شيا-كوي مو, جينغشيان ليو, سيكي تشين, إريك ستورز, أندريه لويز ن. تارغينو دا كوستا, أندرو هيوستن, مايكل سي. ويندل, ريكيا جي. جاياسينغ, مايكل دي. إغليزيا, كونغ, جون م. هيرندون, أوستن ن. ساوثارد-سميث, شينهاو ليو, جاكلين ميد, ألا كاربوفا, أندرو شينكل, س. بيتر غودغيبور, عبد الرحمن طه موسى علي عبد الظاهر, بينغ بو, لورين فولغوم, سامانثا ليفينغستون, متين بالابان, أنجيلا هيل, جوزيف إي. إيبوليتو, فستين ثورسون, جيسون م. هيلد, إيان س. هاجمان, إريك إتش. كيم, بيتر أو. بايغوينوف, ألبرت إتش. كيم, ماري م. مولن, كوريش آي. شوقي, تاو جو, ميليسا أ. رايمرز, كودي وايمهولت, ليانغ-آي كانغ, سيدهارث ف. بورام, ديبورا ج. فايز, راسل باتشينسكي, كاثرين سي. فوه, ميلان ج. تشيدا, ويليام إي. جيلاندرز, رايان سي. فيلدز, بنيامين ج. رافائيل, فنغ تشين & لي دينغ
الملخص
لدراسة التفاعلات المكانية بين خلايا السرطان وغير السرطان, قمنا هنا بفحص مجموعة من 131 مقطع ورمي من 78 حالة عبر 6 أنواع من السرطان باستخدام تقنية النسخ الجيني المكاني (ST). تم دمج ذلك مع 48 عينة من تسلسل RNA أحادي النواة المتطابقة و22 عينة من الكشف المشترك بواسطة الفهرسة (CODEX). لوصف هياكل الورم والموائل، عرفنا ‘المناطق الدقيقة للورم’ على أنها تجمعات خلايا سرطانية متميزة مكانياً تفصلها مكونات دعامية. كانت تختلف في الحجم والكثافة بين أنواع السرطان، مع أكبر المناطق الدقيقة التي لوحظت في العينات النقيلي. قمنا أيضًا بتجميع المناطق الدقيقة ذات التغيرات الجينية المشتركة في ‘النسخ الفرعية المكانية’. أظهرت خمسة وثلاثون مقطع ورمي هياكل فرعية. عرضت النسخ الفرعية المكانية ذات التغيرات المميزة في عدد النسخ والطفرات أنشطة سرطانية مختلفة. حددنا زيادة النشاط الأيضي في المركز وزيادة تقديم المستضد على طول الحواف الرائدة للمناطق الدقيقة. كما لاحظنا تسللات متغيرة لخلايا T داخل المناطق الدقيقة ووجود البلعميات بشكل رئيسي عند حدود الورم. قمنا بإعادة بناء هياكل الورم ثلاثية الأبعاد من خلال تسجيل 48 مقطع ST متسلسل من 16 عينة، مما قدم رؤى حول التنظيم المكاني والتنوع في الأورام. بالإضافة إلى ذلك، باستخدام خوارزمية تعلم عميق غير خاضعة للإشراف ودمج بيانات ST وCODEX، حددنا كل من الأحياء المناعية الساخنة والباردة وعلامات الإرهاق المناعي المعززة المحيطة بالنسخ الفرعية ثلاثية الأبعاد. تسهم هذه النتائج في فهم تطور الورم المكاني من خلال التفاعلات مع البيئة الدقيقة المحلية في الفضاء ثنائي وثلاثي الأبعاد، مما يوفر رؤى قيمة في بيولوجيا الورم.
تظهر النسخ الفرعية المقاومة للعلاج غالبًا في السرطان, ويمكن أن تدفع البيئة الدقيقة للورم (TME) المقاومة من خلال آليات متعددة. لا تحافظ تقنيات الكتلة أو تقنيات الخلايا الفردية على المعلومات المكانية اللازمة لفهم هذه الديناميات، ولكن ST الأدوات، مثل Visium, يمكن أن تحل الهياكل الفرعية للورم. تم دمج بيانات ST مع أنواع بيانات أخرى لفحص الهيكل الكلوني الدقيق وتحديد التفاعلات بين الخلايا (CCIs) مع البيئة الدقيقة. يمكن أن تكمل التصوير المتعدد CODEX طرق ST من خلال تحديد موقع البروتينات مكانيًا.
تظل التطورات الكلونية واحدة من أكثر المشاكل تعقيدًا في السرطان. أي، التكيف المكاني والزماني للورم مع المحفزات البيئية والعلاجية من خلال تراكم الطفرات والاختيار القائم على اللياقة. ركزت الدراسات السابقة
على استنتاج التاريخ التطوري من خلال الطفرات، ولكن التقنيات الأحدث، بما في ذلك تلك المذكورة أعلاه، قد مكنت من تحقيقات أعمق بكثير في الديناميات الكلونية المكانية. إن احتمال تطبيق العديد من هذه التقنيات على مجموعة كبيرة، ذات طاقة جيدة، عبر السرطان للتحقيق بشكل أعمق في هذه الظواهر يحفز العمل الحالي.
هنا نبلغ عن التوصيف الشامل لـ 131 مقطع ST ورمي عبر 6 أنواع مختلفة من السرطان: سرطان الثدي (BRCA)، سرطان القولون والمستقيم (CRC)، سرطان البنكرياس القنوي (PDAC)، سرطان الخلايا الكلوية (RCC)، سرطان بطانة الرحم (UCEC) وسرطان القناة الصفراوية (CHOL). نستخدم نهجًا يجمع بين بيانات ST وCODEX وبيانات التسلسل الكتلي وبيانات التسلسل الخلوي الفردي للعينات المتطابقة لتوصيف مناطق الورم المتميزة مكانيًا
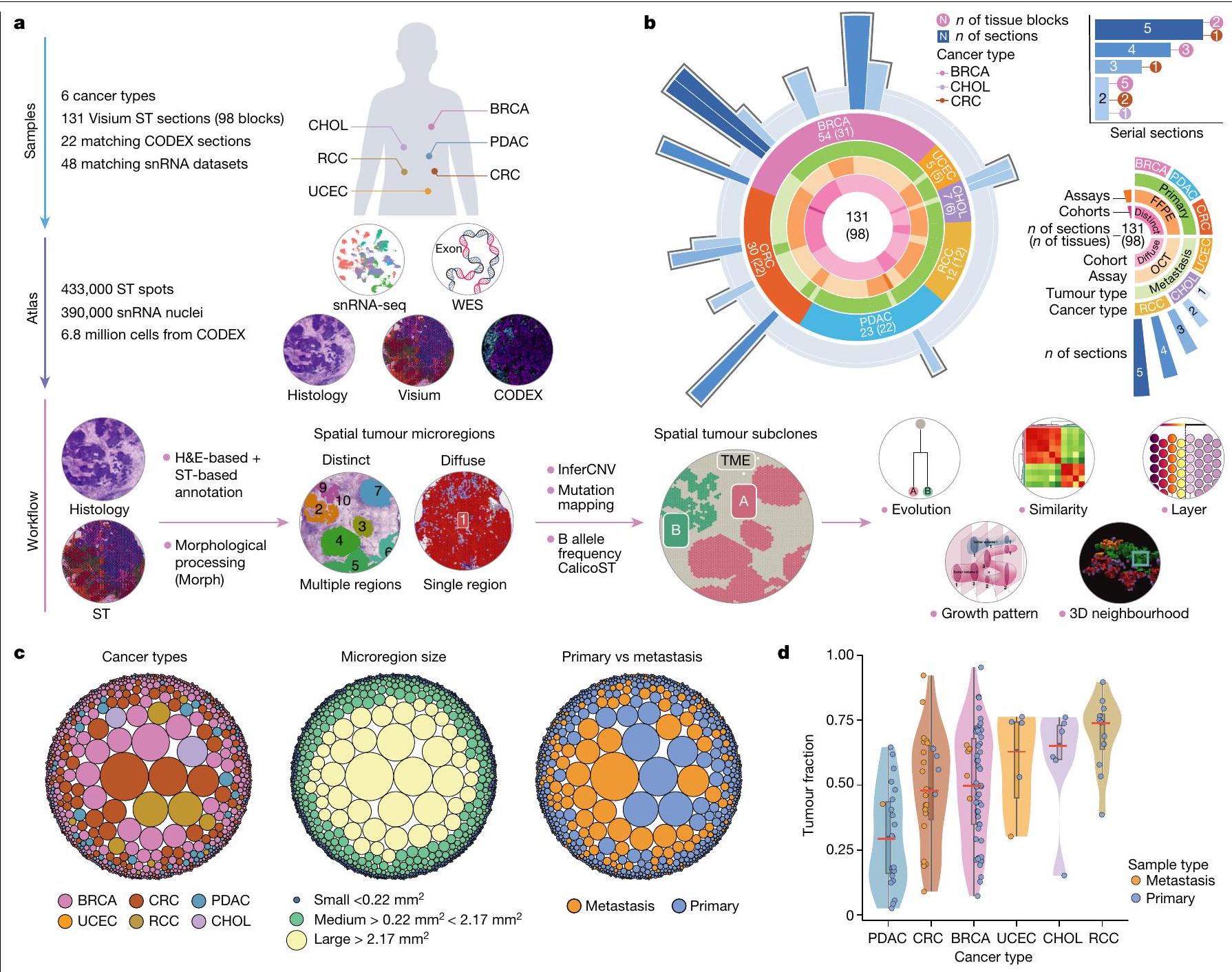
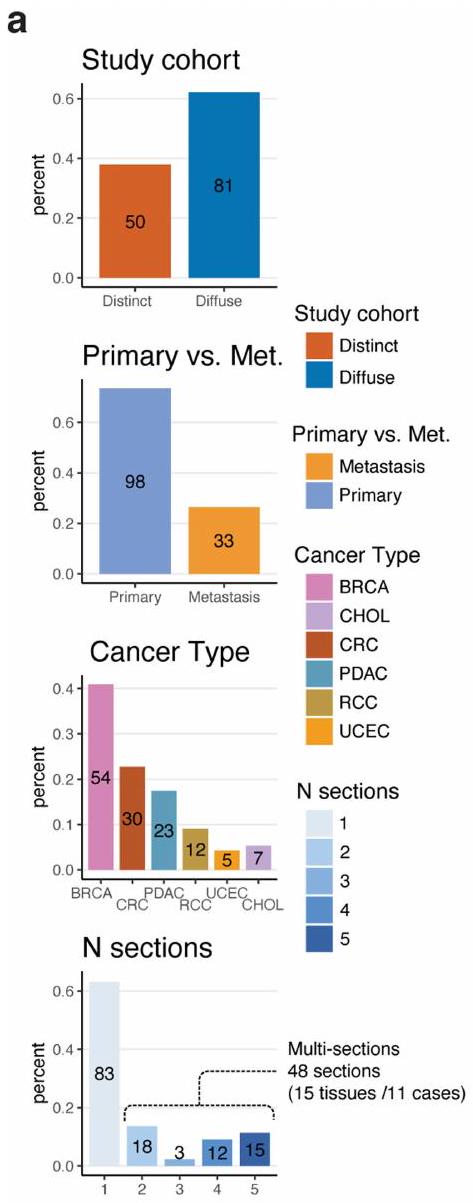
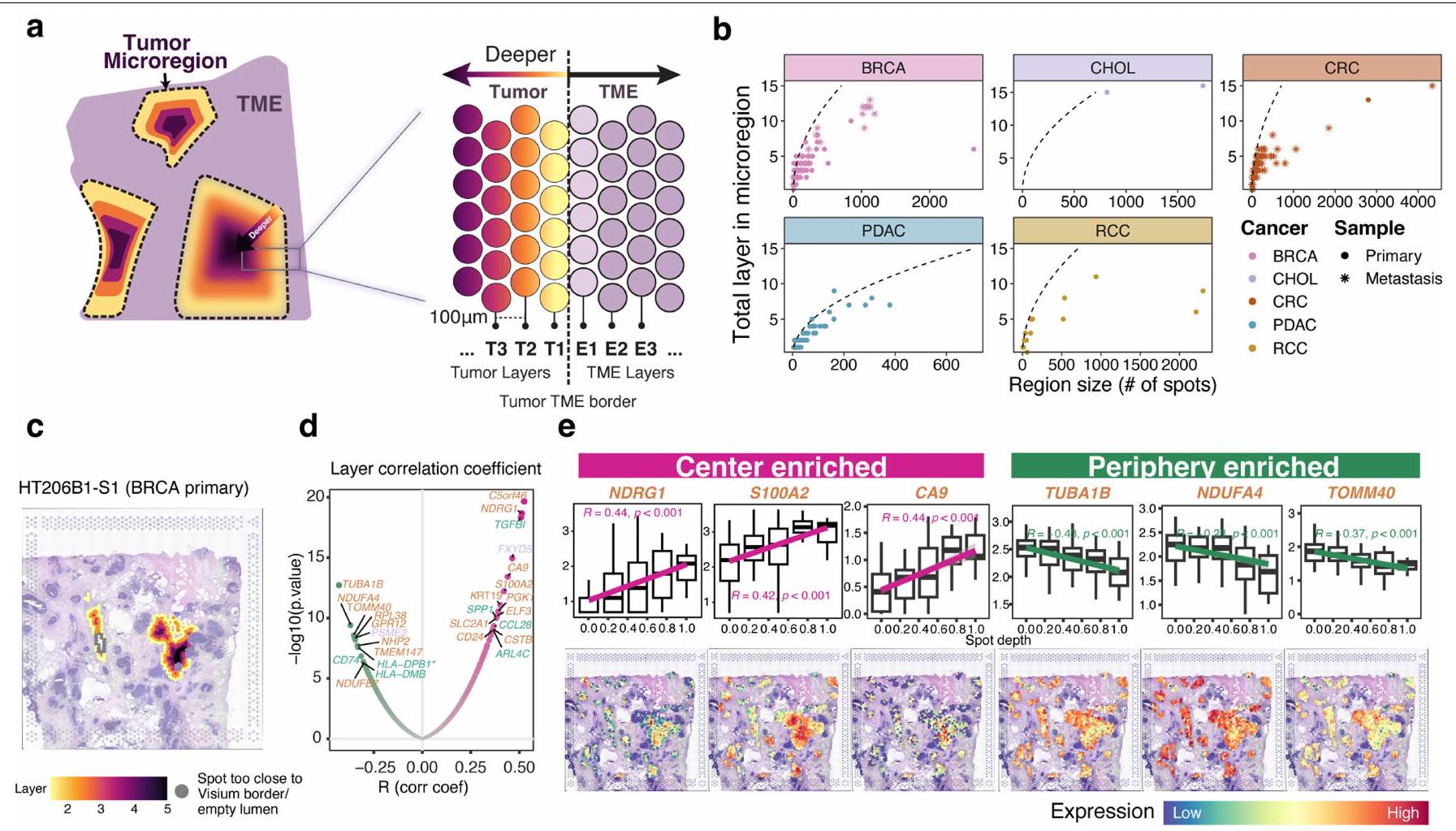
الشكل 1| تعريف المناطق الدقيقة المكانية للورم. أ، عينة، نوع البيانات ونظرة عامة على سير العمل لمجموعة النسخ الفرعية المكانية من 131 مقطع ST من 6 أنواع مختلفة من السرطان مع 22 و48 مجموعة بيانات CODEX وsnRNA المتطابقة على التوالي. تشمل البيانات 54 BRCA، 30 CRC، 23 PDAC، 12 RCC، 5 UCEC و7 CHOL. أسفل، سير العمل لإنشاء المناطق الدقيقة للورم، واستنتاج النسخ الفرعية للورم المكانية وإجراء التحليلات اللاحقة. بناءً على توزيع مناطق الورم، قمنا بفصل العينات إلى مجموعات متميزة مكانيًا ومجموعات مائية. شملت التحليلات تحليل تطور النسخ الفرعية للورم، التشابه النسخي وتفاعلات TME القائمة على الطبقات، بناء نمط نمو الورم، وإعادة بناء الأحياء ثلاثية الأبعاد متعددة المقاطع باستخدام كود مخصص (الطرق). ب، مخطط نظرة عامة دائري على مستوى كتلة الأنسجة. يشير ارتفاع نتوء الحلقة الخارجية إلى عدد المقاطع لكل كتلة نسيجية. تُظهر الإضافة في أعلى اليمين أسطورة،
مع أرقام في فقاعات تشير إلى عدد كتل الأنسجة مع عدد معين من المقاطع المتسلسلة. تشير الحلقات المتعاقبة إلى نوع السرطان مع التعليقات التوضيحية لعدد المقاطع (كتلة نسيجية)، نوع الورم، نوع الاختبار وتعيين المجموعة المكانية. ج، توزيع المناطق الدقيقة في المجموعة المتميزة مكانيًا على مستوى المقطع ملونًا حسب نوع السرطان (يسار)، مجموعة حجم المنطقة الدقيقة (وسط) والأولية مقابل النقيلة (يمين). تشير كل دائرة إلى منطقة دقيقة واحدة. يمثل حجم كل دائرة حجم المنطقة الدقيقة. د، نسبة بقع الورم مقابل بقع المناعية الدعامية عبر أنواع السرطان للمجموعة بأكملها على مستوى المقطع. تمثل كل نقطة عينة، ملونة حسب النوع: أولية ( مقاطع من 60 حالة) أو نقيلية ( مقاطع من 16 حالة). يمثل الخط المركزي في مخطط الصندوق الوسيط، مع الحواف السفلية والعلوية تشير إلى الربع الأول والثالث. تمتد الشعيرات إلى أعلى وأدنى القيم ضمن 1.5 مرة من النطاق بين الربعين (IQR) من الحواف.
بواسطة المكونات الدعامية، والتي نسميها ‘المناطق الدقيقة للورم’. نوضح أن هناك نسخ جينية متميزة داخل هذه المناطق الدقيقة مع تغيرات محددة في عدد النسخ (CNVs) ومع نشاط مختلف داخل المسارات السرطانية، وخاصة مسار MYC. من الجدير بالذكر أننا نظهر أن التفاعلات المناعية-الورمية والدعامية-الورمية تختلف بين هذه المناطق الورمية. بالإضافة إلى ذلك، تسلط دراستنا الضوء على الخصائص المتميزة بين الأورام الأولية والنقيلية، بما في ذلك الاختلافات في أنماط نمو الورم والملفات النسخية. لدعم نتائجنا التي تظهر أن التجمعات المناعية في TME تحيط بمناطق ورمية معينة مكانيًا، نستخدم CODEX وأداة إعادة بناء ثلاثية الأبعاد متعددة النماذج المدربة على مقاطع ST المجاورة. تؤكد النتائج على الاتصال بين النسخ الفرعية والمناطق الدقيقة في مقاطع مختلفة ضمن الفضاء ثلاثي الأبعاد. تبرز هذه الإعادات
المناطق والتفاعلات بين الورم والمناعة. بشكل عام، يوفر هذا النهج المكاني للأوميكس رؤى أعمق في التطور الكلوني والتمييزات المكانية عبر ستة أنواع مختلفة من الأورام الصلبة، مما يمهد الطريق لمزيد من التقدم في فهم آليات مقاومة العلاج في السرطان.
المناطق الدقيقة المكانية عبر السرطانات
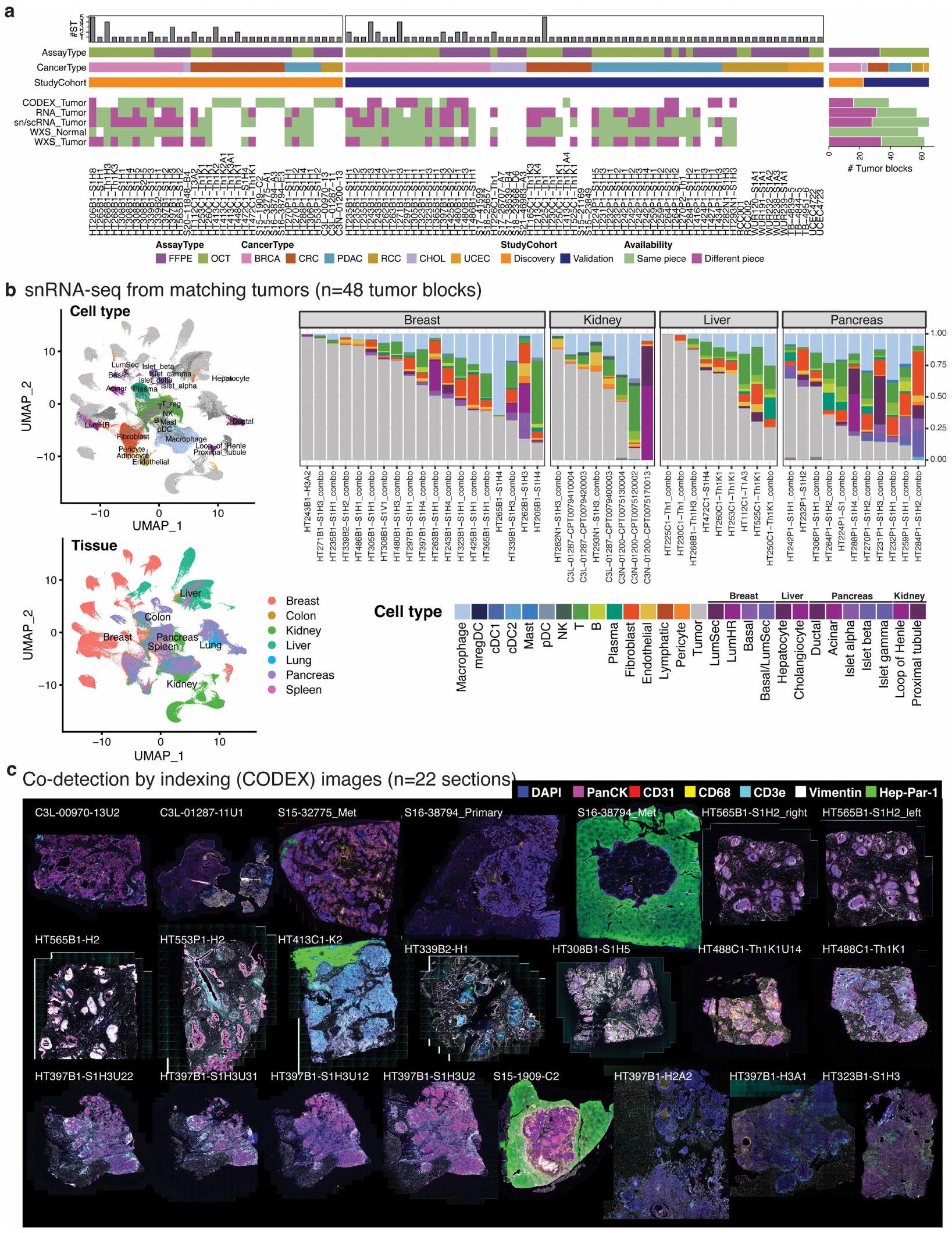
قمنا بتوصيف 131 مقطع ورمي مع بيانات ST من 98 كتلة تغطي 6 أنواع من السرطان كجزء من شبكة أطلس الأورام البشرية التابعة للمعهد الوطني للسرطان: 54 BRCA، 30 CRC، 23 PDAC، 12 RCC، 5 UCEC و7 CHOL (الشكل 1أ، الشكل البياني الممتد 1 والجدول التكميلي 1) من 78 حالة ( و6 حالات، على التوالي). كانت ثلاث عينات من RCC من العيادة
تحليل بروتينات الأورام. باستخدام صبغة الهيماتوكسيلين والإيوسين (H&E) والملفات النسخية، حددنا الميكرو مناطق الورم ككتل خلايا سرطانية متميزة مكانياً مفصولة بمناطق دعامية (الطرق) وخصصنا نقاط فيزيوم كخبيثة أو غير خبيثة. استخدمنا مجموعة أدوات Morph بعد ذلك لتحديد حدود الورم، وتحديد المسافات بين النقاط والحدود وبناء طبقات من النقاط، مع فهرسة أعماقها إلى حدود الورم (الطرق). اخترنا 50 مقطعًا مع مناطق ورمية متعددة كـ ‘مجموعة متميزة مكانياً’ (الشكل البياني الممتد 2) و82 عينة مع مناطق ورمية منتشرة كـ ‘مجموعة منتشرة مكانياً’ (الشكل البياني الممتد 3). كما قمنا بإنتاج مقاطع متسلسلة من 15 كتلة نسيج ورمي، مما أسفر عن 48 مقطعًا مناسبًا لإعادة بناء الورم ثلاثي الأبعاد (الشكل 1b والشكل البياني الممتد 4a).
استنادًا إلى المساحة المقدرة لكل ميكرو منطقة ورمية (الطرق)، قمنا بتصنيف أحجام الميكرو مناطق إلى صغيرة (بقع أو )، متوسط ( بقع أو ) أو كبير ( بقع أو (الشكل 1د). كان لدى CRC مناطق ميكروية أكبر (متوسط 2.9 طبقات) مقارنة بـ BRCA (متوسط 2.1 طبقات؛ معدل الاكتشاف الخاطئويلش-اختبار) و PDAC (متوسط 2.37 طبقة؛ FDRويلش-اختبار). على العكس، كانت أعماق الميكرومنطقة BRCA و PDAC غير قابلة للتمييز إحصائيًا (ويلش-اختبار). كان لدى RCC أعلى نسبة ورم، في حين كان لدى PDAC أدنى نسبة (الشكل 1c)، وهو ما يرجع على الأرجح إلى المحتوى الأعلى من النسيج الداعم وكثافة خلايا الورم الأقل في PDAC.، مما يؤدي بدوره إلى أحجام ميكرو مناطق أصغر. كانت الأورام الأولية عمومًا تحتوي على المزيد من الميكرو مناطق الصغيرة (66.3%) مقارنة بالانتقالات (40.2%)، التي كانت تحتوي على المزيد من الميكرو مناطق المتوسطة الحجم (43.2%) (الشكل 4b، c من البيانات الموسعة). كانت الميكرو مناطق الأكبر حجمًا موجودة بشكل رئيسي في الانتقالات (16.3% مقارنة بـ 3.2% في الأورام الأولية)، التي كانت تحتوي أيضًا على ميكرو مناطق أعمق من الأورام الأولية (3.4 مقارنة بـ 1.9 طبقات؛ ويلش-اختبار FDR ). هذا الاختلاف استمر في أقسام BRCA فقط (FDR )، حيث كانت لدينا بيانات لكل من النقائل (5 مقاطع، 44 ميكرومنطقة، عمق متوسط 4.2) والأورام الأولية (8 مقاطع، 222 ميكرومنطقة، عمق متوسط 1.7). تشير هذه النتائج إلى وجود نمو متباين بين الأورام الأولية والنقيلية وتأثير بيئة الورم المحدد بالعضو على نمو وتنظيم الميكرومنطقة. تشمل الأمثلة عينات HT268B1-Th1H3 (نقيلة كبدية من BRCA) وHT260C1-Th1K1U1 (نقيلة كبدية من CRC)، التي كانت تحتوي على مناطق كبيرة تشغل (400-500 نقطة)، في حين أن العينة HT27OP1-H2U1 (PDAC) كانت أصغر (متوسط منأو 26 موقعًا) ولكن عدد أكبر من الميكرو مناطق ( ) (الشكل البياني الموسعفي المجموعة المكانية المتميزة، كانت العينات ذات أعلى عدد من الميكروregions من كتل BRCA الغنية بسرطان القنوات في الموقع (DCIS) (HT397B1-S1H2، HT339B1-S1H3 و HT206B1-S1؛ الشكل 4d من البيانات الموسعة). قد تعكس هذه التوزيعة ميل خلايا سرطان القنوات للنمو على طول القناة الإفرازية في كلا العضوين، مما قد يفسر ملاحظتنا للعديد من المناطق الصغيرة.
التطور الكلوني البؤري في الميكرو مناطق
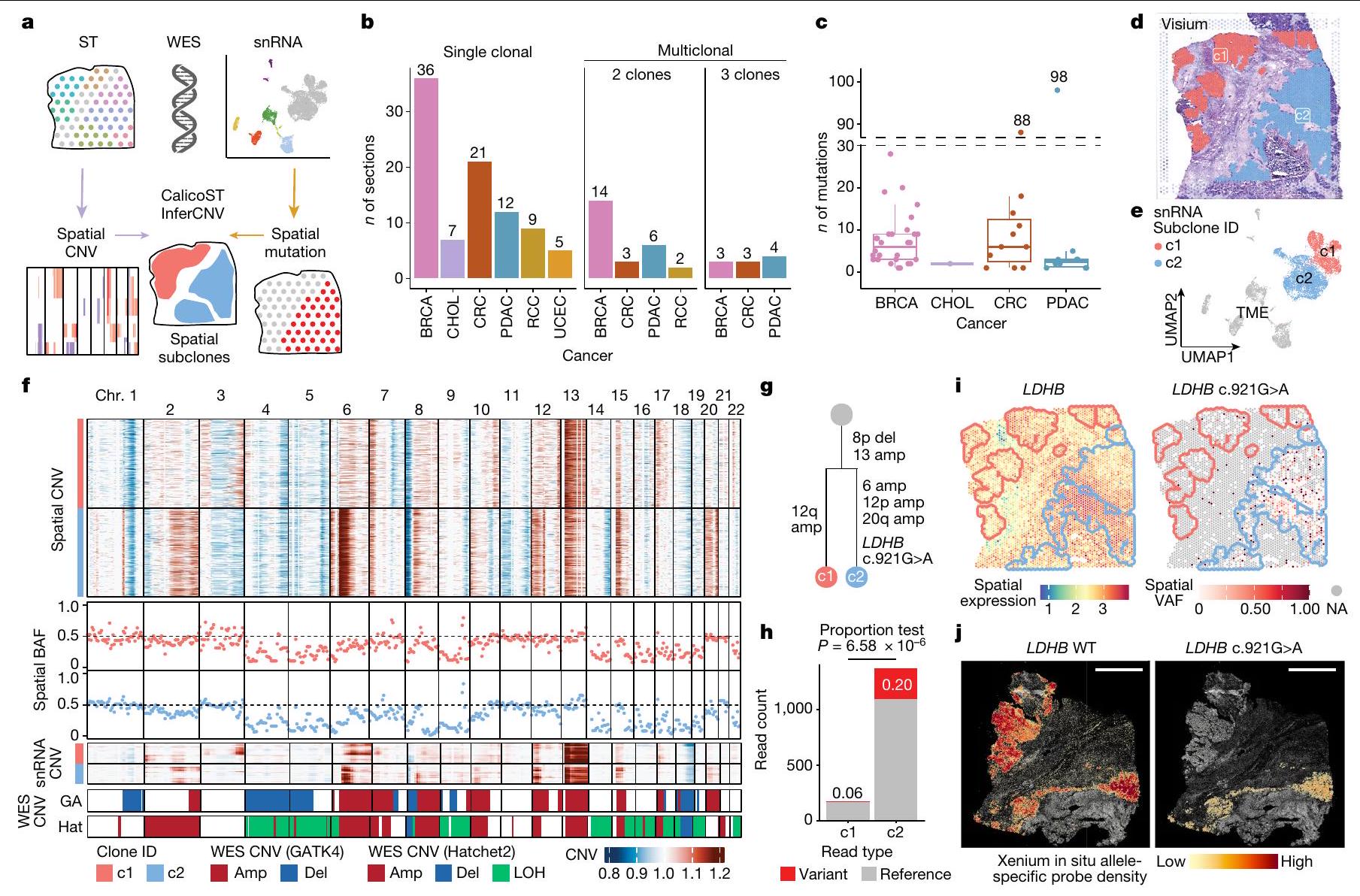
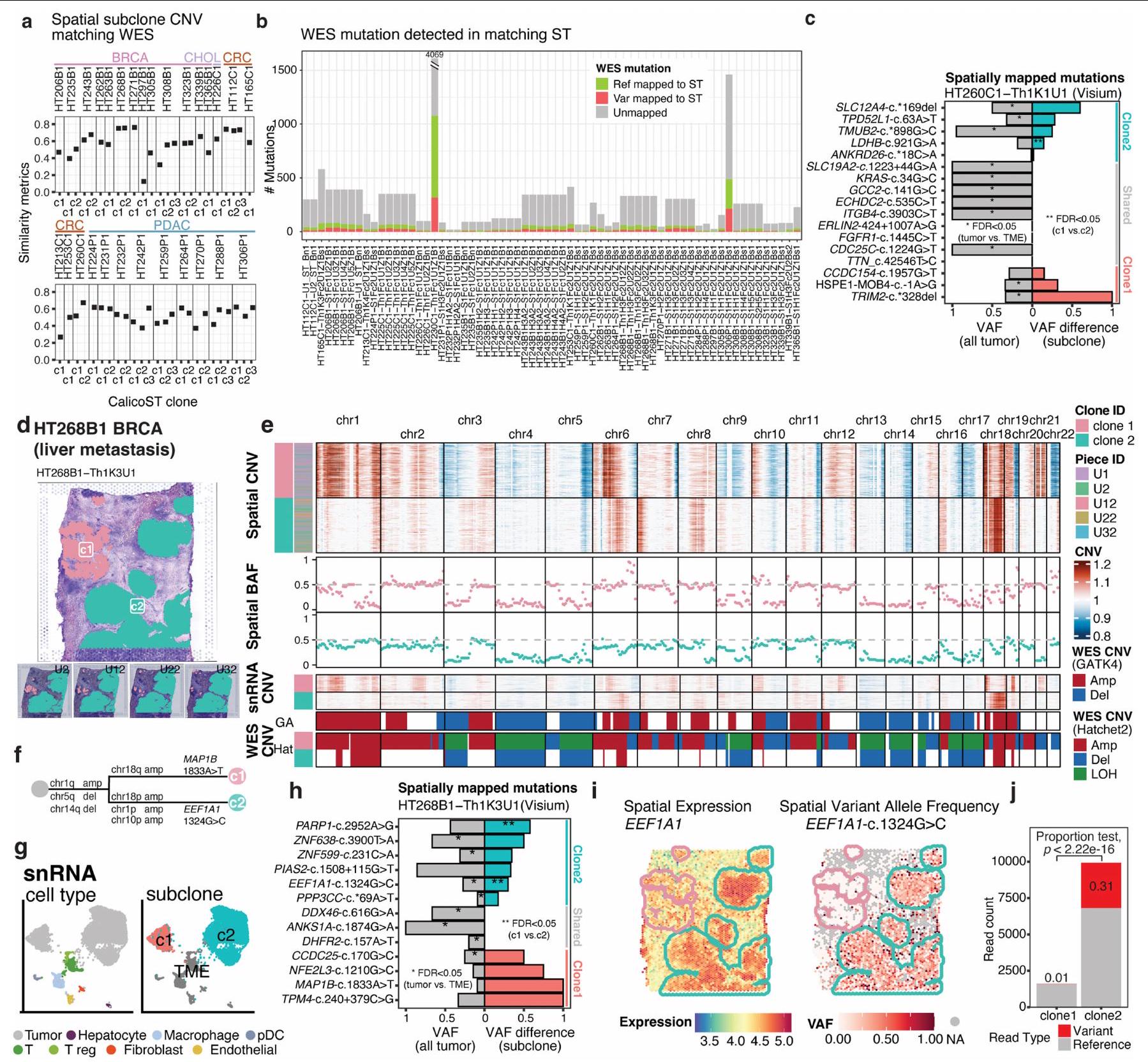
قمنا بتحديد التغيرات العددية في الجينوم على مستوى الجينوم باستخدام CalicoST وInferCNV (الشكل التوضيحي التكميلي 1 والطرق)، مع اختيار الأحداث الموثوقة في كل ميكرو منطقة من خلال تصفية تلك الموجودة في بيانات تسلسل الإكسوم الكامل المتطابقة (الجدول التكميلي 2). ثم قمنا بتجميع الميكرو مناطق إلى تحت مجموعات مكانية بناءً على تشابه التغيرات العددية (الشكل 2a والطرق). اكتشفنا التغيرات العددية المكانية في 125 من أصل 131 مقطعًا، حيث لاحظنا 1-3 تحت مجموعات لكل مقطع (حواليمع نسخة واحدة،مع 2 سلالات فرعية ومع 3 تحت النسخ) (الشكل 2ب). يمكن أن يتكون النسخة الواحدة من تحت النسخ التي لا يمكن لوظيفتنا اكتشافها، مثل تحت النسخ المختلطة في نفس الميكرو منطقة وتحت النسخ المتميزة بالتغيرات الجينية التي لا تغطيها النسخة الجينية لفيزيوم. ضمن هذه القيود، حددنا عدة تحت نسخ مكانية في مقاطع من 4 أنواع من السرطان: BRCA (17 مقطعًا)، PDAC (10 مقاطع)، CRC (6 مقاطع) و RCC (2 مقاطع). تم مقارنة ملفات CNV لتحت النسخ المكانية مع بيانات WES المطابقة وأظهرت تشابهًا عاليًا على مستوى الجينوم (الطرق والشكل الممتد 5أ).
قمنا أيضًا برسم الطفرات الجسدية على مقاطع ST المدمجة في درجة حرارة القطع المثلى (OCT)، حيث أظهرت كل مقطع من 1 إلى 98 طفرة تم رسمها بشكل محدد في مناطق الورم (الشكل 2c والشكل التمديدي 5b).
عينة HT260C1 (نقائل الكبد CRC) احتوت على 12 منطقة ميكروية للورم، والتي تم رسمها إلى 2 من تحت الأنماط المكانية (الشكل 2d). تم تحديد وتأكيد كلا الحدثين النسليين (تكبير الكروموسوم 13 وحذف الكروموسوم 8p)، بالإضافة إلى عدة أحداث تحت نسيلية (تكبيرات في الكروموسومات 6p و12p و20q في النمط c2، وتكبير الكروموسوم 12q في النمط c1)، مع بيانات تسلسل RNA أحادي النواة المتطابقة (snRNA-seq) واستنتاج CNV القائم على WES (الشكل 2e، f). على الرغم من أن علم الأنسجة أشار إلى وجود فصل ليفي بين النمطين الفرعيين، إلا أن العديد من CNVs النسيلية المشتركة اقترحت أصلًا مشتركًا (الشكل 2g). قدمت المتغيرات الجسدية من النسخ مزيدًا من الأدلة الداعمة لنسيلية الورم. بالإضافة إلى ذلك، تم رسم 17 طفرة جسدية قائمة على WES إلى ST (الشكل 5c من البيانات الموسعة). أظهرت عدة طفرات تباينًا في تردد الأليل المتغير (VAF) في مناطق الورم مقارنة بالمناطق الطبيعية (الشكل 5c من البيانات الموسعة، اليسار) وتباينات VAF بين النمطين الفرعيين (الشكل 5c من البيانات الموسعة، اليمين). أظهر كلا النمطين الفرعيين تعبير، وVAF للطفرات ج. كان أعلى بشكل ملحوظ في السلالة الفرعية c2 مقارنة بـ c1 (الشكل 2h,i؛اختبار النسبة ذو الجانبين). باستخدام بيانات زينيوم، قمنا بتحليل كل منالنمط البري (WT) و c.الأليلات في قسم الورم المتطابق، الأليل WT في كلا السلالات الفرعية والأليل الطافر في السلالة الفرعية c2 (الشكل 2j). انحرفت السلالة الفرعية c2 عن c1، مع اكتساب تغييرات جينية فريدة في كل من CNV والطفرات. وبالمثل، أظهر عينة نقيلة كبدية من BRCA، HT268B1، سلالتين فرعيتين متميزتين مكّنت من دعمها بواسطة بيانات snRNA-seq المتطابقة، مع اختلافات في CNV على مستوى الذراع الكروموسومي (الشكل التمديدي 5d-g) وطفرات فرعية (الشكل التمديدي 5h). على سبيل المثال، تم التعبير عن EEF1A1 في كلا السلالتين الفرعيتين، في حين كانت الطفرة EEF1A1 1324G.تمت ملاحظته بشكل خاص في السلالة الفرعية c2 (اختبار النسبة FDR < 0.05؛ الشكل التوضيحي الممتد 5i،j).
عينة PDAC الأولية، على الرغم من مناطقها الدقيقة الأصغر، احتوت أيضًا على عدة تحت سلالات مكانية. أظهرت العينة HT270P1 ثلاث تحت سلالات عبر قسمين من كتلتين ورميتين مع الحفاظ على OCT وFFPE، على التوالي (الشكل التوضيحي التكميلي 2a). أظهرت معظم حالات BRCA الأولية أن جميع الميكرو مناطق الورم تنتمي إلى سلالة جينية واحدة، مثل HT206B1 عبر خمسة أقسام متسلسلة (الشكل التوضيحي التكميلي 1b). في سبع من تسع حالات، شملت سلالة واحدة كل من DCIS و IDC، مما أشار إلى أن الانتقال بينهما يحدث دون تغييرات كبيرة في عدد النسخ (الشكل التوضيحي التكميلي 2b). ومع ذلك، أظهرت العينة BRCA الأولية HT397B1 ثلاث تحت سلالات مكانية عبر أربعة أقسام من كتلتين ورميتين (الشكل التوضيحي التكميلي 2a). أظهرت اثنتان من السلالات كل من أشكال DCIS و IDC، بينما أظهرت السلالة 3 شكل IDC فقط، مما أشار إلى انتقال متوازي من DCIS إلى IDC بين تحت السلالات المكانية (الشكل التوضيحي التكميلي 2b).
تكشف تعدد أشكال النوكليوتيدات المفردة عن فقدان التغايرية المحايدة لعدد النسخ الذي يتم تجاهله فقط من خلال استنتاج عمق القراءة. عينة من نقائل الكبد CRC (HT112C1-Th1) مع انحراف قوي في تردد الأليل B في الكروموسوم 21 المحايد لعدد النسخ تشير إلى تغيير وراثي تحت سلالي في السلالة A (الشكل التوضيحي 2a). تشير هذه الملاحظات مجتمعة إلى أن السلالات الفرعية المكانية داخل قسم الورم ربما تنشأ من سلف مشترك.
التغيرات الجينية تؤدي إلى تفاوت الأورام
لتخفيف تأثير خلايا المناعة والخلايا الداعمة المتسللة، قمنا أولاً باستبعاد جينات علامات نوع الخلايا غير الخبيثة المحددة للأعضاء باستخدام بيانات تسلسل RNA أحادي الخلية (الطرق). بعد ذلك، استخدمنا طريقة قائمة على الانتروبيا، ROGUE، لتحليل التباين النسخي بين الميكرو مناطق الورم (الطرق). كشفت تحليلاتنا أن سرطان البنكرياس القنوي (PDAC) كان لديه أعلى تباين. )، في حين أن BRCA،
الشكل 2 | التغير الجيني يكشف عن التطور الكلوني المكاني. أ، ملخص لعملية العمل لتحديد تحت الأنواع المكانية. ب، أعداد النسخ المكانية المكتشفة في كل قسم ملخصة حسب نوع السرطان. تم ملاحظة أقسام متعددة النسخ (2 أو 3 تحت أنواع مكانية) في BRCA وCRC وPDAC وRCC في هذه المجموعة.أقسام من 74 حالة تم اكتشاف CNV فيها). ج، رسم خرائط الطفرات الجسدية المكانية لكل قسم تم اكتشافه في بيانات OCT ST. الطفرات الجسدية المستمدة من WES التي تظهر VAF أعلى بشكل ملحوظ في مناطق الورم مقارنة بالمناطق غير الورمية (اختبار ثنائي الحدين، FDR<0.05) موضحة (مقاطع OCT من 29 حالة). يمثل الخط المركزي في الرسم البياني الصندوقي الوسيط، مع وجود الحواف السفلية والعلوية التي تشير إلى الربع الأول والثالث. تمتد الشعيرات إلى أعلى وأدنى القيم ضمن 1.5 مرة من IQR من الحواف. د، هـ، عينة من نقائل سرطان القولون والمستقيم في الكبد (HT260C1) تحتوي على 12 منطقة ميكروية للورم تظهر 2 من تحت الورم، c1 و c2، مفصولة مكانيًا (د)، مع دعم من بيانات snRNA-seq المتطابقة (هـ). ف، خريطة حرارية لتقديرات CNVs الجسدية لكل نقطة تظهر كلا من الأحداث المشتركة والفريدة من نوعها في CNV بين النمطين الفرعيين المكانيين. تُظهر ترددات الأليل B (BAFs) في كل نمط فرعي مكاني من نفس النافذة الجينومية في المسارات الوسطى، وحالات CNV المستنتجة من snRNA وWES باستخدام GATK4 (GA) وHatchet2 (Hat) موضحة في المسارات السفلية. g، العلاقة النشوء والتطور المتوقعة لـ c 1 ونسبة التغير الجيني ج. أعلى بشكل ملحوظ في c2 مقارنة بـ c1 (اختبار النسبة ذو الجانبين). i، الطفرة تحت السلالية ج. يتم اكتشافه بشكل فريد في النسخة 2 في النسخ الجزيئي المكاني، بينماالتعبير موجود في كلا النسخ الفرعية المكانية. يظهر كثافة المجس المحدد للأليل في Xenium الأليل البري.في كل من السلالات الفرعية والأليل الطافر بشكل فريد في السلالة الفرعية اليمنى (c2). قضبان القياس، 2 مم. amp، تضخيم؛ del، حذف؛ LOH، فقدان التغايرية؛ NA، غير قابل للتطبيق؛ UMAP، تقريب وطرح متعدد الأبعاد الموحد.
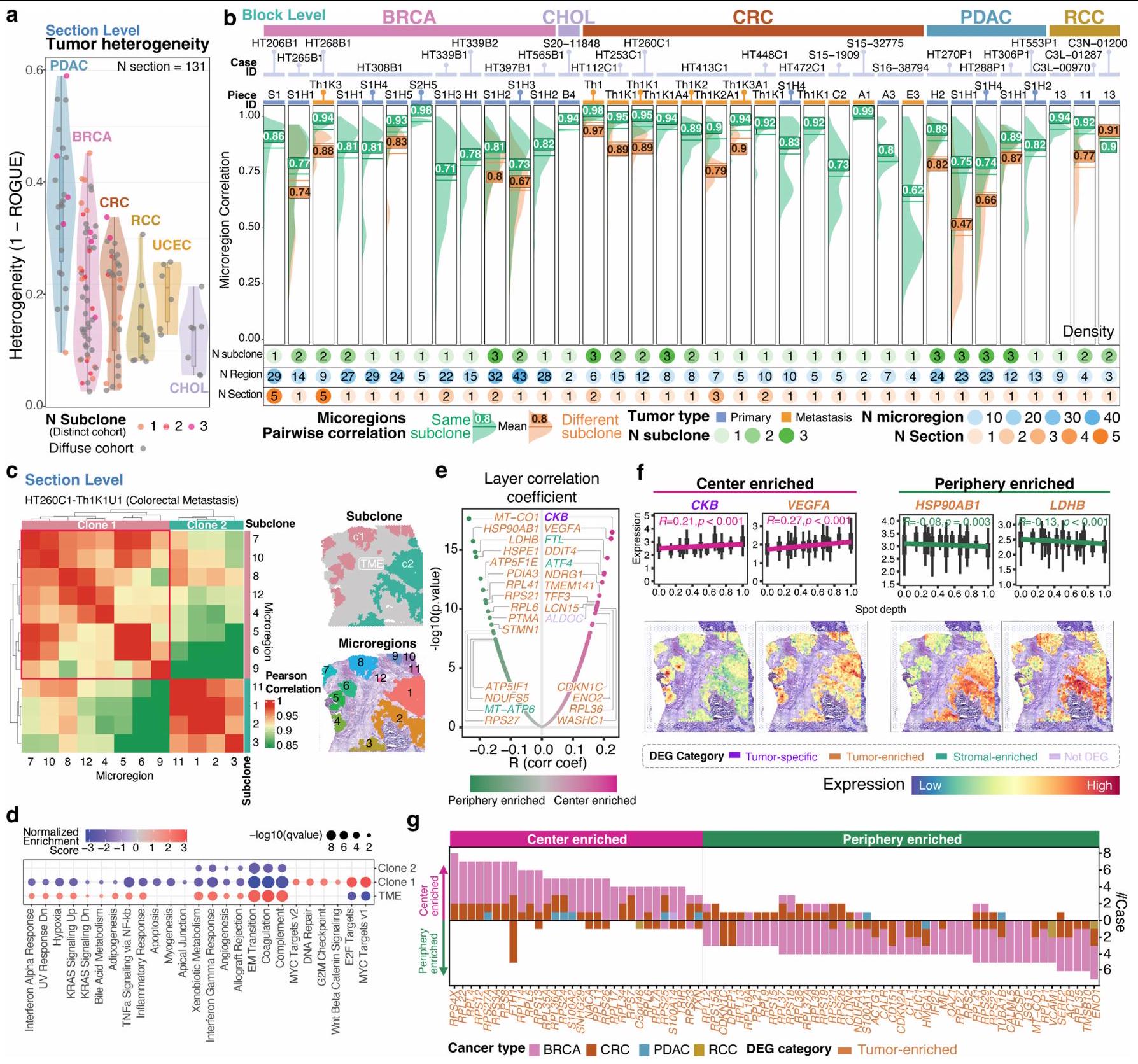
كان لدى CRC وRCC وUCEC مستويات معتدلة (0.05-0.45)، وكان لدى CHOL أدنى المستويات (<0.2) (الشكل 6a من البيانات الموسعة). للتحقيق بشكل أعمق في تأثير التغيرات الجينية والتكيفات البيئية الدقيقة على الملفات التعبيرية، قمنا بتقييم التشابه التعبيري باستخدام ارتباط بيرسون الثنائي بين الميكرو مناطق الورم (الطرق)، مقارنةً داخل وبين مختلف النسخ الجينية. لاحظنا تشابهًا أكبر داخل النسخ الفرعية (الشكل 6b من البيانات الموسعة، باللون الأخضر) مقارنةً بين النسخ الفرعية المختلفة (الشكل 6b من البيانات الموسعة، باللون البرتقالي) عبر جميع العينات من BRCA وCRC وPDAC وRCC (الشكل 6b من البيانات الموسعة والطرق). ظل هذا النمط ثابتًا في كل من العينات الأولية والعينات النقيليّة في BRCA وCRC، مما يبرز الدور المركزي للتكوين الجيني في تشكيل التشابهات التعبيرية عبر الميكرو مناطق.
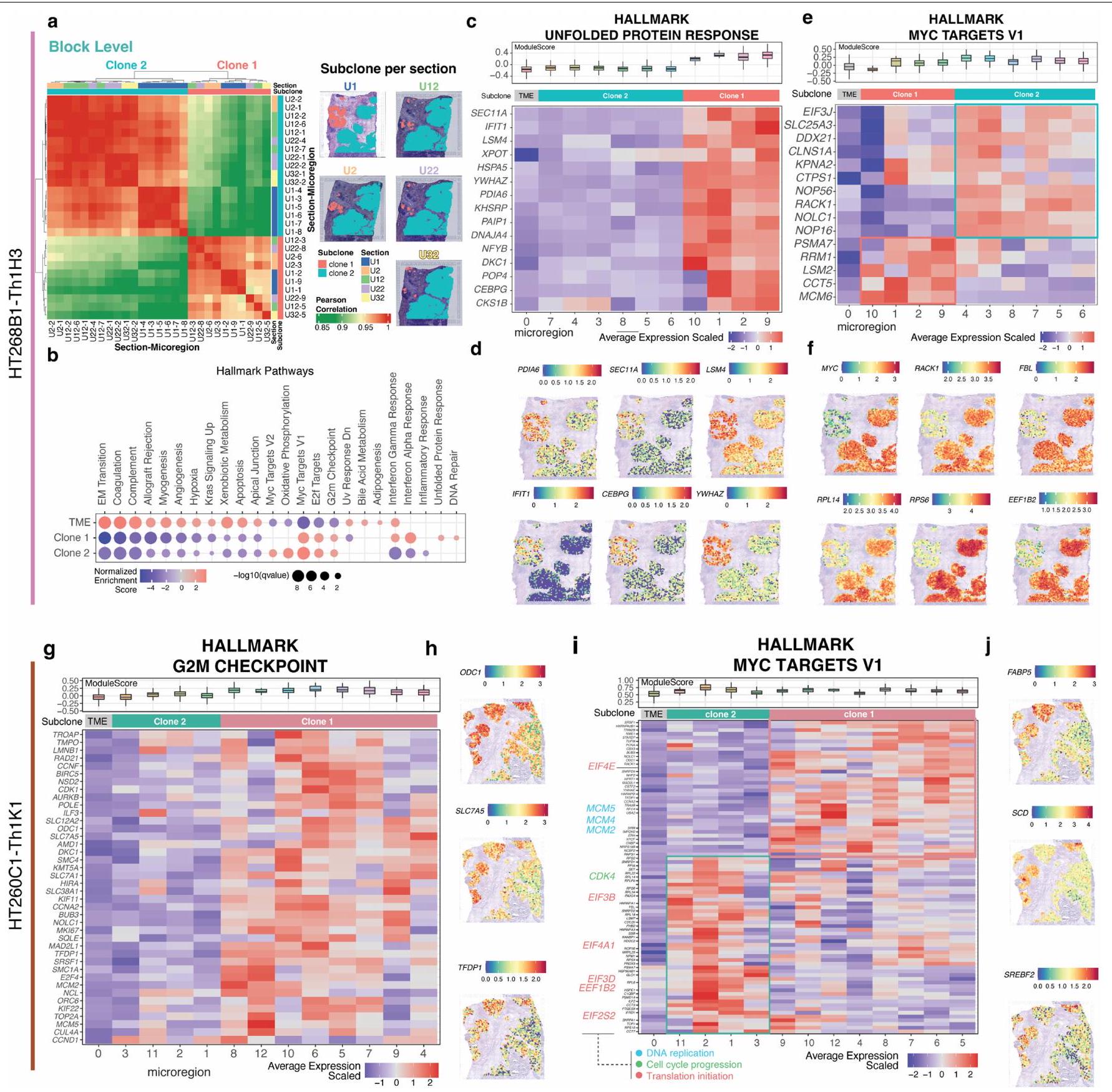
لفهم تفعيل المسارات المسرطنة عبر الميكرو مناطق، قمنا بإجراء تحليل إثراء مجموعة الجينات (GSEA) باستخدام الجينات المعبر عنها بشكل مختلف (DEGs) بين الميكرو مناطق الورم ومناطق السدى (الطرق). حدد تحليلنا مسارات شائعة مثل MYC وE2F المشتركة عبر الميكرو مناطق، ولكنها متميزة. مسارات، مثل استجابة البروتين غير المطوي، كانت محددة لبعض الميكرو مناطق في العينة HT268B1 (نقائل BRCA) (الشكل التمديدي 7a-f). هنا، كانت التغيرات الجينية تتحكم في الملف التعبيري، بينما في عينات أخرى، نشأت اختلافات طفيفة من البيئة الميكروية المحلية أو أحداث جينية غير مكتشفة. على سبيل المثال، أظهرت العينة HT260C1 وجود فرعين فرعيين بملفات عدد نسخ متميزة (الشكل 2d)، بينما كانت الميكرو مناطق داخل الفرع 1 تظهر تشابهات تعبيرية متباينة، على الرغم من وجود CNVs مشابهة (الشكل التمديدي 6c، المشار إليها بالصناديق المنقطة).
عند تطبيق GSEA لتحديد المسارات الخاصة بالنسائل الفرعية، وجدنا مستويات تعبير ميكروإقليمية متباينة ضمن مسار نقطة التفتيش G2M في النسيلة 1 (البيانات الموسعة الأشكال 6d و7g,h، المشار إليها بالصناديق المنقطة) في العينة HT260C1. تم عكس هذا النمط في MYC لكل من عينات النقائل HT260C1 وHT268B1، مع مجموعات متميزة من جينات MYC السفلية بين النسائل الورمية (البيانات الموسعة الشكل 7e,f,i,j والشكل التكميلي 3). تؤكد هذه النتيجة على تعقيد هذا المسار، الذي يتم تنظيمه بشكل غير طبيعي في السرطان ويؤثر على العديد من العمليات المسرطنة.النسخة 2 من HT260C1 كانت زيادة في معقد بدء الترجمة (عائلة eIF) وتقدم G1S (CDK4)، في حين أن النسخة 1 كانت تحتوي على جينات تكرار الحمض النووي (عائلة MCM) (الشكل 7i من البيانات الموسعة).
اختبرت اختبارات الإثراء باستخدام Enrichr مع مجموعة بيانات اضطراب الأدوية LINCS L1000 (الطرق) استجابات علاجية متباينة للنسخ الفرعية (الشكل التوضيحي التكميلي 4). على سبيل المثال، يجب أن تستجيب جميع النسخ الفرعية في HT397B1-S1H3 و HT112C1-Th1 لمثبط المTOR تورين-2، ولكن فقط النسخة 1، وليس النسخة 2، في HT268B1-Th1K3 استجابت. هذه التباينات تبرز أهمية تحليل النسخ الفرعية المكانية.
المسارات الخلوية في مركز الورم وحافته
قمنا بمزيد من التحقيق لمعرفة ما إذا كانت هناك برامج نسخ مختلفة موجودة في مراكز الميكرو منطقة الورم (النوى) مقارنةً بحوافها (الواجهات بين الورم والـ TME-ستروما). لهذا الغرض، استخدمنا Morph لقياس المسافة من كل بقعة ورم إلى أقرب حدود ورم-TME (الطرق والبيانات الموسعة الشكل 8a). العلاقة بين عمق الطبقة الكلي لميكرو المنطقة وحجمها (المساحة الكلية المقاسة بعدد البقع المشغولة) تصف الشكل العام للمنطقة (البيانات الموسعة الشكل 8b). تشير الخط المرجعي المتقطع إلى علاقة العمق-الحجم للمناطق الدائرية المثالية. بالنسبة لجميع أنواع السرطان الخمسة في المجموعة المكانية المتميزة ( )، كانت المناطق الأصغر تميل إلى إظهار أشكال قريبة من الدائرة. ولكن مع زيادة عمق الطبقة، كانت المناطق تميل إلى الانحراف عن شكلها الدائري، وظهرت واجهة موسعة بين خلايا الورم وخلايا غير الورم.
يمكن أيضًا وصف ملفات التعبير الجيني المكاني من حيث عمق الطبقة. لكل نقطة مخصصة لطبقة معينة، قمنا بشكل مستقل بإجراء تحليل انحدار خطي بين التعبير الجيني وعمق النقطة لكل جين. تقديرات نقاء الورم باستخدام طرق RCTD.أو تقديرتم تضمينها كمتغير مشترك لضبط الانخفاضات المحتملة في النقاء نحو حواف القسم (الطرق). تشير العلاقة الإيجابية مع العمق إلى زيادة التعبير الجيني نحو مركز الورم والعكس صحيح. في عينة نقائل CRC HT260C1، كانت الجينات الأكثر غنى في المركز الأعلى ( و تم عرض الجينات الغنية في المركز (HSP90AB1 و LDHB) مع خط الانحدار ونمط التعبير المكاني في الشكل التمديدي 6e و 6f. كما تم عرض الجينات الغنية في المركز العلوي (NDRG1 و S100A2 و CA9) والجينات الغنية في المحيط (TUBA1B و NDUFA4 و TOMM4O) لـ HT206C1 في الشكل التمديدي 8c-e. تم دعم هذه النتائج ببيانات snRNA-seq من عينات الأورام المتطابقة، والتي توضح أن الجينات المشتركة الرئيسية يتم التعبير عنها بشكل رئيسي بواسطة الخلايا الخبيثة، مع مساهمات صغيرة من خلايا المناعة والستروما (الشكل التكميلية 5a-d).
بعد ذلك، حددنا الجينات التي كانت غنية بشكل متكرر في مراكز الأورام وفي الأطراف عبر الحالات (الشكل 6g من البيانات الموسعة). كانت الجينات الأكثر شيوعًا الغنية في المراكز متورطة في تجميع الريبوسوم (جينات عائلة RPL و RPS مثل RPS4X و RPL22 و RPL4)، إلى جانب جينات مثلorf46 (المرجع 21) و RNA غير المشفر الطويل SNHG29 (المرجع 22)، المرتبطين بنمو الأورام في أنواع مختلفة من السرطان. بالمقابل، كانت محيط الورم غنيًا بالجينات التالية: مجموعة مختلفة من جينات الريبوسوم RPL و RPS (RPL35، RPLP1 و RPS27)؛ ENO1، وهو بروتين سرطاني متعدد الوظائف يشارك في التحلل السكري، والغزو، وكبت المناعة.; TMSB10، الذي يعزز التكاثر والغزو في ; و , مما يحفز تكوين البلعميات M2 . تشير هذه العمليات البيولوجية التفاضلية إلى أن الخلايا الخبيثة في المركز تخضع بنشاط لترجمة البروتين، بينما تلك الموجودة على الحواف تشارك في هجرة الورم وتعديل المناعة، متداخلة مع مكونات المناعة والستروما.
تفاعلات الورم-TME المحددة بالنمط النسلي
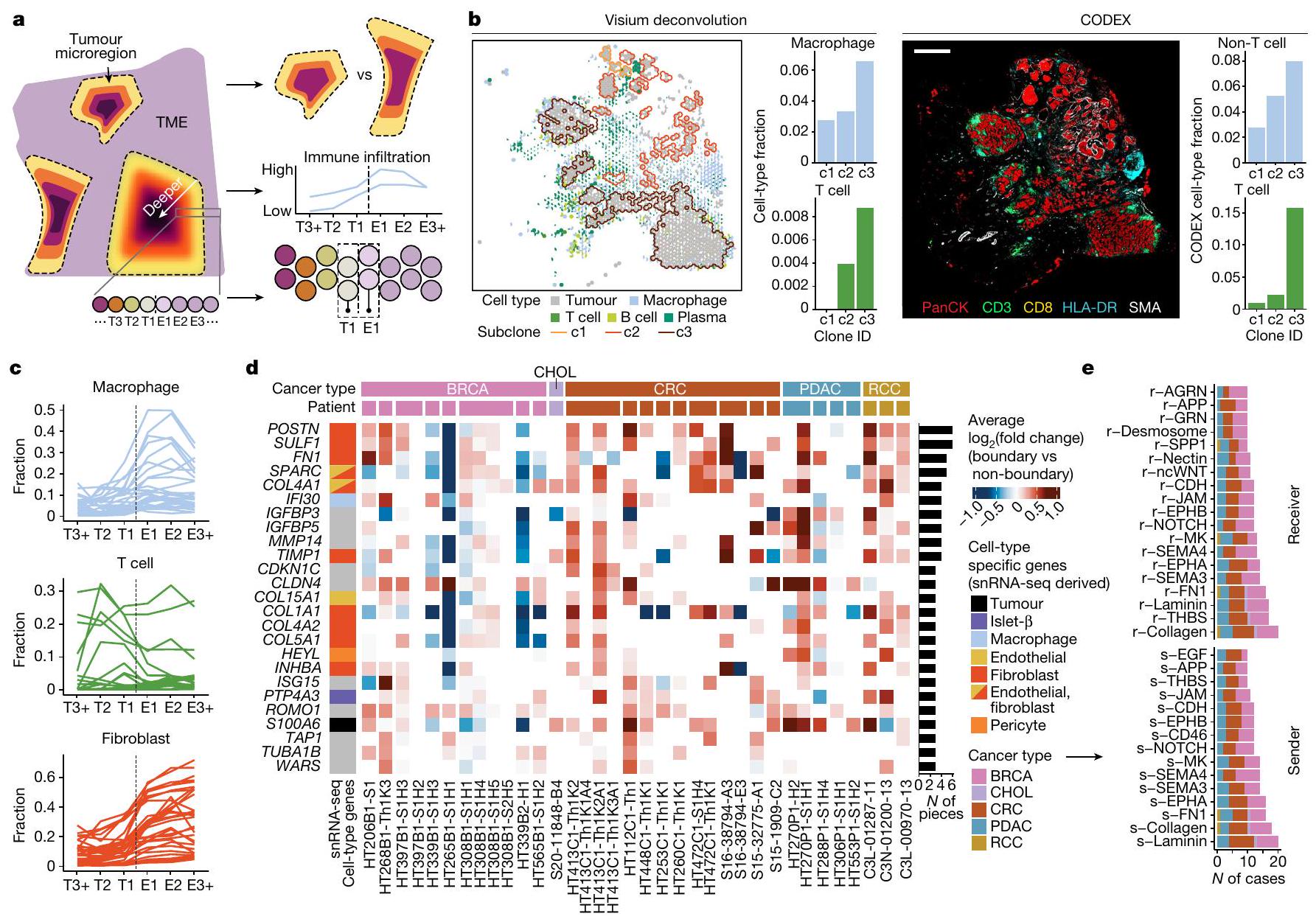
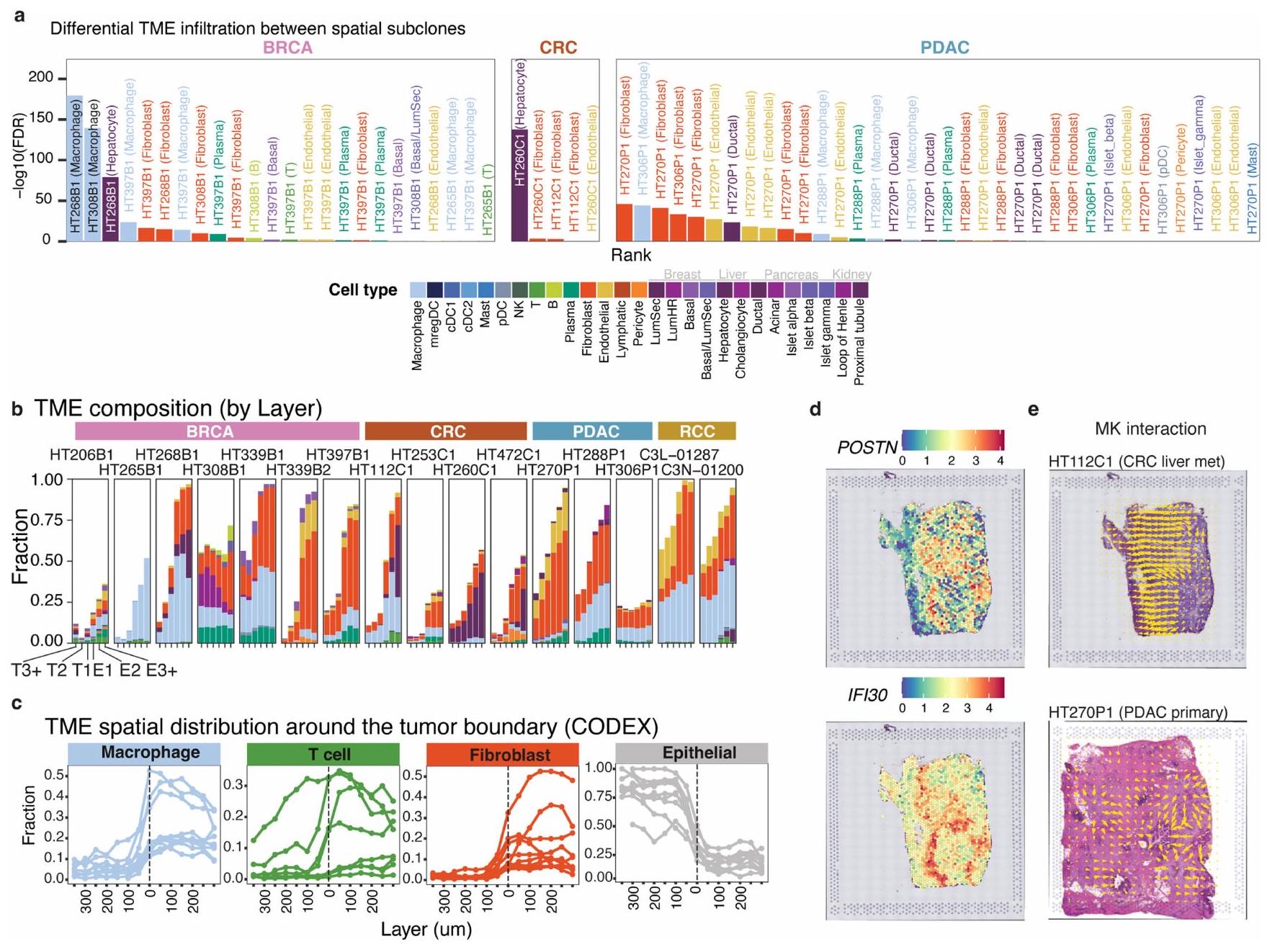
لتحقيق في تكوين TME في مناطق حدود الورم، قمنا بفحص التسلل التفاضلي للخلايا غير الورمية بين الأنماط الفرعية المكانية للورم، وموقع هذا التسلل والجينات
وCCIs الغنية في المناطق الحدودية (الشكل 3أ). استخدمنا بيانات snRNA-seq المطابقة كمرجع لتفكيك نوع الخلايا على مستوى النقاط وأجرينا تحليل التسلل التفاضلي بين جميع الأنماط الفرعية المكانية لنفس العينة. كانت الأنواع الخلوية التفاضلية العليا من حيث التسلل تشمل البلعميات في BRCA، والخلايا الكبدية في CRC والليفية في PDAC (بيانات موسعة الشكل 9أ). عينة BRCA الأساسية، HT397B1، مع ثلاثة أنماط فرعية مكانية أظهرت تسللاً تفاضلياً في كل منالخلايا والبلعميات، مع النمط الفرعييظهر أكبر نسبة من كلاهما (الشكل 3ب). أكدت بيانات CODEX زيادة مستوى علامات خلايا T (CD3 وCD8) وصبغة علامة المناعة غير T (HLA-DR) في النمط الفرعي c3.
استخدمنا التعيينات الطبقية الموصوفة أعلاه لتحديد ما إذا كان التسلل يظهر أنماطاً مكانية، وحددنا ست مناطق مرتبة من مركز الورم إلى TME: T3+، T2، T1، E1، E2 وE3+. أظهرت هذه الاتجاه التنازلي المتوقع لخلايا الورم (بيانات موسعة الشكل 9ب)، ولكن أنماط مختلفة للخلايا غير الورمية. تجمعت البلعميات خارج الورم في E1 وE2، بينما أظهرت خلايا T تسللاً مباشرة خارج (E1 وE2) وداخل الورم (T1 وT2) (الشكل 3ج). كانت كل من نسب البلعميات وخلايا T منخفضة في TME البعيد (الطبقة E3+) حيث تهيمن الخلايا الليفية، وهو ملاحظة مدعومة ببيانات CODEX (بيانات موسعة الشكل 9ج). مع مسافة بين الطبقات تبلغ فقط، تشير ملاحظتنا إلى أن هناك تجنيداً مكانياً قوياً للخلايا المناعية بواسطة الأورام على المستوى المجهري.
قمنا أيضاً بإجراء تحليل التعبير التفاضلي بين المناطق الحدودية T1 وE1 ومع جميع النقاط الأخرى (الشكل 3د). كانت الجينات الحدودية العليا المشتركة عبر العينات وأنواع السرطان تشمل جينات بروتينات مصفوفة خارج الخلية (POSTN و) وبروتينات تنشيط البلعميات المستحثة بواسطة الإنترفيرون (IFI3O) (بيانات موسعة الشكل 9د). أظهرت بيانات snRNA-seq المطابقة أن الجينات الحدودية العليا كانت لها تعبير أعلى بشكل ملحوظ (تم تعديل، تصحيح بونفيروني) عبر المجموعة في أنواع خلايا غير ورمية (POSTN، FN1 وTIMP1 في الخلايا الليفية وIFI3O في البلعميات)، مما يشير إلى أن هناك تفاعلات بين خلايا الورم وغير الورم عند الحدود (الشكل 3د). لتحديد CCIs المكانية، قمنا بتشغيل COMMOT على 18 حالة مع 39 مقطعاً ثم تمييزنا إشارات المرسل-المستقبل التفاضلية بين النقاط داخل وخارج مناطق حدود الورم. كانت CCIs المشتركة العليا في المناطق الحدودية هي مستقبلات مصفوفة خارج الخلية (ECM) (الكولاجين، اللامينين، FN1 وTHBS)، الإشارات المفرزة (SEMA3، SEMA4، ncWNT وMK) والتصاق الخلايا (EPHB وNOTCH) (الشكل 3هـ). كمثال، تم ملاحظة مسار MK في عينات CRC وPDAC وBRCA، حيث تنتقل الإشارة من مناطق الخلايا الخبيثة إلى واجهة TME (بيانات موسعة الشكل 9هـ). شمل المسار تفاعلات بين الليغاند MDK والمستقبلات NCL وSDC4 (مرجع 26). تفرز الخلايا الخبيثة MDK لإنشاء بيئة مثبطة للمناعة وعائية، مما يعزز بدوره نمو الورم. وجدنا أيضاً مسارات ECM التي تنتقل فيها الإشارة من TME نحو مناطق الخلايا الخبيثة. واحدة من التفاعلات العليا، مسار THBS، تصف مكونات ECM THBS1-THBS4 (التي تشفر الثرومبوسبوندين) ترتبط بمستقبلات سطح الخلية CD36 وCD47، مما يعدل بدوره التصاق الخلايا، التكاثر وتكوين الأوعية. توضح زيادة الخلايا المناعية المرتبطة بالورم، الجينات وCCIs داخل-wide المناطق الحدودية التواصل بين الخلايا الخبيثة وبيئتها التي ستكون غير مرئية للتقنيات غير المكانية.
هيكل الورم ثلاثي الأبعاد وتفاعلات TME
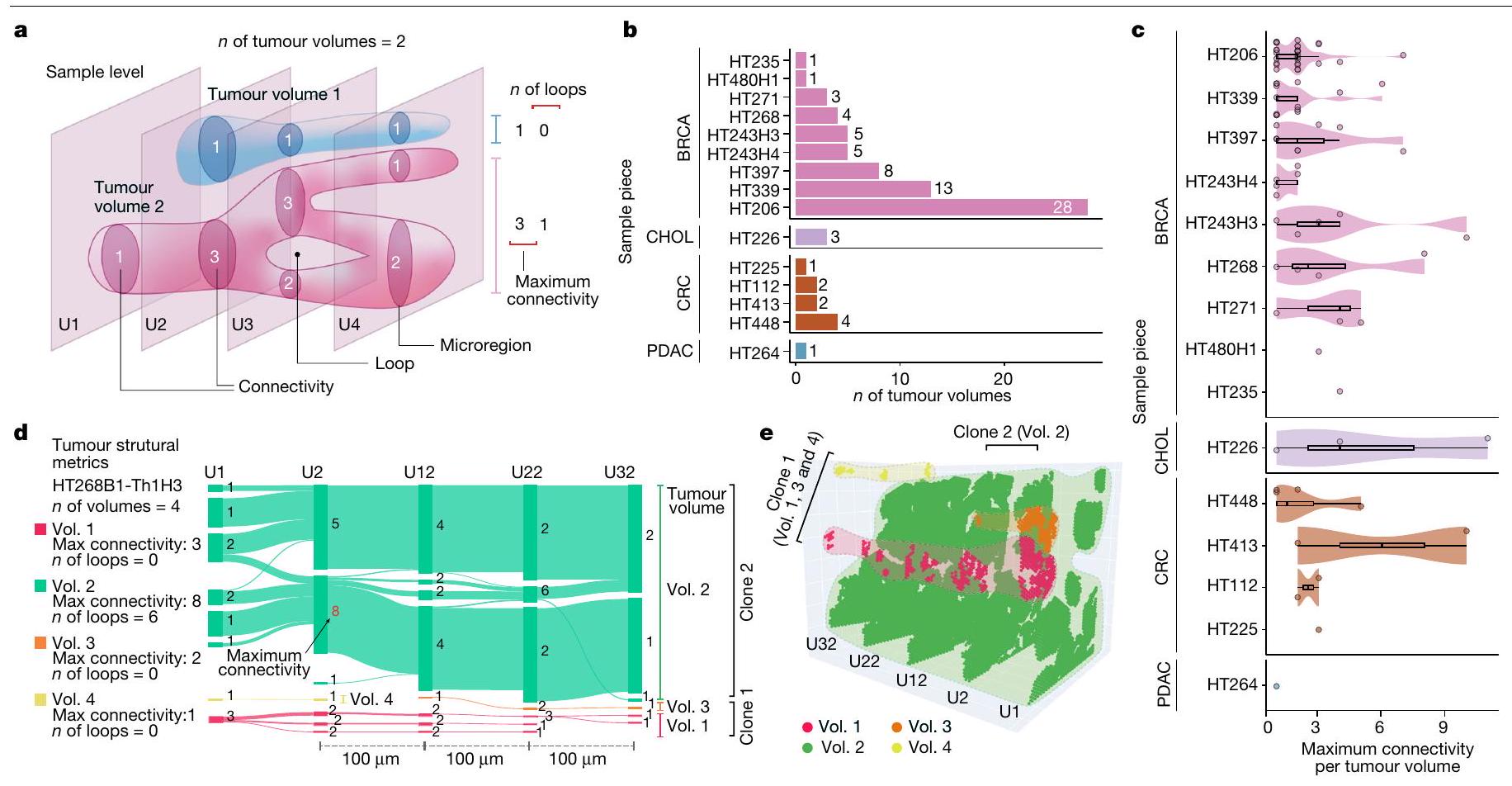
لتحقيق في أنماط نمو الورم وتفاعلات TME في 3D، قمنا بتقطيع الأورام من BRCA وCRC وPDAC وCHOL بشكل متسلسل، وأجرينا ST على 11 عينة وCODEX على عينتين. باستخدام PASTE2، قمنا بتسجيل 48 مقطعاً من 11 عينة ST لبناء أحجام الأورام (الشكل 4أ والطرق). كشفت تحليلاتنا عن اختلافات في أعداد أحجام الأورام بين العينات، حيث أظهرت BRCA أعلى عدد من الأحجام (الشكل 4ب)، خاصة في العينات مع
الشكل 3 | التسلل المناعي والستروما داخل الميكرو مناطق الورم المكانية. أ، تصميم التحليل يركز على مستوى التسلل التفاضلي بين الأنماط الفرعية المكانية وموقع هذا التسلل بالنسبة لحدود الورم-TME. ب، يسار، عينة BRCA الأساسية HT397B1 تظهر مستويات أعلى من البلعميات وخلايا T في النمط الفرعي c 3. يمين، تظهر علامات خلايا T (CD3 وCD8) وعلامة المناعة غير T (HLA-DR) كثافة أعلى في نفس المناطق من بيانات CODEX. شريط القياس، 1 مم. ج، نسبة البلعميات، خلايا T والخلايا الليفية المتوسطة عبر الميكرو مناطق في الطبقات التالية: T3 وما فوق (T3+)، T2، T1، E1، E2، وE3 وما فوق (E3+)، في 14 حالة. د، الجينات الغنية تفاضلياً في مناطق حدود الورم. اللون يمثل(تغيير مضاعف) لتعبير الجين بين المناطق الحدودية والمناطق غير الحدودية، مع مقارنات غير تفاضلية باللون الأبيض (حالات متميزة مكانياً). يمثل كل عمود كتلة ورم (متوسط مقاطع متعددة) وتم تجميع كتل الورم من نفس المريض معاً (بدون فجوات بين الأعمدة). يتم عرض التعبير المحدد بنوع الخلايا المستند إلى snRNA-seq على اليسار، وعدد كتل الورم ذات الزيادة الملحوظة معروض على اليمين (تم تعديل، تصحيح بونفيروني). هـ، CCIs الغنية عند الحدود المشتركة عبر العينات (حالات متميزة مكانياً) بناءً على إشارات المرسل (r)-المستقبل (s).
أنماط نمو الورم الصغيرة البارزة الشبيهة بالقنوات (HT206B1-S1، 1A-1E؛ HT339B1-S1H3، 3D و3E؛ وHT397B1-S1H3، 4B-4D؛ بيانات موسعة الشكل 2). بالمقابل، أظهرت مقاطع CHOL وCRC وPDAC وغيرها من مقاطع BRCA أوراماً أكثر غزارة شكلت هياكل أكبر مستمرة أدت إلى أحجام أقل ولكن أكبر.
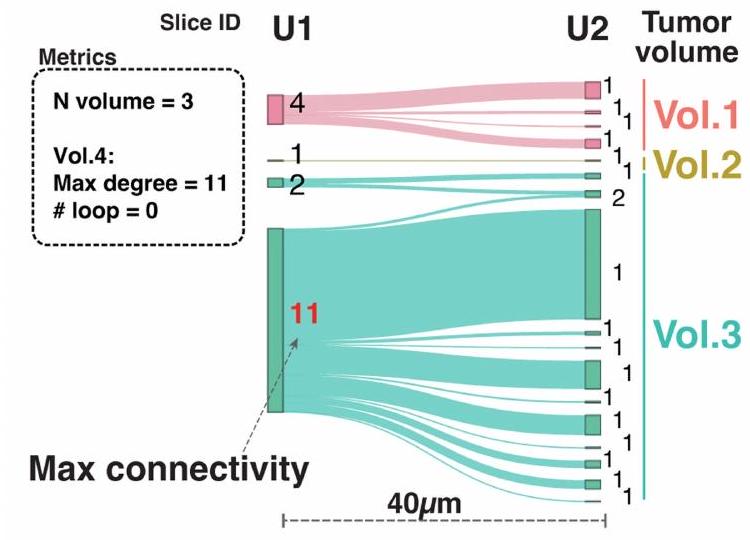
ثم قمنا بتحليل هذه الأحجام الورمية من حيث التعقيد الهيكلي باستخدام مقياسين طوبولوجيين رئيسيين: (1) الاتصال (الدرجة)، الذي يقيس عدد الاتصالات بالمناطق المجاورة، و(2) عدد الحلقات لكل حجم، الذي يشير إلى الحالات التي تنقسم فيها المقاطع المجاورة وتندمج لتشكيل هياكل على شكل كعكة. تعتبر أعلى درجة اتصال مؤشراً على تعقيد الهيكل الورمي. من بين 15 حجم ورم تم تحليله، كان لدى 8 (6 BRCA، 1 CHOL و1 CRC) درجة اتصال قصوى تتجاوز 5، مما يعكس التكوين المتكرر لهياكل متفرعة معقدة في هذه الأورام (الشكل 4ج). تم ملاحظة أعلى درجة اتصال تبلغ 11 في عينة CHOL HT226C1-Th1، والتي نتجت عن حجم مدمج كبير في U1 الذي تفتت إلى ميكرو مناطق أصغر في المقطع المجاور U2 (بيانات موسعة الشكل 10أ، ب). بالإضافة إلى ذلك، احتوت 5 من 81 حجم عبر 15 قطعة ورمية على هياكل حلقة معقدة، مع أعلى عدد حلقات يبلغ 12 موجود في الحجم 14 من العينة
HT206B1 (بيانات موسعة الشكل 10ج). من المحتمل أن تكون هذه الحلقات ناتجة عن نمط النمو الشبيه بـ DCIS المتشابك في الحجم 14، كما تم تأكيده من خلال علم الأنسجة الخاص به (بيانات موسعة الشكل 10د).
اخترنا عينة نقيلة كبدية من BRCA، HT268B1-Th1H3، لإعادة بناء حجم ثلاثي الأبعاد وتحليل هيكلي مفصل. احتوت هذه العينة على أربعة أحجام ورمية (الحجم 1 إلى الحجم 4) (الشكل 4د، هـ). شكلت الأحجام 1 و3 و4 أحجام فرعية منفصلة داخل النمط الفرعي 2، والتي من المحتمل أن تكون متصلة بما يتجاوزتم فحص قسم الأنسجة. كان الحجم 2، وهو أكبر وأعقد حجم، يخص فقط النسخة 1، مع أقصى اتصال يبلغ 8 و6 حلقات، مما يدل على تفرع ودمج كبيرين (الشكل 4d). أكدت الصور النسيجية لـ HT268B1 (البيانات الموسعة الشكل 2a-e) وجود انقسام ودمج مرئيين، مما يوضح أن الأورام حتى بدون هياكل قنوية أو فصية واضحة يمكن أن تظهر أنماط نمو متنوعة وسلوك غازي وتفرع معقد.
بعد ذلك، استخدمنا نهج التعلم العميق غير المراقب لتحديد الأحياء الخلوية ثلاثية الأبعاد في مجموعات بيانات ST المقطعة بالتسلسل (الطرق). بعد تسجيل المقاطع التسلسلية، تم تدريب مشفر رؤية المحولات (ViT) على مقاطع ST وCODEX وH&E. ثم تم استخدام رقع الصور المعلّمة لبناء الأحياء الخلوية ثلاثية الأبعاد.
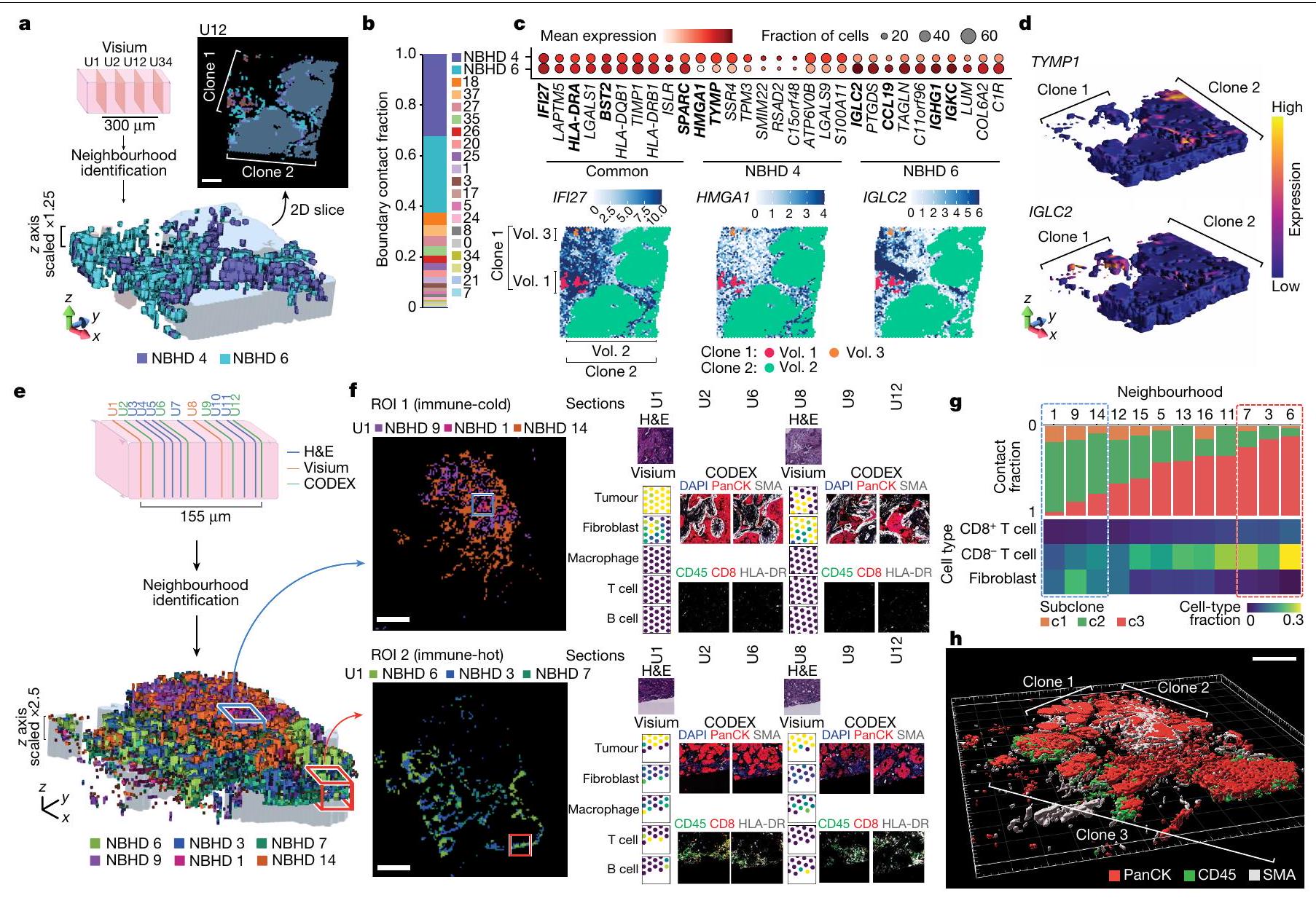
الشكل 4 | إعادة بناء حجم الورم ثلاثي الأبعاد يكشف أنماط نمو ورم متنوعة. أ، مخطط نمط نمو الورم مع حجمين ثلاثيي الأبعاد متميزين من أربعة مقاطع ST. تم توضيح عدد أحجام الأورام، والاتصال، والاتصال الأقصى وعدد الحلقات لتوضيح مفهوم هذه الكميات الهندسية. ب، عدد أحجام الأورام (المكونات) لكل عينة. ج، توزيع الاتصال الأقصى لقطع العينات. كل نقطة تمثل حجم ورم واحد.تمثل الخط المركزي في الرسم البياني الصندوقي الوسيط، مع الجزء السفلي و المفصلات العلوية تشير إلى الربع الأول والثالث. تمتد الشعيرات إلى أعلى وأدنى القيم ضمن 1.5 مرة من نطاق التباين بين الربعين من المفصلات. د، رسم سانكي يوضح اتصالات الميكرو منطقة الورم عبر الأقسام، مع أحجام الورم ثلاثية الأبعاد لكتلة الأنسجة HT268B1-Th1H3 (نقائل BRCA). تم تمييز أقصى اتصال للحجم 2 (الحجم 2) باللون الأحمر. الأرقام بجانب كل ميكرو منطقة تشير إلى اتصالاتها. هـ، توزيع الميكرو منطقة وأحجام الورم ثلاثية الأبعاد على الأقسام ST ل HT268B1-Th1H3. الذي مكن من اكتشاف هذه الأحياء عبر أقسام متعددة (الشكل البياني الإضافي 10e والشكل التوضيحي الإضافي 7b). قمنا بتطبيق هذا النهج على عينة BRCA الأساسية HT397B1 (ستة أقسام H&E، أربعة أقسام CODEX واثنان من أقسام Visium ST) وعينة انتشار الكبد BRCA HT268B1 (أربعة أقسام Visium ST) (الشكل البياني الإضافي 10f والشكل التوضيحي الإضافي 7c,d). في العينة HT268B1، تم تصنيف الأحياء التي تحتوي على تداخل لا يقل عن 60% مع السلالات الفرعية المكانية المحددة مسبقًا (الشكل 2d) على أنها غنية بالأورام، بينما تم تصنيف البقية كأحياء بيئية (الطرق). كان اثنان من هذه الأحياء (4 و 6) في اتصال وثيق مع محيط كلا السلالتين الفرعيتين (الشكل 5a، b). ومن الجدير بالذكر أنه عند مشاهدتها في الفضاء ثلاثي الأبعاد، تحيط الأحياء بشكل كبير بالسلالات الفرعية، باستثناء الجزء العلوي من الكتلة حيث كانت الأحياء أكثر تكسراً. ثم قمنا بت quantifying هذين الحيين بناءً على الجينات المعبر عنها بشكل مشترك وفريد (الشكل 5c)، ووجدنا استجابات مناعية (IFI27 و HLA-DRA) وجينات هيكلية (BST2 و SPARC) في كلاهما. كان الحي 4 غنيًا بـ HMGA1، الذي ينظم بنية الكروماتين وله دور في تقدم الخلايا الخبيثة.، و عامل مرتبط بتكوين الأوعية الدمويةتم إثراء الجوار 6 بالجينات المهمة للتنظيم المناعي (CCL19) وارتباط مستقبلات الأجسام المضادة (IGLC2 وIGHG1 وIGKC)، والتي تعتبر تنبؤية فيفي الأبعاد الثلاثة، استمررنا في رؤية ارتباط واضح لهذه الجينات المعبر عنها بشكل مختلف، وخاصة TYMP1 وIGLC2، مع النسخ 1 و2، على التوالي، في جميع أنحاء حجم الأنسجة (الشكل 5d والشكل الإضافي 10g).
عينة BRCA الرئيسية HT397B1 كانت تحتوي على منطقتين رئيسيتين من مورفولوجيا الورم TME: منطقة باردة مناعياً تحتوي على كل من مورفولوجيا DCIS و IDC تحتوي على النسخ 1 و 2، ومنطقة IDC ساخنة مناعياً تحتوي على النسخة 3 (الأشكال 5e، f و 3b والشكل التكميلي 2b). قمنا بتصنيف جيران TME حسب نسبة الاتصال مع النسخة 3. الذي أبرز الربعين العلوي والسفلي (الشكل 5e، g). وفقًا لتصنيفات نوع الخلايا المستندة إلى CODEX، كانت الأحياء المرتبطة بالنسخة 3 تحتوي على نسبة أعلى من خلايا T ونسبة أقل من خلايا الألياف مقارنة بتلك القريبة من النسختين 1 و 2، وهو نتيجة تتماشى مع النتائج السابقة (الأشكال 5g و 3b). للتحقق بشكل أكبر من حالة المناعة والستروما في هذه الأحياء، اخترنا منطقتين من الاهتمام (ROIs): ROI 1، الواقعة في النسخة 2 الباردة من المناعة، و ROI 2، الواقعة في النسخة 3 الساخنة من المناعة. كانت ROI 1 تحتوي على نسب منخفضة من البلعميات، خلايا T وخلايا B، بينما كانت ROI 2 تحتوي على مستويات أعلى بكثير من هذه الأنواع من الخلايا (الشكل 5f). كانت هذه الاتجاهات واضحة أيضًا في مقاطع CODEX، حيث كانت علامات المناعة أكثر كثافة في ROI 2. بالإضافة إلى ذلك، أظهرت ROI 1 زيادة في نسبة خلايا الألياف، مع تعبير عالٍ عن الأكتين العضلي الملساء (SMA) في العضلات الملساء المحيطة بمناطق الورم، وهو علامة مميزة لـ DCIS. تشير نتائجنا إلى أن هذه الارتباطات بين أنواع الخلايا ونسخ DCIS و IDC تشبه في الأبعاد الثلاثة، استنادًا إلى كثافات أنواع الخلايا المحسوبة حول حجم الورم ثلاثي الأبعاد وتوليد أحجام المناعة والستروما والظهارة (الشكل 5h والشكل الممتد 10h). توضح هذه التحليلات أن إعادة البناء ثلاثي الأبعاد توفر حساسية متزايدة للتحقيق في بيئات الورم الدقيقة المتنوعة.
نقاش
حدد هذه الدراسة مناطق ميكروية متميزة جينومياً ونسخ فرعية موزعة مكانياً في عينات من أنواع الأورام الصلبة. نقترح أن تباين CNV هو محرك رئيسي للاختلافات النسخية التي تُرى في هذه المناطق الميكروية. النسخ الفرعية المكانية المحددة في نفس كتلة الورم تشترك في سلف مشترك، وهو اكتشاف يتماشى مع الدراسات السابقة حول تطور الورم.محرك رئيسي ثانٍ للتغير هو التعرض للبيئة المجهرية للورم، وقد لاحظنا اختلافات واضحة
الشكل 5 | تفاعلات الورم – TME في مناطق الحدود الدقيقة تظهر التباين. أ، بالنسبة لعينة HT268B1، وهي عينة من نقائل BRCA، تم إنشاء أحجام الجوار ثلاثية الأبعاد مع 4 مقاطع Visium ST تمتدتُعرض الأحياء 4 و 6 (NBHD 4 و NBHD 6، على التوالي)، مع أعلى نسبة تواصل مع حدود السلالة الفرعية، في عرض ثلاثي الأبعاد وكخطة ثنائية الأبعاد مغطاة بتعليقات السلالة الفرعية. شريط القياس،، تم عرض الأحياء TME من خلال نسبة الاتصال مع حدود السلالة الفرعية. ج، تم تصنيف DEGs إلى ثلاث مجموعات: فريدة من نوعها للحي 4، فريدة من نوعها للحي 6، ومشتركة. يتم عرض التعبيرات المكانية للجينات المختارة من كل مجموعة (أسفل)، مع تسليط الضوء على الجينات المذكورة في النص الرئيسي بالخط العريض. د، إعادة بناء ثلاثية الأبعاد لمناطق الورم، حيث يتم تلوين شبكة سطح الورم بكثافة النسخ لـ TYMP1 و IGLC2 ضمن نصف قطر موقع معين. على سبيل المثال، بالنسبة لـ HT397B1، وهو عينة BRCA أساسية، تم إنشاء أحجام جيران ثلاثية الأبعاد متكاملة مع 6 عينات H&E و2 عينات Visium ST.
وأربعة أقسام من CODEX تمتدالأحياء التي تم وصفها في TMEيتم عرضها كحجم ثلاثي الأبعاد. تصور منطقتين من الاهتمام في HT397B1، تحديدًا المنطقة الأكثر برودة مناعيًا ROI 1 والمنطقة الأكثر حرارة مناعيًا ROI 2. يتم عرض شرائح ثنائية الأبعاد من الحجم ثلاثي الأبعاد مع مناطق مجاورة مميزة بالربع المرتبطة بكل منطقة من الاهتمام. يتم عرض شرائح Visium ST (U1 و U8) كنسب نوع الخلايا المحسوبة بواسطة RCTD. بالنسبة لشرائح CODEX (U2 و U6 و U9 و U12)، يتم عرض DAPI و pan-cytokeratin (PanCK) و SMA و HLA-DR و CD45 و CD8. شريط القياس، تُعرض أحياء TME كنسبة من الاتصال بحدود كل من الثلاثة تحت الأنواع. بالإضافة إلى ذلك، نسبة نوع الخلية منوتم عرض الخلايا والألياف. تم حساب كسور نوع الخلايا من مقاطع CODEX. تم التأكيد على الربعين العلوي والسفلي من الأحياء بالنسبة لكسرة الاتصال مع السلالة الفرعية 3 كصناديق متقطعة. h، تم إنشاء أحجام السطح الظهاري ثلاثي الأبعاد (PanCK، أحمر)، المناعي (CD45، أخضر) واللحمي (SMA، أبيض) من مقاطع CODEX. مقياس الرسم،. أنماط النسخ المرتبطة بعمق خلايا السرطان من حافة الميكرومنطقة، بالإضافة إلى التعبير الجيني المحدد الغني في خلايا الحافة المجاورة لخلايا المناعة في البيئة المجهرية للورم. أخيرًا، حددت إعادة بناء حجم الورم ثلاثي الأبعاد الجوار ثلاثي الأبعاد لتنوع البيئة المجهرية الإقليمية.
لقد لاحظنا تباينات جينومية وترانسكريبتومية بين الميكرو مناطق الورم في عينات متعددة. على الرغم من أن بعض الأورام كانت متسقة نسبيًا في ملفاتها الترانسكريبتومية، إلا أن البعض الآخر يمكن تقسيمه وفقًا للتعبير الجيني. وقد تم تفسير هذه التباينات جزئيًا من خلال رسم أحداث عدد النسخ إلى مناطق ST. تم رسم توزيعات تحت الأنواع السرطانية ذات التغيرات الجينية عبر الأنسجة.وقد تم إثبات وجود تحت استنساخ قائم على الطفرات في مناطق مكانية داخل ورم واحد. بالإضافة إلى رسم خرائط الطفرات المكانية من النسخ الجيني والتحقق منها باستخدام الهجين الموضعي المحدد بالأليل، قمنا بتوصيف التباين المكاني للأورام وفقًا للتغيرات في التعبير الجيني المتعلقة بقرب أنواع الخلايا المناعية والستروما مقارنةً بـ خلايا السرطان المعزولة داخل الميكرو مناطق المكانية. هناك حاجة إلى فهم أكثر دقة للطريقة التي تشكل بها خلايا السرطان وبيئة الورم بعضها البعض داخل الأورام لاستغلال هذه التفاعلات بشكل أفضل في العلاج.
تطور تحت الخلوية هو محرك رئيسي لمقاومة العلاج، حيث إن ظهور تحت الخلايا المقاومة غالبًا ما يؤدي إلى فشل العلاج.هنا قمنا بتوصيف الهيكل والتوزيع للنسخ الفرعية الورمية المكانية المتميزة في عدة أورام صلبة وأظهرنا أنها يمكن أن تظهر استجابات متفاوتة لمركبات متطابقة من خلال تحليل تداخل مجموعة الجينات المزعزعة. من المحتمل أن تستكشف الأعمال الترجمة المستقبلية النسخ الفرعية تحت الضغوط الانتقائية المتنوعة للعلاجات المضادة للسرطان، مما سيساعد في توجيه تصميم أساليب جديدة، مثل تحسين الجمع بين العلاجات المحلية والنظامية.
هناك عدة قيود على الدراسة. تلتقط التغيرات العددية الجينية المستنتجة من RNA الأحداث الجينية الكبيرة ولكنها لا تلتقط التغيرات العددية البؤرية. توفر خرائط الطفرات المكانية دعمًا إضافيًا في تحديدات تحت الخلوية، ولكن مع القيد المتمثل في أن الطفرات القريبة فقط هي التي يمكن تحديدها.
يتم الكشف عن الطرف 3′ من كل نسخة بشكل تفضيلي. بالإضافة إلى ذلك، فإن مجموعة بيانات ST الخاصة بنا لا تحقق دقة الخلية المفردة مع المنصة الحالية.-قطر البقعة). تم استخدام تسلسل RNA النووي الصغير المتطابق لاستنتاج التعبير بين الورم وغير الورم، وتم استخدام تصوير CODEX للتحقق من نتائجنا حول تركيبة البيئة المجاورة للورم. ومع ذلك، لا يزال التعبير المكاني المباشر من أنواع الخلايا المختلفة مستنتجًا. في الختام، مكنتنا بياناتنا ثلاثية الأبعاد المعاد بناؤها من التحقيق المكاني في بنية الورم، والنسائل الفرعية، والأحياء الخلوية، والبيئة المجاورة للورم. نتوقع أن يتم تأسيس مثل هذا التحليل بسرعة بشكل أوسع ضمن أبحاث السرطان.ستسهل التقدمات القادمة في التكنولوجيا تحليلات أعمق وستعزز الدراسات المستقبلية للأورام.
المحتوى عبر الإنترنت
أي طرق، مراجع إضافية، ملخصات تقارير Nature Portfolio، بيانات المصدر، بيانات موسعة، معلومات تكميلية، شكر وتقدير، معلومات مراجعة الأقران؛ تفاصيل مساهمات المؤلفين والمصالح المتنافسة؛ وبيانات توفر البيانات والرموز متاحة علىhttps://doi.org/10.1038/s41586-024-08087-4.
فو، ت. وآخرون. الهندسة المكانية للبيئة الدقيقة المناعية تنظم مناعة الورم والاستجابة العلاجية. مجلة أمراض الدم والأورام 14، 98 (2021).
شميت، م. و.، لوبي، ل. أ. وسالك، ج. ج. تأثير طفرات المقاومة الفرعية على العلاج المستهدف للسرطان. نات. ريف. كلين. أونكول. 13، 335-347 (2016).
روبر، ن. وآخرون. التطور النسلي والتنوع في آليات مقاومة الأوزيمرتينيب المكتسبة في سرطان الرئة ذو الطفرة في EGFR. تقارير الخلايا. ميد. 1، 100007 (2020).
تريدان، أ.، غالماريني، س. م.، باتيل، ك. وتانوك، إ. ف. مقاومة الأدوية وبيئة الورم الصلب. ج. المعهد الوطني للسرطان. 99، 1441-1454 (2007).
Qu، Y. وآخرون. مقاومة غير ذاتية الخلايا للعلاج المضاد للأورام المدفوعة ببيئة الورم. مول. كانسر 18، 69 (2019).
دينغ، ل. وآخرون. التطور النسلي في اللوكيميا النقوية الحادة المتكررة كما كشفت تسلسلات الجينوم الكامل. ناتشر 481، 506-510 (2012).
يانغ، د. وآخرون. تتبع السلالة يكشف عن الديناميات التطورية، والمرونة، وطرق تطور الورم. خلية 185، 1905-1923.e25 (2022).
ستول، ب. ل. وآخرون. تصور وتحليل تعبير الجينات في مقاطع الأنسجة بواسطة النسخ الجيني المكاني. ساينس 353، 78-82 (2016).
راو، أ.، باركلي، د.، فرانسا، ج. س. وياناي، إ. استكشاف بنية الأنسجة باستخدام النسخ الجيني المكاني. ناتشر 596، 211-220 (2021).
تشوي زو، د. وآخرون. السائقون المقيدون مكانيًا وسكان الخلايا الانتقالية يتعاونون مع البيئة الدقيقة في سرطان البنكرياس غير المعالج والمقاوم للعلاج الكيميائي. نات. جينيت. 54، 1390-1405 (2022).
بلاك، س. وآخرون. تصوير الأنسجة المتعدد باستخدام CODEX مع الأجسام المضادة المرتبطة بالحمض النووي. نات. بروتوك. 16، 3802-3835 (2021).
غريفز، م. ومالاي، س. س. التطور النسلي في السرطان. الطبيعة 481، 306-313 (2012).
دينغ، ل.، رافائيل، ب. ج.، تشين، ف. و ويندل، م. س. تقدمات لدراسة التطور النسلي في السرطان. رسائل السرطان. 340، 212-219 (2013).
إريكسون، أ. وآخرون. التغيرات في عدد النسخ المستنسخة المكانية في الأنسجة الحميدة والخبيثة. ناتشر 608، 360-367 (2022).
دي ماجيو، ف. وإل-شاكينكيري، ك. هـ. التنسج الليفي والفيزياء الحيوية في سرطان القناة البنكرياسية الغدي: هل يمكننا أن نتعلم من سرطان الثدي؟ البنكرياس 49، 313-325 (2020).
تشانغ، إكس. وآخرون. ثيموسين بيتا 10 هو منظم رئيسي لتكون الأورام والانتقال وهو علامة مصل جديدة في سرطان الثدي. أبحاث سرطان الثدي 19، 15 (2017).
Chen، R. H. وآخرون. ISG15 الذي تفرزه خلايا الورم يعزز هجرة خلايا الورم وكبت المناعة من خلال تحفيز نمط ماكروفاج شبيه M2. Front. Immunol. 11، 594775 (2020).
جين، س. وآخرون. استنتاج وتحليل التواصل بين الخلايا باستخدام CellChat. نات. كوميونيك. 12، 1088 (2021).
فيليبو، ب. س.، كاراجيانيس، ج. س. وكونستانتينيدو، أ. عامل النمو ميدكين (MDK): لاعب رئيسي في تقدم السرطان وهدف علاجي واعد. أونكوجين 39، 2040-2054 (2020).
سيك، إ. وآخرون. تنشيط مستقبلات CD47 يسبب تكاثر الأورام الدبقية البشرية ولكن ليس الدبقية الطبيعية عبر مسار يعتمد على Akt. غليا 59، 308-319 (2011).
إيزنبرغ، ج. س.، فريزر، و. أ. وروبرتس، د. د. ثرومبوسبوندين-1: منظم فسيولوجي لإشارات أكسيد النيتريك. خلية. علوم الحياة الجزيئية 65، 728-742 (2008).
جان، أ. وآخرون. تحديد ببتيد TAX2 كعامل مضاد للسرطان غير متوقع جديد. أونكوتارجت 6، 17981-18000 (2015).
وانغ، ي.، هو، ل.، تشنغ، ي. & قوه، ل. HMGA1 في السرطان: تصنيف السرطان حسب الموقع. مجلة الطب الخلوي والجزيئي 23، 2293-2302 (2019).
هوانغ، ر.، هوانغ، د.، داي، و. ويانغ، ف. يرتبط الإفراط في التعبير عن HMGA1 بالحالة الخبيثة وتوقعات سرطان الثدي. مول. سيل. بيكيم. 404، 251-257 (2015).
ميتسيلو، أ. وآخرون. الدور التنبؤي للتعبير عن الفوسفاتاز الثيميدين في مرضى سرطان القولون والمستقيم وارتباطه ببروتينات مرتبطة بتكوين الأوعية الدموية ومكونات المصفوفة خارج الخلوية. في vivo 26، 1057-1067 (2012).
باريس، أ.، فريلي-بيثينكورت، إ.، يوبانكس، ج.، خو، س. وأناند، س. إن إنزيم الفوسفاتاز الثيميدين يسهل عجز الأوعية الدموية المستحث بواسطة الجين I القابل للتحفيز بواسطة حمض الريتينويك. مرض موت الخلايا. 14، 294 (2023).
شميت، م. وآخرون. التأثير التنبؤي لجلوبيولين المناعة كابا C (IGKC) في سرطان الثدي المبكر. السرطانات 13، 3626 (2021).
وانغ، ج. وآخرون. CCL19 لديه القدرة على أن يكون علامة بيولوجية تنبؤية محتملة ومعدل لبيئة المناعة الورمية (TIME) لسرطان الثدي: تحليل شامل استنادًا إلى قاعدة بيانات TCGA. الشيخوخة 14، 4158-4175 (2022).
نيشيمورا، ت. وآخرون. التاريخ التطوري لسرطان الثدي والنسخ المرتبطة به. ناتشر 620، 607-614 (2023).
سيميونوف، ك. ب. وآخرون. تتبع سلالة الخلايا الفردية للسرطان النقيلي يكشف عن اختيار حالات التحول الظهاري المختلط. خلية السرطان 39، 1150-1162.e9 (2021).
تشين، هـ. ن. وآخرون. التطور الجينومي ونماذج متنوعة من النقائل الجهازية في سرطان القولون والمستقيم. غوت 71، 322-332 (2022).
ليونغ، م. ل. وآخرون. تسلسل الحمض النووي على مستوى الخلية الواحدة يكشف نموذج انتشار متأخر في سرطان القولون والمستقيم النقيلي. أبحاث الجينوم. 27، 1287-1299 (2017).
هو، ز. وآخرون. دليل كمي على انطلاق النقائل المبكر في سرطان القولون والمستقيم. نات. جينت. 51، 1113-1122 (2019).
تانغ، ج. وآخرون. تسلسل الإكسوم على مستوى الخلية الواحدة يكشف عن عدة تحت أنواع في سرطان القولون والمستقيم النقيلي. جينوم ميد. 13، 148 (2021).
بوس، أ. م. وآخرون. حل آليات مقاومة العلاج في الورم النقوي المتعدد من خلال تحليل تحت المجموعات متعددة الأوميات. بلود 142، 1633-1646 (2023).
براكسون، أ. م. وآخرون. رسم الخرائط الجينومية ثلاثية الأبعاد يكشف عن تعدد البؤر في الأورام السابقة للسرطان في البنكرياس البشري. ناتشر 629، 679-687 (2024).
ملاحظة الناشر: تظل شركة سبرينغر ناتشر محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
الوصول المفتوح هذه المقالة مرخصة بموجب رخصة المشاع الإبداعي النسبية-غير التجارية-بدون اشتقاقات 4.0 الدولية، التي تسمح بأي استخدام غير تجاري، ومشاركة، وتوزيع، وإعادة إنتاج في أي وسيلة أو صيغة، طالما أنك تعطي الائتمان المناسب للمؤلفين الأصليين والمصدر، وتوفر رابطًا لرخصة المشاع الإبداعي، وتوضح إذا قمت بتعديل المادة المرخصة. ليس لديك إذن بموجب هذه الرخصة لمشاركة المواد المعدلة المشتقة من هذه المقالة أو أجزاء منها. الصور أو المواد الأخرى من طرف ثالث في هذه المقالة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة، ما لم يُشار إلى خلاف ذلك في سطر الائتمان للمادة. إذا لم تكن المادة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة وكان استخدامك المقصود غير مسموح به بموجب اللوائح القانونية أو يتجاوز الاستخدام المسموح به، فستحتاج إلى الحصول على إذن مباشرة من صاحب حقوق الطبع والنشر. لعرض نسخة من هذه الرخصة، قم بزيارة http:// creativecommons.org/licenses/by-nc-nd/4.0/. (ج) المؤلف(ون) 2024
¹قسم الطب، جامعة واشنطن في سانت لويس، سانت لويس، ميزوري، الولايات المتحدة الأمريكية.معهد ماكدونيل للجينوم، جامعة واشنطن في سانت لويس، سانت لويس، ميزوري، الولايات المتحدة الأمريكية.قسم علوم الحاسوب، جامعة برينستون، برينستون، نيو جيرسي، الولايات المتحدة الأمريكية.قسم الجراحة، جامعة واشنطن في سانت لويس، سانت لويس، ميزوري، الولايات المتحدة الأمريكية.مركز سيتيمان للسرطان، جامعة واشنطن في سانت لويس، سانت لويس، ميزوري، الولايات المتحدة الأمريكية.قسم الأشعة، جامعة واشنطن في سانت لويس، سانت لويس، ميزوري، الولايات المتحدة الأمريكية.معهد البيولوجيا النظامية، سياتل، واشنطن، الولايات المتحدة الأمريكية.قسم الأورام الطبية، قسم الطب الباطني، جامعة واشنطن كلية الطب، سانت لويس، ميزوري، الولايات المتحدة الأمريكية.قسم علم الأمراض والمناعة، جامعة واشنطن في سانت لويس، سانت لويس، ميزوري، الولايات المتحدة الأمريكية.قسم التوليد وأمراض النساء، جامعة واشنطن في سانت لويس، سانت لويس، ميزوري، الولايات المتحدة الأمريكية.قسم جراحة المسالك البولية، قسم الجراحة، جامعة واشنطن، سانت لويس، ميزوري، الولايات المتحدة الأمريكية.قسم علوم الأعصاب، جامعة واشنطن كلية الطب، سانت لويس، ميزوري، الولايات المتحدة الأمريكية.قسم جراحة الأعصاب، جامعة واشنطن كلية الطب، سانت لويس، ميزوري، الولايات المتحدة الأمريكية.قسم الأورام النسائية، قسم التوليد وأمراض النساء، جامعة واشنطن، سانت لويس، ميزوري، الولايات المتحدة الأمريكية.قسم علوم الحاسوب والهندسة، جامعة واشنطن في سانت لويس، سانت لويس، ميزوري، الولايات المتحدة الأمريكية.قسم الوراثة، جامعة واشنطن في سانت لويس، سانت لويس، ميزوري، الولايات المتحدة الأمريكية.قسم الأنف والأذن والحنجرة – جراحة الرأس والعنق، جامعة واشنطن كلية الطب، سانت لويس، ميزوري، الولايات المتحدة الأمريكية.قسم التوليد وأمراض النساء، جامعة كاليفورنيا، سان فرانسيسكو، سان فرانسيسكو، كاليفورنيا، الولايات المتحدة الأمريكية.ساهم هؤلاء المؤلفون بالتساوي: تشيا-كوي مو، جينغشيان ليو.هؤلاء المؤلفون أشرفوا بشكل مشترك على هذا العمل: سيكي تشين، إريك ستورز، أندريه لويز ن. تارغينو دا كوستا، أندرو هيوستن.البريد الإلكتروني: gillandersw@wustl.edu; rcfields@wustl.edu; braphael@princeton.edu; fchen@wustl.edu; lding@wustl.edu
طرق
طرق تجريبية
عينات ومعالجة العينات. تم جمع جميع العينات بموافقة مستنيرة في كلية الطب بجامعة واشنطن في سانت لويس. تم جمع العينات من BRCA و PDAC و CRC و CHOL و RCC و UCEC خلال الاستئصال الجراحي وتم التحقق منها بواسطة علم الأمراض القياسي (بروتوكولات مجلس المراجعة المؤسسية 201108117 و 201411135 و 202106166). بعد التحقق،تمت إزالة جزء من الورم، وتصويره، ووزنه، وقياسه. ثم تم تقسيم كل جزء إلى 6-9 قطع، ثم تم تقسيمها مرة أخرى إلى 4 قطع مقطوعة عرضيًا. تم وضع هذه القطع الأربع كل واحدة على حدة في الفورمالين، وتجميدها بسرعة في النيتروجين السائل، وDMEM، وتجميدها بسرعة قبل تضمينها في OCT. كان الغرض من اختيار معالجة الشبكة بدلاً من أخذ عينات باستخدام المثقاب قائمًا على الفائدة، حيث قلل من الأنسجة المتبقية. يمكن العثور على البروتوكولات ذات الصلة فيبروتوكولات.آي أو (https://doi.org/10.17504/protocols.io.bszynf7w) .
تحضير ST والتسلسل. تم قطع الأنسجة المدمجة في OCT أو عينات الأنسجة المحفوظة في الفورمالين (FFPE) ووضعها على شريحة تعبير جيني مكاني من فيزيوم وفقًا لدليل بروتوكولات تحضير الأنسجة من فيزيوم. تم قطع العينات المستخدمة للشرائح المتسلسلة وجمعها بفاصل يتراوح من 5 إلىعند إجراء القطع التسلسلي، تم تسمية القسم الأول بـ U1، تلاه U2، U3، وهكذا. تم تحميل الأقسام المختارة على شرائح فيزيوم وتم تسجيل المسافة بين كل قسم. بالنسبة للعينات المدمجة في OCT، تم وصف الطرق بالتفصيل في منشور سابق.باختصار، تم تغليف عينات الأنسجة الطازجة بمادة OCT في درجة حرارة الغرفة دون أي فقاعات. بعد فحص جودة RNA باستخدام جهاز Tapestation وفحص الشكل باستخدام صبغة H&E لعينات الأنسجة المدمجة في OCT، تم تقطيع الكتل إلى حجم مناسب يتناسب مع مناطق الالتقاط ثم تم قسمتها إلىالمقاطع. تم تثبيت المقاطع في الميثانول، وتم صبغها بصبغة H&E وتم تصويرها فيتم التكبير باستخدام إعداد التصوير في الحقل الساطع على مجهر لايكا DMi8. ثم تم نفاذ عينات الأنسجة لمدة 18 دقيقة وتم بناء مكتبات ST وفقًا لدليل مستخدم مجموعة مواد التعبير الجيني المكاني Visium CG000239 Rev A (10x Genomics). تم عكس النسخ من RNA الرسول المعتمد على البوليمر، والذي تم التقاطه باستخدام بادئات على الشرائح. بعد ذلك، تم تصنيع الشريط الثاني وفصله عن الشريط الأول. ثم تم نقل cDNA الحر من الشرائح إلى الأنابيب لمزيد من التضخيم وبناء المكتبة. تم تسلسل المكتبات على خلية تدفق S4 من نظام Illumina NovaSeq-6000. بالنسبة لعينات FFPE، تم وصف الطرق بالتفصيل في منشور سابق.باختصار، تم إجراء مراقبة الجودة من خلال تقييم DV200 للـ RNA المستخرج من مقاطع الأنسجة المحفوظة في الفورمالين (FFPE) وفقًا لبروتوكول مجموعة Qiagen RNeasy FFPE، ثم تلا ذلك إجراء اختبار التصاق الأنسجة الموصوف في بروتوكول 10x Genomics. المقاطع ( ) تم وضعها على شريحة تعبير الجينات المكاني Visium وفقًا لدليل بروتوكولات Visium Spatial – تحضير الأنسجة (10x Genomics، CG000408 Rev A). بعد التجفيف طوال الليل، تم تحضين الشرائح في لمدة ساعتين. ثم تم إجراء إزالة البارافين وفقًا للبروتوكول الخاص بـ Visium Spatial لإزالة البارافين من العينات المحفوظة في الفورمالين، وصبغ H&E، والتصوير، وإزالة الروابط المتقاطعة (10x Genomics، CG000409 Rev A). تم صبغ الشرائح بـ H&E وتم تصويرها فيالتكبير باستخدام إعداد التصوير في المجال الساطع على ميكروسكوب لايكا DMi8. بعد ذلك، تم إجراء إزالة الروابط على الفور للشرائح الملونة بصبغة H&E. بعد ذلك، تمت إضافة لوحات مجسات النسخ الكامل البشري إلى الأنسجة. بعد أن تزاوجت أزواج المجسات هذه مع جيناتها المستهدفة وترابطت مع بعضها البعض، تم تحرير منتجات الربط بعد معالجة RNase واختراق الغشاء. ثم تم تزاوج المجسات المرتبطة مع الأوليغونوكليوتيدات المشفرة مكانيًا في منطقة الالتقاط. تم إنشاء مكتبات ST من المجسات وتم تسلسلها على خلية تدفق S4 من نظام Illumina NovaSeq 6000. يمكن العثور على البروتوكولات ذات الصلة فيبروتوكولات.آي أو (https://doi.org/10.17504/protocols.io.x54v9d3opg3e/v1 وhttps://doi.org/10.17504/protocols. io.kxygx95ezg8j/v1) .
تحضير CODEX والتصوير. تم شراء أجسام مضادة أحادية أو متعددة النسيلة خالية من الحامل (الجدول التكميلي 3) وتم التحقق منها باستخدام صبغة المناعة الفلورية (IF) في قنوات متعددة. بعد الفحص، تم ربط الأجسام المضادة باستخدام مجموعة ربط الأجسام المضادة من أكويا (Akoya Biosciences، SKU 7000009) مع رمز شريطي (Akoya Biosciences) تم تعيينه وفقًا لنتائج صبغة IF. تم شراء عدة علامات شائعة مباشرة من خلال أكويا. تم إجراء صبغ CODEX والتصوير وفقًا لتعليمات الشركة المصنعة (دليل مستخدم CODEX، الإصدار C). باختصار،تم وضع مقاطع FFPE على شرائح زجاجية مغطاة بـ APTES (سيغما، 440140) وتم خبزها عندبين عشية وضحاها قبل إزالة البارافين. في اليوم التالي، تم تحضين الأنسجة في الزيلين، وإعادة ترطيبها في الإيثانول وغسلها فيقبل استرجاع المستضد باستخدام محلول TE، pH 9 (Genemed، 10-0046) في ماء مغلي لمدة 10 دقائق في طباخ الأرز. ثم تم حجب عينات الأنسجة باستخدام محلول الحجب (مجموعة صبغ CODEX، SKU 7000008) وتم صبغها باستخدام مجموعة الأجسام المضادة المحددة إلى حجملمدة 3 ساعات في درجة حرارة الغرفة في غرفة رطبة. يتم توفير عامل التخفيف لكل جسم مضاد في ورقة معلومات دورة CODEX (الجدول التكميلي 3). تم تصوير تجربة CODEX متعددة الدورات باستخدام ميكروسكوب فلوريسانس من Keyence (النموذج BZ-X810) مزود بعدسة Nikon CFI Plan Apo.أداة CODEX (Akoya Biosciences) ومدير أداة CODEX (Akoya Biosciences). ثم تم تجميع الصور الخام ومعالجتها باستخدام معالج CODEX (Akoya Biosciences). بعد الانتهاء من التصوير المتعدد، تم إجراء صبغ H&E على نفس الأنسجة. جودة الصبغ لكل جسم مضاد في CODEX تظهر كقناة واحدة باللون الأخضر مع DAPI باللون الأزرق في الأشكال التكميلية 10 و 11.
تحضير تعليق النواة المفردة. تقريبًامن المجمد بسرعة أو المسحوق بالتبريد أوتم استرجاع أقسام OCT من الأنسجة من كل عينة وتوزيعها لتحضير النواة لاستخدامها في مجموعة Next GEM Single Cell Multiome ATAC + Gene Expression أو مجموعة Next GEM Single Cell 3′ Kit v.3.1. تم إعادة تعليق العينات في محلول التحلل (10 مليمول تريس- (ثيرمو، 15567027)، 10 مللي مولار NaCl (ثيرمو، AM9759)، 3 مللي مولار MgCl 2 (ثيرمو، AM9530G)، بديل (% v/v) (سيغما، 74385-1L)، 1 مللي مول DTT (سيغما، 646563)، محلول BSA بتركيز 1% ( ) (MACS، 130-091-376)، ماء خالٍ من النوكلياز (Invitrogen، AM9937)، بالإضافة إلى مثبط ناز، معلق ومهجن من خلال الدق، ومصفى من خلالمنخل الخلايا (pluriSelect)، ثم تم تخفيفه بمحلول الغسيل (بساتم جمع الراشح، ثم تم الطرد المركزي عند 500 جرام لمدة 6 دقائق في. ثم تم إعادة تعليق الكريات النووية في محلول غسيل BSA مع مثبط RNase، وتم صبغها بـ 7AAD، وتم تنقية النوى وفرزها بواسطة FACS. يمكن العثور على البروتوكولات ذات الصلة في بروتوكولات.آي أو(https://doi.org/10.17504/protocols.io.14egn7w6zv5d/v1، https://doi.org/10.17504/protocols.io.261gednx7v47/v1) .
تحضير تعليق الخلايا المفردة. حواليتم قطع كل ورم إلى قطع صغيرة باستخدام شفرة. تمت إضافة إنزيمات وكواشف من مجموعة تفكيك الأورام البشرية (Miltenyi Biotec، 130-095-929) إلى نسيج الورم مع 1.75 مل من DMEM. تم تحميل التعليق الناتج في أنبوب gentleMACS (Miltenyi Biotec، 130-093-237) وتم تعريضه لجهاز التفكيك gentleMACS Octo مع السخانات (Miltenyi Biotec، 130-096-427). بعدفي برنامج التفكك الحراري (37h_TDK_1)، تم إزالة العينات من جهاز التفكك وتم تصفيتها من خلالمنخل صغير (PluriSelect، رقم 43-10040-60) أوشبكة نايلون (Fisher Scientific، 22-363-547) إلىأنبوب مخروطي على الثلج. ثم تم تدوير العينة بسرعة 400 ج لمدة 5 دقائق فيبعد إزالة السائل العلوي، عندما أصبح الحبيبة الحمراء مرئية، تم إعادة تعليق حبيبة الخلايا باستخدامإلى 3 مل من محلول تحليل ACK (ثيرمو فيشر، A1049201) لـلإيقاف التفاعل، 10 مل من PBS (كورنينج؛ 21-040-CM) معتم إضافة BSA (Miltenyi Biotec؛ 130-091-376) وتم الطرد عند 400 جرام لمدة 5 دقائق في. بعد إزالة الـ المستخلص، تم إعادة تعليق الخلايا في 1 مل من PBS مع، وتمت رؤية الخلايا الحية والميتة باستخدام صبغة التريبان الزرقاء. وأخيرًا، تم تدوير العينة في جهاز الطرد المركزي عند لمدة 5 دقائق عند وتم إعادة تعليقها فيإلى 1 مل من PBS مع BSA إلى تركيز نهائي من خلايا لكلالبروتوكول متاح علىبروتوكولات.آي أو (https://doi. org/10.17504/protocols.io.bsnqnddw) .
تحضير مكتبة النواة المفردة والتسلسل. تم عزل النوى والخلايا والخرز المشفر في قطرات زيتية باستخدام جهاز 10x Genomics Chromium. تم عد وتعويض تعليقات النواة المفردة لتتراوح بين 500-1,800 نواة لكلتم استخدام جهاز الهيموسيتومتر. تم إجراء النسخ العكسي لاحقًا لإدماج رموز شريطية محددة للخلايا والنصوص. تم تشغيل جميع عينات snRNA-seq باستخدام مجموعة مكتبة الخلايا الفردية 3′ من كروميو Next GEM وكريات الهلام v.3.1 (10x Genomics). بالنسبة لمجموعة المولتيوم، تم استخدام Chromium Next GEM Single Cell Multiome ATAC + تعبير الجينات.الجينوميات). ثم تم إخضاع النوى للبروتوكولات اللاحقة بواسطة 10x (Next GEM Single Cell Multiome ATAC + تعبير الجينات: https://cdn.10xgenomics. com/image/upload/v1666737555/support-documents/CG000338_ ChromiumNextGEM_Multiome_ATAC_GEX_User_Guide_RevF.pdf. مجموعة Next GEM للخلايا المفردة 3′ الإصدار 3.1: https://support.10xgenomics.com/تمت معالجة التعليق الخلوي المفرد وفقًا لبروتوكول مجموعة Next GEM Single Cell 3′ Kit v.3.1. ثم تم تجميع المكتبات المرمزة بالباركود وتسلسلها على نظام Illumina NovaSeq 6000 مع خلايا التدفق المرتبطة.
استخراج الحمض النووي الجينومي. تم الحصول على عينات أنسجة الورم من عينات تم استئصالها جراحيًا. بعد إزالة قطعة للتحضير للخلايا المفردة الطازجة، تم تجميد العينة المتبقية بسرعة في النيتروجين السائل وتخزينها فيقبل استخراج الحمض النووي بكميات كبيرة، تم تجميد العينات وطحنها (Covaris) وتقسيمها لاستخراج الكميات الكبيرة. تم استخراج الحمض النووي الجينومي من عينات الأنسجة باستخدام مجموعة DNeasy Blood and Tissue (Qiagen، 69504) أو مجموعة QIAamp DNA Mini (Qiagen، 51304). تم تنقية الحمض النووي الجينومي من خلايا الدم المحيطية وحيدة النواة المجمدة باستخدام مجموعة QIAamp DNA Mini (Qiagen، 51304) وفقًا لتعليمات الشركة المصنعة (Qiagen). تم تقييم كمية الحمض النووي باستخدام الفلورية باستخدام اختبار Qubit dsDNA HS (Q32854) وفقًا لتعليمات الشركة المصنعة (Thermo Fisher Scientific). البروتوكولات متاحة على protocols.io.https://doi.org/10.17504/protocols.io.bsnhndb6) .
تحليل WES. تم تكسير حوالي 100-250 نانوغرام من الحمض النووي الجينومي على جهاز Covaris LE220 مستهدفًا إدخالات بطول 250 قاعدة. تم بناء مكتبات مؤتمتة ذات فهارس مزدوجة باستخدام مجموعة تحضير مكتبة KAPA Hyper (روش) على منصة SciClone NGS (بيركن إلمر). تم تجميع ما يصل إلى عشرة مكتبات بنسب متساوية من حيث الكتلة قبل الالتقاط الهجين المستهدف.مجموعة مكتبة. تم دمج مجموعات المكتبة باستخدام كاشف xGen Exome Research Panel v.1.0 (IDT Technologies)، الذي يغطيالمنطقة المستهدفة (19,396 جين) من الجينوم البشري. تم تهجين المكتبات لـقبعةتبع ذلك غسل صارم لإزالة شظايا المكتبة المدمجة بشكل زائف. تم استرجاع شظايا المكتبة الغنية وتم إجراء تحسين دورة PCR لمنع التضخيم المفرط. تم تضخيم المكتبات الغنية باستخدام خليط KAPA HiFi (روش) قبل التسلسل. تم تحديد تركيز كل مجموعة مكتبة تم التقاطها من خلال qPCR باستخدام مجموعة قياس مكتبة KAPA وفقًا لبروتوكول الشركة المصنعة (روش) لإنتاج عدد الكتل المناسب لجهاز Illumina NovaSeq-6000. بعد ذلك، تم إنتاج قراءات مزدوجة الطرف تستهدف 12 جيجابايت من التسلسل لتحقيق حواليالتغطية لكل مكتبة.
تحضير مكتبة زينيوم والتصوير. تم قطع كتل البارافين (كتل FFPE) عندوتم وضعها على شرائح زينيوم وفقًا لدليل تحضير الأنسجة FFPE (10x Genomics، CG000578، Rev B). هؤلاء تمت معالجة الشرائح بسلسلة من الغسلات باستخدام الزيلين والإيثانول لإزالة البارافين وفك الروابط، باستخدام معزز الأنسجة FFPE كما هو موضح (10x Genomics، CG000580، Rev B). تم إجراء تهجين البروبي في الموقع طوال الليل باستخدام 379 بروب من لوحة الأنسجة المتعددة البشرية Xenium.الجينوميات، 1000626) بالإضافة إلى 100 مجس مخصص إضافي (الجدول التكميلي 6). بعد ربط المجسات، خضعت العينة لتكبير الدائرة المتدحرجة، وتم إخماد الخلفية باستخدام خليط من الفلورسنت التلقائي. تم صبغ النوى بـ DAPI لتحسين تتبع العينة وتقدير حدود الخلايا (10x Genomics، CG000582، Rev D). تم تحميل هذه العينات، جنبًا إلى جنب مع المحاليل والمواد الاستهلاكية لفك الشفرة، في محلل Xenium (10x Genomics، 1000481). تم بدء التشغيل باستخدام الإرشادات المقدمة (10x Genomics، CG000584، Rev C). تم تصوير هذه المراسلين الفلوريين الذين تم تزاوجهم مع المناطق التكميلية المستهدفة من cDNA الدائري المشفر. تم إجراء صبغ H&E على نفس المنطقة بعد الانتهاء من التشغيل.
طرق تحليلية
التكميم والتحليل الإحصائي. تم إجراء جميع تحليلات البيانات في بيئات R و Python. تم تقديم تفاصيل الوظائف والمكتبات المحددة في أقسام الطرق ذات الصلة أعلاه. تم تحديد الأهمية باستخدام اختبار ويلكوكسون لمجموع الرتب، اختبار النسبة، اختبار الهيبرجيومتري، أو اختبار ارتباط بيرسون، حسب الاقتضاء.القيم < 0.05 اعتُبرت ذات دلالة إحصائية. تفاصيل الاختبارات الإحصائية موضحة في أساطير الأشكال وأقسام الطرق ذات الصلة.
معالجة بيانات WES. تم معالجة ملفات FASTQ باستخدام trimGalore (الإصدار 0.6.7؛ مع المعلمات: –length 36 وجميع المعلمات الأخرى مضبوطة على القيم الافتراضية؛https://github.com/FelixKrueger/TrimGaloreتم محاذاة ملفات FASTQ بعد ذلك إلى الجينوم المرجعي البشري GRCh38 الخاص بمركز بيانات الجينوم (GRCh38.d1.vd1) باستخدام BWA-mem (الإصدار 0.7.17) مع المعامل -M وجميع المعاملات الأخرى مضبوطة على الإعدادات الافتراضية. تم تحويل ملف SAM الناتج إلى ملف BAM باستخدام samtools.https://github.com/samtools/samtools;v.1.14) عرض مع المعلمات -Shb، وجميع الآخرين تم تعيينهم على الإعدادات الافتراضية. تم فرز ملفات BAM وتم وضع علامات على التكرارات باستخدام أداة SortSam من Picard (v.2.6.26) مع المعلمات التالية: CREATE_INDEX=true، SORT_ORDER=coordinate، VALIDATION_STRINGENCY=STRICT، وجميع الآخرين تم تعيينهم على الإعدادات الافتراضية؛ وMarkDuplicates مع المعلمة REMOVE_DUPLICATES=true، وجميع الآخرين تم تعيينهم على الإعدادات الافتراضية. ثم تم فهرسة ملفات BAM النهائية باستخدام samtools (v.1.14) index مع تعيين جميع المعلمات على الإعدادات الافتراضية.
استدعاء الطفرات باستخدام تسلسل الجينوم الكامل. تم استدعاء الطفرات الجسدية من بيانات تسلسل الجينوم الكامل باستخدام خط أنابيب Somaticwrapper (الإصدار 2.2؛https://github“. com/ding-lab/somaticwrapper)، والذي يتضمن أربعة مكالمات مختلفة: Strelka (الإصدار 2.9.10)موتيكت (الإصدار 1.1.7)فار سكان (الإصدار 2.3.8) و Pindel (الإصدار 0.2.5) لقد احتفظنا بالمتغيرات أحادية النوكليوتيد (SNVs) الموجودة في الإكسونات التي تم تحديدها بواسطة أي اثنين من أدوات MUTECT (الإصدار 1.1.7) وVarScan (الإصدار 2.3.8) وStrelka (الإصدار 2.9.10) والإضافات والحذف (indels) التي تم تحديدها بواسطة أي اثنين من أدوات VarScan (الإصدار 2.3.8) وStrelka (الإصدار 2.9.10) وPindel (الإصدار 0.2.5). بالنسبة للمتغيرات أحادية النوكليوتيد والإضافات والحذف المدمجة، قمنا بتطبيق و حد أدنى من التغطية لقصور الورم والأنسجة الطبيعية، على التوالي. كما قمنا بتصفية SNVs و indels بناءً على VAF أدنى قدره 0.05 في الأورام و VAF أقصى قدره 0.02 في العينات الطبيعية. كما قمنا بتصفية أي SNV ضمن 10 قواعد من indel الموجود في نفس عينة الورم. أخيرًا، قمنا بإنقاذ الطفرات النادرة التي تتراوح VAFs الخاصة بها بين 0.015 و 0.05 بناءً على قائمة توافق الجينات المعتمدة.. في خطوة لاحقة، استخدمنا Somaticwrapper لدمج SNVs المتجاورة في تعدد أشكال النوكليوتيدات المزدوجة باستخدام COCOON (https://github.com/ding-lab/COCOONSكما ورد في دراسة سابقة.
تعيين الطفرات إلى بيانات snRNA-seq و ST. استخدمنا أداة داخلية تُدعى scVarScan التي يمكنها تحديد القراءات التي تدعم الأليل المرجعي والأليل المتغير الذي يغطي موقع الطفرة في كل خلية من خلال تتبع معلومات شريط الخلية والجزيئات في snRNA-seq و تسلسل RNA أحادي الخلية (scRNA-seq) أو ملف Visium bam. الأداة متاحة مجانًا على GitHubhttps://github.com/ding-lab/10Xmapping). لاستخدامها في رسم الخرائط، استخدمنا الطفرات الجسدية عالية الثقة من بيانات تسلسل الإكسوم الكامل (WES) التي تم إنتاجها بواسطة Somaticwrapper (الموصوفة أعلاه). تم تصفية قراءات Visium مسبقًا باستخدام العلامة ‘xf:i:25’ للقراءات التي تساهم في حسابات المعرفات الجزيئية الفريدة.
اختبار إحصائي لمعدل الطفرة الجينية (VAF) في الفضاء. لكل قسم من أقسام ST، قمنا بتطبيق مجموعتين من الاختبارات الإحصائية على جميع الطفرات الجسدية المستندة إلى WES التي تم تحديدها في ST. أولاً، بالنسبة لكل طفرة تحتوي على أكثر من 30 قراءة من التغطية على ST عبر جميع النقاط، تم حساب VAF لجميع نقاط منطقة الورم وجميع نقاط المنطقة غير الورمية كعدد القراءات المتغيرة عبر جميع النقاط مقسومًا على عدد القراءات الكلية عبر جميع النقاط. ثم تم إجراء اختبار ثنائي الحدين باستخدام VAF لنقاط غير الورم كخلفية: binom.test(alternative=”greater”). بعد ذلك، تم إجراء اختبار النسبة بين VAFs في أنواع فرعية مكانية مختلفة باستخدام prop.test(alternative=”two.sided”). أخيرًا، تم إجراء تصحيح للاختبارات المتعددة على كلا المجموعتين من الاختبارات باستخدام الدالة p.adjust().
استدعاء CNV باستخدام WES. تم استدعاء CNVs الجسدية باستخدام GATK (الإصدار 4.1.9.0). على وجه التحديد، تم تقسيم الجينوم البشري المرجعي hg38 (بوابة بيانات NCIGDC) إلى فترات مستهدفة باستخدام وظيفة PreprocessIntervals، مع تعيين طول الفترات إلى 1,000 قاعدة واتباع قاعدة دمج الفترات OVERLAPPING_ONLY. ثم تم إنشاء لوحة من العينات الطبيعية باستخدام كل عينة طبيعية كمدخل ووظائف GATK CollectReadCounts مع الوسيطة –interval-merging-rule OVERLAPPING_ONLY، تليها CreateReadCountPanelOfNormals مع الوسيطة –minimum-interval-median-percentile 5.0. بالنسبة لعينات الأورام، تم عد القراءات التي تداخلت مع الفترة المستهدفة باستخدام وظيفة GATK CollectReadCounts. ثم تم توحيد عدد قراءات الأورام وإزالة الضوضاء باستخدام وظيفة GATK DenoiseReadCounts، مع تحديد لوحة العينات الطبيعية بواسطة –count-panel-of-normals. تم إنشاء العد الأليلي للأورام للمتغيرات الموجودة في af-only-gnomad.hg38.vcf وفقًا لأفضل ممارسات GATK (تم تصفية المتغيرات بشكل إضافي إلى أفوتمت معالجة الإدخالات المميزة بـ ‘PASS’ باستخدام وظيفة GATK CollectAllelicCounts. ثم تم نمذجة القطاعات باستخدام وظيفة GATK ModelSegments، مع استخدام نسبة النسخ المنقحة وعدد الأليلات الورمية كمدخلات. ثم تم استدعاء نسب النسخ للقطاعات على مناطق القطاعات باستخدام وظيفة GATK CallCopyRatioSegments.
أدوات السريرتم استخدام التقاطع لرسم نسب عدد النسخ من المقاطع إلى الجينات ولتعيين التضخيمات أو الحذف المبلغ عنها. بالنسبة للجينات التي تتداخل مع مقاطع متعددة، تم استخدام برنامج بايثون مخصص لتصنيف تلك الجينة على أنها مضخمة أو محايدة أو محذوفة بناءً على نسبة عدد النسخ الموزونة المحسوبة من نسب النسخ لكل مقطع متداخل، وأطوال التداخلات وعتبة النقاط المستخدمة في دالة CallCopyRatioSegments. إذا كانت النتيجةكانت قيمة قطع الدرجات ضمن نطاق الافتراضيعتبات النقاط المستخدمة بواسطة CallCopyRatioSegments (الإصدار 0.9، 1.1)، ثم حدود الإعداد الافتراضيتم استخدام عتبات النقاط بدلاً من ذلك (م replicating منطق دالة CallCopyRatioSegments).
معالجة بيانات ST. لكل عينة، حصلنا على مصفوفة الميزات-الباركود غير المفلترة لكل عينة من خلال تمرير ملفات FASTQ المفككة والصورة المرتبطة H&E إلى Space Ranger (الإصدار 1.3.0، 2.0.0 و 2.1.0 باستخدام أمر ‘count’ مع المعلمات الافتراضية مع تمكين إعادة توجيه الصور) ومرجع الجينوم GRCh38 المبني مسبقًا 2020-A (GRCh38 و Ensembl 98). تم استخدام Seurat لجميع التحليلات اللاحقة. قمنا بإنشاء كائن Seurat باستخدام وظيفة Load10X_Spatial لكل شريحة. ثم تم توسيع كل شريحة وتطبيعها باستخدام وظيفة SCTransform لتصحيح تأثيرات الدفعة. أي تحليل مدمج أو تقسيم لاحق للخلايا والعينات لعينة تحتوي على عدة شرائح خضعت لنفس طريقة التوسيع والتطبيع. تم تجميع النقاط باستخدام خوارزمية لوفيان الأصلية، وأبعاد تحليل المكونات الرئيسية الثلاثين العليا باستخدام FindNeighbors. وظائف FindClusters و FindClusters كما هو موضح في كتيب ‘تحليل، تصور، ودمج مجموعات البيانات المكانية باستخدام Seurat’ من Seurathttps://satijalab.org/seurat/articles/spatial_vignette.html).
تم استخدام InferCNV و CalicoST لاستدعاء CNV على بيانات Visium ST. لاكتشاف CNVs الكروموسومية واسعة النطاق باستخدام scRNA-seq و snRNA-seq وبيانات Visium، تم استخدام InferCNV (الإصدار 1.10.1) مع المعلمات الافتراضية الموصى بها لبيانات 10x Genomics.https://github.com/broadinstitute/inferCNV“). تم تشغيل InferCNV على مستوى العينة وفقط باستخدام بيانات مصفاة بعد مراقبة الجودة باستخدام مصفوفة العد الخام. بالنسبة لبيانات snRNA-seq و scRNA-seq، تم استخدام جميع الخلايا غير الخبيثة كمرجع مع التسمية ‘غير ورمي’ وكانت جميع الخلايا الخبيثة تحمل نفس التسمية ‘ورمي’، مع المعلمات التالية: analysis_mode=”subclusters”، –cluster_by_groups=T، –denoise=T، و –HMM=T. بالنسبة لبيانات Visium ST، تم استخدام 200 نقطة تم تصنيفها على أنها ‘غير خبيثة’ مع أدنى درجة نقاء ESTIMATE كمرجع، وكانت النقاط ‘الخبيثة’ تحمل معرف الميكرو منطقة كتصنيف، مع المعلمات التالية: window_length، analysis_mode=”sample”، –cluster_by_groups=T، –denoise=T، و –HMM=T. CalicoST (https:// github.com/raphael-group/CalicoST) تم تشغيله على بيانات Visium ST بنفس توضيح الإدخال (معرف الميكرومنطقة). تم اعتبار جميع النقاط من نفس الميكرومنطقة كأصغر وحدة تحليل. ثم تم تشغيل CalicoST مع المعلمات الافتراضية وتم فحص النتائج يدويًا.
حساب درجة تشابه ملف عدد النسخ. لتحديد التشابه بين ملفي CNV مكانيين، نستخدم درجة تشابه جاكارد المعدلة. تم تعريف ملف CNV على أنه مجموعة من النوافذ الجينومية مع عدد النسخ المحايد (0) أو التضخيم (1) أو الحذف (-1). ثم تمت مقارنة ملفي CNV، وتم تقسيم النوافذ الجينومية المتداخلة بحيث تحتوي كلا الملفين على نفس مجموعات النوافذ (مع الوظيفة المخفضة من حزمة GenomicRanges v.1.46.1). ثم تم تعريف درجة تشابه CNV (Sim) كما يلي:
أينيشير إلى حجم النافذة الجينوميةيشير إلى توضيح CNV (0، 1 أو -1) للبروفايلفي النافذة الجينومية، و يشير إلى توضيح CNV للملف B في النافذة الجينوميةعبر جميع النوافذ الجينومية حيث لا يكون A أو B محايدًا بالنسبة لتغير عدد النسخ.
لتحديد التشابه بين ملف CNV المكاني و CNV المستند إلى WES (المتعلق بالشكل التوضيحي الإضافي 5a)، استخدمنا درجة تشابه تتوسط الحساسية (نسبة CNVs المستندة إلى WES التي تم اكتشافها أيضًا في CNVs المكانية) والخصوصية (نسبة CNVs المكانية التي تتفق مع CNVs المستندة إلى WES). على وجه التحديد،
أينيشير إلى حجم النافذة الجينوميةمن CNV المكاني،يشير إلى حجم النافذة الجينوميةمن CNV قائم على WES،يشير إلى توضيح CNV (0، 1 أو -1) للملف A في النافذة الجينومية.
تحديد تحت الاستنساخ المكاني بناءً على تشابه ملف تعريف CNV. في سير عمل OCT (الشكل التوضيحي التكميلي 1a)، قامت CalicoST بتحديد CNVs في الوقت نفسه وتجميع الميكرو مناطق في تحت الاستنساخ المكاني. في سير عمل FFPE، تم أولاً اختيار أحداث CNV المكاني الموثوقة في كل ميكرو منطقة من خلال مقارنتها مع WES المطابق. ثم، تم حساب درجة تشابه CNV بشكل زوجي عبر جميع الميكرو مناطق الورم. أخيرًا، تم تجميع الميكرو مناطق باستخدام درجات تشابه CNV باستخدام دالة hclust.تشابه CNV، الطريقة=”ward.D2″)، وتم تقسيمها إلى مجموعات باستخدام دالة cutree (تمت مراجعة التعيينات النهائية للفرعيات يدويًا لتجنب التجميع المفرط ولإزالة ملفات CNV الشاذة الصغيرة.
تحديد مناطق الورم الدقيقة وتحديد الطبقات. باستخدام تقنية Visium ST، تم تحديد مناطق الورم الدقيقة من خلال عملية متعددة الخطوات باستخدام صبغة H&E. تم تصنيف كل نقطة ST إما على أنها نسيج داعم أو ورم من خلال مراجعة الشكل الظاهري يدويًا على المقاطع الملونة بصبغة H&E. إذا كان هناك على الأقلإذا كانت بكسلات ضمن شكل خلية خبيثة مغطاة، تم تصنيف البقعة على أنها ورم. خلاف ذلك، تم تصنيفها على أنها نسيج داعم. بعد ذلك، قمنا بتعريف ميكرو مناطق ورمية متميزة باستخدام مجموعة من ثلاث قواعد. القاعدة الأولى تحدد أن بقع الورم المتجاورة مباشرة يتم تمييزها في البداية كمنطقة ورمية واحدة. القاعدة الثانية تنص على أنه إذا كانت منطقتان ورميتان متميزتان تشغلان معًا ما لا يقل عن 50% من بقعة واحدة، يتم تخصيص البقعة للمنطقة الورمية المتميزة ذات النسبة الأعلى المشغولة. أخيرًا، القاعدة الثالثة تحدد أنه إذا كان هناك فرق مورفولوجي واضح بين بقع الورم داخل منطقة ورمية واحدة، يجب فصل المنطقة الميكروية إلى مناطق ميكروية متميزة، واحدة لكل مورفولوجيا واضحة.
بعد ذلك، قمنا بتشغيل مجموعة أدوات Morph (https://github.com/ding-lab/المورف)، الذي يستخدم الشكل الرياضي لتنقيح الميكرو مناطق الورم. أي أنه إذا كان العدد الإجمالي للبقع في منطقة ميكرو أقل من أو يساوي ثلاثة، فإننا نُصنّف جميع هذه البقع على أنها نسيج داعم. أخيرًا، قام المورف بتعيين الطبقة (على سبيل المثال، T1) لكل بقعة من بقع منطقة الورم الميكروية من خلال سلسلة من عمليات الشكل الرياضي الموصوفة في طريقة تحليل ارتباط عمق البقعة، والتي تشير إلى عمق بقعة معينة داخل منطقة ميكرو.
حساب متوسط مساحة البقعة وحجم الميكرومنطقة. لحساب المساحة التي تشغلها كل بقعة، استخدمنا حجم البقعة ( ) والمسافة من مركز إلى مركز بين كل نقطة ( ) مقدمة من الجينومياتhttp://kb.10xgenomics.com/hc/ar/articles/360035487572-ما-هي-دقة-المساحة-وتكوين-منطقة-الالتقاط-لشريحة-تعبير-الجينات-Visium-v1-). كما هو موضح في الشكل التوضيحي الإضافي 6، تشكل بقع فيزيوم شبكة سداسية تغطي العينة. الوحدة المتكررة في هذه الشبكة هي شكل شبه منحرف مركزي عند مركز كل بقعة، وتتكون من ثمانية مثلثات متساوية الأضلاع. كل مثلث له جانب من (نصف المسافة من المركز إلى المركز). باستخدام معادلة مساحة المثلثات المتساوية الأضلاع وضربها في 8، حصلنا على مساحة كل شبه منحرف كـ، وهو متوسط المساحة التي تشغلها كل بقعة. لحساب حجم الميكرومنطقة، قمنا بضرب عدد البقع في 8,660 وقسمنا علىللحصول على الحجم في.
تقدير كثافة الميكرو منطقة. قمنا بتقدير كثافة الميكرو منطقة لكل قسم من خلال اتباع الصيغة: الكثافة لكلالميكرومنطقة لكل حجم قسم (بالنقاط) ثم مقسوم على لكل نقطة. ثم الكثافة لكل الكثافة لكلالميكروإقليم لكل.
“تعيين نوع الخلية. تم تعيين نوع الخلية بناءً على العلامات المعروفة التالية: خلايا B، CD79A، CD79B، CD19، MS4A1، IGHD، CD22 وCD52؛ cDC1، CADM1، XCR1، CLEC9A، RAB32 وC1orf54؛ cDC2، CD1C، FCER1A، CLEC10A وCD1E؛ mregDC، LAMP3، CCR7، FSCN1، CD83 وCCL22؛ pDC، IL3RA، BCL11A، CLEC4C وNRP1؛ البلعميات، CX3CR1، CD80، CD86، CD163 وMSR1؛ خلايا الص mast، HPGD، TPSB2، HDC، SLC18A2، CPA3 وSLC8A3؛ الخلايا البطانية، EMCN، FLT1، PECAM1، VWF، PTPRB، ACTA2 وANGPT2؛ الخلايا الليفية، COL1A1، COL3A1، COL5A1، LUM وMMP2؛ الخلايا المحيطية، RGS5، PLXDC1، FN1 وMCAM؛ خلايا NK، FCGR3A، GZMA وNCAM1؛ خلايا البلازما، CD38، SDC1، IGHG1، IGKC وMZB1؛ خلايا T، IL7R، CD4، CD8A، CD8B، CD3G، CD3D وCD3E؛ وخلايا T التنظيمية، IL2RA، CTLA4، FOXP3، TNFRSF18 وIKZF2. تم تعيين الخلايا الظهارية الطبيعية في الثدي بالعلامات التالية: LumSec، GABRP، ELF5، CL28، KRT15، BARX2 وHS3ST4؛ LumHR، ANKRD3OA، ERBB4، AFF3، TTC6، ESR1، NEK10 وXBP1؛ والأساسية، SAMD5، FBXO32، TP63، RBBP8 وKLHL13. تم تعيين الخلايا الظهارية الطبيعية في الكبد بالعلامات التالية: الخلايا الكبدية، ALB، CYP3A؛ خلايا القناة الصفراوية، SOX9، CFTR و PKD2. خلايا الظهارة الطبيعية في البنكرياس، بما في ذلك القنوات، والحويصلات، وجزرجزيرة صغيرةو جزيرةتمت إضافة تعليقات على الخلايا باستخدام SingleR (الإصدار 1.8.1) باستخدام بيانات مرجعية BaronPancreasData (‘البشر’).
تحليل ارتباط عمق البقع. لقد حددنا ارتباطًا بين تعبير الجينات وعمق البقع في منطقة الورم الدقيقة الخاصة بها. أولاً، تم تعيين عمق لكل بقعة يُعرف بأنه المسافة إلى أقرب بقعة مواجهة للبيئة المجهرية للورم. تم قياس هذا العمق في عدة طبقات من خلال عملية تكرارية حيث تم اعتبار جميع البقع الخبيثة المجاورة مباشرة للبقع غير الخبيثة كطبقة 1، ثم تم اعتبار جميع البقع الخبيثة المجاورة مباشرة للطبقة 1 كطبقة 2، وتم تكرار العملية حتى تم تعيين رقم طبقة لجميع البقع. إذا كانت طبقة البقعة أكبر من أقصر مسافة بين البقعة وأي حدود من حدود Visium (بما في ذلك حافة نافذة التقاط Visium، حافة شريحة الأنسجة وأي بقع فارغة داخل الشريحة)، فإننا استبعدنا مثل هذه البقع، حيث كنا نعرف فقط الحد الأعلى لعمق هذه البقعة. بالإضافة إلى ذلك، تم استبعاد المناطق الدقيقة للورم التي تحتوي على أقل من 3 طبقات أو 50 بقعة من التحليل. تم أخذ المسافة بين الطبقات كمسافة مركز إلى مركز لبقع Visium. ).
لإعطاء نفس الوزن للمناطق الكبيرة والصغيرة، تم تطبيع عمق كل نقطة بشكل إضافي بواسطة الحد الأقصى لعمق الميكرومنطقة التي تنتمي إليها هذه النقطة. ثم قمنا بإجراء اختبارات الارتباط الجزئي بشكل مستقل بين تعبير الجين (تم الكشف عن ما لا يقل عن 1 ترانسكريبت من الجين في أكثر منمن جميع النقاط) وعمق كل نقطة بشكل موحد، مع نقاء الورم كمتغير مصاحب على النحو التالي:
حيث إن كسر الطبقة هو رقم الطبقة مقسومًا على العدد الإجمالي للطبقات في الورم لتطبيع المناطق الدقيقة الكبيرة والصغيرة، و rho هو معامل ارتباط الطبقة، و هو معامل الارتباط لنقاء التغاير. تم استنتاج النقاء باستخدام فك التشفير عندما كانت هناك بيانات متطابقة من تسلسل RNA أحادي الخلية (نسبة الورم المفككة لكل نقطة بواسطة RCTD)، أو باستخدام ESTIMATE (أي، تقدير درجة نقاء الورم لكل نقطة) في الحالات الأخرى. تم التحقق من كل جين مقابل مجموعة من قوائم الجينات غير الخبيثة المستمدة من تسلسل RNA أحادي الخلية لضمان أن التغيير في النسبة لم يكن ناتجًا عن تحول في تركيبة نوع الخلايا. أخيرًا، قمنا بإجراء تعديلات على الاختبارات المتعددة لجميع الاختبارات التي أجريت في كل قسم من أقسام ST.
تحليل إثراء مسار GSEA لعمق النقاط. لتلخيص البرامج البيولوجية الغنية في مركز وأطراف الميكرو مناطق الورم عبر المقاطع، حصلنا أولاً على معامل الارتباط المتوسط على مستوى المجموعة. إذا لم يكن الاختبار ذا دلالة إحصائية (تم تعيين قيمة rho لتكون 0 للإشارة إلى عدم وجود ارتباط. إذا لم يتم إجراء اختبار على قسم ما (<50% من النقاط تحتوي على نص واحد على الأقل)، تم أيضًا تعيين rho كـ 0. عندما كان هناك حالة تحتوي على أقسام متعددة، أخذنا أولاً متوسط rho عبر الأقسام لتجنب التحيز نحو الأورام التي تحتوي على المزيد من الأقسام. ثم تم حساب متوسط rho لكل مجموعة (جميع العينات أو العينات من كل نوع من أنواع السرطان).
بنفس الطريقة، تم حساب إحصائيات الرتبة لكل اختبار كـقيمة“رو” للاختبارات مع. للاختبارات مع أو الجينات التي لم يتم اختبارها، كانت إحصائية الرتبة 0. ثم قمنا بحساب متوسط إحصائيات الرتبة لكل حالة، تليها المتوسط لكل نوع من أنواع السرطان. أخيرًا، مع القائمة الكاملة لإحصائيات الرتبة المحسوبة لجميع الجينات المختبرة، استخدمنا الدالة GSEA (المعلمات: pvalueCutoff=0.5؛ الحزمة: clusterProfiler v.3.18.1) للحصول على درجة الإثراء المنظم لمسارات Hallmark (الحزمة: msigdbr 7.5.1) من MSigDB.. أخيرًا، فقط المسارات التي تحتوي على تم الاحتفاظ بها في النتائج النهائية.
تصنيف الجينات المرتبطة بالورم وغير المرتبطة بالورم. نستخدم التعبير التفاضلي ومرشحات نسبة التعبير، مقارنة التعبير بين أنواع الخلايا في بيانات snRNA-seq المتطابقة لتوصيف الجينات المحددة في تحليل التركيز في المركز والمحيط بشكل أكبر. الخطوات المنفذة في هذه العملية أدت إلى توليد أربعة الفئات: محددة للأورام، محددة للستروما، غنية بالأورام وغنية بالستروما (الشكل التوضيحي التكميلي 8أ، ب). تم تصنيف الجينات التي لم تتجاوز الحد الأدنى المهم في أي تحليل للتعبير التفاضلي بشكل منفصل على أنها ليست جينات تعبير تفاضلي (DEG).
لتمييز هذه المجموعات الأربع، قمنا أولاً بإجراء تحليل جيني تفاضلي لأنواع الخلايا في بيانات snRNA-seq المتطابقة، وتم تصفيتها بواسطة حد دلالة تقليدي. (تغيير الطي) ، تم تعديلهاتصحيح بونفيروني)، للحصول على الجينات المعبر عنها بشكل مختلف (DEGs) (الشكل التوضيحي التكميلي 8a). نظرًا للاختلاف في الأورام، قد توجد بعض الجينات الخاصة بالأورام فقط في مجموعة فرعية من الأورام. لذلك، قمنا أولاً بتقسيم مجموعات الأورام إلى مجموعات فرعية (باستخدام وظيفة Subcluster في Seurat مع دقة 0.5) للحصول على مجموعات فرعية للأورام. ثم قمنا بمقارنة كل مجموعة فرعية مع جميع الخلايا غير الورمية الأخرى. تم اعتبار الجين DEG ورميًا إذا أظهرت مجموعة فرعية واحدة على الأقل من الأورام تعبيرًا كبيرًا مقارنة بالخلايا غير الورمية، والعكس صحيح بالنسبة للجينات غير الورمية DEGs (الشكل التوضيحي التكميلي 8a، b).
بالنسبة للجينات المحددة للورم أو السدى، تم تصنيف DEG على أنه محدد للورم إذا استوفى كلا من المعايير التالية: (1) هو DEG عند مقارنته بجميع أنواع الخلايا غير الورمية من مجموعة فرعية واحدة على الأقل من الأورام؛ و (2) كانت تعبيره في جميع أنواع الخلايا غير الورمية (الشكل التوضيحي 8c).
تم تطبيق العكس على الجينات المعبر عنها بشكل خاص في الخلايا الداعمة للمرشحين. إذا لم يستوفِ الجين المعبر عنه بشكل خاص كلا الشرطين ليكون محددًا للورم أو الخلايا الداعمة، فقد تم تصنيفه إما كغني بالورم أو غني بالخلايا الداعمة بناءً على ما إذا كان مستوى التعبير أعلى في أنواع خلايا الورم أو الخلايا الداعمة (الشكل التوضيحي التكميلي 8أ).
تحليل استجابة العلاج المحددة حسب تحت الاستنساخ المكاني. ركزنا على عشر حالات (تتكون من أربع عينات BRCA، عينتين CRC وأربع عينات PDAC) تحتوي على عدة تحت استنساخ مكاني لهذا التحليل. للحصول على الجينات المعبر عنها بشكل مختلف المحددة حسب تحت الاستنساخ، استخدمنا FindMarkers من الدالة في Seurat مع خيار اختبار ‘wilcox’ للجينات المعبر عنها بشكل مختلف بين كل تحت استنساخ وTME. ثم طبقنا الحد الأدنى للتعديل.متوسط (تغيير الطي) ونسبة التعبير في نوع خلية واحد على الأقل > 0.4 لاختيار الجينات المعبر عنها بشكل ملحوظ. لاستنتاج استجابة العلاج، استخدمنا قاعدة بيانات الاضطراب LINCS L1000 (المرجع 65)، وبالتحديد مجموعة بيانات LINCS_L1000_Chem_Pert_down من Enrichr.لتقييم تداخل مجموعة الجينات بين الجينات المعبر عنها بشكل مرتفع في النسخ الفرعية المكانية والجينات المعبر عنها بشكل منخفض بعد معالجة المركب. لإنشاء الرسم في الشكل التكميلية 4، قمنا بترتيب البيانات حسب ‘نسبة الأودds’ واختيار أفضل المركبات من كل نسخة فرعية. تم الحصول على بيانات المركب المقابلة، بما في ذلك آلية العمل، من CLUE (clue.io‘موارد CMap الموسعة إصدار 2020’) لإضافة تعليق على خريطة الحرارة.
قائمة الجينات المحظورة الخاصة بالأعضاء لأنواع الخلايا غير الخبيثة. لتمييز النسخ المتماثلة التي تنشأ من الخلايا السرطانية مقابل الخلايا السدوية أو المناعية غير الخبيثة، استخدمنا بيانات snRNA-seq المدمجة لكل عضو (الثدي، الكلى، الكبد والبنكرياس) لتحليل علامات نوع الخلية. استخدم هذا التحليل وظيفة FindAllMarkers في Seurat مع خيار اختبار ‘wilcox’. بعد ذلك، قمنا بتنقيح قائمة الجينات من خلال تطبيق عوامل تصفية مثل المتوسط (تغير الطي) > 2، نسبة التعبير في نوع خلية واحد على الأقلوتم تعديلهاقيم لضمان اختيار علامات قوية لكل نوع من الخلايا. القائمة الناتجة للجينات متاحة في الجدول التكميلية 5. كانت هذه القائمة أساسية في استبعاد جينات الخلايا غير السرطانية من التحليلات المتعلقة بأنماط التعبير الخاصة بالسرطان، مثل تحليل تشابه الميكرومنطقة الثنائي. ومن الجدير بالذكر أنه خلال التحليل، لاحظنا خريطة ملحوظة لمختلف أنواع الخلايا الظهارية في مجموعة بيانات مرجعية snRNA-seq لـ BRCA عند استخدام طريقة فك التشفير RCTD. من المحتمل أن تنبع هذه الملاحظة من الأنواع الفرعية المتنوعة لـ BRCA الموجودة في المجموعة. لمعالجة ذلك، اخترنا دمج جميع أنواع الخلايا الظهارية في فئة واحدة أثناء تحديد علامات نوع الخلايا واستبعدناها من القائمة السوداء. بالنسبة للأورام التي تنشأ من أعضاء غير الأربعة المذكورة أعلاه، قمنا بتجميع جميع الجينات الموجودة في قائمة سوداء عبر الأعضاء لتشكيل قائمة سوداء شاملة متعددة الأعضاء، والتي ساعدت في تصفية النسخ غير السرطانية.
تحليل ملف النسخ المجهري للميكرومنطقة. من أجل تنوع الورم العام، اخترنا النقاط المحددة بواسطة Morph ثم قمنا بتشغيل ROGUE (الإصدار 1.0)لقياس التباين كـ 1-ROGUE. ثم قمنا بمقارنة الملفات التعبيرية للميكرو مناطق من خلال اختيار أعلى 500 ميزة متغيرة بعد استبعاد مناطق السدى في عينات ST بعد معالجة Morph. شمل تقييمنا الأولي إجراء اختبارات ارتباط بيرسون لكل زوج من الميكرو مناطق، باستخدام مجموعة من أعلى 250-1,500 جين متغير مع زيادات قدرها 250 (أي، 250،لقد لاحظنا ارتباطات متسقة تقريبًا لجميع القيم بعد استخدام أكثر من 500، مما دفعنا لاختيار أفضل 500 جين لهذا التحليل. قلل هذا الاختيار من خطر اختيار عدد قليل جدًا من الجينات المتغيرة (على سبيل المثال، <250 من أكثر الجينات المتغيرة) مع تجنب أيضًا تضمين العديد من الجينات ذات التأثير الضئيل على الملف التعبيري. تم إجراء تحليل GSEA باستخدام الدالة GSEA (المعلمات: pvalueCutoff = 0.5؛ الحزمة: clusterProfiler v.3.18.1) للحصول على درجة الإثراء المنظم لمسارات Hallmark (الحزمة: msigdbr v.7.5.1) من MSigDB..
حساب درجة الوحدة. تم حساب درجات الوحدة في أعلى كل خريطة حرارية في الشكل البياني الموسع 6 باستخدام وظيفة AddModuleScore من Seurat.باستخدام الجينات المدرجة في كل خريطة حرارية. يمثل هذا الدرجة مستويات التعبير المتوسطة لمجموعة من الجينات. تم حساب الدرجة لكل نقطة وتم استخدام مخطط صندوقي لعرض توزيع درجات الوحدة في كل ميكرو منطقة.
تحليل نوع الخلية في تقنية ST. تم فك تركيب نوع الخلية لكل نقطة باستخدام RCTDمع المعلمات الافتراضية ووضع الدوبلت = ‘متعدد’. كانت المرجعية لكل تجربة هي أنواع الخلايا المعلنة يدويًا من كائن Seurat لعينة snRNA-seq أو Multiome المطابقة. لقياس التوزيع المكاني لكل نوع من أنواع الخلايا، تم حساب متوسط نسبة نوع الخلايا من 6 طبقات (T3 وما فوق، T2، T1، E1، E2، E3 وما فوق) من كل ميكرو منطقة ورمية. لمقارنة تسلل TME التفاضلي بين السلالات الفرعية المكانية، تمت مقارنة نسبة نوع الخلايا من جميع النقاط بين السلالات الفرعية المكانية باستخدام اختبار ويلكوكسون للرتب المزدوجة مع تعديل FDR.
التفاعل الخلوي المكاني عند حدود الورم. قمنا بتقييم التفاعل الخلوي المكاني (CCI) في عينة ST باستخدام COMمع قاعدة بيانات CellChat وحدود المسافة لـ“، مع اتباع نفس العتبة المستخدمة في النشر الأصلي لـ Visium. تم مقارنة الإشارات الوسيطة للمرسل والمستقبل لكل عائلة تفاعل بين جميع نقاط حدود الورم (بما في ذلك طبقة حدود الورم وطبقة حدود البيئة المجاورة للورم) وجميع النقاط غير الحدودية (اختبار ويلكوكسون لرتب المجموعات) على عينة. تعتبر مسارات التفاعل التي تزيد فيها الفروق في الإشارات عن 0.1 و FDR أقل من 0.05 غنية بشكل ملحوظ بالحدود. تم تحديد جينات التعبير التفاضلي الحدودية باستخدام وظيفة Find Markers على ثلاث مجموعات من المقارنات: الحدود/الورم، الحدود/البيئة المجاورة للورم، والحدود/جميع النقاط غير الحدودية. تحتوي جينات التعبير التفاضلي الحدودية على تعديل القيمة 0.25 في اختبار الحدود/غير الحدود، و (تغيير الطي) في الاختبارين الآخرين.
محاذاة الأقسام التسلسلية وحساب عامل التفرع. قمنا بتطبيق PASTE2 (المرجع 70)، أداة المحاذاة المعتمدة على ST المحدثة PASTEلتمكين محاذاة جزئية للصورة. تم محاذاة مقاطع متسلسلة من نفس قطعة الورم بشكل ثنائي باستخدام الإعدادات الافتراضية. حصلت كل نقطة بيانات فيزيوم في كل قسم من أقسام ST على إحداثيات جديدة، تُعرف باسم و ، استنادًا إلى نتائج المحاذاة. ثم حددنا أقرب نقطة في كل قسم مجاور لكل نقطة، موصلين إياها على طول المحور. سهلت هذه العملية ربط النقاط عبر جميع الأقسام علىالمحور. لتقييم ما إذا كانت ميكرومنطقة واحدة متصلة بأخرى في قسم مجاور، قمنا أولاً بإزالة بقع النسيج الداعم ثم قمنا بعدّ البقع المتصلة. إذا كانت أي ميكرومنطقة في قسم واحد متصلة بـ القسم التالي الذي يحتوي على أكثر من ثلاثة مواقع مشتركة، ثم اعتبرنا هذين الميكروتينين، الموجودين في أقسام مختلفة، متصلين في الفضاء ثلاثي الأبعاد ويشكلان نفس حجم الورم. تم تسمية هذا الاتصال بالحجم 1، الحجم 2، وهكذا في الأشكال (الشكل 5d، e و الشكل الإضافي 9a-d).
استخدمنا مقياسين هندسيين لوصف حجم الورم: الاتصال والحلقة. بالنسبة للاتصال (الدرجة)، يقيس هذا المقياس عدد الاتصالات من منطقة ميكرو فردية إلى الأقسام المجاورة. على سبيل المثال، إذا كانت المنطقة الميكرو 2 في القسم 2 متصلة بـ 3 مناطق ميكرو في القسم 1 و2 في القسم 3، فإن اتصالها هو 5. الحد الأقصى من الاتصال في حجم الورم هو أعلى اتصال بين مناطقه الميكرو. بالنسبة للحلقة، تم حساب هذا المقياس كإجمالي عدد الاتصالات ناقص إجمالي عدد المناطق الميكرو زائد واحد، مما يحدد الهياكل المعقدة للحلقات داخل حجم الورم.
تسجيل مقاطع Visium وCODEX وH&E المتسلسلة. قبل التسجيل، خضعت بيانات التصوير للتحولات التالية. تم تحويل الصور المتعددة إلى صور بتدرج الرمادي لشدة DAPI. ثم تم تقليل حجم الصورة بنسبة 5 قبل اختيار النقاط الرئيسية. تم استخدام صور H&E (التي تم تقليل حجمها أيضًا بنسبة 5) لاختيار النقاط الرئيسية مع بيانات Visium.
للتسجيل، استخدمنا BigWarp، الذي تم تعبئته في تطبيق برنامج Fiji/ ImageJ. لتسجيل كل مجموعة من الصور المتسلسلة، استخدمنا القسم التسلسلي الأول كصورة ثابتة والصورة الثانية كصورة متحركة. بعد أن تم تشويه الصورة الثانية لتتناسب مع الصورة الأولى، تم استخدام الصورة الثانية كصورة ثابتة لتحويل الصورة الثالثة. استمر تسجيل النقاط الرئيسية بهذه الطريقة لجميع الصور في تجربة القسم التسلسلي. تم اختيار ما مجموعه 4-20 نقطة رئيسية لكل تحويل صورة. بمجرد اختيار النقاط الرئيسية، تم تصدير حقل متحرك من BigWarp لكل تحويل صورة. ثم تم تكبير هذا الحقل الكثيف للإزاحة بعامل 5 حتى يمكن استخدامه لتشويه بيانات التصوير بدقة كاملة. ثم تم استخدام حقل الإزاحة الكثيف بدقة كاملة لتسجيل بيانات الملتقط أو Visium المقابلة. الكود المستخدم للتسجيل متاح على GitHub (https://github.com/ding-lab/فطر/شجرة/تقديم النسخة الفرعية).
معالجة مدخلات تحديد الحي. بمجرد تسجيل بيانات التصوير، تم معالجتها بالطريقة التالية قبل إدخالها في النموذج.
بالنسبة لبيانات Visium ST، تم تحديد الجينات لتكون الجينات المعبر عنها في حد أدنى من 5% من النقاط عبر جميع الأقسام المتسلسلة وكانت أعداد التعبير تم تحويلها. تم تطبيع بيانات CODEX و Visium ST و H&E عن طريق طرح متوسط التعبير وقسمة الناتج على الانحراف المعياري لكل جين.
تم توليد ملفات التعبير لكل رقعة بشكل مختلف للبيانات الأصلية للصورة (CODEX و H&E) والبيانات المعتمدة على النقاط (Visium). تم حساب التعبيرات لرقع CODEX و H&E كمتوسط كثافات البكسل لكل قناة صورة على جميع البكسلات داخل حدود الرقعة. تم حساب رقع Visium بنفس الطريقة إلى حد كبير؛ ومع ذلك، تم وزن ملف التعبير لكل نقطة داخل الرقعة بشكل خطي حسب مسافتها من مركز الرقعة. ساعد هذا الوزن التفاضلي في حساب التباين في التعبير بسبب عدد النقاط التي تقع ضمن حدود الرقعة.
معمارية نموذج تحديد الحي. يتكون نموذج توضيح الحي من مشفر تلقائي مع هيكل محول بصري (ViT) (الشكل التوضيحي التكميلي 7). باختصار، المشفر التلقائي هو طريقة تدريب غير خاضعة للإشراف حيث يعمل المشفر (مكون التضمين) والمفكك (مكون إعادة البناء) معًا لتعلم كيفية توليد بيانات الإدخال. على وجه التحديد، يستنتج الشبكة تقريبًا،إلى دالة التوليد الخلفية الحقيقية،لإنتاج المخرجات، بناءً على المدخلات. كان المشفر المستخدم غير متماثل، مما يعني أن المشفر والمفكك لم يكونا نسخًا عكسية لبعضهما البعض. كان المشفر يتكون من ViT مشابه الهندسة المعمارية للهندسات المعمارية الموصوفة سابقًا (الجدول التكميلي 4).
تعمل نماذج فيت على رموز الصور كمدخلات. باختصار، رموز الصور هيتمثيلات بعدية لمناطق من صورة الإدخال. خلال التدريب، تم أخذ عينات من بلاطات الصور من توزيع موحد عبر مجموعة الأقسام المدخلة (الشكل التوضيحي التكميلي 7a). ثم تم تقسيم البلاطة المأخوذة إلى مناطق، حيث تم تحديد عدد المناطق بواسطة معلمين فائقين: ارتفاع المنطقة (ph) وعرض المنطقة (pw). ثم تم تسطيح كل منطقة إلىمتجه، حيثهو عدد القنوات في الصورة (في حالة بيانات النسخ الجيني المكاني، هو عدد الجينات). ثم تم دمج الأجزاء غير المطوية في مصفوفة، حيث هو عدد الرقع في صورة البلاطة. كل صف في هذه المصفوفة هو رمز يمثل رقعة في صورة البلاطة. ثم تم إسقاط الرموز بواسطة طبقة خطية لتشكيل ، حيث هو بعد كتل المحولات.
بعد ذلك، تم دمج رمز الشريحة مع رموز التصحيح. كان رمز الشريحة (الذي يمثل الشريحة التي تم اختيار الصورة منها) مفهرسًا من تمثيل قابل للتدريب بحجمشرائح، حيث n_slides هو عدد الشرائح في تجربة القسم التسلسلي. الدافع وراء رمز الشريحة هو أنه مع تمريره عبر كتل المحولات، جنبًا إلى جنب مع رموز الباتش، يمكن تبادل المعلومات بين جميع الرموز، مما يسمح لرمز الشريحة بتعلم الانتباه إلى التمثيلات المفيدة للباتشات. هذه الميزة سمحت للنموذج بأن يكون أكثر قوة تجاه تأثيرات الدفعة بين الأقسام التسلسلية. بعد إضافة رمز الشريحة، تم إضافة تضمينات موضعية لجميع الرموز وتم تمريرها عبر كتل المحولات التي تتكون منها مشفر ViT. جميع المتغيرات المذكورة أعلاه وتفاصيل بنية المحول متاحة في الجدول التكميلية 4.
بعد أن مرت عبر المشفر، تم تمثيل الرقع كتمثيل بحجم. كانت الخطوة التالية في الهيكل المعماري هي تخصيص الأحياء. تم تخصيص الأحياء للقطع بطريقة هرمية، مما يعني أن القطعة تم تصنيفها إلى عدة أحياء تختلف في مستوى التحديد. لكل مستوى من مستويات هرم الأحياء، كانت المستويات اللاحقة تتكون من تقسيمات للأحياء في المستويات السابقة، أي أنه باستثناء المستوى الأول، كانت كل حي مجموعة فرعية من حي في مستوى سابق من الهيكل. من أجل هذه التحليل، أنشأ النموذج ثلاثة مستويات من الأحياء، كل منها لديه القدرة على اكتشاف ما يصل إلى (المستوى 1)، (المستوى 2) و (المستوى 3) الأحياء، على التوالي. بالنسبة لهذا التحليل، جميع الأحياء المعروضة هي أحياء تم توضيحها في مستوى التسلسل الهرمي 3. احتوى النموذج على ثلاثة دفاتر رموز (واحد لكل مستوى) بحجم الأحياء، حيث NBHDs هو عدد الأحياء الممكنة التي يمكن تعيينها للمستوى المعطى. تم إسقاط تجسيدات الباتشات الناتجة عن مشفر ViT بواسطة ثلاثة كتل مستقلة من الطبقات الخطية (واحدة لكل مستوى) التي تخرج احتمال تعيين كل باتش إلى حي معين. ثم تم استخدام هذه الاحتمالات لاسترجاع تجسيدات الأحياء من كتاب الرموز المقابل لمستوى الحي. ثم تم استخدام ثلاثة كتل خطية (واحدة لكل مستوى) لإعادة بناء تجسيدات الباتشات بشكل مستقل عند كل مستوى إلى القيم الأصلية لبكسلات كل باتش. الكود المستخدم لتدريب النموذج متاح على GitHub.https://github.com/ding-lab/فطر/شجرة/تقديم النسخة الفرعية).
دالة خسارة النموذج. تحتوي دالة الخسارة العامة على مساهمتين رئيسيتين: متوسط مربع الخطأ (MSE) في إعادة بناء قطع الإدخال، وخسارة الانتروبيا المتقاطعة على التوزيع المشفر والتوزيع الطبيعي بمتوسط 0 وتباين 1.0.
خلال التدريب، كان المشفر التلقائي يحاول في الوقت نفسه تحسين مهمتين رئيسيتين: إعادة بناء ملف التعبير لكل جزء من الصورة وتوافق تسميات الجوار بين الأقسام المجاورة. أجبرت هاتان الهدفان المتنافسان النموذج على تعلم أنماط التعبير التمثيلية مع الحفاظ أيضًا على توافق الأحياء بين الأقسام المدخلة، مما ساعد لمكافحة الفروق بين الأحياء بسبب تأثيرات الدفعات. تم قياس الفروق في تعبير الرقع بواسطة MSE، بينما تم فرض قرب الأحياء من خلال تقليل الانتروبيا المتقاطعة للرقع المتجاورة مع بعضها البعض فيالتوجيه أثناء التدريب.
تم تعريف دالة الخسارة العامة أدناه:
أين (حد أقصى 0.01) و (مضبوطة على 1.0) هي مقاييس لفقدان الجوار ( ) وخسارة إعادة البناء ( )، على التوالي. خلال التدريب، تم زيادة الخط بشكل خطي من 0 إلى قيمته القصوى.
تدريب النموذج والاستدلال. تم تدريب نموذجين منفصلين لـ HT397B1 (ست شرائح H&E، أربع شرائح CODEX وشرحتان Visium ST) و HT268B1 (أربع شرائح Visium ST). تم تقديم معلمات التدريب الفائقة، مثل حجم الدفعة وعدد خطوات التدريب، في الجدول التكميلي 4. بالنسبة لـ HT268B1، تم تدريب حالة واحدة فقط لأنه كان هناك نوع بيانات واحد فقط. بالنسبة لـ HT397B1، تم تدريب ثلاث حالات نموذجية (واحدة لكل نوع بيانات) وتم دمجها لاحقًا وفقًا للإجراء الموصوف في القسم ‘بناء وتكامل الجوار ثلاثي الأبعاد’.
بعد التدريب، تم تنفيذ استدلال النموذج على بلاطات الصور المتداخلة لكل شريحة باستخدام نافذة منزلقة بحجم 8 وخطوة 2 (أي، 2 رقعة متداخلة بين بلاطات الصور).تم استخراج بقع المركز لكل بلاطة وإعادة ترتيبها لتتناسب مع اتجاه الشريحة الأصلية. كانت كل صورة ‘تضمين بقعة معاد بناؤها’ بدقة 50 بكسل. (أي أن كل قطعة حي تمثل منطقة تكون عرض) باستثناء Visium ST، حيث كانت دقة الباتش 100 بكسل.
بناء وتكامل الأحياء ثلاثية الأبعاد. بعد تعيين الأحياء لكل قسم، تم استيفاء الشرائح لتوليد حجم حي ثلاثي الأبعاد. لهذا، استخدمنا الاستيفاء الخطي لاحتمالات تعيين الأحياء باستخدام مكتبة torchio..
بعد التداخل، قمنا أيضًا بدمج أحجام الأحياء المجاورة لـ HT397B1، والتي تم من خلالها إنشاء أحجام متعددة محددة لنوع البيانات باستخدام نهج التجميع القائم على الرسم البياني. باختصار، تم تحديد جميع تعليقات الفوكسل المجاورة المتداخلة. ثم تم بناء رسم بياني، حيث تمثل العقد كل مجموعة من تقسيمات الأحياء المجاورة، وتكون الحواف هي المسافة (في ملف التعبير) بين هذه المجموعات. ثم تم تجميع هذا الرسم البياني باستخدام خوارزمية تجميع الرسم البياني Leiden لتحديد الأحياء المجاورة المدمجة. تم تقديم المعلمات الفائقة لعملية التجميع أعلاه في الجدول التكميلي 4.3. تم عرض الأحياء المجاورة ثلاثية الأبعاد باستخدام أداة التصور مفتوحة المصدر Napari (https:// github.com/napari/napari).
تحليل وقياس الأحياء المجاورة ثلاثية الأبعاد. ثم تم تعيين الأحياء المجاورة لنقاط Visium ST بالطريقة التالية. تم تعيين كل نقطة بتسمية الحي المجاور الذي يتداخل مع مركز نقطة الحي.
لتركيز على الأحياء المجاورة الأكثر ارتباطًا بعلم الأحياء TME، قمنا بتصفية الأحياء المجاورة التي لديها >50% تداخل مع النسخ الفرعية المعلنة عن عدد النسخ. بالإضافة إلى ذلك، استبعدنا الأحياء المجاورة التي تم رسمها على أقل من عشرة نقاط إجمالية عبر جميع أقسام ST لعينة.
تم تعريف منطقة حدود النسخ الفرعية لنسخ الأورام على أنها اتحاد الطبقة الخارجية من النقاط المعلنة عن النسخ الفرعية والنقاط التي تمتد طبقة واحدة خارجها، مما يمثل منطقة تقريبًا عند واجهة الورم-TME. تم حساب النسب المحددة للنسخ الفرعية كالتداخلات المجاورة مع الطبقة الخارجية لكل نسخة فرعية.
في HT397B1، تم حساب DEGs لجميع الأحياء المجاورة، وليس فقط تلك التي تم تصفيتها لتداخل النسخ الفرعية وعدد النقاط. تم تجميع أعلى 50 DEG لـ
تم تجميع الأحياء المجاورة 4 و 6 في ثلاث فئات: مشتركة، فريدة من نوعها للحي 4 وفريدة من نوعها للحي 6. للعرض في الشكل 5، تم اختيار أعلى 10 لكل مجموعة للعرض بناءً على معايير الفرز التالية. تم حساب متوسط فرق التعبير بين الأحياء المجاورة 4 و 6 لكل جين عن طريق طرح متوسط التعبير في الحي 6 من الحي 4. تم ترتيب DEGs المشتركة بطريقة تصاعدية بناءً على فرق التعبير المطلق لكل جين. تم ترتيب الجينات الفريدة للحي 4 والحي 6 حسب فرق التعبير المتوسط بطريقة تنازلية وتصاعدية، على التوالي.
تسمية نوع الخلية لبيانات تصوير CODEX. تتكون سيرتنا العملية لتسمية الخلايا من أربع خطوات رئيسية: (1) تحويل تنسيق الصورة، (2) تقسيم الخلايا، (3) توليد الميزات المكانية و (4) تصنيف نوع الخلية. أولاً، قمنا بتحويل مخرجات الصورة من منصة CODEX (.qptiff) إلى تنسيق OME-TIFF الشهير مفتوح المصدر. خلال هذه العملية، قمنا أيضًا بإنتاج صورة منفصلة لكل عينة، حيث يتم أحيانًا تضمين أقسام متعددة من الأنسجة في نفس عملية التصوير. ثم استخدمنا نموذج تقسيم النوى + الغشاء المدرب مسبقًا Mesmer في إطار DeepCell لتقسيم النوى والخلايا الكاملة. تم استخدام DAPI كصورة كثافة النوى، وتم تجميع القنوات pan-cytokeratin و HLA-DR و SMA و CD4 و CD45 و Hep-Par-1 و CD31 و E-cadherin و CD68 و CD3e، بالنسبة لتلك الموجودة في صورة معينة، إلى قناة واحدة متوسطة واستخدمت كصورة كثافة الغشاء.
ثم نستخدم إجراء تصنيف لتحديد أنواع الخلايا. أولاً، لمكافحة الاختلافات في توزيعات كثافة البروتين بين عمليات التصوير وأنواع الأنسجة، تم تعيين عتبات يدويًا لجميع قنوات البروتين المستخدمة أثناء تصنيف الخلايا لكل صورة من خلال الفحص البصري. فوق هذه العتبة الكثافة، اعتُبر البكسل إيجابيًا لعلامة معينة، ودونها، اعتُبر البكسل سلبيًا. ثم استخدمنا حدود تقسيم الخلايا من الخطوة السابقة لحساب نسبة البكسلات الإيجابية لجميع علامات تصنيف الخلايا في كل خلية. نتيجة هذه العملية هي مصفوفة ميزات (عدد الخلايا عدد البروتينات) تمثل نسب العلامات الإيجابية لكل بروتين تصنيف خلايا في كل خلية. اعتُبرت الخلية إيجابية لعلامة إذا كانت من بكسلاتها إيجابية لتلك العلامة. ثم تم تصنيف الخلايا باستراتيجية تصنيف محددة لكل عينة. خلال التصنيف، تم إخضاع كل خلية لسلسلة من بوابات AND، حيث إذا اجتازت الخلية جميع المعايير لخطوة معينة، تم تصنيفها كنوع الخلية المحدد لتلك الخطوة، بينما إذا فشلت في المعايير، تم تمريرها إلى الخطوة التالية في استراتيجية التصنيف. تم تقديم استراتيجيات التصنيف المستخدمة للعينات في هذه الورقة في الجدول التكميلي 4.
كانت التسميات التالية هي مجموعة جميع تسميات أنواع الخلايا الممكنة: الظهارية، خلية CD4 T، خلية CD8 T، خلية T تنظيمية، خلية T، بلاعم، بلاعم-M2، خلية B، خلية دندريتية، مناعية، بطانية، ليفية وكبدية. بالنسبة لبعض الصور، لم تكن جميع البروتينات المطلوبة لتصنيف نوع خلية معينة موجودة. على سبيل المثال، لم يكن CD4 موجودًا في كل لوحة صورة وكان متاحًا للاستخدام في تصنيف خلايا CD4 T. في هذه الحالات، تم بناء استراتيجية التصنيف بحيث يمكن تصنيف الخلايا كأنواع خلايا أكثر عمومية إذا لم تكن البروتينات المحددة موجودة (أي، تم تصنيفها بشكل أوسع كخلية T بدلاً من خلية CD4 T). إذا كانت الخلية سلبية لجميع الخطوات في استراتيجية التصنيف، تم تصنيفها على أنها ‘غير مصنفة’. يمكن العثور على الشيفرة لتحويل تنسيق الصورة وتقسيم الخلايا على GitHub (https://github.com/estorrs/ خط أنابيب التصوير المتعدد).
قياس المسافة إلى حدود الورم على CODEX. بعد التسجيل، تم رسم نقاط Visium المسمى كأورام على شرائح CODEX باستخدام إحداثيات الصور المتراصة. كانت إحداثيات مركز كل نقطة في الشريحة المتراصة CODEX هي نفسها نظيرتها في Visium. احتلت كل نقطة في الشريحة المتراصة CODEX منطقة دائرة نصف قطرها 150 بكسل. ثم تم حساب تحويلات المسافة الإقليدية في الشريحة المتراصة CODEX لكل بكسل باستخدام scipy.ndimage.distance_transform_edt من Python. تم حساب كل من المسافات من الميكرو مناطق وداخل الميكرو مناطق.
إعادة بناء حجم الورم ثلاثي الأبعاد وقياس الموقع. تم إنشاء تصورات شبكة السطح لأحجام الأورام لـ HT397B1 و HT268B1 باستخدام الخطوات التالية: (1) اختيار حي الورم، (2) بناء الشبكة و (3) تلوين الشبكة. أولاً، تم اعتبار الأحياء المجاورة المدمجة مع مقاييس الورم (الموصوفة أدناه) التي تتجاوز عتبة معينة جزءًا من حجم الورم. في HT268B1، كانت المقياس المستخدم لقياس الطابع الظهاري هو نسبة النقاط المعلنة عن النسخ الفرعية Visium ST لكل حي. تم اعتبار الأحياء المجاورة التي تحتوي على نقاط النسخ الفرعية ورم. في HT397B1، استخدمنا بدلاً من ذلك نسبة الخلايا الظهارية المعلنة عن CODEX، حيث كانت أقسام CODEX تفوق أقسام Visium ST لتلك العينة. تم اعتبار الأحياء المجاورة التي تحتوي على نسبة الخلايا الظهارية ورم.
ثم تم بناء حجم جديد حيث تم اعتبار الأحياء المجاورة المصنفة كأحياء ورم باستخدام المعايير أعلاه فوكسلات إيجابية للورم، وتم اعتبار جميع الفوكسلات الأخرى سلبية للورم. تم بعد ذلك تنعيم قناع الورم ثلاثي الأبعاد باستخدام نواة غاوسية (سيغما ). ثم تم استخدام القيم الناتجة كمدخلات لخوارزمية المكعبات المتحركة لإنشاء شبكة سطح لحجم الورم. استخدمنا تنفيذ scikit-image (skimage.measure.marching_cubes) لخوارزمية المكعبات المتحركة مع المعلمات الافتراضية.
لتلوين شبكة السطح، قمنا بإنشاء أحجام ميزات ثلاثية الأبعاد (الموصوفة أدناه)، ثم قمنا بتلوين النقاط على شبكة السطح بناءً على قيمة الفوكسل في الموقع المقابل في حجم الميزة. حجم الميزة هو حجم حيث يصف كل فوكسل في الحجم بعض الميزات من مجموعة بيانات الأقسام التسلسلية (على سبيل المثال، تعبير جين معين، نسبة الخلايا، وما إلى ذلك). تم بناء أحجام الميزات المستخدمة في هذا التحليل بالطريقة التالية. أولاً، في الأقسام التسلسلية التي كان فيها ميزة قابلة للتطبيق، تم تجميع الميزة بنفس دقة الأحياء المجاورة ثلاثية الأبعاد (في هذه الحالة). تم بعد ذلك استيفاء الميزة المجمعة فيتوجيه لملء الفجوات بين الأقسام. كان الحجم الناتج بنفس شكل حجم الحي المتكامل، حيث كانت قيمة كل فوكسل هي العدد المجمع للميزات للفوكسل. بالنسبة لـ HT268B1، كانت الميزات المستخدمة هي التعبير المسجل لـ TYMP1 و IGLC2. بالنسبة لـ HT397B1، استخدمنا نسبة الخلايا الليفية وخلايا المناعة. تم توضيح الخلايا كما هو موضح في قسم ‘توضيح نوع الخلايا لبيانات تصوير CODEX’. تم تصور شبكة السطح باستخدام Napari.https://github.com/تم ضبط التباين على أساس حجم إلى حجم. كما قمنا بتصوير حجم نسيج HT397B1 باستخدام منصة Imaris، حيث قمنا بإنشاء أسطح من علامات CODEX التالية: بان-سايتوكيرتين (ظهاري)، CD45 (مناعي) وSMA (دعمي).
تصميم مجس زينيوم. تم تصميم مجسات جينات وزرع زينيوم مخصصة باستخدام مصمم لوحة زينيوم.https://cloud.10xgenomics. com/xenium-panel-designer) اتباع التعليمات الموضحة في تعليمات ‘البدء مع تصميم لوحة زينيوم’ (https:// www.10xgenomics.com/support/in-situ-gene-expression/documenta-tion/steps/panel-design/xenium-panel-getting-started#design-tool). باختصار، تم جمع تسلسلات بطول 21 قاعدة تحيط بموقع المتغير المستهدف من النسخة الكنسية لـ Ensembl (Ensembl v.100). ثم تم تقييم جميع نقاط الربط الأربعة الممكنة (اثنان للأليل البري واثنان للأليل المتغير، وثلاثة في حالة الحذف – اثنان للأليل البري وواحد للأليل المتغير). تم استبعاد المواقع المتغيرة التي كانت متاحة فيها نقاط الربط غير المفضلة فقط (CG، GT، GG وGC). كانت القاعدتان في تسلسل نقطة الربط هما آخر قاعدة من RBD5 (مجال ربط RNA) وأول قاعدة من مجس RBD3. كانت نقاط الربط المفضلة دائمًا ذات أولوية على نقاط الربط المحايدة ما لم يكن من الضروري استخدام نقطة ربط محايدة لتجنب الشعرات، أو مناطق البوليمر المتجانس، أو الديمرات، أو درجة حرارة تزاوج غير مواتية. ثم تم تعديل أطوال المجسات لـ RBD5 وRBD3 من الطول الابتدائي البالغ 21 قاعدة لاستهداف درجة حرارة بين و لكل مسبار (الهدف العام و تم استبعاد المواقع المتغيرة التي تم التنبؤ بأنها ستشكل ثنائيات أو حلقات شعرية بواسطة محلل أوليغو الخاص بشركة IDT.
تم استبعاد المواقع المتغيرة التي تحتوي على مناطق بوليمرية متجانسة من خمسة قواعد متتالية أو أكثر في أي من مجسات RBD5 أو RBD3.
فك تشفير التعبير المكاني. هنا استخدمنا كل من نتائج فك التشفير والتعبيرات المحددة لنوع الخلية في بيانات snRNA-seq لفك تشفير بيانات التعبير في Visium ST (الشكل التوضيحي التكميلي 9). باختصار، بالنسبة لجين معين Gene1، قمنا أولاً بحساب متوسط التعبير لجين1 لكل نوع خلية في بيانات snRNA-seq المتطابقة، ثم قمنا بتصفية تعبير هذه الجينات في أنواع الخلايا التي تحتوي علىمن أعلى متوسط تعبير، ثم قسمة متوسط تعبير كل نوع خلية على مجموع جميع متوسطات التعبير، مما يؤدي إلى إنشاء مصفوفة مساهمة التعبير لكل نوع خلية ( ). ثم بالنسبة لنقطة معينة، تم ضرب المساهمة لكل نوع خلية في نسبة نوع الخلية من نتيجة تطور نوع الخلية (على سبيل المثال، RCTD)، ثم تم تطبيعها إلى 1 لإعطاء مصفوفة المساهمة النهائية للتعبير (WN). على سبيل المثال، في الشكل التكميلية 9a، الجين 1 لديه و المساهمات من أنواع الخلايا المعنيةو C بناءً على تعبير snRNA-seq المصفى. بالنسبة لـ Spot1، حيث يوجد نوع خلية واحد فقط، B، في البقعة،يعطي النهائيمساهمة الجين 1 في نوع الخلية ب في النقطة 1. النقطة 2 تحتوي علىأنواع الخلايا A و B، وبالتالي فإن مساهمة نوع الخلايا المعيارية في النقطة 2 هيلنوع الخلية A، ولنمط الخلية B. تم الحصول على التعبير النهائي المفكك عن طريق ضرب التعبير الأصلي لكل نقطة (5 و 20 في النقطة 1 والنقطة 2) مع المساهمة المستندة إلى نوع الخلية للحصول على القيم النهائية للتعبير المفكك للنقطة 1 – نمط الخلية B.سبوت 2 – نوع الخلية A، وسبوت 2 – نوع الخلية ب8.58.
ملخص التقرير
معلومات إضافية حول تصميم البحث متاحة في ملخص تقارير مجموعة نيتشر المرتبط بهذه المقالة.
يمكن الوصول إلى جميع برامج المعلوماتية الحيوية المستخدمة في هذه الدراسة من مستودع GitHub العام (https://github.com/ding-lab/ST_subclone_publication). الشيفرة الخاصة بتحديد وبناء الجوار ثلاثي الأبعاد متاحة على GitHub (https://github.com/ding-lab/معالجة تصوير متعدد الاستخدامات مستضافة على GitHub (https://github.com/estorrs/خط أنابيب التصوير المتعدد). الشيفرة الخاصة بـ Morph متاحة على GitHub (https://github.com/ding-lab/morph). 46. هيرندون، ج.، فيلدز، ر.، زو، د. س. ودينغ، ل. جمع ومعالجة العينات البيولوجية 2.0.بروتوكولات.آي أوhttps://doi.org/10.17504/protocols.io.bszynf7w (2021). 47. وو، ي. وآخرون. الكشف عن الخصائص الوراثية والترجمة الجينية يكشف عن علامات التقدم والمسارات الأساسية في سرطان الخلايا الكلوية الواضحة. نات. كوميونيك. 14، 1681 (2023). 48. هيوستن، أ.، تشين، س. وتشين، ف. النسخ الجيني المكاني لتقنية OCT باستخدام 10x Genomics Visium.بروتوكولات.آي أوhttps://doi.org/10.17504/protocols.io.x54v9d3opg3e/v1 (2023). 49. هيوستن، أ.، تشين، س. وتشين، ف. النسخ الجيني المكاني للأنسجة المحفوظة باستخدام تقنية 10x Genomics Visium.بروتوكولات.آي أوhttps://doi.org/10.17504/protocols.io.kxygx95ezg8j/v1 (2023). 50. كارافان، و.، جاياسينغه، ر. و الدين، ن. ن. بروتوكول WU sn-prep للأورام الصلبة – بروتوكول snRNA الإصدار 2.8.بروتوكولات.آي أوhttps://doi.org/10.17504/protocols.io.14egn7w6zv5d/v1 (2022). 51. جاياسينغ، ر.، كارافان، و.، هيوستن، أ. وألدين، ن. ن. بروتوكول WU sn-prep للأورام الصلبة – snRNA+ATAC المشترك v2.9.بروتوكولات.آي أوhttps://doi.org/10.17504/protocols. io. 261 جد (2023). 52. جاياسينغ، ر.، دينغ، ل. و تشين، ف. بروتوكول WU sc-prep للأورام الصلبة الإصدار 2.1.بروتوكولات.آي أوhttps://doi.org/10.17504/protocols.io.bsnqnddw (2021). 53. جاياسينغ، ر.، دينغ، ل.، تشين، ف. وساتوك. استخراج الحمض النووي بالجملة (مختبر دينغ).بروتوكولات.آي أوhttps://doi.org/10.17504/protocols.io.bsnhndb6 (2021). 54. كيم، س. وآخرون. ستريلكا2: استدعاء سريع ودقيق للمتغيرات الجرثومية والسمية. نات. ميثودز 15، 591-594 (2018). 55. سيبولسكي، ك. وآخرون. الكشف الحساس عن الطفرات النقطية الجسدية في عينات السرطان غير النقية والمتنوعة. نات. بيوتكنولوجي. 31، 213-219 (2013). 56. كوبولدت، د. س. وآخرون. فارسكان 2: اكتشاف الطفرات الجسدية وتغير عدد النسخ في السرطان من خلال تسلسل الإكسوم. أبحاث الجينوم. 22، 568-576 (2012). 57. يي، ك.، شولتز، م. هـ.، لونغ، ق.، أبويلر، ر. & نينغ، ز. باندل: نهج نمو النمط لاكتشاف نقاط الانكسار للحذف الكبير والإدخالات متوسطة الحجم من قراءات قصيرة مزدوجة النهاية. المعلوماتية الحيوية 25، 2865-2871 (2009). 58. بيلي، م. هـ. وآخرون. التوصيف الشامل لجينات السرطان المحركة والطفرات. خلية 173، 371-385.e18 (2018). 59. لي، ي. وآخرون. بروتوجينوميات السرطان الشامل تربط المحركات السرطانية بالحالات الوظيفية. خلية 186، 3921-3944.e25 (2023). 60. تيرخانوفا، ن. ف. وآخرون. التنظيم الجيني غير الوراثي خلال انتقالات السرطان عبر 11 نوعًا من الأورام. ناتشر 623، 432-441 (2023). 61. مكينا، أ. وآخرون. أداة تحليل الجينوم: إطار عمل MapReduce لتحليل بيانات تسلسل الحمض النووي من الجيل التالي. أبحاث الجينوم. 20، 1297-1303 (2010). 62. كوينلان، أ. ر. وهال، آي. م. BEDTools: مجموعة مرنة من الأدوات لمقارنة الميزات الجينومية. المعلوماتية الحيوية 26، 841-842 (2010). 63. ما، سي. وآخرون. استنتاج الشذوذات في عدد النسخ المحددة بالأليل وجغرافيا تطور الورم من النسخ الجينية المكانية. مسودة على bioRxivhttps://doi.org/10.1101/2024.03.09.584244 (2024). 64. ليبرزون، أ. وآخرون. مجموعة مجموعات الجينات المميزة في قاعدة بيانات التوقيعات الجزيئية (MSigDB). نظام الخلايا. 1، 417-425 (2015). 65. سوبرا مانيان، أ. وآخرون. خريطة الاتصال من الجيل التالي: منصة L1000 وأول 1,000,000 ملف. خلية 171، 1437-1452.e17 (2017). 66. كوليشوف، م. ف. وآخرون. إنريش: خادم ويب شامل لتحليل إثراء مجموعة الجينات، تحديث 2016. أبحاث الأحماض النووية. 44، W90-W97 (2016). 67. ليو، ب. وآخرون. مقياس قائم على الإنتروبيا لتقييم نقاء تجمعات الخلايا الفردية. نات. كوميونيك. 11، 3155 (2020). 68. هاو، ي. وآخرون. تحليل متكامل لبيانات الخلايا الفردية متعددة الأنماط. خلية 184، 3573-3587.e29 (2021). 69. كانغ، ز. وآخرون. فحص التواصل بين الخلايا في النسخ الجيني المكاني عبر النقل الأمثل الجماعي. نات. ميثودز 20، 218-228 (2023). 70. ليو، إكس، زيرا، ر. ورابائيل، ب. ج. محاذاة جزئية لبيانات النسخ الجيني الموزعة مكانياً متعددة الشرائح. أبحاث الجينوم 33، 1124-1132 (2023). 71. بوجوفيتش ج. أ.، هانسلوفسكي ب.، وونغ أ.، سالفيلد س. في مؤتمر IEEE الدولي الثالث عشر لتصوير الطب الحيوي (ISBI) 2016، 1123-1126 (2016) 72. دوسوفيتسكي، أ. وآخرون. الصورة تساويكلمات: المحولات للتعرف على الصور على نطاق واسع. مسودة مسبقة فيhttps://doi.org/10.48550/arXiv.2010.11929 (2021). 73. هو، ك. وآخرون. في مؤتمر IEEE/CVF لعام 2022 حول رؤية الكمبيوتر والتعرف على الأنماط (CVPR) 15979-15988 (IEEE، 2022). 74. بيريز-غارسيا، ف.، سباركس، ر. وأورسلين، س. تورتشIO: مكتبة بايثون لتحميل الصور الطبية بكفاءة، والمعالجة المسبقة، والتعزيز، وأخذ العينات المعتمدة على الرقع في التعلم العميق. طرق الحوسبة والبرامج في الطب الحيوي 208، 106236 (2021). 75. غرينوالد، ن. ف. وآخرون. تقسيم الخلايا الكاملة لصور الأنسجة بأداء على مستوى الإنسان باستخدام توضيح البيانات على نطاق واسع والتعلم العميق. نات. بيوتكنولوجي. 40، 555-565 (2022). 76. لوينر، ت.، لوبيس، هـ.، فييرا، أ. و تافاريس، ج. تنفيذ فعال لحالات مكعبات الزحف مع ضمانات طوبولوجية. مجلة أدوات الرسوم البيانيةhttps://doi.org/10.1080/10867651.2003.10487582 (2012). 77. لورنسن، و. إ. & كلاين، هـ. إ. مكعبات المسيرة: خوارزمية بناء سطح ثلاثي الأبعاد عالية الدقة. ACM SIGGRAPH Comp. Graph. 21، 163-169 (1987).
الشكر والتقدير تم تنفيذ هذا العمل كجزء من شبكة أطلس الأورام البشرية التابعة للمعهد الوطني للسرطان (HTAN). نشكر المرضى والموظفين والعلماء الذين ساهموا في هذه الدراسة؛ والموظفين في مراكز أعضاء HTAN، ومجموعة تحليل البروتينات السريرية للأورام، ومركز سايت مان للسرطان في جامعة واشنطن على دعمهم؛ و I. Strunilin، J. Wang،ي.لي، و.و. ليانغ و ي. لي على ملاحظاتهم حول المخطوطة. دعمت المنح التالية هذا العمل: U2CCA233303 لـ L.D. و R.C.F. و W.E.G.; U54AG075934 لـ L.D. و R.C.F. و F.C.; U24CA210972 لـ L.D.; R01NS107833 و R01NS117149 لـ M.G.C.; R01HG009711 و R01CA260112 لـ L.D. و F.C.; U24CA209837 لـ K.I.S.; K12CA167540 لـ L.-I.K; ومنح NIH/NCI U24CA248453 و U24CA264027 لـ B.J.R. C.M. هو زميل ديمون روني مدعوم من مؤسسة أبحاث السرطان ديمون روني (DRQ-15-22).
مساهمات المؤلفين تصميم الدراسة وتخطيطها: ل.د.، ف.س.، ب.ج.ر.، ر.س.ف. و و.إ.ج. تطوير وتنفيذ التجارب أو جمع البيانات: س.س.، أ.س.، أ. هيوستن، ج.م.، ر.ج.ج.، س.ب.ج.، أ.ت.م.أ.أ.، ب.س.، ج.م. هيرندون، و.إ.ج.، ر.س.ف.، ر.ب.، ف.س. و ل.د. الحسابات والتحليلات الإحصائية: ج.-ك.م.، ج.ل.، إ.س.، أ.ل.ن.ت.د.س.، س.م.، إكس.ل. و م.س.و. تفسير البيانات والتحليلات البيولوجية: ج.-ك.م.، ج.ل.، س.س.، إ.س.، أ.ل.ن.ت.د.س.، أ.ك.، أ.ن.س.-س.، ل.ف.، س.ل.، م.ب.، أ. هيل، ج.إ.آي.، ف.ت.، ج.م.هولد، إ.س.هـ.، إ.هـ.ك.، ب.أ.ب.، أ.هـ.ك.، م.م.م.، ك.ي.س.، س.ف.ب.، ت.ج.، م.أ.ر.، س.و.، ل.-إ.ك.، د.ج.ف.، م.ج.س.، ر.ب.، ك.س.ف.، و.إ.ج.، ر.س.ف.، ر.ج.ج.، ب.ج.ر.، ف.س. و ل.د. الكتابة: ج.-ك.م.، ج.ل.، س.س.، إ.س.، أ.ل.ن.ت.د.س.، م.د.إ.، ل.د. و م.س.و. الإدارة: ف.س. و ل.د.
المصالح المتنافسة يعلن المؤلفون عدم وجود مصالح متنافسة.
معلومات إضافية
معلومات إضافية النسخة الإلكترونية تحتوي على مواد إضافية متاحة فيhttps://doi.org/10.1038/s41586-024-08087-4. يجب توجيه المراسلات والطلبات للحصول على المواد إلى ويليام إي. جيلاندرز، ريان سي. فيلدز، بنيامين ج. رافائيل، فنغ تشين أو لي دينغ. تُعرب مجلة Nature عن شكرها لموريتز جيرستونغ، وآرتيم لوماكين، وكريستيان شورتش، والمراجعين الآخرين المجهولين، على مساهمتهم في مراجعة هذا العمل. معلومات إعادة الطباعة والتصاريح متاحة علىhttp://www.nature.com/reprints.
الشكل 1 من البيانات الموسعة | نظرة عامة على البيانات في مجموعات الأورام المكانية. أ، نظرة عامة على توفر البيانات لمجموعة الأورام المكانية مع أنواع الاختبارات، وأنواع السرطان، ومجموعة الدراسة. ب، 48 تسلسل RNA أحادي الخلية من كتل الأورام المتطابقة عبر 7 أنواع الأنسجة (يسار). توزيع أنواع الخلايا من أنواع الأنسجة متعددة العينات (يمين). ج، 22 الكشف المشترك عن طريق الفهرسة (CODEX) التصوير المناعي المتعدد الفلورية من مقاطع الورم المتطابقة.
الشكل البياني الممتد 2 | علم الأنسجة للفوج المتميز مكانيًا. علم الأنسجة للفوج المتميز مكانيًا ملون حسب نوع السرطان. وردي فاتح BRCA (عدد=23)، بنفسجي فاتحبرتقاليشريحةأزرق فاتحوبني فاتح.
الشكل البياني الموسع 3| علم الأنسجة لمجموعة الانتشار المكاني. علم الأنسجة لمجموعة الانتشار المكاني ملون حسب أنواع السرطان. وردي فاتح BRCA ( )، بنفسجي فاتح برتقاليأزرق فاتحبني فاتح“، وبرتقالي باهت.
توزيع حجم الميكرو منطقة في المجموعة المتميزة
نوع العينة: ورم
حد القطع المتوسط العالي
ن
نسبة_حسب_حجم_المجموعة
نقيلة
كبير
٢٨
٥٨.٣
نقيلة
متوسط
73
٢٧.٤
نقيلة
صغير
68
14.0
أساسي
كبير
20
٤١.٧
أساسي
متوسط
193
72.6
أساسي
صغير
419
٨٦.٠
نسبة_حسب_المتوسط_الأساسي
16.6
٤٣.٢
٤٠.٢
3.2
30.5
66.3
الشكل 4 من البيانات الموسعة | توزيع الميكرو مناطق والخصائص عبر توزيع عدد العينات لكل مجموعة، الأولية مقابل النقائل، نوع السرطان، وعدد الأقسام لكل عينة. توزيع حجم الميكرومنطقة بين العينات الأولية والنقائل (الأوليةأقسام من 60 حالة؛ نقيلةأقسام من 16 حالة). ج، جدول يحتوي على عدد الميكرو مناطق لكل حجم. د، الكثافة المقدرة للميكرو مناطق لكل كتلة نسيج. يتم الإشارة إلى عدد الميكرو مناطق، الأقسام، وحالة الأورام الأولية مقابل النقائل، سرطان كل كتلة على اليسار. هـ، أحجام الميكرو مناطق كمثال،، مخططات توزيع الحجم، والميكروإقليم ومجموعات الحجم المرئية على Visium ST لـ HT268B1-Th1K3U1 (BRCA) و HT260C1-Th1K1U1 (CRC) و HT270P1-H2U1 (PDAC).
البيانات الموسعة الشكل 5|التوصيف الجينومي للفرعيات المكانية. أ، تشابه ملف CNV الجينومي للفرعيات المكانية على مستوى الجينوم مقارنةً مع CNV المستنتج من WES المتطابق. ب، توزيع المتغيرات المرسومة على نسخ ST من جميع الطفرات الجسدية المكتشفة من WES المتطابق. Ref: الأليل المرجعي؛ Var: الأليل المتغير. ج، الطفرات الجسدية المرسومة على HT260C1 مشتركة وفريدة من نوعها لفرعيتين مكانيتيين. لكل طفرة مستمدة من WES، تم مقارنة VAFs بين مناطق الورم وغير الورم (اختبار ثنائي، يسار) وVAFs بين فرعيتين مكانيتيتين (اختبار النسبة، يمين). تم وضع علامة على الطفرات التي لديها FDR<0.05 (تصحيح بنجاميني وهوشبرغ) بـ* و** لفرق VAF وVAF، على التوالي. د، عينة من نقائل سرطان الثدي في الكبد (HT268B1) تظهر فرعيتين مكانيتيتين عبر 5 مقاطع. هـ، في خريطة الحرارة (الأعلى)، تظهر تقديرات التغيرات في عدد النسخ الجسدية (CNV) لكل نقطة. الأحداث المشتركة والفريدة من نوعها في CNV بين السلالات الفرعية المكانية. تُظهر ترددات الأليل-ب (BAF) في كل سلالة فرعية مكانية من نفس النافذة الجينومية في المسارات الوسطى، بينما تُظهر حالة CNV المستنتجة من snRNA وWES لنفس النافذة الجينومية في المسارات السفلية. ف، العلاقة النشوء والتطور المتوقعة بين السلالتين الفرعيتين للورم. ج، UMAP لمطابقة snRNA تُظهر أنواع الخلايا وسلالتين فرعيتين للورم. ح، الطفرات الجسدية المرسومة على HT268B1 مشتركة وفريدة من نوعها لسلالتين فرعيتين مكانيتين. حسابات VAF وتحليلاتها الإحصائية هي نفسها كما في اللوحة ج. ط-ي، الطفرة الفرعية EEF1A1.1324G يتم الكشف عن C بشكل فريد في Clone2 في النسخ الجينية المكانية، بينما يتم التعبير عن EEF1A1 في كلا النسختين الفرعيتين المكانية.اختبار النسبة ذو الجانبين).
الشكل 6 من البيانات الموسعة | نمط التعبير المكاني لمناطق الورم الدقيقة
مدفوع بالتعديل الجيني وعمق الورم. أ، تم تقييم تباين الورم بواسطة نظام تسجيل 1-ROGUE. تشير الدرجات الأعلى إلى تباينات أعلى. كل نقطة تمثل مقطعًا واحدًا وتلون حسب تصنيف المجموعة المعنية وعدد السلالات الفرعية.أقسام من 78 حالة). تمثل الخط المركزي في الرسم البياني الصندوقي الوسيط، مع وجود الحواف السفلية والعلوية التي تشير إلى الربع الأول والثالث. تمتد الشعيرات إلى أعلى وأدنى القيم ضمن 1.5 مرة من نطاق الربعين (IQR) من الحواف. ب، (اللوحة العلوية) توزيع معاملات بيرسون الزوجية بين أزواج الميكرو مناطق ضمن نفس القسم. يتم تقسيم التوزيع حسب تلك التي من نفس فرع الورم (أخضر) وتلك التي من فروع مختلفة (برتقالي). الرقم في الصناديق والخط الصلب يشير إلى متوسط كل توزيع. (اللوحة السفلية) عدد فروع الورم، الميكرو مناطق، والأقسام لكل كتلة نسيج. ج. معامل بيرسون الزوجي للميكرو مناطق بناءً على الأعلى
أكثر 500 جين متغير في القسم U1 من HT260C1-Th1H3. يبرز المربع الأحمر معاملات بيرسون الزوجية للمناطق الدقيقة داخل النسخة 1.d، درجات إثراء المسار للنسخة 1 (c1) والنسخة 2 (c2)، وTME، حيث يمثل حجم الفقاعة القيمة p المصححة (اختبار ويلكوكسون ذو الجانبين مع تعديل FDR). e، معامل الارتباط الجزئي رو (مع نقاء الورم كمتغير مشترك) و –-القيمة) بين مستوى التعبير والطبقة لجميع الجينات في نفس القسم. تشير العلاقة الإيجابية إلى تعبير أعلى في مركز الورم، بينما تشير العلاقة السلبية إلى تعبير أعلى في محيط الورم. يتم تصنيف الجينات باستخدام snRNA-seq المتطابق على النحو التالي: اللون الأرجواني للورم المحدد، والبرتقالي للورم الغني، والأخضر للغني بالستروما، والأرجواني الفاتح لغير DEG.f، الجينات الغنية في المركز والمحيط مع خطوط الارتباط وأنماط التعبير المكاني (ارتباط بيرسون). g، أعلى الجينات المشتركة الغنية في المركز والمحيط عبر أنواع السرطان (FDR و رو أو rho<-0.1) (الارتباط الجزئي مع إجراء بنجاميني-هوشبرغ).
البيانات الموسعة الشكل 7| التباين النسخي وإثراء المسارات في تحت الأورام. أ، معامل الارتباط بيرسون الثنائي بين أعلى 500 جين الأكثر تباينًا في جميع الأقسام الخمسة من الكتلة HT268B1-Th1H3. تم تصفية المناطق الدقيقة التي تحتوي على أقل من 10 نقاط لهذا التحليل. ب، تحليل إثراء مسار GSEA المميز لتحت الأورام مقارنةً ببيئة الورم في القسم الأول (U1) من HT268B1-Th1H3 (اختبار ويلكوكسون ذو الجانبين مع تعديل FDR). متوسط تعبير الجينات للجينات المرتفعة في تحت الأورام من تحليل GSEA ومثال على التعبير المكاني في ج، د، استجابة البروتين غير المطوي، و هـ، و و، مجموعة جينات MYC المستهدفة v1 في القسم الأول (U1). ز، ح، نقطة تفتيش G2M، و
مجموعة جينات الهدف MYC v1. (ط) تم تسليط الضوء على الجينات المشاركة في تكرار الحمض النووي (الأزرق الفاتح)، وتقدم دورة الخلية (الأخضر الفاتح)، وبدء الترجمة (الأحمر الفاتح). متوسط تعبير الجينات التي تم تنظيمها في فرعيات من تحليل GSEA ومثال على التعبير المكاني في (ج، هـ، ز، ط) (اللوحات العلوية) درجة الوحدة، أو مستوى التعبير المتوسط للبرنامج، تم حسابه باستخدام وظيفة Seurat AddModuleScore (الطريقة) باستخدام الجينات المدرجة في كل خريطة حرارية. يمثل الخط المركزي في الرسم البياني الصندوقي الوسيط، مع الإشارات السفلية والعلوية التي تشير إلى الربع الأول والثالث. تمتد الشعيرات إلى أعلى وأدنى القيم ضمن 1.5 مرة من النطاق بين الربعين (IQR) من الإشارات.
الشكل 8 من البيانات الموسعة | عمق الطبقة وتعبير الجينات في الورم
الميكرو مناطق. أ، مخطط لتعيين عمق الورم للمناطق الميكروية للورم التي تم قياسها بعدد الطبقات. بدءًا من حدود الورم/البيئة المجاورة للورم، يتم تعريف الطبقات القريبة من بقع الورم بشكل متكرر على أنها T1، T2، T3، إلخ، ويتم تعريف بقع البيئة المجاورة للورم البعيدة بشكل مشابه على أنها E1، E2، E3، إلخ. ب، العلاقة بين العمق (المقاس بعدد الطبقات الكلي) والحجم (المقاس بعدد البقع) لكل منطقة ميكروية. تمثل الخط المرجعي المتقطع العلاقة المتوقعة بين العمق والحجم للمناطق الدائرية المثالية. ج، عينة من سرطان الثدي الأولي تحتوي على منطقتين ميكرويتين للورم مؤهلتين لتحليل ارتباط الطبقات. تم تمييز البقع المستبعدة من التحليل بسبب قربها من الحافة الفيزيائية للأنسجة باللون الرمادي. د، الارتباط الجزئي. معامل رهو (مع نقاء الورم كمتغير مصاحب) و-قيمةبين مستويات التعبير والطبقات لجميع الجينات. تشير العلاقة الإيجابية إلى زيادة التعبير في مركز الورم، بينما تشير العلاقة السلبية إلى زيادة التعبير في محيط الورم. e، الجينات الغنية في المركز والمحيط مع خطوط الارتباط وأنماط التعبير المكاني (ارتباط بيرسون). يمثل الخط المركزي في الرسم البياني الصندوقي الوسيط، مع وجود الحواف السفلية والعلوية التي تشير إلى الربع الأول والثالث. تمتد الشعيرات إلى أعلى وأدنى القيم ضمن 1.5 مرة من نطاق الربع (IQR) من الحواف. باستخدام طريقة فك تعبير التعبير المكاني لاستنتاج التعبير المحدد لنوع الخلية، تم الحصول على نتائج متطابقة تقريبًا (الشكل التكميلي 9 والطرق).
الشكل البياني الموسع 9 | تسلل تحت الأنواع المكاني وتركيب الخلايا عبر طبقات الورم. أ، إحصائيات FDR لمقارنة مستوى التسلل حسب نوع الخلية بين أي زوجين من تحت الأنواع المكانية في نفس العينة. تم عرض جميع إحصائيات FDR المهمة (<0.05) لنوع الخلية من كل عينة هنا مما يشير إلى ملاحظة التسلل المختلف.حالات متميزة مكانيًا مع إزالة الالتباس المتاحة). ب، تركيبة نوع الخلايا في 6 مناطق محددة بواسطة الطبقات التالية، T 3 وما فوق (T3+)، T2، T1، E1، E2، وE3 و أعلى (E3+)، في 16 حالة. ج، نسبة البلعميات، خلايا T، الألياف، والورم في CODEX عبر 6 مناطق محددة بواسطة الطبقات التالية، T3 وما فوق، T2، T1،، و E 3 وما فوق. كل نقطة بيانات تمثل عينة واحدة ونقاط البيانات من نفس العينة متصلة. د، التعبير المكاني للجينات الغنية بالحدود POSTN و IFI3O. هـ، مثال على التفاعلات بين الخلايا في مسار MK (MDK) في قسمين من الورم، مع اتجاه السهم الذي يشير إلى اتجاه الإشارة وطول السهم الذي يشير إلى قوة الإشارة. اHT226C1-Th1 (سرطان القنوات الصفراوية) HT206B1-S1 (سرطان الثدي الأولي)
تسعى Nature Portfolio إلى تحسين إمكانية تكرار العمل الذي ننشره. يوفر هذا النموذج هيكلًا للاتساق والشفافية في التقرير. لمزيد من المعلومات حول سياسات Nature Portfolio، يرجى الاطلاع على سياسات التحرير وقائمة التحقق من سياسة التحرير.
الإحصائيات
لجميع التحليلات الإحصائية، تأكد من أن العناصر التالية موجودة في أسطورة الشكل، أسطورة الجدول، النص الرئيسي، أو قسم الطرق.
تم التأكيد
X حجم العينة بالضبط ( ) لكل مجموعة/شرط تجريبي، معطاة كرقم منفصل ووحدة قياس إكس بيان حول ما إذا كانت القياسات قد أُخذت من عينات متميزة أو ما إذا كانت نفس العينة قد تم قياسها عدة مرات إكس اختبار(ات) الإحصاء المستخدمة وما إذا كانت أحادية الجانب أو ثنائية الجانب يجب أن تُوصف الاختبارات الشائعة فقط بالاسم؛ واصفًا التقنيات الأكثر تعقيدًا في قسم الطرق. إكس وصف لجميع المتغيرات المشتركة التي تم اختبارها إكس وصف لأي افتراضات أو تصحيحات، مثل اختبارات الطبيعية والتعديل للمقارنات المتعددة إكس وصف كامل للمعلمات الإحصائية بما في ذلك الاتجاه المركزي (مثل المتوسطات) أو تقديرات أساسية أخرى (مثل معامل الانحدار) والتباين (مثل الانحراف المعياري) أو تقديرات عدم اليقين المرتبطة (مثل فترات الثقة) إكس لاختبار الفرضية الصفرية، فإن إحصائية الاختبار (على سبيل المثال، ) مع فترات الثقة، أحجام التأثير، درجات الحرية وقيمة ملحوظة أعطِالقيم كقيم دقيقة كلما كان ذلك مناسبًا. لتحليل بايزي، معلومات حول اختيار القيم الأولية وإعدادات سلسلة ماركوف مونت كارلو للتصاميم الهرمية والمعقدة، تحديد المستوى المناسب للاختبارات والتقارير الكاملة عن النتائج إكس تقديرات أحجام التأثير (مثل حجم تأثير كوهين)بيرسون )، مما يشير إلى كيفية حسابها
تحتوي مجموعتنا على الويب حول الإحصائيات لعلماء الأحياء على مقالات تتناول العديد من النقاط المذكورة أعلاه.
جي إيه تي كيه v4.1.9.0https://github.com/broadinstitute/gatk أدوات السرير 2.30.0،https://github.com/arq5x/bedtools2 سكرابلت v0.2.3،https://github.com/swolock/scrublet Seurat v4.3.0، حزمة R ComplexHeatmap v2.14.0، حزمة R tidyverse v1.3.2، حزمة R ggplot2 v3.3.5، حزمة R RColorBrewer v1.1.3، حزمة R singleR v1.8.1، حزمة R clusterProfiler v3.18.1، حزمة R msigdbr v7.5.1، حزمة R ROGUE v1.0، حزمة R RCTD v1.2.0، حزمة R كوموت،https://github.com/zcang/COMMOT لصق2،https://github.com/raphael-group/paste2 ppcor v1.1، حزمة R stats v4.1.2، حزمة R ggpubr v0.6.0، حزمة R بايثون 3.7.12 و 3.9.6 sklearn v0.24.2، حزمة بايثون ماتplotlib v3.4.2، py39hf3d152e_0 numpy v1.21.1، h49503c6_1_cpython org.Hs.eg.db الإصدار 3.12.0، حزمة R باندا v1.3.1، py39hdeOf152_0 فيجي/إيميج جي، الإصدار 2.14.0 BigWarp، حزمة Figi/ImageJ ناباري v0.4.19 إيماريس v10.1.1 ديب سيل v0.12.9،https://github.com/vanvalenlab/deepcell-tf بالنسبة للمخطوطات التي تستخدم خوارزميات أو برامج مخصصة تكون مركزية في البحث ولكن لم يتم وصفها بعد في الأدبيات المنشورة، يجب أن تكون البرمجيات متاحة للمحررين والمراجعين. نحن نشجع بشدة على إيداع الشيفرة في مستودع مجتمعي (مثل GitHub). راجع إرشادات مجموعة Nature لتقديم الشيفرة والبرمجيات لمزيد من المعلومات.
بيانات
معلومات السياسة حول توفر البيانات
يجب أن تتضمن جميع المخطوطات بيانًا عن توفر البيانات. يجب أن يتضمن هذا البيان المعلومات التالية، حيثما ينطبق:
رموز الانضمام، معرفات فريدة، أو روابط ويب لمجموعات البيانات المتاحة للجمهور
وصف لأي قيود على توفر البيانات
بالنسبة لمجموعات البيانات السريرية أو بيانات الطرف الثالث، يرجى التأكد من أن البيان يتماشى مع سياستنا
البحث الذي يتضمن مشاركين بشريين، بياناتهم، أو مواد بيولوجية
معلومات السياسة حول الدراسات التي تشمل مشاركين بشريين أو بيانات بشرية. انظر أيضًا معلومات السياسة حول الجنس، الهوية/التقديم الجنسي، والتوجه الجنسي والعرق، والعرقية والعنصرية.
التقارير عن الجنس والنوع الاجتماعي
تم جمع معلومات جنس المريض كجزء من هذه الدراسة
التقارير عن العرق أو الإثنية أو غيرها من التجمعات الاجتماعية ذات الصلة
تم جمع معلومات عن عرق أو إثنية المرضى كجزء من هذه الدراسة
خصائص السكان
تتكون مجموعة البيانات لدينا من عينات من 6 أنواع من الأورام، مع أعمار المرضى تتراوح بين 37-82 عامًا. يمكن العثور على توزيع العينات والمعلومات السريرية عبر المجموعات في الجدول التكميلي 1، والشكل البياني الموسع 1a.
التوظيف
تم اختيار المرضى الذين يستوفون المعايير السريرية ووافقوا على الدراسة للإدراج في التحليل الجيني والجزيئي للأورام. 75 حالة من 6 أنواع من السرطان هي جزء من دراسة شبكة أطلس الأورام البشرية (HTAN)، و3 حالات من سرطان الكلى النقيلي (ccRCC) هي من التحليل السريري (اتحاد تحليل البروتينات السرطانية السريرية). لم يكن هناك انحياز في اختيار الذات أو أي انحيازات أخرى في تجنيد المرضى.
رقابة الأخلاقيات
تم جمع جميع العينات بموافقة مستنيرة في كلية الطب بجامعة واشنطن في سانت لويس. سرطان الثدي
تم جمع عينات (BRCA) وعينات سرطان الغدة البنكرياسية (PDAC) وعينات سرطان القولون والمستقيم (CRC) وعينات سرطان القنوات الصفراوية (CHOL) وعينات سرطان الكلى (RCC) وعينات سرطان بطانة الرحم (UCEC) خلال الاستئصال الجراحي وتم التحقق منها بواسطة علم الأمراض القياسي (بروتوكول IRB 201108117 و201411135 و202106166). تم جمع عينات الورم خلال الاستئصال الجراحي وتم التحقق منها بواسطة علم الأمراض القياسي.
يرجى ملاحظة أنه يجب أيضًا تقديم معلومات كاملة حول الموافقة على بروتوكول الدراسة في المخطوطة.
التقارير المتخصصة في المجال
يرجى اختيار الخيار أدناه الذي يناسب بحثك بشكل أفضل. إذا لم تكن متأكدًا، اقرأ الأقسام المناسبة قبل اتخاذ قرارك. علوم الحياةالعلوم السلوكية والاجتماعيةالعلوم البيئية والتطورية والبيئية لنسخة مرجعية من الوثيقة بجميع الأقسام، انظرnature.com/documents/nr-reporting-summary-flat.pdf
تصميم دراسة علوم الحياة
يجب على جميع الدراسات الإفصاح عن هذه النقاط حتى عندما يكون الإفصاح سلبياً.
حجم العينة
تم اختيار أحجام العينات بناءً على توفر العينات المجمعة عبر 6 أنواع من السرطان. لم يتم استخدام أي طرق إحصائية محددة لتحديد حجم العينة مسبقًا. جمعت هذه المجموعة ما مجموعه 132 مقطعًا من 79 عينة لحالة Visium ST، بالإضافة إلى عدة عينات لتسلسل RNA المفصول المقترن (snRNA-seq) وتسلسل الجينوم الكامل (WES) وCODEX. يتماشى حجم العينة هذا أو يتجاوز العديد من بيانات النسخ الجزيئي المكاني لـ Visium المنشورة سابقًا في تاريخ التقديم.
استثناءات البيانات
لم يتم استبعاد أي بيانات من التحليل
التكرار
لضمان إمكانية التكرار، استخدمنا أدوات معلوماتية حيوية متعددة لنفس التحليلات وقارنّا النتائج عبر التقنيات (بما في ذلك التسلسل الكمي، تسلسل الخلايا المفردة، التسلسل المكاني، والتصوير المتعدد) من نفس عينات الورم. لتأكيد التغيرات في عدد النسخ الفرعية في المناطق المكانية للورم، استخدمنا طريقتين مستقلتين لاستنتاج عدد النسخ، CalicoST (github.com/raphael-group/CalicoST) وInferCNV (https://github.com/broadinstitute/infercnv) (الشكل التوضيحي التكميلي 5a). كما قمنا بالتحقق من استنتاج CNV المكاني مع استدعاء CNV المعتمد على WES ولاحظنا توافقًا يتراوح بين 0.43-0.82 في تشابه جاكارد (الطرق) بين السلالة الفرعية المكانية السائدة وWES (الشكل التوضيحي التكميلي 5c). علاوة على ذلك، تم التحقق من الطفرات الفرعية المكانية مع استدعاء الطفرات المعتمد على WES (معتم الكشف عن طفرات WES في النسخ الجينية المكانية، الشكل التوضيحي التكميلي 5d).
لدعم نتائجنا في مناطق مركز الورم والأطراف، استخدمنا تسلسل RNA المفرد المتطابق من نفس عينات الورم وأظهرنا تعبير معظم الجينات الغنية في المركز والغنية في الأطراف في الخلايا الخبيثة (الشكل التوضيحي التكميلي 8a).
تم تأكيد تركيبات بيئة الورم تحت الخلوية باستخدام التلوين المناعي المتعدد مع الكشف المشترك عن طريق الفهرسة (CODEX) (الشكل 5c).
العشوائية
لم يتضمن تصميم الدراسة تخصيص المرضى إلى مجموعات العلاج، لذلك لم تكن إجراءات العشوائية ذات صلة.
عمى
تصميم الدراسة لم يتضمن تخصيص المرضى إلى مجموعات العلاج، لذلك لم تكن إجراءات التعمية ذات صلة.
التقارير عن مواد وأنظمة وطرق محددة
نحتاج إلى معلومات من المؤلفين حول بعض أنواع المواد والأنظمة التجريبية والأساليب المستخدمة في العديد من الدراسات. هنا، يرجى الإشارة إلى ما إذا كانت كل مادة أو نظام أو طريقة مدرجة ذات صلة بدراستك. إذا لم تكن متأكدًا مما إذا كان عنصر القائمة ينطبق على بحثك، يرجى قراءة القسم المناسب قبل اختيار رد.
التحقق الأجسام المضادة المستخدمة في هذا التطبيق قد تم استخدامها بالفعل في مختبرنا وتم تأكيدها بواسطة واحدة أو أكثر من الطرق التالية. أولاً: تقنية الويسترن بلوت تنتج نمط التوزيع الصحيح (الوزن الجزيئي الصحيح، الغياب في الخلايا غير المعدلة أو مستخلصات البروتين من الفئران التي تفتقر للجين ولكنها موجودة في العينات من النوع البري أو العينات التي تم فيها نقل بناء التعبير). ثانياً، يظهر التلوين المناعي النمط الصحيح (خصوصية الأنسجة الصحيحة، الموقع داخل الخلايا، الغياب/الانخفاض في الخلايا غير المعدلة، الحضور/الارتفاع في الخلايا المعدلة، التوضع المشترك مع العلامات للبروتينات المدمجة المعبر عنها في الخلايا). ثالثاً، تظهر اثنان أو أكثر من الأجسام المضادة المستقلة ضد نفس المستضد نفس النمط المتوقع. بالإضافة إلى ذلك، قمنا بتضمين معلومات مفصلة عن الأجسام المضادة في جدول الملحق_04_CODEX_Antibody.xlsx، وصور التحقق الإضافية في 2_Response_letter.pdf و2b_Respons_Ab_IF_testing_0610.pdf
النباتات
مخزونات البذور
تقرير عن مصدر جميع مخزونات البذور أو المواد النباتية الأخرى المستخدمة. إذا كان ذلك مناسبًا، يرجى ذكر مركز مخزون البذور ورقم الفهرس. إذا تم جمع عينات نباتية من الحقل، يرجى وصف موقع الجمع، التاريخ وإجراءات أخذ العينات.
أنماط جينية نباتية جديدة
وصف الطرق التي تم من خلالها إنتاج جميع الأنماط الجينية النباتية الجديدة. يشمل ذلك تلك التي تم إنشاؤها من خلال الأساليب الجينية المتحولة، وتحرير الجينات، والطفرات المعتمدة على المواد الكيميائية/الإشعاع، والتهجين. بالنسبة لخطوط الجينات المتحولة، وصف طريقة التحويل، وعدد الخطوط المستقلة التي تم تحليلها، والجيل الذي أجريت عليه التجارب. بالنسبة لخطوط تحرير الجينات، وصف المحرر المستخدم، والتسلسل الداخلي المستهدف للتحرير، وتسلسل RNA الدليل المستهدف (إذا كان ذلك مناسبًا) وكيفية عمل المحرر.
المصادقة
تم تطبيقه.
وصف أي إجراءات تحقق لكل مخزون بذور مستخدم أو جينوتيب جديد تم إنشاؤه. وصف أي تجارب تم استخدامها لتقييم تأثير الطفرة، وحيثما ينطبق، كيف تم فحص التأثيرات الثانوية المحتملة (مثل إدخالات T-DNA في موقع ثانٍ، التباين، تحرير الجينات خارج الهدف).
Chia-Kuei Mo , Jingxian Liu , Siqi Chen , Erik Storrs , Andre Luiz N. Targino da Costa , Andrew Houston , Michael C. Wendl , Reyka G. Jayasinghe , Michael D. Iglesia , Cong , John M. Herndon , Austin N. Southard-Smith , Xinhao Liu , Jacqueline Mudd , Alla Karpova , Andrew Shinkle , S. Peter Goedegebuure , Abdurrahman Taha Mousa Ali Abdelzaher , Peng Bo , Lauren Fulghum , Samantha Livingston , Metin Balaban , Angela Hill , Joseph E. Ippolito , Vesteinn Thorsson , Jason M. Held , Ian S. Hagemann , Eric H. Kim , Peter O. Bayguinov , Albert H. Kim , Mary M. Mullen , Kooresh I. Shoghi , Tao Ju , Melissa A. Reimers , Cody Weimholt , Liang-I Kang , Sidharth V. Puram , Deborah J. Veis , Russell Pachynski , Katherine C. Fuh , Milan G. Chheda , William E. Gillanders , Ryan C. Fields , Benjamin J. Raphael , Feng Chen & Li Ding
Abstract
To study the spatial interactions among cancer and non-cancer cells , we here examined a cohort of 131 tumour sections from 78 cases across 6 cancer types by Visium spatial transcriptomics (ST). This was combined with 48 matched singlenucleus RNA sequencing samples and 22 matched co-detection by indexing (CODEX) samples. To describe tumour structures and habitats, we defined ‘tumour microregions’ as spatially distinct cancer cell clusters separated by stromal components. They varied in size and density among cancer types, with the largest microregions observed in metastatic samples. We further grouped microregions with shared genetic alterations into ‘spatial subclones’. Thirty five tumour sections exhibited subclonal structures. Spatial subclones with distinct copy number variations and mutations displayed differential oncogenic activities. We identified increased metabolic activity at the centre and increased antigen presentation along the leading edges of microregions. We also observed variable T cell infiltrations within microregions and macrophages predominantly residing at tumour boundaries. We reconstructed 3D tumour structures by co-registering 48 serial ST sections from 16 samples, which provided insights into the spatial organization and heterogeneity of tumours. Additionally, using an unsupervised deep-learning algorithm and integrating ST and CODEX data, we identified both immune hot and cold neighbourhoods and enhanced immune exhaustion markers surrounding the 3D subclones. These findings contribute to the understanding of spatial tumour evolution through interactions with the local microenvironment in 2D and 3D space, providing valuable insights into tumour biology.
Treatment-resistant subclones often arise in cancer , and the tumour microenvironment (TME) can further drive resistance through multiple mechanisms . Neither bulk nor single-cell technologies preserve the spatial information necessary to understand these dynamics, but ST instruments, such as Visium , can resolve tumour substructures. ST data have been integrated with other data types to examine fine-scale clonal structure and to identify cell-cell interactions (CCIs) with the microenvironment . CODEX multiplex imaging can further complement ST methods by spatially localizing proteins.
Clonal evolution remains one of the most intractable problems of cancer . That is, the spatial and temporal adaptation of a tumour to environmental and treatment stimuli through mutation accumulation and fitness-based selection . Previous studies have concentrated
on inferring evolutionary history through mutations, but newer technologies, including those mentioned above, have enabled substantially deeper investigations of spatial clonal dynamics . The prospect of applying several such technologies to a large, well-powered, cross-cancer cohort to further investigate these phenomena motivates the current work.
Here we report the comprehensive characterization of 131 tumour ST sections across 6 different cancers: breast cancer (BRCA), colorectal carcinoma (CRC), pancreatic ductal adenocarcinoma (PDAC), renal cell carcinoma (RCC), uterine corpus endometrial carcinoma (UCEC) and cholangiocarcinoma (CHOL). We use an approach that combines ST, CODEX and bulk sequencing data and single-cell sequencing data of matching samples to profile spatially distinct tumour regions separated
Fig.1|Definition of tumour spatial microregions. a, Sample, data type and workflow overview of the spatial subclone cohort of 131 Visium ST sections from 6 different cancer types with 22 and 48 respective matching CODEX and snRNA datasets. Data encompass 54 BRCA, 30 CRC, 23 PDAC, 12 RCC, 5 UCEC and 7 CHOL samples. Bottom, workflow for generating spatial tumour microregions, inferring spatial tumour subclones and conducting downstream analyses. Based on the distribution of tumour regions, we separated samples into spatially distinct and spatially diffuse cohorts. Analyses included tumour subclone evolution analysis, transcriptional similarity and layer-based TME interactions, tumour growth pattern construction, and multisection 3D neighbourhood reconstructions using custom code (Methods). b, Circular cohort overview plot at the tissue block level. The outcrop height of the outermost ring indicates the number of sections per tissue block. The top right insert shows a legend,
with numbers in bubbles indicating counts of tissue blocks with a given number of serial sections. Successive rings indicate the cancer type annotated with section (tissue block) counts, tumour type, assay type and spatial cohort designation.c, Microregion distribution in the spatially distinct cohort at the section level coloured by cancer type (left), microregion size group (middle) and primary versus metastasis (right). Each circle indicates one microregion. The size of each circle represents the size of the microregion. d, Tumour versus stromal-immune spot fractions across cancer types for the entire cohort at the section level. Each point represents a sample, coloured by type: primary ( sections from 60 cases) or metastatic ( sections from 16 cases). The box plot’s centre line represents the median, with the lower and upper hinges indicating the first and third quartiles. Whiskers extend to the highest and lowest values within 1.5 times the interquartile range (IQR) from the hinges.
by stromal components, which we call ‘tumour microregions’. We demonstrate that there are distinct genetic clones within these microregions with specific copy number variations (CNVs) and with differential activity within oncogenic pathways, particularly the MYC pathway. Notably, we show that immune-tumour and stromal-tumour interactions vary among these tumour regions. Additionally, our study highlights distinct characteristics between primary and metastatic tumours, including differences in tumour growth patterns and transcriptional profiles. To further support our findings showing that immune populations in the TME surround specific spatial tumour regions, we use CODEX and a multimodal 3D reconstruction tool trained on adjacent ST sections. The results confirm the connectivity of subclones and microregions in different sections within 3D space. These reconstructions highlight
tumour-immune interphase niches and interactions. Overall, this spatial omics approach provides deeper insights into clonal evolution and the microregional distinctions across six different solid tumour types, paving the way to continued advances in understanding the mechanisms of therapeutic resistance in cancer.
Spatial microregions across cancers
We profiled 131 tumour sections with ST data from 98 blocks spanning 6 cancer types as part of the NCI’s Human Tumor Atlas Network: 54 BRCA, 30 CRC, 23 PDAC, 12 RCC, 5 UCEC and 7 CHOL (Fig. 1a, Extended Data Fig. 1 and Supplementary Table 1) from 78 cases ( and 6 cases, respectively). Three RCC samples were from the Clinical
Proteomics Tumour Analysis Consortium. Using histological haematoxylin and eosin (H&E) staining and transcriptional profiles, we identified tumour microregions as spatially distinct cancer cell clusters separated by stromal areas (Methods) and designated Visium spots as malignant or non-malignant. We used the Morph toolset to subsequently refine tumour boundaries, determine distances of spots from boundaries and construct layers of spots, indexing their depths to tumour boundaries (Methods). We selected 50 sections with multiple tumour regions as the ‘spatially distinct cohort’ (Extended Data Fig. 2) and 82 samples with diffuse tumour regions as the ‘spatially diffuse cohort’ (Extended Data Fig. 3). We also produced serial sections of 15 tumour tissue blocks, which resulted in 48 sections suitable for 3D tumour reconstruction (Fig. 1b and Extended Data Fig. 4a).
Based on the estimated area per tumour microregion (Methods), we categorized microregion sizes as small ( spots or ), medium ( spots or ) or large ( spots or (Fig.1d). CRC had larger microregions (average of 2.9 layers) than BRCA (average of 2.1 layers; false discovery rate , Welch’s -test) and PDAC (average of 2.37 layers; FDR , Welch’s -test). Conversely, BRCA and PDAC microregion depths were statistically indistinguishable ( , Welch’s -test). RCC had the highest tumour fraction, whereas PDAC had the lowest (Fig. 1c), which is probably due to the higher stromal content and lower tumour cell density in PDAC , which in turn leads to smaller microregion sizes. Primary tumours generally had more small microregions (66.3%) compared to metastases (40.2%), which had more medium-sized microregions (43.2%) (Extended Data Fig. 4b,c). Larger microregions were predominantly found in metastases (16.3% compared with 3.2% in primary), which also had deeper microregions than primary tumours ( 3.4 compared with 1.9 layers; Welch’s -test FDR ). This difference held for BRCA-only sections (FDR ), for which we had data for both metastases ( 5 sections, 44 microregions, mean depth of 4.2 ) and primary tumours ( 8 sections, 222 microregions, mean depth of 1.7). These results suggest that there is divergent growth between primary and metastatic tumours and an organ-specific TME effect on microregion growth and organization. Examples include samples HT268B1-Th1H3 (BRCA liver metastasis) and HT260C1-Th1K1U1 (CRC liver metastasis), which had large regions occupying (400-500 spots), whereas sample HT27OP1-H2U1 (PDAC) had a smaller (mean of or 26 spots) but greater number of microregions ( ) (Extended Data Fig. ). In the spatially distinct cohort, samples with the highest microregion counts were from BRCA blocks rich in ductal carcinoma in situ (DCIS) (HT397B1-S1H2,HT339B1-S1H3 and HT206B1-S1; Extended Data Fig. 4d). This distribution could reflect the tendency of ductal cancer cells to grow along the secretion duct in both organs, which may explain our observation of numerous small regions.
Focal clonal evolution in microregions
We discerned genome-wide CNVs using CalicoST and InferCNV (Supplementary Fig. 1 and Methods), selecting confident events in each microregion by filtering those in matching whole-exome sequencing (WES) data (Supplementary Table 2). We then clustered microregions into spatial subclones based on CNV similarity (Fig. 2a and Methods). We detected spatial CNVs in 125 out of the 131 sections, out of which we observed 1-3 subclones per section (about with a single clone, with 2 subclones and with 3 subclones) (Fig. 2b). A single clone can be composed of subclones that our workflow cannot detect, such as subclones intermixed in the same microregion and subclones differentiated by genetic alterations not covered by the Visium transcriptome. Within these limitations, we identified multiple spatial subclones in sections from 4 cancer types: BRCA ( 17 sections), PDAC (10 sections), CRC (6 sections) and RCC (2 sections). CNV profiles of spatial subclones were compared with matching WES data and showed high genome-wide similarity (Methods and Extended Data Fig. 5a).
We also mapped somatic mutations onto optimal cutting temperature (OCT)-embedded ST sections, for which each section showed 1-98 mutations mapped specifically in tumour regions (Fig. 2c and Extended Data Fig. 5b).
Sample HT260C1(CRC liver metastasis) contained 12 tumour microregions, which mapped to 2 spatial subclones (Fig. 2d). Both clonal events (chromosome 13 amplification and chromosome 8p deletion), as well as several subclonal events (amplifications in chromosomes 6 p , 12 p and 20 q in clone c 2 , and amplification of chromosome 12 q in clone c1), were identified and confirmed with matching single-nucleus RNA sequencing (snRNA-seq) data and WES-based CNV inference (Fig. 2e,f). Although histology indicated a fibrotic separation between the two subclones, multiple shared clonal CNVs suggested a common origin (Fig. 2g). Somatic variants from transcripts provided further supporting evidence for tumour clonality. In addition, 17 WES-based somatic mutations were mapped to ST (Extended Data Fig. 5c). Several mutations showed differential variant allele frequency (VAF) in tumour regions compared with normal regions (Extended Data Fig. 5c, left) and differential VAFs between the two subclones (Extended Data Fig. 5c, right). Both subclones showed expression, and a VAF for mutation c. was significantly higher in subclone c2 than in c1 (Fig. 2h,i; , two-sided proportion test). Using Xenium data, we analysed both wild-type (WT) and c. alleles on a matching tumour section, the WT allele in both subclones and the mutant allele in subclone c2 (Fig. 2j). Subclone c2 diverged from c1, with a gain of unique genetic alterations in both CNV and mutations. Similarly, a BRCA liver metastasis sample, HT268B1, showed two distinct spatial subclones that were supported by matching snRNA-seq data, with chromosomal-arm-level CNV differences (Extended Data Fig. 5d-g) and subclonal mutations (Extended Data Fig. 5h). For example, EEF1A1 was expressed in both spatial clones, whereas mutation EEF1A1 1324G was specifically observed in subclone c2 (proportion test FDR < 0.05; Extended Data Fig. 5i,j).
Primary PDAC samples, despite their smaller microregions, also contained multiple spatial subclones. Sample HT270P1 showed three subclones across two sections from two tumour blocks with OCT and formalin-fixed paraffin-embedded (FFPE) preservation, respectively (Supplementary Fig. 2a). Most primary BRCA cases showed that all tumour microregions belonged to a single genetic clone, such as HT206B1 across five serial sections (Supplementary Fig. 1b). In seven out of nine cases, a single subclone encompassed both DCIS and invasive ductal carcinoma (IDC) morphology, which indicated that the transition between them happens without large copy number alterations (Supplementary Fig. 2b). However, primary BRCA sample HT397B1 showed three spatial subclones across four sections from two tumour blocks (Supplementary Fig. 2a). Two of the clones showed both DCIS and IDC morphologies, whereas clone 3 only showed IDC morphology, which indicated a parallel transition from DCIS to IDC between the spatial subclones (Supplementary Fig. 2b).
Single-nucleotide polymorphisms reveal copy-number-neutral loss of heterozygosity that is missed by read-depth inference alone. A CRC liver metastasis sample (HT112C1-Th1) with strong B allele frequency deviation in copy-number-neutral chromosome 21 indicated a subclonal genetic alteration in clone A (Supplementary Fig. 2a). These observations collectively suggest that spatial subclones within a tumour section probably stem from a common ancestor.
Genetic changes drive tumour disparities
To mitigate the influence of infiltrating immune and stromal cells, we first excluded organ-specific, non-malignant cell-type marker genes using snRNA-seq data (Methods). Subsequently, we used an entropy-based method, ROGUE, to analyse transcriptional heterogeneity among tumour microregions (Methods). Our analysis revealed that PDAC had the highest heterogeneity ( ), whereas BRCA,
Fig. 2 | Genetic alteration reveals spatial clonal evolution. a, Summary of workflow for identifying spatial subclones.b, Numbers of spatial clones detected in each section summarized by cancer type. Multiclonal sections (2 or 3 spatial subclones) are observed in BRCA, CRC, PDAC and RCC in this cohort ( sections from 74 cases with detected CNV).c, Spatial somatic mutation mapping per section detected in OCT ST data. WES-derived somatic mutations with significantly higher VAF in tumour regions than non-tumour regions (binomial test,FDR<0.05) are shown ( OCT sections from 29 cases). The box plot’s centre line represents the median, with the lower and upper hinges indicating the first and third quartiles. Whiskers extend to the highest and lowest values within 1.5 times the IQR from the hinges. d, e, A colorectal cancer liver metastasis sample (HT260C1) with 12 tumour microregions demonstrates 2 tumour subclones, c1 and c2, separated spatially (d), with support from matching snRNA-seq data (e).f, Heatmap of estimated somatic CNVs per spot shows both
shared and unique CNV events between the two spatial subclones. The B allele frequencies (BAFs) in each spatial subclone from the same genomic window are shown in the middle tracks and corresponding snRNA-inferred and WES-inferred CNV statuses using GATK4 (GA) and Hatchet2 (Hat) are shown in the bottom tracks.g, The predicted phylogenetic relationship of c 1 and , The VAF for mutation c. is significantly higher in c2 than c1 ( , two-sided proportion test). i, Subclonal mutation c. is uniquely detected in clone 2 in spatial transcriptomics, whereas expression is in both spatial subclones.j, Xenium in situallele-specific probe density shows the WT allele of in both subclones and the mutant allele uniquely in the right subclone (c2). Scale bars, 2 mm . amp, amplification; del, deletion; LOH , loss of heterozygosity; NA, not applicable; UMAP, uniform manifold approximation and projection.
CRC, RCC and UCEC had moderate levels (0.05-0.45), and CHOL had the lowest levels (<0.2) (Extended Data Fig. 6a). To further investigate the effect of genetic alterations and microenvironmental adaptations on transcriptional profiles, we assessed transcriptional similarity using pairwise Pearson correlation among tumour microregions (Methods), comparing within and between different genetic clones. We observed greater similarity within subclones (Extended Data Fig. 6b, green) compared with between different subclones (Extended Data Fig. 6b, orange) across all samples from BRCA, CRC, PDAC and RCC (Extended Data Fig. 6b and Methods). This pattern remained consistent in both primary and metastatic samples in BRCA and CRC, which underscores the central role of genetic composition in shaping transcriptional similarities across microregions.
To understand the activation of oncogenic pathways across microregions, we performed gene set enrichment analysis (GSEA) with differentially expressed genes (DEGs) between tumour microregions and stroma regions (Methods). Our analysis identified common pathways such as MYC and E2F shared across microregions, but distinct
pathways, such as the unfolded protein response, that was specific to some microregions in sample HT268B1 (BRCA metastasis) (Extended Data Fig. 7a-f). Here genetic alterations controlled the transcriptional profile, whereas in other samples, subtle variations arose from the local microenvironment or undetected genetic events. For example, sample HT260C1 showed two subclones with distinct copy number profiles (Fig. 2d), whereas microregions within clone 1 had varying expression similarities, despite having similar CNVs (Extended Data Fig. 6c, indicated by the dotted boxes).
When applying GSEA to identify subclone-specific pathways, we found varied microregional expression levels within the G2M checkpoint pathway in clone 1 (Extended Data Figs. 6d and 7g,h, indicated by the dotted boxes) in sample HT260C1. This pattern was mirrored in MYC for both metastasis samples HT260C1 and HT268B1, with distinct sets of MYC downstream genes among tumour subclones (Extended Data Fig. 7e,f,i,j and Supplementary Fig. 3). This result underscores the complexity of this pathway, which is commonly dysregulated in cancer and influences many oncogenic processes . Clone 2 of HT260C1 had
an enrichment of translation initiation complex (eIF family) and G1S progression (CDK4), whereas clone 1 had DNA replication genes (MCM family) (Extended Data Fig. 7i).
Enrichment tests using Enrichr with the drug perturbation dataset LINCS L1000 (Methods) predicted varying treatment responses of subclones (Supplementary Fig. 4). For instance, all subclones in HT397B1-S1H3 and HT112C1-Th1 should respond to the mTOR inhibitor torin-2, but only clone 1, not clone 2, in HT268B1-Th1K3 responded. This variation underscores the importance of profiling spatial subclones.
Cellular pathways at the tumour core and edge
We further investigated whether different transcriptional programs exist in tumour microregion centres (cores) compared to their leading edges (interfaces between the tumour and the TME-stroma). To that end, we used Morph to measure the distance of each tumour spot to its nearest tumour-TME border (Methods and Extended Data Fig. 8a). The relationship between the total layer depth of a microregion and its size (total area measured as the number of spots occupied) describes the general shape of the region (Extended Data Fig. 8b). The dashed reference line indicates the depth-size relationship of perfectly circular regions. For all five cancer types in the spatially distinct cohort ( ), smaller regions tended to exhibit near-circular shapes. But as the layer depth increased, regions tended to deviate from their circular shape, and an expanded interface between tumour and non-tumour cells arose.
Spatial gene expression profiles can also be characterized in terms of layer depth. For each layer-assigned spot, we independently performed a linear regression between gene expression and spot depth for each gene. Tumour purity estimates using the methods RCTD or ESTIMATE were included as a covariate to adjust for possible purity decreases towards section edges (Methods). A positive correlation with depth indicates increased gene expression towards the tumour core and vice versa. In the CRC metastasis sample HT260C1, the top centre-enriched genes ( and ) and periphery-enriched genes (HSP90AB1 and LDHB) are shown with the regression line and spatial expression pattern in Extended Data Fig. 6e,f. Also shown are respective top centre-enriched (NDRG1,S100A2 and CA9) and periphery-enriched genes (TUBA1B, NDUFA4 and TOMM4O) for HT206C1 in Extended Data Fig. 8c-e. These results were supported by snRNA-seq data from matching tumour samples, which demonstrates that the top shared genes are mainly expressed by malignant cells, with small contributions from immune and stroma cells (Supplementary Fig. 5a-d).
We subsequently identified genes recurrently enriched in tumour centres and in peripheries across cases (Extended Data Fig. 6g). Top shared centre-enriched genes were involved in ribosome assembly (RPL and RPS family genes such as RPS4X, RPL22 and RPL4), along with genes such as orf46 (ref. 21) and the long non-coding RNA SNHG29 (ref. 22), which are linked to tumour growth in various cancer types. By contrast, the tumour periphery was enriched in the following genes: a different set of ribosomal RPL and RPS genes (RPL35, RPLP1 and RPS27); ENO1, a multifunctional oncoprotein involved in glycolysis, invasion and immunosuppression ; TMSB10, which promotes proliferation and invasion in ; and , which induces the formation of M2 macrophages . These differential biological processes indicate that malignant cells in the core are actively undergoing protein translation, whereas those at the edges are involved in tumour migration and immune modulation, interfacing with immune and stromal components.
Clonal-specific tumour-TME interactions
To investigate TME composition in tumour boundary regions, we examined the differential infiltration of non-tumour cells between tumour spatial subclones, the location of such infiltration and genes
and CCIs enriched in boundary regions (Fig. 3a). We used matching snRNA-seq data as a reference for spot-level cell-type deconvolution and performed pairwise differential infiltration analysis between all spatial subclones of the same sample. Top differential cell types in terms of infiltration included macrophages in BRCA, hepatocytes in CRC and fibroblasts in PDAC (Extended Data Fig. 9a). A primary BRCA sample, HT397B1, with three spatial subclones showed differential infiltration in both cells and macrophages, with subclone showing the largest fraction of both (Fig. 3b). CODEX data validated the increased level of T cell markers (CD3 and CD8) and the non-T cell immune marker (HLA-DR) staining in subclone c3.
We used the above-described layer assignments to ascertain whether infiltration exhibits spatial patterns, and we defined six ordered regions from the tumour core to the TME: T3+, T2, T1, E1, E2 and E3+. These showed the expected decreasing trend for tumour cells (Extended Data Fig. 9b), but various patterns for non-tumour cells. Macrophages clustered outside the tumour in E1 and E2, whereas T cells showed infiltration both immediately outside (E1 and E2) and inside the tumour (T1 and T2) (Fig. 3c). Both macrophage and T cell fractions were decreased in the distant TME (layer E3+) where fibroblasts dominate, an observation supported by CODEX data (Extended Data Fig. 9c). With an inter-layer distance of only , our observation indicates that there is strong spatial recruitment of immune cells by tumours at the microscopic level.
We also performed differential expression analysis between the boundary regions T1 and E1 and with all other spots (Fig. 3d). Top boundary genes shared across samples and cancer types included genes of extracellular matrix proteins (POSTN and ) and interferon-induced macrophage activation proteins (IFI3O) (Extended Data Fig. 9d). Matching snRNA-seq data showed that the top boundary genes had significantly higher expression (adjusted , Bonferroni correction) across the cohort in non-tumour cell types (POSTN, FN1 and TIMP1 in fibroblasts and IFI3O in macrophages), which suggested that there are interactions between tumour and non-tumour cells at the boundary (Fig. 3d). To quantify spatial CCIs, we ran COMMOT on 18 cases with 39 sections and then discerned differential receiver-sender signals between spots within and outside tumour boundary regions. The top shared CCIs in boundary regions were extracellular matrix (ECM) receptors (collagen, laminin, FN1 and THBS), secreted signalling (SEMA3, SEMA4, ncWNT and MK) and cell-cell adhesion (EPHB and NOTCH) (Fig. 3e). As an example, the MK pathway was observed in CRC, PDAC and BRCA samples, for which the signal goes from malignant cell regions to the TME interface (Extended Data Fig. 9e). The pathway included interactions between the ligand MDK and the receptors NCL and SDC4 (ref. 26). Malignant cells secrete MDK to create an immunosuppressive and angiogenic environment , which in turn promotes tumour growth. We also found ECM pathways for which the signal goes from the TME towards malignant cell regions. One of the top interactions, the THBS pathway, describes ECM components THBS1-THBS4 (which encodes thrombospondin) binding to cell surface receptors CD36 and CD47, which in turn modulate cell adhesion, proliferation and angiogenesis . Enrichment of tumour-associated immune cells, genes and CCIs within -wide boundary regions illustrates communication between malignant cells and their environment that would be invisible to spatially agnostic technologies.
3D tumour structure and TME interactions
To investigate tumour growth patterns and TME interactions in 3D, we serially sectioned tumours from BRCA, CRC, PDAC and CHOL, conducting ST on 11 samples and CODEX on 2 samples. Using PASTE2, we co-registered 48 sections from the 11 ST specimens to construct tumour volumes (Fig. 4a and Methods). Our analysis revealed variations in tumour volume numbers among samples, with BRCA showing the highest volume count (Fig. 4b), particularly in samples with
Fig. 3 | Immune and stromal infiltration inside spatial tumour microregions. a, Analysis design focusing on the differential infiltration level between spatial subclones and the spatial location of such infiltration with respect to the tumour-TME border.b, Left, a primary BRCA sample HT397B1 shows higher macrophage and T cell levels in clone c 3 . Right, T cell markers (CD3 and CD8) and the non-T cell immune marker (HLA-DR) show higher intensity in the same regions from CODEX data. Scale bar, 1 mm. c, Fraction of macrophages, T cells and fibroblasts averaged across microregions in the following layers: T3 and above(T3+), T2, T1, E1, E2, and E3 and above(E3+), in 14 cases. d, Genes differentially enriched in the tumour boundary regions. Colour represents the (fold change)
of gene expression between boundary regions and non-boundary regions, with non-differential comparisons in white ( spatially distinct cases). Each column represents a tumour block (multisection averaged) and tumour blocks from the same patient were grouped together (no gaps between columns). snRNA-seq-based cell-type specific expression is shown on the left, and the number of tumour blocks with significant enrichment is shown on the right (adjusted , Bonferroni correction). e, Boundary-enriched CCIs shared across samples ( spatially distinct cases) based on receiver (r)-sender(s) signals.
prominent miniature duct-like tumour growth patterns (HT206B1-S1, 1A-1E;HT339B1-S1H3, 3D and 3E; and HT397B1-S1H3, 4B-4D; Extended Data Fig. 2). By contrast, CHOL, CRC, PDAC and other BRCA sections exhibited more invasive tumours that formed larger, continuous structures that resulted in fewer but larger volumes.
We then analysed these tumour volumes for structural complexity using two topological metrics: (1) connectivity (degree), which measures the number of connections to adjacent microregions, and (2) the number of loops per volume, which indicates instances in which adjacent sections split and merge to form doughnut-shaped structures. The maximum connectivity score serves as an indicator of tumour structural complexity. Of the 15 tumour volumes analysed, 8 (6 BRCA, 1 CHOL and 1 CRC) had a maximum connectivity score exceeding 5, which reflected the frequent formation of complex branching structures in these tumours (Fig. 4c). The highest connectivity score of 11 was observed in CHOL sample HT226C1-Th1, which resulted from a large merged volume in U1 that fragmented into smaller microregions in the adjacent section U2 (Extended Data Fig. 10a,b). Additionally, 5 out of 81 volumes across 15 tumour pieces contained complex loop structures, with the highest loop count of 12 found in volume 14 of sample
HT206B1 (Extended Data Fig. 10c). These loops probably result from the interwoven DCIS-like growth pattern in volume 14, as confirmed by its histology (Extended Data Fig. 10d).
We selected a BRCA liver metastasis sample, HT268B1-Th1H3, for detailed 3D volume reconstruction and structural analysis. This sample contained four tumour volumes (volume 1 to volume 4) (Fig. 4d,e). Volumes 1, 3 and 4 formed separate subvolumes within clone 2 , which probably connected beyond the approximately tissue section examined. Volume 2, the largest and most complex volume, belonged solely to clone 1 , with a maximum connectivity of 8 and 6 loops, which indicated substantial branching and merging (Fig. 4d). Histological images of HT268B1 (Extended Data Fig. 2a-e) confirmed visible splitting and merging, thereby demonstrating that even tumours without clear ductal or lobular structures can exhibit diverse growth patterns, invasive behaviour and complex branching.
We next used an unsupervised deep-learning approach to identify 3D cellular neighbourhoods in serial-sectioned ST datasets (Methods). After registration of serial sections, a vision transformer (ViT) autoencoder was trained on ST, CODEX and H&E sections. Annotated image patches were then used to construct 3D cellular neighbourhoods,
Fig. 4 | 3D tumour volume reconstruction reveals diverse tumour growth patterns. a, Tumour growth pattern diagram with two distinct tumour 3D volumes from four ST sections. The number of tumour volumes, connectivity, maximum connectivity and the number of loops are annotated to illustrate the concept of these geometric quantities. b, Number of tumour volumes (components) for each sample piece.c, Distribution of maximum connectivity of sample pieces. Each dot represents one tumour volume ( sections from 14 cases). The box plot’s centre line represents the median, with the lower and
upper hinges indicating the first and third quartiles. Whiskers extend to the highest and lowest values within 1.5 times the IQR from the hinges. d, Sankey plot showing tumour microregion connections across sections, with 3D tumour volumes for tissue block HT268B1-Th1H3 (BRCA metastasis). The maximum connectivity of volume 2 (Vol. 2) is highlighted in red. Numbers next to each microregion indicate its connectivity. e, Microregion and 3D tumour volume spatial distributions on the ST sections for HT268B1-Th1H3.
which enabled the discovery of these neighbourhoods across multiple sections (Extended Data Fig. 10e and Supplementary Fig. 7b). We applied this approach to a primary BRCA sample HT397B1 (six H&E, four CODEX and two Visium ST sections) and a BRCA liver metastasis sample HT268B1 (four Visium ST sections) (Extended Data Fig. 10f and Supplementary Fig. 7c,d).
In sample HT268B1, neighbourhoods with at least 60% overlap with previously defined spatial subclones (Fig. 2d) were classified as tumour-enriched, with the remainder as TME neighbourhoods (Methods). Two of these neighbourhoods ( 4 and 6 ) were in close contact with the periphery of both subclones (Fig. 5a,b). Notably, when viewed in 3D space, the neighbourhoods largely contiguously surround the subclones, except for the upper portion of the block where the neighbourhoods were more broken. We then quantified these two neighbourhoods based on shared and unique DEGs (Fig. 5c), and we found immune responses (IFI27 and HLA-DRA) and stromal (BST2 and SPARC) genes in both. Neighbourhood 4 was enriched in HMGA1, which regulates chromatin structure and has a role in malignant cell progression , and , a factor involved in angiogenesis . Neighbourhood 6 was enriched for genes important for immunoregulatory (CCL19) and immunoglobulin receptor binding (IGLC2, IGHG1 and IGKC), which are prognostic in . In three dimensions, we continued to see clear association of these DEGs, in particular TYMP1 and IGLC2, with clones 1 and 2, respectively, throughout the tissue volume (Fig. 5d and Extended Data Fig. 10g).
The primary BRCA sample HT397B1 had two main regions of TMEtumour morphology: an immune-cold area with both DCIS and IDC morphologies containing clones 1 and 2, and an immune-hot IDC region that harboured clone 3 (Figs. 5e,f and 3b and Supplementary Fig. 2b). We stratified TME neighbourhoods by their contact fraction with clone 3,
which highlighted the top and bottom quartiles (Fig. 5e,g). According to CODEX-based cell-type annotations, neighbourhoods associated with clone 3 had a higher fraction of T cells and a decreased fraction of fibroblast cells compared with those near clones 1 and 2 , a result that aligned with previous findings (Figs. 5g and 3b). To further verify the immune-stromal status of these neighbourhoods, we selected two regions of interest (ROIs): ROI 1, located in the immune-cold clone 2, and ROI 2, located in the immune-hot clone 3. ROI 1 had low fractions of macrophages, T cells and B cells, whereas ROI 2 had much higher levels of these cell types (Fig. 5f). These trends were also evident in CODEX sections, for which immune markers were more intense in ROI 2. Additionally, ROI 1 showed an increased fibroblast cell fraction, with smooth muscle actin (SMA) highly expressed in the myoepithelium surrounding the tumour regions, a hallmark of DCIS. Our findings indicate that these cell-type associations and DCIS and IDC-like subclones are consistent in three dimensions, based on calculated cell-type densities around the 3D tumour volume and the generation of immune, stromal and epithelial volumes (Fig. 5h and Extended Data Fig. 10h). These analyses demonstrate that 3D reconstruction offers increased sensitivity for investigating heterogeneous tumour microenvironments.
Discussion
This study identified genomically distinct spatial microregions and spatially distributed subclones in samples across solid tumour types. We propose that CNV variability is a major driver of the transcriptional variation seen in these microregions. Spatial subclones identified in the same tumour block shared a common ancestry, a finding congruent with previous studies of tumour evolution . A second major driver of variability is exposure to the TME, and we observed distinct
Fig. 5 | Tumour-TME interactions in microregion boundary regions demonstrate heterogeneity. a, For HT268B1, a BRCA metastasis sample, 3D neighbourhood volumes were generated with 4 Visium ST sections spanning . Neighbourhoods 4 and 6 (NBHD 4 and NBHD 6, respectively), with the highest contact fraction with the subclone boundary, are displayed in 3D and as a 2D plane overlaid with subclone annotations. Scale bar, , TME neighbourhoods displayed by contact fraction with the subclone boundary. c, DEGs were categorized into three groups: unique to neighbourhood 4, unique to neighbourhood 6, and shared. Spatial expressions of selected genes from each group are shown (bottom), with genes mentioned in the main text highlighted in bold. d, 3D reconstruction of tumour regions, where tumour surface mesh is coloured by the transcript density of TYMP1 and IGLC2 within a radius of a given location. e, For HT397B1, a primary BRCA sample, integrated 3D neighbourhood volumes were generated with 6 H&E, 2 Visium ST
and 4 CODEX sections spanning . TME neighbourhoods described in are shown as a 3D volume.f, Visualization of two ROIs in HT397B1, specifically the more immune-cold ROI 1 and immune-hot ROI 2. 2D slices of the 3D volume with quartile-highlighted neighbourhoods associated with each ROI are shown. Visium ST slides (U1 and U8) are shown as RCTD-imputed cell-type fractions. For CODEX slides (U2,U6,U9 and U12), DAPI, pan-cytokeratin (PanCK), SMA, HLA-DR, CD45 and CD8 are displayed. Scale bar, , TME neighbourhoods are displayed as a fraction of contact with the border of each of the three subclones. Additionally, cell type fraction of and cells and fibroblasts are shown. Cell-type fractions were calculated from CODEX sections. The top and bottom quartiles of neighbourhoods with respect to contact fraction with subclone 3 are emphasized as dashed boxes. h, 3D epithelial (PanCK, red), immune (CD45, green) and stromal (SMA, white) surface volumes generated from CODEX sections. Scale bar, .
transcriptional patterns associated with cancer cell depth from the microregion edge, as well as specific enriched gene expression in edge cells adjacent to immune cells of the TME. Finally, 3D tumour volume reconstruction identified 3D neighbourhoods of regional TME variation.
We observed genomic and transcriptomic heterogeneities among tumour microregions in multiple samples. Although some tumours were relatively consistent in their transcriptomic profiles, others could be subdivided according to gene expression. This variability was partially explained by mapping copy number events to ST regions. Distributions of cancer subclones with genetic variations have been mapped across tissues , and mutation-based subclones in spatial regions within a single tumour have been demonstrated . In addition to spatial mutation mapping from transcriptome and validation with allele-specific in situ hybridization, we characterized spatial tumour heterogeneity according to changes in gene expression related to the proximity of immune and stromal cell types compared with more
insulated cancer cells within spatial microregions. A more nuanced understanding of the way cancer and TME cells shape each other within tumours is needed to better exploit these interactions therapeutically.
Subclonal evolution is a major driver of therapeutic resistance, with the emergence of resistant subclones often resulting in treatment failure . Here we characterized the structure and distribution of spatially distinct tumour subclones in multiple solid tumours and showed that they can exhibit varying responses to identical compounds through perturbation gene set overlap analysis. Future translational work will probably investigate subclones under the varying selective pressures of anticancer therapies, which will help to guide the design of new approaches, such as optimizing the combination of local and systemic therapies.
There are several limitations of the study. RNA-inferred spatial CNV captures large genetic events but not focal copy number changes. Spatial mutation mappings provide additional support on subclonal identifications, but with the limitation that only mutations near the
3′ end of each transcript are preferentially detected. Additionally, our ST dataset does not achieve single-cell resolution with the current platform ( -diameter spot). Matching snRNA-seq was used to infer tumour-non-tumour expression and CODEX imaging was used to validate our findings on TME composition. However, direct spatial expression from different cell types of origin remains inferred. In closing, our reconstructed 3D data enabled spatial investigation of tumour architecture, subclones, cellular neighbourhoods and TME. We anticipate that such analysis will rapidly establish itself more broadly within cancer research . Coming advancements in technology will facilitate even deeper analyses and will further empower future tumour studies.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-024-08087-4.
Fu, T. et al. Spatial architecture of the immune microenvironment orchestrates tumor immunity and therapeutic response. J. Hematol. Oncol. 14, 98 (2021).
Schmitt, M. W., Loeb, L. A. & Salk, J. J. The influence of subclonal resistance mutations on targeted cancer therapy. Nat. Rev. Clin. Oncol. 13, 335-347 (2016).
Roper, N. et al. Clonal evolution and heterogeneity of osimertinib acquired resistance mechanisms in EGFR mutant lung cancer. Cell Rep. Med. 1, 100007 (2020).
Trédan, O., Galmarini, C. M., Patel, K. & Tannock, I. F. Drug resistance and the solid tumor microenvironment. J. Natl Cancer Inst. 99, 1441-1454 (2007).
Qu, Y. et al. Tumor microenvironment-driven non-cell-autonomous resistance to antineoplastic treatment. Mol. Cancer 18, 69 (2019).
Ding, L. et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by wholegenome sequencing. Nature 481, 506-510 (2012).
Yang, D. et al. Lineage tracing reveals the phylodynamics, plasticity, and paths of tumor evolution. Cell 185, 1905-1923.e25 (2022).
Ståhl, P. L. et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78-82 (2016).
Rao, A., Barkley, D., França, G. S. & Yanai, I. Exploring tissue architecture using spatial transcriptomics. Nature 596, 211-220 (2021).
Cui Zhou, D. et al. Spatially restricted drivers and transitional cell populations cooperate with the microenvironment in untreated and chemo-resistant pancreatic cancer. Nat. Genet. 54, 1390-1405 (2022).
Black, S. et al. CODEX multiplexed tissue imaging with DNA-conjugated antibodies. Nat. Protoc. 16, 3802-3835 (2021).
Greaves, M. & Maley, C. C. Clonal evolution in cancer. Nature 481, 306-313 (2012).
Ding, L., Raphael, B. J., Chen, F. & Wendl, M. C. Advances for studying clonal evolution in cancer. Cancer Lett. 340, 212-219 (2013).
Erickson, A. et al. Spatially resolved clonal copy number alterations in benign and malignant tissue. Nature 608, 360-367 (2022).
Lomakin, A. et al. Spatial genomics maps the structure, nature and evolution of cancer clones. Nature 611, 594-602 (2022).
Di Maggio, F. & El-Shakankery, K. H. Desmoplasia and biophysics in pancreatic ductal adenocarcinoma: can we learn from breast cancer? Pancreas 49, 313-325 (2020).
Chen, H., Liu, H. & Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal. Transduct. Target Ther. 3, 5 (2018).
Cable, D. M. et al. Robust decomposition of cell type mixtures in spatial transcriptomics. Nat. Biotechnol. 40, 517-526 (2022).
Yoshihara, K. et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 4, 2612 (2013).
Gencheva, R. & Arnér, E. S. J. Thioredoxin reductase inhibition for cancer therapy. Annu. Rev. Pharmacol. Toxicol. 62, 177-196 (2022).
Jiang, Y. et al. A systematic analysis of C5ORF46 in gastrointestinal tumors as a potential prognostic and immunological biomarker. Front. Genet. 13, 926943 (2022).
Han, L., Li, Z., Jiang, Y., Jiang, Z. & Tang, L. SNHG29 regulates miR-223-3p/CTNND1 axis to promote glioblastoma progression via Wnt/ -catenin signaling pathway. Cancer Cell Int. 19, 345 (2019).
Huang, C. K., Sun, Y., Lv, L. & Ping, Y. ENO1 and cancer. Mol. Ther. Oncolytics 24, 288-298 (2022).
Zhang, X. et al. Thymosin beta 10 is a key regulator of tumorigenesis and metastasis and a novel serum marker in breast cancer. Breast Cancer Res 19, 15 (2017).
Chen, R. H. et al. Tumor cell-secreted ISG15 promotes tumor cell migration and immune suppression by inducing the macrophage M2-like phenotype. Front. Immunol. 11, 594775 (2020).
Jin, S. et al. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 12, 1088 (2021).
Filippou, P. S., Karagiannis, G. S. & Constantinidou, A. Midkine (MDK) growth factor: a key player in cancer progression and a promising therapeutic target. Oncogene 39, 2040-2054 (2020).
Sick, E. et al. Activation of CD47 receptors causes proliferation of human astrocytoma but not normal astrocytes via an Akt-dependent pathway. Glia 59, 308-319 (2011).
Isenberg, J. S., Frazier, W. A. & Roberts, D. D. Thrombospondin-1: a physiological regulator of nitric oxide signaling. Cell. Mol. Life Sci. 65, 728-742 (2008).
Jeanne, A. et al. Identification of TAX2 peptide as a new unpredicted anti-cancer agent. Oncotarget 6, 17981-18000 (2015).
Wang, Y., Hu, L., Zheng, Y. & Guo, L. HMGA1 in cancer: cancer classification by location. J. Cell. Mol. Med. 23, 2293-2302 (2019).
Huang, R., Huang, D., Dai, W. & Yang, F. Overexpression of HMGA1 correlates with the malignant status and prognosis of breast cancer. Mol. Cell. Biochem. 404, 251-257 (2015).
Mitselou, A. et al. Predictive role of thymidine phosphorylase expression in patients with colorectal cancer and its association with angiogenesis-related proteins and extracellular matrix components. In Vivo 26, 1057-1067 (2012).
Baris, A., Fraile-Bethencourt, E., Eubanks, J., Khou, S. & Anand, S. Thymidine phosphorylase facilitates retinoic acid inducible gene-I induced endothelial dysfunction. Cell Death Dis. 14, 294 (2023).
Schmidt, M. et al. Prognostic impact of immunoglobulin kappa C (IGKC) in early breast cancer. Cancers 13, 3626 (2021).
Wang, J. et al. CCL19 has potential to be a potential prognostic biomarker and a modulator of tumor immune microenvironment (TIME) of breast cancer: a comprehensive analysis based on TCGA database. Aging 14, 4158-4175 (2022).
Nishimura, T. et al. Evolutionary histories of breast cancer and related clones. Nature 620, 607-614 (2023).
Dang, H. X. et al. The clonal evolution of metastatic colorectal cancer. Sci. Adv. 6, eaay9691 (2020).
Simeonov, K. P. et al. Single-cell lineage tracing of metastatic cancer reveals selection of hybrid EMT states. Cancer Cell 39, 1150-1162.e9 (2021).
Chen, H. N. et al. Genomic evolution and diverse models of systemic metastases in colorectal cancer. Gut 71, 322-332 (2022).
Leung, M. L. et al. Single-cell DNA sequencing reveals a late-dissemination model in metastatic colorectal cancer. Genome Res. 27, 1287-1299 (2017).
Hu, Z. et al. Quantitative evidence for early metastatic seeding in colorectal cancer. Nat. Genet. 51, 1113-1122 (2019).
Tang, J. et al. Single-cell exome sequencing reveals multiple subclones in metastatic colorectal carcinoma. Genome Med. 13, 148 (2021).
Poos, A. M. et al. Resolving therapy resistance mechanisms in multiple myeloma by multiomics subclone analysis. Blood 142, 1633-1646 (2023).
Braxton, A. M. et al. 3D genomic mapping reveals multifocality of human pancreatic precancers. Nature 629, 679-687 (2024).
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http:// creativecommons.org/licenses/by-nc-nd/4.0/.
(c) The Author(s) 2024
¹Department of Medicine, Washington University in St Louis, St Louis, MO, USA. McDonnell Genome Institute, Washington University in St Louis, St Louis, MO, USA. Department of Computer Science, Princeton University, Princeton, NJ, USA. Department of Surgery, Washington University in St Louis, St Louis, MO, USA. Siteman Cancer Center, Washington University in St Louis, St Louis, MO, USA. Department of Radiology, Washington University in St Louis, St Louis, MO, USA. Institute for Systems Biology, Seattle, WA, USA. Division of Medical Oncology, Department of Internal Medicine, Washington University School of Medicine, St Louis, MO, USA. Department of Pathology and Immunology, Washington University in St Louis, St Louis, MO, USA. Department of Obstetrics and Gynecology, Washington University in St Louis, St Louis, MO, USA. Division of Urological Surgery, Department of Surgery, Washington University, St Louis, MO, USA. Department of Neuroscience, Washington University School of Medicine, St Louis, MO, USA. Department of Neurosurgery, Washington University School of Medicine, St Louis, MO, USA. Division of Gynecologic Oncology, Department of Obstetrics and Gynecology, Washington University, St Louis, MO, USA. Department of Computer Science and Engineering, Washington University in St Louis, St Louis, MO, USA. Department of Genetics, Washington University in St Louis, St Louis, MO, USA. Department of Otolaryngology-Head and Neck Surgery, Washington University School of Medicine, St Louis, MO, USA. Department of Obstetrics and Gynecology, University of California, San Francisco, San Francisco, CA, USA. These authors contributed equally: Chia-Kuei Mo, Jingxian Liu. These authors jointly supervised this work: Siqi Chen, Erik Storrs, Andre Luiz N. Targino da Costa, Andrew Houston. e-mail: gillandersw@wustl.edu; rcfields@wustl.edu; braphael@princeton.edu; fchen@wustl.edu; lding@wustl.edu
Methods
Experimental methods
Specimens and sample processing. All samples were collected with informed consent at the Washington University School of Medicine in St Louis. Samples from BRCA, PDAC, CRC, CHOL, RCC and UCEC were collected during surgical resection and verified by standard pathology (institutional review board protocols 201108117, 201411135 and 202106166). After verification, a portion of the tumour was removed, photographed, weighed and measured. Each portion was then subdivided into 6-9 pieces and then further subdivided into 4 transverse-cut pieces. These four pieces were then each respectively placed into formalin, snap-frozen in liquid nitrogen, DMEM and snap-frozen before embedding in OCT. The purpose of choosing grid processing over punch sampling was utility-based, as it minimized remaining tissue. Relevant protocols can be found at protocols.io (https://doi.org/10.17504/protocols.io.bszynf7w) .
ST preparation and sequencing. OCT-embedded tissue or FFPE tissue samples were sectioned and placed on a Visium Spatial Gene Expression Slide following the Visium Spatial Protocols-Tissue Preparation guide. Samples used for serial sections were sectioned and collected with an interval range from 5 to . When doing serial sectioning, the first section was named as U1, followed by U2, U3, and so on. Selected sections were loaded onto Visium slides and the distance between each section was recorded. For OCT-embedded samples, detailed methods have been described in a previous publication . In brief, fresh tissue samples were coated with room temperature OCT without any bubbles. After RNA quality check using a Tapestation and a morphology check using H&E staining for the OCT-embedded tissue samples, blocks were scored into a suitable size that fit the capture areas and then sectioned into sections. Sections were then fixed in methanol, stained with H&E and imaged at magnification using the bright-field imaging setting on a Leica DMi8 microscope. Tissue samples were then permeabilized for 18 min and ST libraries were constructed following the Visium Spatial Gene Expression Reagent kits user guide CG000239 Rev A (10x Genomics). cDNA was reverse transcribed from the poly-adenylated messenger RNA, which was captured using primers on the slides. Next, the second strand was synthesized and denatured from the first strand. Free cDNA was then transferred from slides to tubes for further amplification and library construction. Libraries were sequenced on a S4 flow cell of an Illumina NovaSeq-6000 system. For FFPE samples, detailed methods have been described in a previous publication . In brief, quality control was done by evaluating DV200 of RNA extracted from FFPE tissue sections per the Qiagen RNeasy FFPE Kit protocol, then followed by performing the Tissue Adhesion Test described in the 10x Genomics protocol. Sections ( ) were placed on a Visium Spatial Gene Expression Slide according to the Visium Spatial Protocols-Tissue Preparation guide (10x Genomics, CG000408 Rev A). After overnight drying, slides were incubated at for 2 h . Deparaffinization was then performed following the protocol for Visium Spatial for FFPE-Deparaffinization, H&E staining, Imaging and Decrosslinking (10x Genomics, CG000409 Rev A). Sections were stained with H&E and imaged at magnification using the bright-field imaging setting on a Leica DMi8 microscope. Afterwards, decrosslinking was performed immediately for H&E stained sections. Next, human whole transcriptome probe panels were added to the tissue. After these probe pairs hybridized to their target genes and ligated to one another, the ligation products were released following RNase treatment and permeabilization. The ligated probes were then hybridized to the spatially barcoded oligonucleotides on the capture area. ST libraries were generated from the probes and sequenced on a S4 flow cell of an Illumina NovaSeq 6000 system. Relevant protocols can be found at protocols.io (https://doi.org/10.17504/ protocols.io.x54v9d3opg3e/v1 and https://doi.org/10.17504/protocols. io.kxygx95ezg8j/v1) .
CODEX preparation and imaging. Carrier-free monoclonal or polyclonal anti-human antibodies were purchased (Supplementary Table3) and verified using immunofluorescence (IF) staining in multiple channels. After screening, antibodies were conjugated using an Akoya Antibody Conjugation kit (Akoya Biosciences, SKU 7000009) with a barcode (Akoya Biosciences) assigned according to the IF staining results. Several common markers were directly purchased through Akoya Biosciences. CODEX staining and imaging were performed according to the manufacturer’s instructions (CODEX user manual, Rev C). In brief, FFPE sections were placed on coverslips coated with APTES (Sigma, 440140) and baked at overnight before deparaffinization. The next day, tissues were incubated in xylene, rehydrated in ethanol and washed in before antigen retrieval with TE buffer, pH 9 (Genemed, 10-0046) in boiling water for 10 min in a rice cooker. The tissue samples were then blocked using blocking buffer (CODEX staining kit, SKU 7000008) and stained with the marker antibody panel to a volume of for 3 h at room temperature in a humidified chamber. The dilution factor for each antibody is provided in the CODEX cycle information sheet (Supplementary Table 3). Imaging of the CODEX multicycle experiment was performed using a Keyence fluorescence microscope (model BZ-X810) equipped with a Nikon CFI Plan Apo objective, a CODEX instrument (Akoya Biosciences) and a CODEX instrument manager (Akoya Biosciences). The raw images were then stitched and processed using the CODEX processor (Akoya Biosciences). After multiplex imaging was completed, H&E staining was performed on the same tissue. Staining quality for each antibody in CODEX is shown as a single channel in green with DAPI in blue in Supplementary Figs. 10 and 11.
Single-nucleus suspension preparation. Approximately of flash-frozen or cryopulverized or of OCT sections of tissue from each sample were retrieved and aliquoted for nucleus preparation for use in a Next GEM Single Cell Multiome ATAC + Gene Expression kit or a Next GEM Single Cell 3′ Kit v.3.1 kit. Samples were resuspended in lysis buffer ( 10 mM Tris- (Thermo, 15567027 ), 10 mM NaCl (Thermo, AM9759), 3 mM MgCl 2 (Thermo, AM9530G), substitute (% v/v) (Sigma, 74385-1L), 1 mM DTT (Sigma, 646563), 1% stock BSA solution ( ) (MACS, 130-091-376), nuclease-free water (Invitrogen, AM9937), plus Nase inhibitor), resuspended and homogenized through douncing, and filtered through a cell strainer (pluriSelect), then diluted with wash buffer ( BSA, PBS and RNase inhibitor). The filtrate was collected, then centrifuged at 500 g for 6 min at . The nuclear pellet was then resuspended in BSA wash buffer with RNase inhibitor, stained with 7AAD, and nuclei were purified and sorted by FACS. Relevant protocols can be found at protocols.io(https://doi.org/10.17504/protocols.io.14egn7w6zv5d/v1, https://doi.org/10.17504/protocols.io.261gednx7v47/v1) .
Single-cell suspension preparation. Approximately of each tumour was cut into small pieces using a blade. Enzymes and reagents from a Human Tumour Dissociation kit (Miltenyi Biotec, 130-095-929) were added to the tumour tissue along with 1.75 ml of DMEM. The resulting suspension was loaded into a gentleMACSC-tube (Miltenyi Biotec, 130-093-237) and subjected to the gentleMACS Octo Dissociator with Heaters (Miltenyi Biotec, 130-096-427). After on the heated dissociation programme (37h_TDK_1), samples were removed from the dissociator and filtered through a mini strainer (PluriSelect, no. 43-10040-60) or a nylon mesh (Fisher Scientific, 22-363-547) into a conical tube on ice. The sample was then spun down at 400 g for 5 min at . After removing the supernatant, when a red pellet was visible, the cell pellet was resuspended using to 3 ml ACK lysis solution (Thermo Fisher, A1049201) for . To quench the reaction, 10 ml PBS (Corning; 21-040-CM) with BSA (Miltenyi Biotec; 130-091-376) was added and spun down at 400 g for 5 min at . After removing the
supernatant, the cells were resuspended in 1 ml PBS with , and live and dead cells were visualized using trypan blue. Finally, the sample was spun down at for 5 min at and resuspended in to 1 ml PBS with BSA to a final concentration of cells per . The protocol is available at protocols.io (https://doi. org/10.17504/protocols.io.bsnqnddw) .
Single-nucleus library preparation and sequencing. Nuclei and cells and barcoded beads were isolated in oil droplets using a 10x Genomics Chromium instrument. Single-nucleus suspensions were counted and adjusted to a range of 500-1,800 nuclei per l using a haemocytometer. Reverse transcription was subsequently performed to incorporate cell and transcript-specific barcodes. All snRNA-seq samples were run using a Chromium Next GEM Single Cell 3′ Library and Gel Bead kit v.3.1 (10x Genomics). For the multiome kit, Chromium Next GEM Single Cell Multiome ATAC + Gene Expression was used ( Genomics). Nuclei were then subjected to downstream protocols by 10x (Next GEM Single Cell Multiome ATAC + Gene Expression: https://cdn.10xgenomics. com/image/upload/v1666737555/support-documents/CG000338_ ChromiumNextGEM_Multiome_ATAC_GEX_User_Guide_RevF.pdf. Next GEM Single Cell 3′ Kit v3.1: https://support.10xgenomics.com/ single-cell-gene-expression/library-prep/doc/user-guide-chromium-single-cell-3-reagent-kits-user-guide-v31-chemistry-dual-index). Singlecell suspensions were subject to the Next GEM Single Cell 3′ Kit v.3.1 protocol. Barcoded libraries were then pooled and sequenced on an Illumina NovaSeq 6000 system with associated flow cells.
Genomic DNA extraction. Tumour tissue samples were obtained from surgically resected specimens. After a piece was removed for fresh single-cell preparation, the remaining sample was snap-frozen in liquid nitrogen and stored at . Before bulk DNA extraction, samples were cryopulverized (Covaris) and aliquoted for bulk extraction. Genomic DNA was extracted from tissue samples with either a DNeasy Blood and Tissue kit (Qiagen, 69504) or a QIAamp DNA Mini kit (Qiagen, 51304). Genomic germline DNA was purified from cryopreserved peripheral blood mononuclear cells using a QIAamp DNA Mini kit (Qiagen, 51304) according to the manufacturer’s instructions (Qiagen). The DNA quantity was assessed by fluorometry using a Qubit dsDNA HS assay (Q32854) according to the manufacturer’s instructions (Thermo Fisher Scientific). Protocols are available at protocols. io (https://doi.org/10.17504/protocols.io.bsnhndb6) .
WES analysis. About 100-250 ng of genomic DNA was fragmented on a Covaris LE220 instrument targeting 250-bp inserts. Automated dual-indexed libraries were constructed using a KAPA Hyper library prep kit (Roche) on a SciClone NGS platform (Perkin Elmer). Up to ten libraries were pooled at an equimolar ratio by mass before the hybrid capture targeting a library pool. The library pools were hybridized using xGen Exome Research Panel v.1.0 reagent (IDT Technologies), which spans a target region (19,396 genes) of the human genome. The libraries were hybridized for hat followed by a stringent wash to remove spuriously hybridized library fragments. Enriched library fragments were eluted and PCR cycle optimization was performed to prevent overamplification. The enriched libraries were amplified using KAPA HiFi master mix (Roche) before sequencing. The concentration of each captured library pool was determined through qPCR using a KAPA library Quantification kit according to the manufacturer’s protocol (Roche) to produce cluster counts appropriate for the Illumina NovaSeq-6000 instrument. Next, paired-end reads were generated targeting 12 Gb of sequence to achieve around coverage per library.
Xenium library preparation and imaging. Paraffin blocks (FFPE blocks) were sectioned at and placed on Xenium slides following the FFPE Tissue Preparation guide (10x Genomics, CG000578, Rev B). Those
slides underwent a series of xylene and ethanol washes for deparaffinization and decrosslinking, using the FFPE tissue enhancer as outlined (10x Genomics, CG000580, Rev B). Overnight in situ probe hybridization was performed using 379 probes from the Xenium Human Multi-Tissue Panel ( Genomics, 1000626) plus an additional 100 custom probes (Supplementary Table 6). After hybridization probes were ligated, the sample underwent rolling circle amplification, and the background was quenched using an autofluorescence mixture. Nuclei were stained with DAPI to improve sample tracking and approximate cell boundaries (10x Genomics, CG000582, Rev D). These samples, along with buffers and decoding consumables, were loaded into a Xenium analyzer (10x Genomics, 1000481). The run was initialized using the guidance provided (10x Genomics, CG000584, Rev C). These fluorescent reporters hybridized to targeted complementary regions of the barcoded circularized cDNA were imaged. H&E staining was performed on the same region after the run was complete.
Analytical methods
Quantification and statistical analysis. All data analyses were conducted in R and Python environments. Details of specific functions and libraries are provided in the relevant methods sections above. Significance was determined using the Wilcoxon rank-sum test, proportion test, hypergeometric test or Pearson correlation test, as appropriate. values < 0.05 were considered significant. Details of statistical tests are provided in the figure legends and the relevant methods sections.
WES data processing. FASTQ files were preprocessed using trimGalore (v.0.6.7; with parameters: –length 36 and all other parameters set to default; https://github.com/FelixKrueger/TrimGalore). FASTQ files were then aligned to the GDC’s GRCh38 human reference genome (GRCh38.d1.vd1) using BWA-mem (v.0.7.17) with parameter -M and all others set to default. The output SAM file was converted to a BAM file using the samtools (https://github.com/samtools/samtools; v.1.14) view with parameters -Shb, and all others set to default. BAM files were sorted and duplicates were marked using Picard (v.2.6.26) SortSam tool with the following parameters: CREATE_INDEX=true, SORT_ORDER=coordinate, VALIDATION_STRINGENCY=STRICT, and all others set to default; and MarkDuplicates with parameter REMOVE_ DUPLICATES=true, and all others set to default. The final BAM files were then indexed using the samtools (v.1.14) index with all parameters set to default.
Mutation calling using WES. Somatic mutations were called from WES data using the Somaticwrapper pipeline (v.2.2; https://github. com/ding-lab/somaticwrapper), which includes four different callers: Strelka (v.2.9.10) , MUTECT (v.1.1.7) , VarScan (v.2.3.8) and Pindel (v.0.2.5) . We kept exonic single nucleotide variants (SNVs) called by any two callers among MUTECT (v.1.1.7), VarScan (v.2.3.8) and Strelka (v.2.9.10) and insertions and deletions (indels) called by any two callers among VarScan (v.2.3.8), Strelka (v.2.9.10) and Pindel (v.0.2.5). For the merged SNVs and indels, we applied a and minimal coverage cut-off for tumour and normal tissue, respectively. We also filtered SNVs and indels by a minimal VAF of 0.05 in tumours and a maximal VAF of 0.02 in normal samples. We also filtered any SNV within 10 bp of an indel found in the same tumour sample. Finally, we rescued the rare mutations with VAFs within 0.015 and 0.05 based on an established gene consensus list . In a downstream step, we used Somaticwrapper to combine adjacent SNVs into double-nucleotide polymorphisms usingCOCOON (https://github.com/ding-lab/COCOONS), as reported in a previous study .
Mutation mapping to snRNA-seq and ST data. We applied an in-house tool called scVarScan that can identify reads supporting the reference allele and variant allele covering the variant site in each cell by tracing cell and molecular barcode information in a snRNA-seq and
single-cell RNA sequencing (scRNA-seq) or Visium bam file. The tool is freely available at GitHub (https://github.com/ding-lab/10Xmapping). For mapping, we used high-confidence somatic mutations from WES data produced by Somaticwrapper (described above). Visium reads were prefiltered with the flag ‘xf:i:25’ for reads contributing to unique molecular identifier counts.
Spatial mutation VAF statistical test. For each ST section, we applied two sets of statistical tests to all WES-based somatic mutations mapped to ST. First, for each mutation with greater than 30 reads of coverage on ST across all spots, the VAF was calculated for all tumour region spots and all non-tumour region spots as the number of variant reads across all spots divided by the number of total reads across all spots. A binomial test was then done using VAF of non-tumour spots as the background:binom.test(alterative=”greater”). Then, a proportion test was done between the VAFs in different spatial subclones with prop. test(alternative=”two.sided”). Finally, multiple testing correction was done on both sets of tests with the function p.adjust().
CNV calling using WES. Somatic CNVs were called using GATK (v.4.1.9.0) . Specifically, the hg38 human reference genome (NCIGDC data portal) was binned into target intervals using the PreprocessIntervals function, with the bin length set to 1,000 bp and using the interval-merging-rule of OVERLAPPING_ONLY. A panel of normals was then generated using each normal sample as input and the GATK functions CollectReadCounts with the argument –interval-merging-rule OVERLAPPING_ONLY, followed by CreateReadCountPanelOfNormals with the argument –minimum-interval-median-percentile 5.0. For tumour samples, reads that overlapped the target interval were counted using the GATK function CollectReadCounts. Tumour read counts were then standardized and denoised using the GATK function DenoiseReadCounts, with the panel of normals specified by –count-panel-of-normals. Allelic counts for tumours were generated for variants present in the af-only-gnomad.hg38.vcf according to GATK best practices (variants further filtered to af and entries marked with ‘PASS’) using the GATK function CollectAllelicCounts. Segments were then modelled using the GATK function ModelSegments, with the denoised copy ratio and tumour allelic counts used as inputs. Copy ratios for segments were then called on the segment regions using the GATK function CallCopyRatioSegments.
Bedtools intersection was used to map copy number ratios from segments to genes and to assign the called amplifications or deletions. For genes overlapping multiple segments, a custom Python script was used to call that gene as amplified, neutral or deleted based on a weighted copy number ratio calculated from the copy ratios of each overlapped segment, the lengths of the overlaps and the score threshold used by the CallCopyRatioSegments function. If the resulting score cut-off value was within the range of the default score thresholds used by CallCopyRatioSegments (v.0.9,1.1), then the bounds of the default score threshold were used instead (replicating the logic of the CallCopyRatioSegments function).
ST data processing. For each sample, we obtained the unfiltered feature-barcode matrix per sample by passing the demultiplexed FASTQ files and associated H&E image to Space Ranger (v.1.3.0, v.2.0.0 and v2.1.0 ‘count’ command using default parameters with reorient-images enabled) and the prebuilt GRCh38 genome reference 2020-A (GRCh38 and Ensembl 98). Seurat was used for all subsequent analyses. We constructed a Seurat object using the Load10X_Spatial function for every slide. Each slide was then scaled and normalized with the SCTransform function to correct for batch effects. Any merged analysis or subsequent subsetting of cells and samples for a sample with several slides underwent the same scaling and normalization method. Spots were clustered using the original Louvain algorithm, and the top 30 principal component analysis dimensions using the FindNeighbors
and FindClusters functions as described in the ‘Analysis, visualization, and integration of spatial datasets with Seurat’ vignette from Seurat (https://satijalab.org/seurat/articles/spatial_vignette.html).
InferCNV and CalicoST for CNV calling on Visium ST data. To detect large-scale chromosomal CNVs using scRNA-seq, snRNA-seq and Visium data, InferCNV (v.1.10.1) was used with default parameters recommended for 10x Genomics data (https://github.com/broadinstitute/inferCNV). InferCNV was run at the sample level and only with post-quality control filtered data using the raw counts matrix. For snRNA-seq and scRNA-seq data, all non-malignant cells were used as a reference with the annotation ‘non-tumour’ and all malignant cells had the same annotation ‘tumour’, with the following parameters: analysis_mode=”subclusters”,–cluster_by_groups=T,–denoise=T, and –HMM=T. For Visium ST data, 200 spots annotated as ‘non-malignant’ with the lowest ESTIMATE purity score were used as a reference, and ‘malignant’ spots had their microregion ID as annotation, with the following parameters: window_length , analysis_mode=”sample”, –cluster_by_groups=T, –denoise=T, and –HMM=T. CalicoST (https:// github.com/raphael-group/CalicoST) was run on Visium ST data with the same input annotation (microregion ID). All spots from the same microregions were treated as the smallest unit of analysis. CalicoST was then run with default parameters with results manually inspected.
Copy number profile similarity score calculation. To determine the similarity between two spatial CNV profiles, we use a modified Jaccard similarity score. A CNV profile was defined as a set of genomic windows with annotation copy number neutral ( 0 ), amplification ( 1 ) or deletion ( -1 ). Two CNV profiles were then compared, and overlapping genomic windows were broken down so that both profiles had the same sets of windows (with the function reduced from the package GenomicRanges v.1.46.1). Then, the CNV similarity score (Sim) was defined as follows:
where denotes the size of the genomic window denotes the CNV annotation ( 0,1 or -1 ) for profile in genomic window , and denotes the CNV annotation for profile B in genomic window across all genomic windows where either A or B is not CNV neutral.
To determine the similarity between a spatial CNV profile and WES-based CNV (related to Extended Data Fig. 5a), we used a similarity score averaging the sensitivity (fraction of WES-based CNVs also detected in spatial CNVs) and specificity (fraction of spatial CNVs agreeing with WES-based CNVs). Specifically,
where denotes the size of the genomic window from spatial CNV, denotes the size of the genomic window from a WES-based CNV, denotes the CNV annotation ( 0,1 or -1 ) for profile A in genomic window .
Spatial subclone identification based on CNV profile similarity. In the OCT workflow (Supplementary Fig. 1a), CalicoST simultaneously identified CNVs and groups microregions into spatial subclones. In the FFPE workflow, confident spatial CNV events in each microregion were first selected by comparing them with matching WES. Then, a pairwise CNV similarity score was calculated across all tumour microregions. Finally, microregions were clustered with CNV similarity scores using the function hclust ( CNV similarity, method=”ward.D2″), and divided into clusters with function cutree ( (hclust$height)). Final subclone assignments were manually reviewed to avoid overclustering and to eliminate small outlier CNV profiles.
Tumour microregion annotation and layer determination. Using Visium ST, tumour microregions were determined through a multistep process using H&E. Each ST spot was assigned as either stroma or tumour by manually reviewing the morphology on H&E stained sections. If at least of the pixels within a spot covered malignant cell morphology, the spot was labelled as tumour. Otherwise, it was labelled as stroma. Next, we defined distinct tumour microregions using a set of three rules. The first rule specified that tumour spots immediately adjacent to one another are initially marked as a single tumour microregion. The second rule states that if two distinct tumour regions together occupied at least 50% of one single spot, the spot is assigned to the distinct tumour region with the higher percentage occupied. Finally, the third rule specified that if there was a clear morphological difference of the tumour spots within one tumour microregion, the microregion must be separated into distinct microregions, one per clear morphology.
Afterwards, we ran the Morph toolset (https://github.com/ding-lab/ morph), which uses mathematical morphology to refine the tumour microregions. That is, if the total number of spots in a microregion is less than or equal to three, then we labelled all such spots as stroma. Last, Morph assigned the layer (for example, T1) of each spot of a tumour microregion by a sequence of mathematical morphology operations described in the Spot-depth correlation analysis method, which denotes the depth of a given spot inside a microregion.
Average spot area and microregion size calculation. To calculate the area each spot takes, we used the spot size ( ) and centre-to-centre distance between each spot ( ) provided by Genomics (http://kb.10xgenomics.com/hc/en-us/articles/360035487572-What-is-the-spatial-resolution-and-configuration-of-the-capture-area-of-the-Visium-v1-Gene-Expression-Slide-). As illustrated in Supplementary Fig. 6, the Visium spots form a hexagonal lattice that covers the sample. The repeating unit of this lattice is a trapezoid shape centred at each spot’s centre that is composed of eight equilateral triangles. Each triangle has a side of (half of the spot the centre-to-centre distance). Using the area equation of equilateral triangles and multiplying it by 8, we obtained the area of each trapezoid as , which is the average area occupied by each spot. To calculate the microregion size, we multiplied the spot count by 8,660 and divided by to obtain the size in .
Micoregion density estimation. We estimated microregion density per section by following the formula: density per microregion per section size (in spots) then divided by per spot. Then density per density per microregion per .
Cell-type annotation. Cell-type assignment was done based on the following known markers: B cell, CD79A, CD79B, CD19, MS4A1, IGHD, CD22 and CD52; cDC1, CADM1,XCR1,CLEC9A,RAB32 and C1orf54; cDC2,CD1C, FCER1A, CLEC10A and CD1E; mregDC, LAMP3, CCR7, FSCN1, CD83 and CCL22; pDC, IL3RA, BCL11A, CLEC4C and NRP1; macrophage, CX3CR1, CD80, CD86, CD163 and MSR1; mast cell, HPGD, TPSB2, HDC, SLC18A2, CPA3 and SLC8A3; endothelial,EMCN,FLT1,PECAM1,VWF,PTPRB,ACTA2 and ANGPT2; fibroblast, COL1A1, COL3A1, COL5A1, LUM and MMP2; pericyte, RGS5, PLXDC1, FN1 and MCAM; NK cell, FCGR3A, GZMA and NCAM1; plasma cell, CD38, SDC1, IGHG1, IGKC and MZB1; T cell, IL7R, CD4, CD8A, CD8B, CD3G, CD3D and CD3E; and regulatory T cell, IL2RA, CTLA4, FOXP3, TNFRSF18 and IKZF2. Normal epithelial cells in the breast were annotated with the following markers: LumSec, GABRP, ELF5, CL28, KRT15, BARX2 and HS3ST4; LumHR, ANKRD3OA, ERBB4, AFF3, TTC6, ESR1, NEK10 and XBP1; and basal, SAMD5, FBXO32, TP63, RBBP8 and KLHL13. Normal epithelial cells in the liver were annotated with the following markers: hepatocyte, ALB, CYP3A7, HMGCS1, ACSS2 and ; cholangiocyte, SOX9, CFTR and PKD2. Normal epithelial cells in the pancreas, including ductal, acinar, islet- , islet- and
islet cells, were annotated with singleR (v.1.8.1) using reference data BaronPancreasData(‘human’).
Spot-depth correlation analysis. We identified a correlation between gene expression and spot depth in its tumour microregion. First, each spot was assigned a depth defined as the distance to the closest TME-facing spot in its tumour microregion. This depth was quantified in several layers through an iterative process whereby all the malignant spots immediately adjacent to non-malignant spots were considered layer 1, and then all malignant spots immediately adjacent to layer 1 were considered layer 2, and the process was repeated until all spots were assigned with a layer number. If a spot’s layer was larger than the smallest distance between the spot and any Visium border (including the edge of the Visium capture window, edge of the tissue section and any empty spots inside the section), then we excluded such spots, as we only knew the upper bound of the depth of this spot. Additionally, tumour microregions with fewer than 3 layers or 50 spots were excluded from the analysis. The distance between layers was taken as the centre-to-centre distance of Visium spots ( ).
To give the same weight to bigger and smaller regions, the depth of each spot was further normalized by the maximum depth of the microregion this spot belonged. Then, we performed partial correlation tests independently between gene expression (at least 1 transcript detected from the gene in more than of all spots) and normalized depth of each spot, with tumour purity as a covariate as follows:
where layer fraction is the layer number divided by the total number of layers in a tumour to normalize for large and small microregions, rho is the layer correlation coefficient, and is the correlation coefficient for covariant purity. Purity was inferred with deconvolution when there was matching snRNA-seq data (deconvoluted tumour fraction per spot by RCTD), or with ESTIMATE (that is, tumour purity estimate score per spot) otherwise. Each gene was checked against a set of snRNA-seq-derived non-malignant gene lists to ensure that the change in fraction did not derive from a shift in cell type composition. Finally, we performed multiple-testing adjustments for all tests done in each ST section.
Spot-depth GSEA pathway enrichment analysis. To summarize biological programs enriched in the centre and periphery of tumour microregions across sections, we first obtained the cohort-level average layer correlation coefficient. If a test was not significant ( ), rho was assigned to be 0 to indicate no correlation. If a test was not performed on a section (<50% of the spots have at least one transcript), rho was also assigned as 0 . When a case had multiple sections, we first took the average rho across sections to avoid bias towards tumours with more sections. Then, the average of rho was calculated for each cohort (all samples or samples from each cancer type).
In the same fashion, rank statistics were calculated for each test as value rho for tests with . For tests with or genes not tested, the rank statistic was 0 . We then calculated average rank statistics per case, followed by the average per cancer type. Finally, with the full list of rank statistics calculated for all genes tested, we used the function GSEA (parameters: pvalueCutoff=0.5; package: clusterProfiler v.3.18.1) to obtain the normalized enrichment score of Hallmark pathways (package: msigdbr 7.5.1) from the MSigDB . Finally, only pathways with were kept in the final results.
Tumour intrinsic and non-tumour gene categorization. We use differential expression and per cent expression filters, comparing expression among cell types in the matching snRNA-seq data to further characterize genes identified in the centre and periphery enriched analysis. The steps implemented in this workflow generated four
categories: tumour-specific, stromal-specific, tumour-enriched and stromal-enriched (Supplementary Fig. 8a,b). Genes that did not pass the significant cutoff in any differential expression analysis were labelled separately as not DEG.
To distinguish these four groups, we first performed differential gene analysis of cell types in the matching snRNA-seq data, filtered by a conventional significance cut-off ( (fold change) , adjusted , Bonferroni correction), to obtain DEGs (Supplementary Fig. 8a). Given the heterogeneity in tumours, certain tumour-specific genes might only exist in a subpopulation of tumours. Therefore, we first subclustered the tumour populations (using the Subcluster function in Seurat with a resolution of 0.5) to obtain tumour subclusters. We then compared each subcluster with all other non-tumour cells. A gene was considered a tumour DEG if at least one tumour subcluster showed significant expression compared with the non-tumour cells and vice versa for non-tumour DEGs (Supplementary Fig. 8a,b).
For candidate tumour or stromal-specific genes, a DEG was designated as tumour-specific if it met both of the following criteria: (1) it is a DEG when compared with all non-tumour cell types from at least one tumour subcluster; and (2) its expression was in all non-tumour cell types (Supplementary Fig. 8c).
The reverse applied to candidate stromal-specific DEGs. If a DEG did not meet both of these requirements to be tumour or stromal specific, it was designated as either tumour-enriched or stromal-enriched based on whether the expression level was higher in tumour or stromal cell types (Supplementary Fig. 8a).
Spatial subclone-specific treatment response analysis. We focused on ten cases (comprising four BRCA, two CRC and four PDAC samples) with multiple spatial subclones for this analysis. To obtain subclone-specific DEGs, we used FindMarkers from the function in Seurat with the ‘wilcox’ test option DEGs between each subclone and TME. We then applied the cut-off for adjusted , average (fold change) and per cent expression in at least one cell type > 0.4 to select significant DEGs. To infer treatment response, we used the perturbation database LINCS L1000 (ref. 65), specifically the LINCS_L1000_Chem_Pert_down dataset from Enrichr , to evaluate the gene set overlap between upregulated DEGs in spatial subclones and downregulated genes after compound treatment. To make the plot in Supplementary Fig. 4, we sorted the data by ‘Odd.Ratio’ and selected top compounds from each subclone. The corresponding compound metadata, including mechanism of action, was obtained from CLUE (clue.io, ‘Expanded CMap LINCS Resource 2020 Release’) to add annotation on the heatmap.
Organ-specific gene blacklist for non-malignant cell types. To distinguish transcripts originating from cancerous versus nonmalignant stromal or immune cells, we used merged snRNA-seq data per organ (breast, kidney, liver and pancreas) for cell-type marker analysis. This analysis used the FindAllMarkers function in Seurat with the ‘wilcox’ test option. Subsequently, we refined the gene list by applying filters such as average (fold change) > 2, per cent expression in at least one cell type and adjusted values to ensure robust marker selection for each cell type. The resultant gene list is available in Supplementary Table 5. This list was instrumental in excluding non-cancerous cell genes from analyses pertaining to cancer-specific expression patterns, such as pairwise microregion similarity analysis. Of note, during the analysis, we observed a notable mapping of various epithelial cell types in the snRNA-seq reference dataset for BRCA when using the RCTD deconvolution method. This observation probably stems from the diverse BRCA subtypes present in the cohort. To address this, we opted to combine all epithelial cell types into a single category during the identification of cell-type markers and excluded them from the blacklist. For tumours originating from organs other than the four mentioned above, we aggregated all genes present in the
blacklist across organs to form a comprehensive multiorgan blacklist, which aided in filtering out non-cancerous transcripts.
Microregion transcriptional profile analysis. For overall tumour heterogeneity, we selected Morph-identified spots then ran ROGUE (v.1.0) to measure heterogeneity as 1-ROGUE. We then compared the transcriptional profiles of microregions by selecting the top 500 most variable features after excluding stroma regions in ST samples following Morph processing. Our initial evaluation involved conducting Pearson correlation tests for each pair of microregions, using a range of the top 250-1,500 most variable genes with increments of 250 (that is, 250, . We observed consistent correlations for nearly all values beyond using more than 500, which led us to select the top 500 genes for this analysis. This choice reduced the risk of selecting too few variable genes (for example, <250 most variable genes) while also avoiding the inclusion of numerous genes with minimal effect on the transcriptional profile. GSEA analysis was done using the function GSEA (parameters: pvalueCutoff = 0.5; package: clusterProfiler v.3.18.1) to obtain the normalized enrichment score of Hallmark pathways (package: msigdbr v.7.5.1) from the MSigDB .
Module score calculation. Module scores on top of each heatmap in Extended Data Fig. 6 were calculated with the AddModuleScore function from Seurat using the genes listed in each heatmap. This score represents the average expression levels of a gene set. The score was calculated for each spot and a box plot was used to show the distribution of module scores in each microregion.
ST cell-type decomposition. Cell-type composition per spot was deconvolved using RCTD with default parameters and doublet mode = ‘multi’. The reference for each run was the cell types manually annotated from the Seurat object of the matching snRNA-seq or Multiome sample. To quantify spatial distribution of each cell type, cell type fraction of 6 layers (T3 and above, T2, T1, E1, E2, E3 and above) from each tumour microregion is calculated and averaged in each sample. To compare differential TME infiltration between spatial subclones, cell type fraction from all spots between spatial subclones was compared with pairwise Wilcoxon rank-sum test and FDR adjustment.
Spatial cell-cell interaction at tumour boundary. We evaluated the spatial-based cell-cell interaction (CCI) in the ST sample using COM with CellChat database and distance threshold of , following the same threshold used in the original publication for Visium. The median sender and receiver signals for each interaction family were compared between all tumour boundary spots (including tumour boundary layer and TME boundary layer) and all non-boundary spots(Wilcoxon rank-sum test) on a sample. Interaction pathways with signal difference great than 0.1 and FDR less than 0.05 are considered significantly boundary-enriched. Boundary DEGs were identified with Find Markers function on three sets of comparisons: boundary/tumour, boundary/TME and boundary/all non-boundary. A boundary DEG has adjusted value 0.25 in boundary/non-boundary test, and (fold change) in the other two tests.
Serial section alignment and branching factor calculation. We applied PASTE2 (ref. 70), the updated ST-based alignment tool PASTE , to enable partial image alignment. Serial sections of the same tumour piece were aligned pairwise with default settings. Each Visium data point in every ST section received new coordinates, denoted as and , based on the alignment results. We then identified the nearest spot on each adjacent section for every spot, connecting them along the axis. This process facilitated the linking of spots across all sections on the axis. To assess whether one microregion was connected to another in an adjacent section, we first removed stromal spots and then counted the connected spots. If any microregion on one section connected to
the next section with more than three shared spots, then we considered these two microregions, located on different sections, as connected in 3D space and forming the same tumour volume. This connection was labelled as volume 1, volume 2, and so forth in the figures (Fig. 5d, eand Extended Data Fig. 9a-d).
We used two geometric metrics to describe tumour volume: connectivity and loop. For connectivity (degree), this metric quantifies the number of connections from an individual microregion to adjacent sections. For example, if microregion 2 in section 2 connects to 3 microregions in section 1 and 2 in section 3 , its connectivity is 5 . The maximum connectivity of a tumour volume is the highest connectivity among its microregions. For loop, this metric was calculated as the total number of connections minus the total number of microregions plus one, identifying intricate loop structures within the tumour volume.
Registration of Visium, CODEX and H&E serial sections. Before registration, imaging data underwent the following transformations. Multiplex images were converted to greyscale images of DAPI intensity. The image was then downscaled by a factor of 5 before key point selection. H&E images (also downsampled by a factor of 5) were used for keypoint selection with Visium data.
For registration, we used BigWarp , which was packaged in the Fiji/ ImageJ software application. To register each collection of serial images, we used the first serial section as the fixed image and the second image as the moving image. After the second image was warped to the first image, the second image was used as the fixed image for the transformation of the third image. Key point registration proceeded in this fashion for all images in the serial section experiment. A total of 4-20 key points were selected per image transformation. Once key points were selected, a moving field was exported from BigWarp for each image transformation. This dense displacement field was then upscaled by a factor of 5 so it could be used to warp the full-resolution imaging data. The full-resolution dense displacement field was then used to register its corresponding multiplex or Visium data. The code used for registration is available at GitHub (https://github.com/ding-lab/ mushroom/tree/subclone_submission).
Neighbourhood identification input preprocessing. Once imaging data were registered, they were processed in the following manner before model input.
For Visium ST data, genes were limited to genes expressed in a minimum of 5% of spots across all serial sections and expression counts were transformed. CODEX, Visium ST and H&E data were normalized by subtracting the mean expression and dividing by the standard deviation for each gene.
Expression profiles for each patch were generated differently for image-native data (CODEX and H&E) and point-based data (Visium). Expressions for CODEX and H&E patches were calculated as the average pixel intensities for each image channel over all pixels within the patch bounds. Visium patches were calculated in largely the same manner; however, the expression profile of each spot within the patch was linearly weighted by its distance to the centre of the patch. This differential weight helped to account for variation expression due to the number of spots that fall within patch boundaries.
Neighbourhood identification model architecture. The neighbourhood annotation model consisted of an autoencoder with a vision transformer (ViT) backbone (Supplementary Fig. 7). In brief, an autoencoder is an unsupervised training method for which an encoder (embedding component) and a decoder (reconstruction component) work together to learn how input data are generated. Specifically, the network derives an approximation, , to the true posterior generating function, , for the output, given the input. The autoencoder used was asymmetric, meaning that the encoder and decoder were not inverse copies of one another. The encoder consisted of a ViT with a similar
architecture to previously described architectures (Supplementary Table 4).
ViTs work on image tokens as input. In brief, image tokens are -dimensional representations of patches of the input image. During training, image tiles were sampled from a uniform distribution across the set of input sections (Supplementary Fig. 7a). The sampled tile was then split into patches, for which the number of patches was determined by two hyperparameters: patch height ( ph ) and patch width ( pw ). Each patch was then flattened to vector, where is the number of channels in the image (in the case of spatial transcriptomics data, is the number of genes). The unrolled patches were then concatenated into a matrix, where is the number of patches in the image tile. Each row in this matrix is a token that represents a patch in the image tile. The tokens were then projected by a linear layer to shape , where is the dimension of the transformer blocks.
After this, a slide token was concatenated to patch tokens. The slide token (representing the slide from which the image tile was selected) was indexed from a trainable embedding of size slides , where n_slides is the number of slides in the serial section experiment. The motivation for the slide token is that as it is passed through the transformer blocks, along with the patch tokens, information can be shared across all tokens, allowing the slide token to learn to attend to useful representations of the patches. This feature allowed the model to be more robust to batch effects between serial sections. Following the addition of the slide token, positional embeddings were added to all tokens and passed through the transformer blocks comprising the ViT encoder. All variables above and details of the transformer architecture are available in Supplementary Table 4.
Once passed through the encoder, patches were represented as an embedding of size . The next step of the architecture was neighbourhood assignment. Neighbourhoods were assigned to patches in a hierarchical manner, meaning that a patch was classified into several neighbourhoods that differed in level of specificity. For each level of the neighbourhood hierarchy, the subsequent levels comprised partitions of the previous levels’ neighbourhoods, that is, except for the first level, each neighbourhood was a subset of a neighbourhood in a previous level of the hierarchy. For this analysis, the model generated three levels of neighbourhoods, each with the capacity to discover up to (level 1), (level 2) and (level 3) neighbourhoods, respectively. For this analysis, all neighbourhoods shown are neighbourhoods annotated at hierarchy level 3. The model contained three codebooks (one for each level) that are of size NBHDs , where NBHDs is the number of possible neighbourhoods that can be assigned for the given level. The patch embeddings output by the ViT encoder were projected by three independent blocks of linear layers (one for each level) that output each patch’s probability of assignment to a given neighbourhood. These probabilities were then used to retrieve neighbourhood embeddings from the codebook corresponding to the neighbourhood level. Three linear blocks (one for each level) were then used to independently reconstruct patch embeddings at each level to each patch’s original pixel values. The code used for training the model is available at GitHub (https://github.com/ding-lab/ mushroom/tree/subclone_submission).
Model loss function. The overall loss function has two main contributions: mean squared error (MSE) on the reconstruction of the input patches, and cross-entropy loss on the encoded distribution and the normal distribution with 0 mean and 1.0 variance.
During training, the autoencoder was simultaneously trying to optimize two main tasks: the reconstruction of the expression profile of each image patch embedding and the alignment of neighbourhood labels between adjacent sections. These two competing objectives forced the model to learn representative expression patterns while also keeping neighbourhoods aligned between input sections, which helped
to combat neighbourhood differences due to batch effects. Differences in patch expression were quantified by MSE, whereas neighbourhood adjacency was enforced by minimizing the cross-entropy of patches adjacent to each other in the direction during training.
The overall loss function is defined below:
Where (maximum of 0.01) and (set to 1.0) are scalers for the neighbourhood loss ( ) and reconstruction loss ( ), respectively. During training, for was linearly increased from 0 to its maximum value.
Model training and inference. Two separate runs of the model were trained for HT397B1 (six H&E, four CODEX and two Visium ST slides) and HT268B1 (four Visium ST slides). Training hyperparameters, such as batch size and number of training steps, are provided in Supplementary Table 4. For HT268B1, only one instance was trained because only one data type was present. For HT397B1, three model instances were trained (one for each data type) and were subsequently merged following the procedure described in the section ‘3D neighbourhood construction and integration’.
Following training, the model inference was performed on overlapping image tiles for each slide using a sliding window of size 8 and a stride of 2 (that is, 2 overlapping patches between image tiles). The centre patches of each tile were extracted and retiled to match the original slide orientation. Each reconstructed ‘patch embedding image’ was at a resolution of 50 pixels (that is, each neighbourhood patch represents an area that is wide) with the exception of Visium ST, for which the patch resolution was 100 pixels .
3D neighbourhood construction and integration. After the assignment of neighbourhoods for each section, slides were interpolated to generate a 3D neighbourhood volume. For this, we used linear interpolation of neighbourhood assignment probabilities with the torchio library .
Following interpolation, we also integrated neighbourhood volumes for HT397B1, for which multiple data-type-specific volumes were generated using a graph-based clustering approach. In brief, all overlapping neighbourhood voxel annotations were identified. A graph was then constructed, whereby nodes represented each neighbourhood partition combination, and edges are the distance (in the expression profile) between these partition combinations. This graph was then clustered with the Leiden graph clustering algorithm to identify integrated neighbourhoods. Hyperparameters for the above clustering process are provided in Supplementary Table 4.3D neighbourhoods were displayed using the open-source visualization tool Napari (https:// github.com/napari/napari).
Analysis and quantification of 3D neighbourhoods. Neighbourhoods were then assigned to Visium ST spots in the following manner. Each spot was assigned the neighbourhood label of the neighbourhood overlapping its spot centroid.
To focus on neighbourhoods most related to the TME biology, we filtered out neighbourhoods with >50% overlap with copy number annotated subclones. Additionally, we excluded neighbourhoods that mapped to fewer than ten total spots across allST sections for a sample.
The subclone boundary region for tumour clones was defined as the union of the outermost layer of subclone annotated spots and the spots one layer expanded out from them, representing an area roughly at the tumour-TME interface. Subclone-specific fractions were calculated as the neighbourhood overlaps with the outermost layer of each subclone.
In HT397B1, DEGs were calculated for all neighbourhoods, not only those filtered for subclone overlap and spot count. The top 50 DEGs for
neighbourhoods 4 and 6 were grouped into three categories: shared, unique to neighbourhood 4 and unique to neighbourhood 6. For the display in Fig. 5, the top 10 for each group were selected for display based on the following sorting criteria. The mean expression delta between neighbourhoods 4 and 6 was calculated for each gene by subtracting the mean expression in neighbourhood 6 from neighbourhood 4.Shared DEGs were ordered in ascending fashion based on the absolute expression delta of each gene. Genes unique to neighbourhood 4 and neighbourhood 6 were ordered by mean expression delta in descending and ascending fashion, respectively.
Cell-type annotation of CODEX imaging data. Our workflow for cell annotation consisted of four main steps: (1) image format conversion, (2) cell segmentation, (3) spatial feature generation and (4) cell-type classification. First, we converted image output by the CODEX platform (.qptiff) to the popular open-source OME-TIFF format. During this process, we also produced a separate image for each sample, as multiple sections of tissue are sometimes included on the same imaging run. We then used the Mesmer pre-trained nuclei + membrane segmentation model in the DeepCell framework to segment nuclei and whole cells. DAPI was used as the nuclei intensity image, and the channels pan-cytokeratin, HLA-DR, SMA, CD4, CD45, Hep-Par-1, CD31, E-cadherin, CD68 and CD3e were, for those present in a given image, mean-averaged to a single channel and used as the membrane intensity image.
We then use a gating procedure to identify cell types. First, to combat differences in protein intensity distributions between imaging runs and tissue types, thresholds were manually set for all protein channels used during cell typing for each image by visual inspection. Above this intensity threshold, a pixel was considered positive for a given marker, and below it, a pixel was considered negative. We then used the cell segmentation boundaries from the previous step to calculate the fraction of positive pixels for all cell typing markers in each cell. The result of this process is a feature matrix (num cells num proteins) representing positive marker fractions for each cell typing protein in every cell. A cell was considered positive for a marker if of its pixels were positive for that marker. Cells were then labelled with a gating strategy specific to each sample. During gating, each cell was subjected to a series of AND gates, whereby if a cell passed all criteria for a given step, it was annotated as the cell type specified for that step, whereas if it failed the criteria it was passed on to the next downstream step in the gating strategy. The gating strategies used for the samples in this paper are presented in Supplementary Table 4.
The following labels were the set of all possible cell type annotations: epithelial, CD4 T cell, CD8 T cell, regulatory T cell, T cell, macrophage, macrophage-M2, B cell, dendritic, immune, endothelial, fibroblast and hepatocyte. For some images, not all proteins required to gate a specific cell type were present. For example, CD4 was not in every image panel and available to use in the annotation of CD4 T cells. In these instances, the gating strategy was constructed such that cells can be labelled as more general cell types if specific proteins are not present (that is, labelled more broadly as T cell instead of CD4 T cell). If a cell was negative for all steps in the gating strategy, it was annotated as ‘unlabelled’. The code for image format conversion and cell segmentation can be found at GitHub (https://github.com/estorrs/ multiplex-imaging-pipeline).
Distance to tumour boundary quantification on CODEX. After registration, Visium spots labelled as tumours were mapped to CODEX slides using the coordinates of the aligned images. The coordinates of the centre of each spot in the CODEX-aligned slide were the same as its Visium counterpart. Each spot in the CODEX-aligned slide occupied the area of a circle with a radius of 150 pixels. The Euclidean distance transforms in the CODEX-aligned slide were then calculated for each pixel using Python’s scipy.ndimage.distance_transform_edt. Both the distances from the microregions and within the microregions were calculated.
3D tumour volume reconstruction and location quantification. The surface mesh visualizations of the tumour volumes for HT397B1 and HT268B1 were generated using the following steps: (1) tumour neighbourhood selection, (2) mesh construction and (3) mesh colouring. First, integrated neighbourhoods with tumour metrics (described below) exceeding a given threshold were considered to be part of the tumour volume. In HT268B1, the metric used to quantify epithelial character was the fraction of subclone annotated Visium ST spots per neighbourhood. Neighbourhoods with subclone spots were considered tumour. In HT397B1, we instead used the fraction of CODEX-annotated epithelial cells, as CODEX sections outnumbered the Visium ST sections for that sample. Neighbourhoods with fraction of epithelial cells were considered tumour.
A new volume was then constructed whereby neighbourhoods classified as tumour neighbourhoods using the above criteria were considered tumour-positive voxels, and all other voxels were tumour-negative. This 3D tumour mask was then smoothed with a Gaussian kernel (sigma ). The resulting values were then used as input for the marching cubes algorithm to generate a surface mesh for the tumour volume. We used the scikit-image implementation (skimage.measure.marching_cubes) of the marching cubes algorithm with default parameters.
To colour the surface mesh, we generated 3D feature volumes (described below), and then coloured points on the surface mesh based on the voxel value at the corresponding location in the feature volume.A feature volume is a volume whereby each voxel in the volume describes some feature from the serial section dataset (for example, expression of a given gene, fraction of cells, and so on). Feature volumes used in this analysis were constructed in the following manner. First, in the serial sections for which a feature was applicable, the feature was binned at the same resolution as the 3D neighbourhoods ( in this case). The binned feature was then interpolated in the direction to fill in gaps between sections. The resulting volume was of the same shape as the integrated neighbourhood volume, for which the value of each voxel was the aggregated feature count for the voxel. For HT268B1, the features used were logged expression of TYMP1 and IGLC2. For HT397B1, we used fibroblast and immune cell fraction. Cells were annotated as described in the section ‘Cell-type annotation of CODEX imaging data’. The surface mesh was visualized using Napari (https://github.com/ napari/napari) and contrast was adjusted on a volume-to-volume basis. We also visualized the HT397B1 tissue volume with the Imaris platform, for which we generated surfaces from the following CODEX markers: pan-cytokeratin (epithelial), CD45 (immune) and SMA (stromal).
Xenium probe design. Custom Xenium gene and mutation probes were designed using Xenium Panel Designer (https://cloud.10xgenomics. com/xenium-panel-designer) following instructions outlined in the ‘Getting Started with Xenium Panel Design’ instructions (https:// www.10xgenomics.com/support/in-situ-gene-expression/documenta-tion/steps/panel-design/xenium-panel-getting-started#design-tool). In brief, 21-bp sequences flanking the targeted transcribed variant site were curated from the Ensembl canonical transcript (Ensembl v.100). All four possible ligation junctions (two for the WT allele and two for the variant allele, three in the case of deletions-two for the WT allele and one for the variant allele) were then evaluated. Variant sites for which only non-preferred junctions (CG, GT, GG and GC) were available were excluded. The two bases of the ligation junction sequence were the last base of the RBD5 (RNA binding domain) and the first base of the RBD3 probe. Preferred junctions were always prioritized over neutral junctions unless a neutral junction was necessary to avoid hairpins, homopolymer regions, dimers or an unfavourable annealing temperature. Probe lengths for RBD5 and RBD3 were then adjusted from the 21-bp starting length to target a temperature between and per probe (overall target and ). Variant sites with probes predicted to form dimers or hairpins by IDT’s oligo analyzer were excluded.
Variant sites with homopolymer regions of five consecutive bases or more in either the RBD5 or RBD3 probes were excluded.
Spatial expression deconvolution. Here we used both deconvolution results and cell-type-specific expressions in the snRNA-seq data to deconvolve the Visium ST expression data (Supplementary Fig. 9). In brief, for a given Gene1, we first calculated the average expression of Gene1 per cell type in matched snRNA-seq data, subsequently filtering out the expression of such genes in cell types having of the highest average expression, and then dividing each cell-type average expression from the sum of all average expressions, thereby creating the expression contribution per cell type matrix ( ). Then for a given spot, the contribution per cell type was multiplied by cell type proportion from the cell type devolution result (for example, RCTD), then normalized to 1 to give a final expression contribution matrix (WN). For instance, in Supplementary Fig. 9a, Gene1 has and contributions from respective cell types and C based on the filtered snRNA-seq expression. For Spot1, as there is only 1 cell type, B, in the spot, gives the final contribution of Gene1 to cell type B in Spot1. Spot2 contains A and B cell types, respectively, the normalized cell type contribution in spot 2 is therefore for the cell type A , and for the cell type B. The final deconvolved expression was obtained by multiplying the original expression per spot (5 and 20 in Spot1 and Spot2) with the respective cell-type-based contribution to obtain the final deconvoluted expression values of Spot1 – cell type B , Spot2 – cell type A , and Spot2 – cell type B 8.58.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
All bioinformatics programs used in this study can be accessed from the GitHub public repository (https://github.com/ding-lab/ST_subclone_publication). The code for 3D neighbourhood identification and construction is available at GitHub (https://github.com/ding-lab/ mushroom/tree/subclone-resubmission). The code for multiplex imaging processing is hosted at GitHub (https://github.com/estorrs/ multiplex-imaging-pipeline). The code for Morph is accessible at GitHub (https://github.com/ding-lab/morph).
46. Herndon, J., Fields, R., Zhou, D. C. & Ding, L. Biospecimen collection and processing 2.0. protocols.iohttps://doi.org/10.17504/protocols.io.bszynf7w (2021).
47. Wu, Y. et al. Epigenetic and transcriptomic characterization reveals progression markers and essential pathways in clear cell renal cell carcinoma. Nat. Commun. 14, 1681 (2023).
48. Houston, A., Chen, S. & Chen, F. Spatial transcriptomics for OCT using 10x Genomics Visium. protocols.iohttps://doi.org/10.17504/protocols.io.x54v9d3opg3e/v1 (2023).
49. Houston, A., Chen, S. & Chen, F. Spatial transcriptomics for FFPE utilizing 10x Genomics Visium. protocols.iohttps://doi.org/10.17504/protocols.io.kxygx95ezg8j/v1 (2023).
50. Caravan, W., Jayasinghe, R. & Al Deen, N. N. WU sn-prep protocol for solid tumors-snRNA protocol v2.8. protocols.iohttps://doi.org/10.17504/protocols.io.14egn7w6zv5d/v1 (2022).
51. Jayasinghe, R., Caravan, W., Houston, A. & AlDeen, N. N. WU sn-prep protocol for solid tumors-joint snRNA+ATAC v2.9. protocols.iohttps://doi.org/10.17504/protocols. io. 261 ged (2023).
52. Jayasinghe, R., Ding, L. & Chen, F. WU sc-prep protocol for solid tumors v2.1. protocols.iohttps://doi.org/10.17504/protocols.io.bsnqnddw (2021).
53. Jayasinghe, R., Ding, L., Chen, F. & Satok. Bulk DNA extraction (Ding Lab). protocols.iohttps://doi.org/10.17504/protocols.io.bsnhndb6 (2021).
54. Kim, S. et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods 15, 591-594 (2018).
55. Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213-219 (2013).
56. Koboldt, D. C. et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568-576 (2012).
57. Ye, K., Schulz, M. H., Long, Q., Apweiler, R. & Ning, Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25, 2865-2871 (2009).
58. Bailey, M. H. et al. Comprehensive characterization of cancer driver genes and mutations. Cell 173, 371-385.e18 (2018).
59. Li, Y. et al. Pan-cancer proteogenomics connects oncogenic drivers to functional states. Cell 186, 3921-3944.e25 (2023).
60. Terekhanova, N. V. et al. Epigenetic regulation during cancer transitions across 11 tumour types. Nature 623, 432-441 (2023).
61. McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297-1303 (2010).
62. Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841-842 (2010).
63. Ma, C. et al. Inferring allele-specific copy number aberrations and tumor phylogeography from spatially resolved transcriptomics. Preprint at bioRxiv https://doi.org/10.1101/ 2024.03.09.584244 (2024).
64. Liberzon, A. et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417-425 (2015).
65. Subramanian, A. et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 171, 1437-1452.e17 (2017).
66. Kuleshov, M. V. et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90-W97 (2016).
67. Liu, B. et al. An entropy-based metric for assessing the purity of single cell populations. Nat. Commun. 11, 3155 (2020).
68. Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573-3587.e29 (2021).
69. Cang, Z. et al. Screening cell-cell communication in spatial transcriptomics via collective optimal transport. Nat. Methods 20, 218-228 (2023).
70. Liu, X., Zeira, R. & Raphael, B. J. Partial alignment of multislice spatially resolved transcriptomics data. Genome Res. 33, 1124-1132 (2023).
71. Bogovic J. A., Hanslovsky P., Wong A., Saalfeld S. in 2016 IEEE 13th International Symposium on Biomedical Imaging (ISBI), 1123-1126 (2016)
72. Dosovitskiy, A. et al. An image is worth words: transformers for image recognition at scale. Preprint at https://doi.org/10.48550/arXiv.2010.11929 (2021).
73. He, K. et al. in 2022 IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR) 15979-15988 (IEEE, 2022).
74. Pérez-García, F., Sparks, R. & Ourselin, S. TorchIO: a Python library for efficient loading, preprocessing, augmentation and patch-based sampling of medical images in deep learning. Comput. Methods Programs Biomed. 208, 106236 (2021).
75. Greenwald, N. F. et al. Whole-cell segmentation of tissue images with human-level performance using large-scale data annotation and deep learning. Nat. Biotechnol. 40, 555-565 (2022).
76. Lewiner, T., Lopes, H., Vieira, A. W. & Tavares, G. Efficient implementation of marching cubes’ cases with topological guarantees. J. Graph. Tools https://doi.org/10.1080/ 10867651.2003.10487582 (2012).
77. Lorensen, W. E. & Cline, H. E. Marching cubes: a high resolution 3D surface construction algorithm. ACM SIGGRAPH Comp. Graph. 21, 163-169 (1987).
Acknowledgements This work was performed as part of the NCI Human Tumor Atlas Network (HTAN). We thank the patients, staff and scientists who contributed to this study; staff at HTAN member centres, the Clinical Proteomics Tumor Analysis Consortium and the Siteman Cancer Center at Washington University for their support; and I. Strunilin, J. Wang, Y.Li, W.-W.Liang and Y. Li for their feedback on the manuscript. The following grants supported this work: U2CCA233303 to L.D., R.C.F. and W.E.G.; U54AG075934 to L.D., R.C.F. and F.C.; U24CA210972 to L.D.; R01NS107833 and R01NS117149 to M.G.C.; R01HG009711 and R01CA260112 to L.D. and F.C.; U24CA209837 to K.I.S.; K12CA167540 to L.-I.K; and NIH/NCI grants U24CA248453 and U24CA264027 to B.J.R. C.M. is a Damon Runyon Fellow supported by the Damon Runyon Cancer Research Foundation (DRQ-15-22).
Author contributions Study conception and design: L.D., F.C., B.J.R., R.C.F. and W.E.G. Developed and performed experiments or data collection: S.C., A.S., A. Houston, J.M., R.G.J., S.P.G., A.T.M.A.A., P.B., J.M. Herndon, W.E.G., R.C.F., R.P., F.C. and L.D. Computation and statistical analyses: C.-K.M., J.L., E.S., A.L.N.T.d.C., C.M., X.L. and M.C.W. Data interpretation a biological analyses: C.-K.M., J.L., S.C., E.S., A.L.N.T.d.C., A.K., A.N.S.-S., L.F., S.L., M.B., A. Hill, J.E.I., V.T., J.M.Held, I.S.H., E.H.K., P.O.B., A.H.K., M.M.M., K.I.S., S.V.P., T.J., M.A.R., C.W., L.-I.K., D.J.V., M.G.C., R.P., K.C.F., W.E.G., R.C.F., R.G.J., B.J.R., F.C. and L.D. Writing: C.-K.M., J.L., S.C., E.S., A.L.N.T.d.C., M.D.I., L.D. and M.C.W. Administration: F.C. and L.D.
Competing interests The authors declare no competing interests.
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41586-024-08087-4.
Correspondence and requests for materials should be addressed to William E. Gillanders, Ryan C. Fields, Benjamin J. Raphael, Feng Chen or Li Ding.
Peer review information Nature thanks Moritz Gerstung, Artem Lomakin, Christian Schürch and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Reprints and permissions information is available at http://www.nature.com/reprints.
Extended Data Fig. 1| Data Overview in Spatial Tumor Cohorts. a, Overview of the data availability of the spatial tumor cohort with assay types, cancer types, and study cohort.b, 48 snRNA-seq from matching tumor blocks across 7
tissue types (left). Cell type distribution from multi-sample tissue types (right). c, 22 Co-detection by indexing (CODEX) multiplexed immunofluorescence imaging from matching tumor sections.
Extended Data Fig. 2 | Histology of the Spatially Distinct Cohort. Histology of the spatially distinct cohort colored by cancer type. Light pink BRCA (n=23), light purple , orange slice , light blue , and light brown .
Extended Data Fig. 3| Histology of the Spatially Diffuse Cohort. Histology of the spatially diffuse cohort colored by cancer types. Light pink BRCA ( ), light purple , orange , light blue , light brown , and pale orange .
b Micoregion size distribution in the distinct cohort
sample_type_tumor
mid_high_cutoff
n
percent_by_size_group
Metastasis
large
28
58.3
Metastasis
medium
73
27.4
Metastasis
small
68
14.0
Primary
large
20
41.7
Primary
medium
193
72.6
Primary
small
419
86.0
percent_by_pri_met
16.6
43.2
40.2
3.2
30.5
66.3
Extended Data Fig. 4 | Microregion Distribution and Characteristics across
Cohorts.a, Sample count distribution for each cohort, primary vs. metastasis, cancer type, and count of section per sample.b, microregion size distribution between primary and metastasis samples (primary sections from 60 cases; metastasis sections from 16 cases). c, table with the count of microregions
per size. d, Estimated microregion density per tissue block. The number of microregions, sections, and primary vs metastasis status, cancer of each block is indicated on the left. e, Example microregion sizes, , size distribution histograms, and , microregion and , size groups visualized on Visium ST for HT268B1-Th1K3U1 (BRCA), HT260C1-Th1K1U1 (CRC), and HT270P1-H2U1 (PDAC).
Extended Data Fig. 5|Genomic Profiling of Spatial Subclones. a, Spatial subclone genome-wide CNV profile similarity compared with matching WESinferred CNV.b, Distribution of variants mapped to ST transcripts out of all somatic mutations detected from matching WES. Ref: reference allele; Var: variant allele. c, Somatic mutations mapped to HT260C1 are shared and unique to two spatial subclones. For each WES-derived mutation, VAFs between tumor and non-tumor regions (binomial test, left) and VAFs between two spatial subclones (proportion test, right) were compared. Mutations with FDR<0.05 (Benjamini & Hochberg correction) were annotated with*and** for VAF and VAF differences, respectively. d, A breast cancer liver metastasis sample (HT268B1) showing 2 spatial subclones across 5 sections. e, In the heatmap (top), estimated somatic copy number variations (CNV) per spot show both
shared and unique CNV events between the two spatial subclones. The b-allele frequencies (BAF) in each spatial subclone from the same genomic window are shown in the middle tracks, while the snRNA- and WES-inferred CNV status of the same genomic window is shown in the bottom tracks.f, The predicted phylogenetic relationship of the 2 tumor spatial subclones.g, UMAP of matching snRNA showing cell types and two tumor subclones. h, Somatic mutations mapped to HT268B1 are shared and unique to two spatial subclones. VAF calculations and their statistical analyses are the same as in panel c. i-j, Subclonal mutation EEF1A1.1324G C is uniquely detected in Clone2 in spatial transcriptomics, while EEF1A1 expression is in both spatial subclones ( , two-sided proportion test).
Extended Data Fig. 6 | Spatial Expression Pattern of Tumor Microregions
Driven by Genetic Alteration and Tumor Depth. a, Tumor heterogeneity evaluated by 1-ROGUE scoring. Higher scores indicate higher heterogeneities. Each point represents one section and is colored by its respective cohort designation and number of subclones ( sections from 78 cases). The box plot’s center line represents the median, with the lower and upper hinges indicating the first and third quartiles. Whiskers extend to the highest and lowest values within 1.5 times the interquartile range (IQR) from the hinges. b, (Top panel) Distribution of pairwise Pearson correlations between pairs of microregions within the same section. Distribution is split by those from the same tumor subclone (green) and those from different subclones (orange). The number in the boxes and the solid line indicate the mean of each distribution. (Bottom panel) Number of tumor subclones, microregions, and sections per tissue block.c. Pairwise Pearson correlation of microregions based on the top
500 most variable genes in section U1 of HT260C1-Th1H3. The red box highlights the pairwise Pearson correlations of microregions within Clone 1.d, Pathway enrichment scores for Clone 1 (c1), and Clone 2 (c2), and TME, where bubble size represents corrected p-value (two-sided Wilcoxon rank-sum test FDR adjusted). e, Partial correlation coefficient rho (with tumor purity as a covariate) and – -value) between expression level and layer for all genes in the same section. Positive correlation indicates higher expression in the tumor center and negative correlation indicates higher expression in the tumor periphery. Genes are categorized using matching snRNA-seq as follows: purple for tumorspecific, orange for tumor-enriched, green for stromal-enriched, and light purple for not DEG.f, Center- and periphery-enriched genes with their correlation lines and spatial expression patterns (Pearson correlation). g, Top shared centerand periphery-enriched genes across cancer types (FDR and rho or rho<-0.1) (partial correlation with Benjamini-Hochberg procedure).
Extended Data Fig. 7| Transcriptional Variability and Pathway Enrichment in Tumor Subclones. a, Pairwise Pearson correlation of the top 500 most variable genes in all 5 sections of Block HT268B1-Th1H3. Microregions with less than 10 spots were filtered out for this analysis. b, GSEA hallmark pathway enrichment analysis of tumor subclones compared to TME in the first section (U1) of HT268B1-Th1H3 (Two-sided Wilcoxon rank-sum test FDR adjusted). Average gene expression of upregulated genes in subclones from GSEA analysis and example spatial expression in c, d, Unfolded protein response, and e,f, MYC target v1 gene set in the first section (U1). g, h, G2M checkpoint, and
, MYC target v1 gene set. (i) Genes involved in DNA replication (light blue), cell cycle progression (light green), and translation initiation (light red) are highlighted. Average gene expression of upregulated genes in subclones from GSEA analysis and example spatial expression in c, e, g, i, (top panels) Module score, or average expression level of the program, calculated with Seurat AddModuleScore function (Method) using genes listed in each heatmap. The box plot’s center line represents the median, with the lower and upper hinges indicating the first and third quartiles. Whiskers extend to the highest and lowest values within 1.5 times the interquartile range (IQR) from the hinges.
Extended Data Fig. 8 | Layer Depth and Gene Expression in Tumor
Microregions. a, Schema of tumor depth designation for tumor microregions quantified in the number of layers. Starting from the tumor/TME border, proximal layers of tumor spots are iteratively defined as T1, T2, T3, etc, and distal TME spots are similarly defined as E1, E2, E3, etc. b, Relationship between the depth (measured in a total number of layers) and size (measured in the number of spots) of each microregion. The dashed reference line represents the projected depth-size relationship of perfect circular regions.c, A primary breast cancer sample with two tumor microregions eligible for the layer correlation analysis. Spots excluded from the analysis due to their proximity to the physical edge of the tissue are labeled in gray. d, Partial correlation
coefficient rho (with tumor purity as covariate) and -value between expression levels and layers for all genes. Positive correlation indicates higher expression in the tumor center and negative correlation indicates higher expression in the tumor periphery. e, Top center- and periphery-enriched genes with their correlation lines and spatial expression patterns (Pearson correlation). The box plot’s center line represents the median, with the lower and upper hinges indicating the first and third quartiles. Whiskers extend to the highest and lowest values within 1.5 times the interquartile range (IQR) from the hinges. Using Spatial Expression Deconvolution method to infer cell-type-specific expression yielded nearly identical results (Supplementary Fig. 9 and Methods).
Extended Data Fig. 9 | Spatial Subclone Infiltration and Cell Composition Across Tumor Layers. a, FDR statistics of comparing infiltration level by cell type between any two pairs of spatial subclones on the same sample. All significant FDR (<0.05) of the cell type from each sample was shown here indicating differential infiltration is observed ( spatially distinct cases with available matching deconvolution). b, Cell type composition of 6 regions defined by the following layers, T 3 and above (T3+), T2, T1, E1, E2, and E3 and
above (E3+), in 16 cases. c, Fraction of macrophage, T cell, fibroblast, and tumor in CODEX across 6 regions defined by the following layers, T3 and above, T2, T1, , and E 3 and above. Each data point represents one sample and data points from the same sample are connected.d, Spatial expression of boundary-enriched genes POSTN and IFI3O. e, Example cell-cell interactions in MK pathway (MDK) in two tumor sections, with the arrow direction indicating signal direction and the arrow length indicating signal strength.
a HT226C1-Th1 (Cholangiocarcinoma) HT206B1-S1 (Breast Primary)
b
d
Max connectivity
e
f
natureportfolio
Li Ding, Feng Chen, Benjamin J. Raphael, Ryan
Corresponding author(s): C. Fields, William E. Gillanders
Last updated by author(s): Aug 12, 2024
Reporting Summary
Nature Portfolio wishes to improve the reproducibility of the work that we publish. This form provides structure for consistency and transparency in reporting. For further information on Nature Portfolio policies, see our Editorial Policies and the Editorial Policy Checklist.
Statistics
For all statistical analyses, confirm that the following items are present in the figure legend, table legend, main text, or Methods section.
Confirmed
X The exact sample size ( ) for each experimental group/condition, given as a discrete number and unit of measurement X
A statement on whether measurements were taken from distinct samples or whether the same sample was measured repeatedly X
The statistical test(s) used AND whether they are one- or two-sided
Only common tests should be described solely by name; describe more complex techniques in the Methods section. X
A description of all covariates tested X
A description of any assumptions or corrections, such as tests of normality and adjustment for multiple comparisons X
A full description of the statistical parameters including central tendency (e.g. means) or other basic estimates (e.g. regression coefficient) AND variation (e.g. standard deviation) or associated estimates of uncertainty (e.g. confidence intervals) X
For null hypothesis testing, the test statistic (e.g. ) with confidence intervals, effect sizes, degrees of freedom and value noted Give values as exact values whenever suitable. For Bayesian analysis, information on the choice of priors and Markov chain Monte Carlo settings For hierarchical and complex designs, identification of the appropriate level for tests and full reporting of outcomes X
Estimates of effect sizes (e.g. Cohen’s , Pearson’s ), indicating how they were calculated
Our web collection on statistics for biologists contains articles on many of the points above.
Software and code
Policy information about availability of computer code
GATK v4.1.9.0, https://github.com/broadinstitute/gatk
Bedtools 2.30.0, https://github.com/arq5x/bedtools2
Scrublet v0.2.3, https://github.com/swolock/scrublet
Seurat v4.3.0, R-package
ComplexHeatmap v2.14.0, R-package
tidyverse v1.3.2, R-package
ggplot2 v3.3.5, R-package
RColorBrewer v1.1.3, R-package
singleR v1.8.1, R-package
clusterProfiler v3.18.1, R-package
msigdbr v7.5.1, R-package
ROGUE v1.0, R-package
RCTD v1.2.0, R-package
COMMOT, https://github.com/zcang/COMMOT
PASTE2, https://github.com/raphael-group/paste2
ppcor v1.1, R-package
stats v4.1.2, R-package
ggpubr v0.6.0, R-package
python v3.7.12 and v.3.9.6
sklearn v0.24.2, python-package
matplotlib v3.4.2, py39hf3d152e_0
numpy v1.21.1, h49503c6_1_cpython
org.Hs.eg.db v3.12.0, R-package
pandas v1.3.1, py39hdeOf152_0
Fiji/ImageJ, v2.14.0
BigWarp, Figi/ImageJ-package
Napari v0.4.19
Imaris v10.1.1
DeepCell v0.12.9, https://github.com/vanvalenlab/deepcell-tf
For manuscripts utilizing custom algorithms or software that are central to the research but not yet described in published literature, software must be made available to editors and reviewers. We strongly encourage code deposition in a community repository (e.g. GitHub). See the Nature Portfolio guidelines for submitting code & software for further information.
Data
Policy information about availability of data
All manuscripts must include a data availability statement. This statement should provide the following information, where applicable:
Accession codes, unique identifiers, or web links for publicly available datasets
A description of any restrictions on data availability
For clinical datasets or third party data, please ensure that the statement adheres to our policy
Research involving human participants, their data, or biological material
Policy information about studies with human participants or human data. See also policy information about sex, gender (identity/presentation), and sexual orientation and race, ethnicity and racism.
Reporting on sex and gender
Patient sex information was collected as part of this study
Reporting on race, ethnicity, or other socially relevant groupings
Patient race or ethnicity information was collected as part of this study
Population characteristics
Our dataset comprises samples from 6 tumor types, with patients ages 37-82. The distribution of the samples and clinical information across the cohorts can be found in the Supplementary Table 1, and Extended Data Fig. 1a.
Recruitment
Patients who fit the clinical criteria and consented to the study were selected for inclusion in the genetic and molecular tumor analysis. 75 cases from 6 cancers are part of Human Tumor Atlas Network (HTAN) study, and 3 ccRCC cases are from Clinical (Clinical Proteomic Tumor Analysis Consortium). There was no self-selection bias or other biases in the recruitment of patients
Ethics oversight
All samples were collected with informed consent at the Washington University School of Medicine in St Louis. Breast cancer
(BRCA) samples, pancreatic adenocarcinoma (PDAC) samples, colorectal cancer (CRC) samples, cholangiocarcinoma (CHOL) samples, renal carcinoma (RCC) samples and uterine corpus endometrial carcinoma (UCEC) samples were collected during surgical resection and verified by standard pathology (IRB protocol 201108117, 201411135, and 202106166). Tumor samples were collected during surgical resection and verified by standard pathology.
Note that full information on the approval of the study protocol must also be provided in the manuscript.
Field-specific reporting
Please select the one below that is the best fit for your research. If you are not sure, read the appropriate sections before making your selection.
Life sciences Behavioural & social sciences Ecological, evolutionary & environmental sciences
For a reference copy of the document with all sections, see nature.com/documents/nr-reporting-summary-flat.pdf
Life sciences study design
All studies must disclose on these points even when the disclosure is negative.
Sample size
Sample sizes were chosen based on the availability of samples collected across 6 cancer types. No specific statistical methods were used to predetermine sample size. This cohort collected a total of 132 sections from 79 cases samples for Visium ST, along with several samples for paired snRNA-seq, WES, and CODEX. This sample size aligns with or exceeds that of many previously published Visium spatial transcriptomics data on the date of submission.
Data exclusions
No data were excluded from the analysis
Replication
To ensure reproducibility, we used multiple bioinformatics tools for the same analyses and compared results across technologies (including bulk, single-cell, spatial sequencing, and multiplexed imaging) from the same tumor samples. To confirm subclonal copy number alterations in tumor spatial regions, we used two independent copy number inference methods, CalicoST (github.com/raphael-group/CalicoST) and InferCNV (https://github.com/broadinstitute/infercnv) (Supplementary Fig. 5a). We further validated spatial CNV inference with matching WES-based CNV calling and observed 0.43-0.82 agreement in Jaccard similarity (methods) between the dominant spatial subclone and WES (Supplementary Fig. 5c). Further, spatial subclonal mutations were validated with matching WES-based mutation calling (with WES mutations detected in spatial transcriptomics, Supplementary Fig. 5d).
To support our findings in the tumor center and periphery regions, we used matching snRNA-seq from the same tumor samples and demonstrated expression of most center-enriched and periphery-enriched genes in malignant cells (Supplementary Fig. 8a).
Subclonal tumor microenvironment compositions were confirmed with multiplexed immunostaining with co-detection by indexing (CODEX) (Fig. 5c).
Randomization
The study design didn’t involve the allocation of patients into treatment groups, therefore the randomization procedure was not relevant.
Blinding
The study design didn’t involve the allocation of patients into treatment groups, therefore blinding procedure was not relevant.
Reporting for specific materials, systems and methods
We require information from authors about some types of materials, experimental systems and methods used in many studies. Here, indicate whether each material, system or method listed is relevant to your study. If you are not sure if a list item applies to your research, read the appropriate section before selecting a response.
Validation
Antibodies used in this application have already been used in our laboratory and confirmed by one or more of the following methods. First: Western blotting produces the correct banding pattern (correct molecular weight, absence in non-transfected cells or protein extracts from mice deficient for the gene but present in wild-type samples or samples with transfection of an expression construct). Second, immunostaining shows the correct pattern (correct tissue specificity, intracellular location, absent/low in non-transfected cells, present/high in transfected cells, colocalization with the tags for fusion proteins expressed in cells). Third, 2 or more independent antibodies against the same antigen show the same expected pattern.
In addition, we have included detail antibody information in Supplementary Table_04_CODEX_Antibody.xlsx, further validation images in 2_Response_letter.pdf and 2b_Respons_Ab_IF_testing_0610.pdf
Plants
Seed stocks
Report on the source of all seed stocks or other plant material used. If applicable, state the seed stock centre and catalogue number. If plant specimens were collected from the field, describe the collection location, date and sampling procedures.
Novel plant genotypes
Describe the methods by which all novel plant genotypes were produced. This includes those generated by transgenic approaches, gene editing, chemical/radiation-based mutagenesis and hybridization. For transgenic lines, describe the transformation method, the number of independent lines analyzed and the generation upon which experiments were performed. For gene-edited lines, describe the editor used, the endogenous sequence targeted for editing, the targeting guide RNA sequence (if applicable) and how the editor
Authentication
was applied.
Describe any authentication procedures for each seed stock used or novel genotype generated. Describe any experiments used to assess the effect of a mutation and, where applicable, how potential secondary effects (e.g. second site T-DNA insertions, mosiacism, off-target gene editing) were examined.