التقدير الطولي لبكتيريا بيفيدوباكتيريوم لونغوم تحت النوع إنفانتيس يكشف عن استعمار متأخر في أمعاء الرضع مستقل عن تركيبة حليب الأم من الكربوهيدرات الحليب البشرية Longitudinal quantification of Bifidobacterium longum subsp. infantis reveals late colonization in the infant gut independent of maternal milk HMO composition
التقدير الطولي لبكتيريا بيفيدوباكتيريوم لونغوم تحت النوع إنفانتيس يكشف عن استعمار متأخر في أمعاء الرضع مستقل عن تركيبة حليب الأم من الكربوهيدرات الحليب البشرية
تاريخ الاستلام: 19 يوليو 2023
تاريخ القبول: 15 يناير 2024
تاريخ النشر على الإنترنت: 30 يناير 2024
(4) تحقق من التحديثات
يحتوي حليب الثدي على أوليغوسكاريد حليب الإنسان (HMOs) التي لا يمكن هضمها من قبل الرضع، لكنها تغذي ميكروبيوم الأمعاء النامي لديهم. بينما تُعتبر بيفيدوباكتيريوم من أفضل المستفيدين من HMOs الفردية، فإن دراسة طولية تفحص المجتمع الميكروبي المتطور بدقة عالية مقترنة بتركيبة HMO في حليب الأمهات مفقودة. هنا، طورنا طريقة عالية الإنتاجية لقياس بيفيدوباكتيريوم لونغوم تحت النوع إنفانتيس (BL. إنفانتيس)، وهو مستفيد بارع من HMO، وطبقناها على مجموعة طولية تتكون من 21 ثنائي أم-رضيع. لاحظنا تغييرات كبيرة في ميكروبيوم أمعاء الرضع على مدار عدة أشهر، بينما ظلت تركيبة HMO في حليب الأمهات مستقرة نسبيًا. على الرغم من أن أنواع بيفيدوباكتيريوم أثرت بشكل كبير على تباين العينات، لم تتوافق أي HMO محددة مع وفرة أنواع بيفيدوباكتيريوم. من المدهش أننا وجدنا أن استعمار BL. إنفانتيس بدأ متأخرًا في فترة الرضاعة الطبيعية سواء في مجموعتنا أو في مواقع جغرافية أخرى، مما يبرز أهمية التركيز على ديناميات BL. إنفانتيس في أمعاء الرضع.
يعتبر حليب الثدي التغذية المثالية للرضع خلال الأشهر الستة الأولى من حياتهم . لقد شكلت ملايين السنين من التطور تركيبة حليب الثدي بحيث أن ثالث مكون الأكثر وفرة، أوليغوسكاريد حليب الإنسان (HMOs)، لا يمكن هضمه من قبل الرضيع، ولكنه يعمل كركيزة لبكتيريا أمعاء الرضيع . هناك أنواع مختلفة من HMOs، والتي يمكن تصنيفها بشكل كبير إلى ثلاث مجموعات: مفسوسة، مصلبة، أو محايدة. يتكون كل HMO من 3 إلى 32 مونومر، وعينة حليب واحدة تحتوي عادةً على 50 إلى 200 نوع متميز من . من بين عوامل أخرى، تلعب الجينات الأمومية دورًا في إنتاج HMOs محددة في حليب الثدي .
على سبيل المثال، الأمهات اللاتي لديهن جين الفوكوزيل ترانسفيراز 2 (FUT2) غير النشط، المعروف باسم غير السريين، يفشلن في تشكيل روابط ألفا-1،2 بين الفوكوز واللاكتوز أو هياكل HMO الأخرى، مما يؤدي إلى عدم وجود 2’FL وغيرها من الجليكوزيدات الفوكوزيل ألفا-1،2 . بالإضافة إلى ذلك، يمكن أن تؤثر العوامل البيئية مقترنة بعمر الرضيع على تركيبة HMO .
في المتوسط، يتمتع الرضع بوفرة نسبية أعلى من أنواع بيفيدوباكتيريوم أثناء الرضاعة . لقد أظهرت الدراسات السابقة أن أنواع بيفيدوباكتيريوم قادرة على استخدام عدة , ومع ذلك، تختلف هذه القدرة بين الأنواع وحتى داخل نوع واحد
الأنواع . من بين جميع أنواع بيفيدوباكتيريوم وتحت الأنواع، يُعتبر بيفيدوباكتيريوم لونغوم تحت النوع إنفانتيس (BL. إنفانتيس) هو الأكثر شهرة في استخدام HMO، والذي ينمو بكفاءة على معظم أنواع HMOs , ويمتلك تنوعًا كبيرًا من جينات استخدام HMO . بالمقابل، تمتلك أنواع بيفيدوباكتيريوم الأخرى قدرة أقل على استخدام HMO، على سبيل المثال، سلالات B. بريفي لا يمكنها استخدام 3’SL و6’SL على الإطلاق، ومعظمها لا يمكنها استخدام HMOs المفسوسة . نظرًا لأن HMOs تعمل كغذاء لميكروبيوم الأمعاء، يمكن للمرء أن يفترض أن تركيبات HMO المختلفة في حليب الأمهات تؤثر على المجتمع الميكروبي النامي في الأمعاء.
حتى الآن، ركزت معظم الأبحاث التي تتناول علاقة HMO-البكتيريا في أمعاء الرضع على نقطة زمنية واحدة . هناك حاجة إلى دراسة مجموعة طولية من أجل فحص كيف تؤثر التغيرات في تركيبة HMO على ميكروبيوم أمعاء الرضع مع مرور الوقت.
لقياس وفرة أنواع بيفيدوباكتيريوم المختلفة في مجتمعات الميكروبيوم، يتم استخدام نهجين شائعين: تسلسل 16SrRNA وميتاجينوميات الشوتغن. بينما يسمح تسلسل الميتاجينوم بتصنيف على مستوى الأنواع، يوفر تسلسل 16S-rRNA تصنيفًا فقط على مستوى الجنس لمجتمعات الميكروبيوم. الوحدة الأساسية المعلّمة في تسلسل 16S-rRNA تُعرف بوحدة التصنيف التشغيلية (OTU)، والتي يمكن تعيينها لتصنيف على مستوى الجنس وقد تمثل عدة أنواع من الجنس المعين. يمكن أن يتم تصنيف عدة أنواع على أنها نفس OTU، وبالتالي، فإن استخدام تسلسل 16S-rRNA حتى الآن قدم في الغالب ارتباطات ضعيفة أو لا ارتباطات مع وفرة محددة من . يمكن أن يتضمن OTU واحد عدة أنواع (أو تحت أنواع) بقدرات مختلفة لاستخدام HMO، وبالتالي، هناك حاجة إلى تعريف تصنيفي بدقة أعلى.
يمكن العثور على أكبر تنوع في قدرة استخدام HMO داخل نوع بيفيدوباكتيريوم لونغوم. بشكل عام، يمكن تقسيم هذا النوع إلى نوعين فرعيين موجودين في البشر: B. لونغوم تحت النوع لونغوم (BL. لونغوم) وB. لونغوم تحت النوع إنفانتيس (BL. إنفانتيس). يُوجد BL. لونغوم في كل من الرضع والبالغين، بينما . إنفانتيس فريد من نوعه في أمعاء الرضع. أظهرت الدراسات أن BL. إنفانتيس يمكنه استخدام تقريبًا جميع HMOs , بينما . لونغوم لديه مجموعة محدودة. لدراسة علاقة HMO-الميكروب، مع الأخذ في الاعتبار هذه الاختلافات في استخدام HMO داخل تحت أنواع B. لونغوم، هناك حاجة إلى طريقة عالية الإنتاجية ودقيقة. استخدمت الدراسات السابقة طرقًا مختلفة للتفريق بين BL. إنفانتيس وBL. لونغوم، مثل أو طريقة نسبة بيفيدوباكتيريوم لونغوم-إنفانتيس (BLIR) , ومع ذلك، تتطلب هذه الطرق الحمض النووي الأصلي وليست عالية الإنتاجية. بحث آخرون عن . جينات محددة لإنفانتيس مثل مجموعة H 1 أو مجموعات أخرى من BL. إنفانتيس , ومع ذلك، لا تعطي هذه الطرق نسبة دقيقة بين تحت الأنواع، بل تشير إلى وجودها أو عدم وجودها. يمكن تطبيق الطريقة الجديدة التي نقترحها هنا أيضًا على الكميات الضخمة من البيانات المتاحة في المستودعات العامة.
هنا، نقوم بإنشاء مجموعة جديدة متطابقة من عينات حليب الثدي وبراز الرضع التي تم جمعها على مدار السنة الأولى من الحياة. نطور طريقة تسمح بقياس BL. إنفانتيس من بيانات الميتاجينوم الموجودة، ونطبقها على عينات من مجموعتنا لدراسة العلاقة بين وفرة أنواع بيفيدوباكتيريوم في أمعاء الرضع وتركيبة HMO في حليب الأمهات مع مرور الوقت. أخيرًا، نطبق طريقة قياس تحت أنواع B. لونغوم على مجموعات بيانات أمعاء الرضع الموجودة لفحص توقيت استعمار BL. إنفانتيس عبر المواقع الجغرافية.
النتائج
تصميم المجموعة
لقد أنشأنا مجموعة طولية جديدة وفريدة لاختبار العلاقة بين HMOs في حليب الأمهات وميكروبيوم أمعاء الرضع النامي. تتكون مجموعتنا من 21 ثنائي أم-رضيع مع عينات براز الرضع وعينات حليب الثدي التي تم جمعها في نفس اليوم. بشكل إجمالي، جمعنا 80 عينة براز و50 عينة حليب ثدي مع معلومات التغذية للرضيع
وعلاجات المضادات الحيوية (الشكل التوضيحي التكميلي 1، البيانات التكملية 1). جمعنا هذه العينات بين عمر أسبوعين و41 أسبوعًا، وساهم كل ثنائي بين عينة واحدة إلى ثماني عينات متطابقة.
تسمح الجينات المحددة بعلامات أفضل لقياس تحت أنواع B. لونغوم
بيفيدوباكتيريوم لونغوم تحت النوع إنفانتيس (BL. إنفانتيس) هو الأكثر شهرة في استخدام HMOs , ومع ذلك، فإن الطرق الحالية للتصنيف الضريبي من الميتاجينوم غير قادرة على فصل نوع بيفيدوباكتيريوم لونغوم (B. لونغوم) إلى تحت أنواعه الرئيسية؛ بيفيدوباكتيريوم لونغوم تحت النوع لونغوم (BL. لونغوم) وBL. إنفانتيس . ميتا فليان هو أحد الأدوات الأكثر شيوعًا لتوصيف تركيبة السكان الميكروبي من بيانات الميتاجينوم، باستخدام جينات علامات محددة لكل مجموعة تصنيفية . ومع ذلك، لا يحتوي ميتا فليان على جينات علامات محددة لـ . إنفانتيس وبالتالي يصنف . لونغوم على مستوى التصنيف النوعي. نظرًا للاختلافات بين . تحت أنواع لونغوم في سياق استخدام HMO، هناك حاجة متزايدة إلى طريقة عالية الإنتاجية ستسمح بالتحديد والقياس الدقيق لـ BL. إنفانتيس من بيانات الميتاجينوم.
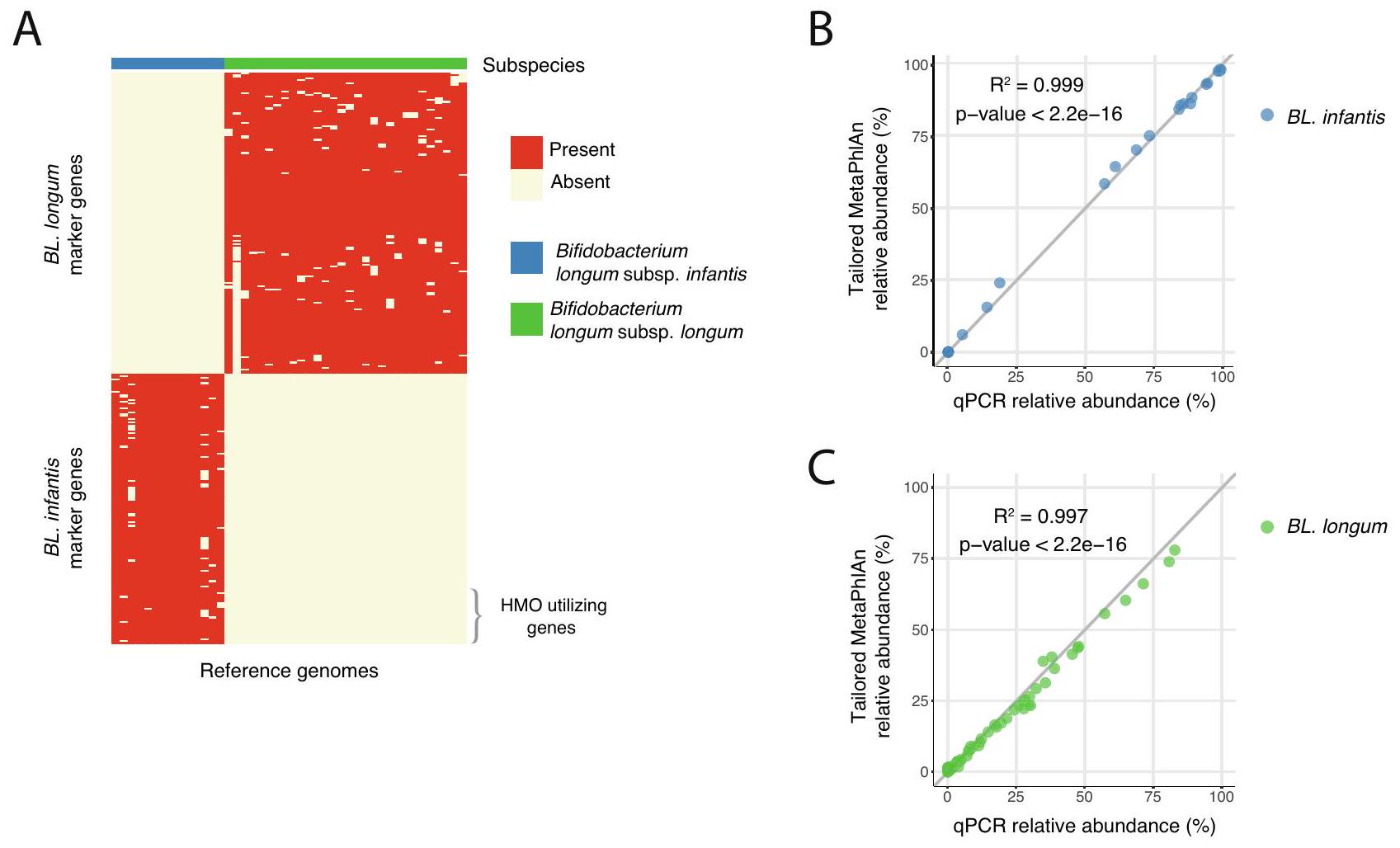
هنا نحدد . علامات تحت أنواع لونغوم المحددة ونستخدمها في ميتا فليان مخصصقاعدة بيانات تسمح بتحديد وفرة نوعين من الأنواع الفرعية لبكتيريا B. longum: BL. infantis و BL. longum (الشكل التكميلي 2A). لبناء مجموعة البيانات الجديدة الخاصة بنا، بحثنا عن جينات علامات فريدة لكل نوع فرعي. تم اختيار جين علامة إذا كان موجودًا في على الأقل من الجينومات المرجعية لنوع فرعي واحد وليس في جينوم واحد من النوع الفرعي الآخر (الشكل 1A، الطرق). اخترنا استبعاد نوعين فرعيين من B. longum: Bifidobacterium longum subsp. suis و Bifidobacterium longum subsp. suillum حيث نادرًا ما توجد في البشر ، وكمية محدودة من الجينومات المرجعية موجودة لهذه الأنواع الفرعية (الطرق).
من أجل التحقق من نتائجنا، طبقنا MetaPhlAn مع مجموعة جديدة من جينات العلامات مقترنة مع qPCR النوع الفرعي لبيانات تسلسل الميتاجينوم من 68 عينة براز للأطفال الرضع. عند مقارنة وفرة BL. infantis و BL. longum في كل طريقة، لاحظنا ارتباطًا قويًا بين نهجنا الحسابي و qPCR (لـ BL. infantis و 0.997 لـ . longum؛ الشكل 1B، C). تؤكد هذه النتيجة خصوصية وطبيعة طريقة لدينا لكل من BL. infantis (الشكل 1B) و BL. longum (الشكل 1C). في بعض العينات، فشل MetaPhlAn في تعيين تصنيف لنسبة صغيرة من . longum وبالتالي تم تصنيفه على أنه غير مصنف . longum (الشكل التكميلي 2B، C).
يظهر ميكروبيوم أمعاء الرضع تغييرات مفرطة بينما يتسم تركيب HMO في حليب الأمهات بالاستقرار النسبي
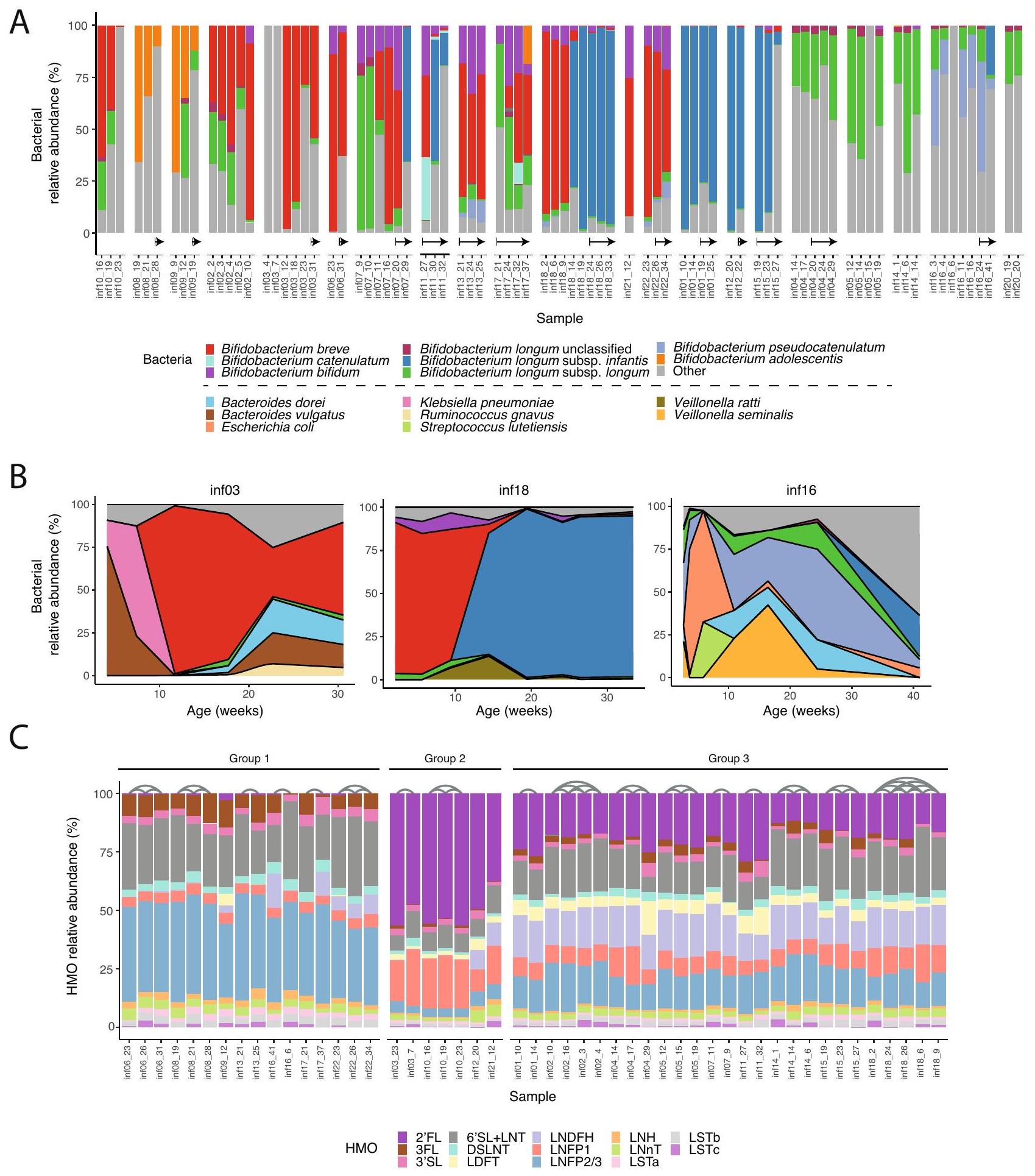
لفحص ديناميات أنواع Bifidobacterium في أمعاء الرضع، أجرينا تسلسل الميتاجينوم وحللنا البيانات باستخدام قاعدة بيانات MetaPhlAn الجديدة لدينا. لاحظنا انتشارًا كبيرًا لبكتيريا Bifidobacterium (على الأقل عينة واحدة مع ) في جميع الرضع (الشكل 2A)، بما يتماشى مع توقعاتنا حيث كان معظم الرضع في دراستنا يرضعون . ظلت Bifidobacterium وفيرة للغاية حتى بعد إدخال الأطعمة الصلبة للأطفال (الشكل 2A، الأسهم). تضمنت الأنواع الوفيرة Bifidobacterium breve و Bifidobacterium bifidum و Bifidobacterium pseudocatenulatum وأنواع B. longum الفرعية (الشكل 2A)، إلى جانب أنواع Bacteroides مثل Bacteroides dorei و Bacteroides vulgatus (الشكل التكميلي 3). من المثير للاهتمام، أن وجود BL. longum و BL. infantis كان متبادلاً، مما يعكس المنافسة المحتملة داخل الأنواع، كما تم اقتراحه سابقًا .
بشكل عام، وجدنا أن ميكروبيوم أمعاء الرضع شهد تغييرات كبيرة على مدار عدة أشهر. بينما كان تركيب البكتيريا يميل إلى الاستقرار على مدار بضعة أسابيع، كانت هناك نقاط زمنية معينة حدث فيها تغيير في التركيب (الشكل 2A، B). في الرضع الذين كانت لدينا عينات منهم من عدة نقاط زمنية، وجدنا أن هذا التغيير يحدث حوالي 10 أسابيع من العمر. على سبيل المثال، في
الشكل 1 | تحديد وفرة الأنواع الفرعية لبكتيريا Bifidobacterium longum باستخدام جينات العلامات يمكّن من الكشف على مستوى الأنواع الفرعية. A تحديد الجينات الفريدة في . infantis (الأزرق) و BL. longum (الأخضر) من الجينومات المرجعية التي يمكن أن تعمل كجينات علامات في تحديد وفرة . longum الأنواع الفرعية. يتم وضع علامات على الجينات المستخدمة في HMO (HUGs) في الأسفل اليمين وتشكل فقط جزءًا صغيرًا من BL. infantis
جينات العلامات.التحقق من صحة النهج الحسابي، مقارنة نتائج تحديد جينات العلامات المخصصة من MetaPhlAn مع نتائج qPCR التجريبية لـ BL. infantis و BL. longum.تم حساب قيمة -value للانحدار باستخدام اختبار – ذو جانبين.
infant03 (inf03) على مدار الأسابيع الـ 15 الأولى، كانت البكتيريا السائدة في الأمعاء تتغير باستمرار، بما في ذلك B. vulgatus و Klebsiella pneumoniae، وأخيرًا . breve؛ كانت أمعاء infant18 مهيمنة في البداية بواسطة B. breve، تليها تغيير كامل إلى BL. infantis؛ وكانت أمعاء infant16 تتغير باستمرار في تركيبها الميكروبي (مهيمنة بواسطة Escherichia coli و Veillonella seminalis و B. pseudocatenulatum؛ الشكل 2B). لم يكن من الواضح دائمًا ما الذي أدى إلى هذه التحولات الميكروبية، ومع ذلك، نظرًا لأن Bifidobacterium وميكروبات الأمعاء الأخرى تستخدم HMOs، افترضنا أن التغيرات في تركيب HMO في حليب الأمهات قد تكون تسبب تغييرات بكتيرية في أمعاء الرضع.
لفحص تأثير تركيب HMO في حليب الأمهات على ميكروبيوم أمعاء الرضع، قمنا بتحديد 16 HMO شائعة في 50 عينة حليب من 20 أم باستخدام كروماتوغرافيا السائل عالية الأداء مع الكشف عن الفلورسنت (HPLC-FLD؛ الطرق). على عكس ميكروبيوم أمعاء الرضع الديناميكي، ظل تركيب HMOs في حليب الأمهات مستقرًا نسبيًا على مدار عدة أشهر، من حيث تركيزها في الحليب (الشكل التكميلي 4A) ووفرة HMOs المحددة (الشكل 2C). قسمنا عينات الحليب إلى ثلاث مجموعات رئيسية، بناءً على تركيب HMO الخاص بها: تلك التي تحتوي على وفرة منخفضة أو لا تحتوي على (المجموعة 1، غير السريين )؛ عينات من الأمهات السريين مع وفرة عالية جدًا من و LNFP1 () وكميات أصغر من HMOs الأخرى (المجموعة 2)؛ وعينات من الأمهات السريين مع وفرة أقل من ، وعدم وجود HMO سائد واضح (المجموعة 3؛ الشكل 2C). لم نجد تغييرات كبيرة بمرور الوقت في وفرة HMOs المحددة، بخلاف LSTc الذي تم تقليله إلى ما يقرب من 0 على مدار أسابيع (الشكل التكميلي 4B، C)، بما يتماشى مع النتائج السابقة . بشكل عام، كانت التغييرات التي وجدت في تركيب HMO في عينات الحليب المتتالية أقل وضوحًا بكثير من تلك الموجودة في
السكان الميكروبي من عينات أمعاء الرضع المتتالية (-test، , الشكل التكميلي 5A).
تشكل الأنواع السائدة من Bifidobacterium ميكروبيوم أمعاء الرضع
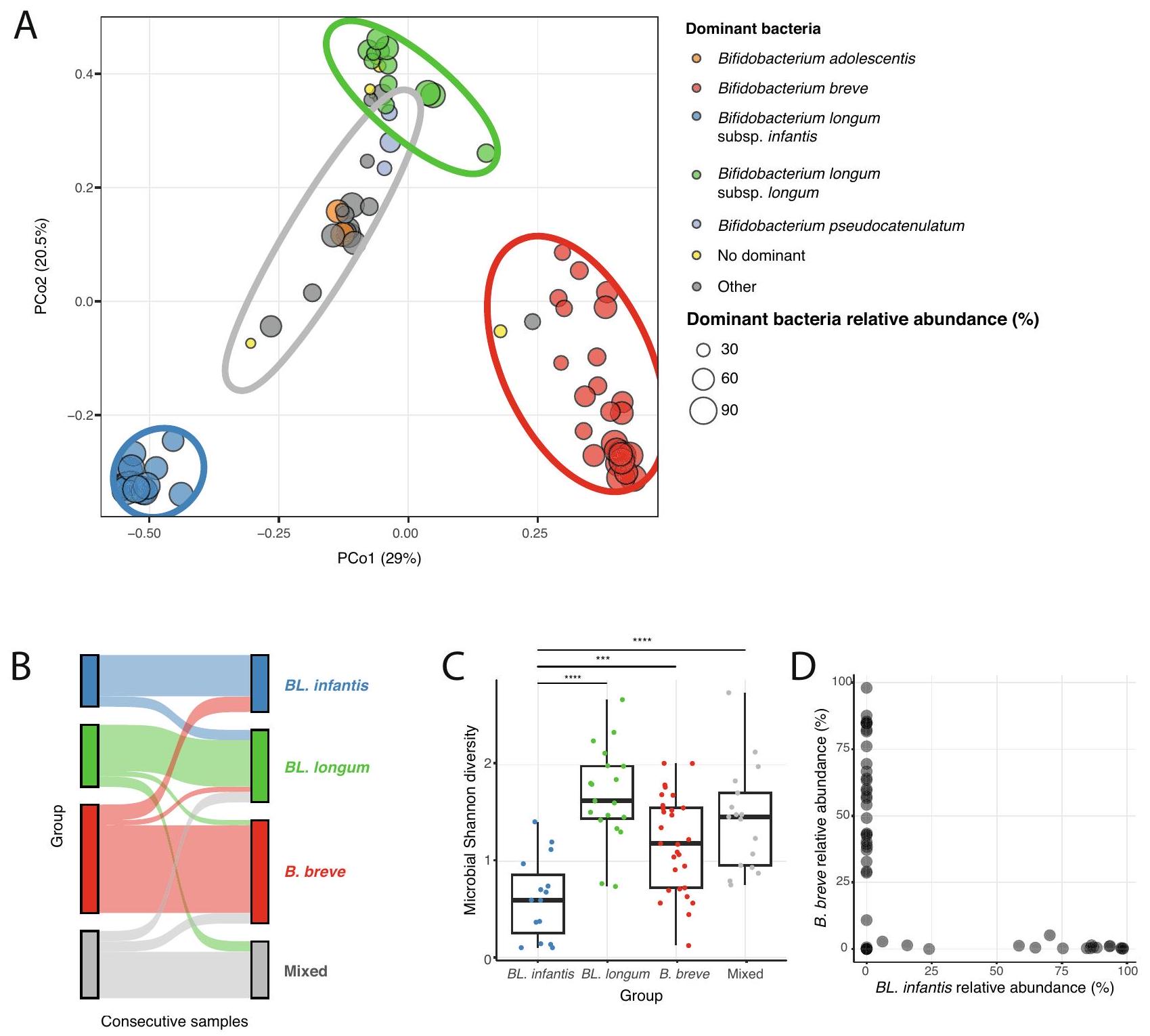
أظهر المقارنة العامة لتركيب ميكروبيوم أمعاء الرضع أن أنواع Bifidobacterium تلعب دورًا كبيرًا في أمعاء الرضع الذين يرضعون (الشكل 2A). بعد ذلك بحثنا عن اختلافات عبر العينات في محاولة لوصف الملفات الميكروبية المختلفة لأمعاء الرضع. قمنا بفحص تنوع عينات أمعاء الرضع في مجموعتنا، باستخدام نهج تقليل الأبعاد (PCoA مع اختلاف BrayCurtis، الطرق)، ووجدنا أن العينات تتجمع في مجموعات متميزة (باستخدام K -means، ). كانت المجموعات الثلاث الأولى تحتوي على عينات مع نوع سائد واحد من Bifidobacterium (مع وفرة نسبية من ): B. breve و BL. longum و BL. infantis، وكانت المجموعة الرابعة تحتوي على عينات مهيمنة إما بنوع مختلف من Bifidobacterium (Bifidobacterium adolescentis، B. pseudocatenulatum) أو أنواع أخرى (تسمى “مختلط”، الشكل 3A). بينما عادةً ما كانت العينات المتتالية من نفس الرضيع تُعطى لنفس المجموعة، لوحظت تبديلات عشوائية في المجموعات، مما يعزز اكتشافنا أن الميكروبيوم يتغير في هذه الفترة الزمنية (الشكل 3B). بشكل عام، تسلط هذه التحليلات الضوء على أهمية أنواع Bifidobacterium في عيناتنا، حيث إنها عوامل رئيسية تؤثر على التباين في تركيب الميكروبات في أمعاء الرضع.
للمزيد من التحقيق في هذه المجموعات الأربعة من عينات أمعاء الرضع، قمنا بفحص تنوع ألفا لسكان الميكروبيوم في العينات الموجودة في كل مجموعة باستخدام مؤشر شانون. وجدنا أن تنوع ألفا للعينات داخل مجموعة BL. infantis كان أقل مقارنة بالعينات من المجموعات الأخرى (-test، , الشكل 3C)، مما يشير إلى أنه عندما توجد BL. infantis، فإنها تهيمن على
الشكل 2 | تركيب وديناميات أنواع Bifidobacterium في أمعاء الرضع و HMOs في حليب الأمهات. A وفرة نسبية لأنواع Bifidobacterium في أمعاء الرضع (ملونة) مع جميع الأنواع الأخرى مصنفة كـ “أخرى” (رمادي). يتم تجميع العينات من نفس الرضيع معًا، مرتبة حسب العمر. تشير الأسهم إلى العينات المأخوذة بعد إدخال الطعام الصلب. B التغيرات الزمنية في الوفرة النسبية للمجتمع الميكروبي في ثلاثة رضع (inf03 و inf16 و inf18) في
الأسابيع 30-40 الأولى من الحياة. البكتيريا الأكثر انتشارًا ملونة، والبقية تشير إليها كـ “أخرى” (رمادي). الألوان كما في (A)، مع ألوان إضافية للأنواع غير Bifidobacterium. C الوفرة النسبية لـ 16 HMO تم قياسها في حليب الأمهات. يتم تصنيف العينات إلى ثلاث مجموعات بناءً على ملفات HMO الخاصة بها، وتربط الأقواس العينات المأخوذة من نفس الأم (الطرق).
المجتمع عند مستويات عالية من الوفرة النسبية مما يترك مكانًا بيئيًا أصغر لبكتيريا أخرى في أمعاء الرضع. بعد ذلك، ركزنا على مجموعة “المختلطة”، وسألنا عما إذا كانت المتغيرات الإضافية قد تلعب دورًا في هذه الملفات الميكروبية. قمنا بفحص الرضاعة الطبيعية، واستخدام المضادات الحيوية من الأم أو الرضيع، وطريقة الولادة،
نوع الرضاعة (مضخة أو مباشرة)، وإدخال الأطعمة الصلبة، ومع ذلك لم نجد أي متغير محدد مرتبط بالملف الميكروبي لمجموعة “المختلطة”. كما هو متوقع، كانت مجموعة . إنفانتيس تتكون فقط من الرضع الذين لم يتلقوا كميات من حليب الأطفال أو كميات منخفضة .
الشكل 3 | تتأثر التباينات عبر العينات بشكل كبير بالأنواع السائدة من بيفيدوباكتيريوم. تحليل المكونات الرئيسية (PCoA) لعينات ميكروبيوم أمعاء الرضع باستخدام عدم التشابه لبراي-كورتيس. يتم تلوين النقاط بناءً على الأنواع السائدة من بيفيدوباكتيريوم في كل عينة، وتكون العينات التي لا تهيمن عليها بيفيدوباكتيريوم ملونة باللون الرمادي. يتم تصنيف العينات التي لا تحتوي على نوع سائد ( الوفرة النسبية) على أنها “لا نوع سائد” (أصفر). يمثل حجم كل نقطة الوفرة النسبية للبكتيريا السائدة في تلك العينة. تشمل البيضاوات أربع مجموعات تم تحديدها باستخدام تجميع k-means. تمثل كل مجموعة نوعًا سائدًا رئيسيًا (مشار إليه بلون البيضاوة): BL. إنفانتيس (أزرق)، BL. لونغوم (أخضر)، B. بريف
لتوصيف العلاقات بين الأنواع السائدة من بيفيدوباكتيريوم في أمعاء الرضع، قمنا بفحص حدوثها ضمن مجموعات لا تكون فيها سائدة. وجدنا أن . إنفانتيس و. بريف متعارضان، مما يتماشى مع دراسة سابقة في رضع هازدا , مما يعني وجود تنافس على نفس المكان في أمعاء الرضع (الشكل 3D). ومع ذلك، لا يزال غير واضح ما هو المكان المحدد الذي يتنافس عليه . بريف و. إنفانتيس، نظرًا لقدرة . بريف المحدودة على استخدام مجموعة متنوعة من HMOs . أخيرًا، لاحظنا BL. لونغوم في بعض العينات التي تهيمن عليها B. بريف (الشكل 2A)، مما يشير إلى أن . بريف قد تعتمد على مشتقات من BL. لونغوم من خلال التغذية المتبادلة في هذه العينات .
HMOs الفردية ليست مرتبطة بأنواع بيفيدوباكتيريوم محددة
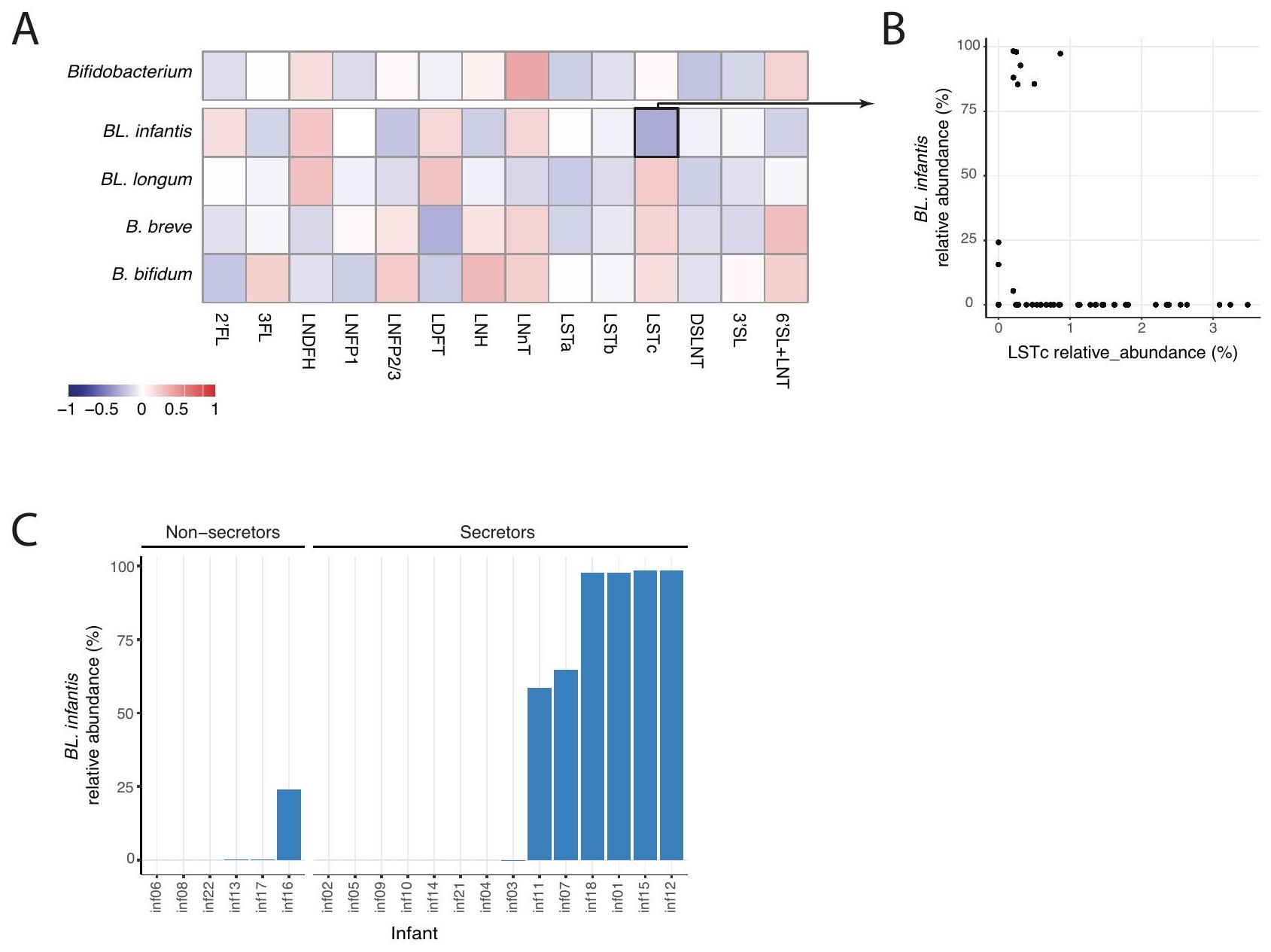
من المعروف جيدًا أن أنواع بيفيدوباكتيريوم المختلفة لديها قدرات مختلفة في استخدام HMOs . لذلك، قد تستفيد HMOs المحددة أنواع بيفيدوباكتيريوم معينة في أمعاء الرضع بناءً على ملفات استخدام HMOs الخاصة بها. ومع ذلك، لم نجد أي ارتباط كبير بين وفرة بيفيدوباكتيريوم بشكل عام والأنواع الرئيسية من بيفيدوباكتيريوم والأنواع الفرعية مع HMOs محددة (الشكل 4A) أو مجموعات HMOs (مركبة، سيلولوزية أو محايدة؛ الشكل التكميلية 5B). بالإضافة إلى ذلك، أظهرت نماذج الارتباط الخطي التي تأخذ في الاعتبار الرضع الفرديين عدم وجود ارتباط كبير بين
الشكل 4 | تركيبة HMO في حليب الأمهات لا تظهر أي ارتباط كبير مع بيفيدوباكتيريوم في أمعاء الرضع. ارتباطات بيرسون بين جميع HMOs المقاسة وجنس بيفيدوباكتيريوم، بالإضافة إلى الأنواع الفردية الرئيسية والأنواع الفرعية (BL. إنفانتيس، BL. لونغوم، B. بريف وB. بيفيدوم). لم يتم العثور على أي من هذه الارتباطات لتكون ذات دلالة إحصائية (-اختبار). مقارنة الوفرة النسبية
لـ LSTc في حليب الأمهات (-المحور) مع الوفرة النسبية لـ . إنفانتيس في ميكروبيوم أمعاء الرضع (-المحور). ج أقصى وفرة نسبية لـ . إنفانتيس في كل رضيع مقسمة إلى مجموعتين من الرضع مع أمهات سريعات وغير سريعات، مما يبرز الوفرة العالية لـ BL. إنفانتيس فقط في الرضع من الأمهات السريعات، ولكن ليس في جميعهم.
HMOs وأنواع بيفيدوباكتيريوم والأنواع الفرعية في أمعاء الرضع (عند الحاجة إلى FDR ). ومع ذلك، وجدنا أن BL. إنفانتيس أظهرت وفرة عالية () حصريًا في الرضع من الأمهات السريعات (الشكل 4B، -اختبار ). بالإضافة إلى ذلك، لاحظنا ارتباطًا سلبيًا متواضعًا وغير ذي دلالة () بين BL. إنفانتيس وLSTc (الشكل 4C). من الجدير بالذكر أهمية أخذ عامل التوقيت في الاعتبار عند تفسير هذه النتائج. قد تساهم الوجود المتأخر لـ BL. إنفانتيس في الأمعاء (الذي سيتم مناقشته بمزيد من التفصيل لاحقًا) والانخفاض التدريجي لـ LSTc مع مرور الوقت (الشكل التكميلية 4B، C) في الارتباط الملحوظ. يشير عدم وجود تباين في تركيبة HMO جنبًا إلى جنب مع عدم وجود ارتباطات بين HMOs والميكروبات إلى أن التحولات الميكروبية، وخاصة داخل أنواع بيفيدوباكتيريوم، لا يمكن تفسيرها بتغيير في تركيبة HMO في حليب الأمهات.
الميتاجينومات مع BL. إنفانتيس تحتوي على المزيد من الجينات المستخدمة في HMO
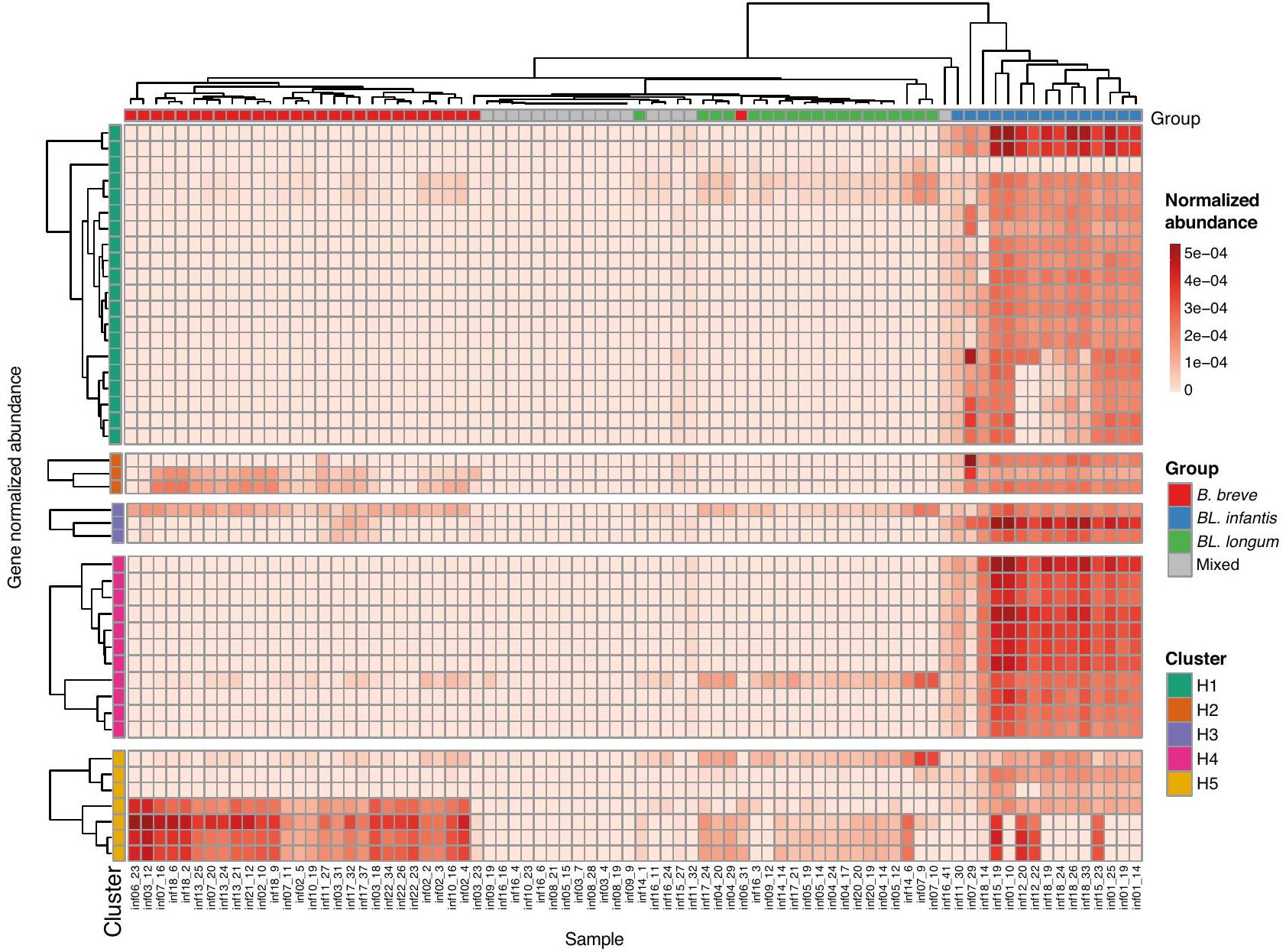
الميتاجينومات التي تم الحصول عليها من نقاط زمنية مختلفة لعدة رضع تحتوي على سلالات وأنواع متميزة، مما يؤدي إلى ملفات وفرة جينية متغيرة يمكن أن تمكن أنماط مختلفة من استخدام HMOs. لتقييم إمكانات استخدام HMOs لأنواع بيفيدوباكتيريوم المحددة في مجموعة بياناتنا، قمنا بالتحقيق في وجود جينات استخدام HMOs (HUGs) منظمة في خمسة تجمعات متميزة (; الشكل 5). لاحظنا أن الأنواع السائدة في كل عينة أثرت بشكل كبير على القدرة النظرية للميتا جينوم في استخدام HMOs. كما هو متوقع، أظهرت العينات التي تهيمن عليها BL. إنفانتيس أعلى وفرة من HUGs، مما يؤكد قدرتها الاستثنائية في استخدام HMOs . من الجدير بالذكر أن بعض هذه العينات أظهرت تباينًا عاليًا في وفرة الجينات من التجمعات H 1 وH 5، مما يشير إلى قدرة محتملة أقل على نقل بعض واستخدام لاكتو-ن-تتراوز (LNT) ولاكتو-ن-نيوتتراوز (LNnT.
من المثير للاهتمام، أن الميتاجينومات التي كانت مهيمنة بواسطة B. بريف أو . لونغوم احتوت أيضًا على جينات من تجمع H5، مما يبرز قدرتها على استخدام HMOs بناءً على لاكتو-ن-بيوز (LNB) . ومع ذلك، أظهرت العينات من مجموعات “المختلطة” الحد الأدنى أو عدم وجود HUGs، مما يشير إلى إما جينات بديلة لاستخدام HMOs أو عدم القدرة على استخدام HMOs تمامًا.
BL. إنفانتيس لا تستعمر أمعاء الرضع في الأسابيع الأولى من الرضاعة الطبيعية
BL. إنفانتيس هو الأكثر كفاءة في استخدام HMOs في أمعاء الرضع ، لذلك توقعنا أن يكون لدى BL. إنفانتيس ميزة لياقة في أمعاء الرضع الذين يرضعون من الثدي منذ الأيام الأولى من الرضاعة. ومع ذلك، على الرغم من أن الغالبية العظمى من الرضع في مجموعاتنا تم إرضاعهم منذ الولادة، تم اكتشاف BL. إنفانتيس بشكل أساسي بدءًا من 10 أسابيع من العمر (الشكل 6A). أظهرت نماذج الارتباط الخطي ارتباطًا إيجابيًا واضحًا بين الوفرة النسبية لـ BL. إنفانتيس والعمر (معامل )، والذي لم يتم العثور عليه لأي نوع آخر من
الشكل 5 | وفرة جينات استخدام HMOs في عينات الميتاجينوم. وفرة الجينات العادية لجينات استخدام HMOs (HUGs) في جميع عينات الرضع. يتم تصنيف الجينات (الصفوف) ووضع علامات عليها وفقًا لمجموعتها الجينية
(التجمع H1-H5). يتم توضيح العينات (الأعمدة) بناءً على مجموعتها المهيمنة المعينة.
أنواع بيفيدوباكتيريوم أو الأنواع الفرعية. بشكل عام، أظهرت BL. إنفانتيس أعلى وفرة عند 10-25 أسبوعًا، تليها انخفاض تدريجي في الوفرة (الشكل 6A).
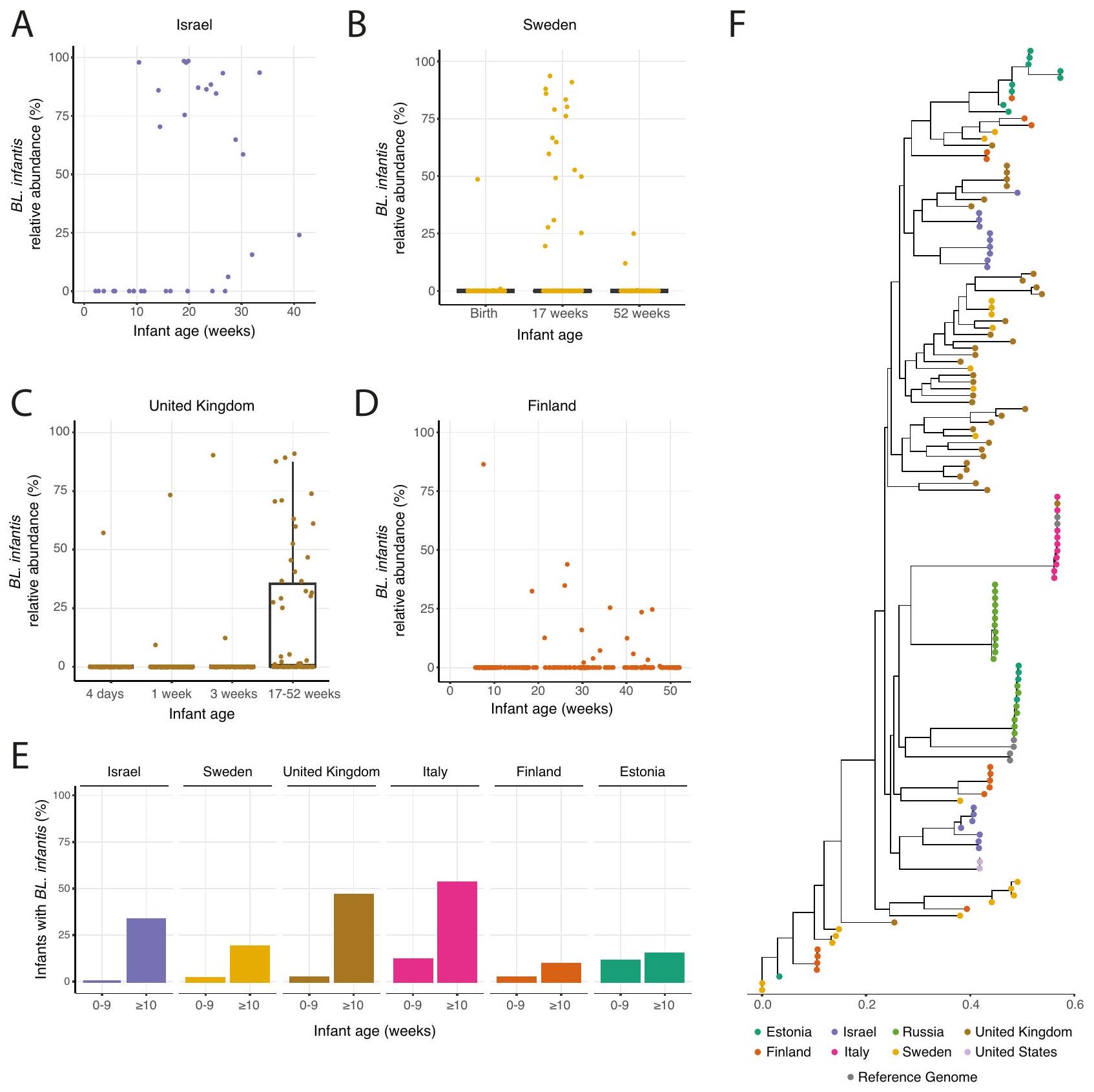
مكنت منهجيتنا الحسابية المبتكرة من استكشاف الاستعمار المتأخر لـ BL. إنفانتيس عبر مناطق جغرافية مختلفة، مما يوفر بوابة لمزيد من التحقيق المتعمق في هذه الظاهرة. وبالتالي، لدعم نتائجنا، قمنا بفحص مجموعات رضع إضافية من السويد ، المملكة المتحدة ، إستونيا ، إيطاليا ، روسيا ، الولايات المتحدة ومجموعتين من فنلندا تضم عينات من إجمالي 1,017 رضيعًا على مدار السنة الأولى من الحياة. عبر جميع المجموعات، لوحظ نمط مشابه من الاستعمار المتأخر لـ BL. إنفانتيس: في السويد، لم يتم ملاحظة BL. إنفانتيس عند الولادة تقريبًا، حيث وصلت ذروتها في الانتشار عند 17 أسبوعًا، تليها انخفاض تدريجي في كل من الانتشار والوفرة النسبية بحلول 52 أسبوعًا (الشكل 6B). في المملكة المتحدة، تم ملاحظة BL. إنفانتيس في أربعة فقط من 178 رضيعًا في الأسابيع الثلاثة الأولى من الحياة، وخلال فترة الرضاعة اللاحقة (17-52 أسبوعًا) تم العثور على BL. إنفانتيس في 28 رضيعًا (الشكل 6C). في فنلندا، كانت مجموعة واحدة تضم 126 رضيعًا تحتوي على رضيع واحد فقط بمستويات قابلة للاكتشاف من BL. إنفانتيس في الأسابيع العشرة الأولى من الحياة، و11 رضيعًا إضافيًا حصلوا عليها لاحقًا (الشكل 6D). في مجموعة فنلندية ثانية، لم يتم ملاحظة BL. إنفانتيس في أي من عينات الرضع (الأعمار أسابيع؛ فنلندا2، الشكل التوضيحي التكميلي 6A). في المجموعات الإيطالية والإستونية، لوحظ نمط مشابه ومع ذلك كانت وفرة BL. infantis النسبية أقل في معظم الرضع (الشكل التوضيحي التكميلي 6B،. كانت المجموعة من الولايات المتحدة تحتوي على عينات فقط من الأسبوعين الأولين من الحياة، مما يكشف عن عدم وجود رضّع مع . الرضع في الأول أسبوع من الحياة، وكان هناك رضيع واحد فقط من بين 77 رضيعًا لديه مستويات قابلة للاكتشاف في الأسبوع الثاني من الحياة (الشكل التوضيحي 6D). أخيرًا، في المجموعة الروسية التي تتكون من عينات تم جمعها من الرضع الذين تتراوح أعمارهم بين 12 أسبوعًا وما فوق، كان BL. infantis قابلاً للاكتشاف في 14 من 69 رضيعًا، حيث أصبح وجوده ملحوظًا فقط عند 20 أسبوعًا (الشكل التوضيحي 6E). بشكل عام، أظهرت جميع المجموعات استعمارًا متأخرًا لـ BL. infantis، والذي يبدأ عادةً عند 10 أسابيع من العمر، أو لاحقًا.-اختبار،; الشكل 6E، الشكل التوضيحي 6F).
لفحص الاختلافات بين سلالات BL. infantis عبر الدول المختلفة، ركزنا بعد ذلك علىتركيب سلالة إنفانتيس على مستوى السلالة، باستخدام ملفات تعريف SNP على “المحدد حديثًاوجدنا أن سلالات BL. infantis داخل نفس الرضيع في مجموعتنا كانت أكثر تشابهًا مع بعضها البعض من السلالات بين الرضع غير المرتبطين.-اختبار،الشكل التوضيحي 6G). علاوة على ذلك، في بعض البلدان (مثل إيطاليا وروسيا)، كانت معظم السلالات متشابهة جدًا، بينما في بلدان أخرى (مثل السويد والمملكة المتحدة) كان هناك تنوع أكبر بكثير بين السلالات (الشكل 6F، الشكل التوضيحي 6H). كانت السلالات من مجموعاتنا الإسرائيلية متجمعة في مجموعتين متميزتين، واحدة أكثر تشابهًا مع السلالات الموجودة في الولايات المتحدة (أرجواني فاتح، الشكل 6F) والأخرى أقرب إلى السلالات الموجودة في المملكة المتحدة (بني، الشكل 6F). على وجه التحديد، كانت السلالات من العينات الإيطالية متميزة جدًا عن جميع السلالات الأخرى، باستثناء سلالة واحدة من المملكة المتحدة. أخيرًا، تم تجميع جينومات BL. infantis المرجعية في ثلاث مجموعات، بعضها مطابق للسلالات الإيطالية، بينما تجمعت أخرى بشكل وثيق مع السلالات الروسية والإستونية (رمادي، الشكل 6F). تتيح لنا هذه النتائج استكشاف التنوع الموجود داخل.
الشكل 6 | الاستعمار المتأخر وانخفاض انتشار BL. infantis في أمعاء الرضع في مواقع جغرافية متعددة. أ. الوفرة النسبية لـ. الرضع في مجموعتنا الإسرائيلية، بين الرضع حيث تم الكشف عن . infantis في أي نقطة زمنية. ب-د وفرة نسبية لـ. الرضع في نقاط زمنية مختلفة في عينات من ب السويد (عينات من 100 رضيع) المملكة المتحدة (عينات من 169 رضيعًا) و د فنلندا (عينات من 108 رضيعًا). بالنسبة للسويد والمملكة المتحدة حيث تم جمع العينات في نقاط زمنية محددة، فإن الرسوم البيانية الصندوقية هي موضحة، مع حدود الصندوق عند النسب المئوية 25 و75، والوسيط مميز. تمثل الشعيرات 1.5 * IQR والنقاط التي تتجاوزها هي القيم الشاذة. E نسبة الرضع في مواقع جغرافية متعددة تحمل BL. infantis في الأسابيع العشرة الأولى من الحياة وفي الأسبوع العاشر وما بعده. F شجرة النشوء والتطور تعرضتم العثور على سلالات من نوع .infantis عبر جميع المجموعات. يتم تمثيل السلالة السائدة من كل عينة، وتُرمز السلالات بالألوان بناءً على المجموعة المعنية، مما يبرز أن بعض الدول (مثل إيطاليا) لديها سلالات متشابهة جدًا في جميع العينات. إنفانتيس وتسليط الضوء على التباين المثير للاهتمام عبر المواقع الجغرافية.
نقاش
في هذه الدراسة قدمنا نهجًا مبتكرًا لقياس. إنفانتيس وتمييزه عن BL. لونغوم في البيانات الميتاجينومية. تتيح طريقتنا للباحثين التركيز على دراسة هذا النوع الفرعي المتميز وعلاقاته مع HMOs من البيانات الميتاجينومية الموجودة. يمكن تكييف هذا النهج لتمييز الأنواع الفرعية في أنواع ميكروبية إضافية، وخاصة داخل B. لونغوم (مثل الأنواع الفرعية BL. سويس و BL. سويلوم) بمجرد توفر أعداد كافية من أصبحت الجينومات المرجعية متاحة. استخدمنا نهجنا لاستكشاف التنوع داخل ميكروبيوم أمعاء الرضع واكتشفنا عدم وجود ارتباطات مع HMOs الفردية الموجودة في حليب الأمهات. كشفت تحليلاتنا أن التباين بين العينات تأثر بشكل كبير بالأنواع السائدة من بيفيدوباكتيريوم في كل عينة.
أشارت الأبحاث السابقة إلى أن استعمار أنواع بيفيدوباكتيريوم في أمعاء الرضع قد يتأثر بتأثيرات الأولوية.. ومع ذلك، كشفت دراستنا عن تغييرات كبيرة في الأنواع السائدة من بيفيدوباكتيريوم داخل نفس الرضيع على مدار عدة أسابيع (الشكل 3A، B). وهذا يشير إلى أنه مع مرور الوقت، هناك عوامل إضافية مسؤولة عن بيفيدوباكتيريوم ازدهار الأنواع، مثل تنافس الأنواع والتغذية المتبادلة. على سبيل المثال، تم الإبلاغ عن أن B. breve، على الرغم من قدرتها المحدودة على استخدام HMOs، يمكن أن تتفوق على منافسين أقوى إذا تم إدخالها مبكرًا في مجتمع ميكروبي.. بالإضافة إلى ذلك،. بريفيدوبكتيريوم لديه القدرة على التغذية المتبادلة على السكريات الأحادية المستمدة من أوليغوسكاريد الحليب بواسطة أنواع أخرى من بريفيدوبكتيريوم. هذا يعني أن . قد تهيمن بريف على السكان في البداية عندما تكون الكربوهيدرات متاحة، لكنها تفقد المنافسة لاحقًا لصالح أنواع أخرى من بيفيدوباكتيريوم بمجرد استنفاد هذه الكربوهيدرات. في بياناتنا، وجدنا أن كان لدى مجموعة بريف dominated مجموعة متنوعة من الكائنات الدقيقة (الشكل 3C)، ربما لأنها تتغذى على مشتقات HMOs من أنواع أخرى. هناك حاجة إلى مزيد من البحث لفهم التغيرات الميكروبية في أمعاء الرضع وتأثير التغذية المتبادلة على الديناميات الميكروبية في أمعاء الرضع.
بينما ظلت HMOs الموجودة في حليب الأم مستقرة نسبيًا على مدى عدة أسابيع، لاحظنا تغييرات ملحوظة في ميكروبيوم أمعاء الرضيع خلال هذه الفترة. وهذا يشير إلى أن حتى التغيرات الطفيفة في تركيبة حليب الثدي قد تؤثر على تطور ميكروبيوم الأمعاء. بدلاً من ذلك، من الممكن أن تلعب مكونات أخرى موجودة في حليب الثدي، مثل السيتوكينات والميكروRNAs والأجسام المضادة، دورًا في التأثير على ميكروبيوم الرضيع.. بالإضافة إلى ذلك، لا يمكننا استبعاد التأثيرات البيئية التي قد تساهم في هذه التغيرات مثل بدء الرعاية النهارية، والتي تحدث عادة في إسرائيل في سن 15-26 أسبوعًا. تشير نتائجنا إلى أنه لم تكن هناك ارتباطات ذات دلالة إحصائية بين HMOs وBifidobacterium، مما يدعم الفكرة القائلة بأن هناك عوامل إضافية تتجاوز HMOs تشارك في تشكيل ميكروبيوم الرضع. الدراسات السابقةلقد قمت بدراسة تركيبة الميكروبيوم وعلاقته بالهيمو أوليغوسكاريد باستخدام تسلسل الأمبليكون 16S-rRNA. وقد أبلغت هذه الدراسات عن نتائج متباينة، حيث لم تجد بعض الدراسات أي ارتباطات ذات دلالة إحصائية، بينما لاحظت دراسات أخرى ارتباطات متواضعة. ومن المثير للاهتمام أن بعض الدراسات حددت ارتباطًا سلبيًا بين وحدات تصنيف بيفيدوباكتيريوم (OTUs) والعديد من الهيمو أوليغوسكاريد.يمكن أن يُعزى ذلك إلى انخفاض وفرة بيفيدوباكتيريوم بشكل عام في أمعاء الرضع مع مرور الوقت، مصحوبًا بزيادة في بعض HMOs المحددة، بما يتماشى مع نتائجنا المتعلقة بـ LSTc و BL. infantis (الشكل 4B). بالإضافة إلى ذلك، من الممكن أن تكون مثل هذه الارتباطات المحددة قابلة للرصد فقط باستخدام مجموعة أكبر.
أخيرًا، وجدنا أن BL. infantis لا يستعمر عادةً أمعاء الرضع في الأسابيع الأولى من الرضاعة الطبيعية وأن ليس جميع الرضع الذين يرضعون من الثدي لديهم مستويات قابلة للاكتشاف من BL. infantis. من المهم، عندماتم العثور على BL. infantis في أمعاء الرضع، وكان غالبًا ما يهيمن على مجتمع الميكروبيوم المعوي. وقد عزز تحليل مجموعات إضافية من الرضع من مواقع جغرافية متعددة نتائجنا بشأن الاستعمار المتأخر لـ BL. infantis في أمعاء الرضع. وقد أفادت دراسات سابقة بوجود انتشار منخفض لـ. الرضع خلال النقاط الزمنية المبكرةومع ذلك، لم تتضمن هذه الدراسات أخذ عينات متكررة في الأشهر الأولى من الحياة، مما يفتقر إلى القدرة على تحديد توقيت وصول BL. infantis بدقة. اقترحت دراسة سابقة أن وصول BL. infantis يتأثر بتاريخ ممارسات الرضاعة الطبيعية في بلد معين.، مما يؤثر على التعرض العام لـسلالات .infantis. من المحتمل أن تكون البلدان التي لديها معدلات رضاعة طبيعية تاريخياً أقل لديها انتشار أقل لـ. الرضع، مما يؤدي إلى اكتساب الرضع . إنفانتيس في مرحلة لاحقة من خلال النقل الأفقي.
على الرغم من أن BL. infantis هو أحد الكائنات المعوية الأكثر دراسة في الرضع، إلا أن الجينومات المرجعية المتاحة للجمهور لا تغطي كل التنوع الجيني لهذا النوع الفرعي (رمادي، الشكل 6F). لتوسيع البحث المتعلق بـ BL. infantis، يجب أن يتم توصيف جينومات مرجعية إضافية بالكامل، من عزلات طبيعية تمثل مواقع جغرافية وأنماط حياة متنوعة. هناك حاجة إلى مزيد من البحث، خاصة أخذ عينات طولية من الرضع والبيئة المحيطة بهم، لتوضيح توقيت ومصادر اكتساب الرضع.. الرضع وفهم الفروق الملحوظة بين البلدان.
طرق
جمع العينات
تم جمع عينات من حليب الثدي والبراز كجزء من مجموعة حليب الثدي للأطفال (BMB) من الأمهات والرضع من الولادة حتى عمر سنة واحدة. تم جمع عينات البراز باستخدام eSwab.مع 1 مل من وسط أميز السائل + 1 مسحة FLOQ العادية (كوبان) من أجل الحفاظ على السكان البكتيرية. تم جمع عينات حليب الثدي بواسطة مضخة أو يدويًا وتخزينها في أنابيب معقمة. تم جمع كلا نوعي العينات من قبل الأمهات في منازلهن وتخزينها فيلمدة تصل إلى 24 ساعة ثم يتم شحنها إلى المختبر وتخزينها على المدى الطويل في.
بناء مكتبة الميتاجينوم وتسلسلها
تم استخراج الحمض النووي من عينات البراز باستخدام مجموعة DNeasy PowerSoil Pro (#47014، QIAGEN). تم إعداد مكتبات تسلسل إيلومينا باستخدام مجموعة إعداد مكتبة الحمض النووي Nextera XT (FC-131-1096، إيلومينا) وفقًا لبروتوكول الشركة المصنعة الموصى به مع نصف الحجم والحمض النووي. تم تسلسل العينات باستخدام تسلسل إيلومينا أحادي النهاية 150 نقطة أساس على جهاز NextSeq 500.
تقدير الأنواع الفرعية من B. longum
لتحديد علامات محددة تحت الأنواع لبكتيريا B. longum بدأنا بـ 116تم تنزيل جينومات مرجعية طويلة من NCBI مع اكتمالوتلوث (البيانات التكميلية 2). تم تصنيف الجينومات المرجعية إلى ب. لونغوم تحت النوع لونغوم (BL. longum)، ب. لونغوم تحت النوع إنفانتيس (BL. infantis) وغير معروف بناءً على توضيح NCBI، مما ترك 30 مرجعًا من BL. longum و16 مرجعًا من BL. infantis. حيث يمكن أن تنتمي بعض المراجع غير المعروفة إلى إما . الرضع أو قمنا بإزالة جميعها من أجل التحديد. قررنا عدم تضمين الأنواع الفرعية BL. suis و BL. suillum في طريقتنا بسبب التوافر المحدود لجينومات المرجع لهذه الأنواع الفرعية مما أدى إلى صعوبة في توليد جينات علامات موثوقة. بالإضافة إلى ذلك، نادراً ما تم الإبلاغ عن هذه الأنواع الفرعية في البشر.لذا فهي أقل صلة في إعداداتنا. لذلك من المهم أن نلاحظ أنه باستخدام طريقتنا،. يمكن أن يتم التعرف على S. suis و BL. suillum بشكل خاطئ كنوع فرعي مختلف أو يمكن التعرف عليه بدلاً من ذلك كـ نوع طويل مع أصناف فرعية غير مصنفة.
بانفلاين3تم استخدامه لتحليل الجينوم الشامل لجميع 46 جينوم مرجعي. كشف تجميع جميع ملفات الحضور/الغياب في الجينوم الشامل عن مجموعتين واضحتين، الأولى تضم 14 سلالة من BL. infantis والثانية تشمل جميع. سلالات طويلة واثنان سلالات BL. infantis (BL. infantis 157 F، BL. infantis CCUG52486؛ الشكل التوضيحي 7). أظهرت التحليلات الإضافية أن هذينجينومات مرجعية للرضع التي تم تجميعها معلم تحتوي جينومات المرجع الطويلة على مجموعة استخدام H1 HMO التي تحدد BL. infantis. بالإضافة إلى ذلك، أظهرت دراسة سابقة أن هذين الجينومين المرجعيين لـ BL. infantis من المحتمل جداً أن يكونا. لونغوم، استنادًا إلى النشوء والتطور لجينوم بانجينوم الأساسي لـ 158 سلالة من ب. لونغومبشكل عام، تم الاشتباه في أن BL. infantis 157 F و BL. infantis CCUG52486157 تم تصنيفهما بشكل خاطئ وتم استبعادهما من التحليل الإضافي.
تم اختيار جينات علامات محددة تحت الأنواع على مرحلتين. أولاً، باستخدام الجينوم الشامل، وجدنا 331 جينًا كانت موجودة فيمن نوع فرعي واحد وغير موجود على الإطلاق في النوع الفرعي الآخر. على سبيل المثال، جين كان موجودًا في 13 من أصل 14 مرجعًا لـ BL. infantis ولم يكن موجودًا في أيتم اختيار مرجع longum ليكون جين علامة لـ. الرضع. بعد ذلك، تم تصفية جميع الجينات المختارة لتكون محددة على مستوى الأنواع إلى. longum ولتأكيد عدم وجودها في أنواع بيفيدوباكتيريوم الأخرى. للقيام بذلك، استخدمنا Blastn 2.12.0 لرسم جميع جينات العلامة إلى قاعدة بيانات النوكليوتيدات. من بين 331 جينًا محتملًا للعلامة، تطابق 84 مع أنواع أخرى (مثل B. breve) معتمت تصفية الجينات بسبب عدم توافقها وتغطيتها لأكثر من 50%. شملت مجموعة الجينات النهائية لدينا 119 علامة من BL. infantis و128 علامة من BL. longum (البيانات التكميلية 3). تم تخصيص قاعدة بيانات MetaPhlAn لتشمل الجينات المحددة حديثًا باستخدام التعليمات الموصوفة لـ MetaPhlAn.https://github.com/biobakery/ميتا فليان/wiki/ميتا فليان-4) ثم تم استخدام ميتا فليان 4 مع معلمات –index و –bowtie2db وقاعدة بيانات الجينات المحددة المخصصة لدينا.
للتحقق من النتائج، تم تحديد الأنواع الفرعية من B. longum باستخدام qPCR مع بادئات محددة للأنواع الفرعية.لـ BL. longum (F: GTGTGGATTACCTGCCTACC، R: GTCGCCAACCTTGACCACTT) و BL. infantis (F: ATGATGCGCTGCCACTGTTA، R: CGGTGAGCGTCAATGTATCT). تم تقييم كفاءة البرايمرات من خلال اختبارها في خمس تخفيفات. تم إجراء qPCR عند لمدة 10 ثوانٍ، تليها 40 دورة من لمدة 10 ثوانٍ و لمدة 30 ثانية. النسبة بين . الرضع وتم حساب .longum باستخدام طريقة دلتا-دلتا Ct.
تمت إزالة قراءات المضيف باستخدام خط أنابيب داخلي عن طريق محاذاة القراءات إلى الجينوم البشري باستخدام Bowtie2. (2.4.5-1). تم تصفية العينات وقصها لملحقات Nextera باستخدام fastq-mcf، ea-utilsتم إجراء التصنيف الضريبي باستخدام MetaPhlAn4مع قاعدة البيانات الفريدة لدينا كما هو موصوف أعلاه. تم إجراء التحليل الوظيفي باستخدام HUMAnN3. عناقتم اختيارها من مجموعات HMO الموصوفة سابقًاتم إجراء تحليل الإجهاد باستخدام StrainPhIAnمع المعلمات الافتراضية و –sample_with_n_markers 50. تم حساب SNPs لعلامات الجينات BL. infantis باستخدام المحاذاة المتعددة التسلسلات (MSA) التي تم إنتاجها بواسطة StrainPhIAn 4 (–معدل الطفرات). تم اعتبار BL. infantis قابلاً للاكتشاف عندما كانت وفرتها النسبيةتم إجراء مزيد من التحليل باستخدام نظام داخلي (4.2.2) سكريبت يستخدم dplyr (1.1.2)، tidyr (1.3.0) و tidyverse (2.0.0). تم إنشاء الرسوم البيانية باستخدام ggplot2 (3.4.2) و ggforce (0.4.1)، تم استخدام الألوان من RColorBrewer (1.1-3) وأصدقاء (1.7). تم إنشاء خرائط الحرارة باستخدام pheatmapتم حساب تنوع ألفا وبيتا باستخدام “التنوع” (مؤشر شانون) و”vegdist” (اختلاف براي-كورتيس) من حزمة فيغانتم إنشاء حزمة (2.6-4) و PCoA باستخدام apeتم إنتاج شجرة النشوء والتطور باستخدام حزمة ggtree (5.7-1). (3.6.2) وتم إنشاء مخططات سانكي باستخدام ggsankey (0.0.99999). تم تنزيل مجموعات إضافية من NCBI Sequence Read Archive كما يلي: السويد (PRJEB6456)، الولايات المتحدة (PRJNA591079)، إيطاليا (PRJNA352475)، المملكة المتحدة (PRJEB32631)، فنلندا، إستونيا وروسيا (PRJNA497734) ومجموعة إضافية من فنلندا (فنلندا2، PRJNA475246).
تحديد HMO
تم شراء معايير HMO المستخدمة في هذه الدراسة من مختبرات ديكسترا، المملكة المتحدة. وشملت هذه 2′-فوكوسيللاكتوز (3-فوكوسيللاكتوز (3FL)-سياليللاكتوز ( SL)، -سياليللاكتوز (SL)، لاكتو-N-تتراوز (LNT)، ديسياليللاكتو-N-تتراوز (DSLNT)، لاكتوديفوكوتتراوز (LDFT)، لاكتو-N-ديفوكوهكساوز 1 (LNDFH)، لاكتو-N-فوكوبنتاوز (LNFP) 1، 2، و3، لاكتو-N-هكساوز (LNH)، لاكتو-N-نيوتتراوز (LNnT) وسايليل-لاكتو-N-تتراوز (LST) أ، ب وج. تم استخدام B6-تريساكاريد الخطي كمعيار داخلي.
تم إجراء قياس HMO كما هو موصوف سابقًاباختصارتم دمج حليب الإنسان مع ثلاثي السكاريد لينير B-6 (مختبرات ديكسترا، المملكة المتحدة) وماء بدرجة HPLC، ثم تم إخضاعه لعمود C18 (ثيرمو فيشر #60108-390) وعمود كاربوجراف (ثيرمو فيشر #60302-606) لإزالة البروتينات والأملاح على التوالي. تم وسم العينات باستخدام 2-أمينوبنزاميد (، سيغما) لمدة ساعتين في زيادةتمت الإزالة باستخدام أعمدة السيليكا (Thermo Scientific، #60300-482). تم فصل العينات بواسطة HPLC مع الكشف عن الفلورية على عمود TSKgel Amide-80 (Tosoh Bioscience، طوكيو، اليابان) مع تدرج خطي لنظام المذيب 50 مليمول من أمونيوم الفورمات/الأسيتونيتريل. تم استخدام أوقات الاحتفاظ للمعايير القياسية المشتراة من HMOs لتوضيح قمم HPLC. لم يكن من الممكن فصل قمم 6’SL و LNT وبالتالي تم حسابها معًا. تم حساب كمية كل HMO فردي بناءً على التطبيع مع المعيار الداخلي (البيانات التكميلية 4). تم تحديد الوفرة النسبية لكل من HMOs الفردية بواسطة
تحديد مجموع 16 أوليغوسكاريد محدد كـ 100% من HMOs الكلية.
التحليل الإحصائي
لم يتم استخدام أي طريقة إحصائية لتحديد حجم العينة مسبقًا. لم يكن الباحثون معزولين عن التخصيص أثناء التجارب وتقييم النتائج. تم إجراء اختبار -test المستقل لاختبار الفرق بين المجموعات عند الإشارة باستخدام دالة R ” -test”. تم إجراء اختبار المقترن بين النسب المئوية للرضع في كل مجموعة الذين كانوا . infantis قبل 10 أسابيع والنسبة المئوية للرضع الذين كانت لديهم مستويات قابلة للكشف من . infantis بعد 10 أسابيع. تم حساب المسافات بين عينات ميكروبيوم أمعاء الرضع المتتالية وبين تركيبات HMO في حليب الثدي باستخدام عدم التشابه Bray-Curtis باستخدام دالة “vegdist” من حزمة vegan (2.6-4). تم إجراء الارتباط بين السكان الميكروبيين وتركيبة HMO باستخدام ارتباط بيرسون مع دالة R “cor”. تم حساب قيم – المعدلة باستخدام “corr.test” من حزمة psych .
نماذج الارتباط الخطي
تم استخدام حزمة “Maaslin2” لإجراء نماذج خطية من أجل العثور على الارتباطات بين 16 HMO وأنواع Bifidobacterium وفي بكتيريا أمعاء الرضع. تم تعيين الفرد كعامل عشوائي لأخذ تأثير كل زوج من الأم والرضيع في الاعتبار. بالإضافة إلى ذلك، تم استخدام “Maaslin2” لإجراء نماذج ارتباط خطية للعمر مقارنة بأنواع Bifidobacterium، مع تعديلها للأفراد.
ملخص التقرير
معلومات إضافية حول تصميم البحث متاحة في ملخص تقرير Nature Portfolio المرتبط بهذه المقالة.
توفر البيانات
تم إيداع بيانات تسلسل الميتاجينوم المصفاة من البشر التي تم إنشاؤها في هذه الدراسة في قاعدة بيانات SRA تحت BioProject PRJNA994433. يتم توفير بيانات التعريف الخاصة بالمجموعة في البيانات التكميلية 1. يتم تقديم نتائج تقدير HMO في البيانات التكميلية 4 ونتائج MetaPhlAn في البيانات التكميلية 5.
Westerfield, K. L., Koenig, K. & Oh, R. Breastfeeding: Common questions and answers. Am. Fam. Physician 98, 368-373 (2018).
Sela, D. A. & Mills, D. A. Nursing our microbiota: molecular linkages between bifidobacteria and milk oligosaccharides. Trends Microbiol. 18, 298-307 (2010).
Marcobal, A. et al. Consumption of human milk oligosaccharides by gut-related microbes. J. Agric. Food Chem. 58, 5334-5340 (2010).
Zivkovic, A. M., German, J. B., Lebrilla, C. B. & Mills, D. A. Human milk glycobiome and its impact on the infant gastrointestinal microbiota. Proc. Natl Acad. Sci. USA 108, 4653-4658 (2011).
Han, S. M. et al. Maternal and infant factors influencing human milk oligosaccharide composition: beyond maternal genetics. J. Nutr. https://doi.org/10.1093/jn/nxab028. (2021)
Lewis, Z. T. et al. Maternal fucosyltransferase 2 status affects the gut bifidobacterial communities of breastfed infants. Microbiome 3, 13 (2015).
Plows, J. F. et al. Longitudinal changes in human milk oligosaccharides (HMOs) over the course of 24 months of lactation. J. Nutr. 876-882 https://doi.org/10.1093/jn/nxaa427. (2021)
Yassour, M. et al. Natural history of the infant gut microbiome and impact of antibiotic treatment on bacterial strain diversity and stability. Sci. Transl. Med. 8, 343ra81 (2016).
Bäckhed, F. et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe 17, 690-703 (2015).
Le Doare, K., Holder, B., Bassett, A. & Pannaraj, P. S. Mother’s Milk: a purposeful contribution to the development of the infant microbiota and immunity. Front. Immunol. 9, 361 (2018).
Sela, D. A. et al. Bifidobacterium longum subsp. infantis ATCC -fucosidases are active on fucosylated human milk oligosaccharides. Appl. Environ. Microbiol. 78, 795-803 (2012).
Underwood, M. A. et al. A comparison of two probiotic strains of bifidobacteria in premature infants. J. Pediatr. 163, 1585-1591.e9 (2013).
Garrido, D., Dallas, D. C. & Mills, D. A. Consumption of human milk glycoconjugates by infant-associated bifidobacteria: mechanisms and implications. Microbiology 159, 649-664 (2013).
Garrido, D. et al. Comparative transcriptomics reveals key differences in the response to milk oligosaccharides of infant gutassociated bifidobacteria. Sci. Rep. 5, 13517 (2015).
Ruiz-Moyano, S. et al. Variation in consumption of human milk oligosaccharides by infant gut-associated strains of bifidobacterium breve. Appl. Environ. Microbiol. 79, 6040-6049 (2013).
Duar, R. M. et al. Comparative genome analysis of bifidobacterium longum subsp. infantis strains reveals variation in human milk oligosaccharide utilization genes among commercial probiotics. Nutrients 12, 3247 (2020).
LoCascio, R. G. et al. Glycoprofiling of bifidobacterial consumption of human milk oligosaccharides demonstrates strain specific, preferential consumption of small chain glycans secreted in early human lactation. J. Agric. Food Chem. 55, 8914-8919 (2007).
Duboux, S., Ngom-Bru, C., De Bruyn, F. & Bogicevic, B. Phylogenetic, functional and safety features of 1950s B. infantis strains. Microorganisms 10, 203 (2022).
Sela, D. A. et al. The genome sequence of bifidobacterium longum subsp. infantis reveals adaptations for milk utilization within the infant microbiome. Proc. Natl Acad. Sci. USA. 105, 18964-18969 (2008).
Derrien, M. et al. Gut microbiome function and composition in infants from rural kenya and association with human milk oligosaccharides. Gut Microbes 15, 2178793 (2023).
Pace, R. M. et al. Variation in human milk composition Is related to differences in milk and infant fecal microbial communities. Microorganisms 9, 1153 (2021).
Borewicz, K. et al. The association between breastmilk oligosaccharides and faecal microbiota in healthy breastfed infants at two, six, and twelve weeks of age. Sci. Rep. 10, 4270 (2020).
Barnett, D. J. M. et al. Human milk oligosaccharides, antimicrobial drugs, and the gut microbiota of term neonates: observations from the KOALA birth cohort study. Gut Microbes 15, 2164152 (2023).
Borewicz, K. et al. Correlating infant faecal microbiota composition and human milk oligosaccharide consumption by microbiota of one-month old breastfed infants. Mol. Nutr. Food Res. 63, e1801214 (2019).
Locascio, R. G. et al. A versatile and scalable strategy for glycoprofiling bifidobacterial consumption of human milk oligosaccharides. Microb. Biotechnol. 2, 333-342 (2009).
Seppo, A. E. et al. Infant gut microbiome is enriched with bifidobacterium longum ssp. infantis in old order mennonites with traditional farming lifestyle. Allergy 76, 3489-3503 (2021).
Young, S. L. et al. Bifidobacterial species differentially affect expression of cell surface markers and cytokines of dendritic cells harvested from cord blood. Clin. Diagn. Lab. Immunol. 11, 686-690 (2004).
Davis, J. C. C. et al. Growth and morbidity of gambian infants are influenced by maternal milk oligosaccharides and infant gut microbiota. Sci. Rep. 7, 40466 (2017).
Taft, D. H. et al. Bifidobacterium species Colonization in Infancy: a global cross-sectional comparison by population history of breastfeeding. Nutrients 14, 1423 (2022).
Tso, L., Bonham, K. S., Fishbein, A., Rowland, S. & Klepac-Ceraj, V. Targeted high-resolution taxonomic Identification of bifidobacterium longum subsp. infantis using human milk oligosaccharide metabolizing genes. Nutrients 13, 2833 (2021).
Casaburi, G. et al. Metagenomic insights of the infant microbiome community structure and function across multiple sites in the United States. Sci. Rep. 11, 1472 (2021).
LoCascio, R. G., Desai, P., Sela, D. A., Weimer, B. & Mills, D. A. Broad conservation of milk utilization genes in bifidobacterium longum subsp. infantis as revealed by comparative genomic hybridization. Appl. Environ. Microbiol. 76, 7373-7381 (2010).
Blanco-Míguez, A. et al. Extending and improving metagenomic taxonomic profiling with uncharacterized species using MetaPhlAn 4. Nat. Biotechnol. https://doi.org/10.1038/s41587-023-01688w (2023)
Yanokura, E. et al. Subspeciation of bifidobacterium longum by multilocus approaches and amplified fragment length polymorphism: description of B longum subsp. suillum subsp. nov., isolated from the faeces of piglets. Syst. Appl. Microbiol. 38, 305-314 (2015).
Yatsunenko, T. et al. Human gut microbiome viewed across age and geography. Nature 486, 222-227 (2012).
Avershina, E. et al. Bifidobacterial succession and correlation networks in a large unselected cohort of mothers and their children. Appl. Environ. Microbiol. 79, 497-507 (2013).
Bode, L. Human milk oligosaccharides: every baby needs a sugar mama. Glycobiology 22, 1147-1162 (2012).
Dai, D. L. Y. et al. Breastfeeding enrichment of B. longum subsp. infantis mitigates the effect of antibiotics on the microbiota and childhood asthma risk. Med ( ) https://doi.org/10.1016/j.medj. 2022.12.002 (2022).
Olm, M. R. et al. Robust variation in infant gut microbiome assembly across a spectrum of lifestyles. Science 376, 1220-1223 (2022).
Ojima, M. N. et al. Priority effects shape the structure of infant-type bifidobacterium communities on human milk oligosaccharides. ISME J. https://doi.org/10.1038/s41396-022-01270-3 (2022)
Asakuma, S. et al. Physiology of consumption of human milk oligosaccharides by infant gut-associated bifidobacteria. J. Biol. Chem. https://doi.org/10.1074/jbc.M111.248138 (2011).
Shao, Y. et al. Stunted microbiota and opportunistic pathogen colonization in caesarean-section birth. Nature 574, 117-121 (2019).
Vatanen, T. et al. Genomic variation and strain-specific functional adaptation in the human gut microbiome during early life. Nat. Microbiol 4, 470-479 (2019).
Ferretti, P. et al. Mother-to-infant microbial transmission from different body sites shapes the developing infant gut microbiome. Cell Host Microbe 24, 133-145.e5 (2018).
Mitchell, C. M. et al. Delivery mode affects stability of early infant gut microbiota. Cell Rep. Med. 1, 100156 (2020).
Yassour, M. et al. Strain-level analysis of mother-to-child bacterial transmission during the first few months of Life. Cell Host Microbe 24, 146-154.e4 (2018).
Egan, M., O’Connell Motherway, M., Ventura, M. & van Sinderen, D. Metabolism of sialic acid by bifidobacterium breve UCC2003. Appl. Environ. Microbiol. 80, 4414-4426 (2014).
Lawson, M. A. E. et al. Breast milk-derived human milk oligosaccharides promote Bifidobacterium interactions within a single ecosystem. ISME J. 14, 635-648 (2020).
Carr, L. E. et al. Role of human milk bioactives on infants’ gut and immune health. Front. Immunol. 12, 604080 (2021).
Collado, M. C. et al. Longitudinal study of cytokine expression, lipid profile and neuronal growth factors in human breast milk from term and preterm deliveries. Nutrients 7, 8577-8591 (2015).
Beghini, F. et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. Elife 10, e65088 (2021).
Díaz, R., Torres-Miranda, A., Orellana, G. & Garrido, D. Comparative genomic analysis of Novel bifidobacterium longum subsp. longum strains reveals functional divergence in the human gut microbiota. Microorganisms 9, 1906 (2021).
Kim, H.-B. et al. Development of real-tme PCR assay to specifically detect 22 bifidobacterium Species and subspecies using comparative genomics. Front. Microbiol. 11, 2087 (2020).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357-359 (2012).
Aronesty, E. ea-utils: Command-line Tools for Processing Biological Sequencing Data. https://www.researchgate.net/publication/ 319159822_ea-utils_Command-line_tools_for_processing_ biological_sequencing_data (2011).
Wickham, H., François, R., Henry, L., Müller, K. & Vaughan, D. dplyr: A Grammar of Data Manipulation. https://dplyr.tidyverse. org/ (2023).
Paradis, E. & Schliep, K. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526-528 (2019).
Xu, S. et al. Ggtree: A serialized data object for visualization of a phylogenetic tree and annotation data. iMeta 1, e56 (2022).
Jantscher-Krenn, E. et al. The human milk oligosaccharide disialyllacto-N-tetraose prevents necrotising enterocolitis in neonatal rats. Gut 61, 1417-1425 (2012).
Jantscher-Krenn, E. et al. Human milk oligosaccharides are present in amniotic fluid and how specific patterns dependent on gestational age. Nutrients 14, 2065 (2022).
Revelle, W. psych: Procedures for Psychological, Psychometric, and Personality Research. https://www.researchgate.net/publication/ 281345624_psych_Procedures_for_Psychological_Psychometric_ and_Personality_Research_R_Package_Version_10-95 (2023).
Mallick, H. et al. Multivariable association discovery in populationscale meta-omics studies. PLoS Comput. Biol. 17, e1009442 (2021).
الشكر والتقدير
تم تمويل هذا العمل جزئيًا من قبل منحة مؤسسة أزريلي لزملاء هيئة التدريس (M.Y.)، ومنحة مؤسسة العلوم الإسرائيلية 2660/18 (D.E. & M.Y.) ومنحة مؤسسة واترلو (D.E. & M.Y.)، ومن قبل صندوق العلوم النمساوي (FWF)، تحت رقم المشروع KLI 784 (E.J.K). M.Y. هو رئيس تطوير هيئة التدريس في أبحاث ما حول الولادة.
مساهمات المؤلفين
D.E. أسس المجموعة، أنشأ بيانات التسلسل، قدر وفرة HMO وأجرى جميع التحليلات. S.S. ساعد في إعداد التجربة. E.J.K. أدار وعلم طريقة تقدير HMO. M.Y. أدار العمل. كتب D.E. وM.Y. المخطوطة.
ملاحظة الناشر تظل Springer Nature محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية. إعلانات الأخلاق وافقت جميع الأمهات على المشاركة في دراستنا، التي تمت الموافقة عليها من قبل مجلس مراجعة المؤسسات في الجامعة العبرية (IRB، رقم الموافقة 20042021)، ووقعن على نماذج الموافقة لأنفسهن ولأطفالهن. لم يتم تقديم أي تعويض للمشاركين.
قسم الميكروبيولوجيا وعلم الوراثة الجزيئية، كلية الطب، الجامعة العبرية في القدس، القدس، إسرائيل.قسم التوليد وأمراض النساء، الجامعة الطبية في غراتس، غراتس، النمسا.مدرسة راشيل وسليم بنين لعلوم الكمبيوتر والهندسة، الجامعة العبرية في القدس، القدس، إسرائيل.البريد الإلكتروني: moranya@mail.huji.ac.il
Longitudinal quantification of Bifidobacterium longum subsp. infantis reveals late colonization in the infant gut independent of maternal milk HMO composition
Received: 19 July 2023
Accepted: 15 January 2024
Published online: 30 January 2024
(4) Check for updates
Breast milk contains human milk oligosaccharides (HMOs) that cannot be digested by infants, yet nourish their developing gut microbiome. While Bifidobacterium are the best-known utilizers of individual HMOs, a longitudinal study examining the evolving microbial community at high-resolution coupled with mothers’ milk HMO composition is lacking. Here, we developed a highthroughput method to quantify Bifidobacterium longum subsp. infantis (BL. infantis), a proficient HMO-utilizer, and applied it to a longitudinal cohort consisting of 21 mother-infant dyads. We observed substantial changes in the infant gut microbiome over the course of several months, while the HMO composition in mothers’ milk remained relatively stable. Although Bifidobacterium species significantly influenced sample variation, no specific HMOs correlated with Bifidobacterium species abundance. Surprisingly, we found that BL. infantis colonization began late in the breastfeeding period both in our cohort and in other geographic locations, highlighting the importance of focusing on BL. infantis dynamics in the infant gut.
Breast milk is considered the ideal nutrition for infants during their first 6 months of life . Millions of years of evolution have shaped breast milk composition such that its third most abundant component, human milk oligosaccharides (HMOs), cannot be digested by the infant, but serves as substrate for the infant’s gut bacteria . There are many different types of HMOs, which can be largely classified into three groups: fucosylated, sialylated, or neutral. Each HMO is composed of 3 to 32 monomers, and a single milk sample typically contains 50 to 200 distinct types of . Among other factors, maternal genetics plays a role in the production of specific HMOs in breast milk .
For example, mothers with an inactive fucosyltransferase 2 (FUT2) gene, termed non-secretors, fail to form alpha-1,2 bonds between fucose and lactose or other HMO backbone structures, resulting in the lack of 2’FL and other alpha-1,2-fucosylated glycans . Additionally, environmental factors coupled with infant age can affect HMO composition .
On average, infants have a higher relative abundance of Bifidobacterium species while they are breastfed . Bifidobacterium species were previously shown capable of utilizing multiple , however this ability varies between species and even within a single
species . Among all Bifidobacterium species and subspecies, the best-known HMO utilizer is Bifidobacterium longum subsp. infantis (BL. infantis) which grows efficiently on most types of HMOs , and possesses a large variability of HMO utilizing genes . In contrast, other Bifidobacterium species have a lower capability of HMO utilization, for example B. breve strains cannot utilize 3’SL and 6’SL at all, and most of them cannot utilize fucosylated HMOs . Since HMOs serve as food for the gut microbiome, one may hypothesize that different HMO compositions in mothers’ milk affects the developing gut microbial community.
To date, most research addressing the HMO-bacteria relationship in the infant gut focused on a single time point . A longitudinal cohort study is needed in order to examine how changes in HMO composition impact the infant gut microbiome over time.
To quantify the abundance of various Bifidobacterium species in microbiome communities two approaches are commonly used: 16SrRNA sequencing and shotgun metagenomics. While metagenomic sequencing allows classification at the species level, 16S-rRNA sequencing provides only genus-level classification of microbiome communities. The basic annotated unit in 16S-rRNA sequencing is referred to as operational taxonomic unit (OTU), which can be assigned to genus-level classification and may represent multiple species of the assigned genus. Multiple species can be annotated as the same OTU, hence using 16S-rRNA sequencing so far provided mostly weak or no associations with abundances of specific . A single OTU can include multiple species (or subspecies) with various HMO utilization capabilities, thus, a higher-resolution taxonomic definition is needed.
The largest variability in HMO-utilization capability can be found within the Bifidobacterium longum species. Overall, this species can be divided into two subspecies found in humans: B. longum subsp. longum (BL. longum) and B. longum subsp. infantis (BL. infantis). BL. longum is found both in infants and adults, while . infantis is unique to the infant gut. Studies have shown that BL. infantis can utilize almost all HMOs , while . longum has a limited repertoire. To study the HMO-microbe relationship, taking into account these differences in HMO-utilization within B. longum subspecies, a high-throughput, higher-resolution method is needed. Past studies have used different methods to differentiate between BL. infantis and BL. longum, such as or the Bifidobacterium Longum-Infantis Ratio (BLIR) method , yet these methods require the original DNA and are not high-throughput. Others have searched for . infantis specific genes such as the H 1 cluster or other BL. infantis clusters , however these methods do not give an exact ratio between the subspecies, rather they indicate their presence or absence. The new method we propose here could be applied also to the massive amounts of data available in public repositories.
Here, we establish a new matched cohort of breast milk and infant stool samples collected longitudinally throughout the first year of life. We develop a method to allow BL. infantis quantification from existing metagenomic data, and apply it to samples from our cohort to study the relationship between the abundance of Bifidobacterium species in the infant gut and HMO composition in mothers’ milk over time. Finally, we apply our B. longum subspecies quantification method to existing infant gut datasets to examine the timing of BL. infantis colonization across geographic locations.
Results
Cohort design
We have established a new and unique longitudinal cohort to test the relationship between HMOs in mothers’ milk and the developing infant gut microbiome. Our cohort consists of 21 mother-infant dyads with matched infant stool samples and breast milk samples collected on the same day. Altogether, we collected 80 stool samples and 50 breast milk samples together with the infant nutritional
information and antibiotic treatments (Supplementary Fig. 1, Supplementary Data 1). We collected these samples between the age of 2 weeks and 41 weeks, and each dyad contributed between one to eight paired samples.
Specific marker genes allow better quantification of B. longum subspecies
Bifidobacterium longum subsp. infantis (BL. infantis) is the best known utilizer of HMOs , however current methods for taxonomic classification from metagenomes are unable to separate the Bifidobacterium longum (B. longum) species into its main subspecies; Bifidobacterium longum subsp. longum (BL. longum) and BL. infantis . MetaPhIAn is one of the most common tools for profiling the composition of microbial population from metagenomic data, by using specific marker genes for each taxonomic group . However MetaPhlAn has no specific marker genes for . infantis and therefore classifies . longum at the species taxonomic level. Due to the differences between . longum subspecies in the context of HMO utilization, there is a rising need for a high-throughput method that will allow specific identification and quantification of BL. infantis from metagenomics data.
Here we define . longum subspecies specific markers and use them in a tailored MetaPhlAn database which allows abundance quantification of two B. longum subspecies: BL. infantis and BL. longum (Supplementary Fig. 2A). To construct our new dataset, we searched for marker genes that are unique to each subspecies. A marker gene was selected if it was present in at least of reference genomes of one subspecies and not in a single genome of the other subspecies (Fig. 1A, Methods). We chose to discard two subspecies of B. longum: Bifidobacterium longum subsp. suis and Bifidobacterium longum subsp. suillum since they are rarely found in humans , and a limited amount of reference genomes exist for these subspecies (Methods).
In order to validate our results, we applied MetaPhlAn with our new set of marker genes coupled with subspecies-specific qPCR to metagenomic sequencing data from 68 infant stool samples. When comparing the relative abundance of BL. infantis and BL. longum in each method, we observed a strong correlation between our computational approach and qPCR ( for BL. infantis and 0.997 for . longum; Fig. 1B, C). This finding confirms our method’s specificity and sensitivity for both BL. infantis (Fig. 1B) and BL. longum (Fig. 1C). In some samples, MetaPhlAn failed to assign a classification to a small percentage of . longum and therefore it was designated as unclassified . longum (Supplementary Fig. 2B, C).
The infant gut microbiome shows excessive changes while HMO composition in mothers’ milk is fairly stable
To examine the dynamics of Bifidobacterium species in the infant gut, we conducted metagenomic sequencing and analyzed the data using our novel MetaPhlAn database. We observed a significant prevalence of Bifidobacterium (at least one sample with ) in all the infants (Fig. 2A), in line with our expectation as most infants in our study were breastfed . Bifidobacterium remained highly abundant even after solid foods were introduced to infants (Fig. 2A, arrows). The abundant species included Bifidobacterium breve, Bifidobacterium bifidum, Bifidobacterium pseudocatenulatum and B. longum subspecies (Fig. 2A), along with Bacteroides species such as Bacteroides dorei and Bacteroides vulgatus (Supplementary Fig. 3). Interestingly, the presence of BL. longum and BL. infantis was mutually exclusive, reflecting potential intra-species competition, as previously suggested .
Overall, we found that the infant gut microbiome underwent significant changes over the course of several months. While the bacterial composition tended to be stable over a few weeks, there were certain time points when a switch in composition occurred (Fig. 2A, B). In infants that we had samples from many time points we found this switch to occur around 10 weeks of life. For example, in
Fig. 1 | Quantification of Bifidobacterium longum subspecies using markergenes enables subspecies-level detection. A Identification of unique genes in . infantis (blue) and BL. longum (green) reference genomes that can serve as markergenes in the quantification of . longum subspecies. HMO utilizing genes (HUGs) are marked on the bottom right and constitute only a small fraction of BL. infantis
marker genes. Validation of the computational approach, comparing the tailored MetaPhlAn marker-gene quantification results to the experimental qPCR results of BL. infantis and BL. longum. -value for regression was calculated using a two-sided -test.
infant03 (inf03) over the course of the first 15 weeks the dominant bacteria in the gut constantly changed, including B. vulgatus, Klebsiella pneumoniae, and finally . breve; the gut of infant18 was initially dominated by B. breve, followed by a complete switch to BL. infantis; and the gut of infant16 constantly changed its microbial composition (dominated by Escherichia coli, Veillonella seminalis and B. pseudocatenulatum; Fig. 2B). It was not always clear what triggered these microbial shifts, however since Bifidobacterium and other gut microbes utilize HMOs, we hypothesized that changes in the HMO composition in mothers’ milk might be causing bacterial changes in the infant gut.
To examine the impact of HMO composition in mothers’ milk on the infant gut microbiome, we quantified 16 common HMOs in 50 milk samples from 20 mothers using high performance liquid chromatography with fluorescence detection (HPLC-FLD; Methods). In contrast to the dynamic infant gut microbiome, the composition of HMOs in mothers’ milk remained relatively stable over the course of months, in terms of both their concentration in milk (Supplementary Fig. 4A) and the relative abundance of specific HMOs (Fig. 2C). We divided the milk samples into three main groups, based on their HMO composition: those with low or no abundance (Group 1, non-secretors ); samples from secretor mothers with very high abundance of and LNFP1 ( ) and smaller amounts of other HMOs (Group 2); and samples from secretor mothers with lower abundance of , and no clear dominant HMO (Group 3; Fig. 2C). We found no major changes over time in the abundance of specific HMOs, other than LSTc which was reduced to almost 0 over the course of weeks (Supplementary Fig. 4B, C), in line with previous findings . Overall, the changes found in the HMO composition in consecutive milk samples were significantly less pronounced than those found in the microbial
population from consecutive infant gut samples ( -test, , Supplementary Fig. 5A).
The dominant Bifidobacterium species shape the infant gut microbiome
General comparison of the infant gut microbiome composition determined that Bifidobacterium species play a significant role in the breastfed infant gut (Fig. 2A). We next searched for differences across samples in an attempt to characterize the various microbial profiles of the infant gut. We examined the diversity of the infant gut samples in our cohort, using a dimension reduction approach (PCoA with BrayCurtis dissimilarity, Methods), and found that samples cluster into distinct clusters (using K -means, ). The first three groups had samples with mostly one main Bifidobacterium dominant species (with relative abundance of ): B. breve, BL. longum and BL. infantis, and the fourth group contained samples dominated by either a different Bifidobacterium species (Bifidobacterium adolescentis, B. pseudocatenulatum) or other species (named “Mixed”, Fig. 3A). While usually consecutive samples from the same infant were assigned to the same cluster, occasional cluster switches were observed, strengthening our finding that the microbiome changes in this time frame (Fig. 3B). Overall, these analyses highlight the importance of Bifidobacterium species in our samples, as these are major factors that impact the variation in the infant gut microbial composition.
To further investigate these four groups of infant gut samples, we examined the alpha diversity of the microbiome population in samples found in each group using Shannon index. We found that the alpha diversity of samples within the BL. infantis group was lower compared to samples from other groups ( -test, , Fig. 3C), indicating that when BL. infantis is found, it dominates the
Fig. 2 | Composition and dynamics of Bifidobacterium species in the infant gut and HMOs in mothers’ milk. A Relative abundance of Bifidobacterium species in the infant gut (colorful) with all other species classified as “other” (gray). Samples from the same infant are grouped together, sorted by age. Arrows indicate samples taken after the introduction of solid food. B Temporal changes in the relative abundance of the microbial community in three infants (inf03, inf16, and inf18) in
the first 30-40 weeks of life. The most prevalent bacteria are colored, and the remaining are indicated as “Other” (gray). Colors as in (A), with additional colors for the non-Bifidobacterium species. C Relative abundance of 16 HMOs measured in mothers’ milk. Samples are categorized into three groups based on their HMO profiles, and arches connect samples obtained from the same mother (Methods).
community at such high levels of relative abundance leaving a smaller ecological niche for other bacteria in the infant gut. We next focused on the “Mixed” group, and asked whether additional variables may play a role in these microbial profiles. We examined breastfeeding, maternal or infant antibiotic use, delivery mode,
breastfeeding type (pumped or direct), and introduction of solid foods, yet we did not find any specific variable that was associated with the microbial profile of the “Mixed” group. As expected, the . infantis group consisted solely of infants who received none-to-low amounts of infant formula .
Fig. 3 | Variation across samples is highly impacted by the dominant Bifidobacterium species. A Principal Coordinate Analysis (PCoA) of infant gut microbiome samples using Bray-Curtis dissimilarity. Points are color-coded based on the dominant Bifidobacterium species in each sample, and nonBifidobacterium dominated samples are colored in gray. Samples without a dominant species ( relative abundance) are labeled as “No dominant” (yellow). The size of each point represents the relative abundance of the dominant bacteria in that sample. Ellipses encompass four groups identified using k-means clustering. Each group represents a primary dominant species (indicated by the ellipse color): BL. infantis (blue), BL. longum (green), B. breve
To characterize the relationships among dominant Bifidobacterium species in the infant gut, we examined their occurrence within groups where they are not dominant. We found that . infantis and . breve are mutually exclusive, consistent with a previous study in Hazda infants , implying competition for the same niche in the infant gut (Fig. 3D). However, it remains unclear what specific niche . breve and . infantis are competing for, given . breve’s limited ability to utilize a variety of HMOs . Finally, we observed BL. longum in some B. breve-dominant samples (Fig. 2A), suggesting that . breve may rely on derivatives from BL. longum through cross-feeding in these samples .
Single HMOs are not associated with specific Bifidobacterium species
It is well established that different Bifidobacterium species have different HMO-utilization capabilities . Therefore, specific HMOs may benefit specific Bifidobacterium species in the infant gut based on their HMO utilization profiles. However, we found no significant correlation between the abundance of Bifidobacterium in general and the main Bifidobacterium species and subspecies with specific HMOs (Fig. 4A) or HMO groups (fucosylated, sialylated or neutral; Supplementary Fig. 5B). In addition, linear association models accounting for individual infants showed no significant association between specific
Fig. 4 | Mothers’ milk HMO composition shows no signieficant correlation with infant gut Bifidobacterium. A Pearson correlations between all measured HMOs and the Bifidobacterium genus, as well as the main individual species and subspecies (BL. infantis, BL. longum, B. breve and B. bifidum). None of these correlations were found to be statistically significant ( -test). Comparison of the relative
abundance of LSTc in mothers’ milk ( -axis) with the relative abundance of . infant is in the infant gut microbiome ( -axis). C Maximum relative abundance of . infantis in each infant divided into two groups of infants with secretor and nonsecretor mothers, highlighting the high abundance of BL. infantis only in infants to secretor mothers, but not in all of them.
HMOs and Bifidobacterium species and subspecies in the infant gut (when requiring FDR ). Nevertheless, we found that BL. infantis exhibited a high abundance ( ) exclusively in infants to secretor mothers (Fig. 4B, -test ). In addition, we observed a modest and non-significant negative correlation ( ) between BL. infantis and LSTc (Fig. 4C). It is worth noting the importance of considering the timing factor in interpreting these findings. The delayed presence of BL. infantis in the gut (which will be discussed in more detail later) and the gradual decrease of LSTc over time (Supplementary Fig. 4B,C) could contribute to the observed correlation. The lack of variation in the HMO composition together with the lack of HMOmicrobes associations indicate that the microbial shifts, specifically within Bifidobacterium species, can not be explained by a change in mothers’ milk HMO composition.
Metagenomes with BL. infantis contain more HMO utilizing genes
Metagenomes obtained from various time points of multiple infants contain distinct strains and species, resulting in variable gene abundance profiles which can enable various patterns of HMO utilization. To assess the HMO utilization potential of specific Bifidobacterium species in our dataset, we investigated the presence of HMO-utilizing genes (HUGs) organized into five distinct clusters ( ; Fig. 5). We observed that the dominant species in each sample significantly
influenced the metagenome’s theoretical capacity for HMO utilization. As expected, BL. infantis-dominated samples exhibited the highest abundance of HUGs, confirming its exceptional capability in utilizing HMOs . Notably, some of these samples displayed high variation in gene abundance from clusters H 1 and H 5 , indicating a potential lower capacity to transport some , and utilize lacto-N-tetraose (LNT) and lacto-N-neotetraose ( LNnT.
Interestingly, metagenomes that were dominated by B. breve or . longum also contained genes from the H5 cluster, emphasizing their ability to utilize HMOs based on lacto-N-biose (LNB) . However, samples from the “Mixed” groups exhibited minimal or no HUGs, suggesting either alternative genes for HMO utilization or a lack of capacity to utilize HMOs altogether.
BL. infantis does not colonize the infant gut in early breastfeeding weeks
BL. infantis is the most proficient HMO-utilizer in the infant gut , thus we expected that BL. infantis will have a fitness advantage in the breastfed infant gut from the initial days of breastfeeding. However, despite the majority of infants in our cohorts that were breastfed since birth, BL. infantis was primarily detected starting only at 10 weeks of age (Fig. 6A). Linear association models showed a clear positive association of the relative abundance of BL. infantis with age (coefficient ), which was not found for any other
Fig. 5 | Abundance of HMO utilization genes in metagenomic samples. Normalized genes abundance of HMO utilization genes (HUGs) in all infant samples. Genes (rows) are categorized and labeled according to their respective gene
cluster (H1-H5). Samples (columns) are annotated based on their assigned dominant-species group.
Bifidobacterium species or subspecies. Overall, BL. infantis exhibited the highest abundance at 10-25 weeks, followed by a gradual decrease in abundance (Fig. 6A).
Our innovative computational methodology enabled the exploration of the delayed colonization of BL. infantis across various geographical regions, providing a gateway for further in-depth investigation into this phenomenon. Thus, to corroborate our findings, we examined additional infant cohorts from Sweden , United Kingdom , Estonia , Italy , Russia , United States and two cohorts from Finland comprising samples from a total of 1,017 infants throughout the first year of life. Across all cohorts, a similar pattern of latecolonization of BL. infantis was observed: In Sweden, BL. infantis was not observed at birth almost at all, reaching its peak prevalence at 17 weeks, followed by a gradual decline in both prevalence and relative abundance by 52 weeks (Fig. 6B). In the UK, BL. infantis was observed in only four out of 178 infants in the first 3 weeks of life, and during the later infancy period (17-52 weeks) BL. infantis was found in 28 infants (Fig. 6C). In Finland, one cohort with 126 infants had only a single infant with detectable levels of BL. infantis in the first 10 weeks of life, and additional 11 infants gained it later on (Fig. 6D). In a second Finnish cohort, BL. infantis was not observed in any infant samples (ages weeks; Finalnd2, Supplementary Fig. 6A). In the Italian and Estonian cohorts, a similar pattern was observed however the relative abundance of BL. infantis was lower in most infants (Supplementary Fig. 6B, . The cohort from the the United states contained samples only from the first 2 weeks of life, revealing no infants with . infantis in the first
week of life, and only one out of 77 infants with detectable levels in the second week of life (Supplementary Fig. 6D). Finally, in the Russian cohort which consisted of samples collected from infants aged 12 weeks and beyond, BL. infantis was detectable in 14/69 infants,with its presence becoming noticeable only at 20 weeks (Supplementary Fig. 6E). Overall, all cohorts exhibited a late-colonization of BL. infantis, commonly starting at 10 weeks of age, or later (paired -test, ; Fig. 6E, Supplementary Fig. 6F).
To examine variations between BL. infantis strains across the different countries we next focused on . infantis strain-level composition, using the SNP profiles on the newly-identified . infantis marker genes (Methods; Fig. 6F). We found that BL. infantis strains within the same infant in our cohort were more similar to each other than strains between unrelated infants ( -test, , Supplementary Fig. 6G). Furthermore, in some countries (i.e., Italy & Russia), most of the strains were very similar, while in other countries (i.e., Sweden & UK) there was a much larger variation between strains (Fig. 6F, Supplementary Fig. 6H). Strains from our Israeli cohorts were clustered in two distinct groups, one more similar to strains found in the US (light purple, Fig. 6F) and the other closer to strains found in the UK (brown, Fig. 6F). Specifically, the strains from the Italian samples were very distinct from all other strains, with the exception of a single UK strain. Finally, BL. infantis reference genomes were clustered into three groups, some identical to the Italian strains, while others clustered closely with Russian and Estonian strains (gray, Fig. 6F). These findings allow us to explore the variation found within .
Fig. 6 | Late colonization and low prevalence of BL. infantis in the infant gut in multiple geographic locations. A Relative abundance of . infantis in our Israeli cohort, among infants where . infantis was detected at any time point. B-D Relative abundance of . infantis at different time points in samples from B Sweden ( samples from 100 infants) C UK ( samples from 169 infants) and D Finland ( samples from 108 infants). For Sweden and the United Kingdom where samples were collected at defined time points box plots are
shown, with box boundaries at the 25th and 75th percentiles, and the median highlighted. Whiskers represent 1.5 * IQR and points past them are outliers. E The percentage of infants in multiple geographical locations harboring BL. infantis in the first 10 weeks of life and at 10 weeks and later. F Phylogenetic tree displaying . infantis strains found across all cohorts. The dominant strain from each sample is represented, and strains are color-coded based on their corresponding cohort, highlighting that some countries (like Italy) have very similar strains in all samples.
infantis and highlight interesting variability across geographic locations.
Discussion
In this study we introduced an innovative approach to quantify . infantis and distinguish it from BL. longum in metagenomic data. Our method enables researchers to concentrate on studying this distinct subspecies and its associations with HMOs from existing metagenomic data. This approach can be adapted to differentiation of subspecies in additional microbial species, and specifically further within B. longum (such as BL. suis & BL. suillum subspecies) once sufficient numbers of
reference genomes become available. We employed our approach to explore the diversity within the infant gut microbiome and discovered the lack of associations with individual HMOs present in mothers’ milk. Our analysis revealed that the variability between samples was greatly influenced by the dominant Bifidobacterium species in each sample.
Previous research has suggested that colonization of Bifidobacterium species in the infant gut may be influenced by priority effects . However, our study revealed substantial changes in the dominant Bifidobacterium species within the same infant over the course of several weeks (Fig. 3A, B). This indicates that over time there are additional factors responsible for Bifidobacterium
species prosperity, such as species competition and crossfeeding. For example, it was reported that B. breve, despite having limited ability to utilize HMOs, can outcompete stronger competitors if introduced early into a microbial community . In addition, . breve has the capacity to cross-feed on monosaccharides derived from HMOs by other Bifidobacterium species . This implies that . breve may initially dominate the population when carbohydrates are available, but subsequently loses the competition to other Bifidobacterium species once these carbohydrates are depleted. In our data we found that the . breve dominated group had a diverse microbial population (Fig. 3C), perhaps since it cross feeds on HMOs derivatives from other species. More research is needed to understand the microbial shifts in the infant gut and the effect cross-feeding has on the microbial dynamics in the infant gut.
While the HMOs present in a mother’s milk remained relatively stable over numerous weeks, we observed notable changes in the infant gut microbiome during this time period. This suggests that even subtle variations in the composition of breast milk may have an impact on the development of the gut microbiome. Alternatively, it is possible that other components present in breast milk, such as cytokines, microRNAs and antibodies, play a role in influencing the infant’s microbiome . In addition, we can not rule out environmental effects that may contribute to these changes such as starting daycare which in Israel typically occurs at 15-26 weeks of age. Our findings indicate that there were no significant correlations between HMOs and Bifidobacterium, further supporting the idea that additional factors beyond HMOs are involved in shaping the infant’s microbiome. Previous studies have examined the composition of the microbiome and its relationship with HMOs using 16S-rRNA amplicon sequencing. These studies have reported varying results, with some finding no significant correlations, while others observed modest correlations. Interestingly, some of the studies identified a negative correlation between Bifidobacterium OTUs and multiple HMOs . This could be attributed to a decrease in the overall Bifidobacterium abundance within the infant gut over time, coupled with an increase in specific HMOs, in line with our findings regarding LSTc and BL. infantis (Fig. 4B). Additionally, it is possible that such specific correlations may be observable only using a larger cohort.
Finally, we found that BL. infantis does not commonly colonize the infant gut in the early weeks of breastfeeding and that not all breastfed infants have detectable levels of BL. infantis. Importantly, when . infantis was found in the infant gut, it commonly dominated the gut microbiome community. Analyzing additional infant cohorts from multiple geographical locations strengthened our findings regarding the late colonization of BL. infantis in infants’ gut. Previous studies have reported a low prevalence of . infantis during early time points , however these studies did not incorporate frequent sampling in the first months of life, thus lacking the ability to precisely determine the timing of BL. infantis arrival. A previous study proposed that the arrival of BL. infantis is influenced by the history of breastfeeding practices of a given country , which impacts the overall exposure to . infantis strains. Countries with historically lower breastfeeding rates are likely to have a lower prevalence of . infantis, resulting in infants acquiring . infantis at a later stage through horizontal transfer .
Although BL. infantis is one of the more-studied infant gut commensals, its publicly-available reference genomes do not span the entire genomic variation of this subspecies (gray, Fig. 6F). To expand the BL. infantis-related research, additional reference genomes should be characterized in full, from natural isolates representing diverse geographic locations and lifestyles. Further research, especially longitudinal sampling of infants and their surroundings, is required to elucidate the timing and sources from which infants acquire . infantis and to comprehend the differences observed between countries.
Methods
Sample collection
Breast milk and stool samples were collected as part of the Breast Milk Baby (BMB) cohort from mothers and infants from birth till 1 year old. Stool samples were collected using eSwab with 1 ml of liquid Amies medium +1 regular FLOQSwabs (Copan) in order to preserve bacterial population. Breast milk samples were collected by pump or manually and stored in sterile tubes. Both sample types were collected by mothers in their homes and stored at for up to 24 h and then shipped to the lab and stored long term at .
Metagenomic library construction and sequencing
DNA was extracted from stool samples using DNeasy PowerSoil Pro Kit (#47014, QIAGEN). Illumina sequencing libraries were prepared using Nextera XT DNA Library Preparation kit (FC-131-1096, Illumina) according to the manufacturer’s recommended protocol with half of the volume and the DNA. Samples were sequenced using Illumina single-end 150 bp sequencing on a NextSeq 500 device.
B. longum subspecies quantification
To identify B. longum subspecies-specific markers we started with 116 . longum reference genomes downloaded from the NCBI with completeness of and contamination of (Supplementary Data 2). Reference genomes were classified to B. longum subsp. longum (BL. longum), B. longum subsp. infantis (BL. infantis) and unknown based on NCBI annotation, leaving 30 BL. longum and 16 BL. infantis references. As some of the unknown references could belong to either . infantis or . longum, we removed them all for specificity. We decided not to include subspecies BL. suis and BL. suillum in our method due to the limited availability of reference genomes for these subspecies leading to difficulty to generate reliable marker genes. In addition, these subspecies were rarely reported in humans , thus are less relevant in our settings. Therefore it is important to note that using our method, . suis and BL. suillum can be misidentified as a different subspecies or alternatively identified as . longum species with unclassified subspecies.
PanPhlan3 was used to analyze the pangenome of all 46 reference genomes. Clustering all presence/absence profiles in the pangenome revealed two clear clusters, the first of 14 BL. infantis strains and the second included all . longum strains and two . infantis strains (BL. infantis 157 F, BL. infantis CCUG52486; Supplementary Fig. 7). Additional analysis showed that these two . infantis reference genomes that were clustered with . longum reference genomes did not contain the H1 HMO utilization cluster which defines BL. infantis . In addition, a previous study showed that these two BL. infantis reference genomes are most probably . longum, based on the phylogeny of the core pangenome of 158 B. longum strains . Taken together, BL. infantis 157 F and BL. infantis CCUG52486157 were suspected as mis-annotated and were excluded from further analysis.
Subspecies-specific marker genes were chosen in two steps. First, using the pangenome we found 331 genes that were present in of one subspecies and not present at all in the other subspecies. For example, a gene that was present in 13 out of the 14 BL . infantis references and not in any . longum references was selected to be a marker gene for . infantis. Next, all selected genes were filtered to be specific at the species level to . longum and to confirm they do not exist in other Bifidobacterium species. To do so, we used Blastn 2.12.0 to map all marker genes to the nucleotide database. Of the 331 putative marker genes, 84 matched other species (such as B. breve) with alignment and over 50% coverage and therefore were filtered out. Our final set of maker genes included 119 BL. infantis and 128 BL. longum markers (Supplementary Data 3). The MetaPhlAn database was customized to include the newly defined marker genes using described MetaPhlAn instructions (https://github.com/biobakery/ MetaPhlAn/wiki/MetaPhlAn-4) and then MetaPhlAn4 was used with
the –index and –bowtie2db parameters and our customized markergene database.
To verify the results, subspecies of B. longum were determined using qPCR with subspecies-specific primers for BL. longum (F: GTGTGGATTACCTGCCTACC, R: GTCGCCAACCTTGACCACTT) and BL. infantis (F: ATGATGCGCTGCCACTGTTA, R: CGGTGAGCGTCAATGTATCT). The efficiency of the primers was assessed by testing them in five dilutions. qPCR was performed at for 10 s , followed by 40 cycles of for 10 s and for 30 s . The ratio between . infantis and . longum was calculated using the delta-delta Ct method.
Host reads were removed using an in house pipeline by aligning reads to the human genome by Bowtie2 (2.4.5-1). Samples were filtered and trimmed for Nextera adapters using fastq-mcf, ea-utils (1.05). Taxonomic profiling was done using MetaPhlAn4 with our unique database as described above. Functional profiling was done using HUMAnN3 . HUGs were selected from previously described HMO clusters . Strain analysis was performed using StrainPhIAn with default parameters and –sample_with_n_markers 50. SNPs for BL. infantis marker-genes were calculated using the multiple-sequence alignment (MSA) produced by StrainPhIAn 4 (–mutation rates parameter). BL. infantis was considered detectable when its relative abundance was . Further analysis was done using an in house (4.2.2) script utilizing dplyr (1.1.2), tidyr (1.3.0) and tidyverse (2.0.0). Plots were created using ggplot2 (3.4.2) and ggforce (0.4.1), colors were used from RColorBrewer (1.1-3) and pals (1.7). Heatmaps were created using pheatmap . Alpha and beta diversity were calculated using “diversity” (Shannon index) and “vegdist” (Bray-Curtis dissimilarity) from the vegan (2.6-4) package and the PCoA was created using the ape (5.7-1) package. Phylogenetic tree was produced using ggtree (3.6.2) and the sankey plots were created using ggsankey (0.0.99999). Additional cohorts were downloaded from NCBI Sequence Read Archive as following: Sweden (PRJEB6456), United states (PRJNA591079), Italy (PRJNA352475), United Kingdom (PRJEB32631), Finland, Estonia and Russia (PRJNA497734) and an additional cohort from finland (Finland2, PRJNA475246).
HMO quantification
HMO standards used in this study were purchased from Dextra Laboratories, United Kingdom. These included 2′-fucosyllactose ( FL), 3 -fucosyllactose (3FL), -sialyllactose ( SL), -sialyllactose ( SL), lacto-N-tetraose (LNT), disialyllacto-N-tetraose (DSLNT), Lactodifucotetraose (LDFT), lacto-N-difucohexaose 1 (LNDFH), lacto-Nfucopentaose (LNFP) 1, 2, and 3, lacto-N-hexaose (LNH), lacto-Nneotetraose (LNnT) and sialyl-lacto-N-tetraose (LST) a, b and c. Linear B6-Trisaccharide was used as an internal standard.
HMO quantification was performed as previously described . Briefly of human milk was combined with Linear B-6 Trisaccharide (Dextra Laboratories, UK) and HPLC grade water, then subjected to C18 columns (Thermo Scientific #60108-390) and carbograph columns (Thermo Scientific #60302-606) to remove proteins and salts respectively. Samples were labeled using 2 -aminobenzamide ( , Sigma) for 2 h at . Excess was removed using Silica columns (Thermo Scientific, #60300-482). Samples were separated by HPLC with fluorescence detection on a TSKgel Amide-80 column (Tosoh Bioscience, Tokyo, Japan) with a linear gradient of a 50 mM ammonium formate/acetonitrile solvent system. Retention times of purchased standard HMOs were used to annotate HPLC peaks. 6’SL and LNT peaks could not be separated and therefore, were calculated together. The amount of each individual HMO was calculated based on normalization to the internal standard (Supplementary Data 4). The relative abundance of each of the individual HMOs was determined by
setting the sum of the 16 identified oligosaccharides as 100% total HMOs.
Statistical analysis
No statistical method was used to predetermine the sample size. The investigators were not blinded to allocation during experiments and outcome assessment. Independent -test was performed to test between groups when mentioned using the R function ” -test”. Paired test was done between the percentages of infants in each cohort that had . infantis prior to 10 weeks and the percentage of infants that had detectable levels of . infantis after 10 weeks. Distances between consecutive infant gut microbiome samples and between breast milk HMO compositions were calculated using Bray-Curtis dissimilarity using the “vegdist” function from the vegan (2.6-4) package. Correlation between the microbial population and HMO composition was performed using Pearson correlation with the “cor” R function. Adjusted -values were calculated using “corr.test” from the psych package .
Linear association models
The “Maaslin2” package was used to perform linear models in order to find associations between 16 HMOs and Bifidobacterium species and in the infant gut bacteria. The individual was set as a random factor to account for the effect of each mother-infant pair. In addition, “Maaslin2” was used to perform linear association models of age compared to Bifidobacterium species, adjusted to individuals.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The Human-filtered metagenomic sequencing data generated in this study has been deposited in the SRA database under BioProject PRJNA994433. Metadata of the cohort is provided in Supplementary Data 1. HMO quantification results are provided in Supplementary Data 4 and MetaPhlAn results in Supplementary Data 5.
Westerfield, K. L., Koenig, K. & Oh, R. Breastfeeding: Common questions and answers. Am. Fam. Physician 98, 368-373 (2018).
Sela, D. A. & Mills, D. A. Nursing our microbiota: molecular linkages between bifidobacteria and milk oligosaccharides. Trends Microbiol. 18, 298-307 (2010).
Marcobal, A. et al. Consumption of human milk oligosaccharides by gut-related microbes. J. Agric. Food Chem. 58, 5334-5340 (2010).
Zivkovic, A. M., German, J. B., Lebrilla, C. B. & Mills, D. A. Human milk glycobiome and its impact on the infant gastrointestinal microbiota. Proc. Natl Acad. Sci. USA 108, 4653-4658 (2011).
Han, S. M. et al. Maternal and infant factors influencing human milk oligosaccharide composition: beyond maternal genetics. J. Nutr. https://doi.org/10.1093/jn/nxab028. (2021)
Lewis, Z. T. et al. Maternal fucosyltransferase 2 status affects the gut bifidobacterial communities of breastfed infants. Microbiome 3, 13 (2015).
Plows, J. F. et al. Longitudinal changes in human milk oligosaccharides (HMOs) over the course of 24 months of lactation. J. Nutr. 876-882 https://doi.org/10.1093/jn/nxaa427. (2021)
Yassour, M. et al. Natural history of the infant gut microbiome and impact of antibiotic treatment on bacterial strain diversity and stability. Sci. Transl. Med. 8, 343ra81 (2016).
Bäckhed, F. et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe 17, 690-703 (2015).
Le Doare, K., Holder, B., Bassett, A. & Pannaraj, P. S. Mother’s Milk: a purposeful contribution to the development of the infant microbiota and immunity. Front. Immunol. 9, 361 (2018).
Sela, D. A. et al. Bifidobacterium longum subsp. infantis ATCC -fucosidases are active on fucosylated human milk oligosaccharides. Appl. Environ. Microbiol. 78, 795-803 (2012).
Underwood, M. A. et al. A comparison of two probiotic strains of bifidobacteria in premature infants. J. Pediatr. 163, 1585-1591.e9 (2013).
Garrido, D., Dallas, D. C. & Mills, D. A. Consumption of human milk glycoconjugates by infant-associated bifidobacteria: mechanisms and implications. Microbiology 159, 649-664 (2013).
Garrido, D. et al. Comparative transcriptomics reveals key differences in the response to milk oligosaccharides of infant gutassociated bifidobacteria. Sci. Rep. 5, 13517 (2015).
Ruiz-Moyano, S. et al. Variation in consumption of human milk oligosaccharides by infant gut-associated strains of bifidobacterium breve. Appl. Environ. Microbiol. 79, 6040-6049 (2013).
Duar, R. M. et al. Comparative genome analysis of bifidobacterium longum subsp. infantis strains reveals variation in human milk oligosaccharide utilization genes among commercial probiotics. Nutrients 12, 3247 (2020).
LoCascio, R. G. et al. Glycoprofiling of bifidobacterial consumption of human milk oligosaccharides demonstrates strain specific, preferential consumption of small chain glycans secreted in early human lactation. J. Agric. Food Chem. 55, 8914-8919 (2007).
Duboux, S., Ngom-Bru, C., De Bruyn, F. & Bogicevic, B. Phylogenetic, functional and safety features of 1950s B. infantis strains. Microorganisms 10, 203 (2022).
Sela, D. A. et al. The genome sequence of bifidobacterium longum subsp. infantis reveals adaptations for milk utilization within the infant microbiome. Proc. Natl Acad. Sci. USA. 105, 18964-18969 (2008).
Derrien, M. et al. Gut microbiome function and composition in infants from rural kenya and association with human milk oligosaccharides. Gut Microbes 15, 2178793 (2023).
Pace, R. M. et al. Variation in human milk composition Is related to differences in milk and infant fecal microbial communities. Microorganisms 9, 1153 (2021).
Borewicz, K. et al. The association between breastmilk oligosaccharides and faecal microbiota in healthy breastfed infants at two, six, and twelve weeks of age. Sci. Rep. 10, 4270 (2020).
Barnett, D. J. M. et al. Human milk oligosaccharides, antimicrobial drugs, and the gut microbiota of term neonates: observations from the KOALA birth cohort study. Gut Microbes 15, 2164152 (2023).
Borewicz, K. et al. Correlating infant faecal microbiota composition and human milk oligosaccharide consumption by microbiota of one-month old breastfed infants. Mol. Nutr. Food Res. 63, e1801214 (2019).
Locascio, R. G. et al. A versatile and scalable strategy for glycoprofiling bifidobacterial consumption of human milk oligosaccharides. Microb. Biotechnol. 2, 333-342 (2009).
Seppo, A. E. et al. Infant gut microbiome is enriched with bifidobacterium longum ssp. infantis in old order mennonites with traditional farming lifestyle. Allergy 76, 3489-3503 (2021).
Young, S. L. et al. Bifidobacterial species differentially affect expression of cell surface markers and cytokines of dendritic cells harvested from cord blood. Clin. Diagn. Lab. Immunol. 11, 686-690 (2004).
Davis, J. C. C. et al. Growth and morbidity of gambian infants are influenced by maternal milk oligosaccharides and infant gut microbiota. Sci. Rep. 7, 40466 (2017).
Taft, D. H. et al. Bifidobacterium species Colonization in Infancy: a global cross-sectional comparison by population history of breastfeeding. Nutrients 14, 1423 (2022).
Tso, L., Bonham, K. S., Fishbein, A., Rowland, S. & Klepac-Ceraj, V. Targeted high-resolution taxonomic Identification of bifidobacterium longum subsp. infantis using human milk oligosaccharide metabolizing genes. Nutrients 13, 2833 (2021).
Casaburi, G. et al. Metagenomic insights of the infant microbiome community structure and function across multiple sites in the United States. Sci. Rep. 11, 1472 (2021).
LoCascio, R. G., Desai, P., Sela, D. A., Weimer, B. & Mills, D. A. Broad conservation of milk utilization genes in bifidobacterium longum subsp. infantis as revealed by comparative genomic hybridization. Appl. Environ. Microbiol. 76, 7373-7381 (2010).
Blanco-Míguez, A. et al. Extending and improving metagenomic taxonomic profiling with uncharacterized species using MetaPhlAn 4. Nat. Biotechnol. https://doi.org/10.1038/s41587-023-01688w (2023)
Yanokura, E. et al. Subspeciation of bifidobacterium longum by multilocus approaches and amplified fragment length polymorphism: description of B longum subsp. suillum subsp. nov., isolated from the faeces of piglets. Syst. Appl. Microbiol. 38, 305-314 (2015).
Yatsunenko, T. et al. Human gut microbiome viewed across age and geography. Nature 486, 222-227 (2012).
Avershina, E. et al. Bifidobacterial succession and correlation networks in a large unselected cohort of mothers and their children. Appl. Environ. Microbiol. 79, 497-507 (2013).
Bode, L. Human milk oligosaccharides: every baby needs a sugar mama. Glycobiology 22, 1147-1162 (2012).
Dai, D. L. Y. et al. Breastfeeding enrichment of B. longum subsp. infantis mitigates the effect of antibiotics on the microbiota and childhood asthma risk. Med ( ) https://doi.org/10.1016/j.medj. 2022.12.002 (2022).
Olm, M. R. et al. Robust variation in infant gut microbiome assembly across a spectrum of lifestyles. Science 376, 1220-1223 (2022).
Ojima, M. N. et al. Priority effects shape the structure of infant-type bifidobacterium communities on human milk oligosaccharides. ISME J. https://doi.org/10.1038/s41396-022-01270-3 (2022)
Asakuma, S. et al. Physiology of consumption of human milk oligosaccharides by infant gut-associated bifidobacteria. J. Biol. Chem. https://doi.org/10.1074/jbc.M111.248138 (2011).
Shao, Y. et al. Stunted microbiota and opportunistic pathogen colonization in caesarean-section birth. Nature 574, 117-121 (2019).
Vatanen, T. et al. Genomic variation and strain-specific functional adaptation in the human gut microbiome during early life. Nat. Microbiol 4, 470-479 (2019).
Ferretti, P. et al. Mother-to-infant microbial transmission from different body sites shapes the developing infant gut microbiome. Cell Host Microbe 24, 133-145.e5 (2018).
Mitchell, C. M. et al. Delivery mode affects stability of early infant gut microbiota. Cell Rep. Med. 1, 100156 (2020).
Yassour, M. et al. Strain-level analysis of mother-to-child bacterial transmission during the first few months of Life. Cell Host Microbe 24, 146-154.e4 (2018).
Egan, M., O’Connell Motherway, M., Ventura, M. & van Sinderen, D. Metabolism of sialic acid by bifidobacterium breve UCC2003. Appl. Environ. Microbiol. 80, 4414-4426 (2014).
Lawson, M. A. E. et al. Breast milk-derived human milk oligosaccharides promote Bifidobacterium interactions within a single ecosystem. ISME J. 14, 635-648 (2020).
Carr, L. E. et al. Role of human milk bioactives on infants’ gut and immune health. Front. Immunol. 12, 604080 (2021).
Collado, M. C. et al. Longitudinal study of cytokine expression, lipid profile and neuronal growth factors in human breast milk from term and preterm deliveries. Nutrients 7, 8577-8591 (2015).
Beghini, F. et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. Elife 10, e65088 (2021).
Díaz, R., Torres-Miranda, A., Orellana, G. & Garrido, D. Comparative genomic analysis of Novel bifidobacterium longum subsp. longum strains reveals functional divergence in the human gut microbiota. Microorganisms 9, 1906 (2021).
Kim, H.-B. et al. Development of real-tme PCR assay to specifically detect 22 bifidobacterium Species and subspecies using comparative genomics. Front. Microbiol. 11, 2087 (2020).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357-359 (2012).
Aronesty, E. ea-utils: Command-line Tools for Processing Biological Sequencing Data. https://www.researchgate.net/publication/ 319159822_ea-utils_Command-line_tools_for_processing_ biological_sequencing_data (2011).
Wickham, H., François, R., Henry, L., Müller, K. & Vaughan, D. dplyr: A Grammar of Data Manipulation. https://dplyr.tidyverse. org/ (2023).
Paradis, E. & Schliep, K. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526-528 (2019).
Xu, S. et al. Ggtree: A serialized data object for visualization of a phylogenetic tree and annotation data. iMeta 1, e56 (2022).
Jantscher-Krenn, E. et al. The human milk oligosaccharide disialyllacto-N-tetraose prevents necrotising enterocolitis in neonatal rats. Gut 61, 1417-1425 (2012).
Jantscher-Krenn, E. et al. Human milk oligosaccharides are present in amniotic fluid and how specific patterns dependent on gestational age. Nutrients 14, 2065 (2022).
Revelle, W. psych: Procedures for Psychological, Psychometric, and Personality Research. https://www.researchgate.net/publication/ 281345624_psych_Procedures_for_Psychological_Psychometric_ and_Personality_Research_R_Package_Version_10-95 (2023).
Mallick, H. et al. Multivariable association discovery in populationscale meta-omics studies. PLoS Comput. Biol. 17, e1009442 (2021).
Acknowledgements
This work was funded in part by the Azrieli Foundation grant for faculty fellows (M.Y.), by the Israel Science Foundation grant 2660/18 (D.E. & M.Y.) by the Waterloo foundation (D.E. & M.Y.), and by the Austrian Science Fund (FWF), under project number KLI 784 (E.J.K). M.Y. is the Rosalind, Paul and Robin Berlin Faculty Development Chair in Perinatal Research.
Author contributions
D.E. established the cohort, generated the sequencing data, quantified the HMO abundance and performed all analyses. S.S. assisted with the experimental setup. E.J.K. guided and taught the HMO-quantification method. M.Y. guided the work. D.E. and M.Y. wrote the manuscript.
Correspondence and requests for materials should be addressed to Moran Yassour.
Peer review information Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.Ethics declarations All mothers have agreed to participate in our study, which was approved by the Hebrew University’s Institutional Review Board (IRB, approval number 20042021), and signed our consent forms for themselves and their infants. No compensation was provided to participants.
Microbiology & Molecular Genetics Department, Faculty of Medicine, The Hebrew University of Jerusalem, Jerusalem, Israel. Department of Obstetrics and Gynecology, Medical University of Graz, Graz, Austria. The Rachel and Selim Benin School of Computer Science and Engineering, The Hebrew University of Jerusalem, Jerusalem, Israel. e-mail: moranya@mail.huji.ac.il