DOI: https://doi.org/10.1007/s11596-024-2832-z
PMID: https://pubmed.ncbi.nlm.nih.gov/38336987
تاريخ النشر: 2024-02-01
أيض النحاس وموات النحاس: الآليات الجزيئية وآفاق العلاج في الأمراض التنكسية العصبية
الملخص
[الملخص] النحاس عنصر أساسي نادر، ويلعب دورًا حيويًا في العديد من العمليات الفسيولوجية داخل جسم الإنسان. خلال الأيض الطبيعي، يحافظ جسم الإنسان على توازن النحاس. يمكن أن يؤثر نقص النحاس أو زيادته سلبًا على وظيفة الخلايا. لذلك، يتم تنظيم توازن النحاس بشكل صارم. تشير الدراسات الحديثة إلى أن النحاس يمكن أن يحفز شكلًا محددًا من موت الخلايا، وهو ما يعرف بالكوبروتوبسيس، الذي يتم تحفيزه بواسطة مستويات مفرطة من النحاس داخل الخلايا. يؤدي الكوبروتوبسيس إلى تجميع البروتينات الميتوكوندرية المليئة بالليبيدات، وفقدان بروتينات مجموعات الحديد والكبريت. في الأمراض التنكسية العصبية، يرتبط حدوث وتقدم الاضطرابات العصبية بتوازن النحاس. تلخص هذه المراجعة التقدم في توازن النحاس والكوبروتوبسيس في الجهاز العصبي والأمراض التنكسية العصبية. وهذا يقدم آفاق بحثية توفر رؤى جديدة حول العلاج المستهدف للأمراض التنكسية العصبية بناءً على الكوبروتوبسيس.
للبروتينات الحاوية على مجموعات الحديد والكبريت، وإجهاد البروتينات السام، وفي النهاية، موت الخلايا
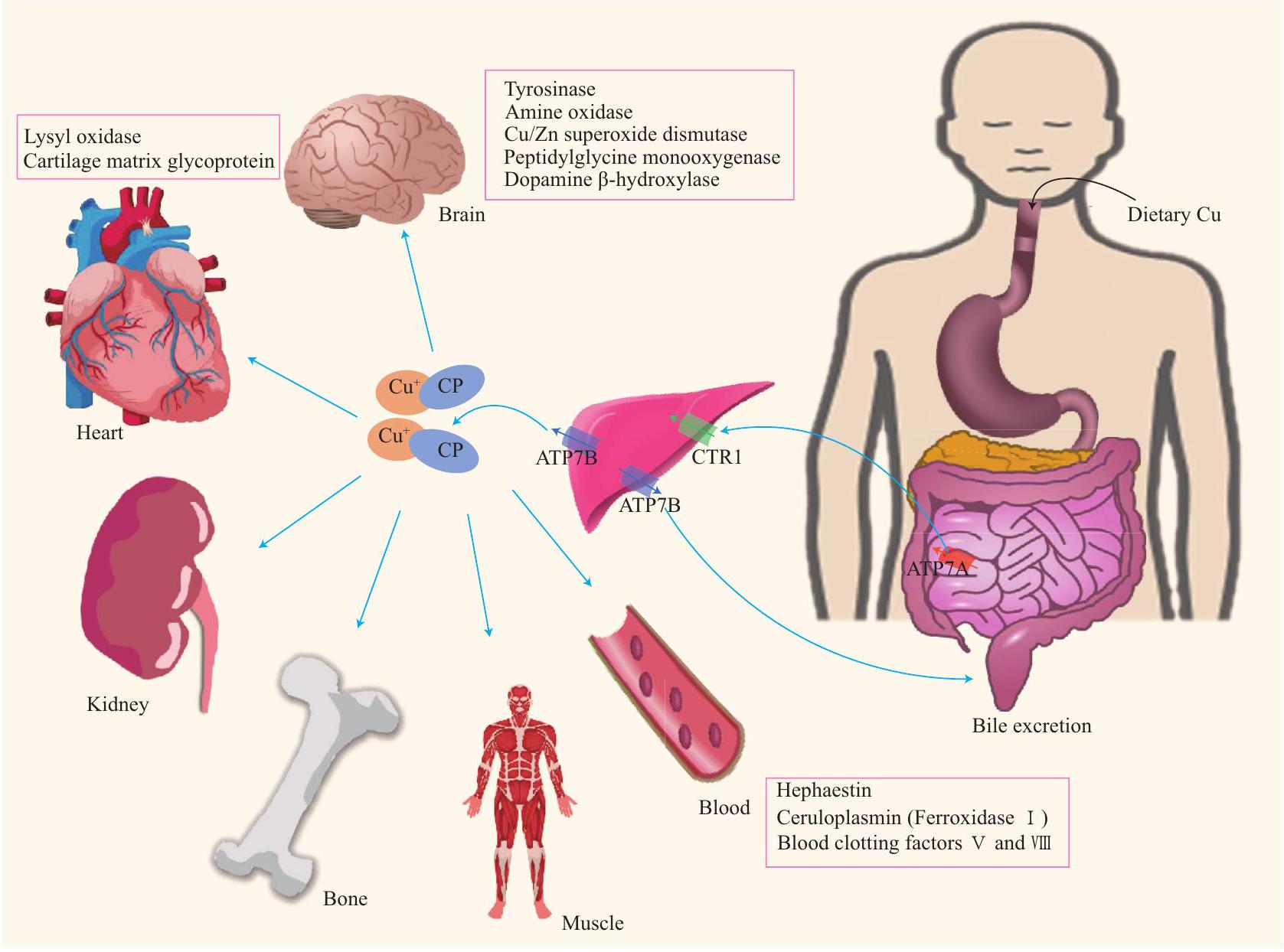
1 الأيض النحاسي النظامي والخلايا في جسم الإنسان
1.1 التمثيل الغذائي للنحاس في الجسم البشري
من الشذوذات الجينية في ATP7A. تؤدي النقل غير السليم للنحاس في خلايا الظهارة المعوية الدقيقة إلى استقلاب غير طبيعي للنحاس.
1.2 استقلاب النحاس الخلوي في جسم الإنسان
1.2.1 الت Chelation
1.2.2 COX17 شابير النحاس COX17 يرتبط
وقد يؤثر SCO2 على سلامة هذه المسار الإشاري
1.2.3 شابيرون النحاس لإنزيم سوبر أكسيد ديسموتاز (CCS) يرتبط النحاس بـ CCS، وينقل النحاس إلى SOD1. هذا يعزز تحلل الأنواع التفاعلية من الأكسجين (ROS)، ويقلل من تراكم ROS، ويحمي الخلايا من أضرار الجذور الحرة. نقص SOD1 سيزيد من الإجهاد التأكسدي.
1.2.4 بروتين مضاد الأكسدة 1 (ATOX1)/ATP7A/B يرتبط ATOX1 بالنحاس، من أجل توصيل النحاس إلى ATP7B على الشبكة الغولجية العبور (ATP7A في خلايا أخرى). هذا يعزز تخليق البروتينات الإنزيمية المسبقة للنحاس، بما في ذلك أكسيداز الليسين، التيروزيناز، والسيرولوبلاسمين.
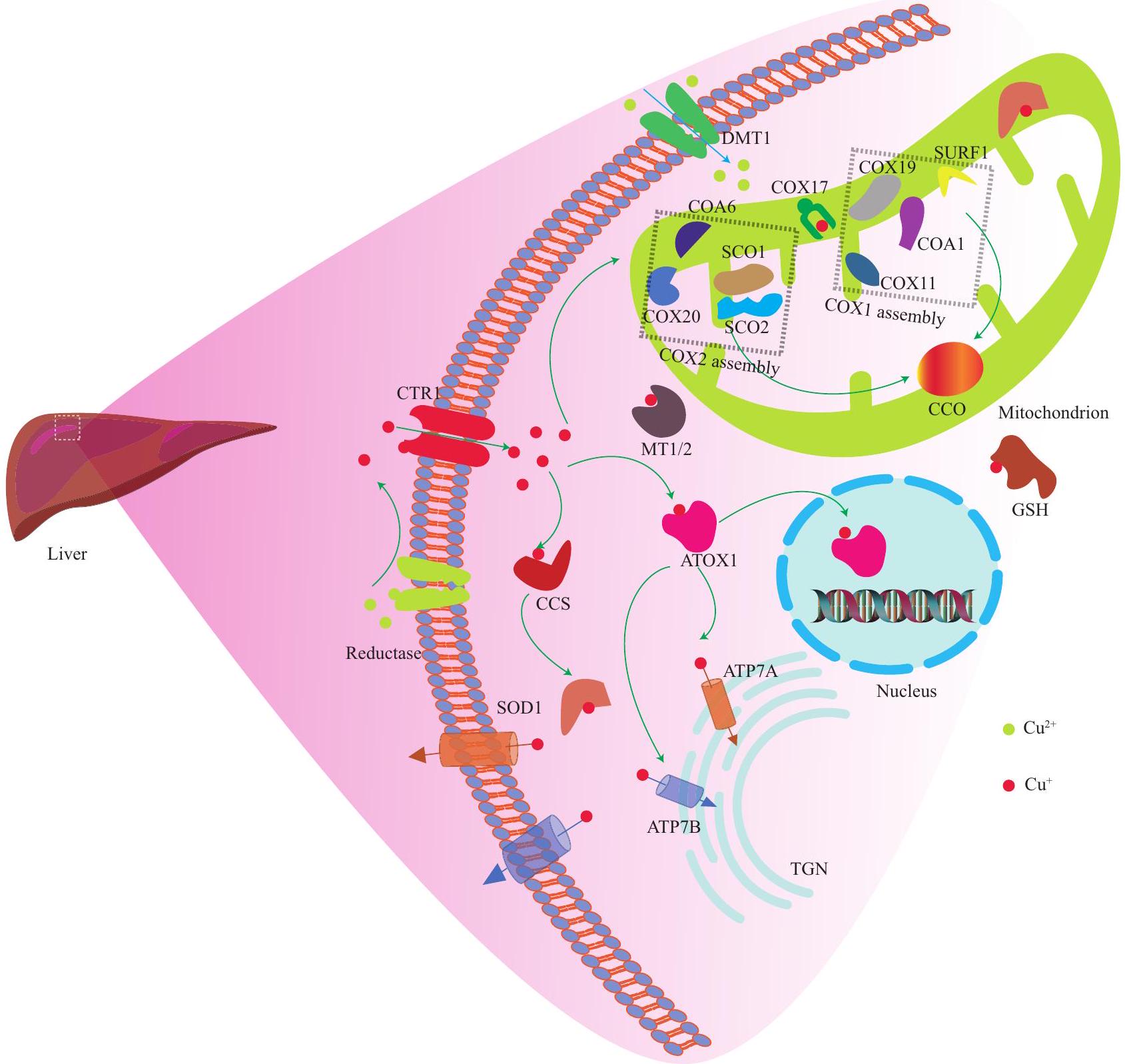
خارجي
2 توازن النحاس في جسم الإنسان
2.1 توازن النحاس في الجهاز العصبي
حاجز الدم-الدماغ (BBB) كأيونات نحاس حرة، ويتم إطلاقه في نسيج الدماغ والسائل الدماغي الشوكي. يبدو أن الحاجز الدموي الدماغي هو الطريق الرئيسي لدخول النحاس إلى نسيج الدماغ، ومن المحتمل أن يحافظ الحاجز الدموي الدماغي وحاجز السائل الدماغي الشوكي على توازن النحاس في الدماغ. علاوة على ذلك، تعبر خلايا كل من الحاجز الدموي الدماغي وحاجز السائل الدماغي الشوكي عن بروتينات تشارك في نقل النحاس. تعبر خلايا الحاجز الدموي الدماغي عن مستويات أعلى من بروتينات ناقل النحاس، بما في ذلك CTR1 وDMT1 وATP7B مقارنة بنسيج الدماغ. يتم نقل النحاس بسهولة أكبر إلى نسيج الدماغ عبر الشعيرات الدموية الدماغية، مقارنة بالنقل عبر الضفيرة المشيمية.
2.2 عدم توازن النحاس في مرض منكس ومرض ويلسون
يؤثر على الدماغ والكبد
2.2.1 مرض منكس
2.2.2 مرض ويلسون
أعراض الحالة، قياس استقلاب النحاس، وتحليل
2.3 توازن النحاس والأمراض التنكسية العصبية
2.3.1 مرض الزهايمر (AD) وتوازن النحاس مرض الزهايمر هو اضطراب عصبي تنكسي شائع قد ينشأ من عوامل متنوعة، مثل العمر والبيئة والوراثة. من المحتمل أن يؤدي زيادة متوسط العمر المتوقع للإنسان في السنوات القادمة إلى زيادة عدد المرضى بـ
تساهم في تلف الأنسجة التأكسدي في دماغ المرضى الذين يعانون من
2.3.2 مرض هنتنغتون (HD) وتوازن النحاس HD هو اضطراب سائد جسديًا في الجهاز العصبي
2.3.3 التصلب الجانبي الضموري (ALS) وتوازن النحاس يؤدي التصلب الجانبي الضموري إلى تدهور انتقائي في الخلايا العصبية الحركية ومن ثم الوفاة
| التدخلات | شرط | مراحل الدراسة | النتائج | الموقع |
| مسح PET GE180 | م | الثاني | تم استخدام GE180 لتحليل الالتهاب الإقليمي والعالمي في دماغ المرضى الذين يعانون من مرض الزهايمر ومرض باركنسون، ووجد أن زيادة GE180 في الدماغ ككل مرتبطة بوظيفة معرفية أضعف، بما في ذلك الفص الجبهي/الحدبي/الجدارية/الصدغي. | نيفادا، الولايات المتحدة الأمريكية |
| قرص زنك سيستين محتجز في المعدة | م.ع | الثاني | كان للمادة المقارنة النشطة المعطاة عن طريق الفم تحمل أفضل، مقارنة بأسيتات الزنك الفموي، وقد أدت إلى تقليل مستويات النحاس غير المرتبط بالسيرولوبلازمين في المصل، وزيادة مستويات الزنك في المصل. | فلوريدا، الولايات المتحدة الأمريكية |
|
|
م.ع | الثاني | تم العثور على تغييرات في الوظيفة الإدراكية، وبيتا-أميلويد في مستشفى جامعة سارلاند، والسائل الدماغي الشوكي، وحجم الدماغ. | |
|
|
التصلب الجانبي الضموري | الثاني | لا يوجد منشور | أريزونا، الولايات المتحدة |
|
|
التصلب الجانبي الضموري | الثاني | لا يوجد منشور | نيو ساوث ويلز/فيكتوريا، أستراليا |
|
|
MS | أنا | لا يوجد منشور | |
|
|
PD | أنا | جرعة الدواء كانت
|
|
| زيت جوز الهند – إبيغالوكاتشين غالات | MS | الثاني | لا يوجد منشور | فالنسيا، إسبانيا |
| برنامج تمارين متعددة الأنماط | PD | لا | لا يوجد منشور | مستشفى تشانغ غونغ التذكاري |
| دراسة رصدية: التعرض للنحاس | PD | لا يوجد منشور | إسرنيا/نابولي، إيطاليا | |
| إدارة التعب: | ||||
| برنامج الأفراد (MFIP) | PD | لا | لا يوجد منشور | نوفا سكوشا، كندا |
| لم يتم البحث عنه | إتش دي | |||
بينما قد تفسر العوامل البيئية، تراكم السموم العصبية، تلف الإجهاد التأكسدي، وعوامل النمو غير الكافية
2.3.4 مرض باركنسون وتوازن النحاس. تشمل الأعراض السريرية الرئيسية لمرض باركنسون الاهتزازات أثناء الراحة، وتعديلات في توتر العضلات، وبطء الحركة، وعدم الاستقرار الوضعي.
تتميز علم الأمراض بتقليل عدد الخلايا العصبية الدوبامينية، وتكوين تجمعات بروتينية تتكون من
3 الكوبروتوسيس في جسم الإنسان
3.1 تعريف الكوبروتوس
توازن النحاس، وأفاد بأن النحاس يرتبط بالبروتينات المليئة بالليبوي في دورة TCA
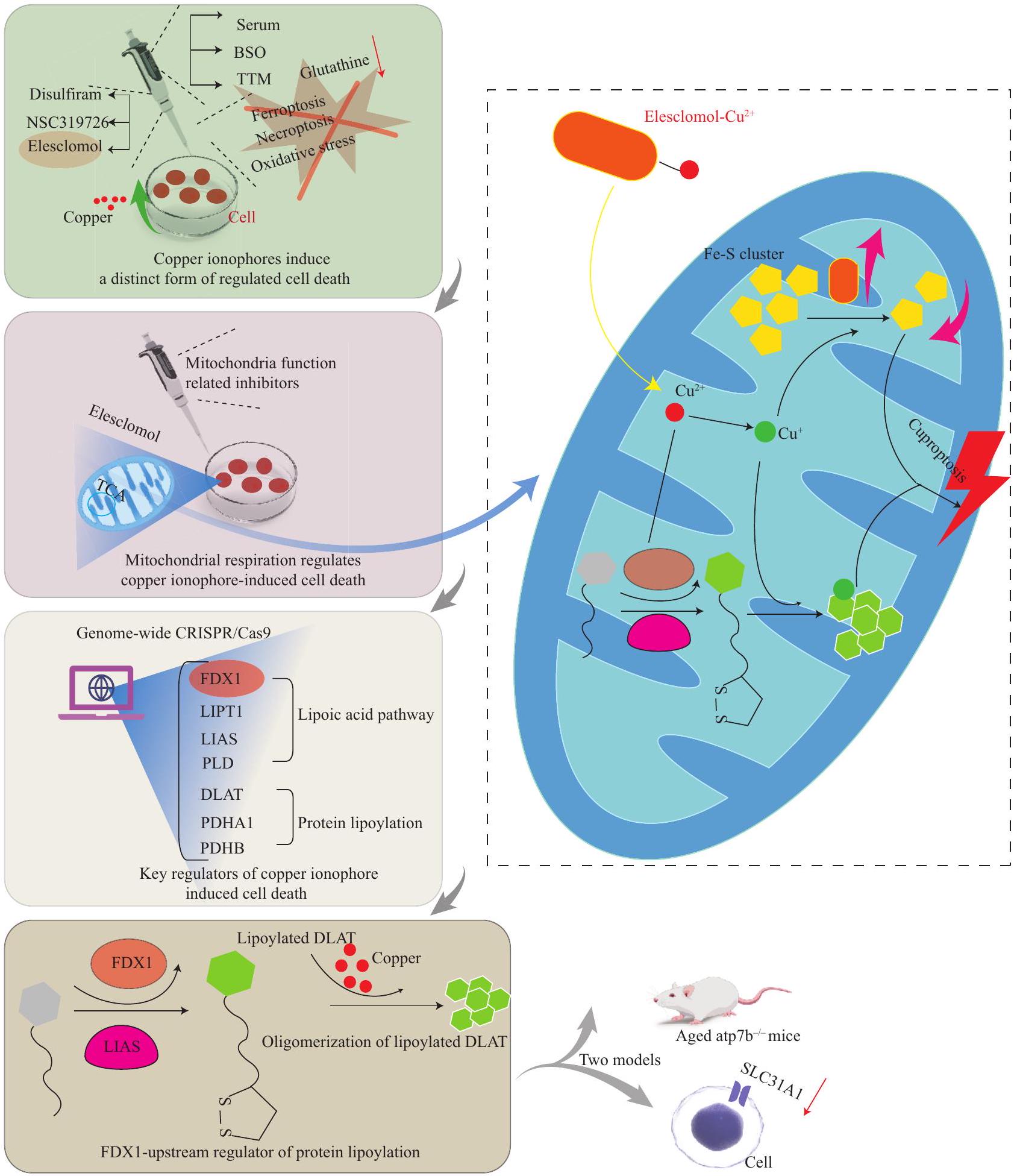
الكبدية من الموت الناتج عن النحاس في أنسجة الكبد للأفراد المصابين بمرض ويلسون، وفي الحيوانات التي تعاني من نقص ATP7B
لاستغلال التأثيرات التآزرية للكوبروبتوسيس والفيروبتوسيس ضد السرطان
3.2 الرابط بين الكوبروبتوسيس والفيروبتوسيس
انخفاض تعبير الجينات المرتبطة بالكوبروبتوسيس والفيروبتوسيس معدلات بقاء أعلى
3.3 الكوبروبتوسيس والأمراض التنكسية العصبية
, و
تم استخدام جزيئات النانو القائمة على النحاس على نطاق واسع في الحياة الإنتاجية بسبب خصائصها المتميزة.
| الوظيفة | المرض | مسار الإشارة المحتمل | المراجع | EREG |
| يمكن أن يؤثر EREG على المناعة والكوبروبتوسيس | الورم الدبقي | يؤثر EREG على تعبير PDL1/المتعلق بالكوبروبتوسيس من خلال التأثير على تعبير FDX1. | [201] | FDX1 |
| مرتبط بالتسلل المناعي | الورم الدبقي | كانت جينات الكوبروبتوسيس المرتبطة بـ FDX1 مرتبطة إيجابيًا بتعبير جينات علامات الالتهام الذاتي LIPT2 و NNAT، والتي تشارك في الترابط، قد تكون العلامة غير المعروفة Atg5 و Atg12 و BECN-1. | [200] | MAP1LC3A |
| منظم الالتهام الذاتي، MAP1LC3A المعروف باسم LC3 | مرض ويلسون | زاد النحاس من تعبير MAP1LC3A. | في خلايا ATP7B المعطلة، يكون mTOR أقل نشاطًا، وينفصل عن الليزوزومات. ينتقل عامل النسخ EB من الركيزة mTOR إلى النواة، ويتم تنشيط جينات الالتهام الذاتي لحماية الخلايا من الموت الناتج عن النحاس. | ATP7 |
| تتواجد بروتينات ATP7A و ATP7B في جهاز جولجي، وتنظم توازن النحاس. | التنكس العصبي | تتطلب سلامة آليات توازن النحاس المعتمدة على جولجي، والتي تتطلب مجمع ATP7 و COG، الحفاظ على سلامة وظيفة الميتوكوندريا. | [233] | ATP7A |
| يمكن أن تسبب طفرة ATP7A مرض منكس بسبب نقص النحاس النظامي. | مرض منكس | يزيد عدم توازن النحاس، بسبب العيوب في ATP7A، من تعبير UCHL1، الذي يتطلب بدوره، من أجل الآلية المرضية لعدم توازن النحاس. | مرض باركنسون | [231] |
| CTR1 | CTR1 هو ناقل نحاس في غشاء الخلية. | السرطان | يعزز النحاس من تكوين الأورام من خلال تنشيط مسار PDK1- Nedd41 الذي ينظم CTR1 سلبًا من خلال ubiquitination AKT والتدهور اللاحق/ Nedd41-CTR1-النحاس-PDK1-AKT. | [196] |
| الفيريتين | الفيريتين هو بروتين تخزين الحديد. | إصابة إزالة الميالين | تشكل Cuprizone النحاس بسرعة وتحرر الحديد من الفيريتين، مما يحفز أكسدة الدهون المعتمدة على الحديد وفقدان الخلايا الدبقية السريع من خلال الفيروبتوسيس. | [222] |
| MTFI | تعتبر عوامل النسخ المرتبطة بالمعادن الكلاسيكية مرتبطة ارتباطًا وثيقًا بتوازن النحاس والكوبروبتوسيس. | تحميل النحاس أو نقصه | يؤدي تحميل النحاس إلى تنشيط النسخ لـ MTF1 الذي يرتبط بـ MRE من CTR1B من خلال مسارات MTF1 و MRE المعتمدة، ويعزز نسخها وتعبيرها، ويعزز التعبير النووي لـ MTF1، الذي بدوره، يقدم النحاس، ويحافظ على توازن النحاس عندما يكون النحاس ناقصًا. | [28] |
| p53 | قد يعزز p53 الكوبروبتوسيس من خلال تثبيط التحلل السكري، وتعزيز الأيض الميتوكوندري. | السرطان | تنظم p53 FDXR الذي يشفر إنزيم اختزال الفيريدوكسين، وهو بروتين صغير مثبط للورم miPEP133، المسؤول عن نقل الإلكترونات من NADPH إلى FDX1/2، الذي يتم تشفيره بواسطة النسخة الأولية ومن ثم إلى السيتوكروم P450 لتجمعات الحديد-كبريت من miR-34a الذي يتم تفعيله بواسطة p53، وقد وُجد أنه يساهم في التكوين الحيوي (تقوم p53 بتحفيز التعبير عن
|
[198] |
| ديisulfiram | دي سولفيرام هو ناقل للنحاس، يتحد مع النحاس لتعزيز موت الخلايا. | سرطان | ديisulfiram مع
|
[256] |
| (مستمر من الصفحة السابقة) | ||||
| منظمات مسارات الإشارة أو الجزيئات المستهدفة بواسطة النحاس | وظيفة | مرض | مسار الإشارة المحتمل | المراجع |
| كوممد10 | COMMD10 هو مثبط للسرطان ومنظم لعملية استقلاب النحاس. | سرطان | COMMD10 يثبط التغذية الراجعة الإيجابية HIF1a/CP COMMD10، HIF1
|
[223] |
| NRF2 | NRF2 مسؤول عن تنظيم استجابة مضادات الأكسدة، ويلعب دورًا حاسمًا في التخفيف من الفيروبتوسيس. | سرطان | يعمل DSF/Cu على تنشيط الفسفرة بشكل كبير. يمكن أن يعزز DSF/Cu السمية الخلوية لـ p62، مما يسهل الارتباط التنافسي مع السورافينيب، ويوقف نمو الورم، سواء في المختبر أو في الجسم الحي، من خلال تثبيط مسار إشارة NRF2 وMAPK كيناز في الوقت نفسه. | [224] |
| جي بي إكس 4 | يلعب GPX4 دورًا رئيسيًا في حجب الفيروبتوز من خلال القضاء على هيدروبيروكسيدات الفوسفوليبيد. | سرطان | النحاس يحفز التحلل الماكروتلقائي لـ GPX4 لقيادة الفيروبتوزيس و TAX1BP1، وهذا يعمل كمتلقي تلقائي لـ GPX4 من خلال الارتباط المباشر ببروتين GPX4 سيستين C107 و C148. التحلل والانهيار اللاحق لـ GPX4 استجابةً لضغط النحاس. | [225] |
| HMGB1 | HMGB1، نمط جزيئي مرتبط بالضرر، يتم إفرازه بواسطة خلايا الكوبروتوز لتInitiate الالتهاب. | تسبب تراكم النحاس في نقص ATP في خلايا الكوبروتوس، حيث تنشط AGERates AMPK لتعزيز فسفرة HMGB1، مما يؤدي إلى إنتاج السيتوكينات الالتهابية المعتمدة، مما ينتج عنه زيادة في إفراز DAMP خارج الخلية. وقد انخفض بشكل كبير. | [199] | |
| GNAQ | يلعب GNAQ دورًا مهمًا في إشارات GPCR. | ميلانوما العنبية | في خلايا تحمل طفرات GNAQ، ينتج Cu-ES أنواع الأكسجين التفاعلية (ROS). بعد ذلك، يعزز هذا فسفرة YAP، ويمنع تراكمه في النواة. إن تعطيل YAP يقلل من تعبير SNAI2، مما يؤدي بدوره إلى قمع هجرة خلايا UM. | [202] |
تنشيط البلعمة الذاتية يمكن أن يحمي خلايا الكبد من الموت الناتج عن النحاس
مع الوظائف الفسيولوجية الطبيعية للأعصاب الطرفية والدماغ. علاوة على ذلك، ستكون استراتيجية العلاج “دواء مركب متعدد الأهداف” مناسبة للتحكم في مرض الزهايمر. الاستراتيجية الحالية لكيميائيي الأدوية في مكافحة مرض الزهايمر هي تصميم والتحقيق في أدوية متعددة الوظائف ذات خصائص مضادة لـ
5 الخاتمة
الوصول المفتوح
mmons.org/licenses/by/4.0/)، الذي يسمح بالاستخدام والمشاركة والتكيف والتوزيع وإعادة الإنتاج في أي وسيلة أو صيغة، طالما أنك تعطي الائتمان المناسب للمؤلفين الأصليين والمصدر، وتوفر رابطًا لرخصة المشاع الإبداعي، وتوضح ما إذا تم إجراء تغييرات. الصور أو المواد الأخرى من طرف ثالث في هذه المقالة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقال، ما لم يُشار إلى خلاف ذلك في سطر الائتمان للمواد. إذا لم تكن المادة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقال وكان استخدامك المقصود غير مسموح به بموجب اللوائح القانونية أو يتجاوز الاستخدام المسموح به، فستحتاج إلى الحصول على إذن مباشرة من صاحب حقوق الطبع والنشر. لعرض نسخة من هذه الرخصة، قم بزيارة http://creativecommons.org/licenses/by/4.0/.
توفر البيانات الداعمة
بيان تضارب المصالح
REFERENCES
2 Festa RA, Thiele DJ. Copper: an essential metal in biology. Curr Biol, 2011,21(21):R877-R883
3 Maung MT, Carlson A, Olea-Flores M, et al. The molecular and cellular basis of copper dysregulation and its relationship with human pathologies. FASEB J, 2021,35(9):e21810
4 Gromadzka G, Tarnacka B, Flaga A, et al. Copper Dyshomeostasis in Neurodegenerative Diseases-Therapeutic Implications. Int J Mol Sci, 2020,21(23):9259
5 Zhao WJ, Fan CL, Hu XM, et al. Regulated Cell Death of Retinal Ganglion Cells in Glaucoma: Molecular Insights and Therapeutic Potentials. Cell Mol Neurobiol, 2023,43(7):3161-3178
6 Zhang Q, Hu XM, Zhao WJ, et al. Targeting Necroptosis: A Novel Therapeutic Option for Retinal Degenerative Diseases. Int J Biol Sci, 2023,19(2):658-674
7 Wan H, Yan YD, Hu XM, et al. Inhibition of mitochondrial VDAC1 oligomerization alleviates apoptosis and necroptosis of retinal neurons following OGD/R injury. Ann Anat, 2023,247:152049
8 Yan WT, Zhao WJ, Hu XM, et al. PANoptosis-like cell death in ischemia/reperfusion injury of retinal neurons. Neural Regen Res, 2023,18(2):357-363
9 Tang D, Kang R, Berghe TV, et al. The molecular machinery of regulated cell death. Cell Res, 2019, 29(5):347-364
10 Yang YD, Li ZX, Hu XM, et al. Insight into Crosstalk Between Mitophagy and Apoptosis/Necroptosis: Mechanisms and Clinical Applications in Ischemic
11 Yan WT, Yang YD, Hu XM, et al. Do pyroptosis, apoptosis, and necroptosis (PANoptosis) exist in cerebral ischemia? Evidence from cell and rodent studies. Neural Regen Res, 2022,17(8):1761-1768
12 Chen XY, Dai YH, Wan XX, et al. ZBP1-Mediated Necroptosis: Mechanisms and Therapeutic Implications. Molecules, 2022,28(1):52
13 Chen J, Wang Y, Li M, et al. Netrin-1 Alleviates Early Brain Injury by Regulating Ferroptosis via the PPAR
14 Zhang Q, Wan XX, Hu XM, et al. Targeting Programmed Cell Death to Improve Stem Cell Therapy: Implications for Treating Diabetes and Diabetes-Related Diseases. Front Cell Dev Biol, 2021,9:809656
15 Hu XM, Zhang Q, Zhou RX, et al. Programmed cell death in stem cell-based therapy: Mechanisms and clinical applications. World J Stem Cells, 2020,12(8):787-802
16 Mou YH, Wang J, Wu JC, et al. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol, 2019,29,12(1):34
17 Tsvetkov P, Coy S, Petrova B, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science, 2022,375(6586):1254-1261
18 An Y, Li S, Huang X, et al. The Role of Copper Homeostasis in Brain Disease. Int J Mol Sci, 2022,23 (22):13850
20 Cobine PA, Brady DC. Cuproptosis: Cellular and molecular mechanisms underlying copper-induced cell death. Mol Cell, 2022,82(10):1786-1787
21 Arredondo M, Núñez MT. Iron and copper metabolism. Mol Aspects Med, 2005,26(4-5):313-327
22 Nutrition classics. The Journal of Biological Chemistry, Vol. LXXV II, 1928: Iron in nutrition. VII. Copper as a supplement to iron for hemoglobin building in the rat. By E.B. Hart, H. Steenbock, J. Waddell, and C.A. Elvehjem. Nutr Rev, 1987,45(6):181-183
23 Khoshbin K, Camilleri M. Effects of dietary components on intestinal permeability in health and disease. Am J Physiol Gastrointest Liver Physiol, 2020,319(5):G589-G608
24 Nose Y, Wood LK, Kim BE, et al. Ctr1 is an apical copper transporter in mammalian intestinal epithelial cells in vivo that is controlled at the level of protein stability. J Biol Chem, 2010,285(42):32385-32392
25 Lane DJR, Bae DH, Merlot AM, et al. Duodenal cytochrome b (DCYTB) in iron metabolism: an update on function and regulation. Nutrients, 2015,7(4):22742296
26 Zimnicka AM, Maryon EB, Kaplan JH. Human copper transporter hCTR1 mediates basolateral uptake of copper into enterocytes: implications for copper homeostasis. J Biol Chem, 2007,282(36):26471-26480
27 Schuchardt JP, Hahn A. Intestinal Absorption and Factors Influencing Bioavailability of Magnesium-An Update. Curr Nutr Food Sci, 2017,13(4):260-278
28 Lutsenko S. Dynamic and cell-specific transport
networks for intracellular copper ions. J Cell Sci, 2021, 134(21):jcs240523
29 Nyasae L, Bustos R, Braiterman L, et al. Dynamics of endogenous ATP7A (Menkes protein) in intestinal epithelial cells: copper-dependent redistribution between two intracellular sites. Am J Physiol Gastrointest Liver Physiol, 2007,292(4):G1181-G1194
30 Liang ZD, Tsai WB, Lee MY, et al. Specificity protein 1 (sp1) oscillation is involved in copper homeostasis maintenance by regulating human high-affinity copper transporter 1 expression. Mol Pharmacol, 2012,81(3):455-464
31 Zhao J, Guo S, Schrodi SJ, et al. Cuproptosis and cuproptosis-related genes in rheumatoid arthritis: Implication, prospects, and perspectives. Front Immunol, 2022,13:930278
32 Linder MC. Ceruloplasmin and other copper binding components of blood plasma and their functions: an update. Metallomics, 2016,8(9):887-905
33 Xie J, Yang Y, Gao Y, et al. Cuproptosis: mechanisms and links with cancers. Mol Cancer, 2023,22(1):46
34 Casareno RL, Waggoner D, Gitlin JD. The copper chaperone CCS directly interacts with copper/zinc superoxide dismutase. J Biol Chem, 1998,273(37):23625-23628
35 Punter FA, Adams DL, Glerum DM. Characterization and localization of human COX17, a gene involved in mitochondrial copper transport. Hum Genet, 2000,107(1):69-74
36 Pierson H, Muchenditsi A, Kim B-E, et al. The Function of ATPase Copper Transporter ATP7B in Intestine. Gastroenterology, 2018,154(1):168-180.e165
37 Telianidis J, Hung YH, Materia S, et al. Role of the P-Type ATPases, ATP7A and ATP7B in brain copper homeostasis. Front Aging Neurosci, 2013,5:44
38 Liu H, Lai W, Liu X, et al. Exposure to copper oxide nanoparticles triggers oxidative stress and endoplasmic reticulum (ER)-stress induced toxicology and apoptosis in male rat liver and BRL-3A cell. J Hazard Mater, 2021,401:123349
39 van Rensburg MJ, van Rooy M, Bester MJ, et al. Oxidative and haemostatic effects of copper, manganese and mercury, alone and in combination at physiologically relevant levels: An ex vivo study. Hum Exp Toxicol, 2019,38(4):419-433
40 Comes G, Fernandez-Gayol O, Molinero A, et al. Mouse metallothionein-1 and metallothionein-2 are not biologically interchangeable in an animal model of multiple sclerosis, EAE. Metallomics, 2019,11(2):327337
41 Leary SC, Winge DR, Cobine PA. “Pulling the plug” on cellular copper: the role of mitochondria in copper export. Biochim Biophys Acta, 2009,1793(1):146-153
42 Horn D, Barrientos A. Mitochondrial copper metabolism and delivery to cytochrome c oxidase. IUBMB Life, 2008,60(7):421-429
43 Pacheu-Grau D, Wasilewski M, Oeljeklaus S, et al. COA6 Facilitates Cytochrome c Oxidase Biogenesis as Thiol-reductase for Copper Metallochaperones in Mitochondria. J Mol Biol, 2020,432(7):2067-2079
44 Robinson NJ, Winge DR. Copper metallochaperones. Annu Rev Biochem, 2010,79:537-562
46 Leary SC. Redox regulation of SCO protein function: controlling copper at a mitochondrial crossroad. Antioxid Redox Signal, 2010,13(9):1403-1416
47 Morgada MN, Abriata LA, Cefaro C, et al. Loop recognition and copper-mediated disulfide reduction underpin meta 1 site assembly of CuA in human cytochrome oxidase. Proc Natl Acad Sci U S A, 2015, 112(38):11771-11776
48 Zhou J, Li XY, Liu YJ, et al. Full-coverage regulations of autophagy by ROS: from induction to maturation. Autophagy, 2022,18(6):1240-1255
49 Bompiani KM, Tsai CY, Achatz FP, et al. Copper transporters and chaperones CTR1, CTR2, ATOX1, and CCS as determinants of cisplatin sensitivity. Metallomics, 2016,8(9):951-962
50 Skopp A, Boyd SD, Ullrich MS, et al. Copper-zinc superoxide dismutase (Sod1) activation terminates interact ion between its copper chaperone (Ccs) and the cytosolic metal-binding domain of the copper importer Ctr1. Biometals, 2019,32(4):695-705
51 Gupta A, Lutsenko S. Human copper transporters: mechanism, role in human diseases and therapeutic potential. Future Med Chem, 2009,1(6):1125-1142
52 Hamza I, Faisst A, Prohaska J, et al. The metallochaperone Atox1 plays a critical role in perinatal copper homeostasis. Proc Natl Acad Sci USA, 2001,98(12):6848-6852
53 Itoh S, Kim HW, Nakagawa O, et al. Novel role of antioxidant-1(Atox1) as a copper-dependent transcription factor involved in cell proliferation. J Biol Chem, 2008,283(14):9157-9167
54 Mattie MD, McElwee MK, Freedman JH. Mechanism of copper-activated transcription: activation of AP-1, and the JNK/SAPK and p38 signal transduction pathways. J Mol Biol, 2008,383(5):1008-1018
55 Satake H, Suzuki K, Aoki T, et al. Cupric ion blocks NF kappa B activation through inhibiting the signalinduced phosphorylation of I kappa B alpha. Biochem Biophys Res Commun, 1995,216(2):568-573
56 Chen L, Li N, Zhang M, et al. APEX2-based Proximity Labeling of Atox1 Identifies CRIP2 as a Nuclear Copperbinding Protein that Regulates Autophagy Activation. Angew Chem Int Ed Engl, 2021,60(48):25346-25355
57 An Y, Li S, Huang X, et al. The Role of Copper Homeostasis in Brain Disease. Int J Mol Sci, 2022, 23(22):13850
58 Tong X, Tang R, Xiao M, et al. Targeting cell death pathways for cancer therapy: recent developments in necroptosis, pyroptosis, ferroptosis, and cuproptosis research. J Hematol Oncol, 2022,15(1):174
59 Tümer Z, Møller LB. Menkes disease. Eur J Hum Genet, 2010,18(5):511-518
60 Członkowska A, Litwin T, Dusek P, et al. Wilson disease. Nat Rev Dis Primers, 2018,4(1):21
61 Bush AI. Metals and neuroscience. Curr Opin Chem Biol, 2000,4(2):184-191
62 DiDonato M, Narindrasorasak S, Forbes JR, et al. Expression, purification, and metal binding properties
of the N-terminal domain from the wilson disease putative copper-transporting ATPase (ATP7B). J Biol Chem, 1997,272(52):33279-33282
63 Lönnerdal B. Intestinal regulation of copper homeostasis: a developmental perspective. Am J Clin Nutr, 2008,88(3):846S-850S
64 Aisen P, Enns C, Wessling-Resnick M. Chemistry and biology of eukaryotic iron metabolism. Int J Biochem Cell Biol, 2001,33(10):940-959
65 Akil M, Schwartz JA, Dutchak D, et al. The psychiatric presentations of Wilson’s disease. J Neuropsychiatry Clin Neurosci, 1991,3(4):377-382
66 Mairet-Coello G, Tury A, Esnard-Feve A, et al. FADlinked sulfhydryl oxidase QSOX: topographic, cellular, and subcellular immunolocalization in adult rat central nervous system. J Comp Neurol, 2004,473(3):334-363
67 Trombley PQ, Horning MS, Blakemore LJ. Interactions between carnosine and zinc and copper: implications for neuromodulation and neuroprotection. Biochemistry (Mosc), 2000,65(7):807-816
68 Johnson KA, Conn PJ, Niswender CM. Glutamate receptors as therapeutic targets for Parkinson’s disease. CNS Neurol Disord Drug Targets, 2009,8(6):475-491
69 D’Ambrosi N, Rossi L. Copper at synapse: Release, binding and modulation of neurotransmission. Neurochem Int, 2015,90:36-45
70 Moriya M, Ho YH, Grana A, et al. Copper is taken up efficiently from albumin and alpha2-macroglobulin by cultured human cells by more than one mechanism. Am J Physiol Cell Physiol, 2008,295(3):C708-C721
71 Montes S, Rivera-Mancia S, Diaz-Ruiz A, et al. Copper and copper proteins in Parkinson’s disease. Oxid Med Cell Longev, 2014,2014:147251
72 Wu W, Ruan X, Gu C, et al. Blood-cerebrospinal fluid barrier permeability of metals/metalloids and its determinants in pediatric patients. Ecotoxicol Environ Saf, 2023,266:115599
73 Scheiber IF, Mercer JFB, Dringen R. Metabolism and functions of copper in brain. Prog Neurobiol, 2014,116:33-57
74 Dringen R, Scheiber IF, Mercer JFB. Copper metabolism of astrocytes. Front Aging Neurosci, 2013,5:9
75 Howell SB, Safaei R, Larson CA, et al. Copper transporters and the cellular pharmacology of the platinum-containing cancer drugs. Mol Pharmacol, 2010,77(6):887-894
76 Garza-Lombó C, Posadas Y, Quintanar L, et al. Neurotoxicity Linked to Dysfunctional Metal Ion Homeostasis and Xenobiotic Metal Exposure: Redox Signaling and Oxidative Stress. Antioxid Redox Signal, 2018,28(18):1669-1703
77 Varela-Nallar L, Toledo EM, Chacón MA, et al. The functional links between prion protein and copper. Biol Res, 2006,39(1):39-44
78 Stuerenburg HJ. CSF copper concentrations, bloodbrain barrier function, and coeruloplasmin synthesis during the treatment of Wilson’s disease. J Neural Transm (Vienna), 2000,107(3):321-329
79 Choi BS, Zheng W. Copper transport to the brain by the blood-brain barrier and blood-CSF barrier. Brain Res, 2009,1248:14-21
80 Kaler SG. ATP7A-related copper transport diseases-
emerging concepts and future trends. Nat Rev Neurol, 2011,7(1):15-29
81 Nishihara E, Furuyama T, Yamashita S, et al. Expression of copper trafficking genes in the mouse brain. Neuroreport, 1998,9(14):3259-3263
82 Barber RG, Grenier ZA, Burkhead JL. Copper Toxicity Is Not Just Oxidative Damage: Zinc Systems and Insight from Wilson Disease. Biomedicines, 2021,9(3):316
83 Mercer JF, Ambrosini L, Horton S, et al. Animal models of Menkes disease. Adv Exp Med Biol, 1999,448:97108
84 Menkes JH, Alter M, Steigleder GK, et al. A sex-linked recessive disorder with retardation of growth, peculiar hair, and focal cerebral and cerebellar degeneration. Pediatrics, 1962,29:764-779
85 Møller LB, Mogensen M, Horn N. Molecular diagnosis of Menkes disease: genotype-phenotype correlation. Biochimie, 2009,91(10):1273-1277
86 Shim H, Harris ZL. Genetic defects in copper metabolism. J Nutr, 2003,133(5 Suppl 1):1527S-1531S
87 Kaler SG, Gahl WA, Berry SA, et al. Predictive value of plasma catecholamine levels in neonatal detection of Menkes disease. J Inherit Metab Dis, 1993,16(5):907908
88 Szauter KM, Cao T, Boyd CD, et al. Lysyl oxidase in development, aging and pathologies of the skin. Pathol Biol (Paris), 2005,53(7):448-456
89 Royce PM, Camakaris J, Danks DM. Reduced lysyl oxidase activity in skin fibroblasts from patients with Menkes’ syndrome. Biochem J, 1980,192(2):579-586
90 Sarkar B, Lingertat Walsh K, Clarke JT. Copper-histidine therapy for Menkes disease. J Pediatr, 1993,123(5):828830
91 George DH, Casey RE. Menkes disease after copper histidine replacement therapy: case report. Pediatr Dev Pathol, 2001,4(3):281-288
92 Kim JH, Lee BH, Kim YM, et al. Novel mutations and clinical outcomes of copper-histidine therapy in Menkes disease patients. Metab Brain Dis, 2015,30(1):75-81
93 Cumings JN. The copper and iron content of brain and liver in the normal and in hepato-lenticular degeneration. Brain, 1948,71(Pt. 4):410-415
94 Przybyłkowski A, Gromadzka G, Chabik G, et al. Liver cirrhosis in patients newly diagnosed with neurological phenotype of Wilson’s disease. Funct Neurol, 2014,29 (1):23-29
96 Horoupian DS, Sternlieb I, Scheinberg IH. Neuropathological findings in penicillamine-treated patients with Wilson’s disease. Clin Neuropathol, 1988,7(2):62-67
97 Bertrand E, Lewandowska E, Szpak GM, et al. Neuropathological analysis of pathological forms of astroglia in Wilson’s disease. Folia Neuropathol, 2001,39(2):73-79
98 Langwińska-Wośko E, Litwin T, Szulborski K, et al. Optical coherence tomography and electrophysiology of retinal and visual pathways in Wilson’s disease. Metab Brain Dis, 2016,31(2):405-415
100 European Association for Study of L. EASL Clinical Practice Guidelines: Wilson’s disease. J Hepatol, 2012,56(3):671-685
101 Barnes DE, Yaffe K. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol, 2011,10(9):819-828
102 Morris GP, Clark IA, Vissel B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol Commun, 2014,2:135
103 Zhang YL, Wang J, Zhang ZN, et al. The relationship between amyloid-beta and brain capillary endothelial cells in Alzheimer’s disease. Neural Regen Res, 2022,17(11):2355-2363
104 Gaggelli E, Kozlowski H, Valensin D, et al. Copper homeostasis and neurodegenerative disorders (Alzheimer’s, prion, and Parkinson’s diseases and amyotrophic lateral sclerosis). Chem Rev, 2006,106 (6):1995-2044
106 Newcombe EA, Camats-Perna J, Silva ML, et al. Inflammation: the link between comorbidities, genetics, and Alzheimer’s disease. J Neuroinflammation, 2018, 15(1):276
107 Lovell MA, Robertson JD, Teesdale WJ, et al. Copper, iron and zinc in Alzheimer’s disease senile plaques. J Neurol Sci, 1998,158(1):47-52
108 Su XY, Wu WH, Huang ZP, et al. Hydrogen peroxide can be generated by tau in the presence of
109 Yu J, Luo X, Xu H, et al. Identification of the key molecules involved in chronic copper exposureaggravated memory impairment in transgenic mice of Alzheimer’s disease using proteomic analysis. J Alzheimers Dis, 2015,44(2):455-469
110 James SA, Volitakis I, Adlard PA, et al. Elevated labile Cu is associated with oxidative pathology in Alzheimer disease. Free Radic Biol Med, 2012,52(2):298-302
111 Yu F, Gong P, Hu Z, et al. Cu ( II ) enhances the effect of Alzheimer’s amyloid-
112 Lu J , Wu Dm, Zheng Yi, et al. Trace amounts of copper exacerbate beta amyloid-induced neurotoxicity in the cholesterol-fed mice through TNF-mediated inflammatory pathway. Brain Behav Immun, 2009,23 (2):193-203
114 Krasemann S, Madore C, Cialic R, et al. The TREM2APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity, 2017,47(3):566-581.e9
115 Chen Y, Chen J, Wei H, et al. Akkermansia muciniphila-Nlrp3 is involved in the neuroprotection
of phosphoglycerate mutase 5 deficiency in traumatic brain injury mice. Front Immunol, 2023,14:1172710
116 Zhou Z, Shang L, Zhang Q, et al. DTX3L induced NLRP3 ubiquitination inhibit R28 cell pyroptosis in OGD/R injury. Biochim Biophys Acta Mol Cell Res, 2023,1870(3):119433
117 He YF, Hu XM, Khan MA, et al. HSF1 Alleviates Brain Injury by Inhibiting NLRP3-Induced Pyroptosis in a Sepsis Model. Mediators Inflamm, 2023,2023:2252255
118 Huang Y, Wang S, Huang F, et al. c-FLIP regulates pyroptosis in retinal neurons following oxygen-glucose deprivation/recovery via a GSDMD-mediated pathway. Ann Anat, 2021,235:151672
119 Liao LS, Lu S, Yan WT, et al. The Role of HSP90
120 Yan WT, Lu S, Yang YD, et al. Research trends, hot spots and prospects for necroptosis in the field of neuroscience. Neural Regen Res, 2021,16(8):16281637
121 Hu XM, Li ZX, Lin RH, et al. Guidelines for Regulated Cell Death Assays: A Systematic Summary, A Categorical Comparison, A Prospective. Front Cell Dev Biol, 2021,9:634690
122 Wakhloo D, Oberhauser J, Madira A, et al. From cradle to grave: neurogenesis, neuroregeneration and neurodegeneration in Alzheimer’s and Parkinson’s diseases. Neural Regen Res, 2022,17(12):2606-2614
123 Banjara M, Ghosh C. Sterile Neuroinflammation and Strategies for Therapeutic Intervention. Int J Inflam, 2017,2017:8385961
124 Scheiber IF, Dringen R. Astrocyte functions in the copper homeostasis of the brain. Neurochem Int, 2013,62(5):556-565
125 Pal A, Vasishta Rk, Prasad R. Hepatic and hippocampus iron status is not altered in response to increased serum ceruloplasmin and serum “free” copper in Wistar rat model for non-Wilsonian brain copper toxicosis. Biol Trace Elem Res, 2013,154(3):403-411
126 Qian Y, Zheng Y, Taylor R, et al. Involvement of the molecular chaperone Hspa5 in copper homeostasis in astrocytes. Brain Res, 2012,1447:9-19
127 Pike CJ, Cummings BJ, Monzavi R, et al. Beta-amyloid-induced changes in cultured astrocytes parallel reactive astrocytosis associated with senile plaques in Alzheimer’s disease. Neuroscience, 1994,63(2):517531
128 DeWitt DA, Perry G, Cohen M, et al. Astrocytes regulate microglial phagocytosis of senile plaque cores of Alzheimer’s disease. Exp Neurol, 1998,149(2):329340
129 Choo XY, Liddell JR, Huuskonen MT, et al. Cu
130 Mandal PK, Saharan S, Tripathi M, et al. Brain glutathione levels–a novel biomarker for mild cognitive impairment and Alzheimer’s disease. Biol psychiatry, 2015,78(10):702-710
131 Uttamsingh V, Keller DA, Anders MW. Acylase I-catalyzed deacetylation of N-acetyl-L-cysteine and S-alkyl-N -acetyl-L-cysteines. Chem Res Toxicol,
132 Ashraf A, So PW. Spotlight on Ferroptosis: IronDependent Cell Death in Alzheimer’s Disease. Front Aging Neurosci, 2020,12:196
133 Derry PJ, Hegde ML, Jackson GR, et al. Revisiting the intersection of amyloid, pathologically modified tau and iron in Alzheimer’s disease from a ferroptosis perspective. Prog Neurobiol, 2020,184:101716
134 Bush AI. Drug development based on the metals hypothesis of Alzheimer’s disease. J Alzheimers Dis, 2008,15(2):223-240
135 Walker FO. Huntington’s disease. Lancet, 2007,369 (9557):218-228
137 Dexter DT, Carayon A, Javoy-Agid F, et al. Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain, 1991,114(Pt4):19531975
138 Tabrizi SJ, Ghosh R, Leavitt BR. Huntingtin Lowering Strategies for Disease Modification in Huntington’s Disease. Neuron, 2019,101(5):801-819
139 Xiao G, Fan Q, Wang X, et al. Huntington disease arises from a combinatory toxicity of polyglutamine and copper binding. Proc Natl Acad Sci U S A, 2013,110(37):14995-15000
140 Fox JH, Kama JA, Lieberman G, et al. Mechanisms of copper ion mediated Huntington’s disease progression. PLoS One, 2007,2(3):e334
141 Pamp K, Bramey T, Kirsch M, et al. NAD(H) enhances the Cu ( II )-mediated inactivation of lactate dehydrogenase by increasing the accessibility of sulfhydryl groups. Free Radic Res, 2005,39(1):31-40
142 Harms L, Meierkord H, Timm G, et al. Decreased N -acetyl-aspartate/choline ratio and increased lactate in the frontal lobe of patients with Huntington’s disease: a proton magnetic resonance spectroscopy study. J Neurol Neurosurg Psychiatry, 1997,62(1):27-30
143 Sheline CT, Choi DW.
144 Cherny RA, Ayton S, Finkelstein DI, et al. PBT2 Reduces Toxicity in a C. elegans Model of polyQ Aggregation and Extends Lifespan, Reduces Striatal Atrophy and Improves Motor Performance in the R6/2 Mouse Model of Huntington’s Disease. J Huntingtons Dis, 2012,1(2):211-219
145 Boillée S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron, 2006,52(1):39-59
146 Swinnen B, Robberecht W. The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol, 2014,10(11):661-670
147 Feldman EL, Goutman SA, Petri S, et al. Amyotrophic lateral sclerosis. Lancet, 2022,400(10360):1363-1380
148 Hardiman O, Al-Chalabi A, Chio A, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers, 2017,3:17071
149 Gil-Bea FJ, Aldanondo G, Lasa-Fernández H, et al. Insights into the mechanisms of copper dyshomeostasis in amyotrophic lateral sclerosis. Expert Rev Mol Med, 2017,19:e7
151 Williams JR, Trias E, Beilby PR, et al. Copper delivery to the CNS by CuATSM effectively treats motor neuron disease in SOD(G93A) mice co-expressing the Copper-Chaperone-for-SOD. Neurobiol Dis, 2016,89:1-9
152 Cabreiro F, Ackerman D, Doonan R, et al. Increased life span from overexpression of superoxide dismutase in Caenorhabditis elegans is not caused by decreased oxidative damage. Free Radic Biol Med, 2011,51(8):1575-1582
153 Enge TG, Ecroyd H, Jolley DF, et al. Longitudinal assessment of metal concentrations and copper isotope ratios in the G93A SOD1 mouse model of amyotrophic lateral sclerosis. Metallomics, 2017,9(2):161-174
154 Roos PM, Vesterberg O, Syversen T, et al. Metal concentrations in cerebrospinal fluid and blood plasma from patients with amyotrophic lateral sclerosis. Biol Trace Elem Res, 2013,151(2):159-170
155 Hilton JB, Kysenius K, White AR, et al. The accumulation of enzymatically inactive cuproenzymes is a CNS-specific phenomenon of the SOD1G37R mouse model of ALS and can be restored by overexpressing the human copper transporter hCTR1. Exp Neurol, 2018,307:118-128
156 Tokuda E, Okawa E, Ono Si. Dysregulation of intracellular copper trafficking pathway in a mouse model of mutant copper/zinc superoxide dismutaselinked familial amyotrophic lateral sclerosis. J Neurochem, 2009,111(1):181-191
157 Hottinger AF, Fine EG, Gurney ME, et al. The copper chelator d-penicillamine delays onset of disease and extend s survival in a transgenic mouse model of familial amyotrophic lateral sclerosis. Eur J Neurosci, 1997,9(7):1548-1551
158 Tokuda E, Ono Si, Ishige K, et al. Ammonium tetrathiomolybdate delays onset, prolongs survival, and slows progression of disease in a mouse model for amyotrophic lateral sclerosis. Exp Neurol, 2008,213(1):122-128
159 Roberts BR, Lim NKH, McAllum EJ, et al. Oral treatment with Cu ( II )(atsm) increases mutant SOD1 in vivo but protects motor neurons and improves the phenotype of a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci, 2014,34(23):8021-8031
160 Hilton JB, Mercer SW, Lim NKH, et al.
161 Tokuda E, Okawa E, Watanabe S, et al. Dysregulation of intracellular copper homeostasis is common to transgenic mice expressing human mutant superoxide dismutase-1s regardless of their copper-binding abilities. Neurobiol Dis, 2013,54:308-319
162 DeLazzari F, Bubacco L, Whitworth AJ, et al. Superoxide Radical Dismutation as New Therapeutic Strategy in Parkinson’s Disease. Aging Dis, 2018,9(4):716-728
163 Reich SG, Savitt JM. Parkinson’s Disease. Med Clin
164 Karimi-Moghadam A, Charsouei S, Bell B, et al. Parkinson Disease from Mendelian Forms to Genetic Susceptibility: New Molecular Insights into the Neurodegeneration Process. Cell Mol Neurobiol, 2018,38(6):1153-1178
165 Tolosa E, Garrido A, Scholz SW, et al. Challenges in the diagnosis of Parkinson’s disease. Lancet Neurol, 2021,20(5):385-397
166 Youdim MB, Ben-Shachar D, Riederer P. Is Parkinson’s disease a progressive siderosis of substantia nigra resulting in iron and melanin induced neurodegeneration? Acta Neurol Scand Suppl, 1989,126:47-54
167 Riederer P, Sofic E, Rausch WD, et al. Transition metals, ferritin, glutathione, and ascorbic acid in parkinsonian brains. J Neurochem, 1989,52(2):515-520
168 Perry TL, Godin DV, Hansen S. Parkinson’s disease: a disorder due to nigral glutathione deficiency? Neurosci Lett, 1982,33(3):305-310
169 Jenner P. Oxidative stress in Parkinson’s disease. Ann Neurol, 2003,53:S26-S36
170 Bisaglia M, Mammi S, Bubacco L. Structural insights on physiological functions and pathological effects of alpha-synuclein. FASEB J, 2009,23(2):329-340
171 Binolfi A, Rasia RM, Bertoncini CW, et al. Interaction of alpha-synuclein with divalent metal ions reveals key differences: a link between structure, binding specificity and fibrillation enhancement. J Am Chem Soc, 2006,128(30):9893-9901
172 Dudzik CG, Walter ED, Millhauser GL. Coordination features and affinity of the
174 Anderson JP, Walker DE, Goldstein JM, et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem, 2006,281(40):2973929752
175 Dikiy I, Eliezer D. N-terminal acetylation stabilizes N-terminal helicity in lipid- and micelle-bound
176 Mason RJ, Paskins AR, Dalton CF, et al. Copper Binding and Subsequent Aggregation of
177 Bisaglia M, Bubacco L. Copper Ions and Parkinson’s Disease: Why Is Homeostasis So Relevant? Biomolecules, 2020,10(2):195
178 Uitti RJ, Rajput AH, Rozdilsky B, et al. Regional metal concentrations in Parkinson’s disease, other chronic neurological diseases, and control brains. Can J Neurol Sci, 1989,16(3):310-314
179 de Freitas LV, da Silva CCP, Ellena J, et al. Structural and vibrational study of 8-hydroxyquinoline-2carboxaldehyde isonicotinoyl hydrazine-a potential metal-protein attenuating compound (MPAC) for the treatment of Alzheimer’s disease. Spectrochim Acta A
180 McAllum EJ, Lim NKH, Hickey JL, et al. Therapeutic effects of
181 Wang M, Wan H, Wang S, et al. RSK3 mediates necroptosis by regulating phosphorylation of RIP3 in rat retinal ganglion cells. J Anat, 2020,237(1):29-47
182 Guo LM, Wang Z, Li SP, et al. RIP3/MLKL-mediated neuronal necroptosis induced by methamphetamine at
183 Wang Z, Guo LM, Wang Y, et al. Inhibition of HSP90
184 Chen J, Li M, Liu Z, et al. Molecular mechanisms of neuronal death in brain injury after subarachnoid hemorrhage. Front Cell Neurosci, 2022,16:1025708
185 Hunsaker EW, Franz KJ. Emerging Opportunities To Manipulate Metal Trafficking for Therapeutic Benefit. Inorg Chem, 2019,58(20):13528-13545
186 Zheng P, Zhou C, Lu L, et al. Elesclomol: a copper ionophore targeting mitochondrial metabolism for cancer therapy. J Exp Clin Cancer Res, 2022,41(1):271
187 Hasinoff BB, Yadav AA, Patel D, et al. The cytotoxicity of the anticancer drug elesclomol is due to oxidative stress indirectly mediated through its complex with Cu ( II ). J Inorg Biochem, 2014, 137: 22-30
188 Yang W, Wang Y, Huang Y, et al. 4-Octyl itaconate inhibits aerobic glycolysis by targeting GAPDH to promote cuproptosis in colorectal cancer. Biomed Pharmacother, 2023,159:114301
189 Tsvetkov P, Detappe A, Cai K, et al. Mitochondrial metabolism promotes adaptation to proteotoxic stress. Nat Chem Biol, 2019,15(7):681-689
190 Solmonson A, DeBerardinis RJ. Lipoic acid metabolism and mitochondrial redox regulation. J Biol Chem, 2018,293(20):7522-7530
191 Lutsenko S. Atp7b-/- mice as a model for studies of Wilson’s disease. Biochem Soc Trans, 2008,36(Pt 6):1233-1238
193 Xue J, Yang G, Ding H, et al. Role of NSC319726 in ovarian cancer based on the bioinformatics analyses. Onco Targets Ther, 2015,8:3757-3765
194 Shimada K, Reznik E, Stokes ME, et al. CopperBinding Small Molecule Induces Oxidative Stress and Cell-Cycle Arrest in Glioblastoma-Patient-Derived Cells. Cell Chem Biol, 2018,25(5):585-594.e587
195 Polishchuk EV, Merolla A, Lichtmannegger J, et al. Activation of Autophagy, Observed in Liver Tissues From Patients With Wilson Disease and From ATP7B-Deficient Animals, Protects Hepatocytes From Copper-Induced Apoptosis. Gastroenterology, 2019,156(4):1173-1189.e1175
196 Guo J, Cheng J, Zheng N, et al. Copper Promotes Tumorigenesis by Activating the PDK1-AKT Oncogenic Pathway in a Copper Transporter 1 Dependent Manner. Adv Sci (Weinh), 2021,8(18):e2004303
197 Chen GH, Lv W, Xu YH, et al. Functional analysis of
198 Hu W, Zhang C, Wu R, et al. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci U S A, 2010,107(16):7455-7460
199 Liu J, Liu Y, Wang Y, et al. HMGB1 is a mediator of cuproptosis-related sterile inflammation. Front Cell Dev Biol, 2022,10:996307
200 Lu H, Zhou L, Zhang B, et al. Cuproptosis key gene FDX1 is a prognostic biomarker and associated with immune infiltration in glioma. Front Med (Lausanne), 2022,9:939776
201 Zhou Y, Xiao D, Jiang X, et al. EREG is the core oncoimmunological biomarker of cuproptosis and mediates the cross-talk between VEGF and CD99 signaling in glioblastoma. J Transl Med, 2023,21(1):28
202 Li Y, Yang J, Zhang Q, et al. Copper ionophore elesclomol selectively targets GNAQ/11-mutant uveal melanoma. Oncogene, 2022,41(27):3539-3553
203 Lv H, Liu X, Zeng X, et al. Comprehensive Analysis of Cuproptosis-Related Genes in Immune Infiltration and Prognosis in Melanoma. Front Pharmacol, 2022,13:930041
204 Xu S, Liu D, Chang T, et al. Cuproptosis-Associated lncRNA Establishes New Prognostic Profile and Predicts Immunotherapy Response in Clear Cell Renal Cell Carcinoma. Front Genet, 2022,13:938259
205 Zhang Z, Zeng X, Wu Y, et al. Cuproptosis-Related Risk Score Predicts Prognosis and Characterizes the Tumor Microenvironment in Hepatocellular Carcinoma. Front Immunol, 2022,13:925618
206 Yan C, Niu Y, Ma L, et al. System analysis based on the cuproptosis-related genes identifies LIPT 1 as a novel therapy target for liver hepatocellular carcinoma. J Transl Med, 2022,20(1):452
207 Bao JH, Lu WC, Duan H, et al. Identification of a novel cuproptosis-related gene signature and integrative analyses in patients with lower-grade gliomas. Front Immunol, 2022,13:933973
208 LiuH, Tang T. Pan-cancer genetic analysis of cuproptosis and copper metabolism-related gene set. Front Oncol, 2022,12:952290
209 Guo B, Yang F, Zhang L, et al. Cuproptosis Induced by ROS Responsive Nanoparticles with Elesclomol and Copper Combined with
210 Xu Y, Liu SY, Zeng L, et al. An Enzyme-Engineered Nonporous Copper(I) Coordination Polymer Nanoplat form for Cuproptosis-Based Synergistic Cancer Therapy. Adv Mater, 2022,34(43):e2204733
211 Li T, Wang D, Meng M, et al. Copper-Coordinated Covalent Organic Framework Produced a Robust Fenton-Like Effect Inducing Immunogenic Cell Death of Tumors. Macromol Rapid Commun, 2023,44(11):e2200929
212 Collins JF, Prohaska JR, Knutson MD. Metabolic crossroads of iron and copper. Nutr Rev, 2010,68(3):133147
213 Jhelum P, David S. Ferroptosis: copper-iron connection
in cuprizone-induced demyelination. Neural Regen Res, 2022,17(1):89-90
214 Gulec S, Collins JF. Molecular mediators governing iron-copper interactions. Annu Rev Nutr, 2014,34:95116
215 Tang D, Chen X, Kang R, et al. Ferroptosis: molecular mechanisms and health implications. Cell Res, 2021,31(2):107-125
216 Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol, 2021,22(4):266-282
217 Fleming MD, Trenor CC, 3rd, Su MA, et al. Microcytic anaemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nat Genet, 1997,16(4):383-386
218 PatelBN, David S.A novel glycosylphosphatidylinositolanchored form of ceruloplasmin is expressed by mammalian astrocytes. J Biol Chem, 1997,272(32): 20185-20190
219 Mastrogiannaki M, Matak P, Keith B, et al. HIF-2alpha, but not HIF-1 alpha, promotes iron absorption in mice. J Clin Invest, 2009,119(5):1159-1166
220 Ravia JJ, Stephen RM, Ghishan FK, et al. Menkes Copper ATPase (Atp7a) is a novel metal-responsive gene in rat duodenum, and immunoreactive protein is present on brush-border and basolateral membrane domains. J Biol Chem, 2005,280(43):36221-36227
221 Ha JH, Doguer C, Collins JF. Consumption of a HighIron Diet Disrupts Homeostatic Regulation of Intestinal Copper Absorption in Adolescent Mice. Am J Physiol Gastrointest Liver Physiol, 2017,313(4):G535-G360
222 Jhelum P, Santos-Nogueira E, Teo W, et al. Ferroptosis Mediates Cuprizone-Induced Loss of Oligodendrocytes and Demyelination. J Neurosci, 2020,40(48):9327-9341
223 Yang M, Wu X, Hu J, et al. COMMD10 inhibits HIF1
224 Ren X, Li Y, Zhou Y, et al. Overcoming the compensatory elevation of NRF2 renders hepatocellular carcinoma cells more vulnerable to disulfiram/copper-induced ferroptosis. Redox Biol, 2021,46:102122
225 Xue Q, Yan D, Chen X, et al. Copper-dependent autophagic degradation of GPX4 drives ferroptosis. Autophagy, 2023,19(7):1982-1996
226 Shen Y, Li D, Liang Q, et al. Cross-talk between cuproptosis and ferroptosis regulators defines the tumor microenvironment for the prediction of prognosis and therapies in lung adenocarcinoma. Front Immunol, 2023,13:1029092
227 Li Y, Wang RY, Deng YJ, et al. Molecular characteristics, clinical significance, and cancer immune interactions of cuproptosis and ferroptosis-associated genes in colorectal cancer. Front Oncol, 2022,12:975859
228 Zhao C, Zhang Z, Jing T. A novel signature of combing cuproptosis-with ferroptosis-related genes for prediction of prognosis, immunologic therapy responses and drug sensitivity in hepatocellular carcinoma. Front Oncol, 2022,12:1000993
229 Gromadzka G, Tarnacka B, Flaga A, et al. Copper Dyshomeostasis in Neurodegenerative Diseases-Therapeutic Implications. Int J Mol Sci, 2020,21(23):9259
231 Zlatic SA, Vrailas-Mortimer A, Gokhale A, et al. Rare Disease Mechanisms Identified by Genealogical Proteomics of Copper Homeostasis Mutant Pedigrees. Cell Syst, 2018,6(3):368-380.e366
232 Bakkar N, Starr A, Rabichow BE, et al. The M1311V variant of ATP7A is associated with impaired trafficking and copper homeostasis in models of motor neuron disease. Neurobiol Dis, 2021,149:105228
233 Hartwig C, Méndez GM, Bhattacharjee S, et al. GolgiDependent Copper Homeostasis Sustains Synaptic Development and Mitochondrial Content. J Neurosci, 2021,41(2):215-233
234 Choi BY, Jang BG, Kim JH, et al. Copper/zinc chelation by clioquinol reduces spinal cord white matter damage and behavioral deficits in a murine MOG-induced multiple sclerosis model. Neurobiol Dis, 2013,54:382391
235 Lai Y, Lin C, Lin X, et al. Identification and immunological characterization of cuproptosis-related molecular clusters in Alzheimer’s disease. Front Aging Neurosci, 2022,14:932676
236 Gawande MB, Goswami A, Felpin FX, et al. Cu and Cu-Based Nanoparticles: Synthesis and Applications in Catalysis. Chem Rev, 2016,116(6):3722-3811
237 Verma N, Kumar N. Synthesis and Biomedical Applications of Copper Oxide Nanoparticles: An Expanding Horizon. ACS Biomater Sci Eng, 2019,5(3):1170-1188
238 Mani VM, Kalaivani S, Sabarathinam S, et al. Copper oxide nanoparticles synthesized from an endophytic fungus Aspergillus terreus: Bioactivity and anti-cancer evaluations. Environ Res, 2021,201:111502
239 Imani SM, Ladouceur L, Marshall T, et al. Antimicrobial Nanomaterials and Coatings: Current Mechanisms and Future Perspectives to Control the Spread of Viruses Including SARS-CoV-2. ACS Nano, 2020,14(10):12341-12369
240 Brewer GJ. Copper-2 Hypothesis for Causation of the Current Alzheimer’s Disease Epidemic Together with Dietary Changes That Enhance the Epidemic. Chem Res Toxicol, 2017,30(3):763-768
241 McCann CJ, Jayakanthan S, Siotto M, et al. Single nucleotide polymorphisms in the human ATP7B gene modify the properties of the ATP7B protein. Metallomics, 2019,11(6):1128-1139
242 Clifford RJ, Maryon EB, Kaplan JH. Dynamic internalization and recycling of a metal ion transporter: Cu homeostasis and CTR1, the human
system. J Cell Sci, 2016,129(8):1711-1721
243 Narindrasorasak S, Kulkarni P, Deschamps P, et al. Characterization and copper binding properties of human COMMD1 (MURR1). Biochemistry, 2007,46(11):3116-3128
244 Hu XM, Zheng SY, Zhang Q, et al. PANoptosis signaling enables broad immune response in psoriasis: From pathogenesis to new therapeutic strategies. Comput Struct Biotechnol J, 2023,23:64-76
245 Yang GJ, Liu H, Ma DL, et al. Rebalancing metal dyshomeostasis for Alzheimer’s disease therapy. J Biol Inorg Chem, 2019,24(8):1159-1170
246 Tulinska J, Mikusova ML, Liskova A, et al. Copper Oxide Nanoparticles Stimulate the Immune Response and Decrease Antioxidant Defense in Mice After SixWeek Inhalation. Front Immunol, 2022,13:874253
247 Stamenković S, Dučić T, Stamenković V, et al. Imaging of glial cell morphology, SOD1 distribution and elemental composition in the brainstem and hippocampus of the ALS hSOD1G93A rat. Neuroscience, 2017,357:37-55
248 Wen MH, Xie X, Tu J, et al. Generation of a genetically modified human embryonic stem cells expressing fluorescence tagged ATOX1. Stem Cell Res, 2019,41:101631
249 Chen H, Xie X, Chen TY. Single-molecule microscopy for in-cell quantification of protein oligomeric stoichiometry. Curr Opin Struct Biol, 2021,66:112-118
250 Gupta D, Bhattacharjee O, Mandal D, et al. CRISPRCas9 system: A new-fangled dawn in gene editing. Life Sci, 2019,232:116636
251 Wang X, Zhou M, Liu Y, et al. Cope with copper: From copper linked mechanisms to copper-based clinical cancer therapies. Cancer Lett, 2023,561:216157
252 Parpura V, Heneka MT, Montana V, et al. Glial cells in (patho)physiology. J Neurochem, 2012,121(1):4-27
253 Xu MB, Rong PQ, Jin TY, et al. Chinese Herbal Medicine for Wilson’s Disease: A Systematic Review and Meta-Analysis. Front Pharmacol, 2019,10:277
254 Simunkova M, Alwasel SH, Alhazza IM, et al. Management of oxidative stress and other pathologies in Alzheimer’s disease. Arch Toxicol, 2019,93(9):24912513
255 Airoldi C, La Ferla B, D Orazio G, et al. Flavonoids in the Treatment of Alzheimer’s and Other Neurodegenerative Diseases. Curr Med Chem, 2018,25(27):3228-3246
256 Wang D, Tian Z, Zhang P, et al. The molecular mechanisms of cuproptosis and its relevance to cardiovascular disease. Biomed Pharmacother, 2023, 163:114830
(Received Nov. 24, 2023, accepted Dec. 17, 2023)
- Xiao-xia BAN, E-mail: xxb19982021@163.com
*Corresponding authors, Kun XIONG, E-mail: xiongkun2001 @ 163.com; Qi ZHANG, E-mail: zhangqi2014@csu.edu.cn *The study was supported by grants from the National Natural Science Foundation of China (No. 81971891, No. 82172196 and No. 82372507), the Natural Science Foundation of Hunan Province (No. 2023JJ40804), and the Key Laboratory of Emergency and Trauma of Ministry of Education (Hainan Medical University, No. KLET-202210).
DOI: https://doi.org/10.1007/s11596-024-2832-z
PMID: https://pubmed.ncbi.nlm.nih.gov/38336987
Publication Date: 2024-02-01
Copper Metabolism and Cuproptosis: Molecular Mechanisms and Therapeutic Perspectives in Neurodegenerative Diseases*
Abstract
[Abstract] Copper is an essential trace element, and plays a vital role in numerous physiological processes within the human body. During normal metabolism, the human body maintains copper homeostasis. Copper deficiency or excess can adversely affect cellular function. Therefore, copper homeostasis is stringently regulated. Recent studies suggest that copper can trigger a specific form of cell death, namely, cuproptosis, which is triggered by excessive levels of intracellular copper. Cuproptosis induces the aggregation of mitochondrial lipoylated proteins, and the loss of iron-sulfur cluster proteins. In neurodegenerative diseases, the pathogenesis and progression of neurological disorders are linked to copper homeostasis. This review summarizes the advances in copper homeostasis and cuproptosis in the nervous system and neurodegenerative diseases. This offers research perspectives that provide new insights into the targeted treatment of neurodegenerative diseases based on cuproptosis.
of iron-sulfur cluster proteins, proteotoxic stress, and ultimately, cell death
1 SYSTEMIC AND CELLULAR COPPER METABOLISM IN THE HUMAN BODY
1.1 Systemic Copper Metabolism in the Human Body
from genetic abnormalities in ATP7A. Defective copper transportation in small intestinal epithelial cells results in abnormal copper metabolism
1.2 Cellular Copper Metabolism in the Human Body
1.2.1 Chelation
1.2.2 COX17 Copper chaperone COX17 binds
and SCO2 may impact the integrity of this signaling pathway
1.2.3 Copper Chaperone of Superoxide Dismutase (CCS) Copper binds to CCS, and delivers copper to SOD1. This promotes the catabolism of reactive oxygen species (ROS), reduces ROS accumulation, and protects cells from free radical damage. The deficiency of SOD1 would increase oxidative stress
1.2.4 Antioxidant Protein 1 (ATOX1)/ATP7A/B ATOX1 binds copper, in order to deliver copper to ATP7B on the trans-Golgi network (ATP7A in other cells). This promotes the synthesis of copper proenzymes, including lysine oxidase, tyrosinase, and ceruloplasmin
Extracellular
2 COPPER HOMEOSTASIS IN THE HUMAN BODY
2.1 Copper Homeostasis in the Nervous System
blood-brain barrier (BBB) as free copper ions, and is released into the brain parenchyma and cerebrospinal fluid. The BBB appears as the primary route for copper entry into the brain parenchyma, and the BBB and blood-cerebrospinal fluid barrier likely maintains copper homeostasis in the brain. Furthermore, both BBB and blood-cerebrospinal fluid barrier cells express proteins involved in copper transport. Cells in the BBB express higher levels of copper transporter proteins, including CTR1, DMT1 and ATP7B than the brain parenchyma. Copper is transported more easily to the brain parenchyma via cerebral capillaries, as compared with transportation via the choroid plexus
2.2 Copper Homeostatic Imbalance in Menkes Disease and Wilson Disease
affects the brain and liver
2.2.1 Menkes Disease
2.2.2 Wilson Disease
cal symptoms, the measurement of copper metabolism, and the analysis of
2.3 Copper Homeostasis and Neurodegenerative Diseases
2.3.1 Alzheimer’s Disease (AD) and Copper Homeostasis AD is a prevalent neurodegenerative disorder that may stem from diverse factors, such as age, environment, and genetics. The increase in human life expectancy in the coming years would likely result in a growth in the number of patients with
contribute to oxidative tissue damage in the brain of patients with
2.3.2 Huntington’s Disease (HD) and Copper Homeostasis HD is an autosomal dominant disorder of the nervous system
2.3.3 Amyotrophic Lateral Sclerosis (ALS) and Copper Homeostasis ALS leads to selective motor neuron degeneration and subsequent death
| Interventions | Condition | Study phases | Results | Location |
| GE180 PET Scan | AD | II | GE180 was used to analyze the regional and global inflammation in the brain of patients with AD and PD , and greater whole brain GE180 was found to be correlated to poorer cognitive function, including the frontal/cingulate/ parietal/temporal lobe. | Nevada, USA |
| Gastro-retentive zinc cysteine tablet | AD | II | The orally administered active comparator material was associated with better tolerability, when compared to oral zinc acetate, and it induced a reduction in serum nonceruloplasmin bound copper levels, and an elevation in serum zinc levels | Florida, USA |
|
|
AD | II | Changes were found in cognitive function, beta-amyloid in University Hospital, Saarland the CSF, and volumetric in the brain. | |
|
|
ALS | II | No posted | Arizona, United States |
|
|
ALS | II | No posted | New South Wales/Victoria, Australia |
|
|
MS | I | No posted | |
|
|
PD | I | The drug dose was
|
|
| Coconut oilepigallocatechin gallate | MS | II | No posted | Valencia, Spain |
| Multimodal exercise program | PD | No | No posted | Chang Gung Memorial Hospital |
| Observational study: copper exposure | PD | No posted | Isernia/Napoli, Italy | |
| Managing fatigue: | ||||
| The Individual program (MFIP) | PD | No | No posted | Nova Scotia, Canada |
| Not researched | HD | |||
while environmental factors, neurotoxin accumulation, oxidative stress damage, and inadequate growth factors may account for
2.3.4 Parkinson’s Disease and Copper Homeostais The primary clinical manifestations of PD include resting tremors, muscle tone modifica-tions, bradykinesia, and postural instability
pathology is characterized by the reduction of dopaminergic neurons, and the formation of protein aggregates that comprise of
3 CUPROPTOSIS IN THE HUMAN BODY
3.1 Definition of Cuproptosis
copper homeostasis, and reported that copper binds to the lipoylated proteins of the TCA cycle
hepatocytes from copper-induced death in liver tissues of individuals with Wilson disease, and in ATP7B-deficient animals
framework can be developed to leverage the synergistic effects of cuproptosis and ferroptosis against cancer
3.2 Link Between Cuproptosis and Ferroptosis
low expression of genes linked to cuproptosis and ferroptosis had higher survival rates
3.3 Cuproptosis and Neurodegenerative Diseases
been reported. In addition, MYT1L, PDE4D, SNAP91,
4 DISCUSSION
| Regulators of signaling pathways or molecules targeted by Cu | Function | Disease | Possible signaling pathway | References |
| EREG | EREG can influence immunity and cuproptosis | Glioblastoma | EREG affects the expression of PDL1/related to cuproptosis by influencing the expression of FDX1. | [201] |
| FDX1 | Associated with immune infiltration | Glioma | Cuproptosis genes related to FDX1 were positively LIPT2 and NNAT, which are involved in correlated to the expression of autophagy marker genes lipoylation, may be the unidentified marker Atg5, Atg12, and BECN-1. genes for cuproptosis. | [200] |
| MAP1LC3A | Autophagy regulator, MAP1LC3A known as LC3 | Wilson disease | Copper increased the expression of MAP1LC3A. | In ATP7B-knockout cells, mTOR is less active, and is dissociated from lysosomes. The mTOR substrate transcription factor EB translocates into the nucleus, and autophagyrelated genes are activated to protect cells from copper-induced death. |
| ATP7 | ATP7A and ATP7B proteins localize to the Golgi, and regulate copper homeostasis. | Neurodegeneration | The integrity of Golgi-dependent copper homeostasis mechanisms, which require the ATP7 and COG complex, is necessary to maintain mitochondria functional integrity. | [233] |
| ATP7A | The mutation of ATP7A can cause Menkes disease from systemic copper depletion. | Menkes disease | Copper dyshomeostasis, due to defects in ATP7A, increases A connection between copper dyshomeostasis the expression of UCHL1, which in turn, is required for the and the UCHL1/PARK5 pathway of Parkinson pathomechanism of copper dyshomeostasis. disease | [231] |
| CTR1 | CTR1 is a copper transporter in the cell membrane. | Cancer | Copper promotes tumorigenesis by activating the PDK1- Nedd41 negatively regulated CTR1 through AKT oncogenic pathway in a CTR1-dependent manner. ubiquitination and subsequent degradation/ Nedd41-CTR1-copper-PDK1-AKT. | [196] |
| Ferritin | Ferritin is an iron storage protein. | Demyelination injury | Cuprizone chelates copper and rapidly mobilizes iron from CZ induces demyelination via the ferroptosisferritin, which triggers iron-mediated lipid peroxidation and mediated rapid loss of oligodendrocytes. oligodendrocyte loss through ferroptosis. | [222] |
| MTFI | Classical metal binding transcription factors are closely related to copper homeostasis and cuproptosis. | Copper loading or deficient | Copper loading induces the transcriptional activation of MTF1 binds to the MRE of CTR1B to MT through the MTF1 and MRE-dependent pathways, and promote its transcription and expression, promotes the nuclear expression of MTF1, which in turn, which introduces copper, and maintains promotes MT expression. copper homeostasis when copper is depleted. | [28] |
| p53 | p53 might promote cuproptosis by inhibiting glycolysis, and enhancing mitochondrial metabolism. | Cancer | p53 regulates FDXR that encodes a ferredoxin reductase Tumor suppressor microprotein miPEP133, responsible for electron transport from NADPH to FDX1/2, which is encoded by the primary transcript and subsequently to cytochrome P450 for Fe-S cluster of miR-34a activated by p53, was found to biogenesis (p53 induces the expression of
|
[198] |
| Disulfiram | Disulfiram is a copper carrier, which combines with copper to promote cell death. | Cancer | Disulfiram combined with
|
[256] |
| (Continued from the previous page) | ||||
| Regulators of signaling pathways or molecules targeted by Cu | Function | Disease | Possible signaling pathway | References |
| COMMD10 | COMMD10 is a cancer suppressor and copper metabolism regulator. | Cancer | COMMD10 inhibits the HIF1a/CP positive feedback COMMD10, HIF1
|
[223] |
| NRF2 | The NRF2 is responsible for the regulation of antioxidant response, and plays a critical role in mitigating ferroptosis. | Cancer | DSF/Cu dramatically activates the phosphorylation DSF/Cu could strengthen the cytotoxicity of of p62, which facilitates the competitive binding of sorafenib, and arrest tumor growth, both in vitro Keap1, thereby prolonging the half-life of NRF2. and in vivo, by simultaneously inhibiting the signal pathway of NRF2 and MAPK kinase. | [224] |
| GPX4 | GPX4 plays a master role in blocking ferroptosis by eliminating phospholipid hydroperoxides. | Cancer | Copper induces the macroautophagic degradation Exogenous copper increases GPX4 ubiquitinaof GPX4 to drive ferroptosis and TAX1BP1, and tion and the formation of GPX4 aggregates this acts as an autophagic receptor for GPX4 by directly binding to GPX4 protein cysteines degradation and subsequent ferroptosis in response C107 and C148. to copper stress. | [225] |
| HMGB1 | HMGB1, a damage-associated molecular pattern, is released by cuproptotic cells to initiate inflammation. | Copper accumulation-induced ATP depletion activ- In HMGB1-deficient cuproptotic cells, AGERates AMPK to promote HMGB1 phosphorylation, dependent inflammatory cytokine production is resulting in increased DAMP extracellular release. greatly reduced. | [199] | |
| GNAQ | GNAQ plays an important role in GPCR signaling. | Uveal melanoma | In GNAQ mutated cells, Cu-ES produces ROS. Subsequently, this promotes YAP phosphorylation, and inhibits its nuclear accumulation. The inactivation of YAP downregulates the expression of SNAI2, which in turn, suppresses the migration of UM cells. | [202] |
activation of autophagy can protect hepatocytes from copper-induced death
with the normal physiological functions of peripheral nerves and the brain. Furthermore, a “combination drug-multitarget” therapeutic strategy would be appropriate for controlling AD. The present strategy of medicinal chemists in combating AD is to design and investigate multifunctional drugs with anti-
5 CONCLUSION
Open Access
mmons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
Availability of Supporting Data
Conflict of Interest Statement
REFERENCES
2 Festa RA, Thiele DJ. Copper: an essential metal in biology. Curr Biol, 2011,21(21):R877-R883
3 Maung MT, Carlson A, Olea-Flores M, et al. The molecular and cellular basis of copper dysregulation and its relationship with human pathologies. FASEB J, 2021,35(9):e21810
4 Gromadzka G, Tarnacka B, Flaga A, et al. Copper Dyshomeostasis in Neurodegenerative Diseases-Therapeutic Implications. Int J Mol Sci, 2020,21(23):9259
5 Zhao WJ, Fan CL, Hu XM, et al. Regulated Cell Death of Retinal Ganglion Cells in Glaucoma: Molecular Insights and Therapeutic Potentials. Cell Mol Neurobiol, 2023,43(7):3161-3178
6 Zhang Q, Hu XM, Zhao WJ, et al. Targeting Necroptosis: A Novel Therapeutic Option for Retinal Degenerative Diseases. Int J Biol Sci, 2023,19(2):658-674
7 Wan H, Yan YD, Hu XM, et al. Inhibition of mitochondrial VDAC1 oligomerization alleviates apoptosis and necroptosis of retinal neurons following OGD/R injury. Ann Anat, 2023,247:152049
8 Yan WT, Zhao WJ, Hu XM, et al. PANoptosis-like cell death in ischemia/reperfusion injury of retinal neurons. Neural Regen Res, 2023,18(2):357-363
9 Tang D, Kang R, Berghe TV, et al. The molecular machinery of regulated cell death. Cell Res, 2019, 29(5):347-364
10 Yang YD, Li ZX, Hu XM, et al. Insight into Crosstalk Between Mitophagy and Apoptosis/Necroptosis: Mechanisms and Clinical Applications in Ischemic
11 Yan WT, Yang YD, Hu XM, et al. Do pyroptosis, apoptosis, and necroptosis (PANoptosis) exist in cerebral ischemia? Evidence from cell and rodent studies. Neural Regen Res, 2022,17(8):1761-1768
12 Chen XY, Dai YH, Wan XX, et al. ZBP1-Mediated Necroptosis: Mechanisms and Therapeutic Implications. Molecules, 2022,28(1):52
13 Chen J, Wang Y, Li M, et al. Netrin-1 Alleviates Early Brain Injury by Regulating Ferroptosis via the PPAR
14 Zhang Q, Wan XX, Hu XM, et al. Targeting Programmed Cell Death to Improve Stem Cell Therapy: Implications for Treating Diabetes and Diabetes-Related Diseases. Front Cell Dev Biol, 2021,9:809656
15 Hu XM, Zhang Q, Zhou RX, et al. Programmed cell death in stem cell-based therapy: Mechanisms and clinical applications. World J Stem Cells, 2020,12(8):787-802
16 Mou YH, Wang J, Wu JC, et al. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol, 2019,29,12(1):34
17 Tsvetkov P, Coy S, Petrova B, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science, 2022,375(6586):1254-1261
18 An Y, Li S, Huang X, et al. The Role of Copper Homeostasis in Brain Disease. Int J Mol Sci, 2022,23 (22):13850
20 Cobine PA, Brady DC. Cuproptosis: Cellular and molecular mechanisms underlying copper-induced cell death. Mol Cell, 2022,82(10):1786-1787
21 Arredondo M, Núñez MT. Iron and copper metabolism. Mol Aspects Med, 2005,26(4-5):313-327
22 Nutrition classics. The Journal of Biological Chemistry, Vol. LXXV II, 1928: Iron in nutrition. VII. Copper as a supplement to iron for hemoglobin building in the rat. By E.B. Hart, H. Steenbock, J. Waddell, and C.A. Elvehjem. Nutr Rev, 1987,45(6):181-183
23 Khoshbin K, Camilleri M. Effects of dietary components on intestinal permeability in health and disease. Am J Physiol Gastrointest Liver Physiol, 2020,319(5):G589-G608
24 Nose Y, Wood LK, Kim BE, et al. Ctr1 is an apical copper transporter in mammalian intestinal epithelial cells in vivo that is controlled at the level of protein stability. J Biol Chem, 2010,285(42):32385-32392
25 Lane DJR, Bae DH, Merlot AM, et al. Duodenal cytochrome b (DCYTB) in iron metabolism: an update on function and regulation. Nutrients, 2015,7(4):22742296
26 Zimnicka AM, Maryon EB, Kaplan JH. Human copper transporter hCTR1 mediates basolateral uptake of copper into enterocytes: implications for copper homeostasis. J Biol Chem, 2007,282(36):26471-26480
27 Schuchardt JP, Hahn A. Intestinal Absorption and Factors Influencing Bioavailability of Magnesium-An Update. Curr Nutr Food Sci, 2017,13(4):260-278
28 Lutsenko S. Dynamic and cell-specific transport
networks for intracellular copper ions. J Cell Sci, 2021, 134(21):jcs240523
29 Nyasae L, Bustos R, Braiterman L, et al. Dynamics of endogenous ATP7A (Menkes protein) in intestinal epithelial cells: copper-dependent redistribution between two intracellular sites. Am J Physiol Gastrointest Liver Physiol, 2007,292(4):G1181-G1194
30 Liang ZD, Tsai WB, Lee MY, et al. Specificity protein 1 (sp1) oscillation is involved in copper homeostasis maintenance by regulating human high-affinity copper transporter 1 expression. Mol Pharmacol, 2012,81(3):455-464
31 Zhao J, Guo S, Schrodi SJ, et al. Cuproptosis and cuproptosis-related genes in rheumatoid arthritis: Implication, prospects, and perspectives. Front Immunol, 2022,13:930278
32 Linder MC. Ceruloplasmin and other copper binding components of blood plasma and their functions: an update. Metallomics, 2016,8(9):887-905
33 Xie J, Yang Y, Gao Y, et al. Cuproptosis: mechanisms and links with cancers. Mol Cancer, 2023,22(1):46
34 Casareno RL, Waggoner D, Gitlin JD. The copper chaperone CCS directly interacts with copper/zinc superoxide dismutase. J Biol Chem, 1998,273(37):23625-23628
35 Punter FA, Adams DL, Glerum DM. Characterization and localization of human COX17, a gene involved in mitochondrial copper transport. Hum Genet, 2000,107(1):69-74
36 Pierson H, Muchenditsi A, Kim B-E, et al. The Function of ATPase Copper Transporter ATP7B in Intestine. Gastroenterology, 2018,154(1):168-180.e165
37 Telianidis J, Hung YH, Materia S, et al. Role of the P-Type ATPases, ATP7A and ATP7B in brain copper homeostasis. Front Aging Neurosci, 2013,5:44
38 Liu H, Lai W, Liu X, et al. Exposure to copper oxide nanoparticles triggers oxidative stress and endoplasmic reticulum (ER)-stress induced toxicology and apoptosis in male rat liver and BRL-3A cell. J Hazard Mater, 2021,401:123349
39 van Rensburg MJ, van Rooy M, Bester MJ, et al. Oxidative and haemostatic effects of copper, manganese and mercury, alone and in combination at physiologically relevant levels: An ex vivo study. Hum Exp Toxicol, 2019,38(4):419-433
40 Comes G, Fernandez-Gayol O, Molinero A, et al. Mouse metallothionein-1 and metallothionein-2 are not biologically interchangeable in an animal model of multiple sclerosis, EAE. Metallomics, 2019,11(2):327337
41 Leary SC, Winge DR, Cobine PA. “Pulling the plug” on cellular copper: the role of mitochondria in copper export. Biochim Biophys Acta, 2009,1793(1):146-153
42 Horn D, Barrientos A. Mitochondrial copper metabolism and delivery to cytochrome c oxidase. IUBMB Life, 2008,60(7):421-429
43 Pacheu-Grau D, Wasilewski M, Oeljeklaus S, et al. COA6 Facilitates Cytochrome c Oxidase Biogenesis as Thiol-reductase for Copper Metallochaperones in Mitochondria. J Mol Biol, 2020,432(7):2067-2079
44 Robinson NJ, Winge DR. Copper metallochaperones. Annu Rev Biochem, 2010,79:537-562
46 Leary SC. Redox regulation of SCO protein function: controlling copper at a mitochondrial crossroad. Antioxid Redox Signal, 2010,13(9):1403-1416
47 Morgada MN, Abriata LA, Cefaro C, et al. Loop recognition and copper-mediated disulfide reduction underpin meta 1 site assembly of CuA in human cytochrome oxidase. Proc Natl Acad Sci U S A, 2015, 112(38):11771-11776
48 Zhou J, Li XY, Liu YJ, et al. Full-coverage regulations of autophagy by ROS: from induction to maturation. Autophagy, 2022,18(6):1240-1255
49 Bompiani KM, Tsai CY, Achatz FP, et al. Copper transporters and chaperones CTR1, CTR2, ATOX1, and CCS as determinants of cisplatin sensitivity. Metallomics, 2016,8(9):951-962
50 Skopp A, Boyd SD, Ullrich MS, et al. Copper-zinc superoxide dismutase (Sod1) activation terminates interact ion between its copper chaperone (Ccs) and the cytosolic metal-binding domain of the copper importer Ctr1. Biometals, 2019,32(4):695-705
51 Gupta A, Lutsenko S. Human copper transporters: mechanism, role in human diseases and therapeutic potential. Future Med Chem, 2009,1(6):1125-1142
52 Hamza I, Faisst A, Prohaska J, et al. The metallochaperone Atox1 plays a critical role in perinatal copper homeostasis. Proc Natl Acad Sci USA, 2001,98(12):6848-6852
53 Itoh S, Kim HW, Nakagawa O, et al. Novel role of antioxidant-1(Atox1) as a copper-dependent transcription factor involved in cell proliferation. J Biol Chem, 2008,283(14):9157-9167
54 Mattie MD, McElwee MK, Freedman JH. Mechanism of copper-activated transcription: activation of AP-1, and the JNK/SAPK and p38 signal transduction pathways. J Mol Biol, 2008,383(5):1008-1018
55 Satake H, Suzuki K, Aoki T, et al. Cupric ion blocks NF kappa B activation through inhibiting the signalinduced phosphorylation of I kappa B alpha. Biochem Biophys Res Commun, 1995,216(2):568-573
56 Chen L, Li N, Zhang M, et al. APEX2-based Proximity Labeling of Atox1 Identifies CRIP2 as a Nuclear Copperbinding Protein that Regulates Autophagy Activation. Angew Chem Int Ed Engl, 2021,60(48):25346-25355
57 An Y, Li S, Huang X, et al. The Role of Copper Homeostasis in Brain Disease. Int J Mol Sci, 2022, 23(22):13850
58 Tong X, Tang R, Xiao M, et al. Targeting cell death pathways for cancer therapy: recent developments in necroptosis, pyroptosis, ferroptosis, and cuproptosis research. J Hematol Oncol, 2022,15(1):174
59 Tümer Z, Møller LB. Menkes disease. Eur J Hum Genet, 2010,18(5):511-518
60 Członkowska A, Litwin T, Dusek P, et al. Wilson disease. Nat Rev Dis Primers, 2018,4(1):21
61 Bush AI. Metals and neuroscience. Curr Opin Chem Biol, 2000,4(2):184-191
62 DiDonato M, Narindrasorasak S, Forbes JR, et al. Expression, purification, and metal binding properties
of the N-terminal domain from the wilson disease putative copper-transporting ATPase (ATP7B). J Biol Chem, 1997,272(52):33279-33282
63 Lönnerdal B. Intestinal regulation of copper homeostasis: a developmental perspective. Am J Clin Nutr, 2008,88(3):846S-850S
64 Aisen P, Enns C, Wessling-Resnick M. Chemistry and biology of eukaryotic iron metabolism. Int J Biochem Cell Biol, 2001,33(10):940-959
65 Akil M, Schwartz JA, Dutchak D, et al. The psychiatric presentations of Wilson’s disease. J Neuropsychiatry Clin Neurosci, 1991,3(4):377-382
66 Mairet-Coello G, Tury A, Esnard-Feve A, et al. FADlinked sulfhydryl oxidase QSOX: topographic, cellular, and subcellular immunolocalization in adult rat central nervous system. J Comp Neurol, 2004,473(3):334-363
67 Trombley PQ, Horning MS, Blakemore LJ. Interactions between carnosine and zinc and copper: implications for neuromodulation and neuroprotection. Biochemistry (Mosc), 2000,65(7):807-816
68 Johnson KA, Conn PJ, Niswender CM. Glutamate receptors as therapeutic targets for Parkinson’s disease. CNS Neurol Disord Drug Targets, 2009,8(6):475-491
69 D’Ambrosi N, Rossi L. Copper at synapse: Release, binding and modulation of neurotransmission. Neurochem Int, 2015,90:36-45
70 Moriya M, Ho YH, Grana A, et al. Copper is taken up efficiently from albumin and alpha2-macroglobulin by cultured human cells by more than one mechanism. Am J Physiol Cell Physiol, 2008,295(3):C708-C721
71 Montes S, Rivera-Mancia S, Diaz-Ruiz A, et al. Copper and copper proteins in Parkinson’s disease. Oxid Med Cell Longev, 2014,2014:147251
72 Wu W, Ruan X, Gu C, et al. Blood-cerebrospinal fluid barrier permeability of metals/metalloids and its determinants in pediatric patients. Ecotoxicol Environ Saf, 2023,266:115599
73 Scheiber IF, Mercer JFB, Dringen R. Metabolism and functions of copper in brain. Prog Neurobiol, 2014,116:33-57
74 Dringen R, Scheiber IF, Mercer JFB. Copper metabolism of astrocytes. Front Aging Neurosci, 2013,5:9
75 Howell SB, Safaei R, Larson CA, et al. Copper transporters and the cellular pharmacology of the platinum-containing cancer drugs. Mol Pharmacol, 2010,77(6):887-894
76 Garza-Lombó C, Posadas Y, Quintanar L, et al. Neurotoxicity Linked to Dysfunctional Metal Ion Homeostasis and Xenobiotic Metal Exposure: Redox Signaling and Oxidative Stress. Antioxid Redox Signal, 2018,28(18):1669-1703
77 Varela-Nallar L, Toledo EM, Chacón MA, et al. The functional links between prion protein and copper. Biol Res, 2006,39(1):39-44
78 Stuerenburg HJ. CSF copper concentrations, bloodbrain barrier function, and coeruloplasmin synthesis during the treatment of Wilson’s disease. J Neural Transm (Vienna), 2000,107(3):321-329
79 Choi BS, Zheng W. Copper transport to the brain by the blood-brain barrier and blood-CSF barrier. Brain Res, 2009,1248:14-21
80 Kaler SG. ATP7A-related copper transport diseases-
emerging concepts and future trends. Nat Rev Neurol, 2011,7(1):15-29
81 Nishihara E, Furuyama T, Yamashita S, et al. Expression of copper trafficking genes in the mouse brain. Neuroreport, 1998,9(14):3259-3263
82 Barber RG, Grenier ZA, Burkhead JL. Copper Toxicity Is Not Just Oxidative Damage: Zinc Systems and Insight from Wilson Disease. Biomedicines, 2021,9(3):316
83 Mercer JF, Ambrosini L, Horton S, et al. Animal models of Menkes disease. Adv Exp Med Biol, 1999,448:97108
84 Menkes JH, Alter M, Steigleder GK, et al. A sex-linked recessive disorder with retardation of growth, peculiar hair, and focal cerebral and cerebellar degeneration. Pediatrics, 1962,29:764-779
85 Møller LB, Mogensen M, Horn N. Molecular diagnosis of Menkes disease: genotype-phenotype correlation. Biochimie, 2009,91(10):1273-1277
86 Shim H, Harris ZL. Genetic defects in copper metabolism. J Nutr, 2003,133(5 Suppl 1):1527S-1531S
87 Kaler SG, Gahl WA, Berry SA, et al. Predictive value of plasma catecholamine levels in neonatal detection of Menkes disease. J Inherit Metab Dis, 1993,16(5):907908
88 Szauter KM, Cao T, Boyd CD, et al. Lysyl oxidase in development, aging and pathologies of the skin. Pathol Biol (Paris), 2005,53(7):448-456
89 Royce PM, Camakaris J, Danks DM. Reduced lysyl oxidase activity in skin fibroblasts from patients with Menkes’ syndrome. Biochem J, 1980,192(2):579-586
90 Sarkar B, Lingertat Walsh K, Clarke JT. Copper-histidine therapy for Menkes disease. J Pediatr, 1993,123(5):828830
91 George DH, Casey RE. Menkes disease after copper histidine replacement therapy: case report. Pediatr Dev Pathol, 2001,4(3):281-288
92 Kim JH, Lee BH, Kim YM, et al. Novel mutations and clinical outcomes of copper-histidine therapy in Menkes disease patients. Metab Brain Dis, 2015,30(1):75-81
93 Cumings JN. The copper and iron content of brain and liver in the normal and in hepato-lenticular degeneration. Brain, 1948,71(Pt. 4):410-415
94 Przybyłkowski A, Gromadzka G, Chabik G, et al. Liver cirrhosis in patients newly diagnosed with neurological phenotype of Wilson’s disease. Funct Neurol, 2014,29 (1):23-29
96 Horoupian DS, Sternlieb I, Scheinberg IH. Neuropathological findings in penicillamine-treated patients with Wilson’s disease. Clin Neuropathol, 1988,7(2):62-67
97 Bertrand E, Lewandowska E, Szpak GM, et al. Neuropathological analysis of pathological forms of astroglia in Wilson’s disease. Folia Neuropathol, 2001,39(2):73-79
98 Langwińska-Wośko E, Litwin T, Szulborski K, et al. Optical coherence tomography and electrophysiology of retinal and visual pathways in Wilson’s disease. Metab Brain Dis, 2016,31(2):405-415
100 European Association for Study of L. EASL Clinical Practice Guidelines: Wilson’s disease. J Hepatol, 2012,56(3):671-685
101 Barnes DE, Yaffe K. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol, 2011,10(9):819-828
102 Morris GP, Clark IA, Vissel B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol Commun, 2014,2:135
103 Zhang YL, Wang J, Zhang ZN, et al. The relationship between amyloid-beta and brain capillary endothelial cells in Alzheimer’s disease. Neural Regen Res, 2022,17(11):2355-2363
104 Gaggelli E, Kozlowski H, Valensin D, et al. Copper homeostasis and neurodegenerative disorders (Alzheimer’s, prion, and Parkinson’s diseases and amyotrophic lateral sclerosis). Chem Rev, 2006,106 (6):1995-2044
106 Newcombe EA, Camats-Perna J, Silva ML, et al. Inflammation: the link between comorbidities, genetics, and Alzheimer’s disease. J Neuroinflammation, 2018, 15(1):276
107 Lovell MA, Robertson JD, Teesdale WJ, et al. Copper, iron and zinc in Alzheimer’s disease senile plaques. J Neurol Sci, 1998,158(1):47-52
108 Su XY, Wu WH, Huang ZP, et al. Hydrogen peroxide can be generated by tau in the presence of
109 Yu J, Luo X, Xu H, et al. Identification of the key molecules involved in chronic copper exposureaggravated memory impairment in transgenic mice of Alzheimer’s disease using proteomic analysis. J Alzheimers Dis, 2015,44(2):455-469
110 James SA, Volitakis I, Adlard PA, et al. Elevated labile Cu is associated with oxidative pathology in Alzheimer disease. Free Radic Biol Med, 2012,52(2):298-302
111 Yu F, Gong P, Hu Z, et al. Cu ( II ) enhances the effect of Alzheimer’s amyloid-
112 Lu J , Wu Dm, Zheng Yi, et al. Trace amounts of copper exacerbate beta amyloid-induced neurotoxicity in the cholesterol-fed mice through TNF-mediated inflammatory pathway. Brain Behav Immun, 2009,23 (2):193-203
114 Krasemann S, Madore C, Cialic R, et al. The TREM2APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity, 2017,47(3):566-581.e9
115 Chen Y, Chen J, Wei H, et al. Akkermansia muciniphila-Nlrp3 is involved in the neuroprotection
of phosphoglycerate mutase 5 deficiency in traumatic brain injury mice. Front Immunol, 2023,14:1172710
116 Zhou Z, Shang L, Zhang Q, et al. DTX3L induced NLRP3 ubiquitination inhibit R28 cell pyroptosis in OGD/R injury. Biochim Biophys Acta Mol Cell Res, 2023,1870(3):119433
117 He YF, Hu XM, Khan MA, et al. HSF1 Alleviates Brain Injury by Inhibiting NLRP3-Induced Pyroptosis in a Sepsis Model. Mediators Inflamm, 2023,2023:2252255
118 Huang Y, Wang S, Huang F, et al. c-FLIP regulates pyroptosis in retinal neurons following oxygen-glucose deprivation/recovery via a GSDMD-mediated pathway. Ann Anat, 2021,235:151672
119 Liao LS, Lu S, Yan WT, et al. The Role of HSP90
120 Yan WT, Lu S, Yang YD, et al. Research trends, hot spots and prospects for necroptosis in the field of neuroscience. Neural Regen Res, 2021,16(8):16281637
121 Hu XM, Li ZX, Lin RH, et al. Guidelines for Regulated Cell Death Assays: A Systematic Summary, A Categorical Comparison, A Prospective. Front Cell Dev Biol, 2021,9:634690
122 Wakhloo D, Oberhauser J, Madira A, et al. From cradle to grave: neurogenesis, neuroregeneration and neurodegeneration in Alzheimer’s and Parkinson’s diseases. Neural Regen Res, 2022,17(12):2606-2614
123 Banjara M, Ghosh C. Sterile Neuroinflammation and Strategies for Therapeutic Intervention. Int J Inflam, 2017,2017:8385961
124 Scheiber IF, Dringen R. Astrocyte functions in the copper homeostasis of the brain. Neurochem Int, 2013,62(5):556-565
125 Pal A, Vasishta Rk, Prasad R. Hepatic and hippocampus iron status is not altered in response to increased serum ceruloplasmin and serum “free” copper in Wistar rat model for non-Wilsonian brain copper toxicosis. Biol Trace Elem Res, 2013,154(3):403-411
126 Qian Y, Zheng Y, Taylor R, et al. Involvement of the molecular chaperone Hspa5 in copper homeostasis in astrocytes. Brain Res, 2012,1447:9-19
127 Pike CJ, Cummings BJ, Monzavi R, et al. Beta-amyloid-induced changes in cultured astrocytes parallel reactive astrocytosis associated with senile plaques in Alzheimer’s disease. Neuroscience, 1994,63(2):517531
128 DeWitt DA, Perry G, Cohen M, et al. Astrocytes regulate microglial phagocytosis of senile plaque cores of Alzheimer’s disease. Exp Neurol, 1998,149(2):329340
129 Choo XY, Liddell JR, Huuskonen MT, et al. Cu
130 Mandal PK, Saharan S, Tripathi M, et al. Brain glutathione levels–a novel biomarker for mild cognitive impairment and Alzheimer’s disease. Biol psychiatry, 2015,78(10):702-710
131 Uttamsingh V, Keller DA, Anders MW. Acylase I-catalyzed deacetylation of N-acetyl-L-cysteine and S-alkyl-N -acetyl-L-cysteines. Chem Res Toxicol,
132 Ashraf A, So PW. Spotlight on Ferroptosis: IronDependent Cell Death in Alzheimer’s Disease. Front Aging Neurosci, 2020,12:196
133 Derry PJ, Hegde ML, Jackson GR, et al. Revisiting the intersection of amyloid, pathologically modified tau and iron in Alzheimer’s disease from a ferroptosis perspective. Prog Neurobiol, 2020,184:101716
134 Bush AI. Drug development based on the metals hypothesis of Alzheimer’s disease. J Alzheimers Dis, 2008,15(2):223-240
135 Walker FO. Huntington’s disease. Lancet, 2007,369 (9557):218-228
137 Dexter DT, Carayon A, Javoy-Agid F, et al. Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain, 1991,114(Pt4):19531975
138 Tabrizi SJ, Ghosh R, Leavitt BR. Huntingtin Lowering Strategies for Disease Modification in Huntington’s Disease. Neuron, 2019,101(5):801-819
139 Xiao G, Fan Q, Wang X, et al. Huntington disease arises from a combinatory toxicity of polyglutamine and copper binding. Proc Natl Acad Sci U S A, 2013,110(37):14995-15000
140 Fox JH, Kama JA, Lieberman G, et al. Mechanisms of copper ion mediated Huntington’s disease progression. PLoS One, 2007,2(3):e334
141 Pamp K, Bramey T, Kirsch M, et al. NAD(H) enhances the Cu ( II )-mediated inactivation of lactate dehydrogenase by increasing the accessibility of sulfhydryl groups. Free Radic Res, 2005,39(1):31-40
142 Harms L, Meierkord H, Timm G, et al. Decreased N -acetyl-aspartate/choline ratio and increased lactate in the frontal lobe of patients with Huntington’s disease: a proton magnetic resonance spectroscopy study. J Neurol Neurosurg Psychiatry, 1997,62(1):27-30
143 Sheline CT, Choi DW.
144 Cherny RA, Ayton S, Finkelstein DI, et al. PBT2 Reduces Toxicity in a C. elegans Model of polyQ Aggregation and Extends Lifespan, Reduces Striatal Atrophy and Improves Motor Performance in the R6/2 Mouse Model of Huntington’s Disease. J Huntingtons Dis, 2012,1(2):211-219
145 Boillée S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron, 2006,52(1):39-59
146 Swinnen B, Robberecht W. The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol, 2014,10(11):661-670
147 Feldman EL, Goutman SA, Petri S, et al. Amyotrophic lateral sclerosis. Lancet, 2022,400(10360):1363-1380
148 Hardiman O, Al-Chalabi A, Chio A, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers, 2017,3:17071
149 Gil-Bea FJ, Aldanondo G, Lasa-Fernández H, et al. Insights into the mechanisms of copper dyshomeostasis in amyotrophic lateral sclerosis. Expert Rev Mol Med, 2017,19:e7
151 Williams JR, Trias E, Beilby PR, et al. Copper delivery to the CNS by CuATSM effectively treats motor neuron disease in SOD(G93A) mice co-expressing the Copper-Chaperone-for-SOD. Neurobiol Dis, 2016,89:1-9
152 Cabreiro F, Ackerman D, Doonan R, et al. Increased life span from overexpression of superoxide dismutase in Caenorhabditis elegans is not caused by decreased oxidative damage. Free Radic Biol Med, 2011,51(8):1575-1582
153 Enge TG, Ecroyd H, Jolley DF, et al. Longitudinal assessment of metal concentrations and copper isotope ratios in the G93A SOD1 mouse model of amyotrophic lateral sclerosis. Metallomics, 2017,9(2):161-174
154 Roos PM, Vesterberg O, Syversen T, et al. Metal concentrations in cerebrospinal fluid and blood plasma from patients with amyotrophic lateral sclerosis. Biol Trace Elem Res, 2013,151(2):159-170
155 Hilton JB, Kysenius K, White AR, et al. The accumulation of enzymatically inactive cuproenzymes is a CNS-specific phenomenon of the SOD1G37R mouse model of ALS and can be restored by overexpressing the human copper transporter hCTR1. Exp Neurol, 2018,307:118-128
156 Tokuda E, Okawa E, Ono Si. Dysregulation of intracellular copper trafficking pathway in a mouse model of mutant copper/zinc superoxide dismutaselinked familial amyotrophic lateral sclerosis. J Neurochem, 2009,111(1):181-191
157 Hottinger AF, Fine EG, Gurney ME, et al. The copper chelator d-penicillamine delays onset of disease and extend s survival in a transgenic mouse model of familial amyotrophic lateral sclerosis. Eur J Neurosci, 1997,9(7):1548-1551
158 Tokuda E, Ono Si, Ishige K, et al. Ammonium tetrathiomolybdate delays onset, prolongs survival, and slows progression of disease in a mouse model for amyotrophic lateral sclerosis. Exp Neurol, 2008,213(1):122-128
159 Roberts BR, Lim NKH, McAllum EJ, et al. Oral treatment with Cu ( II )(atsm) increases mutant SOD1 in vivo but protects motor neurons and improves the phenotype of a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci, 2014,34(23):8021-8031
160 Hilton JB, Mercer SW, Lim NKH, et al.
161 Tokuda E, Okawa E, Watanabe S, et al. Dysregulation of intracellular copper homeostasis is common to transgenic mice expressing human mutant superoxide dismutase-1s regardless of their copper-binding abilities. Neurobiol Dis, 2013,54:308-319
162 DeLazzari F, Bubacco L, Whitworth AJ, et al. Superoxide Radical Dismutation as New Therapeutic Strategy in Parkinson’s Disease. Aging Dis, 2018,9(4):716-728
163 Reich SG, Savitt JM. Parkinson’s Disease. Med Clin
164 Karimi-Moghadam A, Charsouei S, Bell B, et al. Parkinson Disease from Mendelian Forms to Genetic Susceptibility: New Molecular Insights into the Neurodegeneration Process. Cell Mol Neurobiol, 2018,38(6):1153-1178
165 Tolosa E, Garrido A, Scholz SW, et al. Challenges in the diagnosis of Parkinson’s disease. Lancet Neurol, 2021,20(5):385-397
166 Youdim MB, Ben-Shachar D, Riederer P. Is Parkinson’s disease a progressive siderosis of substantia nigra resulting in iron and melanin induced neurodegeneration? Acta Neurol Scand Suppl, 1989,126:47-54
167 Riederer P, Sofic E, Rausch WD, et al. Transition metals, ferritin, glutathione, and ascorbic acid in parkinsonian brains. J Neurochem, 1989,52(2):515-520
168 Perry TL, Godin DV, Hansen S. Parkinson’s disease: a disorder due to nigral glutathione deficiency? Neurosci Lett, 1982,33(3):305-310
169 Jenner P. Oxidative stress in Parkinson’s disease. Ann Neurol, 2003,53:S26-S36
170 Bisaglia M, Mammi S, Bubacco L. Structural insights on physiological functions and pathological effects of alpha-synuclein. FASEB J, 2009,23(2):329-340
171 Binolfi A, Rasia RM, Bertoncini CW, et al. Interaction of alpha-synuclein with divalent metal ions reveals key differences: a link between structure, binding specificity and fibrillation enhancement. J Am Chem Soc, 2006,128(30):9893-9901
172 Dudzik CG, Walter ED, Millhauser GL. Coordination features and affinity of the
174 Anderson JP, Walker DE, Goldstein JM, et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem, 2006,281(40):2973929752
175 Dikiy I, Eliezer D. N-terminal acetylation stabilizes N-terminal helicity in lipid- and micelle-bound
176 Mason RJ, Paskins AR, Dalton CF, et al. Copper Binding and Subsequent Aggregation of
177 Bisaglia M, Bubacco L. Copper Ions and Parkinson’s Disease: Why Is Homeostasis So Relevant? Biomolecules, 2020,10(2):195
178 Uitti RJ, Rajput AH, Rozdilsky B, et al. Regional metal concentrations in Parkinson’s disease, other chronic neurological diseases, and control brains. Can J Neurol Sci, 1989,16(3):310-314
179 de Freitas LV, da Silva CCP, Ellena J, et al. Structural and vibrational study of 8-hydroxyquinoline-2carboxaldehyde isonicotinoyl hydrazine-a potential metal-protein attenuating compound (MPAC) for the treatment of Alzheimer’s disease. Spectrochim Acta A
180 McAllum EJ, Lim NKH, Hickey JL, et al. Therapeutic effects of
181 Wang M, Wan H, Wang S, et al. RSK3 mediates necroptosis by regulating phosphorylation of RIP3 in rat retinal ganglion cells. J Anat, 2020,237(1):29-47
182 Guo LM, Wang Z, Li SP, et al. RIP3/MLKL-mediated neuronal necroptosis induced by methamphetamine at
183 Wang Z, Guo LM, Wang Y, et al. Inhibition of HSP90
184 Chen J, Li M, Liu Z, et al. Molecular mechanisms of neuronal death in brain injury after subarachnoid hemorrhage. Front Cell Neurosci, 2022,16:1025708
185 Hunsaker EW, Franz KJ. Emerging Opportunities To Manipulate Metal Trafficking for Therapeutic Benefit. Inorg Chem, 2019,58(20):13528-13545
186 Zheng P, Zhou C, Lu L, et al. Elesclomol: a copper ionophore targeting mitochondrial metabolism for cancer therapy. J Exp Clin Cancer Res, 2022,41(1):271
187 Hasinoff BB, Yadav AA, Patel D, et al. The cytotoxicity of the anticancer drug elesclomol is due to oxidative stress indirectly mediated through its complex with Cu ( II ). J Inorg Biochem, 2014, 137: 22-30
188 Yang W, Wang Y, Huang Y, et al. 4-Octyl itaconate inhibits aerobic glycolysis by targeting GAPDH to promote cuproptosis in colorectal cancer. Biomed Pharmacother, 2023,159:114301
189 Tsvetkov P, Detappe A, Cai K, et al. Mitochondrial metabolism promotes adaptation to proteotoxic stress. Nat Chem Biol, 2019,15(7):681-689
190 Solmonson A, DeBerardinis RJ. Lipoic acid metabolism and mitochondrial redox regulation. J Biol Chem, 2018,293(20):7522-7530
191 Lutsenko S. Atp7b-/- mice as a model for studies of Wilson’s disease. Biochem Soc Trans, 2008,36(Pt 6):1233-1238
193 Xue J, Yang G, Ding H, et al. Role of NSC319726 in ovarian cancer based on the bioinformatics analyses. Onco Targets Ther, 2015,8:3757-3765
194 Shimada K, Reznik E, Stokes ME, et al. CopperBinding Small Molecule Induces Oxidative Stress and Cell-Cycle Arrest in Glioblastoma-Patient-Derived Cells. Cell Chem Biol, 2018,25(5):585-594.e587
195 Polishchuk EV, Merolla A, Lichtmannegger J, et al. Activation of Autophagy, Observed in Liver Tissues From Patients With Wilson Disease and From ATP7B-Deficient Animals, Protects Hepatocytes From Copper-Induced Apoptosis. Gastroenterology, 2019,156(4):1173-1189.e1175
196 Guo J, Cheng J, Zheng N, et al. Copper Promotes Tumorigenesis by Activating the PDK1-AKT Oncogenic Pathway in a Copper Transporter 1 Dependent Manner. Adv Sci (Weinh), 2021,8(18):e2004303
197 Chen GH, Lv W, Xu YH, et al. Functional analysis of
198 Hu W, Zhang C, Wu R, et al. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci U S A, 2010,107(16):7455-7460
199 Liu J, Liu Y, Wang Y, et al. HMGB1 is a mediator of cuproptosis-related sterile inflammation. Front Cell Dev Biol, 2022,10:996307
200 Lu H, Zhou L, Zhang B, et al. Cuproptosis key gene FDX1 is a prognostic biomarker and associated with immune infiltration in glioma. Front Med (Lausanne), 2022,9:939776
201 Zhou Y, Xiao D, Jiang X, et al. EREG is the core oncoimmunological biomarker of cuproptosis and mediates the cross-talk between VEGF and CD99 signaling in glioblastoma. J Transl Med, 2023,21(1):28
202 Li Y, Yang J, Zhang Q, et al. Copper ionophore elesclomol selectively targets GNAQ/11-mutant uveal melanoma. Oncogene, 2022,41(27):3539-3553
203 Lv H, Liu X, Zeng X, et al. Comprehensive Analysis of Cuproptosis-Related Genes in Immune Infiltration and Prognosis in Melanoma. Front Pharmacol, 2022,13:930041
204 Xu S, Liu D, Chang T, et al. Cuproptosis-Associated lncRNA Establishes New Prognostic Profile and Predicts Immunotherapy Response in Clear Cell Renal Cell Carcinoma. Front Genet, 2022,13:938259
205 Zhang Z, Zeng X, Wu Y, et al. Cuproptosis-Related Risk Score Predicts Prognosis and Characterizes the Tumor Microenvironment in Hepatocellular Carcinoma. Front Immunol, 2022,13:925618
206 Yan C, Niu Y, Ma L, et al. System analysis based on the cuproptosis-related genes identifies LIPT 1 as a novel therapy target for liver hepatocellular carcinoma. J Transl Med, 2022,20(1):452
207 Bao JH, Lu WC, Duan H, et al. Identification of a novel cuproptosis-related gene signature and integrative analyses in patients with lower-grade gliomas. Front Immunol, 2022,13:933973
208 LiuH, Tang T. Pan-cancer genetic analysis of cuproptosis and copper metabolism-related gene set. Front Oncol, 2022,12:952290
209 Guo B, Yang F, Zhang L, et al. Cuproptosis Induced by ROS Responsive Nanoparticles with Elesclomol and Copper Combined with
210 Xu Y, Liu SY, Zeng L, et al. An Enzyme-Engineered Nonporous Copper(I) Coordination Polymer Nanoplat form for Cuproptosis-Based Synergistic Cancer Therapy. Adv Mater, 2022,34(43):e2204733
211 Li T, Wang D, Meng M, et al. Copper-Coordinated Covalent Organic Framework Produced a Robust Fenton-Like Effect Inducing Immunogenic Cell Death of Tumors. Macromol Rapid Commun, 2023,44(11):e2200929
212 Collins JF, Prohaska JR, Knutson MD. Metabolic crossroads of iron and copper. Nutr Rev, 2010,68(3):133147
213 Jhelum P, David S. Ferroptosis: copper-iron connection
in cuprizone-induced demyelination. Neural Regen Res, 2022,17(1):89-90
214 Gulec S, Collins JF. Molecular mediators governing iron-copper interactions. Annu Rev Nutr, 2014,34:95116
215 Tang D, Chen X, Kang R, et al. Ferroptosis: molecular mechanisms and health implications. Cell Res, 2021,31(2):107-125
216 Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol, 2021,22(4):266-282
217 Fleming MD, Trenor CC, 3rd, Su MA, et al. Microcytic anaemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nat Genet, 1997,16(4):383-386
218 PatelBN, David S.A novel glycosylphosphatidylinositolanchored form of ceruloplasmin is expressed by mammalian astrocytes. J Biol Chem, 1997,272(32): 20185-20190
219 Mastrogiannaki M, Matak P, Keith B, et al. HIF-2alpha, but not HIF-1 alpha, promotes iron absorption in mice. J Clin Invest, 2009,119(5):1159-1166
220 Ravia JJ, Stephen RM, Ghishan FK, et al. Menkes Copper ATPase (Atp7a) is a novel metal-responsive gene in rat duodenum, and immunoreactive protein is present on brush-border and basolateral membrane domains. J Biol Chem, 2005,280(43):36221-36227
221 Ha JH, Doguer C, Collins JF. Consumption of a HighIron Diet Disrupts Homeostatic Regulation of Intestinal Copper Absorption in Adolescent Mice. Am J Physiol Gastrointest Liver Physiol, 2017,313(4):G535-G360
222 Jhelum P, Santos-Nogueira E, Teo W, et al. Ferroptosis Mediates Cuprizone-Induced Loss of Oligodendrocytes and Demyelination. J Neurosci, 2020,40(48):9327-9341
223 Yang M, Wu X, Hu J, et al. COMMD10 inhibits HIF1
224 Ren X, Li Y, Zhou Y, et al. Overcoming the compensatory elevation of NRF2 renders hepatocellular carcinoma cells more vulnerable to disulfiram/copper-induced ferroptosis. Redox Biol, 2021,46:102122
225 Xue Q, Yan D, Chen X, et al. Copper-dependent autophagic degradation of GPX4 drives ferroptosis. Autophagy, 2023,19(7):1982-1996
226 Shen Y, Li D, Liang Q, et al. Cross-talk between cuproptosis and ferroptosis regulators defines the tumor microenvironment for the prediction of prognosis and therapies in lung adenocarcinoma. Front Immunol, 2023,13:1029092
227 Li Y, Wang RY, Deng YJ, et al. Molecular characteristics, clinical significance, and cancer immune interactions of cuproptosis and ferroptosis-associated genes in colorectal cancer. Front Oncol, 2022,12:975859
228 Zhao C, Zhang Z, Jing T. A novel signature of combing cuproptosis-with ferroptosis-related genes for prediction of prognosis, immunologic therapy responses and drug sensitivity in hepatocellular carcinoma. Front Oncol, 2022,12:1000993
229 Gromadzka G, Tarnacka B, Flaga A, et al. Copper Dyshomeostasis in Neurodegenerative Diseases-Therapeutic Implications. Int J Mol Sci, 2020,21(23):9259
231 Zlatic SA, Vrailas-Mortimer A, Gokhale A, et al. Rare Disease Mechanisms Identified by Genealogical Proteomics of Copper Homeostasis Mutant Pedigrees. Cell Syst, 2018,6(3):368-380.e366
232 Bakkar N, Starr A, Rabichow BE, et al. The M1311V variant of ATP7A is associated with impaired trafficking and copper homeostasis in models of motor neuron disease. Neurobiol Dis, 2021,149:105228
233 Hartwig C, Méndez GM, Bhattacharjee S, et al. GolgiDependent Copper Homeostasis Sustains Synaptic Development and Mitochondrial Content. J Neurosci, 2021,41(2):215-233
234 Choi BY, Jang BG, Kim JH, et al. Copper/zinc chelation by clioquinol reduces spinal cord white matter damage and behavioral deficits in a murine MOG-induced multiple sclerosis model. Neurobiol Dis, 2013,54:382391
235 Lai Y, Lin C, Lin X, et al. Identification and immunological characterization of cuproptosis-related molecular clusters in Alzheimer’s disease. Front Aging Neurosci, 2022,14:932676
236 Gawande MB, Goswami A, Felpin FX, et al. Cu and Cu-Based Nanoparticles: Synthesis and Applications in Catalysis. Chem Rev, 2016,116(6):3722-3811
237 Verma N, Kumar N. Synthesis and Biomedical Applications of Copper Oxide Nanoparticles: An Expanding Horizon. ACS Biomater Sci Eng, 2019,5(3):1170-1188
238 Mani VM, Kalaivani S, Sabarathinam S, et al. Copper oxide nanoparticles synthesized from an endophytic fungus Aspergillus terreus: Bioactivity and anti-cancer evaluations. Environ Res, 2021,201:111502
239 Imani SM, Ladouceur L, Marshall T, et al. Antimicrobial Nanomaterials and Coatings: Current Mechanisms and Future Perspectives to Control the Spread of Viruses Including SARS-CoV-2. ACS Nano, 2020,14(10):12341-12369
240 Brewer GJ. Copper-2 Hypothesis for Causation of the Current Alzheimer’s Disease Epidemic Together with Dietary Changes That Enhance the Epidemic. Chem Res Toxicol, 2017,30(3):763-768
241 McCann CJ, Jayakanthan S, Siotto M, et al. Single nucleotide polymorphisms in the human ATP7B gene modify the properties of the ATP7B protein. Metallomics, 2019,11(6):1128-1139
242 Clifford RJ, Maryon EB, Kaplan JH. Dynamic internalization and recycling of a metal ion transporter: Cu homeostasis and CTR1, the human
system. J Cell Sci, 2016,129(8):1711-1721
243 Narindrasorasak S, Kulkarni P, Deschamps P, et al. Characterization and copper binding properties of human COMMD1 (MURR1). Biochemistry, 2007,46(11):3116-3128
244 Hu XM, Zheng SY, Zhang Q, et al. PANoptosis signaling enables broad immune response in psoriasis: From pathogenesis to new therapeutic strategies. Comput Struct Biotechnol J, 2023,23:64-76
245 Yang GJ, Liu H, Ma DL, et al. Rebalancing metal dyshomeostasis for Alzheimer’s disease therapy. J Biol Inorg Chem, 2019,24(8):1159-1170
246 Tulinska J, Mikusova ML, Liskova A, et al. Copper Oxide Nanoparticles Stimulate the Immune Response and Decrease Antioxidant Defense in Mice After SixWeek Inhalation. Front Immunol, 2022,13:874253
247 Stamenković S, Dučić T, Stamenković V, et al. Imaging of glial cell morphology, SOD1 distribution and elemental composition in the brainstem and hippocampus of the ALS hSOD1G93A rat. Neuroscience, 2017,357:37-55
248 Wen MH, Xie X, Tu J, et al. Generation of a genetically modified human embryonic stem cells expressing fluorescence tagged ATOX1. Stem Cell Res, 2019,41:101631
249 Chen H, Xie X, Chen TY. Single-molecule microscopy for in-cell quantification of protein oligomeric stoichiometry. Curr Opin Struct Biol, 2021,66:112-118
250 Gupta D, Bhattacharjee O, Mandal D, et al. CRISPRCas9 system: A new-fangled dawn in gene editing. Life Sci, 2019,232:116636
251 Wang X, Zhou M, Liu Y, et al. Cope with copper: From copper linked mechanisms to copper-based clinical cancer therapies. Cancer Lett, 2023,561:216157
252 Parpura V, Heneka MT, Montana V, et al. Glial cells in (patho)physiology. J Neurochem, 2012,121(1):4-27
253 Xu MB, Rong PQ, Jin TY, et al. Chinese Herbal Medicine for Wilson’s Disease: A Systematic Review and Meta-Analysis. Front Pharmacol, 2019,10:277
254 Simunkova M, Alwasel SH, Alhazza IM, et al. Management of oxidative stress and other pathologies in Alzheimer’s disease. Arch Toxicol, 2019,93(9):24912513
255 Airoldi C, La Ferla B, D Orazio G, et al. Flavonoids in the Treatment of Alzheimer’s and Other Neurodegenerative Diseases. Curr Med Chem, 2018,25(27):3228-3246
256 Wang D, Tian Z, Zhang P, et al. The molecular mechanisms of cuproptosis and its relevance to cardiovascular disease. Biomed Pharmacother, 2023, 163:114830
(Received Nov. 24, 2023, accepted Dec. 17, 2023)
- Xiao-xia BAN, E-mail: xxb19982021@163.com
*Corresponding authors, Kun XIONG, E-mail: xiongkun2001 @ 163.com; Qi ZHANG, E-mail: zhangqi2014@csu.edu.cn *The study was supported by grants from the National Natural Science Foundation of China (No. 81971891, No. 82172196 and No. 82372507), the Natural Science Foundation of Hunan Province (No. 2023JJ40804), and the Key Laboratory of Emergency and Trauma of Ministry of Education (Hainan Medical University, No. KLET-202210).