DOI: https://doi.org/10.1038/s41593-023-01558-3
PMID: https://pubmed.ncbi.nlm.nih.gov/38267524
تاريخ النشر: 2024-01-24
ترابط الأيض بين الخلايا الدبقية قليلة التغصن والمحاور يتم بواسطة البوتاسيوم الخارجي K+ ويحافظ على صحة المحاور
تم القبول: 13 ديسمبر 2023
نُشر على الإنترنت: 24 يناير 2024
(أ) التحقق من التحديثات
الملخص
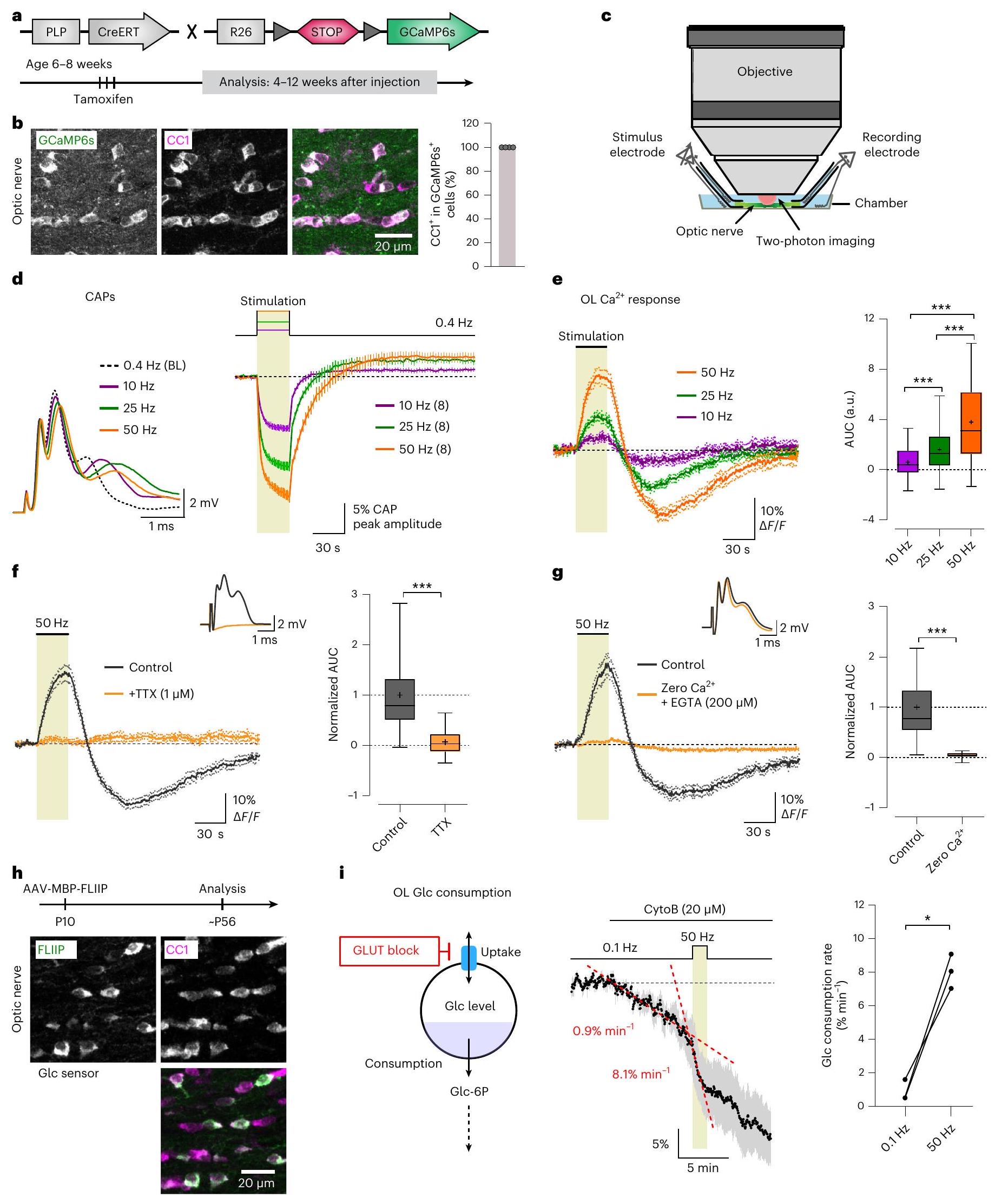
تعتمد سلامة المحاور الميالينية على الدعم الهوموستاتيكي من الخلايا الدبقية قليلة التغصن (OLs). لتحديد كيفية اكتشاف الخلايا الدبقية قليلة التغصن للنبضات المحورية وكيفية تنظيم الاقتران الأيضي السريع بين المحاور والخلايا الدبقية قليلة التغصن في المادة البيضاء، درسنا الكالسيوم المعتمد على النشاط (
يؤدي نقص ناقل أحادي الكربوكسيلات 1 (MCT1) إلى اعتلال المحاور المتأخر، مما يعني أن إفراز اللاكتات و/أو البيروفات من الخلايا الدبقية قليلة التغصن له دور في صحة المحاور.
النتائج
إطلاق النبضات المحورية
لمدة 30 ثانية. قبل وبعد هذه الفترة، تلقت الأعصاب
تكتشف الخلايا الدبقية الشوكية النبضات المحورية من خلال
يظهر انخفاض بنسبة
ال
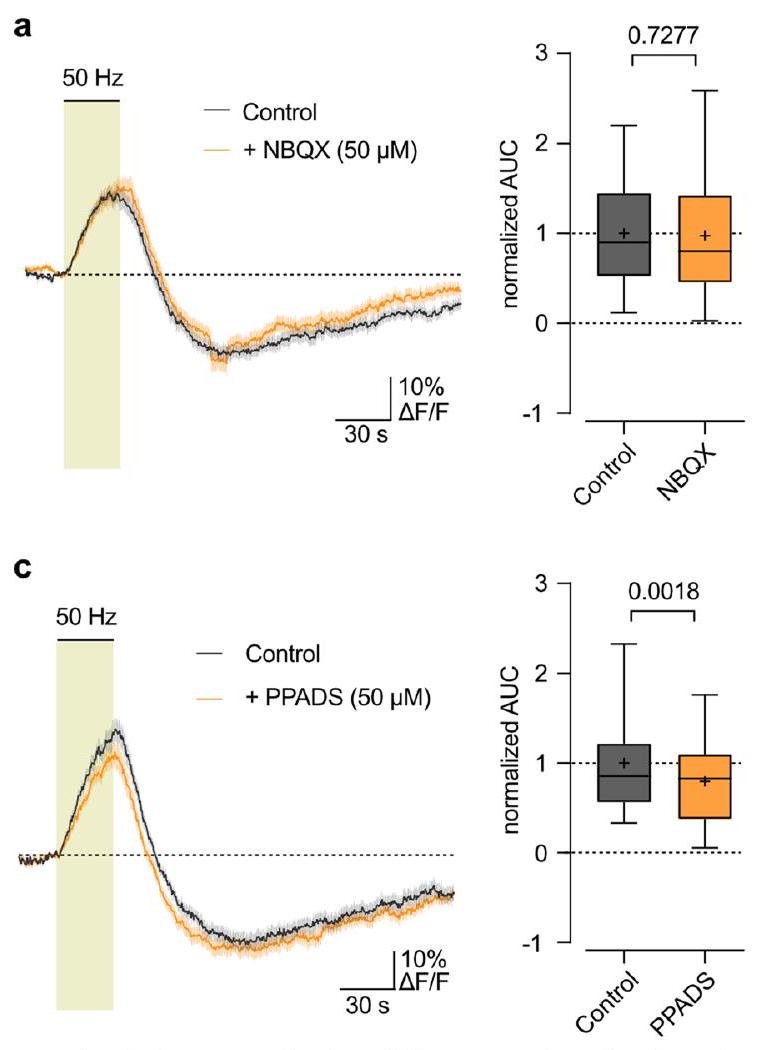
(PPADS;
| أجسام مضادة | نوع المضيف، النوع | الطريقة، التخفيف | المصدر، رقم الكتالوج. |
| مضاد-CC1 | فأر، وحيد النسيلة | إتش آي سي، 1:100 | كالبيوكيم، رقم الكات. OP80 (نسخة 5.24) |
| مضاد GFP | دجاج، متعدد النسائل | إتش آي سي، 1:1,000 | أفيز لابز، رقم الكاتالوج GFP-1020 |
| مضاد-GFAP | دجاج، متعدد النسائل | إتش آي سي، 1:2000 | أبكام، رقم الكات. ab4674 |
| مضاد IBA1 | أرنب، متعدد النسائل | إتش آي سي، 1:1,000 | فوجي فيلم واكو كيميكالز، رقم الكاتالوج 019-19741 |
| مضاد كير4.1 | أرنب، متعدد النسائل | IB، 1:1,000 | ألومون، رقم الكاتالوج APC-035 |
| مضاد MCT1/SLC16A1 | أرنب، متعدد النسائل | IB، 1:500 | من إنتاج كاثرين كوش
|
| مضاد GLUT1 | أرنب، متعدد النسائل | IB، 1:500 | من إنتاج كاثرين كوش
|
| مضاد-CNP | فأر، وحيد النسيلة | IB، 1:1,000 | سيغما، رقم الكاتالوج C 5922 (استنساخ 11-5B) |
| مناهض لـ PLP | أرنب، متعدد النسائل | IB، 1:5,000 | A431 (مرجع 118) |
| مضاد MOG | فأر، وحيد النسيلة | IB، 1:5,000 | كرياتيف بايولابز، رقم الكاتالوج PABZ-152 (النسخة 8-18C5) |
| مضاد ATP1
|
فأر، وحيد النسيلة | IB، 1:1,000 | أبكام، رقم الكات. ab7671 (نسخة 464.6) |
| مضاد ATP1
|
فأر، وحيد النسيلة | IB، 1:1,000 | أبكام، رقم الكات. ab2826 (النسخة XVIF9- G10) |
| أجسام مضادة للفأر IgG HRP | ماعز، متعدد النسائل | IB، 1:10,000 | جاكسون إيمونوريسيرش، رقم الكاتالوج 115-035-003 |
| أجسام مضادة ضد الأرانب IgG HRP | ماعز، متعدد النسائل | IB، 1:10,000 | جاكسون إيمونوريسيرش، رقم الكاتالوج 111-035-003 |
| مضاد الفأر Cy3 | حمار، متعدد النسائل | إتش سي، 1:700 | جاكسون إيمونوريسيرش، رقم الكاتالوج 715-165-151 |
| مضاد الأرنب Cy3 | حمار، متعدد النسائل | إتش سي، 1:700 | جاكسون إيمونوريسيرش، رقم الكاتالوج 711-165-152 |
| أنتي دجاج أليكسا 488 | حمار، متعدد النسائل | إتش سي، 1:700 | جاكسون إيمونوريسيرش، رقم الكاتالوج 711-545-152 |
تحفيز. زيادات مؤقتة في الحمام
إطلاق النار بعد
بواسطة
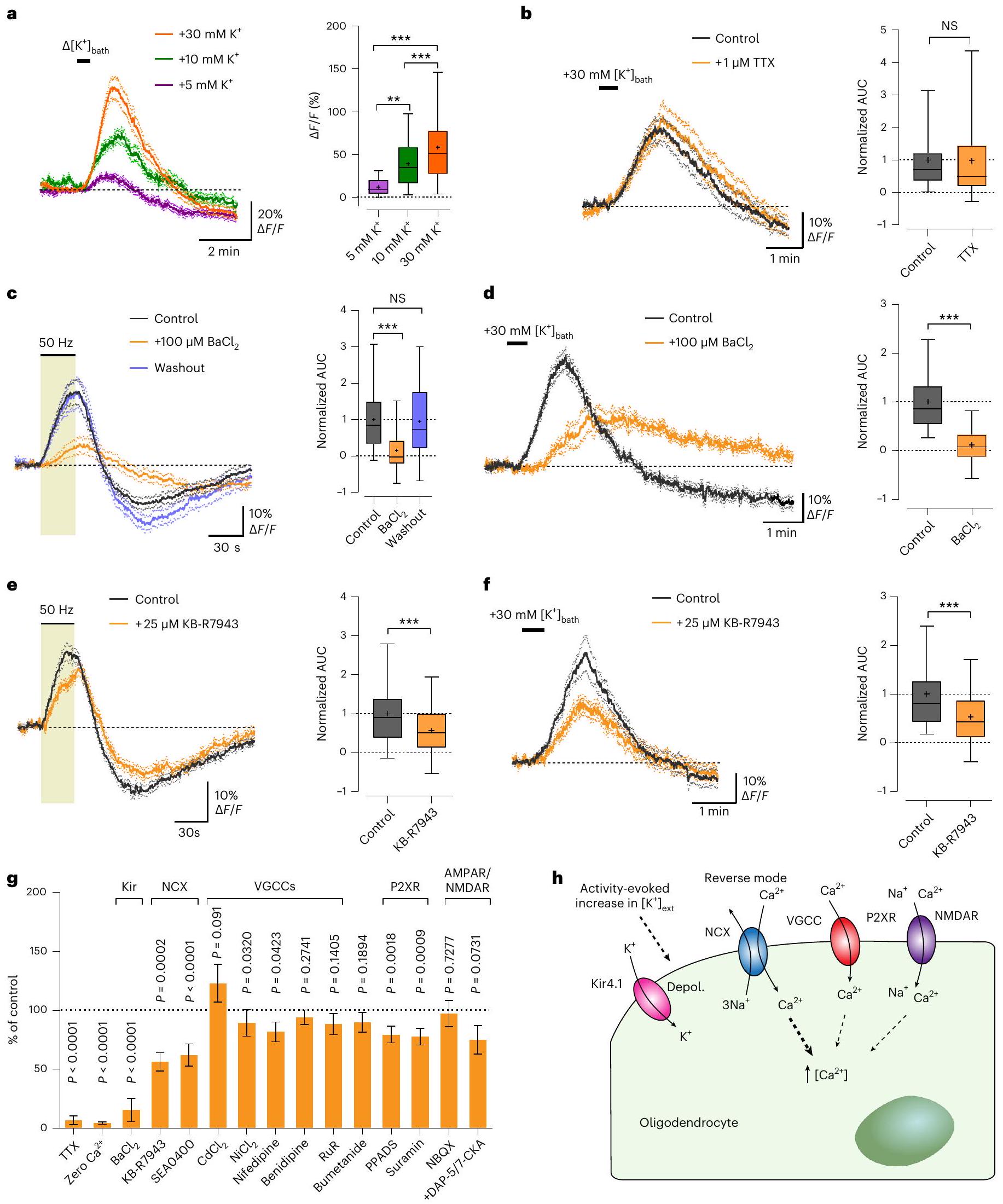
مثبط محدد لـ NKCC1. ومع ذلك،
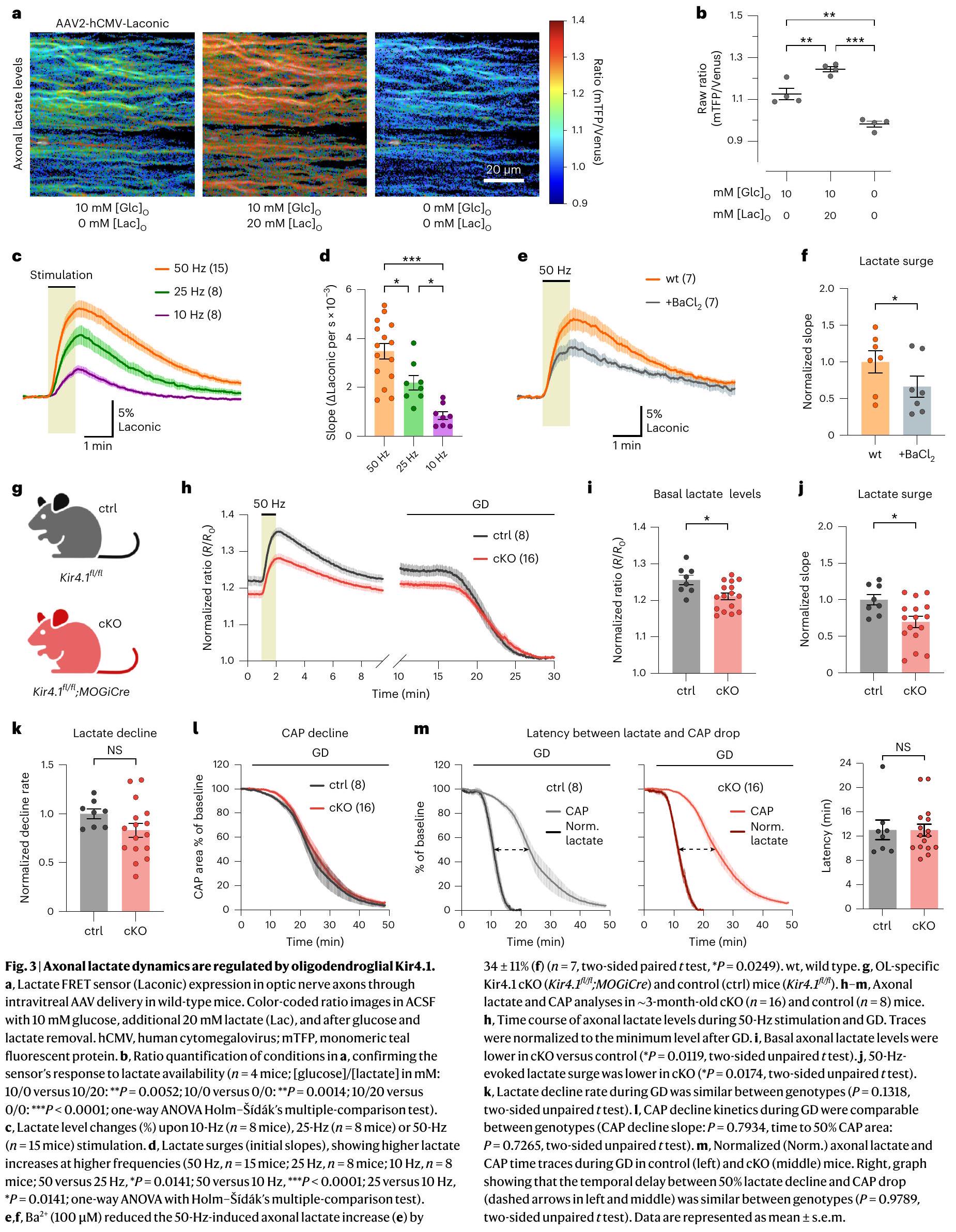
تنظم خلايا أوليجوديندروغليا Kir4.1 ديناميات اللاكتات المحورية
استهلاك اللاكتات. يبدو أن هذا غير محتمل، حيث أن تدهور اللاكتات خلال GD لم يكن أسرع ولكن كان أبطأ قليلاً في الفئران المعدلة وراثياً cKO مقارنةً بالتحكم (الشكل 3h، k). وهذا يت corroborated بمعدل انخفاض CAP المماثل خلال GD (الشكل 3l). أظهرت الجينوتيباتان نفس التأخير بين استنفاد اللاكتات وانخفاض CAP (الشكل 3m). من الجدير بالذكر أن انخفاض CAP لوحظ فقط عندما اقترب اللاكتات المحوري من الاستنفاد (الشكل 3m). بالنظر إلى الانخفاض المتزامن لمستويات CAP و ATP خلال GD
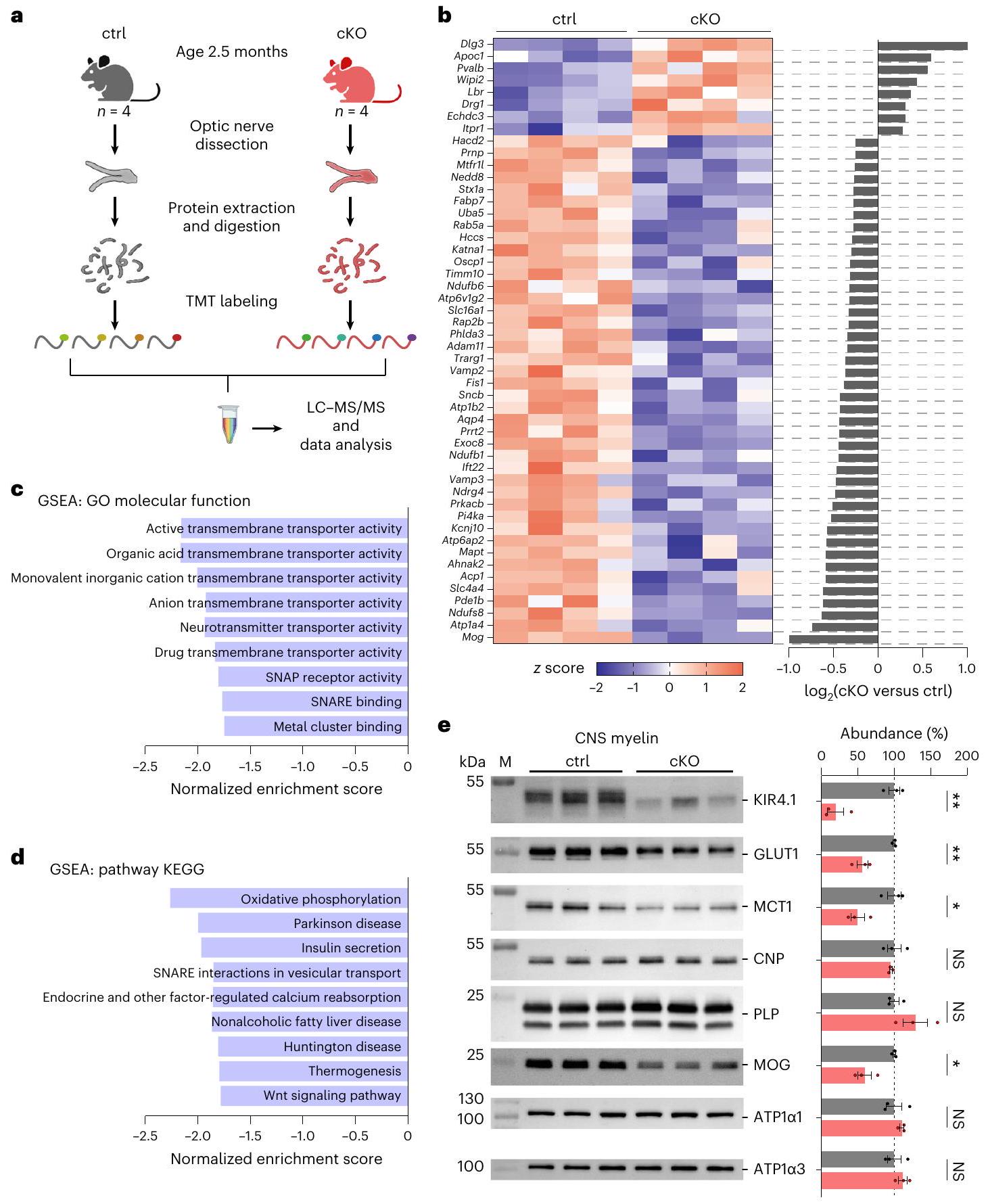
انخفاض MCT1 وGLUT1 في المايلين في الجهاز العصبي المركزي لفئران Kir4.1 cKO
تغييرات طفيفة في ديناميات ATP المحورية في فئران Kir4.1 cKO
من خلالويب جستالت.أورغ، مع تصنيف البروتينات حسب
مستشعر ATP ATeam1.03 (المرجع 52) في المحاور من خلال توصيل AAV داخل الجسم الزجاجي
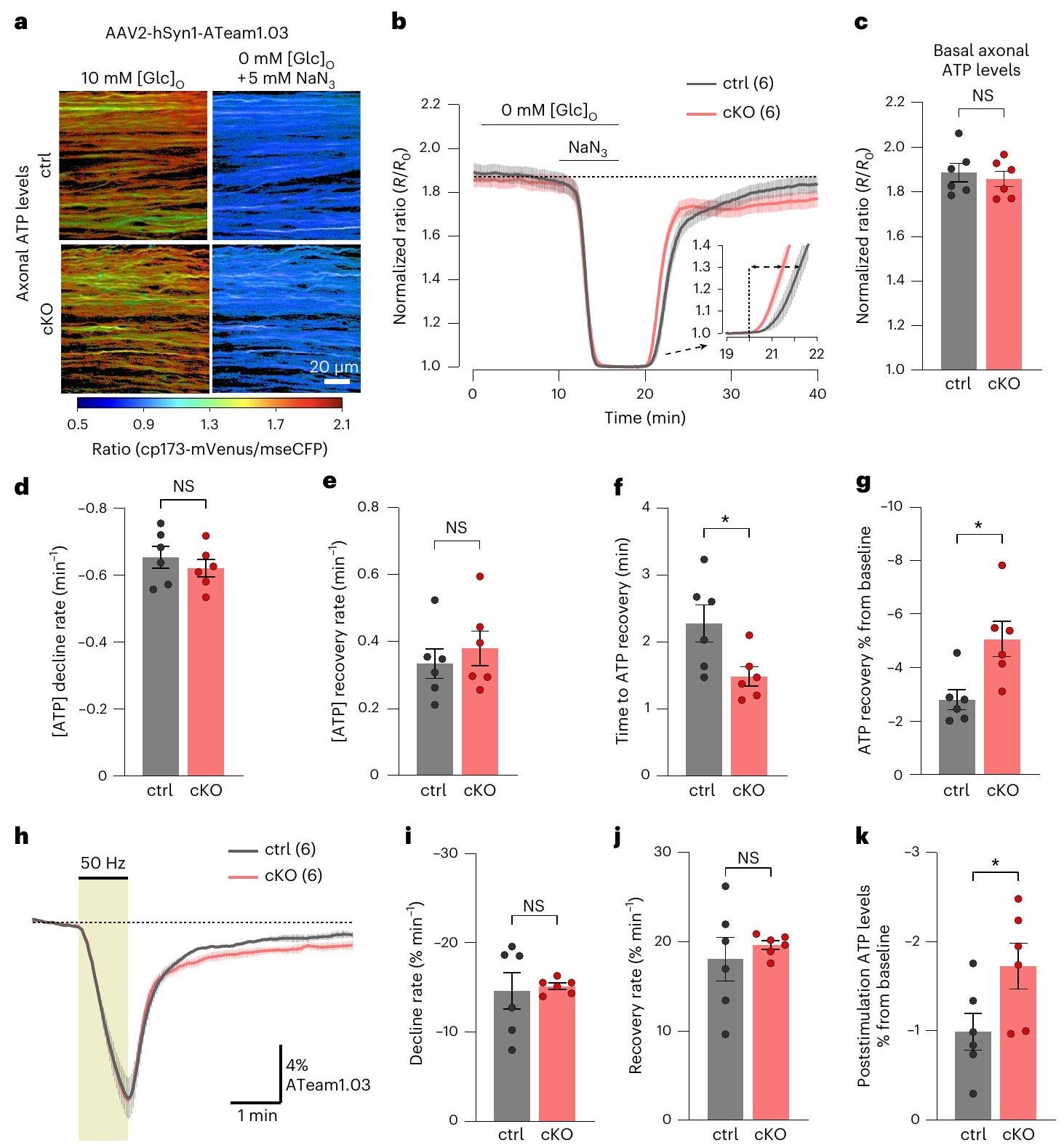
معدل الانخفاض خلال GD + MI كان أيضًا قابلًا للمقارنة بين الأنماط الجينية (الشكل 5b، d)، ومعدل استعادة ATP بعد
بعد GD + MI، كان حظر التوصيل المحوري مشابهًا بين الأنماط الجينية، وبدت بداية واستعادة حركية إطلاق المحاور غير متغيرة (الشكل البياني الممتد 10a). تحليل مساحة CAP الجزئي (pCAP)، الذي يعكس ديناميات ذروتي CAP الأولى والثانية
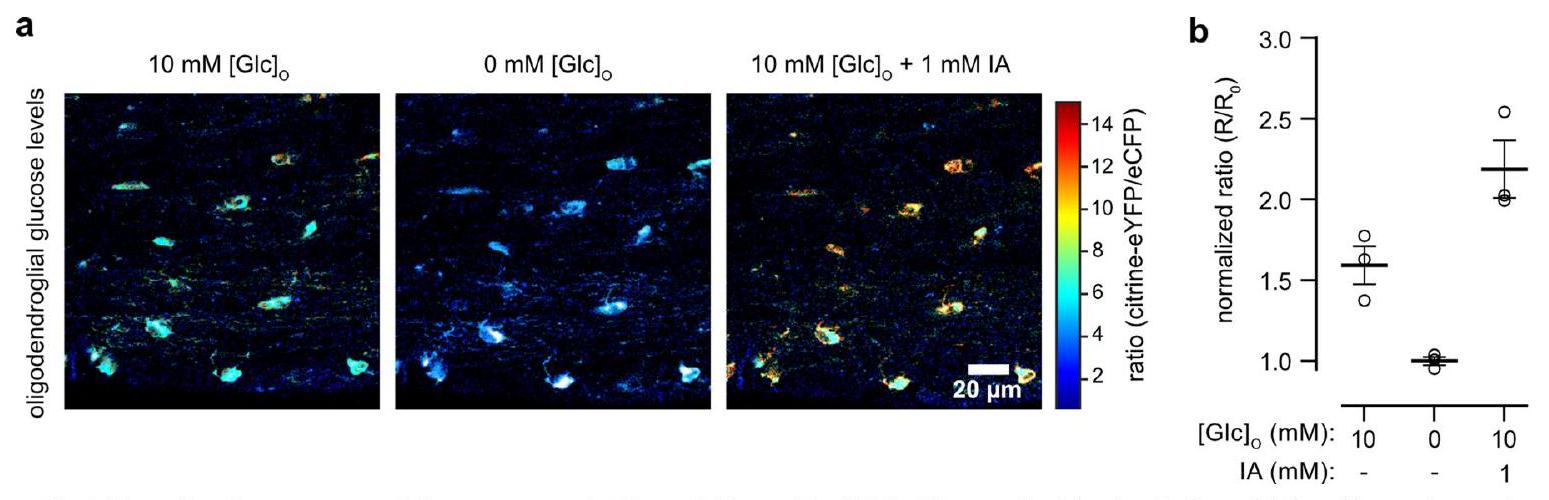
تعبير مستشعر FRET للجلوكوز المعتمد على AAV (FLIIP) في محاور الأعصاب البصرية. تظهر صور النسبة الملونة من الأعصاب الضابطة (الأعلى) و cKO (الأسفل) مستويات الجلوكوز في ACSF مع 10 مللي مول من الجلوكوز وبعد GD. eYFP، بروتين فلوري أصفر معزز؛ eCFP، بروتين فلوري أزرق معزز. ب، مسار الزمن لمستويات الجلوكوز في المحاور.
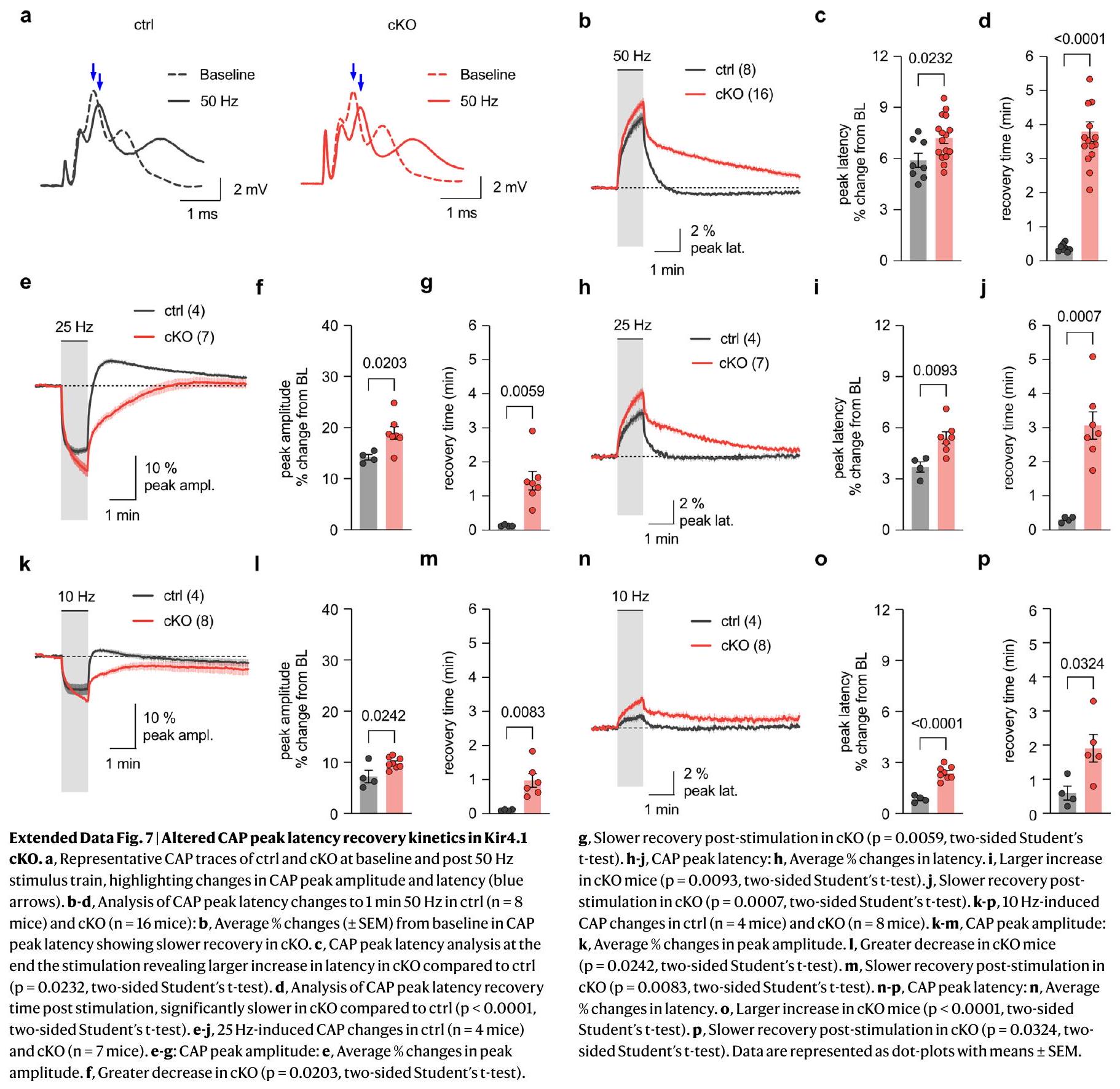
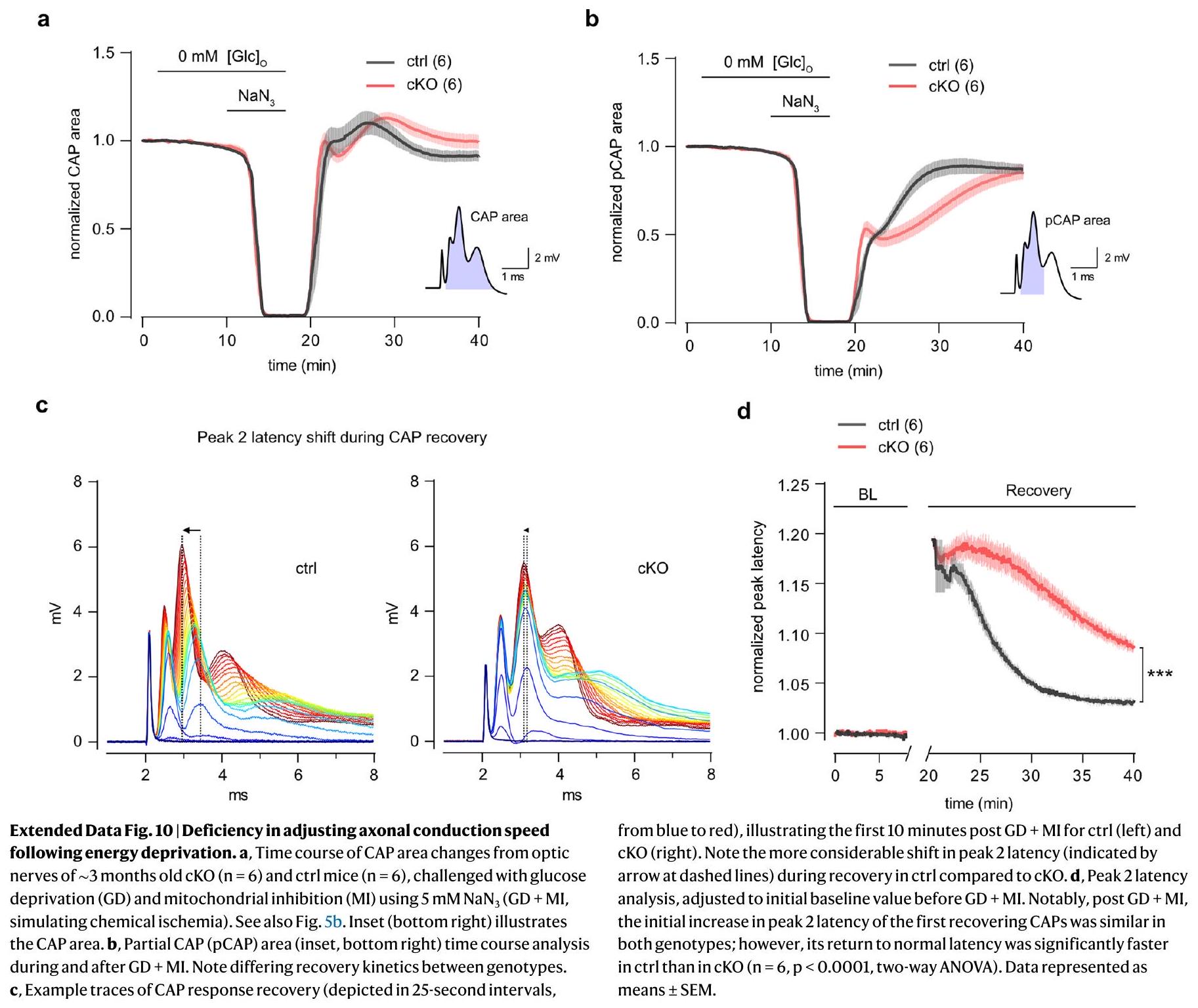
انخفضت الكمون؛ ومع ذلك، تم تقليل هذا التحول في الكمون الذروي (زيادة في سرعة التوصيل) بشكل ملحوظ في أعصاب cKO (الشكل 10d من البيانات الموسعة)، مما يشير إلى عجز في ضبط سرعة التوصيل بعد الحرمان الحاد من الطاقة. نظرًا لأن نقص التروية الكيميائية يزيد
تعديل سرعات التوصيل. قد تسهم هذه القدرة المحدودة على تعديل السرعة في تقليل استعادة ATP في المحاور العصبية cKO بعد نقص التروية الكيميائية (الشكل 5b,g).
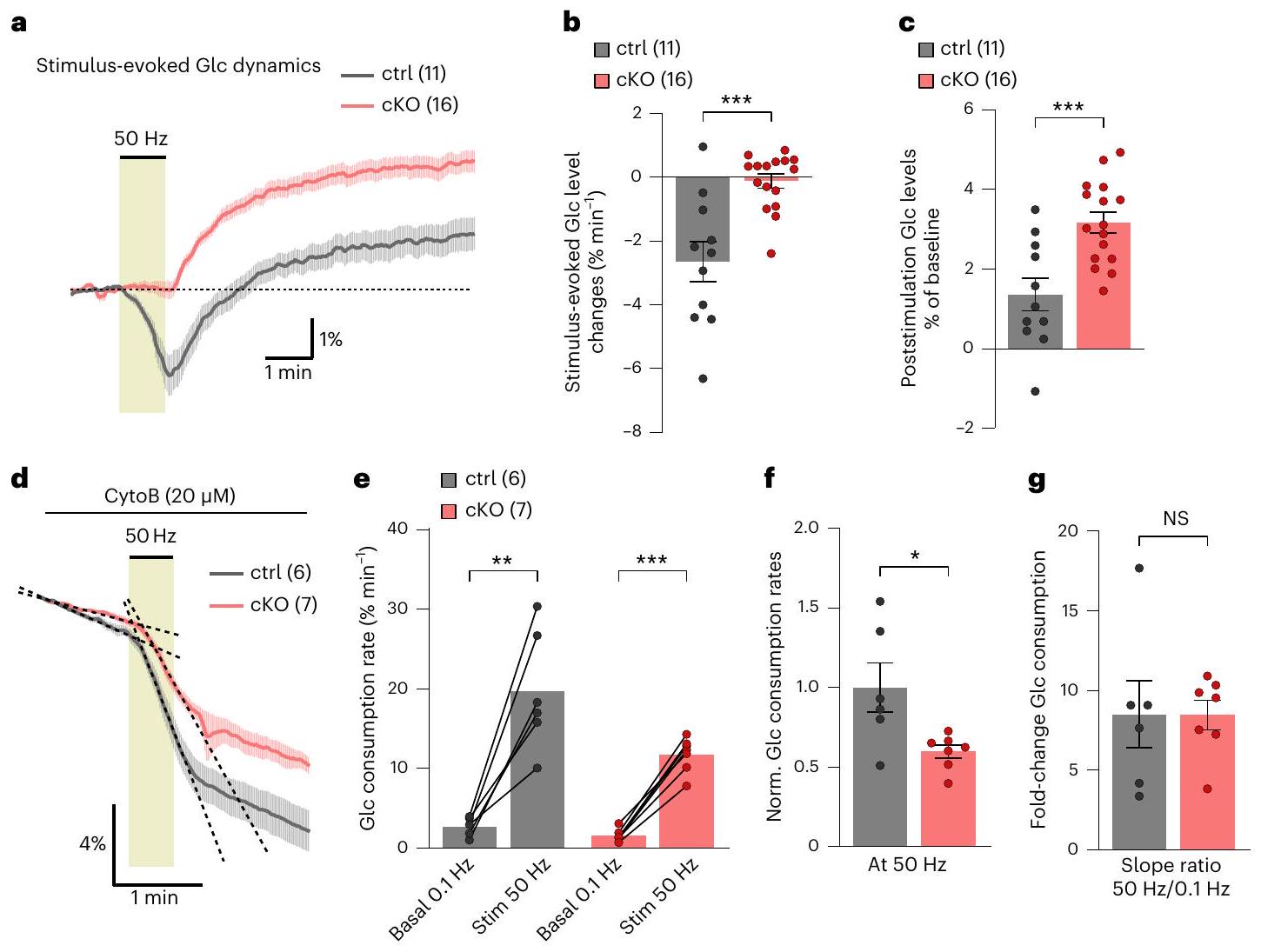
قياس ديناميات ATP خلال وبعد
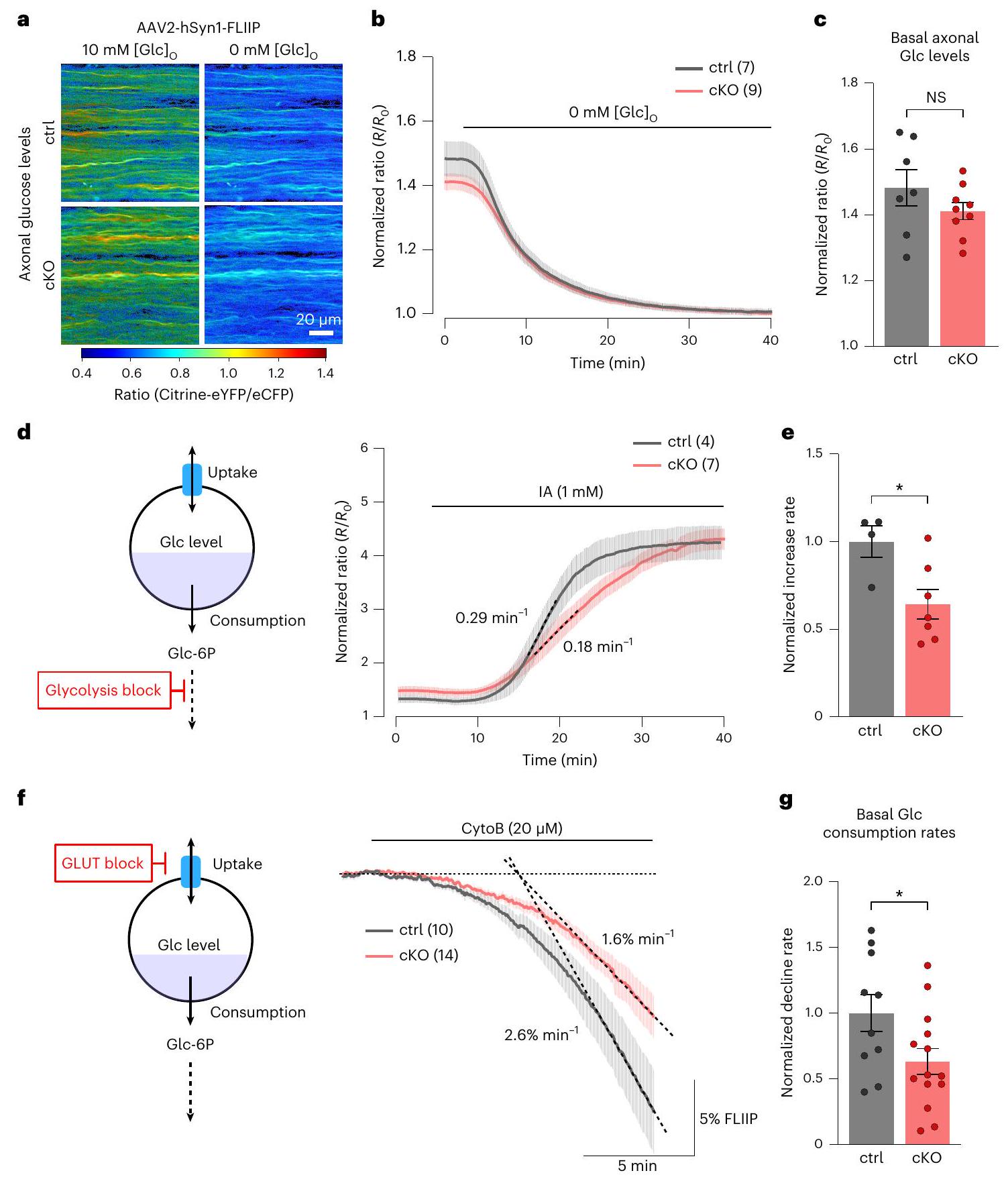
انخفاض في استقلاب الجلوكوز المحوري في فئران Kir4.1 cKO
كان
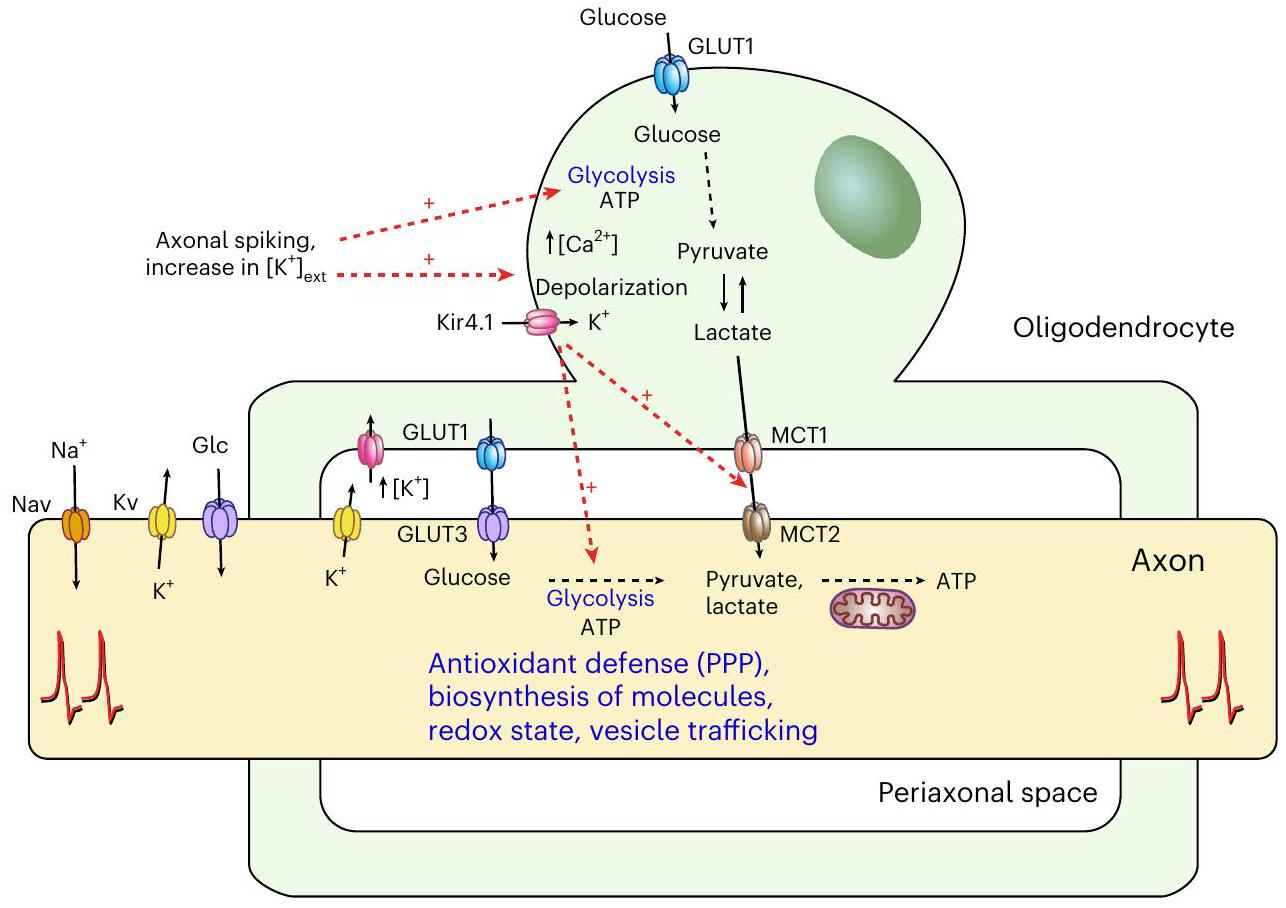
امتصاص الجلوكوز عند عقد رانفييه. الخلايا الدبقية قليلة التغصن
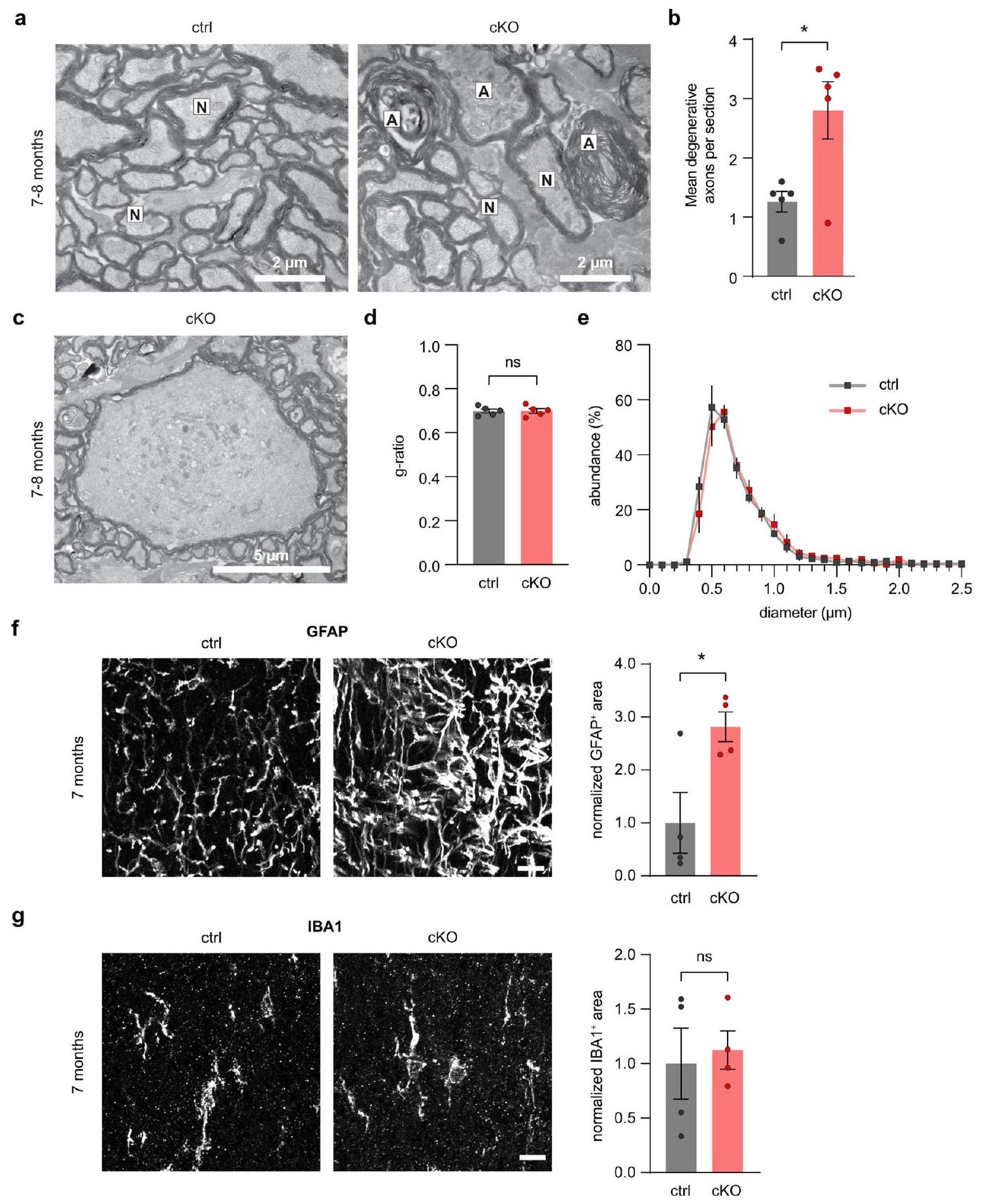
نقاش
المساعدة في تمايز خلايا OL خلال التطور
المحتوى عبر الإنترنت
References
- Salvadores, N., Sanhueza, M., Manque, P. & Court, F. A. Axonal degeneration during aging and its functional role in neurodegenerative disorders. Front. Neurosci. 11, 451 (2017).
- Medana, I. M. & Esiri, M. M. Axonal damage: a key predictor of outcome in human CNS diseases. Brain 126, 515-530 (2003).
- Saab, A. S., Tzvetanova, I. D. & Nave, K.-A. The role of myelin and oligodendrocytes in axonal energy metabolism. Curr. Opin. Neurobiol. 23, 1065-1072 (2013).
- Nave, K.-A. Myelination and the trophic support of long axons. Nat. Rev. Neurosci. 11, 275-283 (2010).
- Philips, T. & Rothstein, J. D. Oligodendroglia: metabolic supporters of neurons. J. Clin. Invest. 127, 3271-3280 (2017).
- Duncan, G. J., Simkins, T. J. & Emery, B. Neuron-oligodendrocyte interactions in the structure and integrity of axons. Front. Cell Dev. Biol. 9, 653101 (2021).
- Xin, W. & Chan, J. R. Myelin plasticity: sculpting circuits in learning and memory. Nat. Rev. Neurosci. 21, 682-694 (2020).
- Fünfschilling, U. et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 485, 517-521 (2012).
- Lee, Y. et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487, 443-448 (2012).
- Saab, A. S. et al. Oligodendroglial NMDA receptors regulate glucose import and axonal energy metabolism. Neuron 91, 119-132 (2016).
- Trevisiol, A. et al. Monitoring ATP dynamics in electrically active white matter tracts. eLife 6, e24241 (2017).
- Tekkök, S. B., Brown, A. M., Westenbroek, R., Pellerin, L. & Ransom, B. R. Transfer of glycogen-derived lactate from astrocytes to axons via specific monocarboxylate transporters supports mouse optic nerve activity. J. Neurosci. Res. 81, 644-652 (2005).
- Philips, T. et al. MCT1 deletion in oligodendrocyte lineage cells causes late-onset hypomyelination and axonal degeneration. Cell Rep. 34, 108610 (2021).
- Edgar, J. M. et al. Río-Hortega’s drawings revisited with fluorescent protein defines a cytoplasm-filled channel system of CNS myelin. J. Anat. 239, 1241-1255 (2021).
- Saab, A. S. & Nave, K.-A. Myelin dynamics: protecting and shaping neuronal functions. Curr. Opin. Neurobiol. 47, 104-112 (2017).
- Snaidero, N. et al. Antagonistic functions of MBP and CNP establish cytosolic channels in CNS myelin. Cell Rep. 18, 314-323 (2017).
- Griffiths, I. et al. Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science 280, 1610-1613 (1998).
- Lüders, K. A. et al. Maintenance of high proteolipid protein level in adult central nervous system myelin is required to preserve the integrity of myelin and axons. Glia 67, 634-649 (2019).
- Edgar, J. M. et al. Oligodendroglial modulation of fast axonal transport in a mouse model of hereditary spastic paraplegia. J. Cell Biol. 166, 121-131 (2004).
- Steyer, A. M. et al. Pathology of myelinated axons in the PLP-deficient mouse model of spastic paraplegia type 2 revealed by volume imaging using focused ion beam-scanning electron microscopy. J. Struct. Biol. 210, 107492 (2020).
- Trevisiol, A. et al. Structural myelin defects are associated with low axonal ATP levels but rapid recovery from energy deprivation in a mouse model of spastic paraplegia. PLoS Biol. 18, e3000943 (2020).
- Mukherjee, C. et al. Oligodendrocytes provide antioxidant defense function for neurons by secreting ferritin heavy chain. Cell Metab. 32, 259-272 (2020).
- Larson, V. A. et al. Oligodendrocytes control potassium accumulation in white matter and seizure susceptibility. eLife 7, e34829 (2018).
- Schirmer, L. et al. Oligodendrocyte-encoded Kir4.1 function is required for axonal integrity. eLife 7, e36428 (2018).
- Kettenmann, H., Sonnhof, U. & Schachner, M. Exclusive potassium dependence of the membrane potential in cultured mouse oligodendrocytes. J. Neurosci. 3, 500-505 (1983).
- Yamazaki, Y. et al. Modulatory effects of oligodendrocytes on the conduction velocity of action potentials along axons in the alveus of the rat hippocampal CA1 region. Neuron Glia Biol. 3, 325-334 (2007).
- Battefeld, A., Klooster, J. & Kole, M. H. P. Myelinating satellite oligodendrocytes are integrated in a glial syncytium constraining neuronal high-frequency activity. Nat. Commun. 7, 11298 (2016).
- Looser, Z. J., Barrett, M. J. P., Hirrlinger, J., Weber, B. & Saab, A. S. Intravitreal AAV-delivery of genetically encoded sensors enabling simultaneous two-photon imaging and electrophysiology of optic nerve axons. Front. Cell. Neurosci. 12, 377 (2018).
- Doerflinger, N. H., Macklin, W. B. & Popko, B. Inducible site-specific recombination in myelinating cells. Genesis 35, 63-72 (2003).
- Madisen, L. et al. Transgenic mice for intersectional targeting of neural sensors and effectors with high specificity and performance. Neuron 85, 942-958 (2015).
- Chen, T.-W. et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295-300 (2013).
- Takanaga, H., Chaudhuri, B. & Frommer, W. B. GLUT1 and GLUT9 as major contributors to glucose influx in HepG2 cells identified by a high sensitivity intramolecular FRET glucose sensor. Biochim. Biophys. Acta 1778, 1091-1099 (2008).
- Bittner, C. X. et al. High resolution measurement of the glycolytic rate. Front. Neuroenergetics 2, 26 (2010).
- Bittner, C. X. et al. Fast and reversible stimulation of astrocytic glycolysis by
and a delayed and persistent effect of glutamate. J. Neurosci. 31, 4709-4713 (2011). - Micu, I. et al. The molecular physiology of the axo-myelinic synapse. Exp. Neurol. 276, 41-50 (2016).
- Micu, I. et al. NMDA receptors mediate calcium accumulation in myelin during chemical ischaemia. Nature 439, 988-992 (2006).
- James, G. & Butt, A. M. P2X and P2Y purinoreceptors mediate ATP-evoked calcium signalling in optic nerve glia in situ. Cell Calcium 30, 251-259 (2001).
- Kirischuk, S., Scherer, J., Kettenmann, H. & Verkhratsky, A. Activation of P2-purinoreceptors triggered
release from InsP3-sensitive internal stores in mammalian oligodendrocytes. J. Physiol. 483, 41-57 (1995). - Matute, C. et al. P2X(7) receptor blockade prevents ATP excitotoxicity in oligodendrocytes and ameliorates experimental autoimmune encephalomyelitis. J. Neurosci. 27, 9525-9533 (2007).
- Stevens, B., Porta, S., Haak, L. L., Gallo, V. & Fields, R. D. Adenosine: a neuron-glial transmitter promoting myelination in the CNS in response to action potentials. Neuron 36, 855-868 (2002).
- Ransom, C. B., Ransom, B. R. & Sontheimer, H. Activity-dependent extracellular
accumulation in rat optic nerve: the role of glial and axonal pumps. J. Physiol. 522, 427-442 (2000). - Bay, V. & Butt, A. M. Relationship between glial potassium regulation and axon excitability: a role for glial Kir4.1 channels. Glia 60, 651-660 (2012).
- Olsen, M. L. & Sontheimer, H. Functional implications for Kir4.1 channels in glial biology: from
buffering to cell differentiation. J. Neurochem. 107, 589-601 (2008). - Boscia, F. et al. Silencing or knocking out the
exchanger-3 (NCX3) impairs oligodendrocyte differentiation. Cell Death Differ. 19, 562-572 (2012). - Casamassa, A. et al. Ncx3 gene ablation impairs oligodendrocyte precursor response and increases susceptibility to experimental autoimmune encephalomyelitis. Glia 64, 1124-1137 (2016).
- Spencer, S. A., Suárez-Pozos, E., Escalante, M., Myo, Y. P. & Fuss, B. Sodium-calcium exchangers of the SLC8 family in oligodendrocytes: functional properties in health and disease. Neurochem. Res. 45, 1287-1297 (2020).
- Friess, M. et al. Intracellular ion signaling influences myelin basic protein synthesis in oligodendrocyte precursor cells. Cell Calcium 60, 322-330 (2016).
- Moyon, S. et al. TET1-mediated DNA hydroxymethylation regulates adult remyelination in mice. Nat. Commun. 12, 3359 (2021).
- Yamazaki, Y., Abe, Y., Fujii, S. & Tanaka, K. F. Oligodendrocytic
co-transporter 1 activity facilitates axonal conduction and restores plasticity in the adult mouse brain. Nat. Commun. 12, 5146 (2021). - San Martín, A. et al. A genetically encoded FRET lactate sensor and its use to detect the Warburg effect in single cancer cells. PLoS ONE 8, e57712 (2013).
- Djukic, B., Casper, K. B., Philpot, B. D., Chin, L.-S. & McCarthy, K. D. Conditional knock-out of
leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J. Neurosci. 27, 11354-11365 (2007). - Imamura, H. et al. Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc. Natl Acad. Sci. USA 106, 15651-15656 (2009).
- Hamilton, N. B., Kolodziejczyk, K., Kougioumtzidou, E. & Attwell, D. Proton-gated
-permeable TRP channels damage myelin in conditions mimicking ischaemia. Nature 529, 523-527 (2016). - Meyer, N. et al. Oligodendrocytes in the mouse corpus callosum maintain axonal function by delivery of glucose. Cell Rep. 22, 2383-2394 (2018).
- Zhang, X. et al. Oligodendroglial glycolytic stress triggers inflammasome activation and neuropathology in Alzheimer’s disease. Sci. Adv. 6, eabb8680 (2020).
- Mot, A. I., Depp, C. & Nave, K.-A. An emerging role of dysfunctional axon-oligodendrocyte coupling in neurodegenerative diseases. Dialogues Clin. Neurosci. 20, 283-292 (2018).
- Kenigsbuch, M. et al. A shared disease-associated oligodendrocyte signature among multiple CNS pathologies. Nat. Neurosci. 25, 876-886 (2022).
- Kaya, T. et al.
cells induce interferon-responsive oligodendrocytes and microglia in white matter aging. Nat. Neurosci. 25, 1446-1457 (2022). - Brasko, C., Hawkins, V., De La Rocha, I. C. & Butt, A. M. Expression of Kir4.1 and Kir5.1 inwardly rectifying potassium channels in oligodendrocytes, the myelinating cells of the CNS. Brain Struct. Funct. 222, 41-59 (2017).
- Papanikolaou, M., Butt, A. M. & Lewis, A. A critical role for the inward rectifying potassium channel Kir7.1 in oligodendrocytes of the mouse optic nerve. Brain Struct. Funct. 225, 925-934 (2020).
- Almeida, R. G. et al. Myelination induces axonal hotspots of synaptic vesicle fusion that promote sheath growth. Curr. Biol. 31, 3743-3754 (2021).
- Micu, I., Plemel, J. R., Caprariello, A. V., Nave, K. A. & Stys, P. K. Axo-myelinic neurotransmission: a novel mode of cell signalling in the central nervous system. Nat. Rev. Neurosci. 19, 49-58 (2018).
- Hines, J. H., Ravanelli, A. M., Schwindt, R., Scott, E. K. & Appel, B. Neuronal activity biases axon selection for myelination in vivo. Nat. Neurosci. 18, 683-689 (2015).
- Wake, H., Lee, P. R. & Fields, R. D. Control of local protein synthesis and initial events in myelination by action potentials. Science 333, 1647-1651 (2011).
- Mensch, S. et al. Synaptic vesicle release regulates myelin sheath number of individual oligodendrocytes in vivo. Nat. Neurosci. 18, 628-630 (2015).
- Kukley, M., Capetillo-Zarate, E. & Dietrich, D. Vesicular glutamate release from axons in white matter. Nat. Neurosci. 10, 311-320 (2007).
- Ziskin, J. L., Nishiyama, A., Rubio, M., Fukaya, M. & Bergles, D. E. Vesicular release of glutamate from unmyelinated axons in white matter. Nat. Neurosci. 10, 321-330 (2007).
- Krasnow, A. M., Ford, M. C., Valdivia, L. E., Wilson, S. W. & Attwell, D. Regulation of developing myelin sheath elongation by oligodendrocyte calcium transients in vivo. Nat. Neurosci. 21, 24-28 (2018).
- Baraban, M., Koudelka, S. & Lyons, D. A.
activity signatures of myelin sheath formation and growth in vivo. Nat. Neurosci. 21, 19-23 (2018). - Battefeld, A., Popovic, M. A., de Vries, S. I. & Kole, M. H. P. High-frequency microdomain
transients and waves during early myelin internode remodeling. Cell Rep. 26, 182-191 (2019). - Kanda, H. et al. TREK-1 and TRAAK are principal
channels at the nodes of Ranvier for rapid action potential conduction on mammalian myelinated afferent nerves. Neuron 104, 960-971 (2019). - Brohawn, S. G. et al. The mechanosensitive ion channel TRAAK is localized to the mammalian node of Ranvier. eLife 8, e50403 (2019).
- Rash, J. E. Molecular disruptions of the panglial syncytium block potassium siphoning and axonal saltatory conduction: pertinence to neuromyelitis optica and other demyelinating diseases of the central nervous system. Neuroscience 168, 982-1008 (2010).
- Cohen, C. C. H. et al. Saltatory conduction along myelinated axons involves a periaxonal nanocircuit. Cell 180, 311-322 (2020).
- Ruminot, I., Schmälzle, J., Leyton, B., Barros, L. F. & Deitmer, J. W. Tight coupling of astrocyte energy metabolism to synaptic activity revealed by genetically encoded FRET nanosensors in hippocampal tissue. J. Cereb. Blood Flow Metab. 39, 513-523 (2019).
- Wang, N. et al. Potassium channel
regulates oligodendrocyte differentiation via intracellular pH regulation. Glia 70, 2093-2107 (2022). - Fernández-Moncada, I. et al. Bidirectional astrocytic GLUT1 activation by elevated extracellular K. Glia 69, 1012-1021 (2021).
- Zuend, M. et al. Arousal-induced cortical activity triggers lactate release from astrocytes. Nat. Metab. 2, 179-191 (2020).
- Sotelo-Hitschfeld, T. et al. Channel-mediated lactate release by
-stimulated astrocytes. J. Neurosci. 35, 4168-4178 (2015). - Köhler, S. et al. Gray and white matter astrocytes differ in basal metabolism but respond similarly to neuronal activity. Glia 71, 229-244 (2023).
- Menichella, D. M. et al. Genetic and physiological evidence that oligodendrocyte gap junctions contribute to spatial buffering of potassium released during neuronal activity. J. Neurosci. 26, 10984-10991 (2006).
- Neusch, C., Rozengurt, N., Jacobs, R. E., Lester, H. A. & Kofuji, P. Kir4.1 potassium channel subunit is crucial for oligodendrocyte development and in vivo myelination. J. Neurosci. 21, 5429-5438 (2001).
- Orthmann-Murphy, J. L., Abrams, C. K. & Scherer, S. S. Gap junctions couple astrocytes and oligodendrocytes. J. Mol. Neurosci. 35, 101-116 (2008).
- Hösli, L. et al. Decoupling astrocytes in adult mice impairs synaptic plasticity and spatial learning. Cell Rep. 38, 110484 (2022).
- Jahn, O. et al. The CNS myelin proteome: deep profile and persistence after post-mortem delay. Front. Cell. Neurosci. 14, 239 (2020).
- Zhang, Y. et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929-11947 (2014).
- Gargareta, V.-I. et al. Conservation and divergence of myelin proteome and oligodendrocyte transcriptome profiles between humans and mice. eLife 11, e77019 (2022).
- Lam, M. et al. CNS myelination requires VAMP2/3-mediated membrane expansion in oligodendrocytes. Nat. Commun. 13, 5583 (2022).
- Leto, D. & Saltiel, A. R. Regulation of glucose transport by insulin: traffic control of GLUT4. Nat. Rev. Mol. Cell Biol. 13, 383-396 (2012).
- Martin, L. B., Shewan, A., Millar, C. A., Gould, G. W. & James, D. E. Vesicle-associated membrane protein 2 plays a specific role in the insulin-dependent trafficking of the facilitative glucose transporter GLUT4 in 3T3-L1 adipocytes. J. Biol. Chem. 273, 1444-1452 (1998).
- Lodhi, I. J. et al. Gapex-5, a Rab31 guanine nucleotide exchange factor that regulates Glut4 trafficking in adipocytes. Cell Metab. 5, 59-72 (2007).
- Dienel, G. A. Brain glucose metabolism: integration of energetics with function. Physiol. Rev. 99, 949-1045 (2019).
- Barros, L. F. et al. Fluid brain glycolysis: limits, speed, location, moonlighting, and the fates of glycogen and lactate. Neurochem. Res. 45, 1328-1334 (2020).
- Herrero-Mendez, A. et al. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat. Cell Biol. 11, 747-752 (2009).
- Zala, D. et al. Vesicular glycolysis provides on-board energy for fast axonal transport. Cell 152, 479-491 (2013).
- Baeza-Lehnert, F. et al. Non-Canonical Control of Neuronal Energy Status by the
Pump. Cell Metab. 29, 668-680.e4 (2019). - Meyer, D. J., Díaz-García, C. M., Nathwani, N., Rahman, M. & Yellen, G. The
pump dominates control of glycolysis in hippocampal dentate granule cells. eLife 11, e81645 (2022). - Frühbeis, C. et al. Oligodendrocytes support axonal transport and maintenance via exosome secretion. PLoS Biol. 18, e3000621 (2020).
- Chamberlain, K. A. et al. Oligodendrocytes enhance axonal energy metabolism by deacetylation of mitochondrial proteins through transcellular delivery of SIRT2. Neuron 109, 3456-3472 (2021).
- Frühbeis, C. et al. Neurotransmitter-triggered transfer of exosomes mediates oligodendrocyte-neuron communication. PLoS Biol. 11, e1001604 (2013).
as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended
use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons. org/licenses/by/4.0/.
© The Author(s) 2024
طرق
الحيوانات
علاج التاموكسيفين
حقن AAV
ناقلات AAV
لديناميكا ATP المحورية، استخدمنا مستشعر FRET ATeam1.03 (المرجع 52) المعبأ في ssAAV-2/2-hSyn1-ATeam1.03-WPRE-hGHp(A)
الكهربائية العصبية للعصب البصري وتصوير الفوتونين
الحلول
تسجيلات وتحليل CAP
من بداية التحفيز إلى الذروة. تم استخدام منطقة pCAP لدمج التغيرات في السعة والزمن في الذروتين الأوليين، والتي تعكس المحاور الكبيرة والمتوسطة الحجم. تضمنت بروتوكولات التحفيز بداية
تصوير الكالسيوم والتحليل
تصوير وتحليل المستقلبات
الكيمياء المناعية
باستخدام مجمد ليكا CM3050 S. تم وضع المقاطع على شرائح SuperFrost Plus (Thermo Fisher Scientific). بالنسبة للكيمياء المناعية، تم معالجة الشرائح أولاً بـ 0.3% Triton X-100 في محلول تريس 50 مليمول (pH 7.4) مع
EM والتحليل
البروتيوميات والتحليل
الانفصالات المفقودة. تم تعيين الحد الأقصى لـ FDR للببتيدات إلى 0.01. تم حساب تغييرات طي البروتين بناءً على قيم الكثافة. تم استخدام مجموعة من الوظائف المطبقة في حزمة R prolfqua
تنقية المايلين والتحليل المناعي
تفاعل البوليميراز المتسلسل الكمي في الوقت الحقيقي
(
التحليلات الإحصائية
ملخص التقرير
توفر البيانات
توفر الشيفرة
References
- Hövelmeyer, N. et al. Apoptosis of oligodendrocytes via Fas and TNF-R1 is a key event in the induction of experimental autoimmune encephalomyelitis. J. Immunol. 175, 5875-5884 (2005).
- Fleischmann, T., Jirkof, P., Henke, J., Arras, M. & Cesarovic, N. Injection anaesthesia with fentanyl-midazolam-medetomidine in adult female mice: importance of antagonization and perioperative care. Lab. Anim. 50, 264-274 (2016).
- Paterna, J.-C., Feldon, J. & Büeler, H. Transduction profiles of recombinant adeno-associated virus vectors derived from serotypes 2 and 5 in the nigrostriatal system of rats. J. Virol. 78, 6808-6817 (2004).
- Snaidero, N. et al. Myelin replacement triggered by single-cell demyelination in mouse cortex. Nat. Commun. 11, 4901 (2020).
- Mezydlo, A. et al. Remyelination by surviving oligodendrocytes is inefficient in the inflamed mammalian cortex. Neuron 111, 1748-1759 (2023).
- Mayrhofer, J. M. et al. Design and performance of an ultra-flexible two-photon microscope for in vivo research. Biomed. Opt. Express 6, 4228-4237 (2015).
- Pologruto, T. A., Sabatini, B. L. & Svoboda, K. Scanlmage: flexible software for operating laser scanning microscopes. Biomed. Eng. Online 2, 13 (2003).
- Stys, P. K., Ransom, B. R. & Waxman, S. G. Compound action potential of nerve recorded by suction electrode: a theoretical and experimental analysis. Brain Res. 546, 18-32 (1991).
- Barrett, M. J. P., Ferrari, K. D., Stobart, J. L., Holub, M. & Weber, B. CHIPS: an extensible toolbox for cellular and hemodynamic two-photon image analysis. Neuroinformatics 16, 145-147 (2018).
- Glück, C. et al. Distinct signatures of calcium activity in brain mural cells. eLife 10, e70591 (2021).
- Möbius, W. et al. Electron microscopy of the mouse central nervous system. Methods Cell Biol. 96, 475-512 (2010).
- Türker, C. et al. B-Fabric: the Swiss army knife for life sciences. In Proceedings of the 13th International Conference on Extending Database Technology (eds Manolescu, I. et al.) 717-720 (ACM, 2010).
- Wolski, W. E., Panse, C., Grossmann, J., D’Errico, M. & Nanni, P. prolfqua-an R package for proteomics label-free quantification. F1000Research https://doi.org/10.7490/f1000research.1118455.1 (2021).
- Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
- Erwig, M. S. et al. Myelin: methods for purification and proteome analysis. Methods Mol. Biol. 1936, 37-63 (2019).
- Stumpf, S. K. et al. Ketogenic diet ameliorates axonal defects and promotes myelination in Pelizaeus-Merzbacher disease. Acta Neuropathol. 138, 147-161 (2019).
- Berghoff, S. A. et al. Blood-brain barrier hyperpermeability precedes demyelination in the cuprizone model. Acta Neuropathol. Commun. 5, 94 (2017).
- Jung, M., Sommer, I., Schachner, M. & Nave, K. A. Monoclonal antibody O10 defines a conformationally sensitive cell-surface epitope of proteolipid protein (PLP): evidence that PLP misfolding underlies dysmyelination in mutant mice. J. Neurosci. 16, 7920-7929 (1996).
شكر وتقدير
مروج MBP، ومركز الجينوميات الوظيفية في زيورخ لدعم البروتيوميات. تم دعم D.E.B. و K.-A.N. من قبل مؤسسة أدلسون للبحوث الطبية. تم دعم L.F.B. من قبل فوندسيكت 1230145. تم دعم B.W. من قبل المؤسسة الوطنية السويسرية للعلوم (31003A_156965). تم دعم A.S.S. من خلال جائزة زمالة مهنية من سينابسيس، مركز علوم الأعصاب في زيورخ، مؤسسة كلويتا والمؤسسة الوطنية السويسرية للعلوم (Eccellenza 187000).
مساهمات المؤلفين
تمويل
المصالح المتنافسة
معلومات إضافية
https://doi.org/10.1038/s41593-023-01558-3.
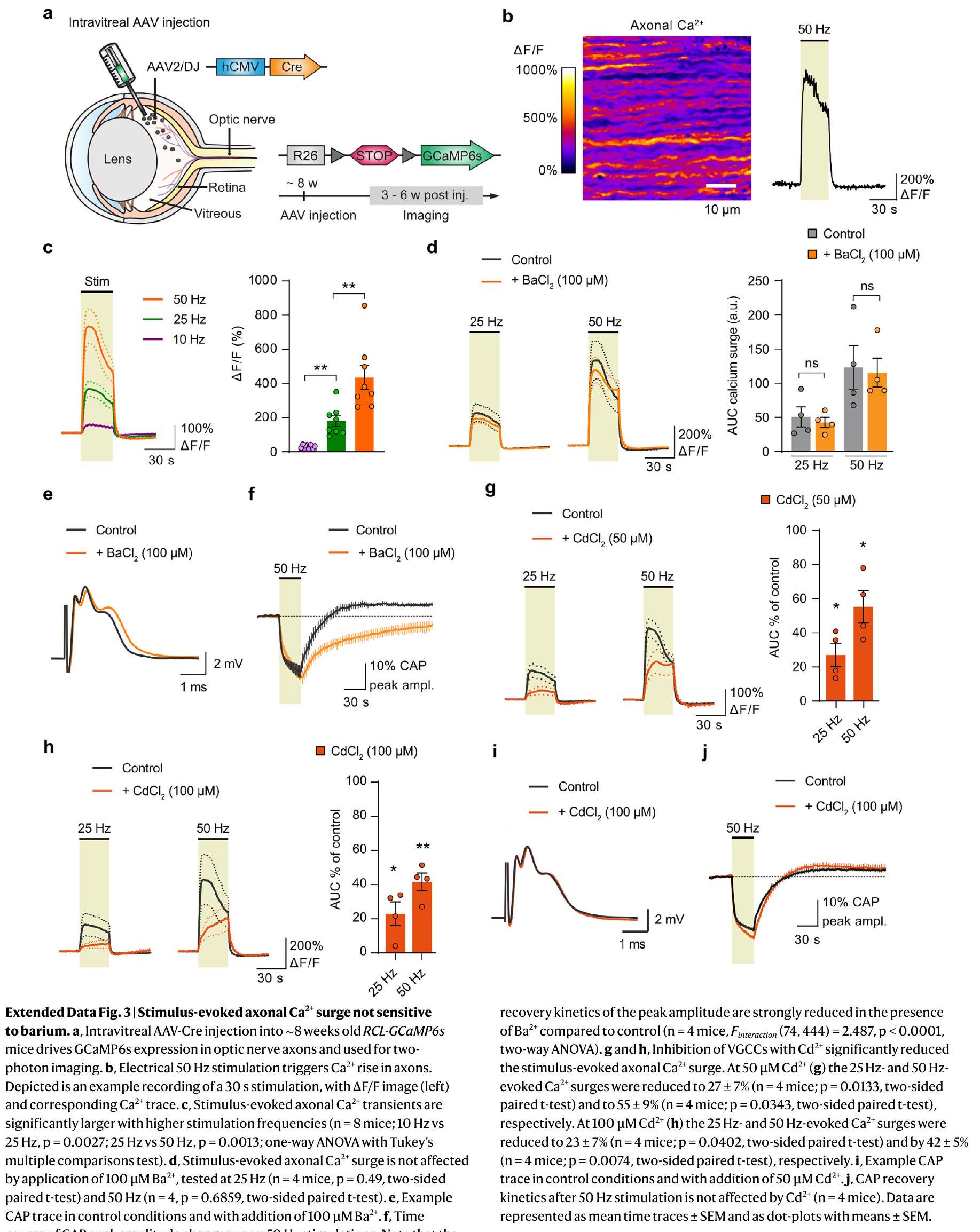
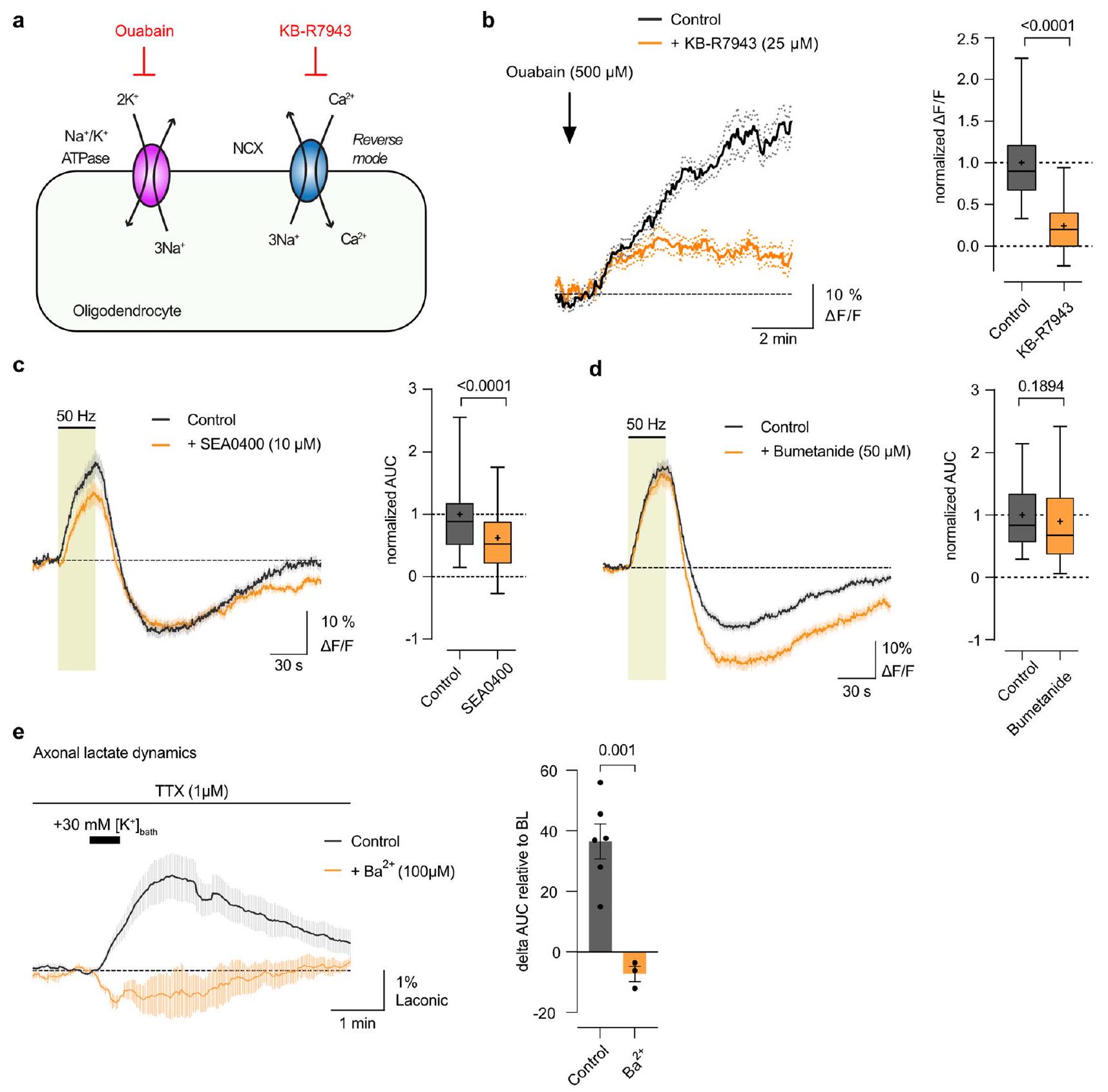
مع مثبط NKCC1 بوميتانيد (
الشكل 6 من البيانات الموسعة | انظر الصفحة التالية للتعليق.
سماكة الغمد (نسبة g) بين الأنماط الجينية
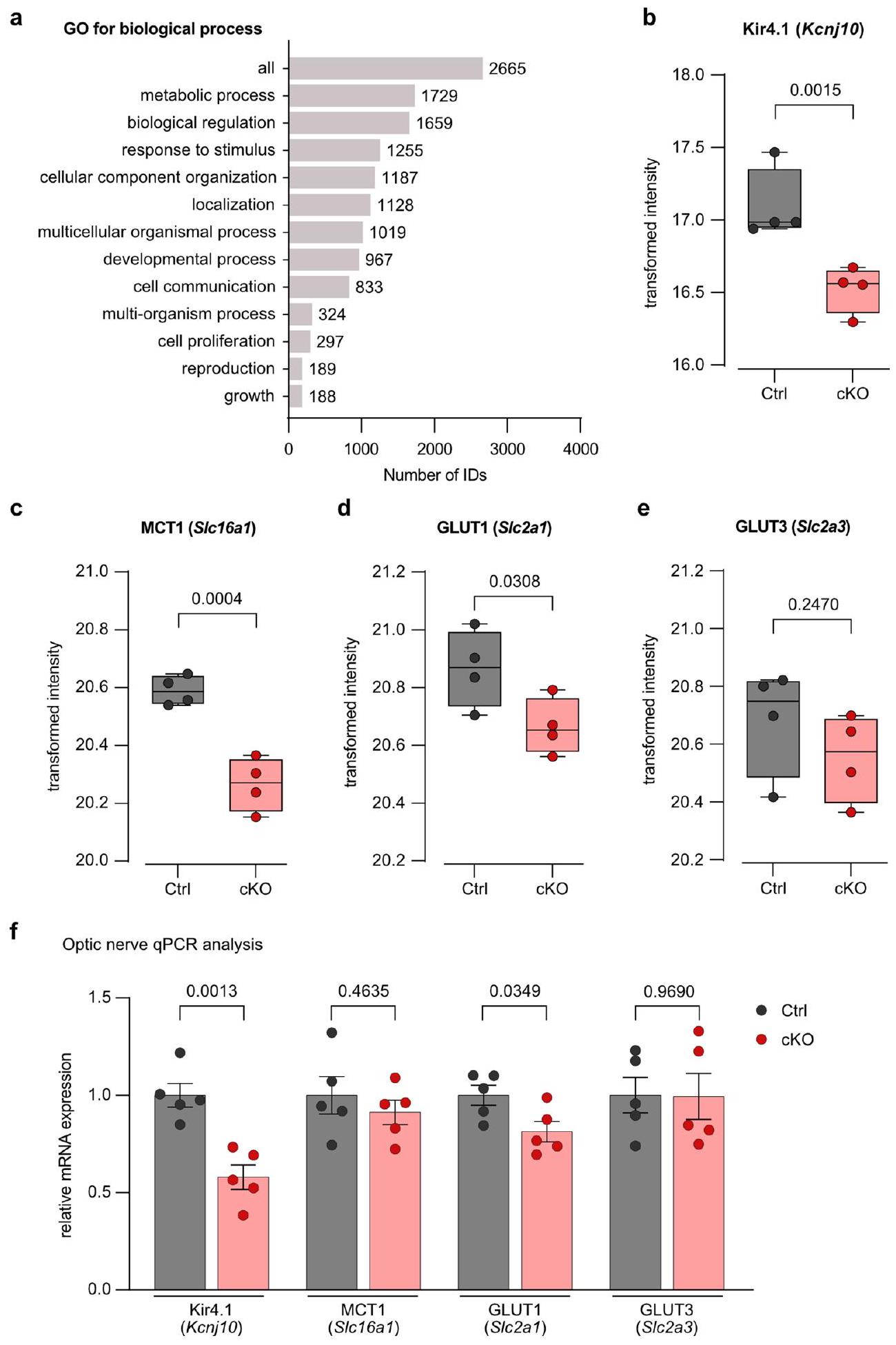
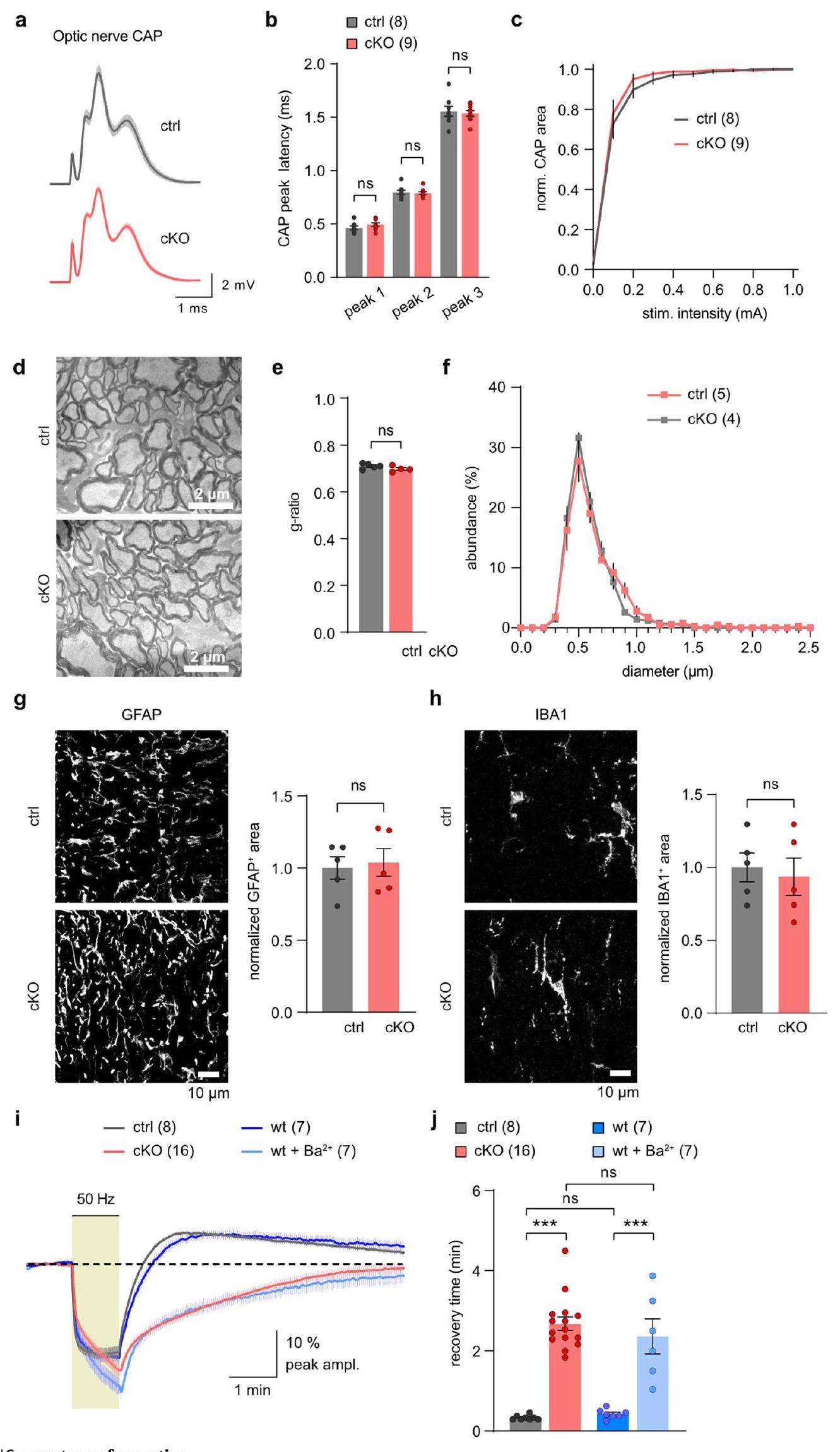
اختبار t). e، وفرة GLUT3 (Slc2a3) لا تتغير بين الأنماط الجينية (
محفظة الطبيعة
أيمن س. صعب
آخر تحديث بواسطة المؤلفين:
20 نوفمبر 2023
ملخص التقرير
الإحصائيات
تم التأكيد
يجب أن تُوصف الاختبارات الشائعة فقط بالاسم؛ واصفًا التقنيات الأكثر تعقيدًا في قسم الطرق.
لتحليل بايزي، معلومات حول اختيار القيم الأولية وإعدادات سلسلة ماركوف مونت كارلو
للتصاميم الهرمية والمعقدة، تحديد المستوى المناسب للاختبارات والتقارير الكاملة عن النتائج
تقديرات أحجام التأثير (مثل حجم تأثير كوهين)
تحتوي مجموعتنا على الإنترنت حول الإحصائيات لعلماء الأحياء على مقالات تتناول العديد من النقاط المذكورة أعلاه.
البرمجيات والشيفرة
معلومات السياسة حول توفر كود الكمبيوتر
جمع البيانات
علم الأنسجة المناعية: تم الحصول على صور متداخلة باستخدام مجهر ليزر ماسح ضوئي متداخل من زيس LSM 700 أو زيس LSM 800 مزود بـ
المجهر الإلكتروني (EM): تم التقاط صور EM باستخدام مجهر إلكتروني Zeiss EM912 (شركة كارل زيس للميكروسكوب، أوبيركوخن، ألمانيا) مزود بنظام على المحور
البروتيوميات: تم استخدام جهاز Orbitrap Fusion Lumos (Thermo Scientific) لتحليل مطيافية الكتلة، والذي كان مزودًا بمصدر Digital PicoView (New Objective) ومتصلاً بجهاز M-Class UPLC (Waters).
التحليل المناعي: تم إجراء الكشف باستخدام الكشف الكيميائي المعزز بالضوء (ECL) وفقًا لتعليمات الشركة المصنعة (Western Lightning Plus-ECL أو SuperSignal West Femto Maximum Sensitive Substrate؛ Thermo Fisher Scientific، سان ليون-روت، ألمانيا). تم مسح المناعية باستخدام ECL Chemostar (Intas Science Imaging، غوتنغن، ألمانيا).
تحليل البيانات
جاري المعالجة.
بيانات
يجب أن تتضمن جميع المخطوطات بيانًا عن توفر البيانات. يجب أن يوفر هذا البيان المعلومات التالية، حيثما ينطبق:
- رموز الانضمام، معرفات فريدة، أو روابط ويب لمجموعات البيانات المتاحة للجمهور
- وصف لأي قيود على توفر البيانات
- بالنسبة لمجموعات البيانات السريرية أو بيانات الطرف الثالث، يرجى التأكد من أن البيان يتماشى مع سياستنا
البحث الذي يتضمن مشاركين بشريين، بياناتهم، أو مواد بيولوجية
غير متوفر
الإشراف الأخلاقي
غير متوفر
غير متوفر
غير متوفر
التقارير الخاصة بالمجال
علوم الحياة
العلوم السلوكية والاجتماعية
العلوم البيئية والتطورية والإيكولوجية
لنسخة مرجعية من الوثيقة بجميع الأقسام، انظرnature.com/documents/nr-reporting-summary-flat.pdf
تصميم دراسة العلوم الحياتية
| حجم العينة | إن توفير مجموعات من الفئران المعدلة وراثيًا المتطابقة في العمر حد من القدرة على تحديد أحجام العينات مسبقًا. لم تُستخدم أي طرق إحصائية لتحديد حجم العينة مسبقًا. أحجام العينات المستخدمة في هذه الدراسة تعادل المعايير المتبعة في هذا المجال. |
| استثناءات البيانات | في بعض الأحيان كانت تعبيرات المستشعر ضعيفة جدًا (على سبيل المثال، بسبب عدم كفاية التعبير الناتج عن كري المدعوم بالتاموكسيفين أو توصيل AAV)، تم استبعاد هذه الحيوانات من التجارب. |
| استنساخ | عدد التكرارات (نقاط البيانات الفردية من كل خلية و/أو حيوان) موضح في الأشكال أو أساطير الأشكال. |
| التوزيع العشوائي | تم اختيار الفئران بناءً على النمط الجيني أو تم طلب فئران من النوع البري من تشارلز ريفر. تم تعيين الفئران عشوائيًا إلى مجموعات تجريبية، اعتمادًا على توفر المجموعات المتحولة. |
| عمى | بشكل عام، خلال جمع البيانات لم يكن الباحثون معزولين عن الظروف. ومع ذلك، كان الباحث الذي جمع مجموعة بيانات EM معزولاً عن الظروف التجريبية. كلما كان ذلك ممكنًا، كان المحققون معزولين لتحليل البيانات، خاصةً لتحليل صور EM. |
| المواد والأنظمة التجريبية | طرق | ||
| غير متوفر | مشارك في الدراسة | غير متوفر | مشارك في الدراسة |
 |
الأجسام المضادة | |
|
|
|
|
|
|
علم الحفريات وعلم الآثار | |
|
|
|||
|
|||
|
|||
|
|||
الأجسام المضادة
| الأجسام المضادة المستخدمة |
|
||||||||||||||||||
| التحقق |
|
الحيوانات وغيرها من الكائنات البحثية
| الحيوانات المخبرية |
|
|||
| الحيوانات البرية | لم يتم استخدام حيوانات برية في الدراسة | |||
| التقارير عن الجنس | تم استخدام كل من ذكور وإناث الفئران في التجارب |
معهد علم الأدوية وعلم السموم، جامعة زيورخ، زيورخ، سويسرا. مركز علوم الأعصاب زيورخ، جامعة زيورخ والمعهد الفيدرالي للتكنولوجيا في زيورخ، زيورخ، سويسرا. قسم علم الوراثة العصبية، معهد ماكس بلانك للعلوم متعددة التخصصات، غوتنغن، ألمانيا. قسم علوم الأعصاب سولومون إتش. سنايدر، جامعة جونز هوبكنز، بالتيمور، ماريلاند، الولايات المتحدة الأمريكية. مركز الدراسات العلمية (CECs)، فالديبيا، تشيلي. كلية الطب والعلوم، جامعة سان سيباستيان، فالديبیا، تشيلي. البريد الإلكتروني: asaab@pharma.uzh.ch - يرجى ملاحظة أنه يجب أيضًا تقديم معلومات كاملة حول الموافقة على بروتوكول الدراسة في المخطوطة.
DOI: https://doi.org/10.1038/s41593-023-01558-3
PMID: https://pubmed.ncbi.nlm.nih.gov/38267524
Publication Date: 2024-01-24
Oligodendrocyte-axon metabolic coupling is mediated by extracellular
Accepted: 13 December 2023
Published online: 24 January 2024
(A) Check for updates
Abstract
The integrity of myelinated axons relies on homeostatic support from oligodendrocytes (OLs). To determine how OLs detect axonal spiking and how rapid axon-OL metabolic coupling is regulated in the white matter, we studied activity-dependent calcium (
of monocarboxylate transporter1(MCT1) leads to late-onset axonopathy, implying that lactate and/or pyruvate release from OLs has a role in axonal health
Results
Axonal spiking triggers
for 30 s . Before and after this period, nerves received
OLs detect axonal spiking through
showing a reduction by
the
(PPADS;
| Antibody | Host species, type | Method, dilution | Source, cat. no. |
| Anti-CC1 | Mouse, monoclonal | IHC, 1:100 | Calbiochem, cat. no. OP80 (clone 5.24) |
| Anti-GFP | Chicken, polyclonal | IHC, 1:1,000 | Aves Labs, cat. no. GFP-1020 |
| Anti-GFAP | Chicken, polyclonal | IHC, 1:2,000 | Abcam, cat. no. ab4674 |
| Anti-IBA1 | Rabbit, polyclonal | IHC, 1:1,000 | FUJIFILM Wako Chemicals, cat. no. 019-19741 |
| Anti-Kir4.1 | Rabbit, polyclonal | IB, 1:1,000 | Alomone, cat. no. APC-035 |
| Anti-MCT1/SLC16A1 | Rabbit, polyclonal | IB, 1:500 | Produced by Kathrin Kusch
|
| Anti-GLUT1 | Rabbit, polyclonal | IB, 1:500 | Produced by Kathrin Kusch
|
| Anti-CNP | Mouse, monoclonal | IB, 1:1,000 | Sigma, cat. no. C 5922 (clone 11-5B) |
| Anti-PLP | Rabbit, polyclonal | IB, 1:5,000 | A431 (ref. 118) |
| Anti-MOG | Mouse, monoclonal | IB, 1:5,000 | Creative Biolabs, cat. no. PABZ-152 (clone 8-18C5) |
| Anti-ATP1
|
Mouse, monoclonal | IB, 1:1,000 | Abcam, cat. no. ab7671 (clone 464.6) |
| Anti-ATP1
|
Mouse, monoclonal | IB, 1:1,000 | Abcam, cat. no. ab2826 (clone XVIF9- G10) |
| Anti-mouse IgG HRP | Goat, polyclonal | IB, 1:10,000 | Jackson ImmunoResearch, cat. no. 115-035-003 |
| Anti-rabbit IgG HRP | Goat, polyclonal | IB, 1:10,000 | Jackson ImmunoResearch, cat. no. 111-035-003 |
| Anti-mouse Cy3 | Donkey, polyclonal | IHC, 1:700 | Jackson ImmunoResearch, cat. no. 715-165-151 |
| Anti-rabbit Cy3 | Donkey, polyclonal | IHC, 1:700 | Jackson ImmunoResearch, cat. no. 711-165-152 |
| Anti-chicken Alexa 488 | Donkey, polyclonal | IHC, 1:700 | Jackson ImmunoResearch, cat. no. 711-545-152 |
stimulation. Transient increases in the bath
firing after
by
a specific NKCC1 blocker. However,
Oligodendroglial Kir4.1 regulates axonal lactate dynamics
lactate consumption. The latter seems unlikely, as lactate decay during GD was not faster but marginally slower in cKO mice than in controls (Fig. 3h,k). This is further corroborated by a similar CAP decline rate during GD (Fig. 3l). The two genotypes showed the same latency between lactate depletion and CAP decline (Fig.3m). Notably, the CAP drop was observed only when axonal lactate neared depletion (Fig.3m). Given the concurrent decline of CAP and ATP levels during GD
Reduced MCT1 and GLUT1 in central nervous system myelin of Kir4.1 cKO mice
Minor changes in axonal ATP dynamics in Kir4.1 cKO mice
through WebGestalt.org, with proteins ranked by
the ATP sensor ATeam1.03 (ref. 52) in axons through intravitreal AAV delivery
decline rate during GD + MI was also comparable between genotypes (Fig. 5b,d), and the ATP recovery rate after
after GD + MI, indicating axonal conduction block, was comparable between genotypes, and the onset and recovery kinetics of axonal firing appeared unchanged (Extended Data Fig. 10a). The analysis of the partial CAP (pCAP) area, which reflects the dynamics of the first and second CAP peaks
a, AAV-mediated glucose FRET sensor (FLIIP) expression in optic nerve axons. Color-coded ratio images from control (top) and cKO (bottom) nerves show glucose levels in ACSF with 10 mM glucose and after GD. eYFP, enhanced yellow fluorescent protein; eCFP, enhanced cyan fluorescent protein.b, Time course of axonal glucose levels in
latency decreased; however, this peak latency shift (increase in conduction velocity) was markedly reduced in cKO nerves (Extended Data Fig. 10d), indicating a deficit in adjusting conduction speed following acute energy deprivation. Given that chemical ischemia increases
adjusting conduction speeds. This impaired speed adjustment might contribute to the reduced ATP recovery in cKO axons after chemical ischemia (Fig. 5b,g).
measured ATP dynamics during and after
Reduced axonal glucose metabolism in Kir4.1 cKO mice
was
glucose uptake at the nodes of Ranvier. Oligodendroglial
Discussion
aiding in OL differentiation during development
Online content
References
- Salvadores, N., Sanhueza, M., Manque, P. & Court, F. A. Axonal degeneration during aging and its functional role in neurodegenerative disorders. Front. Neurosci. 11, 451 (2017).
- Medana, I. M. & Esiri, M. M. Axonal damage: a key predictor of outcome in human CNS diseases. Brain 126, 515-530 (2003).
- Saab, A. S., Tzvetanova, I. D. & Nave, K.-A. The role of myelin and oligodendrocytes in axonal energy metabolism. Curr. Opin. Neurobiol. 23, 1065-1072 (2013).
- Nave, K.-A. Myelination and the trophic support of long axons. Nat. Rev. Neurosci. 11, 275-283 (2010).
- Philips, T. & Rothstein, J. D. Oligodendroglia: metabolic supporters of neurons. J. Clin. Invest. 127, 3271-3280 (2017).
- Duncan, G. J., Simkins, T. J. & Emery, B. Neuron-oligodendrocyte interactions in the structure and integrity of axons. Front. Cell Dev. Biol. 9, 653101 (2021).
- Xin, W. & Chan, J. R. Myelin plasticity: sculpting circuits in learning and memory. Nat. Rev. Neurosci. 21, 682-694 (2020).
- Fünfschilling, U. et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 485, 517-521 (2012).
- Lee, Y. et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487, 443-448 (2012).
- Saab, A. S. et al. Oligodendroglial NMDA receptors regulate glucose import and axonal energy metabolism. Neuron 91, 119-132 (2016).
- Trevisiol, A. et al. Monitoring ATP dynamics in electrically active white matter tracts. eLife 6, e24241 (2017).
- Tekkök, S. B., Brown, A. M., Westenbroek, R., Pellerin, L. & Ransom, B. R. Transfer of glycogen-derived lactate from astrocytes to axons via specific monocarboxylate transporters supports mouse optic nerve activity. J. Neurosci. Res. 81, 644-652 (2005).
- Philips, T. et al. MCT1 deletion in oligodendrocyte lineage cells causes late-onset hypomyelination and axonal degeneration. Cell Rep. 34, 108610 (2021).
- Edgar, J. M. et al. Río-Hortega’s drawings revisited with fluorescent protein defines a cytoplasm-filled channel system of CNS myelin. J. Anat. 239, 1241-1255 (2021).
- Saab, A. S. & Nave, K.-A. Myelin dynamics: protecting and shaping neuronal functions. Curr. Opin. Neurobiol. 47, 104-112 (2017).
- Snaidero, N. et al. Antagonistic functions of MBP and CNP establish cytosolic channels in CNS myelin. Cell Rep. 18, 314-323 (2017).
- Griffiths, I. et al. Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science 280, 1610-1613 (1998).
- Lüders, K. A. et al. Maintenance of high proteolipid protein level in adult central nervous system myelin is required to preserve the integrity of myelin and axons. Glia 67, 634-649 (2019).
- Edgar, J. M. et al. Oligodendroglial modulation of fast axonal transport in a mouse model of hereditary spastic paraplegia. J. Cell Biol. 166, 121-131 (2004).
- Steyer, A. M. et al. Pathology of myelinated axons in the PLP-deficient mouse model of spastic paraplegia type 2 revealed by volume imaging using focused ion beam-scanning electron microscopy. J. Struct. Biol. 210, 107492 (2020).
- Trevisiol, A. et al. Structural myelin defects are associated with low axonal ATP levels but rapid recovery from energy deprivation in a mouse model of spastic paraplegia. PLoS Biol. 18, e3000943 (2020).
- Mukherjee, C. et al. Oligodendrocytes provide antioxidant defense function for neurons by secreting ferritin heavy chain. Cell Metab. 32, 259-272 (2020).
- Larson, V. A. et al. Oligodendrocytes control potassium accumulation in white matter and seizure susceptibility. eLife 7, e34829 (2018).
- Schirmer, L. et al. Oligodendrocyte-encoded Kir4.1 function is required for axonal integrity. eLife 7, e36428 (2018).
- Kettenmann, H., Sonnhof, U. & Schachner, M. Exclusive potassium dependence of the membrane potential in cultured mouse oligodendrocytes. J. Neurosci. 3, 500-505 (1983).
- Yamazaki, Y. et al. Modulatory effects of oligodendrocytes on the conduction velocity of action potentials along axons in the alveus of the rat hippocampal CA1 region. Neuron Glia Biol. 3, 325-334 (2007).
- Battefeld, A., Klooster, J. & Kole, M. H. P. Myelinating satellite oligodendrocytes are integrated in a glial syncytium constraining neuronal high-frequency activity. Nat. Commun. 7, 11298 (2016).
- Looser, Z. J., Barrett, M. J. P., Hirrlinger, J., Weber, B. & Saab, A. S. Intravitreal AAV-delivery of genetically encoded sensors enabling simultaneous two-photon imaging and electrophysiology of optic nerve axons. Front. Cell. Neurosci. 12, 377 (2018).
- Doerflinger, N. H., Macklin, W. B. & Popko, B. Inducible site-specific recombination in myelinating cells. Genesis 35, 63-72 (2003).
- Madisen, L. et al. Transgenic mice for intersectional targeting of neural sensors and effectors with high specificity and performance. Neuron 85, 942-958 (2015).
- Chen, T.-W. et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295-300 (2013).
- Takanaga, H., Chaudhuri, B. & Frommer, W. B. GLUT1 and GLUT9 as major contributors to glucose influx in HepG2 cells identified by a high sensitivity intramolecular FRET glucose sensor. Biochim. Biophys. Acta 1778, 1091-1099 (2008).
- Bittner, C. X. et al. High resolution measurement of the glycolytic rate. Front. Neuroenergetics 2, 26 (2010).
- Bittner, C. X. et al. Fast and reversible stimulation of astrocytic glycolysis by
and a delayed and persistent effect of glutamate. J. Neurosci. 31, 4709-4713 (2011). - Micu, I. et al. The molecular physiology of the axo-myelinic synapse. Exp. Neurol. 276, 41-50 (2016).
- Micu, I. et al. NMDA receptors mediate calcium accumulation in myelin during chemical ischaemia. Nature 439, 988-992 (2006).
- James, G. & Butt, A. M. P2X and P2Y purinoreceptors mediate ATP-evoked calcium signalling in optic nerve glia in situ. Cell Calcium 30, 251-259 (2001).
- Kirischuk, S., Scherer, J., Kettenmann, H. & Verkhratsky, A. Activation of P2-purinoreceptors triggered
release from InsP3-sensitive internal stores in mammalian oligodendrocytes. J. Physiol. 483, 41-57 (1995). - Matute, C. et al. P2X(7) receptor blockade prevents ATP excitotoxicity in oligodendrocytes and ameliorates experimental autoimmune encephalomyelitis. J. Neurosci. 27, 9525-9533 (2007).
- Stevens, B., Porta, S., Haak, L. L., Gallo, V. & Fields, R. D. Adenosine: a neuron-glial transmitter promoting myelination in the CNS in response to action potentials. Neuron 36, 855-868 (2002).
- Ransom, C. B., Ransom, B. R. & Sontheimer, H. Activity-dependent extracellular
accumulation in rat optic nerve: the role of glial and axonal pumps. J. Physiol. 522, 427-442 (2000). - Bay, V. & Butt, A. M. Relationship between glial potassium regulation and axon excitability: a role for glial Kir4.1 channels. Glia 60, 651-660 (2012).
- Olsen, M. L. & Sontheimer, H. Functional implications for Kir4.1 channels in glial biology: from
buffering to cell differentiation. J. Neurochem. 107, 589-601 (2008). - Boscia, F. et al. Silencing or knocking out the
exchanger-3 (NCX3) impairs oligodendrocyte differentiation. Cell Death Differ. 19, 562-572 (2012). - Casamassa, A. et al. Ncx3 gene ablation impairs oligodendrocyte precursor response and increases susceptibility to experimental autoimmune encephalomyelitis. Glia 64, 1124-1137 (2016).
- Spencer, S. A., Suárez-Pozos, E., Escalante, M., Myo, Y. P. & Fuss, B. Sodium-calcium exchangers of the SLC8 family in oligodendrocytes: functional properties in health and disease. Neurochem. Res. 45, 1287-1297 (2020).
- Friess, M. et al. Intracellular ion signaling influences myelin basic protein synthesis in oligodendrocyte precursor cells. Cell Calcium 60, 322-330 (2016).
- Moyon, S. et al. TET1-mediated DNA hydroxymethylation regulates adult remyelination in mice. Nat. Commun. 12, 3359 (2021).
- Yamazaki, Y., Abe, Y., Fujii, S. & Tanaka, K. F. Oligodendrocytic
co-transporter 1 activity facilitates axonal conduction and restores plasticity in the adult mouse brain. Nat. Commun. 12, 5146 (2021). - San Martín, A. et al. A genetically encoded FRET lactate sensor and its use to detect the Warburg effect in single cancer cells. PLoS ONE 8, e57712 (2013).
- Djukic, B., Casper, K. B., Philpot, B. D., Chin, L.-S. & McCarthy, K. D. Conditional knock-out of
leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J. Neurosci. 27, 11354-11365 (2007). - Imamura, H. et al. Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc. Natl Acad. Sci. USA 106, 15651-15656 (2009).
- Hamilton, N. B., Kolodziejczyk, K., Kougioumtzidou, E. & Attwell, D. Proton-gated
-permeable TRP channels damage myelin in conditions mimicking ischaemia. Nature 529, 523-527 (2016). - Meyer, N. et al. Oligodendrocytes in the mouse corpus callosum maintain axonal function by delivery of glucose. Cell Rep. 22, 2383-2394 (2018).
- Zhang, X. et al. Oligodendroglial glycolytic stress triggers inflammasome activation and neuropathology in Alzheimer’s disease. Sci. Adv. 6, eabb8680 (2020).
- Mot, A. I., Depp, C. & Nave, K.-A. An emerging role of dysfunctional axon-oligodendrocyte coupling in neurodegenerative diseases. Dialogues Clin. Neurosci. 20, 283-292 (2018).
- Kenigsbuch, M. et al. A shared disease-associated oligodendrocyte signature among multiple CNS pathologies. Nat. Neurosci. 25, 876-886 (2022).
- Kaya, T. et al.
cells induce interferon-responsive oligodendrocytes and microglia in white matter aging. Nat. Neurosci. 25, 1446-1457 (2022). - Brasko, C., Hawkins, V., De La Rocha, I. C. & Butt, A. M. Expression of Kir4.1 and Kir5.1 inwardly rectifying potassium channels in oligodendrocytes, the myelinating cells of the CNS. Brain Struct. Funct. 222, 41-59 (2017).
- Papanikolaou, M., Butt, A. M. & Lewis, A. A critical role for the inward rectifying potassium channel Kir7.1 in oligodendrocytes of the mouse optic nerve. Brain Struct. Funct. 225, 925-934 (2020).
- Almeida, R. G. et al. Myelination induces axonal hotspots of synaptic vesicle fusion that promote sheath growth. Curr. Biol. 31, 3743-3754 (2021).
- Micu, I., Plemel, J. R., Caprariello, A. V., Nave, K. A. & Stys, P. K. Axo-myelinic neurotransmission: a novel mode of cell signalling in the central nervous system. Nat. Rev. Neurosci. 19, 49-58 (2018).
- Hines, J. H., Ravanelli, A. M., Schwindt, R., Scott, E. K. & Appel, B. Neuronal activity biases axon selection for myelination in vivo. Nat. Neurosci. 18, 683-689 (2015).
- Wake, H., Lee, P. R. & Fields, R. D. Control of local protein synthesis and initial events in myelination by action potentials. Science 333, 1647-1651 (2011).
- Mensch, S. et al. Synaptic vesicle release regulates myelin sheath number of individual oligodendrocytes in vivo. Nat. Neurosci. 18, 628-630 (2015).
- Kukley, M., Capetillo-Zarate, E. & Dietrich, D. Vesicular glutamate release from axons in white matter. Nat. Neurosci. 10, 311-320 (2007).
- Ziskin, J. L., Nishiyama, A., Rubio, M., Fukaya, M. & Bergles, D. E. Vesicular release of glutamate from unmyelinated axons in white matter. Nat. Neurosci. 10, 321-330 (2007).
- Krasnow, A. M., Ford, M. C., Valdivia, L. E., Wilson, S. W. & Attwell, D. Regulation of developing myelin sheath elongation by oligodendrocyte calcium transients in vivo. Nat. Neurosci. 21, 24-28 (2018).
- Baraban, M., Koudelka, S. & Lyons, D. A.
activity signatures of myelin sheath formation and growth in vivo. Nat. Neurosci. 21, 19-23 (2018). - Battefeld, A., Popovic, M. A., de Vries, S. I. & Kole, M. H. P. High-frequency microdomain
transients and waves during early myelin internode remodeling. Cell Rep. 26, 182-191 (2019). - Kanda, H. et al. TREK-1 and TRAAK are principal
channels at the nodes of Ranvier for rapid action potential conduction on mammalian myelinated afferent nerves. Neuron 104, 960-971 (2019). - Brohawn, S. G. et al. The mechanosensitive ion channel TRAAK is localized to the mammalian node of Ranvier. eLife 8, e50403 (2019).
- Rash, J. E. Molecular disruptions of the panglial syncytium block potassium siphoning and axonal saltatory conduction: pertinence to neuromyelitis optica and other demyelinating diseases of the central nervous system. Neuroscience 168, 982-1008 (2010).
- Cohen, C. C. H. et al. Saltatory conduction along myelinated axons involves a periaxonal nanocircuit. Cell 180, 311-322 (2020).
- Ruminot, I., Schmälzle, J., Leyton, B., Barros, L. F. & Deitmer, J. W. Tight coupling of astrocyte energy metabolism to synaptic activity revealed by genetically encoded FRET nanosensors in hippocampal tissue. J. Cereb. Blood Flow Metab. 39, 513-523 (2019).
- Wang, N. et al. Potassium channel
regulates oligodendrocyte differentiation via intracellular pH regulation. Glia 70, 2093-2107 (2022). - Fernández-Moncada, I. et al. Bidirectional astrocytic GLUT1 activation by elevated extracellular K. Glia 69, 1012-1021 (2021).
- Zuend, M. et al. Arousal-induced cortical activity triggers lactate release from astrocytes. Nat. Metab. 2, 179-191 (2020).
- Sotelo-Hitschfeld, T. et al. Channel-mediated lactate release by
-stimulated astrocytes. J. Neurosci. 35, 4168-4178 (2015). - Köhler, S. et al. Gray and white matter astrocytes differ in basal metabolism but respond similarly to neuronal activity. Glia 71, 229-244 (2023).
- Menichella, D. M. et al. Genetic and physiological evidence that oligodendrocyte gap junctions contribute to spatial buffering of potassium released during neuronal activity. J. Neurosci. 26, 10984-10991 (2006).
- Neusch, C., Rozengurt, N., Jacobs, R. E., Lester, H. A. & Kofuji, P. Kir4.1 potassium channel subunit is crucial for oligodendrocyte development and in vivo myelination. J. Neurosci. 21, 5429-5438 (2001).
- Orthmann-Murphy, J. L., Abrams, C. K. & Scherer, S. S. Gap junctions couple astrocytes and oligodendrocytes. J. Mol. Neurosci. 35, 101-116 (2008).
- Hösli, L. et al. Decoupling astrocytes in adult mice impairs synaptic plasticity and spatial learning. Cell Rep. 38, 110484 (2022).
- Jahn, O. et al. The CNS myelin proteome: deep profile and persistence after post-mortem delay. Front. Cell. Neurosci. 14, 239 (2020).
- Zhang, Y. et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929-11947 (2014).
- Gargareta, V.-I. et al. Conservation and divergence of myelin proteome and oligodendrocyte transcriptome profiles between humans and mice. eLife 11, e77019 (2022).
- Lam, M. et al. CNS myelination requires VAMP2/3-mediated membrane expansion in oligodendrocytes. Nat. Commun. 13, 5583 (2022).
- Leto, D. & Saltiel, A. R. Regulation of glucose transport by insulin: traffic control of GLUT4. Nat. Rev. Mol. Cell Biol. 13, 383-396 (2012).
- Martin, L. B., Shewan, A., Millar, C. A., Gould, G. W. & James, D. E. Vesicle-associated membrane protein 2 plays a specific role in the insulin-dependent trafficking of the facilitative glucose transporter GLUT4 in 3T3-L1 adipocytes. J. Biol. Chem. 273, 1444-1452 (1998).
- Lodhi, I. J. et al. Gapex-5, a Rab31 guanine nucleotide exchange factor that regulates Glut4 trafficking in adipocytes. Cell Metab. 5, 59-72 (2007).
- Dienel, G. A. Brain glucose metabolism: integration of energetics with function. Physiol. Rev. 99, 949-1045 (2019).
- Barros, L. F. et al. Fluid brain glycolysis: limits, speed, location, moonlighting, and the fates of glycogen and lactate. Neurochem. Res. 45, 1328-1334 (2020).
- Herrero-Mendez, A. et al. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat. Cell Biol. 11, 747-752 (2009).
- Zala, D. et al. Vesicular glycolysis provides on-board energy for fast axonal transport. Cell 152, 479-491 (2013).
- Baeza-Lehnert, F. et al. Non-Canonical Control of Neuronal Energy Status by the
Pump. Cell Metab. 29, 668-680.e4 (2019). - Meyer, D. J., Díaz-García, C. M., Nathwani, N., Rahman, M. & Yellen, G. The
pump dominates control of glycolysis in hippocampal dentate granule cells. eLife 11, e81645 (2022). - Frühbeis, C. et al. Oligodendrocytes support axonal transport and maintenance via exosome secretion. PLoS Biol. 18, e3000621 (2020).
- Chamberlain, K. A. et al. Oligodendrocytes enhance axonal energy metabolism by deacetylation of mitochondrial proteins through transcellular delivery of SIRT2. Neuron 109, 3456-3472 (2021).
- Frühbeis, C. et al. Neurotransmitter-triggered transfer of exosomes mediates oligodendrocyte-neuron communication. PLoS Biol. 11, e1001604 (2013).
as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended
use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons. org/licenses/by/4.0/.
© The Author(s) 2024
Methods
Animals
Tamoxifen treatment
AAV injections
AAV vectors
axonal ATP dynamics, we used the FRET sensor ATeam1.03 (ref. 52) packaged in ssAAV-2/2-hSyn1-ATeam1.03-WPRE-hGHp(A)
Optic nerve electrophysiology and two-photon imaging
Solutions
CAP recordings and analysis
from stimulus onset to peak. The pCAP area was used to integrate the amplitude and latency changes of the first two peaks, reflective of large and medium-sized axons. Stimulation protocols involved an initial
Calcium imaging and analysis
Metabolite imaging and analysis
Immunohistochemistry
using a Leica CM3050 S cryostat. Sections were placed on SuperFrost Plus slides (Thermo Fisher Scientific). For immunohistochemistry, slides were first treated with 0.3% Triton X-100 in 50 mM Tris buffer ( pH 7.4 ) with
EM and analysis
Proteomics and analysis
missed cleavages. The maximum FDR for peptides was set to 0.01 . Protein fold changes were computed based on intensity values. A set of functions implemented in R package prolfqua
Myelin purification and immunoblotting
Real-time qPCR
(
Statistical analyses
Reporting summary
Data availability
Code availability
References
- Hövelmeyer, N. et al. Apoptosis of oligodendrocytes via Fas and TNF-R1 is a key event in the induction of experimental autoimmune encephalomyelitis. J. Immunol. 175, 5875-5884 (2005).
- Fleischmann, T., Jirkof, P., Henke, J., Arras, M. & Cesarovic, N. Injection anaesthesia with fentanyl-midazolam-medetomidine in adult female mice: importance of antagonization and perioperative care. Lab. Anim. 50, 264-274 (2016).
- Paterna, J.-C., Feldon, J. & Büeler, H. Transduction profiles of recombinant adeno-associated virus vectors derived from serotypes 2 and 5 in the nigrostriatal system of rats. J. Virol. 78, 6808-6817 (2004).
- Snaidero, N. et al. Myelin replacement triggered by single-cell demyelination in mouse cortex. Nat. Commun. 11, 4901 (2020).
- Mezydlo, A. et al. Remyelination by surviving oligodendrocytes is inefficient in the inflamed mammalian cortex. Neuron 111, 1748-1759 (2023).
- Mayrhofer, J. M. et al. Design and performance of an ultra-flexible two-photon microscope for in vivo research. Biomed. Opt. Express 6, 4228-4237 (2015).
- Pologruto, T. A., Sabatini, B. L. & Svoboda, K. Scanlmage: flexible software for operating laser scanning microscopes. Biomed. Eng. Online 2, 13 (2003).
- Stys, P. K., Ransom, B. R. & Waxman, S. G. Compound action potential of nerve recorded by suction electrode: a theoretical and experimental analysis. Brain Res. 546, 18-32 (1991).
- Barrett, M. J. P., Ferrari, K. D., Stobart, J. L., Holub, M. & Weber, B. CHIPS: an extensible toolbox for cellular and hemodynamic two-photon image analysis. Neuroinformatics 16, 145-147 (2018).
- Glück, C. et al. Distinct signatures of calcium activity in brain mural cells. eLife 10, e70591 (2021).
- Möbius, W. et al. Electron microscopy of the mouse central nervous system. Methods Cell Biol. 96, 475-512 (2010).
- Türker, C. et al. B-Fabric: the Swiss army knife for life sciences. In Proceedings of the 13th International Conference on Extending Database Technology (eds Manolescu, I. et al.) 717-720 (ACM, 2010).
- Wolski, W. E., Panse, C., Grossmann, J., D’Errico, M. & Nanni, P. prolfqua-an R package for proteomics label-free quantification. F1000Research https://doi.org/10.7490/f1000research.1118455.1 (2021).
- Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
- Erwig, M. S. et al. Myelin: methods for purification and proteome analysis. Methods Mol. Biol. 1936, 37-63 (2019).
- Stumpf, S. K. et al. Ketogenic diet ameliorates axonal defects and promotes myelination in Pelizaeus-Merzbacher disease. Acta Neuropathol. 138, 147-161 (2019).
- Berghoff, S. A. et al. Blood-brain barrier hyperpermeability precedes demyelination in the cuprizone model. Acta Neuropathol. Commun. 5, 94 (2017).
- Jung, M., Sommer, I., Schachner, M. & Nave, K. A. Monoclonal antibody O10 defines a conformationally sensitive cell-surface epitope of proteolipid protein (PLP): evidence that PLP misfolding underlies dysmyelination in mutant mice. J. Neurosci. 16, 7920-7929 (1996).
Acknowledgements
the MBP promoter, and the Functional Genomics Center Zurich for proteomics support. D.E.B. and K.-A.N. were supported by the Adelson Medical Research Foundation. L.F.B. was supported by Fondecyt 1230145. B.W. was supported by the Swiss National Science Foundation (31003A_156965). A.S.S. was supported by a Synapsis Career Fellowship Award, Neuroscience Center Zurich, the Cloëtta Foundation and the Swiss National Science Foundation (Eccellenza 187000).
Author contributions
Funding
Competing interests
Additional information
https://doi.org/10.1038/s41593-023-01558-3.
with NKCC1 blocker Bumetanide (
Extended Data Fig. 6 | See next page for caption.
sheath thickness ( g -ratio) between genotypes (
t-test). e, Abundance of GLUT3 (Slc2a3) is unchanged between genotypes (
natureportfolio
Aiman S. Saab
Last updated by author(s):
Nov 20, 2023
Reporting Summary
Statistics
Confirmed
Only common tests should be described solely by name; describe more complex techniques in the Methods section.
For Bayesian analysis, information on the choice of priors and Markov chain Monte Carlo settings
For hierarchical and complex designs, identification of the appropriate level for tests and full reporting of outcomes
Estimates of effect sizes (e.g. Cohen’s
Our web collection on statistics for biologists contains articles on many of the points above.
Software and code
Policy information about availability of computer code
Data collection
Immunohistochemistry: Confocal images were acquired with a Zeiss LSM 700 or Zeiss LSM 800 confocal laser scanning microscope equipped with a
Electron microscopy (EM): EM pictures were captured with a Zeiss EM912 electron microscope (Carl Zeiss Microscopy GmbH, Oberkochen, Germany) equipped with an on-axis
Proteomics: For mass spectrometry analysis, an Orbitrap Fusion Lumos (Thermo Scientific) was used, which was equipped with a Digital PicoView source (New Objective) and coupled to a M-Class UPLC (Waters).
Immunoblotting: Detection was carried out using enhanced chemiluminescent detection (ECL) according to the manufacturer’s instructions (Western Lightning Plus-ECL or SUperSignal West Femto Maximum Sensitive Substrate; Thermo Fisher Scientific, St Leon-Rot, Germany). Immunoblots were scanned using ECL Chemostar (Intas Science Imaging, Göttingen, Germany).
Data analysis
processing.
Data
All manuscripts must include a data availability statement. This statement should provide the following information, where applicable:
- Accession codes, unique identifiers, or web links for publicly available datasets
- A description of any restrictions on data availability
- For clinical datasets or third party data, please ensure that the statement adheres to our policy
Research involving human participants, their data, or biological material
N/A
Ethics oversight
N/A
N/A
N/A
Field-specific reporting
Life sciences
Behavioural & social sciences
Ecological, evolutionary & environmental sciences
For a reference copy of the document with all sections, see nature.com/documents/nr-reporting-summary-flat.pdf
Life sciences study design
| Sample size | The provision of age-matched transgenic mouse cohorts limited the ability to determine sample sizes in advance. No statistical methods were used to predetermine sample size. Sample sizes used in this study are equivalent to the standard in the field. |
| Data exclusions | ometimes sensor expression was too weak (e.g. due to insufficient tamoxifen-induced Cre-mediated expression or AAV delivery), these animals were excluded from experiments. |
| Replication | Number of repetitions (individual data points from each cells and/or animal) are indicated in figures or figure legends. |
| Randomization | Selection of mice was based on genotype or wildtype mice were ordered from Charles River. Mice were randomly assigned to experimental groups, depending on availability of transgenic cohorts. |
| Blinding | Overall, during data collection experimenters were not blinded to the conditions. However, experimenter who collected EM dataset was blinded to the experimental conditions. Whenever possible, investigators were blinded for data analysis, especially for EM image analysis. |
| Materials & experimental systems | Methods | ||
| n/a | Involved in the study | n/a | Involved in the study |
|
Antibodies | |
|
|
|
|
|
|
Palaeontology and archaeology | |
|
|
|||
|
|||
|
|||
|
|||
Antibodies
| Antibodies used |
|
||||||||||||||||||
| Validation |
|
Animals and other research organisms
| Laboratory animals |
|
|||
| Wild animals | no wild animals were used in the study | |||
| Reporting on sex | Both male and female mice were used for experiments |
Institute of Pharmacology and Toxicology, University of Zurich, Zurich, Switzerland. Neuroscience Center Zurich, University and ETH Zurich, Zurich, Switzerland. Department of Neurogenetics, Max Planck Institute for Multidisciplinary Sciences, Göttingen, Germany. Solomon H. Snyder Department of Neuroscience, Johns Hopkins University, Baltimore, MD, USA. Centro de Estudios Científicos (CECs), Valdivia, Chile. Facultad de Medicina y Ciencia, Universidad San Sebastián, Valdivia, Chile. e-mail: asaab@pharma.uzh.ch - Note that full information on the approval of the study protocol must also be provided in the manuscript.