DOI: https://doi.org/10.1038/s41420-023-01796-1
PMID: https://pubmed.ncbi.nlm.nih.gov/38218941
تاريخ النشر: 2024-01-13
توازن النحاس والموت الخلوي الناتج عن النحاس في تصلب الشرايين: الأيض، الآليات والاستراتيجيات العلاجية المحتملة
الملخص
النحاس هو عنصر غذائي أساسي يلعب دورًا محوريًا في العديد من العمليات الفسيولوجية في جميع أنواع الخلايا تقريبًا. ومع ذلك، فإن عدم تنظيم توازن النحاس، سواء نحو الزيادة أو النقص، يمكن أن يؤدي إلى تغييرات مرضية، مثل تصلب الشرايين. مع ظهور مفهوم موت الخلايا الناتج عن النحاس، المعروف باسم الكوبروبتوسيس، أصبح الباحثون يركزون بشكل متزايد على الدور المحتمل لعدم توازن النحاس في تصلب الشرايين. في هذه المراجعة، نقدم نظرة عامة شاملة على استقلاب النحاس الخلوي والنظامي. ثم نقوم بتلخيص الأدلة التي تربط عدم توازن النحاس بتصلب الشرايين ونوضح الآليات المحتملة التي تكمن وراء تطور تصلب الشرايين من حيث زيادة النحاس ونقصه. علاوة على ذلك، نناقش الأدلة والآليات المتعلقة بالكوبروبتوسيس، ونتناول تفاعلاته مع أنماط أخرى من موت الخلايا، ونبرز دور خلل الميتوكوندريا المرتبط بالكوبروبتوسيس في تصلب الشرايين. أخيرًا، نستكشف الاستراتيجية العلاجية لاستهداف هذه الصورة الجديدة من موت الخلايا، بهدف تقديم بعض الرؤى لإدارة تصلب الشرايين.
حقائق
- النحاس عنصر أساسي نادر مطلوب في عمليات فسيولوجية مختلفة في جسم الإنسان.
- لقد تم ربط عدم تنظيم توازن النحاس، سواء نحو الزيادة أو النقص، بمشاكل صحية متنوعة، بما في ذلك تصلب الشرايين.
- يُعترف بأن عدم تنظيم توازن النحاس والموت الخلوي الناتج عن النحاس (الكوبروتوبسيس) هما من العوامل المحتملة المساهمة في مسببات تصلب الشرايين.
أسئلة مفتوحة
- ما هو النطاق الآمن لمستويات النحاس لتجنب تطور تصلب الشرايين؟ كيف يمكن تحديد تركيز النحاس المناسب لعلاج تصلب الشرايين؟
- ما هي العلامات البيولوجية المرشحة المحتملة التي يمكن أن تشير بشكل موثوق وحساس إلى حدوث الكوبرتوبيس في سياق تصلب الشرايين؟
- ما هي الأدوار المحتملة غير المكتشفة للنحاس في وظيفة الميتوكوندريا، وهل هناك تفاعل بين النحاس وديناميات الميتوكوندريا أو الميتوفاجي؟
مقدمة
العواقب السريرية، مثل احتشاء عضلة القلب (MI) والسكتة الدماغية، التي تعد من الأسباب الرئيسية للمراضة والوفيات في جميع أنحاء العالم [3]. تتضمن آلية حدوث تصلب الشرايين عوامل وراثية وبيئية واستقلابية متعددة [4].
أيض النحاس
توازن النحاس الجهازي
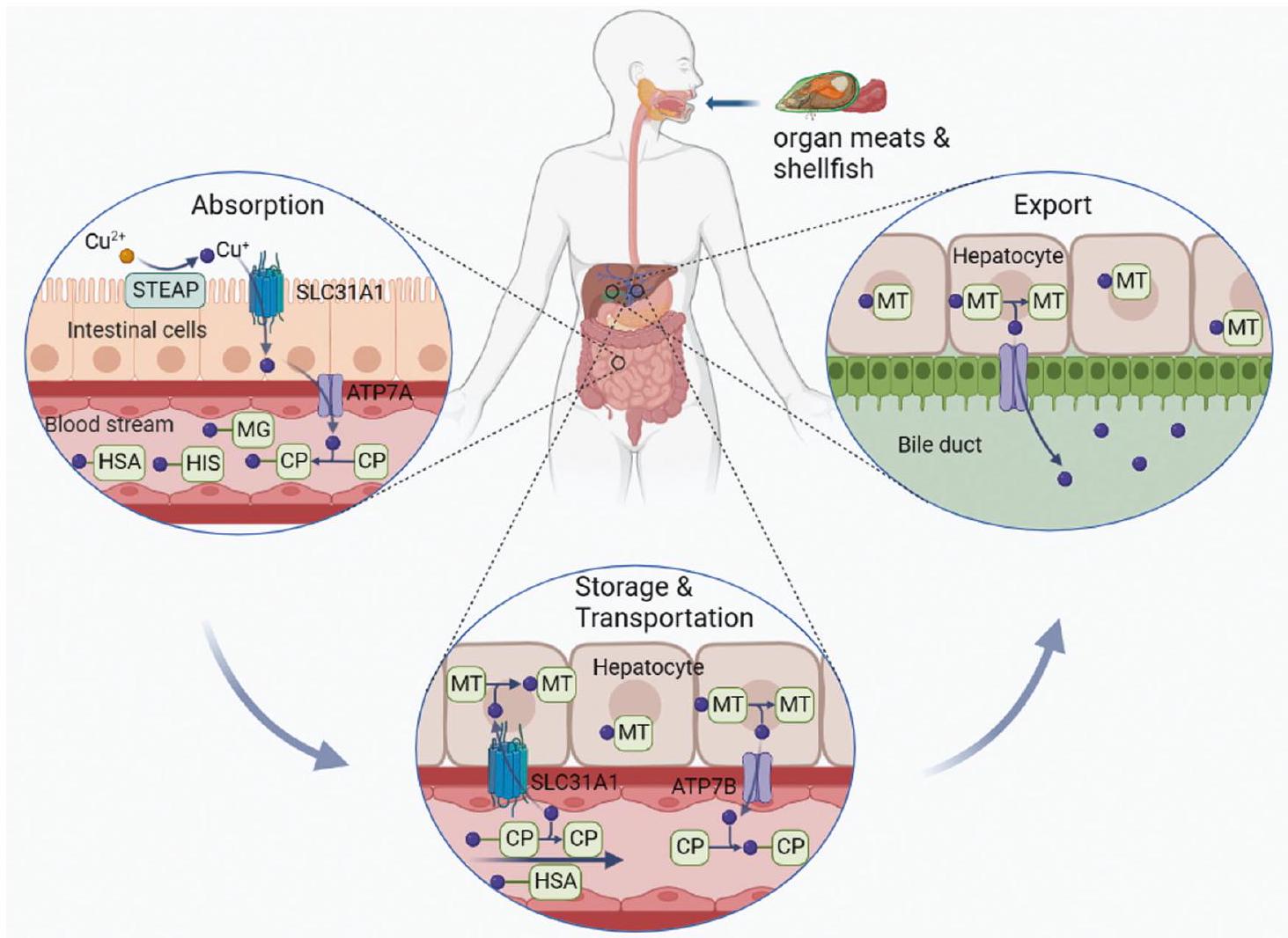
يعود النحاس إلى مجرى الدم. هنا، ترتبط أيونات النحاس بشركائها القابلين للذوبان وتُنقل إلى أنسجة وأعضاء محددة [22].
توازن النحاس الخلوي

كما يعمل كمانح للنحاس داخل الفضاء بين الغشائين الميتوكوندريين (IMS) [37]. وبالتالي، فإن COX17 ضروري للتجميع السليم لـ COX، حيث أظهرت الطفرات في COX17 أنها تقلل من نشاط COX، مما يؤدي إلى خلل في الميتوكوندريا وإجهاد أكسيدي [38].
أدلة تربط عدم توازن النحاس بتصلب الشرايين
| البلد، المؤلف، السنة | طرق وسكان الدراسة | نتيجة |
| فنلندا، ريونانن وآخرون [47] | 230 رجلًا توفوا بسبب أمراض القلب والأوعية الدموية و298 مجموعة متطابقة؛ متابعة لمدة 10 سنوات. | كان الخطر النسبي المعدل لوفيات مرض القلب التاجي بين أعلى وأدنى ثلثين من النحاس في المصل 2.86 (
|
| الولايات المتحدة، فورد، وآخرون [48] | 4574 مشاركًا تتراوح أعمارهم
|
كان هناك
|
| الهولندي، كوك وآخرون [49] | سرطان
|
الأفراد الذين لديهم مستويات مرتفعة من النحاس في المصل (أعلى من
|
| شرق فنلندا، سالونين وآخرون [50] | 1666 رجلًا تم اختيارهم عشوائيًا تتراوح أعمارهم بين 42 و48 و54 أو 60 عامًا دون وجود أعراض مرض القلب الإقفاري. | مستوى النحاس في المصل المرتفع هو عامل خطر لمرض القلب الإقفاري الذي يعمل بشكل مستقل، كما يتضح من زيادة خطر النوبة القلبية الحادة بمعدل يتراوح بين 3.5 إلى 4.0 أضعاف المرتبطة بمستويات النحاس المرتفعة في المصل (1.021.16 ملغ / لتر و
|
| نادينا، روبن وآخرون [51] | مرضى تصلب الشرايين الذين تتراوح أعمارهم
|
كانت مستويات النحاس في الطبقة الداخلية من الآفات أعلى بشكل ملحوظ في مرضى تصلب الشرايين مقارنةً بتلك الموجودة في الأفراد الأصحاء.
|
| بنغلاديش، بيغوم وآخرون [52] | 60 مريضًا تم تشخيصهم بالنوبة القلبية الحادة و60 شخصًا سليمًا. | كان مستوى النحاس في المصل أعلى بشكل ملحوظ في مرضى احتشاء عضلة القلب الحاد مقارنةً بالتحكم.
|
| غريغورز، باربرا وآخرون [53] | 74 مشاركًا من مرضى MI و72 شاهدًا صحيًا. | كان مستوى النحاس في المصل المرتفع مرتبطًا بشكل كبير بالنوبة القلبية (MI)
|
| إيطاليا، تارانتينو وآخرون [54] | 100 مريضًا بدينًا كان لديهم انتشار منخفض للأمراض المصاحبة. | تمت علاقة سلبية بين توافر النحاس المعدل وسماكة جدار الشريان السباتي (IMT)
|
| صربيا، تاسيć وآخرون [55] | 91 مريضًا (متوسط العمر
|
كان لدى المرضى الذين يعانون من لويحات نزفية مستويات أعلى بشكل ملحوظ من النحاس في المصل مقارنةً بأولئك الذين يعانون من لويحات متكلسة؛ قد تساهم تركيزات النحاس العالية في تصلب الشرايين. |
من خلال العديد من الدراسات، تم ربط عدم توازن النحاس والزيادة أو النقص الناتج في النحاس بتصلب الشرايين.
أدلة تربط بين زيادة النحاس وتصلب الشرايين
أدلة تربط نقص النحاس وتصلب الشرايين

آليات محتملة لتطور تصلب الشرايين بسبب عدم توازن النحاس
الإجهاد التأكسدي
يؤدي إلى تلف الأكسدة لجزيئات الدهون. يمكن أن تعزز مستويات النحاس المرتفعة أكسدة الدهون وتؤدي إلى تكوين البروتين الدهني منخفض الكثافة المؤكسد (ox-LDL)، وهو عامل خطر لتصلب الشرايين. علاوة على ذلك، أظهرت الدراسات أن مستويات النحاس المرتفعة تقلل من نشاط إنزيمات مضادات الأكسدة، مثل
نقص النحاس يؤدي إلى انخفاض مستويات أكسيد النيتريك (NO)، مما قد يعزز من خلل وظيفة البطانة، ويقلل من استرخاء الأوعية الدموية، ويزيد من الإجهاد التأكسدي، وبالتالي يساهم في تطور تصلب الشرايين [70]. بالإضافة إلى تأثيره على إنزيمات مضادات الأكسدة، قد يؤدي نقص النحاس أيضًا إلى تقليل نشاط COX ويؤدي إلى التفاعل التأكسدي المعطل للمركب الأول (NADH: أكسيد الكوينون). قد يؤدي هذا التفاعل التأكسدي بعد ذلك إلى زيادة إنتاج الجذور الحرة في الخلايا التي تعاني من نقص النحاس، مما يزيد من الإجهاد التأكسدي الخلوي [74].
التهاب
خلل وظيفة البطانة
تمثيل الدهون
عوامل خطر أخرى

موت الخلايا الناتج عن النحاس وتصلب الشرايين من موت الخلايا الناتج عن النحاس إلى الكوبروتوبسيس
يؤدي إلى تجمع البروتينات الميتوكوندرية المرتبطة بالنحاس والمعدلة بالليبويد، مما يمكن أن يعطل دورة TCA وبالتالي يتداخل مع إنتاج الطاقة الخلوية. العوامل التنظيمية العليا FDX1 و LIAS حاسمة في هذه العملية. تجمع هذه البروتينات والتقليل اللاحق من
التفاعلات المحتملة بين موت الخلايا الناتج عن النحاس ومسارات موت الخلايا الأخرى
تعرض خلايا الميكروغليا في الفئران للنحاس يحفز استجابة التهابية، مما يؤدي إلى زيادة تنظيم بروتينات محور NLRP3/caspase 1/GSDMD وحدوث البيروبتوس. من المحتمل أن تكون هذه التأثيرات ناتجة عن التنشيط المبكر لمسار ROS/NF-кВ والتعطيل اللاحق للميتوفاجي. علاوة على ذلك، لوحظت نتائج مماثلة في البلعميات الفئران المعالجة بجزيئات أكسيد النحاس النانوية. يؤدي تعرض جزيئات أكسيد النحاس النانوية إلى تحفيز الإجهاد التأكسدي وتنشيط إنزيمات NLRP3، مما يؤدي إلى التعبير عن pro-IL-1.
خلل الميتوكوندريا المرتبط بالكوبروتوسيس وتصلب الشرايين
قد disrupt النحاس داخل الخلايا أيضًا وظيفة الميتوكوندريا عن طريق تغيير نشاط العديد من الإنزيمات الرئيسية، مثل تلك المشاركة في دورة حمض ثلاثي الكربوكسيليك (TCA) والفوسفوريلation التأكسدية. لوحظت خلل شديد في الميتوكوندريا وانخفاض في أنشطة العديد من إنزيمات الكبد، بما في ذلك المركب I، المركب II، المركب III، المركب IV، والأكونيتاز، في المرضى الذين يعانون من زيادة النحاس. هذه التأثيرات قد تكون متوسطة من خلال تراكم النحاس في الميتوكوندريا. علاوة على ذلك، يؤدي علاج النحاس إلى تغييرات انتقائية في الإنزيمات الأيضية من خلال الليبويلايشن، وهو تعديل ما بعد الترجمة للّيسين محفوظ بشكل كبير. تحدث بروتينات الليبويلايشن فقط على Dihydrolipoamide S-Acetyltransferase (DLAT)، Dihydrolipoamide S-Succinyltransferase (DLST)، Dihydrolipoamide Branched Chain Transacylase E2 (DBT)، وGlycine Cleavage System Protein H (GCSH)، وكلها تشارك في المجمعات الأيضية التي تنظم نقاط دخول الكربون إلى دورة TCA. بالإضافة إلى ذلك، قد يؤدي زيادة النحاس إلى فتح ثقب الانتقال النفاذية للميتوكوندريا ويسبب إطلاق عوامل مؤيدة للاستماتة، مما يؤدي في النهاية إلى موت الخلايا.
استراتيجيات علاجية محتملة تستهدف الكوبرتوسيس في تصلب الشرايين: مخلبات النحاس
علاج مرض القلب والأوعية الدموية الناتج عن تصلب الشرايين [143]. قد تُعزى النتائج المتناقضة بين هذه الدراسات إلى اختلافات في تصميم البحث و/أو خصائص مجموعات الدراسة. لذلك، هناك حاجة إلى مزيد من البحث قبل أن يمكن التوصية بالاستخدام الروتيني لعلاج الإزالة.
تنظيم تعبير بروتين ناقل النحاس
أيونوفورات النحاس
الاستنتاجات وآفاق المستقبل
REFERENCES
- Libby P. The changing landscape of atherosclerosis. Nature. 2021;592:524-33.
- Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, et al. Atherosclerosis. Nat Rev Dis Prim. 2019;5:56.
- Herrington W, Lacey B, Sherliker P, Armitage J, Lewington S. Epidemiology of atherosclerosis and the potential to reduce the global burden of Atherothrombotic disease. Circ Res. 2016;118:535-46.
- Björkegren JLM, Lusis AJ. Atherosclerosis: recent developments. Cell. 2022;185:1630-45.
- Festa RA, Thiele DJ. Copper: an essential metal in biology. Curr Biol. 2011;21:R877-83.
- Kim BE, Nevitt T, Thiele DJ. Mechanisms for copper acquisition, distribution and regulation. Nat Chem Biol. 2008;4:176-85.
- Chen L, Min J, Wang F. Copper homeostasis and cuproptosis in health and disease. Signal Transduct Target Ther. 2022;7:378.
- Chen X, Cai Q, Liang R, Zhang D, Liu X, Zhang M, et al. Copper homeostasis and copper-induced cell death in the pathogenesis of cardiovascular disease and therapeutic strategies. Cell Death Dis. 2023;14:105.
- Uriu-Adams JY, Keen CL. Copper, oxidative stress, and human health. Mol Asp Med. 2005;26:268-98.
- Fukai T, Ushio-Fukai M, Kaplan JH. Copper transporters and copper chaperones: roles in cardiovascular physiology and disease. Am J Physiol Cell Physiol. 2018;315:C186-c201.
- Tang D, Chen X, Kroemer G. Cuproptosis: a copper-triggered modality of mitochondrial cell death. Cell Res. 2022;32:417-8.
- Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375:1254-61.
- Yang L, Yang P, Lip GYH, Ren J. Copper homeostasis and cuproptosis in cardiovascular disease therapeutics. Trends Pharm Sci. 2023;44:573-85.
- Maung MT, Carlson A, Olea-Flores M, Elkhadragy L, Schachtschneider KM, Navarro-Tito N, et al. The molecular and cellular basis of copper dysregulation and its relationship with human pathologies. FASEB J. 2021;35:e21810.
- Bost M, Houdart S, Oberli M, Kalonji E, Huneau JF, Margaritis I. Dietary copper and human health: current evidence and unresolved issues. J Trace Elem Med Biol. 2016;35:107-15.
- Xie J, Yang Y, Gao Y, He J. Cuproptosis: mechanisms and links with cancers. Mol Cancer. 2023;22:46.
- Nose Y, Kim BE, Thiele DJ. Ctr1 drives intestinal copper absorption and is essential for growth, iron metabolism, and neonatal cardiac function. Cell Metab. 2006;4:235-44.
- Lönnerdal B. Intestinal regulation of copper homeostasis: a developmental perspective. Am J Clin Nutr. 2008;88:846s-50s.
- Lutsenko S. Dynamic and cell-specific transport networks for intracellular copper ions. J Cell Sci. 2021;134:96-114.
- Ramos D, Mar D, Ishida M, Vargas R, Gaite M, Montgomery A, et al. Mechanism of copper uptake from blood plasma ceruloplasmin by mammalian cells. PLoS One. 2016;11:e0149516.
- Santoro A, Calvo JS, Peris-Díaz MD, Krężel A, Meloni G, Faller P. The glutathione/ metallothionein system challenges the design of efficient
-activating copper complexes. Angew Chem Int Ed Engl. 2020;59:7830-5. - La Fontaine S, Ackland ML, Mercer JF. Mammalian copper-transporting P-type ATPases, ATP7A and ATP7B: emerging roles. Int J Biochem Cell Biol. 2010;42:206-9.
- Wijmenga C, Klomp LW. Molecular regulation of copper excretion in the liver. Proc Nutr Soc. 2004;63:31-9.
- Hernandez S, Tsuchiya Y, García-Ruiz JP, Lalioti V, Nielsen S, Cassio D, et al. ATP7B copper-regulated traffic and association with the tight junctions: copper excretion into the bile. Gastroenterology. 2008;134:1215-23.
- Huster D, Kühne A, Bhattacharjee A, Raines L, Jantsch V, Noe J, et al. Diverse functional properties of Wilson disease ATP7B variants. Gastroenterology. 2012;142:947-56.e5.
- Maryon EB, Molloy SA, Ivy K, Yu H, Kaplan JH. Rate and regulation of copper transport by human copper transporter 1 (hCTR1). J Biol Chem. 2013;288:18035-46.
- Liang ZD, Tsai WB, Lee MY, Savaraj N, Kuo MT. Specificity protein 1 (sp1) oscillation is involved in copper homeostasis maintenance by regulating human high-affinity copper transporter 1 expression. Mol Pharm. 2012;81:455-64.
- Kuo YM, Gybina AA, Pyatskowit JW, Gitschier J, Prohaska JR. Copper transport protein (Ctr1) levels in mice are tissue specific and dependent on copper status.
J Nutr. 2006;136:21-6. - Muller PA, Klomp LW. ATOX1: a novel copper-responsive transcription factor in mammals? Int J Biochem Cell Biol. 2009;41:1233-6.
- Prohaska JR, Gybina AA. Intracellular copper transport in mammals. J Nutr. 2004;134:1003-6.
- Yang D, Xiao P, Qiu B, Yu HF, Teng CB. Copper chaperone antioxidant 1: multiple roles and a potential therapeutic target. J Mol Med Berl. 2023;101:527-42.
- Itoh S, Kim HW, Nakagawa O, Ozumi K, Lessner SM, Aoki H, et al. Novel role of antioxidant-1 (Atox1) as a copper-dependent transcription factor involved in cell proliferation. J Biol Chem. 2008;283:9157-67.
- Rosenzweig AC. Copper delivery by metallochaperone proteins. Acc Chem Res. 2001;34:119-28.
- Dong
, Zhang , Zhao J, Lei J, Chen Y, Li X, et al. The rational design of specific SOD1 inhibitors via copper coordination and their application in ROS signaling research. Chem Sci. 2016;7:6251-62. - Elchuri S, Oberley TD, Qi W, Eisenstein RS, Jackson Roberts L, Van Remmen H, et al. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene. 2005;24:367-80.
- Ding Y, Chen Y, Wu Z, Yang N, Rana K, Meng X, et al. SsCox17, a copper chaperone, is required for pathogenic process and oxidative stress tolerance of Sclerotinia sclerotiorum. Plant Sci. 2022;322:111345.
- Horng YC, Cobine PA, Maxfield AB, Carr HS, Winge DR. Specific copper transfer from the Cox17 metallochaperone to both Sco1 and Cox11 in the assembly of yeast cytochrome C oxidase. J Biol Chem. 2004;279:35334-40.
- Takahashi Y, Kako K, Kashiwabara S, Takehara A, Inada Y, Arai H, et al. Mammalian copper chaperone Cox17p has an essential role in activation of cytochrome
oxidase and embryonic development. Mol Cell Biol. 2002;22:7614-21. - Lutsenko S, Barnes NL, Bartee MY, Dmitriev OY. Function and regulation of human copper-transporting ATPases. Physiol Rev. 2007;87:1011-46.
- La Fontaine S, Mercer JF. Trafficking of the copper-ATPases, ATP7A and ATP7B: role in copper homeostasis. Arch Biochem Biophys. 2007;463:149-67.
- Horn N, Wittung SP. et al. ATP7A-regulated enzyme metalation and trafficking in the menkes disease puzzle. Biomedicines. 2021;9:123-142.
- Chen J, Jiang Y, Shi H, Peng Y, Fan X, Li C. The molecular mechanisms of copper metabolism and its roles in human diseases. Pflug Arch. 2020;472:1415-29.
- Wolf D, Ley K. Immunity and inflammation in atherosclerosis. Circ Res. 2019;124:315-27.
- Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. 2011;17:1410-22.
- Campia U, Gerhard-Herman M, Piazza G, Goldhaber SZ. Peripheral artery disease: past, present, and future. Am J Med. 2019;132:1133-41.
- Endres M, Moro MA, Nolte CH, Dames C, Buckwalter MS, Meisel A. Immune pathways in etiology, acute phase, and chronic sequelae of ischemic stroke. Circ Res. 2022;130:1167-86.
- Reunanen A, Knekt P, Marniemi J, Mäki J, Maatela J, Aromaa A. Serum calcium, magnesium, copper and zinc and risk of cardiovascular death. Eur J Clin Nutr. 1996;50:431-7.
- Ford ES. Serum copper concentration and coronary heart disease among US adults. Am J Epidemiol. 2000;151:1182-8.
- Kok FJ, Van Duijn CM, Hofman A, Van der Voet GB, De Wolff FA, Paays CH, et al. Serum copper and zinc and the risk of death from cancer and cardiovascular disease. Am J Epidemiol. 1988;128:352-9.
- Salonen JT, Salonen R, Korpela H, Suntioinen S, Tuomilehto J. Serum copper and the risk of acute myocardial infarction: a prospective population study in men in eastern Finland. Am J Epidemiol. 1991;134:268-76.
- Stadler N, Lindner RA, Davies MJ. Direct detection and quantification of transition metal ions in human atherosclerotic plaques: evidence for the presence of
elevated levels of iron and copper. Arterioscler Thromb Vasc Biol. 2004;24:949-54. - Begum S, Sultana I, Faysal MR, Alam S, Tasnim J, Akter T, et al. Study of changes in serum copper level in patients with acute myocardial infarction. Mymens Med J. 2023;32:39-43.
- Nowicki GJ, Ślusarska B, Prystupa A, Blicharska E, Adamczuk A, Czernecki T, et al. Assessment of concentrations of heavy metals in postmyocardial infarction patients and patients free from cardiovascular event. Cardiol Res Pr. 2021;2021:9546358.
- Tarantino G, Porcu C, Arciello M, Andreozzi P, Balsano C. Prediction of carotid intima-media thickness in obese patients with low prevalence of comorbidities by serum copper bioavailability. J Gastroenterol Hepatol. 2018;33:1511-7.
- Tasić NM, Tasić D, Otašević P, Veselinović M, Jakovljević V, Djurić D, et al. Copper and zinc concentrations in atherosclerotic plaque and serum in relation to lipid metabolism in patients with carotid atherosclerosis. Vojnosanit Pregl. 2015;72:801-6.
- Trumbo
, Yates , Schlicker , Poos M. Dietary reference intakes: vitamin A, vitamin K, arsenic, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. J Am Diet Assoc. 2001;101:294-301. - Rock E, Mazur A, O’Connor JM, Bonham MP, Rayssiguier Y, Strain JJ. The effect of copper supplementation on red blood cell oxidizability and plasma antioxidants in middle-aged healthy volunteers. Free Radic Biol Med. 2000;28:324-9.
- Chen F, Du M, Blumberg JB, Ho Chui KK, Ruan M, Rogers G, et al. Association among dietary supplement use, nutrient intake, and mortality among U.S. adults: a cohort study. Ann Intern Med. 2019;170:604-13.
- Lamb DJ, Avades TY, Ferns GA. Biphasic modulation of atherosclerosis induced by graded dietary copper supplementation in the cholesterol-fed rabbit. Int J Exp Pathol. 2001;82:287-94.
- Wang T, Xiang P, Ha JH, Wang X, Doguer C, Flores SRL, et al. Copper supplementation reverses dietary iron overload-induced pathologies in mice. J Nutr Biochem. 2018;59:56-63.
- Hughes WM Jr, Rodriguez WE, Rosenberger D, Chen J, Sen U, Tyagi N, et al. Role of copper and homocysteine in pressure overload heart failure. Cardiovasc Toxicol. 2008;8:137-44.
- Diaf M, Khaled MB. Associations between dietary antioxidant intake and markers of atherosclerosis in middle-aged women from North-Western Algeria. Front Nutr. 2018;5:29.
- Kim YW, Byzova TV. Oxidative stress in angiogenesis and vascular disease. Blood. 2014;123:625-31.
- Fang Y, Xing C, Wang X, Cao H, Zhang C, Guo X, et al. Activation of the ROS/HO1/NQO1 signaling pathway contributes to the copper-induced oxidative stress and autophagy in duck renal tubular epithelial cells. Sci Total Environ. 2021;757:143753.
- Valko M, Morris H, Cronin MT. Metals, toxicity and oxidative stress. Curr Med Chem. 2005;12:1161-208.
- Dhalla NS, Temsah RM, Netticadan T. Role of oxidative stress in cardiovascular diseases. J Hypertens. 2000;18:655-73.
- Esterbauer H, Gebicki J, Puhl H, Jürgens G. The role of lipid peroxidation and antioxidants in oxidative modification of LDL. Free Radic Biol Med. 1992;13:341-90.
- Ozcelik D, Uzun H. Copper intoxication; antioxidant defenses and oxidative damage in rat brain. Biol Trace Elem Res. 2009;127:45-52.
- Brezová V, Dvoranová D, Zúbor V, Breza M, Mazúr M, Valko M. Photochemical properties of camptothecin in the presence of copper(II) ions: the role of radicals as prospective species in photodynamic therapy. Mol Biotechnol. 2007;37:48-51.
- AI-Bayati MA, Jamil DA, AI-Aubaidy HA. Cardiovascular effects of copper deficiency on activity of superoxide dismutase in diabetic nephropathy. N. Am J Med Sci. 2015;7:41-6.
- Linder MC, Hazegh-Azam M. Copper biochemistry and molecular biology. Am J Clin Nutr. 1996;63:797s-811s.
- Sukalski KA, LaBerge TP, Johnson WT. In vivo oxidative modification of erythrocyte membrane proteins in copper deficiency. Free Radic Biol Med. 1997;22:835-42.
- Bertinato J, Iskandar M, L’Abbé MR. Copper deficiency induces the upregulation of the copper chaperone for
superoxide dismutase in weanling male rats. J Nutr. 2003;133:28-31. - Johnson WT, Thomas AC. Copper deprivation potentiates oxidative stress in HL60 cell mitochondria. Proc Soc Exp Biol Med. 1999;221:147-52.
- Malekahmadi M, Firouzi S, Rezayi M, Ghazizadeh H, Ranjbar G, Ferns GA, et al. Association of zinc and copper status with cardiovascular diseases and their assessment methods: a review study. Mini Rev Med Chem. 2020;20:2067-78.
- Ansteinsson V, Refsnes M, Skomedal T, Osnes JB, Schiander I, Låg M. Zinc- and copper-induced interleukin-6 release in primary cell cultures from rat heart. Cardiovasc Toxicol. 2009;9:86-94.
- Zhou Q, Zhang Y, Lu L, Zhang H, Zhao C, Pu Y, et al. Copper induces microgliamediated neuroinflammation through ROS/NF-кB pathway and mitophagy disorder. Food Chem Toxicol. 2022;168:113369.
- Schuschke DA, Saari JT, Miller FN. Leukocyte-endothelial adhesion is impaired in the cremaster muscle microcirculation of the copper-deficient rat. Immunol Lett. 2001;76:139-44.
- Sakai N, Shin T, Schuster R, Blanchard J, Lentsch AB, Johnson WT, et al. Marginal copper deficiency increases liver neutrophil accumulation after ischemia/ reperfusion in rats. Biol Trace Elem Res. 2011;142:47-54.
- Gordon SA, Lominadze D, Saari JT, Lentsch AB, Schuschke DA. Impaired deformability of copper-deficient neutrophils. Exp Biol Med Maywood. 2005;230:543-8.
- Schuschke DA, Percival SS, Lominadze D, Saari JT, Lentsch AB. Tissue-specific ICAM-1 expression and neutrophil transmigration in the copper-deficient rat. Inflammation. 2002;26:297-303.
- Lominadze D, Saari JT, Percival SS, Schuschke DA. Proinflammatory effects of copper deficiency on neutrophils and lung endothelial cells. Immunol Cell Biol. 2004;82:231-8.
- Lentsch AB, Kato A, Saari JT, Schuschke DA. Augmented metalloproteinase activity and acute lung injury in copper-deficient rats. Am J Physiol Lung Cell Mol Physiol. 2001;281:L387-93.
- Paynter DI, Moir RJ, Underwood EJ. Changes in activity of the Cu-Zn superoxide dismutase enzyme in tissues of the rat with changes in dietary copper. J Nutr. 1979;109:1570-6.
- Förstermann U, Münzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006;113:1708-14.
- Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315-424.
- Starkebaum G, Harlan JM. Endothelial cell injury due to copper-catalyzed hydrogen peroxide generation from homocysteine. J Clin Invest. 1986;77:1370-6.
- Versari D, Daghini E, Virdis A, Ghiadoni L, Taddei S. Endothelial dysfunction as a target for prevention of cardiovascular disease. Diabetes Care. 2009;32:S314-21.
- Kattoor AJ, Kanuri SH, Mehta JL. Role of Ox-LDL and LOX-1 in Atherogenesis. Curr Med Chem. 2019;26:1693-700.
- Craig WY, Poulin SE, Palomaki GE, Neveux LM, Ritchie RF, Ledue TB. Oxidationrelated analytes and lipid and lipoprotein concentrations in healthy subjects. Arterioscler Thromb Vasc Biol. 1995;15:733-9.
- Kazemi-Bajestani SM, Ghayour-Mobarhan M, Ebrahimi M, Moohebati M, Esmaeili HA, Parizadeh MR, et al. Serum copper and zinc concentrations are lower in Iranian patients with angiographically defined coronary artery disease than in subjects with a normal angiogram. J Trace Elem Med Biol. 2007;21:22-8.
- Song YF, Luo Z, Zhang LH, Hogstrand C, Pan YX. Endoplasmic reticulum stress and disturbed calcium homeostasis are involved in copper-induced alteration in hepatic lipid metabolism in yellow catfish Pelteobagrus fulvidraco. Chemosphere. 2016;144:2443-53.
- Morrell A, Tallino S, Yu L, Burkhead JL. The role of insufficient copper in lipid synthesis and fatty-liver disease. IUBMB Life. 2017;69:263-70.
- Ye J, DeBose RA. Regulation of cholesterol and fatty acid synthesis. Cold Spring Harb Perspect Biol. 2011;3:96-105.
- Tang Z, Gasperkova D, Xu J, Baillie R, Lee JH, Clarke SD. Copper deficiency induces hepatic fatty acid synthase gene transcription in rats by increasing the nuclear content of mature sterol regulatory element binding protein 1. J Nutr. 2000;130:2915-21.
- Loyke HF. Copper and zinc in experimental hypertension. Biol Trace Elem Res. 1991;29:45-9.
- Ozumi K, Sudhahar V, Kim HW, Chen GF, Kohno T, Finney L, et al. Role of copper transport protein antioxidant 1 in angiotensin II-induced hypertension: a key regulator of extracellular superoxide dismutase. Hypertension. 2012;60:476-86.
- Halliwell B, Gutteridge JM. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem J. 1984;219:1-14.
- Gaetke LM, Chow CK. Copper toxicity, oxidative stress, and antioxidant nutrients. Toxicology. 2003;189:147-63.
- Tardito S, Bassanetti I, Bignardi C, Elviri L, Tegoni M, Mucchino C, et al. Copper binding agents acting as copper ionophores lead to caspase inhibition and paraptotic cell death in human cancer cells. J Am Chem Soc. 2011;133:6235-42.
- Nagai M, Vo NH, Shin Ogawa L, Chimmanamada D, Inoue T, Chu J, et al. The oncology drug elesclomol selectively transports copper to the mitochondria to induce oxidative stress in cancer cells. Free Radic Biol Med. 2012;52:2142-50.
- Lill R, Freibert SA. Mechanisms of mitochondrial iron-sulfur protein biogenesis. Annu Rev Biochem. 2020;89:471-99.
- Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther. 2021;6:128.
- Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol. 2001;9:113-4.
- Nunes T, de Souza HS. Inflammasome in intestinal inflammation and cancer. Mediat Inflamm. 2013;2013:654963.
- Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16:407-20.
- Aachoui Y, Sagulenko V, Miao EA, Stacey KJ. Inflammasome-mediated pyroptotic and apoptotic cell death, and defense against infection. Curr Opin Microbiol. 2013;16:319-26.
- Tao X, Wan X, Wu D, Song E, Song Y. A tandem activation of NLRP3 inflammasome induced by copper oxide nanoparticles and dissolved copper ion in J774A. 1 macrophage. J Hazard Mater. 2021;411:125134.
- Liu H, Guo H, Deng H, Cui H, Fang J, Zuo Z, et al. Copper induces hepatic inflammatory responses by activation of MAPKs and NF-kB signalling pathways in the mouse. Ecotoxicol Environ Saf. 2020;201:110806.
- Deng H, Zhu S, Yang H, Cui H, Guo H, Deng J, et al. The dysregulation of inflammatory pathways triggered by copper exposure. Biol Trace Elem Res. 2023;201:539-48.
- Liao J, Yang F, Tang Z, Yu W, Han Q, Hu L, et al. Inhibition of caspase-1dependent pyroptosis attenuates copper-induced apoptosis in chicken hepatocytes. Ecotoxicol Environ Saf. 2019;174:110-9.
- Zhang X, Ren Z, Xu W, Jiang Z. Necroptosis in atherosclerosis. Clin Chim Acta. 2022;534:22-8.
- Li M, Wang ZW, Fang LJ, Cheng SQ, Wang X, Liu NF. Programmed cell death in atherosclerosis and vascular calcification. Cell Death Dis. 2022;13:467.
- Gutierrez KD, Davis MA, Daniels BP, Olsen TM, Ralli-Jain P, Tait SW, et al. MLKL activation triggers NLRP3-mediated processing and release of IL-1
independently of gasdermin-D. J Immunol. 2017;198:2156-64. - Miao Y, Liu J, Liu X, Yuan Q, Li H, Zhang Y, et al. Machine learning identification of cuproptosis and necroptosis-associated molecular subtypes to aid in prognosis assessment and immunotherapy response prediction in low-grade glioma. Front Genet. 2022;13:951239.
- Kuriakose T, Man SM, Malireddi RK, Karki R, Kesavardhana S, Place DE, et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol. 2016;1:52-68.
- Jiao H, Wachsmuth L, Kumari S, Schwarzer R, Lin J, Eren RO, et al. Publisher Correction: Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation. Nature. 2020;580:E10.
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060-72.
- Hirschhorn T, Stockwell BR. The development of the concept of ferroptosis. Free Radic Biol Med. 2019;133:130-43.
- Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26:165-76.
- Yang F, Pei R, Zhang Z, Liao J, Yu W, Qiao N, et al. Copper induces oxidative stress and apoptosis through mitochondria-mediated pathway in chicken hepatocytes. Toxicol Vitr. 2019;54:310-6.
- Ren X, Li Y, Zhou Y, Hu W, Yang C, Jing Q, et al. Overcoming the compensatory elevation of NRF2 renders hepatocellular carcinoma cells more vulnerable to disulfiram/copper-induced ferroptosis. Redox Biol. 2021;46:102122.
- Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688-92.
- Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317-31.
- Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91-8.
- Xue Q, Yan D, Chen X, Li X, Kang R, Klionsky DJ, et al. Copper-dependent autophagic degradation of GPX4 drives ferroptosis. Autophagy. 2023;19:1982-96.
- Gao W, Huang Z, Duan J, Nice EC, Lin J, Huang C. Elesclomol induces copperdependent ferroptosis in colorectal cancer cells via degradation of ATP7A. Mol Oncol. 2021;15:3527-44.
- Li F, Wu X, Liu H, Liu M, Yue Z, Wu Z, et al. Copper depletion strongly enhances ferroptosis via mitochondrial perturbation and reduction in antioxidative mechanisms. Antioxid Basel. 2022;11:652-98.
- Arciello M, Rotilio G, Rossi L. Copper-dependent toxicity in SH-SY5Y neuroblastoma cells involves mitochondrial damage. Biochem Biophys Res Commun. 2005;327:454-9.
- Cobine PA, Moore SA, Leary SC. Getting out what you put in: Copper in mitochondria and its impacts on human disease. Biochim Biophys Acta Mol Cell Res. 2021;1868:118867.
- Sheline CT, Choi DW. Cu2+ toxicity inhibition of mitochondrial dehydrogenases in vitro and in vivo. Ann Neurol. 2004;55:645-53.
- Gu M, Cooper JM, Butler P, Walker AP, Mistry PK, Dooley JS, et al. Oxidativephosphorylation defects in liver of patients with Wilson’s disease. Lancet. 2000;356:469-74.
- Solmonson A, DeBerardinis RJ. Lipoic acid metabolism and mitochondrial redox regulation. J Biol Chem. 2018;293:7522-30.
- Rowland EA, Snowden CK, Cristea IM. Protein lipoylation: an evolutionarily conserved metabolic regulator of health and disease. Curr Opin Chem Biol. 2018;42:76-85.
- Su R, Wang R, Cao H, Pan J, Chen L, Li C, et al. High copper levels promotes broiler hepatocyte mitochondrial permeability transition in vivo and in vitro. Biol Trace Elem Res. 2011;144:636-46.
- Chistiakov DA, Shkurat TP, Melnichenko AA, Grechko AV, Orekhov AN. The role of mitochondrial dysfunction in cardiovascular disease: a brief review. Ann Med. 2018;50:121-7.
- Ciccarelli G, Conte S, Cimmino G, Maiorano P, Morrione A, Giordano A, et al. Mitochondrial dysfunction: the hidden player in the pathogenesis of atherosclerosis? Int J Mol Sci. 2023;24:56-68.
- Huynh DTN, Heo KS. Role of mitochondrial dynamics and mitophagy of vascular smooth muscle cell proliferation and migration in progression of atherosclerosis. Arch Pharm Res. 2021;44:1051-61.
- Wei H, Zhang WJ, McMillen TS, Leboeuf RC, Frei B. Copper chelation by tetrathiomolybdate inhibits vascular inflammation and atherosclerotic lesion development in apolipoprotein E-deficient mice. Atherosclerosis. 2012;223:306-13.
- Wei H, Frei B, Beckman JS, Zhang WJ. Copper chelation by tetrathiomolybdate inhibits lipopolysaccharide-induced inflammatory responses in vivo. Am J Physiol Heart Circ Physiol. 2011;301:H712-20.
- Clarke CN, Clarke NE, Mosher RE. Treatment of angina pectoris with disodium ethylene diamine tetraacetic acid. Am J Med Sci. 1956;232:654-66.
- Lamas GA, Goertz C, Boineau R, Mark DB, Rozema T, Nahin RL, et al. Effect of disodium EDTA chelation regimen on cardiovascular events in patients with previous myocardial infarction: the TACT randomized trial. JAMA. 2013;309:1241-50.
- Villarruz-Sulit MV, Forster R, Dans AL, Tan FN, Sulit DV. Chelation therapy for atherosclerotic cardiovascular disease. Cochrane Database Syst Rev. 2020;5:Cd002785.
- Sudhahar V, Shi Y, Kaplan JH, Ushio-Fukai M, Fukai T, et al. Whole-transcriptome sequencing analyses of nuclear antixoxidant-1 in endothelial cells: role in inflammation and atherosclerosis. Cells. 2022;11:881-96.
- Kohno T, Urao N, Ashino T, Sudhahar V, McKinney RD, Hamakubo T, et al. Novel role of copper transport protein antioxidant-1 in neointimal formation after vascular injury. Arterioscler Thromb Vasc Biol. 2013;33:805-13.
- Das A, Sudhahar V, Ushio-Fukai M, Fukai T. Novel interaction of antioxidant-1 with TRAF4: role in inflammatory responses in endothelial cells. Am J Physiol Cell Physiol. 2019;317:C1161-c71.
- Xue Q, Kang R, Klionsky DJ, Tang D, Liu J, Chen X, et al. Copper metabolism in cell death and autophagy. Autophagy. 2023;12:65-104.
- O’Day SJ, Eggermont AM, Chiarion-Sileni V, Kefford R, Grob JJ, Mortier L, et al. Final results of phase III SYMMETRY study: randomized, double-blind trial of elesclomol plus paclitaxel versus paclitaxel alone as treatment for chemotherapy-naive patients with advanced melanoma. J Clin Oncol. 2013;31:1211-8.
- Liu S, Zhao Y, Shen M, Hao Y, Wu X, Yao Y, et al. Hyaluronic acid targeted and pH-responsive multifunctional nanoparticles for chemo-photothermal synergistic therapy of atherosclerosis. J Mater Chem B. 2022;10:562-70.
مساهمات المؤلفين
التمويل
المصالح المتنافسة
معلومات إضافية
مستشفى جوانغ آن، أكاديمية العلوم الطبية الصينية، بكين 100053، الصين. مستشفى شييوان، أكاديمية العلوم الطبية الصينية، بكين 100091، الصين. البريد الإلكتروني: liulongtao1976@126.com; wumin19762000@126.com
DOI: https://doi.org/10.1038/s41420-023-01796-1
PMID: https://pubmed.ncbi.nlm.nih.gov/38218941
Publication Date: 2024-01-13
Copper homeostasis and cuproptosis in atherosclerosis: metabolism, mechanisms and potential therapeutic strategies
Abstract
Copper is an essential micronutrient that plays a pivotal role in numerous physiological processes in virtually all cell types. Nevertheless, the dysregulation of copper homeostasis, whether towards excess or deficiency, can lead to pathological alterations, such as atherosclerosis. With the advent of the concept of copper-induced cell death, termed cuproptosis, researchers have increasingly focused on the potential role of copper dyshomeostasis in atherosclerosis. In this review, we provide a broad overview of cellular and systemic copper metabolism. We then summarize the evidence linking copper dyshomeostasis to atherosclerosis and elucidate the potential mechanisms underlying atherosclerosis development in terms of both copper excess and copper deficiency. Furthermore, we discuss the evidence for and mechanisms of cuproptosis, discuss its interactions with other modes of cell death, and highlight the role of cuproptosis-related mitochondrial dysfunction in atherosclerosis. Finally, we explore the therapeutic strategy of targeting this novel form of cell death, aiming to provide some insights for the management of atherosclerosis.
FACTS
- Copper is an essential trace element required in various physiological processes in the human body.
- Dysregulation of copper homeostasis, whether towards excess or deficiency, has been implicated in various health problems, including atherosclerosis.
- Dysregulation of copper homeostasis and copper-induced cell death (cuproptosis) are acknowledged as potential contributors to the pathogenesis of atherosclerosis.
OPEN QUESTIONS
- What is the safe window for copper levels to avoid the development of atherosclerosis? How can the suitable copper concentration be determined for the treatment of atherosclerosis?
- What are the potential candidate biomarkers that can reliably and sensitively indicate the occurrence of cuproptosis in the context of atherosclerosis?
- What are the potential undiscovered roles of copper in mitochondrial function, and is there interplay between copper and mitochondrial dynamics or mitophagy?
INTRODUCTION
clinical consequences, such as myocardial infarction (MI) and stroke, which are major causes of morbidity and mortality worldwide [3]. The pathogenesis of atherosclerosis involves multiple genetic, environmental, and metabolic factors [4].
COPPER METABOLISM
Systemic copper homeostasis
liver back into the bloodstream. Here, copper ions bind to their soluble partners and are transported to specific tissues and organs [22].
Cellular copper homeostasis
also acts as a copper donor within the mitochondrial intermembrane space (IMS) [37]. COX17 is thus essential for proper COX assembly, with mutations in COX17 shown to further reduce COX activity, resulting in mitochondrial dysfunction and oxidative stress [38].
EVIDENCE LINKING COPPER DYSHOMEOSTASIS TO ATHEROSCLEROSIS
| Country, author, year | Methods and study population | Result |
| Finland, Reunanen et al. [47] | 230 men who died from CVDs and 298 matched controls; 10 years follow up. | The adjusted relative risk of CHD mortality between the highest and lowest tertiles of serum copper were 2.86 (
|
| United States, Ford, et al. [48] | 4,574 participants aged
|
There was an
|
| Dutch, Kok et al. [49] | Cancer (
|
Individuals with elevated serum copper levels (above
|
| Eastern Finland, Salonen et al. [50] | 1666 randomly selected men aged 42,48,54, or 60 years with no symptomatic IHD. | Elevated serum copper level is a risk factor for IHD that acts independently, as shown by a 3.5 -fold to 4.0 -fold increased risk of AMI associated with high serum copper levels (1.021.16 mg /liter and
|
| Nadina, Robyn et al. [51] | Atherosclerotic patients aged
|
Copper levels in the intima of lesions were notably higher in atherosclerotic patients compared to those in healthy controls (
|
| Bangladesh, Begum et al. [52] | 60 patients diagnosed with AMI and 60 healthy controls. | The mean serum copper level was significantly higher in AMI patients compared to that in controls (
|
| Grzegorz, Barbara et al. [53] | 74 participants of MI and 72 healthy controls. | Higher serum copper level was significantly associated with MI (
|
| Italy, Tarantino et al. [54] | 100 obese patients who had a low prevalence of comorbidities. | Altered copper bioavailability was negatively associated with carotid IMT (
|
| Serbia, Tasić et al. [55] | 91 patients (mean age
|
Patients with hemorrhagic plaque had significantly higher serum copper levels compared to those with calcified plaque; high copper concentrations may contribute to atherosclerosis. |
from numerous studies has linked copper dyshomeostasis and the resultant excess or deficiency of copper to atherosclerosis.
Evidence linking copper excess and atherosclerosis
Evidence linking copper deficiency and atherosclerosis
POTENTIAL MECHANISMS OF ATHEROSCLEROSIS DEVELOPMENT DUE TO COPPER DYSHOMEOSTASIS
Oxidative stress
leads to the oxidative damage of lipid molecules. Increased copper levels can promote lipid peroxidation and result in the formation of oxidized low-density lipoprotein (ox-LDL), which is an atherosclerosis risk factor [67]. Furthermore, increased copper levels have been shown to reduce the activity of antioxidant enzymes, such as
deficiency leads to a reduction in NO levels, which may promote endothelial dysfunction, reduce vascular relaxation, and increase oxidative stress, thus ultimately contributing to atherosclerosis development [70]. In addition to affecting antioxidant enzymes, copper deficiency may also reduce COX activity and lead to oxidative inactivation of complex I (NADH: ubiquinone oxidoreductase). This oxidative inactivation may then lead to elevated production of ROS in copper-deficient cells, thereby exacerbating cellular oxidative stress [74].
Inflammation
Endothelial dysfunction
Lipid metabolism
Other risk factors
COPPER-INDUCED CELL DEATH AND ATHEROSCLEROSIS From copper-induced cell death to cuproptosis
leads to the aggregation of copper-bound lipoylated mitochondrial proteins, which can disrupt the TCA cycle and, therefore, interfere with cellular energy production. The upstream regulatory factors FDX1 and LIAS are critical in this process. Aggregation of these proteins and the subsequent reduction of
Potential interactions between copper-induced cell death and other cell death pathways
copper exposure in mouse microglial cells triggers an inflammatory response, resulting in the upregulation of NLRP3/caspase 1/ GSDMD axis proteins and subsequent pyroptosis. These effects are likely mediated by the early activation of the ROS/NF-кВ pathway and subsequent disruption of mitophagy [77]. Moreover, comparable outcomes were observed in murine macrophages treated with copper oxide nanoparticles. Copper oxide nanoparticle exposure induces oxidative stress and activates NLRP3 inflammasomes, leading to the expression of pro-IL-1
Cuproptosis-related mitochondrial dysfunction and atherosclerosis
intracellular copper may also disrupt mitochondrial function by altering the activity of several key enzymes, such as those involved in the tricarboxylic acid (TCA) cycle and oxidative phosphorylation [131]. Severe mitochondrial dysfunction and decreases in the activities of several liver enzymes, including complex I, complex II, complex III, complex IV, and aconitase, were observed in patients with copper overload. These effects are potentially mediated by the accumulation of copper in the mitochondria [132]. Furthermore, copper treatment induces selective changes in metabolic enzymes through lipoylation, which is a highly conserved lysine post-translational modification. Protein lipoylation occurs only on Dihydrolipoamide S-Acetyltransferase (DLAT), Dihydrolipoamide S-Succinyltransferase (DLST), Dihydrolipoamide Branched Chain Transacylase E2 (DBT), and Glycine Cleavage System Protein H (GCSH), all of which are involved in metabolic complexes that regulate carbon entry points to the TCA cycle [133, 134]. Additionally, copper overload may trigger the opening of the mitochondrial permeability transition pore and cause the release of pro-apoptotic factors, ultimately resulting in cell death [135].
POTENTIAL THERAPEUTIC STRATEGIES TARGETING CUPROPTOSIS IN ATHEROSCLEROSIS Copper chelators
treatment of atherosclerotic cardiovascular disease [143]. The contradictory results among these studies may be attributed to variations in research design and/or characteristics of the study populations. Therefore, further research is needed before routine use of chelation therapy can be recommended.
Regulation of copper chaperone protein expression
Copper ionophores
CONCLUSIONS AND FUTURE PERSPECTIVES
REFERENCES
- Libby P. The changing landscape of atherosclerosis. Nature. 2021;592:524-33.
- Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, et al. Atherosclerosis. Nat Rev Dis Prim. 2019;5:56.
- Herrington W, Lacey B, Sherliker P, Armitage J, Lewington S. Epidemiology of atherosclerosis and the potential to reduce the global burden of Atherothrombotic disease. Circ Res. 2016;118:535-46.
- Björkegren JLM, Lusis AJ. Atherosclerosis: recent developments. Cell. 2022;185:1630-45.
- Festa RA, Thiele DJ. Copper: an essential metal in biology. Curr Biol. 2011;21:R877-83.
- Kim BE, Nevitt T, Thiele DJ. Mechanisms for copper acquisition, distribution and regulation. Nat Chem Biol. 2008;4:176-85.
- Chen L, Min J, Wang F. Copper homeostasis and cuproptosis in health and disease. Signal Transduct Target Ther. 2022;7:378.
- Chen X, Cai Q, Liang R, Zhang D, Liu X, Zhang M, et al. Copper homeostasis and copper-induced cell death in the pathogenesis of cardiovascular disease and therapeutic strategies. Cell Death Dis. 2023;14:105.
- Uriu-Adams JY, Keen CL. Copper, oxidative stress, and human health. Mol Asp Med. 2005;26:268-98.
- Fukai T, Ushio-Fukai M, Kaplan JH. Copper transporters and copper chaperones: roles in cardiovascular physiology and disease. Am J Physiol Cell Physiol. 2018;315:C186-c201.
- Tang D, Chen X, Kroemer G. Cuproptosis: a copper-triggered modality of mitochondrial cell death. Cell Res. 2022;32:417-8.
- Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375:1254-61.
- Yang L, Yang P, Lip GYH, Ren J. Copper homeostasis and cuproptosis in cardiovascular disease therapeutics. Trends Pharm Sci. 2023;44:573-85.
- Maung MT, Carlson A, Olea-Flores M, Elkhadragy L, Schachtschneider KM, Navarro-Tito N, et al. The molecular and cellular basis of copper dysregulation and its relationship with human pathologies. FASEB J. 2021;35:e21810.
- Bost M, Houdart S, Oberli M, Kalonji E, Huneau JF, Margaritis I. Dietary copper and human health: current evidence and unresolved issues. J Trace Elem Med Biol. 2016;35:107-15.
- Xie J, Yang Y, Gao Y, He J. Cuproptosis: mechanisms and links with cancers. Mol Cancer. 2023;22:46.
- Nose Y, Kim BE, Thiele DJ. Ctr1 drives intestinal copper absorption and is essential for growth, iron metabolism, and neonatal cardiac function. Cell Metab. 2006;4:235-44.
- Lönnerdal B. Intestinal regulation of copper homeostasis: a developmental perspective. Am J Clin Nutr. 2008;88:846s-50s.
- Lutsenko S. Dynamic and cell-specific transport networks for intracellular copper ions. J Cell Sci. 2021;134:96-114.
- Ramos D, Mar D, Ishida M, Vargas R, Gaite M, Montgomery A, et al. Mechanism of copper uptake from blood plasma ceruloplasmin by mammalian cells. PLoS One. 2016;11:e0149516.
- Santoro A, Calvo JS, Peris-Díaz MD, Krężel A, Meloni G, Faller P. The glutathione/ metallothionein system challenges the design of efficient
-activating copper complexes. Angew Chem Int Ed Engl. 2020;59:7830-5. - La Fontaine S, Ackland ML, Mercer JF. Mammalian copper-transporting P-type ATPases, ATP7A and ATP7B: emerging roles. Int J Biochem Cell Biol. 2010;42:206-9.
- Wijmenga C, Klomp LW. Molecular regulation of copper excretion in the liver. Proc Nutr Soc. 2004;63:31-9.
- Hernandez S, Tsuchiya Y, García-Ruiz JP, Lalioti V, Nielsen S, Cassio D, et al. ATP7B copper-regulated traffic and association with the tight junctions: copper excretion into the bile. Gastroenterology. 2008;134:1215-23.
- Huster D, Kühne A, Bhattacharjee A, Raines L, Jantsch V, Noe J, et al. Diverse functional properties of Wilson disease ATP7B variants. Gastroenterology. 2012;142:947-56.e5.
- Maryon EB, Molloy SA, Ivy K, Yu H, Kaplan JH. Rate and regulation of copper transport by human copper transporter 1 (hCTR1). J Biol Chem. 2013;288:18035-46.
- Liang ZD, Tsai WB, Lee MY, Savaraj N, Kuo MT. Specificity protein 1 (sp1) oscillation is involved in copper homeostasis maintenance by regulating human high-affinity copper transporter 1 expression. Mol Pharm. 2012;81:455-64.
- Kuo YM, Gybina AA, Pyatskowit JW, Gitschier J, Prohaska JR. Copper transport protein (Ctr1) levels in mice are tissue specific and dependent on copper status.
J Nutr. 2006;136:21-6. - Muller PA, Klomp LW. ATOX1: a novel copper-responsive transcription factor in mammals? Int J Biochem Cell Biol. 2009;41:1233-6.
- Prohaska JR, Gybina AA. Intracellular copper transport in mammals. J Nutr. 2004;134:1003-6.
- Yang D, Xiao P, Qiu B, Yu HF, Teng CB. Copper chaperone antioxidant 1: multiple roles and a potential therapeutic target. J Mol Med Berl. 2023;101:527-42.
- Itoh S, Kim HW, Nakagawa O, Ozumi K, Lessner SM, Aoki H, et al. Novel role of antioxidant-1 (Atox1) as a copper-dependent transcription factor involved in cell proliferation. J Biol Chem. 2008;283:9157-67.
- Rosenzweig AC. Copper delivery by metallochaperone proteins. Acc Chem Res. 2001;34:119-28.
- Dong
, Zhang , Zhao J, Lei J, Chen Y, Li X, et al. The rational design of specific SOD1 inhibitors via copper coordination and their application in ROS signaling research. Chem Sci. 2016;7:6251-62. - Elchuri S, Oberley TD, Qi W, Eisenstein RS, Jackson Roberts L, Van Remmen H, et al. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene. 2005;24:367-80.
- Ding Y, Chen Y, Wu Z, Yang N, Rana K, Meng X, et al. SsCox17, a copper chaperone, is required for pathogenic process and oxidative stress tolerance of Sclerotinia sclerotiorum. Plant Sci. 2022;322:111345.
- Horng YC, Cobine PA, Maxfield AB, Carr HS, Winge DR. Specific copper transfer from the Cox17 metallochaperone to both Sco1 and Cox11 in the assembly of yeast cytochrome C oxidase. J Biol Chem. 2004;279:35334-40.
- Takahashi Y, Kako K, Kashiwabara S, Takehara A, Inada Y, Arai H, et al. Mammalian copper chaperone Cox17p has an essential role in activation of cytochrome
oxidase and embryonic development. Mol Cell Biol. 2002;22:7614-21. - Lutsenko S, Barnes NL, Bartee MY, Dmitriev OY. Function and regulation of human copper-transporting ATPases. Physiol Rev. 2007;87:1011-46.
- La Fontaine S, Mercer JF. Trafficking of the copper-ATPases, ATP7A and ATP7B: role in copper homeostasis. Arch Biochem Biophys. 2007;463:149-67.
- Horn N, Wittung SP. et al. ATP7A-regulated enzyme metalation and trafficking in the menkes disease puzzle. Biomedicines. 2021;9:123-142.
- Chen J, Jiang Y, Shi H, Peng Y, Fan X, Li C. The molecular mechanisms of copper metabolism and its roles in human diseases. Pflug Arch. 2020;472:1415-29.
- Wolf D, Ley K. Immunity and inflammation in atherosclerosis. Circ Res. 2019;124:315-27.
- Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. 2011;17:1410-22.
- Campia U, Gerhard-Herman M, Piazza G, Goldhaber SZ. Peripheral artery disease: past, present, and future. Am J Med. 2019;132:1133-41.
- Endres M, Moro MA, Nolte CH, Dames C, Buckwalter MS, Meisel A. Immune pathways in etiology, acute phase, and chronic sequelae of ischemic stroke. Circ Res. 2022;130:1167-86.
- Reunanen A, Knekt P, Marniemi J, Mäki J, Maatela J, Aromaa A. Serum calcium, magnesium, copper and zinc and risk of cardiovascular death. Eur J Clin Nutr. 1996;50:431-7.
- Ford ES. Serum copper concentration and coronary heart disease among US adults. Am J Epidemiol. 2000;151:1182-8.
- Kok FJ, Van Duijn CM, Hofman A, Van der Voet GB, De Wolff FA, Paays CH, et al. Serum copper and zinc and the risk of death from cancer and cardiovascular disease. Am J Epidemiol. 1988;128:352-9.
- Salonen JT, Salonen R, Korpela H, Suntioinen S, Tuomilehto J. Serum copper and the risk of acute myocardial infarction: a prospective population study in men in eastern Finland. Am J Epidemiol. 1991;134:268-76.
- Stadler N, Lindner RA, Davies MJ. Direct detection and quantification of transition metal ions in human atherosclerotic plaques: evidence for the presence of
elevated levels of iron and copper. Arterioscler Thromb Vasc Biol. 2004;24:949-54. - Begum S, Sultana I, Faysal MR, Alam S, Tasnim J, Akter T, et al. Study of changes in serum copper level in patients with acute myocardial infarction. Mymens Med J. 2023;32:39-43.
- Nowicki GJ, Ślusarska B, Prystupa A, Blicharska E, Adamczuk A, Czernecki T, et al. Assessment of concentrations of heavy metals in postmyocardial infarction patients and patients free from cardiovascular event. Cardiol Res Pr. 2021;2021:9546358.
- Tarantino G, Porcu C, Arciello M, Andreozzi P, Balsano C. Prediction of carotid intima-media thickness in obese patients with low prevalence of comorbidities by serum copper bioavailability. J Gastroenterol Hepatol. 2018;33:1511-7.
- Tasić NM, Tasić D, Otašević P, Veselinović M, Jakovljević V, Djurić D, et al. Copper and zinc concentrations in atherosclerotic plaque and serum in relation to lipid metabolism in patients with carotid atherosclerosis. Vojnosanit Pregl. 2015;72:801-6.
- Trumbo
, Yates , Schlicker , Poos M. Dietary reference intakes: vitamin A, vitamin K, arsenic, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. J Am Diet Assoc. 2001;101:294-301. - Rock E, Mazur A, O’Connor JM, Bonham MP, Rayssiguier Y, Strain JJ. The effect of copper supplementation on red blood cell oxidizability and plasma antioxidants in middle-aged healthy volunteers. Free Radic Biol Med. 2000;28:324-9.
- Chen F, Du M, Blumberg JB, Ho Chui KK, Ruan M, Rogers G, et al. Association among dietary supplement use, nutrient intake, and mortality among U.S. adults: a cohort study. Ann Intern Med. 2019;170:604-13.
- Lamb DJ, Avades TY, Ferns GA. Biphasic modulation of atherosclerosis induced by graded dietary copper supplementation in the cholesterol-fed rabbit. Int J Exp Pathol. 2001;82:287-94.
- Wang T, Xiang P, Ha JH, Wang X, Doguer C, Flores SRL, et al. Copper supplementation reverses dietary iron overload-induced pathologies in mice. J Nutr Biochem. 2018;59:56-63.
- Hughes WM Jr, Rodriguez WE, Rosenberger D, Chen J, Sen U, Tyagi N, et al. Role of copper and homocysteine in pressure overload heart failure. Cardiovasc Toxicol. 2008;8:137-44.
- Diaf M, Khaled MB. Associations between dietary antioxidant intake and markers of atherosclerosis in middle-aged women from North-Western Algeria. Front Nutr. 2018;5:29.
- Kim YW, Byzova TV. Oxidative stress in angiogenesis and vascular disease. Blood. 2014;123:625-31.
- Fang Y, Xing C, Wang X, Cao H, Zhang C, Guo X, et al. Activation of the ROS/HO1/NQO1 signaling pathway contributes to the copper-induced oxidative stress and autophagy in duck renal tubular epithelial cells. Sci Total Environ. 2021;757:143753.
- Valko M, Morris H, Cronin MT. Metals, toxicity and oxidative stress. Curr Med Chem. 2005;12:1161-208.
- Dhalla NS, Temsah RM, Netticadan T. Role of oxidative stress in cardiovascular diseases. J Hypertens. 2000;18:655-73.
- Esterbauer H, Gebicki J, Puhl H, Jürgens G. The role of lipid peroxidation and antioxidants in oxidative modification of LDL. Free Radic Biol Med. 1992;13:341-90.
- Ozcelik D, Uzun H. Copper intoxication; antioxidant defenses and oxidative damage in rat brain. Biol Trace Elem Res. 2009;127:45-52.
- Brezová V, Dvoranová D, Zúbor V, Breza M, Mazúr M, Valko M. Photochemical properties of camptothecin in the presence of copper(II) ions: the role of radicals as prospective species in photodynamic therapy. Mol Biotechnol. 2007;37:48-51.
- AI-Bayati MA, Jamil DA, AI-Aubaidy HA. Cardiovascular effects of copper deficiency on activity of superoxide dismutase in diabetic nephropathy. N. Am J Med Sci. 2015;7:41-6.
- Linder MC, Hazegh-Azam M. Copper biochemistry and molecular biology. Am J Clin Nutr. 1996;63:797s-811s.
- Sukalski KA, LaBerge TP, Johnson WT. In vivo oxidative modification of erythrocyte membrane proteins in copper deficiency. Free Radic Biol Med. 1997;22:835-42.
- Bertinato J, Iskandar M, L’Abbé MR. Copper deficiency induces the upregulation of the copper chaperone for
superoxide dismutase in weanling male rats. J Nutr. 2003;133:28-31. - Johnson WT, Thomas AC. Copper deprivation potentiates oxidative stress in HL60 cell mitochondria. Proc Soc Exp Biol Med. 1999;221:147-52.
- Malekahmadi M, Firouzi S, Rezayi M, Ghazizadeh H, Ranjbar G, Ferns GA, et al. Association of zinc and copper status with cardiovascular diseases and their assessment methods: a review study. Mini Rev Med Chem. 2020;20:2067-78.
- Ansteinsson V, Refsnes M, Skomedal T, Osnes JB, Schiander I, Låg M. Zinc- and copper-induced interleukin-6 release in primary cell cultures from rat heart. Cardiovasc Toxicol. 2009;9:86-94.
- Zhou Q, Zhang Y, Lu L, Zhang H, Zhao C, Pu Y, et al. Copper induces microgliamediated neuroinflammation through ROS/NF-кB pathway and mitophagy disorder. Food Chem Toxicol. 2022;168:113369.
- Schuschke DA, Saari JT, Miller FN. Leukocyte-endothelial adhesion is impaired in the cremaster muscle microcirculation of the copper-deficient rat. Immunol Lett. 2001;76:139-44.
- Sakai N, Shin T, Schuster R, Blanchard J, Lentsch AB, Johnson WT, et al. Marginal copper deficiency increases liver neutrophil accumulation after ischemia/ reperfusion in rats. Biol Trace Elem Res. 2011;142:47-54.
- Gordon SA, Lominadze D, Saari JT, Lentsch AB, Schuschke DA. Impaired deformability of copper-deficient neutrophils. Exp Biol Med Maywood. 2005;230:543-8.
- Schuschke DA, Percival SS, Lominadze D, Saari JT, Lentsch AB. Tissue-specific ICAM-1 expression and neutrophil transmigration in the copper-deficient rat. Inflammation. 2002;26:297-303.
- Lominadze D, Saari JT, Percival SS, Schuschke DA. Proinflammatory effects of copper deficiency on neutrophils and lung endothelial cells. Immunol Cell Biol. 2004;82:231-8.
- Lentsch AB, Kato A, Saari JT, Schuschke DA. Augmented metalloproteinase activity and acute lung injury in copper-deficient rats. Am J Physiol Lung Cell Mol Physiol. 2001;281:L387-93.
- Paynter DI, Moir RJ, Underwood EJ. Changes in activity of the Cu-Zn superoxide dismutase enzyme in tissues of the rat with changes in dietary copper. J Nutr. 1979;109:1570-6.
- Förstermann U, Münzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006;113:1708-14.
- Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315-424.
- Starkebaum G, Harlan JM. Endothelial cell injury due to copper-catalyzed hydrogen peroxide generation from homocysteine. J Clin Invest. 1986;77:1370-6.
- Versari D, Daghini E, Virdis A, Ghiadoni L, Taddei S. Endothelial dysfunction as a target for prevention of cardiovascular disease. Diabetes Care. 2009;32:S314-21.
- Kattoor AJ, Kanuri SH, Mehta JL. Role of Ox-LDL and LOX-1 in Atherogenesis. Curr Med Chem. 2019;26:1693-700.
- Craig WY, Poulin SE, Palomaki GE, Neveux LM, Ritchie RF, Ledue TB. Oxidationrelated analytes and lipid and lipoprotein concentrations in healthy subjects. Arterioscler Thromb Vasc Biol. 1995;15:733-9.
- Kazemi-Bajestani SM, Ghayour-Mobarhan M, Ebrahimi M, Moohebati M, Esmaeili HA, Parizadeh MR, et al. Serum copper and zinc concentrations are lower in Iranian patients with angiographically defined coronary artery disease than in subjects with a normal angiogram. J Trace Elem Med Biol. 2007;21:22-8.
- Song YF, Luo Z, Zhang LH, Hogstrand C, Pan YX. Endoplasmic reticulum stress and disturbed calcium homeostasis are involved in copper-induced alteration in hepatic lipid metabolism in yellow catfish Pelteobagrus fulvidraco. Chemosphere. 2016;144:2443-53.
- Morrell A, Tallino S, Yu L, Burkhead JL. The role of insufficient copper in lipid synthesis and fatty-liver disease. IUBMB Life. 2017;69:263-70.
- Ye J, DeBose RA. Regulation of cholesterol and fatty acid synthesis. Cold Spring Harb Perspect Biol. 2011;3:96-105.
- Tang Z, Gasperkova D, Xu J, Baillie R, Lee JH, Clarke SD. Copper deficiency induces hepatic fatty acid synthase gene transcription in rats by increasing the nuclear content of mature sterol regulatory element binding protein 1. J Nutr. 2000;130:2915-21.
- Loyke HF. Copper and zinc in experimental hypertension. Biol Trace Elem Res. 1991;29:45-9.
- Ozumi K, Sudhahar V, Kim HW, Chen GF, Kohno T, Finney L, et al. Role of copper transport protein antioxidant 1 in angiotensin II-induced hypertension: a key regulator of extracellular superoxide dismutase. Hypertension. 2012;60:476-86.
- Halliwell B, Gutteridge JM. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem J. 1984;219:1-14.
- Gaetke LM, Chow CK. Copper toxicity, oxidative stress, and antioxidant nutrients. Toxicology. 2003;189:147-63.
- Tardito S, Bassanetti I, Bignardi C, Elviri L, Tegoni M, Mucchino C, et al. Copper binding agents acting as copper ionophores lead to caspase inhibition and paraptotic cell death in human cancer cells. J Am Chem Soc. 2011;133:6235-42.
- Nagai M, Vo NH, Shin Ogawa L, Chimmanamada D, Inoue T, Chu J, et al. The oncology drug elesclomol selectively transports copper to the mitochondria to induce oxidative stress in cancer cells. Free Radic Biol Med. 2012;52:2142-50.
- Lill R, Freibert SA. Mechanisms of mitochondrial iron-sulfur protein biogenesis. Annu Rev Biochem. 2020;89:471-99.
- Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther. 2021;6:128.
- Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol. 2001;9:113-4.
- Nunes T, de Souza HS. Inflammasome in intestinal inflammation and cancer. Mediat Inflamm. 2013;2013:654963.
- Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16:407-20.
- Aachoui Y, Sagulenko V, Miao EA, Stacey KJ. Inflammasome-mediated pyroptotic and apoptotic cell death, and defense against infection. Curr Opin Microbiol. 2013;16:319-26.
- Tao X, Wan X, Wu D, Song E, Song Y. A tandem activation of NLRP3 inflammasome induced by copper oxide nanoparticles and dissolved copper ion in J774A. 1 macrophage. J Hazard Mater. 2021;411:125134.
- Liu H, Guo H, Deng H, Cui H, Fang J, Zuo Z, et al. Copper induces hepatic inflammatory responses by activation of MAPKs and NF-kB signalling pathways in the mouse. Ecotoxicol Environ Saf. 2020;201:110806.
- Deng H, Zhu S, Yang H, Cui H, Guo H, Deng J, et al. The dysregulation of inflammatory pathways triggered by copper exposure. Biol Trace Elem Res. 2023;201:539-48.
- Liao J, Yang F, Tang Z, Yu W, Han Q, Hu L, et al. Inhibition of caspase-1dependent pyroptosis attenuates copper-induced apoptosis in chicken hepatocytes. Ecotoxicol Environ Saf. 2019;174:110-9.
- Zhang X, Ren Z, Xu W, Jiang Z. Necroptosis in atherosclerosis. Clin Chim Acta. 2022;534:22-8.
- Li M, Wang ZW, Fang LJ, Cheng SQ, Wang X, Liu NF. Programmed cell death in atherosclerosis and vascular calcification. Cell Death Dis. 2022;13:467.
- Gutierrez KD, Davis MA, Daniels BP, Olsen TM, Ralli-Jain P, Tait SW, et al. MLKL activation triggers NLRP3-mediated processing and release of IL-1
independently of gasdermin-D. J Immunol. 2017;198:2156-64. - Miao Y, Liu J, Liu X, Yuan Q, Li H, Zhang Y, et al. Machine learning identification of cuproptosis and necroptosis-associated molecular subtypes to aid in prognosis assessment and immunotherapy response prediction in low-grade glioma. Front Genet. 2022;13:951239.
- Kuriakose T, Man SM, Malireddi RK, Karki R, Kesavardhana S, Place DE, et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol. 2016;1:52-68.
- Jiao H, Wachsmuth L, Kumari S, Schwarzer R, Lin J, Eren RO, et al. Publisher Correction: Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation. Nature. 2020;580:E10.
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060-72.
- Hirschhorn T, Stockwell BR. The development of the concept of ferroptosis. Free Radic Biol Med. 2019;133:130-43.
- Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26:165-76.
- Yang F, Pei R, Zhang Z, Liao J, Yu W, Qiao N, et al. Copper induces oxidative stress and apoptosis through mitochondria-mediated pathway in chicken hepatocytes. Toxicol Vitr. 2019;54:310-6.
- Ren X, Li Y, Zhou Y, Hu W, Yang C, Jing Q, et al. Overcoming the compensatory elevation of NRF2 renders hepatocellular carcinoma cells more vulnerable to disulfiram/copper-induced ferroptosis. Redox Biol. 2021;46:102122.
- Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688-92.
- Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317-31.
- Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91-8.
- Xue Q, Yan D, Chen X, Li X, Kang R, Klionsky DJ, et al. Copper-dependent autophagic degradation of GPX4 drives ferroptosis. Autophagy. 2023;19:1982-96.
- Gao W, Huang Z, Duan J, Nice EC, Lin J, Huang C. Elesclomol induces copperdependent ferroptosis in colorectal cancer cells via degradation of ATP7A. Mol Oncol. 2021;15:3527-44.
- Li F, Wu X, Liu H, Liu M, Yue Z, Wu Z, et al. Copper depletion strongly enhances ferroptosis via mitochondrial perturbation and reduction in antioxidative mechanisms. Antioxid Basel. 2022;11:652-98.
- Arciello M, Rotilio G, Rossi L. Copper-dependent toxicity in SH-SY5Y neuroblastoma cells involves mitochondrial damage. Biochem Biophys Res Commun. 2005;327:454-9.
- Cobine PA, Moore SA, Leary SC. Getting out what you put in: Copper in mitochondria and its impacts on human disease. Biochim Biophys Acta Mol Cell Res. 2021;1868:118867.
- Sheline CT, Choi DW. Cu2+ toxicity inhibition of mitochondrial dehydrogenases in vitro and in vivo. Ann Neurol. 2004;55:645-53.
- Gu M, Cooper JM, Butler P, Walker AP, Mistry PK, Dooley JS, et al. Oxidativephosphorylation defects in liver of patients with Wilson’s disease. Lancet. 2000;356:469-74.
- Solmonson A, DeBerardinis RJ. Lipoic acid metabolism and mitochondrial redox regulation. J Biol Chem. 2018;293:7522-30.
- Rowland EA, Snowden CK, Cristea IM. Protein lipoylation: an evolutionarily conserved metabolic regulator of health and disease. Curr Opin Chem Biol. 2018;42:76-85.
- Su R, Wang R, Cao H, Pan J, Chen L, Li C, et al. High copper levels promotes broiler hepatocyte mitochondrial permeability transition in vivo and in vitro. Biol Trace Elem Res. 2011;144:636-46.
- Chistiakov DA, Shkurat TP, Melnichenko AA, Grechko AV, Orekhov AN. The role of mitochondrial dysfunction in cardiovascular disease: a brief review. Ann Med. 2018;50:121-7.
- Ciccarelli G, Conte S, Cimmino G, Maiorano P, Morrione A, Giordano A, et al. Mitochondrial dysfunction: the hidden player in the pathogenesis of atherosclerosis? Int J Mol Sci. 2023;24:56-68.
- Huynh DTN, Heo KS. Role of mitochondrial dynamics and mitophagy of vascular smooth muscle cell proliferation and migration in progression of atherosclerosis. Arch Pharm Res. 2021;44:1051-61.
- Wei H, Zhang WJ, McMillen TS, Leboeuf RC, Frei B. Copper chelation by tetrathiomolybdate inhibits vascular inflammation and atherosclerotic lesion development in apolipoprotein E-deficient mice. Atherosclerosis. 2012;223:306-13.
- Wei H, Frei B, Beckman JS, Zhang WJ. Copper chelation by tetrathiomolybdate inhibits lipopolysaccharide-induced inflammatory responses in vivo. Am J Physiol Heart Circ Physiol. 2011;301:H712-20.
- Clarke CN, Clarke NE, Mosher RE. Treatment of angina pectoris with disodium ethylene diamine tetraacetic acid. Am J Med Sci. 1956;232:654-66.
- Lamas GA, Goertz C, Boineau R, Mark DB, Rozema T, Nahin RL, et al. Effect of disodium EDTA chelation regimen on cardiovascular events in patients with previous myocardial infarction: the TACT randomized trial. JAMA. 2013;309:1241-50.
- Villarruz-Sulit MV, Forster R, Dans AL, Tan FN, Sulit DV. Chelation therapy for atherosclerotic cardiovascular disease. Cochrane Database Syst Rev. 2020;5:Cd002785.
- Sudhahar V, Shi Y, Kaplan JH, Ushio-Fukai M, Fukai T, et al. Whole-transcriptome sequencing analyses of nuclear antixoxidant-1 in endothelial cells: role in inflammation and atherosclerosis. Cells. 2022;11:881-96.
- Kohno T, Urao N, Ashino T, Sudhahar V, McKinney RD, Hamakubo T, et al. Novel role of copper transport protein antioxidant-1 in neointimal formation after vascular injury. Arterioscler Thromb Vasc Biol. 2013;33:805-13.
- Das A, Sudhahar V, Ushio-Fukai M, Fukai T. Novel interaction of antioxidant-1 with TRAF4: role in inflammatory responses in endothelial cells. Am J Physiol Cell Physiol. 2019;317:C1161-c71.
- Xue Q, Kang R, Klionsky DJ, Tang D, Liu J, Chen X, et al. Copper metabolism in cell death and autophagy. Autophagy. 2023;12:65-104.
- O’Day SJ, Eggermont AM, Chiarion-Sileni V, Kefford R, Grob JJ, Mortier L, et al. Final results of phase III SYMMETRY study: randomized, double-blind trial of elesclomol plus paclitaxel versus paclitaxel alone as treatment for chemotherapy-naive patients with advanced melanoma. J Clin Oncol. 2013;31:1211-8.
- Liu S, Zhao Y, Shen M, Hao Y, Wu X, Yao Y, et al. Hyaluronic acid targeted and pH-responsive multifunctional nanoparticles for chemo-photothermal synergistic therapy of atherosclerosis. J Mater Chem B. 2022;10:562-70.
AUTHOR CONTRIBUTIONS
FUNDING
COMPETING INTERESTS
ADDITIONAL INFORMATION
Guang’an men Hospital, China Academy of Chinese Medical Sciences, Beijing 100053, China. Xiyuan Hospital, China Academy of Chinese Medical Sciences, Beijing 100091, China. email: liulongtao1976@126.com; wumin19762000@126.com