نموذج أساسي لتشخيص الأمراض النسيجية الحاسوبية ذات الجودة السريرية واكتشاف السرطانات النادرة A foundation model for clinical-grade computational pathology and rare cancers detection
يهدف تحليل صور علم الأمراض باستخدام الذكاء الاصطناعي إلى تمكين أنظمة دعم القرار السريري والطب الدقيق. يعتمد نجاح مثل هذه التطبيقات على القدرة على نمذجة الأنماط المتنوعة التي لوحظت في صور علم الأمراض. لهذا الغرض، نقدم فيرتشو، أكبر نموذج أساسي لعلم الأمراض الحسابي حتى الآن. بالإضافة إلى تقييم توقعات العلامات الحيوية وتحديد الخلايا، نوضح أن نموذجًا أساسيًا كبيرًا يمكّن من اكتشاف السرطان الشامل، محققًا 0.95 منطقة تحت منحنى (مؤشر التشغيل المستلم) عبر تسعة أنواع شائعة وسبعة أنواع نادرة من السرطان. علاوة على ذلك، نوضح أنه مع بيانات تدريب أقل، يمكن لجهاز الكشف عن السرطان الشامل المبني على فيرتشو تحقيق أداء مشابه لنماذج الدرجة السريرية المحددة للأنسجة في الإنتاج وتفوق عليها في بعض المتغيرات النادرة من السرطان. تسلط مكاسب أداء فيرتشو الضوء على قيمة نموذج أساسي وتفتح إمكانيات للعديد من التطبيقات ذات التأثير العالي مع كميات محدودة من بيانات التدريب المعلّمة.
يعد التحليل المرضي للأنسجة أمرًا أساسيًا لتشخيص وعلاج السرطان. بشكل متزايد، يتم استبدال التحضيرات التقليدية النسيجية المستخدمة لفحص المجهر الضوئي بنظيراتها الرقمية، المعروفة أيضًا باسم صور الشرائح الكاملة (WSIs)، مما يمكّن من استخدام علم الأمراض الحسابيللانتقال من نقاط إثبات أكاديمية في المقام الأول إلى أدوات روتينية في الممارسة السريرية. يطبق علم الأمراض الحسابي الذكاء الاصطناعي (AI) على WSIs الرقمية لدعم التشخيص، والتوصيف وفهم المرض. ركزت الأعمال الأولية على أدوات دعم القرار السريري
لتحسين سير العمل الحالي, وفي عام 2021 تم إطلاق أول نظام علم أمراض معتمد من إدارة الغذاء والدواء للذكاء الاصطناعي. ومع ذلك، نظرًا للزيادة الهائلة في أداء رؤية الكمبيوتر، وهو فرع من الذكاء الاصطناعي يركز على الصور، تحاول الدراسات الأحدثفتح رؤى جديدة من WSIs الروتينية وكشف النتائج غير المكتشفة مثل التنبؤ والعلاج. إذا كانت ناجحة، ستعزز هذه الجهود من فائدة WSIs الملونة بصبغة الهيماتوكسيلين والإيوزين (H&E) وتقلل من الاعتماد على الاختبارات المناعية المتخصصة وغالبًا ما تكون مكلفة أو الاختبارات الجينومية.
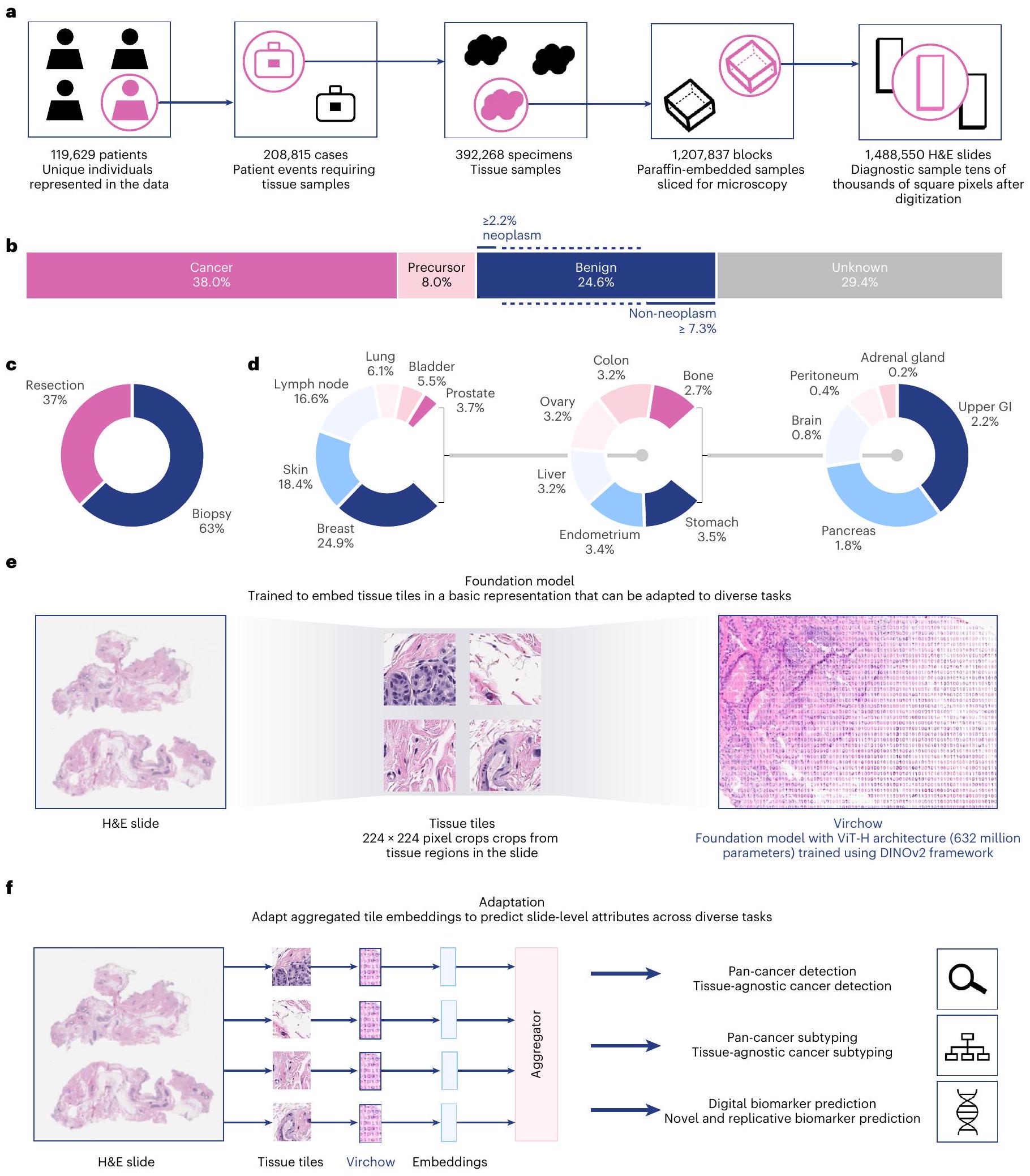
الشكل 1| نظرة عامة على الدراسة. مجموعة بيانات التدريب، خوارزمية التدريب وتطبيق فيرتشو، نموذج أساسي لعلم الأمراض الحسابي.
a، يمكن وصف بيانات التدريب من حيث المرضى، الحالات، العينات، الكتل أو الشرائح، كما هو موضح. ب-د، توزيع الشرائح كدالة لنوع السرطان
الحالة (ب)، الجراحة (ج) ونوع الأنسجة (د). هـ، يتطلب تدفق البيانات أثناء التدريب معالجة الشريحة إلى بلاطات، والتي يتم قصها بعد ذلك إلى وجهات نظر عالمية ومحلية., مخطط لتطبيقات النموذج الأساسي باستخدام نموذج مجمع للتنبؤ بالسمات على مستوى الشريحة. GI، الجهاز الهضمي.
عامل رئيسي في مكاسب أداء نماذج رؤية الكمبيوتر كان إنشاء شبكات عصبية عميقة على نطاق واسع، تُسمى نماذج أساسية. يتم تدريب النماذج الأساسية على مجموعات بيانات ضخمة،
أوامر من حيث الحجم أكبر من أي شيء تم استخدامه تاريخيًا لعلم الأمراض الحسابي-باستخدام عائلة من الخوارزميات، تُعرف بالتعلم الذاتي المراقب (على سبيل المثال، المراجع 22-26)، والتي لا تتطلب
تسميات مُنسقة. تولد النماذج الأساسية تمثيلات بيانات، تُسمى التضمينات، يمكن أن تعمم بشكل جيد على مهام تنبؤية متنوعة. وهذا يوفر ميزة واضحة على الطرق الحالية المحددة للتشخيص في علم الأمراض الحسابي، والتي، مقيدة بمجموعة فرعية من صور علم الأمراض، من غير المرجح أن تعكس الطيف الكامل من التباينات في مورفولوجيا الأنسجة والتحضيرات المخبرية اللازمة للتعميم الكافي في الممارسة. قيمة التعميم من مجموعات بيانات كبيرة أكبر حتى للتطبيقات التي تعاني من كميات غير كافية من البيانات لتطوير نماذج مخصصة، كما هو الحال في اكتشاف أنواع الأورام غير الشائعة أو النادرة، وكذلك للمهام التشخيصية الأقل شيوعًا مثل التنبؤ بالتغيرات الجينومية المحددة، والنتائج السريرية والاستجابة للعلاج. يجب أن يلتقط نموذج أساسي ناجح طيفًا واسعًا من الأنماط، بما في ذلك مورفولوجيا الخلايا، بنية الأنسجة، خصائص الصبغ، مورفولوجيا النواة، الأشكال الانقسامية، النخر، الاستجابة الالتهابية، تكوين الأوعية الدموية وتعبير العلامات الحيوية وبالتالي سيكون مناسبًا جيدًا للتنبؤ بمجموعة متنوعة من خصائص WSIs. إذا تم تدريبه بكمية كافية من WSIs الرقمية في مجال علم الأمراض، يمكن أن يشكل مثل هذا النموذج أساسًا للتنبؤ السريري القوي لكل من السرطانات الشائعة والنادرة، فضلاً عن المهام الحيوية الأخرى مثل تصنيف السرطان، وكمية العلامات الحيوية، وعدّ الحالات الخلوية والأحداث والتنبؤ بالاستجابة للعلاج.
يعتمد أداء النموذج الأساسي بشكل حاسم على حجم مجموعة البيانات وحجم النموذج، كما يتضح من نتائج قانون التوسع. تستخدم النماذج الأساسية الحديثة في مجال الصور الطبيعية ملايين الصور (على سبيل المثال، ImageNet, JFT-300M و LVD-142M) لتدريب نماذج تحتوي على مئات الملايين إلى مليارات المعلمات (على سبيل المثال، محولات الرؤية (ViTs)). على الرغم من التحديات في جمع مجموعات بيانات على نطاق واسع في مجال علم الأمراض، فقد استخدمت الأعمال الرائدة الأخيرة مجموعات بيانات تتراوح من 30,000 إلى 400,000 WSIs لتدريب نماذج أساسية تتراوح في الحجم من 28 مليون إلى 307 مليون معلمة (انظر الملاحظة التكميلية 1 للحصول على ملخص مفصل للنماذج الحديثة). تُظهر هذه الأعمال أن ميزات الصورة الناتجة عن التعلم الذاتي المراقب لصور علم الأمراض تتفوق على ميزات الصورة المدربة على الصور الطبيعية وأن الأداء يتحسن مع الحجم.
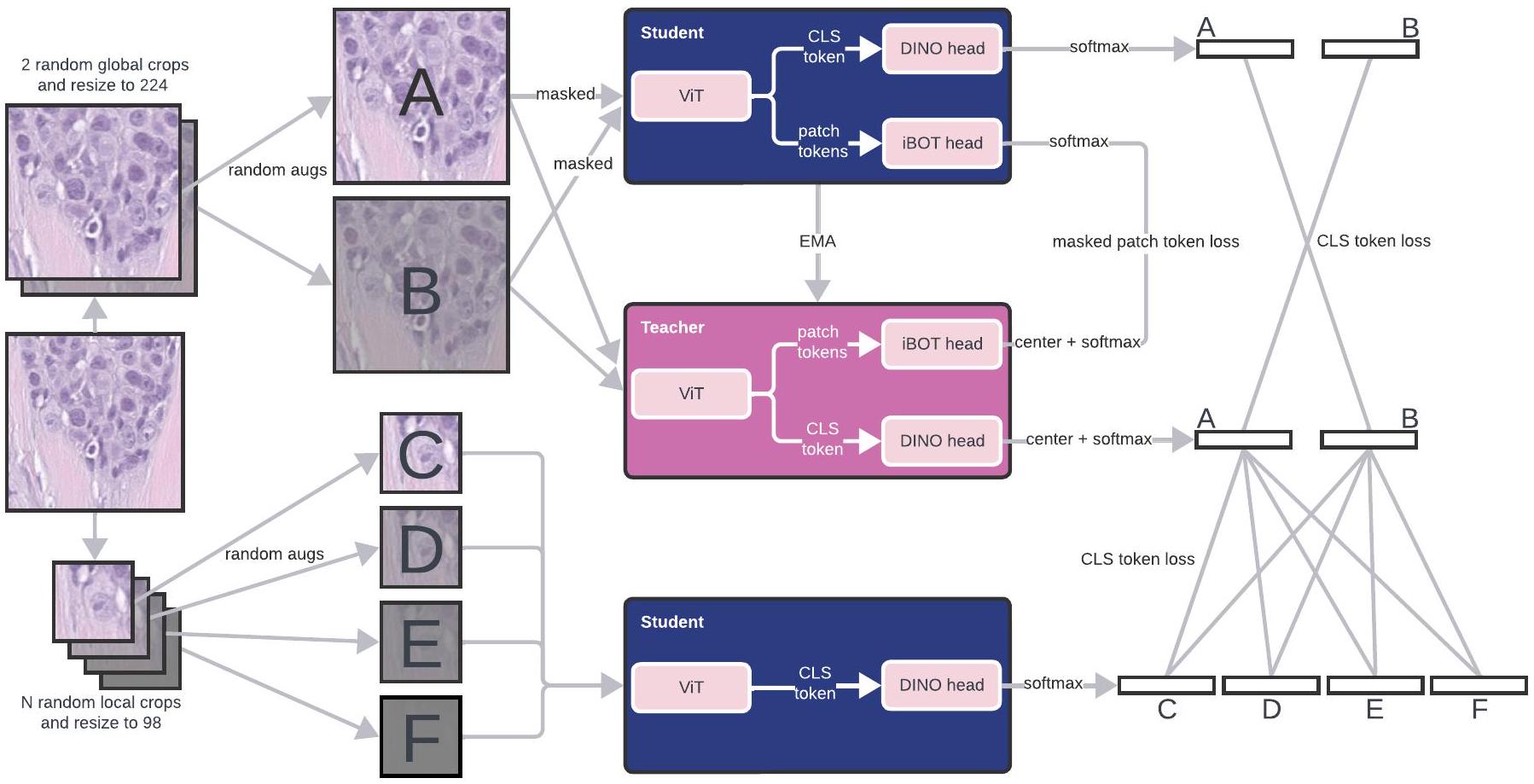
هنا، نقدم نموذجًا أساسيًا لعلم الأمراض على نطاق مليون صورة، فيرتشو، الذي سمي تكريمًا لرودولف فيرتشو، الذي يُعتبر والد علم الأمراض الحديث واقترح أول نظرية لعلم الأمراض الخلوي. يتم تدريب فيرتشو على بيانات من حوالي 100,000 مريض تتوافق مع حوالي 1.5 مليون WSIs ملونة بصبغة H&E تم الحصول عليها من مركز ميموريال سلوان كيترينغ للسرطان (MSKCC)، وهو أكثر من WSIs في مجموعات بيانات التدريب السابقة في علم الأمراض (مفصل في الشكل 1a و ‘مجموعة بيانات التدريب على نطاق المليون’ في الطرق). تتكون بيانات التدريب من أنسجة سرطانية وغير سرطانية، تم جمعها عبر الخزعة (63%) والاستئصال (37%)، من 17 نسيجًا عالي المستوى (الشكل 1ب-د). يتم تدريب فيرتشو، وهو نموذج ViT يحتوي على 632 مليون معلمة، باستخدام خوارزمية DINO v. 2, وهي خوارزمية ذاتية الإشراف للطالب والمعلم متعددة الرؤى (الشكل 1هـ؛ انظر ‘بنية فيرتشو و
التدريب’ في الطرق للحصول على تفاصيل التدريب). تستفيد DINO v. 2 من المناطق العالمية والمحلية لبلاطات الأنسجة لتعلم إنتاج تضمينات لبلاطات WSIs (الشكل 1هـ)، والتي يمكن تجميعها عبر الشرائح واستخدامها لتدريب مجموعة متنوعة من المهام التنبؤية اللاحقة (الشكل 1و).
مدفوعين بتسليط الضوء على التأثير السريري المحتمل لنموذج أساس علم الأمراض، نقوم بتقييم أداء نموذج شامل للسرطان تم تدريبه باستخدام تمثيلات فيرشو لتوقع السرطان على مستوى العينة عبر أنسجة مختلفة. تتفوق تمثيلات فيرشو على جميع نماذج الأساس أو تتطابق معها في جميع أنواع السرطان المختبرة، بما في ذلك السرطانات النادرة والبيانات خارج التوزيع. تُظهر المقارنة الكمية مع ثلاثة منتجات ذكاء اصطناعي متخصصة من الدرجة السريرية أن النموذج الشامل للسرطان يعمل تقريبًا بنفس كفاءة المنتجات السريرية بشكل عام ويتفوق عليها في بعض المتغيرات النادرة من السرطانات. لتقديم دليل على المجالات المحتملة للتركيز في التقدمات المستقبلية في علم الأمراض الحسابي، يتم أيضًا إجراء تحليل نوعي، يصف أنماط الأخطاء حيث يفشل نموذج الذكاء الاصطناعي في تحديد أو تحديد خلايا سرطانية بشكل خاطئ. مدفوعين بتبسيط سير العمل السريري، قمنا بتقييم استخدام تمثيلات فيرشو لتدريب توقع العلامات الحيوية، متفوقين عمومًا على النماذج الأخرى. بشكل عام، توفر نتائجنا دليلًا على أن النماذج الأساسية على نطاق واسع يمكن أن تكون أساسًا لنتائج قوية في حدود جديدة من علم الأمراض الحسابي.
النتائج
تم تقييم تمثيلات نموذج فيرشو على فئتين من تطبيقات علم الأمراض الحاسوبية على مستوى الشرائح: الكشف عن السرطان الشامل (‘يمكن فيرشو من الكشف عن السرطان الشامل’ و ‘نحو أداء بمستوى سريري’) وتوقع العلامات الحيوية (‘الكشف عن العلامات الحيوية في التصوير الروتيني يلغي الحاجة للاختبارات الإضافية’). تتطلب هذه المهام تدريب نموذج مجمع تحت إشراف ضعيف لتجميع تمثيلات البلاط إلى توقعات على مستوى الشريحة. كما تم إجراء سلسلة من المعايير الخطية على مستوى البلاط لتقييم التمثيلات مباشرة على البلاطات النسيجية الفردية (‘تظهر المعايير على مستوى البلاط والتحليل النوعي القابلية للتعميم’).
فيرشو يتيح الكشف عن جميع أنواع السرطان
كان أحد الأهداف الرئيسية لعملنا هو تطوير نموذج واحد للكشف عن السرطان، بما في ذلك السرطانات النادرة (المعرفة من قبل المعهد الوطني للسرطان (NCI) على أنها سرطانات ذات معدل حدوث سنوي في الولايات المتحدة يقل عن 15 شخصًا لكل 100,000 (المرجع 46))، عبر أنسجة مختلفة. يستنتج نموذج الكشف عن السرطان الشامل وجود السرطان باستخدام تمثيلات فيرشو كمدخلات. للتقييم، يتم استخدام شرائح من MSKCC وشرائح مقدمة للاستشارة إلى MSKCC من العديد من المواقع الخارجية عالميًا. يتم الإبلاغ عن الأداء المصنف عبر تسعة أنواع شائعة وسبعة أنواع نادرة من السرطان. التمثيلات التي تم إنشاؤها بواسطة فيرشو، UNIفيكونو CTransPathيتم تقييمها. يتم تدريب مجمعات السرطان الشاملة باستخدام تسميات على مستوى العينة، مع الحفاظ على نفس بروتوكول التدريب لجميع التضمينات (انظر ‘كشف السرطان الشامل’ في الطرق للحصول على تفاصيل البيانات والتدريب).
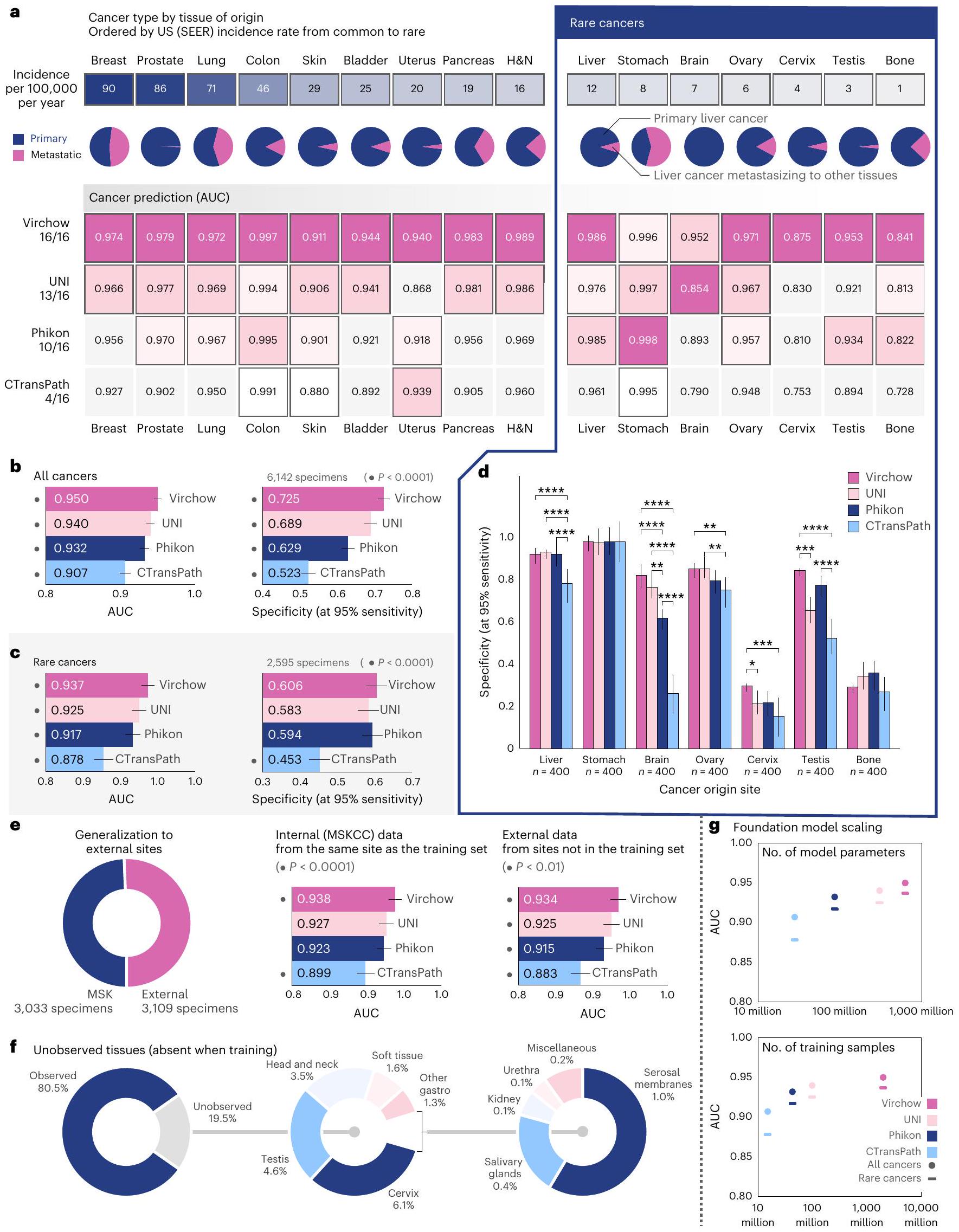
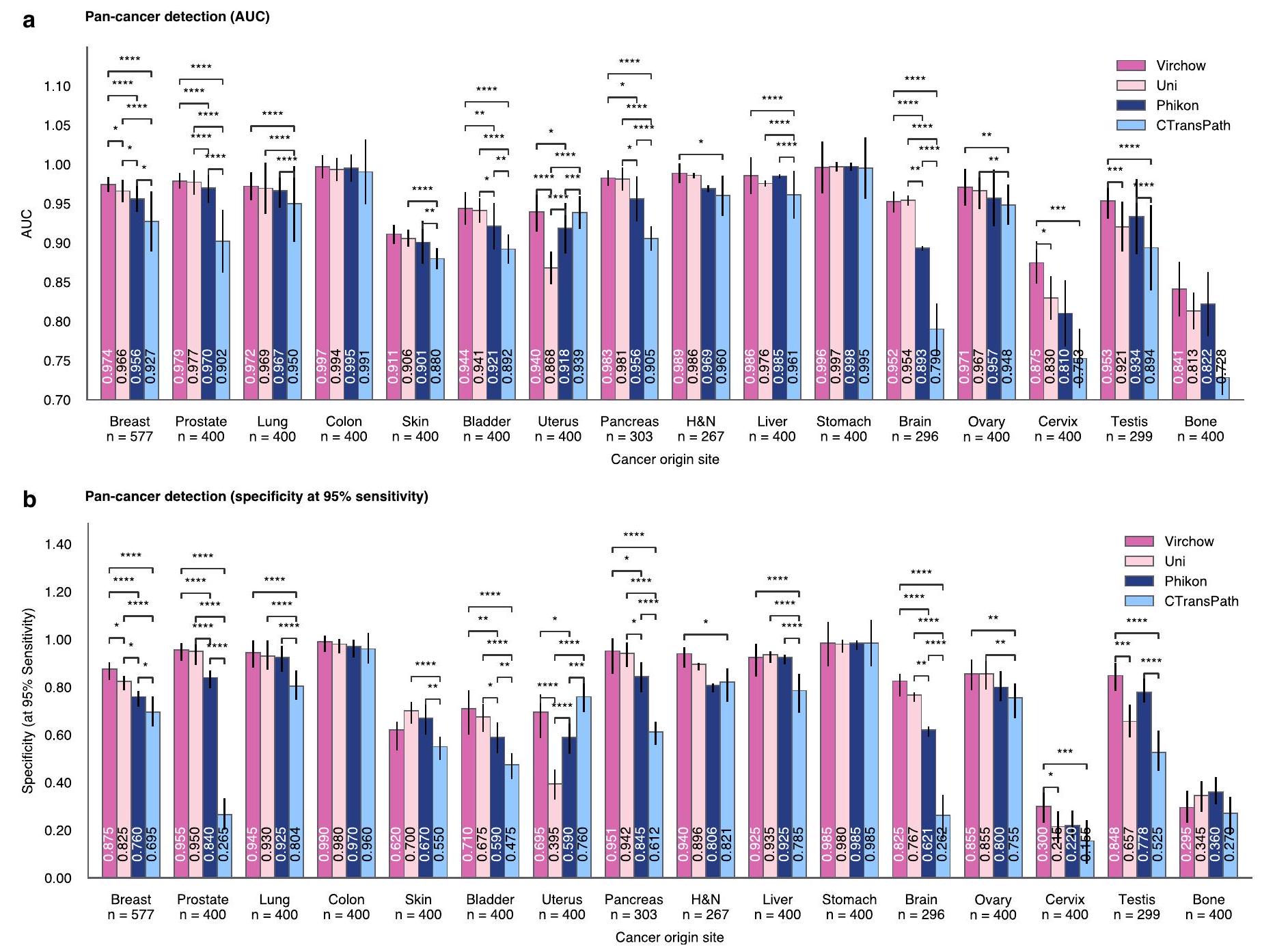
الشكل 2 | فيرشو يمكّن تدريب كاشف شامل للسرطان. نتائج الكشف عن السرطان الشامل. يتم التنبؤ بالكشف على مستوى العينة باستخدام شبكة تجميع تم تدريبها مع فيرشو، UNI، Phikon أو CTransPath كمدخلات. أ، أداء الكشف عن السرطان (AUC) مقسم حسب نوع السرطان كما تحدده الأنسجة الأصلية. يتم عرض معدل الحدوث ونسبة الانبثاث لكل نوع من أنواع السرطان. تمكّن تجسيدات فيرشو أفضل أداء للكشف عن السرطان عبر جميع أنواع السرطان، بما في ذلك السرطانات النادرة. لكل نوع من أنواع السرطان، يتم عرض AUC المقابل بشكل إحصائي ملحوظ.تُبرز التضمينات ذات الأداء العالي باللون الأرجواني. عندما يكون هناك أكثر من AUC واحد ليس رماديًا، فإن الأداء يُعتبر ‘مربوطًا’ (لا يوجد فرق ذو دلالة إحصائية). النموذج الأساسي المستخدم لإنتاج التضمينات للموحد موضح في الهامش على اليسار، مع عدد أنواع السرطان التي حقق فيها الموحد المقابل (أو تعادل مع) أعلى AUC. جميع الدلالات الإحصائية (أ-هـ) تُحسب باستخدام اختبار دي لون الثنائي لـ AUC واختبار كوكرا.اختبار متبوع باختبار مك نيمار للخصوصية، كلاهما مصحح لـ مقارنات متعددة باستخدام طريقة هولم. ب، ج، أداء الكشف عن السرطان ملخص لجميع أنواع السرطان (ب) وللسرطانات النادرة (ج). تظهر أشرطة الخطأ (ب-هـ) فترة الثقة الثنائية الجانبين 95% المحسوبة باستخدام طريقة دي لونغ لـ AUC وطريقة ويلسون للخصوصية؛ – تشير إلى الفروق التي تعتبر ذات دلالة إحصائية مقارنة بالبقية. ). د، الحساسية عند الخصوصية لاكتشاف السرطان النادر ). e، الكشف عن السرطان القائم على فيرشو يتعمم بشكل جيد على البيانات من المؤسسات الخارجية التي لم تكن ممثلة في مجموعة التدريب؛ تم تدريب جميع المجمعات وفيرشو فقط على بيانات من MSKCC. نصف العينات فقط في مجموعة اختبار السرطان الشامل من MSKCC. ف، تحتوي خمس العينات المستخدمة لتقييم نموذج السرطان الشامل على أنسجة لم يتم ملاحظتها في مجموعات تدريب فيرشو أو المجمعات الشاملة للسرطان. ج، أداء الكشف عن السرطان يتناسب مع حجم النموذج الأساسي وعدد عينات التدريب (البلاط) المستخدمة لتدريبه. H&N، الرأس والعنق.
أنتجت تمثيلات فيرشو أفضل أداء في اكتشاف السرطان على جميع أنواع السرطان (الشكل 2أ). حقق اكتشاف السرطان الشامل باستخدام تمثيلات UNI أداءً مشابهًا إحصائيًا ( ) لثمانية من تسعة أنواع شائعة من السرطان وخمسة من سبعة أنواع نادرة من السرطان؛ ومع ذلك، في جميع الحالات باستثناء حالة واحدة، كانت المنطقة المحددة تحت منحنى (مؤشر التشغيل المستلم) (AUC) أقل. بشكل عام، حقق نموذج بان-السرطان AUC قدره 0.950 مع تضمينات فيرشو، و0.940 مع تضمينات UNI، و0.932 مع تضمينات فيكون، و0.907 مع تضمينات CTransPath (الشكل 2b؛ جميعها تختلف بشكل ملحوظ مع انظر الشكل 3 من البيانات الموسعة لمزيد من التفاصيل حول مقاييس AUC والخصوصية، مصنفة حسب نوع السرطان.
أداء الكشف عن السرطان النادر ملحوظ بشكل خاص. بالمقارنة مع AUC المذكور سابقًا والذي بلغ 0.950 بشكل عام، حققت تمثيلات فيرشو AUC قدره 0.937 في حالات السرطان النادر (الشكل 2c)، مما يدل على القدرة على التعميم على البيانات النادرة. ومع ذلك، كان الأداء عبر أنواع السرطان النادر الفردية غير متساوٍ، حيث كان الكشف عن سرطانات عنق الرحم والعظام أكثر تحديًا (AUC < 0.9) بغض النظر عن التمثيلات المستخدمة (الشكل 2a,d). حسنت تمثيلات فيرشو الكشف عن عنق الرحم إلى AUC قدره 0.875 مقارنةً بـأو 0.753 عند استخدام تمثيلات UNI أو Phikon أو CTransPath، على التوالي. وبالمثل، حققت تمثيلات Virchow قيمة AUC تبلغ 0.841 لاكتشاف سرطان العظام، مقارنةً بـ و 0.728 مع UNI و Phikon و CTransPath، على التوالي. عند الحساسية، نوضح أن نموذج الكشف عن السرطان الشامل باستخدام تمثيلات فيرشو يمكن أن يحققالخصوصية، مقارنة بـ62.9% أو 52.3% باستخدام UNI أو Phikon أو CTransPath، على التوالي، تم تدريبها على بيانات أقل (الشكل 2ب).
تم تقييم قوة تمثيلات فيرشو للبيانات المأخوذة من مجموعة سكانية مختلفة عن مجموعة التدريب (بيانات OOD) مباشرة باستخدام بيانات من مؤسسات غير MSKCC (حيث تم تدريب كل من فيرشو ومجمع السرطان الشامل فقط على بيانات من MSKCC) وغير مباشرة من خلال تضمين بيانات من أنسجة لم يتم ملاحظتها أثناء التدريب (الشكل 2e، f). نظرًا لأن مقاييس AUC لا يمكن مقارنتها بدقة عبر مجموعات بيانات مختلفة (بسبب اختلاف نسب العينات الإيجابية إلى السلبية)، فإننا نبلغ عن AUC لجميع نماذج السرطان الشامل على جميع البيانات أو السرطانات النادرة (الشكل 2b)، وكذلك على البيانات الداخلية أو الخارجية (الشكل 2e)، ونظهر أن الفروق في AUC عبر النماذج تظل متسقة في كل مجموعة فرعية. وهذا يدل على أن تمثيلات فيرشو تعمم بشكل جيد على بيانات جديدة أو نادرة وتتفوق على الآخرين باستمرار. على الرغم من أنه لا يمكن مقارنة AUC بدقة عبر مجموعات البيانات، يمكننا ملاحظة أن جميع النماذج تحقق AUC مشابهًا على كل من البيانات الداخلية والخارجية، مما يشير إلى أنها تعمم بشكل جيد حيث يمكن أن تكون البيانات الخارجية تحديًا لأنها تُقدم إلى MSKCC للاستشارة. علاوة على ذلك، فإن عنق الرحم، والخصية، والرأس والعنق (H&N) هي أنسجة لم تُرَ أثناء التدريب، ولا تزال تمثيلات فيرشو تتفوق على النماذج المنافسة. بشكل عام، فإن اكتشاف السرطان الشامل يعمم عبر أنواع السرطان، بما في ذلك السرطانات النادرة، وكذلك على بيانات OOD عند استخدام تمثيلات نموذج الأساس.
يكشف مقارنة أداء السرطان الشامل بناءً على تضمينات نماذج الأساس المختلفة أن الأداء يتناسب مع حجم نموذج الأساس وحجم بيانات التدريب (الشكل 2g). وُجد أن اكتشاف السرطان يتناسب تقريبًا لوغاريتميًا مع عدد معلمات النموذج (الشكل 2g، الأعلى)؛ على الرغم من أن الأداء يتناسب مع عدد عينات بلاط التدريب، إلا أن الاتجاه (الشكل 2g، الأسفل) يشير إلى عوائد متناقصة. على الرغم من أن مجموعات بيانات التدريب، وهياكل النماذج، واستراتيجيات التحسين تختلف عبر فيرشو، يوني، فيكون، وCTransPath، إلا أن هناك تشابهات كافية
الشكل 3 | approaches اكتشاف السرطان الشامل وأحيانًا يتجاوز أداء المنتجات السريرية، باستخدام بيانات أقل. أ،ب، الأداء كما تم قياسه بواسطة AUC لثلاثة منتجات سريرية مقارنة بنموذج السرطان الشامل المدرب على تضمينات فيرشو، على المتغير النادر (أ) ومجموعات بيانات اختبار المنتج (ب). يحقق كاشف السرطان الشامل، المدرب على تضمينات نموذج الأساس فيرشو، أداءً مشابهًا للمنتجات ذات الجودة السريرية بشكل عام ويتفوق عليها في المتغيرات النادرة للسرطانات. ج، تم تدريب كاشف السرطان الشامل على عدد أقل من العينات المعلّمة مقارنة بنماذج البروستاتا والثدي وBLN السريرية، لتحفيز تحليل التوسع. جميع النماذج تعتمد على المحولات: يستخدم CTransPath محول Swin، والباقي يستخدم ViTsبأحجام مختلفة. تم تدريب فيكون باستخدام خوارزمية iBOT، وتم تدريب كل من فيرشو ويوني باستخدام خوارزمية DINO v. 2مع معلمات مشابهة. iBOT وDINO v. 2 هما نهجان مرتبطان حيث أن الأخير يبني على اقتراح نمذجة الصورة المقنعة للأول. يتميز CTransPath من حيث خوارزمية التدريب حيث استخدم خوارزمية تعلم تبايني تعتمد على MoCov3 (المرجع 48). لمعرفة تأثير حجم مجموعة البيانات بشكل مستقل عن حجم النموذج، نوجه القارئ إلى الدراسة في المرجع 41.
نحو أداء ذو جودة سريرية
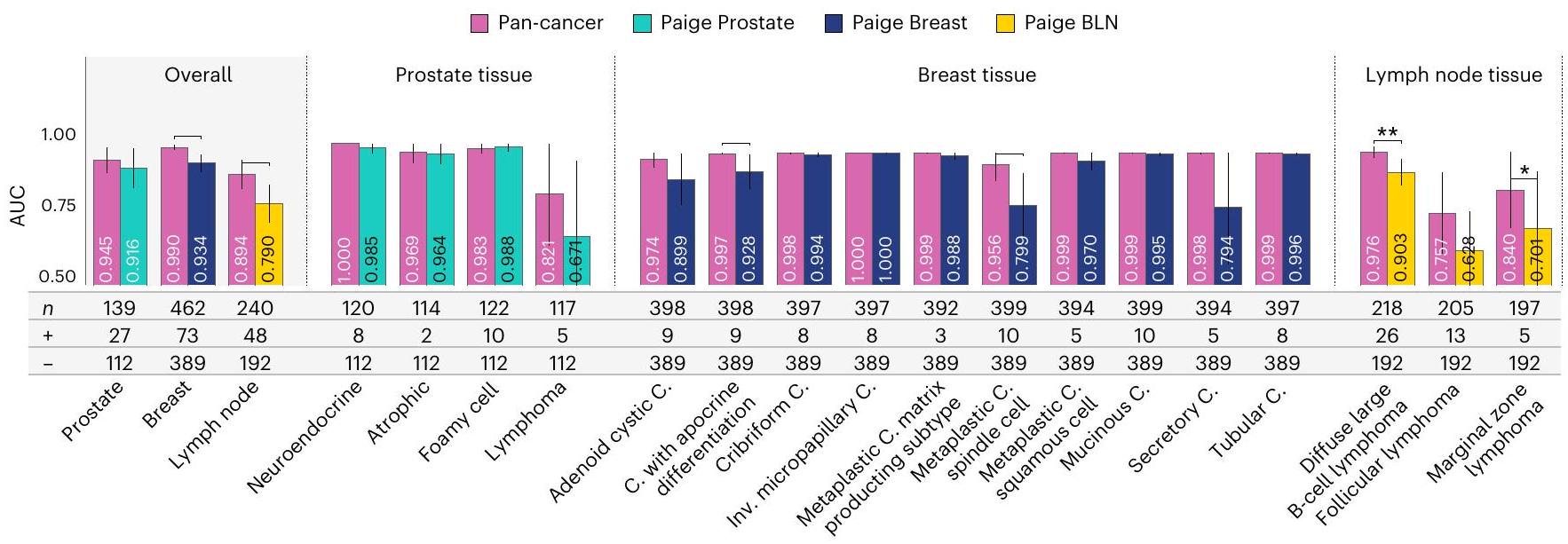
وعد نماذج الأساس هو تحسين التعميم؛ ومع ذلك، فإن هذا الادعاء يصعب التحقق منه دون الوصول إلى نماذج متخصصة مدربة ومختبرة بدقة. لهذا الغرض، أجرينا تحليلًا مقارنًا بين نموذج اكتشاف السرطان الشامل القائم على فيرشو ونماذج تجارية متخصصة، تحديدًا Paige Prostate وPaige Breast وPaige Breast Lymph Node (BLN). يركز المقارنة على AUC لاكتشاف السرطان، تحديدًا لسرطان البروستاتا، وسرطان الثدي الغازي، وانتقالات سرطان الثدي في العقد اللمفاوية. تم تدريب هذه النماذج التجارية باستخدام تعلم ضعيف الإشراف متعدد الحالات كما هو موصوف في المراجع 14،49 تحديدًا لاكتشاف السرطان. تم إجراء التقييم في إعدادين: (1) مجموعات بيانات اختبار المنتج و(2) مجموعات بيانات المتغيرات النادرة للسرطان في الأنسجة المعنية (الشكل 3ب-د).
يؤدي نموذج اكتشاف السرطان الشامل القائم على فيرشو، المدرب على السرطانات عبر العديد من الأنسجة، تقريبًا بنفس جودة نماذج المتخصصين السريرية للبروستاتا والثدي وBLN (الشكل 3ج) بينما يتفوق عليها في العديد من المتغيرات النادرة للسرطانات (الشكل 3د). من المهم ملاحظة أن مجموعة تدريب السرطان الشامل لم تستفد من نفس التحسين مثل مجموعات تدريب المنتجات، مثل تعزيز الفئات الفرعية ومراقبة جودة العلامات. علاوة على ذلك، تم تدريب نموذج السرطان الشامل على عدد أقل من العينات المحددة للأنسجة مقارنة بالنماذج السريرية (الشكل 3 والشكل الممتد 4). بشكل ملموس، تم تدريب Paige Prostate على 66,713 كتلة، وتم تدريب Paige Breast على 44,588 عينة، وBLN على 8,150 عينة، بينما تم تدريب السرطان الشامل (باستخدام تضمينات فيرشو) على 35,387 مجموعة من الشرائح (كتل أو عينات) في المجموع، منها 2,829 هي بروستاتا، 1,626 هي ثدي و1,441 هي عقدة لمفاوية. يحقق نموذج السرطان الشامل AUC قدرهو0.971 على البروستاتا والثدي وBLN، على التوالي. يقترب هذا الأداء من أداء النماذج التجارية؛ ومع ذلك، لا يزال يتفوق عليه نموذج Paige Prostate المعتمد من إدارة الغذاء والدواء (0.980 مقابل 0.995 AUC،) ونموذج Paige Breast (0.985 مقابل. من ناحية أخرى، فإنه أفضل إحصائيًا بشكل ملحوظ في اكتشاف النقائل الكبيرة مقارنة بنموذج Paige BLN (0.999 مقابل 0.994 AUC،). علاوة على ذلك، لا يوجد فرق إحصائي ملحوظ () في المقارنات الأخرى لـ BLN أو بعض المقارنات المصنفة لسرطان الثدي (الشكل 3ج).
بالإضافة إلى الاقتراب من نماذج المتخصصين من حيث AUC العامة، يتطابق نموذج السرطان الشامل أو يتفوق على هذه النماذج في المتغيرات النادرة للسرطانات، كما هو موضح في الشكل 3د. في أنسجة البروستاتا والعقد اللمفاوية، يكون نموذج السرطان الشامل قادرًا على اكتشاف اللمفوما. هذا ملحوظ بشكل خاص لأن أيًا من النماذج لم يتم تدريبها في الأورام الخبيثة اللمفاوية. نظرًا لخطها النسب المختلف (تنشأ السرطانات من خلايا الظهارة، بينما تنشأ اللمفوما من الأنسجة اللمفاوية) فإن مظهرها المورفولوجي يميل إلى أن يكون مختلفًا تمامًا.
في اثنين من أربعة متغيرات اللمفوما، يتفوق نموذج السرطان الشامل على النموذج المتخصص. إن تحسين اكتشاف اللمفوما الكبيرة المنتشرة ملحوظ حيث أن هذا المتغير عدواني بشكل خاص. في أنسجة الثدي، يتفوق نموذج السرطان الشامل على
نموذج Paige Breast بشكل عام وخاصة في بعض المتغيرات النادرة الهيستولوجية، بما في ذلك سرطان الغدة اللعابية الكيسي، وسرطان الغدة مع تمايز أبوقريني ()، وسرطان الغدة المتحولة الخلوية ()، وسرطان الغدة المتحولة الخلوية الحرشفية والخاصة جدًا
توقع المتغيرات النادرة: يتفوق نموذج السرطان الشامل على المنتجات ذات الجودة السريرية في بعض المتغيرات النادرة لسرطانات البروستاتا والثدي والعقد اللمفاوية
أ
ب
ج
أحجام مجموعة التدريب: العينات
د
سرطان غير متمايز بشكل سيء في البنكرياس
تركيز محطم مع سرطان الغدة في الصفاق
تغيرات ليفية التهابية تفاعلية في موقع الخزعة السابقة
خلايا لمفاوية محطمة وطية نسيجية
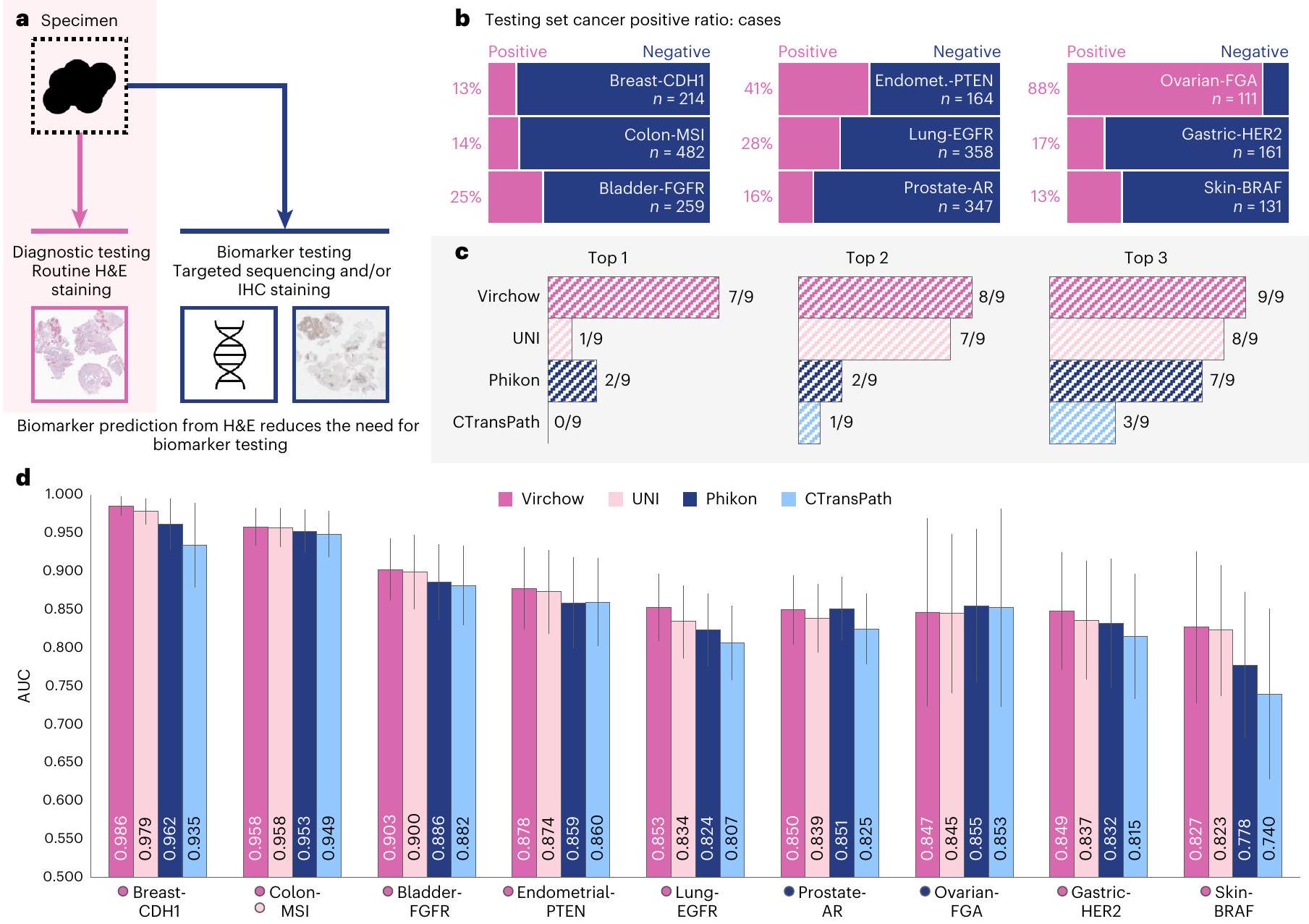
الشكل 4 | نتائج توقع العلامات الحيوية. أ، تساعد تضمينات فيرشو في توقع العلامات الحيوية مباشرة منالشرائح، مما يقلل من الحاجة إلى التسلسل المستهدف أو صبغ IHC. ب، نسبة الحالات الإيجابية في كل مجموعة بيانات اختبار العلامات الحيوية. ج، عدد العلامات الحيوية التي تم استخدام تضمينات فيرشو، يوني، فيكون أو CTransPath لتدريب مجمع أنتج AUC في الأعلى. لا تأخذ هذه الترتيبات في الاعتبار الأهمية الإحصائية عبر النماذج لكل علامة حيوية بسبب القوة الإحصائية المنخفضة؛ بدلاً من ذلك، تعتمد على اعتبار
سرطان إفرازي. نلاحظ أنه بسبب ندرة هذه المتغيرات من السرطانات، فإن توقع المتغيرات النادرة يفتقر إلى القوة الإحصائية لمجموعات بيانات المنتجات.
لفهم أنماط الخطأ لنموذج السرطان الشامل عبر الأنسجة المختلفة، قام طبيب الأمراض بفحص حالات الخطأ ضمن مجموعة مختارة من تقييم WSIs (انظر ‘معيار المنتج الشامل’ في قسم ‘مجموعات بيانات التقييم السريري’ في الطرق). تم اختيار نقطة التشغيل لكل نسيج لتحقيق تقريبًاحساسية وخصوصية على مجموعة بيانات ضبط. تم توثيق هذه الأنماط الخطأ باستخدام نص حر أولاً، والذي تم تصنيفه لاحقًا لتوفير ملخص شامل. نفترض أن هذه الأنماط قد تكون مفيدة لدراسات اكتشاف السرطان المماثلة، مما يوفر رؤى قيمة لتحسين نماذج الأساس المستقبلية وتطبيقات الذكاء الاصطناعي السريرية. تم تحليل أنماط الإيجابيات الكاذبة والسلبيات الكاذبة بشكل منفصل، كما هو موضح في الشكل 3e.
عند تحليل حالات الإيجابيات الكاذبة والسلبيات الكاذبة، تم التوصل إلى أن نسبة كبيرة يمكن أن تُعزى إلى نتائج محددة. شكلت التحضيرات الهيستولوجية التي تحتوي على بؤر سرطانية صغيرة فقط الغالبية () من السلبيات الكاذبة. كانت بعض الأورام، التي لم يتم اكتشافها كسرطان (11.9%)، ذات إمكانات خبيثة حدودية، مثل الأورام السليلة المعوية أو الأورام الحدودية السائلة للأورام. كانت أخرى (9.5%)، مثل الورم الدبقي منخفض الدرجة، تظهر فقط ميزات هيستولوجية دقيقة جدًا من الخباثة. كانت آثار العلاج، والتموت الواسع، وعيوب الأنسجة التي تحجب السرطان مسؤولة عن بعض السلبيات الكاذبة. في 11 حالة (26.2%)، كان هناك أكثر من سرطان بسيط داخل العينة، ولم يكن بالإمكان تفسير النتيجة السلبية للنموذج.
شكلت الغالبية العظمى من حالات الإيجابيات الكاذبة فئتين. كانت الآفات السابقة في العينات التي تفتقر إلى السرطان الغازي تشكل معظم (53.2%) من الإيجابيات الكاذبة. وقد وُجدت هذه الحالات بشكل متكرر في المثانة والثدي وعنق الرحم والجلد (خلل التنسج الحرشفي) والمريء. أظهرت معظم الآفات السابقة المكتشفة خلل تنسج عالي الدرجة، مع ميزات خلوية تشبه تلك الخاصة بالسرطان الغازي، على الرغم من أنه تم اكتشاف بعض بؤر الخلل التنموي منخفض الدرجة أيضًا في تقاطع المريء والمعدة والجلد. كانت ثاني أكثر الأسباب شيوعًا ( ) للإيجابيات الكاذبة هي عيوب الأنسجة، وخاصة عيوب الضغط (حيث يتم سحق الخلايا غير الورمية جسديًا أثناء إعداد العينة، مما يؤدي إلى تأثير تدفق مميز للنوى)، وطيات الأنسجة والمناطق غير الواضحة. كانت التغيرات التفاعلية داخل السدى أو المكونات اللمفاوية، التي تشكل ، وفي الأنسجة الظهارية غير الورمية، تمثل ، مسؤولة أيضًا عن النتائج الإيجابية الكاذبة. عدد من هذه النتائج، مثل تغييرات موقع الخزعة،
خلل التنسج الظهاري التفاعلي، ضمور الغدد والمخاط السدي الخالي من الخلايا، هي مقلدات خبيثة معروفة تتحدى علماء الأمراض أيضًا. كانت هناك ثلاث حالات (3.2%) أورام حميدة تم التعرف عليها بشكل خاطئ على أنها سرطان. وشملت هذه الأورام الأورام السدوية المعوية الحميدة، والأورام الوعائية العضلية الكبدية، والأكياس الكيسية المصلية للبنكرياس.
تجنب الكشف عن العلامات الحيوية في التصوير الروتيني الاختبارات الإضافية
يمكن أن يقلل التنبؤ بالعلامات الحيوية من الصور الملونة القياسية H&E من الاعتماد على الاختبارات باستخدام طرق إضافية والتأخيرات الكبيرة المرتبطة بإعادة النتائج للمرضى (انظر الشكل 4أ). يتم التنبؤ بحالة علامة حيوية في عينة باستخدام شبكة تجميع مع تضمينات نموذج الأساس كمدخلات. تلعب هذه العلامات الحيوية دورًا حاسمًا في تشخيص وعلاج أنواع مختلفة من السرطانات، ويتم وصف كل منها بمزيد من التفاصيل في ‘الكشف عن العلامات الحيوية’ في الطرق (انظر أيضًا الجدول التكميلي 3.1 والشكل 4ب). تتكون مجموعات بيانات الكشف عن العلامات الحيوية من WSIs من الأقسام النسيجية المطابقة للكتل المستخدمة لاستخراج الحمض النووي وتحليل الطفرات المتكاملة MSK للأهداف القابلة للتنفيذ (تسلسل MSK-IMPACT) ، حيث تم تحليل الأخير لتحديد حالة التغيرات الجينية وتأسيس علامة ثنائية تشير إلى وجود أو عدم وجود المتغيرات: أي، العلامة الحيوية (انظر الشكل 4أ). مشابهًا لتقييم السرطان الشامل، يتم استخدام النماذج المتاحة للجمهور UNI ، Phikon وCTransPath كنماذج أساسية للمقارنات.
نلاحظ أن نتائج التنبؤ بالعلامات الحيوية تفتقر إلى القوة الإحصائية الكافية لتقييم الفروق الإحصائية الهامة عبر النماذج؛ بدلاً من ذلك، نستنتج أداء النموذج النسبي من تقييم العديد من التنبؤات المختلفة للعلامات الحيوية. في تحليلنا المقارن الموضح في الشكل 4ج، أظهرت تضمينات فيرشو أداءً متفوقًا في سبعة من تسعة علامات حيوية رقمية تم تقييمها، محققة درجات AUC التي تجاوزت تلك الخاصة بأقرب نماذج أساسية. يبرز هذا الأداء قوة تضمينات فيرشو عبر مجموعة متنوعة من العلامات الحيوية. حتى في فئات مستقبلات الأندروجين البروستاتية (AR) ونسبة الجينوم المعدل (FGA) المبيض، حيث لم تحقق فيرشو المركز الأول، ظلت منافسًا قويًا، مع درجات AUC تبلغ 0.849 و0.847 على التوالي. تؤكد هذه النتائج على إمكانية تضمينات فيرشو في تمثيل الأنماط النسيجية H&E بدقة، مما يوفر رؤى تنبؤية حول العلامات الحيوية التي يتم التعرف عليها تقليديًا من خلال استخراج الحمض النووي وتسلسل MSK-IMPACT.
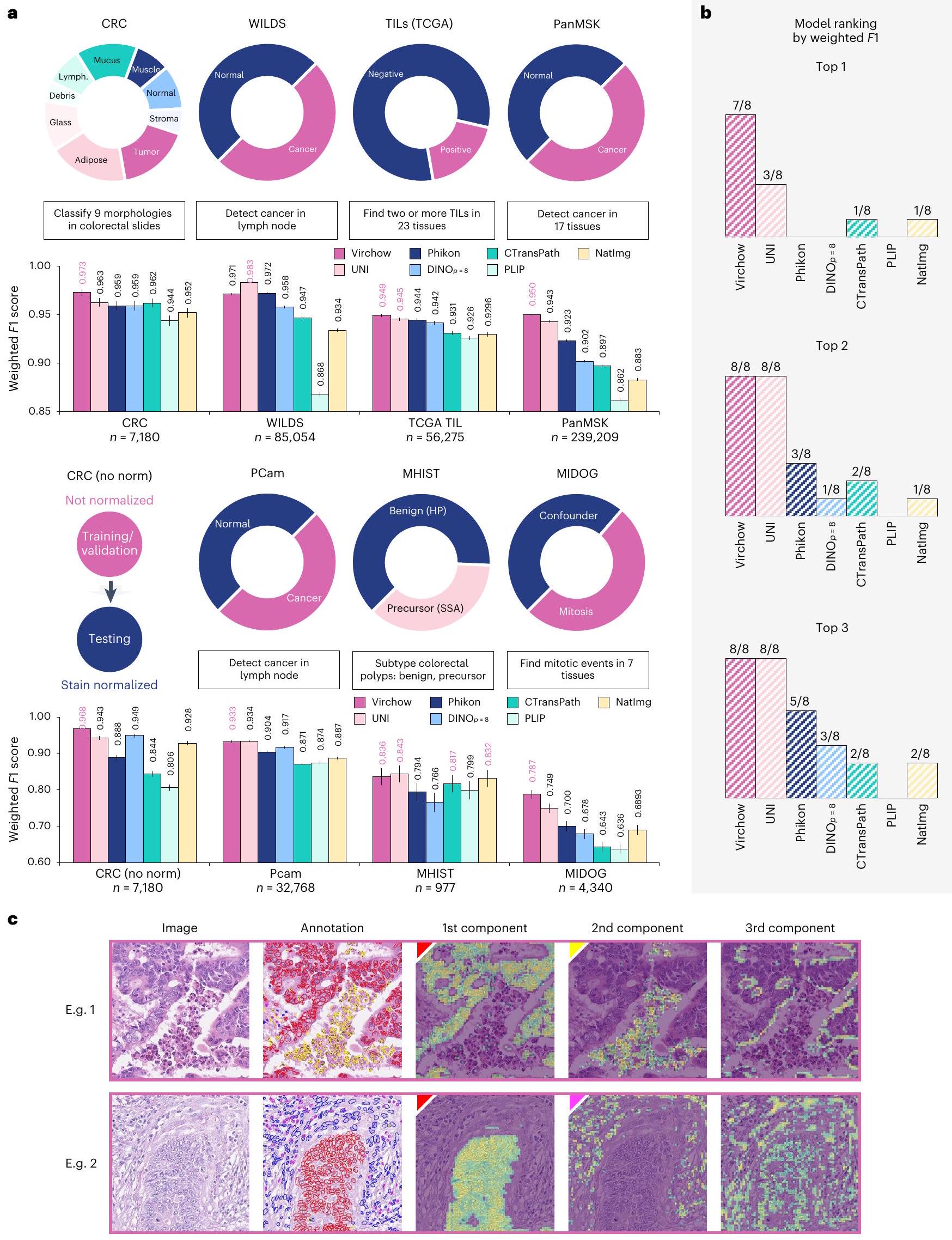
تظهر المعايير على مستوى البلاط والتحليل النوعي القابلية للتعميم
لتقييم تضمينات مستوى البلاط مباشرة دون تأثير تدريب شبكة التجميع، قمنا بتقييم أداء فيرشو على مجموعة من المعايير على مستوى البلاط من خلال الاستكشاف الخطي. يهدف تقييم الاستكشاف الخطي إلى قياس جودة وقابلية فصل التمثيلات التي تعلمها نموذج ذاتي الإشراف. نقارن تضمينات فيرشو بتضمينات نموذج الأساس من خلال تطبيق نفس بروتوكول الاستكشاف الخطي لكل نموذج، باستخدام نفس بيانات التدريب والتحقق والاختبار (انظر ‘معايير مستوى البلاط’ في الطرق لمزيد من التفاصيل). يتم إجراء التحليل على كل من مجموعات البيانات العامة ومجموعة بيانات MSKCC الداخلية للكشف عن السرطان الشامل.
تعتبر مجموعة البيانات الداخلية متعددة الأنسجة للكشف عن السرطان الشامل على مستوى البلاط (المشار إليها باسم PanMSK) معيارًا ضمن التوزيع، حيث تتكون من تعليقات على مجموعة من المرضى عبر مجموعة متنوعة من مجموعات الأنسجة المختارة للتدريب (انظر الشكل 1د).
تعتبر مجموعات البيانات العامة معايير خارج التوزيع وتوصف في قسم ‘معايير مستوى البلاط’ في الطرق. بالإضافة إلى UNI ، Phikon وCTransPath ، يتم تقييم DINO (نموذج مكون من 39 مليون معلمة تم تدريبه باستخدام أطلس جينوم السرطان (TCGA) ومجموعة بيانات داخلية)، وPLIP (نموذج مكون من 87 مليون معلمة تم تدريبه باستخدام أزواج صور النصوص المرضية) وNatImg (نموذج مكون من 1.1 مليار معلمة تم تدريبه على 142 مليون صورة طبيعية).
كما هو موضح في الشكل 5أ، ج، تتطابق أو تتجاوز تضمينات فيرشو أداء التضمينات الأخرى في سبعة من ثمانية مهام معيارية (انظر الشكل 5أ، ب؛ انظر الجدول التكميلي 4.2 لمقاييس إضافية). النماذج المنافسة الأقرب هي UNI وPhikon، حيث سجلت UNI في المراتب الثلاثة الأولى ثلاث مرات وفي المراتب الثانية لجميع المهام وسجلت Phikon في المراتب الثانية ثلاث مرات. تظهر فيرشو أداءً قويًا خارج التوزيع كما تم قياسه بواسطة مهام WILDS و’CRC (بدون معيار)’. يتم الحصول على بيانات اختبار WILDS من مستشفى لم يتم مواجهته في مجموعة التدريب. تقدم مهمة ‘CRC (بدون معيار)’ تحولًا في التوزيع من مجموعة التدريب المعتمدة على الصبغة من خلال تجنب تطبيع الصبغة على مجموعة الاختبار. بدون تطبيع، ينخفض أداء فيرشو بمقدار -0.005 فقط في درجة الموزونة، مما يشير إلى القوة في مواجهة التغيرات في معالجة البيانات.
لتقييم نوعي ما إذا كانت التضمينات التي تعلمها فيرشو تميل إلى فصل الصورة إلى مجموعات ميزات ذات معنى دلالي، قمنا بإجراء تحليل ميزات غير إشرافي مشابه للإجراء في المرجع 33 باستخدام مجموعة بيانات CoNSeP ، التي تحتوي على شرائح ملونة H&E من أدينوكارسينوما القولون (مفصلة تحت ‘تحليل الميزات النوعية’ في الطرق).
نلاحظ تقريبًا تقسيمًا دلاليًا لأنواع الخلايا في صور CoNSeP (انظر الشكل 5د). في كلا المثالين، أبرز المكون الرئيسي الأول الخلايا الظهارية الخبيثة (الحمراء). بينما أبرز المكون الرئيسي الثاني، على التوالي، الخلايا المتنوعة (الصفراء) والخلايا الالتهابية (المغنطية). أظهر DINO الإصدار 2 أنه تعلم فصل ميزات دلالية مماثلة على الصور الطبيعية، مما يسمح بفصل المقدمة/الخلفية (على سبيل المثال، التمييز بين حافلة أو طائر من الخلفية) بالإضافة إلى توضيح الأجزاء (على سبيل المثال، العجلات مقابل النوافذ في حافلة) . هنا، نوضح أن هذه الخاصية الناشئة للنموذج تنتقل إلى مجال الأمراض. تدعم هذه النتيجة المشجعة توقعنا بأن الميزات غير الإشرافية التي تعلمها فيرشو ذات معنى وقابلة للتفسير لمجموعة واسعة من المهام اللاحقة.
النقاش
تتمثل قيمة نموذج الأساس في علم الأمراض في جانبين: القابلية للتعميم وكفاءة بيانات التدريب. في دراستنا، نوضح كلا هذين الفائدتين. تم تعميم التنبؤ بالسرطان الشامل القائم على فيرشو بشكل جيد على أنواع الأنسجة أو الشرائح المقدمة من المؤسسات التي لم يتم ملاحظتها في بيانات التدريب. تم اكتشاف الأنواع الفرعية النسيجية النادرة من السرطان تقريبًا بنفس جودة الأنواع الشائعة. تم إظهار أن نفس نموذج الكشف عن السرطان الشامل يطابق تقريبًا أداء النماذج ذات الجودة السريرية بشكل عام (AUC من 0.001 إلى 0.007 خلف المنتجات السريرية، ) وتجاوزتهم في اكتشاف بعض المتغيرات النادرة من السرطانات، على الرغم من التدريب باستخدام عدد أقل من التسميات الخاصة بالأنسجة. هذه النتيجة أكثر إثارة للإعجاب عند ملاحظة أن مجموعة بيانات التدريب لنموذج السرطان الشامل، كدليل على المفهوم، تفتقر إلى مراقبة الجودة وإثراء البيانات والتسميات الفرعية التي يتم القيام بها عادةً لنماذج الذكاء الاصطناعي المتاحة تجارياً. أخيراً، نلاحظ أن تمثيلات فيرشو لم يتم ضبطها بدقة، واستخدمت النماذج هياكل تجميع بسيطة لإجراء التنبؤات. هذه النتائج تعزز الثقة في أن
الشكل 5 | ملخص لاستكشاف المستوى الفرعي. أ، وصف لكل معيار مستوى فرعي (أعلى) مع النتائج المقابلة لتمثيلات نماذج الأساس المختلفة (أسفل). لكل مهمة، يتم تمييز النتيجة الأعلى بخط عريض ومميز باللون الأرجواني. يتم تمييز نتائج متعددة عندما لا يوجد فرق ذو دلالة إحصائية بينها (; اختبار مك نيمار). تشير أشرطة الخطأ إلى فترات الثقة الثنائية الجانبين بنسبة 95% تم حسابها باستخدام 1,000
دورة إعادة التقدير. ب، عدد المهام التي سجلت فيها كل نموذج في القمة. يمكن أن تتساوى النماذج في الرتبة اعتمادًا على الدلالة الإحصائية (). ج، ميزات تمثيل فيرشو تتعلم هياكل ذات معنى. الخلايا في مجموعة بيانات CoNSeP المميزة بواسطة مكونات التمثيل الرئيسية: الظهارة الخبيثة (أحمر)، متنوع (أصفر) والتهاب (أرجواني).
مع وجود نطاق كافٍ، ستعمل نماذج الأساس ككتل بناء للتطوير المستقبلي لمجموعة واسعة من المهام اللاحقة.
هناك بعض المجالات التي نتوقع أن يكون لها تأثير ذو قيمة عالية بشكل خاص. في الممارسة السريرية، حيث تكون معظم عينات الخزعة حميدة، يمكن لنظام اكتشاف السرطان الشامل أن يعطي الأولوية للحالات للمساعدة في تقليل زمن التشخيص. مع تقليل متطلبات بيانات التدريب، يمكن تطوير منتجات ذات جودة سريرية للسرطانات الأقل شيوعًا. ستزيد توقعات العلامات الحيوية باستخدام صور H&E الروتينية من معدلات الفحص؛ وتقلل من الاختبارات التدخلية المدمرة للأنسجة؛ وتوفر بسرعة البيانات اللازمة لاتخاذ قرارات علاجية أكثر استنارة. أظهرت تمثيلات فيرشو أداءً مرتفعًا بما يكفي لتشير إلى أن هذه الأدوات قابلة للتحقيق. في الواقع، يفتح فيرشو القدرة على اكتشاف المتغيرات النسيجية غير العادية للسرطان بدقة ووضوح بالإضافة إلى حالة العلامة الحيوية، وهو شيء يصعب تحقيقه مع التدريب الخاص بالسرطان أو العلامات الحيوية بسبب الكمية المحدودة من بيانات التدريب المرتبطة.
على الرغم من التحسينات الملحوظة، لا تزال هناك جوانب من تطوير فيرشو تستحق مزيدًا من المناقشة. تختلف بيانات علم الأمراض النسيجية عن بيانات الصور الطبيعية بطرق رئيسية: توزيع الكيانات المرضية والهياكل النسيجية ذات الذيل الطويل، نقص تنوع مقاييس الأجسام، والفضاء اللوني المقيد. تحاول خوارزميات التعلم الذاتي الإشراف مطابقة التحيزات الاستقرائية لخوارزمية التعلم مع توزيع البيانات؛ ومع ذلك، في هذا العمل، كما في العديد من الأعمال الأخرى في التعلم الذاتي الإشراف لعلم الأمراض الحاسوبي، تعتمد إعدادات الخوارزمية والتدريب إلى حد كبير على ما كان ناجحًا في مجال الصور الطبيعية. قد تكشف الدراسات الإضافية أن تغيير هذه الخيارات التصميمية سيحسن الأداء في مجال علم الأمراض.
لا يزال سؤالًا مفتوحًا في أي نقطة يتم تشبع نموذج وحجم البيانات. وجدنا أن أداء اكتشاف السرطان الشامل يتناسب مع حجم النموذج ومجموعة البيانات (الشكل 2g)، وهو ما يتماشى مع الملاحظات السابقة لنماذج الأساس في مجالات أخرى. يبدو أن التحسين في الأداء بالنسبة لحجم النموذج لا يزال في نطاق تقريبي لوغاريتمي خطي؛ ومع ذلك، لوحظت اتجاهات تحت اللوغاريتمية كدالة لبيانات التدريب. قد تكون الاتجاهات في حجم بيانات التدريب مبسطة بشكل مفرط لأنها لا تلتقط التوازن بين زيادة عدد صور الشرائح مقابل البلاط. الإعداد معقد للغاية لرسم استنتاجات دقيقة حول تأثير تنوع مجموعة البيانات، على الرغم من أننا نفترض أن زيادة التنوع تساعد في تعلم ميزات قوية ونادرة. في الواقع، لقد أظهر أنه يمكن أن يحسن تدريب نموذج على أنسجة متعددة أو متغيرات سرطانية أداء الاكتشاف لكل سرطان, حيث يتم ملاحظة العديد من الميزات الشكلية عبر السرطانات من طوبوغرافيات مختلفة. بشكل عام، تشير تحقيقاتنا في سلوك التوسع إلى أن زيادة عدد معلمات النموذج تظل محورًا بارزًا للاستكشاف.
لعملنا عدة قيود. تم الحصول على مجموعة بيانات التدريب من مركز واحد مع أنواع ماسحات ضوئية محدودة. كما هو الحال مع معظم نماذج علم الأمراض النسيجية ذات الإشراف الذاتي، يتم إنشاء التمثيلات على مستوى البلاط باستخدام تكبير (0.5 مpp) بدلاً من مستوى الشريحة وبالتالي تتطلب تدريب نموذج تجميع. على الرغم من أن زيادة حجم نموذج الأساس على مستوى البلاط قد تحسن الأداء، فمن المحتمل أن هذه النماذج يجب أن تمتد إلى مستوى الشريحة لتحقيق الكفاءة البيانية المطلوبة للمهام ذات البيانات المنخفضة مثل توقع العلامات الحيوية، استجابة العلاج أو النتائج السريرية. إن التحقيق العميق في هياكل التجميع وإجراءات التدريب يتجاوز نطاق هذا العمل. كما هو الحال مع جميع النماذج التي تهدف إلى التطبيق السريري، يتطلب الأمر تحققًا دقيقًا من الأداء. علاوة على ذلك، يجب أخذ اعتبارات الأجهزة في الاعتبار تجاه نشر نماذج بحجم فيرشو أو أكبر؛ قد يكون تقطير النموذج مناسبًا لبعض المهام. بسبب حجم التدريب، لم تتمكن دراستنا من استكشاف فعالية استراتيجيات توازن البيانات والتقطير بشكل كامل. لا يزال التحدي المتمثل في تنسيق بيانات التدريب التي تحافظ على الميزات النادرة مع تقليل التكرار سؤالًا مفتوحًا. بالنظر إلى توزيع الذيل الطويل في علم الأمراض الرقمية، نتساءل عن ملاءمة طرق تقطير البيانات المعتمدة على التجميع
مثل تلك المستخدمة في نموذج DINO الأصلي v. 2 للصور الطبيعية.
لقد دعمت التقدمات الأخيرة في علم الأمراض الحاسوبي زيادة حجم مجموعة البيانات وتقليل الاعتماد على التسميات. باستخدام التعلم متعدد الحالات مع التسميات على مستوى مجموعات الشرائح، تم تمكين تشخيصات ذات صلة سريريًا من خلال التوسع إلى مجموعات بيانات تدريب بحجم. عادةً ما كانت هذه الأعمال السابقة تبدأ مع تهيئة معلمات تمثيل النموذج باستخدام أوزان نموذج مدرب مسبقًا، وغالبًا ما تكون تلك المدربة على ImageNet في إعداد إشرافي. كانت هذه العملية، المسماة التعلم الانتقالي، مدفوعة بالملاحظة أن أداء النموذج يعتمد بشكل حاسم على قدرة النموذج على التقاط ميزات الصورة. لم يكن التعلم الانتقالي داخل المجال ممكنًا نظرًا لتوفر مجموعات بيانات علم الأمراض المعلّمة بشكل محدود. الآن، يمكّن التعلم الذاتي الإشراف من النقل داخل المجال من خلال إزالة متطلبات التسميات، مما يدفع موجة ثانية من التوسع إلى عشرات الآلاف من صور الشرائح لإبلاغ تمثيل الصورة. يمثل فيرشو زيادة كبيرة في حجم بيانات التدريب إلى 1.5 مليون صورة شريحة – حجم بيانات يزيد عن 3,000 مرة عن ImageNet كما تم قياسه بواسطة العدد الإجمالي للبكسلات. هذا الحجم الكبير من البيانات بدوره يحفز نماذج كبيرة يمكنها التقاط تنوع ميزات الصورة في صور الشرائح. في هذا العمل، أظهرنا أن هذه الطريقة يمكن أن تشكل الأساس لنماذج ذات جودة سريرية في علم الأمراض السرطاني.
المحتوى عبر الإنترنت
أي طرق، مراجع إضافية، ملخصات تقارير Nature Portfolio، بيانات المصدر، بيانات موسعة، معلومات إضافية، شكر وتقدير، معلومات مراجعة الأقران؛ تفاصيل مساهمات المؤلفين والمصالح المتنافسة؛ وبيانات توفر البيانات والرمز متاحة علىhttps://doi.org/10.1038/s41591-024-03141-0.
References
Deng, S. et al. Deep learning in digital pathology image analysis: a survey. Front. Med. 14, 470-487 (2020).
Srinidhi, C. L., Ciga, O. & Martel, A. L. Deep neural network models for computational histopathology: a survey. Med. Image Anal. 67, 101813 (2021).
Cooper, M., Ji, Z. & Krishnan, R. G. Machine learning in computational histopathology: challenges and opportunities. Genes Chromosomes Cancer 62, 540-556 (2023).
Song, A. H. et al. Artificial intelligence for digital and computational pathology. Nat. Rev. Bioeng. 1, 930-949 (2023).
Fuchs, T. J. & Buhmann, J. M. Computational pathology: challenges and promises for tissue analysis. Comput. Med. Imaging Graph. 35, 515-530 (2011).
Abels, E. et al. Computational pathology definitions, best practices, and recommendations for regulatory guidance: a white paper from the digital pathology association. J. Pathol. 249, 286-294 (2019).
Fuchs, T. J., Wild, P. J., Moch, H. & Buhmann, J. M. Computational pathology analysis of tissue microarrays predicts survival of renal clear cell carcinoma patients. In Proc. Medical Image Computing and Computer-Assisted Intervention (eds Metaxas, D. et al.) 1-8 (Springer, 2008).
Kong, J. et al. Computer-aided evaluation of neuroblastoma on whole-slide histology images: classifying grade of neuroblastic differentiation. Pattern Recognit. 42, 1080-1092 (2009).
Bejnordi, B. E. et al. Diagnostic assessment of deep learning algorithms for detection of lymph node metastases in women with breast cancer. JAMA 318, 2199-2210 (2017).
Raciti, P. et al. Clinical validation of artificial intelligenceaugmented pathology diagnosis demonstrates significant gains in diagnostic accuracy in prostate cancer detection. Arch. Path. Lab. Med. 147, 1178-1185 (2022).
da Silva, L. M. et al. Independent real-world application of a clinical-grade automated prostate cancer detection system. J. Pathol. 254, 147-158 (2021).
Perincheri, S. et al. An independent assessment of an artificial intelligence system for prostate cancer detection shows strong diagnostic accuracy. Mod. Pathol. 34, 1588-1595 (2021).
Raciti, P. et al. Novel artificial intelligence system increases the detection of prostate cancer in whole slide images of core needle biopsies. Mod. Pathol. 33, 2058-2066 (2020).
Campanella, G. et al. Clinical-grade computational pathology using weakly supervised deep learning on whole slide images. Nat. Med. 25, 1301-1309 (2019).
Reis-Filho, J. S. et al. Abstract pd11-01: an artificial intelligencebased predictor of cdh1 biallelic mutations and invasive lobular carcinoma. Cancer Res. https://doi.org/10.1158/1538-7445. SABCS21-PD11-01 (2022).
Wagner, S. J. et al. Transformer-based biomarker prediction from colorectal cancer histology: a large-scale multicentric study. Cancer Cell 41, 1650-1661 (2023).
Coudray, N. et al. Classification and mutation prediction from non-small cell lung cancer histopathology images using deep learning. Nat. Med. 24, 1559-1567 (2018).
Kather, J. N. et al. Deep learning can predict microsatellite instability directly from histology in gastrointestinal cancer. Nat. Med. 25, 1054-1056 (2019).
Bilal, M. et al. Development and validation of a weakly supervised deep learning framework to predict the status of molecular pathways and key mutations in colorectal cancer from routine histology images: a retrospective study. Lancet Digit. Health 3, e763-e772 (2021).
Xie, C. et al. Computational biomarker predicts lung ICI response via deep learning-driven hierarchical spatial modelling from H&E. Preprint at https://doi.org/10.21203/rs.3.rs-1251762/v1 (2022).
Kacew, A. J. et al. Artificial intelligence can cut costs while maintaining accuracy in colorectal cancer genotyping. Frontiers in Oncology https://doi.org/10.3389/fonc.2021.630953 (2021).
Chen, T., Kornblith, S., Norouzi, M. & Hinton, G. A simple framework for contrastive learning of visual representations. In Proc. 37th International Conference on Machine Learning (eds Daumé, H. & Singh, A.) 1597-1607 (JMLR.org, 2020).
Caron, M. et al. Unsupervised learning of visual features by contrasting cluster assignments. In Proc. 34th International Conference on Neural Information Processing Systems (eds Larochelle, H. et al.) 9912-9924 (Curran Associates, 2020).
Caron, M. et al. Emerging properties in self-supervised vision transformers. In Proc. IEEE/CVF International Conference on Computer Vision 9630-9640 (IEEE, 2021).
He, K. et al. Masked autoencoders are scalable vision learners. In Proc. IEEE/CVF Conference on Computer Vision and Pattern Recognition 15979-15988 (IEEE, 2022).
Zhai, X., Kolesnikov, A., Houlsby, N. & Beyer, L. Scaling vision transformers. In Proc. IEEE/CVF Conference on Computer Vision and Pattern Recognition 12094-12103 (IEEE, 2022).
OpenAI. GPT-4 technical report. Preprint at https://doi.org/ 10.48550/arXiv. 2303.08774 (2023).
Deng, J. et al. Imagenet: a large-scale hierarchical image database. In Proc. IEEE Conference on Computer Vision and Pattern Recognition 248-255 (IEEE, 2009).
Sun, C., Shrivastava, A., Singh, S. & Gupta, A. Revisiting unreasonable effectiveness of data in deep learning era. In Proc. IEEE International Conference on Computer Vision 843-852 (IEEE, 2017).
Oquab, M. et al. DINOv2: Learning Robust Visual Features without Supervision. In Transactions on Machine Learning Research 2835-8856 (TMLR, 2024).
Dosovitskiy, A. et al. An image is worth words: transformers for image recognition at scale. In The Ninth International Conference on Learning Representations https://openreview.net/ forum?id=YicbFdNTTy (OpenReview.net, 2021).
Wang, X. et al. Transformer-based unsupervised contrastive learning for histopathological image classification. Med. Image Anal. 81, 102559 (2022).
Ciga, O., Xu, T. & Martel, A. L. Self supervised contrastive learning for digital histopathology. Mach. Learn. Appl. 7, 100198 (2022).
Filiot, A. et al. Scaling self-supervised learning for histopathology with masked image modeling. Preprint at https://doi.org/10.1101/ 2023.07.21.23292757 (2023).
Azizi, S. et al. Robust and data-efficient generalization of self-supervised machine learning for diagnostic imaging. Nat. Biomed. Eng. 7, 1-24 (2023).
Kang, M., Song, H., Park, S., Yoo, D. & Pereira, S. Benchmarking self-supervised learning on diverse pathology datasets. In Proc. IEEE/CVF Conference on Computer Vision and Pattern Recognition 3344-3354 (IEEE, 2023).
Dippel, J. et al. RudolfV: A foundation model by pathologists for pathologists. Preprint at https://doi.org/10.48550/ arXiv.2401.04079 (2024).
Chen, R. J. et al. Towards a general-purpose foundation model for computational pathology. Nat. Med. 30, 850-862 (2024).
Campanella, G. et al. Computational pathology at health system scale-self-supervised foundation models from three billion images. Preprint at https://doi.org/10.48550/arXiv.2310.07033 (2023).
Schultz, M. Rudolf Virchow. Emerg. Infect. Dis. 14, 1480-1481 (2008).
Reese, D. M. Fundamentals-Rudolf Virchow and modern medicine. West. J. Med. 169, 105 (1998).
Virchow, R. & Chance, F. Cellular Pathology as Based Upon Physiological and Pathological Histology: Twenty Lectures Delivered in the Pathological Institute of Berlin During the Months of February, March and April, 1858 (De Witt, 1860).
Liu, Z. et al. Swin transformer: hierarchical vision transformer using shifted windows. In Proc. IEEE/CVF International Conference on Computer Vision 9992-10002 (IEEE, 2021).
Chen, X., Xie, S. & He, K. An empirical study of training self-supervised vision transformers. In Proc. IEEE/CVF International Conference on Computer Vision 9620-9629 (IEEE, 2021).
Casson, A. et al. Joint breast neoplasm detection and subtyping using multi-resolution network trained on large-scale H&E whole slide images with weak labels. In Proc. Medical Imaging with Deep Learning (eds Oguz, I. et al.) 18-38 (JMLR, 2024).
Zehir, A. et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 23, 703-713 (2017).
Huang, Z., Bianchi, F., Yuksekgonul, M., Montine, T. J. & Zou, J. A visual-language foundation model for pathology image analysis using medical twitter. Nat. Med. 29, 2307-2316 (2023).
Graham, S. et al. Hover-net: simultaneous segmentation and classification of nuclei in multi-tissue histology images. Med. Image Anal. 58, 101563 (2019).
Cheerla, A. & Gevaert, O. Deep learning with multimodal representation for pancancer prognosis prediction. Bioinformatics 35, i446-i454 (2019).
Noorbakhsh, J. et al. Deep learning-based cross-classifications reveal conserved spatial behaviors within tumor histological images. Nat. Commun. 11, 6367 (2020).
Ilse, M., Tomczak, J. & Welling, M. Attention-based deep multiple instance learning. In Proc. 35th International Conference on Machine Learning (eds Dy, J. & Krause, A.) 2127-2136 (JMLR, 2018).
Chen, R. J. et al. Scaling vision transformers to gigapixel images via hierarchical self-supervised learning. In Proc. IEEE/CVF Conference on Computer Vision and Pattern Recognition 1612316134 (IEEE, 2022).
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
(c) The Author(s) 2024
طرق
مجموعة بيانات تدريب بحجم مليون
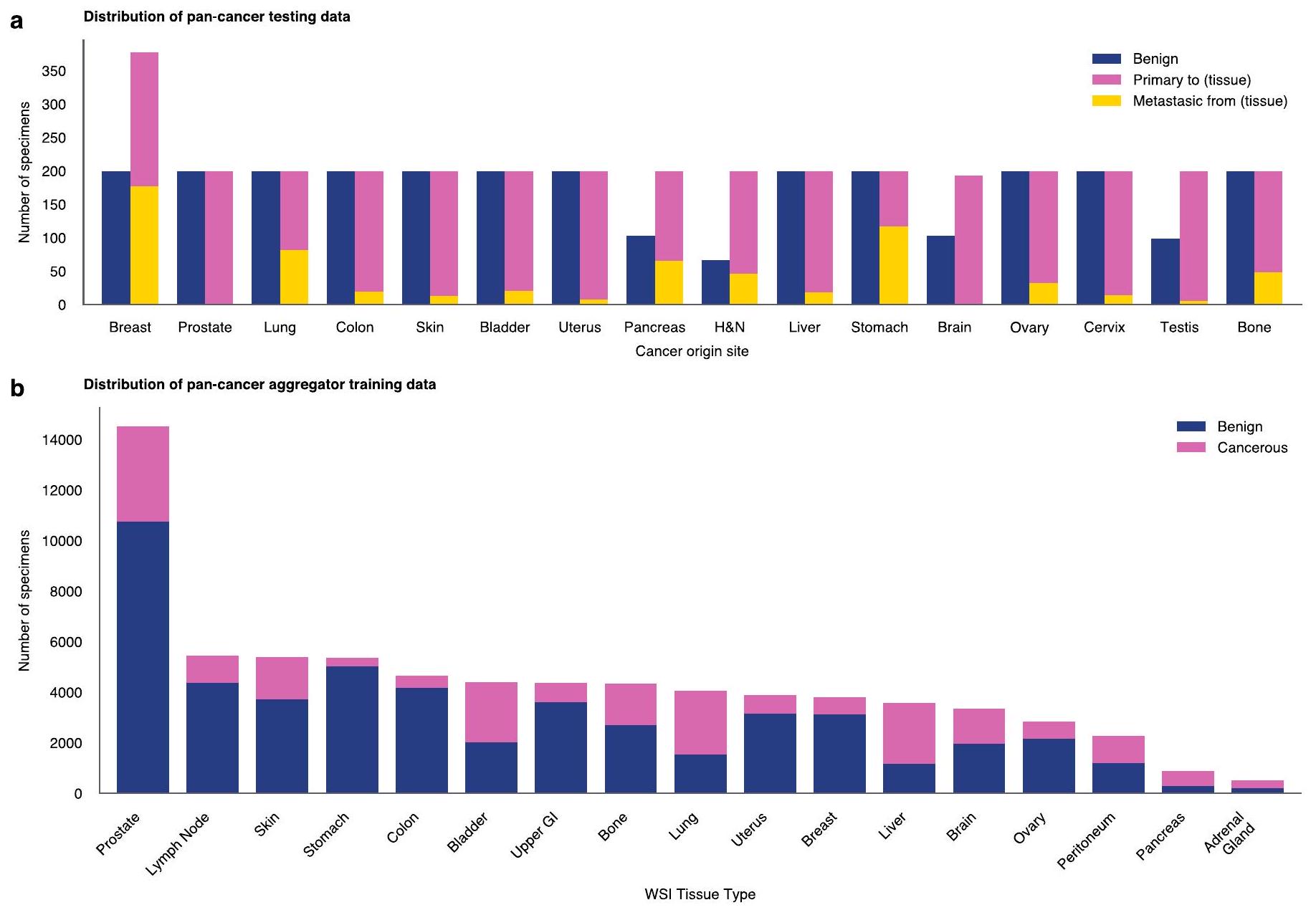
لم يكن مراجعة مجلس المراجعة المؤسسية قابلاً للتطبيق على البحث الموصوف في هذه الدراسة. تم إجراء هذه الدراسة البحثية بأثر رجعي من بيانات غير محددة تم ترخيصها لـPaige.AI، شركة من MSKCC. البيانات المستخدمة في هذه الدراسة تم جمعها جميعها في الأصل للاستخدام السريري من قبل MSKCC في بيئة الممارسة، وبالتالي تعتبر بيانات ثانوية. تم استخدام البيانات التي تم إلغاء تعريفها مسبقًا فقط من قبل MSKCC في التحليل، وتمت إزالة معرفات المرضى الفريدة تمامًا من مجموعة البيانات التحليلية. على حد علمنا، لم تقم MSKCC بنقل أي بيانات لم يوافق عليها المريض المعني أو لم يوافق على إشعار ممارسات الخصوصية الخاص بـ MSKCC أو إشعار أو تنازل أو موافقة مشابهة بشكل كبير. تتكون مجموعة بيانات تدريب علم الأمراض الرقمية من 1,488,550 صورة شاملة (WSIs) مستمدة من 119,629 مريضًا. جميع هذه الصور ملونة بصبغة H&E، وهي صبغة روتينية تصبغ النوى باللون الأزرق والمصفوفة خارج الخلوية والسيتوبلازم باللون الوردي. تم مسح الصور الشاملة عند دقة أو 0.5 مليمتر لكل بكسل باستخدام ماسحات لايكا. تشمل السبع عشرة مجموعة نسيجية عالية المستوى، كما هو موضح في الشكل 1c.
تبلغ أحجام صور الشرائح الرقمية (WSIs) جيجابكسل، ومن الصعب استخدامها مباشرة أثناء التدريب. بدلاً من ذلك، تم تدريب فيرتشو على قطع الأنسجة التي تم أخذ عينات منها من الأنسجة الأمامية في كل صورة شريحة رقمية. لاكتشاف الأنسجة الأمامية، تم تقليل دقة كل صورة شريحة رقمية. باستخدام الاستيفاء الثنائي، وتم تقييم كل بكسل من الصورة المنقوصة لمعرفة ما إذا كانت درجة لونه، تشبعه وقيمته ضمن [90،180]، [8،255] و [103،255]، على التوالي. جميع غير المتداخلين بلاط يحتوي على الأقلتم جمع الأنسجة حسب المنطقة. تم تدريب فيرشو على 2 مليار بلاطة تم أخذ عينات منها عشوائيًا مع الاستبدال من حوالي 13 مليار بلاطة أنسجة متاحة.
معمار فيرشو والتدريب
يستخدم فيرشو معمارية فيت ‘هائلة’ (ViT-H/14)، وهي فيتمع 632 مليون معلمة تم تدريبها باستخدام خوارزمية التعلم الذاتي DINO v. 2 (المرجع 33)، كما هو موضح في الشكل الممتد 1. يعد ViT تكييفًا لنموذج المحول لتحليل الصور، حيث يتم التعامل مع الصورة كسلسلة من القطع. يتم تضمين هذه القطع ومعالجتها من خلال مشفر المحول الذي يستخدم آليات الانتباه الذاتي. تتيح هذه الطريقة لـ ViT التقاط العلاقات المكانية المعقدة عبر الصورة. يعتمد DINO v. 2 على نموذج الطالب-المعلم: بالنظر إلى شبكة الطالب وشبكة المعلم، كل منهما يستخدم نفس الهيكل، يتم تدريب الطالب لمطابقة تمثيل المعلم. شبكة الطالب محدودة المعلومات، حيث يتم تدريبها باستخدام تباينات ضوضائية من بلاطات الإدخال. شبكة المعلم هي متوسط متحرك أسي يتم تحديثه ببطء لشبكات الطلاب السابقة؛ إن مطابقة المعلم تحقق تأثيرًا مشابهًا للتجميع على توقعات الطلاب السابقة.يتعلم الطالب تمثيلاً عالمياً للصورة من خلال مطابقة رمز الفئة الخاص بالمعلم، بالإضافة إلى التمثيلات المحلية من خلال مطابقة رموز الباتش الخاصة بالمعلم. يتم مطابقة رموز الباتش فقط لمجموعة مختارة من الرموز التي تم إخفاؤها عشوائياً من صورة الإدخال (بالنسبة للطالب)، كما هو الحال في نمذجة الصورة المخفية.يساعد التنظيم الإضافي نماذج DINO v. 2 على التفوق على النسخة السابقة من DINO..
تم استخدام المعلمات الفائقة الافتراضية لتدريب نموذج DINO v. 2 لـ Virchow كما هو موضح في المرجع 33 مع التغييرات التالية: جدول درجة حرارة المعلم من 0.04-0.07 في 186,000 تكرار وجدول معدل التعلم الجذري المعكوس مع تسخين لمدة 495,000 تكرار (بدلاً من 100,000) وتبريد خطي إلى 0.0 لآخر 819,200 تكرار.. تم تدريب فيرشو باستخدام آدم دبليو (، بدقة float16. لاحظ أنه مع ViT-H، استخدمنا 131,072 نموذجًا (وبالتالي رؤوس إسقاط بُعد 131,072). خلال التدريب الموزع، تم أخذ عينة من كل دفعة صغيرة عن طريق اختيار عشوائي لوحدة صورة واحدة لكل وحدة معالجة رسومات و256 بلاطة أمامية لكل وحدة صورة.
كشف السرطان الشامل
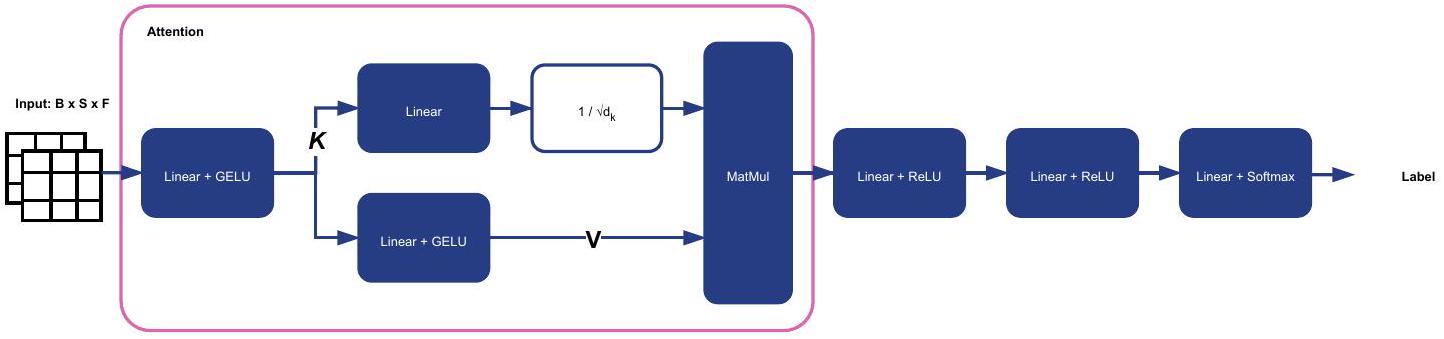
يتطلب الكشف عن السرطان على مستوى العينة نموذجًا يجمع تمثيلات النموذج الأساسي من جميع البلاطات الأمامية لجميع الصور النسيجية الكاملة في عينة للكشف عن وجود السرطان. جميع نماذج الكشف عن السرطان الشامل المدربة في هذا العمل تستخدم Agataنموذج المجمع، مشرف بشكل ضعيف مع التعلم متعدد الحالات (انظر الشكل 2 في البيانات الموسعة لتفاصيل الهيكل).
توليد التضمين. من أجلصورة الإدخال، يتم تعريف تضمين فيرتشو على أنه تجميع رمز الفئة والمتوسط عبر جميع الرموز المتوقعة الـ 256 الأخرى. ينتج عن ذلك حجم التضمينبالنسبة لـ Phikon، يتم استخدام رمز الفئة فقط، كما هو موصى به في المرجع 37. بالنسبة لـ CTransPath، يتم استخدام متوسط جميع الرموز حيث لا يوجد رمز فئة.
بيانات التدريب. لتدريب نموذج المجمع، قمنا بإعداد مجموعة فرعية من مجموعة بيانات التدريب المستخدمة لتدريب فيرشو (انظر ‘مجموعة بيانات التدريب على نطاق المليون’ في الطرق للحصول على التفاصيل)، مع دمجها مع تسميات على مستوى العينة (على مستوى الكتلة لنسج البروستاتا) تشير إلى وجود أو عدم وجود سرطان مستخرجة من التقارير التلخيصية والتشخيصية. تتكون مجموعات بيانات التدريب والتحقق المدمجة من 89,417 شريحة عبر 40,402 عينة. انظر الشكل 4b من البيانات الموسعة لتوزيع بيانات التدريب، مصنفة حسب نوع نسيج WSI وحالة السرطان.
تدريب المجمع. تم تدريب مجمع أغاتا كما هو موضح في الشكل 2 من البيانات الموسعة. نظرًا لأن التسمية تكون على مستوى العينة، يجب تجميع جميع البلاطات التي تنتمي إلى نفس العينة أثناء التدريب. يعتبر التدريب باستخدام التضمينات لجميع البلاطات لعينة ما مكلفًا من حيث الذاكرة بشكل كبير. لذلك، نقوم باختيار الشريحة ذات أعلى احتمال متوقع للإصابة بالسرطان لكل عينة ونعيد توجيه التدرجات فقط لتلك الشريحة.
تم أيضًا تدريب مجمعات البيانات باستخدام تمثيلات Phikon وCTransPath كخطوط أساسية. تم تدريب جميع المجمعات لمدة 25 دورة باستخدام خسارة الانتروبيا المتقاطعة وAdamW.محسن بمعدل تعلم أساسي قدره 0.0003. خلال كل جولة تدريب، تم اختيار نقطة التحقق التي حققت أعلى AUC في التحقق للتقييم.
مجموعة بيانات الاختبار. يتم تقييم نماذج الكشف عن السرطان الشامل على مجموعة من البيانات المستمدة من MSKCC ومؤسسات خارجية. لم يتم رؤية أي من المرضى في مجموعة التقييم خلال التدريب. تحتوي مجموعة البيانات على 22,932 شريحة من 6,142 عينة عبر 16 نوعًا من السرطان. نفترض أنه كلما زادت البيانات التي تم تدريب النموذج الأساسي عليها، زادت أداء المهام اللاحقة، خاصة في المهام التي تعاني من قيود في البيانات. لاختبار هذه الفرضية، نقوم بتصنيف أنواع السرطان إلى مجموعات سرطانية شائعة أو نادرة. وفقًا للمعهد الوطني للسرطان، يتم تعريف السرطانات النادرة على أنها تلك التي تحدث لأقل من 15 شخصًا من بين 100,000 كل عام في الولايات المتحدة.استنادًا إلى هذا التعريف، يتكون السرطان الشائع من 14,179 شريحة من 3,547 عينة مصدرها الثدي، البروستاتا، الرئة، القولون، الجلد، المثانة، الرحم، البنكرياس ورأس والعنق، بينما يتكون السرطان النادر من 8,753 شريحة من 2,595 عينة مصدرها الكبد، المعدة، الدماغ، المبيض، عنق الرحم، الخصية والعظام. لاحظ أن كل نوع من أنواع السرطان يتم تحديده من خلال نسيجه الأصلي وبالتالي قد يظهر في أي نسيج (كسرطان أولي أو نقيل). من ناحية أخرى، تم أخذ عينات من العينات الحميدة لكل نوع من أنواع السرطان فقط من النسيج الأصلي. على سبيل المثال، يحتوي طبقة الكبد على 182 عينة كبد مع سرطان الكبد (أولي)، 18 عينة غير كبدية مع سرطان الكبد (نقيلي) و200 عينة كبد حميدة. لكل نوع من أنواع السرطان، توضح الشكل 2a التوزيع بين السرطان الأولي والنقيلي، ويظهر الشكل الإضافي 4a أيضًا عدد العينات الحميدة.
تتضمن مجموعة بيانات الاختبار 15,622 شريحة من 3,033 عينة تم جمعها في MSKCC (المشار إليها بـ ‘داخلي’ في الشكل 2b)، بالإضافة إلى 7,310 شرائح (3109 عينات) أُرسلت إلى MSKCC من مؤسسات حول العالم (‘خارجي’ في الشكل 2b). انظر الشكل 4a من البيانات الموسعة لتوزيع بيانات الاختبار، مقسماً حسب نوع السرطان (للعينات التي تحتوي على سرطان) أو حسب نوع الأنسجة (للعينات الحميدة).
استخراج التسميات. لتحديد تشخيص السرطان السريري على مستوى العينة، تم استخدام نظام معالجة اللغة الطبيعية القائم على القواعد. يقوم هذا النظام بتفكيك التقارير على مستوى الحالة إلى مستوى العينة ويحلل التقارير السريرية المرتبطة بكل عينة، مما يوفر فهماً شاملاً لكل حالة.
التحليل الإحصائي. يتم مقارنة أداء النماذج الثلاثة باستخدام مقياسين: AUC والخصوصية عندالحساسية. AUC هو مقياس عام مناسب لأنه لا يتطلب اختيار عتبة لمخرجات احتمالات النموذج، وهو ما قد يحتاج إلى ضبط لمجموعات البيانات الفرعية المختلفة. الخصوصية عند 95% حساسية هي معلوماتية لأن النظام السريري يجب أن يكون حساسًا وليس فقط دقيقًا في الممارسة. بالنسبة لـ AUC، فإن اختبار DeLong الثنائي.بطريقة هولميتم تطبيق التصحيح للتحقق من الأهمية الإحصائية. بالنسبة للخصوصية، أولاً يتم استخدام اختبار كوكرا.اختباريتم تطبيقه، ثم يتم استخدام اختبار مك نماريتم تطبيقه بعد الحدث لجميع الأزواج باستخدام طريقة هولم للتصحيح. الجانبانتم حساب فترات الثقة في الشكل 2ب-هـ والشكل الإضافي 3 باستخدام طريقة دي لونغلـ AUC وطريقة ويلسونلزيادة الدقة. بالإضافة إلى التحليل العام، يتم أيضًا إجراء تحليل مصنف لكل نوع من أنواع السرطان.
مجموعات بيانات التقييم السريري
لإجراء تقييم شامل لنموذج الكشف عن السرطان الشامل القائم على فيرشو، نستخدم سبعة مجموعات بيانات إضافية (انظر الجدول التكميلية 2.1 للحصول على التفاصيل). واحدة من هذه المجموعات هي مجموعة بيانات شاملة للأنسجة، والباقي هي مجموعات بيانات أحادية الأنسجة تحتوي على أنسجة تمتلك Paige منتجات سريرية لها: أي البروستاتا والثدي والعقد اللمفاوية.
معيار منتج الأنسجة الشامل. تحتوي مجموعة البيانات هذه على 2419 شريحة عبر 18 نوعًا من الأنسجة (الجدول التكميلي 2.2). يتم فحص كل شريحة بشكل فردي بواسطة أخصائي علم الأمراض وتسمية وفقًا لوجود سرطان غازي. الفرق المهم بين مجموعة البيانات الاختبارية في ‘كشف السرطان الشامل’ وهذه المجموعة هو أن الأولى مصنفة وفقًا لنوع الأنسجة في العينات السرطانية، بينما الثانية مصنفة وفقًا لنوع الأنسجة لجميع الشرائح، حيث أن ذلك أكثر صلة في الإعدادات السريرية. نستخدم هذه المجموعة من البيانات لتحديد أنماط الفشل لنموذج كشف السرطان الشامل.
معيار منتج البروستاتا. تحتوي مجموعة البيانات هذه على 2,947 كتلة (3,327 شريحة) من خزعات إبرة البروستاتا الأساسية (الجدول التكميلية 2.7). تم استخراج التسميات للكتل من التقارير الملخصة التي تم جمعها في مركز ميموريال سلون كيترينج للسرطان. تم تنسيق مجموعة البيانات هذه لتقييم الأداء المستقل لنموذج Paige Prostate Detect، وهو نموذج سريري خاص بالأنسجة. نستخدم مجموعة البيانات هذه لمقارنة نموذج الكشف عن السرطان الشامل مع Paige Prostate Detect.
معيار المتغيرات النادرة في البروستاتا. تحتوي هذه المجموعة على 28 شريحة تحتوي على متغيرات نادرة من سرطان البروستاتا (ورم عصبي صماوي، ضمور، لمفومة ليمفاوية صغيرة الخلايا، سرطان الخلايا الرغوية، لمفومة جريبية) و112 شريحة حميدة (الجدول التكميلية 2.8). تم تنسيق الشرائح السرطانية ووضع علامات عليها بواسطة أخصائي علم الأمراض، وتم إلحاقها بشرائح من كتل حميدة تم تحديدها من التقارير الملخصة التي تم جمعها في مركز ميموريال سلون كيترينغ للسرطان.
معيار منتج الثدي. تحتوي مجموعة البيانات هذه على 190 شريحة تحتوي على سرطان غازي و1,501 شريحة حميدة، تم تصنيفها بشكل فردي بواسطة أخصائي علم الأمراض وفقًا لوجود فرط تنسج قنوي غير نمطي، فرط تنسج فصي غير نمطي، سرطان فصي في الموقع، سرطان قنوي في الموقع، سرطان قنوي غازي، سرطان فصي غازي و/أو أنواع فرعية أخرى (الجدول التكميلي 2.5). تم تنسيق مجموعة البيانات هذه لتقييم الأداء المستقل لنموذج Paige Breast، وهو نموذج خاص بالأنسجة وعالي الجودة سريريًا. نستخدم معلومات الأنواع الفرعية للتحليل الطبقي.
معيار المتغيرات النادرة في الثدي. تحتوي مجموعة البيانات هذه على 23 حالة من سرطان القنوات الغازية الغازي أو سرطان الغدد اللبنية الغازي (كحالة تحكم)، و75 حالة من المتغيرات النادرة (سرطان الغدد اللعابية الكيسية، سرطان مع تمايز إفرازي، سرطان شبكي، سرطان ميكروبابي غازي، سرطان ميتابلاستيكي (نوع منتج للمصفوفة، خلايا مغزلية وخلايا حرشفية)، سرطان مخاطي، سرطان إفرازي وسرطان أنبوبي) و392 حالة حميدة (إجمالي 5,031 شريحة). تم تنسيق الحالات السرطانية بواسطة أخصائي علم الأمراض، وتمت إضافتها إلى الحالات الحميدة المحددة من التقارير الملخصة التي تم جمعها في مركز ميموريال سلوان كيترينغ للسرطان. انظر الجدول التكميلي 2.6 للحصول على التفاصيل.
BLN. تحتوي مجموعة البيانات هذه على 458 شريحة من العقد اللمفاوية مع سرطان الثدي النقيلي و295 شريحة من العقد اللمفاوية الحميدة (الجدول التكميلي 2.3). تم تصنيف كل شريحة بواسطة أخصائي علم الأمراض وفقًا لوجود سرطان غازي، ويتم قياس أكبر ورم على الشريحة لتصنيف الورم إلى نقائل كبيرة، نقائل صغيرة أو خلايا ورمية مت infiltrating. نستخدم الفئات للتقييم الطبقي.
معيار المتغيرات النادرة في العقد اللمفاوية. تحتوي هذه المجموعة على 48 عينة من المتغيرات النادرة للأورام (سرطان الغدد اللمفاوية الكبيرة المنتشرة، سرطان الغدد اللمفاوية الجريبية، سرطان الغدد اللمفاوية الهامشية، سرطان هودجكين) تم اختيارها بواسطة أخصائي علم الأمراض و192 عينة حميدة تم تحديدها من التقارير الملخصة التي تم جمعها في مركز ميموريال سلون كيترينغ للسرطان (الجدول التكميلي 2.4).
كشف العلامات الحيوية
قمنا بصياغة كل مهمة توقع العلامات الحيوية كمشكلة تصنيف حالة مرضية ثنائية، حيث تشير العلامة الإيجابية إلى وجود العلامة الحيوية. تتكون كل حالة من شريحة واحدة أو أكثر من شرائح H&E التي تشترك في نفس العلامة الثنائية. قمنا بتقسيم كل مجموعة بيانات بشكل عشوائي إلى مجموعات تدريب واختبار، مع ضمان عدم وجود تداخل بين المرضى، كما هو موضح في الجدول التكميلي 3.1. يتم وصف الأهمية السريرية لكل علامة حيوية أدناه.
قولون-ميكروساتلايت. تحدث عدم استقرار الميكروساتلايت (MSI) عندما تتعرض مناطق الحمض النووي ذات التسلسلات القصيرة المتكررة (الميكروساتلايت) للاضطراب بسبب طفرات أحادية النوكليوتيد، مما يؤدي إلى تباين في هذه التسلسلات عبر الخلايا. عادةً، تقوم جينات إصلاح عدم التطابق (MMR) (MSH1، MSH2، MSH6، PMS2) بتصحيح هذه الطفرات، مما يحافظ على التناسق في الميكروساتلايت. ومع ذلك، فإن تعطيل أي جين من جينات MMR (من خلال طفرة في الخلايا الجرثومية، طفرة جسدية أو كتم جيني) يؤدي إلى زيادة معدل الطفرات غير المصححة عبر الجينوم. يتم الكشف عن MSI باستخدام تفاعل البوليميراز المتسلسل أو تسلسل الجيل التالي، الذي يحدد عددًا كبيرًا من الطفرات غير المصلّحة في الميكروساتلايت، مما يدل على عجز في إصلاح عدم التطابق (dMMR). يشير عدم استقرار الميكروساتلايت العالي (MSI-H) إلى dMMR في الخلايا، ويمكن التعرف عليه عبر IHC، الذي يظهر عدم وجود صبغة لبروتينات MMR. يتواجد MSI-H في حوالي 15% من سرطانات القولون والمستقيم (CRCs)، وغالبًا ما يرتبط بطفرات في الخلايا الجرثومية التي تزيد من خطر الإصابة بالسرطان الوراثي. وبالتالي، يُوصى بإجراء فحص روتيني لـ MSI أو dMMR القائم على IHC لجميع عينات سرطان القولون والمستقيم الأولية. تتضمن مجموعة بيانات قولون-MSI، التي تتكون من 2,698 عينة CRC مع 288 حالة إيجابية لـ MSI-H/dMMR، استخدام كل من IHC وتسلسل MSK-IMPACT للكشف عن dMMR وMSI-H، مع إعطاء الأولوية لنتائج IHC عندما تكون نتائج الاختبارين متاحة.
سرطان الثدي-CDH1. يرتبط الفقد الثنائي الأليل لجين الكاديرين 1 (CDH1) (الذي يشفر E-cadherin) ارتباطًا قويًا بسرطان الثدي القنوي ونمط نسيجي مميز وسلوك بيولوجي مختلف.تم اعتبار الطفرات المعطلة لجين CDH1 المرتبطة بفقدان التغايرية أو طفرة فقدان وظيفة جسمية ثانية كما تحددها نتائج اختبار تسلسل MSK-IMPACT كـ ‘طفرات ثنائية الأليل لجين CDH1’. تتضمن مجموعة بيانات CDH1 ما مجموعه 1,077 عينة من سرطان الثدي الأولي إيجابي مستقبلات الاستروجين (ER+)، حيث كانت 139 إيجابية و918 سلبية. تم استبعاد 20 عينة المتبقية التي تحتوي على أنواع أخرى من الطفرات – أي الطفرات أحادية الأليل.
المثانة – FGFR. يتم ترميز مستقبل عامل نمو الألياف (FGFR) بواسطة أربعة جينات (FGFR1، FGFR2، FGFR3، FGFR4). يسمح فحص تغييرات جينات FGFR في سرطان المثانة بتحديد المرضى القابلين للاستهداف بواسطة مثبطات FGFR. تشير التجارب القصصية من علماء الأمراض إلى أنه قد يكون هناك إشارة شكلية لتغييرات FGFR.. يركز التصنيف الثنائي لـ FGFR على طفرات FGFR3 p.S249C و p.R248C و p.Y373C و p.G37OC، واندماجات FGFR3-TACC3، وطفرات FGFR2 p.N549H و p.N549K و p.N549S و p.N549T استنادًا إلى بيانات من مجموعة MSK-IMPACT. من إجمالي 1,038 عينة (1,087 صورة شاملة)، لديتعديلات.
رئة-EGFR. الـفحص الطفرات المسرطنة في سرطان الرئة غير صغير الخلايا ضروري لتحديد الأهلية للعلاجات المستهدفة في مراحل متقدمة من سرطان الرئة غير صغير الخلايا.الحالة المسرطنة لـتم تحديد الطفرة بناءً على توضيح OncoKB. تم تعريف طفرات EGFR ذات أي تأثير ورمي (بما في ذلك المتوقعة/المحتملة) على أنها علامة إيجابية، وتم استبعاد الطفرات ذات الحالة الورمية غير المعروفة.
سرطان البروستاتا-AR. تم العثور على تضخيم/زيادة التعبير في 50% من سرطانات البروستاتا المقاومة للإخصاء وكان مرتبطًا بمقاومة العلاج بالحرمان من الأندروجين. في مجموعة بيانات AR، تم تحديد تضخيم عدد النسخ لـباستخدام اختبار تسلسل MSK-IMPACT، حيث كانت نسبة التغيير أكبر من اثنين.
سرطان المعدة-HER2. يعتبر التعبير المفرط لمستقبل عامل نمو البشرة البشري 2 (HER2) و/أو التضخيم أكثر تنوعًا بكثير في سرطان المعدة مقارنة بسرطان الثدي. تم العثور على حوالي 20% من مرضى سرطان المعدة مرتبطين بالتعبير المفرط لـ HER2/التضخيم العالي المستوى، ومن المحتمل أن يستفيدوا من العلاج باستخدام علاج الأجسام المضادة المضادة لـ HER2. هنا، تم اعتبار نتيجة IHC لـ HER2 بمقدار 2+، مؤكدة إيجابية مع التهجين الموضعي بالفلوريسين (FISH) أو نتيجة IHC بمقدار 3+ على أنها تضخيم HER2.
سرطان الرحم-PTEN. PTEN هو أكثر جينات كابح الورم تعرضًا للطفرات في سرطان الرحم. أظهرت وجود طفرة PTEN ارتباطًا كبيرًا بتوقعات أسوأ في البقاء وعودة المرض. تم تحديد الحالة الورمية لطفرة PTEN بناءً على تسلسل MSK-IMPACT وتوضيح OncoKB. تم تعريف المتغيرات المرتبطة بأي تأثير ورمي (بما في ذلك المتوقعة و/أو المحتملة) على أنها علامة إيجابية لطفرات PTEN، وتم استبعاد المتغيرات ذات الحالة الورمية غير المعروفة.
سرطان الغدة الدرقية-RET. كانت طفرات RET مرتبطة بشكل كبير بسرطان الغدة الدرقية النخاعي، الذي يمثل حواليمن جميع سرطانات الغدة الدرقية. يلعب فحص طفرات RET الورمية دورًا مهمًا في تشخيص وتوقع سرطان الغدة الدرقية النخاعي. تم تحديد العلامة الإيجابية لطفرات RET الورمية بناءً على تسلسل MSK-IMPACT وتوضيح OncoKB.
سرطان الجلد-BRAF. BRAF هو واحد من أكثر الجينات تعرضًا للطفرات في الميلانوما، وطفرات V600E هي الأكثر شيوعًا، مما يؤدي إلى تنشيط مستمر لمسار إشارة BRAF/MEK/ERK. أظهرت العلاجات المستهدفة باستخدام مثبطات BRAF نتائج بقاء أفضل في المرضى الذين يعانون منميلانوما متحورة V600. لذلك، يساعد اكتشاف طفرات BRAF V600 في الميلانوما على تحديد استراتيجيات العلاج. في مجموعة بيانات BRAF، تم تحديد حالة الطفرة الورمية ووجود المتغير V600E بناءً على مجموعة MSK-IMPACT وتوضيح OncoKB.
سرطان المبيض-FGA. يتميز سرطان المبيض عالي الدرجة بارتفاع انتشار طفرات TP53 وعدم استقرار الجينوم مع تغييرات جينية واسعة النطاق. تم تحديد نسبة الجينوم المتغيرة (FGA) من بيانات تسلسل MSK-IMPACT، حيث تم اعتبار FGA
كعلامة إيجابية. تم تحديد حد لـ FGA الذي زاد من طفرات TP53 في توزيع حالات سرطان المبيض.
تدريب المجمع. لتوقع العلامات الحيوية تحت إشراف ضعيف، استخدمنا التضمينات وAgata, كما في ‘كشف السرطان الشامل’، لتحويل مجموعة من البلاطات المستخرجة من WSIs التي تنتمي إلى نفس الحالة إلى علامة الهدف على مستوى الحالة. يتم استخدام Virchow لتوليد تضمينات على مستوى البلاطة في جميع مجموعات البيانات التي تم تقييمها بدقةعندالتكبير. لمقارنة جودة التضمينات بشكل شامل، قمنا بتدريب مجمع لمعدلات التعلم في,ونبلغ عن أفضل درجات AUC للاختبار الملاحظة في الشكل 4b. نظرًا لصغر حجم مجموعات بيانات العلامات الحيوية، لم يتم اختيار معدل التعلم على مجموعة التحقق لتقييم التعميم؛ بل، تعتبر هذه بمثابة معيار عبر الأنواع المختلفة من تضمينات البلاطة (Virchow، UNI، Phikon وCTransPath)، مما يعطي تقديرًا لأفضل أداء ممكن للعلامات الحيوية لكل نوع.
التحليل الإحصائي. يتم استخدام AUC لمقارنة النماذج دون الحاجة إلى اختيار عتبة على قيم الاحتمال المتوقعة للنماذج، والتي قد تختلف حسب مجموعة البيانات الفرعية. تم حساب فترات الثقة ذات الجانبينفي الشكل 4b باستخدام طريقة DeLong.
تقييم مستوى البلاطة
لتقييم Virchow على الصور بحجم البلاطة، يتم وصف بروتوكول الاستكشاف الخطي، بالإضافة إلى أوصاف مجموعة البيانات والتحليل الإحصائي أدناه. تم تلخيص تفاصيل مجموعة البيانات، بما في ذلك تقسيمات التدريب والتحقق والاختبار، أيضًا في الجدول التكميلي 4.1.
بروتوكول الاستكشاف الخطي. لكل تجربة، قمنا بتدريب مصنف بلاطة خطي بحجم دفعة 4,096 باستخدام مُحسِّن الانحدار العشوائي مع جدول معدل تعلم جيبي، من 0.01 إلى 0، لمدة 12,500 تكرار، على قمة التضمينات التي تم إنشاؤها بواسطة مشفر مجمد. العدد الكبير من التكرارات يهدف إلى السماح لأي مصنف خطي بالتقارب بقدر ما يمكن في كل خطوة من خطوات معدل التعلم على طول جدول معدل التعلم. تم تطبيع جميع التضمينات بواسطة-التسجيل قبل التصنيف. لم تستخدم تجارب الاستكشاف الخطي زيادة البيانات. لتقييم مجموعة الاختبار، تم اختيار نقطة تفتيش المصنف التي حققت أقل خسارة على مجموعة التحقق. تم استخدام مجموعة تحقق لجميع المهام. إذا لم يتم توفير واحدة مع مجموعة البيانات العامة، قمنا بتقسيم عشوائيمن بيانات التدريب لإنشاء مجموعة تحقق.
PanMSK. من أجل معيار شامل داخل التوزيع، تم الاحتفاظ بـ 3,999 شريحة عبر 17 نوعًا من الأنسجة في الشكل 1d من مجموعة بيانات التدريب المجمعة من MSKCC. من بين هذه، احتوت 1,456 على سرطان تم توضيحه جزئيًا أو بشكل شامل مع أقنعة تقسيم من قبل أطباء الأمراض. تم استخدام هذه التوضيحات لإنشاء مجموعة بيانات على مستوى البلاطة لتصنيف السرطان مقابل غير السرطان، والتي نشير إليها باسم PanMSK. جميع الصور في PanMSK هيبلاطات بكسل عند 0.5 مpp. انظر الملاحظة التكميلية 5 لمزيد من التفاصيل.
CRC. تحتوي مجموعة بيانات التصنيف العامة CRCعلى 100,000 صورة للتدريب (منها اخترنا عشوائيًا 10,000 للتحقق) و7,180 صورة للاختبار (بكسل) عندالتكبير مصنفة إلى تسع فئات مورفولوجية. يتم إجراء التحليل باستخدام كل من النسخ المعدلة (NCT-CRC-HE-100K) وغير المعدلة (NCT-CRC-HE-100K-NONORM) من مجموعة البيانات. يجب ملاحظة أن مجموعة التدريب تم تطبيعها في كلا الحالتين، وأن مجموعة الاختبار فقط هي غير المعدلة في النسخة الأخيرة. وبالتالي، تتضمن النسخة غير المعدلة من CRC تحولًا في التوزيع من التدريب إلى الاختبار.
WILDS. تتكون مجموعة بيانات Camelyon17-WILDS العامة من 455,954 صورة، كل منها بدقةبكسل، تم التقاطها عندالتكبير وتم تقليل حجمها من. تم اشتقاق هذه المجموعة من مجموعة بيانات Camelyon17 الأكبر وتركز على النقائل اللمفية
. كل صورة في مجموعة البيانات موضحة بعلامة ثنائية تشير إلى وجود أو عدم وجود ورم داخل منطقة البكسل المركزية. تم تصميمها بشكل فريد لاختبار تعميم OOD، تتكون مجموعة التدريب (335,996 صورة) من بيانات من ثلاثة مستشفيات مختلفة، بينما تنشأ مجموعة التحقق الفرعية (34,904 صورة) ومجموعة الاختبار الفرعية (85,054 صورة) كل منهما من مستشفيات منفصلة غير ممثلة في بيانات التدريب.
MHIST. تحتوي مجموعة بيانات التصنيف العامة للزوائد القولونية (MHIST) على 3,152 صورة (بكسل) تعرض إما زوائد مفرطة أو أدينومات مسننة جالسة عندالتكبير (تم تقليل حجمها منلزيادة مجال الرؤية). تحتوي هذه المجموعة على 2,175 صورة في مجموعة التدريب الفرعية (منها اخترنا عشوائيًا 217 للتحقق) و977 صورة في مجموعة الاختبار.
TCGA TIL. تتكون مجموعة بيانات TCGA TIL العامة من 304,097 صورة (بكسل) عندالتكبير، مقسمة إلى 247,822 صورة تدريب، 38,601 صورة تحقق و56,275 صورة اختبار. تعتبر الصور إيجابية لليمفاويات المتسللة للورم إذا كان هناك على الأقل اثنان من TILs موجودين ويتم تصنيفها سلبية بخلاف ذلك. قمنا بزيادة حجم الصور إلىلاستخدامها مع Virchow.
PCam. تتكون مجموعة بيانات PatchCamelyon (PCam) العامة من 327,680 صورة (بكسل) عندالتكبير، تم تقليل حجمها منلزيادة مجال الرؤيةتم تقسيم البيانات إلى مجموعة تدريب (262,144 صورة)، ومجموعة تحقق (32,768 صورة)، ومجموعة اختبار (32,768 صورة). تم تصنيف الصور إما كسرطانية أو حميدة. قمنا بزيادة عدد الصور إلىبكسلات للاستخدام مع فيرشو.
MIDOG. تتكون مجموعة بيانات MIDOG العامة من 21,806 حدثًا انقسامياً وغير انقسامي تم تصنيفها علىمناطق WSI من عدة أنواع من الأورام والأنواع وأجهزة المسحتم تحويل البيانات إلى مهمة تصنيف ثنائية من خلال توسيع كلت annotation بكسل إلىالمناطق ثم التحول عشوائيًا في المناطق الأفقية والعمودية بحيث لا يكون الحدث مركزيًا في البلاطة. تم إزالة جميع الحالات السلبية التي تداخلت مع الحالات الإيجابية من مجموعة البيانات. تتكون مجموعة البيانات الناتجة من مجموعات التدريب والتحقق والاختبار مع 13,107 و 4,359 و 4,340 صورة، على التوالي (منهاو2222 تحتوي على أحداث انقسام خلوي، على التوالي، والباقي يحتوي على عوامل مشوشة تحاكي أحداث الانقسام الخلوي).
تصنيف TCGA CRC-MSI. تتكون مجموعة البيانات العامة لتصنيف TCGA CRC-MSI منالمناطق المأخوذة فيتكبير يعرض عينات من سرطان الغدد المعويةتم استخراج العينات وتوضيحها من TCGA. تم تصنيف المناطق إما على أنها غير مستقرة ميكروساتلايت أو مستقرة ميكروساتلايت. قمنا بتقليل عدد العينات في المناطق إلىلاستخدامه مع فيرشو.
التحليل الإحصائي. يتم استخدام درجة F1 (الموزونة) لمقارنة النماذج حيث أن هذه المقياس قوي أمام عدم توازن الفئات. كما يتم حساب الدقة والدقة المتوازنة، كما هو موضح في الملاحظة التكميلية 4. الجانبانتم حساب فترات الثقة في الشكل 5 والجدول التكميلي 4.2 باستخدام 1000 تكرار للتقنية المعروفة باسم البوتستراب على المقاييس في مجموعة الاختبار دون إعادة تدريب المصنف. تم استخدام اختبار مك نمار لتحديد الدلالة الإحصائية. ) الفروقات بين النتائج.
تحليل الميزات النوعية
قمنا بإجراء تحليل ميزات غير خاضع للإشراف مشابه للإجراء في المرجع 33، باستخدام مجموعة بيانات CoNSeP.شرائح ملونة بصبغة H&E مع سرطان الغدد المعوية. يوفر CoNSeP تعليقات نووية للخلايا في الفئات السبع التالية: الظهارة الطبيعية، الظهارة الخبيثة/المشوهة، الخلايا الليفية، العضلات، الالتهابية، البطانية ومختلفة (بما في ذلك النخرية، الانقسام الخلوي والخلايا التي لم يمكن تصنيفها). لأن صور CoNSeP بحجم 1000و
فيرشو يأخذ صورًا بحجمقمنا بتغيير حجم الصور إلىوقسمتهم إلىشبكة غير متداخلةالصور الفرعية قبل استخراج ميزات مستوى البلاط. بالنسبة لصورة معينة، استخدمنا تحليل المكونات الرئيسية (PCA) على جميع ميزات البلاط من الصور الفرعية، وقمنا بتطبيع المكونين الرئيسيين الأول والثاني إلى قيم ضمنوتم تحديد العتبة عند 0.5. الشكل 5 د يظهر بعض الأمثلة على فصل الميزات غير المراقب الذي تم تحقيقه بهذه الطريقة.
برمجيات
لجمع البيانات، استخدمنا بايثون (الإصدار 3.10.11) مع باندا (الإصدار 2.2.2) لفهرسة البيانات والبيانات الوصفية المستخدمة في التدريب المسبق وتقييم الأداء. تم استخدام OpenSlide (الإصدار 1.3.1) وPillow (الإصدار 10.0.0) لمعالجة صور البلاطات للاختبار. حيثما كان ذلك مناسبًا، قمنا باستخراج تسميات لكل عينة من التقارير السريرية باستخدام DBT (الإصدار 1.5.0). استخدمنا بايثون (الإصدار 3.10.11) لجميع التجارب والتحليلات في الدراسة، والتي يمكن تكرارها باستخدام مكتبات مفتوحة المصدر كما هو موضح أدناه. للتدريب المسبق الذاتي، استخدمنا PyTorch (الإصدار 2.0.1) وTorchvision (الإصدار 0.15.1). تم نقل كود DINO الإصدار 2 من المستودع الرسمي.https://github.com/facebookresearch/ dinov2) وتم تكييفه مع PyTorch Lightning (الإصدار 1.9.0). تم إجراء جميع معالجة WSI أثناء التدريب المسبق عبر الإنترنت وكانت مدعومة من cucim (الإصدار 23.10.0) وtorchvision (الإصدار 0.16.1). لاختبار المهام اللاحقة، نستخدم scikit-learn (الإصدار 1.4.2) للانحدار اللوجستي وحساب المقاييس. تم الحصول على تنفيذات أخرى لمشفرات الصور المدربة مسبقًا التي تم تقييمها في الدراسة من الروابط التالية: UNI (https://huggingface.co/MahmoodLab/UNI), فيكون (https://huggingface.co/owkin/phikon), DINOp=8 (https://github. com/lunit-io/benchmark-ssl-pathology),PLIP(https://huggingface.co/فينيد/بليب)، سيترانس باث (https://github.com/Xiyue-Wang/TransPath) وصورة الطبيعة الأصلية المدربة مسبقًا باستخدام DINO v. 2 (https://github.com/facebookresearch/dinov2).
ملخص التقرير
معلومات إضافية حول تصميم البحث متاحة في ملخص تقارير مجموعة نيتشر المرتبط بهذه المقالة.
توفر البيانات
لم تقم هذه الدراسة بجمع بيانات المرضى بشكل محدد. استخدمت التحليل الرجعي شرائح علم الأمراض الرقمية الكاملة غير المعروفة الهوية والبيانات الوصفية المرتبطة بها التي تم ترخيصها حصريًا بواسطةبايج.إيه آي، شركة من MSKCC. يجب تقديم طلبات البيانات إلى Paige AI (https://paige.ai/contact-us/) وتم تقييمها بواسطة Paige AI و MSKCC على أساس كل حالة على حدة. سيتم منح جميع الطلبات التي تتوافق مع اللوائح الداخلية بشأن خصوصية البيانات وحقوق الملكية الفكرية. استخدمت هذه الدراسة أيضًا مجموعات البيانات المتاحة للجمهور التالية للتقييم اللاحق: CRC (NCT-CRC-HE-100K و NCT-CRC-HE-100K-NONORM، المتاحة عبر Zenodo في https://zenodo.org/records/1214456 (مرجع 77))، WILDS (Camelyon17؛ https://wilds.stanford.edu/get_started), بيكام (https://github.com/basveeling/pcam),MHIST(https://bmirds.github. io/MHIST)، TCGA TIL (متاح عبر زينودو في https://zenodo.org/السجلات/6604094 (المرجع 71))، MIDOG (https://midog.deepmicroscopy. org/download-dataset/) و TCGA CRC-MSI (متاح عبر زينودو على https://zenodo.org/records/3832231 (مرجع 76)).
توفر الشيفرة
يمكن الوصول إلى النموذج لأغراض البحث غير التجاري وتكرار النتائج المبلغ عنها في هذه المخطوطة علىhttps://huggingface. co/paige-ai/Virchow. لمساعدة الباحثين في استخدام نموذجنا، يتوفر مجموعة أدوات تطوير البرمجيات العامة للاستفادة من تضمينات نموذج الأساس لتطوير تطبيقات WSI في الأسفل على https://github. com/Paige-Al/paige-ml-sdk. لقد وثقنا جميع التجارب بتفاصيل كافية في قسم الطرق لدينا لتمكين التكرار المستقل. على الرغم من أنه لا يمكن مشاركة قاعدة الشيفرة الكاملة بسبب الاعتماد على المكتبات الملكية وتكوينات الأجهزة المحددة، مثل الكتل الموزعة والتخزين، فإن المكونات الأساسية لعملنا اعتمد على مستودعات المصادر المفتوحة. تشمل هذه بنية نموذج DINO الإصدار 2 المستخدمة للتدريب الذاتي.https://github.com/facebookresearch/dinov2)، بالإضافة إلى إطار تدريب PyTorch Lightning (https://github.com/Lightning-Al/pytorch-lightning) ومكتبة التورش الأساسية (https://github.com/pytorch/pytorch ) المستخدمة للتدريب والاستدلال مع هذا النموذج.
References
Tarvainen, A. & Valpola, H. Mean teachers are better role models: weight-averaged consistency targets improve semi-supervised deep learning results. In Proc. 31st Conference on Neural Information Processing Systems (eds von Luxburg, U. et al.) 1195-1204 (ACM, 2017).
Xie, Z. et al. Simmim: a simple framework for masked image modeling. In Proc. IEEE/CVF Conference on Computer Vision and Pattern Recognition 9643-9653 (IEEE, 2022).
DeLong, E. R., DeLong, D. M. & Clarke-Pearson, D. L. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics 44, 837-845 (1988).
Holm, S. A simple sequentially rejective multiple test procedure. Scand. J. Statist. 6, 65-70 (1979).
Cochran, W. G. The comparison of percentages in matched samples. Biometrika 37, 256-266 (1950).
McNemar, Q. Note on the sampling error of the difference between correlated proportions or percentages. Psychometrika 12, 153-157 (1947).
Wilson, E. B. Probable inference, the law of succession, and statistical inference. J. Am. Stat. Assoc. 22, 209-212 (1927).
Berx, G. & Van Roy, F. The E-cadherin/catenin complex: an important gatekeeper in breast cancer tumorigenesis and malignant progression. Breast Cancer Res. 3, 289-293 (2001).
Al-Ahmadie, H. A. et al. Somatic mutation of fibroblast growth factor receptor-3 (FGFR3) defines a distinct morphological subtype of high-grade urothelial carcinoma. J. Pathol. 224, 270-279 (2011).
Kalemkerian, G. P. et al. Molecular testing guideline for the selection of patients with lung cancer for treatment with targeted tyrosine kinase inhibitors: American Society of Clinical Oncology endorsement of the College of American Pathologists/ international Association for the Study of Lung Cancer/Association for Molecular Pathology Clinical Practice guideline update. J. Clin. Oncol. https://doi.org/10.1200/JCO.2017.76.7293 (2018).
Chakravarty, D. et al. OncoKB: a precision oncology knowledge base. JCO Precis. Oncol. 1, 1-16 (2017).
Kather, J. N. et al. Predicting survival from colorectal cancer histology slides using deep learning: a retrospective multicenter study. PLoS Med. 16, e1002730 (2019).
Wei, J. et al. A petri dish for histopathology image analysis. In Proc. Artificial Intelligence in Medicine (eds Tucker, A. et al) 11-24 (2021).
Kaczmarzyk, J. R., Abousamra, S., Kurc, T., Gupta, R. & Saltz, J. Dataset for tumor infiltrating lymphocyte classification (304,097 image patches from TCGA). Zenodo https://doi.org/10.5281/ zenodo. 6604093 (2022).
Abousamra, S. et al. Deep learning-based mapping of tumor infiltrating lymphocytes in whole slide images of 23 types of cancer. Front. Oncol. 11, 806603 (2022).
Saltz, J. et al. Spatial organization and molecular correlation of tumor-infiltrating lymphocytes using deep learning on pathology images. Cell Rep. 23, 181-193 (2018).
Veeling, B. S., Linmans, J., Winkens, J., Cohen, T. & Welling, M. Rotation equivariant CNNs for digital pathology. In Proc. Medical Image Computing and Computer Assisted Intervention (eds Frangi, A. F. et al.) 210-218 (2018).
Aubreville, M. et al. A comprehensive multi-domain dataset for mitotic figure detection. Sci. Data 10, 484 (2023).
Kather, J. N. Histological image tiles for TCGA-CRC-DX, color-normalized, sorted by MSI status, train/test split. Zenodo https://doi.org/10.5281/zenodo. 3832231 (2020).
Kather, J. N., Halama, N. & Marx, A. 100,000 histological images of human colorectal cancer and healthy tissue. Zenodo https://doi.org/ 10.5281/zenodo. 1214455 (2018).
شكر وتقدير
نشكر P. Rosenfield من مايكروسوفت و D. Dierov من Paige على مساهماتهما في جعل هذا التعاون ممكنًا. تم دعم البحث المبلغ عنه في هذه المنشورة جزئيًا من خلال منحة دعم مركز السرطان من المعاهد الوطنية للصحة / المعهد الوطني للسرطان (رقم المنحة P30CA008748).
مساهمات المؤلفين
ساهم كل من T.J.F. و S.L. و B.R. و R.Y. و W.A.M. و N.F. و K.S. في تصميم الدراسة وتنسيقها، وبناء شراكة عبر الشركات وفريق بحث والحصول على اتفاقيات مشاركة البيانات. ساهم كل من T.J.F. و B.R. و D.S.K. و C.K. و N.F. كمستشارين بحثيين. قدم كل من D.S.K. و G.O. و E.M. و M.H. و E.Y. و H.W. و J.A.R. إرشادات سريرية. كتب كل من E.V. و A.B. و A.C. و G.S. و M.Z. و S.L. و K.S. و E.Z. و J.H. و N.T. و P.M. و A.v.E. و D.L. و J.V. و E.R. الشيفرة، وطوروا البنية التحتية، ودربوا النماذج طوال فترة الدراسة. عمل كل من E.V. و A.B. و A.C. و G.S. و M.Z. و K.S. و D.L. و Y.K.W. و M.C.H.L. و J.H.B. و R.A.G. و G.O. و E.M. و M.H. و E.Y. و H.W. و J.R. و D.S.K. و S.L. على إعداد البيانات. قام كل من E.V. و A.B. و A.C. و G.S. و M.Z. و K.S. و E.Z. و D.L. و J.V. و E.R. و Y.K.W. و J.D.K. و M.C.H.L. و J.H.B. و R.A.G. و J.R. و S.L. بإجراء التقييم والتحليل. عمل كل من E.V. و A.B. و A.C. و G.S. و M.Z. و K.S. و D.L. و Y.K.W. و J.D.K. و M.C.H.L. و J.H.B. و R.A.G. و C.K. و D.S.K. و S.L. و T.J.F. على صياغة وتنقيح المخطوطة.
المصالح المتنافسة
إي.في، آي.بي، آي.سي، جي.إس، إم.زد، بي.إم، آي.في.إي، دي.إل، جي.في، إي.آر، واي.كي.دبليو، جي.دي.كي، إم.سي.إتش.إل، جي.إتش.بي، آر.إيه.جي، جي.أو، جي.إيه.آر، واي.إيه.إم، آر.واي، دي.كي، إس.إل وتي.جي.إف هم موظفون ومساهمون فيبايج.إيه آي. عمل E.W. و M.H. و C.K. و B.R. كمستشارين لـبايج.إيه آيلقد تلقت D.S.K. تعويضًا عن التحدث والاستشارات من Merck. K.S. وE.Z. وJ.H. وN.T. وN.F. هم موظفون في Microsoft. تحتفظ Memorial Sloan Kettering (MSK) بمصالح مالية وملكية فكرية فيبايج.إيه آيالتي تتعلق بالبحث المقدم في هذه المخطوطة. S.L. و E.V. و A.B. و G.S. و M.Z. و A.C. و J.B. و M.L. و R.G. و T.F. و B.R. هم مخترعون في براءة اختراع أمريكية مؤقتة (رقم الطلب 18/521903) تم تقديمها تتعلق بالجوانب المنهجية لهذا العمل. يعلن المؤلفون الآخرون عن عدم وجود مصالح متنافسة.
الشكل التوضيحي للبيانات الموسعة 1 | مخطط لمجمع أغاتا. أغاتا
يتعلم المجمع الانتباه إلى البلاط الذي يساهم في قرار التسمية باستخدام الانتباه المتقاطع. يتم تعريف العملية باستخدام الاستعلام Q والمفتاح K ومصفوفة القيمة V: سوفتماكس، حيث هو بعد المصفوفة الرئيسية. على عكس آلية الانتباه الذاتي النموذجية حيث يتم توقعها من المدخلات، يتم تحديد Q مباشرة بواسطة النموذج لتقليل استهلاك ذاكرة GPU. عند التجميع عبر عشرات أو مئات الآلاف من البلاطات في عينة، يتطلب الانتباه الكامل الكثير من ذاكرة GPU. يمكن تفسير هذا الانتباه المبسط على أنه مجموع مرجح متعلم لجميع ميزات مستوى البلاطة.
بالفعل، فإن الانتباه الذاتي الكامل يتطلب ذاكرة تربيعية بالنسبة لعدد البلاطات، في حين أن الانتباه في أغاتا خطي. يتم الحصول على K و V من خلال طبقتين متتاليتين من طبقات الإسقاط Gaussian Error Linear Unit (GELU) كما يلي: و حيث x هو تضمين البلاط، و هي معلمات الوزن والانحياز لطبقات الإسقاط. في تجاربنا،يُنتج مفاتيح بُعدها 256يُنتج قيمًا بُعدها 512، ونتجاهل التقييس بواسطةبعد خطوة الانتباه، يتم استخدام طبقتين خطيتين مع تفعيل غير خطي (ReLU) تليهما طبقة خطية نهائية مع تفعيل سوفت ماكس.
الشكل البياني الممتد 2 | مخطط روتين تدريب DINOv2. استخدم فيرشو معمارية ViT-H، تم تدريبها باستخدام DINOv2. من بلاطة واحدة، يتم إنشاء 2 من المحاصيل العالمية و8 من المحاصيل المحلية، جميعها مع تحسينات عشوائية. يتم قناع المحاصيل العالمية بشكل عشوائي وتغذيتها إلى نموذج الطالب، بينما يتم تغذية النسخ غير المقنعة إلى نموذج المعلم. يحاول الطالب إنتاج تمثيل عالمي للمناظر (عبر رمز CLS) يتطابق مع تمثيل المعلم. وجهة نظر معاكسة. يحاول الطالب أيضًا إنتاج تمثيلات لرموز الصورة المخبأة تتطابق مع تمثيلات المعلم لنفس الرموز ولكن غير المخبأة. يتم إمداد الطالب فقط بالمحاصيل المحلية التي تحاول إنتاج تمثيل يتطابق مع تمثيلات المعلم للمحاصيل العالمية. المعلم هو نسخة من الطالب تعتمد على المتوسط المتحرك الأسي (EMA).
الشكل البياني الموسع 3 | نتائج الكشف عن جميع أنواع السرطان لكل موقع منشأ للسرطان. أ. المساحة تحت منحنى (مؤشر التشغيل المستلم) (AUC)؛ ب. الخصوصية عندالحساسية.هو الرأس والعنق. في كلا الرسمين، يتم حساب مقارنة ثنائية للمعنى الإحصائي باستخدام اختبار دي لونغ الثنائي لمنطقة تحت المنحنى (AUC) و
اختبار كوكhran Q يليه اختبار مك نمار للخصوصية، كلاهما مصحح لمقارنات متعددة باستخدام طريقة هولم (*, **** ). تظهر أشرطة الخطأ الجانبين فترة الثقة المحسوبة باستخدام طريقة دي لونغ لـ AUC وطريقة ويلسون للخصوصية.
الشكل 4 من البيانات الموسعة | توزيع مجموعة بيانات السرطان الشامل. أ. عدد العينات حسب موقع أصل السرطان في مجموعة بيانات اختبار السرطان الشامل (هو الرأس والعنق). ب. عدد العينات لكل نوع من الأنسجة في مجموعة بيانات تدريب مجمع السرطان الشامل.
محفظة الطبيعة
المؤلف(المؤلفون) المراسلون:
آخر تحديث بواسطة المؤلفين: 3 يونيو 2024
ملخص التقرير
تسعى Nature Portfolio إلى تحسين إمكانية تكرار العمل الذي ننشره. يوفر هذا النموذج هيكلًا للاتساق والشفافية في التقرير. لمزيد من المعلومات حول سياسات Nature Portfolio، يرجى الاطلاع على سياسات التحرير وقائمة مراجعة سياسة التحرير.
الإحصائيات
لجميع التحليلات الإحصائية، تأكد من أن العناصر التالية موجودة في أسطورة الشكل، أسطورة الجدول، النص الرئيسي، أو قسم الطرق.
تم التأكيد
حجم العينة بالضبط ( ) لكل مجموعة/شرط تجريبي، معطاة كرقم منفصل ووحدة قياس بيان حول ما إذا كانت القياسات قد أُخذت من عينات متميزة أو ما إذا كانت نفس العينة قد تم قياسها عدة مرات إكس اختبار(ات) الإحصاء المستخدمة وما إذا كانت أحادية الجانب أو ثنائية الجانب يجب أن تُوصف الاختبارات الشائعة فقط بالاسم؛ واصفًا التقنيات الأكثر تعقيدًا في قسم الطرق. وصف لجميع المتغيرات المشتركة التي تم اختبارها وصف لأي افتراضات أو تصحيحات، مثل اختبارات الطبيعية والتعديل للمقارنات المتعددة إكس وصف كامل للمعلمات الإحصائية بما في ذلك الاتجاه المركزي (مثل المتوسطات) أو تقديرات أساسية أخرى (مثل معامل الانحدار) والتباين (مثل الانحراف المعياري) أو تقديرات مرتبطة بعدم اليقين (مثل فترات الثقة) إكس لاختبار الفرضية الصفرية، فإن إحصائية الاختبار (على سبيل المثال، ) مع فترات الثقة، أحجام التأثير، درجات الحرية وقيمة ملحوظة أعطِالقيم كقيم دقيقة كلما كان ذلك مناسبًا. لتحليل بايزي، معلومات حول اختيار القيم الأولية وإعدادات سلسلة ماركوف مونت كارلو للتصاميم الهرمية والمعقدة، تحديد المستوى المناسب للاختبارات والتقارير الكاملة عن النتائج تقديرات أحجام التأثير (مثل حجم تأثير كوهين)بيرسون )، مما يشير إلى كيفية حسابها تحتوي مجموعتنا على الويب حول الإحصائيات لعلماء الأحياء على مقالات تتناول العديد من النقاط المذكورة أعلاه.
البرمجيات والشيفرة
معلومات السياسة حول توفر كود الكمبيوتر
جمع البيانات
لجمع البيانات، استخدمنا بايثون (3.10.11) مع باندا (2.2.2) لفهرسة البيانات والبيانات الوصفية المستخدمة في التدريب المسبق وتقييم الأداء. تم استخدام OpenSlide (1.3.1) وPillow (10.0.0) لمعالجة صور البلاطات للاختبار. حيثما كان ذلك مناسبًا، قمنا باستخراج تسميات لكل عينة من التقارير السريرية باستخدام DBT (1.5.0).
بالنسبة للمخطوطات التي تستخدم خوارزميات أو برامج مخصصة تكون مركزية في البحث ولكن لم يتم وصفها بعد في الأدبيات المنشورة، يجب أن تكون البرمجيات متاحة للمحررين والمراجعين. نحن نشجع بشدة على إيداع الشيفرة في مستودع مجتمعي (مثل GitHub). راجع إرشادات مجموعة Nature لتقديم الشيفرة والبرمجيات لمزيد من المعلومات.
بيانات
معلومات السياسة حول توفر البيانات
يجب أن تتضمن جميع المخطوطات بيانًا حول توفر البيانات. يجب أن يوفر هذا البيان المعلومات التالية، حيثما ينطبق:
رموز الوصول، المعرفات الفريدة، أو الروابط الإلكترونية لمجموعات البيانات المتاحة للجمهور
وصف لأي قيود على توفر البيانات
بالنسبة لمجموعات البيانات السريرية أو بيانات الأطراف الثالثة، يرجى التأكد من أن البيان يتماشى مع سياستنا
معلومات السياسة حول الدراسات التي تشمل المشاركين في الأبحاث البشرية والجنس والنوع في البحث.
التقارير حول الجنس والنوع
لم يتم تضمين الجنس أو النوع كمتغير مصاحب في أي مرحلة من مراحل تحليلنا التجريبي.
خصائص السكان
لم نقم بجمع أو استخدام أي متغيرات تتعلق بخصائص السكان في أي مرحلة من مراحل الدراسة.
التوظيف
لم يكن هناك حاجة لتوظيف مرضى لاستخدام صور الشرائح الكاملة لعلم الأنسجة بشكل رجعي.
الإشراف الأخلاقي
لم يكن مراجعة مجلس المراجعة المؤسسية (IRB) قابلة للتطبيق على البحث الموصوف في هذه الدراسة. تم إجراء هذه الدراسة البحثية بشكل رجعي من بيانات غير معروفة الهوية مرخصة لـ Paige.AI، Inc. من مركز ميموريال سلوان كيترينغ للسرطان (MSKCC). تم جمع البيانات المستخدمة في هذه الدراسة جميعها في الأصل للاستخدام السريري من قبل MSKCC في بيئة الممارسة، وبالتالي تعتبر بيانات ثانوية. تم استخدام البيانات التي تم التعرف عليها مسبقًا فقط من قبل MSKCC في التحليل، وتمت إزالة المعرفات الفريدة للمرضى تمامًا من مجموعة البيانات التحليلية.
يرجى ملاحظة أنه يجب أيضًا تقديم معلومات كاملة حول الموافقة على بروتوكول الدراسة في المخطوطة.
التقارير الخاصة بالمجالات
يرجى اختيار الخيار أدناه الذي يناسب بحثك بشكل أفضل. إذا لم تكن متأكدًا، اقرأ الأقسام المناسبة قبل اتخاذ قرارك.
علوم الحياة العلوم السلوكية والاجتماعية العلوم البيئية والتطورية والبيئية
لنسخة مرجعية من الوثيقة بجميع الأقسام، انظر nature.com/documents/nr-reporting-summary-flat.pdf
تصميم دراسة علوم الحياة
يجب على جميع الدراسات الإفصاح عن هذه النقاط حتى عندما يكون الإفصاح سلبيًا.
حجم العينة
لم يتم إجراء أي حسابات لحجم العينة.
تم جمع ما مجموعه 1,488,550 صورة شريحة كاملة لعلم الأمراض H&E لتدريب نموذج الأساس. تشير الأداء المتفوق لنموذجنا المدرب مسبقًا مقارنة بجميع المعايير الأخرى إلى أن حجم العينة كان كافيًا. لمزيد من المعلومات حول مجموعات البيانات اللاحقة، يرجى الرجوع إلى قسم مجموعات البيانات والتقييم في قسم الطرق من المخطوطة.
استبعاد البيانات
لم يتم إجراء أي استبعاد بيانات معين.
التكرار
كانت محاولات التكرار ناجحة للنتائج المبلغ عنها للنموذج. يمكن تطبيق النموذج مفتوح المصدر على https://huggingface.co/ paige-ai/Virchow. يمكن الوصول إلى SDK لتكرار تجارب المجمع على https://github.com/Paige-Al/paige-ml-sdk
العشوائية
لتقييم البيانات اللاحقة التي تتضمن إنشاء تقسيمات التدريب والتحقق والاختبار، استخدمنا التقسيمات الرسمية المقدمة من المحققين الأصليين لكل مجموعة بيانات كلما كانت متاحة. عندما لم تكن هذه التقسيمات متاحة، قمنا بإنشائها عشوائيًا. بشكل عام، قمنا بإنشاء تقسيمات عشوائية مصنفة حسب الفئة، مع ضمان نسب فئات مماثلة عبر التقسيمات، وإذا كان ذلك ممكنًا، على مستوى المرضى، مع ضمان بقاء الشرائح من نفس المريض ضمن نفس التقسيم. كانت البذور العشوائية ثابتة، وتم توثيق التقسيمات لضمان إمكانية التكرار.
التعمية
التعمية ليست ضرورية لدراستنا.
التقارير عن المواد والأنظمة والأساليب المحددة
نحتاج إلى معلومات من المؤلفين حول بعض أنواع المواد والأنظمة التجريبية والأساليب المستخدمة في العديد من الدراسات. هنا، حدد ما إذا كانت كل مادة أو نظام أو طريقة مدرجة ذات صلة بدراستك. إذا لم تكن متأكدًا مما إذا كان عنصر القائمة ينطبق على بحثك، اقرأ القسم المناسب قبل اختيار رد.
المواد والأنظمة التجريبية
الطرق
غير متاح
مشارك في الدراسة
غير متاح
مشارك في الدراسة
X
X
X
علم الحفريات وعلم الآثار
X
البيانات السريرية
(T) تحقق من التحديثات
¹Paige، نيويورك، نيويورك، الولايات المتحدة. ²Microsoft Research، كامبريدج، ماساتشوستس، الولايات المتحدة. مركز ميموريال سلوان كيترينغ للسرطان، نيويورك، نيويورك، الولايات المتحدة. NSW Health Pathology، مستشفى سانت جورج، سيدني، نيو ساوث ويلز، أستراليا. جامعة روتشستر، روتشستر، نيويورك، الولايات المتحدة. ساهم هؤلاء المؤلفون بالتساوي: يوجين فورونتسوف، أليكان بوزكورت، آدم كاسون، جورج شايكوفسكي، ميشال زيلتشوفسكي، كريستين سيفرسون. البريد الإلكتروني: siqi.liu@paige.ai
بما في ذلك جزء صغير من أنسجة البروستاتا (أزرق)، والثدي (أزرق) وBLN (أصفر) التي تم تدريب هذه النماذج السريرية عليها. د، تصنيف نماذج الفشل لنموذج السرطان الشامل وأربعة أمثلة نموذجية على الأنواع الرئيسية من الفشل. في جميع اللوحات، * يستخدم للإشارة إلى الأهمية الإحصائية الزوجية (*P<0.05، **P<0.01، ***P<0.001، ****P<0.0001؛ اختبار DeLong الزوجي). تشير أشرطة الخطأ إلى فترة الثقة 95% ذات الجانبين، المقدرة بطريقة DeLong. C.، سرطان. Inv.، غازية.
The analysis of histopathology images with artificial intelligence aims to enable clinical decision support systems and precision medicine. The success of such applications depends on the ability to model the diverse patterns observed in pathology images. To this end, we present Virchow, the largest foundation model for computational pathology to date. In addition to the evaluation of biomarker prediction and cell identification, we demonstrate that a large foundation model enables pan-cancer detection, achieving 0.95 specimen-level area under the (receiver operating characteristic) curve across nine common and seven rare cancers. Furthermore, we show that with less training data, the pan-cancer detector built on Virchow can achieve similar performance to tissue-specific clinical-grade models in production and outperform them on some rare variants of cancer. Virchow’s performance gains highlight the value of a foundation model and open possibilities for many high-impact applications with limited amounts of labeled training data.
Pathologic analysis of tissue is essential for the diagnosis and treatment of cancer. Increasingly, the traditional histological preparations used for light microscopy examination are being replaced by their digital counterparts, also known as whole-slide images (WSIs), which enables the use of computational pathology to move from primarily academic proof points to routine tools in clinical practice. Computational pathology applies artificial intelligence (AI) to digitized WSIs to support the diagnosis, characterization and understanding of disease . Initial work has focused on clinical decision support tools
to enhance current workflows , and in 2021 the first Food and Drug Administration-approved AI pathology system was launched . However, given the incredible gains in performance of computer vision, a subfield of AI focused on images, more recent studies attempt to unlock new insights from routine WSIs and reveal undiscovered outcomes such as prognosis and therapeutic response . If successful, such efforts would enhance the utility of hematoxylin and eosin (H&E)-stained WSIs and reduce reliance on specialized and often expensive immunohistochemistry (IHC) or genomic testing .
Fig. 1| Overview of the study. The training dataset, training algorithm and application of Virchow, a foundation model for computational pathology.
a, The training data can be described in terms of patients, cases, specimens, blocks or slides, as shown. b-d, The slide distribution as a function of cancer
status (b), surgery (c) and tissue type (d). e, The dataflow during training requires processing the slide into tiles, which are then cropped into global and local views. , Schematic of applications of the foundation model using an aggregator model to predict attributes at the slide level. GI, gastrointestinal.
A major factor in the performance gains of computer vision models has been the creation of large-scale deep neural networks, termed foundation models. Foundation models are trained on enormous
datasets-orders of magnitude greater than any used historically for computational pathology-using a family of algorithms, referred to as self-supervised learning (for example, refs. 22-26), which do not require
curated labels. Foundation models generate data representations, called embeddings, that can generalize well to diverse predictive tasks . This offers a distinct advantage over current diagnostic-specific methods in computational pathology, which, limited to a subset of pathology images, are less likely to reflect the full spectrum of variations in tissue morphology and laboratory preparations necessary for adequate generalization in practice. The value of generalization from large datasets is even greater for applications with inadequate quantities of data to develop bespoke models, as is the case for the detection of uncommon or rare tumor types, as well as for less common diagnostic tasks such as the prediction of specific genomic alterations, clinical outcomes and therapeutic response. A successful pathology foundation model should capture a broad spectrum of patterns, including cellular morphology, tissue architecture, staining characteristics, nuclear morphology, mitotic figures, necrosis, inflammatory response, neovascularization and biomarker expression and therefore would be well-suited to predicting a wide variety of WSI characteristics. If trained with a sufficiently large quantity of digitized WSIs in the pathology domain, such a model could form the basis for clinically robust prediction of both common and rare cancers, as well as for other critical tasks such as subtyping of cancer, quantification of biomarkers, counting of cellular instances and events and the prediction of therapeutic response.
Foundation model performance crucially depends on dataset and model size, as demonstrated by scaling law results . Modern foundation models in the natural image domain use millions of images (for example, ImageNet , JFT-300M and LVD-142M ) to train models with hundreds of millions to billions of parameters (for example, vision transformers (ViTs) ). Despite the challenges in collecting large-scale datasets in the pathology domain, recent pioneering works have utilized datasets ranging from 30,000 to 400,000 WSIs to train foundation models ranging in size from 28 million to 307 million parameters (see Supplementary Note 1 for a detailed summary of recent models). These works demonstrate that image features produced with self-supervised learning of pathology images outperform image features trained on natural images and that performance improves with scale.
Here, we present a million-image-scale pathology foundation model, Virchow, named in honor of Rudolf Virchow, who is regarded as the father of modern pathology and proposed the first theory of cellular pathology . Virchow is trained on data from approximately 100,000 patients corresponding to approximately 1.5 million H&E stained WSIs acquired from Memorial Sloan Kettering Cancer Center (MSKCC), which is more WSIs than in prior training datasets in pathology (detailed in Fig. 1a and ‘Million-scale training dataset’ in Methods). The training data are composed of cancerous and benign tissues, collected via biopsy (63%) and resection (37%), from 17 high-level tissues (Fig. 1b-d). Virchow, a 632 million parameter ViT model, is trained using the DINO v. 2 algorithm , a multiview student-teacher self-supervised algorithm (Fig. 1e; see ‘Virchow architecture and
training’ in Methods for training details). DINO v. 2 leverages global and local regions of tissue tiles to learn to produce embeddings of WSI tiles (Fig. 1e), which can be aggregated across slides and used to train a variety of downstream predictive tasks (Fig.1f).
Motivated by highlighting the potential clinical impact of a pathology foundation model, we assess the performance of a pan-cancer model trained using the Virchow embeddings to predict specimen-level cancer across different tissues. Virchow embeddings outperform or match all baseline models on all tested cancer types, notably including rare cancers and out-of-distribution (OOD) data. Quantitative comparison to three specialized clinical-grade AI products demonstrates that the pan-cancer model performs nearly as well as the clinical products in general and outperforms them on some rare variants of cancers. To provide evidence for potential focus areas for future advances in computational pathology, qualitative analysis is also performed, characterizing the error patterns where the AI model fails to identify or falsely identifies cancerous cells. Motivated by simplifying clinical workflows, we evaluated the use of Virchow embeddings to train biomarker prediction, generally outperforming other models. Overall, our results provide evidence that large-scale foundation models can be the basis for robust results in a new frontier of computational pathology.
Results
The Virchow model embeddings were evaluated on two categories of slide-level computational pathology applications: pan-cancer detection (‘Virchow enables pan-cancer detection’ and ‘Towards clinical-grade performance’) and biomarker prediction (‘Biomarker detection in routine imaging obviates additional testing’). These tasks require training a weakly supervised aggregator model to group tile embeddings to slide-level predictions. A series of tile-level linear probing benchmarks were also performed to directly assess the embeddings on individual tissue tiles (‘Tile-level benchmarks and qualitative analysis demonstrate generalizability’).
Virchow enables pan-cancer detection
A key aim of our work was to develop a single model to detect cancer, including rare cancers (defined by the National Cancer Institute (NCI) as cancers with an annual incidence in the United States of fewer than 15 people per 100,000 (ref. 46)), across various tissues. The pan-cancer detection model infers the presence of cancer using Virchow embeddings as input. For evaluation, slides from MSKCC and slides submitted for consultation to MSKCC from numerous external sites globally are used. Stratified performance across nine common and seven rare cancer types is reported. Embeddings generated by Virchow, UNI , Phikon and CTransPath are evaluated. Pan-cancer aggregators are trained using specimen-level labels, maintaining the same training protocol for all embeddings (see ‘Pan-cancer detection’ in Methods for data and training details).
Fig. 2 | Virchow enables training a robust pan-cancer detector. Pan-cancer detection results. Detection is predicted at the specimen level using an aggregator network trained with Virchow, UNI, Phikon or CTransPath tile embeddings as input.a, Cancer detection performance (AUC) stratified by cancer type as determined by origin tissue. The incidence rate and proportion of metastasis of each cancer are shown. Virchow embeddings enable the best cancer detection performance across all cancer types, including rare cancers. For each cancer type, the AUC corresponding to the statistically significantly ( ) top-performing embeddings is highlighted in magenta. When more than one AUC is not gray, performance is ‘tied’ (no statistically significant difference). The foundation model used to produce tile embeddings for the aggregator is shown in the margin on the left, along with the number of cancer types for which the corresponding aggregator achieved (or tied for) the top AUC. All statistical significance (a-e) is computed using the pairwise DeLong’s test for AUC and Cochran’s test followed by McNemar’s test for specificity, both corrected for
multiple comparisons with Holm’s method. b,c, Cancer detection performance summarized for all cancers (b) and for rare cancers (c). Error bars (b-e) show the two-sided 95% confidence interval computed with DeLong’s method for AUC and Wilson’s method for specificity; the – denotes the differences that are statistically significant from the rest ( ). d, Sensitivity at specificity for rare cancer detection ( ). e, Virchowbased cancer detection generalizes well to data from external institutions that were not represented in the training set; all aggregators and Virchow were trained only on data from MSKCC. Only half of the specimens in the pan-cancer testing set are from MSKCC.f, One-fifth of the specimens used for pan-cancer model evaluation contained tissues that were not observed in the training sets of Virchow or the pan-cancer aggregators. g, Cancer detection performance scales with the size of the underlying foundation model and the number of training samples (tiles) used to train it. H&N, head and neck.
Virchow embeddings yielded the best cancer detection performance on all cancer types (Fig. 2a). Pan-cancer detection using UNI embeddings achieved statistically similar performance ( ) for eight of the nine common cancer types and five of the seven rare cancer types; nevertheless, in all but one case, the specific area under (the receiver operating characteristic) curve (AUC) score was lower. Overall the pan-cancer model achieved an AUC of 0.950 with Virchow embeddings, 0.940 with UNI embeddings, 0.932 with Phikon embeddings and 0.907 with CTransPath embeddings (Fig. 2b; all significantly different with ). See Extended Data Fig. 3 for more detailed AUC and specificity metrics, stratified by cancer type.
Rare cancer detection performance is particularly noteworthy. Compared to the aforementioned AUC of 0.950 overall, Virchow embeddings yielded an AUC of 0.937 on rare cancers (Fig. 2c), demonstrating generalization to rare data. Performance across the individual rare cancers was, however, non-uniform, with detection of cervical and bone cancers proving more challenging (AUC < 0.9) irrespective of the embeddings used (Fig. 2a,d). Virchow embeddings improved cervix detection to 0.875 AUC compared with or 0.753 when using UNI, Phikon or CTransPath embeddings, respectively. Similarly, Virchow embeddings yielded 0.841 AUC for bone cancer detection, compared to and 0.728 with UNI, Phikon and CTransPath, respectively. At sensitivity, we show that a pan-cancer detection model using Virchow embeddings can achieve specificity, compared to , 62.9% or 52.3% using UNI, Phikon or CTransPath embeddings, respectively, trained on less data (Fig. 2b).
The robustness of Virchow embeddings to data sampled from a different population than the training set (OOD data) is evaluated directly with data from institutions other than MSKCC (both Virchow and the pan-cancer aggregator were trained only on data from MSKCC) and indirectly by including data from tissues which were not observed during training (Fig. 2e,f). As AUC measures cannot be exactly compared across different data subsets (due to different positive to negative sample ratios), we report AUC for all pan-cancer models on all data or rare cancers (Fig. 2b), as well as on internal or external data (Fig. 2e), and demonstrate that the AUC differences across models remain consistent in each subpopulation. This demonstrates that Virchow embeddings generalize well to new or rare data and outperform the others consistently. Although AUC cannot be exactly compared across data subsets, we can observe that all models achieve a similar AUC on both internal and external data, suggesting that they generalize well as external data can be challenging because it is submitted to MSKCC for consultation. Furthermore, cervix, testis and head and neck (H&N) are tissues not seen during training, and Virchow embeddings still outperform competing models. Overall, pan-cancer detection generalizes across cancer types, including rare cancers, as well as on OOD data when using foundation model embeddings.
The comparison of pan-cancer performance based on different foundation model embeddings reveals that performance scales with the size of the foundation model and the size of the training data (Fig. 2g). Cancer detection was found to scale approximately logarithmically with the number of model parameters (Fig. 2g, top); although performance scaled with the number of training tile samples, the trend (Fig. 2g, bottom) suggests diminishing returns. Although the training datasets, model architectures and optimization strategies differ across Virchow, UNI, Phikon and CTransPath, there are enough similarities
Fig. 3 | Pan-cancer detection approaches and sometimes surpasses clinical product performance, using less data. a,b, Performance as measured by AUC of three clinical products compared to the pan-cancer model trained on Virchow embeddings, on the rare variant (a) and product testing datasets (b). The pancancer detector, trained on Virchow foundation model embeddings, achieves similar performance to clinical-grade products in general and outperforms them on rare variants of cancers. c, The pan-cancer detector was trained on fewer labeled specimens than the Prostate, Breast and BLN clinical models,
to motivate the scaling analysis. All models are transformer-based: CTransPath uses a Swin transformer , and the rest use ViTs of different sizes. Phikon was trained using the iBOT algorithm , and both Virchow and UNI were trained using the DINO v. 2 algorithm with similar hyperparameters. iBOT and DINO v. 2 are related approaches as the latter builds on the masked image modeling proposal of the former. CTransPath is differentiated in terms of training algorithm as it used a contrastive learning algorithm based on MoCov3 (ref. 48). To learn about the effect of dataset size independent of model size, we direct the reader to the study in ref. 41.
Toward clinical-grade performance
A promise of foundation models is improved generalization; however, this claim is difficult to verify without access to rigorously trained and tested tissue-specific specialist models. To this end, we conducted a comparative analysis between the Virchow-based pan-cancer detection model and specialist commercial models, specifically Paige Prostate, Paige Breast and Paige Breast Lymph Node (BLN). The comparison focuses on the AUC for cancer detection, specifically for prostate cancer, invasive breast cancer and metastases of breast cancer in lymph nodes. These commercial models were trained using multiple-instance weakly supervised learning as described in refs. 14,49 specifically for cancer detection. The evaluation was performed in two settings: (1) product testing datasets and (2) rare cancer variant datasets in the respective tissues (Fig. 3b-d).
The Virchow-based pan-cancer detection model, trained on cancers across numerous tissues, performs nearly as well as the prostate, breast and BLN clinical specialist models (Fig. 3c) while outperforming them on many rare variants of cancers (Fig.3d). It is important to note that the pan-cancer training set did not benefit from the same refinement as the product training sets, such as enrichment for subpopulations and label quality control. Furthermore, the pan-cancer model was trained on fewer tissue-specific specimens than the clinical models (Fig. 3 and Extended Data Fig. 4). Concretely, Paige Prostate was trained on 66,713 blocks, Paige Breast was trained on 44,588 specimens and BLN on 8150 specimens, whereas pan-cancer (using Virchow embeddings) was trained on only 35,387 groups of slides (blocks or specimens) in total, of which 2,829 are prostate, 1,626 are breast and 1,441 are lymph node. The pan-cancer model achieves an AUC of and 0.971 on prostate, breast and BLN, respectively. This performance approaches that of commercial models; however, it is still surpassed by the Food and Drug Administration-approved Paige Prostate model ( 0.980 versus 0.995 AUC, ) and the Paige Breast model ( 0.985 versus . On the other hand, it is statistically significantly better at detecting macrometastases than Paige BLN ( 0.999 versus 0.994 AUC, ). Furthermore, there is no statistically significant difference ( ) in the other BLN comparisons or some of the stratified breast cancer comparisons (Fig. 3c).
In addition to approaching the specialist models in terms of overall AUCs, the pan-cancer model matches or outperforms these models on rare variants of cancers, as shown in Fig. 3d. In prostate and lymph node tissues, the pan-cancer model is capable of detecting lymphoma. This is particularly noteworthy because none of the models were trained in hematolymphoid malignancies. Owing to their different lineage (carcinomas originate from epithelial cells, whereas lymphomas arise from lymphoid tissue) their morphologic appearance tends to be quite
different. In two of the four lymphoma variants, the pan-cancer model outperforms the specialized model. Improved detection of diffused large B-cell lymphoma is noteworthy as this variant is particularly aggressive. In breast tissue, the pan-cancer model outperforms the
Paige Breast model overall and especially on some rare histological variants, including adenoid cystic carcinoma, carcinoma with apocrine differentiation ( ), metaplastic carcinoma spindle cell ( ), metaplastic carcinoma squamous cell and the exceptionally unusual
Rare variants prediction: pan-cancer model outperforms clinical-grade products on some rare variants of prostate, breast and lymph node cancers
a
b
C
Training set sizes: specimens
d
Poorly differentiated carcinoma in pancreas
Crushed focus with adenocarcinoma in peritoneum
Reactive fibroinflammatory changes in prior biopsy site
Crushed lymphocytes and a tissue fold
Fig. 4 | Biomarker prediction results. a, Virchow embeddings help predict biomarkers directly from slides, reducing the need for targeted sequencing or IHC staining. b, The fraction of positive cases in each biomarker testing dataset. c, The number of biomarkers on which using Virchow, UNI, Phikon or CTransPath embeddings to train an aggregator produced an AUC in the top . This ranking does not consider statistical significance across models for each biomarker due to low statistical power; instead, it relies on considering
secretory carcinoma. We note that due to the rarity of these variants of cancers, rare variants prediction lacks the statistical power of the product datasets.
To comprehend the error patterns of the pan-cancer model across various tissues, a pathologist examined the error cases within a curated set of evaluation WSIs (see ‘Pan-tissue product benchmark’ in the section ‘Clinical evaluation datasets’ in Methods). The operating point for each tissue was selected to achieve approximately sensitivity and specificity on a tuning dataset. These error patterns were documented using free text first, which was subsequently categorized to provide a comprehensive summary. We posit that these patterns could be beneficial to similar cancer detection studies, providing valuable insights for the enhancement of future foundational models and clinical AI applications. The false positive and false negative patterns were analyzed separately, as depicted in Fig. 3e.
Upon analysis of the false positive and false negative cases, it was discerned that a substantial proportion could be attributed to specific findings. Histological preparations that contained only small tumoral foci constituted the majority ( ) of the false negatives. Certain neoplasms, undetected as cancer (11.9%), were of borderline malignant potential, such as gastrointestinal stromal tumors or borderline serous
neoplasm of the ovary. Others (9.5%), such as low-grade astrocytoma, exhibited only very subtle histologic features of malignancy. Treatment effects, extensive necrosis and tissue artifacts obscuring the cancer accounted for a few false negatives. In 11 cases (26.2%), there was more than minimal cancer within the specimen, and the negative result of the model could not be explained.
The majority of the false positive cases fell into two categories. Precursor lesions in specimens lacking invasive cancer constituted most (53.2%) of the false positives. These were found most frequently in the bladder, breast, cervix, skin (squamous dysplasia) and esophagus. Most detected precursors exhibited high-grade dysplasia, with cytologic features resembling those of invasive carcinoma, although some foci of low-grade dysplasia were also detected in the gastroesophageal junction and skin. The second most common ( ) cause of false positive results was tissue artifacts, especially crush artifacts (in which non-neoplastic cells are physically crushed during sample preparation, resulting in a characteristic streaming effect of the nuclei), tissue folds and out-of-focus regions. Reactive alterations within the stroma or lymphoid components, constituting , and in non-neoplastic epithelial tissue, representing , were also responsible for false positive results. A number of these findings, such as biopsy site changes,
reactive epithelial atypia, glandular atrophy and acellular stromal mucin, are well-recognized malignant mimics that challenge pathologists as well. Three cases (3.2%) were benign neoplasms misidentified as cancer. These included benign gastrointestinal stromal tumors, hepatic angiomyolipomas and serous cystadenomas of the pancreas.
Biomarker detection in routine imaging obviates additional testing
The prediction of biomarkers from standard H&E stained images can reduce the reliance on testing using additional methods and the associated substantial delays in returning results to patients (Fig. 4a). The status of a biomarker in a specimen is predicted using an aggregator network with the foundation model embeddings as input. These biomarkers play a crucial role in the diagnosis and treatment of various cancers, and each is described in further detail in ‘Biomarker detection’ in Methods (see also Supplementary Table 3.1 and Fig. 4b). The biomarker detection datasets consist of WSIs from the histological sections matching the blocks used for DNA extraction and MSK-integrated mutation profiling of actionable targets (MSK-IMPACT) sequencing , the latter of which was analyzed to determine the status of genetic alterations and establish a binary label indicating the presence or absence of the variants: that is, the biomarker (Fig. 4a). Similar to the pan-cancer evaluation, the publicly available UNI , Phikon and CTransPath models are used as baseline models for comparisons.
We note that the biomarker prediction results lacked sufficient statistical power to assess statistically significant differences across models; instead, we conclude relative model performance from evaluating many different biomarker predictions. In our comparative analysis shown in Fig. 4c, Virchow embeddings demonstrated superior performance in seven of the nine evaluated digital biomarkers, achieving AUC scores that exceeded those of the nearest baseline foundation models. This performance underscores the robustness of Virchow embeddings across a diverse range of biomarkers. Even in the categories of prostate-androgen receptor (AR) and ovarian-fraction of genome altered (FGA), where Virchow did not secure the top position, it remained a strong contender, with AUCs of 0.849 and 0.847 , respectively. These findings underscore the potential of Virchow embeddings to accurately represent H&E histologic phenotypes, offering predictive insights into biomarkers that are traditionally identified through DNA extraction and MSK-IMPACT sequencing.
Tile-level benchmarks and qualitative analysis demonstrate generalizability
To directly evaluate tile-level embeddings without the confounder of training an aggregator network, we evaluated Virchow performance on a set of tile-level benchmarks by linear probing. Linear probe evaluation aims to gauge the quality and separability of representations learned by a self-supervised model. We compare Virchow embeddings to baseline model embeddings by applying the same linear probing protocol for each model, using the same training, validation and testing data splits (see ‘Tile-level benchmarking’ in Methods for further details). The analysis is performed both on public datasets and on an internal MSKCC pan-cancer dataset.
The internal multitissue dataset for pan-cancer detection at the tile level (referred to as PanMSK) is an in-distribution benchmark, as it is composed of annotations on a held out set of patients across the entire diverse set of tissue groups selected for training (Fig. 1d).
The public datasets are OOD benchmarks and are described in the ‘Tile-level benchmarking’ section in Methods. In addition to UNI , Phikon and CTransPath , DINO (ref. 39) ( 49 million parameter model trained using The Cancer Genome Atlas (TCGA) and an internal dataset), PLIP ( 87 million parameter model trained using pathology image-text pairs) and NatImg (1.1 billion parameter model trained on 142 million natural images) are evaluated.
As shown in Fig. 5a,c, Virchow embeddings match or surpass the performance of other embeddings in seven of the eight benchmark tasks (Fig. 5a,b; see Supplementary Table 4.2 for additional metrics). The closest competing models are UNI and Phikon, with UNI scoring in the top1 three times and in the top2 for all tasks and Phikon scoring in among the top 2 three times. Virchow demonstrates strong OOD performance as measured by the WILDS and ‘CRC (no norm)’ tasks. The WILDS test data is sourced from a hospital that is not encountered in the training set. The ‘CRC (no norm)’ task introduces a distribution shift from the stain-normalized training set by avoiding stain normalization on the testing set. Without normalization, Virchow’s performance declines by only -0.005 in weighted score, indicating robustness to variations in data preprocessing.
To qualitatively evaluate whether the embeddings learned by Virchow tend to separate the image into semantically meaningful clusters of features, we performed an unsupervised feature analysis similar to the procedure in ref. 33 using the CoNSeP dataset , which contains H&E stained slides of colorectal adenocarcinoma (detailed under ‘Qualitative feature analysis’ in Methods).
We observe approximate semantic segmentation of the cell types in the CoNSeP images (Fig. 5d). In both examples, the first principal component highlighted malignant epithelium (red) cells. The second principal component, respectively, highlighted miscellaneous cells (yellow) and inflammatory (magenta) cells. DINO v. 2 was shown to learn a similar semantic feature separation on natural images, allowing foreground/background separation (for example, discriminating a bus or a bird from the background) as well as part annotation (for example, wheels versus windows in a bus) . Here, we show that this emerging property of the model carries over to the pathology domain. This encouraging result supports our expectation that the unsupervised features learned by Virchow are meaningful and interpretable for a wide range of downstream tasks.
Discussion
The value of a pathology foundation model is twofold: generalizability and training data efficiency. In our study, we demonstrate both of these benefits. Virchow-based pan-cancer prediction generalized well to tissue types or slides submitted from institutions not observed in the training data. Rare histological subtypes of cancer were detected nearly as well as common variants. The same pan-cancer detection model was shown to almost match the performance of clinical-grade models overall (AUC from 0.001 to 0.007 behind clinical products, ) and surpassed them in the detection of some rare variants of cancers, despite training with fewer tissue-specific labels. This result is even more impressive when noting that the training dataset of the pan-cancer model, as a proof of concept, lacks the quality control and subpopulation enrichment of data and labels that are typically done for commercially available AI models. Finally, we note that Virchow embeddings were not fine-tuned, and models used simple aggregator architectures to make predictions. These results build confidence that,
Fig. 5 | A summary of tile-level linear probing. a, A description of each tile-level benchmark (top) along with the corresponding results for the embeddings of different foundation models (bottom). For each task, the top result is bolded and highlighted in magenta. Multiple results are highlighted when there is no statistically significant difference between them ( ; McNemar’s test). Error bars denote two-sided 95% confidence intervals computed using 1,000
bootstrapping iterations.b, The number of tasks in which each model scored in the top . Models can tie for a rank depending on statistical significance ( ). c, Virchow embedding features learn meaningful structures. Cells in the CoNSeP dataset highlighted by embedding principal components: malignant epithelium (red), miscellaneous (yellow) and inflammatory (magenta).
with sufficient scale, foundation models will serve as the building blocks for the future development of a wide variety of downstream tasks.
There are a few areas in which we anticipate particularly high-value impact. In clinical practice, where most biopsy samples are benign, a pan-cancer detection system can prioritize cases to help reduce diagnostic turnaround. With decreasing training data requirements, clinical-grade products for less common cancers could be developed. Biomarker prediction using routine H&E WSIs would increase screening rates; reduce intrusive, tissue-destructive testing; and rapidly provide the data needed to make more informed treatment decisions. Virchow embeddings demonstrated sufficiently high performance to suggest these tools are achievable. Indeed, Virchow unlocks the ability to accurately and precisely detect unusual histological variants of cancer as well as biomarker status, something that is difficult to achieve with cancer- or biomarker-specific training due to the limited amount of associated training data.
Despite the observed improvements, there are still aspects of Virchow’s development that merit further discussion. Histopathology data differs from natural image data in key ways: the long-tailed distribution of pathologic entities and histological structures, the lack of object scale diversity and the restricted color space. Self-supervised learning algorithms attempt to match the inductive biases of the learning algorithm to the data distribution; however, in this work, as in many other works in self-supervised learning for computational pathology, algorithmic and training settings are largely based on what was successful in the natural image domain. Further study may reveal that altering these design choices will further improve performance in the pathology domain.
It remains an open question at what point the model and data scale are saturated. We found that pan-cancer detection performance scales with model and dataset size (Fig. 2g), which is consistent with observations of prior foundation models in other domains . The improvement in performance with respect to model size appears to still be in an approximately log-linear range; however, sub-log-linear trends were observed as a function of training data. Trends in training data size may be oversimplified as they do not capture the tradeoff between increasing the number of WSIs versus tiles. The setting is too complex to draw precise conclusions about the effect of dataset diversity, although we posit that increased diversity helps to learn robust and rare features. Indeed, it has been shown that training a model on multiple tissues or cancer variants can improve detection performance for each cancer , as many morphological features are observed across cancers from different topographies . Overall, our investigation into scaling behavior suggests that increasing the number of model parameters remains a salient axis to explore.
Our work has several limitations. The training dataset is acquired from one center with limited scanner types. As with most histopathology self-supervised models, embeddings are generated at the tile level using magnification ( 0.5 mpp ) as opposed to the slide level and therefore require training an aggregation model. Although scaling up the size of a tile-level foundation model may improve performance, it is likely that such models must be extended to the slide level to achieve the data efficiency required for low-data tasks such as the prediction of biomarkers, treatment response or clinical outcome. A deep investigation of aggregator architectures and training procedures is beyond the scope of this work. As is the case for all models aiming for clinical application, thorough stratified performance validation is required. Furthermore, hardware considerations must be made toward the deployment of models the size of Virchow or larger; model distillation may be appropriate for some tasks. Due to the scale of training, our study has not been able to fully explore the effectiveness of data-balancing and -distillation strategies. The challenge of curating training data that preserves rare features while reducing redundancy remains an open question. Considering the long-tail distribution in digital pathology, we question the suitability of clustering-based data
distillation methods such as those used in the original DINO v. 2 model for natural images .
Recent advances in computational pathology have been supported by increased dataset scale and reduced reliance on labels. Using multiple-instance learning with labels at the level of groups of slides has enabled clinically relevant diagnostics by scaling to training datasets on the order of . These earlier works typically initialized the model’s embedding parameters using pretrained model weights, often those trained on ImageNet in a supervised setting. This process, called transfer learning, was motivated by the observation that model performance critically depends on the model’s ability to capture image features. In-domain transfer learning was not possible given the limited availability of labeled pathology datasets. Now self-supervised learning is enabling in-domain transfer by removing the label requirement, driving a second wave of scaling to tens of thousands of WSIs to inform image representation . Virchow marks a major increase in training data scale to 1.5 million WSIs-a volume of data that is over 3,000 times the size of ImageNet as measured by the total number of pixels. This large scale of data in turn motivates large models that can capture the diversity of image features in WSIs. In this work, we have demonstrated that this approach can form the foundation for clinical-grade models in cancer pathology.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41591-024-03141-0.
References
Deng, S. et al. Deep learning in digital pathology image analysis: a survey. Front. Med. 14, 470-487 (2020).
Srinidhi, C. L., Ciga, O. & Martel, A. L. Deep neural network models for computational histopathology: a survey. Med. Image Anal. 67, 101813 (2021).
Cooper, M., Ji, Z. & Krishnan, R. G. Machine learning in computational histopathology: challenges and opportunities. Genes Chromosomes Cancer 62, 540-556 (2023).
Song, A. H. et al. Artificial intelligence for digital and computational pathology. Nat. Rev. Bioeng. 1, 930-949 (2023).
Fuchs, T. J. & Buhmann, J. M. Computational pathology: challenges and promises for tissue analysis. Comput. Med. Imaging Graph. 35, 515-530 (2011).
Abels, E. et al. Computational pathology definitions, best practices, and recommendations for regulatory guidance: a white paper from the digital pathology association. J. Pathol. 249, 286-294 (2019).
Fuchs, T. J., Wild, P. J., Moch, H. & Buhmann, J. M. Computational pathology analysis of tissue microarrays predicts survival of renal clear cell carcinoma patients. In Proc. Medical Image Computing and Computer-Assisted Intervention (eds Metaxas, D. et al.) 1-8 (Springer, 2008).
Kong, J. et al. Computer-aided evaluation of neuroblastoma on whole-slide histology images: classifying grade of neuroblastic differentiation. Pattern Recognit. 42, 1080-1092 (2009).
Bejnordi, B. E. et al. Diagnostic assessment of deep learning algorithms for detection of lymph node metastases in women with breast cancer. JAMA 318, 2199-2210 (2017).
Raciti, P. et al. Clinical validation of artificial intelligenceaugmented pathology diagnosis demonstrates significant gains in diagnostic accuracy in prostate cancer detection. Arch. Path. Lab. Med. 147, 1178-1185 (2022).
da Silva, L. M. et al. Independent real-world application of a clinical-grade automated prostate cancer detection system. J. Pathol. 254, 147-158 (2021).
Perincheri, S. et al. An independent assessment of an artificial intelligence system for prostate cancer detection shows strong diagnostic accuracy. Mod. Pathol. 34, 1588-1595 (2021).
Raciti, P. et al. Novel artificial intelligence system increases the detection of prostate cancer in whole slide images of core needle biopsies. Mod. Pathol. 33, 2058-2066 (2020).
Campanella, G. et al. Clinical-grade computational pathology using weakly supervised deep learning on whole slide images. Nat. Med. 25, 1301-1309 (2019).
Reis-Filho, J. S. et al. Abstract pd11-01: an artificial intelligencebased predictor of cdh1 biallelic mutations and invasive lobular carcinoma. Cancer Res. https://doi.org/10.1158/1538-7445. SABCS21-PD11-01 (2022).
Wagner, S. J. et al. Transformer-based biomarker prediction from colorectal cancer histology: a large-scale multicentric study. Cancer Cell 41, 1650-1661 (2023).
Coudray, N. et al. Classification and mutation prediction from non-small cell lung cancer histopathology images using deep learning. Nat. Med. 24, 1559-1567 (2018).
Kather, J. N. et al. Deep learning can predict microsatellite instability directly from histology in gastrointestinal cancer. Nat. Med. 25, 1054-1056 (2019).
Bilal, M. et al. Development and validation of a weakly supervised deep learning framework to predict the status of molecular pathways and key mutations in colorectal cancer from routine histology images: a retrospective study. Lancet Digit. Health 3, e763-e772 (2021).
Xie, C. et al. Computational biomarker predicts lung ICI response via deep learning-driven hierarchical spatial modelling from H&E. Preprint at https://doi.org/10.21203/rs.3.rs-1251762/v1 (2022).
Kacew, A. J. et al. Artificial intelligence can cut costs while maintaining accuracy in colorectal cancer genotyping. Frontiers in Oncology https://doi.org/10.3389/fonc.2021.630953 (2021).
Chen, T., Kornblith, S., Norouzi, M. & Hinton, G. A simple framework for contrastive learning of visual representations. In Proc. 37th International Conference on Machine Learning (eds Daumé, H. & Singh, A.) 1597-1607 (JMLR.org, 2020).
Caron, M. et al. Unsupervised learning of visual features by contrasting cluster assignments. In Proc. 34th International Conference on Neural Information Processing Systems (eds Larochelle, H. et al.) 9912-9924 (Curran Associates, 2020).
Caron, M. et al. Emerging properties in self-supervised vision transformers. In Proc. IEEE/CVF International Conference on Computer Vision 9630-9640 (IEEE, 2021).
He, K. et al. Masked autoencoders are scalable vision learners. In Proc. IEEE/CVF Conference on Computer Vision and Pattern Recognition 15979-15988 (IEEE, 2022).
Zhai, X., Kolesnikov, A., Houlsby, N. & Beyer, L. Scaling vision transformers. In Proc. IEEE/CVF Conference on Computer Vision and Pattern Recognition 12094-12103 (IEEE, 2022).
OpenAI. GPT-4 technical report. Preprint at https://doi.org/ 10.48550/arXiv. 2303.08774 (2023).
Deng, J. et al. Imagenet: a large-scale hierarchical image database. In Proc. IEEE Conference on Computer Vision and Pattern Recognition 248-255 (IEEE, 2009).
Sun, C., Shrivastava, A., Singh, S. & Gupta, A. Revisiting unreasonable effectiveness of data in deep learning era. In Proc. IEEE International Conference on Computer Vision 843-852 (IEEE, 2017).
Oquab, M. et al. DINOv2: Learning Robust Visual Features without Supervision. In Transactions on Machine Learning Research 2835-8856 (TMLR, 2024).
Dosovitskiy, A. et al. An image is worth words: transformers for image recognition at scale. In The Ninth International Conference on Learning Representations https://openreview.net/ forum?id=YicbFdNTTy (OpenReview.net, 2021).
Wang, X. et al. Transformer-based unsupervised contrastive learning for histopathological image classification. Med. Image Anal. 81, 102559 (2022).
Ciga, O., Xu, T. & Martel, A. L. Self supervised contrastive learning for digital histopathology. Mach. Learn. Appl. 7, 100198 (2022).
Filiot, A. et al. Scaling self-supervised learning for histopathology with masked image modeling. Preprint at https://doi.org/10.1101/ 2023.07.21.23292757 (2023).
Azizi, S. et al. Robust and data-efficient generalization of self-supervised machine learning for diagnostic imaging. Nat. Biomed. Eng. 7, 1-24 (2023).
Kang, M., Song, H., Park, S., Yoo, D. & Pereira, S. Benchmarking self-supervised learning on diverse pathology datasets. In Proc. IEEE/CVF Conference on Computer Vision and Pattern Recognition 3344-3354 (IEEE, 2023).
Dippel, J. et al. RudolfV: A foundation model by pathologists for pathologists. Preprint at https://doi.org/10.48550/ arXiv.2401.04079 (2024).
Chen, R. J. et al. Towards a general-purpose foundation model for computational pathology. Nat. Med. 30, 850-862 (2024).
Campanella, G. et al. Computational pathology at health system scale-self-supervised foundation models from three billion images. Preprint at https://doi.org/10.48550/arXiv.2310.07033 (2023).
Schultz, M. Rudolf Virchow. Emerg. Infect. Dis. 14, 1480-1481 (2008).
Reese, D. M. Fundamentals-Rudolf Virchow and modern medicine. West. J. Med. 169, 105 (1998).
Virchow, R. & Chance, F. Cellular Pathology as Based Upon Physiological and Pathological Histology: Twenty Lectures Delivered in the Pathological Institute of Berlin During the Months of February, March and April, 1858 (De Witt, 1860).
Liu, Z. et al. Swin transformer: hierarchical vision transformer using shifted windows. In Proc. IEEE/CVF International Conference on Computer Vision 9992-10002 (IEEE, 2021).
Chen, X., Xie, S. & He, K. An empirical study of training self-supervised vision transformers. In Proc. IEEE/CVF International Conference on Computer Vision 9620-9629 (IEEE, 2021).
Casson, A. et al. Joint breast neoplasm detection and subtyping using multi-resolution network trained on large-scale H&E whole slide images with weak labels. In Proc. Medical Imaging with Deep Learning (eds Oguz, I. et al.) 18-38 (JMLR, 2024).
Zehir, A. et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 23, 703-713 (2017).
Huang, Z., Bianchi, F., Yuksekgonul, M., Montine, T. J. & Zou, J. A visual-language foundation model for pathology image analysis using medical twitter. Nat. Med. 29, 2307-2316 (2023).
Graham, S. et al. Hover-net: simultaneous segmentation and classification of nuclei in multi-tissue histology images. Med. Image Anal. 58, 101563 (2019).
Cheerla, A. & Gevaert, O. Deep learning with multimodal representation for pancancer prognosis prediction. Bioinformatics 35, i446-i454 (2019).
Noorbakhsh, J. et al. Deep learning-based cross-classifications reveal conserved spatial behaviors within tumor histological images. Nat. Commun. 11, 6367 (2020).
Ilse, M., Tomczak, J. & Welling, M. Attention-based deep multiple instance learning. In Proc. 35th International Conference on Machine Learning (eds Dy, J. & Krause, A.) 2127-2136 (JMLR, 2018).
Chen, R. J. et al. Scaling vision transformers to gigapixel images via hierarchical self-supervised learning. In Proc. IEEE/CVF Conference on Computer Vision and Pattern Recognition 1612316134 (IEEE, 2022).
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
(c) The Author(s) 2024
Methods
Million-scale training dataset
Institutional review board review was not applicable for the research described in this study. This research study was conducted retrospectively from deidentified data licensed to Paige.AI, Inc. from MSKCC. The data used in this study were all collected originally for clinical use by MSKCC in the practice setting and are therefore considered secondary data. Only data previously deidentified by MSKCC were utilized in the analysis, and unique patient identifiers were completely removed from the analytical dataset. To the best of our knowledge, MSKCC has not transferred any data for which the applicable patient has not consented to or otherwise agreed to MSKCC’s Notice of Privacy Practices or a substantially similar notice, waiver or consent. The training digital pathology dataset comprises 1,488,550 WSIs derived from 119,629 patients. These WSIs are all stained with H&E, a routine stain that stains the nuclei blue and the extracellular matrix and cytoplasm pink. The WSIs are scanned at resolution or 0.5 mpp using Leica scanners. Seventeen high-level tissue groups are included, as illustrated in Fig. 1c.
WSIs are gigapixels in size and are challenging to use directly during training. Instead, Virchow was trained on tissue tiles that were sampled from foreground tissue in each WSI. To detect foreground, each WSI was downsampled with bilinear interpolation, and every pixel of the downsampled image was evaluated as to whether its hue, saturation and value were within [90,180], [8,255] and [103,255], respectively. All non-overlapping tiles containing at least tissue by area were collected. Virchow was trained on 2 billion tiles sampled randomly with replacement from approximately 13 billion available tissue tiles.
Virchow architecture and training
Virchow employs the ViT ‘huge’ architecture (ViT-H/14), a ViT with 632 million parameters that was trained using the DINO v. 2 (ref. 33) self-supervised learning algorithm, as illustrated in Extended Data Fig. 1. The ViT is an adaptation of the transformer model for image analysis, treating an image as a sequence of patches. These patches are embedded and processed through a transformer encoder that uses self-attention mechanisms. This approach allows ViT to capture complex spatial relationships across the image. DINO v. 2 is based on a student-teacher paradigm: given a student network and a teacher network, each using the same architecture, the student is trained to match the representation of the teacher. The student network is information-limited, as it is trained using noisy variations of input tiles. The teacher network is a slowly updated exponential moving average of past student networks; matching the teacher achieves an effect similar to ensembling over prior student predictions . The student learns a global representation of an image by matching the teacher’s class token, as well as local representations by matching the teacher’s patch tokens. Patch tokens are only matched for a select subset of tokens that are randomly masked out of an input image (for the student), as done in masked image modeling . Additional regularization helps DINO v. 2 trained models outperform the earlier DINO variant .
The default hyperparameters for training the DINO v. 2 model were used for Virchow as detailed in ref. 33 with the following changes: a teacher temperature schedule of 0.04-0.07 in 186,000 iterations and a reciprocal square root learning rate schedule with a warmup of 495,000 iterations (instead of 100,000) and linear cooldown to 0.0 for the last 819,200 iterations . Virchow was trained using AdamW ( , ) with float16 precision. Note that with ViT-H, we used 131,072 prototypes (and thus 131,072-dimensional projection heads). During distributed training, each mini-batch was sampled by randomly selecting one WSI per graphics processing unit and 256 foreground tiles per WSI.
Pan-cancer detection
Specimen-level pan-cancer detection requires a model that aggregates foundation model embeddings from all foreground tiles of all WSIs in
a specimen to detect the presence of cancer. All pan-cancer detection models trained in this work use an Agata aggregator model, weakly supervised with multiple-instance learning (see Extended Data Fig. 2 for architecture details).
Embedding generation. For a input tile image, a Virchow embedding is defined as the concatenation of the class token and the mean across all 256 of the other predicted tokens. This produces an embedding size of . For Phikon, only the class token is used, as recommended by ref. 37. For CTransPath, the mean of all tokens is used as there is no class token.
Training data. To train the aggregator model, we prepared a subset of the training dataset used for training Virchow (see ‘Million-scale training dataset’ in Methods for details), combined with specimenlevel labels (block-level for prostate tissue) indicating the presence or absence of cancer extracted from synoptic and diagnostic reports. The training and validation datasets combined consist of 89,417 slides across 40,402 specimens. See Extended Data Fig. 4b for the training data distribution, stratified by WSI tissue type and cancer status.
Aggregator training. The Agata aggregator was trained as described in Extended Data Fig. 2. Because the label is at the level of the specimen, all tiles belonging to the same specimen need to be aggregated during training. Training using embeddings for all tiles of a specimen is prohibitively memory-intensive. We thus select the slide with the highest predicted cancer probability per specimen and backpropagate the gradients only for that slide.
As baselines, aggregators using Phikon and CTransPath embeddings were also trained. All aggregators were trained for 25 epochs using the cross-entropy loss and the AdamW optimizer with a base learning rate of 0.0003 . During each training run, the checkpoint with the highest validation AUC was selected for evaluation.
Testing dataset. The pan-cancer detection models are evaluated on a combination of data sourced from MSKCC and external institutions. None of the patients in the evaluation set were seen during training. The dataset contains 22,932 slides from 6,142 specimens across 16 cancer types. We hypothesize that the more data the foundation model is trained on, the better the downstream task performance, especially on data-constrained tasks. To test this hypothesis, we categorize cancer types into common or rare cancer groups. According to the NCI, rare cancers are defined as those occurring in fewer than 15 people out of 100,000 each year in the United States . Based on this definition, common cancer comprises 14,179 slides from 3,547 specimens originating in breast, prostate, lung, colon, skin, bladder, uterus, pancreas and H&N, and rare cancer comprises 8,753 slides from 2,595 specimens originating in liver, stomach, brain, ovary, cervix, testis and bone. Note that each cancer type is determined by its tissue of origin and thus may appear in any tissue (as primary or metastatic cancer). On the other hand, benign specimens for each cancer type were sampled only from the tissue of origin. For example, the liver stratum contains 182 liver specimens with liver cancer (primary), 18 non-liver specimens with liver cancer (metastatic) and 200 benign liver specimens. For each cancer type, Fig. 2a shows the distribution between primary and metastatic cancer, and Extended Data Fig. 4a additionally shows the number of benign specimens.
The testing dataset includes 15,622 slides from 3,033 specimens collected at MSKCC (denoted as ‘Internal’ in Fig. 2b), in addition to 7,310 slides ( 3109 specimens) sent to MSKCC from institutions around the world (‘External’ in Fig. 2b). See Extended Data Fig. 4a for the testing data distribution, stratified by cancer type (for specimens with cancer) or by tissue type (for benign specimens).
Label extraction. To establish the clinical cancer diagnosis at the specimen level, a rule-based natural language processing system was employed. This system decomposes case-level reports to the specimen level and analyzes the associated clinical reports with each specimen, thereby providing a comprehensive understanding of each case.
Statistical analysis. The performance of the three models is compared using two metrics: AUC and specificity at sensitivity. AUC is a suitable general metric because it does not require selecting a threshold for the model’s probability outputs, something that may need tuning for different data subpopulations. Specificity at 95% sensitivity is informative because a clinical system must be not only sensitive but also specific in practice. For AUC, the pairwise DeLong’s test with Holm’s method for correction is applied to check for statistical significance. For specificity, first Cochran’s test is applied, and then McNemar’s test is applied post hoc for all pairs with Holm’s method for correction. The two-sided confidence intervals in Fig. 2b-e and Extended Data Fig. 3 were calculated using DeLong’s method for AUC and Wilson’s method for specificity. In addition to overall analysis, stratified analysis is also conducted for each cancer type.
Clinical evaluation datasets
To perform an extensive evaluation of the Virchow-based pan-cancer detection model, we employ seven additional datasets (see Supplementary Table 2.1 for details). One of these datasets is pan-tissue, and the rest are single-tissue datasets containing tissues for which Paige has clinical products: that is, prostate, breast and lymph node.
Pan-tissue product benchmark. This dataset contains 2,419 slides across 18 tissue types (Supplementary Table 2.2). Each slide is individually inspected by a pathologist and labeled according to presence of invasive cancer. An important distinction between the testing dataset in ‘Pan-cancer detection’ and this dataset is that the former is stratified according to origin tissue in cancerous specimens, whereas the latter is stratified according to tissue type for all slides, as it is more relevant in a clinical setting. We use this dataset to identify failure modes of the pan-cancer detection model.
Prostate product benchmark. This dataset contains 2,947 blocks ( 3,327 slides) of prostate needle core biopsies (Supplementary Table 2.7). Labels for the blocks are extracted from synoptic reports collected at MSKCC. This dataset has been curated to evaluate the standalone performance of Paige Prostate Detect, which is a tissue-specific, clinical-grade model. We use this dataset to compare the pan-cancer detection model to Paige Prostate Detect.
Prostate rare variants benchmark. This dataset contains 28 slides containing rare variants of prostate cancer (neuroendocrine tumor, atrophic, small lymphocytic lymphoma, foamy cell carcinoma, follicular lymphoma) and 112 benign slides (Supplementary Table 2.8). Cancerous slides are curated and labeled by a pathologist, and are appended with slides from benign blocks determined from synoptic reports collected at MSKCC.
Breast product benchmark. This dataset contains 190 slides with invasive cancer and 1,501 benign slides, labeled individually by a pathologist according to presence of atypical ductal hyperplasia, atypical lobular hyperplasia, lobular carcinoma in situ, ductal carcinoma in situ, invasive ductal carcinoma, invasive lobular carcinoma and/or other subtypes (Supplementary Table 2.5). This dataset has been curated to evaluate the standalone performance of Paige Breast, which is a tissue-specific, clinical-grade model. We use the subtype information for stratified analysis.
Breast rare variants benchmark. This dataset contains 23 cases of invasive ductal carcinoma or invasive lobular carcinoma (as control), 75 cases of rare variants (adenoid cystic carcinoma, carcinoma with apocrine differentiation, cribriform carcinoma, invasive micropapillary carcinoma, metaplastic carcinoma (matrix producting subtype, spindle cell and squamous cell), mucinous carcinoma, secretory carcinoma and tubular carcinoma) and 392 benign cases (total 5,031 slides). Cancerous cases are curated by a pathologist, and are appended with benign cases determined from synoptic reports collected at MSKCC. See Supplementary Table 2.6 for details.
BLN. This dataset contains 458 lymph node slides with metastasized breast cancer and 295 benign lymph node slides (Supplementary Table 2.3). Each slide has been labeled by a pathologist according to presence of invasive cancer, and the largest tumoron the slide is measured to categorize the tumor into macrometastasis, micrometastasis or infiltrating tumor cells. We use the categories for stratified evaluation.
Lymph node rare variants benchmark. This dataset contains 48 specimens of rare variants of cancers (diffused large B-cell lymphoma, follicular lymphoma, marginal zone lymphoma, Hodgkin’s lymphoma) selected by a pathologist and 192 benign specimens determined from synoptic reports collected at MSKCC (Supplementary Table 2.4).
Biomarker detection
We formulated each biomarker prediction task as a binary pathology case classification problem, where a positive label indicates the presence of the biomarker. Each case consists of one or more H&E slides that share the same binary label. We randomly split each dataset into training and testing subsets, ensuring no patient overlap, as shown in Supplementary Table 3.1. The clinical importance of each biomarker is described below.
Colon-MSI. Microsatellite instability (MSI) occurs when DNA regions with short, repeated sequences (microsatellites) are disrupted by single nucleotide mutations, leading to variation in these sequences across cells. Normally, mismatch repair (MMR) genes (MSH1, MSH2, MSH6, PMS2) correct these mutations, maintaining consistency in microsatellites. However, inactivation of any MMR gene (through germline mutation, somatic mutation or epigenetic silencing) results in an increased rate of uncorrected mutations across the genome. MSI is detected using polymerase chain reaction or next-generation sequencing, which identifies a high number of unrepaired mutations in microsatellites, indicative of deficient mismatch repair (dMMR). Microsatellite instability high (MSI-H) suggests dMMR in cells, identifiable via IHC, which shows absent staining for MMR proteins. MSI-H is present in approximately 15% of colorectal cancers (CRCs), often linked to germline mutations that elevate hereditary cancer risk. Consequently, routine MSI or IHC-based dMMR screening is recommended for all primary colorectal carcinoma samples. The Colon-MSI dataset, comprising 2,698 CRC samples with 288 MSI-H/dMMR positive cases, uses both IHC and MSK-IMPACT sequencing for dMMR and MSI-H detection, prioritizing IHC results when both test outcomes are available.
Breast-CDH1. The biallelic loss of cadherin 1 (CDH1) gene (encoding E-cadherin) is strongly correlated with lobular breast cancer and a distinct histologic phenotype and biologic behavior . CDH1 inactivating mutations associated with loss of heterozygosity or a second somatic loss-of-function mutation as determined by MSK-IMPACT sequencing test results were considered as ‘CDH1 biallelic mutations’. The CDH1 dataset comprises a total of 1,077 estrogen receptor-positive (ER+) primary breast cancer samples, in which 139 were positive and 918 were negative. The remaining 20 samples with other types of variants-that is, monoallelic mutations-were excluded.
Bladder-FGFR. The fibroblast growth factor receptor (FGFR) is encoded by four genes (FGFR1, FGFR2, FGFR3, FGFR4). FGFR gene alterations screening in bladder carcinoma allows the identification of patients targetable by FGFR inhibitors. Anecdotal experience from pathologists suggested there may be a morphological signal for FGFR alterations . The FGFR binary label focuses on FGFR3 p.S249C, p.R248C,p.Y373C,p.G37OC mutations,FGFR3-TACC3 fusions and FGFR2 p.N549H,pN549K,p.N549S,p.N549T mutations based on data from the MSK-IMPACT cohort. From the total of 1,038 samples ( 1,087 WSIs), have alterations.
Lung-EGFR. The oncogenic mutation screening in non-small cell lung cancer is essential to determine eligibility for targeted therapies in late stage non-small cell lung cancer . The oncogenic status of mutation was determined based on OncoKB annotation . EGFR mutations with any oncogenic effect (including predicted/likely oncogenic) were defined as positive label, and mutation with unknown oncogenic status were excluded.
Prostate-AR. The amplification/overexpression was found in 50% of castration resistant prostate cancers and was associated with resistance to androgen deprivation therapy. In the AR dataset, the copy number amplification of was determined by MSK-IMPACT sequencing test, for which the fold change was greater than two.
Gastric-HER2. Human epidermal growth factor receptor 2 (HER2) overexpression and/or amplification are much more heterogeneous in gastric cancer compared to breast cancer. Approximate 20% of gastric cancer patients are found to correlate with HER2 overexpres-sion/high-level amplification, and they would be likely to benefit from treatment with an anti-HER2 antibody therapy. Here, a HER2 IHC result of 2+, confirmed positive with fluorescence in situ hybridization (FISH) or an IHC result of 3+ were considered HER2 amplification.
Endometrial-PTEN. PTEN is the most frequently mutated tumor suppressor gene in endometrial cancer. The presence of PTEN mutation showed to be significantly associated with poorer prognosis in survival and disease recurrence. The oncogenic status of PTEN mutation was determined based on MSK-IMPACT sequencing and OncoKB annotation . The variants associated with any oncogenic effect (including predicted and/or likely oncogenic) were defined as positive label for PTEN mutations, and variants with unknown oncogenic status were excluded.
Thyroid-RET. RET mutations were highly associated with medullary thyroid cancer, which accounts for about of all thyroid cancer. Screening RET oncogenic mutations plays an important role in diagnosis and prognosis of medullary thyroid cancer. The positive label for RET oncogenic mutation was determined by MSK-IMPACT sequencing and OncoKB annotation .
Skin-BRAF.BRAF is one of the most frequently mutated genes in melanoma, and V600E mutation is the most common variant, which leads to constitutive activation of the BRAF/MEK/ERK signaling pathway. Targeted therapy with BRAF inhibitors showed better survival outcome in patients with V600-mutated melanoma. Therefore, the detection of BRAF V600 mutations in melanoma helps to determine treatment strategies. In the BRAF dataset, the oncogenic mutation status and the presence of V600E variant were determined based on the MSK-IMPACT cohort and OncoKB annotation .
Ovarian-FGA. High-grade serous ovarian cancer is characterized by high prevalence of TP53 mutations and genome instability with widespread genetic alteration. The fraction of genome altered (FGA) was determined from MSK-IMPACT sequencing data, where FGA
was treated as a positive label. A cut-off for FGA was established that enriched for TP53 mutations in the distribution of ovarian cancer cases.
Aggregator training. For weakly supervised biomarker prediction, we used embeddings and Agata , as in ‘Pan-cancer detection’, to transform a set of tiles extracted from WSIs that belong to the same case to the case-level target label. Virchow is used to generate tile-level embeddings on all the evaluated datasets with resolution at magnification. To thoroughly compare the quality of the embeddings, we trained an aggregator for learning rates in , and report the best observed test AUC scores in Fig. 4b. Due to the small biomarker dataset sizes, the learning rate was not chosen on a validation set to evaluate generalization; rather, this serves as a benchmark across the different types of tile embeddings (Virchow, UNI, Phikon and CTransPath), yielding an estimate of the best possible biomarker performance for each type.
Statistical analysis. AUC is used to compare models without having to select a threshold on the models’ predicted probability values, which may differ by data subpopulation. The two-sided confidence intervals in Fig. 4b are calculated using DeLong’s method .
Tile-level benchmarking
For evaluating Virchow on tile-sized images, the linear probing protocol, as well as dataset descriptions and the statistical analysis, are described below. Dataset details, including training, validation, and testing splits, are also summarized in Supplementary Table 4.1.
Linear probing protocol. For each experiment, we trained a linear tile classifier with a batch size of 4,096 using the stochastic gradient descent optimizer with a cosine learning rate schedule, from 0.01 to 0 , for 12,500 iterations, on top of embeddings generated by a frozen encoder. The large number of iterations is intended to allow any linear classifier to converge as far as it can at each learning rate step along the learning rate schedule. All embeddings were normalized by -scoring before classification. Linear probing experiments did not use data augmentation. For testing set evaluation, the classifier checkpoint that achieved the lowest loss on the validation set was selected. A validation set was used for all tasks. If one was not provided with the public dataset, we randomly split out of the training data to make a validation set.
PanMSK. For a comprehensive in-distribution benchmark, 3,999 slides across the 17 tissue types in Fig. 1d were held out from the training dataset collected from MSKCC. Of these, 1,456 contained cancer that was either partially or exhaustively annotated with segmentation masks by pathologists. These annotations were used to create a tile-level dataset of cancer versus non-cancer classification, which we refer to as PanMSK. All images in PanMSK are pixel tiles at 0.5 mpp . See Supplementary Note 5 for further details.
CRC. The CRC classification public dataset contains 100,000 images for training (from which we randomly selected 10,000 for validation) and 7,180 images for testing ( pixels) at magnification sorted into nine morphological classes. Analysis is performed with both the Macenko-stain-normalized (NCT-CRC-HE-100K) and unnormalized (NCT-CRC-HE-100K-NONORM) variants of the dataset. It should be noted that the training set is normalized in both cases, and only the testing test is unnormalized in the latter variant. Thus, the unnormalized variant of CRC involves a distribution shift from training to testing.
WILDS. The Camelyon17-WILDS dataset is a public dataset comprising 455,954 images, each with a resolution of pixels, taken at magnification and downsampled from . This dataset is derived from the larger Camelyon17 dataset and focuses on lymph
node metastases. Each image in the dataset is annotated with a binary label indicating the presence or absence of a tumor within the central pixel region. Uniquely designed to test OOD generalization, the training set (335,996 images) is composed of data from three different hospitals, whereas the validation subset ( 34,904 images) and testing subset (85,054 images) each originate from separate hospitals not represented in the training data.
MHIST. The colorectal polyp classification public dataset (MHIST ) contains 3,152 images ( pixels) presenting either hyperplastic polyp or sessile serrated adenoma at magnification (downsampled from to increase the field of view). This dataset contains 2,175 images in the training subset (of which we randomly selected 217 for validation) and 977 images in the testing subset.
TCGA TIL. The TCGA TIL public dataset is composed of 304,097 images ( pixels) at magnification , split into 247,822 training images, 38,601 validation images and 56,275 testing images. Images are considered positive for tumor-infiltrating lymphocytes if at least two TILs are present and labeled negative otherwise. We upsampled the images to to use with Virchow.
PCam. The PatchCamelyon (PCam) public dataset consists of 327,680 images ( pixels) at magnification, downsampled from to increase the field of view . The data is split into a training subset (262,144 images), a validation subset (32,768 images), and a testing subset ( 32,768 images). Images are labeled as either cancer or benign. We upsampled the images to pixels to use with Virchow.
MIDOG. The MIDOG public dataset consists of 21,806 mitotic and non-mitotic events labeled on WSI regions from several tumor, species and scanner types . Data was converted into a binary classification task by expanding each pixel annotation to regions and then randomly shifting in the horizontal and vertical regions such that the event is not centered in the tile. All negative instances that overlapped with positive instances were removed from the dataset. The resulting dataset consists of training, validation and testing subsets with 13,107, 4,359 and 4,340 images, respectively (of which and 2,222 have mitotic events, respectively, and the rest contain confounders that mimic mitotic events).
TCGA CRC-MSI. The TCGA CRC-MSI classification public dataset consists of regions taken at magnification presenting colorectal adenocarcinoma samples . Samples were extracted and annotated from TCGA. Regions were labeled either as microsatellite-instable or microsatellite-stable. We downsampled regions to to use with Virchow.
Statistical analysis. The (weighted) F1 score is used to compare models as this metric is robust to class imbalance. Accuracy and balanced accuracy are also computed, as described in Supplementary Note 4. The two-sided confidence intervals in Fig. 5 and Supplementary Table 4.2 were computed with 1,000 bootstrapping iterations over the metrics on the testing set without retraining the classifier. McNemar’s test was used to determine statistically significant ( ) differences between results.
Qualitative feature analysis
We performed an unsupervised feature analysis similar to the procedure in ref. 33, using the CoNSeP dataset of H&E stained slides with colorectal adenocarcinoma. CoNSeP provides nuclear annotations of cells in the following seven categories: normal epithelial, malignant/ dysplastic epithelial, fibroblast, muscle, inflammatory, endothelial and miscellaneous (including necrotic, mitotic and cells that couldn’t be categorized). Because CoNSeP images are of size 1,000 and
Virchow takes in images of size , we resized images to and divided them into a grid of non-overlapping subimages before extracting tile-level features. For a given image, we used principal component analysis (PCA) on all the tile features from the subimages, normalized the first and second principal components to values within and thresholded at 0.5 . Figure 5 d shows some examples of the unsupervised feature separation achieved in this way.
Software
For data collection, we used Python (v.3.10.11) along with Pandas (v.2.2.2) for indexing the data and metadata used for pretraining and benchmarking. OpenSlide (v.1.3.1) and Pillow (v.10.0.0) were used for preprocessing the image tiles for the benchmark. Where appropriate, we extracted per-specimen labels from clinical reports using DBT (v.1.5.0). We used Python (v.3.10.11) for all experiments and analyses in the study, which can be replicated using open-source libraries as outlined below. For self-supervised pretraining, we used PyTorch (v.2.0.1) and Torchvision (v.0.15.1). The DINO v. 2 code was ported from the official repository (https://github.com/facebookresearch/ dinov2) and adapted to PyTorch Lightning (v.1.9.0). All WSI processing during pretraining was performed online and was supported by cucim (v.23.10.0) and torchvision (v.0.16.1). For downstream task benchmarking, we use scikit-learn (v.1.4.2) for logistic regression and metrics computation. Implementations of other pretrained visual encoders benchmarked in the study were obtained from the following links: UNI (https://huggingface.co/MahmoodLab/UNI), Phikon (https://huggingface.co/owkin/phikon), DINOp=8 (https://github. com/lunit-io/benchmark-ssl-pathology),PLIP(https://huggingface.co/ vinid/plip), CTransPath (https://github.com/Xiyue-Wang/TransPath) and the original natural image pretrained DINO v. 2 (https://github.com/ facebookresearch/dinov2).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
This study did not specifically collect patient data. The retrospective analysis utilized proprietary deidentified digital pathology whole slides and associated metadata were exclusively licensed by Paige.AI, Inc. from MSKCC. Requests for data need to be submitted to Paige AI (https://paige.ai/contact-us/) and evaluated by Paige AI and MSKCC on a case-by-case basis. All requests complying with internal regulations on data privacy and intellectual property will be granted. This study also utilized the following publicly available datasets for downstream benchmarking: CRC (NCT-CRC-HE-100K and NCT-CRC-HE-100K-NONORM, available via Zenodo at https://zenodo.org/records/1214456 (ref. 77)), WILDS (Camelyon17; https://wilds.stanford.edu/get_started), PCam (https://github.com/basveeling/pcam),MHIST(https://bmirds.github. io/MHIST), TCGA TIL (available via Zenodo at https://zenodo.org/ records/6604094 (ref. 71)), MIDOG (https://midog.deepmicroscopy. org/download-dataset/) and TCGA CRC-MSI (available via Zenodo at https://zenodo.org/records/3832231 (ref.76)).
Code availability
The model can be accessed for non-commercial research and replication of the results reported in this manuscript at https://huggingface. co/paige-ai/Virchow. To help researchers use our model, a public software development kit for leveraging foundation model embeddings to develop downstream WSI applications is available at https://github. com/Paige-Al/paige-ml-sdk. We have documented all experiments with sufficient details in our Methods section to enable independent replication. Although the full codebase cannot be shared due to dependencies on proprietary libraries and specific hardware configurations, such as distributed clusters and storage, the core components of our work
rely on open-source repositories. These include the DINO v. 2 model architecture used for self-supervised pretraining (https://github.com/ facebookresearch/dinov2), as well as the PyTorch Lightning training framework (https://github.com/Lightning-Al/pytorch-lightning) and the underlying torch library (https://github.com/pytorch/pytorch) used for training and inference with this model.
References
Tarvainen, A. & Valpola, H. Mean teachers are better role models: weight-averaged consistency targets improve semi-supervised deep learning results. In Proc. 31st Conference on Neural Information Processing Systems (eds von Luxburg, U. et al.) 1195-1204 (ACM, 2017).
Xie, Z. et al. Simmim: a simple framework for masked image modeling. In Proc. IEEE/CVF Conference on Computer Vision and Pattern Recognition 9643-9653 (IEEE, 2022).
DeLong, E. R., DeLong, D. M. & Clarke-Pearson, D. L. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics 44, 837-845 (1988).
Holm, S. A simple sequentially rejective multiple test procedure. Scand. J. Statist. 6, 65-70 (1979).
Cochran, W. G. The comparison of percentages in matched samples. Biometrika 37, 256-266 (1950).
McNemar, Q. Note on the sampling error of the difference between correlated proportions or percentages. Psychometrika 12, 153-157 (1947).
Wilson, E. B. Probable inference, the law of succession, and statistical inference. J. Am. Stat. Assoc. 22, 209-212 (1927).
Berx, G. & Van Roy, F. The E-cadherin/catenin complex: an important gatekeeper in breast cancer tumorigenesis and malignant progression. Breast Cancer Res. 3, 289-293 (2001).
Al-Ahmadie, H. A. et al. Somatic mutation of fibroblast growth factor receptor-3 (FGFR3) defines a distinct morphological subtype of high-grade urothelial carcinoma. J. Pathol. 224, 270-279 (2011).
Kalemkerian, G. P. et al. Molecular testing guideline for the selection of patients with lung cancer for treatment with targeted tyrosine kinase inhibitors: American Society of Clinical Oncology endorsement of the College of American Pathologists/ international Association for the Study of Lung Cancer/Association for Molecular Pathology Clinical Practice guideline update. J. Clin. Oncol. https://doi.org/10.1200/JCO.2017.76.7293 (2018).
Chakravarty, D. et al. OncoKB: a precision oncology knowledge base. JCO Precis. Oncol. 1, 1-16 (2017).
Kather, J. N. et al. Predicting survival from colorectal cancer histology slides using deep learning: a retrospective multicenter study. PLoS Med. 16, e1002730 (2019).
Wei, J. et al. A petri dish for histopathology image analysis. In Proc. Artificial Intelligence in Medicine (eds Tucker, A. et al) 11-24 (2021).
Kaczmarzyk, J. R., Abousamra, S., Kurc, T., Gupta, R. & Saltz, J. Dataset for tumor infiltrating lymphocyte classification (304,097 image patches from TCGA). Zenodo https://doi.org/10.5281/ zenodo. 6604093 (2022).
Abousamra, S. et al. Deep learning-based mapping of tumor infiltrating lymphocytes in whole slide images of 23 types of cancer. Front. Oncol. 11, 806603 (2022).
Saltz, J. et al. Spatial organization and molecular correlation of tumor-infiltrating lymphocytes using deep learning on pathology images. Cell Rep. 23, 181-193 (2018).
Veeling, B. S., Linmans, J., Winkens, J., Cohen, T. & Welling, M. Rotation equivariant CNNs for digital pathology. In Proc. Medical Image Computing and Computer Assisted Intervention (eds Frangi, A. F. et al.) 210-218 (2018).
Aubreville, M. et al. A comprehensive multi-domain dataset for mitotic figure detection. Sci. Data 10, 484 (2023).
Kather, J. N. Histological image tiles for TCGA-CRC-DX, color-normalized, sorted by MSI status, train/test split. Zenodo https://doi.org/10.5281/zenodo. 3832231 (2020).
Kather, J. N., Halama, N. & Marx, A. 100,000 histological images of human colorectal cancer and healthy tissue. Zenodo https://doi.org/ 10.5281/zenodo. 1214455 (2018).
Acknowledgements
We thank P. Rosenfield from Microsoft and D. Dierov from Paige for their contributions in making this collaboration possible. Research reported in this publication was supported in part by a Cancer Center Support Grant of the National Institutes of Health/National Cancer Institute (grant no. P30CA008748).
Author contributions
T.J.F., S.L., B.R., R.Y., W.A.M., N.F. and K.S. contributed to study conception and coordination, building a cross-company partnership and research team and getting data sharing agreements. T.J.F., B.R., D.S.K., C.K. and N.F. contributed as research advisors. D.S.K., G.O., E.M., M.H., E.Y., H.W. and J.A.R. provided clinical guidance. E.V., A.B., A.C., G.S., M.Z., S.L., K.S., E.Z., J.H., N.T., P.M., A.v.E., D.L., J.V. and E.R. wrote code, developed infrastructure and trained models throughout the study. E.V., A.B., A.C., G.S., M.Z., K.S., D.L., Y.K.W., M.C.H.L., J.H.B., R.A.G., G.O., E.M., M.H., E.Y., H.W., J.R., D.S.K. and S.L. worked on data preparation. E.V., A.B., A.C., G.S., M.Z., K.S., E.Z., D.L., J.V., E.R., Y.K.W., J.D.K., M.C.H.L., J.H.B., R.A.G., J.R. and S.L. performed evaluation and analysis. E.V., A.B., A.C., G.S., M.Z., K.S., D.L., Y.K.W., J.D.K., M.C.H.L., J.H.B., R.A.G., C.K., D.S.K., S.L. and T.J.F. worked on drafting and revising the manuscript.
Competing interests
E.V., A.B., A.C., G.S., M.Z., P.M., A.v.E., D.L., J.V., E.R., Y.K.W., J.D.K., M.C.H.L., J.H.B., R.A.G., G.O., J.A.R., W.A.M., R.Y., D.K., S.L. and T.J.F. are employees and equity holders of Paige.AI. E.W., M.H., C.K. and B.R. served as consultants for Paige.AI. D.S.K. has received compensation for speaking and consulting from Merck. K.S., E.Z., J.H., N.T. and N.F. are employees of Microsoft. Memorial Sloan Kettering (MSK) maintains financial and intellectual property interests in Paige.AI that are pertinent to the research presented in this manuscript. S.L., E.V., A.B., G.S., M.Z., A.C., J.B., M.L., R.G., T.F. and B.R. are inventors on a provisional US patent (application no. 18/521903) filed corresponding to the methodological aspects of this work. The remaining authors declare no competing interests.
Correspondence and requests for materials should be addressed to Siqi Liu.
Peer review information Nature Medicine thanks Francesco
Ciompi, Lee Cooper and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Lorenzo Righetto, in collaboration with the Nature Medicine team.
Extended Data Fig. 1 | Schematic of the Agata aggregator. The Agata
aggregator learns to attend to tiles that contribute toward the label decision using cross-attention. The operation is defined using query Q , key K , and value matrix V: softmax , where is the output dimension of the key matrix. In contrast to the typical self-attention mechanism where are projected from the inputs, Q is parameterized directly by the model to reduce GPU memory consumption. When aggregating across the tens or hundreds of thousands of tiles in a specimen, full attention requires too much GPU memory. This simplified attention can be interpreted as a learned weighted sum of all tile-level features.
Indeed, full self-attention is quadratic in memory with respect to the number of tiles whereas the attention in Agata is linear. K and V are obtained with two consecutive Gaussian Error Linear Unit (GELU) projection layers as: and , where x is the tile embedding, and are the weight and bias parameters for the projection layers. In our experiments, produces 256 -dimensional keys, produces 512 -dimensional values, and we omit scaling by . After the attention step, two linear layers with non-linear activation (ReLU) are used followed by a final linear layer with softmax activation.
Extended Data Fig. 2 | Schematic of the DINOv2 training routine. Virchow used a ViT-H architecture, trained with DINOv2. From a single tile, 2 global crops and 8 local crops all with random augmentations are created. The global crops are randomly masked and fed to the student model, and the unmasked versions are fed to the teacher model. The student tries to produce a global representation of the views (via the CLS token) that matches the teacher’s representation of the
opposite view. The student also tries to produce representations of the masked image tokens that match the teacher’s representations of the same tokens but unmasked. The local crops are only fed to the student which tries to produce a representation that matches the teacher’s representations of the global crops. The teacher is an exponential moving average (EMA) copy of the student.
Extended Data Fig. 3 | Pan-cancer detection results for every cancer origin site. a. Area under (the receiver operator characteristic) curve (AUC); b. specificity at sensitivity. is head and neck. In both plots, a pairwise comparison of statistical significance is computed using the pairwise DeLong’s test for AUC and
Cochran’s Q test followed by McNemar’s test for specificity, both corrected for multiple comparisons with Holm’s method (* , **** ). Error bars show the two-sided confidence interval computed with DeLong’s method for AUC and Wilson’s method for specificity.
Extended Data Fig. 4 | Pan-cancer dataset distribution. a. Specimen counts per cancer origin site in the pan-cancer testing dataset ( is head and neck). b. Specimen counts per tissue type in the pan-cancer aggregator training dataset.
natureportfolio
Corresponding author(s):
Last updated by author(s): Jun 3, 2024
Reporting Summary
Nature Portfolio wishes to improve the reproducibility of the work that we publish. This form provides structure for consistency and transparency in reporting. For further information on Nature Portfolio policies, see our Editorial Policies and the Editorial Policy Checklist.
Statistics
For all statistical analyses, confirm that the following items are present in the figure legend, table legend, main text, or Methods section.
Confirmed
The exact sample size ( ) for each experimental group/condition, given as a discrete number and unit of measurement A statement on whether measurements were taken from distinct samples or whether the same sample was measured repeatedly X
The statistical test(s) used AND whether they are one- or two-sided
Only common tests should be described solely by name; describe more complex techniques in the Methods section. A description of all covariates tested A description of any assumptions or corrections, such as tests of normality and adjustment for multiple comparisons X
A full description of the statistical parameters including central tendency (e.g. means) or other basic estimates (e.g. regression coefficient) AND variation (e.g. standard deviation) or associated estimates of uncertainty (e.g. confidence intervals) X
For null hypothesis testing, the test statistic (e.g. ) with confidence intervals, effect sizes, degrees of freedom and value noted Give values as exact values whenever suitable. For Bayesian analysis, information on the choice of priors and Markov chain Monte Carlo settings For hierarchical and complex designs, identification of the appropriate level for tests and full reporting of outcomes Estimates of effect sizes (e.g. Cohen’s , Pearson’s ), indicating how they were calculated
Our web collection on statistics for biologists contains articles on many of the points above.
Software and code
Policy information about availability of computer code
Data collection
For data collection, we used Python (3.10.11) along with Pandas (2.2.2) for indexing the data and metadata used for pretraining and benchmarking. OpenSlide (1.3.1) and Pillow (10.0.0) were used for preprocessing the image tiles for the benchmark. Where appropriate, we extracted per-specimen labels from clinical reports using DBT (1.5.0).
Data analysis
We used Python (3.10.11) for all experiments and analyses in the study, which can be replicated using open-source libraries as outlined below. For self-supervised pretraining, we used Pytorch (2.0.1) and Torchvision (0.15.1). The DINOv2 code was ported from the official repository (https://github.com/facebookresearch/dinov2) and adapted to Pytorch-lightning (1.9.0). All WSI processing during pretraining was performed online and was supported by cucim (23.10.0) and torchvision (0.16.1). For downstream task benchmarking, we use scikit-learn (1.4.2) for logistic regression and metrics computation. The baseline foundation models were obtained from the following links: Implementations of other visual pre-trained encoders benchmarked in the study are found at the following links: UNI (https://huggingface.co/ MahmoodLab/UNI), Phikon (https://huggingface.co/owkin/phikon), DINOp=8 (https://github.com/lunit-io/benchmark-ssl-pathology), PLIP (https://huggingface.co/vinid/plip), CTransPath (https://github.com/Xiyue-Wang/TransPath) and the original natural image pre-trained DINOv2 (https://github.com/facebookresearch/dinov2).
For manuscripts utilizing custom algorithms or software that are central to the research but not yet described in published literature, software must be made available to editors and reviewers. We strongly encourage code deposition in a community repository (e.g. GitHub). See the Nature Portfolio guidelines for submitting code & software for further information.
Data
Policy information about availability of data
All manuscripts must include a data availability statement. This statement should provide the following information, where applicable:
Accession codes, unique identifiers, or web links for publicly available datasets
A description of any restrictions on data availability
For clinical datasets or third party data, please ensure that the statement adheres to our policy
Policy information about studies involving human research participants and Sex and Gender in Research.
Reporting on sex and gender
Gender or sex was not included as a covariate at any stage of our experimental analysis.
Population characteristics
We did not collect or use any covariates pertaining to population characteristics at any stage of the study.
Recruitment
No patient recruitment was necessary for using histology whole slide images retrospectively.
Ethics oversight
Institutional review board (IRB) review was not applicable for the research described in this study. This research study was conducted retrospectively from de-identified data licensed to Paige.AI, Inc. from Memorial Sloan Kettering Cancer Center (MSKCC). The data used in this study were all collected originally for clinical use by MSKCC in the practice setting and are therefore considered secondary data. Only data previously de-identified by MSKCC were utilized in the analysis, and unique patient identifiers were completely removed from the analytical dataset.
Note that full information on the approval of the study protocol must also be provided in the manuscript.
Field-specific reporting
Please select the one below that is the best fit for your research. If you are not sure, read the appropriate sections before making your selection.
Life sciences Behavioural & social sciences Ecological, evolutionary & environmental sciences
For a reference copy of the document with all sections, see nature.com/documents/nr-reporting-summary-flat.pdf
Life sciences study design
All studies must disclose on these points even when the disclosure is negative.
Sample size
No sample size calculations were conducted.
A total of 1,488,550 H&E whole histopathology slide images were gathered for training the foundation model. The superior performance of our pretrained model compared to all other baselines indicates that the sample size was sufficient. For information on downstream datasets, please refer to the datasets and evaluation subsection in the Methods section of the manuscript.
Data exclusions
No particular data exclusion was performed.
Replication
Attempts at replication were successful for the reported model results. The open-sourced model can be applied at https://huggingface.co/ paige-ai/Virchow. The SDK for replicating the aggregator experiments can be accessed at https://github.com/Paige-Al/paige-ml-sdk
Randomization
For downstream evaluation involving the creation of training, validation, and test splits, we utilized the official splits provided by the original investigators of each dataset whenever available. When such splits were not available, we created them randomly. Generally, we created random splits stratified by class, ensuring similar class proportions across splits, and, if possible, at the patient level, ensuring that slides from the same patient were kept within the same split. The random seeds were fixed, and the splits were documented to ensure replicability.
Blinding
Blinding is not necessary for our study.
Reporting for specific materials, systems and methods
We require information from authors about some types of materials, experimental systems and methods used in many studies. Here, indicate whether each material, system or method listed is relevant to your study. If you are not sure if a list item applies to your research, read the appropriate section before selecting a response.
Materials & experimental systems
Methods
n/a
Involved in the study
n/a
Involved in the study
X
X
X
Palaeontology and archaeology
X
Clinical data
(T) Check for updates
¹Paige, New York, NY, US. ²Microsoft Research, Cambridge, MA, US. Memorial Sloan Kettering Cancer Center, New York, NY, US. NSW Health Pathology, St George Hospital, Sydney, New South Wales, Australia. University of Rochester, Rochester, NY, US. These authors contributed equally: Eugene Vorontsov, Alican Bozkurt, Adam Casson, George Shaikovski, Michal Zelechowski, Kristen Severson. e-mail: siqi.liu@paige.ai
including a small fraction of the prostate (teal), breast (blue) and BLN (yellow) tissue specimens that these clinical models were respectively trained on. d, A categorization of failure models of the pan-cancer model and four canonical examples of the primary types of failures. In all panels,* is used to indicate pairwise statistical significance (*P<0.05, **P<0.01, ***P<0.001, ****P<0.0001; pairwise DeLong’s test). Error bars denote the two-sided 95% confidence interval, estimated with DeLong’s method. C., carcinoma. Inv., invasive.