the high crystallinity and robustness of this kind of MOF. is isomorphic with pristine MOF 1, crystallizing in the chiral space group, and exhibits a chiral three-dimensional strontium(II)-copper(II) network, featuring hexagonal channels of approximately 0.6 nm (Supplementary Fig. 4). The underlying net is represented by the acs uninodal sixfold-connected motif ( ) and is built from trans-oxamidato-bridged dicopper(II) units of {Cu” [( )mecysmox]} (Fig. 1b and Supplementary Fig. 3b). The dimethyl thioether chains ensure the linkage and stabilization of the dimers, which are thus uniformly distributed within the channels of the MOF (Fig. 1b-d and Supplementary Figs. 2-4).
Palladium atoms are linked by sulfur binding sites with a Pd-Sbond distance of 2.41 (2) (Fig. 1e and Supplementary Fig. 5), longer than previously observed bond distances . The thioether chains from the MOF are arranged in their most stable conformation in which one of the two crystallographically distinct moieties is stabilized in a bent conformation and thereby holds the palladium. In so doing, the terminal methyl groups are oriented towards the smaller interstitial voids, which are generated along the axis (Fig. 1b and Supplementary Fig. 3). The other dimethyl thioether chain is stable in a more distended conformation, pointing towards the centre of the pores (Supplementary Fig. 3). These hexagonal pores allocate gold atoms situated at 3.21 Å to palladium atoms, an interatomic distance shorter than the sum of the van der Waals radii, justifying the classification as a dimer, and agreeing with the tendency for palladiumgold attraction due to the net charge transfer from palladium to gold.
Fig. 1|X-ray crystal structure of @1.a,b, Formation of dimers showing the structures of and determined by SCXRD. , A single channel of in the and plane. Copper and strontium atoms from the network are represented by cyan and purple polyedra, respectively, in , whereas they are represented by grey sticks in c,d. Organic ligands are depicted as grey sticks and thioether groups as yellow
sticks in all cases. Orange and blue spheres represent gold and palladium atoms, respectively, in dimers. Blue, orange and red dotted lines represent the and interactions, respectively. e, Details of X -ray crystal structure showing the main host-guest interactions and related structural parameters of dimers.
The gold atoms are stabilized by interactions involving oxygen atoms from the oxamate moieties, belonging to the MOF core, and delimiting the walls of the MOF at a distance of . It is worth emphasizing a further distance of that, although longer than the bond length, can be considered a very effective stabilizing interaction (Fig. 1e and Supplementary Fig. 5) as supported by extended X-ray absorption fine structure (EXAFS) measurements (see below). The gold atoms are stabilized by interactions involving oxygen atoms from the oxamate moieties, belonging to the MOF core and delimiting the walls of the MOF and weak sulfur binding at distances of 2.91 and Å, respectively (Fig. 1e and Supplementary Fig. 5). No examples of a crystallographically precise dimer appear to have been reported previously. Nevertheless, both the and distances are comparable to those previously reported for palladium and gold nanoclusters . Interpretation of the SCXRD offers a description of the palladium-gold environment with palladium atoms tetra-coordinated by and expected (not detected by maps as predictable for solvent molecules in porous crystals, but confirmed by EXAFS experiments) in a highly distorted environment, and with gold only linked to Pd and , with a coordination number of 2, in a would-be linear geometry, substantially distorted, mainly due to
the effectiveness of the weak Au…S interactions described above. The geometric constraints imposed by the MOF topology and the intrinsic flexibility of the methylcysteine moieties, all possible conformations of which have been averaged in one unit cell by crystallography, contribute to promote distortion from linearity.
Density functional theory (DFT) calculations (see below) support the SCXRD data. The experimental crystal structure of @1 (Fig. 1e) shows that the palladium atom is linked directly to one sulfur atom from the thioether chain at and resides at a longer distance, of , from a second sulfur atom, and that the gold atom achieved stability at a slightly longer distance of and a considerably longer separation of . In contrast, the theoretical calculations indicate that the most stable system is where the palladium atom is directly linked to two sulfur atoms at similar distances of , and one thioether group at a longer Au-S distance ( ) stabilizes the gold atom. The apparent deviation between experimental (SCXRD) and theoretical (DFT calculations) results provides, however, the most realistic answer to the proposed structure of @1. The intrinsic and extraordinary flexibility of the thioether chains confined in pores, detected by thermal and dynamic disorder , and expected in porous crystals, acts synergistically to enable the approach of the second
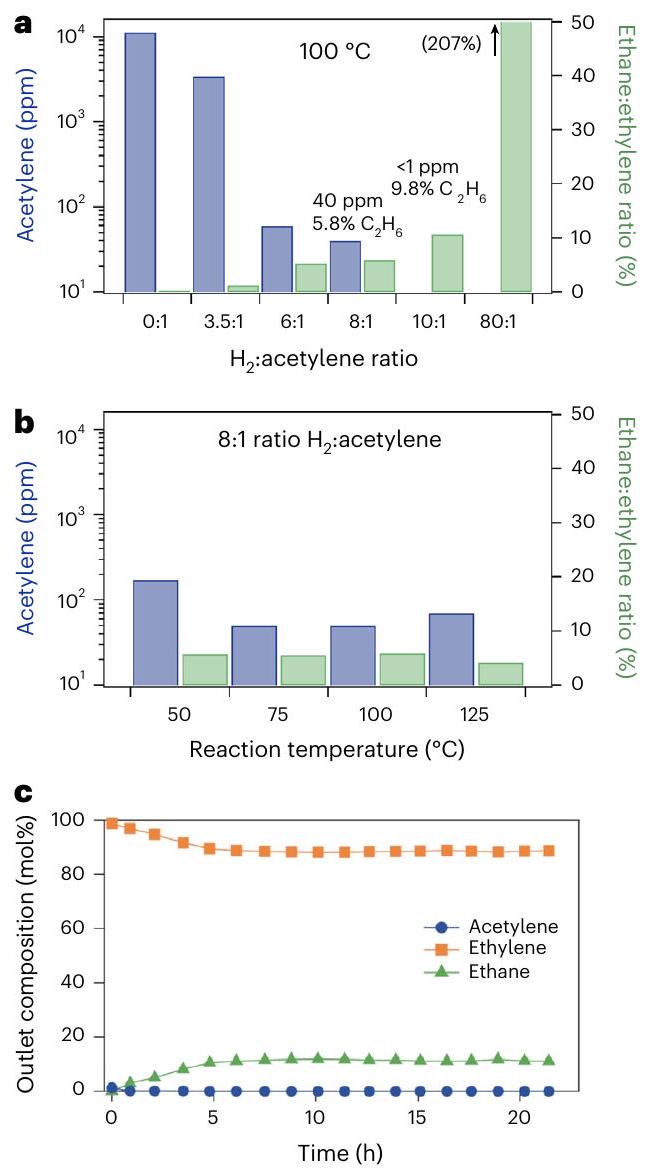
Fig. 2 | Catalytic experiments for the semihydrogenation reaction. a,b, Effect of :acetylene stoichiometry (a) and temperature (b) on the selective acetylene hydrogenation with @1. c, Stability over time for @1 under optimal reaction conditions: :acetylene ratio, of acetylene in ethylene, 24 h reaction time and reaction temperature.
sulfur atom towards the palladium atom. As far as gold is concerned, it is plausible to consider a similar mechanism in which the flexibility of the methylcysteine moieties ensures the gold atoms approach the sulfur atoms, moving from a distance of 3.66 to . This movement is supported by a simultaneous break of the interaction, which serves to first stabilize the loaded gold ions, which are then reduced to and finally stabilized by highly dynamic thioether chains (Supplementary Fig. 5).
EXAFS measurements confirm the bonding of the metal atoms to the sulfur thioether chains (Supplementary Fig. 6). The palladium EXAFS signals (Supplementary Fig. 6a) and the corresponding relative Fourier transform (Supplementary Fig. 6b) of the PdAu-MOF, before and after a partial reduction process (involving just two additions), show the formation of a new bond compatible with and , easily determined by comparison with Pd and PdO references. The formation of the new bonds is more visible in the lower range and results in a slight change of the main peak in the Fourier transform, which is also seen in the crystallographic model for the first-shell fit of palladium (Supplementary Fig. 7). The numeric results of the fit (Supplementary Table 3) show that the typical tetra-coordination found for monometallic palladium complexes is also found in @1, somewhat distorted, with sulfur (and perhaps oxygen) as the first-shell coordinated atoms. The gold EXAFS measurements were not so clear regarding the formation of bonds because the latter are less significant in @1 and tiny amounts of unavoidably formed gold nanoparticles masked the results (Supplementary Fig. 8). However,
it can be seen that @1 contains significant amounts of signals compatible with Au-S bonds, in contrast to gold foil.
To unveil the oxidation states of palladium and gold in the MOF, X-ray absorption near-edge structure (XANES), X-ray photoelectron spectroscopy (XPS), diffuse reflectance infrared Fourier transform spectroscopy with CO probe (DRIFTS) and solid-state magic-angle spinning nuclear magnetic resonance with probe ( ss-MAS-NMR) analyses were carried out.
The palladium XANES analysis of @1 after a partial reduction process (involvingjust two additions; Supplementary Figs. 9 and 10 and Supplementary Table 4) shows the formation of an oxidized monometallic site. The corresponding gold XANES analysis is not included here because the formation of some gold nanoparticles masks the results. The XPS spectra of before and after complete reduction with (Supplementary Fig. 11a,b, respectively) show that the line before reduction is a doublet with binding energies for the and of 337.5 and 342.8 eV , respectively, which are typical of cations and similar to other reported values . On the other hand, the deconvolution of the Au region shows three doublets for and transitions at 86.4 and 90.5 eV (first doublet), 85.3 and 89.5 eV (second), and 84.1 and 87.7 eV (third), which are attributed to and metallic , respectively , and suggests that cations are already partially reduced before treating the MOF with . After reduction, the situation drastically changes for palladium and the observed doublet at and 340.8 eV ( ) can be attributed to slightly positive species . Similarly, the region shows a doublet at and 87.1 eV (Au ) that is indicative of fully reduced gold . These binding energies are in full agreement with those observed for other reported AuPd bimetallic alloys . Moreover, binding energies assigned to and Au in the dimer ( 83.5 and 87.1 eV , respectively) are slightly downshifted compared with those observed, for the same regions, before reduction ( 84.1 and 87.7 eV , respectively). This fact suggests that gold atoms gain electronic density from palladium atoms in the dimer formation process . The and binding energies, and the binding energies from the and regions, are collected in Supplementary Table 5.
The DRIFTS with CO probe spectrum of @1 (Supplementary Fig. 12, top) shows two main signals at 1,975 and , which can be assigned to significantly reduced and species, respectively . A small signal at can also be observed, which can be assigned to a small fraction of single atoms remaining in the material, by comparison with @1 (Supplementary Fig. 12, bottom). A ss-MAS-NMR analysis of @1, unreduced AuPd-MOF and @1 was carried out under an atmosphere of , for 8 h at room temperature. This technique makes it possible to qualitatively distinguish between partially reduced cationic sites (Lewis acid) and reduced metal sites because the intensity of CO coordinated to the former is 1 -fold higher . Furthermore, in the particular case of palladium and gold metals, we deliberately left a small amount of air during the measurement to obtain if gold metal sites were present. The results (Supplementary Fig. 13) show that both @1 and the unreduced AuPd-MOF contain significant amounts of cationic sites (signals between 150 and 180 ppm ) and barely generate (signal at 122 ppm) but, in contrast, @1 contains nearly the half amount of cationic sites and forms significant amounts of . In other words, half of the metal content has been completely reduced to metallic valence in @1, probably gold, while the other half retains some cationic charge. These results, together, match well with the proposed structure of @1.
In addition to the structural characterization, the chemical nature of @1 was also established by various characterization techniques. Elemental analyses ( ) and inductively coupled plasma mass spectrometry (Supplementary Table 6) confirmed the proposed chemical composition. The experimental powder X-ray
Fig. 3 | Experimental mechanistic evidence for @1 and @1.a, Apparent activation energy of the overall acetylene hydrogenation reaction.b, Measured KIE ( ) on the acetylene hydrogenation reaction. Filled symbols, ; empty symbols, . , Hydrogen-splitting activation energy obtained from the HD formation rates. d, Activation energy of the hydrogen-cleavage process.
diffraction pattern of @1 (Supplementary Fig. 14) matches the theoretical one, confirming both the purity and homogeneity of the bulk sample. Thermogravimetric analysis confirms the solvent content of @1 (Supplementary Fig. 15) and makes it possible to establish the proposed chemical formula. The total potential accessible voids in @1 amount to 1,274.0 , accounting for of the unit cell volume . The virtual diameter of the channels decreases from in the pristine MOF 1 to @1 (Supplementary Fig. 3), due to the presence of the dimers within the channels, and adsorption isotherms at 77 K confirm the permanent porosity of (Supplementary Fig. 16, top). It is worthy to notice here that this small pore size is convenient to perform the semihydrogenation of acetylene without competing coupling by-reactions, since a 0.56 nm pore size allows the entrance and diffusion of molecules but hampers the formation and diffusion of bigger molecules (Catalytic results). Competitive adsorption experiments of acetylene and ethylene at show that just of acetylene in pure ethylene produces a significant decrease in the adsorption of ethylene (Supplementary Fig. 16, middle), which suggests a stronger affinity of the MOF sites for acetylene. The Fourier transform infrared spectrum of the treated @1 sample shows the signals associated with acetylene adsorption (Supplementary Fig.16, bottom).
High-angle annular dark-field scanning transmission electron microscopy was employed to analyse the @1 material (Supplementary Fig. 17). The results show that both palladium and gold are homogeneously distributed across the MOF according to energy-dispersive X-ray measurements, together with the other elements of the MOF (that is, sulfur and strontium). The observed weight fractions for palladium and gold are in accordance with the calculated molecular formula for @1 (Supplementary Fig. 18).
Catalytic results
was mixed with powder ( 300 mg ) and loaded into a fixed-bed tubular reactor; different combinations of flow rates/ :hydrocarbon mixtures ( ) and temperatures were screened to find the optimal operational conditions for the acetylene semihydrogenation reaction (Fig. 2a,b; see Supplementary Table 7 for numerical values). The reaction temperature does not have a significant impact on acetylene conversion, at least from 35 to , which suggests a very low activation energy for the reaction on the @1 catalyst (Fig. 3). Dimerization peaks were not observed up to , indicative of a lack of polymerization. Elemental analysis and extractions of the used catalyst with organic solvents (Methods) confirm the lack of formation of products, green oils or coke. Changes in the flow rate revealed that the optimal stoichiometry for :acetylene was 10:1, which corresponds to typical front-end acetylene hydrogenation conditions . The achieved throughput under these conditions, in terms of WHSV, was , and the acetylene left was , the detection limit of the gas chromatograph used, which corresponds to yield, with typically selectivity. To assess the concentration (parts per ) of acetylene remaining, a ten-times bigger loop was installed in the gas chromatograph; however, we were not able to assess accurately the amount of acetylene beyond values of , due to free induction decay signal saturation and peak overlapping. In any case, the conversion, selectivity and productivity values obtained for the catalyst fall within industrial specifications. The stability over time of @1 shows a constant conversion of acetylene and selectivity after a period of stabilization (Fig. 2c).
The palladium and gold metal content in @1 is 2.9 and , respectively (Supplementary Table 8). This very high metal content in the solid provides an evident advantage for the throughput of the in-flow reaction in terms of productivity per mass of catalyst. For the sake of comparison, MOFs containing only one type of metal atoms,
here called @1 (ref. 33) and @1, were also prepared and tested as catalysts (Supplementary Table 7), and since the noble metal content remains similar for these MOFs (Supplementary Table 8), the experiments can be considered comparable. The acetylene hydrogenation reaction was then performed with and/or of Au@1 to maintain the amount of palladium and gold constant, respectively. @1 did not catalyse the acetylene hydrogenation at high flow rates , as does, and requires a residence time and a :acetylene stoichiometry 3-fold higher (Supplementary Table 9). Under these optimized reaction conditions, @1 achieved a 99.6% acetylene conversion (with 40 ppm remaining) and a WHSV of (gas hourly space velocity, ; Supplementary Fig. 19), which are values very different to the catalytic activity of did not show any catalytic activity under all the reaction conditions tested (Supplementary Table 9) and did not change the catalytic activity of @1 when physically mixed with it. The stability/recyclability of @1 was confirmed by XPS and powder X-ray diffraction: performing these analyses after the catalytic experiments showed no significant difference with analyses performed before catalysis (Supplementary Figs. 20 and 21).
We investigated how the catalytic behaviour of the microporous @1 material varied with pore size. Because the preparation of a similar MOF with different-sized channels is not straightforward (the particular amino-acid-based building blocks used drive the formation of this particular MOF and not others), we prepared and tested structurally related microporous zeolites with a similar chemical composition but slightly different pore sizes. Specifically, we prepared different Pd-Au-MS materials (MS, molecular sieves) with pore sizes of 3,5 and . The 3 and pore size materials correspond to typical drying agents, which contain and as countercations, and the pore size corresponds to zeolite -zeolites were avoided to circumvent any possible Brønsted-acid-catalysed process. The palladium and gold metals were introduced by the wet impregnation method (Methods), in this metal order, and the structural integrity of the zeolite framework was confirmed by X-ray diffraction (Supplementary Fig. 22). It should be mentioned here that the metal content in the zeolites can be adjusted (from 1 to ) to avoid metal agglomeration, and that this metal amount can be significantly lower than in @1 (7.1 wt% palladium). Nevertheless, the total metal amount in reaction was equalized to that in the MOF by just adding more zeolite.
The palladium- and gold-containing zeolites showed a different catalytic activity depending on the pore size. Reaction conditions were tuned to have a 90% conversion of acetylene for the @1 catalyst to better compare the catalytic activity of the different materials. The and materials achieved complete acetylene conversion, while Pd-Au-MS 3 Å showed a conversion of (Supplementary Fig. 23). The dimensions of the smaller MS support material cause limitations in achieving a complete acetylene conversion because channels that are too small in diameter can hinder the diffusion and transport of reactants and products within the catalyst structure. Consequently, acetylene molecules are less accessible to the catalytic active sites, resulting in incomplete conversions.
The ethane/ethylene ratio also increased as the pore size increased from 3 to , with the ratio stabilizing at 7 Å pore size. Green oils were not found under any conditions. The material ( channel) shows an intermediate catalytic behaviour, similar to the MS zeolite. It should be noted here that a direct catalytic comparison of the zeolites and the MOF by just considering the pore size is not entirely valid because zeolites have interconnected three-dimensional channels with cavities. It should also be noted here that the catalytic activity of @1 is much higher than that of any zeolite when the reaction conditions were pushed to achieve maximum production rates, even after optimizing the reaction conditions, metal content, order of incorporation (gold before palladium) or feed ratio in the zeolite (the study was done for the MS 5 Å material; Supplementary Figs. 24
and 25). These results support the hypothesis that the small pore size of the MOF combined with the formation of a high-density-number of catalytic species is behind its extremely high catalytic activity.
Mechanistic studies
Kinetic and isotopic experiments were carried out. Figure 3 shows that the measured activation energy for acetylene hydrogenation with as a catalyst is outstandingly low, . The lowest value we could find in the literature for any catalyst in this reaction is (ref. 8) for a alloy material. The activation energy of silver-alloyed palladium single atoms, which is more similar to the industrial catalyst, is (ref. 12). For the sake of comparison, silver-, gold- and copper-alloyed palladium single atoms have been found to have activation energies ranging from 37.3 to (ref.5). It has been reported that thioether additives increase the selectivity for acetylene , and hence the coordinative environment of the alloy must play a role.
Kinetic isotope effects (KIEs) were also measured (Fig. 3b). The results show that changing from to does not produce any significant change in the reaction rate. A very mild primary KIE ( ) was recorded when @1 was used as a catalyst, which could imply that the splitting process is not the limiting step, although this cannot be asserted with certainty due to the low apparent activation energy observed. The fact that dissociation occurs easily and @1 does not catalyse the reaction points to palladium as the active metal atom during the reaction because gold often requires agglomeration or strong ligands to dissociate . When @1 was used, a mild primary was measured, which indicates that the presence of gold facilitates dissociation, which is in good agreement with the observed larger activation energy (Fig. 3a). exchange experiments were performed to accurately study the limiting nature of the dissociation process (Fig. 3c). The isotope exchange experiments revealed that the activation energy for the hydrogen-splitting process is for (Fig.3d), equal within error to the global apparent activation energy, and therefore indicating experimentally that the dissociation step limits the hydrogenation reaction: a quasi-spontaneous process. Despite using three times more sample than for @1 during the exchange measurement, dissociation information was not obtained with @1 due to the very low HD formation, which we correlate to the relatively higher flows required for acetylene hydrogenation over the @1 catalyst (Supplementary Table 9).
Theoretical calculations
Periodic DFT calculations of the dimer within the MOF, in six different configurations, support the crystallographic characterization of @1 and made it possible to select a realistic model of the active site for use in the mechanistic study (Supplementary Fig. 26 and Supplementary Table 10). dimers remain stable in most of the locations considered, with optimized bond lengths of , and with optimized and distances of . The palladium atoms are always slightly positively charged whereas the gold atoms remain almost neutral. The most stable system obtained, labelled F , shows that the palladium atom is directly interacting with two sulfur atoms, and the gold atom is stabilized by one sulfur atom.
Once the catalytic metal site had been optimized, the mechanism of the semihydrogenation reaction of acetylene was studied with a representative fragment of the system . The first step considered was the dissociation of molecular , which takes place on the palladium atom. The most favourable binding site for is opposite to the gold atom, forming structure 1-A (Supplementary Fig. 27). However, the calculated activation energy for the bond breaking in this configuration is extremely high, , which contradicts the experimental results. Alternatively, can also bind palladium by displacing one of
Fig.4|Theoretical calculations. The most plausible computed intermediates and proposed mechanism for acetylene hydrogenation with catalyst. Gold, palladium, copper, sulfur, oxygen, nitrogen, carbon and hydrogen atoms are depicted as gold, cyan, green, yellow, red, blue, grey and white spheres, respectively.
the thioether groups to form structure 1-B. Although the adsorption complex is less stable, the activation energy necessary to dissociate following this pathway is significantly lower, (see energy profile in Supplementary Fig. 28a), and this route can be favoured in the presence of additional molecules (pathway C in Supplementary Figs. 27 and 28).
In all cases, the product of the first elementary step is a structure, named 2 , with one of the hydrogen atoms bridged between palladium and gold, and the other one either monocoordinated to palladium as in structures 2-A and 2-C, or bridged between palladium and gold as in structure 2-B (Supplementary Fig. 27). The gold atom acts as a modifier of the palladium atom, to increase the catalytic activity and selectivity , and also assists during the dissociation event.
The adsorption of acetylene on structure 2-A requires the detachment of one of the thioether groups from palladium (Supplementary Fig. 29) and the transfer of the hydrogen bonded to palladium to one of the carbon atoms through transition state TS34-A, to produce intermediate , with an activation energy of . The pathway requires the participation of a second molecule (structure 5-A in
Supplementary Fig. 29), which dissociates on palladium through TS5-6, forming adsorbed ethylene together with a hydrogen atom on palladium (structure 6-A). The activation energy obtained for this step, , is again too high compared with the experimental data. In contrast, a similar pathway starting from structure 2-B is energetically favoured, with no reaction intermediates that are too stable, and with activation energies of only 17 and for the two elementary steps considered (see optimized geometries in Supplementary Fig. 30 and the energy profile in Supplementary Fig. 28b, orange line). The reason is that the initial detachment of one of the thioether groups from palladium facilitates the adsorption of and acetylene along the pathway. Interestingly, desorption of ethylene from structure 6-B regenerates the active intermediate 2-B with two adsorbed hydrogen atoms, and hence the initial and energetically demanding dissociation of on palladium is no longer necessary in the most plausible catalytic cycle (Fig. 4).
The competitive hydrogenation of ethylene to produce ethane was also computationally investigated. Starting from structure 6-B, the first hydrogen transfer to produce intermediate 7-B involves a low activation
energy of only (Supplementary Figs. 31 and 32). However, the intermediate species obtained is not too stable, the necessary coadsorption of a second molecule is endothermic, and the additional activation energy necessary to dissociate and simultaneously generate ethane is . Comparison of the energy profiles for the acetylene (orange) and ethylene (purple) hydrogenation reactions on the same active site (Supplementary Fig. 28c) shows the energetic difference between the two reactions, with ethane formation being clearly disfavoured.
It could be argued here that the cluster model used in these computational simulations cannot accurately simulate the periodic structure of the MOF, with the main drawback being the neglect of dispersion interactions. Nevertheless, the cluster approach has been widely used to model catalytic processes in MOFs where the active sites are isolated and well defined due to its lower computational cost. Therefore, we also performed the calculations by using a cluster that models accurately the main features of the active site to compare the different reaction mechanisms at a reasonable computational cost. The results confirm the accuracy of the cluster model used after recalculating the dissociation of with a periodic model of the MOF and the Vienna Ab initio Simulation Package code because both approaches lead to the same two possible pathways A and B , which differ in the mode of binding to the PdAu dimer (Supplementary Fig. 33). In pathway A, binds to the palladium atom on the opposite side of gold, forming a more stable complex 1-A that requires a higher activation energy for dissociation of the bond (blue lines in Supplementary Fig. 33), whereas in pathway B , the binding of to palladium displaces one of the thioether groups resulting in a less stable system 1-B that clearly facilitates the dissociation (orange lines in Supplementary Fig. 33). Comparison of periodic (full lines) with cluster (dashed lines) energy profiles shows that all intermediates and transition states are better stabilized in the more realistic periodic model, but since this effect is quite constant, the relative energies do not differ significantly. The two computational approaches lead to the conclusion that dissociation following pathway A requires overcoming a prohibitive activation energy barrier of for the periodic model and for the cluster approach), while the initial displacement of a thioether group in pathway B clearly facilitates H-H rupture, with calculated activation energies of 5 and for the periodic and cluster approaches, respectively. The similarity of geometries, elementary steps and energies confirms that the cluster model extracted from the periodic structure is adequate to perform the mechanistic study, and that saturating nitrogen atoms with hydrogen instead of does not alter significantly the results obtained.
The optimal experimental conditions for the semihydrogenation of acetylene correspond to a hydrogen-rich environment, with a :acetylene ratio of . Therefore, the probability of finding two molecules close to the cluster as described in pathway C for dissociation is high. Under these reaction conditions, the possibility that the hydrogen atoms located at the bridge sites could participate in the hydrogenation reaction needs to be further addressed. For that, new computational models were performed (Supplementary Fig. 34). The models showed that, after the dissociation of the first molecule, at least one of the two hydrogen atoms occupies a stable position at the bridge sites (see structures 2-A and 2-B). Then, acetylene adsorbs bicoordinated to the palladium atom (structures 3-A and 3-B) and the first hydrogen-transfer step occurs, leading to formation of a species monocoordinated to palladium, while the other hydrogen atom remains at the bridge sites (structures 4-A and 4-B). At this point, all our efforts to find a transition state for the attack of the bridged hydrogen atom to the species failed, using either the cluster approach or the periodic models. In contrast, coadsorption of a second molecule close to or on the palladium atom (structures 5-A and 5-B) and its subsequent dissociation forming directly adsorbed ethylene (structures 6-A and 6-B) is straightforward and energetically
feasible. After ethylene desorption, unreacted structures 2-A or 2-B with the bridged hydrogen atom are regenerated, ready to interact with another acetylene molecule and start a new catalytic cycle.
Additionally, to also consider the effect of temperature on the energy barrier of the reaction, we constructed continuous energy and Gibbs free-energy profiles under standard state conditions (1 atm and 298 K ) for hydrogen dissociation and acetylene semihydrogenation. For that, we follow pathways A and B (Supplementary Fig. 35). As expected, the Gibbs free energies (dashed lines) are higher than the electronic energies (full lines) and follow the same trends when comparing the two proposed pathways. Pathway A involves more stable minima and higher activation barriers, whereas pathway is smoother, thus facilitating the catalytic cycle. The absolute activation energies and Gibbs free energies confirm the feasibility of pathway B (Supplementary Table11).
The large difference in the calculated activation energies for dissociation following pathways and is due to the different way of interaction of with palladium, which leads to an easier rupture of the bond in pathway B . The three-dimensional arrangement of the active site in the MOF makes it difficult to observe the geometry differences in the images in Fig. 4 or Supplementary Fig. 34, and therefore, to facilitate this analysis, the reactants (1-A and 1-B), transition states (TS12-A and TS12-B) and products (2-A and 2-B) are schematized in Supplementary Fig. 36, and the most important optimized distances are summarized in Supplementary Table 12. It should be noted that similar geometric parameters are obtained using cluster and periodic models. For the reactant interacts with the palladium atom at the opposite side of the gold atom, so that one of the two hydrogen atoms migrates a long way to reach the bridge position between gold and palladium in product . In the transition state TS12-A, the migrating hydrogen atom is perpendicularly attached to a tetra-coordinated palladium atom, far from the other hydrogen atom ( ) and from gold (2.7-2.8 ). The low stability of this transition state is explained by the fact that the migrating hydrogen atom is only stabilized by one bond with palladium. In contrast, in reactant 1-B, interacts with palladium in the neighbourhood of the bond, leading to an improved activation of the bond (optimized bond distance in 1-A and in 1-B). In addition, dissociation through transition state TS12-B is facilitated by closer interactions between the migrating hydrogen atom and the other hydrogen atom (optimized and in the cluster and periodic models) and with gold (1.55 and in the cluster and periodic models). Such additional interactions make TS12-B clearly more stable than TS12-A. Additional amplified views of 2-A, 3-A, 2-B and 3-B depicted in Supplementary Fig. 37 show that the relative orientation of the hydrogen atom on palladium, the two sulfur atoms and the adsorbed acetylene in 3-A come directly from 2-A, while adsorption of acetylene in 2-B produces the displacement of one bridged hydrogen to its position in 3-B.
Regarding the adsorption of acetylene and ethylene on the sites, because this is occurring in an olefin-rich environment, a preferential desorption or hydrogenation of ethylene has to be considered. To clarify this issue, we performed calculations using the periodic approach, including the whole structure of the MOF and all relevant interactions present in the environment of the active site. Taking into account the mechanism proposed, two different models of the active site were considered: a clean PdAu dimer present only at the beginning of the reaction; and structure 2-B with two adsorbed hydrogen atoms, that is, the initial and final state of the true catalytic cycle. Since the absolute value of the computed van der Waals interactions depends on the total number of atoms of the system, all calculated interaction energies are systematically larger for ethylene than for acetylene; thus, to make a reliable comparison, we took the initial state of acetylene or ethylene placed in the interstitial region of the MOF as a reference, close to the active site but not yet interacting with it (Supplementary Fig. 38). Attempts to adsorb acetylene or ethylene on the gold atom of the active site led to this same reference initial state, confirming that it is stable,
and suitable as a reference state. Adsorption of acetylene and ethylene on the palladium atom led to stable structures in which the molecule is always -bonded to the palladium atom (Supplementary Fig. 38c,d). The calculated interaction energies relative to the reference state are larger on the clean dimer than on the 2-B site, and in both cases slightly larger for acetylene than for ethylene (Supplementary Table 13), confirming a preferential adsorption of acetylene on the active sites.
The transfer of electrons from palladium to gold is also justified by analysis of the Bader atomic charges on palladium and gold obtained from periodic DFT calculations of different catalyst models (Supplementary Table 10). The net atomic charges on palladium and gold in single-atom @1 and @1 catalyst models, with the palladium and gold atoms placed in two different positions within the MOF (in the channels and in the interstitial regions), show a slightly positive charge, for palladium and for gold, whereas the computed charges for six different locations of the PdAu dimer (labelled A-F) give a net positive charge on palladium, increased between and , while the gold atoms become close to neutral or even slightly negatively charged in some cases. The only exception is the structure , in which the dimer is broken ( ) and the two metal atoms behave as individual species, both positively charged.
Conclusions
The semihydrogenation reaction of acetylene in ethylene streams under simulated industrial (front-end) conditions proceeds with yield ( of acetylene remaining) and up to selectivity with a well-defined catalyst composed of a molecular dimer prepared and supported, in situ, on a thioether-functionalized MOF. The performance of this material is remarkable in comparison to existing catalysts, since it works under a nearly barrierless activation energy ( ), at temperatures ranging from 35 to , with ethane as the only by-product. A combined experimental and computational mechanistic study reveals that the palladium atom acts as the main catalytic site while the gold atom and the thioether moieties of the MOF assist during the dissociation step. This study provides insights into the mechanism of this industrial reaction and join the concepts of single isolated catalytic sites and alloying on a heterometallic catalyst at the atomic level.
Methods
Materials
All chemicals were of reagent-grade quality. They were purchased from commercial sources and used as received. Crystals/polycrystalline samples of were prepared as previously reported (Supplementary Methods).
Well-formed green prisms of , suitable for X-ray diffraction, were obtained by following a dual-step postsynthetic process. First, crystals of were suspended, alternately, in and solutions ( 0.2 mmol of the corresponding gold or palladium salt per mmol of ) for . The process was repeated five times and the resulting solid was then, in a second postsynthetic step, suspended in 5 ml of another solution to which an excess of , divided into 12 fractions (each fraction consisting of 1 mole of per mole of ), was added progressively over 72 h . After each addition, the mixture was allowed to react for 1.5 h . After this period, samples were gently washed with an solution and filtered on paper. Analysis: calculated (%) for : C, 19.48; H, 3.49; S, 10.40; N, 4.54. Found: , 2,961 and .
A gramme-scale procedure, for catalytic experiments, was also carried out by using the same synthetic procedure but with greater
amounts of a powder sample of and equivalent amounts of and with the same successful results and a very high yield ( ). Analysis: calculated (%) for , 4.54. Found: C,19.29; H, 3.29; S, 10.40; N, 4.57. Infrared (KBr): , 2,954 and .
Further details of the material preparation can be found in the Supplementary Methods.
SCXRD
Crystal data for , hexagonal, space group (2) K, (11) (9) , . Further details can be found in the Supplementary Information.
Competitive adsorption of acetylene/ethylene
High-resolution isotherms were measured using a Micromeritics ASAP 2010 volumetric instrument. Approximately 50 mg of in powder form was employed for these experiments. The sample was placed within a glass sample holder and immersed in a liquid circulation thermostatic bath to ensure precise temperature control. Prior to the adsorption experiments, the sample underwent overnight outgassing at 373 K under high vacuum. Isotherms were measured using pure ethylene on one hand, while a gas mixture comprising 1% acetylene in ethylene was utilized on the other hand. The adsorption isotherm was then obtained at 298 K while maintaining the sample at a constant temperature throughout the experiment. Further details can be found in the Supplementary Methods.
Microscopy measurements
High-angle annular dark-field scanning transmission electron microscopy measurements were performed on a double-aberration-corrected, monochromated, FEI Titan3 Themis 60-300 microscope working at 300 kV . Further details can be found in the Supplementary Methods.
XPS measurements
Samples were prepared by sticking, without sieving, the MOF onto a molybdenum plate with cellophane tape, followed by air drying. Measurements were performed on a K-Alpha XPS system using a monochromatic AI K source ( ). Further details can be found in the Supplementary Methods.
ss-MAS-NMR with a probe
C ss-MAS-NMR spectra were recorded on a Bruker AVIII HD 400 WB spectrometer using a 3.2 mm probe spinning the sample at 15 kHz , using pulses for , with decoupling during acquisition and 5 s as recycle delay. Further details can be found in the Supplementary Methods.
X-ray adsorption spectroscopy techniques
XANES and EXAFS measurements were carried out on the NOTOS beamline at the ALBA Synchrotron Light Source, Barcelona, Spain. Further details can be found in the Supplementary Methods.
Catalytic experiments
Hydrogenation reactions. The gas-phase hydrogenation was studied for acetylene (1%) in ethylene (99%). The reactions were carried out in a packed bed reactor with of MOF catalyst, at a total flow rate of , without any inert carrier gas. Hydrogen was fed at stoichiometries of 80:1 to 1:30 with respect to the total moles of ethylene fed to the reactor, but usually performed at stoichiometries of 1:10-1:30. The reactions were followed by online gas chromatography, performed in a column suitable for the separation of C1-C6 hydrocarbons. The reactor was purged for 30 min with after loading the catalyst, and then heated to the reaction temperature.
The reaction temperatures studied ranged from to . Before starting the reactions, the reagent was flowed through the reactor bypass and the areas were calibrated on the gas chromatograph. The catalyst was placed in the centre of the reactor tube, packed between glass wool. was used as a filler and refractory material for the tubular reactor; blank experiments show that any conversion occurs in its presence.
Cleaning of @1 by Soxhlet extraction. After using the material to catalyse the acetylene semihydrogenation reaction, the packed bed was recovered from the reactor and sieved to remove the glass wool. The recovered catalyst ( ) was placed on filter paper and introduced into a Soxhlet set-up for extraction with dichloromethane, which was placed in a 250 ml flask on top of a stir plate programmed at a temperature of . The extraction was left running for 1 h (approximately ten fill and wash cycles in the Soxhlet set-up). The contents of the extraction were analysed by gas chromatography-mass spectrometry. isotope exchange. A fixed-bed reactor was loaded with 2 mg of @1. After maintaining an argon flow rate of for and were added to the argon stream (total flow rate, ). This mixture of and was flowed for 15 min through the reactor bypass to calibrate the signal and then for approximately 15 min through the fixed bed to obtain the measurement. The pressure was kept constant at 1 bar, and the temperature was maintained at throughout the experiment. The same experimental procedure was repeated with a 6 mg sample of . The compounds at the reactor exit ( ) were quantified by online mass spectrometry ( and 3 , respectively).
Data availability
All data supporting the findings of this work are available from the corresponding authors on request. Crystallographic data for the structure reported in this paper have been deposited in the Cambridge Structural Database with CCDC number 2249968.
References
Martín, A. J., Mondelli, C., Jaydev, S. D. & Pérez-Ramírez, J. Catalytic processing of plastic waste on the rise. Chem 7, 1487-1533 (2021).
Shittu, T. D. & Ayodele, O. B. Catalysis of semihydrogenation of acetylene to ethylene: current trends, challenges, and outlook. Front. Chem. Sci. Eng. 16, 1031-1059 (2022).
Szesni, N. et al. Catalyst composition for selective hydrogenation with improved characteristics. US patent 13/276,403 (2013).
Zou, S. et al. Grafting nanometer metal/oxide interface towards enhanced low-temperature acetylene semihydrogenation. Nat. Commun. 12, 5770 (2021).
Pei, G. X. et al. Performance of Cu-alloyed Pd single-atom catalyst for semihydrogenation of acetylene under simulated front-end conditions. ACS Catal. 7, 1491-1500 (2017).
Pei, G. X. et al. Promotional effect of Pd single atoms on Au nanoparticles supported on silica for the selective hydrogenation of acetylene in excess ethylene. New J. Chem. 38, 2043-2051 (2014).
Pei, G. X. et al. Ag alloyed Pd single-atom catalysts for efficient selective hydrogenation of acetylene to ethylene in excess ethylene. ACS Catal. 5, 3717-3725 (2015).
Li, R. et al. Selective hydrogenation of acetylene over Pd-Sn catalyst: identification of intermetallic alloy and crystal plane-dependent performance. Appl. Catal. B 279, 119348 (2020).
Pachulski, A., Schödel, R. & Claus, P. Performance and regeneration studies of catalysts for the selective hydrogenation of acetylene. Appl. Catal. A 400, 14-24 (2011).
Hu, M. et al. MOF-confined sub- 2 nm atomically ordered intermetallic PdZn nanoparticles as high-performance catalysts for selective hydrogenation of acetylene. Adv. Mater. 30, 201801878 (2018).
Huang, F. et al. Atomically dispersed Pd on nanodiamond/ graphene hybrid for selective hydrogenation of acetylene. J. Am. Chem. Soc. 140, 13142-13146 (2018).
Qin, C., Guo, Q., Guo, J. & Chen, P. Atomically dispersed Pd atoms on a simple MgO support with an ultralow loading for selective hydrogenation of acetylene to ethylene. Chem. Asian J. 16, 1225-1228 (2021).
Tejeda-Serrano, M. et al. Isolated sites catalyze the hydrogenation of acetylene in ethylene flows under front-end industrial conditions. J. Am. Chem. Soc. 140, 8827-8832 (2018).
Wang, J. et al. Au@Pt nanotubes within CoZn-based metal-organic framework for highly efficient semihydrogenation of acetylene. iScience 23, 101233 (2020).
Bu, J. et al. Selective electrocatalytic semihydrogenation of acetylene impurities for the production of polymer-grade ethylene. Nat. Catal. 4, 557-564 (2021).
Guo, Y. et al. Photo-thermo semihydrogenation of acetylene on Pd/TiO single-atom catalyst. Nat. Commun. 13, 2648 (2022).
Yang, Z. et al. microrods-supported Pd catalysts for semihydrogenation of acetylene: acidic properties tuned reaction kinetics behaviors. Chem. Eng. J. 445, 136681 (2022).
Zhang, W. et al. Bismuth-modulated surface structural evolution of intermetallic alloy catalysts for selective propane dehydrogenation and acetylene semihydrogenation. ACS Catal. 12, 10531-10545 (2022).
. et al. Anchoring Pd species over defective alumina to achieve high atomic utilization and tunable electronic structure for semihydrogenation of acetylene. Appl. Catal. A 642, 118690 (2022).
Ru, W. Control of local electronic structure of Pd single atom catalyst by adsorbate induction. Small 18, 2103852 (2022).
Luo, Q. et al. Alloyed PdCu nanoparticles within siliceous zeolite crystals for catalytic semihydrogenation. ACS Mater. Au 2, 313-320 (2022).
Delgado, J. A. et al. Controlled one-pot synthesis of PdAg nanoparticles and their application in the semihydrogenation of acetylene in ethylene-rich mixtures. ChemNanoMat 8, e202200058 (2022).
Liu, Y. et al. Polyoxometalate-based metal-organic framework as molecular sieve for highly selective semihydrogenation of acetylene on isolated single Pd atom sites. Angew. Chem. Int. Ed. 60, 22522-22528 (2021).
Song, Y. et al. Understanding the role of coordinatively unsaturated Al sites on nanoshaped Al for creating uniform Ni-Cu alloys for selective hydrogenation of acetylene. ACS Catal. 13, 1952-1963 (2023).
Wang, J., Xu, H., Che, C., Zhu, J. & Cheng, D. Rational design of PdAg catalysts for acetylene selective hydrogenation via structural descriptor-based screening strategy. ACS Catal. 13, 433-444 (2023).
Kley, K. S., De Bellis, J. & Schueth, F. Selective hydrogenation of highly concentrated acetylene streams over mechanochemically synthesized PdAg supported catalysts. Catal. Sci. Technol 13, 119-131 (2023).
Huang, F. et al. Low-temperature acetylene semihydrogenation over the Pd-Cu dual-atom catalyst. J. Am. Chem. Soc. 144, 18485-18493 (2022).
Jian, M., Liu, J.-X. & Li, W.-X. Hydroxyl improving the activity, selectivity and stability of supported Ni single atoms for selective semihydrogenation. Chem. Sci 12, 10290-10298 (2021).
Feng, H. et al. Machine-learning-assisted catalytic performance predictions of single-atom alloys for acetylene semihydrogenation. ACS Appl. Mater. Interfaces 14, 25288-25296 (2022).
Furukawa, H., Cordova, K. E., O’Keeffe, M. & Yaghi, O. M. The chemistry and applications of metal-organic frameworks. Science 341, 974 (2013).
Kitagawa, S. & Matsuda, R. Chemistry of coordination space of porous coordination polymers. Coord. Chem. Rev. 251, 2490-2509 (2007).
Escamilla, P. et al. Metal-organic frameworks as chemical nanoreactors for the preparation of catalytically active metal compounds. Chem. Commun. 59, 836-851 (2023).
Tiburcio, E. et al. Soluble/MOF-supported palladium single atoms catalyze the ligand-, additive-, and solvent-free aerobic oxidation of benzyl alcohols to benzoic acids. J. Am. Chem. Soc. 143, 2581-2592 (2021).
Rivero-Crespo, M. A. et al. Confined water clusters in a MOF catalyze the low-temperature water-gas shift reaction with both oxygen atoms coming from water. Angew. Chem. Int. Ed. 57, 17094-17099 (2018).
Mon, M. et al. Synthesis of densely packaged, ultrasmall clusters within a thioether-functionalized MOF: catalytic activity in industrial reactions at low temperature. Angew. Chem. Int. Ed. 57, 6186-6191 (2018).
Tiburcio, E. et al. Highly efficient MOF-driven silver subnanometer clusters for the catalytic Büchner ring expansion reaction. Inorg. Chem. 61, 11796-11802 (2022).
Fortea-Pérez, F. R. et al. The MOF-driven synthesis of supported palladium clusters with catalytic activity for carbene-mediated chemistry. Nat. Mater. 16, 760-766 (2017).
Reina, M., Wallace, W. T., Wyrwas, R. B., Whetten, R. L. & Martínez, A. Binding of multiple molecules to small gold cluster anions ( ). Int. J. Quantum Chem. 119, e25987 (2019).
Dobrzyńska, J., Dąbrowska, M., Olchowski, R. & Dobrowolski, R. An ion-imprinted thiocyanato-functionalized mesoporous silica for preconcentration of gold(III) prior to its quantitation by slurry sampling graphite furnace AAS. Microchim. Acta 185, 564 (2018).
Oliver-Meseguer, J. et al. Generation and reactivity of electron-rich carbenes on the surface of catalytic gold nanoparticles. J. Am. Chem. Soc. 140, 3215-3218 (2018).
Silva, T. A. G. et al. Restructuring of gold-palladium alloyed nanoparticles: a step towards more active catalysts for oxidation of alcohols. ChemCatChem 11, 4021-4027 (2019).
Nishimura, S., Ikeda, N. & Ebitani, K. Selective hydrogenation of biomass-derived 5-hydroxymethylfurfural (HMF) to 2,5-dimethylfuran (DMF) under atmospheric hydrogen pressure over carbon supported PdAu bimetallic catalyst. Catal. Today 232, 89-98 (2014).
Cerezo-Navarrete, C. et al. Ruthenium nanoparticles canopied by heptagon-containing saddle-shaped nanographenes as efficient aromatic hydrogenation catalysts. Chem. Sci. 13, 13046-13059 (2022).
Hyun, K. et al. Tailoring a dynamic metal-polymer interaction to improve catalyst selectivity and longevity in hydrogenation. Angew. Chem. Int. Ed. 60, 12482-12489 (2021).
Dong, J., Robinson, J. R., Gao, Z.-H. & Wang, L.-S. Selective semihydrogenation of polarized alkynes by a gold hydride nanocluster. J. Am. Chem. Soc. 144, 12501-12509 (2022).
Campos, J. Dihydrogen and acetylene activation by a gold(I)/ platinum(0) transition metal-only frustrated Lewis pair. J. Am. Chem. Soc. 139, 2944-2947 (2017).
Dasgupta, A. et al. Atomic control of active-site ensembles in ordered alloys to enhance hydrogenation selectivity. Nat. Chem. 14, 523-529 (2022).
Gao, R. et al. with electronic coupling single-site pair sites for low-temperature semihydrogenation of alkynes. J. Am. Chem. Soc. 144, 573-581 (2022).
Acknowledgements
Financial support by the projects PID2020-115100GB-I00, PID2020112590 GB-C21, PID2019-104778GB-IO0, CEX2021-001230-S and CEX2019-000919-M (funded by the Spanish MCIINN) is acknowledged. We also acknowledge the Generalitat Valenciana (SEJI/2020/034 and PROMETEO/2021/054), the Ministero dell’Istruzione, dell’Università e della Ricerca (project code PE0000021, ‘Network 4 Energy Sustainable Transition – NEST’) and the European Research Council (814804, MOF-reactors). J.F.-S. thanks the Ramón y Cajal programme (RYC2019-027940-I). N.M. and M.M. thank the Juan de la Cierva programme for a contract (MCIINN, FJC2018-035455-I and FJC2019-040523-I, respectively). M.B. thanks the Instituto de Tecnología Química for the concession of a contract. High-resolution scanning transmission electron microscopy was performed at DME-UCA, Cadiz University with financial support from FEDER/MINECO (PID2019-110018GA-I00 and PID2019-107578GA-IO0). We also thank Diamond Light Source for the award of beamtime (I19 beamline) and provision of synchrotron radiation facilities (proposal CY28808-1). Thanks are also extended for a scholarship (ID 100010434) LCF/BQ/DI19/11730029 (J.B.-S) from ‘La Caixa’ Foundation. Computations were performed on the Tirant III cluster of the Servei d’Informatica of the University of Valencia. This study forms part of the Advanced Materials programme (MFA/2022/048) and was supported by MCIINN with funding from European Union NextGenerationEU (PRTR-C17.11) and by Generalitat Valenciana. We are grateful to A. Vidal and M. Palomino for performing the NMR and acetylene adsorption experiments, respectively.
Author contributions
J.B.-S. performed and interpreted the flow reactions and the experimental mechanistic experiments. N.M. and E.T. carried out the synthesis of the MOF materials. M.B. carried out the flow reactions and prepared the zeolites. M.M. performed some of the material characterizations. J.C.H.-G. carried out and interpreted the microscopy measurements. C.M. carried out the X-ray adsorption spectroscopy experiments. M.B. carried out and supervised the computational studies. J.F.-S. designed the MOF synthesis and interpreted the results. D.A. performed, resolved and interpreted the SCXRD experiments. E.P. supervised the synthesis and characterization of the material and wrote the paper, and A.L.-P. supervised the catalytic part and wrote the paper. All authors contributed to writing the paper.
Competing interests
A patent application has been filed and presented in Spain, with application number P202330601; J.B.-S., M.B., J.F.-S., E.P. and A.L.-P. appear as inventors. The other authors declare no competing interests.
Correspondence and requests for materials should be addressed to Donatella Armentano, Emilio Pardo or Antonio Leyva-Pérez.
Peer review information Nature Catalysis thanks Dingsheng Wang, Xin Zhang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Instituto de Tecnología Química, Universidad Politècnica de València-Consejo Superior de Investigaciones Científicas, Valencia, Spain. Departamento de Química Inorgánica, Instituto de Ciencia Molecular, Universidad de Valencia, Paterna, Spain. Departamento de Ciencia de los Materiales e Ingeniería Metalúrgica y Química Inorgánica, Facultad de Ciencias, Universidad de Cádiz, Campus Universitario Puerto Real, Puerto Real, Spain. CELLS-ALBA Synchrotron, Barcelona, Spain. Dipartimento di Chimica e Tecnologie Chimiche, Università della Calabria, Rende, Cosenza, Italy. These authors contributed equally: Jordi Ballesteros-Soberanas, Nuria Martín, Matea Bacic. e-mail: donatella.armentano@unical.it; emilio.pardo@uv.es; anleyva@itq.upv.es