DOI: https://doi.org/10.1038/s41557-023-01416-6
PMID: https://pubmed.ncbi.nlm.nih.gov/38225269
تاريخ النشر: 2024-01-15
تحديد الطبقات السطحية لهيكل الماء الواجهى في محاليل الإلكتروليت البسيطة
تم القبول: 7 ديسمبر 2023
نُشر على الإنترنت: 15 يناير 2024
(أ) التحقق من التحديثات
الملخص
توزيع الأيونات عند واجهة الهواء/الماء يلعب دورًا حاسمًا في العديد من العمليات الطبيعية. وقد أفادت عدة دراسات أن الأيونات الأكبر تميل إلى أن تكون نشطة على السطح، مما يعني أن الأيونات تقع على سطح الماء، مما يؤدي إلى توليد مجالات كهربائية تحدد بنية الماء عند الواجهة. هنا نتحدى هذه الرؤية من خلال دمج تقنية الكشف عن الترددات الاهتزازية المحددة للسطح مع محاكاة الديناميكا الجزيئية من أولى المبادئ المدعومة بالشبكات العصبية. تظهر نتائجنا أن الأيونات في محاليل الإلكتروليت النموذجية تقع في الواقع في منطقة تحت السطح، مما يؤدي إلى تقسيم هذه الواجهات إلى طبقتين مائيتين متميزتين. السطح الخارجي خالٍ من الأيونات، بينما الطبقة تحت السطح غنية بالأيونات. هذا التقسيم السطحي هو عنصر رئيسي في تفسير إعادة تنظيم الماء الناتجة عن الأيونات عند الواجهة الخارجية للهواء/الماء.
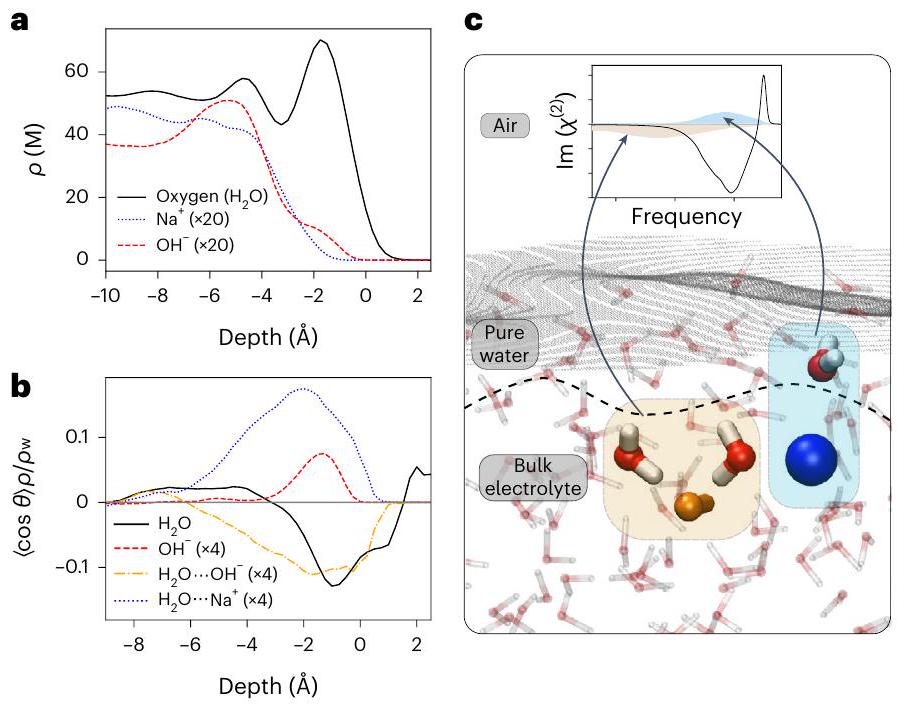
لقد لعب تشكيل طبقة مزدوجة كهربائية عند واجهات الهواء/السائل دورًا أساسيًا في وصف أسطح الإلكتروليتات المائية.
النتائج والمناقشة
نقاط الضعف في نموذج EDL لواجهات الهواء/الإلكتروليت
مرتبط بروابط هيدروجينية
حالة واجهة الهواء/ NaOH (مائي)
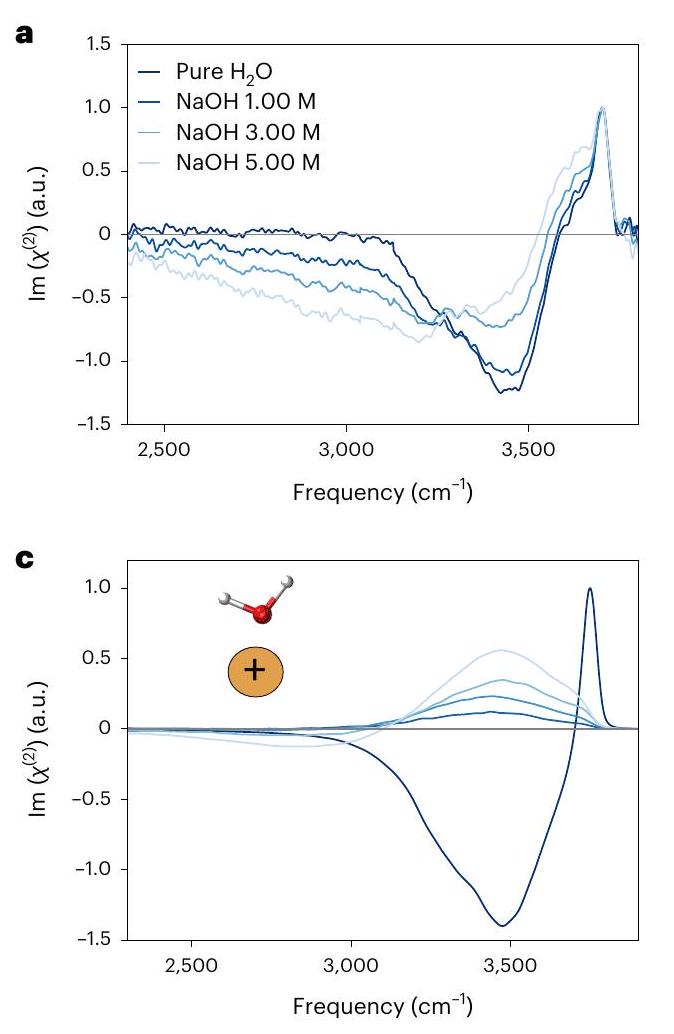
الناشئة من وضع الشد غير المتناظر في جزيئات الماء، التي تتبرع برابطي هيدروجين وتقبل واحدًا
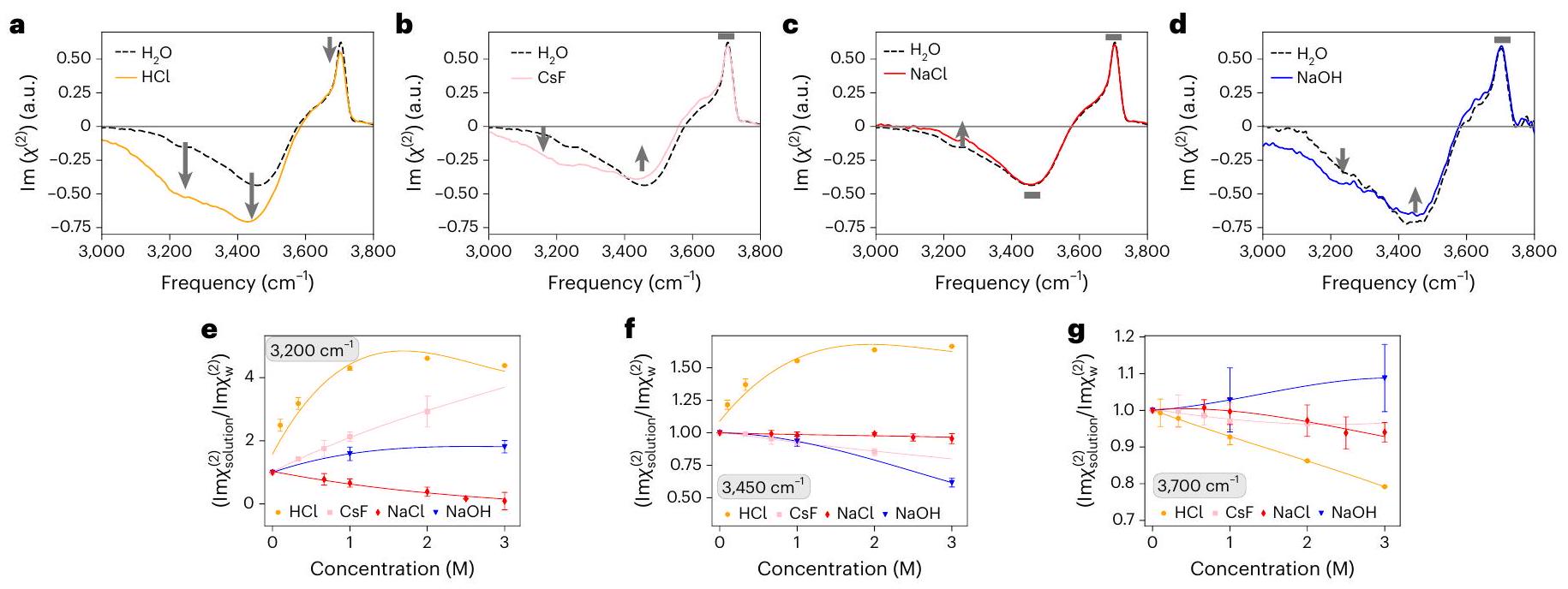
تعميم صورة التدرج إلى الإلكتروليتات البسيطة
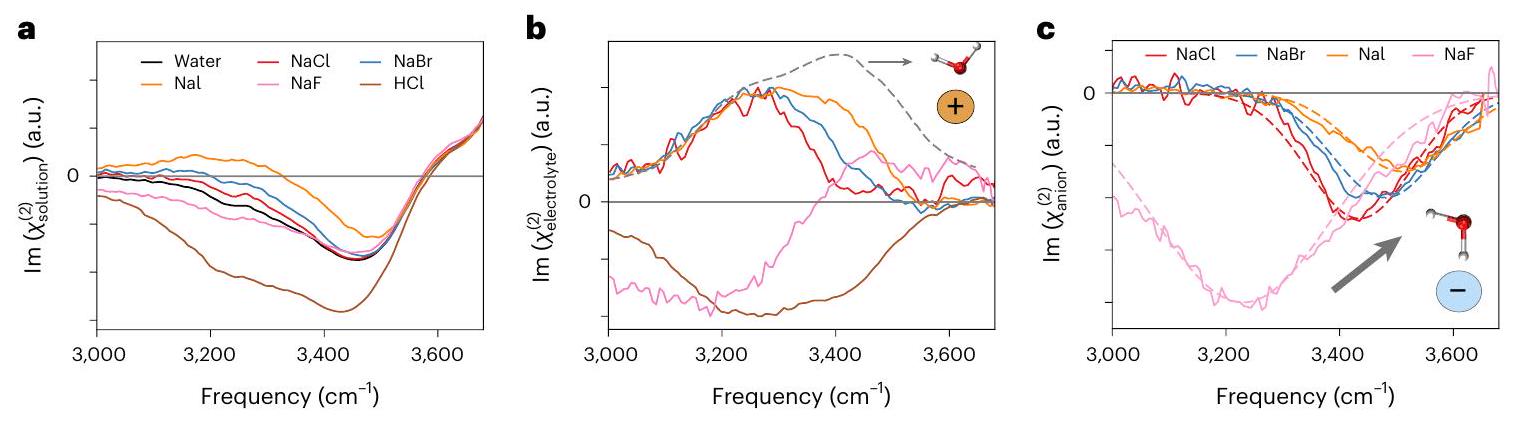
مجال استقطاب الماء أو تفاعل جزيئات الماء مع الكاتيونات، ولكنها مرتبطة بالهيدروجين بجزيئات ماء أخرى، فإن طيف تمدد O-H في الماء يتحكم فيه التفاعلات بين جزيئات الماء.
تظهر القمم السلبية من الماء المنسق مع الكاتيونات والأنيونات، على التوالي. إن فك التداخل لـ
الإلكتروليتات، جنبًا إلى جنب مع الدقة العالية للقياسات (الشكل التكميلي 26)، مكنت من ملاحظة تفاصيل تفاعلات الماء والأيونات، ومكنت من مراجعة نقدية لنطاق تطبيق صورة EDL المعتمدة. بشكل أكثر تحديدًا، بافتراض تكوين EDL، من المتوقع أن تستقطب جزيئات الماء داخل EDL في اتجاه واحد – إما لأعلى أو لأسفل. على الرغم من أنه من الممكن التمييز بين تأثيرات الماء المتفاعل مع الأيونات ذات الشحنات المتنوعة، فإن هذه المساهمات ستظهر باستمرار نفس القطبية. في المقابل، يأخذ النموذج الطبقي في الاعتبار الاستقطاب المزدوج، حيث يمكن للماء أن يتجه في نفس الوقت لأعلى ولأسفل، بما يتماشى مع الملاحظات التجريبية. توسيع صورة التصنيف وأهمية تفاعلات الماء والأيونات التي تم توضيحها في هذه المقالة بشكل كبير على الأوصاف الحالية في الكتب الدراسية، مما يوفر رؤى قوية نحو حل لغز واجهة الهواء/الماء وفهم التفاعلية الكيميائية عند هذه الواجهة الشائعة
المحتوى عبر الإنترنت
References
- Oum, K. W., Lakin, M. J., DeHaan, D. O., Brauers, T. & Finlayson-Pitts, B. J. Formation of molecular chlorine from the photolysis of ozone and aqueous sea-salt particles. Science 279, 74-76 (1998).
- Knipping, E. M. et al. Experiments and simulations of ion-enhanced interfacial chemistry on aqueous NaCl aerosols. Science 288, 301-306 (2000).
- Andreae, M. O. & Crutzen, P. J. Atmospheric aerosols: biogeochemical sources and role in atmospheric chemistry. Science 276, 1052-1058 (1997).
- Geiger, F. M. Second harmonic generation, sum frequency generation and
: dissecting environmental interfaces with a nonlinear optical Swiss army knife. Annu. Rev. Phys. Chem. 60, 61-83 (2009). - Björneholm, O. et al. Water at interfaces. Chem. Rev. 116, 7698-7726 (2016).
- Laage, D., Elsaesser, T. & Hynes, J. T. Water dynamics in the hydration shells of biomolecules. Chem. Rev. 117, 10694-10725 (2017).
- Gonella, G. et al. Water at charged interfaces. Nat. Rev. Chem. 5, 466-485 (2021).
- Shultz, M. J., Schnitzer, C., Simonelli, D. & Baldelli, S. Sum frequency generation spectroscopy of the aqueous interface: ionic and soluble molecular solutions. Int. Rev. Phys. Chem. 19, 123-153 (2000).
- Jungwirth, P. & Tobias, D. J. Molecular structure of salt solutions: a new view of the interface with implications for heterogeneous atmospheric chemistry. J. Phys. Chem. B 105, 10468-10472 (2001).
- Jungwirth, P. & Tobias, D. J. Specific ion effects at the air/water interface. Chem. Rev. 106, 1259-1281 (2006).
- Petersen, P. B. & Saykally, R. J. On the nature of ions at the liquid water surface. Annu. Rev. Phys. Chem. 57, 333-364 (2006).
- Ishiyama, T. & Morita, A. Molecular dynamics study of gas-liquid aqueous sodium halide interfaces. I. Flexible and polarizable molecular modeling and interfacial properties. J. Phys. Chem. C 111, 721-737 (2007).
- Liu, D., Ma, G., Levering, L. M. & Allen, H. C. Vibrational spectroscopy of aqueous sodium halide solutions and air-liquid interfaces: observation of increased interfacial depth. J. Phys. Chem. B 108, 2252-2260 (2004).
- Hua, W., Verreault, D., Huang, Z., Adams, E. M. & Allen, H. C. Cation effects on interfacial water organization of aqueous chloride solutions. I. Monovalent cations:
and . J. Phys. Chem. B 118, 8433-8440 (2014). - Garrett, B. C. Ions at the air/water interface. Science 303, 1146-1147 (2004).
- Tian, C., Ji, N., Waychunas, G. A. & Shen, Y. R. Interfacial structures of acidic and basic aqueous solutions. J. Am. Chem. Soc. 130, 13033-13039 (2008).
- Tian, C., Byrnes, S. J., Han, H.-L. & Shen, Y. R. Surface propensities of atmospherically relevant ions in salt solutions revealed by phase-sensitive sum frequency vibrational spectroscopy. J. Phys. Chem. Lett. 2, 1946-1949 (2011).
- Verreault, D. & Allen, H. C. Bridging the gap between microscopic and macroscopic views of air/aqueous salt interfaces. Chem. Phys. Lett. 586, 1-9 (2013).
- Brown, E. C., Mucha, M., Jungwirth, P. & Tobias, D. J. Structure and vibrational spectroscopy of salt water/air interfaces: predictions from classical molecular dynamics simulations. J. Phys. Chem. B 109, 7934-7940 (2005).
- Petersen, P. B., Johnson, J. C., Knutsen, K. P. & Saykally, R. J. Direct experimental validation of the Jones-Ray effect. Chem. Phys. Lett. 397, 46-50 (2004).
- Raymond, E. A. & Richmond, G. L. Probing the molecular structure and bonding of the surface of aqueous salt solutions. J. Phys. Chem. B 108, 5051-5059 (2004).
- Baer, M. D. & Mundy, C. J. Toward an understanding of the specific ion effect using density functional theory. J. Phys. Chem. Lett. 2, 1088-1093 (2011).
- Tang, F. et al. Molecular structure and modeling of water-air and ice-air interfaces monitored by sum-frequency generation. Chem. Rev. 120, 3633-3667 (2020).
- Das, S. et al. Nature of excess hydrated proton at the water-air interface. J. Am. Chem. Soc. 142, 945-952 (2020).
- Chiang, K.-Y., Dalstein, L. & Wen, Y.-C. Affinity of hydrated protons at intrinsic water/vapor interface revealed by ion-induced water alignment. J. Phys. Chem. Lett. 11, 696-701 (2020).
- Tse, Y.-L. S., Chen, C., Lindberg, G. E., Kumar, R. & Voth, G. A. Propensity of hydrated excess protons and hydroxide anions for the air-water interface. J. Am. Chem. Soc. 137, 12610-12616 (2015).
- Ji, N., Ostroverkhov, V., Tian, C. S. & Shen, Y. R. Characterization of vibrational resonances of water-vapor interfaces by phase-sensitive sum-frequency spectroscopy. Phys. Rev. Lett. 100, 96102 (2008).
- Nihonyanagi, S., Yamaguchi, S. & Tahara, T. Ultrafast dynamics at water interfaces studied by vibrational sum frequency generation spectroscopy. Chem. Rev. 117, 10665-10693 (2017).
- Nihonyanagi, S. et al. Accurate determination of complex
spectrum of the air/water interface. J. Chem. Phys. 143, 124707 (2015). - Ohto, T., Usui, K., Hasegawa, T., Bonn, M. & Nagata, Y. Toward ab initio molecular dynamics modeling for sum-frequency generation spectra; an efficient algorithm based on surface-specific velocity-velocity correlation function. J. Chem. Phys. 143, 124702 (2015).
- Behler, J. & Parrinello, M. Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett. 98, 146401 (2007).
- Behler, J. First principles neural network potentials for reactive simulations of large molecular and condensed systems. Angew. Chem. Int. Ed. 56, 12828-12840 (2017).
- Stiopkin, I. V. et al. Hydrogen bonding at the water surface revealed by isotopic dilution spectroscopy. Nature 474, 192-195 (2011).
- Imamura, T., Ishiyama, T. & Morita, A. Molecular dynamics analysis of NaOH aqueous solution surface and the sum frequency generation spectra: is surface
detected by SFG spectroscopy? J. Phys. Chem. C 118, 29017-29027 (2014). - Mandal, A., Ramasesha, K., de Marco, L. & Tokmakoff, A. Collective vibrations of water-solvated hydroxide ions investigated with broadband 2DIR spectroscopy. J. Chem. Phys. 140, 204508 (2014).
- Hellstróm, M. & Behler, J. Structure of aqueous NaOH solutions: insights from neural-network-based molecular dynamics simulations. Phys. Chem. Chem. Phys. 19, 82-96 (2017).
- Chen, B. et al. First-principles study of aqueous hydroxide solutions. J. Am. Chem. Soc. 124, 8534-8535 (2002).
- Crespo, Y. & Hassanali, A. Unveiling the Janus-like properties of
. J. Phys. Chem. Lett. 6, 272-278 (2015). - Crespo, Y. & Hassanali, A. Characterizing the local solvation environment of
in water clusters with AIMD. J. Chem. Phys. 144, 74304 (2016). - Wen, Y.-C. et al. Unveiling microscopic structures of charged water interfaces by surface-specific vibrational spectroscopy. Phys. Rev. Lett. 116, 16101 (2016).
- Ohno, P. E., Wang, H., Paesani, F., Skinner, J. L. & Geiger, F. M. Second-order vibrational lineshapes from the air/water interface. J. Phys. Chem. A 122, 4457-4464 (2018).
- Nagata, Y., Pool, R. E., Backus, E. H. G. & Bonn, M. Nuclear quantum effects affect bond orientation of water at the water-vapor interface. Phys. Rev. Lett. 109, 226101 (2012).
- Schaefer, J., Backus, E. H. G., Nagata, Y. & Bonn, M. Both inter- and intramolecular coupling of O-H groups determine the vibrational response of the water/air interface. J. Phys. Chem. Lett. 7, 4591-4595 (2016).
- Wei, F., Urashima, S., Nihonyanagi, S. & Tahara, T. Elucidation of the pH -dependent electric double layer structure at the silica/ water interface using heterodyne-detected vibrational sum frequency generation spectroscopy. J. Am. Chem. Soc. 145, 8833-8846 (2023).
- Max, J.-J. & Chapados, C. IR spectroscopy of aqueous alkali halide solutions: pure salt-solvated water spectra and hydration numbers. J. Chem. Phys. 115, 2664-2675 (2001).
- Robertson, W. H. & Johnson, M. A. Molecular aspects of halide ion hydration: the cluster approach. Annu. Rev. Phys. Chem. 54, 173-213 (2003).
- Robertson, W. H., Diken, E. G., Price, E. A., Shin, J.-W. & Johnson, M. A. Spectroscopic determination of the
solvation shell in the clusters. Science 299, 1367-1372 (2003). - Knorke, H., Li, H., Warneke, J., Liu, Z.-F. & Asmis, K. R. Cryogenic ion trap vibrational spectroscopy of the microhydrated sulfate dianions
. Phys. Chem. Chem. Phys. 22, 27732-27745 (2020). - Thaunay, F. et al. Hydration of the sulfate dianion in size-selected water clusters: From
to . Int. J. Mass Spectrom. 418, 15-23 (2017). - Tielrooij, K. J., Garcia-Araez, N., Bonn, M. & Bakker, H. J. Cooperativity in ion hydration. Science 328, 1006-1009 (2010).
- Seki, T. et al. Ions speciation at the water-air interface. J. Am. Chem. Soc. 145, 10622-10630 (2023).
- Devlin, S. W., Benjamin, I. & Saykally, R. J. On the mechanisms of ion adsorption to aqueous interfaces: air-water vs. oil-water. Proc. Natl Acad. Sci. USA 119, e2210857119 (2022).
- Netz, R. R. & Horinek, D. Progress in modeling of ion effects at the vapor/water interface. Annu. Rev. Phys. Chem. 63, 401-418 (2012).
- Duignan, T. T., Parsons, D. F. & Ninham, B. W. Ion interactions with the air-water interface using a continuum solvent model. J. Phys. Chem. B 118, 8700-8710 (2014).
- Levin, Y., dos Santos, A. P. & Diehl, A. lons at the air-water interface: an end to a hundred-year-old mystery? Phys. Rev. Lett. 103, 257802 (2009).
- Ghosal, S. et al. Electron spectroscopy of aqueous solution interfaces reveals surface enhancement of halides. Science 307, 563-566 (2005).
- Petersen, P. B. & Saykally, R. J. Confirmation of enhanced anion concentration at the liquid water surface. Chem. Phys. Lett. 397, 51-55 (2004).
- Kusaka, R., Nihonyanagi, S. & Tahara, T. The photochemical reaction of phenol becomes ultrafast at the air-water interface. Nat. Chem. 13, 306-311 (2021).
- Lesnicki, D., Wank, V., Cyran, J. D., Backus, E. H. G. & Sulpizi, M. Lower degree of dissociation of pyruvic acid at water surfaces than in bulk. Phys. Chem. Chem. Phys. 24, 13510-13513 (2022).
- Auer, B. M. & Skinner, J. L. IR and Raman spectra of liquid water: theory and interpretation. J. Chem. Phys. 128, 224511 (2008).
- Corcelli, S. A. & Skinner, J. L. Infrared and Raman line shapes of dilute HOD in liquid
and from 10 to . J. Phys. Chem. A 109, 6154-6165 (2005).
© The Author(s) 2024
طرق
محاكاة من البداية
تدريب NN
محاكاة MD لـ NN
وتم الحصول عليها من خلال متوسط 40 مسارًا مستقلًا بمدة 200 بيكوثانية تم تنفيذها من إحداثيات أولية غير مترابطة مستخرجة من مسارات NVT. كانت خطوة الزمن المستخدمة في تكامل معادلة الحركة في جميع الحالات 0.5 فيكوتانية. ما لم يُذكر خلاف ذلك، فإن التصنيف كـ
تحضير العينة
طيفية SFG عالية الدقة
عرض نطاق
توفر البيانات
توفر الشيفرة
References
- Kühne, T. D. et al. CP2K: an electronic structure and molecular dynamics software package-Quickstep: efficient and accurate electronic structure calculations. J. Chem. Phys. 152, 194103 (2020).
- Ohto, T. et al. Accessing the accuracy of density functional theory through structure and dynamics of the water-air interface. J. Phys. Chem. Lett. 10, 4914-4919 (2019).
- Singraber, A., Morawietz, T., Behler, J. & Dellago, C. Parallel multistream training of high-dimensional neural network potentials. J. Chem. Theory Comput. 15, 3075-3092 (2019).
- Singraber, A., Behler, J. & Dellago, C. Library-based LAMMPS implementation of high-dimensional neural network potentials. J. Chem. Theory Comput. 15, 1827-1840 (2019).
- Schran, C. et al. Machine learning potentials for complex aqueous systems made simple. Proc. Natl Acad. Sci. USA 118, e2110077118 (2021).
- Schran, C., Brezina, K. & Marsalek, O. Committee neural network potentials control generalization errors and enable active learning. J. Chem. Phys. 153, 104105 (2020).
- Niblett, S. P., Galib, M. & Limmer, D. T. Learning intermolecular forces at liquid-vapor interfaces. J. Chem. Phys. 155, 164101 (2021).
- Gao, A. & Remsing, R. C. Self-consistent determination of long-range electrostatics in neural network potentials. Nat. Commun. 13, 1572 (2022).
- Cox, S. J. Dielectric response with short-ranged electrostatics. Proc. Natl Acad. Sci. USA 117, 19746-19752 (2020).
- Hellström, M. & Behler, J. Concentration-dependent proton transfer mechanisms in aqueous NaOH Solutions: from acceptor-driven to donor-driven and back. J. Phys. Chem. Lett. 7, 3302-3306 (2016).
- Hellström, M., Ceriotti, M. & Behler, J. Nuclear quantum effects in sodium hydroxide solutions from neural network molecular dynamics simulations. J. Phys. Chem. B 122, 10158-10171 (2018).
- Kapil, V. et al. i-PI 2.0: a universal force engine for advanced molecular simulations. Comput. Phys. Commun. 236, 214-223 (2019).
- Thompson, A. P. et al. LAMMPS-a flexible simulation tool for particle-based materials modeling at the atomic, meso and continuum scales. Comput. Phys. Commun. 271, 108171 (2022).
- Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 14101 (2007).
- Willard, A. P. & Chandler, D. Instantaneous liquid interfaces. J. Phys. Chem. B 114, 1954-1958 (2010).
- Vanselous, H. & Petersen, P. B. Extending the capabilities of heterodyne-detected sum-frequency generation spectroscopy: probing any interface in any polarization combination. J. Phys. Chem. C 120, 8175-8184 (2016).
شكر وتقدير
مساهمات المؤلفين
تمويل
المصالح المتنافسة
معلومات إضافية
معهد ماكس بلانك لبحوث البوليمرات، ماينز، ألمانيا. قسم الكيمياء يوسف حامد، جامعة كامبريدج، كامبريدج، المملكة المتحدة.
ساهم هؤلاء المؤلفون بالتساوي: يائير ليتمان، كوو-يانغ تشيانغ. البريد الإلكتروني: yl899@cam.ac.uk; bonn@mpip-mainz.mpg.de
DOI: https://doi.org/10.1038/s41557-023-01416-6
PMID: https://pubmed.ncbi.nlm.nih.gov/38225269
Publication Date: 2024-01-15
Surface stratification determines the interfacial water structure of simple electrolyte solutions
Accepted: 7 December 2023
Published online: 15 January 2024
(A) Check for updates
Abstract
The distribution of ions at the air/water interface plays a decisive role in many natural processes. Several studies have reported that larger ions tend to be surface-active, implying ions are located on top of the water surface, thereby inducing electric fields that determine the interfacial water structure. Here we challenge this view by combining surface-specific heterodyne-detected vibrational sum-frequency generation with neural network-assisted ab initio molecular dynamics simulations. Our results show that ions in typical electrolyte solutions are, in fact, located in a subsurface region, leading to a stratification of such interfaces into two distinctive water layers. The outermost surface is ion-depleted, and the subsurface layer is ion-enriched. This surface stratification is a key element in explaining the ion-induced water reorganization at the outermost air/water interface.
of the formation of an EDL at air/liquid interfaces has played a fundamental role in describing aqueous electrolyte surfaces
Results and discussion
Shortcomings of the EDL model for air/electrolyte interfaces
hydrogen-bonded
The case of the air/ NaOH (aq.) interface
arising from the asymmetric stretching mode in water molecules, which donate two hydrogen bonds and accept one
Generalization of the stratification picture to simple electrolytes
field polarizing water or water molecules interacting with cations, but hydrogen-bonded to other water molecules, the water O-H stretch spectrum is governed by the intermolecular water-water interactions
negative peaks arise from water coordinated to the cations and anions, respectively. The deconvolution of the
of electrolytes, together with the high accuracy of the measurements (Supplementary Fig. 26), enabled the observation of the subtleties of the water-ion interactions, and enabled a critical revision of the domain of applicability of the established EDL picture. More specifically, assuming the formation of an EDL, water molecules within the EDL are predicted to polarize in a single direction-either pointing upwards or downwards. Although it is possible to differentiate the effects of water interacting with ions of varying charges, these contributions would consistently exhibit the same polarity. In contrast, the stratified model accounts for dual polarization, where water can simultaneously orient both upwards and downwards, in alignment with the experimental observations. The stratification picture and the importance of water-ion interactions demonstrated in this Article substantially expand on current textbook descriptions, providing powerful insights towards resolving the air/water interface puzzle and understanding chemical reactivity at this ubiquitous interface
Online content
References
- Oum, K. W., Lakin, M. J., DeHaan, D. O., Brauers, T. & Finlayson-Pitts, B. J. Formation of molecular chlorine from the photolysis of ozone and aqueous sea-salt particles. Science 279, 74-76 (1998).
- Knipping, E. M. et al. Experiments and simulations of ion-enhanced interfacial chemistry on aqueous NaCl aerosols. Science 288, 301-306 (2000).
- Andreae, M. O. & Crutzen, P. J. Atmospheric aerosols: biogeochemical sources and role in atmospheric chemistry. Science 276, 1052-1058 (1997).
- Geiger, F. M. Second harmonic generation, sum frequency generation and
: dissecting environmental interfaces with a nonlinear optical Swiss army knife. Annu. Rev. Phys. Chem. 60, 61-83 (2009). - Björneholm, O. et al. Water at interfaces. Chem. Rev. 116, 7698-7726 (2016).
- Laage, D., Elsaesser, T. & Hynes, J. T. Water dynamics in the hydration shells of biomolecules. Chem. Rev. 117, 10694-10725 (2017).
- Gonella, G. et al. Water at charged interfaces. Nat. Rev. Chem. 5, 466-485 (2021).
- Shultz, M. J., Schnitzer, C., Simonelli, D. & Baldelli, S. Sum frequency generation spectroscopy of the aqueous interface: ionic and soluble molecular solutions. Int. Rev. Phys. Chem. 19, 123-153 (2000).
- Jungwirth, P. & Tobias, D. J. Molecular structure of salt solutions: a new view of the interface with implications for heterogeneous atmospheric chemistry. J. Phys. Chem. B 105, 10468-10472 (2001).
- Jungwirth, P. & Tobias, D. J. Specific ion effects at the air/water interface. Chem. Rev. 106, 1259-1281 (2006).
- Petersen, P. B. & Saykally, R. J. On the nature of ions at the liquid water surface. Annu. Rev. Phys. Chem. 57, 333-364 (2006).
- Ishiyama, T. & Morita, A. Molecular dynamics study of gas-liquid aqueous sodium halide interfaces. I. Flexible and polarizable molecular modeling and interfacial properties. J. Phys. Chem. C 111, 721-737 (2007).
- Liu, D., Ma, G., Levering, L. M. & Allen, H. C. Vibrational spectroscopy of aqueous sodium halide solutions and air-liquid interfaces: observation of increased interfacial depth. J. Phys. Chem. B 108, 2252-2260 (2004).
- Hua, W., Verreault, D., Huang, Z., Adams, E. M. & Allen, H. C. Cation effects on interfacial water organization of aqueous chloride solutions. I. Monovalent cations:
and . J. Phys. Chem. B 118, 8433-8440 (2014). - Garrett, B. C. Ions at the air/water interface. Science 303, 1146-1147 (2004).
- Tian, C., Ji, N., Waychunas, G. A. & Shen, Y. R. Interfacial structures of acidic and basic aqueous solutions. J. Am. Chem. Soc. 130, 13033-13039 (2008).
- Tian, C., Byrnes, S. J., Han, H.-L. & Shen, Y. R. Surface propensities of atmospherically relevant ions in salt solutions revealed by phase-sensitive sum frequency vibrational spectroscopy. J. Phys. Chem. Lett. 2, 1946-1949 (2011).
- Verreault, D. & Allen, H. C. Bridging the gap between microscopic and macroscopic views of air/aqueous salt interfaces. Chem. Phys. Lett. 586, 1-9 (2013).
- Brown, E. C., Mucha, M., Jungwirth, P. & Tobias, D. J. Structure and vibrational spectroscopy of salt water/air interfaces: predictions from classical molecular dynamics simulations. J. Phys. Chem. B 109, 7934-7940 (2005).
- Petersen, P. B., Johnson, J. C., Knutsen, K. P. & Saykally, R. J. Direct experimental validation of the Jones-Ray effect. Chem. Phys. Lett. 397, 46-50 (2004).
- Raymond, E. A. & Richmond, G. L. Probing the molecular structure and bonding of the surface of aqueous salt solutions. J. Phys. Chem. B 108, 5051-5059 (2004).
- Baer, M. D. & Mundy, C. J. Toward an understanding of the specific ion effect using density functional theory. J. Phys. Chem. Lett. 2, 1088-1093 (2011).
- Tang, F. et al. Molecular structure and modeling of water-air and ice-air interfaces monitored by sum-frequency generation. Chem. Rev. 120, 3633-3667 (2020).
- Das, S. et al. Nature of excess hydrated proton at the water-air interface. J. Am. Chem. Soc. 142, 945-952 (2020).
- Chiang, K.-Y., Dalstein, L. & Wen, Y.-C. Affinity of hydrated protons at intrinsic water/vapor interface revealed by ion-induced water alignment. J. Phys. Chem. Lett. 11, 696-701 (2020).
- Tse, Y.-L. S., Chen, C., Lindberg, G. E., Kumar, R. & Voth, G. A. Propensity of hydrated excess protons and hydroxide anions for the air-water interface. J. Am. Chem. Soc. 137, 12610-12616 (2015).
- Ji, N., Ostroverkhov, V., Tian, C. S. & Shen, Y. R. Characterization of vibrational resonances of water-vapor interfaces by phase-sensitive sum-frequency spectroscopy. Phys. Rev. Lett. 100, 96102 (2008).
- Nihonyanagi, S., Yamaguchi, S. & Tahara, T. Ultrafast dynamics at water interfaces studied by vibrational sum frequency generation spectroscopy. Chem. Rev. 117, 10665-10693 (2017).
- Nihonyanagi, S. et al. Accurate determination of complex
spectrum of the air/water interface. J. Chem. Phys. 143, 124707 (2015). - Ohto, T., Usui, K., Hasegawa, T., Bonn, M. & Nagata, Y. Toward ab initio molecular dynamics modeling for sum-frequency generation spectra; an efficient algorithm based on surface-specific velocity-velocity correlation function. J. Chem. Phys. 143, 124702 (2015).
- Behler, J. & Parrinello, M. Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett. 98, 146401 (2007).
- Behler, J. First principles neural network potentials for reactive simulations of large molecular and condensed systems. Angew. Chem. Int. Ed. 56, 12828-12840 (2017).
- Stiopkin, I. V. et al. Hydrogen bonding at the water surface revealed by isotopic dilution spectroscopy. Nature 474, 192-195 (2011).
- Imamura, T., Ishiyama, T. & Morita, A. Molecular dynamics analysis of NaOH aqueous solution surface and the sum frequency generation spectra: is surface
detected by SFG spectroscopy? J. Phys. Chem. C 118, 29017-29027 (2014). - Mandal, A., Ramasesha, K., de Marco, L. & Tokmakoff, A. Collective vibrations of water-solvated hydroxide ions investigated with broadband 2DIR spectroscopy. J. Chem. Phys. 140, 204508 (2014).
- Hellstróm, M. & Behler, J. Structure of aqueous NaOH solutions: insights from neural-network-based molecular dynamics simulations. Phys. Chem. Chem. Phys. 19, 82-96 (2017).
- Chen, B. et al. First-principles study of aqueous hydroxide solutions. J. Am. Chem. Soc. 124, 8534-8535 (2002).
- Crespo, Y. & Hassanali, A. Unveiling the Janus-like properties of
. J. Phys. Chem. Lett. 6, 272-278 (2015). - Crespo, Y. & Hassanali, A. Characterizing the local solvation environment of
in water clusters with AIMD. J. Chem. Phys. 144, 74304 (2016). - Wen, Y.-C. et al. Unveiling microscopic structures of charged water interfaces by surface-specific vibrational spectroscopy. Phys. Rev. Lett. 116, 16101 (2016).
- Ohno, P. E., Wang, H., Paesani, F., Skinner, J. L. & Geiger, F. M. Second-order vibrational lineshapes from the air/water interface. J. Phys. Chem. A 122, 4457-4464 (2018).
- Nagata, Y., Pool, R. E., Backus, E. H. G. & Bonn, M. Nuclear quantum effects affect bond orientation of water at the water-vapor interface. Phys. Rev. Lett. 109, 226101 (2012).
- Schaefer, J., Backus, E. H. G., Nagata, Y. & Bonn, M. Both inter- and intramolecular coupling of O-H groups determine the vibrational response of the water/air interface. J. Phys. Chem. Lett. 7, 4591-4595 (2016).
- Wei, F., Urashima, S., Nihonyanagi, S. & Tahara, T. Elucidation of the pH -dependent electric double layer structure at the silica/ water interface using heterodyne-detected vibrational sum frequency generation spectroscopy. J. Am. Chem. Soc. 145, 8833-8846 (2023).
- Max, J.-J. & Chapados, C. IR spectroscopy of aqueous alkali halide solutions: pure salt-solvated water spectra and hydration numbers. J. Chem. Phys. 115, 2664-2675 (2001).
- Robertson, W. H. & Johnson, M. A. Molecular aspects of halide ion hydration: the cluster approach. Annu. Rev. Phys. Chem. 54, 173-213 (2003).
- Robertson, W. H., Diken, E. G., Price, E. A., Shin, J.-W. & Johnson, M. A. Spectroscopic determination of the
solvation shell in the clusters. Science 299, 1367-1372 (2003). - Knorke, H., Li, H., Warneke, J., Liu, Z.-F. & Asmis, K. R. Cryogenic ion trap vibrational spectroscopy of the microhydrated sulfate dianions
. Phys. Chem. Chem. Phys. 22, 27732-27745 (2020). - Thaunay, F. et al. Hydration of the sulfate dianion in size-selected water clusters: From
to . Int. J. Mass Spectrom. 418, 15-23 (2017). - Tielrooij, K. J., Garcia-Araez, N., Bonn, M. & Bakker, H. J. Cooperativity in ion hydration. Science 328, 1006-1009 (2010).
- Seki, T. et al. Ions speciation at the water-air interface. J. Am. Chem. Soc. 145, 10622-10630 (2023).
- Devlin, S. W., Benjamin, I. & Saykally, R. J. On the mechanisms of ion adsorption to aqueous interfaces: air-water vs. oil-water. Proc. Natl Acad. Sci. USA 119, e2210857119 (2022).
- Netz, R. R. & Horinek, D. Progress in modeling of ion effects at the vapor/water interface. Annu. Rev. Phys. Chem. 63, 401-418 (2012).
- Duignan, T. T., Parsons, D. F. & Ninham, B. W. Ion interactions with the air-water interface using a continuum solvent model. J. Phys. Chem. B 118, 8700-8710 (2014).
- Levin, Y., dos Santos, A. P. & Diehl, A. lons at the air-water interface: an end to a hundred-year-old mystery? Phys. Rev. Lett. 103, 257802 (2009).
- Ghosal, S. et al. Electron spectroscopy of aqueous solution interfaces reveals surface enhancement of halides. Science 307, 563-566 (2005).
- Petersen, P. B. & Saykally, R. J. Confirmation of enhanced anion concentration at the liquid water surface. Chem. Phys. Lett. 397, 51-55 (2004).
- Kusaka, R., Nihonyanagi, S. & Tahara, T. The photochemical reaction of phenol becomes ultrafast at the air-water interface. Nat. Chem. 13, 306-311 (2021).
- Lesnicki, D., Wank, V., Cyran, J. D., Backus, E. H. G. & Sulpizi, M. Lower degree of dissociation of pyruvic acid at water surfaces than in bulk. Phys. Chem. Chem. Phys. 24, 13510-13513 (2022).
- Auer, B. M. & Skinner, J. L. IR and Raman spectra of liquid water: theory and interpretation. J. Chem. Phys. 128, 224511 (2008).
- Corcelli, S. A. & Skinner, J. L. Infrared and Raman line shapes of dilute HOD in liquid
and from 10 to . J. Phys. Chem. A 109, 6154-6165 (2005).
© The Author(s) 2024
Methods
Ab initio simulations
NN training
NN MD simulations
and obtained by averaging 40 independent 200-ps trajectories carried out from uncorrelated initial coordinates extracted from the NVT trajectories. The time step employed for the integration of the equation of motions was in all cases 0.5 fs. Unless stated otherwise, the classification as either
Sample preparation
HD SFG spectroscopy
a bandwidth of
Data availability
Code availability
References
- Kühne, T. D. et al. CP2K: an electronic structure and molecular dynamics software package-Quickstep: efficient and accurate electronic structure calculations. J. Chem. Phys. 152, 194103 (2020).
- Ohto, T. et al. Accessing the accuracy of density functional theory through structure and dynamics of the water-air interface. J. Phys. Chem. Lett. 10, 4914-4919 (2019).
- Singraber, A., Morawietz, T., Behler, J. & Dellago, C. Parallel multistream training of high-dimensional neural network potentials. J. Chem. Theory Comput. 15, 3075-3092 (2019).
- Singraber, A., Behler, J. & Dellago, C. Library-based LAMMPS implementation of high-dimensional neural network potentials. J. Chem. Theory Comput. 15, 1827-1840 (2019).
- Schran, C. et al. Machine learning potentials for complex aqueous systems made simple. Proc. Natl Acad. Sci. USA 118, e2110077118 (2021).
- Schran, C., Brezina, K. & Marsalek, O. Committee neural network potentials control generalization errors and enable active learning. J. Chem. Phys. 153, 104105 (2020).
- Niblett, S. P., Galib, M. & Limmer, D. T. Learning intermolecular forces at liquid-vapor interfaces. J. Chem. Phys. 155, 164101 (2021).
- Gao, A. & Remsing, R. C. Self-consistent determination of long-range electrostatics in neural network potentials. Nat. Commun. 13, 1572 (2022).
- Cox, S. J. Dielectric response with short-ranged electrostatics. Proc. Natl Acad. Sci. USA 117, 19746-19752 (2020).
- Hellström, M. & Behler, J. Concentration-dependent proton transfer mechanisms in aqueous NaOH Solutions: from acceptor-driven to donor-driven and back. J. Phys. Chem. Lett. 7, 3302-3306 (2016).
- Hellström, M., Ceriotti, M. & Behler, J. Nuclear quantum effects in sodium hydroxide solutions from neural network molecular dynamics simulations. J. Phys. Chem. B 122, 10158-10171 (2018).
- Kapil, V. et al. i-PI 2.0: a universal force engine for advanced molecular simulations. Comput. Phys. Commun. 236, 214-223 (2019).
- Thompson, A. P. et al. LAMMPS-a flexible simulation tool for particle-based materials modeling at the atomic, meso and continuum scales. Comput. Phys. Commun. 271, 108171 (2022).
- Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 14101 (2007).
- Willard, A. P. & Chandler, D. Instantaneous liquid interfaces. J. Phys. Chem. B 114, 1954-1958 (2010).
- Vanselous, H. & Petersen, P. B. Extending the capabilities of heterodyne-detected sum-frequency generation spectroscopy: probing any interface in any polarization combination. J. Phys. Chem. C 120, 8175-8184 (2016).
Acknowledgements
Author contributions
Funding
Competing interests
Additional information
Max Planck Institute for Polymer Research, Mainz, Germany. Yusuf Hamied Department of Chemistry, University of Cambridge, Cambridge, UK.
These authors contributed equally: Yair Litman, Kuo-Yang Chiang. e-mail: yl899@cam.ac.uk; bonn@mpip-mainz.mpg.de