الفيروبتوز هو شكل من أشكال موت الخلايا المنظم الذي يحدث نتيجة لتراكم الهيدروبيروكسيدات الدهنية المعتمدة على الحديد. يقوم بروتين السيلينوبروتين جلاوتاثيون بيروكسيداز 4 (GPX4) بتثبيط الفيروبتوز من خلال إزالة سمية الهيدروبيروكسيدات الدهنية عبر بقايا السيلينوسيستين (Sec) الحفازة. Sec، هو الأحماض الأمينية، يتم تخليقها حيوياً من مانح سيلينيوم تفاعلي على tRNA الخاص بهايُعتقد أن السيلينيوم داخل الخلايا يجب أن يتم توصيله ‘بأمان’ و’بكفاءة’ بواسطة بروتين ناقل نظرًا لتفاعليته العالية وتركيزاته المنخفضة جدًا. هنا، حددنا بيروكسيريدكسين 6 (PRDX6) كعامل جديد لتخليق السيلينوبروتين. يؤدي فقدان PRDX6 إلى تقليل تعبير السيلينوبروتينات ويحفز الفيروبتوز عبر تقليل GPX4. من الناحية الميكانيكية، يزيد PRDX6 من كفاءة استخدام السيلينيوم داخل الخلايا من خلال نقل السيلينيوم بين البروتينات داخل السيلينوسيستيل-tRNA.آلية التخليق، مما يؤدي إلى تخليق فعال لـ سيلينوسيستيل- tRNAتسلط هذه النتائج الضوء على أنظمة الأيض السيلينيوم التي لم يتم التعرف عليها من قبل وتوفر رؤى جديدة حول الفيروبتوز.
فيريوبتوسيس، نوع من الموت الخلوي المبرمج الذي يتم تحفيزه بواسطة أكسدة الدهون الناتجة عن الحديد، مرتبط بالعديد من الحالات المرضية مثل السرطان، والأمراض التنكسية العصبية، وإصابة نقص التروية-إعادة التروية؛ وبالتالي، تم دراسته بشكل مكثف.تمتلك الخلايا آليتين رئيسيتين للدفاع ضد الفيروبتوز: GPX4 وبروتين مثبط الفيروبتوز 1 (FSP1). يستخدم GPX4 الجلوتاثيون (GSH) لقمع الفيروبتوز من خلال إزالة سمية الهيدروبيروكسيدات الدهنية وتحويلها إلى كحوليات دهنية غير سامة.، و FSP1 هو أكسيدوريدوكتاز يقوم بتقليل اليوبكوينون (CoQ) إلى يوبكوينول (مضاد أكسدة لالتقاط الجذور الحرة.
GPX4 هو بروتين سيلين يحتوي على حمض الأميني سيك يقيم في الموقع النشطيتم دمج السيلينيوم (Sec) في كودون UGA عندما يكون عنصر تسلسل إدخال السيلينيوم (SECIS) موجودًا في-منطقة غير مترجمة من“. على عكس الأحماض الأمينية الأخرى، فإن ناقل RNA السيلينوسيستين لسيك (Sec-tRNA) ) يتم تخليقه حيوياً من مانح السيلينيوم التفاعلي السيلينيد على tRNA الخاص به من خلال تفاعلات إنزيمية معقدةيتم فوسفات سيلينيد بواسطة سينثيتاز سيلينوفوسفات 2 (SEPHS2)ثم تم دمجه لاحقًا في Sec-tRNAتخليق السيلينوبروتينات ضروري للحياة، كما أظهرت الدراسات التي تصف الوفاة الجنينية في الفئران التي تفتقر إلى تخليق السيلينوبروتينات.على الرغم من أهميتها، لا يزال مسار تخليق السيلينوبروتين غير واضح. على وجه الخصوص، يُفترض أن السيلينيدات شديدة التفاعل تُنقل بأمان وكفاءة بواسطة بروتين ناقل إلى SEPHS2؛ ومع ذلك، لم يتم التعرف عليها بعد..
هنا، قمنا بإجراء فحص شامل للجينوم لتحفيز الفيروبتوز من خلال الحديد وحددنا PRDX6 كعامل جديد لتخليق السيلينوبروتين. أدى فقدان PRDX6 إلى تقليل كبير في تعبير السيلينوبروتينات، بما في ذلك GPX4، وأدى إلى تحفيز الفيروبتوز من خلال تقليل مستويات GPX4. من الناحية الميكانيكية، يعمل PRDX6 كحامل للسيلينيد، حيث ينقل السيلينيد إلى SEPHS2 لتسهيل الاستخدام الفعال للسيلينيوم.
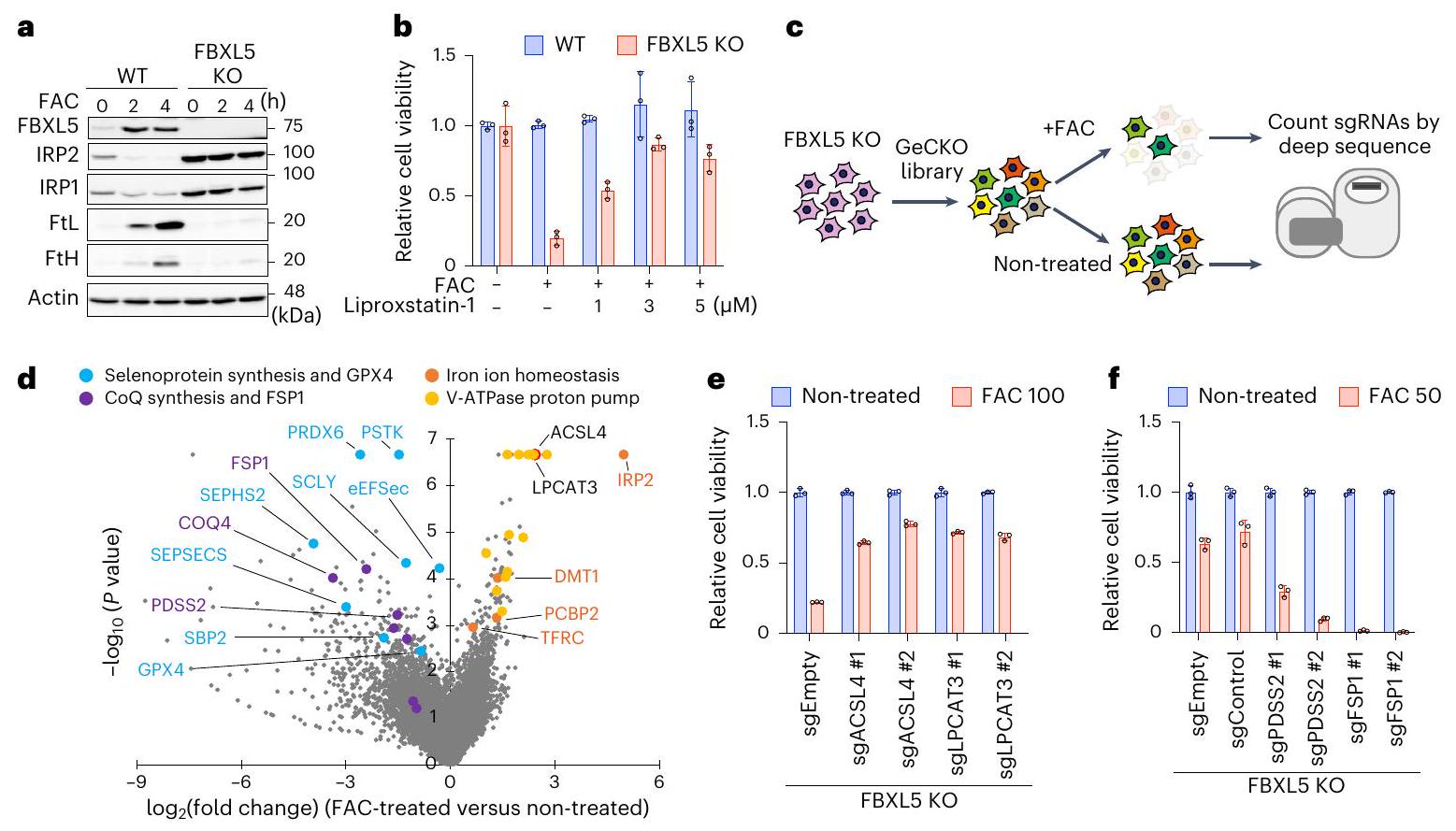
الشكل 1 | فحص CRISPR على مستوى الجينوم لاستجابة الفيروبتوزيس المحفزة بالحديد. أ، تم معالجة خلايا MEFs WT أو FBXL5 KO بـ FAC ( ) للفترات المحددة، وتم تحليل lysates الخلوية بواسطة التحليل المناعي. البيانات تمثل ثلاث تجارب مستقلة. ب، بقاء WT أو FBXL5 KO MEFs المعالجة لمدة 48 ساعة مع FAC ( ) في وجود ليبروكستاتين – أو . ج، مخطط يوضح استراتيجية فحص CRISPR-Cas9. د، مخططات بركانية تظهر الجينات التنظيمية المعنية بالفيروبتوز الناتج عن الحديد.قيمة تم توليد التغير في الطي بواسطة اختبار MAGeCK. قابلية بقاء خلايا FBXL5 KO التي تعبر عن sgEmpty أو sgRNAs المستهدفة لـ ACSL4 أو LPCAT3؛ تم معالجة الخلايا بـ FAC ( ) لـ قابلية خلايا FBXL5 KO التي تعبر عن sgEmpty أو sgControl أو sgRNAs المستهدفة لـ PDSS2 أو FSP1؛ تم معالجة الخلايا بـ FACلمدة 30 ساعة. تُعرض بيانات القابلية للحياة (في ب، هـ، و) كمتوسطالانحراف المعياري لثلاث نسخ بيولوجية.
زيادة استخدام السيلينيوم تقمع الفيروبتوز وتساهم في تقدم السرطانوجدنا أن المرضى الذين يحملون سرطانات ذات تعبير عالٍ عن PRDX6 لديهم توقعات سيئة. توفر هذه النتائج رؤى حول نظام الأيض السيلينيوم غير المعروف سابقًا في الخلايا بالإضافة إلى إمكانيته كهدف لعلاج السرطان.
النتائج
فحص الفيروبتوزيس الناتج عن الحديد
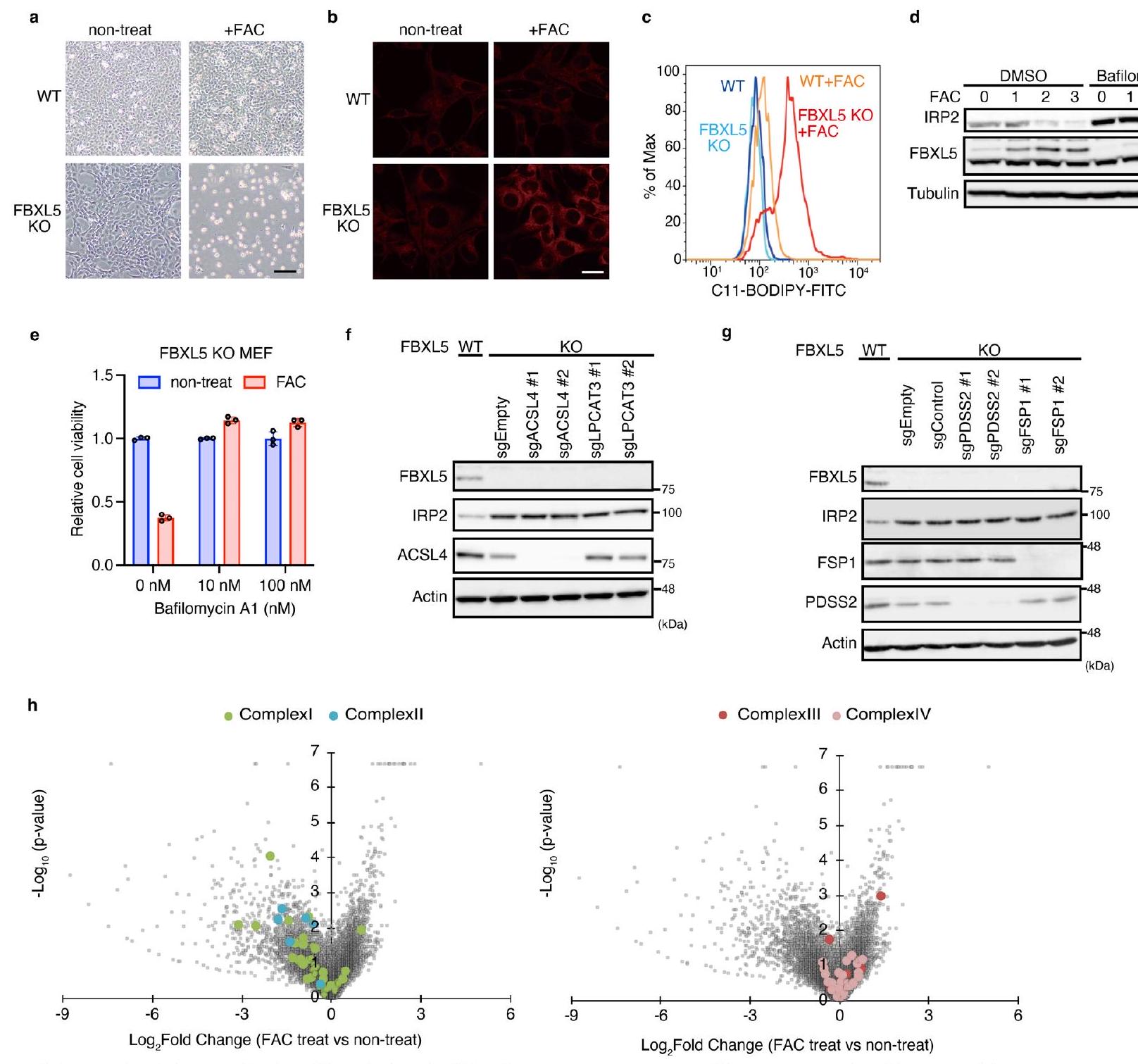
البادئة ‘فيرو’ تشير إلى أن الفيروبتوزيس يجب أن يتم تصنيفه كشكل من أشكال سمية الحديد؛ ومع ذلك، في معظم دراسات الفيروبتوزيس، لا يتم تحفيز موت الخلايا عن طريق إدارة الحديد. بل، يتم تحفيزه عن طريق تثبيط المزيلات لهيبيروكسيد الدهون مثل مثبط GPX4 RSL3 أو مثبط ناقل السيستين إيراستين.لإعادة تحليل الفيروبتوز من منظور سمية الحديد ولتحديد منظمات جديدة، قمنا بتطوير نظام لتحفيز الفيروبتوز يعتمد فقط على إضافة الحديد. يعمل بروتين تنظيم الحديد 2 (IRP2) ونظام بروتين F-box والغني بالتكرارات الليوسينية 5 (FBXL5) كمنظم رئيسي لتوازن الحديد الخلوي من خلال قمع امتصاص الحديد وزيادة تخزين الحديد من خلال تحلل IRP2 بواسطة FBXL5 (المراجع 19-21). لذلك، لتحفيز سمية الحديدقمنا بإزالة FBXL5 من الخلايا الليفية الجنينية للفئران (MEFs) باستخدام نظام CRISPR-Cas9 (الشكل 1a). أدى العلاج بالسترات الأمونيوم الحديدي (FAC) إلى قتل خلايا FBXL5 المعطلة (KO) بشكل انتقائي من خلال زيادة تركيز الحديد داخل الخلايا، كما تم الكشف عنه بواسطة مجس فلوري محدد لـ (الشكل 1ب والشكل البياني الموسع 1أ،ب) يُعتقد أن الفيروبتوزيس يتم تحفيزه بواسطة الهيدروبركسيد من الأحماض الدهنية غير المشبعة (PUFAs) في غشاء الخلية بواسطة الحديد النشط بالأكسدة.كشف صبغ BODIPY 581/591 C11 عن زيادة في الهيدروبيروكسيد في خلايا FBXL5 KO المعززة بالحديد (الشكل 1c من البيانات الموسعة). بدا أن موت خلايا FBXL5 KO الناتج عن الحديد يحدث من خلال الفيروبتوزيس لأنه تم منعه بواسطة مثبط الفيروبتوزيس ليبروكستاتين-1 (الشكل 1b). لتحديد الجينات المشاركة في الفيروبتوزيس الناتج عن الحديد، قمنا بإجراء دراسة جينومية شاملة.
فحص CRISPR حيث تم إصابة خلايا KO لـ FBXL5 بمكتبة GeCKOv2، تلاها زراعة مع أو بدون FAC (الشكل 1c). باستخدام حد قطعأظهر الفحص تحديد حوالي 150 جينًا ترمز لمثبطات أو منشطات الفيروبتوسيس المستحثة بالحديد (الشكل 1د). كما هو متوقع، كان أحد الجينات البارزة التي ترمز لمنشطات الفيروبتوسيس هو IRP2؛ في الواقع، أدى تثبيت IRP2 الناتج عن فقدان FBXL5 إلى تراكم الحديد النشط من الناحية الحمراء في الخلايا. (الشكل 1أ). شملت النتائج البارزة الأخرى منظمات توازن الحديد مثل مستقبل ترانسفيرين 1 (TFRC) وناقل المعادن ثنائية التكافؤ 1 (DMT1) ومجمع مضخة البروتون V-ATPase، والتي تتطلب جميعها لامتصاص الحديد. (الشكل 1د). استخدمنا مثبط V-ATPase بافيلوميسين A1 لتأكيد أن حموضة العضيات ضرورية لامتصاص الحديد والفيروبتوز الناتج عن الحديد (بيانات موسعة الشكل 1د، هـ). والأهم من ذلك، أن قائمة النتائج ذات الثقة العالية تضمنت المنظمات المعروفة للفيروبتوز مثل أسيل-CoA سينثيتاز عضو عائلة السلسلة الطويلة 4 (ACSL4) وليسوفوسفوليديل كولين أسيل ترانسفيراز 3 (LPCAT3). (الشكل 1د)، وكلاهما مطلوب لإدخال الأحماض الدهنية المتعددة غير المشبعة في الفوسفوليبيدات الغشائية. أدى حذف ACSL4 أو LPCAT3 من خلايا FBXL5 KO إلى استعادة حيوية الخلايا (الشكل 1هـ والشكل التمديدي 1و)، مما يشير بشكل أكبر إلى أن الموت الخلوي الناتج عن الحديد يتم أيضًا بواسطة أكسدة الأحماض الدهنية المتعددة غير المشبعة. بالإضافة إلى ذلك، حددنا جينات مثبطة، وآلية تخليق CoQ وFSP1 (الشكل 1د)حذف وحدة الديفوسفات سينثاز 2 (PDSS2)، وهي مكون من آلية تخليق CoQ، أو FSP1 زاد بشكل ملحوظ من موت خلايا FBXL5 KO (الشكل 1f والشكل التمديدي 1g)؛ وبالتالي، فإن حبس الجذور الدهنية بواسطةمشارك أيضًا في قمع الفيروبتوزيس الناتج عن الحديد. من المثير للاهتمام أن الجينات المثبطة التي تشفر معقد سلسلة نقل الإلكترون الميتوكوندري المعقد I و II (CoQ أوكسيدوريدوكتاز)، ولكن ليس المعقد III و IV، كانت غنية (الشكل البياني الممتد 1h)، مما يشير إلى أن في الميتوكوندريا أيضًا يثبط الفيروبتوز. لذلك، فإن فحص الفيروبتوز المستحث بالحديد لدينا، الذي لا يعتمد على المحفزات التقليدية للفيروبتوز مثل RSL3 أو إيراستين، مفيد في تحديد ليس فقط منشطات الفيروبتوز ولكن أيضًا مثبطاته.
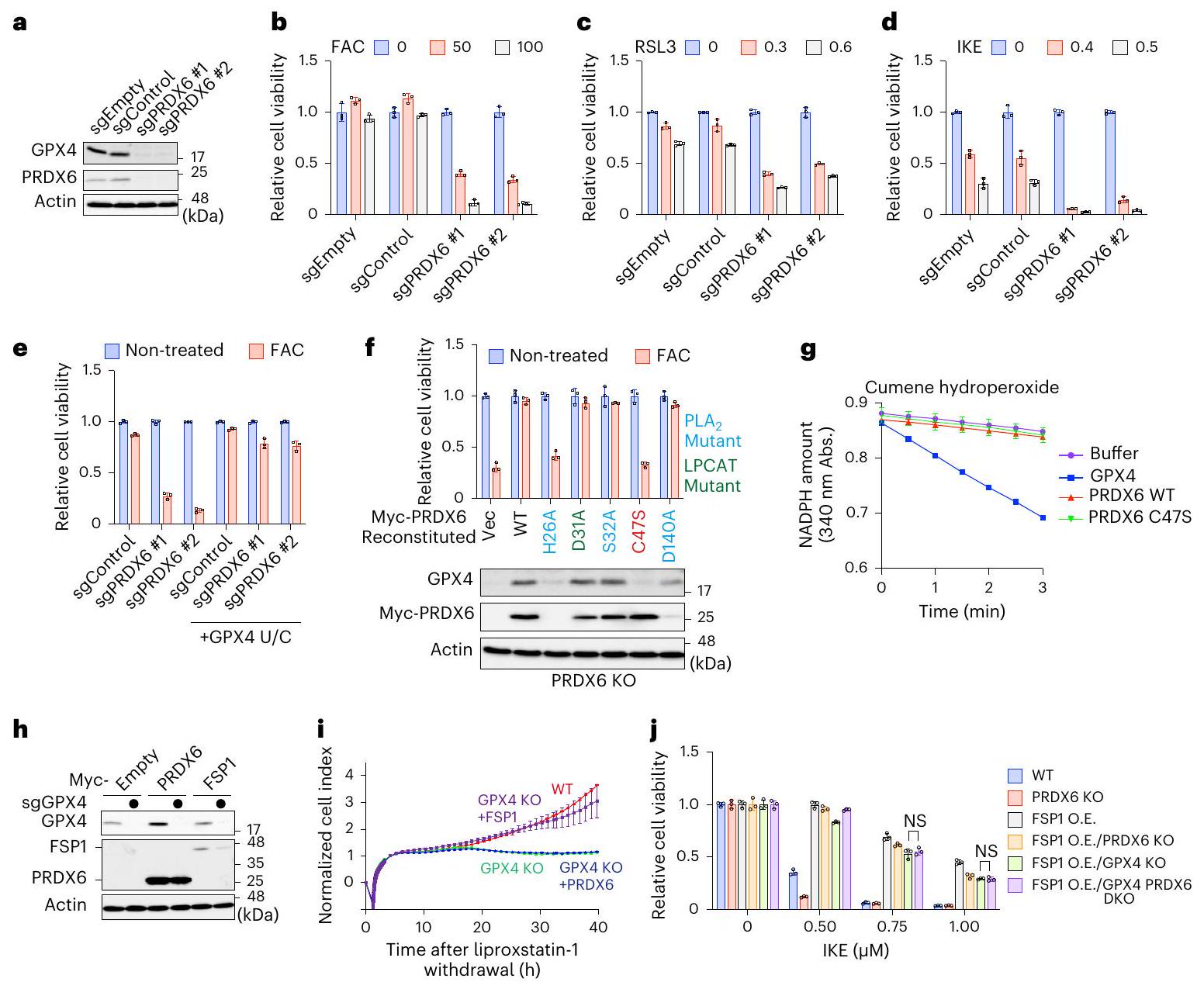
الشكل 2 | PRDX6 يثبط الفيروبتوزيس المحفز بالحديد بشكل غير مباشر من خلال زيادة تعبير GPX4. أ، تحليل المناعية للبروتينات من مستخلصات خلايا تعبر عن sgEmpty أو sgControl أو sgRNAs تستهدف PRDX6. البيانات تمثل ثلاث تجارب مستقلة. ب-د، حيوية خلايا التحكم أو خلايا PRDX6 KO المعالجة بـ FAC (50 أو ) لـ RSL3 (0.3 أو ) لـ أو IKE (0.4 أو ) لـ جدوى خلايا التحكم أو خلايا PRDX6 KO التي تعبر بشكل مستقر عن الطفرة GPX4U/C؛ تم حضن الخلايا مع FAC ( ) لـ جدوى خلايا PRDX6 KO التي تعبر بشكل مستقر عن الطفرات المحددة لـ PRDX6 المربوطة في وجود FAC ) لمدة 48 ساعة. كما يتم عرض تحليل المناعية للعينات من الخلايا المحددة. PLA فوسفوليباز A2. ج، قياس نشاط GPX باستخدام البروتينات المؤتلفة البروتينات. تم استخدام بيروكسيد الكومين كركيزة. تُعرض البيانات كمتوسط s.e.m من ثلاثة تجارب مستقلة. h. تحليل المناعية للبروتينات من مستخلصات خلايا التحكم أو خلايا GPX4 KO التي تعبر بشكل مستقر عن Myc-PRDX6 أو FSP1. البيانات تمثل تجربتين مستقلتين. i، المراقبة المستمرة لمدى حيوية خطوط الخلايا المحددة بعد سحب الليبروكستاتين-1. البيانات مقدمة كمتوسط.الانحراف المعياري للمتوسط لثلاث نسخ بيولوجية. ج، قابلية الخلايا المشار إليها في وجود التركيزات المشار إليها من IKE لمدة 24 ساعة. غير مهم؛ايك; إيك ); تحليل التباين ثنائي الاتجاه. بيانات القابلية للحياة (في ) يتم تقديمها كالمتوسط الانحراف المعياري لثلاث نسخ بيولوجية.
تحديد PRDX6 كمنظم لتعبير GPX4
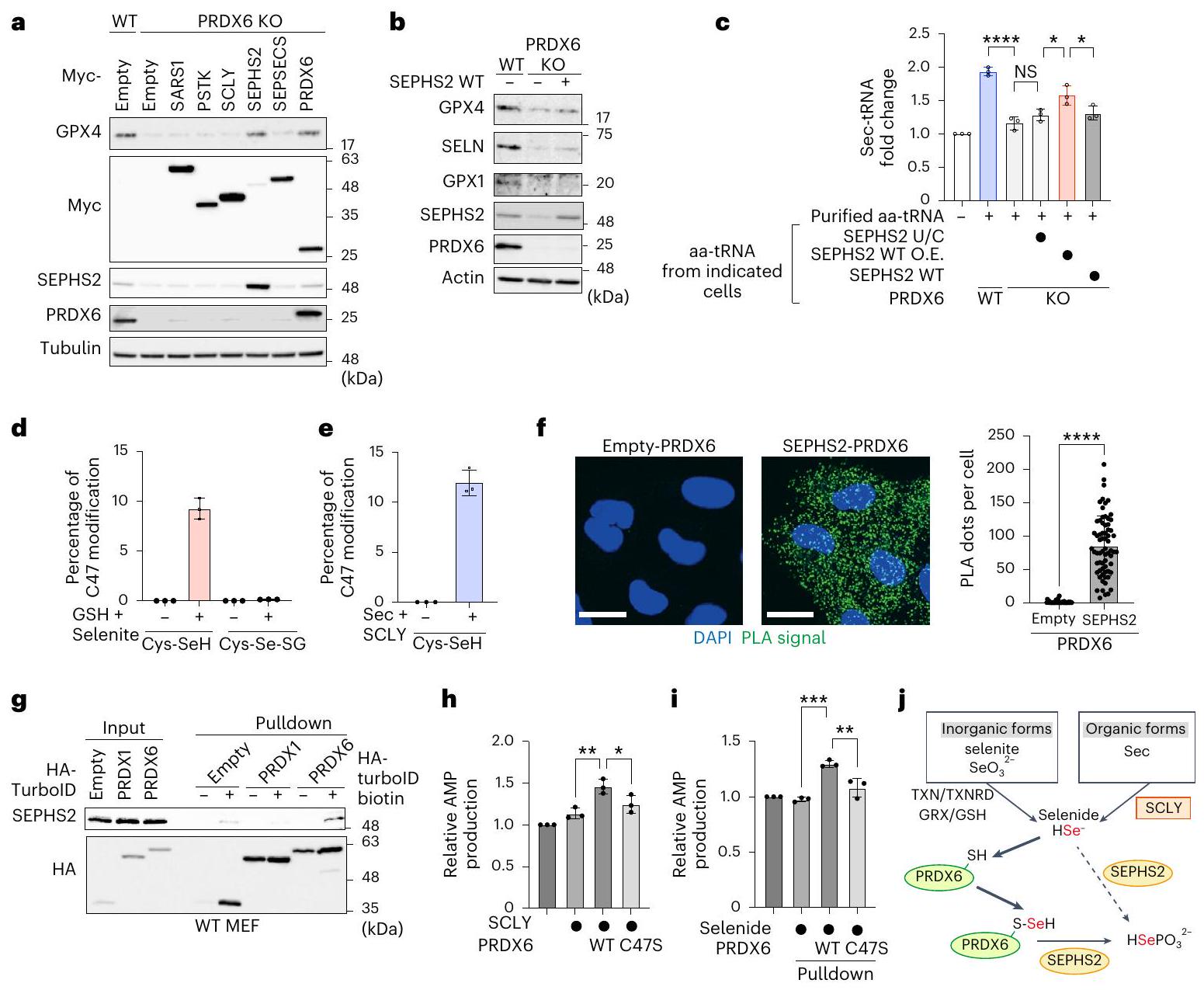
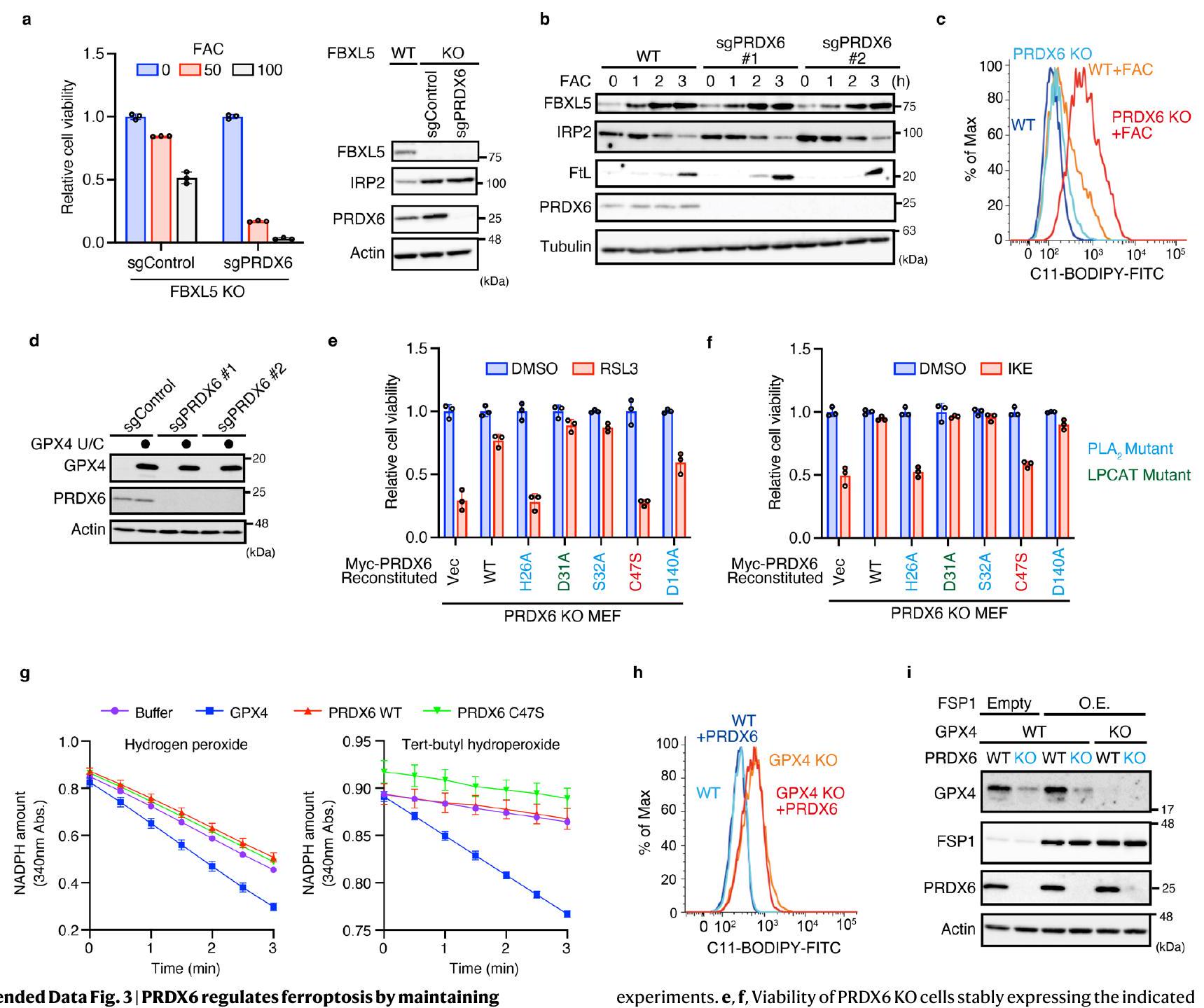
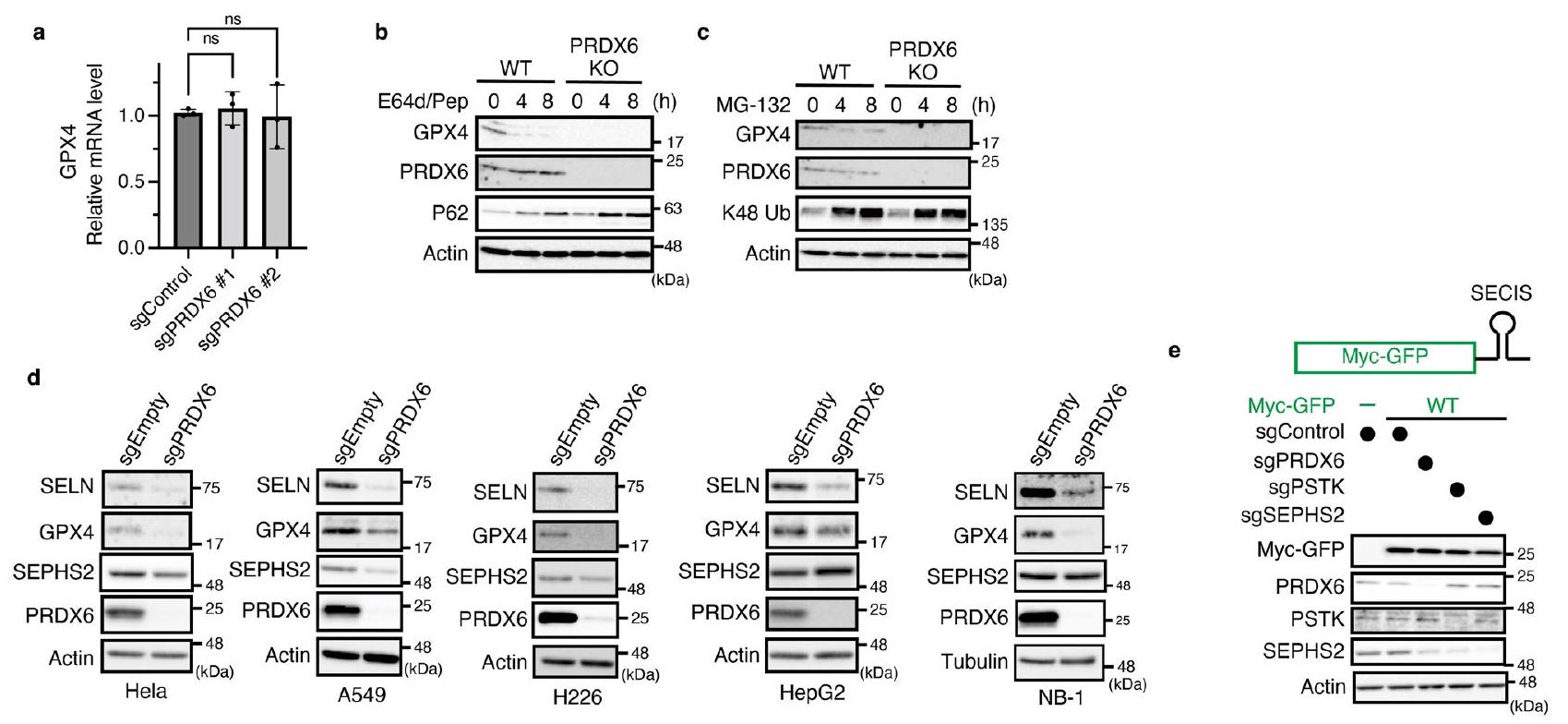
أظهر تحليل علم الأحياء الجيني لمثبطات الفيروبتوز الناتجة عن الحديد أن عوامل تخليق السيلينوبروتين كانت المجموعة الأكثر غنى (الشكل 2a من البيانات الموسعة). قائمة المثبطات ذات الثقة العالية أثارت احتمال أن فقدان عوامل تخليق السيلينوبروتين يجعل الخلايا أكثر حساسية للفيروبتوز الناتج عن الحديد (الشكل 1d). من بين السيلينوبروتينات، تم تصنيف المنظم الرئيسي للفيروبتوز GPX4 بشكل عالٍ كمثبط (الشكل 2b من البيانات الموسعة). لذلك، اشتبهنا في أن منظمات جديدة لـ GPX4 قد تكون مدرجة في قائمة المثبطات لدينا (الشكل 2c من البيانات الموسعة)، وأجرينا فحصًا ثانويًا باستخدام وفرة GPX4 كقراءة (الشكل 2d من البيانات الموسعة). بشكل غير متوقع، وجدنا أن حذف PRDX6 قلل بشكل كبير من تعبير GPX4 (الشكل 2d من البيانات الموسعة)، وهو ما أكدناه باستخدام نوعين مختلفين من RNA الدليلية (gRNAs) (الشكل 2a). من المدهش أن فقدان PRDX6 جعل خلايا FBXL5 KO (الشكل 3a من البيانات الموسعة) وأيضًا خلايا النوع البري (WT) (الشكل 2b) أكثر حساسية للفيروبتوز الناتج عن الحديد، في حين أن حذف PRDX6 لم يكن له تأثير على توازن الحديد لأن استقرار FBXL5 الناتج عن FAC وتدهور IRP2 كان مشابهًا لذلك في خلايا WT (البيانات الموسعة
الشكل 3ب). هذا الاكتشاف استبعد الاحتمال البديل بأن فقدان PRDX6 يمنع الفيروبتوزيس الناتج عن الحديد من خلال تعطيل استقلاب الحديد في الخلايا. أشار زيادة أكسدة الدهون الناتجة عن الحديد في خلايا PRDX6 KO إلى أن PRDX6 هو مثبط أساسي للفيروبتوزيس المحفز بالحديد (الشكل 3c من البيانات الموسعة). بالإضافة إلى ذلك، وجدنا أن PRDX6 يشارك أيضًا في تثبيط الفيروبتوزيس الذي يتوسطه إيميدازول كيتون إيراستين. (IKE) و RSL3 (الشكل 2c، d). لمعرفة ما إذا كان الانخفاض في GPX4 مسؤولاً عن حساسية الفيروبتوز في خلايا PRDX6 KO، قمنا بتعبير عن طافرة GPX4 نشطة جزئيًا، حيث تم استبدال Sec(U)46 بالسيستين (GPX4 U/C) في خلايا PRDX6 KO (بيانات موسعة الشكل 3d).تعبير الطفرة GPX4 U/C أنقذ خلايا PRDX6 KO من الموت الخلوي الناتج عن الحديد (الشكل 2e)، مما يشير إلى أن PRDX6 يثبط موت الخلايا من خلال الحفاظ على تعبير GPX4.
ينتمي PRDX6 إلى عائلة إنزيمات مضادات الأكسدة PRDXs.. من بينها، يتميز PRDX6 بخصيصة فريدة حيث يحتوي على بقايا سيستين محفوظة واحدة (C47)، في حين تحتوي الأعضاء الأخرى من العائلة على اثنتينيظهر PRDX6 ثلاث أنشطة إنزيمية: نشاط بيروكسيداز الجلوتاثيون (GPX) (C47)، والفوسفوليباز A2 نشاط (H26 و S32 و D140) ونشاط LPCAT (D31) (تظهر البقايا الأساسية لكل نشاط بين قوسين)لفحص النشاط المطلوب للحفاظ على وفرة GPX4، قمنا بإعادة تكوين خلايا PRDX6 KO باستخدام PRDX6 WT أو الطفرات (الشكل 2f). على الرغم من أن الطفرة H26A فشلت في استعادة مستويات GPX4 بسبب ضعف التعبير، إلا أن الطفرات S32A و D140A استعادتهما. لذلك، استنتجنا أن نشاط الفوسفوليباز A2 غير ضروري. وجدنا أن الطفرة C47S فشلت أيضًا في استعادة تعبير GPX4 (الشكل 2f). كما أن الطفرة C47S حسّست الخلايا ليس فقط تجاه الحديد ولكن أيضًا تجاه الفيروبتوسيس التقليدي (الشكل 2f والشكل الإضافي 3e، f). وقد أشارت التقارير السابقة إلى أن PRDX6 يظهر نشاط GPX عند C47 (المرجع 33)؛ ومع ذلك، تم الإبلاغ عن نتائج متناقضة أيضًا.وجدنا أن PRDX6 المنقى ليس له نشاط GPX (الشكل 2g والشكل الإضافي 3g)، مما يشير إلى أن السيستين المحفوظ في PRDX6 له وظيفة مختلفة عما كان يُعتقد سابقًا. بعد ذلك، تساءلنا عما إذا كان PRDX6 يقلل من الهيدروبيروكسيدات الدهنية في الخلايا بشكل مباشر. أدى الإفراط في التعبير عن PRDX6 إلى زيادة تعبير GPX4 في خلايا WT (الشكل 2h)، مما يؤكد أن PRDX6 ضروري لتعبير GPX4. حيث أن فقدان GPX4 يجعل الخلايا حساسة بشدة للفيروبتوز.قمنا بزراعة خلايا GPX4 KO في وسط ثقافي يحتوي على مثبط الفيروبتوزيس ليبروكستاتين-1. أدى غسل ليبروكستاتين-1 إلى تثبيط نمو خلايا GPX4 KO (الشكل 2i). أعاد التعبير عن FSP1، الذي يثبط الفيروبتوزيس بشكل مستقل عن GPX4، تكاثر خلايا GPX4 KO، كما تم الإبلاغ عنه سابقًا.; ومع ذلك، فإن تعبير PRDX6 فشل في استعادة تكاثر خلايا GPX4 KO (الشكل 2i). علاوة على ذلك، لم يقلل تعبير PRDX6 من تراكم الهيدروبيروكسيدات الدهنية في خلايا GPX4 KO (بيانات موسعة الشكل 3h)، مما يشير بقوة إلى أن PRDX6 لا يثبط الهيدروبيروكسيد الدهني أو الفيروبتوسيس بشكل مباشر؛ بل إنه يثبطهما عن طريق زيادة كمية GPX4. كما قمنا أيضًا بإنشاء خلايا ذات تعبير مفرط لـ FSP1 (O.E.)/GPX4 و PRDX6 مزدوجة النقص (DKO) لتأكيد نتائجنا (بيانات موسعة الشكل 3i). لم يؤد فقدان PRDX6 إلى زيادة الفيروبتوسيس الناتج عن IKE في خلايا FSP1 O.E./GPX4 KO (الشكل 2j)، مما يدعم فرضيتنا بأن PRDX6 يثبط الفيروبتوسيس بشكل غير مباشر من خلال الحفاظ على تعبير GPX4.
مشاركة PRDX6 في تخليق السيلينوبروتين
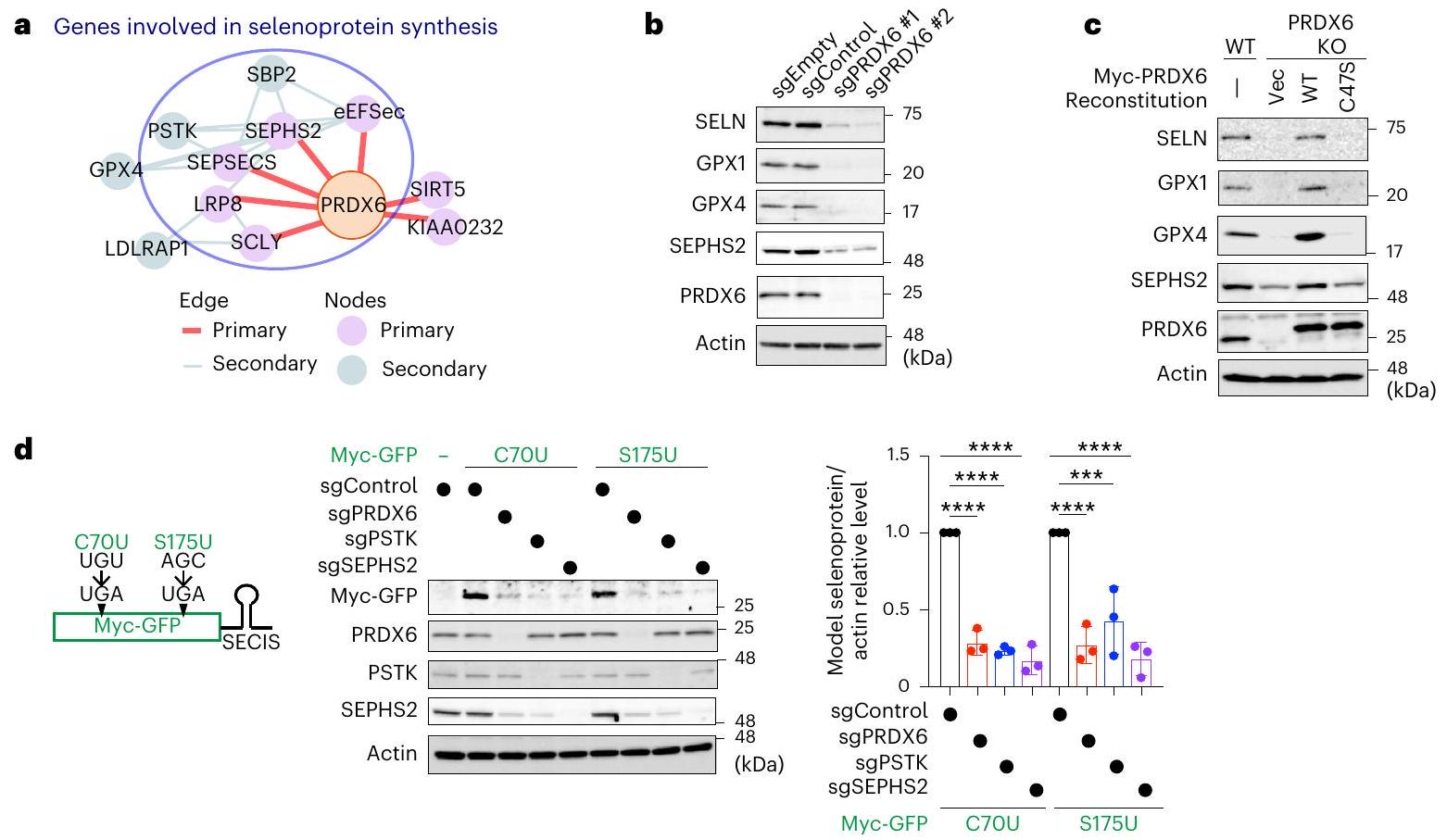
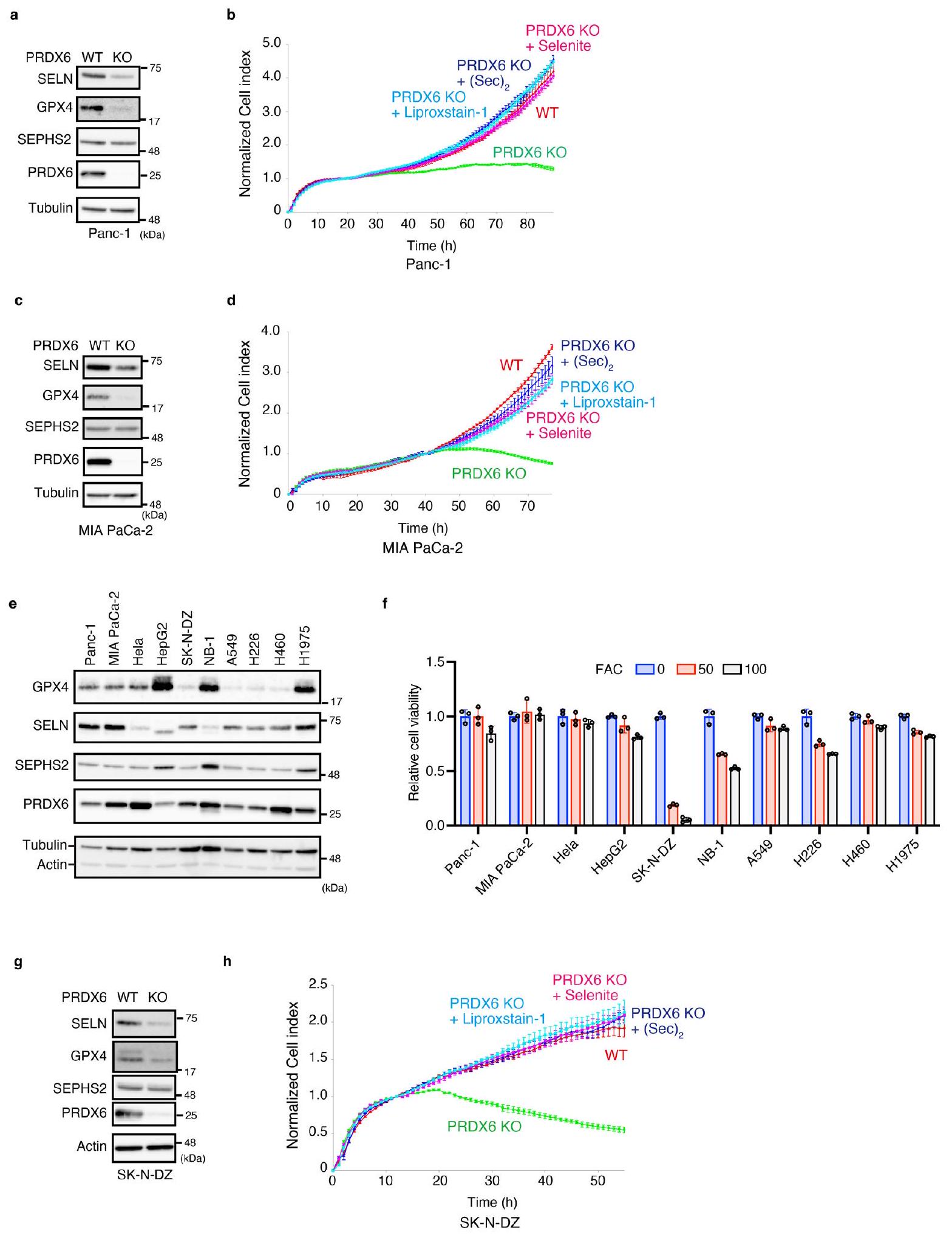
بعد ذلك، قمنا بتفكيك الآلية الكامنة وراء زيادة تعبير GPX4 التي يسببها PRDX6. لم نجد أي اختلافات واضحة في مستويات mRNA لـ GPX4 بين خلايا PRDX6 KO وخلايا WT (الشكل 4a من البيانات الموسعة)، كما أن مثبطات البروتيازوم أو الليزوزوم لم تستعد مستويات GPX4 في خلايا PRDX6 KO (الشكل 4b،c من البيانات الموسعة). أظهر تحليل علم الأحياء الجيني غنى عوامل تخليق السيلينوبروتين (الشكل 2a من البيانات الموسعة). علاوة على ذلك، وجدنا أن أدوات قاعدة بيانات التوازي غير المتحيزكما أشار إلى وجود ارتباط وظيفي قوي بين PRDX6 والجينات المشاركة في تخليق السيلينوبروتين (الشكل 3أ). لذلك، استنتجنا أن PRDX6 يشارك في تخليق السيلينوبروتين. في الواقع، أدى حذف PRDX6 إلى تقليل تعبير السيلينوبروتينات الأخرى، بما في ذلك السيلينوبروتين N (SELN)، GPX1 وSEPHS2 (الشكل 3ب). إن Cys47 المحفوظ في PRDX6 ضروري لتعبير السيلينوبروتينات لأن تعبير السيلينوبروتين تم إنقاذه من خلال إعادة تعبير PRDX6 WT، ولكن ليس من طفرات C47S، في خلايا PRDX6 KO (الشكل 3ج). علاوة على ذلك، أدى فقدان PRDX6 إلى تقليل كمية السيلينوبروتينات في عدة خطوط خلايا سرطانية بشرية (بيانات موسعة الشكل 4د)، مما يشير إلى أن دور PRDX6 محفوظ عبر العديد من أنواع السرطان.
لتأكيد دور PRDX6 في تخليق السيلينوبروتين، قمنا بإنشاء cDNAs التي تشفر سيلينوبروتينات نموذجية تحتوي على كودون UGA في الإطار عند C70 أو S175 من GFP (C70U و S175U)، بالإضافة إلى عنصر 3’UTR SECIS من GPX4 (الشكل 3d). كما هو متوقع، فإن فقدان الفوسفو سيريل-tRNAكيناز (PSTK) أو SEPHS2، وكلاهما ضروري لتخليق Sec-tRNA (المرجع 38)، تم تقليل التعبير عن كل من GFP C70U و S175U بشكل كبير (الشكل 3d). كما أن فقدان PRDX6 قلل أيضًا من التعبير عن كلا البروتينين السيلينوبروتين النموذجيين (الشكل 3d)، في حين أن فقدان PRDX6 لم يؤثر على التعبير عن GFP WT (الشكل التمديدي 4e). تشير هذه النتائج بوضوح إلى أن PRDX6 له دور حاسم في إدخال السيلينيوم في كودون UGA.
PRDX6 يعزز الاستخدام الفعال للسيلينيوم
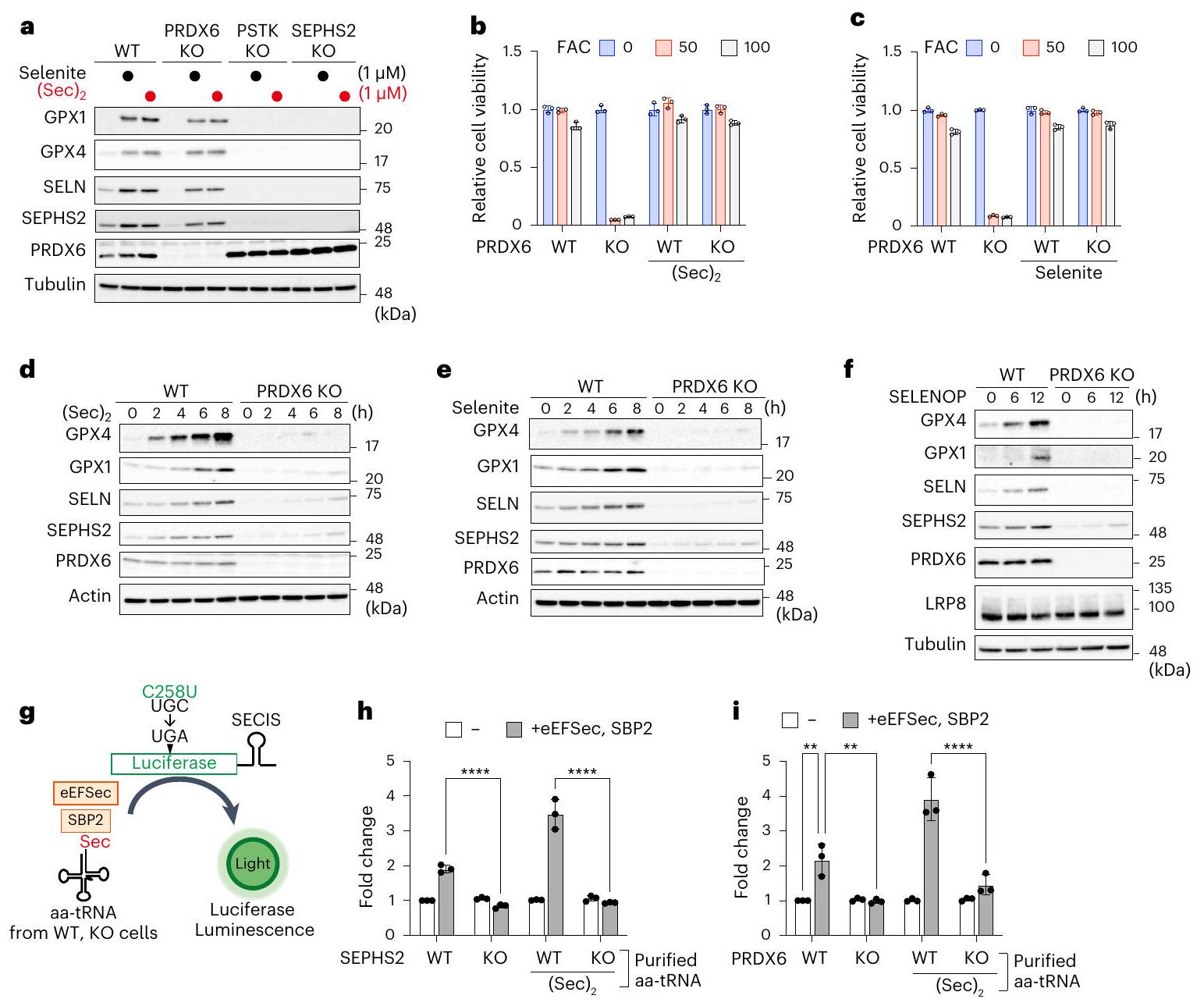
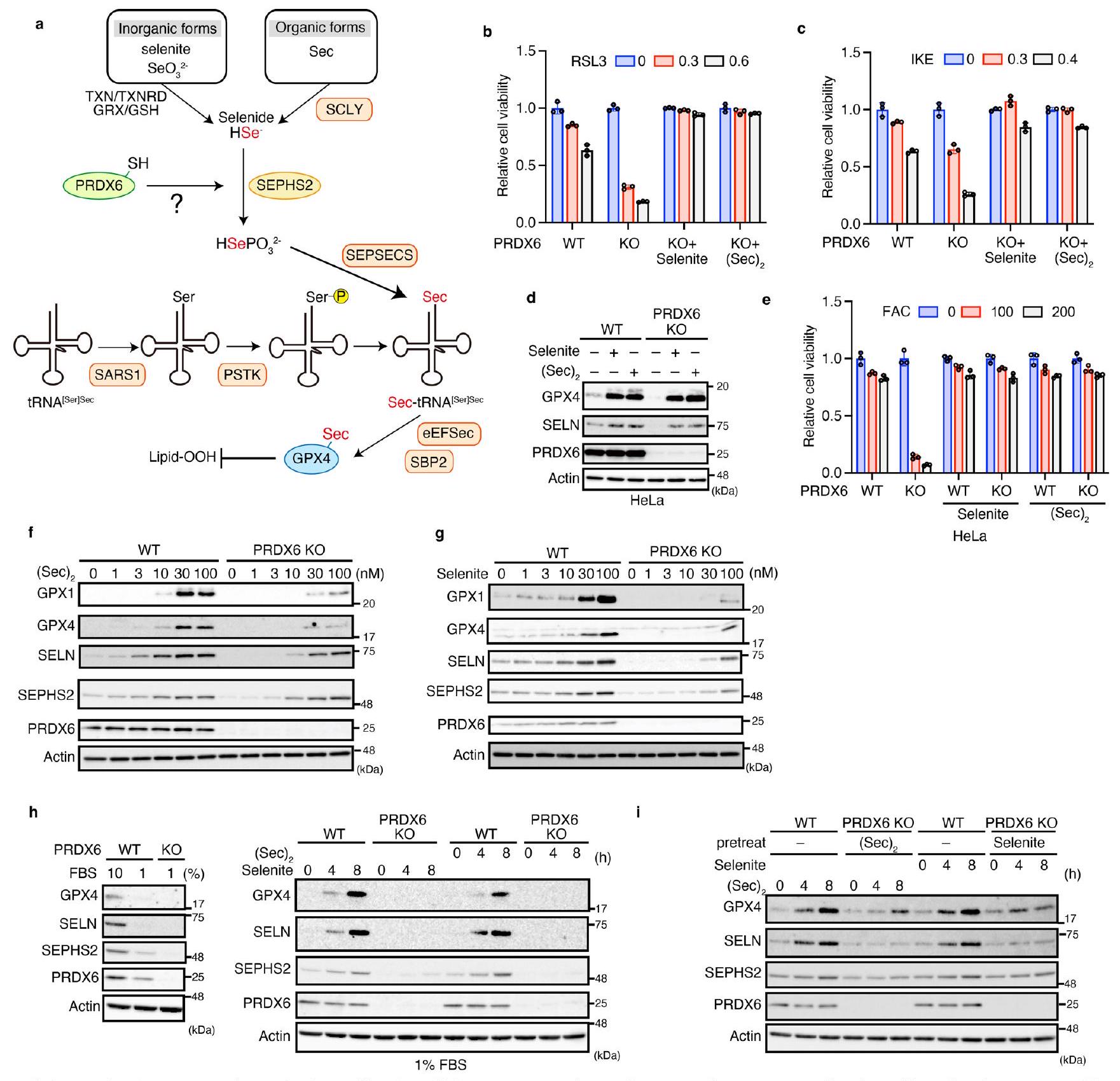
بعد ذلك، استكشفنا دور PRDX6 في تخليق السيلينوبروتين. يمكن استخدام كل من الأشكال غير العضوية والعضوية للسيلينيوم لتخليق Sec-tRNA. (الشكل 5a من البيانات الموسعة). يتم اختزال السيلينيت غير العضوي بواسطة GSH ونظام الثيوريدوكسين، بينما يتم تحلل السيلينوسيستين العضوي بواسطة إنزيم سيلينوسيستين لاييز (SCLY)، مما يؤدي إلى إنتاج مانح السيلينيوم النشط الشائع السيلينيد، والذي يعتبر مهمًا لـ Sec-tRNA.التركيب (الشكل 5أ من البيانات الموسعة)إن إضافة كميات زائدة من السيلينيت أو السيلينوسيستين ((Sec)2) إلى ثقافات خلايا WT زادت بشكل ملحوظ من تعبير السيلينوبروتين، مما يشير إلى أن إمداد السيلينيوم محدود تحت ظروف الثقافة العادية (الشكل 4a). على الرغم من أن أي من مصدر السيلينيوم فشل في تحفيز تعبير السيلينوبروتينات في الخلايا التي تفتقر إلى PSTK أو SEPHS2 (الشكل 4a)، وكلاهما ضروري لـ Sec-tRNA.تركيبوجدنا أن إضافة السيلينيوم الزائد زادت من كمية السيلينوبروتينات في خلايا PRDX6 KO، بغض النظر عن مصدر السيلينيوم (الشكل 4a). إضافة السيلينيت أو (Sec)كما حمت MEFs التي تفتقر إلى PRDX6 من الفيروبتوسيس الناتج عن الحديد والفيروبتوسيس الكلاسيكي (الشكل 4ب، ج والشكل الإضافي 5ب، ج). علاوة على ذلك، زاد إضافة السيلينيوم من تعبير السيلينوبروتين وقمع الفيروبتوسيس الناتج عن الحديد في خلايا HeLa التي تفتقر إلى PRDX6 (الشكل الإضافي 5د، هـ). تظهر هذه النتائج أن إضافة كميات زائدة من السيلينيوم يمكن أن تعوض عن فقدان PRDX6، مما يثير احتمال أن السيلينيوم لا يُستخدم بكفاءة من قبل خلايا PRDX6 KO. في الواقع، عندما تم استخدام السيلينيت أوتمت إضافته إلى الثقافات بطريقة تعتمد على الجرعة أو الوقت، وجدنا أن الزيادات التدريجية في مستويات السيلينوبروتين كانت أقل كفاءة بكثير في خلايا PRDX6KO مقارنة بخلايا WT (الشكل 4d، e والشكل الإضافي 5f، g). كما أن PRDX6 يشارك أيضًا في الاستخدام الفعال للسيلينيوم بعد العلاج بـ SELENOP، وهو مصدر أكثر فسيولوجية للسيلينيوم. (الشكل 4f). لذلك، لتقييم الفرضية التي تفيد بأن PRDX6 متورط في كفاءة استخدام السيلينيوم بشكل أكثر دقة، قمنا بضبط المستوى الأولي للبروتينات السيلينوية في خلايا WT و PRDX6 KO ثم أضفنا السيلينيوم؛ وذلك لأن التعبير الأولي للبروتينات السيلينوية كان منخفضًا بشكل كبير في خلايا PRDX6 KO (الشكل 3b). وجدنا أن تعبير البروتينات السيلينوية في خلايا WT المزروعة في وسط يحتوي على تركيز منخفض من مصل الجنين البقري (FBS) انخفض بشكل ملحوظ؛ حيث أن FBS هو المصدر الوحيد للسيلينيوم في هذا النظام الثقافي (بيانات موسعة الشكل 5h). بعد ذلك، أضفنا السيلينيوم إلى وسط الثقافة الذي يحتوي على 1% FBS ووجدنا أن تخليق البروتينات السيلينوية كان أكثر كفاءة بكثير في خلايا WT مقارنة بخلايا KO (بيانات موسعة الشكل 5h). علاوة على ذلك، حتى عندما تم معالجة خلايا PRDX6 KO مسبقًا بالسيلينيوم ثم تم تغذيتها بسيلينيوم إضافي، كان تخليق البروتينات السيلينوية أقل في خلايا PRDX6 KO مقارنة بخلايا WT (بيانات موسعة الشكل 5i). تدعم هذه النتائج بقوة فرضيتنا بأن PRDX6 متورط في الاستخدام الفعال للسيلينيوم لـ Sec-tRNA.التركيب. بعد ذلك، لتقييم كمية Sec-tRNAفي الخلايا، أنشأنا نظامًا إعادة بناء حساس للغاية في المختبر لتخليق السيلينوبروتين من خلال تعديل اختبار تقرير اللوكيفيراز الذي يتجاوز كودون سيلينوسيستين UGA.نظرًا لأن جنين القمح يفتقر إلى آلية إدماج السيلينيومعامل الإطالة حقيقي النواة Sec-tRNAتم إضافة بروتين ربط SECIS 2 (SBP2) و eEFSec المحدد إلى مستخلص جنين القمح مع tRNA سيلينيوم.مصدر لإنشاء نظام إعادة التكوين (الشكل 4g). إضافة الأحماض الأمينية المرتبطة بـ tRNAs (aa-tRNAs) المنقاة من خلايا WT أو خلايا SEPHS2 KO (الأولى، ولكن ليس الأخيرة، تحتوي على Sec-tRNA ) كشفت أن الأحماض الأمينية الناقلة من خلايا WT زادت من تخليق السيلينوبروتين في وجود eEFSec و SBP2، كما تم تقييمه من خلال قياس توهج اللوكيفيراز (الشكل 4h). علاوة على ذلك، فإن الأحماض الأمينية الناقلة من خلايا WT المزروعة مع تم تعزيز تخليق السيلينوبروتين بشكل أكبر. ومع ذلك، لم تُظهر الأحماض الأمينية-نقل (aa-tRNAs) المنقاة من خلايا الفأر الجنينية الليفية (MEFs) التي تفتقر إلى SEPHS2 زيادة في التخليق، حتى عندما تم زراعة خلايا KO مع (Sec). (الشكل 4h)، مما يؤكد صحة النظام لتقييم كمية Sec-tRNAفي مصادر aa-tRNA. بعد ذلك، قمنا بتنقية aa-tRNAs من خلايا WT أو خلايا PRDX6 KO المزروعة مع أو بدون (Sec) (الشكل 4i). تم تنقية aa-tRNAs من خلايا PRDX6 KO المزروعة في غياب فشل في زيادة تخليق السيلينوبروتين، و(Sec)العلاج زادها قليلاً فقط. تشير هذه النتائج بوضوح إلى أن PRDX6 متورط في الاستخدام الفعال للسيلينيوم لـ Sec-tRNA.تركيب.
الشكل 3 | PRDX6 متورط في تخليق السيلينوبروتين. أ، تحليل شبكة التداخل الضروري لـ PRDX6 باستخدام FIREWORKS. ب، تحليل المناعية للعينات من خلايا تعبر عن sgEmpty أو sgControl أو sgRNAs تستهدف PRDX6. البيانات تمثل ثلاث تجارب مستقلة. ج، تحليل المناعية للعينات من خلايا التحكم أو خلايا PRDX6 KO التي تعبر بشكل مستقر عن PRDX6 WT أو الطفرة C47S. البيانات تمثل ثلاث تجارب مستقلة.
د، مخطط يوضح تراكيب البروتينات السيلينوية النموذجية (يسار). تحليل المناعية للعينات من الخلايا التي تعبر بشكل مستقر عن Myc-GFP C70U أو S175U في خلايا التحكم أو الخلايا التي تم فيها حذف الجينات المحددة (وسط). قياس وفرة البروتينات السيلينوية (يمين). يتم تقديم البيانات كمتوسط الانحراف المعياري لثلاث تجارب مستقلة.; **** ; تحليل التباين الأحادي.
PRDX6 يعزز استخدام السيلينيوم كحامل للسيلينيد
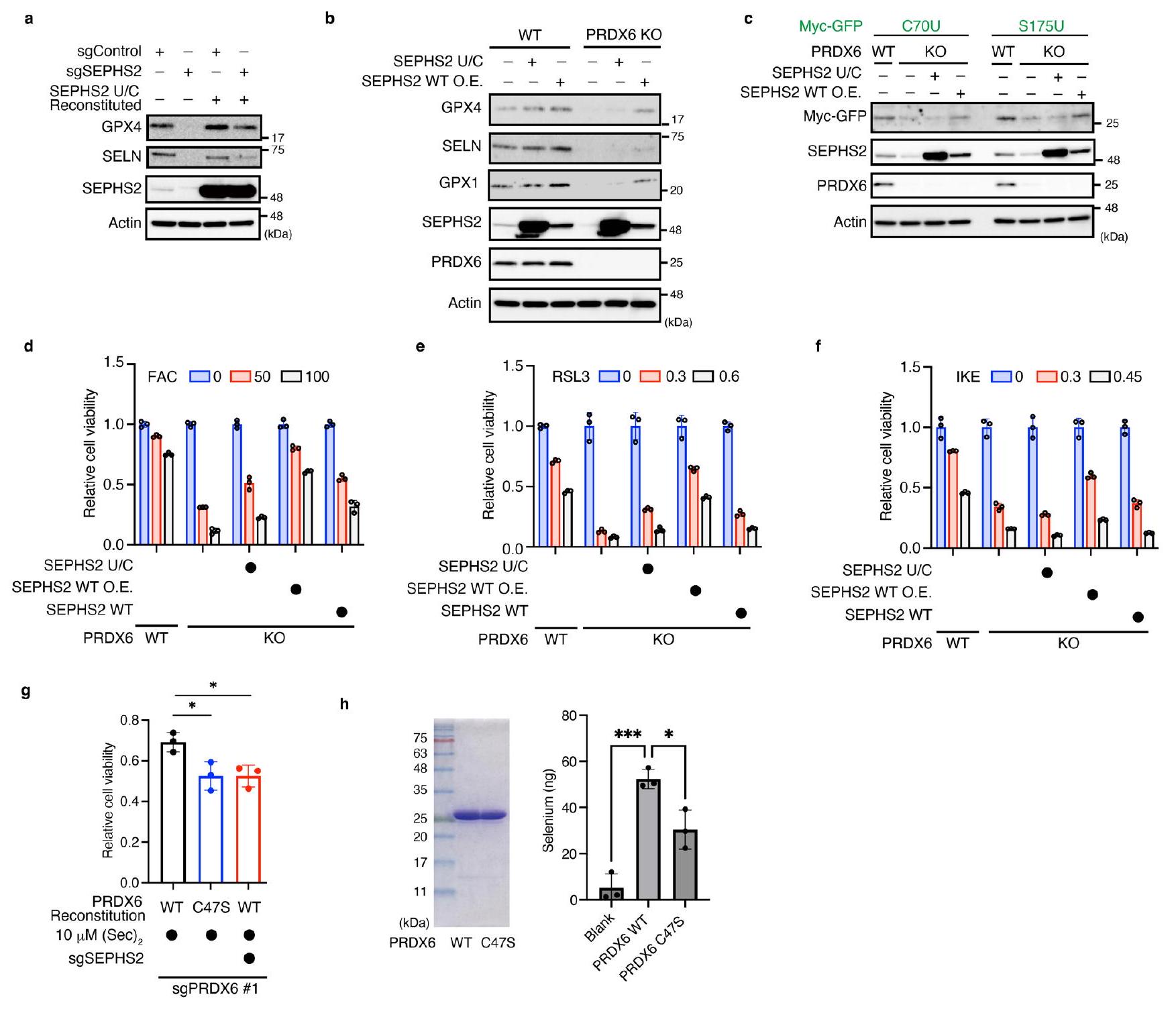
بعد ذلك، استكشفنا الآلية (الآليات) التي من خلالها يزيد PRDX6 من كفاءة استخدام السيلينيوم لـ Sec-tRNA.التركيب. أولاً، قمنا بزيادة التعبير عن الإنزيمات المشاركة في Sec-tRNA التركيب في خلايا PRDX6 KO (الشكل 5a من البيانات الموسعة) ووجدنا أن الإفراط في التعبير (باستثناء SEPHS2 و PRDX6) فشل في استعادة تعبير GPX4 (الشكل 5a). وبالتالي، فإن SEPHS2، وهو بروتين سيليني يقوم بفوسفات السيلينيد لـ Sec-tRNAتخليق Secيبدو أن له دورًا حاسمًا في تعبير GPX4 في خلايا PRDX6 KO. أظهرت الدراسات السابقة أن طفرات SEPHS2 التي تم فيها استبدال السيلينيوم بالسيستين (SEPHS2 U/C) قد أنقذت النمط الظاهري الناقص لـ SEPHS2. (الشكل البياني الممتد 6a). لذلك، قمنا بزيادة التعبير عن SEPHS2U/C أو WT في خلايا PRDX6 KO ووجدنا أن SEPHS2WT، وليس الطفرة U/C، أعاد التعبير عن GPX4 بشكل ملحوظ (الشكل البياني الممتد 6b). ومع ذلك، وجدنا أن زيادة التعبير عن SEPHS2 WT في PRDX6 KO فشلت في إعادة التعبير الكامل عن بروتينات السيلينيوم الأخرى مثل SELN و GPX1 (الشكل البياني الممتد 6b). كما وجدنا أن زيادة التعبير عن SEPHS2 WT أعادت أيضًا التعبير عن نماذج بروتينات السيلينيوم (الشكل البياني الممتد 6c) وحمت الخلايا من الفيروبتوسيس الناتج عن الحديد والفيروبتوسيس الكلاسيكي (الشكل البياني الممتد 6d-f). تثير هذه النتائج احتمال أن فقدان PRDX6 يثبط التعبير عن بروتينات السيلينيوم من خلال تقليل SEPHS2، وليس عن طريق تسهيل استخدام السيلينيوم. لاستبعاد هذه الاحتمالية، قمنا بالتعبير عن SEPHS2 WT بمستويات داخلية في خلايا PRDX6 KO لأن كمية SEPHS2 في الخلايا التي تعبر عن نفسها كانت أعلى بكثير من تلك الموجودة في الخلايا التي تعبر عن SEPHS2 الداخلي (الشكل 5a). وجدنا أن مستوى SEPHS2 WT الداخلي فشل في عكس فقدان التعبير عن بروتينات السيلينيوم في خلايا PRDX6 KO (الشكل 5b) ولم يثبط الفيروبتوسيس الناتج عن الحديد والفيروبتوسيس الكلاسيكي (الشكل البياني الممتد 6d-f). بالإضافة إلى ذلك، عندما تم معالجة خلايا PRDX6 KO مسبقًا بالسيلينيت أولتعديل تعبير SEPHS2 في خلايا KO إلى مستويات قابلة للمقارنة مع تلك الموجودة في خلايا WT قبل تجربة الزمن، لاحظنا أن خلايا WT استخدمت السيلينيوم بشكل أكثر كفاءة من خلايا PRDX6 KO. (الشكل 5i من البيانات الموسعة). تظهر هذه النتائج بوضوح أن PRDX6 ضروري للاستخدام الفعال للسيلينيوم لإنتاج Sec-tRNA.في الخلايا التي تعبر عن مستويات داخلية من SEPHS2. من المهم أننا وجدنا أيضًا أن Sec-tRNAكانت المستويات لا تزال أقل في خلايا PRDX6 KO التي تعبر عن SEPHS2 أكثر من خلايا WT (الشكل 5c)، مما يشير إلى أن التعبير المفرط عن SEPHS2 لا يمكن أن يعوض تمامًا عن الكفاءة المنخفضة لاستخدام السيلينيوم بسبب فقدان PRDX6. بشكل جماعي، تشير البيانات إلى أن السيلينيوم الزائد بالإضافة إلى التعبير المفرط عن SEPHS2 WT، ولكن ليس الطفرة U/C، يعوضان عن فقدان PRDX6، وإن لم يكن ذلك بشكل فعال جداً. وبالتالي، تشير هذه النتائج إلى أن SEPHS2 نفسه يمكن أن يحصل على السيلينيوم من خلال بقايا Sec في SEPHS2؛ ومع ذلك، فإن PRDX6 يسهل بشكل كبير استخدام السيلينيوم بواسطة SEPHS2.
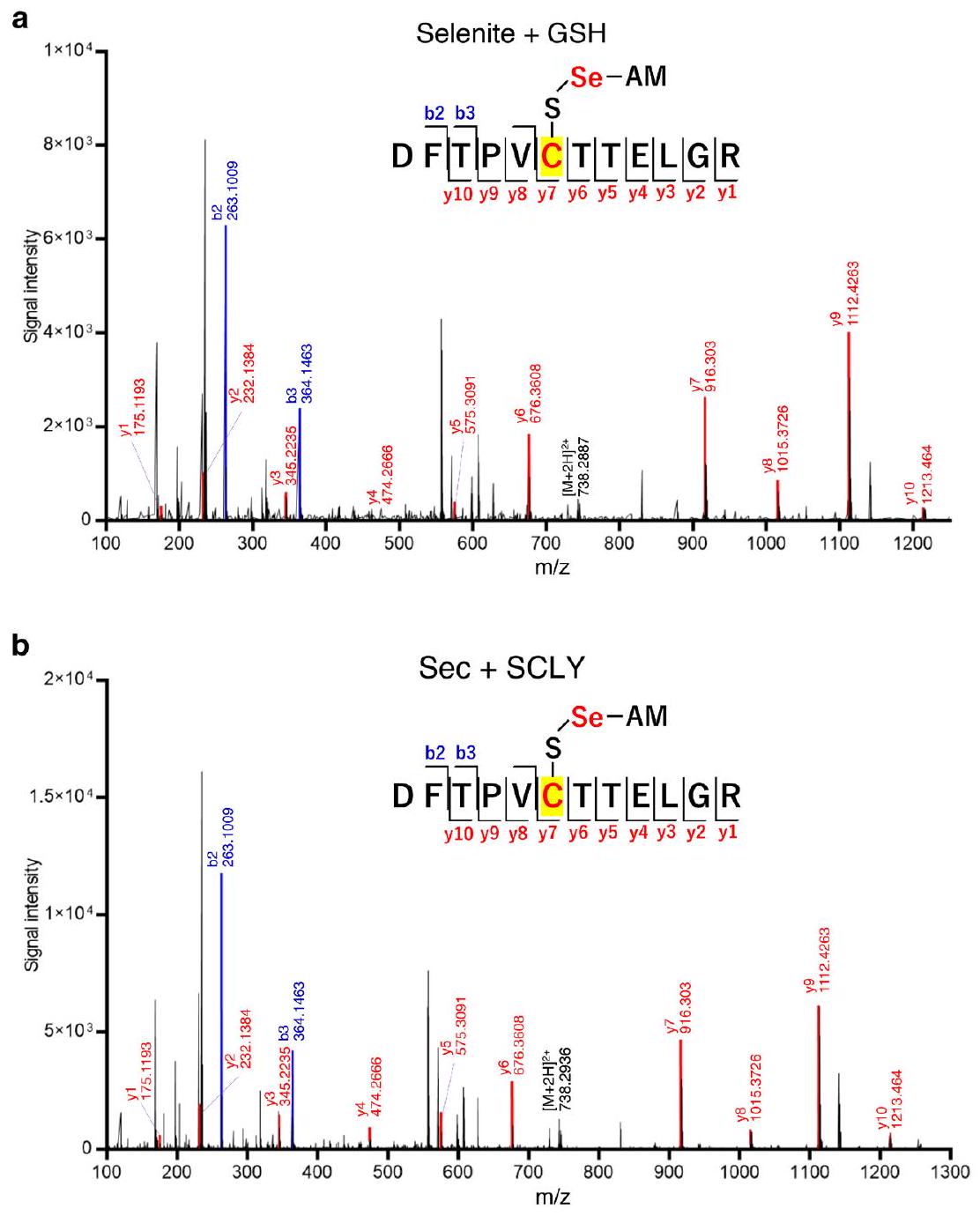
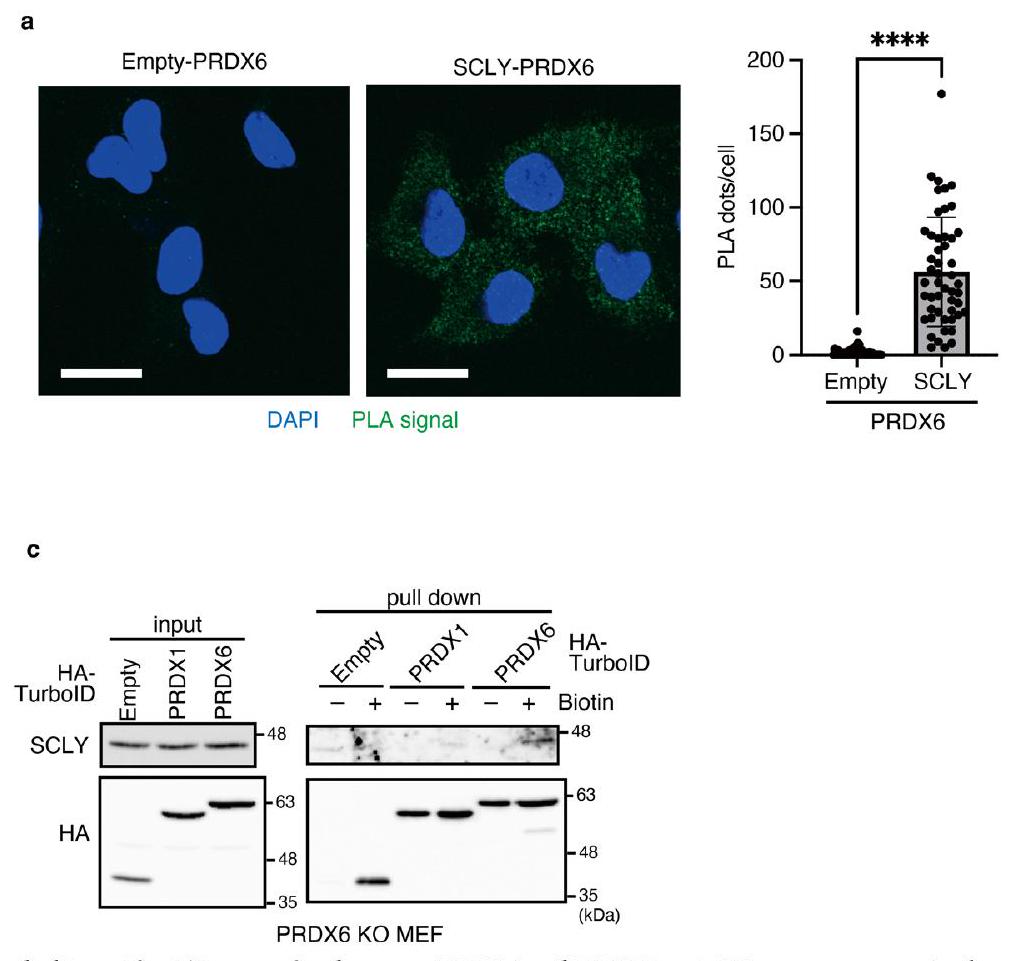
نظرًا لرد فعله العالي، كان يُعتقد أن السيلينيد يجب أن يرتبط ببروتين ناقل غير محدد من خلال بقايا السيستين لنقل فعال إلى SEPHS2 (المراجع 11، 16). إضافة فائض من (Sec)كان سامًا للخلايا؛ ومع ذلك، وجدنا أن PRDX6، الذي يعتبر C47 حاسمًا فيه، يثبط (Sec)السُميّة (الشكل 6g من البيانات الموسعة). بالنظر إلى أن C47 من PRDX6 يلعب أيضًا دورًا حاسمًا في التخليق الفعال للبروتينات السيلينومية (الأشكال 2f و3c)، فمن المحتمل جدًا أن يكون PRDX6 ناقلًا محتملًا للسيلينيد. أظهر تقرير سابق أن الرودانيز يشكل برسيلينيد (-S-SeH) عند بقايا السيستين في وجود السيلينيت وGSH.. لذلك، قمنا بتربية بروتينات PRDX6 WT أو C47S المعاد تركيبها مع السيلينيت وGSH، تلاها الكشف عن السيلينيوم المرتبط بـ PRDX6 بواسطة مطيافية الكتلة المتصلة بالتحريض (ICP-MS) (الشكل التمديدي 6h). وجدنا أن PRDX6 WT يرتبط بالسيلينيوم بشكل أكثر فعالية من الطفرة C47S. بعد ذلك، قمنا بإجراء تحليلات مطيافية الكتلة لتوصيف وسيط PRDX6-selenium بشكل أكبر. أظهرت النتائج أن C47 من PRDX6 يشكل رابطة برسيلينيد عند التفاعل مع GSH والسيلينيت، وكذلك مع SCLY وSec (الشكل التمديدي 7). علاوة على ذلك، وجدنا أن حوالي 10% من PRDX6 C47 شكلت رابطة برسيلينيد في تفاعل مدته 5 دقائق مع السيلينيت وGSH، أو مع SCLY وSec (الشكل 5d,e). بروتينات ناقلة للسيلينيد
الشكل 4 | PRDX6 يعزز الاستخدام الفعال للسيلينيوم. أ، تحليل المناعية للعينات من خلايا التحكم أو خلايا KO المحددة المزروعة لمدة 32 ساعة في وجود سيلينيت الصوديوم. أو . البيانات تمثل تجربتين مستقلتين. ب، ج، تم معالجة خلايا WT أو PRDX6 KO مسبقًا لمدة يومين بـأو سيلينيت الصوديوم. ثم تم معالجة الخلايا لمدة 48 ساعة باستخدام FACفي حضورأو سيلينيت الصوديوم (100 نانومول) (ج)، وتم قياس الحيوية. تُعرض بيانات الحيوية كمتوسطانحراف معياري لثلاث نسخ بيولوجية. د-و، مسار زمني للتعبير عن السيلينوبروتين بواسطة خلايا MEFs من النوع البري أو KO PRDX6 في وجودسيلينيت الصوديوم (e) أوSELENOP (أنثى). البيانات تمثل اثنين (في f) أو ثلاثة (في d و e) تجارب مستقلة. g، مخطط يوضح نظام إعادة البناء في المختبر المستخدم لتقييم كمية Sec-tRNA. h,i, aa-tRNAs، التي تم تنقيتها من الخلايا المحددة المعالجة (أو لا) لمدة 105 دقيقة مع ، تليها معالجة لمدة 15 دقيقة مع السيكلوهكسيميد، تم إضافتها إلى مستخلص جنين القمح في وجود أو غياب eEFSec و SBP2. تُظهر البيانات الخاصة بـ aa-tRNAs من خلايا WT أو خلايا SEPHS2 KO (h)، وخلايا WT أو خلايا PRDX6 KO (i). تُعرض البيانات كمتوسط الانحراف المعياري لثلاث تجارب مستقلة. (WT eEFSec و SBP2،; PRDX6 WT مقابل KO، ); ; تحليل التباين ثنائي الاتجاه. يجب أن تستقبل SCLY السيلينيد ثم تنقله إلى SEPHS2 للفسفرة (الشكل 5a من البيانات الموسعة). كشفت اختبارات الربط القريب (PLAs) أن PRDX6 الداخلي ارتبط بكل من SCLY وSEPHS2 بشكل فعال (الشكل 5f والشكل 8a من البيانات الموسعة). لتأكيد ارتباط PRDX6 بـ SCLY وSEPHS2، قمنا بتعبير HA-TurbolD-PRDX6 أو HA-TurbolD-PRDX1 (وهو نظير لـ PRDX6) في خلايا MEFs WT أو PRDX6 KO. كما هو موضح في الشكل 5g والشكل 8b وc من البيانات الموسعة، ارتبط PRDX6 بشكل محدد بـ SEPHS2 وSCLY.
يتم توصيل السيلينيد إلى SEPHS2 ويتم فوسفوريته لإنتاج سيلينوفوسفات لـ Sec-tRNA.تركيبنظرًا لأن السيلينوفوسفات هو منتج غير مستقرقمنا بتقييم إنتاج AMP، وهو منتج ثانوي لعملية تخليق السيلينوفوسفات، لتقييم تسريع تخليق السيلينوفوسفات بواسطة PRDX6. استخدمنا بروتين SEPHS2 U/C المؤتلف الذي يحتفظ بنشاط الإنزيم لتخليق السيلينوفوسفات.بدلاً من SEPHS2 WT لأن تعبير SEPHS2 WT منخفض للغاية (الشكل 5a) ومن الصعب تنقيته. في تفاعل تخليق السيلينوفوسفات الذي يتم فيه استخدام تحلل Sec بواسطة SCLY كمصدر للسيلينيوم، أدى إضافة PRDX6 WT إلى زيادة فعالة في التخليق لـ سيلينوفوسفات، بينما لم يفعل C47S (الشكل 5h). عندما تم تحضين PRDX6 WT أو C47S مسبقًا مع السيلينيد لتوليد معقد PRDX6-selenide كمصدر للسيلينيوم، تسارع PRDX6 WT إنتاج AMP بواسطة SEPHS2 U/C (الشكل 5i). بشكل جماعي، تشير هذه البيانات إلى أن PRDX6 يسهل Sec-tRNAالتركيب من خلال العمل ك-protein ناقل للسيلينيد (الشكل 5j).
PRDX6 والسرطان
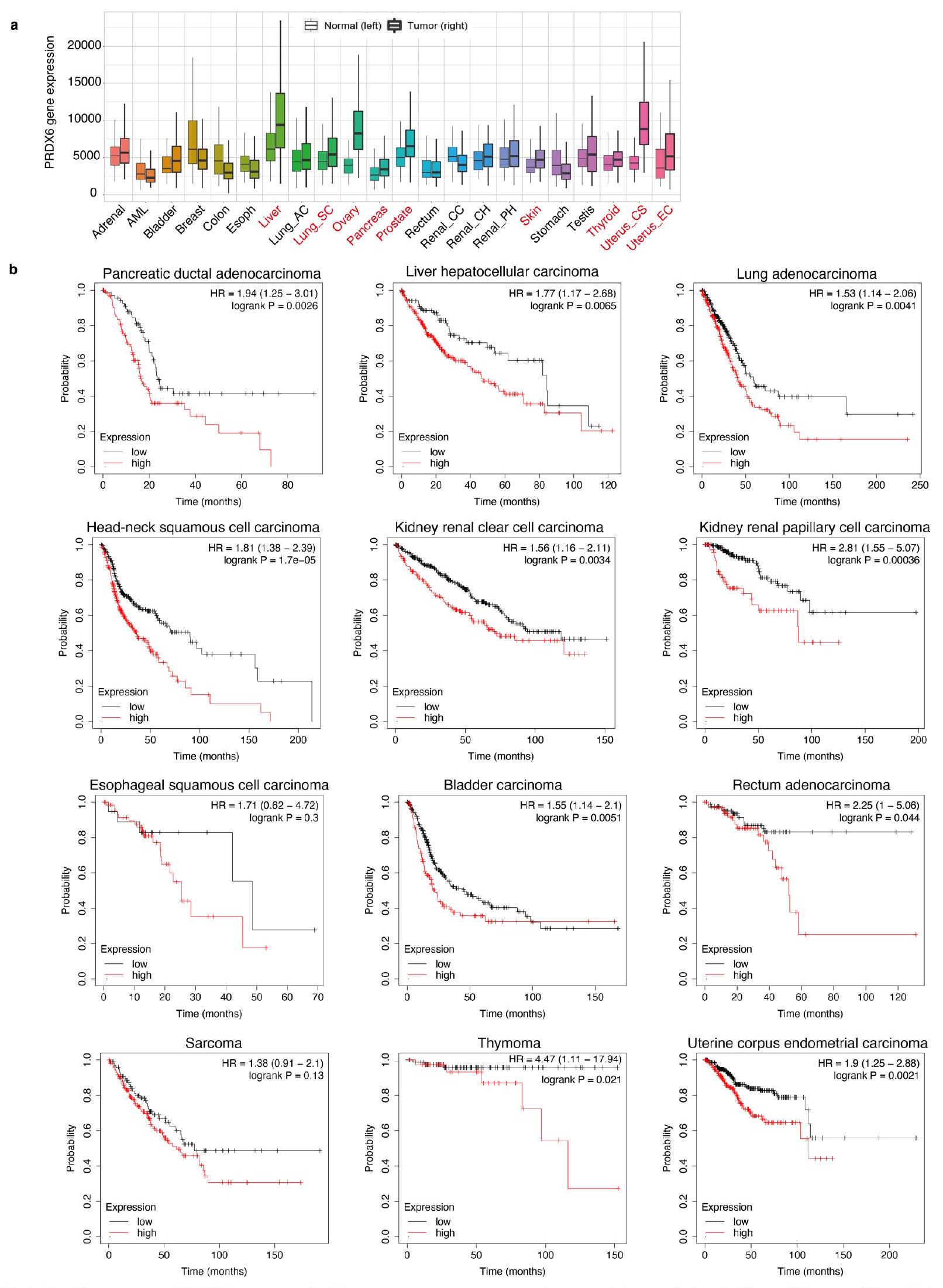
تلعب معظم السيلينوبروتينات دورًا في الحفاظ على توازن الأكسدة والاختزال من خلال تقليل الأضرار التأكسدية، وترتبط التعبيرات العالية للسيلينوبروتينات بدرجة الخباثة.يتماشى مع العلاقة بين السيلينوبروتينات والسرطان، كشفت تحليلات قاعدة البيانات أن تعبير PRDX6 أعلى في الأنسجة السرطانية مقارنة بالأنسجة الطبيعية. (الشكل 9a من البيانات الموسعة) وأن التعبير العالي عن PRDX6 يرتبط بتشخيص سيء لمجموعة متنوعة من السرطانات (الشكل 9b من البيانات الموسعة). أظهر تقرير سابق أن خلايا سرطان البنكرياس المستعصية حساسة للفيروبتوسيس.كما كشفت تحليل قاعدة البيانات أن تعبير PRDX6 مرتفع في سرطان البنكرياس وأن التعبير العالي يرتبط بتشخيص سيء (الشكل 9 من البيانات الموسعة).
الشكل 5 | PRDX6 هو بروتين ناقل للسيلينيد يمكّن من استخدام السيلينيوم بكفاءة. أ، تحليل المناعية للعينات من خلايا التحكم أو خلايا PRDX6 KO التي تعبر بشكل مستقر عن الإنزيمات المشاركة في Sec-tRNAالتركيب. البيانات تمثل ثلاث تجارب مستقلة. ب، تحليل المناعية للعينات من خلايا التحكم أو خلايا PRDX6 KO التي تعبر بشكل مستقر عن SEPHS2 WT بمستويات داخلية. البيانات تمثل ثلاث تجارب مستقلة. ج، تم إضافة aa-tRNAs، المنقاة من الخلايا المحددة المعالجة بالسيكلوهكسيميد لمدة 15 دقيقة، إلى مستخلص جنين القمح بحضور eEFSec و SBP2. البيانات معبر عنها كمتوسط. الانحراف المعياري لثلاث تجارب مستقلة. (SEPHS2 U/C مقابل SEPHS2 WT O.E.,; SEPHS2 WT O.E. مقابل SEPHS2 WT، ); NS ( ); تحليل التباين الأحادي. د، هـ، نسبة تعديل C47 كما تم حسابها بواسطة تحليل مطيافية الكتلة. تم حضانة PRDX6 مع GSH وسيلينيت الصوديوم (د)، أو مع Sec وSCLY (هـ) لمدة 5 دقائق قبل تحليلات مطيافية الكتلة. يتم تقديم البيانات كمتوسط الانحراف المعياري لثلاث نسخ بيولوجية.الارتباط الجسدي تم الكشف عن العلاقة بين PRDX6 و SEPHS2 في PLA. شريط القياس،البيانات تمثل ثلاث تجارب مستقلة. كما يتم عرض تقدير نقاط PLA. تُعرض البيانات كمتوسط. س.د. ( خلايا (فارغة-PRDX6)، الخلايا (SEPHS2-PRDX6) التي تم فحصها،غير متزاوج ذو جانبين-اختبار. تم زراعة MEFs التي تعبر عن HA-TurbolD-فارغ، -PRDX1 أو -PRDX6 لمدة 30 دقيقة مع DMSO أو البيوتين. تم تحليل lysates الخلوية وعينات السحب بواسطة التحليل المناعي. البيانات تمثل تجربتين مستقلتين. h، i، تم تقييم تعزيز PRDX6 لتخليق SEPHS2 بواسطة السيلينوفوسفات من خلال قياس منتج AMP. تم تقديم البيانات كمتوسط.الانحراف المعياري لثلاث تجارب مستقلة تم فيها استخدام السيلينوسيستين وSCLY كمصدر للسيلينيوم؛; ; تم استخدام تحليل التباين الأحادي (h) أو السيلينيوم المرتبط بـ PRDX6 كمصدر للسيلينيوم؛** ( ); ( ); تحليل التباين الأحادي (i).j، مخطط يوضح دور PRDX6 كحامل للسيلينيد.
وجدنا أن فقدان PRDX6 من خطي خلايا سرطان البنكرياس قد قمع النمو، وهو ظاهرة تم عكسها بإضافة السيلينيوم أو ليبروكستاتين-1 (الشكل التمديدي 10a-d). لتعزيز نتائجنا، حصلنا على عدة خلايا سرطانية وقيمنا حساسيتها للفيروبتوز الناتج عن الحديد (الشكل التمديدي 10e,f). أظهرت النتائج أن خلايا الورم العصبي المعززة بـ MYC-N (SK-N-DZ و NB-1) كانت حساسة للفيروبتوز الناتج عن الحديد (الشكل التمديدي 10e,f). كما وجدنا أن فقدان PRDX6 من خلايا SK-N-DZ قد قمع نموها بشكل ملحوظ (الشكل التمديدي 10g,h). أبلغت دراسة سابقة أن MYC-N يزيد من الحديد داخل الخلايا من خلال زيادة تعبير TFRC، وهو بروتين مطلوب لامتصاص الحديد.. بالإضافة إلى ذلك، فإن خلايا السرطان عادة ما تكون مدمنة على الحديد، مما يدفع إلى التكاثر. وبالتالي، قد يكون تثبيط PRDX6 استراتيجية فعالة لجعل خلايا السرطان حساسة بشكل خاص للفيروبتوز الناتج عن الحديد.
نقاش
هنا، قمنا بتطوير نظام لتحفيز الفيروبتوز الذي يمكن أن يقتل الخلايا عند إضافة الحديد إلى وسط الثقافة (الشكل 1c، d). استخدمنا هذا النظام لإجراء فحص شامل للجينوم باستخدام تقنية كريسبر وحددنا PRDX6 كعامل لتخليق السيلينوبروتين. على الرغم من أن PRDX6 كان يُعتبر لفترة طويلة إنزيمًا مضادًا للأكسدة.نظهر هنا أنه يمارس تأثيرات مضادة للأكسدة بشكل غير مباشر من خلال تسهيل التعبير عن السيلينوبروتينات. على الرغم من أن السيلينيوم العضوي وغير العضوي يتم استقلابه إلى سيلينيد، الذي يُستخدم بعد ذلك في تخليق السيلينوبروتينات.لم يتم إثبات المسار الأيضي بشكل قاطع بعد. تشير نتائجنا إلى أن PRDX6 هو بروتين ناقل سيلينيد غير معروف يسهل الاستخدام الفعال للسيلينيد بواسطة SEPHS2 (الأشكال 4 و 5).
من عائلة PRDX، التي تتكون من ستة بروتينات (PRDX1-PRDX6)، تحتوي بروتينات السيستين الثنائية PRDX1-PRDX5 على رابطة ثنائية الكبريت. الرابطة بين بقايا السيستين المحفوظة، والتي يتم تقليلها بواسطة نظام الثيوريدوكسين. بروتين PRDX6 ذو السيستين الواحدة فريد من نوعه حيث يُعتقد أنه يتم تقليله بواسطة GSH وله نشاط GPX.. ومع ذلك، أظهرنا بشكل قاطع في هذه الدراسة أن PRDX6 لا يمتلك نشاط GPX (الشكل 2g والشكل الإضافي 3g). بدلاً من ذلك، وجدنا أن الطفرة C47S من PRDX6 تقلل بشكل كبير من كمية السيلينوبروتينات، بما في ذلك GPX1 و GPX4 (الشكل 3c)، وكلاهما بروتينات معروفة جيدًا تظهر نشاط GPX. نظرًا لأن وظيفة GPX لـ PRDX6 قد تم تقييمها بشكل رئيسي باستخدام خلايا PRDX6 المنخفضة أو KOقد يكون النشاط المنخفض لـ GPX في خلايا غير المحتوية على PRDX6 ناتجًا عن انخفاض في GPX1 و GPX4. يتماشى ذلك مع نتائجنا (الشكل 2 ج والشكل الإضافي 3 ج)، حيث استخدمت تقارير أخرى بروتينات نقية لإظهار أن PRDX6 ليس له نشاط GPX.لذلك، نستنتج أن PRDX6 ليس إنزيمًا مضادًا للأكسدة مثل PRDX1-PRDX5 ولكنه يعمل كحامل بروتين سيلينيد. وقد تم تأكيد ذلك من خلال اكتشافنا أن PRDX1، وهو إنزيم مضاد للأكسدة يُعتبر متماثلًا لـ PRDX6، لم يستعد تعبير GPX4 في خلايا MEFs المعدلة وراثيًا PRDX6 KO (الشكل 8b من البيانات الموسعة).
يعمل كل من السيلينيوم غير العضوي والعضوي كمصادر لـ SEPHS2 لإنتاج السيلينوبروتينات.على الرغم من أن الآلية التي تكمن وراء توصيل السيلينيوم إلى SPEHS2 لا تزال غير معروفة، فقد أظهرنا بوضوح أن PRDX6 يسهل تخليق السيلينوبروتينات (الشكل 4d-f والشكل الممتد 5f-i) ويشكل رابطة بيرسيلينيد عند C47 من كلا مصدرَي السيلينيوم (الشكل 5d,e والشكل الممتد 7). كما وجدنا أن PRDX6 يرتبط بكل من SEPHS2 وSCLY؛ والأخيرة هي إنزيم يحرر السيلينيد من Sec. وبالتالي، يبدو أن PRDX6 يلعب دورًا حاسمًا في توصيل السيلينيد من السيلينيوم العضوي من خلال قبول السيلينيد من SCLY وتوصيله إلى SEPHS2. على الرغم من أن الآليات التي تكمن وراء استقلاب السيلينيوم غير العضوي وتوصيله إلى SEPHS2 لا تزال غير معروفة، فإن نتائجنا تشير بقوة إلى أن PRDX6 له أيضًا دور في توصيل السيلينيد إلى SEPHS2، حتى من السيلينيوم غير العضوي. لذلك، فإن تحديد PRDX6 كبروتين ناقل للسيلينيوم يفتح آفاقًا جديدة للبحث في ديناميات السيلينيوم داخل الخلايا.
من الجدير بالذكر أن التعبير المفرط عن SEPHS2 WT أعاد بشكل كبير التعبير عن السيلينوبروتينات في خلايا PRDX6 KO (الشكل 5a والشكل الإضافي 6b،c). تشير هذه الملاحظات إلى أن SEPHS2 يمكن أن يرتبط مباشرة بالسيلينيد لإنتاج السيلينوفوسفات في غياب PRDX6، وإن كان بكفاءة أقل بكثير؛ في الواقع، فشلت مستويات SEPHS2 الداخلية في استعادة تعبير السيلينوبروتين. نظرًا لأن الطفرة U/C في SEPHS2 لم تتمكن من استعادة تعبير السيلينوبروتينات في خلايا PRDX6 KO (الشكل الإضافي 6b،c)، يبدو أن بقايا Sec في SEPHS2 تسهل استخدام السيلينيد، على الرغم من أن الآلية الجزيئية الدقيقة لا تزال غير محلولة. بقايا Sec وC47 من PRDX6 لها مستوى منخفض منوارتفاع التفاعللذلك، نشك في أن البقايا شديدة التفاعل قد تكون مهمة للتفاعل مع السيلينيد في الخلايا.
لقد لاحظنا أيضًا أن إضافة كميات زائدة من السيلينيوم (بكلا الشكلين العضوي وغير العضوي) أعادت كمية السيلينوبروتينات في خلايا PRDX6 KO (الشكل 4 أ-ج). لذلك، قد تكون قدرة SEPHS2، التي تتفاعل مباشرة مع السيلينيد لتحفيز تخليق بعض السيلينوبروتينات، هي السبب وراء وجود كميات ضئيلة من السيلينوبروتينات في خلايا PRDX6 KO. تبدو هذه الملاحظات متسقة مع الاختلافات في الأنماط الظاهرية لفئران PRDX6 KO والفئران التي تفتقر إلى تخليق السيلينوبروتين.تم الإبلاغ عن سلالتين من الفئران المعدلة وراثيًا PRDX6كلاهما قابل للحياة وخصب، ولا يظهر أي منهما نمط ظاهري واضح، على الرغم من أنهما شديدا الحساسية لمختلف الضغوط التأكسدية. بالمقابل، فإن الفئران التي تفتقر إلى تخليق السيلينوبروتين تكون مميتة في مرحلة الجنين.إن الملاحظة التي تفيد بأن فقدان PRDX6 لا يثبط تمامًا تخليق السيلينوبروتين قد تكون ميزة فيما يتعلق بتطوير مثبطات PRDX6 كأدوية مضادة للسرطان. يُعتقد أن المحفزات الفعالة للفيروبتوز هي استراتيجية واعدة للعلاج المضاد للسرطان، ويعتبر GPX4 هدفًا مناسبًا.. ومع ذلك، قد يكون لتثبيط GPX4 آثار جانبية خطيرة لأن الفئران التي تفتقر إلى GPX4 تموت في مرحلة الجنين.تشير النتائج الحالية مجتمعة إلى أن PRDX6 قد يكون هدفًا بديلاً للأدوية المضادة للسرطان، مع آثار جانبية أقل.
المحتوى عبر الإنترنت
أي طرق، مراجع إضافية، ملخصات تقارير Nature Portfolio، بيانات المصدر، بيانات موسعة، معلومات إضافية، شكر وتقدير، معلومات مراجعة الأقران؛ تفاصيل مساهمات المؤلفين والمصالح المتنافسة؛ وبيانات توفر البيانات والرموز متاحة علىhttps://doi.org/10.1038/s41594-024-01329-z.
References
Jiang, X., Stockwell, B. R. & Conrad, M. Ferroptosis: mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 22, 266-282 (2021).
Lei, G., Zhuang, L. & Gan, B. Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 22, 381-396 (2022).
Yang, W. S. et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317-331 (2014).
Ingold, I. et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell 172, 409-422.e21 (2018).
Friedmann Angeli, J. P. et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 16, 1180-1191 (2014).
Doll, S. et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693-698 (2019).
Bersuker, K. et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688-692 (2019).
Davis, C. D., Tsuji, P. A. & Milner, J. A. Selenoproteins and cancer prevention. Annu. Rev. Nutr. 32, 73-95 (2012).
Zhang, Y. et al. Role of selenoproteins in redox regulation of signaling and the antioxidant system: a review. Antioxidants 9, 383 (2020).
Tsuji, P. A., Santesmasses, D., Lee, B. J., Gladyshev, V. N. & Hatfield, D. L. Historical roles of selenium and selenoproteins in health and development: the good, the bad and the ugly. Int. J. Mol. Sci. 23, 5 (2021).
Manta, B., Makarova, N. E. & Mariotti, M. The selenophosphate synthetase family: a review. Free Radic. Biol. Med. 192, 63-76 (2022).
Veres, Z. et al. Synthesis of 5-methylaminomethyl-2-selenouridine in tRNAs: 31P NMR studies show the labile selenium donor synthesized by the seld gene product contains selenium bonded to phosphorus. Proc. Natl Acad. Sci. USA 89, 2975-2979 (1992).
Veres, Z., Kim, I. Y., Scholz, T. D. & Stadtman, T. C. Selenophosphate synthetase. Enzyme properties and catalytic reaction. J. Biol. Chem. 269, 10597-10603 (1994).
Kumaraswamy, E. et al. Selective removal of the selenocysteine tRNA gene (Trsp) in mouse mammary epithelium. Mol. Cell. Biol. 23, 1477-1488 (2003).
Seeher, S. et al. Secisbp2 is essential for embryonic development and enhances selenoprotein expression. Antioxid. Redox Signal. 21, 835-849 (2014).
Tobe, R. & Mihara, H. Delivery of selenium to selenophosphate synthetase for selenoprotein biosynthesis. Biochim. Biophys. Acta Gen. Subj. 1862, 2433-2440 (2018).
Alborzinia, H. et al. LRP8-mediated selenocysteine uptake is a targetable vulnerability in MYCN-amplified neuroblastoma. EMBO Mol. Med. 15, e18014 (2023).
Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060-1072 (2012).
Salahudeen, A. A. et al. An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science 326, 722-726 (2009).
Vashisht, A. A. et al. Control of iron homeostasis by an iron-regulated ubiquitin ligase. Science 326, 718-721 (2009).
Hentze, M. W., Muckenthaler, M. U., Galy, B. & Camaschella, C. Two to tango: regulation of mammalian iron metabolism. Cell 142, 24-38 (2010).
Iwai, K. Regulation of cellular iron metabolism: iron-dependent degradation of IRP by SCF ubiquitin ligase. Free Radic. Biol. Med. 133, 64-68 (2019).
Hirayama, T., Niwa, M., Hirosawa, S. & Nagasawa, H. High-throughput screening for the discovery of iron homeostasis modulators using an extremely sensitive fluorescent probe. ACS Sens. 5, 2950-2958 (2020).
Moroishi, T., Nishiyama, M., Takeda, Y., Iwai, K. & Nakayama, K. I. The FBXL5-IRP2 axis is integral to control of iron metabolism in vivo. Cell Metab. 14, 339-351 (2011).
Schneider, L. S. et al. Vacuolar-ATPase inhibition blocks iron metabolism to mediate therapeutic effects in breast cancer. Cancer Res. 75, 2863-2874 (2015).
Dixon, S. J. et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem. Biol. 10, 1604-1609 (2015).
Doll, S. et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 13, 91-98 (2017).
Zhang, Y. et al. Imidazole ketone erastin induces ferroptosis and slows tumor growth in a mouse lymphoma model. Cell Chem. Biol. 26, 623-633.e9 (2019).
Bolduc, J. et al. Peroxiredoxins wear many hats: factors that fashion their peroxide sensing personalities. Redox Biol. 42, 101959 (2021).
Perkins, A., Nelson, K. J., Parsonage, D., Poole, L. B. & Karplus, P. A. Peroxiredoxins: guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem. Sci. 40, 435-445 (2015).
Fisher, A. B. Peroxiredoxin 6: a bifunctional enzyme with glutathione peroxidase and phospholipase activities. Antioxid. Redox Signal. 15, 831-844 (2011).
Fisher, A. B. et al. A novel lysophosphatidylcholine acyl transferase activity is expressed by peroxiredoxin 6. J. Lipid Res. 57, 587-596 (2016).
Fisher, A. B., Dodia, C., Manevich, Y., Chen, J. W. & Feinstein, S. I. Phospholipid hydroperoxides are substrates for non-selenium glutathione peroxidase. J. Biol. Chem. 274, 21326-21334 (1999).
Kang, S. W., Baines, I. C. & Rhee, S. G. Characterization of a mammalian peroxiredoxin that contains one conserved cysteine. J. Biol. Chem. 273, 6303-6311 (1998).
Peshenko, I. V. et al. Identification of a 28 kDa secretory protein from rat olfactory epithelium as a thiol-specific antioxidant. Free Radic. Biol. Med. 25, 654-659 (1998).
Peshenko, I. V., Singh, A. K. & Shichi, H. Bovine eye 1-Cys peroxiredoxin: expression in E. coli and antioxidant properties. J. Ocul. Pharmacol. Ther. 17, 93-99 (2001).
Amici, D. R. et al. FIREWORKS: a bottom-up approach to integrative coessentiality network analysis. Life Sci. Alliance 4, e202000882 (2021).
Kang, D. et al. The role of selenium metabolism and selenoproteins in cartilage homeostasis and arthropathies. Exp. Mol. Med. 52, 1198-1208 (2020).
Saito, Y. Selenium transport mechanism via selenoprotein P-its physiological role and related diseases. Front. Nutr. 8, 685517 (2021).
Dobosz-Bartoszek, M. et al. Crystal structures of the human elongation factor eEFSec suggest a non-canonical mechanism for selenocysteine incorporation. Nat. Commun. 7, 12941 (2016).
Shetty, S. P., Shah, R. & Copeland, P. R. Regulation of selenocysteine incorporation into the selenium transport protein, selenoprotein P. J. Biol. Chem. 289, 25317-25326 (2014).
Xu, X. M. et al. Selenophosphate synthetase 2 is essential for selenoprotein biosynthesis. Biochem. J. 404, 115-120 (2007).
Carlisle, A. E. et al. Selenium detoxification is required for cancer-cell survival. Nat. Metab. 2, 603-611 (2020).
Ogasawara, Y., Lacourciere, G. & Stadtman, T. C. Formation of a selenium-substituted rhodanese by reaction with selenite and glutathione: possible role of a protein perselenide in a selenium delivery system. Proc. Natl Acad. Sci. USA 98, 9494-9498 (2001).
Suzuki, N., Verdugo, M., Hatakeyama, T. & Ogra, Y. Structural analysis of chemically synthesized selenophosphate, a donor for selenocysteine biosynthesis. Metallomics Res. 1, 20-25 (2021).
Abe, K., Mihara, H., Nishijima, Y., Kurokawa, S. & Esaki, N. Functional analysis of two homologous mouse selenophosphate synthetases. Biomed. Res. Trace Elem. 19, 76-79 (2008).
Bartha, Á. & Györffy, B. TNMplot.com: a web tool for the comparison of gene expression in normal, tumor and metastatic tissues. Int. J. Mol. Sci. 22, 2622 (2021).
Nagy, Á., Munkácsy, G. & Győrffy, B. Pancancer survival analysis of cancer hallmark genes. Sci. Rep. 11, 6047 (2021).
Badgley, M. A. et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 368, 85-89 (2020).
Lu, Y. et al. MYCN mediates TFRC-dependent ferroptosis and reveals vulnerabilities in neuroblastoma. Cell Death Dis. 12, 511 (2021).
Liu, G. et al. Comparison of glutathione peroxidase 1 and peroxiredoxin 6 in protection against oxidative stress in the mouse lung. Free. Radic. Biol. Med. 49, 1172-1181 (2010).
Yun, H. M. et al. PRDX6 promotes lung tumor progression via its GPx and iPLA activities. Free. Radic. Biol. Med. 69, 367-376 (2014).
Chen, C. et al. Identification of peroxiredoxin 6 as a direct target of withangulatin A by quantitative chemical proteomics in non-small cell lung cancer. Redox Biol. 46, 102130 (2021).
Hall, A., Nelson, K., Poole, L. B. & Karplus, P. A. Structure-based insights into the catalytic power and conformational dexterity of peroxiredoxins. Antioxid. Redox Signal. 15, 795-815 (2011).
Wang, X. et al. Mice with targeted mutation of peroxiredoxin 6 develop normally but are susceptible to oxidative stress. J. Biol. Chem. 278, 25179-25190 (2003).
Mo, Y. et al. 1-Cys peroxiredoxin knock-out mice express mRNA but not protein for a highly related intronless gene. FEBS Lett. 555, 192-198 (2003).
Imai, H. et al. Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene. Biochem. Biophys. Res. Commun. 305, 278-286 (2003).
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons. org/licenses/by/4.0/.
(c) The Author(s) 2024
طرق
RT-PCR والبلسميدات
تم تضخيم إطارات القراءة المفتوحة لـ PRDX6 و PRDX1 و SEPHS2 و GPX4 (الصيغة السيتوسولية) و SARS1 و PSTK و SEPSECS و SCLY و FSP1 من الفأر (m) و PRDX6 و SEPHS2 و SCLY و eEFSec و SBP2 و SEPHS2-3’UTR(SECIS) من الإنسان (h) بواسطة RT-PCR. تم إنشاء تراكيب مقاومة لـ RNA الموجه المفرد (sgRNA) لـ mPRDX6 و mSEPHS2، وطرز من mPRDX6 (H26A و D31A و S32A و C47S و D140A) و hPRDX6 C47S و mGPX4 U46C و mSEPHS2 U/C و hSEPHS2 U/C بواسطة PCR من خطوتين. تم إنشاء GFP-3’SECIS (WT و C70U و S175U) من إطارات القراءة المفتوحة المضخمة لـ GFP و-SECIS من GPX4. تم ربط الحمض النووي بتسلسلات العلامات المناسبة ثم تم استنساخه في ناقلات pMXs-IP و pMXs-IRES-Bsr و pMXs-neo و pT7-7. تم إنتاج لوكفيراز C258U-3’SECIS من إطار القراءة المفتوحة للوكفيراز و-تم الحصول على SECIS من GPX4 ثم تم ربطه في متجه pEU. تم اختيار تسلسلات sgRNA الخاصة بـ CRISPR من CHOPCHOP أو Benchling. كانت تسلسلات الأوليغونيوكليوتيد التي تسبق دافع البروتوسبايسر كما يلي: mFBXL5، GAGGAAGATGGGTTATACCTG؛ mACSL4 #1، GGTTCTACGGGCCGCCCCAA؛ mACSL4 #2، GACCGATCACAATCTCACCTC؛ mLPCAT3 #1، GCCGGTGACTACGATATCAAGTGG؛ mLPCAT3 #2، GTAATAACGGCAGTGACGGTGCGG؛ mPDSS2 #1، GATGCCGGCTGTCGTGCACGA؛ mPDSS2#2، GCGGCATAACCTACAACTGCG؛ mFSP1#1، GCCAGCGCTCACAATTCATCG؛ mFSP1 #2، GCGCTCACAATTCATCGTGG؛ mPRDX6 #1، GATAGAACTATACCTTGCTCC؛ mPRDX6 #2، GCTCACCACACGGGCCGTCAC؛ mPSTK، GAAAGTCGACTTTCCGGCCGC؛ mSEPHS2، GAACCCGTGGATTATCATCGG؛ mMETAP1، GTGTACCGATAGC-CTGCCCA؛ mSNX27، GTTGACAATGCGCACGACCCG؛ mUBE2N، GTGGGGACCACTTATCTATGA؛ mCINP، GGCCCCCTCTATTTCACACG؛ mCENPE، GCGCTATTTATCAGAGCGATG؛ mSTX5A، GTATGCGACTCGATGATCCCG؛ mDDX10، GAGCGATTAAAGCTCCGCACC؛ mKLF5، GACCTGAGGACTCATACGGGT؛ mSCLY، GTAATGAGACCGGCGTCATCA؛ mGPX4، GACGATGCACACGAAACCCC؛ controlgRNA، GCGAGGTATTCGGCTCCGCG؛ hPRDX6، GATGCGGCCGACGGTGGTAT. تم إدخال تسلسلات الدليل في pX459 (ref.58) أو lentiCRISPRv2 (ref.59) (puro، bsr أو hygro).
تم استخدام الكواشف التالية أيضًا: سترات الأمونيوم الحديدي (سيغما-ألدريتش، F5879)، ليبروكستاتين-1 (سيلك، S7699)، بافيلوميسين A1 (سيلك، S1413)، IKE (سيلك، S8877)، RSL3 (سيلك، S8155)، E64d (معهد الببتيد، 4321-v)، بيبستاتين A (معهد الببتيد، 4397-v)، سيلينيت الصوديوم (سيغما-ألدريتش، 214485)، سيلينوسيستين (صناعة الكيمياء طوكيو، E1368)، سيكلوهكسيميد (ألبيوكيم، 239764)، بيروكسيد الهيدروجين (صناعة الكيمياء سانتوكو، 18412)، بيروكسيد التيرت-بيوتيل (سيغما-ألدريتش، 458139)، تريس(2-كربوكسي إيثيل) فوسفين (TCEP) (ناكالاي، 07277)، بيريدوكسال-5-فوسفات (PLP) (سيغما-ألدريتش، P9255) والبيوتين (واكو، 021-08712).
خطوط الخلايا وزراعة الخلايا
تم إنتاج MEFs داخليًا. تم إهداء HepG2 من قبل K. Nakajima (جامعة أوساكا سيتي)، والتي تم شراؤها في الأصل من ATCC (HepG2: HB-8065). تم إهداء HEK293T من قبل E. Nakamura (جامعة كيوتو)، والتي تم شراؤها في الأصل من RIKRN RBC (293T: RCB2202). تم إهداء PLATE من قبل T. Kitamura (جامعة طوكيو). تم إهداء A549 و H226 و H460 و H1975 من قبل A. Sato (جامعة كيوتو)، والتي تم شراؤها في الأصل من ATCC (A549: CCL-185، H226: CRL-5826، H460: HTB-177، H1975: CRL5908). تم شراء خلايا SK-N-DZ و HeLa من ATCC (SK-N-DZ: CRL-2149، HeLa: CCL-2). تم شراء خلايا PANC-1 و MIA Paca-2 من RIKEN RBC (PANC-1: RCB2095، MIA Paca-2: RCB2094). تم شراء خلايا NB-1 من JCRB (NB-1:JCRB0621). تم زراعة جميع الخلايا في حاضنة رطبة عند و تم زراعة خلايا MEFs و HEK293T و PLATE و A549 و HeLa و PANC-1 و MIA Paca-2 و HepG2 في DMEM المدعوم بـFBSالبنسلين وستربتوميسين. تم زراعة خلايا SK-N-DZ في وسط DMEM المدعوم بـFBS،الأحماض الأمينية غير الأساسية (جيبكو، 11140050)،البنسلين وستربتوميسين. تم الحفاظ على خلايا GPX4 و SEPHS2 و PSTK KO MEF في وسط يحتوي علىمن ليبروكستاتين-1. تم زراعة خلايا H226 و H460 و H1975 في وسط معهد روسويل بارك التذكاري (RPMI) 1640 المدعوم بـFBS،البنسلين وستربتوميسين. تم زراعة خلايا NB-1 فيRPMI ووسط MEM مدعوم بـFBS،البنسلين وستربتوميسين.
توليد خلايا الفأر الجنينية المشتقة من الخلايا الجذعية (MEFs) المعطلة لجين FBXL5
لإنتاج خلايا MEFs KO لـ FBXL5، تم استخدام جهاز NEPA21 للإلكتروبورتيشن (NEPAGENE) لإلكتروبورت خلايا MEFs باستخدام بلازميد pX459 الذي يحتوي على تسلسل sgRNA المحدد لـ mFBXL 5. ثم تم معالجة الخلايا بالبوروميسين لمدة يومين. بعد الاختيار، تم زراعة الخلايا بكثافة منخفضة وتم اختيار مستعمرات معزولة. لتحديد خلايا FBXL5 KO، تم تحليل تعبير FBXL5 بواسطة التحليل المناعي.
إنتاج الفيروسات القهقرية وتوليد خطوط خلايا KO المعتمدة على CRISPR-Cas9
تم إنتاج الفيروسات القهقرية عن طريق نقل الجينات المشترك لخلايا HEK293T باستخدام تسلسلات الدليل المحتوية على LentiCRISPRv2 (puro أو bsr أو hygro) ، و البلازميد التعبوي psPAX2 و البلازميد الغلاف VSV-G باستخدام مادة نقل PEI MAX (Polysciences). في اليوم التالي، تم استبدال الوسط بوسط جديد. بعد يومين من الثقافة، تم جمع السائل الفائق المحتوي على الفيروسات القهقرية واستخدامه لإصابة الخلايا المستهدفة طوال الليل في وجودبوليبريدين (ميرك). تم اختيار الخلايا المصابة باستخدام البيروميسين أو البلاسيتيدين أو الهيغرومايسين.
التعبير الفيروسي العكسي
تم نقل pMXs-IP أو pMXs-neo أو pMXs-IRES-Bsr التي تحتوي على الإدخالات المناسبة إلى خلايا التعبئة PLATE أو خلايا GP2-293 مع بلازميد pVSV-G. تم استخدام الفيروسات الناتجة لإصابة خلايا الهدف في وجودبوليبريد. تم اختيار الخلايا التي تم نقلها بشكل مستقر باستخدام البيروميسين أو G-418 أو بلاستيسيدين.
تحلل الخلايا وبلوت الغربي
تم غسل الخلايا بمحلول PBS وتم تحللها بمحلول التحلل ( 50 مليمول / لتر من تريس-هيدروكلوريد ( الرقم الهيدروجيني 7.5 )،ترايتون X-100، 1 مللي مول من PMSF، ومزيج مثبطات البروتياز (سيغما-ألدريتش). تم توضيح المستخلصات عن طريق الطرد المركزي عندلمدة 20 دقيقة عندتم تحديد تركيزات البروتين باستخدام اختبار برادفورد (ناكالاي تيسك)، وتم خلط كميات متساوية من البروتين مع محلول عينة SDS. تم تسخين العينات لمدة 5 دقائق عند، تم فصلها بواسطة SDS-PAGE ونقلها إلى أغشية PVDF (Millipore). بعد الحجب في محلول ملحي مخفف بالترس (TBS) يحتوي علىتويين-20حليب جاف خالي من الدسم، تم تحضين الغشاء مع الأجسام المضادة الأولية المناسبة تليها الأجسام المضادة الثانوية المناسبة. تم تصور الأغشية باستخدام الكيمياء الضوئية المعززة وتم تحليلها على جهاز LAS4000mini أو LAS3000 (GE Healthcare).
اختبار حيوية الخلايا
تم زراعة الخلايا في أطباق 96 بئر (بثلاث نسخ وبكثافة من)خلايا لكل بئر) في وجود أو غياب FAC أو RSL3 أو IKE. بعد الزراعة لمدةتم إضافة مجموعة عد الخلايا-8 (DOJINDO) لمدة 1-2 ساعة. تم قياس الامتصاص عند 450 نانومتر باستخدام جهاز قراءة الميكرو بلايت SpectraMax M5 (Molecular Devices). تم التعبير عن حيوية الخلايا، التي تم مراقبتها باستمرار باستخدام نظام iCELLigence أو xCELLigence (ACEA Bioscience)، كمؤشر خلايا قائم على المقاومة. تم زراعة خلايا GPX4 KO (4500 خلية لكل بئر) على E-Plate L8 PET (ACEA Bioscience)، وتم زراعة خلايا سرطان الغدة البنكرياسية (3000 خلية لكل بئر) وخلايا SK-N-DZ (6000 خلية لكل بئر) على E-Plate16 PET (ACEA Bioscience). تم زراعة خلايا SK-N-DZ في DMEM مضاف إليهFBS،الأحماض الأمينية غير الأساسية و0.25 مللي مول من L-جلوتامين (فوجي فيلم). تم مراقبة مؤشر الخلايا بشكل مستمر. تم تحليل بيانات التسجيل بواسطة برنامج RTCA software lite v.2.2.1.
تلطيخ الخلايا باستخدام BODIPY 581/591 C11، وتلوين الحديد
لتلوين BODIPY 581/591 C11 (Invitrogen)، تم زرع الخلايا في طبق بستة آبار وتم معالجتها بـ FAC. بعدتم إزالة الوسط وتم وسم الخلايا بـ DMEM المحتوي علىبوديبيفيلمدة 30 دقيقة. ثم تم غسل الخلايا ثلاث مرات بمحلول PBS وتم فصلها عن الصفيحة باستخدام التربسين. تم استخدام تصنيف مجموعة الخلايا الأولية (FSC-Area مقابل FSC-height) لضمان استبعاد الثنائيات، وتم قياس الفلورية الخضراء باستخدام FACS CantolI (BD Biosciences) وبرنامج FACS Diva الإصدار 6.1.2 (Becton Dickinson). تم تحليل البيانات باستخدام برنامج FlowJo (الإصدار 9.9.6). بالنسبة لصبغة الحديد، تم زراعة الخلايا فيطبق زجاجي القاع ومعالج بـتمت معالجة الخلايا لمدة 4 ساعات. ثم تم غسل الخلايا ثلاث مرات بمحلول HBSS وتم وسمها بـ لمدة 30 دقيقة مع فيرو أورانج (دو جيندو) متبوعًا بالت visualization تحت مجهر فوف1000 المجهري (أوليمبوس).
فحص CRISPR على مستوى الجينوم
تمت إصابة خلايا MEFs المفقودة لـ FBXL5 بمكتبة GeCKO v. 2 الخاصة بالفئران.عند مضاعفية عدوى تبلغ 0.3 وتم اختيارها باستخدام البيروميسين لمدة أسبوع واحد بعد العدوى. ثم تم معالجة الخلايا بـتمت معالجة الخلايا بـ FAC لمدة 48 ساعة وزرعها لمدة 24 ساعة أخرى في وسط جديد. تم تحلل الخلايا في محلول NTE (15 مليمول من Tris-HCl (pH 7.5)، 150 مليمول من NaCl و 1 مليمول من EDTA)، وتم إعداد الحمض النووي الجيني من الخلايا غير المعالجة والخلايا المعالجة بـ FAC عن طريق استخراج الفينول-كلوروفورم وترسيب الإيزوبروبانول. تم تضخيم تسلسلات sgRNA من الحمض النووي الجيني باستخدام PCR مع بوليميراز DNA هيركولاز II فيوجن. تم استخراج الأمبليكون الناتج من الجل وخضع لتسلسل الحمض النووي على جهاز تسلسل Novaseq 6000 (Illumina). تم تحليل بيانات التسلسل باستخدام خط أنابيب MAGeCK.
جيش التحرير الشعبي
تم إجراء PLA باستخدام مجموعة الربط القريبة (SigmaAldrich). تم زراعة خلايا A549، إما فارغة أو تعبر عن Myc-SEPHS2 أو Myc-SCLY، على شرائح زجاجية دقيقة في طبق 6 آبار. تم غسل الخلايا بمحلول PBS وثبتت لمدة 20 دقيقة معالفورمالديهايد في PBS عند درجة حرارة الغرفة ). ثم تم غسل الخلايا مرتين بمحلول PBS وتم اختراقها بـ ترايتون X-100 في PBS عند درجة حرارة الغرفة لمدة 10 دقائق. تم غسل الخلايا مرتين بـ PBS ثم تم حضنها فيلمدة ساعة واحدة مع محلول حجب Duolink. ثم تم حضن الخلايا في درجة حرارة الغرفة لمدة ساعة واحدة مع الأجسام المضادة الأولية المستهدفة PRDX6 (أجسام مضادة بوليكلونالية من الأرانب؛ 1:100) و Myc (أجسام مضادة وحيدة النسيلة من الفئران؛ 1:200). تم غسل الخلايا مرتين لمدة 5 دقائق مع محلول غسل Duolink A في درجة حرارة الغرفة ثم تم حضنها مع جسم مضاد ثانوي (مضاد للفئران سالب ومضاد للأرانب موجب) فيلمدة ساعة واحدة. تم غسل الخلايا مرتين في درجة حرارة الغرفة لمدة 5 دقائق باستخدام محلول غسيل Duolink A ثم تم تحضينها بمحلول الليغاز.لمدة 30 دقيقة. بعد ذلك، تم غسل الخلايا مرتين بمحلول غسيل Duolink A وتم تحضينها مع البوليميراز في محلول تكبير.لمدة 100 دقيقة. تم غسل الخلايا مرتين في درجة حرارة الغرفة لمدة 10 دقائق باستخدام محلول غسل Duolink B، تلا ذلكمحلول غسيل دوولينك B لمدة دقيقة واحدة. أخيرًا، الغطاء تم تثبيت النظارات باستخدام وسط تثبيت Duolink PLA مع DAPI. تم تصور تفاعلات البروتين-بروتين تحت مجهر Fv1000 المجهري (أوليمبوس). تم عد بؤر PLA بواسطة ImageJ (الإصدار 2.3.0).
سحب TurboID
تم معالجة الخلايا (أو لم تُعالج) لمدة 30 دقيقة بـبيوتين، مغسول بمحلول PBS ومحلول باستخدام محلول التحلل (50 مM تريس- HCl (pH 8.0)، 150 مM NaCl،ترايتون X-100 و 1 مللي مول من PMSF). تم توضيح المستخلصات عن طريق الطرد المركزي فيلمدة 20 دقيقة عندتم تحديد تركيزات البروتين باستخدام اختبار برادفورد، وتم تحضين كميات متساوية من البروتين وستربتافيدين سيفاروز (سايتيفا) overnight.بالدور. تم غسل الكريات ثلاث مرات بمحلول التحلل ومرة واحدة بمحلول PBS. تم استرجاع البروتينات من الكريات باستخدام محلول عينة SDS المضاف إليه 2 مللي مول من البيوتين ثم تم تحليلها بواسطة تقنية الويسترن بلوت.
تنقية البروتين
تم التعبير عن His-TEV-PRDX6 WT أو C47S، His-TEV-hSCLY، hSEPHS2-His، فأر His-TEV-eEFSec وHis-TEV-SBP2 في سلالة E. coli BL21-CodonPlus (DE3)-RIPL (Agilent Technologies). تم تحفيز التعبير عن His-TEV-PRDX6 WT أو C47S عن طريق إضافة 0.2 مللي مول من IPTG، واستمر النمو الثقافي عندلمدة 3 ساعات. تم جمع الخلايا عن طريق الطرد المركزي وتجميدها بسرعة. بعد ذلك، تم إعادة تعليق الخلايا فيلمدة 30 دقيقة في محلول يحتوي على 50 مللي مول من تريس-، -ميركابتوإيثانول (ناكالاي تيسك)كلوريد الليزوزيم (ناكالاي تيسك) DNase (روش)، 2 مللي مول PMSF وخلطة مثبطات البروتياز (روش)، تليها التحلل عند لمدة 20 دقيقة في وجودترايتون X-100. تم إزالة المواد غير القابلة للذوبان عن طريق الطرد المركزي عندلمدة 20 دقيقة عند. تم تنقية -TEV PRDX6 WT أو C47S من السائل الفائق باستخدام حبات Ni-NTA (QIAGEN). تم تحضين العينات المستخلصة لمدة 30 دقيقة في مع 10 مللي مول من ثيوثريتال (DTT) ثم تم إزالة الأملاح في محلول 20 مللي مول من تريس- HCl (pH 7.5) على عمود PD10.
تم تحفيز تعبير His-TEV-hSCLY و hSEPHS2-His بإضافة 0.2 مللي مول من IPTG، واستمرت الثقافة طوال الليل فيتم جمع الخلايا عن طريق الطرد المركزي وتجميدها بسرعة. بعد ذلك، تم إعادة تعليق الخلايا في محلول يحتوي على 50 مللي مول من تريس- HCl (رقم الهيدروجيني 8.0).-ميركابتوإيثانول، 2 مللي مول من PMSF وخلطة مثبطات البروتياز ثم تم تحللها بواسطة الموجات فوق الصوتية فيتمت إزالة المواد غير القابلة للذوبان عن طريق الطرد المركزي فيلمدة 20 دقيقة عندتم تنقية البروتينات باستخدام حبات Ni-NTA. تم إزالة الأملاح من العينات المستخلصة في 20 مليمول من تريس-ومحلول 1 مليمول DTT على عمود PD10.
تم تحفيز تعبير His-TEV-eEFSec وHis-TEV-SBP2 بإضافة 0.3 مللي مولار IPTG، واستمر النمو الثقافي عندلمدة 4 ساعات. تم جمع الخلايا عن طريق الطرد المركزي وتجميدها بسرعة. بعد ذلك، تم إعادة تعليق الخلايا في محلول يحتوي على 20 مللي مول من تريس- HCl (رقم الهيدروجيني 8.0)،إيميدازولالجليسرول، 10 مللي مول 2-ميركابتوإيثانول، 1 مللي مول PMSF وخلطة مثبطات البروتياز ثم تم تحللها بواسطة الموجات فوق الصوتية عندتمت إزالة المواد غير القابلة للذوبان عن طريق الطرد المركزي فيلمدة 20 دقيقة عندتم تنقية البروتينات باستخدام حبات Ni-NTA. تم إزالة الأملاح من العينات المستخرجة على عمود PD10. تم تخزين eEFsec في محلول يحتوي على 20 مللي مول من تريس-NaCl و 1 مللي مول من DTT، وتم تخزين SBP2 في محلول يحتوي على 10 مللي مول من تريس-HCl (pH 7.5)،الجليسرول و 1 مللي مول من DTT.
اختبار GPX
تم قياس نشاط GPX باستخدام مجموعة اختبار مثبط GPX4 (Cayman Chemical). أولاً،بروتين GPX4 (المضمن في المجموعة) أوبروتين PRDX6 تم إذابته فيمن محلول عازلة اختبار GPX4 (المتضمن في المجموعة). ثم،تم إضافة مزيج GSH/GSH المختزل (المتضمن في المجموعة) وخلطه. بعد ذلك،تم إضافة محلول NADPH (المتضمن في المجموعة) وخلطه. أخيرًا،من بيروكسيد الكومين (الموجود في المجموعة)، بيروكسيد الهيدروجين (التركيز النهائي، ) أو بيروكسيد الهيدروجين التيرت-بيوتيلي (التركيز النهائي، ) تم إضافته وخلطه، وامتصاصية تم قياسه عند 340 نانومتر باستخدام جهاز قراءة الميكرو بلايت SpectraMax M5 (Molecular Devices).
اختبار سينثاز السيلينوفوسفات
تم إجراء اختبار لإنزيم سينثاز السيلينوفوسفات باستخدام SCLY والسيلينوسيستين كمصدر للسيلينيوم في محلول منقّى يحتوي على 50 مللي مول من تريس- HCl (رقم الهيدروجيني 7.0).ATPدي تي تيSEPHS2-His، PRDX6 WT أو C47S، SCLY، PLP (سيغما-ألدريتش) وفيلمدة 45 دقيقة. تم قياس منتج AMP الناتج في اختبار AMP-Glo (بروماجا) باستخدام قارئ لوحات نيفو (بيركين إلمر). تم إجراء اختبار سينثاز السيلينوفوسفات باستخدام PRDX6 المرتبط بالسيلينيد كمصدر للسيلينيوم عن طريق الحضانة من PRDX6 WT أو C47S مع 2 مللي مول من سيلينيت الصوديوم و 20 مللي مول من DTT في محلول 20 مللي مول من تريس- HCl (pH 7.5) منزوع الغاز في درجة حرارة الغرفة لمدة 30 دقيقة. ثم، تم تخفيف المحلول 20 مرة في محلول يحتوي على 20 مللي مول من تريس- HCl (pH 7.5) و 150 مللي مول من NaCl وترايتون X-100، وتم إضافة كريات نانوية مغناطيسية Ni-NTA. بعد الدوران عندلمدة ساعة واحدة، تم غسل الحبيبات ثلاث مرات وتم إزالتها باستخدام 300 مللي مولار إيميدازول في محلول 20 مللي مولار تريس- HCl (pH 7.5). تم حضن العينات المستخرجة مع خليط من المحلول يحتوي على 50 مللي مولار تريس-ATPدي تي تي وSEPHS2-His تحت طبقة من زيت المعدن عندلمدة 30 دقيقة. تم قياس منتج AMP الناتج في اختبار AMP-Glo.
RT-qPCR
تم عزل RNA باستخدام مجموعة RNeasy Mini Kit (QIAGEN) وتم تحويله عكسيًا باستخدام مجموعة RNA-to-cDNA عالية السعة (Applied Biosystems). تم الحصول على بيانات RT-qPCR بواسطة نظام ABI ViiA7 PCR في الوقت الحقيقي (Applied Biosystems) وتم تحليلها بواسطة برنامج ViiA7 RUO الإصدار 1.2.3. تم إجراء RT-PCR لجين GPX4 باستخدام البرايمرات التالية:
mGPX4_Fwd،-CCTCTGCTGCAAGAGCCTCCC-3′ و mGPX4_Rev،-CTTATCCAGGCAGACCATGTGC-كانت البادئات الضابطة -mActin_Fwd،-ATGGATGACGATATCGCTC-3′ و-mActin_Rev،-جاتتكاتا سي سي كاجغا غاغ-.
عزل الأحماض الأمينية الناقلة
تم عزل aa-tRNAs من خلايا WT و PRDX6KO و SEPHS2 KO MEF. باختصار، تم معالجة الخلايا (أو عدم معالجتها) بـلمدة 105 دقائق، تليها إضافةسيكلوهكسيميد لمدة 15 دقيقة. ثم تم غسل الخلايا ثلاث مرات في PBS وتم فصلها باستخدام التربسين. بعد الطرد المركزي عندمن مياه DPEC، 0.8 مل من Tbuffer ( 50 مليمول NaOAc ،وتم إضافة 5 مل من EDTA و 2 مل من حمض الفينول pH 4.2 (Nippon gene) إلى كريات الخلايا بهذا الترتيب. تم خلط المحلول لفترة قصيرة ثم تم الطرد المركزي عند لمدة 5 دقائق عند تم نقل الطور المائي إلى أنبوب آخر وإعادة استخراجه بـ 2 مل من حمض الفينول (pH 4.2). تم ترسيب RNA باستخدام 2.5 حجم منالإيثانول، مغسول مرتين بـتم غسلها بالإيثانول وتجفيفها في الهواء لمدة 5 دقائق. تم إعادة تعليق راسب aa-tRNA في محلول 5 مليمول من NaOAc.
اختبار تقرير لوكفيراز Sec UGA لـ Sec-tRNAتقييم
كل تفاعل ( ) يحتوي على مستخلص جنين القمح ( ; بروميجا)، خلطة الأحماض الأمينية، 8 وحدات من مثبط ريبونوكلياز RNasin Plus (بروماجا)، 52.5 مللي مولار من أسيتات البوتاسيوم،من mRNA لمراسل لوكفيراز ومن aa-tRNAs في وجود أو عدم وجود من eEFSec و من SBP2. تم تحضين التفاعلات في تم قياس نشاط اللوسيفيراز لمدة 90 دقيقة في جهاز لومات لومي نومتر (بيرثولد).
ICP-MS
رد الفعل ( ) يحتوي على سيلينيت الصوديومجي إس إتش و (أو لا) بروتين PRDX6 WT أو C47S المعاد التركيب في محلول منقّى يحتوي على 1 مللي مول من EDTA و 50 مللي مول من Tris-HCl (رقم هيدروجيني 7.0). تم تحضين التفاعلات فيلمدة 5 دقائق. تم إزالة أي سيلينيوم لم يرتبط ببروتين PRDX6 باستخدام عمود NAP-5. كانت محلول البروتين ثم تم التركيز باستخدام جهاز التركيز Amicon 3 K. تم أخذ عينة صغيرةتم وضع 0.1 مل من محلول البروتين في أنبوب اختبار زجاجي، وتم تحلل البروتينات بإضافةتليها التسخين عندلمدة ساعتين. تم تخفيف العينات المهضومة بمياه ميلي-كيو، وتم قياس تركيز السيلينيوم بواسطة ICP-MS (Agilent 8800 ICP-MS/MS؛ تقنيات أجيلنت).تم استخدام وضع تغيير الكتلة لمراقبة شدة الإشارة لـتم قياس تركيزات السيلينيوم من منحنى المعايرة القياسي، مع زمن إقامة قدره 100 مللي ثانية.
تحليل مطيافية الكتلة
تفاعل يحتوي على بروتين PRDX6 WT المؤتلف ) ، سيلينيت الصوديوم ( ) و GSH ( ) في محلول منقّى يحتوي على 50 مللي مول من تريس – ، أو PRDX6 WTتم تقليلها مسبقًا بكمية متساوية من TCEP)، SCLY ( ) و PLP ( ) في محلول منقّى يحتوي على 50 مللي مول من تريس- HCl ( الرقم الهيدروجيني 7.0 ). تم حضانة التفاعلات في لمدة 5 دقائق. ثم، تم ألكلة PRDX6 بـ 10 مللي مول من يودوأستاميد فيعامل السطح بروتيازماكس فيلمدة 10 دقائق، تليها عملية الهضم بـتريبسين جولد فيلمدة 3 ساعات. تم تحليل المنتجات باستخدام الكروماتوغرافيا السائلة-الاستشعار الأيوني بالرش-الكتلة الرباعية الزمنية للطيران (LC-ESI-Q-TOF MS/MS). تم إجراء تحليل LC-ESI-Q-TOF باستخدام جهاز 6545XT AdvanceBio LC-Q-TOF (Agilent Technologies) المتصل بنظام HPLC من Agilent. تم تحليل التعديلات على مركز النشاط السيستين (Cys47) باستخدام برنامج Agilent MassHunter BioConfirm الإصدار 10.0. تم الكشف عن مستويات التعديل في ببتيد DFTPVCTTELGR، الذي يتضمن بقايا السيستين في مركز النشاط، من خلال المراقبة عند (لـ CysS-AM (AM، مركب يودوأسيتاميد)) (لـ CysSSe-AM) و 862.3172 (لـ CysSSe-SG). تم تطبيع كفاءة هضم البروتين بواسطة التربسين باستخدام ببتيد FHDFLGDSWGILFSHPR (تم تقييم مستوى تعديل السيلينيوم في الجزء الذي يحتوي على مركز النشاط السيستين من خلال تحديد النسبة النسبية بين شدة الببتيد-CysSSe-AM وشدة الببتيد-CysS-AM. تم استخدام عينات من الببتيد-CysS-AM التي خضعت لهضم التربسين تلاها اختزال باستخدام TCEP وعمليات ألكلة لاحقة باستخدام اليودوأسيتاميد لقياس الببتيدات التي تت correspond إلى الببتيد-CysSSe-AM.
التحليل الإحصائي وإمكانية التكرار
تُعرض البيانات كمتوسط الانحراف المعياري أو المتوسط تم استخدام برنامج GraphPad Prism9 الإصدار 9.4.0 لحسابالقيم باستخدام اختبار الطالب-اختبار، تحليل التباين أحادي الاتجاه أو تحليل التباين ثنائي الاتجاه، يتبعه اختبار المقارنات المتعددة لتوكاي.تم تقديم القيم في أساطير الأشكال. تم تكرار جميع التجارب على الأقل مرتين، كل منها مع نتائج مشابهة (كانت فحوصات التعبير الموضحة في البيانات الموسعة الأشكال 1g و3a تجارب فردية).
ملخص التقرير
معلومات إضافية حول تصميم البحث متاحة في ملخص تقارير مجموعة نيتشر المرتبط بهذه المقالة.
توفر البيانات
تم الحصول على بيانات مخطط الصندوق للتعبير الجيني التفاضلي في الأنسجة الورمية والطبيعية من TNMplothttps://tnmplot.com/analysis). جميع البيانات متاحة في المقال والمعلومات التكميلية، ومن المؤلفين المقابلين عند الطلب المعقول. يتم توفير بيانات المصدر مع هذه الورقة.
References
Ran, F. A. et al. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281-2308 (2013).
Sanjana, N. E., Shalem, O. & Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783-784 (2014).
شكر وتقدير
نشكر أعضاء مختبر إيوائي على الاقتراحات المفيدة؛ وY. سايتو (جامعة توهوكو) وT. توياما (جامعة توهوكو) على تزويدهم بلطف بـ SELENOP؛ وF. زانغ (معهد ماساتشوستس للتكنولوجيا) على توفير مكتبة GeCKO للفئران وغيرها من متجهات CRISPR. تم دعم هذا العمل من قبل جمعية اليابان لتعزيز العلوم (JSPS) KAKENHI: أرقام المنح 20H05505، 22H04810 و22K06222 (إلى H.F.); من قبل JSPS KAKENHI أرقام المنح 23K20040، 21H05263، 18H05277 و22K19397 (إلى T.A.); من قبل وكالة اليابان للعلوم والتكنولوجيا CREST رقم المنحة JPMJCR2024 (إلى T.A.); من قبل JSPS KAKENHI رقم المنحة 19 H 05772 (إلى Y.O.); ومن قبل JSPS KAKENHI رقم المنحة 24112002 ومؤسسة تاكيدا للعلوم (K.I.). لم يكن للممولين أي دور في تصميم الدراسة، جمع البيانات وتحليلها، اتخاذ القرار للنشر أو إعداد المخطوطة.
مساهمات المؤلفين
قام H.F. و K.I. بتصور وتصميم المشروع. قام H.F. بإجراء معظم التجارب. قامت Y.T. بإجراء تحليل ICP-MS. قامت S.O. بإجراء تحليل الطيف الكتلي. قدم N.S. و S.K. و U.B. و T.A. و Y.O. نصائح بشأن تصميم التجارب. كتب H.F. و K.I. المخطوطة، مع مساهمات من جميع المؤلفين الآخرين.
الشكل التوضيحي الممتد 1 | توصيف موت الخلايا الناتج عن الحديد في FBXL5 خلايا KO. أ، صور بتباين الطور للخلايا بعد المعالجة بـ FAC ( ) لمدة 48 ساعة. قضبان القياس، . البيانات تمثل ثلاث تجارب مستقلة. ب، صور مجهرية فلورية للخلايا الملونة بمؤشر FerroOrange بعد العلاج لمدة 4 ساعات مع FAC ( ). قضبان القياس، . البيانات تمثل ثلاثة تجارب مستقلة. ج، تحليل تدفق الخلايا لمستويات هيدروبيروكسيد الدهون في خلايا WT أو FBXL5 KO بواسطة صبغة C11-BODIPY بعد المعالجة بـ FAC ( ) لمدة 24 ساعة. البيانات تمثل تجربتين مستقلتين. د، تم معالجة خلايا MEFs البرية لمدة 3 ساعات مع بافيلوميسين A1 ( 100 نانومتر )، تليها FAC ( ) للموضح تم تحليل lysates الخلوية بواسطة التحليل المناعي. البيانات تمثل تجربتين مستقلتين. e، بقاء خلايا FBXL5 KO MEFs المعالجة لمدة 24 ساعة مع FAC ( ) في وجود بافيلوميسين A1 ( ). تُعرض البيانات كمتوسط الانحراف المعياري لثلاث نسخ بيولوجية.تحليل المناعية للغشاء من المستخلصات من الخلايا المحددة. البيانات (f) تمثل تجربتين مستقلتين. فحص التعبير في (g) هو تجربة واحدة.مخططات البركان لفحص الفيروبتوزيس المحفز بالحديد، مع التركيز على سلسلة نقل الإلكترون الميتوكوندرية (المجمع I و II: اليسار، المجمع III و IV: اليمين).تم توليد قيمة وتغير الطي بواسطة اختبار MAGeCK.
أ
ب
SEPHS2 = عامل تخليق السيلينوبروتين
ترتيب MAGeCK (جين مثبط)
رتبة
اسم الجين
قيمة p
سجلتغيير الطي
1
ميتاف1
-7.382
2
PRDX6
2.20 × 10^-7
-2.5705
٣
PSTK
-1.4595
٤
SNX27
2.20 × 10^-7
-2.5145
٥
UBE2N
2.86E-06
-2.8148
٦
CINP
9.46E-06
-2.1364
٧
SEPHS2
-3.9336
٨
سينبي
٢.٤٤ × ١٠^-٥
-4.3363
9
STX5A
2.93E-05
-2.2028
10
دي دي إكس 10
٤.٢٩ × ١٠^-٥
-5.4515
11
KLF5
٤.٤٢ × ١٠^-٥
-2.5021
12
SCLY
-1.2627
عامل تخليق السيلينوبروتين
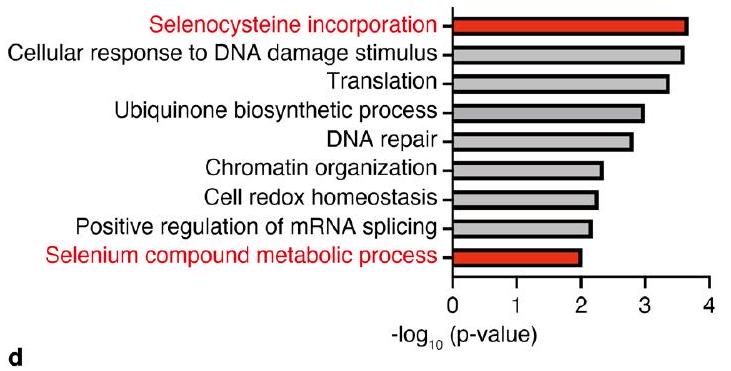
الشكل البياني الموسع 2 | التحقق من جينات الضرب، وتحديد PRDX6 كمنظم لـ GPX4. أ، جينات المثبط الضاربة [حد قطع قيمة p -value ( )] تم فحصها من خلال تحليل علم الأحياء الجيني. تم توليد القيمة بواسطة اختبار ديفيد لجينات الأونتولوجيا. ب، تصنيف النقاط السلبية للبروتينات السيلين. قائمة الفحص. ج، ترتيب MAGeCK لجينات المثبط.تم توليد قيمة وتغير الطي بواسطة اختبار MAGeCK. د، تحليل المناعية للعينات من الخلايا التي تعبر عن sgRNAs المستهدفة للجينات المحددة. البيانات تمثل تجربتين مستقلتين.
الشكل البياني الموسعتنظم PRDX6 عملية الفيروبتوزيس من خلال الحفاظ على تعبير GPX4. أ، حيوية خلايا FBXL5 KO أو خلايا FBXL5/PRDX6 المزدوجة المعطلة التي تم علاجها لمدة 24 ساعة مع FAC (50 أو )، وتحليل المناعية للعينات من الخلايا المحددة. تُعرض بيانات البقاء على قيد الحياة كمتوسط انحراف معياري لثلاث نسخ بيولوجية. ب، تم معالجة خلايا MEFs من النوع البري أو PRDX6 KO بـ FAC (” ) للأوقات المحددة، وتم تحليل lysates الخلوية بواسطة التحليل المناعي. البيانات تمثل تجربتين مستقلتين. ج، تحليل تدفق الخلايا لمستويات هيدروبيروكسيد الدهون في خلايا WT و PRDX6 KO، كما تم تقييمه بواسطة صبغة C11-BODIPY بعد المعالجة بـ FAC ( ) لمدة 90 دقيقة. البيانات تمثل ثلاث تجارب مستقلة. د، تحليل المناعية للعينات من خلايا PRDX6 KO الضابطة التي تعبر عن الطفرة GPX4 U/C. البيانات تمثل اثنين من الطفرات المستقلة PRDX6 بعد الحضانة لمدة 24 ساعة في وجود RSL3. (هـ) أو IKE ( تُعرض بيانات القابلية للحياة كمتوسطانحراف معياري لثلاث نسخ بيولوجية. ج، قياس نشاط GPX باستخدام بروتينات معاد تركيبها. تم استخدام بيروكسيد الهيدروجين (يسار) أو بيروكسيد التيرت-بيوتيل (يمين) كركيزة. تُعرض البيانات كمتوسط.انحراف معياري للمتوسط من ثلاثة تجارب مستقلة. هـ، تحليل تدفق الخلايا لعملية أكسدة الدهون بعد صبغ C11-BODIPY لخلايا WT أو GPX4 KO التي تعبر بشكل مستقر عن PRDX6 بعد إزالة liiproxstatin-1 لمدة 24 ساعة. البيانات تمثل تجربتين مستقلتين. و، تحليل المناعية للعينات من الخلايا المحددة. البيانات تمثل تجربتين مستقلتين.
الشكل 4 من البيانات الموسعة | ينظم PRDX6 تعبير السيلينوبروتينات.
أ، قياس مستويات mRNA لـ GPX4 بواسطة RT-qPCR. تُعبر البيانات عن المتوسطانحراف معياري لثلاث نسخ بيولوجية. غير ذي دلالة إحصائية؛ (sgControl مقابل sgPRDX6 #1 = 0.9523؛ sgControl مقابل sgPRDX6 #2 = 0.9607)؛ تحليل ANOVA أحادي الاتجاه. ب، ج، تحليل المناعية للعينات من خلايا التحكم أو خلايا PRDX6 KO التي تم حضنها لمدة 4 أو 8 ساعات في وجود E64d/pepأو MG-132. تحليل المناعية باستخدام تقنية البلوت من مستخلصات خلايا التحكم أو خلايا PRDX6 KO المأخوذة من خطوط خلايا السرطان البشرية المحددة (Hela، A549، H226، HepG2، NB-1). تحليل المناعية باستخدام تقنية البلوت من مستخلصات خلايا التحكم أو خلايا ذات KO للجينات المحددة والتي تعبر بشكل مستقر عن Myc-GFP WT. البيانات (b-e) تمثل تجربتين مستقلتين.
الشكل البياني الممتدتنظم PRDX6 كفاءة استخدام السيلينيوم.
أ، مخطط يوضح استقلاب السيلينيوم وطريقه تخليق السيلينوبروتين. ب، ج، تم معالجة خلايا WT أو PRDX6 KO مسبقًا لمدة يومين بـأو سيلينيت الصوديوم. ثم تم معالجة الخلايا لمدة 24 ساعة مع RSL3 (0.3 أو ) ( ) أو IKE ( 0.3 أو ) ( ) في وجود 100 نانومتر أو 100 نانومتر من سيلينيت الصوديوم. د، تحليل المناعية للعينات من خلايا هيلا الضابطة أو KO PRDX6 المزروعة لمدة 48 ساعة في وجود سيلينيت الصوديوم. أو أو تم معالجة خلايا هيلا PRDX6 KO مسبقًا بـأو سيلينيت الصوديوم (100 نانومتر) لمدة يومين. ثم تم معالجة الخلايا لمدة 48 ساعة مع FAC (100 أو ) في حضور ( قسمأو سيلينيت الصوديوم (100 نانومول)، وتم قياس حيوية الخلايا.تحليل المناعية مستخلصات من خلايا التحكم أو خلايا PRDX6 KO المزروعة لمدة 32 ساعة في وجود التركيزات المحددة منأو سيلينيت الصوديومتم تحضين خلايا WT أو خلايا PRDX6 KO لمدة 24 ساعة مع FBS في وجود ليبروكستاتين-1. ثم تم معالجة الخلايا بـأو 100 نانومتر من سيلينيت الصوديوم لمدة الأوقات المحددة. تم تحليل lysates الخلوية بواسطة التحليل المناعي. i، تم تحضين خلايا PRDX6 KO لمدة 36 ساعة معأو 40 نانومول من سيلينيت الصوديوم. ثم، تم تعريض خلايا WT أو PRDX6 KO المعالجة مسبقًا بالسيلينيوم إلى 50 نانومولأو 200 نانومتر من سيلينيت الصوديوم لفترات محددة وتم تحليل المستخلصات بواسطة التحليل المناعي. البيانات تمثل حالتين ( ) أو ثلاثة ( تجارب مستقلة. تُعرض بيانات البقاء (ب، ج، هـ) كمتوسطالانحراف المعياري لثلاث نسخ بيولوجية.
الشكل 6 من البيانات الموسعة | التعبير المفرط عن SEPHS2 WT ينقذ نمط ظاهرة PRDX6 KO. أ، تحليل المناعية للبروتينات من مستخلصات خلايا التحكم أو خلايا SEPHS2 KO التي تعبر بشكل مستقر عن الطفرة SEPHS2U/C. ب، تحليل المناعية للبروتينات من مستخلصات خلايا التحكم أو خلايا PRDX6 KO التي تعبر بشكل مستقر عن SEPHS2 WT أو الطفرة U/C. ج، تحليل المناعية للبروتينات من مستخلصات خلايا تعبر بشكل مستقر عن Myc-GFP C70U أو S175U في خلايا التحكم أو خلايا PRDX6 KO التي تعبر بشكل مستقر عن SEPHS2 WT أو الطفرة U/C. البيانات (أ-ج) تمثل تجربتين مستقلتين. د-و، حيوية الخلايا المحددة في وجود FAC (50 أو ) لـ ، أو RSL3 (0.3 أو ) (e) أو IKE ( 0.3 أو ) (f) لمدة 24 ساعة. القابلية للحياة تُعرض البيانات كمتوسطانحراف معياري لثلاث نسخ بيولوجية. ج، قابلية الخلايا المحددة المعالجة لمدة 48 ساعة مع وليبروكستاتين-تُعرض بيانات القابلية للحياة كمتوسط الانحراف المعياري لثلاث نسخ بيولوجية. * PRDX6 WT مقابل C47S = 0.0243؛ WT مقابل sgSEPHS2 = 0.0238 ; تحليل ANOVA أحادي الاتجاه، هلام SDS-PAGE الملون بكوماسي يظهر البروتين المؤتلف PRDX6 WT أو الطفرة C47S المستخدمة في تحليل ICP-MS. تحليل ICP-MS لكمية السيلينيوم المرتبط بالبروتين المؤتلف PRDX6 WT أو C47S. تُعرض البيانات كمتوسط الانحراف المعياري لثلاث تجارب مستقلة.، *** ; تحليل التباين الأحادي.
الشكل البياني الموسع 7 | تحليل طيف الكتلة لتعديل PRDX6 C47. أ، ب، طيف الكتلة لـ PRDX6 C47. PRDX6 (تم حضنه مع GSHوسيلينيت الصوديوم، أو مع و SCLY لمدة 5 دقائق قبل تحليلات مطيافية الكتلة.
الشكل 8 من البيانات الموسعة | التفاعل بين PRDX6 و SEPHS2 أو SCLY.
تم الكشف عن الارتباط الفيزيائي بين PRDX6 و SCLY في اختبار PLA. شريط القياس،البيانات تمثل ثلاث تجارب مستقلة. كما يتم عرض تقدير نقاط PLA. تُعرض البيانات كمتوسط. س.د. [ الخلايا (فارغة-PRDX6)،الخلايا (SCLY-PRDX6) التي تم فحصها،؛
ب
اختبار t ذو الجانبين غير المقترن. تم زراعة خلايا MEFs المفقودة PRDX6 التي تعبر عن HA-TurboID-فارغ، -PRDX1، أو -PRDX6 لمدة 30 دقيقة مع DMSO أوالبيوتين. تم تحليل مستخلصات الخلايا وعينات السحب بواسطة التحليل المناعي. تم تحليل العلاقة بين PRDX6 و SEPHS2 (ب) أو بين PRDX6 و SCLY (ج). البيانات تمثل تجربتين (ج) أو ثلاث تجارب (ب) مستقلة.
الشكل 9 من البيانات الموسعة | يرتبط تعبير PRDX6 بتشخيص سيء لعدة أنواع من السرطان. أ، ملف تعبير PRDX6 في عدة أنواع من الأنسجة الطبيعية والورمية. تم الحصول على البيانات من TNMplothttps://tnmplot. com/analysis/)، وهو خادم ويب يخزن بيانات تتعلق بتعبير الجينات الطبيعية والمرتبطة بالسرطان. أولئك الذين لديهم قيم أعلى في نسيج الورم مقارنةً بالأنسجة الطبيعية ومع وجود اختلافات كبيرة (اختبار مان-ويتني U) تم تمييزها باللون الأحمر ( ). ب، منحنيات بقاء كابلان-ماير تقارن عينات السرطان التي تظهر تعبيرًا عاليًا عن PRDX6 مع العينات التي تظهر تعبيرًا منخفضًا عن PRDX6 (تم بناء المنحنيات باستخدام مخطط كابلان-ماير).
الشكل 10 من البيانات الموسعة | تعبير PRDX6 مهم لبقاء خطوط خلايا السرطان. أ، تحليل المناعية للبروتينات من مستخلصات خلايا Panc-1 WT أو PRDX6 KO. ب، المراقبة المستمرة لمدى حيوية خلايا Panc-1 WT أو PRDX6 KO في وجود أو غياب سيلينيت الصوديوم (100 نانومتر)، (Sec)أو ليبروكستاتين-1. ج، تحليل المناعية للعينات من خلايا MIA PaCa-2 WT أو PRDX6 KO. د، المراقبة المستمرة لمدى حيوية خلايا MIA PaCa-2 WT أو PRDX6 KO في وجود أو غياب سيلينيد الصوديوم (100 نانومتر)، أو ليبروكستاتين-1. e، تحليل المناعية للغشاء من خطوط الخلايا المشار إليها.جدوى خطوط الخلايا المشار إليها في وجود FAC (50 أو ) لمدة 48 ساعة. تُعرض بيانات البقاء على قيد الحياة كمتوسطانحراف معياري لثلاث نسخ بيولوجية. ج، تحليل المناعية للعينات من خلايا WT أو PRDX6 KO SK-N-DZ.المراقبة المستمرة لجدوى خلايا WT أو PRDX6 KO SK-N-DZ في وجود أو غياب سيلينيت الصوديومأو ليبروكستاتين-1 (بيانات الجدوى ) يتم تقديمها كمتوسط s.e.m من ثلاث نسخ بيولوجية. البيانات (أ، ج، هـ، ج) تمثل تجربتين مستقلتين.
natureportfolio
المؤلف(المؤلفون) المعنيون: هيرواكي فوجيتا، كازوهيرو إيوي
آخر تحديث من قبل المؤلف(المؤلفين): 22 فبراير 2024
ملخص التقرير
تسعى Nature Portfolio لتحسين قابلية إعادة إنتاج العمل الذي ننشره. يوفر هذا النموذج هيكلًا للاتساق والشفافية في التقرير. لمزيد من المعلومات حول سياسات Nature Portfolio، راجع سياسات التحرير وقائمة مراجعة سياسة التحرير.
الإحصائيات
لجميع التحليلات الإحصائية، تأكد من أن العناصر التالية موجودة في أسطورة الشكل، أسطورة الجدول، النص الرئيسي، أو قسم الطرق. الحجم الدقيق للعينة ( ) لكل مجموعة/شرط تجريبي، معطى كرقم منفصل ووحدة قياس X بيان حول ما إذا كانت القياسات قد أُخذت من عينات متميزة أو ما إذا كانت نفس العينة قد تم قياسها عدة مرات الاختبار(الاختبارات) الإحصائية المستخدمة وما إذا كانت أحادية أو ثنائية الجانب
يجب وصف الاختبارات الشائعة فقط بالاسم؛ وصف تقنيات أكثر تعقيدًا في قسم الطرق.
وصف لجميع المتغيرات التي تم اختبارها وصف لأي افتراضات أو تصحيحات، مثل اختبارات الطبيعية والتعديل للمقارنات المتعددة
وصف كامل للمعلمات الإحصائية بما في ذلك الاتجاه المركزي (مثل المتوسطات) أو تقديرات أساسية أخرى (مثل معامل الانحدار) والتباين (مثل الانحراف المعياري) أو تقديرات عدم اليقين المرتبطة (مثل فترات الثقة)
لاختبار الفرضية الصفرية، إحصائية الاختبار (مثل ) مع فترات الثقة، أحجام التأثير، درجات الحرية و القيمة المذكورة أعطِ القيم كقيم دقيقة كلما كان ذلك مناسبًا. لمعلومات التحليل البايزي، معلومات حول اختيار الأوليات وإعدادات سلسلة ماركوف مونت كارلو للتصاميم الهرمية والمعقدة، تحديد المستوى المناسب للاختبارات والتقارير الكاملة للنتائج تقديرات أحجام التأثير (مثل كوهين ، بيرسون )، مشيرًا إلى كيفية حسابها
تحتوي مجموعتنا على الإنترنت حول الإحصائيات لعلماء الأحياء على مقالات حول العديد من النقاط أعلاه.
البرمجيات والرموز
معلومات السياسة حول توفر رمز الكمبيوتر
جمع البيانات
تم قياس تحليل Western blotting بواسطة جهاز LAS4000mini أو LAS3000 (GE Healthcare). تم قياس اختبار حيوية الخلايا بواسطة SpectraMax M5 (Molecular Device). تم جمع صور المناعية باستخدام مجهر Fv1000 (Olympus). تم قياس تركيزات DNA و RNA بواسطة Nanodrop2000 (Thermo). تم قياس تركيز البروتين بواسطة SpectraMax M5 (molecular device) أو مقياس الطيف V750 (JASCO). تم قياس اختبار GPX (الامتصاص عند 340 نانومتر) بواسطة SpectraMax M5 (Molecular Devices). تم الحصول على بيانات RT-qPCR بواسطة نظام ABI ViiA7 PCR في الوقت الحقيقي (Applied Biosystems). تم قياس تقرير اللوكيفيراز بواسطة مقياس لومي Lumat (Berthold). تم قياس منتج AMP بواسطة قارئ الألواح Nivo (PerkinElmer). تم الحصول على بيانات بقاء الخلايا على نظام iCelligence باستخدام برنامج RTCA iCelligence (ACEA Biosciences). تم إجراء ICP-MS بواسطة Agilent 8800 ICP-MS/MS (Agilent Technologies). تم إجراء تحليل LC-ESI-Q-TOF باستخدام 6545XT AdvanceBio LC/Q-TOF (Agilent Technologies) المتصل بنظام HPLC من Agilent. تم جمع بيانات تحليل التدفق باستخدام FACS Canto II (BD Biosciences) وبرنامج FACS Diva الإصدار 6.1.2 (Becton Dickinson).
تحليل البيانات
تم استخدام GraphPad Prism9 الإصدار 9.4.0 لإجراء التحليل الإحصائي. تم تحليل بيانات فحص CRISPR بواسطة خط أنابيب MAGeCK. تم استخدام Microsoft Exel الإصدار 16.72 لتحليل البيانات. تم استخدام Image Gauge الإصدار 4.22 (FUJIFILM) للصور الكيميائية المتلألئة. تم استخدام برنامج ViiA7 RUO الإصدار 1.2.3 (Thermo Fisher Scientific) لتحليل تعبير RNA. تم تحليل بيانات تسجيل بقاء الخلايا باستخدام نظام iCelligence بواسطة برنامج RTCA Software Lite الإصدار 2.2.1 (ACEA Biosciences). تم تحليل صور المناعية بواسطة ImageJ (الإصدار 2.3.0). تم استخدام برنامج FlowJo (Tomy Digital Biology، الإصدار 9.9.6) لجميع تحليلات بيانات تحليل التدفق. تم إجراء تحليل التعديل لمركز السيستين النشط (Cys47) باستخدام برنامج MassHunter BioConfirm الإصدار 10.0 (Agilent Technologies). تم ترتيب الأشكال في المخطوطة وتحويلها إلى ملفات PDF باستخدام برنامج illustrator الإصدار 27.4.1 (Adobe).
للمخطوطات التي تستخدم خوارزميات أو برمجيات مخصصة تكون مركزية للبحث ولكن لم يتم وصفها بعد في الأدبيات المنشورة، يجب أن تكون البرمجيات متاحة للمحررين و
البيانات
معلومات السياسة حول توفر البيانات
يجب أن تتضمن جميع المخطوطات بيانًا حول توفر البيانات. يجب أن يوفر هذا البيان المعلومات التالية، حيثما ينطبق:
رموز الوصول، معرفات فريدة، أو روابط ويب لمجموعات البيانات المتاحة للجمهور
وصف لأي قيود على توفر البيانات
بالنسبة لمجموعات البيانات السريرية أو بيانات الطرف الثالث، يرجى التأكد من أن البيان يتماشى مع سياستنا
تم الحصول على بيانات التعبير الجيني التفاضلي في الأنسجة الورمية والطبيعية من TNMplot (https://tnmplot.com/analysis/). جميع البيانات متاحة في المقالة والمعلومات التكميلية، ومن المؤلفين المعنيين عند الطلب المعقول.
المشاركون في البحث البشري
معلومات السياسة حول الدراسات التي تشمل المشاركين في البحث البشري والجنس والنوع في البحث.
التقرير عن الجنس والنوع
غير متاح
خصائص السكان
غير متاح
التجنيد
غير متاح
الإشراف الأخلاقي
غير متاح
يرجى ملاحظة أنه يجب أيضًا تقديم معلومات كاملة حول الموافقة على بروتوكول الدراسة في المخطوطة.
التقرير الخاص بالمجال
يرجى اختيار الخيار أدناه الذي يناسب بحثك بشكل أفضل. إذا لم تكن متأكدًا، اقرأ الأقسام المناسبة قبل اتخاذ قرارك.
علوم الحياة العلوم السلوكية والاجتماعية العلوم البيئية والتطورية والبيئية
يجب على جميع الدراسات الإفصاح عن هذه النقاط حتى عندما يكون الإفصاح سلبيًا.
حجم العينة
لم يتم استخدام اختبارات إحصائية لتحديد حجم العينة. تم تحديد حجم العينة بناءً على الدراسات السابقة في هذا المجال باستخدام نماذج تجريبية مماثلة [PMID: 34031600 و 31634899]، ولتقديم قيم كافية لإجراء اختبارات إحصائية قياسية.
استبعاد البيانات
لم يتم استبعاد أي بيانات من التحليلات.
التكرار
تم تكرار جميع التجارب على الأقل مرتين، كل منها مع نتائج مشابهة (تم عرض فحوصات التعبير في الأشكال الموسعة 1 ج و 3 أ كتجارب فردية).
العشوائية
غير قابل للتطبيق حيث لم يتم إجراء دراسات حيوانية.
التعمية
لم يتم إجراء التعمية لأن الباحث يحتاج إلى معرفة مجموعات العلاج من أجل إجراء التجارب.
التقرير عن المواد والأنظمة والأساليب المحددة
نطلب معلومات من المؤلفين حول بعض أنواع المواد والأنظمة التجريبية والأساليب المستخدمة في العديد من الدراسات. هنا، حدد ما إذا كانت كل مادة أو نظام أو طريقة مدرجة ذات صلة بدراستك. إذا لم تكن متأكدًا مما إذا كان عنصر القائمة ينطبق على بحثك، اقرأ القسم المناسب قبل اختيار رد.
تم التحقق من الأجسام المضادة الأولية في هذه الدراسة أو تم استخدامها في مقالات سابقة لتحليل البروتين الغربي (WB) لعينة من الفأر أو الإنسان.
تم التحقق من الأجسام المضادة ضد PRDX6 (Proteintech، 13585-1-AP)، SEPHS2 (Proteintech، 14109-1-AP)، GPX4 (Proteintech، 67763-1-AP) و(Santa Cruz، sc-166570)، GPX1/2 (Santa Cruz، sc-133160)، PSTK (Santa Cruz، sc-373991)، FBXL5 (Santa Cruz، sc-390102)، ACSL4 (Santa Cruz، sc-365230)، PDSS2 (Santa Cruz، sc-515137) من خلال knockout للجينات الذاتية باستخدام CRISPR/Cas9.
تم التحقق من الأجسام المضادة Anti-FTH1 (Santa Cruz، sc-376594) وanti-ferritin (Sigma-Aldrich، F6136) وAnti-IRP2 (داخلية) في منشور سابق (PMID: 35318808)
تم التحقق من صحة مضاد FSP1 (بروتين تك، 20886) في منشور سابق (PMID: 31634900).
تم التحقق من الأجسام المضادة ضد SELN (سانتا كروز، sc-365824) وضد LRP8 (أبكام، ab108208) في منشورات سابقة (PMID: 35637349).
تفاصيل التحقق للأجسام المضادة التجارية الأخرى موجودة على موقع الشركة المصنعة المرفق كما يلي.
مضاد SCLY (بروتين تك، 67606-1-lg)؛ https://www.ptglab.com/products/SCLY-Antibody-67606-1-lg.htm؛ وقد تم التحقق منه أيضًا من خلال تعبير بناء SCLY المسمى.
أجسام مضادة مرتبطة بـ HRP ضد IgG الفأر (Cell Signaling، #7076)؛ https://www.cellsignal.com/products/secondary-antibodies/anti-mouse-igg-hrp-linked-antibody/7076?country=JP&language=en
الأجسام المضادة المرتبطة بـ HRP ضد الأرانب IgG (GE Healthcare، NA934)؛ https://www.cytivalifesciences.com/en/us/shop/protein-analysis/blotting-and-detection/blotting-standards-and-reagents/amersham-ecl-hrp-conjugated-antibodies-p-06260
خطوط خلايا حقيقية النواة
معلومات السياسة حول خطوط الخلايا والجنس والنوع في البحث
مصدر(s) خط الخلايا
تم إنتاج الخلايا الليفية الجنينية للفئران (MEFs) داخليًا. تم إهداء خلايا HepG2 من قبل الدكتور كويتشي ناكاجيما (جامعة مدينة أوساكا)، والتي تم شراؤها في الأصل من ATCC (HepG2 : HB-8065). تم إهداء خلايا HEK293T من قبل الدكتور إيجيرو ناكامورا (جامعة كيوتو)، والتي تم شراؤها في الأصل من RIKRN RBC (293T : RCB2202). تم إهداء خلايا PLATE من قبل الدكتور توشيو كيتامورا (جامعة طوكيو). تم إهداء خلايا A549 و H226 و H460 و H1975 من قبل الدكتور أتشا ياسو ساتو (جامعة كيوتو)، والتي تم شراؤها في الأصل من ATCC (A549 : CCL-185، H226 : CRL-5826، H460 : HTB-177، H1975 : CRL-5908). تم شراء خلايا SK-N-DZ و HeLa من ATCC (SK-N-DZ : CRL-2149، HeLa : CCL-2). تم شراء خلايا PANC-1 و MIA Paca-2 من RIKEN RBC (PANC-1 : RCB2095، MIA Paca-2 : RCB2094). تم شراء خلايا NB-1 من JCRB (NB-1 : JCRB0621).
المصادقة
لم يتم التحقق من أي من خطوط الخلايا المستخدمة.
تلوث الميكوبلازما
جميع خطوط الخلايا التي تم اختبارها كانت سلبية لوجود تلوث بالميكوبلازما.
الخطوط التي يتم التعرف عليها بشكل خاطئ بشكل شائع (انظر سجل ICLAC)
لم يتم استخدام أي خط خلوي يتم التعرف عليه بشكل خاطئ بشكل شائع.
تدفق الخلايا
المؤامرات
أكد أن:
توضح تسميات المحاور العلامة والفوروشروم المستخدم (مثل CD4-FITC). المقاييس على المحاور مرئية بوضوح. قم بتضمين الأرقام على المحاور فقط للرسم البياني في أسفل اليسار من المجموعة (المجموعة هي تحليل للعلامات المتطابقة). جميع الرسوم البيانية هي رسوم بيانية متساوية الارتفاع مع نقاط شاذة أو رسوم بيانية بالألوان الزائفة. تم توفير قيمة عددية لعدد الخلايا أو النسبة المئوية (مع الإحصائيات).
المنهجية
تحضير العينة
تم زراعة الخلايا في طبق 6 آبار وتم معالجتها بـ FAC. بعد 1.5-24 ساعة، تمت إزالة الوسط، وتم وسم الخلايا بـ DMEM المحتوي على 10 ميكرومول من BODIPY.فيلمدة 30 دقيقة. ثم تم غسل الخلايا ثلاث مرات بمحلول PBS وتم فصلها عن الصفيحة باستخدام التربسين، وتم قياس الفلورية الخضراء بواسطة قياس التدفق الخلوي باستخدام جهاز BD FACSCanto II.
آلة
FACSCanto II (بي دي بيوساينس) لتحليلات السيتومترية التدفقية
برمجيات
برنامج FACS Diva الإصدار 6.1.2 (بيكتون ديكنسون) لجمع البيانات. برنامج FlowJo الإصدار 9.9.6 لتحليل البيانات.
وفرة تجمع الخلايا
تم تحليل ما لا يقل عن 5000 خلية لكل عينة.
استراتيجية البوابة
تم استخدام منطقة FSC مقابل منطقة SSC لتحديد الكتل السكانية الكبيرة من الخلايا. تم اعتماد تحديد الكتلة السكانية الأولية للخلايا (منطقة FSC مقابل ارتفاع FSC) للتأكد من استبعاد الثنائيات واستخدام الخلايا الفردية فقط للتحليل.
قم بتحديد هذا المربع لتأكيد أنه تم تقديم رقم يوضح استراتيجية البوابة في المعلومات التكميلية.
قسم الفيزيولوجيا الجزيئية والخلوية، كلية الطب بجامعة كيوتو، كيوتو، اليابان.مختبر السموم وصحة البيئة، كلية الدراسات العليا للعلوم الصيدلانية، جامعة تشيبا، تشيبا، اليابان.قسم الطب البيئي وعلم السموم الجزيئي، كلية الطب بجامعة توهوكو، سنداي، اليابان.العنوان الحالي: قسم علاج الأورام بالإشعاع، جامعة نيويورك لانغون للصحة، نيويورك، نيويورك، الولايات المتحدة الأمريكية.ساهم هؤلاء المؤلفون بالتساوي: يوكِي تاناكا، سيريُو أوغاتا.البريد الإلكتروني: فوجيسان@mcp.med.kyoto-u.ac.jp; kiwai@mcp.med.kyoto-u.ac.jp
Ferroptosis is a form of regulated cell death induced by iron-dependent accumulation of lipid hydroperoxides. Selenoprotein glutathione peroxidase 4 (GPX4) suppresses ferroptosis by detoxifying lipid hydroperoxides via a catalytic selenocysteine (Sec) residue. Sec, the genetically encoded amino acid, is biosynthesized from a reactive selenium donor on its cognate tRNA . It is thought that intracellular selenium must be delivered ‘safely’ and ‘efficiently’ by a carrier protein owing to its high reactivity and very low concentrations. Here, we identified peroxiredoxin 6 (PRDX6) as a novel selenoprotein synthesis factor. Loss of PRDX6 decreases the expression of selenoproteins and induces ferroptosis via a reduction in GPX4. Mechanistically, PRDX6 increases the efficiency of intracellular selenium utilization by transferring selenium between proteins within the selenocysteyl-tRNA synthesis machinery, leading to efficient synthesis of selenocysteyl-tRNA . These findings highlight previously unidentified selenium metabolic systems and provide new insights into ferroptosis.
Ferroptosis, a type of programmed cell death triggered by iron-induced lipid hydroperoxidation, is involved in numerous pathological conditions such as cancer, neurodegenerative diseases and ischemia-reperfusion injury; as such, it has been studied extensively . Cells have two main defense mechanisms against ferroptosis: GPX4 and ferroptosis suppressor protein1 (FSP1). GPX4 uses glutathione (GSH) to suppress ferroptosis by detoxifying lipid hydroperoxides into nontoxic lipid alcohols , and FSP1 is an oxidoreductase that reduces ubiquinone ( CoQ ) to ubiquinol ( ), a radical-trapping antioxidant .
GPX4 is a selenoprotein in which the highly reactive amino acid Sec resides at the active site . Sec is incorporated into the UGA codon when the Sec-insertion sequence (SECIS) element is present in the -untranslated region of . Unlike other amino acids, selenocysteyl-transfer RNA for Sec (Sec-tRNA ) is biosynthesized from reactive selenium donor selenide on its cognate tRNA
through intricate enzymatic reactions . Selenide is phosphorylated by selenophosphate synthetase 2 (SEPHS2) and subsequently incorporated into Sec-tRNA . Synthesis of selenoproteins is essential for life, as shown by studies describing embryonic lethality in mice lacking selenoprotein synthesis . Despite its importance, however, the selenoprotein synthesis pathway remains unclear. In particular, highly reactive selenides are hypothesized to be delivered safely and efficiently by a carrier protein(s) to SEPHS2; however, they have not yet been identified .
Here, we conducted a genome-wide iron-triggered ferroptosis screen and identified PRDX6 as a novel selenoprotein synthesis factor. Loss of PRDX6 greatly decreased the expression of selenoproteins, including GPX4, and triggered ferroptosis through a reduction in GPX4 levels. Mechanistically, PRDX6 functions as a selenide carrier, transferring selenide to SEPHS2 to facilitate efficient selenium utilization.
Fig. 1 | Genome-wide CRISPR screening for iron-triggered ferroptosis. a, WT or FBXL5 KO MEFs were treated with FAC ( ) for the indicated periods, and cell lysates were analyzed by immunoblotting. Data are representative of three independent experiments. b, Viability of WT or FBXL5 KO MEFs treated for 48 h with FAC ( ) in the presence of liproxstatin- or . c, Schematic showing the CRISPR-Cas9 screening strategy. d, Volcano plots showing the regulatory genes involved in iron-triggered ferroptosis. The value
and fold change were generated by the MAGeCK test.e, Viability of FBXL5 KO cells expressing sgEmpty or sgRNAs targeting ACSL4 or LPCAT3; cells were treated with FAC ( ) for , Viability of FBXL5 KO cells expressing sgEmpty, sgControl or sgRNAs targeting PDSS2 or FSP1; cells were treated with FAC for 30 h . Viability data (in b, e, f) are presented as the mean s.d. of three biological replicates.
Increased selenium utilization suppresses ferroptosis and contributes to cancer progression . We found that patients harboring cancers with high expression of PRDX6 have a poor prognosis. These findings provide insight into the previously unknown selenium metabolic system in cells as well as its potential as a target for cancer therapy.
Results
Iron-induced ferroptosis screen
The prefix ‘ferro’ suggests that ferroptosis should be categorized as a form of iron toxicity; however, in most studies of ferroptosis, cell death is not induced by iron administration. Rather, it is induced by inhibiting erasers for lipid hydroperoxidation such as the GPX4 inhibitor RSL3 or the cystine transporter inhibitor erastin . To re-analyze ferroptosis from the perspective of iron toxicity and to identify new regulators, we developed a ferroptosis induction system based on iron addition alone. The iron regulatory protein 2 (IRP2) and F-box and leucine-rich repeat protein 5 (FBXL5) system acts as a major regulator of cellular iron homeostasis by suppressing iron uptake and increasing iron storage by FBXL5-mediated degradation of IRP2 (refs. 19-21). Therefore, to induce iron toxicity , we knocked out FBXL5 from mouse embryonic fibroblasts (MEFs) using the CRISPR-Cas9 system (Fig. 1a). Treatment with ferric ammonium citrate (FAC) selectively killed FBXL5 knockout (KO) cells by increasing the cellular iron concentration, as detected by a fluorescent probe specific for (Fig. 1b and Extended Data Fig. 1a,b) . Ferroptosis is thought to be triggered by hydroperoxidation of polyunsaturated fatty acids (PUFAs) in the cell membrane by redox-active iron . BODIPY 581/591 C11 staining to detect hydroperoxidation of PUFAs revealed increased hydroperoxidation in iron-repleted FBXL5 KO cells (Extended Data Fig. 1c). Iron-triggered death of FBXL5 KO cells appeared to occur through ferroptosis because it was prevented by the ferroptosis inhibitor liproxstatin-1(Fig.1b). To identify genes involved in iron-triggered ferroptosis, we performed an unbiased genome-wide
CRISPR screen in which FBXL5 KO cells were infected with a GeCKOv2 library, followed by cultivation with or without FAC (Fig. 1c). Using a cutoff of , screening identified around 150 genes encoding iron-triggered ferroptosis suppressors or activators (Fig. 1d). As expected, one of the top hit genes encoding ferroptosis activators was IRP2; indeed, stabilization of IRP2 caused by loss of FBXL5 led to accumulation of redox-active iron in cells (Fig. 1a). Other prominent hits included iron homeostasis regulators such as transferrin receptor 1 (TFRC), divalent metal transporter 1 (DMT1) and the V-ATPase proton pump complex, all of which are required for iron uptake (Fig. 1d). We used the V-ATPase inhibitor bafilomycin A1 to confirm that organelle acidification is essential for iron uptake and iron-triggered ferroptosis (Extended Data Fig. 1d,e). More importantly, the high-confidence hit list included the known ferroptosis regulators acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) (Fig. 1d), both of which are required for insertion of PUFAs into membrane phospholipids. Deletion of ACSL4 or LPCAT3 from FBXL5 KO cells restored cell viability (Fig. 1e and Extended Data Fig. 1f), further indicating that iron-triggered ferroptosis is also mediated by oxidation of PUFAs. In addition, we identified suppressor genes, the CoQ synthesis machinery and FSP1 (Fig. 1d) . Deletion of diphosphate synthase subunit 2 (PDSS2), a component of the CoQ synthesis machinery, or FSP1 markedly increased the death of FBXL5 KO cells (Fig. 1f and Extended Data Fig. 1g); thus, lipid radical trapping by is also involved in suppressing iron-triggered ferroptosis . Interestingly, suppressor genes encoding mitochondrial electron transport chain complex I and II (CoQ oxidoreductase), but not complex III and IV, were enriched (Extended Data Fig. 1h), suggesting that in mitochondria also suppresses ferroptosis. Therefore, our iron-triggered ferroptosis screen, which does not rely on conventional ferroptosis inducers such as RSL3 or erastin, is useful for identifying not only ferroptosis activators but also suppressors.
Fig. 2 | PRDX6 suppresses iron-triggered ferroptosis indirectly by augmenting expression of GPX4. a, Immunoblot analysis of lysates from cells expressing sgEmpty, sgControl or sgRNAs targeting PRDX6. Data are representative of three independent experiments. b-d, Viability of control cells or PRDX6 KO cells treated with FAC ( 50 or ) for , RSL3 ( 0.3 or ) for or IKE ( 0.4 or ) for , Viability of control cells or PRDX6 KO cells stably expressing the GPX4U/C mutant; cells were incubated with FAC ( ) for , Viability of PRDX6 KO cells stably expressing the indicated PRDX6 mutants incubated in the presence of FAC ( ) for 48 h . Immunoblot analysis of lysates from the indicated cells is also shown. PLA , phospholipase A2. g, Measurement of GPX activity using recombinant
proteins. Cumene hydroperoxide was used as a substrate. Data are presented as the mean s.e.m of three independent experiments. h. Immunoblot analysis of lysates from control or GPX4 KO cells stably expressing the Myc-PRDX6 or FSP1. Data are representative of two independent experiments. i, Continuous monitoring of the viability of the indicated cell lines after withdrawal of liproxstatin-1. Data are presented as the mean s.e.m of three biological replicates.j, Viability of the indicated cells in the presence of the indicated concentrations of IKE for 24 h . NS; not significant; IKE ; IKE ); two-way ANOVA. Viability data (in ) are presented as the mean s.d. of three biological replicates.
Identification of PRDX6 as a regulator of GPX4 expression
Gene ontology analysis of the iron-triggered ferroptosis suppressors indicated that selenoprotein synthesis factors were the most enriched group (Extended Data Fig. 2a). The high-confidence suppressor list raised the possibility that loss of selenoprotein synthesis factors sensitizes cells to iron-triggered ferroptosis (Fig.1d). Among selenoproteins, the master ferroptosis regulator GPX4 was ranked highly as a suppressor (Extended Data Fig. 2b). Therefore, we suspected that new GPX4 regulators might be included in our suppressor list (Extended Data Fig. 2c), and we performed a secondary screen using the abundance of GPX4 as a readout (Extended Data Fig. 2d). Unexpectedly, we found that deletion of PRDX6 greatly reduced expression of GPX4 (Extended Data Fig. 2d), which we confirmed using two different guide RNAs (gRNAs) (Fig. 2a). Surprisingly, loss of PRDX6 sensitized not only FBXL5 KO cells (Extended Data Fig. 3a) but also wild-type (WT) cells (Fig. 2b) to iron-triggered ferroptosis, whereas deletion of PRDX6 had no effect on iron homeostasis because FAC-induced stabilization of FBXL5 and degradation of IRP2 was similar to that in WT cells (Extended Data
Fig. 3b) . This finding ruled out the alternative possibility that loss of PRDX6 inhibits iron-induced ferroptosis by disrupting iron metabolism in cells. Increased iron-induced lipid hydroperoxidation in PRDX6 KO cells indicated that PRDX6 is an essential suppressor of iron-triggered ferroptosis (Extended Data Fig. 3c). In addition, we found that PRDX6 is also involved in suppressing ferroptosis mediated by imidazole ketone erastin (IKE) and RSL3 (Fig. 2c,d). To address whether the decrease in GPX4 is responsible for ferroptosis sensitivity in PRDX6 KO cells, we expressed a partially active GPX4 mutant, in which Sec(U)46 is substituted by cysteine (GPX4 U/C) in PRDX6 KO cells (Extended Data Fig. 3d) . Expression of the GPX4 U/C mutant rescued PRDX6 KO cells from iron-triggered ferroptosis (Fig. 2e), suggesting that PRDX6 suppresses cell death by maintaining expression of GPX4.
PRDX6 belongs to the PRDXs family of antioxidant enzymes . Among them, PRDX6 has a unique characteristic in that it contains a single conserved cysteine (C47) residue, whereas the other family members contain two . PRDX6 exhibits three enzymatic activities: glutathione peroxidase (GPX) (C47) activity, phospholipase A2
(H26, S32 and D140) activity and LPCAT (D31) activity (the residues essential for each activity are shown in parentheses) . To examine which activity is required to maintain GPX4 abundance, we reconstituted PRDX6 KO cells with the WT or mutant PRDX6 (Fig. 2f). Although the H26A mutant failed to restore GPX4 levels owing to poor expression, the S32A and D140A mutants did restore them. Therefore, we concluded that phospholipase A2 activity is dispensable. We found that the C47S mutant failed to restore GPX4 expression (Fig. 2f). The C47S mutant also sensitized cells to not only iron-triggered but also canonical ferroptosis (Fig. 2f and Extended Data Fig. 3e,f). Previous reports have indicated that PRDX6 exhibits GPX activity at C47 (ref. 33); however, contrasting results have also been reported . We found that purified PRDX6 has no GPX activity (Fig. 2g and Extended Data Fig. 3g), indicating that the conserved cysteine of PRDX6 has a different function than previously thought. Next, we asked whether PRDX6 reduces lipid hydroperoxides in cells directly. Overexpression of PRDX6 increased the expression of GPX4 in WT cells (Fig. 2h), confirming that PRDX6 is critical for the expression of GPX4. As loss of GPX4 severely sensitizes cells to ferroptosis , we maintained GPX4 KO cells in culture medium containing the ferroptosis inhibitor liproxstatin-1. Washout of liproxstatin-1 inhibited the growth of GPX4 KO cells (Fig. 2i). Expression of FSP1, which suppresses ferroptosis independently of GPX4, restored proliferation of GPX4 KO cells, as previously reported ; however, expression of PRDX6 failed to restore proliferation of GPX4 KO cells (Fig. 2i). Furthermore, expression of PRDX6 did not reduce accumulation of lipid hydroperoxides in GPX4 KO cells (Extended Data Fig. 3h), strongly indicating that PRDX6 does not suppress lipid hydroperoxidation or ferroptosis directly; rather, it suppresses them by increasing the amount of GPX4. We also generated FSP1-overexpressing (O.E.)/GPX4 and PRDX6 double knockout (DKO) cells to confirm our findings (Extended Data Fig. 3i). Loss of PRDX6 did not increase IKE-triggered ferroptosis of FSP1 O.E./GPX4 KO cells (Fig. 2j), further supporting our hypothesis that PRDX6 suppresses ferroptosis indirectly by maintaining expression of GPX4.
Involvement of PRDX6 in selenoprotein synthesis
Next, we dissected the mechanism underlying the PRDX6-mediated increase in GPX4 expression. We found no obvious differences in the levels of GPX4 mRNA between PRDX6 KO and WT cells (Extended Data Fig. 4a), nor did proteasomal or lysosomal inhibitors restore GPX4 levels in PRDX6 KO cells (Extended Data Fig. 4b,c). Gene ontology analysis revealed enrichment of selenoprotein synthesis factors (Extended Data Fig. 2a). Moreover, we found that unbiased co-essentiality database tools also indicated a strong functional connection between PRDX6 and genes involved in selenoprotein synthesis (Fig. 3a). Therefore, we reasoned that PRDX6 is involved in selenoprotein biosynthesis. Indeed, deletion of PRDX6 reduced expression of other selenoproteins, including selenoprotein N (SELN), GPX1 and SEPHS2 (Fig. 3b). The conserved Cys47 of PRDX6 is essential for expression of selenoproteins because selenoprotein expression was rescued by re-expression of PRDX6 WT, but not that of the C47S mutant, in PRDX6 KO cells (Fig. 3c). Moreover, loss of PRDX6 reduced the amount of selenoproteins in several human cancer cell lines (Extended Data Fig. 4d), indicating that the role of PRDX6 is conserved across many cancer types.
To confirm the involvement of PRDX6 in selenoprotein synthesis, we constructed cDNAs encoding model selenoproteins harboring an in-frame UGA codon at C70 or S175 of GFP (C70U and S175U), as well as the 3’UTR SECIS element of GPX4 (Fig. 3d). As expected, loss of phosphoseryl-tRNA kinase (PSTK) or SEPHS2, both of which are essential for synthesis of Sec-tRNA (ref. 38), greatly reduced expression of both GFP C70U and S175U (Fig. 3d). Loss of PRDX6 also reduced expression of both model selenoproteins (Fig. 3d), whereas loss of PRDX6 did not affect expression of GFP WT (Extended Data Fig. 4e). These results clearly indicate that PRDX6 has a crucial role in Sec insertion into the UGA codon.
PRDX6 augments efficient utilization of selenium
Next, we explored the role of PRDX6 in selenoprotein synthesis. Both inorganic and organic forms of selenium can be used to synthesize Sec-tRNA (Extended Data Fig. 5a). Inorganic selenite is reduced by GSH and the thioredoxin system, whereas organic Sec is decomposed by selenocysteine lyase (SCLY), resulting in generation of the common active selenium donor selenide, which is important for Sec-tRNA synthesis (Extended Data Fig. 5a) . Addition of excess selenite or selenocystine ((Sec)2) to cultures of WT cells increased selenoprotein expression markedly, suggesting that the supply of selenium is limited under normal culture conditions (Fig. 4a). Although either selenium source failed to induce expression of selenoproteins in cells lacking PSTK or SEPHS2 (Fig. 4a), both of which are essential for Sec-tRNA synthesis , we found that addition of excess selenium increased the amount of selenoproteins in PRDX6 KO cells, regardless of the selenium source (Fig. 4a). Addition of selenite or (Sec) also protected MEFs lacking PRDX6 from iron-triggered and canonical ferroptosis (Fig. 4b,c and Extended Data Fig. 5b,c). Furthermore, addition of selenium increased selenoprotein expression and suppressed iron-triggered ferroptosis in HeLa cells lacking PRDX6 (Extended Data Fig. 5d,e). These results show that addition of excess selenium can compensate for loss of PRDX6, raising the possibility that selenium is not used efficiently by PRDX6 KO cells. Indeed, when selenite or was added to cultures in a dose-dependent or time-dependent manner, we found that the incremental increases in selenoprotein levels were much less efficient in PRDX6KO than in WT cells (Fig. 4d,e and Extended Data Fig. 5f,g). PRDX6 is also involved in efficient utilization of selenium after treatment with SELENOP, a more physiological source of selenium (Fig. 4f). Therefore, to more rigorously evaluate the hypothesis that PRDX6 is involved in the efficiency of selenium utilization, we adjusted the initial level of selenoproteins in WT and PRDX6 KO cells and then added selenium; this is because initial expression of selenoproteins was greatly reduced in PRDX6 KO cells (Fig. 3b). We found that expression of selenoproteins in WT cells cultivated in medium with a reduced concentration of fetal bovine serum (FBS) fell markedly; FBS is the sole source of selenium in this culture system (Extended Data Fig. 5h). Next, we added selenium to culture medium containing 1% FBS and found that selenoprotein synthesis was much more efficient in WT cells than in KO cells (Extended Data Fig. 5h). Furthermore, even when PRDX6 KO cells were pretreated with selenium and then fed additional selenium, selenoprotein synthesis was lower in PRDX6 KO cells than in WT cells (Extended Data Fig. 5i). These results strongly support our hypothesis that PRDX6 is involved in efficient utilization of selenium for Sec-tRNA synthesis. Next, to evaluate the amount of Sec-tRNA in cells, we established a highly sensitive in vitro reconstructive system for selenoprotein synthesis by modifying a Sec UGA readthrough luciferase reporter assay . Given that wheat germ lacks the Sec incorporation machinery , eukaryotic elongation factor Sec-tRNA -specific (eEFSec) and SECIS binding protein 2 (SBP2) were added to the wheat germ lysate together with a Sec-tRNA source to establish the reconstitution system (Fig. 4g). Addition of aminoacyl-tRNAs (aa-tRNAs) purified from WT or SEPHS2 KO cells (the former, but not the latter, contains Sec-tRNA ) revealed that aa-tRNAs from WT cells increased selenoprotein synthesis in the presence of eEFSec and SBP2, as evaluated by measuring luciferase luminescence (Fig. 4h). Moreover, aa-tRNAs from WT cells cultivated with further enhanced selenoprotein synthesis. However, aa-tRNAs purified from MEFs lacking SEPHS2 did not show increased synthesis, even when KO cells were cultivated with (Sec) (Fig. 4h), confirming the validity of the system for evaluating the amount of Sec-tRNA in aa-tRNA sources. Next, we purified aa-tRNAs from WT or PRDX6 KO cells cultured with or without (Sec) (Fig. 4i). Purified aa-tRNAs from PRDX6 KO cells cultivated in the absence of failed to increase selenoprotein synthesis, and (Sec) treatment increased it only slightly. These results clearly indicate that PRDX6 is involved in the efficient use of selenium for Sec-tRNA synthesis.
Fig. 3 | PRDX6 is involved in selenoprotein synthesis. a, Co-essentiality network analysis of PRDX6 using FIREWORKS. b, Immunoblot analysis of lysates from cells expressing sgEmpty, sgControl or sgRNAs targeting PRDX6. Data are representative of three independent experiments. c, Immunoblot analysis of lysates from control or PRDX6 KO cells stably expressing the PRDX6 WT or C47S mutant. Data are representative of three independent experiments.
d, Schematic showing the constructs of model selenoproteins (left). Immunoblot analysis of lysates from cells stably expressing Myc-GFP C70U or S175U in control cells or cells in which the indicated genes have been knocked out (middle). Quantification of the abundance of selenoproteins (right). Data are presented as the mean s.d. of three independent experiments. ; **** ; one-way ANOVA.
PRDX6 augments selenium utilization as a selenide carrier
Next, we explored the mechanism(s) by which PRDX6 increases the efficiency of selenium utilization for Sec-tRNA synthesis. First, we overexpressed enzymes involved in Sec-tRNA synthesis in PRDX6 KO cells (Extended Data Fig. 5a) and found that overexpression (with the exception of SEPHS2 and PRDX6) failed to restore GPX4 expression (Fig. 5a). Thus, SEPHS2, a selenoprotein that phosphorylates selenide for Sec-tRNA Sec synthesis , appears to have a critical role in GPX4 expression in PRDX6 KO cells. Previous studies have shown that a SEPHS2 mutant in which Sec is replaced by cysteine (SEPHS2 U/C) rescued the SEPHS2-deficient phenotype (Extended Data Fig. 6a). Therefore, we overexpressed SEPHS2U/C or WT in PRDX6 KO cells and found that SEPHS2WT, but not the U/C mutant, restored GPX4 expression markedly (Extended Data Fig. 6b). However, we found that overexpression of SEPHS2 WT in PRDX6 KO failed to fully restore expression of other selenoproteins such as SELN and GPX1 (Extended Data Fig. 6b). We additionally found that overexpression of SEPHS2 WT also restored expression of model selenoproteins (Extended Data Fig. 6c) and protected cells from iron-triggered and canonical ferroptosis (Extended Data Fig. 6d-f). These results raise the possibility that loss of PRDX6 suppresses the expression of selenoproteins through a reduction of SEPHS2, not by facilitating utilization of selenium. To rule out this possibility, we expressed SEPHS2 WT at endogenous levels in PRDX6 KO cells because the amount of SEPHS2 in overexpressing cells was much higher than that in cells expressing endogenous SEPHS2 (Fig. 5a). We found that the level of endogenous SEPHS2 WT failed to reverse loss of selenoprotein expression in PRDX6 KO cells (Fig. 5b) and did not suppress iron-triggered and canonical ferroptosis (Extended Data Fig. 6d-f). In addition, when PRDX6 KO cells were pretreated with selenite or to adjust SEPHS2 expression in KO cells to levels comparable to those in WT cells before the time course experiment, we observed that WT cells utilized selenium more efficiently than PRDX6 KO cells
(Extended Data Fig. 5i). These results show clearly that PRDX6 is necessary for effective utilization of selenium to generate Sec-tRNA in cells expressing endogenous levels of SEPHS2. Importantly, we also found that Sec-tRNA levels were still lower in PRDX6 KO cells overexpressing SEPHS2 than in WT cells (Fig. 5c), suggesting that overexpression of SEPHS2 cannot compensate fully for the reduced efficiency of selenium utilization owing to loss of PRDX6. Collectively, the data suggest that excess selenium as well as overexpression of SEPHS2 WT, but not the U/C mutant, compensates for the loss of PRDX6, albeit not very effectively. Thus, these results imply that SEPHS2 itself can acquire selenium through the Sec residue of SEPHS2; however, PRDX6 greatly facilitates the utilization of selenium by SEPHS2.
Owing to its high reactivity, it was thought that selenide must associate with an unidentified carrier protein through a cysteine residue for efficient transfer to SEPHS2 (refs. 11,16). Addition of excess (Sec) was toxic to cells; however, we found that PRDX6, in which C47 is critical, suppresses (Sec) toxicity (Extended Data Fig. 6g). Considering that C47 of PRDX6 also has a crucial role in the efficient synthesis of selenoproteins (Figs. 2f and 3c), it is highly likely that PRDX6 is a potential selenide carrier. A previous report showed that rhodanese forms perselenide (-S-SeH) at cysteine residues in the presence of selenite and GSH . Therefore, we incubated recombinant PRDX6 WT or C47S proteins with selenite and GSH, followed by the detection of PRDX6-bound selenium by inductively coupled plasma mass spectrometry (ICP-MS) (Extended Data Fig. 6h). We found that PRDX6 WT binds to selenium more effectively than the C47S mutant. Next, we performed mass spectrometry analyses to further characterize the PRDX6-selenium intermediate. The results showed that C47 of PRDX6 forms a perselenide bond when reacting with GSH and selenite, as well as with SCLY and Sec (Extended Data Fig. 7). Moreover, we found that about 10% of PRDX6 C47 formed a perselenide bond in a 5 min reaction with selenite and GSH, or with SCLY and Sec (Fig. 5d,e). Selenide carrier proteins
Fig. 4 | PRDX6 augments efficient selenium utilization. a, Immunoblot analysis of lysates from control or the indicated KO cells cultured for 32 h in the presence of sodium selenite or . Data are representative of two independent experiments. b,c, WT or PRDX6 KO cells were pretreated for 2 days with or sodium selenite . Then, cells were treated for 48 h with FAC in the presence of or sodium selenite ( 100 nM ) (c), and viability was measured. Viability data are presented as the mean s.d. of three biological replicates. d-f, Time course of selenoprotein expression by WT or PRDX6 KO MEFs in the presence of sodium selenite (e) or SELENOP (f). Data are representative of two
(in f) or three (in d and e) independent experiments. g, Schematic showing the in vitro reconstruction system used to evaluate the amount of Sec-tRNA . h,i, aa-tRNAs, which were purified from the indicated cells treated (or not) for 105 min with , followed by treatment for 15 min with cycloheximide, were added to wheat germ extract in the presence or absence of eEFSec and SBP2. Data of aa-tRNAs from WT or SEPHS2 KO cells (h), and WT or PRDX6 KO cells (i) are shown. Data are presented as the mean s.d. of three independent experiments. (WT eEFSec and SBP2, ; PRDX6 WT versus KO, ); ; two-way ANOVA.
should receive selenide from SCLY and then transfer it to SEPHS2 for phosphorylation (Extended Data Fig. 5a). Proximity ligation assays (PLAs) revealed that endogenous PRDX6 bound to both SCLY and SEPHS2 effectively (Fig. 5f and Extended Data Fig. 8a). To confirm binding of PRDX6 to SCLY and SEPHS2, we expressed HA-TurbolD-PRDX6 or HA-TurbolD-PRDX1 (a paralog of PRDX6) in WT or PRDX6 KO MEFs. As shown in Fig. 5 g and Extended Data Fig. 8b,c,PRDX6 bound specifically to SEPHS2 and SCLY.
Selenide is delivered to and phosphorylated by SEPHS2 to generate selenophosphate for Sec-tRNA synthesis . Given that selenophosphate is an unstable product , we evaluated the production of AMP, a by-product of selenophosphate synthesis, to evaluate the acceleration of selenophosphate synthesis by PRDX6. We used a recombinant SEPHS2 U/C protein that retains enzyme activity for selenophosphate synthesis instead of SEPHS2 WT because expression of SEPHS2 WT is extremely low (Fig. 5a) and it is difficult to purify. In the selenophosphate synthesis reaction in which SCLY-mediated decomposition of Sec is used as a selenium source, addition of PRDX6 WT efficiently increased the synthesis of
selenophosphate, whereas C47S did not (Fig. 5h). When PRDX6 WT or C47S was preincubated with selenide to generate the PRDX6-selenide complex as a selenium source, PRDX6 WT accelerated SEPHS2 U/C-mediated production of AMP (Fig. 5i). Collectively, these data suggest that PRDX6 facilitates Sec-tRNA synthesis by functioning as a selenide carrier protein (Fig. 5j).
PRDX6 and cancer
Most selenoproteins have a role in maintaining redox balance by reducing oxidative insults, and high expression of selenoproteins is associated with malignancy grade . Consistent with the relationship between selenoproteins and cancer, database analyses revealed that expression of PRDX6 is higher in cancerous tissues than in normal tissues (Extended Data Fig. 9a) and that high expression of PRDX6 correlates with a poor prognosis for various cancers (Extended Data Fig. 9b). A previous report showed that intractable pancreatic cancer cells are sensitive to ferroptosis . Database analysis also revealed that expression of PRDX6 is high in pancreatic cancer and that high expression correlates with a poor prognosis (Extended Data Fig. 9).
Fig. 5 | PRDX6 is a selenide carrier protein that enables efficient selenium utilization. a, Immunoblot analysis of lysates from control or PRDX6 KO cells stably expressing enzymes involved in Sec-tRNA synthesis. Data are representative of three independent experiments. b, Immunoblot analysis of lysates from control or PRDX6 KO cells stably expressing SEPHS2 WT at endogenous levels. Data are representative of three independent experiments. c, aa-tRNAs, purified from the indicated cells treated with cycloheximide for 15 min , were added to wheat germ extract in the presence of eEFSec and SBP2. Data are expressed as the mean s.d. of three independent experiments. (SEPHS2 U/C versus SEPHS2 WT O.E., ; SEPHS2 WT O.E. versus SEPHS2 WT, ); NS ( ); one-way ANOVA. d,e, Percentage of C47 modification as calculated by mass spectrometry analysis. PRDX6 was incubated with GSH and sodium selenite (d), or with Sec and SCLY (e) for 5 min before mass spectrometry analyses. Data are presented as the mean s.d of three biological replicates. , The physical association
between PRDX6 and SEPHS2 was detected in a PLA. Scale bars, . Data are representative of three independent experiments. Quantification of the PLA dots is also shown. Data are presented as the mean s.d. ( cells (empty-PRDX6), cells (SEPHS2-PRDX6) examined), ; unpaired two-sided -test. g, MEFs expressing HA-TurbolD-empty, -PRDX1 or -PRDX6 were cultured for 30 min with DMSO or biotin. Cell lysates and pulldown samples were analyzed by immunoblotting. Data are representative of two independent experiments. h,i, PRDX6-mediated enhancement of selenophosphate-mediated synthesis of SEPHS2 was evaluated by measuring the AMP product. Data are presented as the mean s.d. of three independent experiments in which selenocysteine and SCLY were used as the selenium source; ; ; one-way ANOVA (h) or selenium bound to PRDX6 was used as the selenium source;** ( ); ( ); one-way ANOVA (i).j, Schematic showing the role of PRDX6 as a selenide carrier.
We found that loss of PRDX6 from two pancreatic cancer cell lines suppressed growth, a phenomenon reversed by the addition of selenium or liproxstatin-1 (Extended Data Fig. 10a-d). To consolidate our findings, we obtained several cancer cells and evaluated their sensitivity to iron-triggered ferroptosis (Extended Data Fig. 10e,f). The results showed that MYC-N-amplified neuroblastoma cells (SK-N-DZ and NB-1) were sensitive to iron-induced ferroptosis (Extended Data Fig. 10e,f). We also found that loss of PRDX6 from SK-N-DZ cells suppressed their growth markedly (Extended Data Fig.10g,h). A previous study reported that MYC-N increases intracellular iron by upregulating the expression of TFRC, a protein required for iron uptake . In addition, cancer cells are generally addicted to iron, which drives proliferation. Thus, inhibiting PRDX6 may be an effective strategy for sensitizing cancer cells specifically to iron-triggered ferroptosis.
Discussion
Here, we developed a ferroptosis induction system that can kill cells upon addition of iron to the culture medium (Fig. 1c,d). We used this system to conduct a genome-wide CRISPR screen and identified PRDX6 as a selenoprotein synthesis factor. Although PRDX6 has long been considered an antioxidant enzyme , we show here that it exerts antioxidant effects indirectly by facilitating the expression of selenoproteins. Although organic and inorganic selenium are metabolized to selenide, which is then used for selenoprotein synthesis , the metabolic pathway has not yet been shown conclusively. Our results indicate that PRDX6 is an unidentified selenide carrier protein that facilitates the efficient utilization of selenide by SEPHS2 (Figs. 4 and 5).
Among the PRDX family, which comprises six proteins (PRDX1PRDX6), the two-cysteine proteins PRDX1-PRDX5 harbor a disulfide
bond between the two conserved cysteine residues, which is reduced by the thioredoxin system. The one-cysteine protein PRDX6 is unique in that it is thought to be reduced by GSH and to have GPX activity . However, we showed conclusively in this study that PRDX6 does not possess GPX activity (Fig. 2g and Extended Data Fig. 3g). Instead, we found that a C47S mutant of PRDX6 profoundly reduces the amount of selenoproteins, including GPX1 and GPX4 (Fig. 3c), both of which are well-known proteins that exhibit GPX activity. Given that the GPX function of PRDX6 has been evaluated mainly using PRDX6 knockdown or KO cells , the reduced GPX activity in PRDX6-deficient cells might be caused by a reduction in GPX1 and GPX4. Consistent with our results (Fig. 2 g and Extended Data Fig. 3 g ), other reports used purified proteins to show that PRDX6 has no GPX activity . Therefore, we conclude that PRDX6 is not an antioxidant enzyme like PRDX1-PRDX5 but it does function as a selenide carrier protein. This was confirmed by our finding that PRDX1, an antioxidant enzyme that is considered to be a paralog of PRDX6, did not restore GPX4 expression in PRDX6 KO MEFs (Extended Data Fig. 8b).
Both inorganic and organic selenium function as sources for SEPHS2 to generate selenoproteins . Although the mechanism underlying delivery of selenium to SPEHS2 remains unknown, we clearly showed that PRDX6 facilitates the synthesis of selenoproteins (Fig. 4d-f and Extended Data Fig. 5f-i) and forms a perselenide bond at the C47 of both selenium sources (Fig. 5d,e and Extended Data Fig. 7). We also found that PRDX6 binds to both SEPHS2 and SCLY; the latter is an enzyme that releases selenide from Sec. Thus, PRDX6 appears to have a crucial role in delivering selenide from organic selenium by accepting selenide from SCLY and delivering it to SEPHS2. Although the mechanisms underlying the metabolism of inorganic selenium and its delivery to SEPHS2 remain unknown, our results strongly suggest that PRDX6 also has a role in the delivery of selenide to SEPHS2, even from inorganic selenium. Therefore, the identification of PRDX6 as a selenium carrier protein opens new avenues of research into intracellular selenium dynamics.
It is noteworthy that overexpression of SEPHS2 WT substantially restored the expression of selenoproteins in PRDX6 KO cells (Fig. 5a and Extended Data Fig. 6b,c). These observations imply that SEPHS2 can bind directly to selenide to produce selenophosphate in the absence of PRDX6, albeit much less efficiently; indeed, endogenous levels of SEPHS2 failed to restore selenoprotein expression. Given that the SEPHS2 U/C mutant could not restore expression of selenoproteins in PRDX6 KO cells (Extended Data Fig. 6b,c), the Sec residue in SEPHS2 seems to facilitate utilization of selenide, although the precise molecular mechanism remains unsolved. The Sec residue and C47 of PRDX6 have a low and high reactivity ; therefore, we suspect that highly reactive residues may be important for the reaction with selenide in cells.
We also observed that addition of excess selenium (both organic and inorganic forms) restored the amount of selenoproteins in PRDX6 KO cells (Fig. 4a-c). Therefore, the ability of SEPHS2, which reacts directly with selenide to induce the synthesis of some selenoproteins, may underlie the finding of trace amounts of selenoproteins in PRDX6 KO cells. These observations appear to be consistent with differences in the phenotypes of PRDX6 KO mice and mice lacking selenoprotein synthesis . Two lines of PRDX6 KO mice have been reported . Both are viable and fertile, and neither shows an overt phenotype, although they are hyper-sensitive to various oxidative stresses. By contrast, mice lacking selenoprotein synthesis are embryonic lethal . The observation that loss of PRDX6 does not completely attenuate selenoprotein synthesis may be advantageous with respect to the development of PRDX6 inhibitors as anti-cancer drugs. Effective inducers of ferroptosis are believed to be a promising strategy for anti-cancer therapy, and GPX4 is regarded as a suitable target . However, inhibition of GPX4 may have severe side effects because GPX4 KO mice are embryonic lethal . Taken together, the present results suggest that PRDX6 may be an alternative target for anti-cancer drugs, with fewer side effects.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41594-024-01329-z.
References
Jiang, X., Stockwell, B. R. & Conrad, M. Ferroptosis: mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 22, 266-282 (2021).
Lei, G., Zhuang, L. & Gan, B. Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 22, 381-396 (2022).
Yang, W. S. et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317-331 (2014).
Ingold, I. et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell 172, 409-422.e21 (2018).
Friedmann Angeli, J. P. et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 16, 1180-1191 (2014).
Doll, S. et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693-698 (2019).
Bersuker, K. et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688-692 (2019).
Davis, C. D., Tsuji, P. A. & Milner, J. A. Selenoproteins and cancer prevention. Annu. Rev. Nutr. 32, 73-95 (2012).
Zhang, Y. et al. Role of selenoproteins in redox regulation of signaling and the antioxidant system: a review. Antioxidants 9, 383 (2020).
Tsuji, P. A., Santesmasses, D., Lee, B. J., Gladyshev, V. N. & Hatfield, D. L. Historical roles of selenium and selenoproteins in health and development: the good, the bad and the ugly. Int. J. Mol. Sci. 23, 5 (2021).
Manta, B., Makarova, N. E. & Mariotti, M. The selenophosphate synthetase family: a review. Free Radic. Biol. Med. 192, 63-76 (2022).
Veres, Z. et al. Synthesis of 5-methylaminomethyl-2-selenouridine in tRNAs: 31P NMR studies show the labile selenium donor synthesized by the seld gene product contains selenium bonded to phosphorus. Proc. Natl Acad. Sci. USA 89, 2975-2979 (1992).
Veres, Z., Kim, I. Y., Scholz, T. D. & Stadtman, T. C. Selenophosphate synthetase. Enzyme properties and catalytic reaction. J. Biol. Chem. 269, 10597-10603 (1994).
Kumaraswamy, E. et al. Selective removal of the selenocysteine tRNA gene (Trsp) in mouse mammary epithelium. Mol. Cell. Biol. 23, 1477-1488 (2003).
Seeher, S. et al. Secisbp2 is essential for embryonic development and enhances selenoprotein expression. Antioxid. Redox Signal. 21, 835-849 (2014).
Tobe, R. & Mihara, H. Delivery of selenium to selenophosphate synthetase for selenoprotein biosynthesis. Biochim. Biophys. Acta Gen. Subj. 1862, 2433-2440 (2018).
Alborzinia, H. et al. LRP8-mediated selenocysteine uptake is a targetable vulnerability in MYCN-amplified neuroblastoma. EMBO Mol. Med. 15, e18014 (2023).
Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060-1072 (2012).
Salahudeen, A. A. et al. An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science 326, 722-726 (2009).
Vashisht, A. A. et al. Control of iron homeostasis by an iron-regulated ubiquitin ligase. Science 326, 718-721 (2009).
Hentze, M. W., Muckenthaler, M. U., Galy, B. & Camaschella, C. Two to tango: regulation of mammalian iron metabolism. Cell 142, 24-38 (2010).
Iwai, K. Regulation of cellular iron metabolism: iron-dependent degradation of IRP by SCF ubiquitin ligase. Free Radic. Biol. Med. 133, 64-68 (2019).
Hirayama, T., Niwa, M., Hirosawa, S. & Nagasawa, H. High-throughput screening for the discovery of iron homeostasis modulators using an extremely sensitive fluorescent probe. ACS Sens. 5, 2950-2958 (2020).
Moroishi, T., Nishiyama, M., Takeda, Y., Iwai, K. & Nakayama, K. I. The FBXL5-IRP2 axis is integral to control of iron metabolism in vivo. Cell Metab. 14, 339-351 (2011).
Schneider, L. S. et al. Vacuolar-ATPase inhibition blocks iron metabolism to mediate therapeutic effects in breast cancer. Cancer Res. 75, 2863-2874 (2015).
Dixon, S. J. et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem. Biol. 10, 1604-1609 (2015).
Doll, S. et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 13, 91-98 (2017).
Zhang, Y. et al. Imidazole ketone erastin induces ferroptosis and slows tumor growth in a mouse lymphoma model. Cell Chem. Biol. 26, 623-633.e9 (2019).
Bolduc, J. et al. Peroxiredoxins wear many hats: factors that fashion their peroxide sensing personalities. Redox Biol. 42, 101959 (2021).
Perkins, A., Nelson, K. J., Parsonage, D., Poole, L. B. & Karplus, P. A. Peroxiredoxins: guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem. Sci. 40, 435-445 (2015).
Fisher, A. B. Peroxiredoxin 6: a bifunctional enzyme with glutathione peroxidase and phospholipase activities. Antioxid. Redox Signal. 15, 831-844 (2011).
Fisher, A. B. et al. A novel lysophosphatidylcholine acyl transferase activity is expressed by peroxiredoxin 6. J. Lipid Res. 57, 587-596 (2016).
Fisher, A. B., Dodia, C., Manevich, Y., Chen, J. W. & Feinstein, S. I. Phospholipid hydroperoxides are substrates for non-selenium glutathione peroxidase. J. Biol. Chem. 274, 21326-21334 (1999).
Kang, S. W., Baines, I. C. & Rhee, S. G. Characterization of a mammalian peroxiredoxin that contains one conserved cysteine. J. Biol. Chem. 273, 6303-6311 (1998).
Peshenko, I. V. et al. Identification of a 28 kDa secretory protein from rat olfactory epithelium as a thiol-specific antioxidant. Free Radic. Biol. Med. 25, 654-659 (1998).
Peshenko, I. V., Singh, A. K. & Shichi, H. Bovine eye 1-Cys peroxiredoxin: expression in E. coli and antioxidant properties. J. Ocul. Pharmacol. Ther. 17, 93-99 (2001).
Amici, D. R. et al. FIREWORKS: a bottom-up approach to integrative coessentiality network analysis. Life Sci. Alliance 4, e202000882 (2021).
Kang, D. et al. The role of selenium metabolism and selenoproteins in cartilage homeostasis and arthropathies. Exp. Mol. Med. 52, 1198-1208 (2020).
Saito, Y. Selenium transport mechanism via selenoprotein P-its physiological role and related diseases. Front. Nutr. 8, 685517 (2021).
Dobosz-Bartoszek, M. et al. Crystal structures of the human elongation factor eEFSec suggest a non-canonical mechanism for selenocysteine incorporation. Nat. Commun. 7, 12941 (2016).
Shetty, S. P., Shah, R. & Copeland, P. R. Regulation of selenocysteine incorporation into the selenium transport protein, selenoprotein P. J. Biol. Chem. 289, 25317-25326 (2014).
Xu, X. M. et al. Selenophosphate synthetase 2 is essential for selenoprotein biosynthesis. Biochem. J. 404, 115-120 (2007).
Carlisle, A. E. et al. Selenium detoxification is required for cancer-cell survival. Nat. Metab. 2, 603-611 (2020).
Ogasawara, Y., Lacourciere, G. & Stadtman, T. C. Formation of a selenium-substituted rhodanese by reaction with selenite and glutathione: possible role of a protein perselenide in a selenium delivery system. Proc. Natl Acad. Sci. USA 98, 9494-9498 (2001).
Suzuki, N., Verdugo, M., Hatakeyama, T. & Ogra, Y. Structural analysis of chemically synthesized selenophosphate, a donor for selenocysteine biosynthesis. Metallomics Res. 1, 20-25 (2021).
Abe, K., Mihara, H., Nishijima, Y., Kurokawa, S. & Esaki, N. Functional analysis of two homologous mouse selenophosphate synthetases. Biomed. Res. Trace Elem. 19, 76-79 (2008).
Bartha, Á. & Györffy, B. TNMplot.com: a web tool for the comparison of gene expression in normal, tumor and metastatic tissues. Int. J. Mol. Sci. 22, 2622 (2021).
Nagy, Á., Munkácsy, G. & Győrffy, B. Pancancer survival analysis of cancer hallmark genes. Sci. Rep. 11, 6047 (2021).
Badgley, M. A. et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 368, 85-89 (2020).
Lu, Y. et al. MYCN mediates TFRC-dependent ferroptosis and reveals vulnerabilities in neuroblastoma. Cell Death Dis. 12, 511 (2021).
Liu, G. et al. Comparison of glutathione peroxidase 1 and peroxiredoxin 6 in protection against oxidative stress in the mouse lung. Free. Radic. Biol. Med. 49, 1172-1181 (2010).
Yun, H. M. et al. PRDX6 promotes lung tumor progression via its GPx and iPLA activities. Free. Radic. Biol. Med. 69, 367-376 (2014).
Chen, C. et al. Identification of peroxiredoxin 6 as a direct target of withangulatin A by quantitative chemical proteomics in non-small cell lung cancer. Redox Biol. 46, 102130 (2021).
Hall, A., Nelson, K., Poole, L. B. & Karplus, P. A. Structure-based insights into the catalytic power and conformational dexterity of peroxiredoxins. Antioxid. Redox Signal. 15, 795-815 (2011).
Wang, X. et al. Mice with targeted mutation of peroxiredoxin 6 develop normally but are susceptible to oxidative stress. J. Biol. Chem. 278, 25179-25190 (2003).
Mo, Y. et al. 1-Cys peroxiredoxin knock-out mice express mRNA but not protein for a highly related intronless gene. FEBS Lett. 555, 192-198 (2003).
Imai, H. et al. Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene. Biochem. Biophys. Res. Commun. 305, 278-286 (2003).
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons. org/licenses/by/4.0/.
(c) The Author(s) 2024
Methods
RT-PCR and plasmids
The open reading frames of mouse (m) PRDX6, PRDX1, SEPHS2, GPX4 (cytosolic form), SARS1, PSTK, SEPSECS, SCLY and FSP1, and human (h) PRDX6, SEPHS2, SCLY, eEFSec, SBP2 and SEPHS2-3’UTR(SECIS) were amplified by RT-PCR. Single-guide RNA (sgRNA) resistant constructs of mPRDX6 and mSEPHS2, and mutants of mPRDX6 (H26A, D31A, S32A, C47S and D140A), hPRDX6 C47S, mGPX4 U46C, mSEPHS2 U/C and hSEPHS2 U/C were generated by two-step PCR. GFP-3’SECIS (WT, C70U and S175U) was generated from the amplified open reading frames of GFP and the -SECIS of GPX4. DNAs were ligated into the appropriate epitope-tag sequences and then cloned into pMXs-IP, pMXs-IRES-Bsr, pMXs-neo and pT7-7 vectors. Luciferase C258U-3’SECIS was generated from the luciferase open reading frame and the -SECIS of GPX4 and then ligated into the pEU vector. CRISPR sgRNA sequences were selected from CHOPCHOP or Benchling. The oligonucleotide sequences preceding the protospacer motif were as follows:
mFBXL5, GAGGAAGATGGGTTATACCTG; mACSL4 #1, GGTTCTACGGGCCGCCCCAA; mACSL4 #2, GACCGATCACAATCTCACCTC; mLPCAT3 #1, GCCGGTGACTACGATATCAAGTGG; mLPCAT3 #2, GTAATAACGGCAGTGACGGTGCGG; mPDSS2 #1, GATGCCGGCTGTCGTGCACGA;mPDSS2#2,GCGGCATAACCTACAACTGCG;mFSP1#1, GCCAGCGCTCACAATTCATCG; mFSP1 #2,GCGCTCACAATTCATCGTGG; mPRDX6 #1, GATAGAACTATACCTTGCTCC; mPRDX6 #2, GCTCACCACACGGGCCGTCAC; mPSTK, GAAAGTCGACTTTCCGGCCGC; mSEPHS2,GAACCCGTGGATTATCATCGG;mMETAP1,GTGTACCGATAGC-CTGCCCA;mSNX27,GTTGACAATGCGCACGACCCG;mUBE2N,GTGGGGACCACTTATCTATGA;mCINP,GGCCCCCTCTATTTCACACG;mCENPE, GCGCTATTTATCAGAGCGATG, mSTX5A, GTATGCGACTCGATGATCCCG; mDDX10, GAGCGATTAAAGCTCCGCACC; mKLF5, GACCTGAGGACTCATACGGGT; mSCLY, GTAATGAGACCGGCGTCATCA; mGPX4, GACGATGCACACGAAACCCC; controlgRNA, GCGAGGTATTCGGCTCCGCG; hPRDX6, GATGCGGCCGACGGTGGTAT. Guide sequences were inserted into pX459 (ref.58) or lentiCRISPRv2 (ref.59) (puro, bsr or hygro).
The following reagents were also used: ferric ammonium citrate (Sigma-Aldrich, F5879), liproxstatin-1 (Selleck, S7699), bafilomycin A1 (Selleck, S1413), IKE (Selleck, S8877), RSL3 (Selleck, S8155), E64d (Peptide Institute, 4321-v), pepstatin A (Peptide Institute, 4397-v), sodium selenite (Sigma-Aldrich, 214485), selenocystine (Tokyo Chemical Industry, E1368), cycloheximide (ALBIOCHEM, 239764), hydrogen peroxide (Santoku Chemical Industry, 18412), tert-butyl hydroperoxide (Sigma-Aldrich, 458139), Tris(2-carboxyethyl)phosphine (TCEP) (Nacalai, 07277), pyridoxal-5-phosphate (PLP) (Sigma-Aldrich, P9255) and biotin (WAKO, 021-08712).
Cell lines and cell culture
MEFs were generated in-house. HepG2 was gifted by K. Nakajima (Osaka City University), originally purchased from ATCC (HepG2: HB-8065). HEK293T was gifted by E. Nakamura (Kyoto University), originally purchased from RIKRN RBC (293T: RCB2202). PLATE was gifted by T. Kitamura (Tokyo University). A549, H226, H460 and H1975 were gifted by A. Sato (Kyoto University), originally purchased from ATCC (A549: CCL-185, H226: CRL-5826, H460: HTB-177, H1975: CRL5908). SK-N-DZ and HeLa cells were purchased from ATCC (SK-N-DZ: CRL-2149, HeLa: CCL-2). PANC-1 and MIA Paca-2 cells were purchased from RIKEN RBC (PANC-1: RCB2095, MIA Paca-2: RCB2094). NB-1 cells were purchased from JCRB (NB-1:JCRB0621). All cells were cultured in a humidified incubator at and . MEFs, HEK293T, PLATE, A549, HeLa, PANC-1, MIA Paca-2 and HepG2 cells were grown in DMEM supplemented with FBS, penicillin and streptomycin. SK-N-DZ cells were grown in DMEM supplemented with FBS, non-essential amino acids (Gibco, 11140050), penicillin and streptomycin. GPX4, SEPHS2 and PSTK KO MEF cells were maintained in medium containing of liproxstatin-1. H226, H460 and H1975 cells were grown in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with FBS, penicillin and streptomycin. NB-1 cells were grown in RPMI and MEM medium supplemented with FBS, penicillin and streptomycin.
Generation of FBXL5 KO MEFs
To generate FBXL5 KO MEFs, a NEPA21 electroporator (NEPAGENE) was used to electroporate MEFs with the pX459 plasmid containing an sgRNA sequence specific for mFBXL 5 . Cells were then treated with puromycin for 2 days. Following selection, the cells were seeded at a low density and isolated colonies were picked. To identify FBXL5 KO cells, expression of FBXL5 was analyzed by immunoblotting.
Lentivirus production and generation of CRISPR-Cas9-mediated KO cell lines
Lentivirus was produced by co-transfecting HEK293T cells with the LentiCRISPRv2 (puro, bsr or hygro)-containing guide sequences, the psPAX2 packaging plasmid and the VSV-G envelope plasmid using PEI MAX (Polysciences) transfection reagent. On the following day, the medium was replaced with fresh medium. After 2 days of culture, lentivirus-containing supernatant was collected and used to infect target cells overnight in the presence of polybrene (Merck). Infected cells were selected with puromycin, blasticidin or hygromycin.
Retroviral expression
pMXs-IP, pMXs-neo or pMXs-IRES-Bsr containing appropriate inserts were transfected into PLATE packaging cells or GP2-293 cells along with the pVSV-G plasmid. The resultant viruses were used to infect target cells in the presence of polybrene. Stably transduced cells were selected using puromycin, G-418 or blasticidin.
Cell lysis and western blotting
Cells were washed with PBS and lysed with lysis buffer ( 50 mM Tris-HCl ( pH 7.5 ), Triton X-100, 1 mM PMSF , and protease inhibitor cocktail (Sigma-Aldrich)). The lysates were clarified by centrifugation at for 20 min at . Protein concentrations were determined using the Bradford assay (Nacalai Tesque), and equal amounts of protein were mixed with SDS sample buffer. Samples were heated for 5 min at , separated by SDS-PAGE and transferred onto PVDF membranes (Millipore). After blocking in Tris-buffered saline (TBS) containing Tween-20 and nonfat dry milk, the membrane was incubated with the appropriate primary antibodies followed by appropriate secondary antibodies. The membranes were visualized using enhanced chemiluminescence and analyzed on a LAS4000mini or LAS3000 instrument (GE Healthcare).
Cell viability assay
Cells were plated in 96-well plates (in triplicate and at a density of cells per well) in the presence or absence of FAC, RSL3 or IKE. After culture for of Cell Counting Kit-8 (DOJINDO) was added for 1-2 h. Absorbance at 450 nm was measured using a SpectraMax M5 microplate reader (Molecular Devices). Cell viability, monitored continuously using the iCELLigence or xCELLigence system (ACEA Bioscience), was expressed as an impedance-based cell index. GPX4 KO cells (4,500 cells per well) were plated onto an E-Plate L8 PET (ACEA Bioscience), and pancreatic ductal adenocarcinoma cells (3,000 cells per well) and SK-N-DZ cells (6,000 cells per well) were plated on an E-Plate16 PET (ACEA Bioscience). SK-N-DZ cells were cultured in DMEM supplemented with FBS, non-essential amino acids and 0.25 mM l-glutamine (Fujifilm). The cell index was monitored continuously. Recording data were analyzed by RTCA software lite v.2.2.1.
Cell staining using BODIPY 581/591 C11, and iron staining
For BODIPY 581/591 C11 (Invitrogen) staining, cells were plated in a six-well plate and treated with FAC. After , the medium was removed and the cells were labeled with DMEM containing BODIPY at for 30 min . Cells were then washed three times with PBS and detached from the plate using trypsin. Initial cell population gating (FSC-Area versus FSC-height) was used to ensure doublet exclusion, and green fluorescence was measured by using FACS CantolI (BD Biosciences) and FACS Diva software v.6.1.2 (Becton Dickinson). Data were analyzed using FlowJo software(v.9.9.6). For iron staining, cells were plated in a glass bottom dish and treated with FAC for 4 h . Cells were then washed three times with HBSS and labeled at for 30 min with FerroOrange (DOJINDO) followed by visualization under a Fv1000 confocal microscope(Olympus).
Genome-wide CRISPR screening
FBXL5 KO MEFs were infected with the mouse GeCKO v. 2 library at a multiplicity of infection of 0.3 and selected with puromycin for 1 week post infection. The cells were then treated with FAC for 48 h and cultured for a further 24 h in fresh medium. The cells were lysed in NTE buffer ( 15 mM Tris- HCl ( pH 7.5 ), 150 mM NaCl and 1 mM EDTA), and genomic DNA from non-treated and FAC-treated cells was prepared by phenol-chloroform extraction and isopropanol precipitation. sgRNA sequences were amplified from genomic DNA by PCR using Herculase II Fusion DNA polymerase. The resultant amplicons were gel-extracted and subjected to DNA sequencing on a Novaseq 6000 (Illumina) sequencer. Sequence data were analyzed using the MAGeCK pipeline.
PLA
The PLA was conducted using the Proximity Ligation Kit (SigmaAldrich). A549 cells, either empty or expressing Myc-SEPHS2 or Myc-SCLY, were cultured on micro cover glass slips in a 6-well plate. Cells were washed with PBS and fixed for 20 min with formaldehyde in PBS at room temperature ( ). Cells were then washed twice with PBS and permeabilized with Triton X-100 in PBS at room temperature for 10 min . Cells were washed twice with PBS and then incubated at for 1 h with Duolink blocking solution. Cells were then incubated at room temperature for 1 h with primary antibodies targeting PRDX6 (rabbit polyclonal; 1:100) and Myc (mouse monoclonal; 1:200). Cells were washed twice for 5 min with Duolink wash buffer A at room temperature and then incubated with a secondary antibody (anti-mouse minus and anti-rabbit plus) at for 1 h . Cells were washed twice at room temperature for 5 min with Duolink wash buffer A and then incubated with ligase solution at for 30 min . Next, the cells were washed twice with Duolink wash buffer A and incubated with polymerase in amplification buffer at for 100 min . Cells were washed twice at room temperature for 10 min with Duolink wash buffer B, followed by Duolink wash buffer B for 1 min . Finally, the cover
glasses were mounted with Duolink PLA mounting medium with DAPI. Protein-protein interactions were visualized under an Fv1000 confocal microscope(Olympus). PLA foci were counted by ImageJ (v.2.3.0).
TurboID pulldown
Cells were treated (or not) for 30 min with biotin, washed with PBS and lysed with lysis buffer ( 50 mM Tris- HCl ( pH 8.0 ), 150 mM NaCl , Triton X-100 and 1 mM PMSF). The lysates were clarified by centrifugation at for 20 min at . Protein concentrations were determined using the Bradford assay, and equal amounts of protein and Streptavidin Sepharose (Cytiva) were incubated overnight at by rotation. The beads were washed three times with lysis buffer and one time with PBS. Proteins were eluted from beads using SDS sample buffer supplemented with 2 mM biotin and then analyzed by western blotting.
Protein purification
His-TEV-PRDX6 WT or C47S, His-TEV-hSCLY, hSEPHS2-His, mouse His-TEV-eEFSec and His-TEV-SBP2 were expressed in E. coli strain BL21-CodonPlus (DE3)-RIPL (Agilent Technologies). Expression of His-TEV-PRDX6 WT or C47S was induced by the addition of 0.2 mM IPTG, and culture was continued at for 3 h . Cells were collected by centrifugation and frozen rapidly. Subsequently, cells were resuspended at for 30 min in buffer containing 50 mM Tris- , -mercaptoethanol (Nacalai Tesque), lysozyme chloride (Nacalai Tesque), DNase (Roche), 2 mM PMSF and a protease inhibitor cocktail (Roche), followed by lysis at for 20 min in the presence of Triton X-100. Insoluble material was removed by centrifugation at for 20 min at . His -TEV PRDX6 WT or C47S were purified from the supernatant using Ni-NTA beads (QIAGEN). The eluted samples were incubated for 30 min at with 10 mM dithiothreitol (DTT) and then desalted in 20 mM Tris- HCl (pH 7.5) buffer on a PD10 column.
Expression of His-TEV-hSCLY and hSEPHS2-His was induced by the addition of 0.2 mM IPTG, and culture was continued overnight at . Cells were collected by centrifugation and frozen rapidly. Subsequently, the cells were resuspended in buffer containing 50 mM Tris- HCl ( pH 8.0 ), -mercaptoethanol, 2 mM PMSF and a protease inhibitor cocktail and then lysed by sonication at . Insoluble material was removed by centrifugation at for 20 min at . Proteins were purified using Ni-NTA beads. The eluted samples were desalted in 20 mM Tris- and 1 mM DTT buffer on a PD10 column.
Expression of His-TEV-eEFSec and His-TEV-SBP2 was induced by the addition of 0.3 mM IPTG, and culture was continued at for 4 h . Cells were collected by centrifugation and frozen rapidly. Subsequently, the cells were resuspended in buffer containing 20 mM Tris- HCl ( pH 8.0 ), imidazole, glycerol, 10 mM 2-mercaptoethanol, 1 mM PMSF and a protease inhibitor cocktail and then lysed by sonication at . Insoluble material was removed by centrifugation at for 20 min at . Proteins were purified using Ni-NTA beads. Eluted samples were desalted on a PD10 column. eEFsec was stored in buffer containing 20 mM Tris- NaCl and 1 mM DTT, and SBP2 was stored in buffer containing 10 mM Tris-HCl (pH 7.5), glycerol and 1 mM DTT .
GPX assay
GPX activity was measured using a GPX4 Inhibitor Screening Assay Kit (Cayman Chemical). First, of GPX4 protein (included in the kit) or of PRDX6 protein was dissolved in of GPX4 assay buffer (included in the kit). Then, lof the GSH/GSH reductase mix (included in the kit) was added and mixed. Next, of the NADPH solution (included in the kit) was added and mixed. Finally, of cumene hydroperoxide (included in the kit), hydrogen peroxide (final concentration, ) or tert-butyl hydroperoxide (final concentration, ) was added and mixed, and absorbance
at 340 nm was measured using SpectraMax M5 microplate reader (Molecular Devices).
Selenophosphate synthetase assay
A selenophosphate synthetase assay using SCLY and selenocysteine as a selenium source was performed in degassed buffer containing 50 mM Tris- HCl ( pH 7.0 ), ATP, DTT, SEPHS2-His, PRDX6 WT or C47S, SCLY, PLP (Sigma-Aldrich) and at for 45 min . The resultant AMP product was measured in an AMP-Glo assay (Promega) using a Nivo plate reader (PerkinElmer). A selenophosphate synthetase assay using selenide-bound PRDX6 as a selenium source was performed by incubating of PRDX6 WT or C47S with 2 mM sodium selenite and 20 mM DTT in degassed 20 mM Tris- HCl ( pH 7.5 ) buffer at room temperature for 30 min . Then, the solution was diluted 20 times in buffer containing 20 mM Tris- HCl ( pH 7.5 ), 150 mM NaCl and Triton X-100, and of Ni-NTA magnetic beads was added. After rotation at for 1 h , beads were washed three times and eluted with 300 mM imidazole in 20 mM Tris- HCl ( pH 7.5 ) buffer. The eluted samples were incubated with a buffer mixture containing 50 mM Tris- ATP, DTT and SEPHS2-His under a layer of mineral oil at for 30 min . The resultant AMP product was measured in an AMP-Glo assay.
RT-qPCR
RNA was isolated using the RNeasy Mini Kit (QIAGEN) and reverse transcribed using a high-capacity RNA-to-cDNA Kit (Applied Biosystems). RT-qPCR data were obtained by ABI ViiA7 Real-Time PCR system (Applied Biosystems) and analyzed by ViiA7 RUO Software v.1.2.3. RT-PCR of GPX4 was performed using the following primers:
mGPX4_Fwd, -CCTCTGCTGCAAGAGCCTCCC-3′ and mGPX4_Rev, -CTTATCCAGGCAGACCATGTGC- .Control primers were -mActin_Fwd, -ATGGATGACGATATCGCTC-3′ and -mActin_Rev, -GATTCCATACCCAGGAAGG- .
Isolation of aa-tRNAs
aa-tRNAs were isolated from WT, PRDX6KO and SEPHS2 KO MEF cells. In brief, cells were treated (or not) with for 105 min , followed by addition of cycloheximide for 15 min . Cells were then washed three times in PBS and detached using trypsin. After centrifugation at of DPEC water, 0.8 ml of Tbuffer ( 50 mM NaOAc , and 5 mM EDTA) and 2 ml of acid-phenol pH 4.2 (Nippon gene) were added to the cell pellets in that order. The solution was mixed briefly and centrifuged at for 5 min at . The aqueous phase was transferred to another tube and re-extracted with 2 ml of acid-phenol ( pH 4.2 ). RNA was precipitated with 2.5 volumes of ethanol, washed twice with ethanol and air-dried for 5 min . The aa-tRNA pellet was resuspended in 5 mM NaOAc buffer.
The luciferase reporter Sec UGA assay for Sec-tRNA evaluation
Each reaction ( ) contained wheat germ lysate ( ; Promega), amino acid mix, 8 U of RNasin Plus ribonuclease inhibitor (Promega), 52.5 mM potassium acetate, of luciferase reporter mRNA and of aa-tRNAs in the presence or absence of of eEFSec and of SBP2. Reactions were incubated at for 90 min . Luciferase activity was measured in a Lumat Luminometer (Berthold).
ICP-MS
The reaction ( ) contained sodium selenite, GSH and (or not) recombinant PRDX6 WT or C47S in degassed buffer containing 1 mM EDTA and 50 mM Tris- HCl ( pH 7.0 ). Reactions were incubated at for 5 min . Any selenium that did not bind to PRDX6 protein was removed using a NAP-5 column. The protein solution was
then concentrated with an Amicon 3 K concentrator. A small aliquot of the protein solution was put into a glass test tube, and the proteins were decomposed by addition of 0.1 ml of followed by heating at for 2 h . The digested samples were diluted with Milli-Q water, and the concentration of selenium was measured by ICP-MS (Agilent 8800 ICP-MS/MS; Agilent Technologies). The mass-shift mode was used to monitor the signal intensity of , with a dwell time of 100 ms . Selenium concentrations were measured from a standard calibration curve.
Mass spectrometry analysis
The reaction contained recombinant PRDX6 WT ( ), sodium selenite ( ) and GSH ( ) in degassed buffer containing 50 mM Tris- , or PRDX6 WT was pre-reduced with an equimolar amount of TCEP), SCLY ( ) and PLP ( ) in degassed buffer containing 50 mM Tris- HCl ( pH 7.0 ). Reactions were incubated at for 5 min . Then, PRDX6 was alkylated with 10 mM iodoacetamide in ProteaseMAX surfactant at for 10 min , followed by digestion with Trypsin Gold at for 3 h . Products were analyzed using liquid chromatography-electrospray ionization-quadrupole time-of-flight tandem mass spectrometry (LC-ESI-Q-TOF MS/MS). LC-ESI-Q-TOF analysis was performed using a 6545XT AdvanceBio LC-Q-TOF apparatus (Agilent Technologies) connected to the Agilent HPLC system. Analysis of the modifications to the active center cysteine (Cys47) was performed using Agilent MassHunter BioConfirm software v.10.0. The modification levels in DFTPVCTTELGR peptide, which includes the active center cysteine residue, were detected by monitoring at (for CysS-AM (AM, iodoacetamide adduct)), (for CysSSe-AM) and 862.3172 (for CysSSe-SG). The efficiency of trypsin-mediated protein digestion was normalized using the FHDFLGDSWGILFSHPR peptide ( ). The level of selenium modification in the fragment containing the active center cysteine was assessed by determining the relative ratio between the intensity of peptide-CysSSe-AM and that of peptide-CysS-AM. Samples of peptide-CysS-AM subjected to trypsin digestion followed by reduction with TCEP and subsequent alkylation with iodoacetamide were used to measure peptides corresponding to peptide-CysSSe-AM.
Statical analysis and reproducibility
Data are presented as the mean s.d. or mean s.e.m. GraphPad Prism9 v.9.4.0 was used to calculate values using Student’s -test, one-way ANOVA or two-way ANOVA, followed by Tukey’s multiple comparison test. The values are presented in the figure legends. All experiments were reproduced at least twice, each with similar results (the expression checks shown in Extended Data Figs. 1g and 3a were single experiments).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Boxplot data of differential gene expression in tumor and normal tissues were obtained from TNMplot (https://tnmplot.com/analysis). All data are available in the article and the supplementary information, and from the corresponding authors upon reasonable request. Source data are provided with this paper.
References
Ran, F. A. et al. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281-2308 (2013).
Sanjana, N. E., Shalem, O. & Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783-784 (2014).
Acknowledgements
We thank the members of the Iwai laboratory for helpful suggestions; Y. Saito (Tohoku University) and T. Toyama (Tohoku University) for kindly providing SELENOP; and F. Zhang (Massachusetts Institute of Technology) for providing the mouse GeCKO library and other CRISPR vectors. This work was supported by the Japan Society for the Promotion of Sciences (JSPS) KAKENHI: Grant Numbers 20H05505, 22H04810 and 22K06222 (to H.F.); by JSPS KAKENHI Grant Numbers 23K20040, 21H05263, 18H05277 and 22K19397 (to T.A.); by the Japan Science and Technology Agency CREST Grant Number JPMJCR2024 (to T.A.); by JSPS KAKENHI Grant Number 19 H 05772 (to Y.O.); and by JSPS KAKENHI Grant Number 24112002 and the Takeda Science Foundation (K.I.). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author contributions
H.F. and K.I. conceived and designed the project. H.F. performed most of the experiments. Y.T. performed ICP-MS analysis. S.O. performed mass spectrometry analysis. N.S., S.K., U.B., T.A. and Y.O. provided advice regarding experimental design. H.F. and K.I. wrote the manuscript, with contributions from all other authors.
Correspondence and requests for materials should be addressed to Hiroaki Fujita or Kazuhiro Iwai.
Peer review information Nature Structural & Molecular Biology thanks José Pedro Friedmann Angeli and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Dimitris Typas, in collaboration with the Nature Structural & Molecular Biology team. Peer reviewer reports are available.
Extended Data Fig. 1 | Characterization of iron-induced cell death in FBXL5
KO cells. a, Phase-contrast images of cells after treatment with FAC ( ) for 48 h . Scale bars, . Data are representative of three independent experiments. b, Fluorescence microscopy images of cells stained with a FerroOrange probe after treatment for 4 h with FAC ( ). Scale bars, . Data are representative of three independent experiments. c, Flow cytometry analysis of lipid hydroperoxidation levels in WT or FBXL5 KO cells by C11-BODIPY staining after treatment with FAC ( ) for 24 h . Data are representative of two independent experiments. d, WT MEFs were pretreated for 3 h with bafilomycin A1 ( 100 nM ), followed by FAC ( ) for the indicated
times. Cell lysates were analyzed by immunoblotting. Data are representative of two independent experiments. e, Viability of FBXL5 KO MEFs treated for 24 h with FAC ( ) in the presence of bafilomycin A1 ( ). Data are presented as the mean s.d. of three biological replicates. , Immunoblot analysis of lysates from the indicated cells. Data (f) are representative of two independent experiments. Expression check of (g) is a single experiment. , Volcano plots of iron-triggered ferroptosis screen, focusing on the mitochondrial electron transport chain (complex I and II: left, complex III and IV: right). The value and fold change were generated by the MAGeCK test.
a
b
SEPHS2 = Selenoprotein synthesis factor
MAGeCK rank (suppressor gene)
Rank
Gene name
p-value
Log Fold Change
1
METAP1
-7.382
2
PRDX6
2.20 E -07
-2.5705
3
PSTK
-1.4595
4
SNX27
2.20 E -07
-2.5145
5
UBE2N
2.86E-06
-2.8148
6
CINP
9.46E-06
-2.1364
7
SEPHS2
-3.9336
8
CENPE
2.44 E -05
-4.3363
9
STX5A
2.93E-05
-2.2028
10
DDX10
4.29E-05
-5.4515
11
KLF5
4.42E-05
-2.5021
12
SCLY
-1.2627
Selenoprotein synthesis factor
Extended Data Fig. 2 | Validation of hit genes, and identification of PRDX6 as a GPX4 regulator. a, Hit suppressor genes [ p -value cut off ( )] were examined by gene ontology analysis. The value was generated by the David gene ontology test.b, Negative score ranking of selenoproteins from the
screening list. c, MAGeCK rank of suppressor genes. The value and fold change were generated by the MAGeCK test. d, Immunoblot analysis of lysates from cells expressing sgRNAs targeting the indicated genes. Data are representative of two independent experiments.
Extended Data Fig. PRDX6 regulates ferroptosis by maintaining expression of GPX4. a, Viability of FBXL5 KO cells or FBXL5/PRDX6 double knockout cells treated for 24 h with FAC ( 50 or ), and immunoblot analysis of lysates from the indicated cells. Viability data are presented as the mean s.d. of three biological replicates.b, WT or PRDX6 KO MEFs were treated with FAC ( ) for the indicated times, and cell lysates were analyzed by immunoblotting. Data are representative of two independent experiments. c, Flow cytometry analysis of lipid hydroperoxidation levels in WT and PRDX6 KO cells, as assessed by C11-BODIPY staining after treatment with FAC ( ) for 90 min . Data are representative of three independent experiments. d, Immunoblot analysis of lysates from control of PRDX6 KO cells expressing the GPX4 U/C mutant. Data are representative of two independent PRDX6 mutants after incubation for 24 h in the presence of RSL3 (e) or IKE ( ) (f). Viability data are presented as the mean s.d. of three biological replicates. g, Measurement of GPX activity using recombinant proteins. Hydrogen peroxide (left) or tert-butyl hydroperoxide (right) were used as a substrate. Data are presented as the mean s.e.m of three independent experiments. h, Flow cytometry analysis of lipid hydroperoxidation after C11-BODIPY staining of WT or GPX4 KO cells stably expressing PRDX6 after removal of liiproxstatin-1 for 24 h . Data are representative of two independent experiments. i, Immunoblot analysis of lysates from the indicated cells. Data are representative of two independent experiments.
Extended Data Fig. 4 | PRDX6 regulates expression of selenoproteins.
a, Quantification of GPX4 mRNA levels by RT-qPCR. Data are expressed as the mean s.d of three biological replicates. ns; not significant; (sgControl vs. sgPRDX6 #1 = 0.9523; sgControl vs. sgPRDX6 #2 = 0.9607); one-way ANOVA. b, c, Immunoblot analysis of lysates from control or PRDX6 KO cells incubated for 4 or 8 h in the presence of E64d/pep or MG-132 .
d, Immunoblot analysis of lysates from control or PRDX6 KO cells derived from the indicated human cancer cell lines (Hela, A549, H226, HepG2, NB-1). e, Immunoblot analysis of lysates from control cells or cells with KO of the indicated genes and stably expressing Myc-GFP WT. Data (b-e) are representative of two independent experiments.
Extended Data Fig. PRDX6 regulates selenium utilization efficiency.
a, Schematic showing selenium metabolism and the selenoprotein synthesis pathway. b, c, WT or PRDX6 KO cells were pretreated for 2 days with or sodium selenite . Then, cells were treated for 24 h with RSL3 ( 0.3 or ) ( ) or IKE ( 0.3 or ) ( ) in the presence of 100 nM or 100 nM sodium selenite. d, Immunoblot analysis of lysates from control or PRDX6 KO Hela cells cultured for 48 h in the presence of sodium selenite or or PRDX6 KO Hela cells were pretreated with or sodium selenite ( 100 nM ) for 2 days. Then, cells were treated for 48 h with FAC ( 100 or ) in the presence of ( Sec or sodium selenite ( 100 nM ), and cell viability was measured. , Immunoblot analysis of
lysates from control or PRDX6 KO cells cultured for 32 h in the presence of the indicated concentrations of or sodium selenite , WT or PRDX6 KO cells were incubated for 24 h with FBS in the presence of liproxstatin-1. Then, cells were treated with or 100 nM sodium selenite for the indicated times. Cell lysates were analyzed by immunoblotting. i, PRDX6 KO cells were preincubated for 36 h with or 40 nM sodium selenite. Then, WT or PRDX6 KO cells pretreated with selenium were exposed to 50 nM or 200 nM sodium selenite for the indicated periods and the lysates were analyzed by immunoblotting. Data are representative of two ( ) or three ( ) independent experiments. Viability data (b, c, e) are presented as the mean s.d. of three biological replicates.
Extended Data Fig. 6| Overexpression of SEPHS2 WT rescues the PRDX6 KO phenotype.a, Immunoblot analysis of lysates from control or SEPHS2 KO cells stably expressing the SEPHS2U/C mutant.b, Immunoblot analysis of lysates from control or PRDX6 KO cells stably expressing the SEPHS2 WT or U/C mutant. c, Immunoblot analysis of lysates from cells stably expressing Myc-GFP C70U or S175U in control or PRDX6 KO cells stably expressing the SEPHS2 WT or U/ C mutant. Data (a-c) are representative of two independent experiments. d-f, Viability of the indicated cells in the presence of FAC ( 50 or ) for , or RSL3 ( 0.3 or ) (e) or IKE ( 0.3 or ) (f) for 24 h . Viability
data are presented as the mean s.d. of three biological replicates. g, Viability of the indicated cells treated for 48 h with and liproxstatin- . Viability data are presented as the mean s.d. of three biological replicates. * PRDX6 WT vs. C47S = 0.0243; WT vs. sgSEPHS2 = 0.0238 ; one-way ANOVA.h, Coomassie-stained SDS-PAGE gel showing the recombinant PRDX6 WT or C47S mutant used for ICP-MS analysis. ICP-MS analysis of the amount of selenium bound to recombinant PRDX6 WT or C47S. Data are presented as the mean s.d. of three independent experiments. , *** ; one-way ANOVA.
Extended Data Fig. 7 | Mass spectrometry analysis of PRDX6 C47 modification. a, b, Mass spectrum of PRDX6 C47. PRDX6 ( ) was incubated with GSH and sodium selenite , or with and SCLY for 5 min prior to mass spectrometry analyses.
Extended Data Fig. 8 | Interaction between PRDX6 and SEPHS2 or SCLY.
a, The physical association between PRDX6 and SCLY was detected in a PLA assay. Scale bars, . Data are representative of three independent experiments. Quantification of the PLA dots is also shown. Data are presented as the mean s.d. [ cells (Empty-PRDX6), cells (SCLY-PRDX6) examined], ;
b
unpaired two-sided t-test.b,c PRDX6 KO MEFs expressing HA-TurboID-empty, -PRDX1, or-PRDX6 were cultured for 30 min with DMSO or biotin. Cell lysates and pulldown samples were analyzed by immunoblotting. The association between PRDX6 and SEPHS2 (b) or between PRDX6 and SCLY (c) was analyzed. Data are representative of two (c) or three (b) independent experiments.
Extended Data Fig. 9 | Expression of PRDX6 is associated with a poor prognosis for several cancers. a, Expression profile of PRDX6 in several types of normal and tumor tissue. Data were obtained from TNMplot (https://tnmplot. com/analysis/), which is a web server that stores data regarding expression of normal and cancer-related genes. Those with higher values in tumor tissue
compared to normal tissue and with significant differences (Mann-Whitney U test) are marked in red ( ). b, Kaplan-Meier survival curves comparing cancer samples showing high PRDX6 expression with samples showing low PRDX6 expression (curves were constructed using Kaplan-Meier plotter).
Extended Data Fig. 10 | Expression of PRDX6 is important for survival of cancer cell lines. a, Immunoblot analysis of lysates from WT or PRDX6 KO Panc-1 cells. b, Continuous monitoring of the viability of WT or PRDX6 KO Panc-1 cells in the presence or absence of sodium selenite ( 100 nM ), (Sec) , or liproxstatin-1 . c, Immunoblot analysis of lysates from WT or PRDX6 KO MIA PaCa-2 cells. d, Continuous monitoring of the viability of WT or PRDX6 KO MIA PaCa-2 cells in the presence or absence of sodium selenite ( 100 nM ), , or liproxstatin-1 . e, Immunoblot analysis of lysates from
indicated cell lines. , Viability of indicated cell lines in the presence of FAC ( 50 or ) for 48 h . Viability data are presented as the mean s.d. of three biological replicates. g, Immunoblot analysis of lysates from WT or PRDX6 KO SK-N-DZ cells. , Continuous monitoring of the viability of WT or PRDX6 KO SK-N-DZ cells in the presence or absence of sodium selenite , or liproxstatin-1 ( ). Viability data ( ) are presented as the mean s.e.m of three biological replicates. Data (a, c, e, g) are representative of two independent experiments.
natureportfolio
Corresponding author(s): Hiroaki Fujita, Kazuhiro Iwai
Last updated by author(s): February 22, 2024
Reporting Summary
Nature Portfolio wishes to improve the reproducibility of the work that we publish. This form provides structure for consistency and transparency in reporting. For further information on Nature Portfolio policies, see our Editorial Policies and the Editorial Policy Checklist.
Statistics
For all statistical analyses, confirm that the following items are present in the figure legend, table legend, main text, or Methods section. The exact sample size ( ) for each experimental group/condition, given as a discrete number and unit of measurement X A statement on whether measurements were taken from distinct samples or whether the same sample was measured repeatedly The statistical test(s) used AND whether they are one- or two-sided
Only common tests should be described solely by name; describe more complex techniques in the Methods section.
A description of all covariates tested A description of any assumptions or corrections, such as tests of normality and adjustment for multiple comparisons
A full description of the statistical parameters including central tendency (e.g. means) or other basic estimates (e.g. regression coefficient) AND variation (e.g. standard deviation) or associated estimates of uncertainty (e.g. confidence intervals)
For null hypothesis testing, the test statistic (e.g. ) with confidence intervals, effect sizes, degrees of freedom and value noted Give values as exact values whenever suitable. For Bayesian analysis, information on the choice of priors and Markov chain Monte Carlo settings For hierarchical and complex designs, identification of the appropriate level for tests and full reporting of outcomes Estimates of effect sizes (e.g. Cohen’s , Pearson’s ), indicating how they were calculated
Our web collection on statistics for biologists contains articles on many of the points above.
Software and code
Policy information about availability of computer code
Data collection
Western blotting anaylysis was measured by a LAS4000mini or LAS3000 instrument (GE Healthcare).Cell viability assay was measured by SpectraMax M5(Molecular Device). Immunofluorescence images were collected using Fv1000 microscopy (Olympus). DNA and RNA concentrations were measured by Nanodrop2000 (Thermo). Protein concentration was measured by SpectraMax M5(molecular device) or V750 spectrophotometer (JASCO). GPX assay (absorbance at 340 nm ) was measured by SpectraMax M5 (Molecular Devices).RT-qPCR data were obtained by ABI ViiA7 Real-Time PCR system (Applied Biosystems). Luciferase reporter was measured by a Lumat Luminometer (Berthold). The AMP product was measured by plate reader Nivo (PerkinElmer). Cell survival data on iCelligence system were acquired by using RTCA iCelligence Software (ACEA Biosciences). ICP-MS was done by Agilent 8800 ICP-MS/MS (Agilent Technologies). LC-ESI-Q-TOF analysis was performed using 6545XT AdvanceBio LC/Q-TOF (Agilent Technologies) connected to the Agilent HPLC system. Flow cytometry data was collected by using FACS Canto II (BD Biosciences) and FACS Diva software ver 6.1.2 (Becton Dickinson).
Data analysis
GraphPad Prism9 version 9.4.0 was used to perform statistical analysis. CRISPR screen data was analyzed by MAGeCK pipeline. Microsoft Exel ver16.72 was used for data analysis. Image Gauge ver4.22 (FUJIFILM) was used for chemiluminescent images. ViiA7 RUO Software ver1.2.3 (Thermo Fisher Scientific) was used for RNA expression analysis. Recording data of cell survival using iCelligence system were analyzed by RTCA Software Lite version2.2.1(ACEA Biosciences). Immunofluorescence images were analyzed by ImageJ (ver2.3.0). FlowJo software (Tomy Digital Biology, version 9.9.6) was used for all analyses of flow cytometry data. The modification analysis of the active center cysteine (Cys47) was performed using MassHunter BioConfirm software ver10.0 (Agilent Technologies). Figures in the manuscript were arranged and converted into PDF files by using illustrator version 27.4.1 (Adobe).
For manuscripts utilizing custom algorithms or software that are central to the research but not yet described in published literature, software must be made available to editors and
Data
Policy information about availability of data
All manuscripts must include a data availability statement. This statement should provide the following information, where applicable:
Accession codes, unique identifiers, or web links for publicly available datasets
A description of any restrictions on data availability
For clinical datasets or third party data, please ensure that the statement adheres to our policy
Data of differential gene expression in tumor and normal tissues were obtained from TNMplot (https://tnmplot.com/analysis/). All data are available in the article and the supplementary information, and from the corresponding authors upon reasonable request.
Human research participants
Policy information about studies involving human research participants and Sex and Gender in Research.
Reporting on sex and gender
N/a
Population characteristics
N/a
Recruitment
N/a
Ethics oversight
N/a
Note that full information on the approval of the study protocol must also be provided in the manuscript.
Field-specific reporting
Please select the one below that is the best fit for your research. If you are not sure, read the appropriate sections before making your selection.
Life sciences Behavioural & social sciences Ecological, evolutionary & environmental sciences
All studies must disclose on these points even when the disclosure is negative.
Sample size
No statistical tests were used to determine sample size. Sample size was determined on the previous studies in the field using similar experiment paradigms [PMID: 34031600 and 31634899], and to give sufficient values to conduct standard statistical tests.
Data exclusions
No data were excluded from the analyses.
Replication
All experiments were reproduced at least twice, each with similar results (the expression checks shown in extended data Figures 1 g and 3 a were single experiments).
Randomization
Not applicable as no animal studies were performed.
Blinding
Blinding was not done because the investigator needs to know the treatment groups in order to perform the experiments.
Reporting for specific materials, systems and methods
We require information from authors about some types of materials, experimental systems and methods used in many studies. Here, indicate whether each material, system or method listed is relevant to your study. If you are not sure if a list item applies to your research, read the appropriate section before selecting a response.
Primary antibodies were validated in this study or used in previous articles for WB of mouse or human sample.
Anti-PRDX6 (Proteintech, 13585-1-AP), anti-SEPHS2 (Proteintech, 14109-1-AP), Anti-GPX4 (Proteintech, 67763-1-AP) and (Santa Cruz, sc-166570), anti-GPX1/2 (Santa Cruz, sc-133160), anti-PSTK (Santa Cruz, sc-373991), anti-FBXL5 (Santa Cruz, sc-390102), anti-ACSL4 (Santa Cruz, sc-365230), anti-PDSS2 (Santa Cruz, sc-515137), have been validated by knockout of the endogenous genes with CRISPR/Cas9.
Anti-FTH1 (Santa Cruz, sc-376594), anti-ferritin (Sigma-Aldrich, F6136) and Anti-IRP2 (in-house) have been validated in previous publication (PMID: 35318808)
Anti-FSP1 (Proteintech, 20886) has been validated in previous publication (PMID: 31634900).
Anti-SELN (Santa Cruz, sc-365824) and anti-LRP8 (abcam, ab108208) have been validated in previous publication (PMID: 35637349).
Validation detail for the other commercial antibodies is on the manufacture’s website that is attached as following.
Anti-SCLY (Proteintech, 67606-1-lg);https://www.ptglab.com/products/SCLY-Antibody-67606-1-lg.htm; and has been also validated by expression of tagged-SCLY construct.
Policy information about cell lines and Sex and Gender in Research
Cell line source(s)
Mouse embryonic fibroblasts (MEFs) were generated in-house. HepG2 was gifted by Dr. Koichi Nakajima (Osaka City University), originally purchased from ATCC (HepG2 : HB-8065). HEK293T was gifted by Dr. Eijiro Nakamura (Kyoto University), originally purchased from RIKRN RBC (293T : RCB2202). PLATE was gifted by Dr. Toshio Kitamura (Tokyo University). A549, H226, H460 and H1975 were gifted by Dr. Atsuyasu Sato (Kyoto University), originally purchased from ATCC (A549 : CCL-185, H226 : CRL-5826, H460 : HTB-177, H1975 : CRL-5908). SK-N-DZ and HeLa cells were purchased from ATCC (SK-N-DZ : CRL-2149, Hela : CCL-2). PANC-1 and MIA Paca-2 cells were purchased from RIKEN RBC (PANC-1 : RCB2095, MIA Paca-2 : RCB2094). NB-1 cells were purchased from JCRB (NB-1 : JCRB0621).
Authentication
None of the cell lines used were not authenticated.
Mycoplasma contamination
All cell lines tested negative for mycoplasma contamination.
Commonly misidentified lines (See ICLAC register)
No commonly misidentified cell line was used.
Flow Cytometry
Plots
Confirm that:
The axis labels state the marker and fluorochrome used (e.g. CD4-FITC).
The axis scales are clearly visible. Include numbers along axes only for bottom left plot of group (a ‘group’ is an analysis of identical markers).
All plots are contour plots with outliers or pseudocolor plots.
A numerical value for number of cells or percentage (with statistics) is provided.
Methodology
Sample preparation
Cells were plated in a 6-well plate and treated with FAC. After 1.5-24 h, the medium was removed, and the cells were labeled with DMEM containing 10 uM BODIPY at for 30 min . Cells were then washed three times with PBS and detached from the plate using trypsin, and green fluorescence was measured by flow cytometry using a BD FACSCanto II cytometer
Instrument
FACSCanto II (BD Biosciences) for flow cytometric analyses
Software
FACS Diva software ver 6.1.2 (Becton Dickinson) for data collection. FlowJo version 9.9.6 for data analysis.
Cell population abundance
At least 5000 cells were analyzed for each sample.
Gating strategy
FSC-area vs SSC-area was used to gate for the bulk populations of cells. Initial cell population gating (FSC-Area vs FSC-height) was adopted to make sure doublet exclusion and only single cell was used for analysis.
Tick this box to confirm that a figure exemplifying the gating strategy is provided in the Supplementary Information.
Department of Molecular and Cellular Physiology, Kyoto University School of Medicine, Kyoto, Japan. Laboratory of Toxicology and Environmental Health, Graduate School of Pharmaceutical Sciences, Chiba University, Chiba, Japan. Department of Environmental Medicine and Molecular Toxicology, Tohoku University Graduate School of Medicine, Sendai, Japan. Present address: Department of Radiation Oncology, New York University Langone Health, New York, NY, USA. These authors contributed equally: Yu-ki Tanaka, Seiryo Ogata. e-mail: fujisan@mcp.med.kyoto-u.ac.jp; kiwai@mcp.med.kyoto-u.ac.jp